
Submitted:
17 December 2024
Posted:
18 December 2024
You are already at the latest version
Abstract
Biallelic rare pathogenic loss-of-function (LOF) variants in lipoprotein lipase (LPL) cause familial chylomicronemia syndrome (FCS). Heterozygosity for these same variants is associated with a highly variable plasma triglyceride (TG) phenotype ranging from normal to severe hypertriglyceridemia (HTG), with longitudinal variation of phenotype severity seen often in a given carrier. Here we provide an updated overview of genetic variation in LPL in the context of HTG, with a focus on disease-causing and/or disease-associated variants. We provide a curated list of 300 disease-causing variants discovered in LPL, as well as an exon-by-exon breakdown of the LPL gene and protein, highlighting the impact of variants and the various functional residues of domains of the LPL protein. We also provide a curated list of variants of unknown or uncertain significance, many of which may be upgraded to pathogenic/likely pathogenic classification should additional case and/or segregation data be reported. Finally, we also review the association between benign/likely benign variants in LPL, many of which are common polymorphisms, and TG phenotype.
Keywords:
lipoprotein lipase
; chylomicronemia
; hypertriglyceridemia
; complex trait
; human genetics
; genomic disorders
1. Introduction
Lipoprotein lipase (LPL) is the major enzyme responsible for regulating plasma triglyceride (TG) levels in many animal species, including humans [1]. Biallelic loss-of-function (LOF) variants in the LPL gene are the predominant cause of familial chylomicronemia syndrome (FCS) cases worldwide, accounting for ~80% of all cases [2]. This form of FCS is synonymous with LPL deficiency or the former Frederickson hyperlipoproteinemia type 1. FCS is characterized by severely compromised if not completely absent plasma lipolytic activity, leading to the pathogenic accumulation of predominantly intestinally-derived chylomicrons (CMs) which produces refractory, severe hypertriglyceridemia (HTG; severe HTG is defined as fasting plasma TG ≥ 10 mmol/L), characteristic physical findings (such as eruptive xanthomas, lipemia retinalis, etc.), failure to thrive in infants, and most seriously, significantly increased lifetime risk for developing acute pancreatitis [2].
The phenotype associated with monoallelic LOF variants in LPL is less appreciated and familiar. We previously found that patients possessing a single affected LPL copy present with highly variable fasting plasma TG levels, both within and between patients longitudinally, ranging from normal TG levels to severe HTG, with secondary factors likely playing a major role in modulating the TG phenotype expressed by carriers of monoallelic LOF variants in the LPL gene [3]. Prior to our report, this variability was not widely known nor reported, and our results may have seemed counterintuitive to previously held assumptions about the TG phenotype associated with heterozygosity for LOF variants in LPL, the potential severity of which may have been underestimated.
Here, we synthesize this new understanding of the phenotype of monoallelic LOF variants in LPL with recent findings regarding LPL structure, isoforms, regulation, and functionality alongside previously well-established findings to provide an up-to-date characterization of the LPL variants as they relate to HTG.
2. Synthesis and Expression of LPL
LPL is predominantly synthesized by the parenchymal cells of adipose, skeletal muscle, and cardiac muscle tissues, and ultimately expressed in the vascular lumen of capillaries supplying these tissues, bound to the surface of endothelial cells [4,5].
LPL expression is highly regulated and complex, with varying physiological states, such as fasting and fed states, invoking tissue-specific differential LPL expression and regulation patterns. The synthesis, secretion, and expression of mature LPL have been previously reviewed by Wu, Kersten and Qi in 2021 [6]. Upon translation of the signal peptide sequence, the nascent LPL peptide is targeted to and transported into the endoplasmic reticulum (ER) via the signal peptide.
Once in the ER and after cleavage of the signal peptide, the nascent LPL peptide is completely translated and then undergoes two major post-translational processes, N-linked glycosylation at two sites, Asn70 and Asn386 (note: amino acid numbering throughout this review includes the signal peptide residues as residues 1-27), and the formation of five intra-molecular disulfide bridges, Cys54-Cys67, Cys243-Cys266, Cys291-Cys302, Cys305-Cys310, and Cys445-Cys465. Glycosylation of Asn70 has been previously shown to be necessary for LPL activity and proper intracellular trafficking [7]. Similarly, the Cys243-Cys266, Cys291-Cys302, and Cys305-Cys310 disulfide bridges have also been shown to be essential for LPL activity [8].
Either as part of the process of facilitating these post-translational modifications, and/or directly following them, the action of lipase maturation factor (LMF1), one of the four essential cofactors of LPL in which biallelic LOF variants have also been found to cause FCS [2], and Sel-1 suppressor of Lin-12-Like 1 (SEL1L) , both found on the ER membrane, ensure proper folding and maturation of the nascent LPL peptide. However, the exact roles of both LMF1 and SEL1L in the LPL maturation process are not well defined and remain somewhat unclear.
Some evidence indicates that LMF1 may help facilitate and promote proper formation of the five disulfide bridges in LPL by interacting with several other ER-resident chaperone proteins such as Erp72, Erp44, ERdj5, and thioredoxin [9]. SEL1L is best known for its role as an obligatory co-factor for the E3 ligase HRD1 involved in ER-associated degradation (ERAD), a critical mechanism that specifically targets ER proteins for proteasomal degradation in the cytosol [10]. It has been suggested that SEL1L may facilitate and promote LPL maturation through both SEL1L-HRD1 ERAD-related and -independent pathways.
It has been observed in mice that high molecular weight aggregates of LPL peptide form within the ER in the absence of SEL1L. Given this, it has been speculated that nascent LPL peptide may be prone to misfolding and SEL1L-HRD1 ERAD may play a critical role in clearing misfolded peptide from the ER to prevent aggregate formation and allow proper LPL maturation to take place [6]. Additionally, there is some in vitro evidence indicating that SEL1L physically interacts with both nascent LPL peptide and LMF1 and promotes their interaction within the ER, indicating that SEL1L has an additional role in LPL maturation independent of the SEL1L-HRD1 ERAD mechanism [11]. Ultimately, after the action of LMF1 and SEL1L, LPL is secreted from the ER to the Golgi apparatus for packaging and transport to its secretory pathway.
Within the Golgi apparatus, LPL monomers are further modified with the addition of complex oligosaccharide groups [12]. LPL is then targeted to one of two different fates within the trans-Golgi network: 1) if the mature LPL binds to the heparan sulfate chain of Syndecan-1 (SDC-1), an integral membrane heparan sulfate proteoglycan (HSPG), it is preferentially sorted to exocytic vesicles of the sphingomyelin secretory pathway [13]; or 2) if mature LPL binds to sortilin-related receptor (SorLA-1), which has been observed to occur in neutral and acidic conditions within vesicular structures, LPL is targeted to a lysosomal degradation pathway [14]. It has been noted that LPL is still constitutively secreted even in the absence of SDC-1 [15], likely through the “bulk-flow” mechanism [16] thought to underlie constitutive secretion of many soluble secretory proteins, although, to the best of our knowledge, it currently remains unknown whether there are any secretion rate differences between the SDC-1-mediated secretion pathway and the “bulk-flow” mechanism for LPL [15].
Within secretory vesicles, LPL may form, in a concentration-dependent manner, catalytically inactive dihedral LPL dimers that assemble into helical oligomers, hypothesized to be a form of LPL storage prior to release and secretion of the catalytically active form in response to substrate/nutritional signaling [17]. In response to substrate availability/nutritional signaling, LPL is released and secreted into the interstitial space, where it binds to negatively-charged HSPGs on the parenchymal cell surface via a positively-charged region, where LPL eventually interacts with/is captured by glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 (GPIHBP1) (another one of the four essential cofactors of LPL in which biallelic LOF variants have also been found to cause FCS [2]) expressed on the basal surface of endothelial cells [18,19,20]. The newly formed LPL-GPIHBP1 complex transcytoses from the interstitial space to the capillary luminal surface of the endothelial cell [21], where LPL is anchored to catalyze the hydrolysis of circulating TGs from various lipoprotein species, primarily CMs and very-low-density lipoproteins (VLDLs) [1].
3. Physiological Roles of LPL in Lipoprotein Metabolism and Their Regulation
LPL has two primary functions related to plasma lipid metabolism: 1) catalysis of the rate-limiting step of plasma TG clearance, specifically of the hydrolysis of circulating plasma TGs, and 2) mediating tissue uptake of several circulating lipoprotein species via mechanisms independent of its catalytic activity.
The lipolytic function of what we now know to be LPL was first observed by Paul Hahn in 1943, when he found that injection of heparinized whole blood or plasma from donor dogs into lipemic dogs abolished their lipemia [22]. However, the specific heparin-induced clearing factor was not isolated and characterized as “lipoprotein lipase” until 1955 by Edward Korn [23,24]. Since then, a rich body of research has accumulated characterizing the biochemistry of LPL-mediated lipolysis.
The basic series of events that leads to LPL-mediated hydrolysis of TGs has been reviewed previously [25,26]. Briefly, the likely sequence of events begins with circulating TG-rich lipoproteins (TRLs), such as VLDLs and/or CMs, bind to endothelial cell membrane bound LPL-GPIHBP1 unit(s), leading to a confirmational change in LPL that alters the lid region from a closed configuration that blocks substrate access to the active site to an open configuration [25,26,27,28]. This interaction is mediated by TRL-bound apolipoprotein (apo) C-II, an essential activator of LPL, binding to and stabilizing the lid region of LPL (discussed in more detail in the Protein Structure of LPL section of this review) [29,30] and TRL-bound apo A-V, another essential cofactor of LPL, binding to GPIHBP1 [31,32] which enhances the association of apo A-V containing lipoproteins with the endothelial cell surface features associated with LPL.
We have previously reviewed apo A-V (APOA5) genetic variation and its roles in plasma lipid metabolism [33]. Either simultaneously or following this confirmational change upon TRL binding, the β5 loop of the N-terminal domain of LPL likely folds back, bringing the oxyanion hole into a catalytically competent position [25]. The sum of these conformational changes is the exposure of hydrophobic residues within and lining the active site, which attracts the FA chains of TG molecules to the catalytic triad and enables the access of the glycerol backbone of TG molecules to the oxyanion hole, enabling hydrolysis of the TG molecule to two FFAs and one 2-monoacylglycerol molecule [25].
Interaction of hydrophobic residues of the lid region and the C-terminal lipid binding domain found on the same face of the 3-dimensional structure of LPL (discussed in more detail in the Protein Structure of LPL section of this review) with lipid substrate have been suggested as a potential mechanism by which LPL binds lipid substrate with the correct orientation to facilitate substrate access and entry to the active site [28]. As membrane bound LPL hydrolyzes TGs to produce FFAs, their accumulation locally triggers the dissociation of LPL from GPIHBP1 [34], releasing the LPL monomer into the plasma where it has been recently proposed to undergo a tail-to-tail homodimerization interaction forming a circulating LPL homodimer [35] that binds to TRLs and their remnants, further hydrolyzing their TG content as they circulate [36], eventually being absorbed by the liver via the action of hepatic LDL receptor-related protein 1 (LRP1), which has been shown to interact with LPL homodimers to enhance hepatic uptake of LDL remnants [37,38].
Rather interestingly, the lipolytic activity of LPL has been associated with several additional physiological roles/effects. First, it has been shown that, at least partially through its enzymatic activity, LPL promotes the proliferation of vascular smooth muscle cells [39,40]. Second, it has been shown that increased LPL lipolytic activity is associated with reduced coronary heart disease and diabetes risk in humans [41,42], with increased insulin sensitization being observed in patients with mutations inactivating angiopoietin-like protein 3 (ANGPTL3), an inhibitor of LPL, and in ANGPTL3 knockout mice [43]. Third, LPL activity has been shown to be necessary for hematopoietic stem progenitor cell maintenance during definitive hematopoiesis in a zebrafish model by regulating and maintaining LPL-mediated release of the essential fatty acid docosahexaenoic acid [44].
LPL catalytic activity and expression are regulated by two primary groups of proteins: 1) the angiopoietin-like (ANGPTL) protein family members ANGPTL3, ANGPTL4, and ANGPTL8 [4,6,45,46,47,48], and 2) several apolipoproteins, most significantly apo A-V, apo C-I, apo C-II, apo C-III, and apo E [4,6,29,45,49,50,51,52,53,54,55,56,57].
The ANGPTLs listed all inhibit LPL activity and are involved in differential tissue expression of LPL in fed versus fasted states [45,58,59,60,61]. Specifically, increased insulin signaling in the fed state induces hepatic and adipose tissue ANGPTL8 expression while downregulating hepatic apo A-V production and adipose tissue ANGPTL4 expression. This leads to an increase in circulating ANGPTL3/8 complex uninhibited by apo A-V, leading to the suppression of LPL activity in oxidative tissues. In turn, LPL-mediated TG hydrolysis is increased in adipose tissue as ANGPTL8 expressed in adipose tissue complexes with remaining ANGPTL4, leading to formation of ANGPTL4/8 complex which has reduced LPL-inhibitory ability than ANGPTL4 alone. Additionally, ANGPTL4/8 complex binding to LPL in adipose tissue prevents circulating ANGPTL4 and ANGPTL3/8 complex from binding them instead.
The net effect of this is that most circulating plasma TG in the fed state are selectively hydrolyzed and taken up by adipose tissues for energy storage. The opposite occurs in the fasted state, where increased apo A-V levels due to lack of insulin signaling enables apo A-V to block the inhibitory effects of circulating ANGPTL3/8 complex and the reduced adipose tissue ANGPTL8 expression leads to increased ANGPTL4-mediated inhibition of adipose tissue LPL. With respect to the second group of regulators, the action of apo C-II and apo A-V have already been discussed (and apo C-II binding to LPL is further discussed in the Protein Structure of LPL section) but the action of apo C-I, apo C-III, and apo E have not been.
While the exact molecular mechanism is unknown, both apo C-I and apo C-III, which are bound to circulating lipoproteins, act as inhibitors of LPL activity, with some evidence suggesting they do so by preventing binding of LPL to the lipid/water interface of TRLs [54]. Interestingly, it has been observed that apo C-III exerts greater inhibitory effect on GPIHBP1-bound LPL than on free LPL [62]. This is unexpected given that GPIHBP1 typically protects LPL from such effects by stabilizing it as noted by the authors of this report on apo C-III [62]. Finally, the role of apo E in regulating LPL activity is isoform dependent, with apo E3 and apo E4 being found to limit LPL-mediated TG hydrolysis [63].
Independent of its lipolytic functionality, LPL has been observed to be involved in tissue uptake of lipoproteins and their remnants by interacting with various cell surface features. Specifically, LPL has been found to 1) mediate the cellular uptake of LDL particles by interacting with both the LDL receptor (LDLR) and proteoglycans on the endothelial cell surface [64], 2) mediate hepatocyte selective uptake of high-density lipoprotein (HDL) associated cholesteryl esters in a manner dependent on hepatocyte cell surface heparan sulfate proteoglycans but independent of LDLR and LRP1 mediated endocytosis pathways [65], and 3) mediate TRL and TRL-remnant uptake in hepatocytes by interacting with LRP1 [66,67,68].
4. Protein Structure of LPL
To this day, the molecular structure of LPL in isolation has not been definitively characterized, due primarily to the fact that the catalytic hydrolase domain of LPL is inherently unstable and prone to spontaneous unfolding that corresponds to loss of catalytic functionality [71]. However, the finding that binding of the intrinsically disordered acidic domain of GPIHBP1 to LPL stabilizes the catalytic domain of LPL [71] has enabled two teams to characterize the crystal structure of the LPL-GPIHBP1 complex instead of LPL in isolation, allowing for new insights into the functional unit of plasma TG metabolism [27,28]. Prior to this, much of our knowledge of LPL structure was derived from inference, primarily based on homology between LPL and pancreatic lipase, a related protein in the same lipase protein superfamily [72,73].
Following the cleavage of the 27 amino acid signal peptide, mature LPL is composed of 448 amino acids with molecular mass of ~55 kDa [74]. Both crystallography studies characterizing the LPL-GPIHBP1 crystal structure confirmed that LPL, as predicted by homology with pancreatic lipase [72], has two primary functional domains: 1) a N-terminal α/β-hydrolase domain containing a catalytic triad and 2) a C-terminal lipid-binding domain formed by a β-barrel which are connected by a hinge region [27,28].
