
Submitted:
31 December 2024
Posted:
02 January 2025
You are already at the latest version
Abstract
The current paradigm of low-T combustion and autoignition of hydrocarbons is based on the sequential two-step oxygenation of fuel radicals. The addition of the first oxygen molecule forms a peroxy radical RO2, which isomerizes to a hydroperoxyalkyl radical (QOOH). The key chain-branching occurs when the second oxygenation adduct (OOQOOH) is isomerized releasing an OH radical and forming a key ketohydroperoxide (KHP) intermediate, O=POOH. Subsequent homolytic dissociation of relatively weak O-O bond in KHP generates two more radicals in the oxidation chain leading to ignition/explosion. Thus, the formation and consumption of KHPs is a key controlling process.We recently introduced a new type of intramolecular isomerization mechanism involving self-catalyzed migration of H-atoms relevant to keto-enol and other isomerization processes designated as “catalytic hydrogen atom transfer - CHAT” (J. Phys. Chem. 2024, 128, 2169), more adequately abbreviated here as SCI-HAT. On this basis, we have identified a new general unimolecular decomposition channel for the formation of enol hydroperoxides (EHP) - the classical isomers of KHPs using first-principles modeling and potential energy surface analysis. Even though the enols are currently involved in various combustion/flame chemistry models, their actual contribution in combustion processes mostly remains neglected, due to the high computed barriers for classical (“direct”) keto-enol tautomerization. Remarkably, the novel SCI-HAT mechanism dramatically reduces activation barriers for such a conversion in the case of EHPs. Here, we present detailed mechanistic and kinetic analysis of the SCI-HAT-facilitated pathways involving some models of n-hexane, n-heptane, and specifically n-pentane as a prototype molecule for gasoline, diesel and hybrid rocket fuels (HRF). We particularly examined the formation and subsequent dissociation kinetics of γ-enol-hydroperoxide (γ-EHP) isomer of the γ-KHP (γ-C5-KHP), the most abundant isomer of the pentane-derived ketohydroperoxides observed experimentally. The novel self-catalyzed bond-exchange mechanism can be regarded as an intramolecular version of the intermolecular relay transfer of H-atoms mediated by an external molecule (molecular catalyst), such as dihydrogen, water, acids, and even radicals. Earlier, we proposed a general systematization of such intermolecular processes illustrated in the simplest case of the H2-mediated reactions termed “dihydrogen catalysis” (Catal. Rev. - Sci. Eng. 2014, 56, 403). Following this systematization, the SCI-HAT catalysis can be assigned to the category of relay-transfer of H-atoms. To gain molecular level insight into the SCI-HAT catalysis, we have additionally explored the role of the catalytic moiety on SCI-HAT reactivity using selected small models. All applied models demonstrated significant reduction of the H-transfer barriers, primarily due to the decreased ring strain in transition states. The electronic and steric factors affecting reactivity and allowing this path to circumvent the geometric disadvantages of the uncatalyzed (direct) H-transfer processes, are also discussed. Depending on the dimensions and specific molecular parameters of the SCI-HAT catalytic moieties, the longer-range and sequential H-migration processes are also identified to extend the role of the new mechanism in combustion of large alkanes and paraffin-wax hybrid rocket fuels. Such processes are particularly illustrated by a combined double keto-enol conversion of heptane-2,6-diketo-4-hydroperoxide introducing a long-range H-migration as a potential chain-branching model.To assess the possible impact of the SCI-HAT channels on global fuel combustion characteristics, we present a detailed kinetic analysis of isomerization and decomposition of pentane 2,4-ketohydroperoxide comparing SCI-HAT with key alternative reactions, including direct dissociation and Korcek channels. Calculated rate parameters were implemented into a modified version of the n-pentane kinetic model developed earlier using RMG automated model generation software (ACS Omega, 2023, 8, 4908). Simulation of ignition delay times using such models revealed significant effects of the new pathways suggesting an important role of the SCI-HAT pathway in low-temperature combustion of large alkanes.
Keywords:
Intramolecular Catalysis
; Ketohydroperoxides
; Hydrogen atom transfer
; Autoignition
; Chain-Branching
; Hydrogen atoms relay transfer
; Dihydrogen catalysis
1. Introduction
Low-temperature combustion of hydrocarbons (< 900 K) is a complex process primarily controlled by fuel-specific oxidation reactions, as opposed to the high temperature processes, which are mostly governed by the breakdown of fuel molecules into small radical fragments prior to oxidation [1,2,3,4,5,6,7]. The key reactions of low-temperature autoignition of hydrocarbon-air mixtures involve formation and decomposition of ketohydroperoxide (KHP) intermediates [3,5,8,9,10,11,12,13,14,15,16]. KHPs also play a central role in tropospheric oxidation and aerosol (SOA) formation processes [17,18]. Whereas much is known about the mechanism of low-T combustion processes in small models, more specific reaction channels are currently emerging for the practically important larger systems providing new insights into the overall mechanisms. One of these pathways actively studied in recent years includes the formation of highly oxygenated moleculess (HOMs) via a third- and higher-degree oxygenation reactions [17,18,19,20]). These and other prospective mechanisms can be particularly important in understanding the combustion mechanism of large and extra-large alkanes - paraffin wax, which is an important hybrid rocket fuel (HRF) [21,23,24,25,26]. Commercial paraffin waxes typically comprise a mixture of extra-large linear alkanes, with small amounts of branched alkanes (paraffin oil). The understanding of their detailed oxidation chemistry is important for improving hybrid rocket performance [24,26].
Scheme 1 provides the current view of the low temperature combustion of large alkanes, involving sequential double oxygenation reactions of fuel radicals accounting for autoignition via formation of key ketohydroperoxide (KHP) intermediate, O=POOH. KHPs further undergo homolytic dissociation of relatively weak O-O bond to generate two active radicals in the oxidation chain to complete the main chain-branching event (vide infra). Scheme 1 also includes two recently discovered alternative unimolecular decomposition channels for KHPs - the Korcek mechanism [27] and highlighted in blue SCI-HAT mechanism [8,15]. Both channels include an intricate isomerization of KHPs. In the first case it leads to the formation and dissociation of a peroxycyclic intermediate, whereas the second case occurs via self-catalyzed keto-enol conversion (SCI-HAT pathway in Scheme 2) and decomposition of enol counterparts.
1.1. SCI-HAT Mechanism. The keto-enol conversion depicted in Scheme 1, is a particular case of the general mechanism we have recently introduced [8,15]. It represents a new type of intramolecular isomerization reactions, which occurs via synchronized (self-catalyzed) migration of hydrogen atoms, initially designated simply as “catalytic hydrogen atom transfer” - CHAT. Because the short CHAT acronym we employed previously is too general and often mistaken with metallocomplex - catalyzed processes, here we employ its more complete definition, viz., the “self-catalyzed intramolecular catalytic hydrogen atom transfer” abbreviated as SCI-HAT, which seems to represent the mechanism uniquely and adequately. An adjective “intramolecular” is also highlighted in the full version because the mere “self-catalysis” term (synonym of autocatalysis), again, is typically used in bimolecular processes, through which a product catalyzes reactions to convert substrates into products. In our case, however, one intramolecular H-migration act catalyzes another H-migration act making it a double H-transfer within the same molecule. This is due to the” compensatory” H-migration phenomenon described in ref.8.
Scheme 2 illustrates the SCI-HAT mechanism applied to pentane 2,4-ketohydroperoxide (2,4-KHP, γ-C5-KHP, or γ-KHP). The catalyst moiety (here, an encircled peroxy group) transfers its H atom to the carbonyl group simultaneously accepting a hydrogen atom of the skeletal α-methylene group, thereby mediating a tautomerization process, forming 2,4-enolhydroperoxide (γ-EHP) and regenerating the catalyst moiety. In contrast to the classical tautomerization via direct H-transfer, that occurs via a strained 4-member-ring TS and faces a high activation barrier of 61 kcal/mol (top part of the Scheme 2), the SCI-HAT mechanism, in contrast, occurs through two enlarged (5 and 6- membered) fused - ring TS structure, thus markedly reducing the conversion barrier due to the split. Therefore, the new mechanism can be characterized as a relay-transfer of H-atoms, consistent with the general systematization of the molecular catalysis processes provided earlier [28,29], albeit it occurs in an intramolecular manner.
Generally, the SCI-HAT mechanism belongs to the intramolecular catalysis class of reactions following the IUPAC guidelines: “Intramolecular Catalysis is the acceleration of a chemical transformation at one site of a molecular entity through the involvement of another functional ('catalytic') group in the same molecular entity, without that group appearing to have undergone change in the reaction product.” [30] The novel mechanism, indeed, occurs via intramolecular transfer of hydrogen atoms mediated by a functional group (sci-hat-group, moiety), such as -CH2OH-, HOO-, -COOH, -SH, -NH2. However, it strongly occurs in a molecular entity, without involvement of a second-party agent/co-reagent, as normally occurs in traditional intramolecular processes involving bimolecular reactions mediated by e.g., water molecules in intramolecular hydrolysis [31] or acids in some isomerization processes [32], with the catalyst group being in the molecule-substrate. In contrary, SCI-HAT is a strictly unimolecular intramolecular process, proceeding without involvement of any external agent or co-reagent; instead the embedded catalyst-group catalyzes the reaction, as illustrated in the bottom portion of Scheme 2.
The sci-hat catalyst group promotes various interconversion processes, such as keto-enol and imino-amino tautomerization, double-bond shift, and cyclization, while recovering itself, as shown in Scheme 2 representing reactions of γ-KHPs, where the hydroperoxy and carbonyl functionalities are separated by a methylene linkage. Thus, SCI-HAT is a general process that proceeds via much lower energy barriers than alternative uncatalyzed (direct) isomerization reactions due primarily to the eased ring strains in transition states and can be relevant for a variety of systems and conditions. Some characteristic examples of the SCI-HAT facilitated processes with varied electronic and steric characteristics (polarity and non-rigidity), are provided in Section 3.1. (simple model results are combined in Table 1).
Self-catalyzed bond-exchange process can be regarded as an intramolecular version of the intermolecular relay transfer of H-atoms mediated by an external single molecule (molecular catalyst), e.g., dihydrogen, water, NH3, various carboxylic (primarily formic and acetic) and inorganic acids, as well as radicals [8].
Asatryan and Ruckenstein have classified these intermolecular catalysis processes into five major reaction categories, illustrated in the simplest possible case of the H2-mediated reactions, called dihydrogen catalysis (DHC) [28,29]. The five suggested categories include: (A) dihydrogen-assisted relay transfer of H-atoms, (B) dihydrogen-assisted stepwise-relay transport of H-atoms/free valence, (C) dihydrogen-assisted proton transport, (D) dihydrogen-assisted dehydrogenation / hydrogenation, and (E) pre-activated dehydrogenation [28,29]. Following this systematization, the SCI-HAT -catalysis belongs within category A - relay transfer of hydrogen atoms (H-atom switch), since the sci-hat-catalyst(moiety) simultaneously acts as a relay H-atom-donor and acceptor, although in an intramolecular manner. Note that two additional subcategories of the relay transfer mechanism of category A have also recently been identified [8].
Note also that a broader systematization of molecular catalysis processes was provided by Francisco et al. considering only double H-bond transfer (viz. H-atom relay) processes in atmospheric chemistry [34]. It should be emphasized, however, that there is a variety of other mechanisms, closely related to category A suggested in the literature, such as a proton-relay mechanism widely employed in biochemistry, concerted biprotonic transfer, and proton pumps for transport of protons across membranes, long known and intensively studied and speculated [35,36,37,38]. The key pieces of evidence for the popular water- and carboxylic (acetic) acid- catalyzed proton-relay keto-enol transformations, for instance, have been identified in the classical works by Bernasconi [37], and Song [38], respectively. However, in contrast to all these bimolecular and multi-molecular catalytic processes, SCI-HAT is a genuine unimolecular and purely intramolecular process with no external source of H-atoms/protons being involved.
Thus, we have identified a novel unimolecular decomposition pathway for the key combustion intermediate KHPs leading to the formation of enol hydroperoxides (EHP) - the classical isomers of KHPs. We have tested their relevance in global combustion processes (Section 3.5). Even though the enols have recently gained much attention in the combustion community, the evaluation of the contribution of EHPs remains unknown and challenging due to the coupling of PESs for two unimolecular alternative reactions, as well as the high computed barriers for direct isomerization of corresponding KHPs. On the other hand, the novel, SCI-HAT-based keto-enol tautomerization mechanism fundamentally increases the conversion rates of the KHPs.
In addition, SCI-HAT process can ” spread over” within a large molecule given that other reactive centers, such as a carbonyl group, are available in the vicinity, because the catalyst moiety is regenerated/reinstated each time, as detailed in Section 3.2. Apparently, such a consecutive act can primarily occur in pre-activated systems (chemically or photochemically) as in the case of ketohydroperoxide intermediates formed from double chemical activation of fuel radicals by two O2 molecules (Scheme 1). Perhaps, some other “double activation” and non-Boltzmann processes, can also promote such reactions [39,40,41]. Furthermore, a longer-range SCI-HAT processes can also occur relevant to combustion of large hydrocarbons depending on the dimension and electronic properties of the SCI-HAT -catalyst groups and the H-donor/acceptor centers.
1.2. Low-T Chain Branching. The currently accepted mechanism of low-T combustion and autoignition of hydrocarbons (large alkanes) illustrated in Scheme 1, is based on the sequential two-step oxygenation of fuel radicals accounting for autoignition. Scheme 1 also includes the Korcek mechanism, as well as the new SCI-HAT reaction mechanism, highlighted in blue, as alternative unimolecular decomposition reactions of KHPs [8,15,27]. The addition of the first O2 molecule to the fuel radical (R), typically produced via a radical attack on the fuel molecule forms a peroxy radical RO2. The peroxy radical per se can abstract a hydrogen atom from an H-donor (typically a fuel molecule) to form a hydroperoxide, ROOH, or undergo an intramolecular H-migration - abstraction process (tail-biting isomerization) to generate a carbon-centered hydroperoxyalkyl radical commonly denoted as QOOH. The direct dissociation of QOOH is a chain-propagating process producing OH radical and QO molecular fragment (cyclic ether or olefin + carbonyl).
Early investigations assumed that the dissociation of the peroxy bond in ROOH (Cycle 1 in Scheme 1) to form two active radicals RO and OH is the key low T chain-branching process [1,2,3,4,5,6,7,42,43,44,45,46]. However, the initial step in this sequence (H-abstraction from a fuel molecule by an alkylperoxy radical), is typically too slow to explain experimental observations, as argued by Taatjes and co-workers (2009) providing fundamental evidence for the olden-days hypothesized (see, e.g., [1,6,8,22,57] alternative second oxygenation pathways, based on the experiments on the cyclohexyl+O2 reaction system [47]. Because the PES for O2QOOH had not yet been explored at the time, the offered evidence was limited to the interpretation of time-resolved experiments suggesting the existence of the very low-lying exit channels, however, supported by the emerging first-principles based predictions on such low-lying branching channels during double oxygenation reactions of the pentyl radicals [48,49] (see also ref.11), as highlighted by the authors [47] and noted by others [7].
