Submitted:
18 December 2024
Posted:
19 December 2024
You are already at the latest version
Abstract
Six novel Microbacterium phages belonging to the Tectiviridae family were isolated using Microbacterium testaceum as a host. Phages MuffinTheCat, Badulia, DesireeRose, Bee17, SCoupsA, and LuzDeMundo were purified from environmental samples by students participating in the SEA-PHAGES program at Alliance University, New York. The phages have linear dsDNA genomes 15,438-15,636bp with 112-120bp inverted terminal repeats. Transmission electron microscopy (TEM) imaging analysis revealed that the six novel phages have six-sided icosahedral double-layered capsids with an internal lipid membrane that occasionally forms protruding nanotubules. Annotation analysis determined that the novel Microbacterium phages all have 32-34 protein-coding genes and no tRNAs. Like other Tectiviridae, the phage genomes are arranged into two segments and include three highly conserved family genes that encode a DNA polymerase, double jelly-roll major capsid protein, and packaging ATPase. Although the novel bacteriophages have 91.6 to 97.5% nucleotide sequence similarity to each other, they are at most 58% similar to previously characterized Tectiviridae genera. Consequently, these novel Microbacterium phages expand the diversity of the Tectiviridae family, and we propose they form the sixth genus, Zetatectivirus.
Keywords:
Microbacterium
; tectivirus
; actinobacteria
; jelly-roll capsid
1. Introduction
Microbacterium spp. are high G+C%, gram-positive rod-shaped aerobes belonging to the order Actinomycetales found throughout the environment in air, food, soil, plants, and water [1,2,3,4]. They are often non-harmful or even beneficial to plants [1,5]. Once thought to be rare in humans, Microbacterium infections have been identified in increasing frequency in patients [6]. Microbacterium spp. have been associated with bacteremia in immunosuppressed patients [7,8,9,10] and were even isolated from the sputum of a patient with cystic fibrosis, leading to speculation that the species could be an emerging pathogen for cystic fibrosis patients [11,12]. Although CRISPR-Cas9 evolved naturally as a defense mechanism in bacteria and archaea, it has been reported that Microbacterium spp. do not use CRISPR-Cas9 systems [13,14]. Other defense mechanisms such as alternative restriction modification systems [15,16] have been identified.
Microbacterium testaceum is an endophytic bacterium that can be found in potato, rice, corn and other agriculturally important crops [17]. Many gram-negative plant pathogens produce N-acylhomoserine lactone (AHL), a quorum sensing signaling compound and strains of M. testaceum have shown early promise as a biocontrol agent with the ability to quench AHL-dependent quorum sensing by plant pathogens [17].
A diverse group of phages infect the Microbacterium spp., with genome sizes from 15 kb to almost 195 kb, typically with high GC content ranging from 50.1% to 71.4%, which is expected given their high GC content host (average 67%) (https://phagesdb.org) [1]. The list of phages that infect Microbacterium spp. is growing quickly; the first Microbacterium spp. phage was discovered in 2007 [18] and at current count, there are 5752 distinct phages that infect Microbacterium spp., most of which have a lytic lifestyle [1]. Of these,11 phages have been isolated using M. testaceum as a host (https://phagesdb.org).
Over 26,000 bacteriophages have been isolated using bacterial hosts belonging to the order Actinomycetales by students participating in the Science Education Alliance-Phage Hunters Advancing Genomics and Evolutionary Science (SEA-PHAGES) program, an inclusive research community (iREC) with programmatic and scientific support provided by Howard Hughes Medical Institute (HHMI), the University of Pittsburgh, and James Madison University [19]. As course-embedded research projects, undergraduate students isolate, name, and characterize novel bacteriophages mostly from soil samples [19,20,21]. Phages infecting bacterial hosts within the phylum Actinobacteria are genetically highly diverse, but the vast majority belong to the class Caudoviricetes having double-stranded DNA genomes, isometric capsids and tails. Of these, most have siphoviral morphology with long, flexible non-contractile tails [1]. Very few podoviral bacteriophages have been isolated which have short, non-contractile tails. Even rarer numbers of tailless phages have been isolated and therefore do not belong to the class Caudoviricetes.
The Tectiviridae family is a diverse group of non-enveloped, tailless DNA phages belonging to the class Tectiliviricetes with both lytic and lysogenic members, including the well-characterized Enterobacteria phage PRD1 [22]. Their genomes are linear, double-stranded DNA approximately 15 kb in length [23]. Hallmarks of the Tectiviridae family genomes include approximately 100 bp inverted terminal repeats (ITR) [24] that flank around 30 open reading frames, including three conserved genes that encode (1) a DNA polymerase, (2) a double jelly-roll major capsid protein, and (3) a packaging ATPase [23,25,26,27]. Tectiviridae replicate their genomes using a protein-primed, strand-displacement mechanism [28] and a family B DNA polymerase [29]. The major capsid protein forms around a lipid membrane acquired from the host during assembly, creating an internal lipid membrane. Upon receptor binding on a new cell, this lipid membrane forms a proteo-lipidic tube, which allows passage of the phage genome into the host cell [30,31]. During egress, an empty capsid forms, into which the phage genome is injected by a FtsK-HerA packaging ATPase integrated at one vertex [31].
Tectiviridae are structurally similar to several eukaryotic and archaeal viruses [32]. PRD1 is very similar to adenoviruses, members of the family Adenoviridae, which are non-enveloped, linear, double-stranded DNA tailless viruses that typically cause respiratory illnesses in vertebrates and belong to the class Tectiliviricetes (https://ictv.global/taxonomy). Adenoviruses are best known for their potential as vectors and were used as SARS-CoV-2 vaccine vehicles [33]. Both viruses have similar major coat proteins and capsid architecture in addition to linear genomes with inverted terminal repeats [34]. Tectiviridae are also very similar to archaeaviruses. Due to the similarity, the Skuldviridae, a new family of archaeaviruses was placed in the class Tectiliviricetes (https://ictv.global/taxonomy) [35]. The similarity of tectiviruses to both eukaryotic and archaeal viruses suggest the viruses may have a common ancestor and tectiviruses may have played a very important role evolutionarily as the precursors of large eukaryotic transposons known as Polintons which are thought to have evolved into most eukaryotic dsDNA viruses [36,37].
Currently, the Tectiviridae family includes five genera and members (and proposed members) that infect both gram-positive and gram-negative bacteria. Tectivirus hosts include bacteria from the phyla Pseudomonatoda (infected by Alpha- and Gammatectivirus), Firmicutes (infected by Betatectivirus), and Actinobacteria (infected by Deltatectivirus and Epsilontectivirus) (https://ictv.global/taxonomy) [26,27]. Most of the phages belonging to the Betatectiviridae, Deltatectiviridae, and Epsilontectiviridae that infect gram-positive hosts are temperate [26,27,38] and presumably have a lysogenic lifestyle.
While lipid membrane-containing phages were once thought to be rare [39,40], advances in isolation and characterization have elucidated the pivotal role of these phages in microbial ecology, playing roles in the food chain and as vehicles for gene transfer [41,42,43]. Yet, in relation to other types of phages, they remain underrepresented, potentially due to the historical use of chloroform in culture-based studies to prevent bacterial contamination [43]. Lipid membrane-containing bacteriophages are sensitive to chloroform; in fact, sensitivity to chloroform is typically used as the first indicator of a lipid membrane. Recent evidence suggests that they are more abundant in the environment than previously believed and understanding them will pave the way for new innovations and applications as well as provide further insight into viral lineages and evolution [22].
This study presents the isolation and characterization of six rare new tailless Tectiviridae members isolated from host M. testaceum, by undergraduate students at Alliance University, NY (formerly known as Nyack College) in the SEA-PHAGES program. These novel tailless phages make up a monophyletic group which we propose represents a new sixth genus of Tectiviridae: Zetatectivirus. Previously, all the Microbacterium phages identified were members of the class Caudoviricetes and these are the first reported tectivirus phages isolated and characterized from Microbacterium spp. With an average genome size of 15.498bp, they are currently the smallest phages known to infect Microbacterium spp. (https://phagesdb.org).
2. Materials and Methods
2.1. Bacterial Strains
All the bacterial strains used in the study were obtained from the Agricultural Research Service (https://nrrl.ncaur.usda.gov/) and grown in peptone-yeast extract-calcium (PYCa) medium supplemented with 0.1% dextrose at 28°C.
2.2. Isolation and Purification of Phages
Soil samples were collected in Nyack, NY, Baldwin, NY, Haskell, NJ, and Seabrook, MD. To isolate the phage, the samples were washed with PYCa media (1.0 g yeast extract, 15 g peptone, 2.5 mL 40% dextrose, and 4.5 mL 1M CaCl2 per liter) supplemented with 0.1% dextrose and filtered through a 0.22 μm filter. The filtered soil extracts were inoculated with mixed cultures including late exponential phase M. testaceum NRRL B-59317 and incubated at 28°C with shaking for 48 hours. After incubation, the cultures were filtered, and the filtrates plated in PYCa top agar lawn with M. testaceum. The plates were incubated for 2 days at 28°C following which isolated phages were picked into phage buffer with glycerol (10 mM Tris-HCl [pH 7.5], 10 mM MgSO4, 1 mM CaCl2, 68 mM NaCl, and 10% glycerol) to plaque purify. Crude lysates were obtained after three rounds of purification as previously described [44,45,46].
2.3. Phage Amplification and Production of High Titer Lysates
Crude phage stocks were diluted and plated on a lawn of the bacterial host to produce plates with near-confluent lysis. Plates were flooded with 8 mL phage buffer, and incubated overnight at 4°C. To purify, the lysates were harvested and filtered through a 0.22 μm filter.