The N-terminal α/β-hydrolase domain spans residues 28-340 [28] and includes several notable functional domains and residues. Ser159, Asp183, and His268 form a serine protease like catalytic triad which is responsible for the catalytic functionality of LPL [27,28], with Trp82 and Leu160 forming an oxyanion hole that the glycerol backbone of TGs is thought to interact with for hydrolysis [25,28,75]. Related to these active site features are a number of hydrophobic residues whose side chains were found to line the active site cleft in the 3-dimensional structure of the LPL-GPIHBP1 complex, namely Trp82, Val84, Trp113, Tyr121, Tyr158, Leu160, Ala185, Pro187, Phe212, Ile221, Phe239, Val260, Val264, and Lys265, and have been stated to be involved in van der Waals interactions that likely guide and/or stabilize the hydrophobic tails of lipid substrate (i.e. TG) in the active site [27]. It should be noted that two of these residues identified by Birrane and colleagues in their study are in fact the residues forming the oxyanion hole described above (Trp82 and Leu160).
Also related to the active site is the lid region, which is thought to regulate substrate availability and specificity to the active site by adopting open or closed confirmations [27,28]. The lid region is formed by the loop of residues extending from the Cys243-Cys266 (with at least one review specifically identifying residues 245-265 as the lid region [6]) disulfide bridge in mature LPL [27,28]. A number of the lid region residues are also included in the above discussed list of hydrophobic residues lining the active site cleft outlined by Birrane and colleagues in their study [27]. As noted by Arora and colleagues, in the open confirmation, several lid region hydrophobic residues (Ile245, Ile249, Val251, Ile252, Leu257, Val-260, Leu263, and Val264) in one of the structures they analyzed created a hydrophobic patch on the surface of the 3D structure of LPL which was on the same face as a second hydrophobic patch formed by several residues (Tyr414, Phe415, Trp417, Trp-420, and Trp421) in the C-terminal Trp-rich lipid binding domain [28]. This was posited by Arora and colleagues as a potential mechanism by which LPL binds to TRL substrates with the correct orientation to facilitate entry of the substrate to the active site [28].
Another key structural component of LPL found within the N-terminal domain are five residues (Ala194, Arg197, Ser199, Asp201, and Asp202) that coordinate a calcium (Ca2+) ion necessary for proper folding of the mature LPL peptide [27]. Recent work has identified that the essential cofactor of LPL, apo C-II, likely exerts a significant part of its enhancing effect on LPL activity by augmenting the thermal stability of LPL (i.e., stabilizing the architecture of the catalytic pocket) and by interacting with and stabilizing the peptide sequences anchoring the lid region of LPL, most likely enabling efficient substrate entry into the catalytic pocket by keeping the lid in open confirmation [30].
Apo C-II was found to interact with four regions within the N-terminal domain of LPL, specifically residues 77-87 (corresponding to the segment connecting β-strand 2 and α-helix 2), 114-126 (corresponding to the segment connecting β-strand 3 and α-helix 3), 212-244 (corresponding to β-strand 7 and the segment connecting it to the lid region), and 245-263 (residues of the lid region) [30]. Interestingly, Kumari and colleagues observed that, with the exception of residues 212-244, these regions correspond with previously identified binding sites for ANGPTL4, a well-known inhibitor of LPL [30,76]. The binding of ANGPTL4 to these regions of LPL has been observed to have an opposite effect to the binding of apo C-II to them, leading to allosteric changes in LPL that destabilize and ultimately lead to the irreversible unfolding of the N-terminal α/β hydrolase domain [30,76].
As noted in the above section on synthesis and expression of LPL, one of the two N-linked glycosylation sites is found within the N-terminal domain, namely Asn70, with the other being located within the C-terminal domain at Asn386 [27,28]. Finally, four of the five disulfide bridges (Cys54-Cys67, Cys243-Cys266, Cys291-Cys302, and Cys305-Cys310) are formed by residues within the N-terminal domain, which were also previously described in the synthesis and expression of LPL section [27,28].
The smaller C-terminal domain has two major sets of functional domains and/or residues: 1) a Trp-rich lipid binding domain and 2) residues involved in interactions with and/or binding to GPIHBP1 [27,28]. The Trp-rich lipid binding domain is contained within residues 412-422 [27,28,77]. This domain is responsible for substrate recognition and has been posited (as discussed above) to be involved in guiding the substrate to the active site in concert with hydrophobic side chains of lid region residues [28]. Finally, many residues in the C-terminal domain of LPL have been found to interact with GPIHBP1. GPIHBP1 interacts with the C-terminal domain of LPL via a Ly6/uPAR (LU) domain with a 3-fingered fold conformation stabilized by five disulfide bonds in GPIHBP1 [27,78,79,80]. LPL residues 443-447 and 465-466 interact with finger 1, residues 447-448 and 463-467 interact with finger 2, and residues 367, 369, 374, 403-406, 447, and 464 interact with finger 3 [27].
Additionally, numerous other residues in the C-terminal domain of LPL are involved in hydrophobic interactions stabilizing the LPL-GPIHBP1 complex [27]. There are also two specific LPL residues found to be involved in hydrogen bonding between LPL and GPIHBP1, Arg447 and Glu384 [27]. GPIHBP1 also binds to an “interdomain interface” formed by LPL residues 306-317 via its intrinsically disordered acidic domain, which is an interaction that seems to be responsible for the ability of GPIHBP1 to stabilize and mitigate the spontaneous unfolding of the N-terminal α/β-hydrolase domain [71]. As noted previously, the second N-linked glycosylation site (Asn386) and the final disulfide bridge forming residues (Cys445-Cys465) are also found within the C-terminal domain of LPL.
With respect to structural isoforms of LPL, historically it was thought that LPL monomers were catalytically inactive/non-functional and head-to-tail homodimerization, previously thought to be facilitated by the action of LMF1 in the ER, was required to produce a catalytically active LPL unit [74,81,82,83]. This head-to-tail homodimer structure was also observed in both of the recent crystallographic studies of the LPL-GPIHBP1 complex [27,28].
However, as discussed at length in both reports of the crystal structure of the LPL-GPIHBP1 complex, logically the head-to-tail homodimer configuration produced by the intercalation of the Trp-rich lipid binding domain of one LPL monomer with the active site of the opposite monomer would prevent substrate binding and catalytic activity [27,28]. Indeed, Arora and colleagues determined that the head-to-tail homodimer structure they observed and studied was likely not physiologically relevant as it was an artifact of the crystal packing interaction utilized in their study [28]. This is consistent with recent observations of catalytically active LPL, both alone and complexed to GPIHBP1, challenging the historical view of the head-to-tail homodimer as the sole functional unit [74].
However, the picture has been further complicated by the recent discovery of two new LPL isoforms, including a new homodimer scheme. First, as previously discussed in the synthesis and expression of LPL section, it was found that LPL may form catalytically inactive helical polymers within secretory vesicles prior to their secretion, likely as a form of storage, indicating a new physiologically relevant non-active form of the protein [17]. Then, even more recently, a new LPL homodimer scheme was discovered.
Specifically, it was found in an in vitro model that the C-terminals of LPL monomers can interface (i.e., a tail-to-tail homodimer) to produce a catalytically active LPL unit [35]. Given the tail-to-tail interaction, it should be noted that this homodimer does not appear to be bound to the endothelial cell surface and instead likely circulates through the bloodstream, further hydrolyzing TG in TRLs and their remnants as it does so [35]. In fact, this finding, while in vitro, is consistent with previous findings of dimeric LPL bound to circulating TRLs that continues to hydrolyze them as they circulate [36,84]. Analysis of the structure of this novel LPL homodimer revealed a hydrophobic pore adjacent to the active site, which was demonstrated to be able to accommodate TG fatty acid chains [35]. It was hypothesized that this hydrophobic pore was responsible for substrate specificity and may also be responsible for facilitating a unidirectional release of FFA from TG hydrolysis, which is rather interesting considering that membrane bound LPL-GPIHBP1 complex releases FFA following TG hydrolysis in a bidirectional manner [35].
5. Genomic Structure of LPL
The LPL gene is located on the short arm of chromosome 8 (8p21.3) and is comprised of 10 exons and 9 introns spanning ~30 kb [25,85]. Exons 1-9 are all roughly similar in size (ranging from 105 to 276 bp in length), with exon 10 being considerably larger at 1950 bp in length, as it encodes the entirety of the 3’ UTR region [25,85]. Given the structural similarities between LPL and other members of the lipase superfamily of proteins, namely hepatic lipase and pancreatic lipase, it seems likely that they derive from a common ancestral gene [25]. As discussed in previous sections, LPL is primarily expressed in adipose tissue, skeletal muscle, and cardiac muscle tissues [4].
6. A Curated Assembly of LPL Variants
Here we report a curated assembly of LPL variants that have been reported in the literature and/or in variant databases as being causative for and/or associated with HTG (Supplementary Tables S1 and S2). We have subdivided this assembly into two tables based on variant pathogenicity determined according to the American College of Medical Genetics and Genomic (ACMG) guidelines [86], with Pathogenic/Likely Pathogenic variants in Supplementary Table S1 and variants of uncertain significance (VUS) in Supplementary Table S2. Supplementary Table S2 additionally contains several variants that may be upgraded from VUS to Likely Pathogenic upon the reporting of new case and/or segregation data and are listed as VUS* to indicate them. Except where the molecular defect is obvious (i.e., premature truncation variants, loss of large number of nucleotides, etc.), detailed notes are provided regarding the evidence and molecular defect(s) associated with each variant (Supplementary Table S1 and S2). Simple notes are provided where the associated defects are obvious. A brief overview and summary of these data are provided in Table 1, Table 2 and Table 3.
Additionally, we have also compiled a non-comprehensive list of common polymorphisms in LPL that have been associated with plasma TG levels (Supplementary Table S3). This list is non-comprehensive as the focus of this review is rare disease-causing variants, not common polymorphisms. We also provide notes regarding the evidence for any molecular effect associated with these variants (Supplementary Table S3).
This curated list of LPL variants was obtained via a similar methodology to that used in our previous review of variants in APOA5 in hypertriglyceridemia [33]. Briefly, we began by compiling LPL variants listed as disease-causing or associated variants in the Human Gene Mutation Database (HGMD) [87], ClinVar [88], and the Leiden Open Variation Database 3.0 (LOVD3) [89] and double-checking the reporting of these variants in the literature where available. We then independently assessed the pathogenicity of these variants using our laboratory pipeline as we have previously described [3,90,91], utilizing the Franklin by Genoox tool (https://franklin.genoox.com) followed by manual curation to determine the pathogenicity classifications for all variants according to the ACMG guidelines [86]. We have also included variants found in our own clinical testing at the Lipid Genetics Clinic, London, Ontario, Canada, if they were considered pathogenic or likely pathogenic under the ACMG guidelines and have not been previously reported in the literature.
We also included several variants from ClinVar and LOVD3 without literature references if they were reported as pathogenic/likely pathogenic in those sources prior to our manual curation. Upon our manual curation, a number of these variants were downgraded to VUS or VUS* (variants of uncertain significance for which additional case and/or segregation data may allow for the ACMG classification to be upgraded to likely pathogenic and/or pathogenic in the future) (Supplementary Table S2). All together, we report a total of 75 variants in our curated assembly without literature references (Supplementary Tables S1 and S2) from our clinic, ClinVar, and LOVD3. We feel that inclusion of these variants is warranted given that they either a) produce obvious molecular defects (i.e., premature truncation, splice site disruption, etc.), b) have multiple forms of indirect evidence supporting a pathogenic effect (i.e., impacting a known or purported functional domain, multiple other variants reported as pathogenic impacting the same residue, etc.), or c) have been observed/reported in clinical testing results as the cause of HTG.
We have excluded variants reported in these sources from our curated assembly based on one or more of the following criteria: a) large scale variants extending to genes other than LPL, b) synonymous variants with no evidence indicating association with TG phenotype, and c) LPL variants reported in association with non-TG related diseases such as neurological and/or developmental disorders. Additionally, we have excluded 183 variants reported in HGMD from analysis and review as at the time of writing, the only literature reference for these variants is a brief report from Deshotels and colleagues regarding the association of these variants and a polygenic risk score constructed utilizing (in part) these variants with HTG in academic lipid clinics [92]. The case data for the carriers of these 180 variants is unclear and/or unavailable, and to the best of our knowledge, they have only been reported in this report from Deshotels and colleagues. Thus, even if some of the variants reported by Deshotels and colleagues are likely deleterious, we felt there is insufficient evidence to warrant including them for detailed analysis in our review, so they have been excluded from analysis and review. We include them in Supplementary Table S4 for readers’ reference.
In total, we compiled a total of 358 unique variants in LPL reported in the literature and/or in variant databases as causative for and/or associated with HTG and related phenotypes (Supplementary Tables S1 and S2). Specifically, we observed 300 pathogenic/likely pathogenic variants (Supplementary Table S1) and 58 VUS/VUS* variants (Supplementary Table S2).
Of the 300 pathogenic/likely pathogenic variants we compiled, 144 are missense variants, 36 are nonsense variants, 52 are small deletions, 21 are small insertions, five are small indels, 29 are splicing variants, 10 are gross deletions, two are gross insertions, one is a complex rearrangement, and one is regulatory region variant (Table 1, Table 2 and Table 3). Of the 58 VUS variants, 55 are missense variants, one is a small deletion, one is a small indel, and one is a regulatory region variant (Table 1 and Table 2).
Several mechanisms of disease have been reported with respect to pathogenic variants in LPL. Some nonsense variants have been hypothesized to trigger nonsense-mediated mRNA decay [93,94,95,96,97]. The principal mechanism of disease, however, appears to be disruption of functionally and/or structurally important domains and/or residues of the LPL peptide, particularly domains and residues involved in the catalytic functionality of the protein specifically. Additionally, catalytically active LPL can have its secretion prevented by variants enhancing the susceptibility of the mature LPL peptide to endoproteolytic cleavage, leading to loss of the C-terminal lipid binding domain [98]. Finally, it has also been shown that it is possible for the immune system to produce antibodies against LPL, leading to loss of their functionality [99]. More details regarding all these mechanisms and their related variants are presented in the proceeding sections.
This is demonstrated by the concentration of pathogenic/likely pathogenic variants observed in the exons encoding these regions compared to the rest of the LPL gene. Specifically, not including large scale variants and variants in non-coding regions of the LPL gene (regulatory and splicing region variants), there are 254 pathogenic/likely pathogenic variants impacting the coding sequence of LPL (Table 1). The distribution of these pathogenic variants across the coding sequence is as follows: 12 (4.72%) are in exon 1, 17 (6.69%) are in exon 2, 38 (14.96%) are in exon 3, 16 (6.30%) are in exon 4, 67 (26.38%) are in exon 5 , 72 (28.35%) are in exon 6, five (1.97%) are in exon 7, 23 (9.06%) are in exon 8, and the final four (1.57%) are in exon 9. The vast majority of these impact exons 2-6 (82.68%) which together encode the N-terminal α/β-hydrolase domain of mature LPL [28], with exons 5 and 6 accounting for a particularly large proportion of the coding region pathogenic/likely pathogenic variants observed at 54.72%. T
he increased concentration of pathogenic/likely pathogenic variants specifically in exons 5 and 6 likely stems from the fact that these two exons encode a large number of functionally and structurally important residues in mature LPL, including but not limited to 1) the majority of the residues forming the active site, including two of the catalytic triad residues (Asp183 and H268), 2) the entirety of the lid region (residues between Cys243 and Cys266), 3) all five calcium ion coordination residues (Ala194, Arg197, Ser199, Asp201, and Asp202), and 4) three disulfide bridge forming residues known to be essential (Cys243–Cys266, Cys291–Cys302, and Cys305–Cys310) (see Protein Structure of LPL section for more details on these domains). Exons 2-4 also encode many residues that line the active site and are likely involved in several functions such as apo C-II binding and lipid substrate interactions as previously discussed in the Protein Structure of LPL section.
We have summarized the coding sequence variants resulting in amino acid changes alongside the relative positions of major functional domains of LPL in Figures 1 to 9, with each figure presenting the region of the LPL peptide encoded by exons 1 to 9, respectively. The nucleic acid changes for noncoding region variants and large-scale variants (i.e., gross deletions, gross insertions, complex rearrangements, etc.) are summarized in Figure 10. A discussion and analysis of the variants summarized in these figures is presented below, with notes on some specific variants of particular interest. Detailed notes for each variant can be found in Supplementary Tables S1 and S2. We also discuss briefly the roles and associations of some benign and likely benign variants for which specific notes for each variant can be found in Supplementary Table S3.
