Submitted:
20 December 2024
Posted:
20 December 2024
You are already at the latest version
Abstract
In multiple sclerosis (MS), there is significant evidence indicating that both progression independent of relapse activity (PIRA) and relapse-related worsening events contribute to the accumulation of progressive disability from the onset of the disease and throughout its course. Understanding the compartmentalized pathophysiology in MS would enhance comprehension of disease progression mechanisms, overcoming the traditional distinction in phenotypes. Smoldering MS activity is thought to be maintained by a continuous interaction between the parenchymal chronic processes of neuroinflammation and neurodegeneration and the intrathecal compartment. This review provides a comprehensive and up-to-date overview of the neuropathological and immunological evidence related to the mechanisms underlying PIRA phenomena in MS, with a focus on studies investigating the impact of currently available therapies on these complex mechanisms.
Keywords:
multiple sclerosis
; PIRA
; smouldering disease
1. Introduction
For decades, neurologists have struggled to assess and anticipate the unpredictable and variable clinical course of multiple sclerosis (MS), much like a challenging riddle that defies an easy resolution. What is the basis of smoldering MS activity? How can the continuum of neurodegeneration and neuroinflammation be described and treated? Recently, the literature has been moving towards solving these questions.
MS is traditionally classified as relapsing-remitting (RR) or primary progressive (PP), based on the initial disease course; over time of disability accrual, a secondary progressive (SP) MS phenotype can be developed. [1,2] MS patients often continue to report the worsening of symptoms even while the peripheral immune response is well controlled with disease-modifying treatments (DMTs) [3,4]: deciphering compartmentalized pathophysiology in MS would therefore aid a deeper comprehension of mechanisms of disease progression, overcoming the classic distinction in phenotypes. [5,6,7,8] There is a mutual contribution of progression independent of relapse activity (PIRA) and relapse-related worsening (RAW) events to progressive disability accrual since the disease onset and throughout the disease. [9,10,11]
These assessments are suggested by the most recent, harmonized definition of PIRA: (a) an increased Expanded Disability Status Scale (EDSS) score at least three months after and one month prior to the onset of an investigator-reported relapse; (b) a new baseline score following each relapse; (c) a confirmation score at least three months following the initial disability increase and one month before the onset of an investigator-reported relapse. [12,13] Debate has surrounded this clinically based definition, which has both strengths and limitations [13], and is heterogeneously described in real-world cohorts. [14]
The research into the etiological mechanisms of PIRA is currently spurred by the finding that it might manifest early in MS patients. [15] PIRA constitutes the main driver of disability accrual, and it is associated with higher disability in the long term. [15,16,17] RAW represents a less frequent phenomenon, likely due to the effectiveness of treatments on neuroinflammation-related phenomena. [18] MS patients presenting with PIRA after a first demyelinating event have an unfavorable long-term prognosis, with a higher probability and faster rate to disability milestones, especially if PIRA occurs early in the disease course. [19] Heterogeneous biomarkers of progression in MS have been proposed over the last few years. [20] In MS, inflammation may be the initial cause of neurodegeneration, which is then sustained by the ongoing compartmentalized inflammation. [6] Smoldering MS activity is supposed to be sustained by an ongoing dynamic between the intrathecal compartment, comprising cerebrospinal fluid (CSF) and meninges, and the parenchymal chronic processes of inflammation and neurodegeneration. This persistent interaction promotes glial activation and neuron loss, caused by a crucial imbalance between damage, repair and brain functional reserve. [21,22,23]
With emphasis on the evidence regarding the impact of currently available therapies on these multifaceted mechanisms, this review attempts to give a broad overview of current understandings of neuropathological and immunological evidence regarding the mechanisms causing PIRA phenomena in MS.
2. Methodology
A comprehensive and up-to-date literature research has been conducted in the two primary academic databases, PubMed and Google Scholar, covering studies on PIRA and smouldering disease in MS. We have integrated in this article the latest molecular studies, original articles, illustrious reviews, research of disease registers and real-world cohorts on therapeutic targets in MS in terms of neuroinflammation and neurodegeneration. Original studies presented at the most recent international congresses were also included. We carefully examined the reference lists of significant papers found throughout the search.
3. Smouldering Biology in Multiple Sclerosis
Even with steady inflammatory parameters, people with MS frequently see a decline in their motor and cognitive impairment. Smouldering-associated worsening is thought to be caused by a confluence of degenerative and inflammatory mechanisms, including anterograde and retrograde axonal degeneration, as well as the breakdown of compensatory processes such as neuronal plasticity and remyelination. [24] PIRA can be considered an epiphenomenon of smouldering MS disease, but it is limited to well-validated MS outcome measures in clinical trials and real-world studies. [25]
The field of MS biomarkers has changed considerably over the last years due to significant improvements in assay technologies and a focused attempt to identify molecules that may provide information on these pathogenic mechanisms. [6,26] Recent positron emission tomography (PET) studies employing radioligands for innate immunity assessment revealed that an unexpectedly high proportion of MS lesions have a smoldering component, which predicts cortical atrophy and EDSS progression. [27] Absinta et al. defined the primary role, in the neurodegenerative programming, of “microglia inflamed in MS” (MIMS) and “astrocytes inflamed in MS”. The “MIMS-foamy” pattern showed an upregulation of genes related to foam-cell differentiation and lipid storage, lipoprotein particle responses, lysosome metabolism and regulation of inflammatory response. In parallel, “MIMS-iron” showed upregulation of genes encoding ribosomal proteins, leading to MHC class II protein complex activation and expression. MIMS-iron analysis also revealed an upregulation of ferritin, immunoglobulin Fcγ and complement component C1 complex, alongside the highest expression of the inflammatory cytokine gene IL1B. The expression of MHC-related and inflammatory markers - such as SOD1, iron-related genes (FTH1, FTL), and complement C1-complex genes - was higher in MIMS-iron when compared directly to MIMS-foamy, suggesting a fundamental role in antigen presentation and spreading of inflammatory damage at the lesion edge. [28] An unbiased in silico approach was able to identify a convergence of cross-regional transcriptomic reactivity in MS oligodendrocytes leading to an upregulation of the myelin-encoding gene MAG and downregulation of the muscarinic receptor-encoding gene CHRM5, contributing to an unresolved self-sustaining inflammation. [29] According to neuro-molecular research on progressive MS, chemokines and cytokines generated by circulating immune cells and meningeal tertiary lymphoid structures may permeate the CSF and enter the cortex, activating microglia and causing damage. [30,31]
4. Radiological Expression of PIRA and Molecular Correlates
Chronic active lesions (CAL) are a crucial sign of chronic inflammation. A recent consensus established biomarkers of CAL, including paramagnetic rim lesions (PRL) identified on susceptibility-sensitive magnetic resonance imaging (MRI), MRI-defined slowly expanding lesions (SEL), and 18-kDa translocator protein (TSPO)-positive lesions on PET. The biomarker that has the strongest histological support is PRL. [32] Histologically, the cellular substrate of iron-positive rim lesions is iron-loaded activated myeloid cells and related pathways. [33] Hofman et al. observed the upregulation of the CD163-HMOX1-HAMP axis at the rims of chronic active lesions, indicating that haptoglobin-bound hemoglobin is a vital source of MC-associated iron uptake, also confirmed by the strong association between PRL levels in MS and CSF-associated sCD163. [34] Additionally, the C1QA, HMOX1, and HAMP genes were activated in those cells, a molecular sign of a proinflammatory profile. On the other hand, IL10 mRNA was elevated by perilesional myeloid cells, which may indicate tissue-regulating and anti-inflammatory properties in different areas of the rim. [34]