Thus, the critical chain branching events occur through the addition of the second O2 to QOOH radical forming an oxygen-centered hydroperoxyalkyl peroxy OOQOOH radical adduct (Scheme 1). Subsequent decomposition of OOQOOH via intramolecular H-abstraction by peroxy radical center (second tail biting event), preferably at the proximal carbon atom bearing the hydroperoxy group, generates the first OH-radical in the oxidation chain and forms a key chain-branching agent - ketohydroperoxide, O=POOH (P has one less H-atom than Q). The nearly instant dissociation of the metastable dihydroperoxy alkyl radical-intermediate is due to the formation of the essentially unbound OH-group [11]. On the other hand, the energetically less preferred H-abstraction from an alternative C-center produces relatively more stable dihydroperoxy alkyl radicals [3,10,11] (HOOPOOH in Scheme 1), which can further add O2 and lead to the formation of highly oxygenated molecules (HOM) actively studied in recent years, particularly in the context of atmospheric aerosol formation [17,19]. Further cleavage of the relatively weak O-O bond in ketohydroperoxides (BDE of 40-45 kcal/mol [13,14,43]) releases a second OH radical and forms another open-shell reactive species - keto-alkoxy radical, O=PO●, thus concluding the low-T chain branching process. Overall, three radicals produced in the oxidation chain per one OH-radical consumed to produce an initial fuel radical, sharply increases the fuel reactivity and triggers ignition/explosion. The fundamental aspects of these processes based on the analysis of the combined R+O2 and QOOH+O2 potential energy surfaces (PES) have been initially reported for model pentyl and propyl radicals [10,11,12,48,49] which showed that O2QOOH isomerization to KHP + OH is the dominant chain branching reaction at low temperatures (below 1000K). Subsequent theoretical studies brought in understanding of the various aspects of the intramolecular isomerization of OOQOOH [50-53,58a], formation of highly oxygenated molecules (HOM) [17,18,19,20], as well as bimolecular reactions involving KHPs [56].
Ketohydroperoxides (KHP) were identified and characterized for the first time in the early 1990s by Sahetchian and coworkers [9,54,55,57,59,60,61] in hexane, heptane and dodecane oxidation by gas chromatography and direct-injection mass spectrometry in a reactor and CFR engine. This study showed that they are formed by isomerization reactions [9,53,57,59,61] and play a key role in combustion chemistry and tropospheric oxidation. However, these key intermediates have gained a fast-growing interest on around 2010 prompted by emerged new detection techniques primarily by Battin-Leclerc and Taatjes and coworkers performing breakthrough studies [3,5,62,63,64,65,66,67,68]. These experiments on formation and decomposition of diverse KHPs were also stemmed from the above noted first principles-based fundamental theoretical predictions [10,11,12,48,49,50].
Subsequently, ketohydroperoxides have been observed in a variety of low-T combustion processes involving hydrocarbons and oxygenated fuels [69,70,71,72,73]. Notably, the KHPs have recently been applied as markers of low temperature kinetics by Dryer, Avedisian and coworkers to demonstrate multistage cool flame behavior of primary reference fuel (PRF) droplets [74]. This study seems to be the most relevant analysis of the combustion of paraffin wax as a hybrid rocket fuel (HRF), since the combustion of HRFs occurs (at variable conditions) via liquid layer formation, fuel entrainment, droplet formation and evaporation [22,23,24,25,26].
In conventional alkane oxidation models, the simple O-O bond fission had been the only dissociation pathway for KHPs numerical simulations [2,3,6,10,11]. However, recent studies have shown that other channels can compete with this chain-branching event, such as the bimolecular reactions of KHP [34,67,68], as well as the unimolecular Korcek reaction [27], in which the decomposition of KHP forms carboxylic acid and carbonyl products. The Korcek pathway was the most significant recent contribution to the mechanism of low-T combustion (Scheme 1) theoretically described by Jalan et al. [27]. It is an alternative to the simple unimolecular dissociation of KHP and occurs via its isomerization to a peroxy-cyclic intermediate. The relevance of the Korcek pathway has recently been evidenced (corroborated) by time-resolved experiments by Taatjes and co-workers confirming the formation of correlated “Korcek pairs” using isotopically labeled n-butane [75]. The authors, however, concluded that even though the Korcek mechanism explains formation of the significant part of the experimentally observed carboxylic acids and carbonyl products, it is not a chain-branching process that can affect autoignition. This is at variance with our SCH-HAT mechanism for decomposition of KHPs (highlighted in blue in Scheme 1), which suggests formation of more diverse products including key mid-temperature chain-branching agent H2O2.
Overall, the SCH-HAT is a new intramolecular isomerization mechanism to provide low energy pathways for formation and decomposition of enol hydroperoxide (EHP) isomers of KHPs.
Here, we explore in detail the role of the novel intramolecular catalytic hydrogen atom transfer (SCI-HAT) mechanism in combustion processes. The unimolecular decomposition of large model KHPs, such as n-heptane, n-hexane along with n-pentane are of special importance for understanding of combustion of non-rigid large hydrocarbons, such as paraffin-wax hybrid-rocket fuels with a multitude of stereochemical transformation channels. We also provide more extended kinetic analysis of sci-hat reactions to study their relevance in combustion of n-alkanes represented by a model n-pentane ketohydroperoxide – γ-C5-KHP, which is the most abundant isomer of the KHPs derived from pentane, in which C=O and HOO groups are separated by a methylene linkage.
To assess the importance of SCH-HAT reactions in combustion processes, we have incorporated calculated in Sec.3.4 rate parameters into a modified in this work version of the chemical kinetic model for combustion of n-pentane generated by RMG automated model generation software [21]. The simulation results on ignition delay times (IDT) compared with literature data are discussed in Sec.3.5, showing the significant role of the SCH-HAT pathways in low-T combustion of large alkanes.
In the next section, we will briefly describe methods used for the first principles modeling and analysis of the potential energy surfaces (PES) of the self-catalyzed intramolecular hydrogen atom transfer (SCI-HAT) pathways, as well as the calculation of rate parameters, along with the generation of modified kinetic models and simulation results. A detailed analysis of the several characteristic small model reactions facilitated by this mechanism is further provided.
2. Methodological Details
A detailed potential energy surface (PES) analysis of reaction pathways was performed using the generalized-gradient approximation (GGA) M06-2X hybrid density functional theory from the Truhlar (Minnesota) group [76], in conjunction with Dunning’s correlation consistent aug-cc-pVTZ basis set [77]. Smaller basis sets were employed for detailed screening of the PES for SCI-HAT rearrangements and classical H-transfer reaction barriers. The M06-2X method is well tested in literature in the same domain, including our previous studies [78,79,80,81,82]. It has been particularly recommended for tautomerization processes by Acevedo and coworkers in predicting experimental gas-phase free energies for various keto-enol tautomerization processes, including the γ-diketo pentane to 2-keto-4-hydoxy-3-pentene [82], which is closely related to the processes studies here. For benchmarking the results, we have also performed selected single point CCSD(T)/6-311+(2d,p) calculations for key models to validate the DFT results. For larger models and initial PES analyses a moderate 6-31+G(d,p) Pople-type basis set augmented with diffuse and polarization functions was employed as recommended by Truhlar et al. as the best affordable basis set for exploration of reaction barriers in large molecular systems [83]. All results presented here are at the M06-2X/aug-cc-pVTZ level, unless otherwise stated.
The first order saddle points were characterized as having only one negative eigenvalue of their Hessian matrices. The absence of imaginary frequencies verified that structures were true minima at their respective levels of theory. The intrinsic reaction coordinate (IRC) analysis was performed to ensure proper connectivity of stationary points. All PES calculations were performed using the Gaussian 16 program (revision A.03) [84].
High-pressure rate constants, k(T), were calculated for the 300–2000 K temperature range using canonical transition state theory. Temperature and pressure-dependent rate constants, k(T,P) were calculated with the automated reaction kinetics and network exploration software (Arkane) using the Rice-Ramsperger-Kassel-Marcus (RRKM) method as implemented in Arkane code [85] as part of the Reaction Mechanism Generator (RMG v3.2.0) program complex [86,87,88].
3. Results and Discussion
Keto-enol tautomerization plays essential roles in combustion chemistry due to the different reactivity of tautomers. Enols are significant combustion intermediates important for flame chemistry [91,92,93,94,95]. The keto tautomer (isomers of carbonyl compounds - ketones/aldehydes) is usually strongly favored, especially in polar liquids [96,97,98,99,100,101,102].
SCI-HAT is a new mechanism for energetically more facile formation of typically less stable enols to explain their roles and unexpected occurrence in combustion (in flame [91,92,93,94] and atmospheric processes (missing enols [103]).
3.1. Keto-Enol Tautomerization and Skeletal Double Bond Shift Isomerization
The uncatalyzed (direct) H-transfer (keto-enol tautomerization) in the gas phase is typically a high-energy process encountering a sizable activation barrier due to the formation of the small (here, 4-membered) strained-ring transition states (top part of the Scheme 2). However, various acid-base catalysts, biocatalysts (enzymes), and molecular catalysts are known to assist tautomerization and reduce the barrier. Solvents can also affect these processes in the condensed phase depending on the polarity of the TS. Polar media typically favors the keto form (see, e.g., [95,104]), due to the higher polarity of keto forms in polar liquids. The di-keto form of acetylacetone, for instance, predominates both in water and DMSO, while enol form is more stable in non-polar CCl4 in accord with corresponding dipole moments [105]. At very low temperatures, in liquids and the solid state, tunneling also significantly impacts the rate parameters [100,101].
A general schematic of the SCI-HAT-processes involving two specific, topologically close processes - the tautomerization and carbon-carbon double bond shift (DBS) isomerization - is provided in Scheme 3. Only forward vibration modes in TS are shown for these reversible reactions, for simplicity. A double-bond shift process facilitated by SCI-HAT catalysis can occur when A is a methylene group.
Apparently, DBS can even be more relevant at elevated temperatures and in flames, where PAHs and soot particles are formed because of its relatively higher homolytic dissociation barrier and thus stability. Tautomerization is known to occur with polar molecules and ions containing functional groups that are at least weakly acidic. Even though the carbon-chain isomerization via a double-bond shift (Scheme 3) is formally identical to that for the sci-hat keto-enol tautomerization (when A is CH2 group versus O-atom), it encounters a significantly higher barrier of activation mainly due to the lack of the electrostatic stabilizing interactions in the TS. On the other hand, the π-electrons involved in the TS make the intramolecular DBS reaction also allowed, as in the case of intermolecular (bimolecular) molecular catalysis processes [28,29].
Effect of the Catalyst Moiety. The SCI-HAT mechanism can potentially involve a variety of “sci-hat-catalyst” groups, such as OOH, -CH2OH, -COOH, and -SH. Therefore, we also explored their catalytic efficiency. Some groups are relatively less thermostable than others and prone to dissociate at lower temperatures such as a peroxy group containing relatively weak O-O bond. Therefore, the SCI-HAT mechanism can potentially operate at different temperature ranges depending on the nature of the catalyst moiety. However, the catalyst group might also somewhat lack the steric flexibility and “stretchability” of the peroxy group as a sci-hat-catalyst. Replacing the hydroperoxy moiety with hydroxymethyl (-CH2OH) group with an added methylene linkage to maintain steric accessibility and flexibility of the OH-group, for instance, does not introduce any significant changes in reaction barriers compared to that of the HOO catalyst (vide infra), yet it is more stable to possible direct dissociation at lower temperatures. Apparently, this does not necessarily mean that the OH-dissociation threshold in KHPs containing OOH-groups is too low. In fact, it is still substantially higher (by 6-8 kcal/mol) than the barrier height for the SCI-HAT conversion, which makes the latter channel competitive (see details in Section 3.4).
Based on the above discussion, a series of simple SCI-HAT reaction models were constructed for the bond-exchange processes to model larger systems involving different XH groups maintaining the C3-backbones. The results are presented in Table 1.
The smallest relevant model of the KHP that undergoes SCI-HAT conversion is 1-hydroperoxy-2-ethanone (HOOCH2CHO) - the hydroperoxy derivative of acetaldehyde. However, it does not contain a CH2 linkage and thus it is not included in Table 1. The catalytic conversion barrier, as expected from the increased strain of the small TS ring, is much higher than those for the analogous extended models provided in Table 1 at ca. 57.7 kcal/mol (vs.75.5 kcal/mol for the direct H-transfer).
Table 1 provides some energetic and electronic characteristics of models, such as activation Gibbs energy barriers for SCI-HAT-facilitated (ΔG#sci-hat) and direct (ΔG#dir) reactions, as well as reaction energies (ΔGr), along with Pauling electronegativity of the acceptor centers (χA), and the partial Mulliken charges on reactive A, X, and carbon centers (q) of the keto forms (here, formal reagents). In Table 1, q(C) is the net charge on the carbon atom connected to the reaction center, which undergoes enolization.
A few general conclusions can be observed by inspection of Table 1.
- 1)
- Formally, a sci-hat group consists of two reactive (double-centered) bonds such as O-O-H, or CH2-O-O involved in the TS to provide steric flexibility and orbital overlaps, except when a single bond is significantly longer to provide access to the acceptor site, as occurs in the case of the S-H bond. For instance, the hydroxymethyl (CH2OH) group as a sci-hat-agent is sterically and energetically almost as effective as OOH (energy profiles are similar, and the barrier heights are very close: 39.61 vs. 40.17 kcal/mol, respectively), suggesting that the ring-strain indeed is the dominant factor in SCI-HAT processes.
- 2)
- The decrease of ring strain in sci-hat TS, relative to the TS for the direct isomerization, is primarily due to the splitting of a small TS-ring of the uncatalyzed (direct) reaction into two larger rings of the catalyzed reaction, as shown in Scheme 2. In addition, one intramolecular H-bond between a pair of donor and acceptor centers (XH…A) is converted into two H-bonded donor-acceptor motifs (AH…X and XH…A, Scheme 3). Therefore, the barrier height dependence on the electronic characteristics is not straightforward; rather it varies with the nature of the different constituent rings. The electronic structure (judging from partial atomic charges or relative electronegativities) of the two H-acceptor and donating centers (χA and χX) is a priori expected to play an important role, and their competition can be a key factor. Therefore, analyzing these factors can be useful in understanding the specific interactions during relay H-atom transfer.
- 3)
- As seen from Table 1, the Pauling electronegativity of the A acceptor centers (χA) correlates with the partial charges on donor atoms. Surprisingly, the partial negative charges (electronegativity) of the A centers are inversely related with the barrier heights (comparisons are made among systems involving the same, here OOH, sci-hat-groups, for consistency); Comparisons among other sci-hat-groups presented in Table 1 containing the same A-center, also confirms this conjecture: when χX decreased – the barrier increased. This suggests that the simple electrostatic theory one could expect to be dominant in single H-bonding pairs is not sufficient to make definitive conclusions.
- 4)
- Notably, the migrating H atom is more positively charged in enols than in the keto ground state, revealing the polar character of the H-OO bond as opposed to the C -H bond, which is more difficult to split. Thus, the more influential ring is the one involving fission of the stronger C-H bond depending on the ability of the X-center to abstract corresponding H-atom.
- 5)
- When a sterically more flexible and polar group (OOH) is combined with a longer double bond of the acceptor site such as C=S, the barrier is reduced. This is in accord with conclusions from Francisco and coworkers on intermolecular H-migration processes, where longer S=O bond forms stronger H-bonds [113].
- 6)
- The barrier heights correlate with the topological properties of PESs. Particularly, an increase of the imaginary frequency in the TS correlates with the barrier heights among systems possessing the same XH sci-hat-catalyst group, e.g., OOH and SH in Table 1.
- 7)
3.2. Long-Range and Sequential SCI-HAT Catalysis
As noted above, the keto-enol tautomerization may also occur between distant centers via the extended saddle-point structures provided by the sci-hat-catalysis. The extent to which this occurs depends on the steric and electronic properties of the catalytic center and accessibility of the other involved reactive centers. Generally, the process can also span the inter-chain regions (areas) as well. Moreover, the SCI-HAT process can potentially occur more than once within the same molecule. Below are some examples for illustration.
Sequential (Double) SCI-HAT Models.SCI-HAT pathway can occur multiple times in a sequential manner. provided that another similar reactive center is available in the vicinity to undergo isomerization, because the sci-hat-moiety is recovered in time, after each elementary act. Multifunctional hydroperoxides with zero, one, two or more carbonyl groups have been shown to form as key species during oxidation of large alkanes (e.g., n-dodecane) in reactors and engines [61]. They were observed in the troposphere [68], cool flames [18], and combustion media [18,19,20]. There is also a variety of multi-carbonyl systems - natural products, such as alkyl malonates and poly(β-oxo)carboxylic acids (polyketides) important for pharmacology, for which keto-enol tautomerization is also a key issue (see, e.g., [95]).