2.4. DNA Sequencing, Annotation and Sequence Analysis
DNA was isolated from a high titer lysate purified by using Promega Wizard DNA extraction kits (http://www.promega.com/) or phenol-chloroform extraction using Phase-Lock Gel (http://www.quantabio.com/) and resuspended in ddH2O. The phage genomes were sequenced at the Pittsburgh Bacteriophage Institute where sequencing libraries were prepared from double-stranded phage genomic DNA using NEB Ultra II FS Kits. These were run on an Illumina MiSeq using 150-cycle v3 Reagent Cartridges yielding 150-base single-end reads representing between 106- and 4572-fold coverage of each genome (Table 1). The raw reads were quality-controlled using Consed version 29 and assembled using Newbler version 2.9. The assemblies were checked for completeness, accuracy, orientation, and genomic termini as previously described [47]. Phage genomes were annotated as described previously [48] using DNA Master (http://cobamide2.bio.pitt.edu), PECAAN (v2021-2024) (https://discover.kbrinsgd.org), Glimmer 3.0 [49], GeneMark 2.5 [50], NCBI BLAST 2.7 and Conserved Domain Database at NCBI [51], Starterator (http://phages.wustl.edu/starterator/) [52], Aragorn version 1.2.38 [53], tRNA-ScanSE 2.0 [54], HHPred (databases: PDB mmCIF70, Pfam-A, and NCBI Conserved Domain databases) [55,56], SOSUI [57], TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) [58], DeepTMHMM [59], DNAbind [60] and Phamerator using database Actinobacteriophage_2422 [61]. EMBOSS was used to verify the terminal repeat sequences initially identified by sequencing [62]. Pairwise average nucleotide identities (ANI) were calculated using the Sequence Demarcation tool [63]. Default parameters were used for all software. All genome sequences are publicly available at phagesdb.org and in GenBank (Table 1).
2.5. Host Range Analysis
High titer lysates were diluted tenfold in phage buffer with glycerol and 3 μL of each dilution were spotted on lawns of Microbacterium spp (M. testaceum NRRL B-59317, M. arborescens NRRL B-59342, M. radiodurans NRRL B-24799, M. foliorum NRRL B-24224, M. saperdae NRRL B-14833). Plates were incubated at 28 oC for 2 days and observed for plaque formation.
2.6. Electron Microscopy
High titer phage lysates were concentrated by centrifugation as described previously [44,45,46]. For negative staining analysis, phage particles were spotted onto formvar and carbon-coated 400 mesh copper grids, rinsed with distilled water and stained with 1% uranyl acetate. Grids were imaged with a JEOL JEM 1400 Transmission Electron Microscope at 100 kV acceleration voltage (JEOL, USA, Ltd, Peabody, MA). Images were captured on an EMSIS Veleta side-mounted 2K × 2K CCD camera (Olympus-SIS, Munich, Germany).
2.7. Phylogenetic Reconstruction
Whole genome comparison was carried out using tectivirus complete genome nucleotide sequences retrieved from NCBI (accession numbers listed in Table 1) and Forthebois was included as an outgroup (MK620900.1). Tectivirus protein sequences (DNA polymerase, major capsid, and DNA packaging ATPase) were retrieved from NCBI Virus [64] omitting those sequences that have not been assigned a genus or classified as a particular protein. A T4 bacteriophage protein was included in each multiple alignment for an outgroup (Table 2).
Within MEGA7 [65], MUSCLE [66] was used to construct multiple alignments of the complete genome nucleotide sequences using the following parameters: gap open penalty -400, gap extend penalty 0, max memory 4095 MB, max iterations 8, UPGMB clustering method, min diag length 24. This alignment was used to generate a UPGMA tree using the following parameters: bootstrap test of phylogeny (500 replicates) [67], amino acid substitution type nucleotide, maximum composite likelihood method, substitutions including transitions and transversions, rate of variation was calculated using a gamma distribution shape parameter = 1, homogeneous pattern among lineages with a complete deletion and including all codon positions. The tree was based on 13,980 positions.
MEGA7 [65] and MUSCLE [66] were used to construct multiple alignments of the DNA polymerase, major capsid, and DNA packaging ATPase protein sequences using the following parameters: gap open penalty -2.9, gap extend penalty 0, hydrophobicity multiplier 1.2, max memory in MB 4095, max iterations 8, clustering method UPGMB, minimum diag length 24. MUSCLE alignments were used to construct phylogenetic trees using the UPGMA method [68] using the following parameters: bootstrap test of phylogeny (500 replicates) [67], amino acid substitution using the Poisson correction method [69], rate of variation was calculated using a gamma distribution shape parameter = 1, complete deletion. The DNA polymerase tree was based on 418 positions, the major capsid tree was based on 276 positions, and DNA packaging ATPase tree was based on 150 sites. All positions containing gaps and missing data were eliminated. Clades were collapsed by genus. Sequences found in each condensed clade can be found in Table 2.
2.8. Genome Comparisons
Comparative genome maps were constructed using Phamerator [61, https://phamerator.org] to display blastn alignments (Figure 3) or an Observable notebook (https://observablehq.com/@cresawn-labs/phamerator-map) to display tblastx alignments (Figure 5).
3. Results
3.1. Isolation and Characterization of Microbacterium testaceum Phages
Microbacterium phages MuffinTheCat, Badulia, DesireeRose, Bee17, SCoupsA, and LuzDeMundo were isolated from environmental samples using host M. testaceum NRRL B-59317 by undergraduate students participating in the SEA-PHAGES program in 2019 and 2021. The phages were isolated from enriched soil samples collected at four discrete locations in the Eastern United States (Table 1). After 48 hrs at 28°C, they form 2-3mm slightly turbid plaques on bacterial lawns of their isolation host.
3.2. Host Range Determination
The host range of the novel Microbacterium phages was determined by spotting ten-fold dilutions of high titer lysate on lawns of several Microbacterium spp including M. arborescens NRRL B-59342, M. radiodurans NRRL B-24799, M. foliorum NRRL B-24224, M. saperdae NRRL B-14833 in addition to the isolation host, M. testaceum NRRL B-59317 (Figure 1). Two sequenced Microbacterium phages with siphovirus-like morphology, Cece isolated using M. radiodurans NRRL B-24799 (Genbank Accession # OP068343) and Paschalis, isolated using M. foliorum NRRL B-24224 (Genbank Accession # MH155873) were used as controls. Host range analyses demonstrate that the novel Microbacterium phages have a narrow host specificity and only infects M. testaceum NRRL B-59317 (Figure 1). Although Cece and Paschalis were unable to infect M. testaceum, both infected their respective isolation hosts as expected. Interestingly, Cece also infects M. arborescens NRRL B-59342 with an EOP of 100%.
3.3. Electron Microscopy
Transmission electron microscopy (TEM) imaging analysis revealed that the six novel Microbacterium phages have six-sided icosahedral double-layered capsids with an average diameter of 60.9nm (n=9) (Figure 2, black arrows). Several of the virion particles have nanotubes that protrude from a capsid vertex (Figure 2D, white arrow). Members of the Tectiviridae family are non-enveloped viruses that have double-layered capsids with an internal lipid membrane and form protruding nanotubules at a low frequency [26]. Taken together, the TEM analysis of the novel Microbacterium phages suggests that they are new members of the Tectiviridae family.
3.4. Genome Sequencing
To genetically characterize these phages, genomic DNA was extracted from high-titer lysates, resuspended in ddH2O, and sequenced at the Pittsburgh Bacteriophage Institute. Sequencing was performed using an Illumina MiSeq Next Generation Sequencer with coverage ranging from 106 to 4572. Visualizing the assembled genomes revealed that the reads of all the genomes ended at a specific base without any circularizing reads. No finishing Sanger sequencing was required. The phage genomes are double-stranded and linear, ranging from 15,438 to 15,636 bp in length. All the genomes have 112-120 bp perfect ITRs (Table 1). Taken together, these data suggest that the phages are likely to have covalently-bound terminal proteins and are similar in length to other members of the Tectiviridae family [70,71]. The GC content of the genomes ranges from 54.6 to 55.1% with an average of 54.9% (Table 1). The phages are unlike any other actinobacteriophage in PhagesDB (www.phagesDB.org) and therefore, they are the founding members of the GE phage cluster.
3.5. Annotation
Annotation analysis determined that the novel Microbacterium phages, MuffinTheCat, Badulia, DesireeRose, Bee17, SCoupsA, and LuzDeMundo all have 32-34 protein coding genes (Table 1, Figure 3). No tRNAs were identified using Aragorn version 1.2.38 [53] and tRNA-ScanSE 3.0 [54]. Similar to other members of the Tectiviridae family, the highly conserved DNA polymerase, DNA packaging ATPase and major capsid genes were identified. In addition, the function of three other genes could be ascertained with a high degree of probability using bioinformatic analysis: a peptidase, phage membrane DNA delivery protein and endolysin. Six transmembrane membrane proteins and a DNA binding protein were also identified. The other 19-21 genes have an undetermined function (Figure 3, Table 2). All the genes in the genomes are homologs and there are no other members of each pham besides the six novel Tectiviridae. Bee17 has two genes not present or closely related to any other known genes, indicated by the numbered white boxes (Figure 3). No integrase gene has been identified and it is unclear if the phages can form lysogens. Other tectiviruses such as Betatectivirus pGIL01 and pGIL16 that form lysogens do not contain integrases and are maintained in the host as extrachromosomal replicating sequences [72].
Figure 3.
Phamerator genome maps of the six novel bacteriophages. The genomes are represented by horizontal marker bars, and the predicted ORFs are shown as numbered colored boxes either above (transcribed rightwards) or below (transcribed leftwards) the genome. Bee17 has two orphans, gp4 and gp34, genes not present in any other genome. The ORFS with predicted functions are labeled. Predicted transmembrane proteins are indicated by the red stars and DNA binding protein by the blue polygon. Purple indicates regions of high blastn similarity between phage genomes, whereas the white areas indicate sequences not shared across genomes. The other colored regions between genomes indicate areas with intermediate similarity.
Figure 3.
Phamerator genome maps of the six novel bacteriophages. The genomes are represented by horizontal marker bars, and the predicted ORFs are shown as numbered colored boxes either above (transcribed rightwards) or below (transcribed leftwards) the genome. Bee17 has two orphans, gp4 and gp34, genes not present in any other genome. The ORFS with predicted functions are labeled. Predicted transmembrane proteins are indicated by the red stars and DNA binding protein by the blue polygon. Purple indicates regions of high blastn similarity between phage genomes, whereas the white areas indicate sequences not shared across genomes. The other colored regions between genomes indicate areas with intermediate similarity.