PRLs have been linked to a more severe course of the disease [35], and elevated levels of serum neurofilament light chain (sNfL) [36], without a clear correlation with their topographical distribution. [37] The number of PRL was also associated with the number of leptomeningeal contrast enhancement foci on T2-FLAIR and real-reconstruction inversion recovery, linking leptomeninges to mechanisms related to the sustaining chronic inflammation. [38]
SELS represent the subset of non-enhancing chronic lesions showing radial and linear expansion over 1–2 years. The proportion of SELs has been correlated with MS progression after 9 years, considering in addition severe SEL microstructural abnormalities as a predictor of EDSS scores worsening and SPMS conversion. [39]
5. Meningeal Inflammation, Subpial Cortical Damage and Focus on Microglia and Diffuse White Matter Pathology
One of the major contributors to the pathophysiology of cortical demyelination in multiple sclerosis is believed to be compartmentalized meningeal inflammation. [43] A change in the balance of tumor necrosis factor (TNF) signaling from TNFR1/TNFR2 and NFkB-mediated anti-apoptotic pathways to TNFR1- and RIPK3-mediated pro-apoptotic/pro-necroptotic signaling in the grey matter (GM) was linked to increased meningeal inflammation. In the grey matter of the MS cortex, TNFR1 was shown to be mainly expressed in neurons and oligodendrocytes, while TNFR2 was primarily expressed by microglia and astrocytes. [44]
A recent study characterized the cortical and meningeal translocator protein (TSPO) expression in vivo and ex vivo: MS patients exhibited abnormally increased TSPO signal in the cortex and meningeal tissue, diffusively in progressive and localized in relapsing phenotypes. In post-mortem SPMS, immunohistochemistry revealed increased TSPO expression in the meninges and adjacent subpial cortical lesions, related to meningeal inflammation. Translocator protein immunostaining was detected on meningeal MHC-class II+ macrophages and cortical-activated MHC-class II+ TMEM119+ microglia. [45]
Activated microglia and infiltrating macrophages may become iron-loaded in acute, actively demyelinating lesions MS lesions. [46] Post-mortem results suggest that early lesion formation may be attributed to MS microglia nodules. In the MS microglia nodules environment, an upregulation of genes related to phagocytosis, adaptive and innate immune responses, lymphocyte activation, lipid metabolism, and metabolic stress has been observed. Van den Bosch et al. also found significant activation of complement, IgG transcription, and MAC formation, constituting a hypermetabolic state with elevated pro-inflammatory cytokines and expression of ROS genes. Gene expression analysis indicates close proximity of activated lymphocytes that are influencing and being influenced by the microglia nodules: NCKAP1L, CASP3, JAK3, TCIRG1, CORO1A, GRB2, IRF8, TLR2, IL18. [47]
MS patients had higher innate inflammation in normal-appearing white matter (NAWM) and cortex than healthy controls. [27,48] Therefore, the accumulation of focal lesions and microstructural tissue abnormalities in cognitively linked WM tracts, the occurrence of focal and diffuse damage in important GM regions, and the presence of functional brain network abnormalities are the causes of the so-called "disconnection syndrome", whose clinical correlation is impaired cognition and fatigue. [49]
A recent review by Zhan and colleagues [50] outlined the role of ectopic lymphoid follicles in MS, stressing the correlation with cortical [51] and even spinal cord [52] pathology. The inflammatory meningeal and perivascular infiltrates were shown to contain a large number of CXCR5+ cells, cytoplasmic nuclear factor of activated T-cell-positive (NFATc1+) cells, enriched CD3+CD27+ memory cells, and CD4+CD69+ tissue-resident cells. [53]
The main biological processes described, with a focus on CAL, paramagnetic rim and meningeal and cortical inflammation mechanisms, are shown in Figure 1.
6. Adaptive Immunity and PIRA: Role of T Cells and B Cells
In MS pathogenesis, B cells are crucial not only for antibody-dependent mechanisms but also through the abnormal production of cytokine and chemokine and an antigen-presenting function that activates T cells and drives autoproliferation of brain-homing T cells, exacerbating the non-resolving neuroinflammation. [54,55] Furthermore, the contribution of B-cells to the formation of ectopic lymphoid aggregates in the meninges is described. [50] B cell-depleting therapies have significantly contributed to modifying the natural history of MS. [56,57]
The importance of regulatory molecules and their associated receptors in peripheral T-cell tolerance, T-cell function, and their activation pathways in MS have been extensively outlined in recent years. [58] What regarding the relation between the adaptative immune response and PIRA? High-throughput T cell receptor β-chain variable gene (TRBV) sequencing (-seq) of genomic (g)DNA in SPMS white matter demyelinating lesions revealed an important sharing of clonally expanded T cells with identical TRBV sequence (clonotypes) across MS lesions independently of their proximity or inflammatory activity, alongside a clonal expansion inside the brain or specific brain homing of CD4+ but not CD8+ T cells. [59] Greater immune cell accumulation was linked to bigger subpial lesion regions, and meningeal accumulations of T and B cells, but not myeloid cells, were geographically near subpial cortical lesions. In patients with more meningeal inflammation, the percentage of active and mixed active/inactive WM lesions was higher, while the percentage of inactive and remyelinated WM lesions was generally lower. [60]
Fransen et al. found an increased number of T cells clustering in the perivascular space and, in active/inactive MS lesions, a tissue-resident memory phenotype of CD8+ T cells with expression of CD69, CD103, CD44, CD49a, PD-1 and absence of S1P1, with upregulated markers for homing (CXCR6), reactivation (Ki-67), and cytotoxicity (GPR56). [61]
7. Fluid Biomarkers and PIRA Phenomena
CSF biomarkers linked to immune-related pathways predict future impairment and correlate with clinical and imaging outcomes of MS severity. [6,62]
A variety of immunoassays can be used to quantify NfL in both CSF and blood, which is released in the CNS's interstitial space when axonal damage occurs. [63] In addition to a clear correlation with disease activity parameters [64], Abdelhak et al. recently documented the occurrence of NfL elevation in advance of confirmed disability worsening independent of clinical relapses. [65]
A new triggering role of parvalbumin, a calcium homeostasis-regulating protein expressed by particular subsets of fast-spiking GABAergic interneurons, emerged over the last years. In postmortem MS cases, parvalbumin levels in the CSF indicate the loss of cortical neurons and correlate at baseline with the cortical atrophy of specific brain regions that are known to be particularly impacted by cortical disease. [66] Parvalbumin levels in the CSF at the time of diagnosis were proposed as potential prognostic neurodegeneration biomarker of GM damage. A correlation has been found with the volume of the inferior frontal and postcentral gyrus, frontal pole, transverse temporal gyrus, cerebellar cortex and with higher atrophy in the right thalamus, pericalcarine cortex, lingual gyrus, and medial frontal gyrus, in parallel with a clinical correlation with cognitive impairment, physical disability, and fatigue. [67]
Cross et al. research aimed to identify markers of nonrelapsing progressive biology disentangling them from markers of acute inflammatory relapse biology. Glial fibrillary acidic protein (GFAP), a protein highly expressed by astrocytes, levels correlated with SEL count, a greater proportion of T2 lesion volume from SELs, and lower T1-weighted intensity within SELs but not with acute inflammatory measures, and neurofilament heavy chain was correlated with SEL count and lower T1-weighted intensity within SELs. [68] Differently from serum levels of NfL, serum GFAP concentration does not typically elevate during acute inflammation but reflects accelerated GM brain volume loss and can be considered a prognostic biomarker for future PIRA. [69]
8. Therapies and PIRA
Determining how DMTs affect PIRA can help unravel the complexity of silent progression pathways. A decrease in relapse activity and, thus, RAW, as well as a reduction of the formation of new MRI lesions, which may eventually reduce the risk of PIRA, were the main explanations for therapeutic benefit in trials investigating PIRA. [9,13,72] PIRA events can be unmasked during treatment with high-efficacy (HE) DMTs, which completely reduce clinical and radiological disease activity. [73] The time interval between disease onset and the first DMT start is a well-recognized strong predictor of disability accumulation, independent of relapse activity, over the long term. [74] A delayed DMT initiation [15] and a shorter treatment exposure [75] are associated with a higher risk of both PIRA and RAW events.