Scheme 4 provides an example of the sequential double SCI-HAT reactions applied to a ketohydroperoxide model heptane-2,6-diketo-4-hydroperoxide (aka 4-hydroxymethyl-2,6-heptane-dione). The hydroperoxy group (sci-hat-agent) transfers its H atom to the oxygen atom of the carbonyl group, and concurrently accepts an H-atom from the CH2-group to mediate tautomerization. A similar rearrangement occurs with the second carbonyl group, again, regenerating the catalyst moiety. PES analysis shows that the barriers to classical (direct) H-transfer processes are very high. The first step enthalpy barrier, for instance, is as high as ΔG#(dir)=66.0 kcal/mol, whereas SCI-HAT catalysis reduces it significantly to as low as ΔG#(sci-hat)=39.64 kcal/mol for the first step, and 39.45 kcal/mol for the second step.
Apparently, the multistep processes can effectively occur when sufficient internal energy is available to overcome the barriers, typically when the molecule is activated chemically or photochemically as in the case of energized KHP intermediates produced during combustion of conventional fuels via dual oxygenation of fuel radicals [3,10,11].
A sequential SCI-HAT process is modeled for different 2,6-diketo-4XH derivatives of the primary reference fuel heptane, where XH= -CH2OH, -COOH, or –SH. To explore the possible reactions of alcohol-derivatives, here we also calculated such a process in case of the CH2OH as a sci-hat-agent, calculated at the M06-2X/cc-pVTZ level of theory. Scheme 4 describes a double- SCI-HAT process catalyzed by OH with the same pentenone backbone in which the hydroperoxy group –OOH in γ-C5-KHP is replaced by CH2OH sci-hat-group. Because the TS with only OH as a catalyst in pentane hydroxy-2,4-KHP- could not be located, we added a methylene group to properly extend the TS-rings and reduce the ring-strains comparable to the OOH group. The results for the CH2OH-catalyzed process provided below are basically like those described above.
Notably, the overall energetics changed insignificantly when the hydroperoxy OOH group was replaced by CH2OH. This reaction can also be considered as a new type of poly-hydroxides formation reaction, particularly facilitated at elevated temperatures.
Long-Range and Combined Models.Various longer-range intramolecular catalytic processes may also occur via extended transition states depending on the size and structure of the molecules and dimension and electronic characteristics of the sci-hat-groups and the acceptor sites. Figure 1 provides an example of such keto-enol tautomerization involving a model 2,4-diketo-6-hydroxy hexane molecule encountering significantly low sequential barriers of activation. Combustion literature provides a variety of examples of such structures as noted above (e.g., [18,61]). Highly oxidized multifunctional carbonyl compounds are well present even in the troposphere [68], with some being identical to those formed during cool flame combustion [18].
As one can see, the hydroxymethyl CH2OH-group we have “added” to the 2,4-pentane-dione (acetylacetone) to create a relevant model, indeed, can be formally considered as a sci-hat-catalyst (instead of the actual catalyst OH-group) owing to the extra methylene linkage allowing steric accessibility of OH to the reaction centers. Thus, in the first step, the OH-group transfers its H-atom to the distant carbonyl oxygen and accepts an H-atom from the proximal carbon backbone to produce enol-keto-hydroxide product through ae barrier of 28.4 kcal/mol (Figure 1). Then, the same catalyst OH- group at C6 transfers its H-atom to the remaining proximal carbonyl group obtaining H-from newly created enol group (shown by arrows in Figure 1) Both TS rings are 6-membered in contrast to the reactions discussed above that involve 4 and 5- membered rings, thus reducing the ring-strains and, hence the corresponding barriers.
We note that a simplified version of the reagent where the internal (central) C=O group is replaced by a simple methylene linkage, also undergoes similar transformation, however, via a substantially higher barrier of 36.54 kcal/mol – almost 8 kcal/mol higher than the original one. This demonstrates the important role of both the electronic and steric factors in determining barrier heights.
Enols can also be responsible for carboxylic acid balance via bimolecular and chemical activation reactions as suggested by Taatjes and coworkers [94] considering the impact of the enols on atmospheric processes, after discovery of enols as important intermediates in combustion chemistry and flames [91,92,93,94,103]. They suggested that the atmospheric carboxylic acids can be formed not only via Korcek mechanism, but also through reactions with enols (both in the atmosphere and in combustion media).
3.3. An Outlook and Possible Implications of SCI-HAT Mechanism
SCI-HAT-enabled formation of enols opens a new avenue for a variety of processes. Tautomerization can play an important role in the chain-branching events in low-and mid - temperature combustion processes because further decomposition of the KHP-derived enols generates various reactive intermediates (reaction 1), among others H2O2– a key chain-branching agent in mid and high temperature combustion. The formation of H2O2 is facilitated by the proximity of the two hydroxyl groups in the enol form of the γ-KHPs, as shown in Scheme 2 and Figure 2 in Sec.3.4. There is some analogy with the Korcek reaction (2) also leading to the unimolecular decomposition of KHPs with a similar energy profile.
KHP → ENOL → H2O2, Pentenone, Criegee Intermediate, and others
KHP → Peroxy-Cycle → Carboxylic Acids + Carbonyls
However, in contrast to the Korcek pathways, which only produce carboxylic acids and carbonyls, the sci-hat-mechanism can generate a variety of compounds including chain-branching agents (such as H2O2 and Criegee intermediate), which can alter global combustion characteristics of the fuels (ignition delay times and flame speeds), dissociating more effectively in the higher temperature regime as a consequential reaction (see, e.g., ref.117. Some other pathways leading to both species already observed in experiments and new products, were also identified for decomposition of γ-C5-KHP, such as methyl-Criegee Intermediate, vinyl hydroperoxide, acetone, diketone, and others, which will be reported in a separate publication.
Note that H2O2 has been found to be the most abundant peroxide during the oxidation of n-pentane at temperatures below 1000K (exhibiting two zone behavior – with maxima below and above 800K) in SVUV-PIMS experiments by Battin-Leclerc and coworkers [65]. Perhaps, such behavior can partly be explained by the operation of the SCI-HAT mechanism requesting further exploration.
At intermediate temperatures, the small peroxide species H2O2 contributes to chain branching by decomposing into two OH radicals [16]. The first reliable quantification of H2O2 formed during low-T combustion was presented by Bahrini et al. for the oxidation of n-butane [106]. Hydrogen peroxide itself is stable up to 1100 K and is typically considered to be formed by H-abstraction reactions of HO2 [3]. However, traditional pathways included in the model by Bugler et al [107] failed to explain experimental results by Rodriguez et al [65]. Perhaps, H2O2 can be formed via other channels including also the novel SCH-HAT pathways for KHP decomposition. Moreover, the new pathway could explain the formation of pentanone products observed in experiment and not explained properly their origin. The pentenone, as the second counterpart of the 2,4-enol decomposition generating H2O2 (Table 3), has been identified during low-T oxidation of n-pentane by GC-MS experiments with the onset of detection at 590K [66].
The simplified version of the combined SCH-HAT reaction described in Figure 1 can also be considered as a new type of the poly-hydroxides formation reaction, particularly facilitated at elevated temperatures. Perhaps enzymes also could employ similar pathways via lower energy ionic versions supported by solvation and tunneling effects.
We also note that more stable SCI-HAT catalytic systems, such as the substituted di-ketone hydroxymethyl, containing a more robust CH2OH group instead of a hydroperoxyl group with weaker O-OH bond described in Scheme 2, can allow the sci-hat mechanism to operate at much higher temperatures relevant to flames.
The reactions of enols contribute significantly towards atmospheric carboxylic acid concentrations in the gas-phase, as noted above [41,75]. The sci-hat pathways could further fortify this conclusion.
We thus hypothesize that there should be many other processes facilitated by SCI-HAT pathways. The novel mechanism may have important ramifications in a variety of processes, including waste oil degradation, and aerosol formation and degradation.
It is important to note that experimentally distinguishing between the direct and SCI-HAT intramolecular mechanisms is challenging compared to distinguishing between direct vs. bimolecular (intermolecular catalysis) processes. To isolate a direct unimolecular keto-enol tautomerization pathway from alternative bimolecular molecular-catalysis processes involving an extra-molecule as a catalyst, such as water and carboxylic acids typically dilute conditions are used, as stressed by Labbe and coworkers [98], such that the chemistry is not clouded by subsequent bimolecular chemistry. On the other hand, the emerging additional unimolecular pathway (SCI-HAT), which would not be altered by diluting, can certainly complicate the diagnostics of the classical reactions and even cast some doubt on ambiguity of the experimental identification of bimolecular molecular-catalysis processes when a sci-hat-channels are available.
Thus, designing experiments to distinguish two alternative intramolecular pathways - a SCI-HAT pathway vs. uncatalyzed (direct) pathway will be quite challenging. Perhaps, indirect evaluation of secondary reactions or isotopic labeling could be of help, albeit in both cases labile O-H and “unwastable” C-H skeletal bonds are combined in the reaction schemes. The exploration of coupled PES could shed light since the results can differ significantly from those calculated on separate PESs. Part of the “hot” KHPs could certainly dissociate via new channels.
3.4. Kinetic Analysis of a SCI-HAT Process Employed for Model Generation
A set of thermochemical properties were calculated for KHP and important species pertaining to our SCI-HAT mechanism, oxy and hydroxyl radical formation via direct dissociation, and Korcek pathways using M06-2X/aug-cc-pVTZ method. High-pressure rate constants were calculated using the Arkane (Automated Reaction Kinetics and Network Exploration) [85] program as distributed in the Reaction Mechanism Generator-Py (RMG v3.2.0) program [86,87,88]. The rate coefficients were calculated using the canonical transition state theory and are fitted to the three-parameter form of the Arrhenius equation. Temperature and pressure-dependent rate coefficients are calculated using the Rice-Ramsperger-Kassel-Marcus (RRKM) theory to solve the master equation with the modified strong collision approximation.
Substituted hydroperoxide groups, such as KHP, predominately dissociate to generate oxy and hydroxyl radicals due to the relatively weak oxygen-oxygen bonds, to participate in subsequent chain branching reactions. Our SCI-HAT -enabled formation of enols should be able to compete at lower temperatures along with propionic acid and acetaldehyde formation via the Korcek decomposition.
To show the relative barrier heights to some comparable KHP pathways, a potential energy diagram is shown in Figure 2 for γ-C5-KHP (here, KHP for simplicity). As discussed above in Scheme 2, the barrier height of the SCI-HAT pathway to create the enol-hydroperoxide ENOL is approximately 39 kcal/mol higher relative to KHP while the classic (direct) keto-enol tautomerization of KHP is almost 22 kcal/mol higher than this at 61 kcal/mol. A breakdown pathway for ENOL is then possible via hydrogen peroxide loss over a 31 kcal/mol TSenol barrier where a hydrogen transfer from the newly created hydroxyl group to the hydroperoxide group generates cis-pentenone. KHP is also shown to form a cyclic peroxide followed by subsequent carbonyl and carboxylic acid products via the Korcek mechanism with barriers of 32 and 52 kcal/mol for TSkorcek-1 and TSkorcek-2. For comparison, Jalan et al [27] determined barriers of approximately 35 and 29-32 kcal/mol for cyclic peroxide and subsequent product formation for the three-carbon ketohydroperoxide. The dissociation limit for KHP is 48 kcal/mol for oxygen-oxygen bond breaking forming hydroxyl and oxy radical.
Calculated rate constants for the species in Figure 2 are summarized in Table 2 below and sample pressure and temperature values are included in Figure 3. The temperatures and pressures in Figure 3 provide a larger range to foundationally gauge the representative nature of our SCI-HAT pathway and how it can compare to two other important known oxidation decomposition pathways. Overall, in the top portion of Figure 3 for 1.0 and 10 atm pressures, the Korcek pathway is favored from 300 K to approximately 350 K at which point the dissociation overtakes it while SCI-HAT is consistently lower across all the temperatures up to 800 K. As the temperature increases, Korcek and SCI-HAT begin to converge together albeit away from dissociation simultaneously. Also, pressure shows a slight increase of the rate constant above approximately 600 K and more pronounced for the dissociation and SCI-HAT pathways. In the bottom portion of Figure 3, rate constants are shown from 1.0 to 10 atm at temperatures of 300 and 800 K. Rate constants are almost constant throughout this region with the SCI-HAT pathway the lowest. At 300 K Korcek is favored while at 800 K dissociation occurs faster. At both temperatures, the SCI-HAT mechanism is comparable to dissociation, at 300K, and Korcek, at 800 K.
In this initial overview comparison of our SCI-HAT mechanism to two important oxidation pathways, at the temperatures and pressures considered for KHP, SCI-HAT has the potential to be an important pathway to consider. A future, more expansive, analysis will serve as a basis to put our SCI-HAT pathway in greater perspective to see its potential impact in the low temperature oxidation mechanism for hydroperoxide chemistry and mid- to higher temperature processes via other more stable compounds, such as multi hydroxy-aldehydes (ketones), described in Scheme 4 and Figure 1.
We again emphasize that the SCI-HAT mechanism generates species that can serve as chain-branching agents, such as H2O2 provided in Figure 2, and Criegee intermediate (not shown here), to alter global fuel characteristics – IDT and flame speeds, as opposed to the Korcek pathway, which forms only acids and carbonyls as primary products – leading to chain termination and removal of KHP. Thus, even a relatively small flux through the SCI-HAT mechanism could have non-trivial impact on ignition.
3.5. Chemical Kinetic Model Generation Using RMG and Simulation of IDT
To explore the impact of the new reaction pathways on combustion of large alkanes, we have generated a set of chemical kinetic models for oxidation of n-pentane, as the smallest relevant model of the large alkanes, using Reaction Mechanism Generation (RMG) software [86,87,88]. The simulation of their performance for predicting ignition delay times (IDTs) at given conditions was performed using CHEMKIN [89,90]. The main mechanism denoted here as PN-ML-m-v1 and reported earlier [21], was compared with newly generated alternative mechanisms, particularly the one supplemented by SCI-HAT reaction channels (PN-ML-m-v2).
RMG constructs kinetic models using libraries of known reactions and kinetics, generalized chemical knowledge, rate rules, and group additivity (GA) techniques [86,87,88]. It uses a set of “reaction families” to generate all possible reactions that a given chemical species can undergo in the presence of other species in the mechanism. Each reaction family represents a particular type of elementary reaction, such as radical recombination or addition to a double bond. There are currently 74 reaction families defined in RMG [86,87,88], with three new ones added in this work described below. RMG employs a rate-based algorithm to determine which species and reactions are included in the model. Species and their associated reactions are added iteratively until the production rates of all species drop below a specified termination threshold. Thermochemical data for species and reaction rate parameters are sourced from a hierarchy, beginning with experimental values in libraries, and utilizing GA and rate rules when experimental data and fundamental calculation results are unavailable. RMG is one of the most advanced and widely used software tools in the field, featuring a stable and robust architecture for developing extensible, modular code.
In this study, RMG v3.2.0 was utilized as the primary tool to generate detailed kinetic models. A simple reactor model was further employed relevant to the gas phase processes.
To test the model predictions (our simulation results), we used experimental data from National University of Ireland, Galway (NUIG) for combustion of pentane isomers, as well as the manually constructed model of the NUIG as a benchmark [108]. The latter model has been tested and validated against rapid compression machine (RCM) and shock tube (ST) data from NUIG [108]
To incorporate SCH-HAT pathways in the model generations, we have developed three new reaction families viz., 2,4-1,3_CHAT and 2,5_CHAT for catalyzed keto-enol conversions of 2,4-, 1,3- and 2,5- KHPs via SCI-HAT, as well as ENOL family to describe dissociation of 1,3- and 2,4- enol products to form 2-pentenal, cis-pentenone, and H2O2 (Table 1), and incorporated them into RMG. To add these new pathways to the RMG family database, we used the Subgraph Isomorphic Decision Tree (SIDT) algorithm [109].
Following the procedures described in [109], we created a root for these new families and by providing training reaction parameters calculated in the kinetic part of this study (Section 3.4), as listed in the Table 3, we calculated branches based on decision tree making to be generalized for similar cases.