3.6. Genome Organization
Genome organization of the Tectiviridae family is highly conserved and it has been previously demonstrated that there is a family-wide conservation of three proteins: a major capsid protein, DNA packaging ATPase and DNA polymerase [23,25,26,27]. The genomes of the six novel phages are organized with the first 9-10 genes on the left arm being leftwards-transcribed with the remainder rightwards-transcribed (Table 2, Figure 3). This two-segment genome organization is most similar to the Deltatectivirus (Wheeheim and Forthebois) and Epsilontectivirus (Toil) which are also tectivirus actinobacteriophages [26,71]. The genome of Gammatectivirus (GC1) also has two segments with the first three genes on the left arm being leftwards-transcribed. In these genomes, the highly conserved DNA polymerase is present on the left arm and transcribed leftwards (Figure 4). The right arm contains structural genes and genes involved in cell lysis, such as the major capsid protein and endolysin. Both genes are homologs of the PRD1 major capsid protein and a LysM-like endolysin, respectively. Also present on the right arm is a homology of PRD1 DNA delivery protein, which assists with delivery of DNA to the host [23]. The similarity of the genome organization to the Deltatectivirus and Epsilontectivirus along with the presence of several homologs of the well-studied Alphatectivirus PRD1 supports the claim that the novel phages are tectiviruses.
3.7. Nucleotide Sequence Similarity
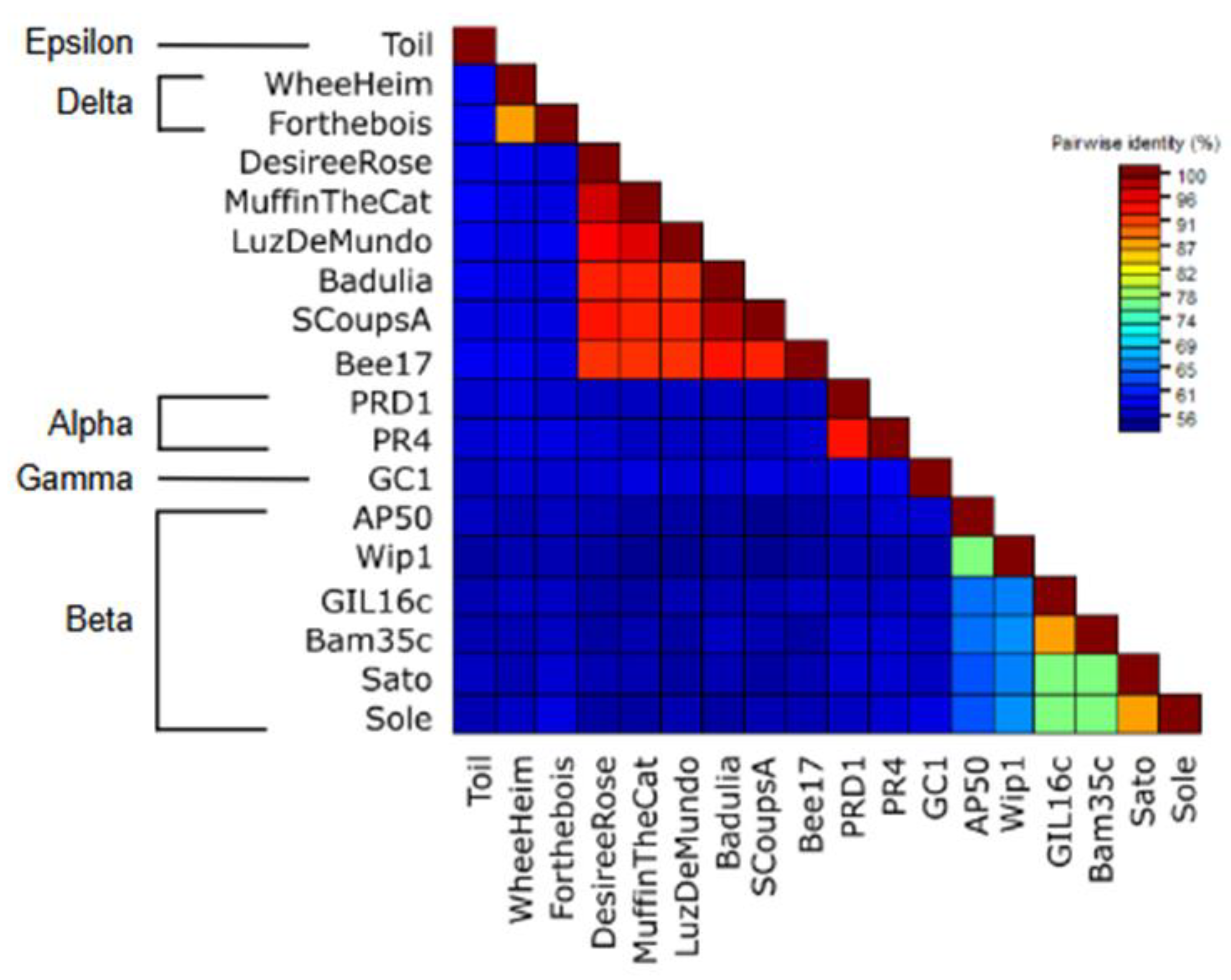
Whole genome comparisons using average nucleotide identity (ANI) analysis is standard in species classification. This method is used by NCBI to evaluate the taxonomy of genomes submitted to GenBank and to determine relatedness between species [73]. Similarly, ANI can be used to determine virus taxon demarcation with a high degree of certainty [74]. ANI calculations show that the six novel bacteriophages have 91.6 to 97.5% nucleotide sequence similarity to each other (Figure 5). They are at most 58% similar to species within each of the current Tectiviridae genera. Therefore, the novel bacteriophages are most similar to each other and distinct from the other tectiviruses. This meets the criteria of these phages being a new species and also belonging to a new genus separate from the known tectiviruses.
Figure 5.
The average nucleotide identities calculated for the six novel Microbacterium phages and representative species from each genus.
Figure 5.
The average nucleotide identities calculated for the six novel Microbacterium phages and representative species from each genus.
3.8. Sequence Analysis and Phylogenetic Reconstruction
To determine the evolutionary history of the six novel phages, the full nucleotide genome sequences of Badulia, SCoupsA, Bee17, DesireeRose, LuzDeMundo, and MuffinTheCat were aligned and compared using phylogenetic reconstruction. Although these phages are closely related with over 90% pairwise nucleotide identity (Figure 5), they formed with high confidence two distinct clades; clade 1 contained DesireeRose, LuzDeMundo, and MuffinTheCat, while clade 2 contained Bee17, Badulia, and SCoupsA (Figure 6).
We further compared each of the three conserved proteins (DNA polymerase, major capsid, and packaging ATPase) of the six novel phages Badulia, SCoupsA, Bee17, DesireeRose, LuzDeMundo, and MuffinTheCat, to previously characterized Tectiviridae, using multiple alignment of amino acid sequences (see Table 1 and Table 3 for accession numbers) and phylogenetic reconstruction (Figure 7A-C). Each of the three trees showed clustering of previously characterized tectiviruses into their expected genera (Alpha-, Beta-, Delta-, Gamma-, and Epsilontectivirus). The six novel phages, clustered together in a distinct clade for each of the three proteins. In each tree, this clade grouped most closely with Delta- and Epsilontectivirus which were also isolated from Actinobacteria species. As known clades were retained and bootstrap values were high, we have high confidence in the phylogenetic reconstruction. There is a clear delineation between the novel phages and the previously characterized genera, which, combined with the ANI data, suggests this clade should be considered a new genus.
4. Conclusions
This work demonstrates that the six novel Microbacterium testaceum phages MuffinTheCat, Badulia, DesireeRose, Bee17, SCoupsA, and LuzDeMundo are new members of the Tectiviridae family. Despite the lack of sequence similarity with other tectiviruses, the novel phages are similar in genome size and organization. The morphological and genomic characterization show that they are distinct from phages in the five previously described genera and therefore, merits the formation of a sixth genera, Zetatectivirus. This expands not only the Tectivirdae family, but also the diversity of actinobacteriophages.