7. Coding Region Variants
7.1. Exon 1 – Signal Peptide
Exon 1 encodes the entirety of the signal peptide and the first few residues of the mature LPL peptide. Within our curated assembly, all variants reported within exon 1 impact codons of the signal peptide, though several do lead to frameshifts that lead to premature termination codon downstream of the signal peptide region (Figure 1). Additionally, all pathogenic variants reported within exon 1 are predicted to lead to a prematurely truncated protein (Figure 1).
This includes four variants eliminating the normal translation initiation codon. Of these, the most interesting is the LPL:c.3G>C:p.Met1Ile variant, as two of the in vitro characterizations of LPL activity and mass for the variant peptide have some contradictions. Specifically, the first report of this variant found that the variant had near completely abolished activity at just 2% of the WT level and protein mass at ~3% of WT level [100]. The second study that analyzed this variant in vitro found that the variant has an activity level 55.93 ± 3.28% with completely abolished protein mass [101], indicating a potential secretion defect. The residual LPL mass level detected in the initial report of this variant may be because it is possible for non-AUG translation initiation codons to be used to initiate translation, though this typically occurs at a reduced efficiency compared to normal translation initiation codon [102,103]. The protein activity level discrepancy is far more surprising. In theory, if the protein was synthesized and secreted appropriately then it should not have an activity defect. Further investigation is needed to verify what the true molecular defect is, as currently, an explanation for this discrepancy is not obvious.
The single VUS variant we identified, LPL:c.26T>G:p.Leu9Arg, is quite interesting. The variant has been reported in the heterozygous state in an individual with severe HTG and the variant was found to be associated with mildly reduced LPL mass [104]. While no direct experimental evidence is available, it is plausible that this variant has an associated true molecular defect as the mutated residue seems to by part of the hydrophobic core of the signal peptide, with the non-polar Leu at this position considered important in humans and other mammals [105,106]. Thus, the substitution of a charged amino acid like Arg at this position may disrupt normal functioning and/or folding of the signal peptide. In fact, in at least one analysis [106], it has been demonstrated that polar residues, and likely charged residues by extension, are not found within wild-type h-motifs and it seems that this variant lies within an h-motif of the LPL signal peptide. However, experimental confirmation would be needed to state for sure whether this variant is truly pathogenic or not.
7.2. Exons 2-6 – N-Terminal α/β-Hydrolase Domain
Exon 2 encodes several key functional motifs, including two disulfide bond forming residues (Cys54-Cys67) [27,28], the first N-linked glycosylation site (Asn70) which is considered essential for proper LPL folding and functionality [7,107], a portion of a motif that has been demonstrated to interact with both apo C-II and ANGPTL4 [30,76], and the first oxyanion hole forming residue (Trp82) (Figure 2). All but four of the pathogenic variants found in exon 2 are premature truncating variants, which have obvious molecular consequences.
Of the four remaining pathogenic variants, the molecular defect has only been determined for two. First, the LPL:c.188C>T:p.Ser63Phe variant was observed in compound heterozygosity with a pathogenic exon 5 variant LPL:c.662T>C:p.Ile221Thr in a patient with lipemic plasma and an in vitro study in HEK 293 cells determined this variant decreased LPL activity by ~62%, which was mirrored by ~54% reduction in post-heparin plasma LPL activity in the patient compared to wild-type [101]. The second is LPL:c.209A>G:p.Asn70Ser which eliminates the first N-linked glycosylation site by altering Asn70 to Ser (Figure 2). This variant was observed in homozygous state in an LPL deficient patient and it has been shown that glycosylation of this residue is essential for proper LPL folding and function [7,107].
Exon 3 begins with encoding the remainder of the apo C-II and ANGPTL4 binding motif encoded in exon 2 and encodes an additional purported apo C-II and ANGPTL4 binding motif [30,76] (Figure 3). Exon 3 contains 38 pathogenic/likely pathogenic coding variants, with 20 of these being premature truncation variants with obvious consequences. The remaining 18 pathogenic variants consist of missense variants, with 7 impacting residues in the 2nd apo C-II and ANGPTL4 binding motif (Figure 3).
While it may appear that a number of these pathogenic missense variants do not directly impact functionally important residues, this is not the case. As previously noted by Birrane and colleagues in their structural characterization of the LPL-GPIHBP1 complex, there are a number of hydrophobic residues lining the active site, including several encoded in exon 3, that are purported to be involved in van der Waals interactions with the hydrophobic tails of lipid substrate in the active site [27].
Generally, the pathogenic missense variants of this exon seem to produce one or more of the following molecular defects (as determined by in vitro functional studies): 1) reduced catalytic activity, 2) reduced protein secretion, 3) reduced protein mass, and/or 4) reduced protein stability (Supplementary Table S1). Perhaps most interesting of these variants is LPL:c.429G>T:p.Glu143Asp. The molecular defect of this variant most likely arises from the nucleotide change and not from the amino acid change, as this variant impacts the final nucleotide of exon 3 which places it in the canonical donor splice site. In silico analyses performed by the original reporters of this variant found that this variant is predicted to decrease functionality of the adjacent donor splice site, and in turn may activate an alternate donor splice site in intron 3, leading to altered splicing of intron 3 [108]. Thus, while the amino acid change may not be deleterious in and of itself, the DNA change likely is.
Exon 4 encodes three primary sets of functional residues: 1) Ser159 is the first catalytic triad residue [27,28], 2) Leu160 is the final oxyanion hole residue [25,28,75], and 3) many of the other residues in this portion of the protein are hydrophobic residues lining the active site that are purported to be involved in van der Waals interactions with lipid substrate entering the active site [27] (Figure 4).
At the time of writing, only one pathogenic variant of the catalytic triad codon has been reported, LPL:c.476G>C:p.Ser159Thr [109]. The molecular defect of this variant is rather obvious given that it alters a highly conserved key residue directly involved in the catalytic functionality of LPL [109]. Specifically, it was found that this variant, reported in two homozygous siblings presenting with FCS, was associated with normal pre-heparin LPL mass, but heparin administration showed no increase in plasma LPL mass, indicating a loss of catalytic function [109].
Similarly, only one pathogenic variant has been reported directly affecting the codon for the final oxyanion hole residue, LPL:c.478C>T:p.Leu160Phe [110]. This variant was reported in homozygous state in a neonate presenting with FCS [110]. While functional data are not available, it is likely that this variant eliminates the functionality of the oxyanion hole, severely hampering catalytic functionality of the protein.
Like the LPL:c.429G>T:p.Glu143Asp variant in exon 3, two variants in exon 4, LPL:c.541G>C:p.Gly181Arg and LPL:c.541G>A:p.Gly181Ser, have been observed to impact the final nucleotide. Interestingly, while LPL:c.541G>C:p.Gly181Arg is predicted to result in alternative splicing, LPL:c.541G>A:p.Gly181Ser was shown to not impact splicing at all [111].
The remaining pathogenic coding sequence variants in this exon effect residues that form the active site (Figure 4).
With respect to the VUS variants located within exon 4, the LPL:c.451G>A:p.Asp151Asn is interesting as it was observed to co-segregate with familial combined hyperlipidemia (FCHL) in a pedigree from Iran and was absent in unaffected individuals [112]. Unfortunately, functional characterization data are not available making it difficult to determine if this variant has a deleterious effect itself or it is linked to another variant that does.
Exon 5 accounts for a large proportion of all pathogenic coding variants observed in LPL (Figure 5). This seems to be due to the large concentration of functional residues and domains encoded by exon 5 as well as the fact that most of the residues encoded by exon 5 form part of the structure of the active site [27,28]. Exon 5 encodes 1) the 2nd catalytic triad residue, Asp183 [27,28], 2) all five residues involved in coordinating a Ca2+ into the 3D LPL structure [27], 3) an apo C-II binding motif [30], 4) the lid region [27,28], 5) an ANGPTL4 binding motif [76], and 6) a cysteine residue involved in forming a disulfide bridge [8,27,28].
Notably, most pathogenic/likely pathogenic variants we observed in this exon appear to directly alter functional domains and/or residues. Three separate variants have been reported altering the 2nd catalytic triad residue, of which only LPL:c.547G>A:p.Asp183Asn and LPL:c.548A>G:p.Asp183Gly have been functionally characterized. The former was found to be normally secreted but had essentially zero catalytic activity [113] while the latter was observed in two separate in vitro studies to result in reduced LPL mass and completely abolished activity [113,114]. This difference is most likely explained by the differences in size, structure, and/or electrochemical properties between Asn and Gly. While both variants eliminate the negatively charged Asp, Asn is much more similar in size to Asp whereas Gly is much smaller, which may explain why LPL:c.548A>G:p.Asp183Gly has an additional mass defect associated with it compared to the LPL:c.547G>A:p.Asp183Asn which for which only catalytic activity is reduced.
Eight variants were found to directly alter residues involved in the coordination of Ca2+ ion into the structure of LPL (Figure 5). Of these, four impact residue 197, two impact residue 199, one impacts residue 201, and one impacts residue 202. Based on functional characterizations performed for three of these variants (LPL:c.590G>A:p.Arg197His, LPL:c.596C>G:p.Ser199Cys, and LPL:c.602A>T:p.Asp201Val), it appears that variants altering these Ca2+ coordinating residues all lead to reduced or completely abolished secretion of protein [27,115,116], with additional defects observed based on the specific variant (Supplementary Table S1). Supporting this observation is further in vitro functional characterization of alterations to Asp202, which found that LPL:Asp202Glu is not secreted [27]. Interestingly though, it was shown that LPL:Asp201Glu would be tolerated as while Asp interfaces with the Ca2+ ion indirectly via an intervening water molecule, Glu at the 201 residue can interface directly with the Ca2+ ion by displacing the water molecule [27].
The frameshift variant impacting one of the Ca2+ coordinating residues, LPL:c.596delC:p.Ser199Phefs*8, is particularly interesting as it was discovered in a heterozygous patient with no other TG-elevating variants suffering from recurrent HTG-linked pancreatitis episodes and it was found that this variant was uniquely associated with an anti-LPL antibody that partially inhibited wild-type LPL activity in vitro and immunosuppressive therapy via azathioprine treatment significantly reduced plasma TG levels in this patient [99]. As the authors of this report note, it is not known whether the presence of prematurely truncated LPL peptide due to the variant triggered the autoimmune response against LPL [99], but autoantibodies against LPL have been found in patients with autoimmune diseases such as systemic lupus erythematosus [117,118,119,120]. However, the heterozygous patient carrying the LPL:c.596delC:p.Ser199Phefs*8 variant had no personal history of autoimmune disease but their father had vitiligo [99]. Furthermore, as Pruneta-Deloche and colleagues note, the autoantibodies against LPL observed in patients with autoimmune disease seem to only result in a ~10% elevation in plasma TG levels, which on its own would be insufficient to induce the severe HTG required to cause pancreatitis [99]. Consequently, they hypothesized that in heterozygous LPL deficient conditions, like those produced by heterozygosity for the premature truncation variant p.Ser199Phefs*8, anti-LPL autoantibodies may induce severe HTG by inhibiting the lower amount of wild-type LPL available [99]. This presents a unique disease mechanism and is consistent with our findings of secondary factors often being necessary to induce severe HTG in heterozygosity for LOF variants in LPL [3].
Interestingly, while functional data are unavailable, the variant affecting the final nucleotide of exon 5, LPL:c.775G>A:p.Asp259Asn, is predicted to result in alternative splicing by both MES and SpliceAI (Supplementary Table S1) as the final nucleotide of exon 5 lies within the canonical donor splice site for intron 5.
As previously noted, among all 9 coding exons of LPL, exon 6 contains the largest number of pathogenic coding variants (Figure 6). Exon 6 encodes several key functional residues/domains: 1) a portion of the lid region [27,28], 2) several disulfide bridge forming residues [8,27,28], and 3) the final catalytic triad residue, His268 [27,28]. The other residues encoded in this exon appear to contribute to active site structure [27,28].
Two variants, LPL:c.802C>T:p.His268Tyr and LPL:c.804C>A:p.His268Gln, have been observed impacting the final catalytic triad residue (Figure 6). Direct functional data are not available for these variants, but analysis of LPL protein expression in a proband homozygous for LPL:c.802C>T:p.His268Tyr found abolished protein expression [121].
Like the variant affecting the final nucleotide of exon 5, the variant affecting the final nucleotide of exon 6, LPL:c.1018G>T:p.Val340Phe, is also predicted to result in alternative splicing (Supplementary Table S2). However, unlike the variant in exon 5, this variant is currently considered VUS due to a paucity of evidence that can be used to assess its pathogenicity.
Most variants in exon 6 do not directly affect residues forming functional domains (Figure 6), but functional characterizations for many of these variants (summarized in Supplementary Table S1 with references) consistently found that variants in this region result in reduced or completely abolished catalytic activity, with additional defects in synthesis and secretion also observed for some variants. This is consistent with the fact that exon 6 encodes a large portion of the active site of mature LPL.
7.3. Exons 7-9 – C-Terminal LIPID binding Domain
Exon 7 has very few pathogenic variants at five total (Figure 7). This is perhaps in line with the fact that exon 7 only contains a small number of directly functionally important residues, namely residues 367, 369, and 374 which are involved in the LPL-GPIHBP1 interaction, specifically with finger 3 of the GPIHBP1 LU domain [27].
Of the five pathogenic coding variants reported in this exon, three are premature truncating variants with rather obvious molecular consequences. The remaining two are missense variants (LPL:c.1051G>A:p.Gly351Arg and LPL:c.1081G>A:p.Ala361Thr) but functional data is not available for either of them. They are considered pathogenic as both have been reported to produce FCS in the homozygous state [122,123,124].
Functional data are unavailable for all but one VUS variant, LPL:c.1094C>T:p.Ser365Phe. While this variant was observed in a patient with normal post-heparin plasma LPL activity and mass, an in vitro experiment found that this variant mildly impaired protein secretion, but simultaneously increased specific activity of the enzyme [125]. This increased specific activity was concluded to be sufficient to compensate for the reduced protein secretion [125]. However, it is plausible that additional factors elevating plasma TGs and/or impacting LPL functionality may overwhelm this compensatory effect, leading to a net deleterious impact for this variant.
Exon 8 contains the bulk of the functional residues and domains of the C-terminal lipid binding domain, including the titular lipid binding domain (Figure 8). Exon 8 encodes 1) several GPIHBP1 interacting residues, including a residue that forms a hydrogen bond with GPIHBP1 and residues involved in interacting with finger 3 of the GPIHBP1 LU domain [27], 2) the final N-linked glycosylation site, Asn386 [27,28], and 3) the Trp-rich lipid binding domain formed by residues 412-422 [27,28,77].
Most pathogenic variants in this exon are premature truncating variants, with only five pathogenic missense variants observed (Figure 8). Of these five, functional data supporting a deleterious effect is available for LPL:c.1187A>T:p.Glu396Val [126], LPL:c.1211T>G:p.Met404Arg [95], LPL:c.1309G>A:p.Glu437Lys [127], and LPL:c.1310A>T:p.Glu437Val [98]. LPL:c.1310A>T:p.Gly437Val is particularly interesting as it was found that this variant renders the LPL peptide susceptible to cleavage by the endoproteolytic enzyme furin at residue 324, a known furin cleavage site in LPL [98]. This presents a potential post-translational mechanism of disease which is supported by the finding of FCS in a homozygote for this variant, in which LPL activity was reduced to 11% of wild-type level [128].
Exon 9 has the fewest pathogenic variants observed in any exon at four total (Figure 9). The major functional features encoded in exon 9 are a number of GPIHBP1 interacting residues as well as the final two disulfide bridge forming residues (Cys445-Cys465) [27,28]. All four pathogenic variants are missense variants. However, of these, only the LPL:c.1334G>A:p.Cys445Tyr and LPL: c.1342G>A:p.Glu448Lys variants have been previously reported in the literature (Supplementary Table S1). It has been demonstrated that both of these variants abolish the ability of LPL to bind to GPIHBP1 while retaining the functionality of the N-terminal α/β hydrolase domain [129]. While other studies have observed a reduction in LPL activity due to these variants, a similar reduction in LPL mass was also observed [130,131], which would indicate that the specific activity of LPL is unchanged, which supports the finding that functionality of the N-terminal α/β hydrolase domain is retained. As a result of the loss of GPIHBP1 binding, LPL transport to the apical surface is eliminated [129].