The rate parameters employed to parameterize these new reaction families were those described in the previous section. The fundamentally based thermochemical data, included in a separate RMG library, was provided for eight added species included in the new families. The calculation of the thermochemical properties for those species is based on M06-2X/aug-cc-pVTZ method using ARM-2 atomization approach [110] to be consistent with calculated rate parameters described above.
To evaluate the ignition delay times (IDT), we used Chemkin Pro 2023 model provided by ANSYS [89,90]. Calculations were performed for a homogeneous constant-pressure batch reactor, where the ignition delay time was defined as the time required for the temperature to increase by 400 K. In generating the n-pentane oxidation mechanism, our approach prioritized experimentally verified and first-principles-based data sources as much as possible, while relying on rate-based criteria to expand the mechanism.
Figure 4 shows the IDT performance for a wide range of temperature from T = 625K to 1350K at P = 10 atm and fuel-rich conditions (ϕ = 2); ϕ is the equivalence ratio of fuel to oxidizer. The simulation results for fuel lean and stoichiometric conditions are provided in Supporting Information.
In overall, we have generated 5 different models. In addition to those described above (PN-ML-m-v1 and PN-ML-m-v2), a simpler model was generated (“from the scratch”) based only on estimation methods, rate rules for reaction kinetics and group additivity for thermochemistry provided in RMG, denoted as PN-ES. Despite reproducing the NTC behavior, the predicted IDTs are too low and shifted to higher temperatures compared to experimental data for this selected condition, as seen from Figure 4. In another case, to further support pentane combustion modeling, we included only the manually developed NUIG mechanism by Bugler et al. [108], implemented in RMG as the CurranPentane library (CPL). CPL provides a whole range of thermochemical and kinetic parameters for pentane combustion. CPL in RMG mainly serves as a source for both thermochemistry (primarily for C5 and some C4 species) and reaction kinetics for pentane isomers. It is partly based on high fidelity experimental and fundamental data, and partly estimated and tuned to their experiments [108]. It should be emphasized that the complete NUIG mechanism (comprising 675 species and 3065 reactions) is too large to serve as a basic seed mechanism for generating combustion and pyrolysis models for larger paraffins while maintaining a manageable mechanism size. Therefore, we employed CPL as a library to provide RMG with data derived from the NUIG mechanism [108]. As can be seen from the Figure 4, the performance of PN-CPL is better than that from purely estimation methods (PN-ES), however the system remains less reactive for the negative temperature coefficient region, compared with experimental data and the full NUIG model predictions.
Furthermore, we utilized a set of other reaction and thermochemistry libraries available in RMG, primarily focusing on high fidelity data for small-species chemistry, denoted as PN-ML (Multiple libraries). For H₂/ O₂ reactions we used the Burke sub-model for H₂ combustion at high pressures [111]. As a source of C1-C2 chemistry, we used the Klippenstein-Glarborg model-library, derived from the recent mechanism published by Hashemi et al. [112]. Additionally, thermochemistry data for C3–C4 species were provided using updated databases available in RMG. Specifically, the thermochemistry was sourced from a database by Goldsmith et al., which includes refined QCISD(T) energies for numerous combustion-relevant small species (“DFT_QCI_thermo”), along with composite-level predictions from CBS-QB3 (“CBS_QB3_1dHR”) and G4MP2 methods [150]. To improve PN-ML model performance, we have previously created and applied a modified reaction and thermochemistry library described in detail in ref. 21. The mechanism denoted here (PN-ML-m-v1) represents the first (basic) version of the modified PN-ML-m model.
The final version of this mechanism (PN-ML-m-v2) additionally involves all three new families and trained reactions from Table 1.
As can be seen from Figure 4, adding the modified library in PM-ML-m-v1 improved the model performance at the defined conditions -here, high-pressure and fuel-rich conditions more relevant to engine and HRF combustion (see also Supporting Information). The performance of both versions of PN-ML-m model is significantly improved compared to the original PN-ML model where we only relied on the databases from RMG, as well as PN-CPL using only CPL database. Further addition of catalytic SCI-HAT isomerization and ENOL dissociation pathways via new families (PN-ML-m-v2) improves even more the model predictions.
To conclude, the novel SCI-HAT pathways, along with enol dissociation reactions are important new pathways to be added to alkane combustion mechanisms to significantly affect the model performance. It is particularly due to the formation of chain-branching H2O2 as the most abundant peroxide in low-T combustion of n-pentane, as shown by Bourgalais et.al by coupling a jet-stirred reactor with an electron/ion coincidence spectrometer, not fully explained through traditional pathways [66].
4. Summary and Conclusions
A novel unimolecular decomposition mechanism involving “self-catalyzed intramolecular catalytic transfer of hydrogen atoms” (SCI-HAT), has been analyzed and applied to certain combustion processes. Low energy pathways are identified for keto-enol tautomerization of the model chain branching intermediates – ketohydroperoxides ranging from small reaction models to larger n-pentane, n-hexane and n-heptane. The newly formed enol-hydroperoxide isomers of pentane derived ketohydroperoxide (γ-C5-KHP) were shown to easily eliminate another chain branching agent, H2O2, and to form experimentally observed pentenone co-products.
The rate parameters for these processes are calculated and shown to compete with two main, alternative KHP decomposition channels – direct dissociation of OH-radicals (main chain branching event in traditional fuel ignition), and Korcek decomposition to form acid and aldehyde. The implementation of rate parameters in a kinetic model developed previously to examine its role in prediction of global combustion characteristics was presented.
Overall, we find that the O-O bond fission remains the dominant process of KHP decomposition due to the higher entropy gain despite the noticeably higher O-O bond-breaking energy of γ-KHP compared to the transition state barriers for the novel sci-hat-tautomerization and the Korcek reactions. The keto-enol pathway is somewhat similar to the Korcek mechanism in terms of the formation and decomposition of an isomer of KHP, yet it leads to the formation of more diverse products, including chain-branching agents. The formation of γ-EHP in the simple case of the γ-KHP encounters an activation barrier somewhat higher than the first step Korcek reaction, however its further decomposition versus the similar second Korcek step is evidently more viable.
Longer-range and sequential SCI-HAT processes were also shown to occur depending on the dimension and electronic characteristics of the sci-hat-group and the proton acceptor site.
Simulation of ignition delay times based on RMG-generated models with and without including new pathways showed the significant effect of the SCH-HAT mechanism on low-temperature combustion of traditional fuels.
Acknowledgements
This research is funded by the United States Department of Energy’s (DoE) National Nuclear Security Administration (NNSA) under the Predictive Science Academic Alliance Program III(PSAAP III) at the University at Buffalo, under contract number DE-NA00039617.
References
- Walker, R.W.; Morley, C. Comprehensive Chemical Kinetics; Chapter 1; Pilling, M.J., Ed.; Elsevier; Volume 35, pp. 1–124. [CrossRef]
- Westbrook, C.K.; Mehl, M.; Pitz, W.J.; Kukkadapu, G.; Wagnon, S.; Zhang, K. Multi-fuel surrogate chemical kinetic mechanisms for real world applications, Phys. Chem. Chem. Phys., 2018, 20, 10588. [Google Scholar] [CrossRef] [PubMed]
- Zádor, J.; Taatjes, C.A.; Fernandes, R.X. Kinetics of elementary reactions in low-temperature autoignition chemistry. Progress in Energy and Combustion Science 2011, 37, 371–421. [Google Scholar] [CrossRef]
- Simmie, J.M. Detailed chemical kinetic models for the combustion of hydrocarbon fuels, Prog. Energy Combust. Sci., 2003, 29, 599–634. [Google Scholar] [CrossRef]
- Wang, Z.; Herbinet, O.; Battin-Leclerc, F.; Hansen, N. Exploring hydroperoxides in combustion: History, recent advances and perspectives, Prog. Energy Combust. Sci. 2019, 73, 132–181. [Google Scholar] [CrossRef]
- Curran, H.J.; Gaffuri, P.; Pitz, W.J.; Westbrook, C.K. A Comprehensive Modeling Study of Iso-Octane Oxidation. Combust. Flame 2002, 129, 253–280. [Google Scholar] [CrossRef]
- Pilling, M.J. From Elementary Reactions to Evaluated Chemical Mechanisms for Combustion Models. Proc. Combust. Inst. 2009, 32, 27–44. [Google Scholar] [CrossRef]
- Asatryan, R.; Hudzik, J.; Swihart, M. Intramolecular Catalytic Hydrogen Atom Transfer (CHAT). J. Phys. Chem. A, 2024, 128, 2169–2190. [Google Scholar] [CrossRef]
- Sahetchian, K.A.; Rigny, R.; Circan, S. Identification of the hydroperoxides formed by isomerization reactions during the oxidation of n-heptane in a reactor and CFR engine. Combust. Flame 1991, 85, 511–514. [Google Scholar] [CrossRef]
- Klippenstein, S.J. From theoretical reaction dynamics to chemical modeling of combustion. Proc. Comb. Inst. 2017, 36, 77–111. [Google Scholar] [CrossRef]
- Asatryan, R.; Bozzelli, J.W. Chain Branching and Termination in the Low-temperature Combustion of n-Alkanes: 2-Pentyl Radical + O2, Isomerization and Association of the Second O2, J. Phys. Chem. A 2010, 114, 7693–7708. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, C.F.; Green, W.H.; Klippenstein, S.J. Role of O2 + QOOH in low-temperature ignition of propane. 1. Temperature and pressure dependent rate coefficients, J. Phys. Chem. A 2012, 116, 3325–3346. [Google Scholar] [PubMed]
- Liu, B.; Di, Q.; Lailliau, M.; Belhadj, N.; Dagaut, P.; Wang, Z. Experimental and kinetic modeling study of low-temperature oxidation of n-pentane. Combustion and Flame 2023, 254, 112813. [Google Scholar] [CrossRef]
- Sahetchian, K.; Heiss, A.; Rigny, R.; Ben-Aïm, R. Determination of the gas-phase decomposition rate constants of heptyl-1 and heptyl-2 hydroperoxides C7H15OOH, Int. J. Chem. Kinet. 1982, 14, 1325–1337. [Google Scholar] [CrossRef]
- Asatryan, R.; Amiri, V.; Hudzik, J.; Swihart, M. Intramolecular Catalytic Hydrogen Atom Transfer (CHAT): A Novel Mechanism Relevant to the Combustion of Traditional Fuels, 2023 AIChE Annual Meeting, Nov 5-10, Orlando, FL.
- Westbrook, C.K. Chemical kinetics of hydrocarbon ignition in practical combustion systems. Proc. Combust. Inst. 2000, 28, 1563–1577. [Google Scholar] [CrossRef]
- Wang, Z.; Popolan-Vaida, D.M.; Chen, B.; Moshammer, K.; Mohamed, S.Y.; Wang, H.; Sioud, S.; Raji, M.A.; Kohse-Höinghaus, K.; Hansen, N.; et al. Unraveling the structure and chemical mechanisms of highly oxygenated intermediates in oxidation of organic compounds. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 13102–13107. [Google Scholar] [CrossRef] [PubMed]
- Belhadj, N.; Benoit, R.; Dagaut, P.; Lailliau, M. Experimental characterization of n-heptane low-temperature oxidation products including keto-hydroperoxides and highly oxygenated organic molecules (HOMs). Combust. and Flame, 2021, 224, 83–93. [Google Scholar] [CrossRef]
- Belhadj, N.; Lailliau, M.; Benoit, R.; Dagaut, P. Experimental and kinetic modeling study of n-pentane oxidation at 10 atm, Detection of complex low-temperature products by Q-Exactive Orbitrap. Combustion and Flame, 2022, 235, 111723. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, L.; Moshammer, K.; Popolan-Vaida, D.M.; Shankar, V.S.B.; Lucassen, A.; Hemken, C.; Taatjes, C.A.; Leone, S.R.; Kohse-Höinghaus, K.; Hansen, N.; Dagaut, P.; Sarathy, S.M. Additional chain-branching pathways in the low-temperature oxidation of branched alkanes. Combustion and Flame 2016, 164, 386–396. [Google Scholar] [CrossRef]
- Amiri, V.; Asatryan, R.; Swihart, M. Automated Generation of a Compact Chemical Kinetic Model for n-Pentane Combustion. ACS Omega 2023, 8, 49098–49114. [Google Scholar] [CrossRef]
- Cox, R.A.; Cole, J.A. Chemical aspects of the autoignition of hydrocarbon-air mixtures. Combust. Flame 1985, 60, 109–123. [Google Scholar] [CrossRef]
- Jens, E.T.; Cantwell, B.J.; Hubard, G.S. Hybrid Rocket Propulsion for outer planet exploration. Acta Astronautica 2016, 128, 119–130. [Google Scholar] [CrossRef]
- Petrarolo, A.; Kobald, M.; Schlechtriem, S. Optical analysis of the liquid layer combustion of paraffin-based hybrid rocket fuels. Acta Astronautica 2019, 158, 313–322. [Google Scholar] [CrossRef]
- Leccese, G.; Cavallini, E.; Pizzarelli, M. State of Art and Current Challenges of the Paraffin-Based Hybrid Rocket Technology. In: AIAA Propulsion and Energy Forum, American Institute of Aeronautics and Astronautics, 2019.
- Amiri, V.; Hudzik, J.; Asatryan, R.; Wagnon, S.W.; Swihart, M. Chemical Kinetic Mechanism for Extra-Large n-Alkanes (C>20). Energy Fuels to be submitted. 2025. [Google Scholar]
- Jalan, A.; Alecu, I.M.; Meana-Pañeda, R.; Aguilera-Iparraguirre, J.; Yang, K.R.; Merchant, S.S.; Truhlar, D.G.; Green, W.H. New Pathways for Formation of Acids and Carbonyl Products in Low-Temperature Oxidation: The Korcek Decomposition of γ-Ketohydroperoxides. J. Am. Chem. Soc. 2013, 135, 11100–11114. [Google Scholar] [CrossRef]
- Asatryan, R.; Ruckenstein, E. Dihydrogen Catalysis: A Remarkable Avenue in the Reactivity of Molecular Hydrogen. Catalysis Reviews 2014, 56, 403–475. [Google Scholar] [CrossRef]
- Asatryan, R.; Bozzelli, J.W.; Ruckenstein, E. Dihydrogen Catalysis: A Degradation Mechanism for N2-Fixation Intermediates. J. Phys. Chem. A 2012, 116, 11618–11642. [Google Scholar] [CrossRef] [PubMed]
- IUPAC. Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. [CrossRef]
- Fersht, A.R.; Kirby, A.J. Intramolecular Nucleophilic Catalysis of Ester Hydrolysis by the Ionized Carboxyl Group. The Hydrolysis of 3,5-Dinitroaspirin Anion, J. Am. Chem. Soc. 1968, 90, 5818–5826. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.; Young, V.G., Jr.; Lectka, T. Intramolecular Catalysis of Amide Isomerization. J. Am. Chem. Soc. 1997, 119, 2307–2308. [Google Scholar] [CrossRef]
- Dryer, F.L.; Brezinsky, K. A flow reactor study of the oxidation of normal-octane and isooctane. Combust. Sci. Technol. 1986, 45, 199–212. [Google Scholar] [CrossRef]
- Kumar, M.; Sinha, A.; Francisco, J.S. ; Role of double hydrogen atom transfer reactions in atmospheric chemistry. Accounts Chem. Res. 2016, 49, 877e883. [Google Scholar] [CrossRef] [PubMed]
- Kasha, M. Proton-transfer Spectroscopy. Perturbation of the Tautomerization Potential. J. Chem. Soc., Faraday Trans. 2 1986, 82, 2379–2392. [Google Scholar] [CrossRef]
- Wu, C.-C.; Lien, M.-H. Ab initio study on the substituent effect in the transition state of keto-enol tautomerism of acetyl derivatives. J. Phys. Chem. 1996, 100, 594–600. [Google Scholar] [CrossRef]
- Bernasconi, C.F.; Fairchild, D.E.; Murray, C.J. Kinetic solvent isotope effect and proton inventory study of the carbon protonation of amine adducts of benzylidene Meldrum's acid and other Meldrum's acid derivatives. Evidence for concerted intramolecular proton transfer. J. Am. Chem. Soc. 1987, 109, 3409–3415. [Google Scholar] [CrossRef]
- Song P-S. ; Sun, M.; Koziolawa, A.; Koziol, J. Phototautomerism of lumichromes and alloxazines, J. Am. Chem. Soc., 1974, 96, 4319–4323.