Author Contributions
Conceptualization, J.W.; methodology, J.W. and H.B.; software, M.B. and S.C.; formal analysis, J.W. and H.B.; investigation, J.W., H.B., A.D., M.B., S.C. and E.G.; resources J.W., H.B., A.D., M.B., S.C. and E.G.; data curation, J.W., H.B., M.B, and S.C.; writing—original draft preparation, J.W. and H.B.; writing—review and editing, J.W., H.B. and E.G.; visualization, J.W., H.B., M.B. and S.C.; supervision, J.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors wish to thank the students at Alliance University who participated in the SEA-PHAGES program especially Marguerite Slavin, Dan Hennessy, Desiree Vargas, Saidy Lopez and Darcy Reimer for the isolation and initial characterization of the novel Microbacterium phages. The authors also wish to thank Mariana Pereira Moraes and Maridalia Herrera for lab support. TEM imaging was performed by Juan Jimenez, Research Specialist and Leona Cohen-Gould, Core Director at the Electron Microscopy & Histology services of the Weill Cornell Medicine Microscopy & Image Analysis Core where the equipment was purchased with funds from a NIH Shared Instrumentation Grant (S10RR027699) for Shared Resources. We are also grateful to John Dennehy for thoughtful comments and feedback on the manuscript. Lastly, this work would not be possible without the support of the Howard Hughes Medical Institute SEA-PHAGES program, Deborah Jacobs-Sera, Daniel R. Russell, Rebecca Garlena and Graham Hatfull of the University of Pittsburgh.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jacobs-Sera, D.; Abad, L.A.; Alvey, R.M.; Anders, K.R.; Aull, H.G.; Bhalla, S.S.; Blumer, L.S.; Bollivar, D.W.; Bonilla, J.A.; Butela, K.A.; et al. Genomic diversity of bacteriophages infecting Microbacterium spp. PLoS ONE 2020, 15, e0234636. [Google Scholar] [CrossRef] [PubMed]
- Zlamala, C.; Schumann, P.; Kämpfer, P.; Valens, M.; Rosselló-Mora, R.; Lubitz, W.; Busse, H.J. Microbacterium aerolatum sp. nov., isolated from the air in the 'Virgilkapelle' in Vienna. Int J Syst Evol Microbiol 2002, 52 Pt 4, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Lott, T.T.; Martin, N.H.; Dumpler, J.; Wiedmann, M.; Moraru, C.I. Microbacterium represents an emerging microorganism of concern in microfiltered extended shelf-life milk products. J Dairy Sci 2023, 106, 8434–8448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xi, L.; Ruan, J.; Huang, Y. Microbacterium marinum sp. nov., isolated from deep-sea water. Syst Appl Microbiol 2012, 35, 81–85. [Google Scholar] [CrossRef]
- Sun, W.; Xiong, Z.; Chu, L.; Li, W.; Soares, M.A.; White, J.F.; Li, H. Bacterial communities of three plant species from Pb-Zn contaminated sites and plant-growth promotional benefits of endophytic Microbacterium sp. (strain BXGe71). J Hazard Mater 2019, 370, 225–231. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Woo, G.K.; Yuen, K.Y. Catheter-related Microbacterium bacteremia identified by 16S rRNA gene sequencing. J Clin Microbiol 2002, 40, 2681–2685. [Google Scholar] [CrossRef]
- Laffineur, K.; Avesani, V.; Cornu, G.; Charlier, J.; Janssens, M.; Wauters, G.; Delmée, M. Bacteremia due to a novel Microbacterium species in a patient with leukemia and description of Microbacterium paraoxydans sp. nov. J Clin Microbiol 2003, 41, 2242–2246. [Google Scholar] [CrossRef]
- Amano, J.; Hase, R.; Otsuka, Y.; Tsuchimochi, T.; Noguchi, Y.; Igarashi, S. Catheter-related bloodstream infection by Microbacterium paraoxydans in a pediatric patient with B-cell precursor acute lymphocytic leukemia: A case report and review of literature on Microbacterium bacteremia. J Infect Chemother 2019, 25, 806–810. [Google Scholar] [CrossRef]
- Chorost, M.S.; Smith, N.C.; Hutter, J.N.; Ong, A.C.; Stam, J.A.; McGann, P.T.; Hinkle, M.K.; Schaecher, K.E.; Kamau, E. Bacteraemia due to. JMM Case Rep 2018, 5, e005169. [Google Scholar] [CrossRef]
- Yusuf, E.; Wybo, I.; François, K.; Pipeleers, L.; Echahidi, F.; Covens, L.; Piérard, D. Microbacterium spp. as a cause of peritoneal-dialysis-related peritonitis in two patients. J Microbiol Immunol Infect 2016, 49, 464–465. [Google Scholar] [CrossRef]
- Sharma, P.; Diene, S.M.; Gimenez, G.; Robert, C.; Rolain, J.M. Genome sequence of Microbacterium yannicii, a bacterium isolated from a cystic fibrosis patient. J Bacteriol 2012, 194, 4785. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Diene, S.M.; Thibeaut, S.; Bittar, F.; Roux, V.; Gomez, C.; Reynaud-Gaubert, M.; Rolain, J.M. Phenotypic and genotypic properties of Microbacterium yannicii, a recently described multidrug resistant bacterium isolated from a lung transplanted patient with cystic fibrosis in France. BMC Microbiol 2013, 13, 97. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 2007, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.A.; Garlena, R.A.; Hatfull, G.F. Complete Genome Sequence of Microbacterium foliorum NRRL B-24224, a Host for Bacteriophage Discovery. Microbiol Resour Announc 2019, 8. [Google Scholar] [CrossRef]
- Striebel, H.M.; Schmitz, G.G.; Kaluza, K.; Jarsch, M.; Kessler, C. MamI, a novel class-II restriction endonuclease from Microbacterium ammoniaphilum recognizing 5'-GATNN decreases NNATC-3'. Gene 1990, 91, 95–100. [Google Scholar] [CrossRef]
- Striebel, H.M.; Seeber, S.; Jarsch, M.; Kessler, C. Cloning and characterization of the MamI restriction-modification system from Microbacterium ammoniaphilum in Escherichia coli. Gene 1996, 172, 41–46. [Google Scholar] [CrossRef]
- Morohoshi, T.; Wang, W.Z.; Someya, N.; Ikeda, T. Genome sequence of Microbacterium testaceum StLB037, an N-acylhomoserine lactone-degrading bacterium isolated from potato leaves. J Bacteriol 2011, 193, 2072–2073. [Google Scholar] [CrossRef]
- Akimkina, T.; Venien-Bryan, C.; Hodgkin, J. Isolation, characterization and complete nucleotide sequence of a novel temperate bacteriophage Min1, isolated from the nematode pathogen Microbacterium nematophilum. Res Microbiol 2007, 158, 582–590. [Google Scholar] [CrossRef]
- Hanauer, D.I.; Graham, M.J.; Betancur, L.; Bobrownicki, A.; Cresawn, S.G.; Garlena, R.A.; Jacobs-Sera, D.; Kaufmann, N.; Pope, W.H.; Russell, D.A.; et al. An inclusive Research Education Community (iREC): Impact of the SEA-PHAGES program on research outcomes and student learning. Proc Natl Acad Sci U S A 2017, 114, 13531–13536. [Google Scholar] [CrossRef]
- Jordan, T.C.; Burnett, S.H.; Carson, S.; Caruso, S.M.; Clase, K.; DeJong, R.J.; Dennehy, J.J.; Denver, D.R.; Dunbar, D.; Elgin, S.C.; et al. A broadly implementable research course in phage discovery and genomics for first-year undergraduate students. mBio 2014, 5, e01051–e01013. [Google Scholar] [CrossRef]
- LaCourse, W.R.; Sutphin, K.L.; Ott, L.E.; Maton, K.I.; McDermott, P.; Bieberich, C.; Farabaugh, P.; Rous, P. Think 500, not 50! A scalable approach to student success in STEM. BMC Proc 2017, 11 (Suppl. 12), 24. [Google Scholar] [CrossRef] [PubMed]
- Mäntynen, S.; Sundberg, L.R.; Oksanen, H.M.; Poranen, M.M. Half a Century of Research on Membrane-Containing Bacteriophages: Bringing New Concepts to Modern Virology. Viruses 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Saren, A.M.; Ravantti, J.J.; Benson, S.D.; Burnett, R.M.; Paulin, L.; Bamford, D.H.; Bamford, J.K. A snapshot of viral evolution from genome analysis of the tectiviridae family. J Mol Biol 2005, 350, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Savilahti, H.; Bamford, D.H. Protein-primed DNA replication: Role of inverted terminal repeats in the Escherichia coli bacteriophage PRD1 life cycle. J Virol 1993, 67, 4696–4703. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Bäckström, D.; Ettema, T.J.G.; Krupovic, M.; Koonin, E.V. Vast diversity of prokaryotic virus genomes encoding double jelly-roll major capsid proteins uncovered by genomic and metagenomic sequence analysis. Virol J 2018, 15, 67. [Google Scholar] [CrossRef]