8. Non-Coding and Large-Scale Variants
Figure 10 outlines the non-coding and large scale (gross deletions, gross insertions, and large-scale complex rearrangements) variants reported in LPL to be associated with and/or produce disease. To the best of our knowledge, only one regulatory region variant in LPL reported in the literature is not considered benign or likely benign according to the ACMG guidelines, LPL:c.-227T>C [132]. This variant was reported in compound heterozygous state with LPL:c.-281T>G in a patient diagnosed with familial combined hyperlipidemia and reduced post-heparin plasma LPL activity, with this variant specifically impacting an Oct-1 transcription factor binding site leading to reduction in transcriptional activity of the variant promoter to less than 15% of wild type [132].
Figure 10.
Gene map of reported LPL noncoding sequence and large-scale variants. Gene map of LPL annotated with variants discovered in regulatory regions 5’ and 3’ untranslated regions (UTRs), promoter region, etc., splice donor and acceptor sites, and introns. Numbering underneath boxes indicates exons. Black boxes indicate untranslated sequences, blue box indicates the sequence encoding the LPL signal peptide, and green boxes indicates coding sequences. For large scale variants, arrows below gene map represent the approximate region of the gene affected with variant annotations above the arrows. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign.
Figure 10.
Gene map of reported LPL noncoding sequence and large-scale variants. Gene map of LPL annotated with variants discovered in regulatory regions 5’ and 3’ untranslated regions (UTRs), promoter region, etc., splice donor and acceptor sites, and introns. Numbering underneath boxes indicates exons. Black boxes indicate untranslated sequences, blue box indicates the sequence encoding the LPL signal peptide, and green boxes indicates coding sequences. For large scale variants, arrows below gene map represent the approximate region of the gene affected with variant annotations above the arrows. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign.
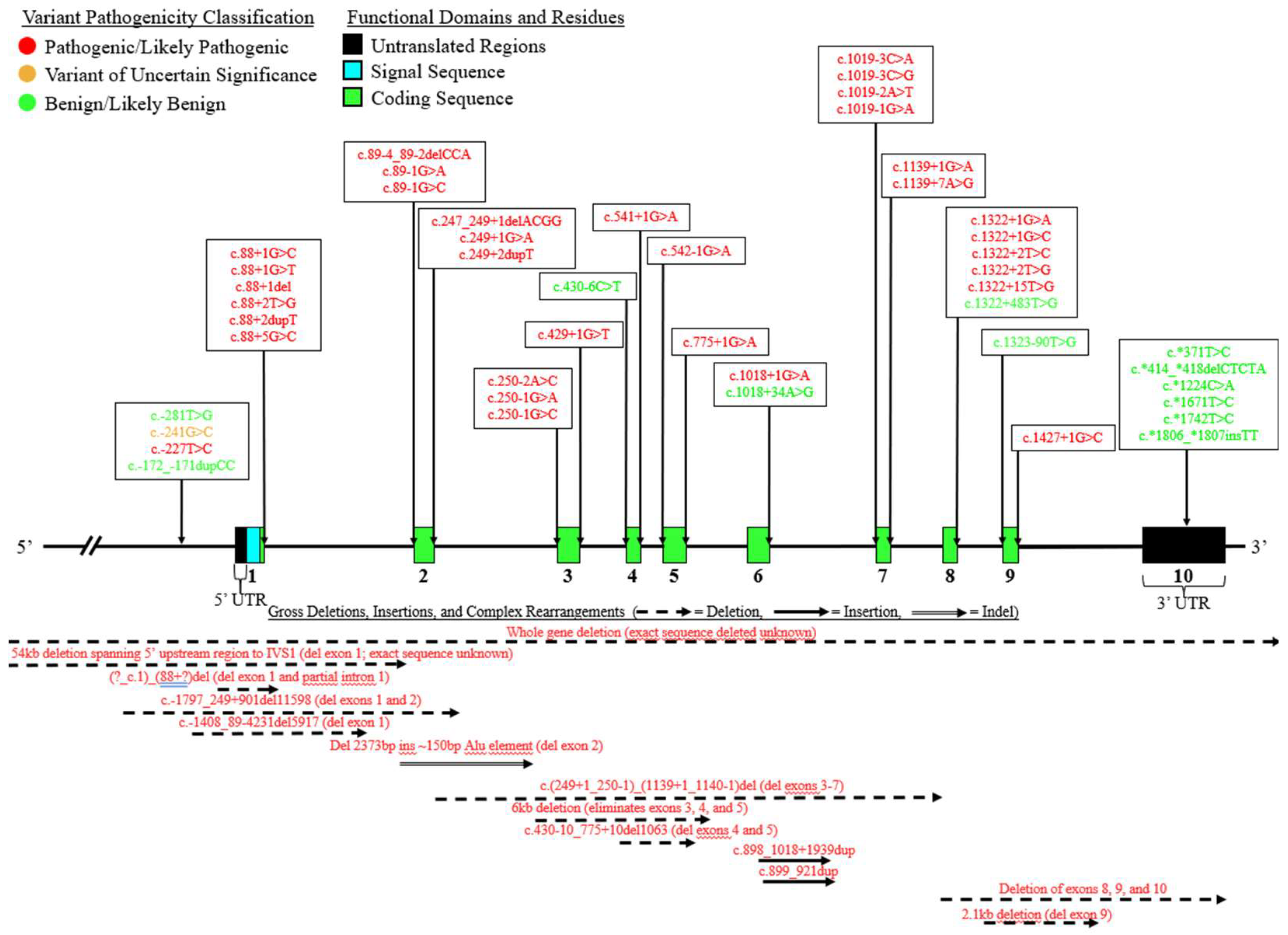
With respect to splicing variants, of the 29 pathogenic variants found to affect either the donor or acceptor splice sites in LPL, the most affected intron is intron 1 with 6 variants impacting the donor splice site and three affecting the acceptor splice site (Figure 10). All pathogenic/likely pathogenic splicing variants have been demonstrated or strongly predicted to abolish the affected splice site functionality, leading to alternative pre-mRNA splicing (Supplementary Table S1).
With respect to large scale variants, there are 10 gross deletions, two gross insertions, and one complex rearrangement for a total of 13 included in our curated list (Figure 10). Apart from the two gross insertions variants, all of these variants lead to loss of one or more exons. Rather interesting is the complex rearrangement variant which, to the best of our knowledge, presents a novel mutational mechanism by which variants in LPL may occur. Specifically, a novel variant leading to LPL deficiency was reported by Okubo and colleagues resulting from Alu retro transposition producing a complex large indel [94]. The major consequence of this variant was the deletion of exon 2, which contains a number of functionally important residues including the essential Asn70 N-linked glycosylation site [7,107], and the beginning portion of the first apo C-II and ANGPTL4 binding motif [30,76] (Figure 10). While this appears to be the only LPL variant reported due to such a mechanism, it is nevertheless evidence of a novel mechanism by which LPL deficiency may occur.
9. Benign/Likely Benign Variants: Deleterious and Gain-of-Function Variants
We include here a discussion of 21 select benign/likely benign variants for the purpose of highlighting the role of these common variants and various features of the LPL gene in determining plasma TG phenotype (Supplementary Table S3).
Most of these variants are common, with only three variants considered to be rare. The first of these, LPL:c.182C>T:p.Ala61Val, was found to likely be in linkage disequilibrium with the pathogenic splicing variant LPL:c.250-1G>C [2]. The second, LPL:c.1134C>G:p.Phe378Leu, was shown to have no effect on mass nor activity in in vitro studies but it was observed in a heterozygote with severe HTG [133]. The third, LPL:c.*414_*418delCTCTA, was shown to modestly reduce LPL gene expression and very slightly reduce translation, likely due to the fact that this small deletion in the 3’UTR disrupts a putative insulin response element [134].
While several of the common benign/likely benign variants we list have been associated with HTG and/or functional defects, such as reduced promoter activity, catalytic activity, etc., the fact that these variants are relatively common in population databases indicates that they are not true LOF variants (Supplementary Table S3). For example, LPL:c.953A>G:p.Asn318Ser has been shown in multiple in vitro studies to reduce LPL catalytic activity [135,136,137] and has also been reported to be modestly associated with HTG [42,137,138,139]. However, the variant is extremely common in population databases and 36 homozygous individuals have been reported in gnomAD [140], which indicates that even if there is a deleterious effect for this variant, it is small and not enough to qualify as a LOF variant.
Nevertheless, several of these common benign/likely benign variants associated with a deleterious effect are quite interesting, such as LPL:c.-281T:G and LPL:c.1322+483T>G. The promoter variant LPL:c.-281T>G was shown to reduce the activity of the LPL promoter in in vitro experiments [132,141]. Interestingly though, LPL:c.-281T>G was later associated with a protective effect on plasma TG levels during the third trimester of pregnancy in women of African American descent [142]. The intron 8 variant LPL:c.1322+483T>G was shown to be associated with a ~20% reduction in gene transcription, likely due to this variant altering a transcription factor binding site in intron 8 [143]. This, alongside the discovery of a common gain-of-function (GOF) variant in intron 8, LPL:c.1323-90T>G, that increases gene expression by ~1.7 times the wild-type level [144] demonstrate the importance of intron 8 in regulating LPL expression.
While our list of variants is not necessarily comprehensive, there is an interesting trend in that nearly all common GOF variants in our list are concentrated toward the end of the coding region of the LPL gene, with most located in exons 9 and 10, the latter of which encodes the 3’ UTR (Figure 10 and Supplementary Table S3).
Perhaps the most notable of these GOF variants is LPL:1421C>G:p.Ser474Term, a common GOF nonsense variant. This variant has been observed in an extremely large number of studies [42,138,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190] and has been reported to increase LPL mass and activity [191] and reduce inhibition of translation [167]. Interestingly, it has also been shown that this variant is in linkage disequilibrium with a separate TG-lowering haplotype which exerts its effect by abolishing miRNA-mediated post-transcription inhibition of LPL [192,193]. It has been suggested that, at least in part, the association of LPL:1421C>G:p.Ser474Term with this haplotype explains its TG-lowering effect [192]. Several of the common 3’ UTR variants we include in our list are part of this haplotype (Supplementary Table S3).
10. Conclusion
LPL is an extremely important regulator of plasma TG metabolism. Here we have summarized the current understanding of the regulation, expression, structure, and physiological roles of LPL as they relate to plasma TG metabolism, and we have provided a comprehensive overview of genetic variation in LPL as it pertains to HTG. We consider the curation and archiving of LPL DNA variants here to be the first draft for future efforts to create a public reference database for both research and clinical purposes. With respect to the latter, there is growing interest in the potential clinical relevance of LPL gene variants detected by clinical next-generation DNA sequencing efforts in patients with various HTG phenotypes, particularly those who are suspected to have FCS. We hope our present curation effort will serve as a bridge towards a definitive LPL variant reference list assembled via a sanctioned and recognized variant curation expert panel and process.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Supplementary Table S1: Curated List of Pathogenic/Likely Pathogenic Variants in LPL.; Supplementary Table S2: Curated List of Variants of Uncertain Significance in LPL.; Supplementary Table S3: Select Benign/Likely Benign Variants Reported in Association with Various TG Phenotypes; Supplementary Table S4: LPL Variants Reported in HGMD Excluded from Analysis and Review to Unavailable Case Data.
Author Contributions
Conceptualization, S.D.P. and R.A.H.; methodology, S.D.P., J.W, A.D.M. and R.A.H.; software, S.D.P., J.W.; validation, S.D.P., J.W, and A.D.M..; formal analysis, S.D.P., J.W, A.D.M. and R.A.H.; investigation, S.D.P. and R.A.H.; resources, S.D.P. and R.A.H.; data curation, S.D.P.; writing—original draft preparation, S.D.P. and R.A.H.; writing—review and editing, , S.D.P., J.W, A.D.M. and R.A.H.; visualization, S.D.P.; supervision, R.A.H project administration, R.A.H.; funding acquisition, R.A.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research and the APC were funded by the Jacob J. Wolfe Distinguished Research Chair in Human Gene Function.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Additional data are available by contacting R.A.H.
Acknowledgments
Not applicable
Conflicts of Interest
The authors declare no conflicts of interest related to the subject matter of this manuscript.