Compartmentalized inflammation in the CNS, reflected by the presence of PRLs, seems to be stubbornly resistant to DMTs currently in use. [35,76] The different pathways of action of DMTs used in clinical practice, however, may have a molecular and biological, and hence clinical, impact on the mechanisms associated with "silent progression," as some data in the literature has demonstrated. Considering a clinical perspective, results of a study from the database of the Italian Multiple Sclerosis Register [77] demonstrated that both ocrelizumab and natalizumab strongly suppress RAW events and have a similar impact on PIRA in naïve RRMS patients. [78] PIRA was never observed in pediatric-onset MS and was reported in a small percentage of adult-onset MS patients during the follow-up (40.0 ± 25.9 months) of a natalizumab-treated cohort in a recent study. [79] Comparing the efficacy of natalizumab with platform therapies in SPMS, the proportion of patients who developed PIRA at 48 months was significantly higher in the interferon beta-1b group compared to natalizumab-treated cohort (72.4% versus 40.2%, p = 0.01). [80] The molecular effectiveness of anti-CD20 monoclonal antibodies [81] is also reflected in CSF measures of lymphocyte biology (sTACI, sCD27, sBCMA) and chemokines (CXCL10, CXCL12). [68]
The effect of DMTs on neurodegenerative mechanisms can be unraveled by observing variations in brain volume loss and CAL, epiphenomena of persistent compartmentalized inflammation, and neurodegeneration. [82] Compared to fingolimod, ocrelizumab-treated patients in this study experienced fewer new white matter lesions and lower deep GM volume loss, lower global cortical thickness change, and reduced cortical thinning/volume loss in several regions of interest. Using a new post-processing MRI method called T1/T2 ratio of iron rim lesions, Eisele et al. found that patients on fingolimod, dimethyl fumarate, and ocrelizumab had a significantly lower 2-year follow-up rate than those not on DMTs. Their findings suggest that DMTs might have a slightly beneficial long-term effect on smoldering MS lesions. [83] Ocrelizumab reduced longitudinal measures of chronic lesion activity such as T1 hypointense lesion volume accumulation and mean normalized T1 signal intensity decrease both in slowly expanding/evolving and non-slowly expanding/evolving lesions. [84] Despite predicted effects on inflammatory networks related to microglia in CAL, anti-CD20 monoclonal antibodies failed to fully resolve paramagnetic rim lesions after a 2-year MRI follow-up, probably due to the paucity of B-cells in CAL, ineffective transit of anti-CD20 antibodies across the blood-brain barrier, and limited tissue turnover of B-cells. [85] Furthermore, exploring the effect of DMTs on SELs, Preziosa et al. recently found that the effects of natalizumab and fingolimod on SEL occurrence were modest, with natalizumab being slightly more effective. [86]
Analyzing the comparison of patients started on low-to-moderate efficacy therapies (LM-DMT) and those who received first-line HE-DMT in the Swedish MS Register, Spelman et al. found a significantly higher unadjusted rate of CDW events in the LM-efficacy group. The HE-DMT group also had a lower rate of PIRA, although the difference between the two groups on this measure was not statistically significant. [87] The relative contributions of PIRA and RAW to the evolution of EDSS in individuals diagnosed and treated at different times were examined in a recent Italian research of 1,405 patients followed for an average of 14.3 years, emphasizing a deceleration of MS course throughout the years determined not only by fewer RAW events, but also by a reduction in PIRA, as results of DMT use. In patients diagnosed in 1980-1996 and 1997-2008, PIRA's average contribution to the overall advancement of EDSS was already significant; however, in patients diagnosed in subsequent years, this contribution substantially increased. [88] Considering also the most recently approved DMTs, ofatumumab markedly reduced the risk of PIRA in early RMS patients in the pooled ASCLEPIOS population, compared to teriflunomide. [89] Subjects treated with cladribine exhibited an effect on neurodegenerative MRI biomarkers, with a markedly reduced annualized brain atrophy [90] and GM volume loss [91] rates compared to placebo. [92] The references mentioned in this paragraph are better characterized in Table 1.
BTK inhibitors (BTKi) represent a potentially effective therapeutic strategy to prevent PIRA and smoldering MS. [93] BTK activity is essential for B cell maturation and function as well as for B cell and myeloid cell intracellular signaling, including microglial pathways. Consequently, BTK inhibition causes peripheral B cell regulation, maturation, proliferation, and autoantibody and cytokine production, along with a decrease in macrophage and microglial activity in the central nervous system. [94,95] Tolebrutinib, which demonstrated greater CNS penetration compared to other BTKi, reduced the volume of SELs [96], modulated microglial genes and had non-cell autonomous impacts on neurons and astrocytes during stimulation of fragment crystallizable gamma receptors. [97]
It is evident from the molecular pathways previously revealed and currently being investigated that "thinking outside the box" is necessary to find new treatments for MS. [98] Novel therapies that improve the function of oligodendrocytes and other glial cells may be useful to prevent neurodegeneration and repair structural damage, in addition to altering or enhancing several metabolic pathways.
9. Conclusions and Future Perspectives
Neurologists and researchers are constantly trying to solve the pathologic puzzle of PIRA, which is essential to understanding and tackling neurodegeneration in MS [99]. Some research grounds are still to be explored. Abnormalities in the glymphatic system, a CNS ‘waste clearance’ system, have been described in several neurodegenerative conditions. Impaired glymphatic function in MS was associated with measures of neurodegeneration and demyelination, in a proposed bidirectional relation of MS-related inflammation affecting the glymphatic system, and glymphatic dysfunction exacerbating MS symptoms [100,101]. A solution to halting non-relapse-related progression in MS patients could lie in understanding how aging affects immune and brain cell activity [102]. Further studies are needed in these directions [103,104].