- Caruso, S.M.; deCarvalho, T.N.; Huynh, A.; Morcos, G.; Kuo, N.; Parsa, S.; Erill, I. A Novel Genus of Actinobacterial Tectiviridae. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Jalasvuori, M.; Koskinen, K. Extending the hosts of Tectiviridae into four additional genera of Gram-positive bacteria and more diverse Bacillus species. Virology 2018, 518, 136–142. [Google Scholar] [CrossRef]
- Bamford, D.H.; Mindich, L. Characterization of the DNA-protein complex at the termini of the bacteriophage PRD1 genome. J Virol 1984, 50, 309–315. [Google Scholar] [CrossRef]
- Knopf, C.W. Evolution of viral DNA-dependent DNA polymerases. Virus Genes 1998, 16, 47–58. [Google Scholar] [CrossRef]
- Peralta, B.; Gil-Carton, D.; Castaño-Díez, D.; Bertin, A.; Boulogne, C.; Oksanen, H.M.; Bamford, D.H.; Abrescia, N.G. Mechanism of membranous tunnelling nanotube formation in viral genome delivery. PLoS Biol 2013, 11, e1001667. [Google Scholar] [CrossRef]
- Santos-Pérez, I.; Oksanen, H.M.; Bamford, D.H.; Goñi, F.M.; Reguera, D.; Abrescia, N.G.A. Membrane-assisted viral DNA ejection. Biochim Biophys Acta Gen Subj 2017, 1861, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Abrescia, N.G.; Cockburn, J.J.; Grimes, J.M.; Sutton, G.C.; Diprose, J.M.; Butcher, S.J.; Fuller, S.D.; San Martín, C.; Burnett, R.M.; Stuart, D.I.; et al. Insights into assembly from structural analysis of bacteriophage PRD1. Nature 2004, 432, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, J.; Pérez-Illana, M.; Martín-González, N.; San Martín, C. Adenovirus Structure: What Is New? Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Benson, S.D.; Bamford, J.K.; Bamford, D.H.; Burnett, R.M. Viral evolution revealed by bacteriophage PRD1 and human adenovirus coat protein structures. Cell 1999, 98, 825–833. [Google Scholar] [CrossRef]
- Medvedeva, S.; Sun, J.; Yutin, N.; Koonin, E.V.; Nunoura, T.; Rinke, C.; Krupovic, M. Three families of Asgard archaeal viruses identified in metagenome-assembled genomes. Nat Microbiol 2022, 7, 962–973. [Google Scholar] [CrossRef]
- Yutin, N.; Shevchenko, S.; Kapitonov, V.; Krupovic, M.; Koonin, E.V. A novel group of diverse Polinton-like viruses discovered by metagenome analysis. BMC Biol 2015, 13, 95. [Google Scholar] [CrossRef]
- Krupovic, M.; Koonin, E.V. Polintons: A hotbed of eukaryotic virus, transposon and plasmid evolution. Nat Rev Microbiol 2015, 13, 105–115. [Google Scholar] [CrossRef]
- Gillis, A.; Mahillon, J. Phages preying on Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis: Past, present and future. Viruses 2014, 6, 2623–2672. [Google Scholar] [CrossRef]
- Ackermann, H.W.; Prangishvili, D. Prokaryote viruses studied by electron microscopy. Arch Virol 2012, 157, 1843–1849. [Google Scholar] [CrossRef]
- Atanasova, N.S.; Senčilo, A.; Pietilä, M.K.; Roine, E.; Oksanen, H.M.; Bamford, D.H. Comparison of lipid-containing bacterial and archaeal viruses. Adv Virus Res 2015, 92, 1–61. [Google Scholar] [CrossRef]
- Silander, O.K.; Weinreich, D.M.; Wright, K.M.; O'Keefe, K.J.; Rang, C.U.; Turner, P.E.; Chao, L. Widespread genetic exchange among terrestrial bacteriophages. Proc Natl Acad Sci U S A 2005, 102, 19009–19014. [Google Scholar] [CrossRef] [PubMed]
- O'Keefe, K.J.; Silander, O.K.; McCreery, H.; Weinreich, D.M.; Wright, K.M.; Chao, L.; Edwards, S.V.; Remold, S.K.; Turner, P.E. Geographic differences in sexual reassortment in RNA phage. Evolution 2010, 64, 3010–3023. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.M.; Hussain, F.A.; Yang, J.; Arevalo, P.; Brown, J.M.; Chang, W.K.; VanInsberghe, D.; Elsherbini, J.; Sharma, R.S.; Cutler, M.B.; et al. A major lineage of non-tailed dsDNA viruses as unrecognized killers of marine bacteria. Nature 2018, 554, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.H.; Mavrich, T.N.; Garlena, R.A.; Guerrero-Bustamante, C.A.; Jacobs-Sera, D.; Montgomery, M.T.; Russell, D.A.; Warner, M.H.; (SEA-PHAGES); Hatfull, G.F.; Bacteriophages of Gordonia spp. Display a Spectrum of Diversity and Genetic Relationships. mBio 2017, 8. [CrossRef]
- Klyczek, K.K.; Bonilla, J.A.; Jacobs-Sera, D.; Adair, T.L.; Afram, P.; Allen, K.G.; Archambault, M.L.; Aziz, R.M.; Bagnasco, F.G.; Ball, S.L.; et al. Tales of diversity: Genomic and morphological characteristics of forty-six Arthrobacter phages. PLoS ONE 2017, 12, e0180517. [Google Scholar] [CrossRef]
- Pope, W.H.; Jacobs-Sera, D.; Russell, D.A.; Peebles, C.L.; Al-Atrache, Z.; Alcoser, T.A.; Alexander, L.M.; Alfano, M.B.; Alford, S.T.; Amy, N.E.; et al. Expanding the diversity of mycobacteriophages: Insights into genome architecture and evolution. PLoS ONE 2011, 6, e16329. [Google Scholar] [CrossRef]
- Russell, D.A. Sequencing, Assembling, and Finishing Complete Bacteriophage Genomes. Methods Mol Biol 2018, 1681, 109–125. [Google Scholar] [CrossRef]
- Pope, W.H.; Jacobs-Sera, D. Annotation of Bacteriophage Genome Sequences Using DNA Master: An Overview. Methods Mol Biol 2018, 1681, 217–229. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res 2005, 33, W451–W454. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI's conserved domain database. Nucleic Acids Res 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed]
- Pacey, M. Starterator Guide. University of Pittsburgh: 2016.
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J Mol Biol 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hirokawa, T.; Boon-Chieng, S.; Mitaku, S. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics 1998, 14, 378–379. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Hallgren, J.; Tsirigos, K.; Pedersen, M.D.; Almagro Armenteros, J.J.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv: 2022.
- Szilágyi, A.; Skolnick, J. Efficient prediction of nucleic acid binding function from low-resolution protein structures. J Mol Biol 2006, 358, 922–933. [Google Scholar] [CrossRef]
- Cresawn, S.G.; Bogel, M.; Day, N.; Jacobs-Sera, D.; Hendrix, R.W.; Hatfull, G.F. Phamerator: A bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics 2011, 12, 395. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource - improved response to emergent viral outbreaks. Nucleic Acids Res 2017, 45, D482–D490. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Sneath, P.H.A.; Sokal, R.R. Numerical Taxonomy; WF Freeman & Co., 1973.
- Zuckerkandl, E.; Pauling, L. Evolutionary Divergence and Convergence in Proteins. In Evolving Genes and Proteins, Bryson, V., Vogel, H.J. Eds.; Academic Press, 1965; pp 97-166.
- Bamford, J.K.; Hänninen, A.L.; Pakula, T.M.; Ojala, P.M.; Kalkkinen, N.; Frilander, M.; Bamford, D.H. Genome organization of membrane-containing bacteriophage PRD1. Virology 1991, 183, 658–676. [Google Scholar] [CrossRef]
- Gill, J.J.; Wang, B.; Sestak, E.; Young, R.; Chu, K.H. Characterization of a Novel Tectivirus Phage Toil and Its Potential as an Agent for Biolipid Extraction. Sci Rep 2018, 8, 1062. [Google Scholar] [CrossRef]
- Gillis, A.; Mahillon, J. Influence of lysogeny of Tectiviruses GIL01 and GIL16 on Bacillus thuringiensis growth, biofilm formation, and swarming motility. Appl Environ Microbiol 2014, 80, 7620–7630. [Google Scholar] [CrossRef]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int J Syst Evol Microbiol 2018, 68, 2386–2392. [Google Scholar] [CrossRef]
- Deng, Z.; Xia, X.; Deng, Y.; Zhao, M.; Gu, C.; Geng, Y.; Wang, J.; Yang, Q.; He, M.; Xiao, Q.; et al. ANI analysis of poxvirus genomes reveals its potential application to viral species rank demarcation. Virus Evol 2022, 8, veac031. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A 2004, 101, 11030–11035. [Google Scholar] [CrossRef]
Figure 1.
Host Range analysis of novel tectiviruses. Tenfold dilutions of crude high titer lysate of each phage were spotted on lawns of Microbacterium spp. Microbacterium phages Cece and Paschalis were isolated using M. radiodurans and M. foliorum, respectively were used as controls.
Figure 1.
Host Range analysis of novel tectiviruses. Tenfold dilutions of crude high titer lysate of each phage were spotted on lawns of Microbacterium spp. Microbacterium phages Cece and Paschalis were isolated using M. radiodurans and M. foliorum, respectively were used as controls.
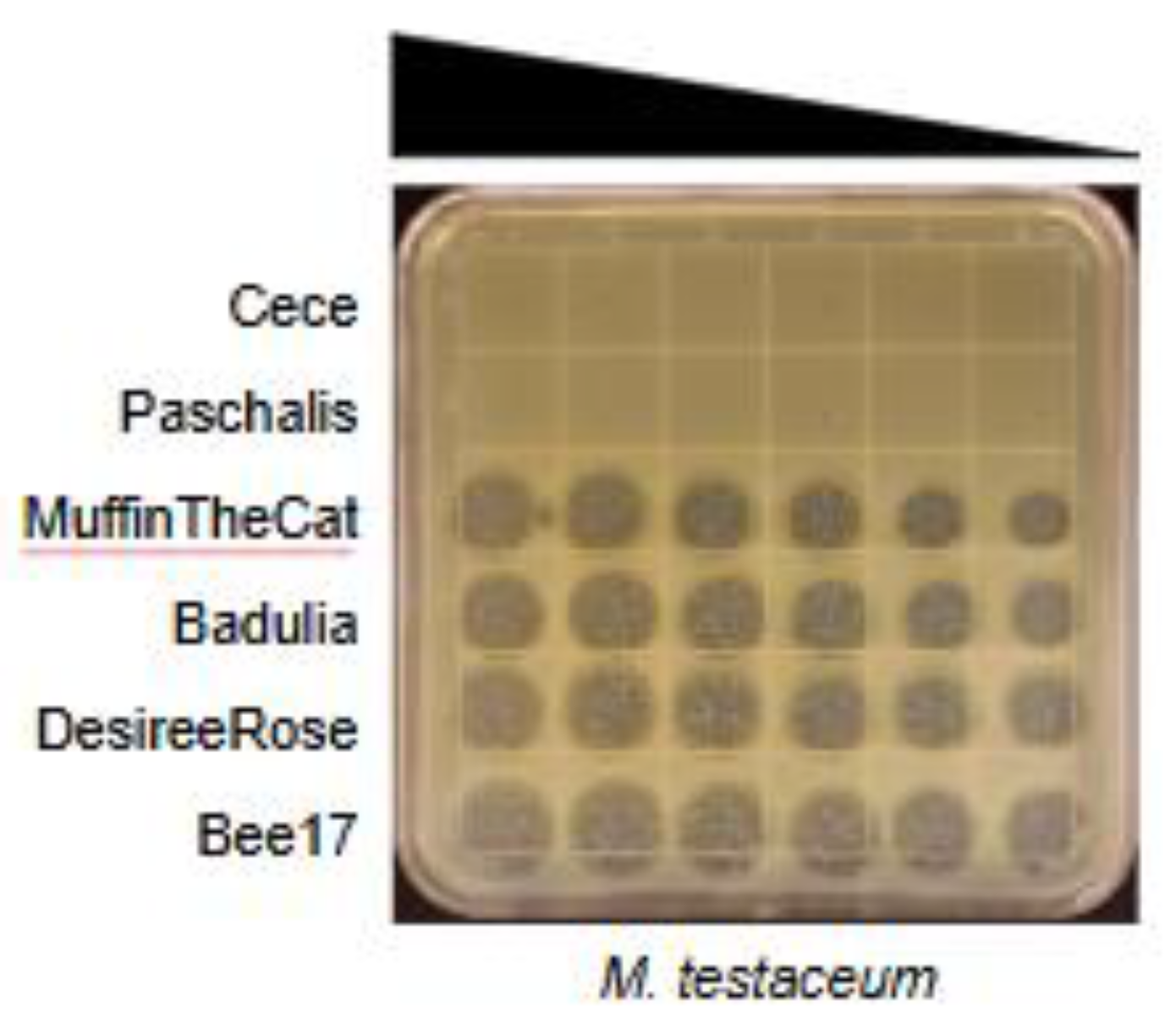

Figure 2.