References
- Young, S.G.; Zechner, R. Biochemistry and Pathophysiology of Intravascular and Intracellular Lipolysis. Genes Dev 2013, 27, 459–484. [Google Scholar] [CrossRef]
- Hegele, R.A.; Berberich, A.J.; Ban, M.R.; Wang, J.; Digenio, A.; Alexander, V.J.; D’Erasmo, L.; Arca, M.; Jones, A.; Bruckert, E.; et al. Clinical and Biochemical Features of Different Molecular Etiologies of Familial Chylomicronemia. Journal of Clinical Lipidology 2018, 12, 920–927.e4. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.D.; Wang, J.; McIntyre, A.D.; Dron, J.S.; Hegele, R.A. The Longitudinal Triglyceride Phenotype in Heterozygotes with LPL Pathogenic Variants. Journal of Clinical Lipidology 2023, 17, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Physiological Regulation of Lipoprotein Lipase. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2014, 1841, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Goulbourne, C.N.; Gin, P.; Tatar, A.; Nobumori, C.; Hoenger, A.; Jiang, H.; Grovenor, C.R.M.; Adeyo, O.; Esko, J.D.; Goldberg, I.J.; et al. The GPIHBP1–LPL Complex Is Responsible for the Margination of Triglyceride-Rich Lipoproteins in Capillaries. Cell Metabolism 2014, 19, 849–860. [Google Scholar] [CrossRef]
- Wu, S.A.; Kersten, S.; Qi, L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol Metab 2021, 32, 48–61. [Google Scholar] [CrossRef]
- Ben-Zeev, O.; Stahnke, G.; Liu, G.; Davis, R.C.; Doolittle, M.H. Lipoprotein Lipase and Hepatic Lipase: The Role of Asparagine-Linked Glycosylation in the Expression of a Functional Enzyme. J Lipid Res 1994, 35, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.Y.; Smith, L.C.; Chan, L. Lipoprotein Lipase:Role of Intramolecular Disulfide Bonds in Enzyme Catalysis. Biochemical and Biophysical Research Communications 1995, 206, 266–271. [Google Scholar] [CrossRef]
- Roberts, B.S.; Babilonia-Rosa, M.A.; Broadwell, L.J.; Wu, M.J.; Neher, S.B. Lipase Maturation Factor 1 Affects Redox Homeostasis in the Endoplasmic Reticulum. EMBO J 2018, 37, e97379. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR Pathways. Trends Biochem Sci 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Sha, H.; Sun, S.; Francisco, A.; Ehrhardt, N.; Xue, Z.; Liu, L.; Lawrence, P.; Mattijssen, F.; Guber, R.; Panhwar, M.S.; et al. The ER-Associated Degradation Adaptor Protein Sel1L Regulates LPL Secretion and Lipid Metabolism. Cell Metab 2014, 20, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Ailhaud, G. Cellular and Secreted Lipoprotein Lipase Revisited. Clinical Biochemistry 1990, 23, 343–347. [Google Scholar] [CrossRef]
- Sundberg, E.L.; Deng, Y.; Burd, C.G. Syndecan-1 Mediates Sorting of Soluble Lipoprotein Lipase with Sphingomyelin-Rich Membrane in the Golgi Apparatus. Developmental Cell 2019, 51, 387–398.e4. [Google Scholar] [CrossRef] [PubMed]
- Klinger, S.C.; Glerup, S.; Raarup, M.K.; Mari, M.C.; Nyegaard, M.; Koster, G.; Prabakaran, T.; Nilsson, S.K.; Kjaergaard, M.M.; Bakke, O.; et al. SorLA Regulates the Activity of Lipoprotein Lipase by Intracellular Trafficking. Journal of Cell Science 2011, 124, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Bankaitis, V.A.; Wang, Y. Lipoprotein Lipase Sorting: Sphingomyelin and a Proteoglycan Show the Way. Trends in Cell Biology 2020, 30, 170–172. [Google Scholar] [CrossRef]
- Pfeffer, S.R.; Rothman, J.E. BIOSYNTHETIC PROTEIN TRANSPORT AND SORTING BY THE ENDOPLASMIC RETICULUM AND GOLGI. Annual Review of Biochemistry 1987, 56, 829–852. [Google Scholar] [CrossRef] [PubMed]
- Gunn, K.H.; Roberts, B.S.; Wang, F.; Strauss, J.D.; Borgnia, M.J.; Egelman, E.H.; Neher, S.B. The Structure of Helical Lipoprotein Lipase Reveals an Unexpected Twist in Lipase Storage. Proc Natl Acad Sci U S A 2020, 117, 10254–10264. [Google Scholar] [CrossRef]
- Allan, C.M.; Larsson, M.; Jung, R.S.; Ploug, M.; Bensadoun, A.; Beigneux, A.P.; Fong, L.G.; Young, S.G. Mobility of “HSPG-Bound” LPL Explains How LPL Is Able to Reach GPIHBP1 on Capillaries. Journal of Lipid Research 2017, 58, 216–225. [Google Scholar] [CrossRef]
- Davies, B.S.J.; Beigneux, A.P.; Barnes, R.H.; Tu, Y.; Gin, P.; Weinstein, M.M.; Nobumori, C.; Nyrén, R.; Goldberg, I.; Olivecrona, G.; et al. GPIHBP1 Is Responsible for the Entry of Lipoprotein Lipase into Capillaries. Cell Metab 2010, 12, 42–52. [Google Scholar] [CrossRef]
- Young, S.G.; Fong, L.G.; Beigneux, A.P.; Allan, C.M.; He, C.; Jiang, H.; Nakajima, K.; Meiyappan, M.; Birrane, G.; Ploug, M. GPIHBP1 and Lipoprotein Lipase, Partners in Plasma Triglyceride Metabolism. Cell Metabolism 2019, 30, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.S.J.; Goulbourne, C.N.; Barnes, R.H.; Turlo, K.A.; Gin, P.; Vaughan, S.; Vaux, D.J.; Bensadoun, A.; Beigneux, A.P.; Fong, L.G.; et al. Assessing Mechanisms of GPIHBP1 and Lipoprotein Lipase Movement across Endothelial Cells. J Lipid Res 2012, 53, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Hahn, P.F. Abolishment of Alimentary Lipemia Following Injection of Heparin. Science 1943, 98, 19–20. [Google Scholar] [CrossRef]
- Korn, E.D. CLEARING FACTOR, A HEPARIN-ACTIVATED LIPOPROTEIN LIPASE. Journal of Biological Chemistry 1955, 215, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Korn, E.D. CLEARING FACTOR, A HEPARIN-ACTIVATED LIPOPROTEIN LIPASE. Journal of Biological Chemistry 1955, 215, 15–26. [Google Scholar] [CrossRef]
- Mead, J.R.; Irvine, S.A.; Ramji, D.P. Lipoprotein Lipase: Structure, Function, Regulation, and Role in Disease. J Mol Med 2002, 80, 753–769. [Google Scholar] [CrossRef]
- Santamarina-Fojo, S.; Brewer, H.B. Lipoprotein Lipase: Structure, Function and Mechanism of Action. Int J Clin Lab Res 1994, 24, 143–147. [Google Scholar] [CrossRef]
- Birrane, G.; Beigneux, A.P.; Dwyer, B.; Strack-Logue, B.; Kristensen, K.K.; Francone, O.L.; Fong, L.G.; Mertens, H.D.T.; Pan, C.Q.; Ploug, M.; et al. Structure of the Lipoprotein Lipase–GPIHBP1 Complex That Mediates Plasma Triglyceride Hydrolysis. Proc Natl Acad Sci U S A 2019, 116, 1723–1732. [Google Scholar] [CrossRef]
- Arora, R.; Nimonkar, A.V.; Baird, D.; Wang, C.; Chiu, C.-H.; Horton, P.A.; Hanrahan, S.; Cubbon, R.; Weldon, S.; Tschantz, W.R.; et al. Structure of Lipoprotein Lipase in Complex with GPIHBP1. Proc Natl Acad Sci U S A 2019, 116, 10360–10365. [Google Scholar] [CrossRef] [PubMed]
- Wolska, A.; Dunbar, R.L.; Freeman, L.A.; Ueda, M.; Amar, M.J.; Sviridov, D.O.; Remaley, A.T. Apolipoprotein C-II: New Findings Related to Genetics, Biochemistry, and Role in Triglyceride Metabolism. Atherosclerosis 2017, 267, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Grønnemose, A.L.; Kristensen, K.K.; Winther, A.-M.L.; Young, S.G.; Jørgensen, T.J.D.; Ploug, M. Inverse Effects of APOC2 and ANGPTL4 on the Conformational Dynamics of Lid-Anchoring Structures in Lipoprotein Lipase. Proceedings of the National Academy of Sciences 2023, 120, e2221888120. [Google Scholar] [CrossRef] [PubMed]
- Gin, P.; Yin, L.; Davies, B.S.J.; Weinstein, M.M.; Ryan, R.O.; Bensadoun, A.; Fong, L.G.; Young, S.G.; Beigneux, A.P. The Acidic Domain of GPIHBP1 Is Important for the Binding of Lipoprotein Lipase and Chylomicrons∗. Journal of Biological Chemistry 2008, 283, 29554–29562. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Nelbach, L.; Weinstein, M.M.; Burgess, B.L.; Beckstead, J.A.; Young, S.G.; Ryan, R.O.; Forte, T.M. Intravenous Injection of apoA-V Reconstituted HDL Decreases Hypertriglyceridemia in Apoav-/- Mice and Requires GPIHBP1. Arterioscler Thromb Vasc Biol 2010, 30, 2504–2509. [Google Scholar] [CrossRef]
- Perera, S.D.; Hegele, R.A. Genetic Variation in Apolipoprotein A-V in Hypertriglyceridemia. Current Opinion in Lipidology 2024, 35, 66. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.K.; Walzem, R.L.; Davies, B.S.J. A Novel NanoBiT-Based Assay Monitors the Interaction between Lipoprotein Lipase and GPIHBP1 in Real Time[S]. Journal of Lipid Research 2020, 61, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Gunn, K.H.; Neher, S.B. Structure of Dimeric Lipoprotein Lipase Reveals a Pore Adjacent to the Active Site. Nat Commun 2023, 14, 2569. [Google Scholar] [CrossRef]
- Zambon, A.; Schmidt, I.; Beisiegel, U.; Brunzell, J.D. Dimeric Lipoprotein Lipase Is Bound to Triglyceride-Rich Plasma Lipoproteins. Journal of Lipid Research 1996, 37, 2394–2404. [Google Scholar] [CrossRef]
- Krapp, A.; Zhang, H.; Ginzinger, D.; Liu, M.S.; Lindberg, A.; Olivecrona, G.; Hayden, M.R.; Beisiegel, U. Structural Features in Lipoprotein Lipase Necessary for the Mediation of Lipoprotein Uptake into Cells. Journal of Lipid Research 1995, 36, 2362–2373. [Google Scholar] [CrossRef] [PubMed]
- Nykjaer, A.; Bengtsson-Olivecrona, G.; Lookene, A.; Moestrup, S.K.; Petersen, C.M.; Weber, W.; Beisiegel, U.; Gliemann, J. The Alpha 2-Macroglobulin Receptor/Low Density Lipoprotein Receptor-Related Protein Binds Lipoprotein Lipase and Beta-Migrating Very Low Density Lipoprotein Associated with the Lipase. Journal of Biological Chemistry 1993, 268, 15048–15055. [Google Scholar] [CrossRef]
- Mamputu, J.C.; Desfaits, A.C.; Renier, G. Lipoprotein Lipase Enhances Human Monocyte Adhesion to Aortic Endothelial Cells. Journal of Lipid Research 1997, 38, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Mamputu, J.-C.; Levesque, L.; Renier, G. Proliferative Effect of Lipoprotein Lipase on Human Vascular Smooth Muscle Cells. Arteriosclerosis, Thrombosis, and Vascular Biology 2000, 20, 2212–2219. [Google Scholar] [CrossRef]
- Lotta, L.A.; Gulati, P.; Day, F.R.; Payne, F.; Ongen, H.; van de Bunt, M.; Gaulton, K.J.; Eicher, J.D.; Sharp, S.J.; Luan, J.; et al. Integrative Genomic Analysis Implicates Limited Peripheral Adipose Storage Capacity in the Pathogenesis of Human Insulin Resistance. Nat Genet 2017, 49, 17–26. [Google Scholar] [CrossRef]
- Khera, A.V.; Won, H.-H.; Peloso, G.M.; O’Dushlaine, C.; Liu, D.; Stitziel, N.O.; Natarajan, P.; Nomura, A.; Emdin, C.A.; Gupta, N.; et al. Association of Rare and Common Variation in the Lipoprotein Lipase Gene With Coronary Artery Disease. JAMA 2017, 317, 937–946. [Google Scholar] [CrossRef]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N Engl J Med 2017, 377, 211–221. [Google Scholar] [CrossRef]
- Liu, C.; Han, T.; Stachura, D.L.; Wang, H.; Vaisman, B.L.; Kim, J.; Klemke, R.L.; Remaley, A.T.; Rana, T.M.; Traver, D.; et al. Lipoprotein Lipase Regulates Hematopoietic Stem Progenitor Cell Maintenance through DHA Supply. Nat Commun 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Chen, Y.Q.; Konrad, R.J. The Regulation of Triacylglycerol Metabolism and Lipoprotein Lipase Activity. Advanced Biology 2022, 6, 2200093. [Google Scholar] [CrossRef]
- Oldoni, F.; Cheng, H.; Banfi, S.; Gusarova, V.; Cohen, J.C.; Hobbs, H.H. ANGPTL8 Has Both Endocrine and Autocrine Effects on Substrate Utilization. JCI Insight 2020, 5, e138777–138777. [Google Scholar] [CrossRef] [PubMed]
- Sylvers-Davie, K.L.; Davies, B.S.J. Regulation of Lipoprotein Metabolism by ANGPTL3, ANGPTL4, and ANGPTL8. Am J Physiol Endocrinol Metab 2021, 321, E493–E508. [Google Scholar] [CrossRef]
- Chi, X.; Britt, E.C.; Shows, H.W.; Hjelmaas, A.J.; Shetty, S.K.; Cushing, E.M.; Li, W.; Dou, A.; Zhang, R.; Davies, B.S.J. ANGPTL8 Promotes the Ability of ANGPTL3 to Bind and Inhibit Lipoprotein Lipase. Molecular Metabolism 2017, 6, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Kong, Y.; Peng, D. New Insights into Apolipoprotein A5 in Controlling Lipoprotein Metabolism in Obesity and the Metabolic Syndrome Patients. Lipids Health Dis 2018, 17, 174. [Google Scholar] [CrossRef] [PubMed]
- Wolska, A.; Reimund, M.; Remaley, A.T. Apolipoprotein C-II: The Re-Emergence of a Forgotten Factor. Current Opinion in Lipidology 2020, 31, 147. [Google Scholar] [CrossRef] [PubMed]
- Ramms, B.; Gordts, P.L.S.M. Apolipoprotein C-III in Triglyceride-Rich Lipoprotein Metabolism. Current Opinion in Lipidology 2018, 29, 171. [Google Scholar] [CrossRef] [PubMed]
- Berbée, J.F.P.; Hoogt, C.C. van der; Sundararaman, D.; Havekes, L.M.; Rensen, P.C.N. Severe Hypertriglyceridemia in Human APOC1 Transgenic Mice Is Caused by apoC-I-Induced Inhibition of LPL. Journal of Lipid Research 2005, 46, 297–306. [Google Scholar] [CrossRef]
- Westerterp, M.; Haan, W. de; Berbeée, J.F.P.; Havekes, L.M.; Rensen, P.C.N. Endogenous apoC-I Increases Hyperlipidemia in apoE-Knockout Mice by Stimulating VLDL Production and Inhibiting LPL. Journal of Lipid Research 2006, 47, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Vorrsjö, E.; Talmud, P.; Lookene, A.; Olivecrona, G. Apolipoproteins C-I and C-III Inhibit Lipoprotein Lipase Activity by Displacement of the Enzyme from Lipid Droplets. J Biol Chem 2013, 288, 33997–34008. [Google Scholar] [CrossRef]