- Asatryan, R.; da Silva, G.; Bozzelli, J.W. Quantum chemical study of the acrolein (CH2CHCHO) + OH + O reactions. J. Phys. Chem. A, 2010, 114, 8302–8311. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.P.; Goldsmith, C.F.; Georgievskii, Y.; Klippenstein, S.J. Towards a quantitative understanding of the role of non-Boltzmann reactant distributions in low temperature oxidation. Proceedings of the Combustion Institute 2015, 35, 205–213. [Google Scholar] [CrossRef]
- Goldsmith, C.F.; Burke, M.P.; Georgievskii, Y.; Klippenstein, J.S. Effect of non-thermal product energy distributions on ketohydroperoxide decomposition kinetics. Proceedings of the Combustion Institute 2015, 35, 283–290. [Google Scholar] [CrossRef]
- DeSain, J.D.; Taatjes, C.A.; Miller, J.A.; Klippenstein, S.J.; Hahn, D.K. Infrared frequency-modulation probing of product formation in alkyl + O2 reactions. Part IV. Reactions of propyl and butyl radicals with O2, Faraday Discussions 2002, 101. [Google Scholar] [CrossRef]
- Sahetchian, K.A.; Rigny, R.; Tardieu de Maleissye, J.; Batt, L.; Anwar Khan, M.; Mathews, S. The pyrolysis of organic hydroperoxides (ROOH). Proc. Combust. Inst. 1992, 24, 637–643. [Google Scholar] [CrossRef]
- Knyazev, V.D.; Slagle, I.R. Thermochemistry of the R-O2 Bond in Alkyl and Chloroalkyl Peroxy Radicals. J. Phys. Chem. A 1998, 102, 1770–1778. [Google Scholar] [CrossRef]
- Hughes, K.J.; Lightfoot, P.D.; Pilling, M.J. Direct measurements of the peroxy - hydroperoxy radical isomerisation, a key step in hydrocarbon combustion, Chem. Phys. Lett., 1992, 191, 581. [Google Scholar]
- Knox, J.H.; Kinnear, C.G. The mechanism of combustion of pentane in the gas phase between 250° and 400°C. Symposium (International) on Combustion 1971, 13, 217–227. [Google Scholar] [CrossRef]
- Fernandes, R.X.; Zádor, J.; Jusinski, L.E.; Miller, J.A.; Taatjes, C.A. Formally direct pathways and low-temperature chain branching in hydrocarbon autoignition: the cyclohexyl + O2 reaction at high pressure. Phys. Chem. Chem. Phys., 2009, 11, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Asatryan, R.; Bozzelli, J.W. Chain branching and termination paths in oxidation of n-alkanes: Comprehensive Complete Basis Set-QB3 study on the association of n-pentyl radical with O2, isomerization and addition of second oxygen molecule Eastern States Section Combust Inst. Meeting, Charlottesville, VA, 2007. Also reported in: 20th Int. Symp. on Gas Kinetics, Manchester, U.K.; 2008, and 235th ACS National Meeting, New Orleans, LA, 2008.
- Asatryan, R.; Bozzelli, J.W. Chain Branching and Termination in Low Temperature Combustion of n-Alkanes: n-Pentan-2-yl Radical Plus O2, Isomerization and Addition of Second O2, 32nd Int. Symp. Combust., Montreal, Canada, 2008.
- Sharma, S.; Raman, S.; Green, W.H. Intramolecular Hydrogen Migration in Alkylperoxy and Hydroperoxyalkylperoxy Radicals: Accurate Treatment of Hindered Rotors. J. Phys. Chem. A 2010, 114, 5689–5701. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, A. Systematic computational study on the unimolecular reactions of alkylperoxy (RO2), hydroperoxyalkyl (QOOH), and hydroperoxyalkylperoxy (O2QOOH) radicals. J. Phys. Chem. A 2011, 115, 3301–3325. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Bao, J.; Wang, Z.; Wang, X.; Truhlar, D.G. Hydrogen shift isomerizations in the kinetics of the second oxidation mechanism of alkane combustion. Reactions of the hydroperoxypentylperoxy OOQOOH radical, Combustion and Flame 2018, 197, 88–101. [Google Scholar] [CrossRef]
- Davis, M.M.; Weidman, J.D.; Abbott, A.S.; Douberly, G.E.; Turney, J.M.; Schaefer III, H.F. J. J. Chem. Phys. 2019, 151, 124302. [Google Scholar]
- Sahetchian, K.; Champoussin, J.C.; Brun, M.; Levy, N.; Blin-Simiand, N.; Aligrot, C.; Jorand, F.; Socoliuc, M.; Heiss, A.; Guerassi, N. Experimental study and modeling of dodecane ignition in a diesel engine, Combust. Flame 1995, 103, 207–220. [Google Scholar] [CrossRef]
- (a) Blin-Simiand, N.; Rigny, R.; Viossat, V.; Circan, S.; Sahetchian, K.; Autoignition of hydrocarbon/Air Mixtures in a CFR Engine: Experimental and Modeling Study, Combust. Sci. and Tech. 1993, 88, 329–348. (b) Sahetchian, K.; Rigny, R.; Blin, N. Evaluation of Hydroperoxide Concentrations During the Delay of Autoignition in an Experimental Four Stroke Engine: Comparison with Cool Flame Studies in a Flow System, Combust. Sci. Technol. 1988, 60, 117–124.
- Xing, L.; Bao, J.L.; Wang, Z.; Zhang, F.; Truhlar, D.G. Degradation of carbonyl hydroperoxides in the atmosphere and in combustion. J. Am. Chem. Soc. 2017, 139, 15821–15835. [Google Scholar] [CrossRef]
- Blin-Simiand, N.; Jorand, F.; Keller, K.; Fiderer, M.; Sahetchian, K. Ketohydroperoxides and ignition delay in internal combustion engines, Combust. Flame 1998, 112, 278–282. [Google Scholar] [CrossRef]
- Hansen, A.S.; Bhagde, T.; Qian, Y.; Cavazos, A.; Huchmala, R.M.; Boyer, M.A.; Gavin-Hanner, C.F.; Klippenstein, S.J.; McCoy, A.B.; Lester, M.I. Infrared spectroscopic signature of a hydroperoxyalkyl radical (●QOOH). J. Chem. Phys. 2022, 156, 014301. [Google Scholar] [CrossRef]
- Jorand, F.; Hess, A.; Perrin, O.; Sahetchian, K.; Kerhoas, L.; Einhorn, J. Isomeric hexyl-ketohydroperoxides formed by reactions of hexoxy and hexylperoxy radicals in oxygen, Int. J. Chem. Kinet. 2003, 35, 354–366. [Google Scholar] [CrossRef]
- Sahetchian, K.A.; Blin, N.; Rigny, R.; Seydi, A.; Murat, M. The oxidation of n-butane and n-heptane in a CFR engine. Isomerization reactions and delay of autoignition, Combust. Flame 1990, 79, 242–249. [Google Scholar] [CrossRef]
- Blin-Simiand, N.; Jorand, F.; Sahetchian, K.; Brun, M.; Kerhoas, L.; Malosse, C.; Ein-horn, J. Hydroperoxides with zero, one, two or more carbonyl groups formed during the oxidation of n-dodecane. Combust. Flame 2001, 126, 1524–1532. [Google Scholar] [CrossRef]
- Eskola, A.J.; Zador, J. t, Antonov, I.O.; Sheps, L.; Savee, J.D.; Osborn, D.L.; Taatjes, C.A. Probing the Low-Temperature Chain-Branching Mechanism for n-Butane Autoignition Chemistry via Time-Resolved Measurements of Ketohydroperoxide Formation in Photolytically Initiated n-C4H10 Oxidation. Proc. Combust. Inst. 2015, 35, 291–298. [Google Scholar]
- Eskola, A.J.; Antonov, I.O.; Sheps, L.; Savee, J.D.; Osborn, D.L.; Taatjes, C.A. Time-resolved measurements of product formation in the low-temperature (550–675 K) oxidation of neopentane: a probe to investigate chain-branching mechanism. Phys. Chem. Chem. Phys. 2017, 19, 13731–13745. [Google Scholar] [CrossRef] [PubMed]
- Battin-Leclerc, F.; Herbinet, O.; Glaude, P.-A.; Fournet, R.; Zhou, Z.; Deng, L.; Guo, H.; Xie, M.; Qi, F. New experimental evidences about the formation and consumption of ketohydroperoxides. Proc. Combust. Inst., 2011, 33, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Herbinet, O.; Wang, Z.; Qi, F.; Fittschen, C.; Westmoreland, P.R.; Battin-Leclerc, F. Measuring hydroperoxide chain-branching agents during n-pentane low-temperature oxidation. Proc. Combust. Inst., 2017, 36, 333–342. [Google Scholar] [CrossRef]
- Bourgalais, J.; Gouid, Z.; Herbinet, O.; Garcia, G.A.; Arnoux, P.; Wang, Z.; Tran, L.-S.; Vanhove, G.; Hochlaf, M.; Nahon, L.; Battin-Leclerc, F. Isomer-sensitive characterization of low temperature oxidation reaction products by coupling a jet-stirred reactor to an electron/ion coincidence spectrometer: case of n-pentane. Phys. Chem. Chem. Phys. 2020, 22, 1222–1241. [Google Scholar] [CrossRef] [PubMed]
- Pelucchi, M.; Bissoli, M.; Cavallotti, C.; Cuoci, A.; Faravelli, T.; Frassoldati, A.; Ranzi, E.; Stagni, A. Improved Kinetic Model of the Low-Temperature Oxidation of n-Heptane. Energy Fuels 2014, 28, 7178–7193. [Google Scholar] [CrossRef]
- Ranzi, E.; Cavallotti, C.; Cuoci, A.; Frassoldati, A.; Pelucchi, M.; Faravelli, T. New reaction classes in the kinetic modeling of low temperature oxidation of n-alkanes. Combust. Flame 2015, 162, 1679–1691. [Google Scholar] [CrossRef]
- Hansen, N.; Moshammer, K.; Jasper, A.W. Isomer-Selective Detection of Keto-Hydroperoxides in the Low-Temperature Oxidation of Tetrahydrofuran. J. Phys. Chem. A 2019, 123, 8274–8284. [Google Scholar] [CrossRef]
- Moshammer, K.; Jasper, A.W.; Popolan-Vaida, D.M.; Lucassen, A.; Diévart, P.; Selim, H.; Eskola, A.J.; Taatjes, C.A.; Leone, S.R.; Sarathy, S.M.; Ju, Y.; Dagaut, P.; Kohse-Höinghaus, K.; Hansen, N. Detection and Identification of the Keto-Hydroperoxide (HOOCH2OCHO) and Other Intermediates during Low-Temperature Oxidation of Dimethyl Ether. J. Phys. Chem. A 2015, 119, 7361–7374. [Google Scholar] [CrossRef] [PubMed]
- Moshammer, K.; Jasper, A.W.; Popolan-Vaida, D.M.; Wang, Z.D.; Shankar, V.S. B.; Ruwe, L.; Taatjes, C.A.; Dagaut, P.; Hansen, N. Quantification of the Keto-Hydroperoxide (HOOCH2OCHO) and Other Elusive Intermediates during Low-Temperature Oxidation of Dimethyl Ether. J. Phys. Chem. A 2016, 120, 7890–7901. [Google Scholar] [CrossRef] [PubMed]
- Mutzel, A.; Poulain, L.; Berndt, T.; Iinuma, Y.; Rodigast, M.; Böge, O.; Richters, S.; Spindler, G.; Sipilä, M.; Jokinen, T.; Kulmala, M.; and Herrmann, H. Highly Oxidized Multifunctional Organic Compounds Observed in Tropospheric Particles: A Field and Laboratory Study. Environmental Science & Technology 2015, 49, 7754–7761. [Google Scholar]
- Battin-Leclerc, F.; Rodriguez, A.; Husson, B.; Herbinet, O.; Glaude, P.A.; Wang, Z.; Cheng, Z.; Qi, F. Products from the oxidation of linear isomers of hexene. J. Phys. Chem. A 2014, 118, 673–683. [Google Scholar] [CrossRef]
- Farouk, T.I.; Xu, Y.; Avedisian, C.T.; Dryer, F.L. Combustion characteristics of primary reference fuel (PRF) droplets: Single stage high temperature combustion to multistage “Cool Flame” behavior. Proc. Combust. Inst. 2017, 36, 2585–2594. [Google Scholar] [CrossRef]
- Popolan-Vaida, D.M.; Eskola, A.J.; Rotavera, B.; Lockyear, J.F.; Wang, Z.; Sarathy, S.M.; Caravan, R.L.; Zador, J.; Sheps, L.; Lucassen, A.; Moshammer, K.; Dagaut, P.; Osborn, D.L.; Hansen, N.; Leone, S.R.; Taatjes, C.A. Formation of Organic Acids and Carbonyl Compounds in n-Butane Oxidation via γ-Ketohydroperoxide Decomposition. Angew. Chem. Int. Ed. 2022, 61, e202209168. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. ; The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts 2008, 120, 215–241. [Google Scholar]
- Dunning, T.H.; Jr Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Asatryan, R.; Pal, Y.; Hachmann, J.; Ruckenstein, E. Roaming-like Mechanism for Dehydration of Diol Radicals. J. Phys. Chem. A 2018, 122, 9738–9754. [Google Scholar] [CrossRef] [PubMed]
- Asatryan, R.; Hudzik, J.M.; Bozzelli, J.W. Khachatryan, L. ; Ruckenstein, E. OH-Initiated Reactions of p-Coumaryl Alcohol Relevant to the Lignin Pyrolysis. Part, I. Potential Energy Surface Analysis, J. Phys. Chem. A 2019, 123, 2570–2585. [Google Scholar]
- Pieniazek SN, Clemente FR, Houk, K. N.; et al. Sources of error in DFT computations of C–C bond formation thermochemistries: π→σ transformations and error cancellation by DFT methods. Angew. Chem. Int. Ed Engl. 2008, 47, 7746–7749. [Google Scholar] [CrossRef]
- Hohenstein EG, Chill ST, Sherrill CD. Assessment of the performance of the M05-2X and M06-2X exchange-correlation functionals for noncovalent interactions in biomolecules. J. Chem. Theory Comput. 2008, 4, 1996–2000. [Google Scholar] [CrossRef]
- McCann, B.W.; McFarland, S.; Acevedo, O. Benchmark continuum solvent models for keto-enol tautomerizations. J. Phys. Chem. A, 2015, 119, 8724–8733. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xu, X.; Truhlar, D.G. ; Minimally augmented Karlsruhe basis sets. Theoretical Chemistry Accounts 2011, 128, 295–305. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, revision A.03; Gaussian, Inc.: Wallingford, CT, 2016.
- Dana, A.G.; Johnson, M.S.; Allen, J.W.; et al. Automated reaction kinetics and network exploration (Arkane): A statistical mechanics, thermodynamics, transition state theory, and master equation software. Int. J. Chem. Kinet. 2023, 55, 300–323. [Google Scholar] [CrossRef]
- Gao, C.W.; Allen, J.W.; Green, W.H.; West, R.H. Reaction Mechanism Generator: Automatic construction of chemical kinetic mechanisms. Computer Physics Communications 2016, 203, 212–225. [Google Scholar] [CrossRef]
- Liu, M.; Grinberg Dana, A. S. Johnson., Goldman, M.J.; Jocher, A.; A.M. Payne, Grambow, C.A.; Han, K.; Yee, N.W.; Mazeau, E.J.; Blondal, K.; West, R.H.; Goldsmith, C.F.; Green, W.H. Reaction Mechanism Generator v3.0: Advances in Automatic Mechanism Generation. Journal of Chemical Information and Modeling 2021, 61, 2686–2696. [Google Scholar] [CrossRef]
- Johnson, M.S.; Dong, X.; Grinberg Dana, A.; Chung, Y.; Farina, D.; Gillis, R.J.; Liu, M.; Yee, N.W.; Blondal, K.; Mazeau, E.; Grambow, C.A.; Payne, A.M.; Spiekermann, K.A.; Pang, H.-W.; Goldsmith, C.F.; West, R.H.; Green, W.H. The RMG Database for Chemical Property Prediction. Chemical Information 2022, 62, 4906–4915. [Google Scholar] [CrossRef]
- Kee, R.J.; Rupley, F.M.; Miller, J.A. Chemkin-II: A Fortran chemical kinetics package for the analysis of gas-phase chemical kinetics; United States, 1989. https://www.osti.gov/biblio/5681118 https://www.osti.gov/servlets/purl/5681118.