Transmission Electron Microscopy images of representative phage as observed after negative staining with uranyl acetate and images taken with JEOL JEM 1400 TEM at 100 kV acceleration voltage. A. Bee17 B. DesireeRose C. LuzDeMondo D. MuffinTheCat. Black arrow indicates the internal lipid membrane. White arrow indicates the tail tube.
Figure 2.
Transmission Electron Microscopy images of representative phage as observed after negative staining with uranyl acetate and images taken with JEOL JEM 1400 TEM at 100 kV acceleration voltage. A. Bee17 B. DesireeRose C. LuzDeMondo D. MuffinTheCat. Black arrow indicates the internal lipid membrane. White arrow indicates the tail tube.
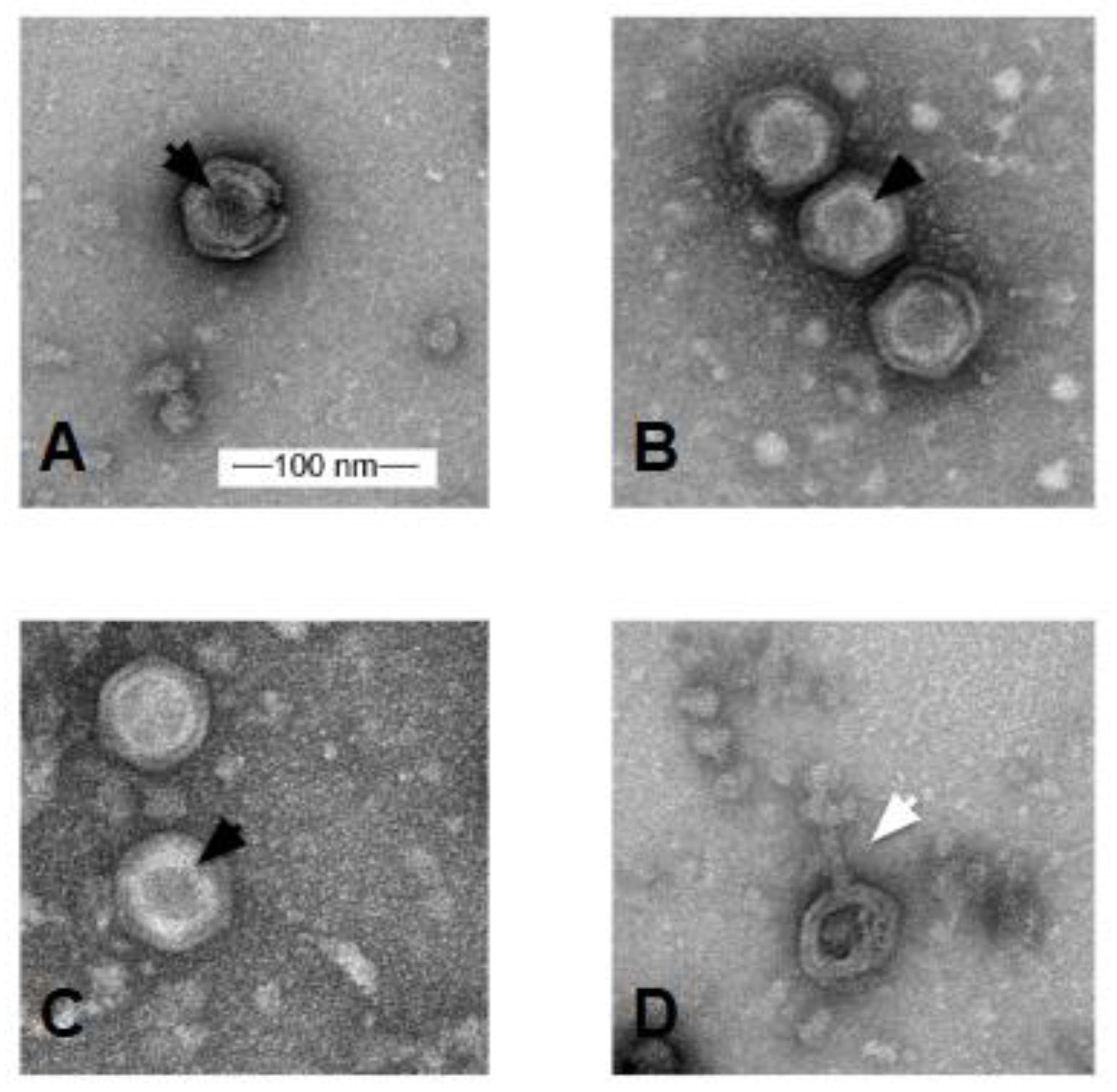
Figure 4.
Genome maps of novel bacteriophages compared to representatives of each genus. The genomes are separated by gray borders, and the predicted ORFs are shown as numbered colored boxes either positioned towards the top of the gray border (transcribed rightward) or near the bottom of the gray border (transcribed leftwards). Genes are color-coded by phamily as in Figure 3. Gene phamilies with a single member are white. The colored regions between the genomes correspond to tblastx hits and are color-coded according to e-value.
Figure 4.
Genome maps of novel bacteriophages compared to representatives of each genus. The genomes are separated by gray borders, and the predicted ORFs are shown as numbered colored boxes either positioned towards the top of the gray border (transcribed rightward) or near the bottom of the gray border (transcribed leftwards). Genes are color-coded by phamily as in Figure 3. Gene phamilies with a single member are white. The colored regions between the genomes correspond to tblastx hits and are color-coded according to e-value.
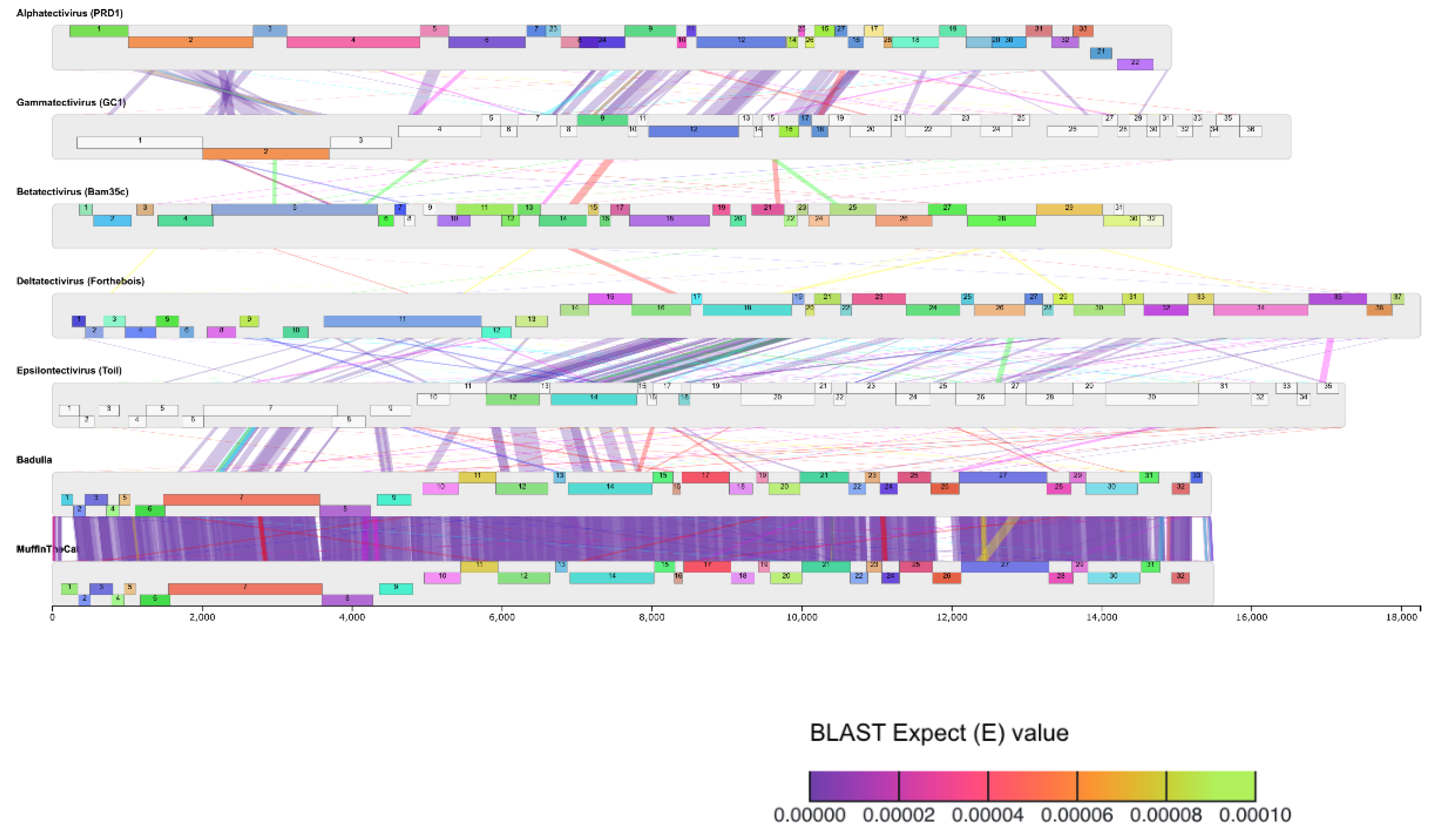
Figure 6.
Phylogenetic reconstruction of novel tectivirus complete genome nucleotide sequences. Trees were constructed using MEGA7 [65]. The UPGMA method [68] was used based on multiple alignments generated by MUSCLE [66]. Bootstrap values are based on 500 replicates and are indicated next to the branches [67]. The evolutionary distances were computed using the Maximum Composite Likelihood method [75] and are in the units of the number of base substitutions per site. The rate variation among sites was modeled with a gamma distribution (shape parameter = 1). First, second, third and non-coding codon positions were included and all positions containing gaps and missing data were eliminated. See Table 1 for accession numbers. Deltatectivirus Forthebois was included as an outgroup (MK620900.1).