- Rensen, P.C.N.; Berkel, T.J.C. van Apolipoprotein E Effectively Inhibits Lipoprotein Lipase-Mediated Lipolysis of Chylomicron-like Triglyceride-Rich Lipid Emulsions in Vitro and in Vivo*. Journal of Biological Chemistry 1996, 271, 14791–14799. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C. Apolipoprotein E Isoforms and Lipoprotein Metabolism. IUBMB Life 2014, 66, 616–623. [Google Scholar] [CrossRef]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure Determines Function, from Atherosclerosis to Alzheimer’s Disease to AIDS. J Lipid Res 2009, 50, S183–S188. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, D.; Schroeder, O.; Russell, A.M.; Fitchett, J.R.; Austin, A.K.; Beyer, T.P.; Chen, Y.Q.; Day, J.W.; Ehsani, M.; Heng, A.R.; et al. An Anti-ANGPTL3/8 Antibody Decreases Circulating Triglycerides by Binding to a LPL-Inhibitory Leucine Zipper-like Motif. Journal of Lipid Research 2022, 63. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Q.; Pottanat, T.G.; Zhen, E.Y.; Siegel, R.W.; Ehsani, M.; Qian, Y.-W.; Konrad, R.J. ApoA5 Lowers Triglyceride Levels via Suppression of ANGPTL3/8-Mediated LPL Inhibition. Journal of Lipid Research 2021, 62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, K. An Updated ANGPTL3-4-8 Model as a Mechanism of Triglyceride Partitioning between Fat and Oxidative Tissues. Progress in lipid research 2022, 85, 101140. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Pottanat, T.G.; Siegel, R.W.; Ehsani, M.; Qian, Y.-W.; Zhen, E.Y.; Regmi, A.; Roell, W.C.; Guo, H.; Luo, M.J.; et al. Angiopoietin-like Protein 8 Differentially Regulates ANGPTL3 and ANGPTL4 during Postprandial Partitioning of Fatty Acids [S]. Journal of Lipid Research 2020, 61, 1203–1220. [Google Scholar] [CrossRef]
- Larsson, M.; Allan, C.M.; Jung, R.S.; Heizer, P.J.; Beigneux, A.P.; Young, S.G.; Fong, L.G. Apolipoprotein C-III Inhibits Triglyceride Hydrolysis by GPIHBP1-Bound LPL [S]. Journal of Lipid Research 2017, 58, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Whitacre, B.E.; Howles, P.; Street, S.; Morris, J.; Swertfeger, D.; Davidson, W.S. Apolipoprotein E Content of VLDL Limits LPL-Mediated Triglyceride Hydrolysis. Journal of Lipid Research 2022, 63. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, B.; Heeren, J.; Blaeser, M.; Radner, H.; Kayser, D.; Aydin, B.; Merkel, M. Lipoprotein Lipase-Facilitated Uptake of LDL Is Mediated by the LDL Receptor. Journal of Lipid Research 2007, 48, 288–298. [Google Scholar] [CrossRef]
- Rinninger, F.; Kaiser, T.; Mann, W.A.; Meyer, N.; Greten, H.; Beisiegel, U. Lipoprotein Lipase Mediates an Increase in the Selective Uptake of High Density Lipoprotein-Associated Cholesteryl Esters by Hepatic Cells in Culture. Journal of Lipid Research 1998, 39, 1335–1348. [Google Scholar] [CrossRef]
- Beisiegel, U.; Weber, W.; Bengtsson-Olivecrona, G. Lipoprotein Lipase Enhances the Binding of Chylomicrons to Low Density Lipoprotein Receptor-Related Protein. Proceedings of the National Academy of Sciences 1991, 88, 8342–8346. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.A.; Fry, G.L.; Waknitz, M.A.; Muhonen, L.E.; Pladet, M.W.; Iverius, P.H.; Strickland, D.K. Lipoprotein Lipase Induces Catabolism of Normal Triglyceride-Rich Lipoproteins via the Low Density Lipoprotein Receptor-Related Protein/Alpha 2-Macroglobulin Receptor in Vitro. A Process Facilitated by Cell-Surface Proteoglycans. Journal of Biological Chemistry 1993, 268, 14168–14175. [Google Scholar] [CrossRef]
- Kounnas, M.Z.; Chappell, D.A.; Strickland, D.K.; Argraves, W.S. Glycoprotein 330, a Member of the Low Density Lipoprotein Receptor Family, Binds Lipoprotein Lipase in Vitro. Journal of Biological Chemistry 1993, 268, 14176–14181. [Google Scholar] [CrossRef]
- Raulin, A.-C.; Martens, Y.A.; Bu, G. Lipoproteins in the Central Nervous System: From Biology to Pathobiology. Annu Rev Biochem 2022, 91, 731–759. [Google Scholar] [CrossRef] [PubMed]
- Cruciani-Guglielmacci, C.; Magnan, C. Brain Lipoprotein Lipase as a Regulator of Energy Balance. Biochimie 2017, 143, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Mysling, S.; Kristensen, K.K.; Larsson, M.; Beigneux, A.P.; Gårdsvoll, H.; Fong, L.G.; Bensadouen, A.; Jørgensen, T.J.; Young, S.G.; Ploug, M. The Acidic Domain of the Endothelial Membrane Protein GPIHBP1 Stabilizes Lipoprotein Lipase Activity by Preventing Unfolding of Its Catalytic Domain. Elife 2016, 5, e12095. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Nakajima, T.; Inoue, I. Molecular Modeling of the Dimeric Structure of Human Lipoprotein Lipase and Functional Studies of the Carboxyl-Terminal Domain. European Journal of Biochemistry 2002, 269, 4701–4710. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F.K.; D’Arcy, A.; Hunziker, W. Structure of Human Pancreatic Lipase. Nature 1990, 343, 771–774. [Google Scholar] [CrossRef]
- Beigneux, A.P.; Allan, C.M.; Sandoval, N.P.; Cho, G.W.; Heizer, P.J.; Jung, R.S.; Stanhope, K.L.; Havel, P.J.; Birrane, G.; Meiyappan, M.; et al. Lipoprotein Lipase Is Active as a Monomer. Proc Natl Acad Sci U S A 2019, 116, 6319–6328. [Google Scholar] [CrossRef] [PubMed]
- van Tilbeurgh, H.; Roussel, A.; Lalouel, J.M.; Cambillau, C. Lipoprotein Lipase. Molecular Model Based on the Pancreatic Lipase x-Ray Structure: Consequences for Heparin Binding and Catalysis. Journal of Biological Chemistry 1994, 269, 4626–4633. [Google Scholar] [CrossRef] [PubMed]
- Leth-Espensen, K.Z.; Kristensen, K.K.; Kumari, A.; Winther, A.-M.L.; Young, S.G.; Jørgensen, T.J.D.; Ploug, M. The Intrinsic Instability of the Hydrolase Domain of Lipoprotein Lipase Facilitates Its Inactivation by ANGPTL4-Catalyzed Unfolding. Proc Natl Acad Sci U S A 2021, 118, e2026650118. [Google Scholar] [CrossRef] [PubMed]
- Lookene, A.; Groot, N.B.; Kastelein, J.J.P.; Olivecrona, G.; Bruin, T. Mutation of Tryptophan Residues in Lipoprotein Lipase: EFFECTS ON STABILITY, IMMUNOREACTIVITY, AND CATALYTIC PROPERTIES *. Journal of Biological Chemistry 1997, 272, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gårdsvoll, H.; Yuan, C.; Lin, L.; Ploug, M.; Huang, M. Crystal Structure of the Urokinase Receptor in a Ligand-Free Form. Journal of Molecular Biology 2012, 416, 629–641. [Google Scholar] [CrossRef]
- Huang, Y.; Fedarovich, A.; Tomlinson, S.; Davies, C. Crystal Structure of CD59: Implications for Molecular Recognition of the Complement Proteins C8 and C9 in the Membrane-Attack Complex. Acta Cryst D 2007, 63, 714–721. [Google Scholar] [CrossRef]
- Gin, P.; Beigneux, A.P.; Voss, C.; Davies, B.S.J.; Beckstead, J.A.; Ryan, R.O.; Bensadoun, A.; Fong, L.G.; Young, S.G. Binding Preferences for GPIHBP1, a GPI-Anchored Protein of Capillary Endothelial Cells. Arterioscler Thromb Vasc Biol 2011, 31, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, M.H.; Ehrhardt, N.; Péterfy, M. Lipase Maturation Factor 1 (Lmf1): Structure and Role in Lipase Folding and Assembly. Curr Opin Lipidol 2010, 21, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Lookene, A.; Zhang, L.; Hultin, M.; Olivecrona, G. Rapid Subunit Exchange in Dimeric Lipoprotein Lipase and Properties of the Inactive Monomer*. Journal of Biological Chemistry 2004, 279, 49964–49972. [Google Scholar] [CrossRef]
- Osborne, J.C.Jr.; Bengtsson-Olivecrona, G.; Lee, N.S.; Olivecrona, T. Studies on Inactivation of Lipoprotein Lipase: Role of the Dimer to Monomer Dissociation. Biochemistry 1985, 24, 5606–5611. [Google Scholar] [CrossRef] [PubMed]
- Reimund, M.; Kovrov, O.; Olivecrona, G.; Lookene, A. Lipoprotein Lipase Activity and Interactions Studied in Human Plasma by Isothermal Titration Calorimetry. J Lipid Res 2017, 58, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Deeb, S.S.; Peng, R. Structure of the Human Lipoprotein Lipase Gene. Biochemistry 1989, 28, 4131–4135. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing Its Use in a Clinical Diagnostic or Research Setting. Hum Genet 2020, 139, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Research 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next Generation in Gene Variant Databases. Human Mutation 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Perera, S.D.; Wang, J.; McIntyre, A.D.; Hegele, R.A. Variability of Longitudinal Triglyceride Phenotype in Patients Heterozygous for Pathogenic APOA5 Variants. Journal of Clinical Lipidology 2023, 17, 659–665. [Google Scholar] [CrossRef]
- Dron, J.S.; Wang, J.; McIntyre, A.D.; Iacocca, M.A.; Robinson, J.F.; Ban, M.R.; Cao, H.; Hegele, R.A. Six Years’ Experience with LipidSeq: Clinical and Research Learnings from a Hybrid, Targeted Sequencing Panel for Dyslipidemias. BMC Med Genomics 2020, 13, 23. [Google Scholar] [CrossRef]
- Deshotels, M.R.; Hadley, T.D.; Roth, M.; Agha, A.M.; Pulipati, V.P.; Nugent, A.K.; Virani, S.S.; Nambi, V.; Moriarty, P.M.; Davidson, M.H.; et al. Genetic Testing for Hypertriglyceridemia in Academic Lipid Clinics: Implications for Precision Medicine-Brief Report. Arterioscler Thromb Vasc Biol 2022, 42, 1461–1467. [Google Scholar] [CrossRef]
- Laurie, A.D.; Kyle, C.V. A Novel Frameshift Mutation in the Lipoprotein Lipase Gene Is Rescued by Alternative Messenger RNA Splicing. Journal of Clinical Lipidology 2017, 11, 357–361. [Google Scholar] [CrossRef]
- Okubo, M.; Horinishi, A.; Saito, M.; Ebara, T.; Endo, Y.; Kaku, K.; Murase, T.; Eto, M. A Novel Complex Deletion–Insertion Mutation Mediated by Alu Repetitive Elements Leads to Lipoprotein Lipase Deficiency. Molecular Genetics and Metabolism 2007, 92, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Lepore, S.M.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Valenti, L.; Berge, K.E.; Retterstøl, K.; Leren, T.P.; Wiklund, O.; et al. Identification and Characterization of Two Novel Mutations in the LPL Gene Causing Type I Hyperlipoproteinemia. Journal of Clinical Lipidology 2016, 10, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Q.; Shi, X.; Chen, W.; Pu, N.; Li, W.; Li, J. Compound but Non-Linked Heterozygous p.W14X and p.L279 V LPL Gene Mutations in a Chinese Patient with Long-Term Severe Hypertriglyceridemia and Recurrent Acute Pancreatitis. Lipids Health Dis 2018, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Suehiro, T.; Yasuoka, N.; Yamamoto, M.; Ito, H.; Yamano, T.; Hashimoto, K. A Novel Nonsense Mutation in Exon 1 and a Transition in Intron 3 of the Lipoprotein Lipase Gene. J Atheroscler Thromb 1996, 3, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Gin, P.; Goulbourne, C.N.; Adeyo, O.; Beigneux, A.P.; Davies, B.S.J.; Tat, S.; Voss, C.V.; Bensadoun, A.; Fong, L.G.; Young, S.G. Chylomicronemia Mutations Yield New Insights into Interactions between Lipoprotein Lipase and GPIHBP1. Human Molecular Genetics 2012, 21, 2961–2972. [Google Scholar] [CrossRef]
- Pruneta-Deloche, V.; Marçais, C.; Perrot, L.; Sassolas, A.; Delay, M.; Estour, B.; Lagarde, M.; Moulin, P. Combination of Circulating Antilipoprotein Lipase (Anti-LPL) Antibody and Heterozygous S172 fsX179 Mutation of LPL Gene Leading to Chronic Hyperchylomicronemia. The Journal of Clinical Endocrinology & Metabolism 2005, 90, 3995–3998. [Google Scholar] [CrossRef]
- Yu, X.-H.; Zhao, T.-Q.; Wang, L.; Liu, Z.-P.; Zhang, C.-M.; Chen, R.; Li, L.; Liu, G.; Hu, W.-C. A Novel Substitution at the Translation Initiator Codon (ATG-->ATC) of the Lipoprotein Lipase Gene Is Mainly Responsible for Lipoprotein Lipase Deficiency in a Patient with Severe Hypertriglyceridemia and Recurrent Pancreatitis. Biochem Biophys Res Commun 2006, 341, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cheng, Y.; Shi, Y.; Zhao, W.; Gao, L.; Fang, L.; Jin, X.; Han, X.; Sun, Q.; Li, G.; et al. Identification and Characterization of Two Novel Compounds: Heterozygous Variants of Lipoprotein Lipase in Two Pedigrees With Type I Hyperlipoproteinemia. Front Endocrinol (Lausanne) 2022, 13, 874608. [Google Scholar] [CrossRef] [PubMed]
- Diaz de Arce, A.J.; Noderer, W.L.; Wang, C.L. Complete Motif Analysis of Sequence Requirements for Translation Initiation at Non-AUG Start Codons. Nucleic Acids Research 2018, 46, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Hecht, A.; Glasgow, J.; Jaschke, P.R.; Bawazer, L.A.; Munson, M.S.; Cochran, J.R.; Endy, D.; Salit, M. Measurements of Translation Initiation from All 64 Codons in E. Coli. Nucleic Acids Res 2017, 45, 3615–3626. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Nomura, A.; Okada, H.; Nakahashi, T.; Nozue, T.; Hayashi, K.; Nohara, A.; Yagi, K.; Inazu, A.; Michishita, I.; et al. Clinical Whole Exome Sequencing in Severe Hypertriglyceridemia. Clinica Chimica Acta 2019, 488, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Owji, H.; Nezafat, N.; Negahdaripour, M.; Hajiebrahimi, A.; Ghasemi, Y. A Comprehensive Review of Signal Peptides: Structure, Roles, and Applications. European Journal of Cell Biology 2018, 97, 422–441. [Google Scholar] [CrossRef] [PubMed]
- Duffy, J.; Patham, B.; Mensa-Wilmot, K. Discovery of Functional Motifs in H-Regions of Trypanosome Signal Sequences. Biochemical Journal 2010, 426, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Inadera, H.; Fujita, Y.; Talley, G.; Morisaki, N.; Yoshida, S.; Saito, Y.; Fojo, S.S.; Brewer, H.B. A Naturally Occurring Mutation at the Second Base of Codon Asparagine 43 in the Proposed N-Linked Glycosylation Site of Human Lipoprotein Lipase: In Vivo Evidence That Asparagine 43 Is Essential for Catalysis and Secretion. Biochemical and Biophysical Research Communications 1994, 205, 506–515. [Google Scholar] [CrossRef]
- Rabacchi, C.; Pisciotta, L.; Cefalù, A.B.; Noto, D.; Fresa, R.; Tarugi, P.; Averna, M.; Bertolini, S.; Calandra, S. Spectrum of Mutations of the LPL Gene Identified in Italy in Patients with Severe Hypertriglyceridemia. Atherosclerosis 2015, 241, 79–86. [Google Scholar] [CrossRef]