In light of the first insights into the new diagnostic criteria for MS proposed at ECTRIMS 2024 [105], disease biomarkers play the main role in defining and estimating the biological damage, especially for radiologically isolated syndrome. A radical change in the approach to the disease is also necessary in clinical terms. Neurologists should concentrate on identifying subtle neurological impairments in cognitive and physical functioning, expanding the limitations of EDSS. Management focused on raising knowledge of the disease progression, which starts at the time of MS diagnosis, and how progressive neurodegeneration phenomena are linked to a disease course only apparently stable should also be a priority. Outcomes in clinical trials should be improved by including the presence of PIRA-related biological correlations, for example, the evolution/resolution of PRLs. Assessing meningeal inflammation, as indicated by various analytes, whenever feasible, may suggest the possibility of therapeutic benefit [76]. Disease registries should be expanded to include variables derived from the study of more recent metabolic patterns or should be more easily linked to genetic and histopathological databases [106].
In conclusion, novel perspectives on the biology and progression of MS have helped to promote and confirm the idea of a smouldering MS, caused by an inextricable tangle of acute peripheral inflammation, chronic neuroinflammation, and neurodegeneration mechanisms. A "biological profiling" of the patient will be necessary to be implemented in parallel with the increasing abolition of classic MS disease phenotypes.
Author Contributions
Conceptualization, writing—original draft preparation, writing—review and editing: T.G. and P.I.; supervision: P.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
NA.
Informed Consent Statement
NA.
Data Availability Statement
NA.
Acknowledgments
NA.
Conflicts of Interest
The authors report no conflicts of interest with respect to the contents of the current review, but note that the authors have received advisory board, membership, speakers honoraria, travel support, research grants, consulting fees, or clinical trial support from the manufacturers of those drugs, including Actelion, Allergan, Almirall, Alexion, Bayer Schering, Biogen, Celgene, Excemed, Genzyme, Forward Pharma, Ipsen, Medday, Merck, Mylan, Novartis, Sanofi, Roche, Teva, and their local affiliates.
Abbreviations Not Included in the Main Text
CASP, cysteine-aspartic acid protease; CHRM5, Cholinergic Receptor Muscarinic 5; CXCR, C-X-C chemokine receptor; CORO1A, Coronin 1A; FTH1, Ferritin heavy chain; FTL, ferritin light chain; GPR, Adhesion G-protein coupled receptor G1; GRB2, growth factor receptor-bound protein 2; MAG, myelin Associated Glycoprotein; HAMP, Hepcidin Antimicrobial Peptide; HMOX1, Heme Oxygenase 1; IRF8, Interferon regulatory factor 8; MAC, membrane attack complex; MHC, Major Histocompatibility Complex; NCKAP1L, NCK Associated Protein 1 Like; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; RIPK3, receptor-interacting serine/threonine-protein kinase 3; SOD1, superoxide dismutase type 1; TCIRG1, T cell immune regulator 1; TMEM119, transmembrane protein 119; JAK, Janus kinase.
References
- Jakimovski D, Bittner S, Zivadinov R, et al. Multiple sclerosis. Lancet. 2024;403(10422):183-202.
- Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
- Comi G, Dalla Costa G, Moiola L. Newly approved agents for relapsing remitting multiple sclerosis: how real-world evidence compares with randomized clinical trials?. Expert Rev Neurother. 2021;21(1):21-34. [CrossRef]
- Lublin FD, Häring DA, Ganjgahi H, et al. How patients with multiple sclerosis acquire disability. Brain. 2022;145(9):3147-3161. [CrossRef]
- Kuhlmann T, Moccia M, Coetzee T, et al. Multiple sclerosis progression: time for a new mechanism-driven framework. Lancet Neurol. 2023;22(1):78-88. [CrossRef]
- Calabrese M, Preziosa P, Scalfari A, et al. Determinants and Biomarkers of Progression Independent of Relapses in Multiple Sclerosis. Ann Neurol. 2024;96(1):1-20. [CrossRef]
- Sorensen PS, Sellebjerg F, Hartung HP, Montalban X, Comi G, Tintoré M. The apparently milder course of multiple sclerosis: changes in the diagnostic criteria, therapy and natural history. Brain. 2020;143(9):2637-2652. [CrossRef]
- Portaccio, E.; Magyari, M.; Havrdova, E.K.; Ruet, A.; Brochet, B.; Scalfari, A.; Di Filippo, M.; Tur, C.; Montalban, X.; Amato, M.P. Multiple sclerosis: Emerging epidemiological trends and redefining the clinical course. Lancet Reg. Health Eur. 2024, 44, 100977. [CrossRef]
- Kappos L, Wolinsky JS, Giovannoni G, et al. Contribution of relapse-independent progression vs relapse-associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA Neurol. 2020;77(9):1132-1140. [CrossRef]
- Giovannoni G, Popescu V, Wuerfel J, et al. Smouldering multiple sclerosis: the ‘real MS’. Ther Adv Neurol Disord. 2022;15:17562864211066751.
- Hauser SL, Cree BAC. Treatment of multiple sclerosis: a review. Am J Med. 2020;133(12):1380-1390.e2. [CrossRef]
- Müller J, Cagol A, Lorscheider J, et al. Harmonizing definitions for progression independent of relapse activity in multiple sclerosis: a systematic review. JAMA Neurol. 2023;80(11):1232-1245.
- Ciccarelli O, Barkhof F, Calabrese M, et al. Using the Progression Independent of Relapse Activity Framework to Unveil the Pathobiological Foundations of Multiple Sclerosis. Neurology. 2024;103(1):e209444. [CrossRef]
- Sharrad D, Chugh P, Slee M, Bacchi S. Defining progression independent of relapse activity (PIRA) in adult patients with relapsing multiple sclerosis: A systematic review. Mult Scler Relat Disord. 2023;78:104899. [CrossRef]
- Iaffaldano P, Portaccio E, Lucisano G, et al. Multiple Sclerosis Progression and Relapse Activity in Children [published correction appears in JAMA Neurol. 2024 Jan 1;81(1):88]. JAMA Neurol. 2024;81(1):50-58.
- Portaccio E, Betti M, De Meo E, et al. Progression independent of relapse activity in relapsing multiple sclerosis: impact and relationship with secondary progression [published correction appears in J Neurol. 2024 Oct;271(10):7066-7068. J Neurol. 2024;271(8):5074-5082.
- Simone M, Lucisano G, Guerra T, et al. Disability trajectories by progression independent of relapse activity status differ in pediatric, adult and late-onset multiple sclerosis. J Neurol. 2024;271(10):6782-6790. [CrossRef]
- Prosperini L, Ruggieri S, Haggiag S, Tortorella C, Gasperini C. Disability patterns in multiple sclerosis: A meta-analysis on RAW and PIRA in the real-world context. Multiple Sclerosis Journal. 2024;30(10):1309-1321. [CrossRef]
- Tur C, Carbonell-Mirabent P, Cobo-Calvo Á, et al. Association of Early Progression Independent of Relapse Activity With Long-term Disability After a First Demyelinating Event in Multiple Sclerosis. JAMA Neurol. 2023;80(2):151-160. [CrossRef]
- Comi, G., Dalla Costa, G., Stankoff, B. et al. Assessing disease progression and treatment response in progressive multiple sclerosis. Nat Rev Neurol 20, 573–586 (2024). [CrossRef]
- Absinta M, Lassmann H, Trapp BD. Mechanisms underlying progression in multiple sclerosis. Curr Opin Neurol. 2020;33(3):277-285. [CrossRef]
- Monaco S, Nicholas R, Reynolds R, Magliozzi R. Intrathecal inflammation in progressive multiple sclerosis. Int J Mol Sci 2020; 21: 1–11. [CrossRef]
- University of California, San Francisco MS-EPIC Team , Cree BAC, Hollenbach JA, et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann Neurol. 2019;85(5):653-666.