- CHEMKIN-PRO 15112 Reaction Design: San Diego; 2023.
- Taatjes, C.A.; Hansen, N.; McIlroy, A.; Miller, J.A.; Senosiain, J.P.; Klippenstein, S.J.; Qi, F.; Sheng, L.; Zhang, Y.; Cool, T.A.; Wang, J.; Westmoreland, P.R.; Law, M.E.; Kasper, T.; Kohse-Höinghaus, K. Enols Are Common Intermediates in Hydrocarbon Oxidation. Science 2005, 308, 1887–1889. [Google Scholar] [CrossRef] [PubMed]
- Taatjes, C.A.; Hansen, N.; Miller, J.A.; Cool, T.A.; Wang, J.; Westmoreland, P.R.; Law, M.E.; Kasper, T.; Kohse-Höinghaus, K. Combustion Chemistry of Enols: Possible Ethenol Precursors in Flames. J. Phys. Chem. A 2006, 110, 3254. [Google Scholar] [CrossRef] [PubMed]
- Huynh, L.K.; Zhang, H.R.; Zhang, S.; Eddings, E.; Sarofim, A.; Law, M.E.; Westmoreland, P.R.; Truong, T.N. Kinetics of Enol Formation from Reaction of OH with Propene. J. Phys. Chem. A 2009, 113, 3177–3185. [Google Scholar] [CrossRef]
- Archibald, A.T.; McGillen, M.R.; Taatjes, C.A.; Percival, C.J.; Shallcross, D.E. Atmospheric transformation of enols: A potential secondary source of carboxylic acids in the urban troposphere. Geophys. Res. Lett. 2007, 34, L21801. [Google Scholar] [CrossRef]
- Erfle, S.; Reim, S.; Michalik, D.; Jiao, H.; Langer, P. Experimental and Theoretical Study of the Keto–Enol Tautomerization of 3,5-Dioxopimelates, Eur. J. Org. Chem. 2011, 4367–4372. [Google Scholar]
- Antonov, L. (Ed.), Tautomerism: Concepts and Applications in Science and Technology; Wiley-VCH, 2016.
- Perez, P.; Toro-Labbe, A. Characterization of Keto-Enol Tautomerism of Acetyl Derivatives from the Analysis of Energy, Chemical Potential, and Hardness. J. Phys. Chem. A 2000, 104, 1557–1562. [Google Scholar] [CrossRef]
- Couch, D.E.; Nguyen, Q.L. D.; Liu, A.; Hickstein, D.D.; Kapteyn, H.C.; Murnane, M.M.; Labbe, N.J. Detection of the keto-enol tautomerization in acetaldehyde, acetone, cyclohexanone, and methyl vinyl ketone with a novel VUV light source. Proc. Comb. Inst. 2021, 368, 1737–1744. [Google Scholar] [CrossRef]
- Ali ST, Antonov L, Fabian WMF. Phenol–quinone tautomerism in (Arylazo)naphthols and the analogous Schiff bases: benchmark calculations. J Phys. Chem. A. 2014, 118, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Jana, K.; Ganguly, B. DFT Study to Explore the Importance of Ring Size and Effect of Solvents on the Keto−Enol Tautomerization Process of α- and β-Cyclodiones. ACS Omega 2018, 3, 8429–8439. [Google Scholar] [CrossRef]
- Jana, K.; Ganguly, B. DFT studies on quantum mechanical tunneling in tautomerization of three-membered rings. Phys. Chem. Chem. Phys. 2018, 20, 28049. [Google Scholar] [CrossRef] [PubMed]
- Mehrani, S.; Tayyari, S.F. ; M.M. Heravi, Morsali, A. Theoretical investigation of solvent effect on the keto–enol tautomerization of pentane-2,4-dione and a comparison between experimental data and theoretical calculations. Can. J. Chem. 2021, 99, 411–424. [Google Scholar] [CrossRef]
- Vereecken, L.; Francisco, J.S. Theoretical studies of atmospheric reaction mechanisms in the troposphere. Chem. Soc. Rev. 2012, 41, 6259–6293. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Nagaoka, K.; Morikawa, K.; Mawatari, K.; Kitamori, T. Keto-Enol Tautomeric Equilibrium of Acetylacetone Solution Confined in Extended Nanospaces. J. Phys. Chem. B 2015, 119, 14750–14755. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Biswas, S.; Pramanik, A.; Sarkar, P. Computational Studies on the Keto-Enol Tautomerism of Acetylacetone. Int. J. Res. Soc. Nat. Sci. 2017, 2, 2455–5916. [Google Scholar]
- Bahrini, C.; Herbinet, O.; Glaude, P.-A.; Schoemaecker C, Fittschen, C. ; Battin-Leclerc, F. Quantification of hydrogen peroxide during the low-temperature oxidation of alkanes. J Am Chem Soc. 2012, 134, 11944–11947. [Google Scholar] [CrossRef]
- Bugler, J.; Rodriguez, A.; Herbinet, O.; Battin-Leclerc, F.; Togbe´, C.; Dayma, G.; Dagaut, P.; Curran, H.J. An experimental and modelling study of n-pentane oxidation in two jet-stirred reactors: The importance of pressure-dependent kinetics and new reaction pathways. Proc. Comb. Inst. 2017, 36, 441–448. [Google Scholar] [CrossRef]
- Bugler, J.; Marks, B.; Mathieu, O.; Archuleta, R.; Camou, A.; Grégoire, C.; Heufer, K.A.; Petersen, E.L.; Curran, H.J. An ignition delay time and chemical kinetic modeling study of the pentane isomers. Combustion and Flame 2016, 163, 138–156. [Google Scholar] [CrossRef]
- Johnson, M.S.; Green, W.H. A machine learning based approach to reaction rate estimation. Reaction Chemistry & Engineering 2024, 9, 1364–1380. [Google Scholar] [CrossRef]
- Asatryan, R.; Bozzelli, J.W.; Simmie, J.M. Thermochemistry of methyl and ethyl nitro, RNO2, and nitrite, RONO, organic compounds. J. Phys. Chem. A 2008, 112, 3172–3185. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.P.; Chaos, M.; Ju, Y.; Dryer, F.L.; Klippenstein, S.J. Comprehensive H2/O2 kinetic model for high-pressure combustion. International Journal of Chemical Kinetics 2012, 44, 444–474. [Google Scholar] [CrossRef]
- Hashemi, H.; Jacobsen, J.G.; Rasmussen, C.T.; Christensen, J.M.; Glarborg, P.; Gersen, S.; van Essen, M.; Levinsky, H.B.; Klippenstein, S.J. High-pressure oxidation of ethane. Combustion and Flame 2017, 182, 150–166. [Google Scholar] [CrossRef]
- Kumar, M.; Francisco, J.S. Hydrogen Sulfide Induced Carbon Dioxide Activation by Metal-Free Dual Catalysis. Chem. Eur. J. 2016, 22, 4359–4363. [Google Scholar] [CrossRef] [PubMed]
- (a) Evans, M.G.; Polanyi, M. Further Considerations on the Thermodynamics of Chemical Equilibria and Reaction Rates. Trans. Faraday Soc. 1938, 32, 1333–1360. [Google Scholar] [CrossRef]
- (a) Bell, R.P. The Theory of Reactions Involving Proton Transfers. Proc. R. Soc. A 1973, 154, 414–429. [Google Scholar]
- Semenov, N.N. Some Problems in Chemical Kinetics and Reactivity, Volume 1, Princeton University Press, 1958, pp.254. [CrossRef]
- Alarcon, J.F.; Ajo, S.; Morozov, A.N.; Mebel, A.M. Theoretical study on the mechanism and kinetics of the oxidation of allyl radical with atomic and molecular oxygen. Combustion and Flame 2023, 257, 112388. [Google Scholar] [CrossRef]
Scheme 1.
Oxidation model for large alkanes including two novel KHP decomposition pathways – sequences of Korcek and keto-enol tautomerization reactions, highlighted in blue. Q, P, and E core structures have correspondingly 1, 2, and 3 H atoms less than R; for pentane derivatives, they represent correspondingly C5H11-, C5H10-, C5H9-, and C5H8- groups.
Scheme 1.
Oxidation model for large alkanes including two novel KHP decomposition pathways – sequences of Korcek and keto-enol tautomerization reactions, highlighted in blue. Q, P, and E core structures have correspondingly 1, 2, and 3 H atoms less than R; for pentane derivatives, they represent correspondingly C5H11-, C5H10-, C5H9-, and C5H8- groups.
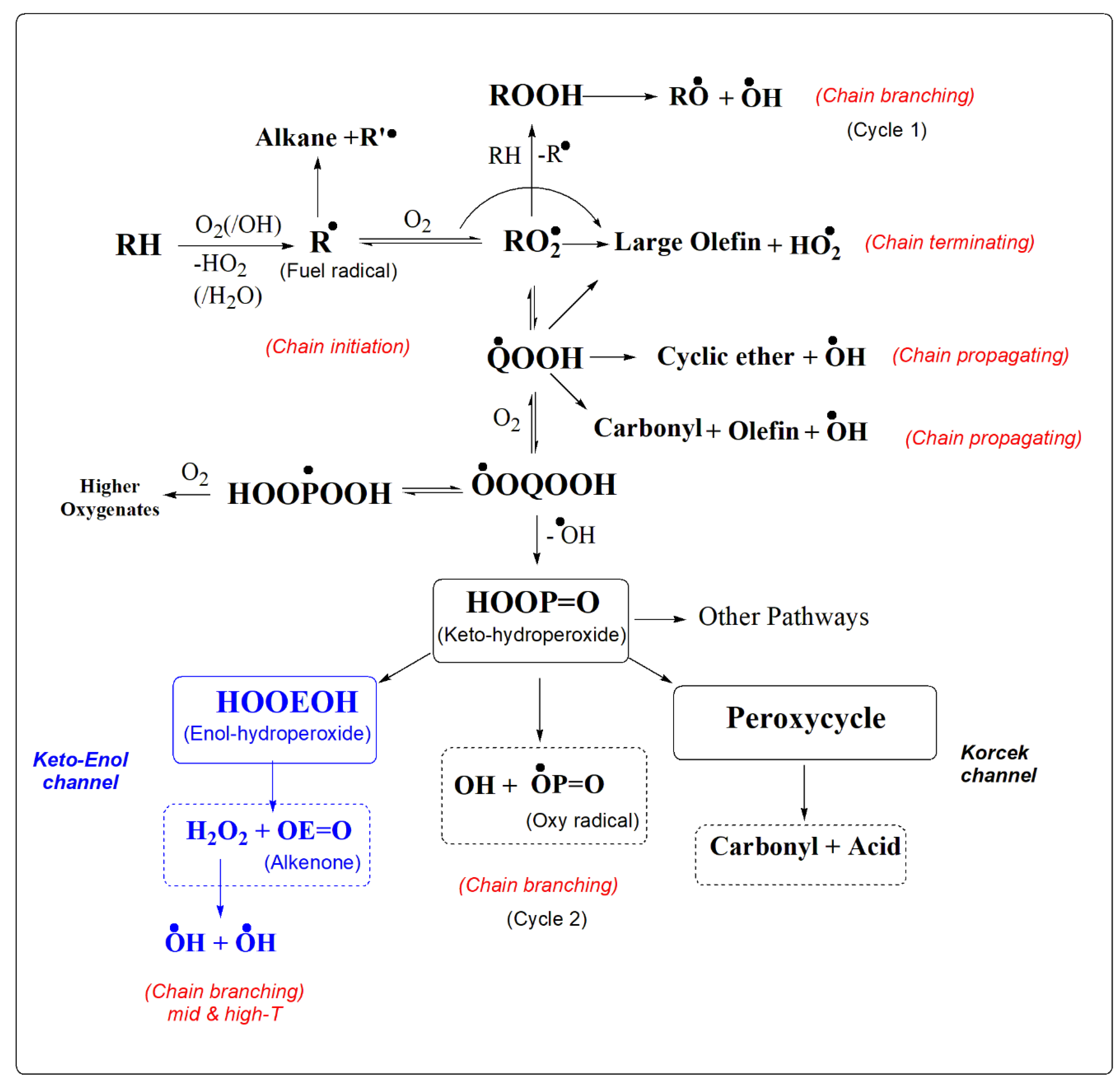
Scheme 2.
Self-catalyzed intramolecular hydrogen atom transfer (SCI-HAT) mechanism versus Direct keto-enol tautomerization of the model pentane-γ-ketohydroperoxide (γ-KHP) - a key chain-branching agent of the combustion of traditional fuels. Relative enthalpies are provided in parenthesis. SCI-HAT reduces the barrier by as much as 22 kcal/mol.
Scheme 2.
Self-catalyzed intramolecular hydrogen atom transfer (SCI-HAT) mechanism versus Direct keto-enol tautomerization of the model pentane-γ-ketohydroperoxide (γ-KHP) - a key chain-branching agent of the combustion of traditional fuels. Relative enthalpies are provided in parenthesis. SCI-HAT reduces the barrier by as much as 22 kcal/mol.
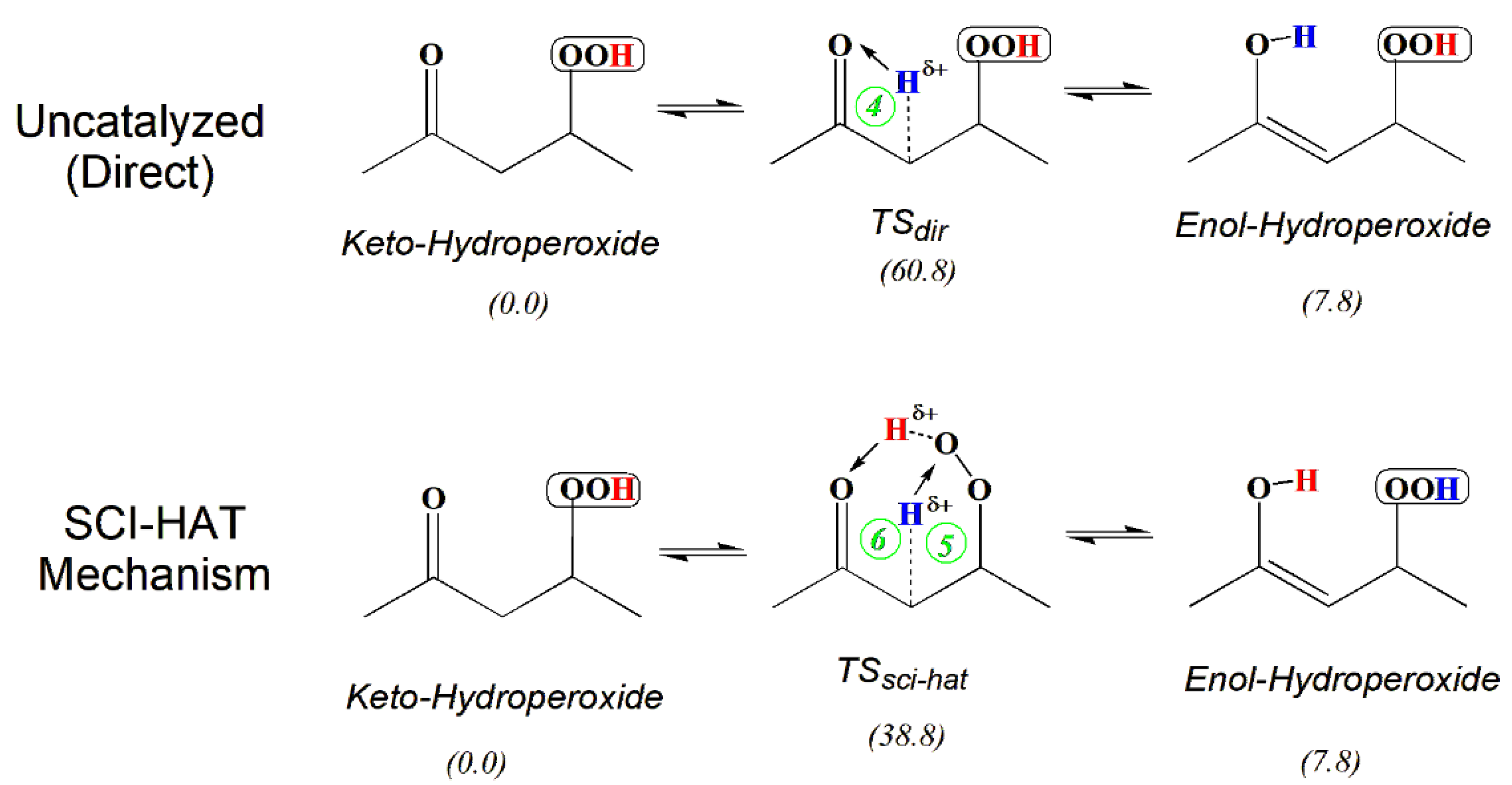
Scheme 3.