- Kolářová, H.; Tesařová, M.; Švecová, Š.; Stránecký, V.; Přistoupilová, A.; Zima, T.; Uhrová, J.; Volgina, S.Y.; Zeman, J.; Honzík, T. Lipoprotein Lipase Deficiency: Clinical, Biochemical and Molecular Characteristics in Three Patients with Novel Mutations in the LPL Gene. Fol. Biol. 2014, 60, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Roshan, R.; Desai, R.; Kadam, S. Neonatal Hyperlipidemia with Pancreatitis: Novel Gene Mutation of Lipoprotein Lipase. J Postgrad Med 2018, 64, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Bruin, T.; Tuzgöl, S.; Van Diermen, D.E.; Hoogerbrugge-van Der Linden, N.; Brunzell, J.D.; Hayden, M.R.; Kastelein, J.J.P. Recurrent Pancreatitis and Chylomicronemia in an Extended Dutch Kindred Is Caused by a Gly154–>Ser Substitution in Lipoprotein Lipase. Journal of Lipid Research 1993, 34, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, E.; Ghayour-Mobarhan, M.; Ferns, G.A.; Pasdar, A. A Novel Variant in LPL Gene Is Associated with Familial Combined Hyperlipidemia. BioFactors 2020, 46, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.H.; Bruin, T.; Tuzgol, S.; Wilson, B.I.; Roederer, G.; Liu, M.S.; Davignon, J.; Kastelein, J.J.; Brunzell, J.D.; Hayden, M.R. Two Naturally Occurring Mutations at the First and Second Bases of Codon Aspartic Acid 156 in the Proposed Catalytic Triad of Human Lipoprotein Lipase. In Vivo Evidence That Aspartic Acid 156 Is Essential for Catalysis. Journal of Biological Chemistry 1992, 267, 1918–1923. [Google Scholar] [CrossRef] [PubMed]
- Faustinella, F.; Chang, A.; Van Biervliet, J.P.; Rosseneu, M.; Vinaimont, N.; Smith, L.C.; Chen, S.H.; Chan, L. Catalytic Triad Residue Mutation (Asp156—-Gly) Causing Familial Lipoprotein Lipase Deficiency. Co-Inheritance with a Nonsense Mutation (Ser447—-Ter) in a Turkish Family. Journal of Biological Chemistry 1991, 266, 14418–14424. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, M.S.; Ginzinger, D.; Frohlich, J.; Brunzell, J.D.; Hayden, M.R. Gene-Environment Interaction in the Conversion of a Mild-to-Severe Phenotype in a Patient Homozygous for a Ser172-->Cys Mutation in the Lipoprotein Lipase Gene. J Clin Invest 1993, 91, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Zheng, Y.; Gan, Z.; Guo, Y.; Jiang, S.; Yang, F.; Xiong, F.; Zheng, H. A Heterozygous LMF1 Gene Mutation (c.1523C>T), Combined With an LPL Gene Mutation (c.590G>A), Aggravates the Clinical Symptoms in Hypertriglyceridemia. Front Genet 2022, 13, 814295. [Google Scholar] [CrossRef]
- Glueck, C.J.; Levy, R.I.; Glueck, H.I.; Gralnick, H.R.; Greten, H.; Fredrickson, D.S. Acquired Type I Hyperlipoproteinemia with Systemic Lupus Erythematosus, Dysglobulinemia and Heparin Resistance. The American Journal of Medicine 1969, 47, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Pauciullo, P.; De Simone, B.; Rubba, P.; Mancini, M. A Case of Association between Type I Hyperlipoproteinemia and Systemic Lupus Erythematosus (SLE). Effects of Steroid Treatment. J Endocrinol Invest 1986, 9, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Reichlin, M.; Fesmire, J.; Quintero-Del-Rio, A.I.; Wolfson-Reichlin, M. Autoantibodies to lipoprotein lipase and dyslipidemia in systemic lupus erythematosus. Arthritis & Rheumatism 2002, 46, 2957–2963. [Google Scholar] [CrossRef]
- de Carvalho, J.F.; Borba, E.F.; Viana, V.S.T.; Bueno, C.; Leon, E.P.; Bonfá, E. Anti–Lipoprotein Lipase Antibodies: A New Player in the Complex Atherosclerotic Process in Systemic Lupus Erythematosus? Arthritis & Rheumatism 2004, 50, 3610–3615. [Google Scholar] [CrossRef]
- Wang, M.; Zhou, Y.; He, X.; Deng, C.; Liu, X.; Li, J.; Zhou, L.; Li, Y.; Zhang, Y.; Liu, H.; et al. Two Novel Mutations of the LPL Gene in Two Chinese Family Cases with Familial Chylomicronemia Syndrome. Clinica Chimica Acta 2021, 521, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Chokshi, N.; Blumenschein, S.D.; Ahmad, Z.; Garg, A. Genotype-Phenotype Relationships in Patients with Type I Hyperlipoproteinemia. Journal of Clinical Lipidology 2014, 8, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, R.; Tada, H.; Kawashiri, M.; Nohara, A.; Nakahashi, T.; Konno, T.; Inazu, A.; Mabuchi, H.; Yamagishi, M.; Hayashi, K. Molecular and Functional Characterization of Familial Chylomicronemia Syndrome. Atherosclerosis 2018, 269, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Sasaki, N.; Tashiro, J.; Inadera, H.; Saito, Y.; Yoshida, S. A Missense Mutation (Ala334⇒Thr) in Exon 7 of the Lipoprotein Lipase Gene in a Case with Type I Hyperlipidemia. Biochemical and Biophysical Research Communications 1993, 191, 1046–1054. [Google Scholar] [CrossRef]
- Chan, L.Y.S.; Lam, C.-W.; Mak, Y.-T.; Tomlinson, B.; Tsang, M.-W.; Baum, L.; Masarei, J.R.L.; Pang, C.-P. Genotype-Phenotype Studies of Six Novel LPL Mutations in Chinese Patients with Hypertriglyceridemia. Human Mutation 2002, 20, 232–233. [Google Scholar] [CrossRef]
- Lun, Y.; Sun, X.; Wang, P.; Chi, J.; Hou, X.; Wang, Y. Severe Hypertriglyceridemia Due to Two Novel Loss-of-Function Lipoprotein Lipase Gene Mutations (C310R/E396V) in a Chinese Family Associated with Recurrent Acute Pancreatitis. Oncotarget 2017, 8, 47741–47754. [Google Scholar] [CrossRef]
- Wiebusch, H.; Funke, H.; Bruin, T.; Bucher, H.; von Eckardstein, A.; Kastelein, J.J.P.; Assmann, G. Compound Heterozygosity for a Known (D250N) and a Novel (E410K) Missense Mutation in the C-Terminal Domain of Lipoprotein Lipase Causes Familial Chylomicronemia. Human Mutation 1996, 8, 381–383. [Google Scholar] [CrossRef]
- Previato, L.; Guardamagna, O.; Dugi, K.A.; Ronan, R.; Talley, G.D.; Santamarina-Fojo, S.; Brewer, H.B. A Novel Missense Mutation in the C-Terminal Domain of Lipoprotein Lipase (Glu410–>Val) Leads to Enzyme Inactivation and Familial Chylomicronemia. Journal of Lipid Research 1994, 35, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Voss, C.V.; Davies, B.S.J.; Tat, S.; Gin, P.; Fong, L.G.; Pelletier, C.; Mottler, C.D.; Bensadoun, A.; Beigneux, A.P.; Young, S.G. Mutations in Lipoprotein Lipase That Block Binding to the Endothelial Cell Transporter GPIHBP1. Proceedings of the National Academy of Sciences 2011, 108, 7980–7984. [Google Scholar] [CrossRef]
- Henderson, H.E.; Hassan, F.; Marais, D.; Hayden, M.R. A New Mutation Destroying Disulphide Bridging in the C-Terminal Domain of Lipoprotein Lipase. Biochemical and Biophysical Research Communications 1996, 227, 189–194. [Google Scholar] [CrossRef]
- Henderson, H.; Leisegang, F.; Hassan, F.; Hayden, M.; Marais, D. A Novel Glu421Lys Substitution in the Lipoprotein Lipase Gene in Pregnancy-Induced Hypertriglyceridemic Pancreatitis. Clinica Chimica Acta 1998, 269, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Nevin, D.N.; Peng, R.; Brunzell, J.D.; Deeb, S.S. A Mutation in the Promoter of the Lipoprotein Lipase (LPL) Gene in a Patient with Familial Combined Hyperlipidemia and Low LPL Activity. Proceedings of the National Academy of Sciences 1995, 92, 4462–4466. [Google Scholar] [CrossRef]
- Zhang, Q.; Cavallero, E.; Hoffmann, M.M.; Cavanna, J.; Kay, A.; Charles, A.; Braschi, S.; Marz, W.; Perlemuter, L.; Jacotot, B.; et al. Mutations at the Lipoprotein Lipase Gene Locus in Subjects with Diabetes Mellitus, Obesity and Lipaemia. Clinical Science 1997, 93, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-X.; Razzaghi, H.; Hokanson, J.E.; Kamboh, M.I. Identification and Characterization of a Novel 5 Bp Deletion in a Putative Insulin Response Element in the Lipoprotein Lipase Gene. Biochim Biophys Acta 2009, 1791, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Reymer, P.W.A.; Gagné, E.; Groenemeyer, B.E.; Zhang, H.; Forsyth, I.; Jansen, H.; Seidell, J.C.; Kromhout, D.; Lie, K.E.; Kastelein, J.; et al. A Lipoprotein Lipase Mutation (Asn291Ser) Is Associated with Reduced HDL Cholesterol Levels in Premature Atherosclerosis. Nat Genet 1995, 10, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Buscà, R.; Peinado, J.; Vilella, E.; Auwerx, J.; Deeb, S.S.; Vilaró, S.; Reina, M. The Mutant Asn291 → Ser Human Lipoprotein Lipase Is Associated with Reduced Catalytic Activity and Does Not Influence Binding to Heparin. FEBS Letters 1995, 367, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Syvänne, M.; Antikainen, M.; Ehnholm, S.; Tenkanen, H.; Lahdenperä, S.; Ehnholm, C.; Taskinen, M.R. Heterozygosity for Asn291–>Ser Mutation in the Lipoprotein Lipase Gene in Two Finnish Pedigrees: Effect of Hyperinsulinemia on the Expression of Hypertriglyceridemia. Journal of Lipid Research 1996, 37, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Sagoo, G.S.; Tatt, I.; Salanti, G.; Butterworth, A.S.; Sarwar, N.; van Maarle, M.; Jukema, J.W.; Wiman, B.; Kastelein, J.J.P.; Bennet, A.M.; et al. Seven Lipoprotein Lipase Gene Polymorphisms, Lipid Fractions, and Coronary Disease: A HuGE Association Review and Meta-Analysis. American Journal of Epidemiology 2008, 168, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, M.J.V.; Bredie, S.J.H.; Boomsma, D.I.; Reymer, P.W.A.; Kastelein, J.J.P.; de Knijff, P.; Demacker, P.N.M.; Stalenhoef, A.F.H.; Havekes, L.M.; Frants, R.R. The Lipoprotein Lipase (Asn291 → Ser) Mutation Is Associated with Elevated Lipid Levels in Families with Familial Combined Hyperlipidaemia. Atherosclerosis 1996, 119, 159–167. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Nevin, D.N.; Iwasaki, L.; Peng, R.; Brown, B.G.; Brunzell, J.D.; Deeb, S.S. Regulatory Mutations in the Human Lipoprotein Lipase Gene in Patients with Familial Combined Hyperlipidemia and Coronary Artery Disease. Journal of Lipid Research 1996, 37, 2627–2637. [Google Scholar] [CrossRef]
- Schmella, M.J.; Ferrell, R.E.; Gallaher, M.J.; Lykins, D.L.; Althouse, A.D.; Roberts, J.M.; Hubel, C.A. The -93T/G LPL Promoter Polymorphism Is Associated With Lower Third-Trimester Triglycerides in Pregnant African American Women. Biol Res Nurs 2015, 17, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Razzaghi, H.; Demirci, F.Y.; Kamboh, M.I. Functional Significance of Lipoprotein Lipase HindIII Polymorphism Associated with the Risk of Coronary Artery Disease. Atherosclerosis 2008, 200, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.P.; Palmen, J.; Putt, W.; Talmud, P.J.; Humphries, S.E.; Drenos, F. Application of Statistical and Functional Methodologies for the Investigation of Genetic Determinants of Coronary Heart Disease Biomarkers: Lipoprotein Lipase Genotype and Plasma Triglycerides as an Exemplar. Human Molecular Genetics 2010, 19, 3936–3947. [Google Scholar] [CrossRef]
- Rodrigues, R.; Artieda, M.; Tejedor, D.; Martínez, A.; Konstantinova, P.; Petry, H.; Meyer, C.; Corzo, D.; Sundgreen, C.; Klor, H.U.; et al. Pathogenic Classification of LPL Gene Variants Reported to Be Associated with LPL Deficiency. J Clin Lipidol 2016, 10, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Yang, Z.; Guo, H.; Zhang, J.; Tang, J.; Rui, D.; Ma, R. [Association of lipoprotein lipase gene Hind III and S447X polymorphisms in metabolic syndrome patients among Kazakh and Han ethnics from Xinjiang]. Zhonghua Liu Xing Bing Xue Za Zhi 2010, 31, 992–996. [Google Scholar]
- Hata, A.; Robertson, M.; Emi, M.; Lalouel, J.M. Direct Detection and Automated Sequencing of Individual Alleles after Electrophoretic Strand Separation: Identification of a Common Nonsense Mutation in Exon 9 of the Human Lipoprotein Lipase Gene. Nucleic Acids Res 1990, 18, 5407–5411. [Google Scholar] [CrossRef] [PubMed]
- Elbein, S.C.; Yeager, C.; Kwong, L.K.; Lingam, A.; Inoue, I.; Lalouel, J.M.; Wilson, D.E. Molecular Screening of the Lipoprotein Lipase Gene in Hypertriglyceridemic Members of Familial Noninsulin-Dependent Diabetes Mellitus Families. The Journal of Clinical Endocrinology & Metabolism 1994, 79, 1450–1456. [Google Scholar] [CrossRef]
- Matern, D.; Seydewitz, H.; Niederhoff, H.; Wiebusch, H.; Brandis, M. Dyslipidaemia in a Boy with Recurrent Abdominal Pain, Hypersalivation and Decreased Lipoprotein Lipase Activity. Eur J Pediatr 1996, 155, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Gagné, S.; Larson, M.; Pimstone, S.; Schaefer, E.; Kastelein, J.; Wilson, P.; Ordovas, J.; Hayden, M. A Common Truncation Variant of Lipoprotein Lipase (Ser447X) Confers Protection against Coronary Heart Disease: The Framingham Offspring Study. Clinical Genetics 1999, 55, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, C.; Stabouli, S.; Roumeliotou, K.; Traeger-Synodinos, J.; Kavazarakis, E.; Gourgiotis, D.; Lambrou, J.; Kanavakis, E. Severe Hypertriglyceridaemia in Diabetic Ketoacidosis: Clinical and Genetic Study. Diabetic Medicine 2004, 21, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Li, L.; Cao, W.; Shan, S.; Lu, J.; Guo, X.; Hu, Y. [The association of S447X and Hind III polymorphism in the lipoprotein lipase gene with dyslipidemia of the metabolic syndrome in patients with essential hypertension]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2005, 22, 151–157. [Google Scholar] [PubMed]
- Guan, G.; Xu, E.; Wang, X.; Xu, Y.; Qiu, S. Associations between Ser447Ter Gene Polymorphism of Lipoprotein Lipase and Atherosclerotic Cerebral Infarction. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics 2006, 23, 519–522. [Google Scholar] [PubMed]
- Herbeth, B.; Gueguen, S.; Leroy, P.; Siest, G.; Visvikis-Siest, S. The Lipoprotein Lipase Serine 447 Stop Polymorphism Is Associated with Altered Serum Carotenoid Concentrations in the Stanislas Family Study. J Am Coll Nutr 2007, 26, 655–662. [Google Scholar] [CrossRef]
- Komurcu-Bayrak, E.; Onat, A.; Poda, M.; Humphries, S.E.; Acharya, J.; Hergenc, G.; Coban, N.; Can, G.; Erginel-Unaltuna, N. The S447X Variant of Lipoprotein Lipase Gene Is Associated with Metabolic Syndrome and Lipid Levels among Turks. Clinica Chimica Acta 2007, 383, 110–115. [Google Scholar] [CrossRef]