- Scalfari A, Traboulsee A, Oh J, et al. Smouldering-Associated Worsening in Multiple Sclerosis: An International Consensus Statement on Definition, Biology, Clinical Implications, and Future Directions. Ann Neurol. 2024;96(5):826-845.
- Giovannoni, Gavin et al. Smouldering-associated worsening or SAW: the next therapeutic challenge in managing multiple sclerosis. Multiple Sclerosis and Related Disorders, Volume 0, Issue 0, 106194 (2024). [CrossRef]
- Di Filippo M, Gaetani L, Centonze D, et al. Fluid biomarkers in multiple sclerosis: from current to future applications. Lancet Reg Health Eur. 2024;44:101009. Published 2024 Aug 22. [CrossRef]
- Hamzaoui M, Garcia J, Boffa G, et al. Positron Emission Tomography with [18 F]-DPA-714 Unveils a Smoldering Component in Most Multiple Sclerosis Lesions which Drives Disease Progression. Ann Neurol. 2023;94(2):366-383. [CrossRef]
- Absinta, M., Maric, D., Gharagozloo, M. et al. A lymphocyte–microglia–astrocyte axis in chronic active multiple sclerosis. Nature 597, 709–714 (2021).
- Trobisch T, Zulji A, Stevens NA, et al. Cross-regional homeostatic and reactive glial signatures in multiple sclerosis. Acta Neuropathol. 2022;144(5):987-1003. [CrossRef]
- Magliozzi R, Howell OW, Nicholas R, Cruciani C, Castellaro M, Romualdi C, Rossi S, Pitteri M, Benedetti MD, Gajofatto A, et al. Inflammatory intrathecal profiles and cortical damage in multiple sclerosis. Ann Neurol. 2018;83(4):739–55. [CrossRef]
- Mazziotti, V., Crescenzo, F., Turano, E. et al. The contribution of tumor necrosis factor to multiple sclerosis: a possible role in progression independent of relapse?. J Neuroinflammation 21, 209 (2024). [CrossRef]
- Bagnato F, Sati P, Hemond CC, et al. Imaging chronic active lesions in multiple sclerosis: a consensus statement. Brain. 2024;147(9):2913-2933. [CrossRef]
- Ludwig N, Cucinelli S, Hametner S, Muckenthaler MU, Schirmer L. Iron scavenging and myeloid cell polarization. Trends Immunol. 2024;45(8):625-638. [CrossRef]
- Hofmann A, Krajnc N, Dal-Bianco A, et al. Myeloid cell iron uptake pathways and paramagnetic rim formation in multiple sclerosis. Acta Neuropathol. 2023;146(5):707-724. [CrossRef]
- Absinta M, Sati P, Masuzzo F, Nair G, Sethi V, Kolb H, Ohayon J, Wu T, Cortese ICM, Reich DS. Association of chronic active multiple sclerosis lesions with disability in vivo. JAMA Neurol. 2019. [CrossRef]
- Maggi P, Kuhle J, Schädelin S, et al. Chronic White Matter Inflammation and Serum Neurofilament Levels in Multiple Sclerosis. Neurology. 2021;97(6):e543-e553. [CrossRef]
- Wittayer M, Weber CE, Platten M, Schirmer L, Gass A, Eisele P. Spatial distribution of multiple sclerosis iron rim lesions and their impact on disability. Mult Scler Relat Disord. 2022;64:103967. [CrossRef]
- Okar SV, Dieckhaus H, Beck ES, et al. Highly Sensitive 3-Tesla Real Inversion Recovery MRI Detects Leptomeningeal Contrast Enhancement in Chronic Active Multiple Sclerosis. Invest Radiol. 2024;59(3):243-251. [CrossRef]
- Preziosa P, Pagani E, Meani A, et al. Slowly Expanding Lesions Predict 9-Year Multiple Sclerosis Disease Progression. Neurol Neuroimmunol Neuroinflamm. 2022;9(2):e1139. Published 2022 Feb 1. [CrossRef]
- Cagol A, Benkert P, Melie-Garcia L, et al. Association of spinal cord atrophy and brain paramagnetic rim lesions with progression independent of relapse activity in people with MS. Neurology. 2024;102:e207768. [CrossRef]
- Wenzel N, Wittayer M, Weber CE, Platten M, Gass A, Eisele P. Multiple sclerosis iron rim lesions are linked to impaired cervical spinal cord integrity using the T1/T2-weighted ratio. J Neuroimaging. 2023;33:240–246.
- Weber CE, Kramer J, Wittayer M, Gregori J, Randoll S, Weiler F, Heldmann S, Rossmanith C, Platten M, Gass A, et al. Association of iron rim lesions with brain and cervical cord volume in relapsing multiple sclerosis. Eur Radiol. 2021. [CrossRef]
- Magliozzi R, Howell OW, Calabrese M, Reynolds R. Meningeal inflammation as a driver of cortical grey matter pathology and clinical progression in multiple sclerosis. Nat Rev Neurol. 2023;19(8):461-476. [CrossRef]
- Magliozzi R, Howell OW, Durrenberger P, et al. Meningeal inflammation changes the balance of TNF signaling in cortical grey matter in multiple sclerosis. J Neuroinflammation. 2019;16(1):259. Published 2019 Dec 7. [CrossRef]
- Herranz E, Treaba CA, Barletta VT, et al. Characterization of cortico-meningeal translocator protein expression in multiple sclerosis. Brain. 2024;147(7):2566-2578. [CrossRef]
- Tham, M. Iron heterogeneity in early active multiple sclerosis lesions Ann. Neurol. 2021; 89:498-510.