Generalized sketch of the SCI-HAT bond-exchange mechanism relevant to conventional (e.g., keto-enol and imino-amino tautomerization, where A is O or NH, respectively; cf. Scheme 2), and skeletal double-bond shift isomerization (A ≡ CH2). A symbolizes the adjacent H-acceptor group of the molecule undergoing conversion. XH represents an electronegative heteroatom containing the H-donor/acceptor ambivalent group (basically, the core of the sci-hat catalyst), such as -OOH, -SH, -COOH, -OH, or -NH2. The broader catalyst moiety responsible for accessibility to A is encircled.
Scheme 3.
Generalized sketch of the SCI-HAT bond-exchange mechanism relevant to conventional (e.g., keto-enol and imino-amino tautomerization, where A is O or NH, respectively; cf. Scheme 2), and skeletal double-bond shift isomerization (A ≡ CH2). A symbolizes the adjacent H-acceptor group of the molecule undergoing conversion. XH represents an electronegative heteroatom containing the H-donor/acceptor ambivalent group (basically, the core of the sci-hat catalyst), such as -OOH, -SH, -COOH, -OH, or -NH2. The broader catalyst moiety responsible for accessibility to A is encircled.
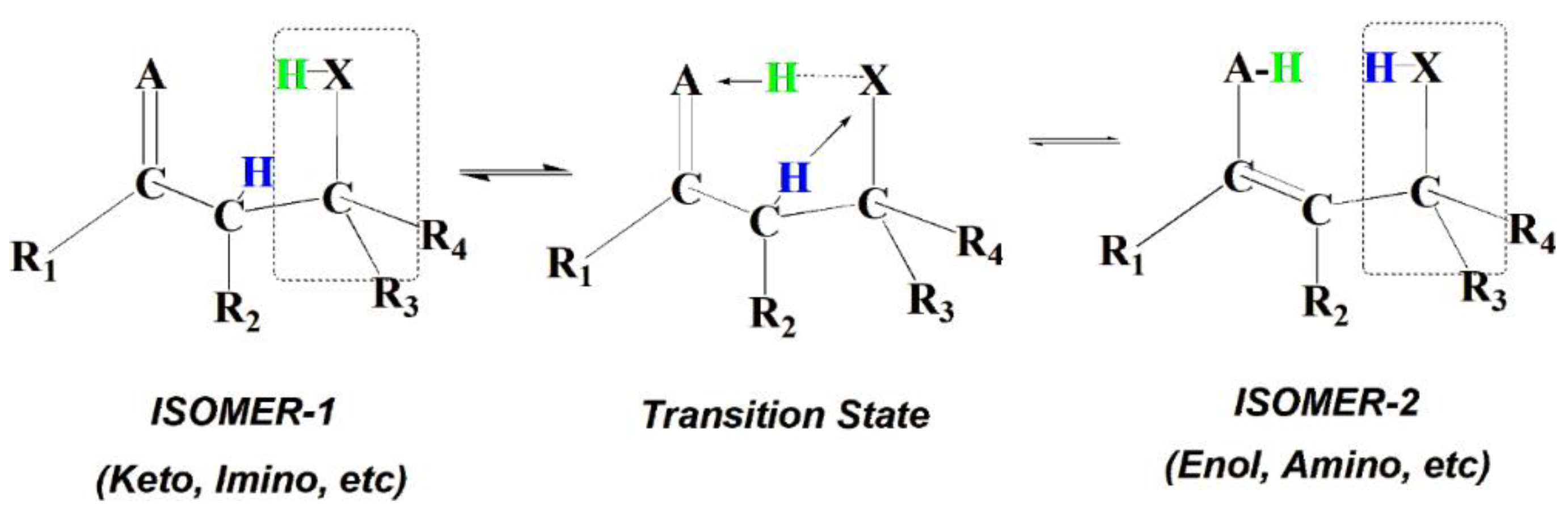
Scheme 4.
Double-SCI-HAT tautomerization of the 2,6-diketo-4-hydroxymethyl heptane (4-hydroxymethyl -2,6-heptane-dione) - a KHP-model of the n-heptane combustion. The sci-hat-catalyst (OH of the encircled CH2OH group) relay-transfers its H atom to the first carbonyl group to catalyze its tautomerization and recovers itself by receiving H-atom from the α-carbon backbone; then the same process occurs with the second carbonyl oxygen, again recovering the hydroxymethyl catalyst group. .
Scheme 4.
Double-SCI-HAT tautomerization of the 2,6-diketo-4-hydroxymethyl heptane (4-hydroxymethyl -2,6-heptane-dione) - a KHP-model of the n-heptane combustion. The sci-hat-catalyst (OH of the encircled CH2OH group) relay-transfers its H atom to the first carbonyl group to catalyze its tautomerization and recovers itself by receiving H-atom from the α-carbon backbone; then the same process occurs with the second carbonyl oxygen, again recovering the hydroxymethyl catalyst group. .
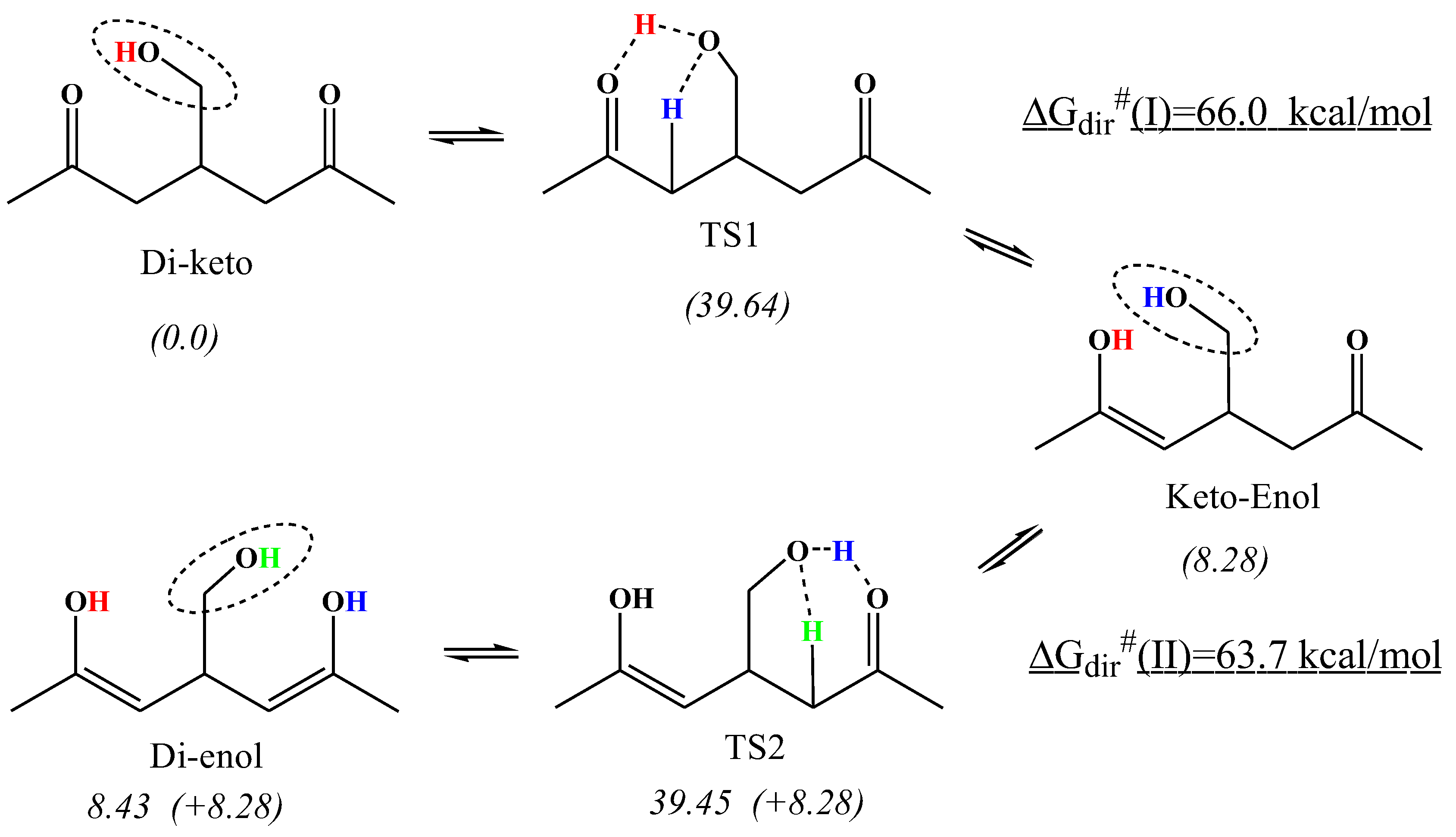
Figure 1.
An illustrative example of the more complex long-range and sequential keto-enol tautomerization of a model 2,4-diketo-6-hydroxy hexane - an intermediate of the n-heptane oxidation. The sci-hat catalyst (OH, or rather encircled CH2OH - group), transfers one of its H-atoms to the distant carbonyl oxygen and concomitantly accepts an H-atom from the α-methylene group to produce the 2- enol-4-keto-6-hydroxy hexane product. Another SCI-HAT rearrangement of regular type provided in Scheme 2, can occur with the second carbonyl group, again, regenerating the catalyst moiety and forming the 2-keto-4-enol-6-hydroxy hexane isomer. .
Figure 1.
An illustrative example of the more complex long-range and sequential keto-enol tautomerization of a model 2,4-diketo-6-hydroxy hexane - an intermediate of the n-heptane oxidation. The sci-hat catalyst (OH, or rather encircled CH2OH - group), transfers one of its H-atoms to the distant carbonyl oxygen and concomitantly accepts an H-atom from the α-methylene group to produce the 2- enol-4-keto-6-hydroxy hexane product. Another SCI-HAT rearrangement of regular type provided in Scheme 2, can occur with the second carbonyl group, again, regenerating the catalyst moiety and forming the 2-keto-4-enol-6-hydroxy hexane isomer. .
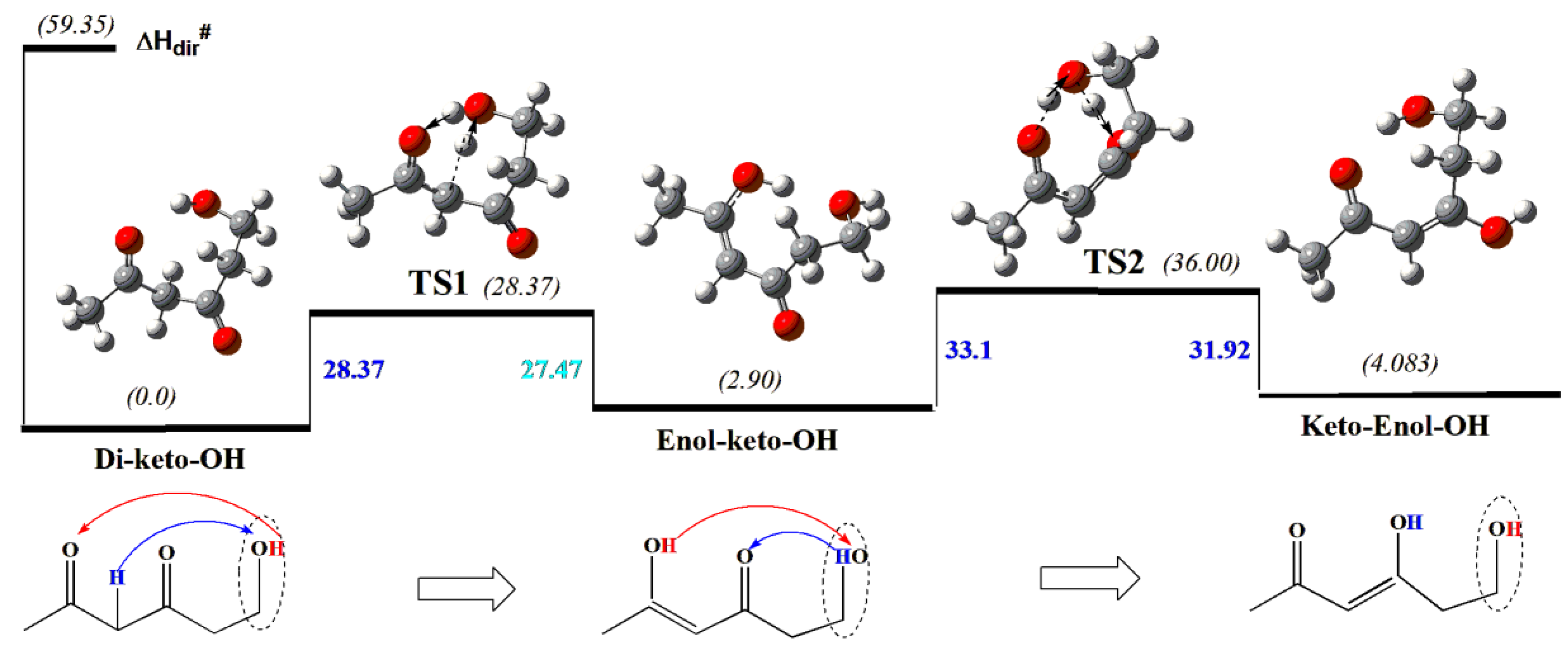
Figure 2.
Potential energy diagram for SCI-HAT, Korcek, and OH dissociation KHP pathways. Enthalpies (kcal/mol) are relative to the KHP baseline.
Figure 2.
Potential energy diagram for SCI-HAT, Korcek, and OH dissociation KHP pathways. Enthalpies (kcal/mol) are relative to the KHP baseline.