Figure 6.
Phylogenetic reconstruction of novel tectivirus complete genome nucleotide sequences. Trees were constructed using MEGA7 [65]. The UPGMA method [68] was used based on multiple alignments generated by MUSCLE [66]. Bootstrap values are based on 500 replicates and are indicated next to the branches [67]. The evolutionary distances were computed using the Maximum Composite Likelihood method [75] and are in the units of the number of base substitutions per site. The rate variation among sites was modeled with a gamma distribution (shape parameter = 1). First, second, third and non-coding codon positions were included and all positions containing gaps and missing data were eliminated. See Table 1 for accession numbers. Deltatectivirus Forthebois was included as an outgroup (MK620900.1).
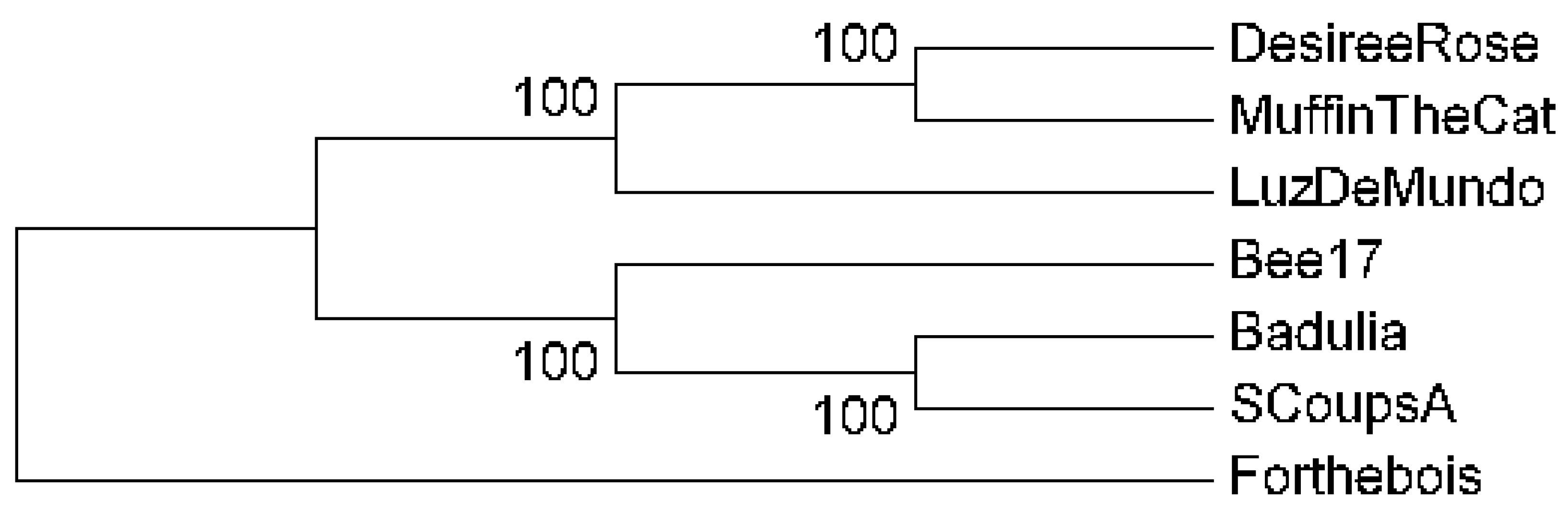
Figure 7.
Phylogenetic reconstruction of tectivirus (a) DNA polymerase, (b) major capsid, and (c) DNA packaging ATPase protein sequences. Trees were constructed using the UPGMA method [68] MEGA7 based on multiple alignments generated using MUSCLE [66]. Bootstrap values are based on 500 replicates and are indicated next to the branches [67]. Branch lengths indicate evolutionary distances, with units in amino acid substitutions per site, as computed using the Poisson correction method [69]. The rate variation was calculated using a gamma distribution shape parameter = 1. All positions containing gaps and missing data were eliminated. Clades were collapsed by genus. Sequences found in each condensed clade can be found in Table 3.
Figure 7.
Phylogenetic reconstruction of tectivirus (a) DNA polymerase, (b) major capsid, and (c) DNA packaging ATPase protein sequences. Trees were constructed using the UPGMA method [68] MEGA7 based on multiple alignments generated using MUSCLE [66]. Bootstrap values are based on 500 replicates and are indicated next to the branches [67]. Branch lengths indicate evolutionary distances, with units in amino acid substitutions per site, as computed using the Poisson correction method [69]. The rate variation was calculated using a gamma distribution shape parameter = 1. All positions containing gaps and missing data were eliminated. Clades were collapsed by genus. Sequences found in each condensed clade can be found in Table 3.
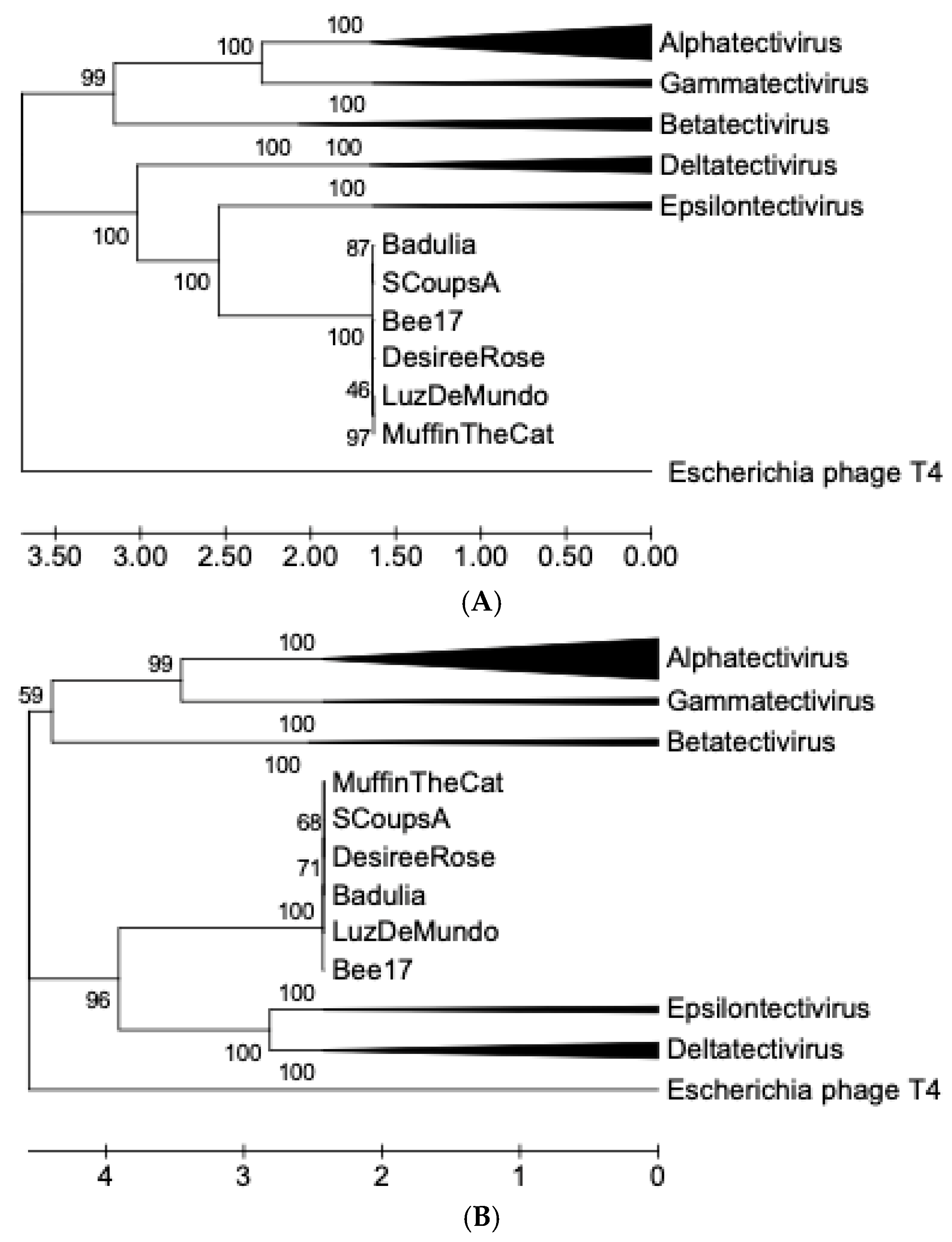
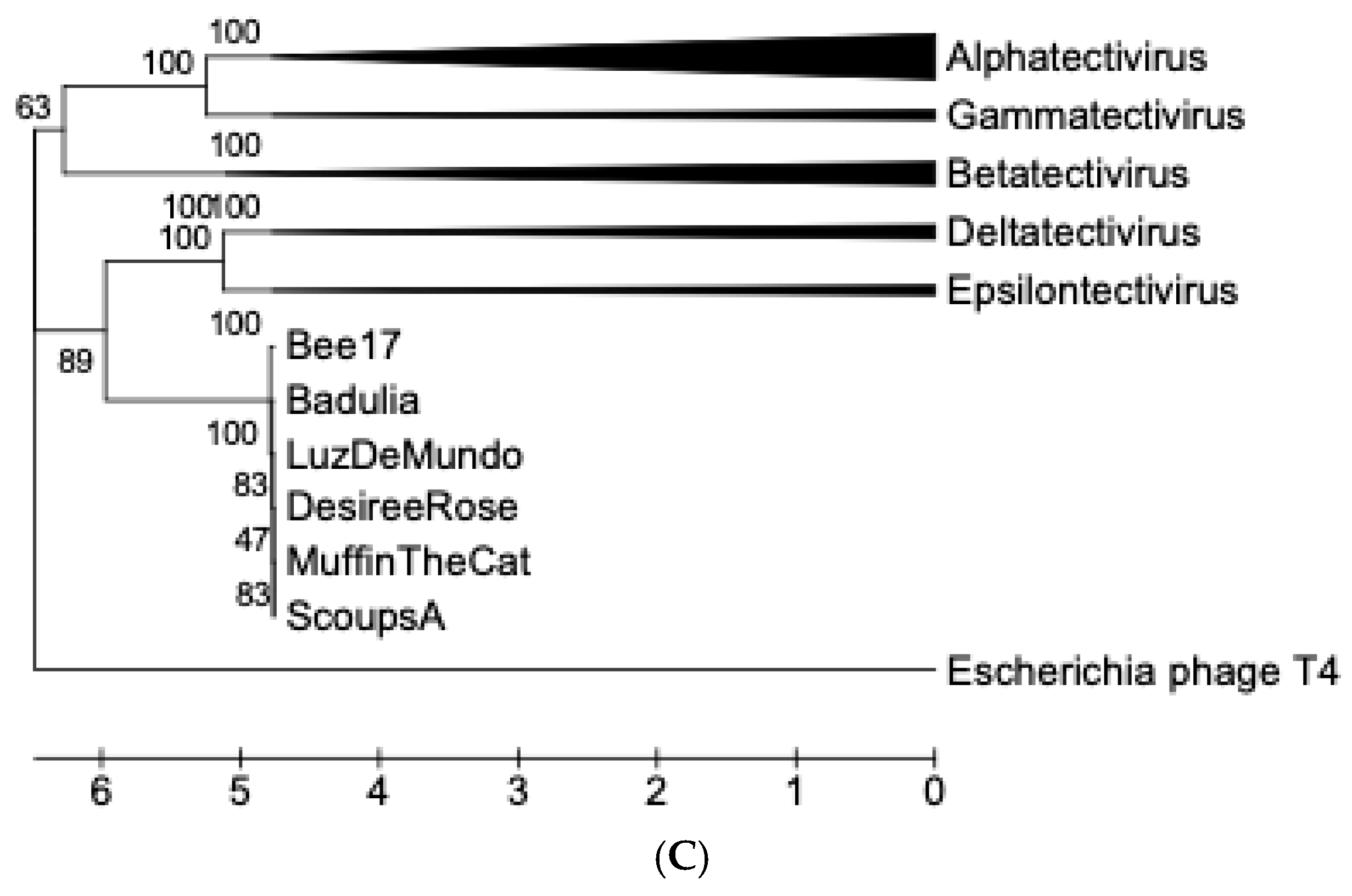

Table 1.