- van den Bosch, A.M.R., van der Poel, M., Fransen, N.L. et al. Profiling of microglia nodules in multiple sclerosis reveals propensity for lesion formation. Nat Commun 15, 1667 (2024). [CrossRef]
- Gallego-Delgado P, James R, Browne E, et al. Neuroinflammation in the normal-appearing white matter (NAWM) of the multiple sclerosis brain causes abnormalities at the nodes of Ranvier. PLoS Biol. 2020;18(12):e3001008. Published 2020 Dec 14. [CrossRef]
- Preziosa P, Pagani E, Meani A, et al. Chronic Active Lesions and Larger Choroid Plexus Explain Cognition and Fatigue in Multiple Sclerosis. Neurol Neuroimmunol Neuroinflamm. 2024;11(2):e200205. [CrossRef]
- Zhan J, Kipp M, Han W, Kaddatz H. Ectopic lymphoid follicles in progressive multiple sclerosis: From patients to animal models. Immunology. 2021;164(3):450-466. [CrossRef]
- Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010; 68: 477–93. [CrossRef]
- Reali C, Magliozzi R, Roncaroli F, Nicholas R, Howell OW, Reynolds R. B cell rich meningeal inflammation associates with increased spinal cord pathology in multiple sclerosis. Brain Pathol. 2020; 30: 779–93. [CrossRef]
- Bell L, Lenhart A, Rosenwald A, Monoranu CM, Berberich-Siebelt F. Lymphoid aggregates in the CNS of progressive multiple sclerosis patients lack regulatory T cells. Front Immunol. 2019; 10: 3090. [CrossRef]
- Cencioni MT, Mattoscio M, Magliozzi R, Bar-Or A, Muraro PA. B cells in multiple sclerosis - from targeted depletion to immune reconstitution therapies. Nat Rev Neurol. 2021;17(7):399-414. [CrossRef]
- Comi G, Bar-Or A, Lassmann H, et al. Role of B Cells in Multiple Sclerosis and Related Disorders. Ann Neurol. 2021;89(1):13-23. [CrossRef]
- Margoni M, Preziosa P, Filippi M, Rocca MA. Anti-CD20 therapies for multiple sclerosis: current status and future perspectives. J Neurol. 2022;269(3):1316-1334. [CrossRef]
- Krajnc N, Bsteh G, Berger T, Mares J, Hartung HP. Monoclonal Antibodies in the Treatment of Relapsing Multiple Sclerosis: an Overview with Emphasis on Pregnancy, Vaccination, and Risk Management. Neurotherapeutics. 2022;19(3):753-773. [CrossRef]
- Afshar B, Khalifehzadeh-Esfahani Z, Seyfizadeh N, Rezaei Danbaran G, Hemmatzadeh M, Mohammadi H. The role of immune regulatory molecules in multiple sclerosis. J Neuroimmunol. 2019;337:577061. [CrossRef]
- Planas R, Metz I, Martin R, Sospedra M. Detailed Characterization of T Cell Receptor Repertoires in Multiple Sclerosis Brain Lesions. Front Immunol. 2018;9:509. Published 2018 Mar 19. [CrossRef]
- Ahmed SM, Fransen NL, Touil H, et al. Accumulation of meningeal lymphocytes correlates with white matter lesion activity in progressive multiple sclerosis. JCI Insight. 2022;7(5):e151683. Published 2022 Mar 8. [CrossRef]
- Fransen NL, Hsiao CC, van der Poel M, et al. Tissue-resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain. 2020;143(6):1714-1730. [CrossRef]
- Kosa P, Barbour C, Varosanec M, Wichman A, Sandford M, Greenwood M, Bielekova B. Molecular models of multiple sclerosis severity identify heterogeneity of pathogenic mechanisms. Nat Commun. 2022;13(1):7670. [CrossRef]
- Gaetani, L., Blennow, K., Calabresi, P. et al. Neurofilament light chain as a biomarker in neurological disorders J Neurol Neurosurg Psychiatry. 2019; 90:870-881.
- Toscano S, Oteri V, Chisari CG, et al. Cerebrospinal fluid neurofilament light chains predicts early disease-activity in Multiple Sclerosis. Mult Scler Relat Disord 2023; 80:105131. [CrossRef]
- Abdelhak A, Benkert P, Schaedelin S, et al. Neurofilament light chain elevation and disability progression in multiple sclerosis. JAMA Neurol 2023; 80: 1317. [CrossRef]
- Magliozzi R, Pitteri M, Ziccardi S, et al. . CSF parvalbumin levels reflect interneuron loss linked with cortical pathology in multiple sclerosis. Ann Clin Transl Neurol. 2021;8(3):534-547. [CrossRef]
- Ziccardi S, Tamanti A, Ruggieri C, et al. CSF Parvalbumin Levels at Multiple Sclerosis Diagnosis Predict Future Worse Cognition, Physical Disability, Fatigue, and Gray Matter Damage. Neurol Neuroimmunol Neuroinflamm. 2024;11(6):e200301. [CrossRef]
- Cross AH, Gelfand JM, Thebault S, et al. Emerging Cerebrospinal Fluid Biomarkers of Disease Activity and Progression in Multiple Sclerosis. JAMA Neurol. 2024;81(4):373–383. [CrossRef]
- Meier S, Willemse EAJ, Schaedelin S, et al. Serum glial fibrillary acidic protein compared with neurofilament light chain as a biomarker for disease progression in multiple sclerosis. JAMA Neurol 2023; 80: 287–297. [CrossRef]
- Pezzini F, Pisani A, Mazziotti V, et al. Intrathecal versus Peripheral Inflammatory Protein Profile in MS Patients at Diagnosis: A Comprehensive Investigation on Serum and CSF. Int J Mol Sci. 2023;24(4):3768. Published 2023 Feb 13. [CrossRef]
- Magliozzi R, Scalfari A, Pisani AI, et al. The CSF Profile Linked to Cortical Damage Predicts Multiple Sclerosis Activity. Ann Neurol. 2020;88(3):562-573.
- Gärtner J, Hauser SL, Bar-Or A, et al. Efficacy and safety of ofatumumab in recently diagnosed, treatment-naive patients with multiple sclerosis: results from ASCLEPIOS I and II. Mult Scler. 2022;28(10):1562-1575. [CrossRef]
- Graf J, Leussink VI, Soncin G, et al. Relapse-independent multiple sclerosis progression under natalizumab. Brain Commun. 2021;3(4):fcab229. Published 2021 Oct 9.
- Iaffaldano P, Lucisano G, Butzkueven H, et al. Early treatment delays long-term disability accrual in RRMS: Results from the BMSD network. Mult Scler. 2021;27(10):1543-1555. [CrossRef]
- Portaccio E, Bellinvia A, Fonderico M, et al. Progression is independent of relapse activity in early multiple sclerosis: a real-life cohort study. Brain. 2022;145(8):2796-2805. [CrossRef]
- Ransohoff RM. Multiple sclerosis: role of meningeal lymphoid aggregates in progression independent of relapse activity. Trends Immunol. 2023;44(4):266-275. [CrossRef]
- Mosconi P, Guerra T, Paletta P, et al. Data monitoring roadmap. The experience of the Italian Multiple Sclerosis and Related Disorders Register. Neurol Sci. 2023;44(11):4001-4011. [CrossRef]
- Iaffaldano P, Lucisano G, Guerra T, et al. A comparison of natalizumab and ocrelizumab on disease progression in multiple sclerosis. Ann Clin Transl Neurol. 2024;11(8):2008-2015. [CrossRef]
- Puthenparampil M, Gaggiola M, Ponzano M, et al. High NEDA and No PIRA in Natalizumab-Treated Patients With Pediatric-Onset Multiple Sclerosis. Neurol Neuroimmunol Neuroinflamm. 2024;11(5):e200303. [CrossRef]
- Chisari CG, Aguglia U, Amato MP, et al. Long-term effectiveness of natalizumab in secondary progressive multiple sclerosis: A propensity-matched study. Neurotherapeutics. 2024;21(4):e00363. [CrossRef]
- de Sèze J, Maillart E, Gueguen A, et al. Anti-CD20 therapies in multiple sclerosis: From pathology to the clinic. Front Immunol. 2023;14:1004795. Published 2023 Mar 23. [CrossRef]
- Bajrami, A., Tamanti, A., Peloso, A. et al. Ocrelizumab reduces cortical and deep grey matter loss compared to the S1P-receptor modulator in multiple sclerosis. J Neurol 271, 2149–2158 (2024). [CrossRef]
- Eisele P, Wittayer M, Weber CE, Platten M, Schirmer L, Gass A. Impact of disease-modifying therapies on evolving tissue damage in iron rim multiple sclerosis lesions. Mult Scler. 2022;28(14):2294-2298. [CrossRef]
- Elliott C, Belachew S, Wolinsky JS, et al. Chronic white matter lesion activity predicts clinical progression in primary progressive multiple sclerosis. Brain. 2019;142(9):2787-2799. [CrossRef]
- Maggi P, Bulcke CV, Pedrini E, et al. B cell depletion therapy does not resolve chronic active multiple sclerosis lesions. EBioMedicine. 2023;94:104701. [CrossRef]
- Preziosa P, Pagani E, Moiola L, Rodegher M, Filippi M, Rocca MA. Occurrence and microstructural features of slowly expanding lesions on fingolimod or natalizumab treatment in multiple sclerosis. Mult Scler. 2021;27(10):1520-1532. [CrossRef]
- Spelman T, Glaser A, Hilert J. Immediate high-efficacy treatment in multiple sclerosis is associated with long-term reduction in progression independent of relapse activity (PIRA) compared to low-moderate efficacy treatment – a Swedish MS Registry study. ECTRIMS 2024. P842/178. Mult Scler J. 2024; 30:(3S):651.