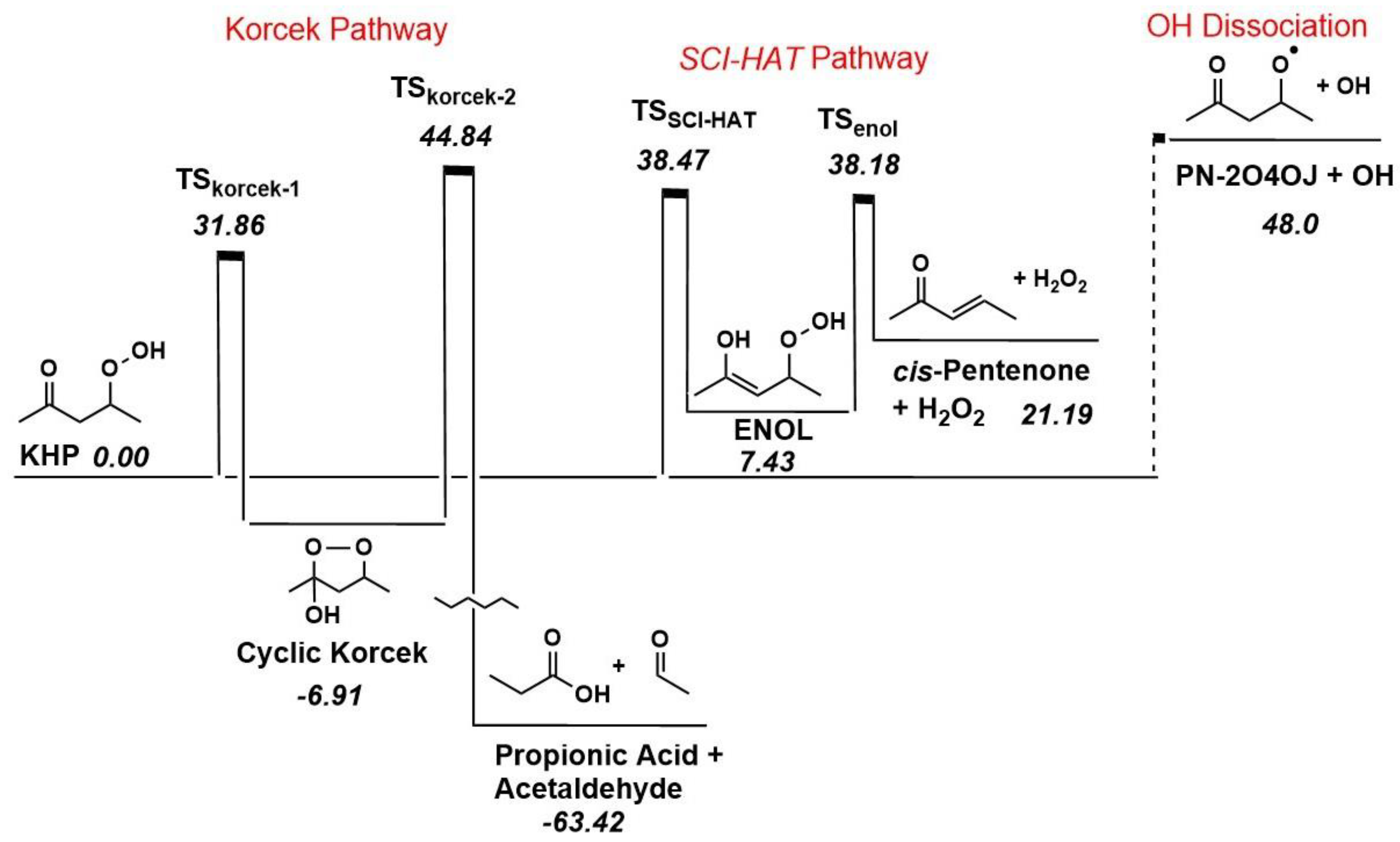
Figure 3.
Rate constants vs. pressure (1.0 and 10 atm) and vs. temperature (300 and 800 K) for KHP pathways. .
Figure 3.
Rate constants vs. pressure (1.0 and 10 atm) and vs. temperature (300 and 800 K) for KHP pathways. .
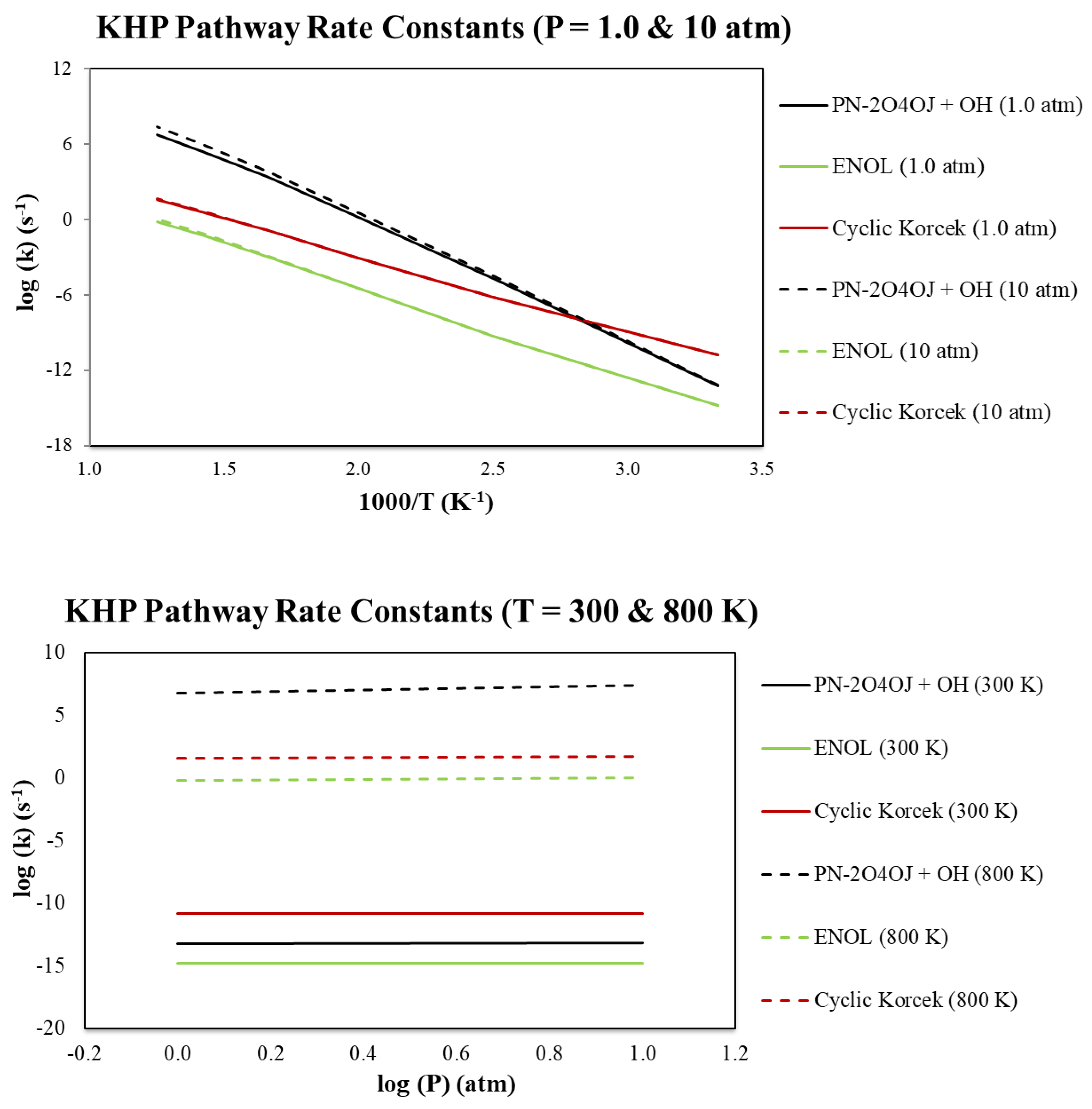
Figure 4.
Effect of SCI-HAT reactions (new kinetic families and thermochemical data) on PN-kinetic model performance for ϕ = 2 and P = 10 atm conditions. Models were generated using: (a) no libraries, only estimation methods (PN-ES), (b) only the Curran Pentane as a Library (PN-CPL), (c) higher fidelity data via multiple libraries (PN-ML), (d) using multiple libraries supplemented by our modified reaction library (PM-ML-m-v1), from our previous study [21], and (e) multiple libraries and our modified reaction library supplemented by three new kinetic families for the SCI-HAT mechanism and new thermochemistry data (PN-ML-m-v2). Symbols represent experimental data; lines are model predictions.
Figure 4.
Effect of SCI-HAT reactions (new kinetic families and thermochemical data) on PN-kinetic model performance for ϕ = 2 and P = 10 atm conditions. Models were generated using: (a) no libraries, only estimation methods (PN-ES), (b) only the Curran Pentane as a Library (PN-CPL), (c) higher fidelity data via multiple libraries (PN-ML), (d) using multiple libraries supplemented by our modified reaction library (PM-ML-m-v1), from our previous study [21], and (e) multiple libraries and our modified reaction library supplemented by three new kinetic families for the SCI-HAT mechanism and new thermochemistry data (PN-ML-m-v2). Symbols represent experimental data; lines are model predictions.
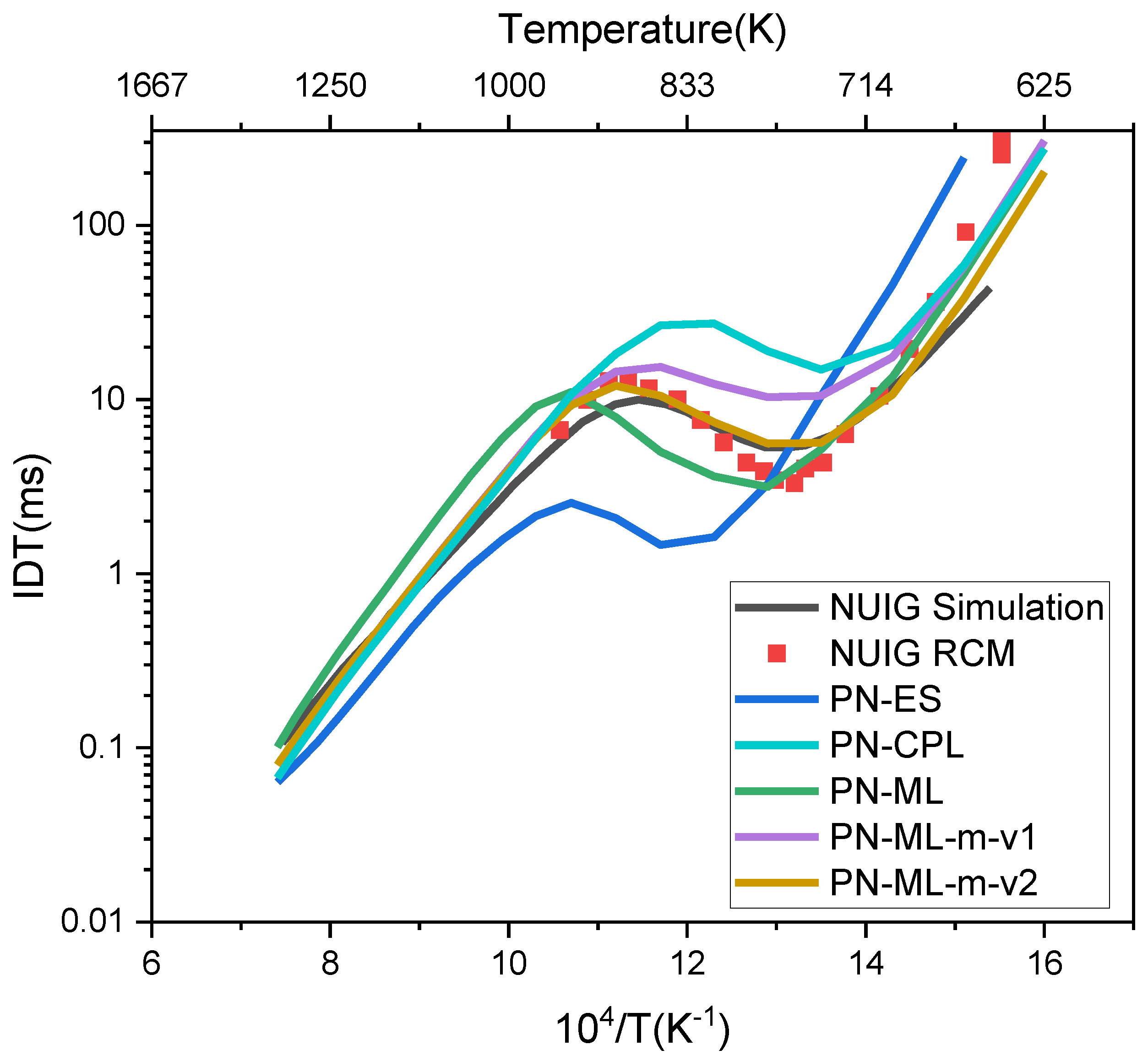

Table 1.
Intramolecular SCI-HAT - Catalyzed Tautomerization of Truncated Tri-Carbon Models.
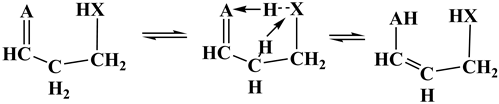 | ||||||||||
| =A | χAa) | XH | q(A)b) | q(X)b) | ΔG#sci-hat |
ν1c), cm-1 |
ΔG#dir | ΔGr | q(H) b) | q(C) b) |
| S | 2.58 | OOH | -0.287 | -0.332 | 34.25 | -1162.2 | 55.97 | -1.95 | 0.261 | -0.333 |
| NH | 3.04 | OOH | -0.673 | -0.488 | 37.57 | -1083.8 | 65.97 | -0.14 | 0.358 | -0.043 |
| NH | 3.04 | SH | -0.757 | -0.302 | 38.30 | -1151.5 | 65.87 | -2.14 | 0.173 | -0.137 |
| O | 3.44 | OOH | -0.728 | -0.457 | 39.61 | -1481.4 | 68.10 | 6.07 | 0.356 | -0.06 |
| O | 3.44 | CH2OH | -0.755 | -0.657 | 40.17 | -1397.0 | 69.17 | 5.56 | 0.276 | -0.345 |
| O | 3.44 | COOH | -0.701 | -0.544 | 45.41 | -1456.1 | 70.73 | 7.83 | 0.375 | -0.354 |
| O | 3.44 | SH | -0.702 | -0.298 | 49.06 | -1295.9 | 70.75 | 6.17 | 0.200 | -0.239 |
| CH2 | 2.55 | OOH | -0.714 | -0.451 | 59.35 | -1681.2 | 75.75 | -1.08 | 0.330 | -0.025 |
a) Pauling Electronegativity of H-acceptor atom A, b) Mulliken partial charges on A, X and migrating H atoms, c) Imaginary frequency for TSsci-hat. Gibbs energies are in kcal/mol, partial charges in electrons.
Table 2.
Fitted Modified Arrhenius Equation Parameters for High-Pressure Rate Constants for SCI-HAT, Korcek, and OH Dissociation KHP Pathways.
Table 2.
Fitted Modified Arrhenius Equation Parameters for High-Pressure Rate Constants for SCI-HAT, Korcek, and OH Dissociation KHP Pathways.
| Reaction Pathway | A (s-1) | n | E (kcal/mol) |
| KHP → Cyclic Korcek | 8.46 ×10-3 | 3.70 | 24.99 |
| Cyclic Korcek → Propionic Acid + Acetaldehyde | 1.89 ×10-10 | 1.17 | 51.50 |
| KHP → ENOL | 2.37 ×10-5 | 4.57 | 30.06 |
| ENOL → cis-Pentenone + H2O2 | 5.53 ×108 | 1.20 | 29.64 |
| KHP → PN-2O4OJ + OH | 4.33 ×1019 | 0.80 | 47.80 |
Table 3.
Rate constants for SCI-HAT and Enol decomposition training reactions (Sec.3.4) employed to create a decision tree for three new reaction families.
Table 3.
Rate constants for SCI-HAT and Enol decomposition training reactions (Sec.3.4) employed to create a decision tree for three new reaction families.
| Reactions | A (s-1) | n | E (kJ/mol) |
| KETO_24 = ENOL_24 | 2.37 ×10-5 | 4.57 | 125.8 |
| KETO_13 = ENOL_13 | 416 | 2.53 | 137.8 |
| KETO_25 = ENOL_25 | 8.42 ×1012 | 6.53 | 111.2 |
| ENOL_13 = 2-pentenal + H2O2 | 1.194 ×1011 | 0.482 | 115.9 |
| ENOL_24 = H2O2 + cis-Pentenone | 5.531 ×108 | 1.199 | 124.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