Phage discovery locations and general characterization of the novel tectiviruses.
| Phage | Accession # | Yr Isolated | Location | Sequence Coverage | Genome size (bp) | %GC | Terminal Repeats (bp) | # Genes |
|---|---|---|---|---|---|---|---|---|
| DesireeRose | OL455892 | 2019 | Nyack, NY | 4572 | 15488 | 55 | 112 | 32 |
| MuffinTheCat | MT952848 | 2019 | Nyack, NY | 824 | 15494 | 55.1 | 114 | 32 |
| LuzDeMundo | OP068334 | 2021 | Baldwin, NY | 537 | 15471 | 55 | 113 | 32 |
| Bee17 | OQ709213 | 2019 | Haskell, NJ | 3019 | 15636 | 54.6 | 119 | 34 |
| Badulia | MZ150790 | 2019 | Nyack, NY | 4407 | 15460 | 54.6 | 120 | 33 |
| SCoupsA | OQ709206 | 2021 | Seabrook, MD | 106 | 15438 | 54.8 | 120 | 33 |
Table 2.
Assigned functions of genes in the genomes of the novel tectiviruses. The genes not included have undetermined functions.
Table 2.
Assigned functions of genes in the genomes of the novel tectiviruses. The genes not included have undetermined functions.
|
Product Functions |
Direction | Desiree Rose | Muffin TheCat | LuzDe Mundo | Bee17 | Badulia | SCoupsA |
|---|---|---|---|---|---|---|---|
| membrane protein | R | gp4 | gp4 | gp4 | gp5 | gp4 | gp4 |
| DNA binding protein | R | gp5 | gp5 | gp5 | gp6 | gp5 | gp5 |
| DNA primase/polymerase | R | gp7 | gp7 | gp7 | gp8 | gp7 | gp7 |
| DNA packaging ATPase | F | gp12 | gp12 | gp12 | gp13 | gp12 | gp12 |
| membrane protein | F | gp13 | gp13 | gp13 | gp14 | gp13 | gp13 |
| major capsid protein | F | gp14 | gp14 | gp14 | gp15 | gp14 | gp14 |
| membrane protein | F | gp15 | gp15 | gp15 | gp16 | gp15 | gp15 |
| membrane protein | F | gp16 | gp16 | gp16 | gp17 | gp16 | gp16 |
| peptidase | F | gp17 | gp17 | gp17 | gp18 | gp17 | gp17 |
| membrane protein | F | gp18 | gp18 | gp18 | gp19 | gp18 | gp18 |
| phage membrane DNA delivery protein | F | gp19 | gp19 | gp19 | gp20 | gp19 | gp19 |
| endolysin | F | gp30 | gp30 | gp30 | gp31 | gp30 | gp30 |
| membrane protein | F | gp32 | gp32 | gp32 | gp33 | gp32 | gp32 |
Table 3.
NCBI accession numbers for phylogenetic reconstruction, retrieved from NCBI Virus [64] and Genome Database (https://www.ncbi.nlm.nih.gov/genome/). The bacterial strain each phage was originally isolated from is listed; this does not represent its entire host range. When multiple strains have been sequenced, median GC content is listed. Organized according to NCBI Taxonomy Browser: https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?mode=Undef&id=10656&lvl=3&lin=f&keep=1&srchmode=1&unlock.
Table 3.
NCBI accession numbers for phylogenetic reconstruction, retrieved from NCBI Virus [64] and Genome Database (https://www.ncbi.nlm.nih.gov/genome/). The bacterial strain each phage was originally isolated from is listed; this does not represent its entire host range. When multiple strains have been sequenced, median GC content is listed. Organized according to NCBI Taxonomy Browser: https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?mode=Undef&id=10656&lvl=3&lin=f&keep=1&srchmode=1&unlock.
|
Subspecies |
Phage (%GC) | Isolation host (%GC) | DNA polymerase | DNA packaging ATPase |
major capsid |
| Alphatectivirus | |||||
| Alphatectivirus PR4 | PR4 (48.3) | Pseudomonas aeruginosa (66.2) | AAX45594.1, YP_337983.1 | YP_337992.1, AAX45618.1 | AAA32442.1, AAX45607.1, YP_337995.1 |
| Alphatectivirus PRD1 | L17 (48.3) | Aeromonas hydrophila (61.1) / Escherichia coli (50.6) | AAX45532.1 | AAX45556.1 | AAX45545.1 |
| PR3 (48.3) | Pseudomonas aeruginosa (66.2) | AAX45563.1 | AAX45587.1 | AAX45576.1 | |
| PR5 (48.3) |
Escherichia coli (50.4-50.8%) |
AAX45625.1 | AAX45649.1 | ||
| PR772 (48.3) | Proteus mirabilis (38.8) | AAX45656.1, AAR99740.1 | AAX45680.1 AAR99751.1 https://www.ncbi.nlm.nih.gov/nuccore/AY848688 |
AAX45669.1 AAR99754.1 |
|
| PRD1 (48.1) |
Pseudomonas sp. (aeruginosa 66.2%) |
AAA32450.1, AAA32452.1, AAX45903.1, YP_009639956.1 | YP_009639965.1, AAX45556.1, AAX45649.1, AAX45680.1, AAX45927.1, AAR99751.1, P27381.3 | UDY80299.1, AAA32445.1, AAX45916.1, YP_009639968.1, P22535.2 | |
| Betatectivirus | |||||
| Betatectivirus AP50 | AP50 (38.7) | Bacillus anthracis (35.2) | YP_002302517.1 | YP_002302526.1, ACB54913.1 | YP_002302529.1 |
| Betatectivirus Bam35 | Bam35c (39.7) | Bacillus thuringiensis (34.9) | NP_943751.1 | NP_943760.1 | NP_943764.1 |
| pGIL02 (39.7) | Bacillus thuringiensis (34.9) | AND28856.1 | Uncharacterized | AND28851.1 | |
| Betatectivirus GIL16 | GIL16c (39.7) | Bacillus thuringiensis (34.9) | YP_224103.1 | AAW33576.1, YP_224111.1 | YP_224110.1 |
| Betatectivirus Wip1 | Wip1 (36.9) | Bacillus anthracis (35.2) | YP_008433325.1* | Uncharacterized | AGT13371.1 |
| Deltatectivirus | |||||
| Deltatectivirus forthebois | Forthebois (53.6) | Streptomyces scabiei (71.3) | YP_010084034.1, QBZ72843.1 | QBZ72848.1 |
QBZ72850.1, YP_010084041.1 |
| Deltatectivirus wheeheim | WheeHeim (54.6) | Streptomyces scabiei (71.3) | YP_010084070.1, QAX92919.1 | QAX92924.1 | QAX92926.1, YP_010084077.1 |
| Unclassified | RickRoss (54.4) | Streptomyces scabiei (71.3) | UYL87501.1 | UYL87506.1 | UYL87508.1 |
| Epsilontectivirus | |||||
| Epsilontectivirus toil | Toil (54.5) | Rhodococcus opacus (67.1) | YP_010084103.1, ARK07690.1 | YP_010084108.1, ARK07695.1 | (coat) ARK07697.1, YP_010084110.1 |
| Gammatectivirus | |||||
| Gammatectivirus GC1 | GC1 (50.5) | Gluconobacter cerinus (55.6) | YP_009620046.1, ATS92570.1 | YP_009620053.1, ATS92577.1 | ATS92580.1, YP_009620056.1 |
| Outgroup | |||||
| T4 (35.3) | Escherichia coli (50.6) | NP_049662.1 | NP_049775.1 | AAA32503.1 | |
*There is an annotation error for Wip1; the accession number listed for the DNA polymerase is not the polymerase protein. Wip1 was therefore omitted from the DNA polymerase tree.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