- Montobbio N, Cordioli C, Signori A, Bovis F, Capra R, Sormani MP. Relapse-Associated and Relapse-Independent Contribution to Overall Expanded Disability Status Scale Progression in Multiple Sclerosis Patients Diagnosed in Different Eras. Ann Neurol. Published online October 9, 2024. [CrossRef]
- Kappos, L., Montalban, X., et al. (2021). Ofatumumab reduces disability progression independent of relapse activity in patients with relapsing multiple sclerosis (2299). Neurology, 96(15_supplement), 2299. [CrossRef]
- De Stefano N, Giorgio A, Battaglini M, De Leucio A, Hicking C, Dangond F, et al. Reduced brain atrophy rates are associated with lower risk of disability progression in patients with relapsing multiple sclerosis treated with cladribine tablets. Mult Scler. 2018;24:222–226. [CrossRef]
- Cortese R, Battaglini M, Sormani MP, Luchetti L, Gentile G, Inderyas M, et al. Reduction in grey matter atrophy in patients with relapsing multiple sclerosis following treatment with cladribine tablets. Eur J Neurol. 2023;30:179–186. [CrossRef]
- Cortese R, Testa G, Assogna F, De Stefano N. Magnetic Resonance Imaging Evidence Supporting the Efficacy of Cladribine Tablets in the Treatment of Relapsing-Remitting Multiple Sclerosis. CNS Drugs. 2024;38(4):267-279. [CrossRef]
- Geladaris A, Torke S, Weber MS. Bruton’s Tyrosine Kinase Inhibitors in Multiple Sclerosis: Pioneering the Path Towards Treatment of Progression? CNS Drugs. 2022; 36(10): 1019–1030.
- García-Merino A. Bruton’s Tyrosine Kinase Inhibitors: A New Generation of Promising Agents for Multiple Sclerosis Therapy. Cells. 2021; 10(10): 2560. [CrossRef]
- Niedziela N, Kalinowska A, Kułakowska A, et al. Clinical and therapeutic challenges of smouldering multiple sclerosis. Neurol Neurochir Pol. 2024;58(3):245-255. [CrossRef]
- Zurmati BM, Khan J, Reich DS, et al. Tolebrutinib Phase 2b Study Group. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: a phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021; 20(9): 729–738. [CrossRef]
- Gruber R, Blazier A, Lee L, et al. Evaluating the Effect of BTK Inhibitor Tolebrutinib in Human Tri-culture (P1-1.Virtual). Neurology. 2022; 98(18_supplement). [CrossRef]
- Bierhansl L, Hartung HP, Aktas O, Ruck T, Roden M, Meuth SG. Thinking outside the box: non-canonical targets in multiple sclerosis. Nat Rev Drug Discov. 2022;21(8):578-600. [CrossRef]
- Tur C, Rocca MA. Progression Independent of Relapse Activity in Multiple Sclerosis: Closer to Solving the Pathologic Puzzle. Neurology. 2024;102(1):e207936.
- Carotenuto A, Cacciaguerra L, Pagani E, Preziosa P, Filippi M, Rocca MA. Glymphatic system impairment in multiple sclerosis: relation with brain damage and disability. Brain. 2022;145(8):2785-2795; [CrossRef]
- Alghanimy A, Work LM, Holmes WM. The glymphatic system and multiple sclerosis: An evolving connection. Mult Scler Relat Disord. 2024;83:105456. [CrossRef]
- Graves JS, Krysko KM, Hua LH, Absinta M, Franklin RJM, Segal BM. Ageing and multiple sclerosis. Lancet Neurol. 2023;22(1):66-77.
- Pukoli D, Vécsei L. Smouldering Lesion in MS: Microglia, Lymphocytes and Pathobiochemical Mechanisms. Int J Mol Sci. 2023;24(16):12631. Published 2023 Aug 10. [CrossRef]
- Yong VW. Microglia in multiple sclerosis: Protectors turn destroyers. Neuron. 2022;110(21):3534-3548. [CrossRef]
- 2024; 105. European Committee for Treatment and Research in Multiple Sclerosis, Montalban X "2024 revisions of the McDonald criteria" ECTRIMS 2024.
- Trojano M, Kalincik T, Iaffaldano P, Amato MP. Interrogating large multiple sclerosis registries and databases: what information can be gained?. Curr Opin Neurol. 2022;35(3):271-277. [CrossRef]
Figure 1.
Smouldering biology in multiple sclerosis: focus on chronic active lesions, meningeal inflammation and subpial cortical damage.
Figure 1.
Smouldering biology in multiple sclerosis: focus on chronic active lesions, meningeal inflammation and subpial cortical damage.
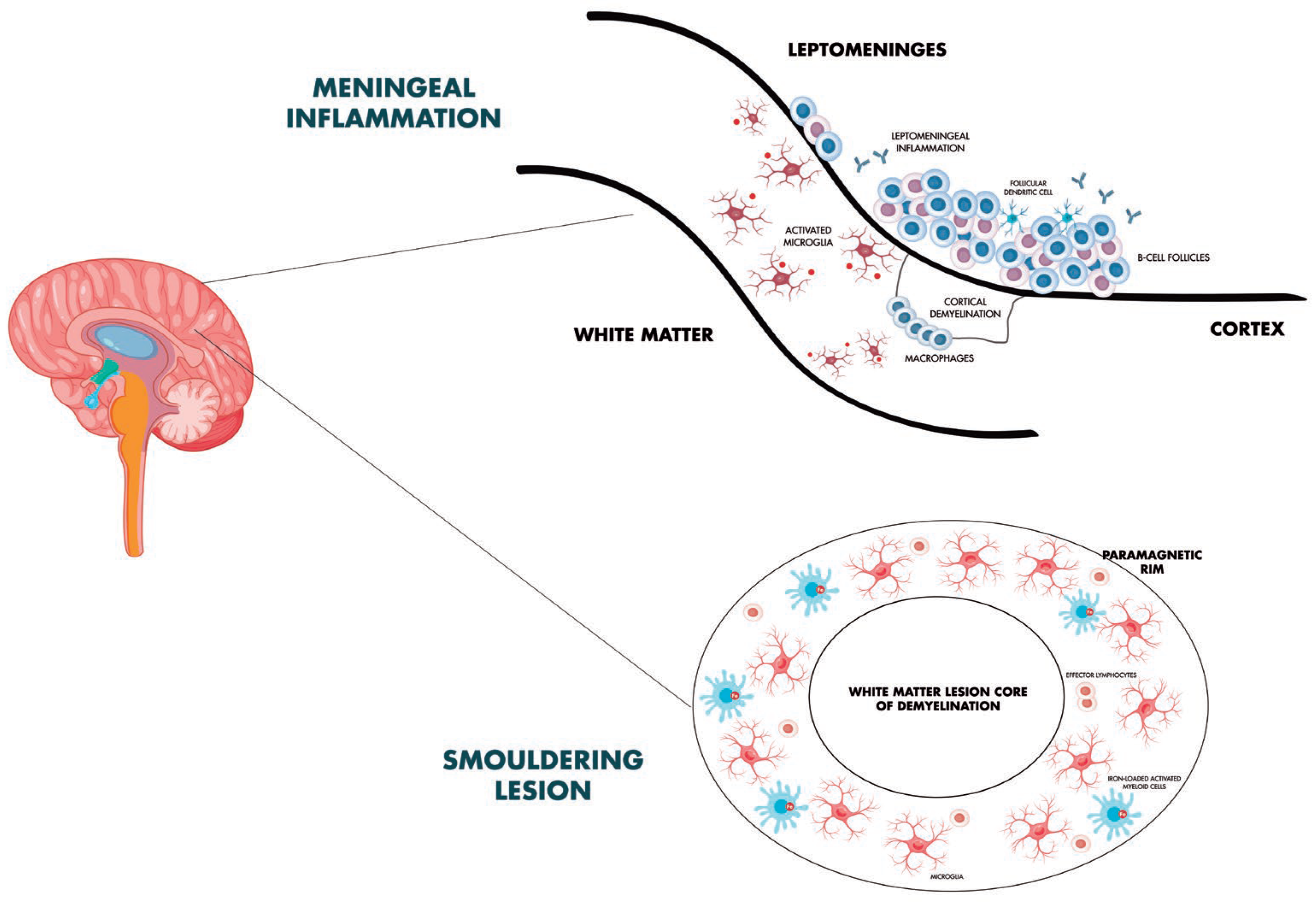

Table 1.
Investigations into how disease-modifying therapies affect PIRA.
| Authors | Study design | Population | Interpretation of results |
|---|---|---|---|
| Graf et al. [73] | Retrospective chart review study | 184 RRMS patients | Patients who are started on natalizumab early in the course of their disease, typically to treat an aggressive clinical presentation, are more likely to experience early confirmed progression independent of relapse activity. |
| Iaffaldano et al. [74] | Retrospective cohort study | 11,871 MS patients (BMSD) |
DMTs should be commenced within 1.2 years from the disease onset to reduce the risk of disability accumulation over the long term. |
| Portaccio et al. [75] | Retrospective cohort study | 5,169 MS patients (CIS, RRMS) (RISM) |
Longer exposure to DMT is associated with a lower risk of both progression independent of relapse activity and relapse-associated worsening events. |
| Iaffaldano et al. [78] | Retrospective cohort study | Total population: 770 MS patients. Matched cohort: 195 patients treated with ocrelizumab, 195 with natalizumab (RISM) |
Natalizumab and ocrelizumab strongly suppress RAW events and, in the short term, the risk of achieving PIRA events, EDSS 4.0 and 6.0 disability milestones is not significantly different. |
| Puthenparampil et al. [79] | Observational retrospective study | Total population: 160 MS patients. Matched cohort: 32 patients pediatric-onset MS and 64 with adult-onset MS |
In naïve patients treated with natalizumab, PIRA was never observed in pediatric-onset MS, while a small percentage of adult-onset MS (12.5%) had PIRA events during the follow-up. |
| Chisari et al. [80] | Retrospective cohort study | Total population: 5,321 SPMS patients. Matched cohort: 421 MS patients treated with natalizumab and 353 with interferon-beta 1b (RISM) |
The proportion of patients who developed PIRA at 48 months is significantly higher in interferon beta-1b group compared to the natalizumab-treated cohort. Patients treated with IFNb-1b are 1.64 times more to likely to develop PIRA |
| Cross et al. [68] | Cohort study assessed data from 2 prospective MS cohorts | Test cohort: 131 MS patients Confirmation cohort: 68 MS patients. | Ocrelizumab reduced CSF measures of acute inflammation, including lymphocyte measures sTACI, sCD27, sBCMA, and chemokine/cytokine measures CXCL13 and CXCL10. Neuroaxonal injury measure NfH and glial measures sTREM2 and YKL-40 resulted modestly reduced. |
| Bajrami et al. [82] | Observational, prospective, longitudinal study | 95 RRMS | Compared to fingolimod, ocrelizumab-treated patients experience fewer new white matter lesions and lower deep grey matter volume loss, lower global cortical thickness change, and reduced cortical thinning/volume loss in several regions of interest. |
| Eisele et al. [83] | Retrospective study | 27 MS patients | Patients on fingolimod, dimethyl fumarate, and ocrelizumab have a considerably lower 2-year follow-up rate of T1/T2 ratio of iron rim lesions. than those not taking DMTs. |
| Elliott et al. [84] | PPMS study population of the ORATORIO trial | ITT population (n = 732); SEL analytical population (n = 555) | Ocrelizumab reduces longitudinal measures of chronic lesion activity such as T1 hypointense lesion volume accumulation and mean normalized T1 signal intensity decrease both in slowly expanding/evolving and non-slowly expanding/evolving lesions. |
| Maggi et al. [85] | Retrospective analysis and imaging, laboratory, and clinical data prospectively collected | 72 MS patients | Despite predicted effects on inflammatory networks related to microglia in CAL, anti-CD20 monoclonal antibodies failed to fully resolve paramagnetic rim lesions after a 2-year MRI follow-up |
| Preziosa et al [86] | Single centre, prospective, longitudinal, open-label, non-randomized cohort study | 52 MS patients | Higher SEL number and volume is observed in the fingolimod vs natalizumab group. Longitudinally, non-SEL MTR increased in both treatment groups. T1 signal intensity decreased in SELs with both treatments and increased in natalizumab non-SELs. |
| Montobbio et al. [88] | Retrospective study | 1,405 MS patients | Across ages, patients diagnosed in more recent times had lower PIRA and RAW than those diagnosed in earlier periods. Patients diagnosed in later years had a significantly higher contribution of PIRA in EDSS progression. |
| Cortese et al. [91] |
MRI data from the CLARITY study | Treatment group: 267 MS patients Placebo group: 265 MS patients |
In the first six months of treatment, patients on cladribine experienced more GM and WM volume loss than those on placebo, most likely as a result of pseudoatrophy. Nonetheless, GM volume loss was considerably less in cladribine-treated patients than in placebo-treated group throughout the course of 6–24 months. |
Abbreviations: BMSD, Big Multiple Sclerosis Data Network; DMT, disease-modifying therapies; GM, gray matter; MS, multiple sclerosis; MTR, magnetization transfer ratio; MRI, magnetic resonance imaging; PIRA, progression independent of relapse activity; RISM, Italian Multiple Sclerosis Register; ITT, intention to treat; RAW, relapse associated worsening; SEL, slowly expanding lesions; WM, white matter.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