- Talmud, P.J.; Flavell, D.M.; Alfakih, K.; Cooper, J.A.; Balmforth, A.J.; Sivananthan, M.; Montgomery, H.E.; Hall, A.S.; Humphries, S.E. The Lipoprotein Lipase Gene Serine 447 Stop Variant Influences Hypertension-Induced Left Ventricular Hypertrophy and Risk of Coronary Heart Disease. Clin Sci (Lond) 2007, 112, 617–624. [Google Scholar] [CrossRef]
- Yang, Y.; Mu, Y.; Zhao, Y.; Liu, X.; Zhao, L.; Wang, J.; Xie, Y. Genetic Screening of the Lipoprotein Lipase Gene for Mutations in Chinese Subjects with or without Hypertriglyceridemia. Journal of Genetics and Genomics 2007, 34, 381–391. [Google Scholar] [CrossRef]
- Aydogan, H.Y.; Isbi̇r, S.; Kurnaz, O.; Gormus, U.; Isbi̇r, T. Associations of Lipoprotein Lipase S447X and Apolipoprotein E Genotypes with Low-Density Lipoprotein Subfractions in Turkish Patients with Coronary Artery Disease. In Vivo 2009, 23, 155–161. [Google Scholar]
- Chang, Y.-T.; Chang, M.-C.; Su, T.-C.; Liang, P.-C.; Su, Y.-N.; Kuo, C.-H.; Wei, S.-C.; Wong, J.-M. Lipoprotein Lipase Mutation S447X Associated with Pancreatic Calcification and Steatorrhea in Hyperlipidemic Pancreatitis. J Clin Gastroenterol 2009, 43, 591–596. [Google Scholar] [CrossRef]
- Deo, R.C.; Reich, D.; Tandon, A.; Akylbekova, E.; Patterson, N.; Waliszewska, A.; Kathiresan, S.; Sarpong, D.; Taylor, H.A.; Wilson, J.G. Genetic Differences between the Determinants of Lipid Profile Phenotypes in African and European Americans: The Jackson Heart Study. PLoS Genet 2009, 5, e1000342. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Kotani, K.; Sano, Y.; Matsuoka, Y.; Tsuzaki, K.; Domichi, M.; Kajii, E.; Sakane, N. S447X Polymorphism in the Lipoprotein Lipase Gene and the Adiponectin Level in the General Population: Results from the Mima Study. JAT 2009, 16, 188–193. [Google Scholar] [CrossRef]
- Jensen, M.K.; Rimm, E.B.; Rader, D.; Schmidt, E.B.; Sørensen, T.I.A.; Vogel, U.; Overvad, K.; Mukamal, K.J. S447X Variant of the Lipoprotein Lipase Gene, Lipids, and Risk of Coronary Heart Disease in 3 Prospective Cohort Studies. Am Heart J 2009, 157, 384–390. [Google Scholar] [CrossRef]
- Salah, A.; Khan, M.; Esmail, N.; Habibullah, S.; Al Lahham, Y. Genetic Polymorphism of S447X Lipoprotein Lipase (LPL) and the Susceptibility to Hypertension. J Crit Care 2009, 24, e11–14. [Google Scholar] [CrossRef]
- van Hoek, M.; Dallinga-Thie, G.M.; Steyerberg, E.W.; Sijbrands, E.J.G. Diagnostic Value of Post-Heparin Lipase Testing in Detecting Common Genetic Variants in the LPL and LIPC Genes. Eur J Hum Genet 2009, 17, 1386–1393. [Google Scholar] [CrossRef]
- Webster, R.J.; Warrington, N.M.; Weedon, M.N.; Hattersley, A.T.; McCaskie, P.A.; Beilby, J.P.; Palmer, L.J.; Frayling, T.M. The Association of Common Genetic Variants in the APOA5, LPL and GCK Genes with Longitudinal Changes in Metabolic and Cardiovascular Traits. Diabetologia 2009, 52, 106–114. [Google Scholar] [CrossRef]
- Wang, C.; Sun, T.; Li, H.; Bai, J.; Li, Y. Lipoprotein Lipase Ser447Ter Polymorphism Associated with the Risk of Ischemic Stroke: A Meta-Analysis. Thromb Res 2011, 128, e107–112. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, G.; Unal, R.; Pokrovskaya, I.D.; Tripathi, P.; Rotter, J.I.; Goodarzi, M.O.; Kern, P.A. The Lipoprotein Lipase (LPL) S447X Gain of Function Variant Involves Increased mRNA Translation. Atherosclerosis 2011, 221, 143. [Google Scholar] [CrossRef]
- Takeuchi, F.; Isono, M.; Katsuya, T.; Yokota, M.; Yamamoto, K.; Nabika, T.; Shimokawa, K.; Nakashima, E.; Sugiyama, T.; Rakugi, H.; et al. Association of Genetic Variants Influencing Lipid Levels with Coronary Artery Disease in Japanese Individuals. PLoS ONE 2012, 7, e46385. [Google Scholar] [CrossRef]
- Brownstein, C.A.; Towne, M.C.; Luquette, L.J.; Harris, D.J.; Marinakis, N.S.; Meinecke, P.; Kutsche, K.; Campeau, P.M.; Yu, T.W.; Margulies, D.M.; et al. Mutation of KCNJ8 in a Patient with Cantú Syndrome with Unique Vascular Abnormalities – Support for the Role of K(ATP) Channels in This Condition. European Journal of Medical Genetics 2013, 56, 678–682. [Google Scholar] [CrossRef]
- Velapasamy, S.; Alex, L.; Chahil, J.K.; Lye, S.H.; Munretnam, K.; Nor Hashim, N.A.; Ramzi, N.H.; Mohd Nordin, N.; Visvalingam, V.; Ler, L.W. Influences of Multiple Genetic Polymorphisms on Ovarian Cancer Risk in Malaysia. Genetic Testing and Molecular Biomarkers 2013, 17, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, Y.; Wang, L.; Liao, Q.; Chang, L.; Xu, L.; Huang, Y.; Ye, H.; Xu, L.; Chen, C.; et al. Meta-Analyses of 8 Polymorphisms Associated with the Risk of the Alzheimer’s Disease. PLoS One 2013, 8, e73129. [Google Scholar] [CrossRef]
- Bentley, A.R.; Chen, G.; Shriner, D.; Doumatey, A.P.; Zhou, J.; Huang, H.; Mullikin, J.C.; Blakesley, R.W.; Hansen, N.F.; Bouffard, G.G.; et al. Gene-Based Sequencing Identifies Lipid-Influencing Variants with Ethnicity-Specific Effects in African Americans. PLoS Genet 2014, 10, e1004190. [Google Scholar] [CrossRef] [PubMed]
- Turlo, K.; Leung, C.S.; Seo, J.J.; Goulbourne, C.N.; Adeyo, O.; Gin, P.; Voss, C.; Bensadoun, A.; Fong, L.G.; Young, S.G.; et al. Equivalent Binding of Wild-Type Lipoprotein Lipase (LPL) and S447X-LPL to GPIHBP1, the Endothelial Cell LPL Transporter. Biochimica et biophysica acta 2014, 1841, 963. [Google Scholar] [CrossRef] [PubMed]
- Zambrano Morales, M.; Fernández Salgado, E.; Balzán Urdaneta, L.; Labastidas, N.; Aranguren-Méndez, J.; Connell, L.; Molero Paredes, T.; Rojas, A.; Panunzio, A. [Lack of association between the S447X variant of the lipoprotein lipase gene and plasma lipids. A preliminary study]. Invest Clin 2014, 55, 133–141. [Google Scholar]
- Emamian, M.; Avan, A.; Pasdar, A.; Mirhafez, S.R.; Sadeghzadeh, M.; Moghadam, M.S.; Parizadeh, S.M.R.; Ferns, G.A.; Ghayour-Mobarhan, M. The Lipoprotein Lipase S447X and Cholesteryl Ester Transfer Protein Rs5882 Polymorphisms and Their Relationship with Lipid Profile in Human Serum of Obese Individuals. Gene 2015, 558, 195–199. [Google Scholar] [CrossRef]
- Dewey, F.E.; Murray, M.F.; Overton, J.D.; Habegger, L.; Leader, J.B.; Fetterolf, S.N.; O’Dushlaine, C.; Van Hout, C.V.; Staples, J.; Gonzaga-Jauregui, C.; et al. Distribution and Clinical Impact of Functional Variants in 50,726 Whole-Exome Sequences from the DiscovEHR Study. Science 2016, 354, aaf6814. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Li, J.; Li, H.; Chen, Y.; Wang, L.; He, M.; Wang, Y.; Sun, L.; Hu, Y.; Huang, J.; et al. Coding-Sequence Variants Are Associated with Blood Lipid Levels in 14,473 Chinese. Human Molecular Genetics 2016, 25, 4107–4116. [Google Scholar] [CrossRef]
- Ren, L.; Ren, X. Meta-Analyses of Four Polymorphisms of Lipoprotein Lipase Associated with the Risk of Alzheimer’s Disease. Neuroscience Letters 2016, 619, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Shatwan, I.M.; Minihane, A.-M.; Williams, C.M.; Lovegrove, J.A.; Jackson, K.G.; Vimaleswaran, K.S. Impact of Lipoprotein Lipase Gene Polymorphism, S447X, on Postprandial Triacylglycerol and Glucose Response to Sequential Meal Ingestion. Int J Mol Sci 2016, 17, 397. [Google Scholar] [CrossRef] [PubMed]
- Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators; Stitziel, N. O.; Stirrups, K.E.; Masca, N.G.D.; Erdmann, J.; Ferrario, P.G.; König, I.R.; Weeke, P.E.; Webb, T.R.; Auer, P.L.; et al. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med 2016, 374, 1134–1144. [Google Scholar] [CrossRef]
- Verma, A.; Verma, S.S.; Pendergrass, S.A.; Crawford, D.C.; Crosslin, D.R.; Kuivaniemi, H.; Bush, W.S.; Bradford, Y.; Kullo, I.; Bielinski, S.J.; et al. eMERGE Phenome-Wide Association Study (PheWAS) Identifies Clinical Associations and Pleiotropy for Stop-Gain Variants. BMC Med Genomics 2016, 9 Suppl 1, 32. [Google Scholar] [CrossRef]
- Hayne, C.K.; Lafferty, M.J.; Eglinger, B.J.; Kane, J.P.; Neher, S.B. Biochemical Analysis of the Lipoprotein Lipase Truncation Variant, LPLS447X, Reveals Increased Lipoprotein Uptake. Biochemistry 2017, 56, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.J.; Peloso, G.M.; Yu, H.; Butterworth, A.S.; Wang, X.; Mahajan, A.; Saleheen, D.; Emdin, C.; Alam, D.; Alves, A.C.; et al. Exome-Wide Association Study of Plasma Lipids in >300,000 Individuals. Nat Genet 2017, 49, 1758–1766. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Peloso, G.M.; Liu, D.J.; Wu, Y.; Zhang, H.; Zhou, W.; Li, J.; Tang, C.S.-M.; Dorajoo, R.; Li, H.; et al. Exome Chip Meta-Analysis Identifies Novel Loci and East Asian-Specific Coding Variants That Contribute to Lipid Levels and Coronary Artery Disease. Nat Genet 2017, 49, 1722–1730. [Google Scholar] [CrossRef]
- Wang, J.; Du, S.; Wang, J.; Zhu, M.; Wen, X.; Yang, W. Association of the Lipoprotein Lipase Gene Ser447Ter Polymorphism with Hypertension and Blood Pressure Variation: Evidence from an Updated Meta-Analysis. Clinical and Experimental Hypertension 2017, 39, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Wessel, J.; Willems, S.M.; Zhao, W.; Robertson, N.R.; Chu, A.Y.; Gan, W.; Kitajima, H.; Taliun, D.; Rayner, N.W.; et al. Refining the Accuracy of Validated Target Identification through Coding Variant Fine-Mapping in Type 2 Diabetes. Nat Genet 2018, 50, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wu, Y.; Wen, Y.; Guo, M.; Zhang, H. The Association of the S447X Mutation in LPL with Coronary Artery Disease: A Meta-Analysis. Minerva Cardioangiol 2019, 67, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Marmontel, O.; Rollat-Farnier, P.A.; Wozny, A.-S.; Charrière, S.; Vanhoye, X.; Simonet, T.; Chatron, N.; Collin-Chavagnac, D.; Nony, S.; Dumont, S.; et al. Development of a New Expanded Next-Generation Sequencing Panel for Genetic Diseases Involved in Dyslipidemia. Clinical Genetics 2020, 98, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.B.; Rom, O.; Surakka, I.; Graham, S.E.; Zhou, W.; Roychowdhury, T.; Fritsche, L.G.; Gagliano Taliun, S.A.; Sidore, C.; Liu, Y.; et al. Loss-of-Function Genomic Variants Highlight Potential Therapeutic Targets for Cardiovascular Disease. Nat Commun 2020, 11, 6417. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, M.S.; Li, X.; Li, Z.; Pampana, A.; Zhang, D.Y.; Park, J.; Aslibekyan, S.; Bis, J.C.; Brody, J.A.; Cade, B.E.; et al. Whole Genome Sequence Analysis of Blood Lipid Levels in >66,000 Individuals. Nat Commun 2022, 13, 5995. [Google Scholar] [CrossRef] [PubMed]
- Rip, J.; Nierman, M.C.; Ross, C.J.; Jukema, J.W.; Hayden, M.R.; Kastelein, J.J.P.; Stroes, E.S.G.; Kuivenhoven, J.A. Lipoprotein Lipase S447X: A Naturally Occurring Gain-of-Function Mutation. Arterioscler Thromb Vasc Biol 2006, 26, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Charrière, S.; Meirhaeghe, A.; Dallongeville, J.; Lefai, E.; Rome, S.; Cuerq, C.; Euthine, V.; Delay, M.; Marmontel, O.; et al. Multiple microRNA Regulation of Lipoprotein Lipase Gene Abolished by 3’UTR Polymorphisms in a Triglyceride-Lowering Haplotype Harboring p.Ser474Ter. Atherosclerosis 2016, 246, 280–286. [Google Scholar] [CrossRef]
- Goodarzi, M.O.; Wong, H.; Quiñones, M.J.; Taylor, K.D.; Guo, X.; Castellani, L.W.; Antoine, H.J.; Yang, H.; Hsueh, W.A.; Rotter, J.I. The 3′ Untranslated Region of the Lipoprotein Lipase Gene: Haplotype Structure and Association with Post-Heparin Plasma Lipase Activity. The Journal of Clinical Endocrinology & Metabolism 2005, 90, 4816–4823. [Google Scholar] [CrossRef]
Figure 1.
Protein map of reported LPL coding sequence variants encoded in exon 1. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Black box indicates the residues forming the signal peptide. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
Figure 1.
Protein map of reported LPL coding sequence variants encoded in exon 1. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Black box indicates the residues forming the signal peptide. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
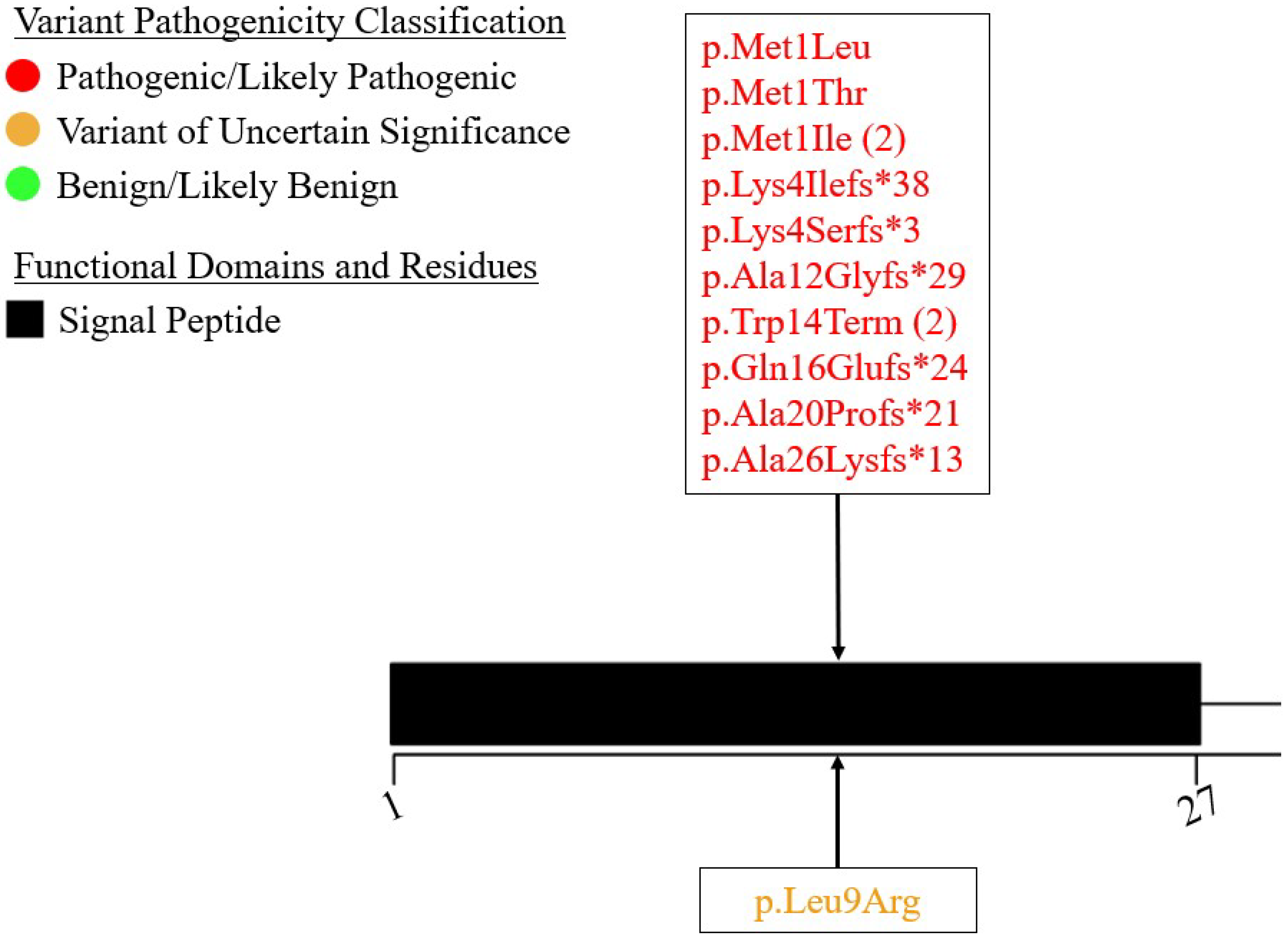
Figure 2.
Protein map of reported LPL coding sequence variants encoded in exon 2. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Light-green box indicates cysteine residues involved in the formation of intramolecular disulfide bonds. Pink box represents asparagine residues at which N-linked glycosylation has been found to occur. Yellow boxes represent residues involved in apo C-II binding. Blue boxes represent residues involved in ANGPTL4 binding. Brown box represents residues involved in the formation of an oxyanion hole in mature protein. Where residues have been found to be involved in multiple functions, multiple boxes are used to represent the overlap. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
Figure 2.
Protein map of reported LPL coding sequence variants encoded in exon 2. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Light-green box indicates cysteine residues involved in the formation of intramolecular disulfide bonds. Pink box represents asparagine residues at which N-linked glycosylation has been found to occur. Yellow boxes represent residues involved in apo C-II binding. Blue boxes represent residues involved in ANGPTL4 binding. Brown box represents residues involved in the formation of an oxyanion hole in mature protein. Where residues have been found to be involved in multiple functions, multiple boxes are used to represent the overlap. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
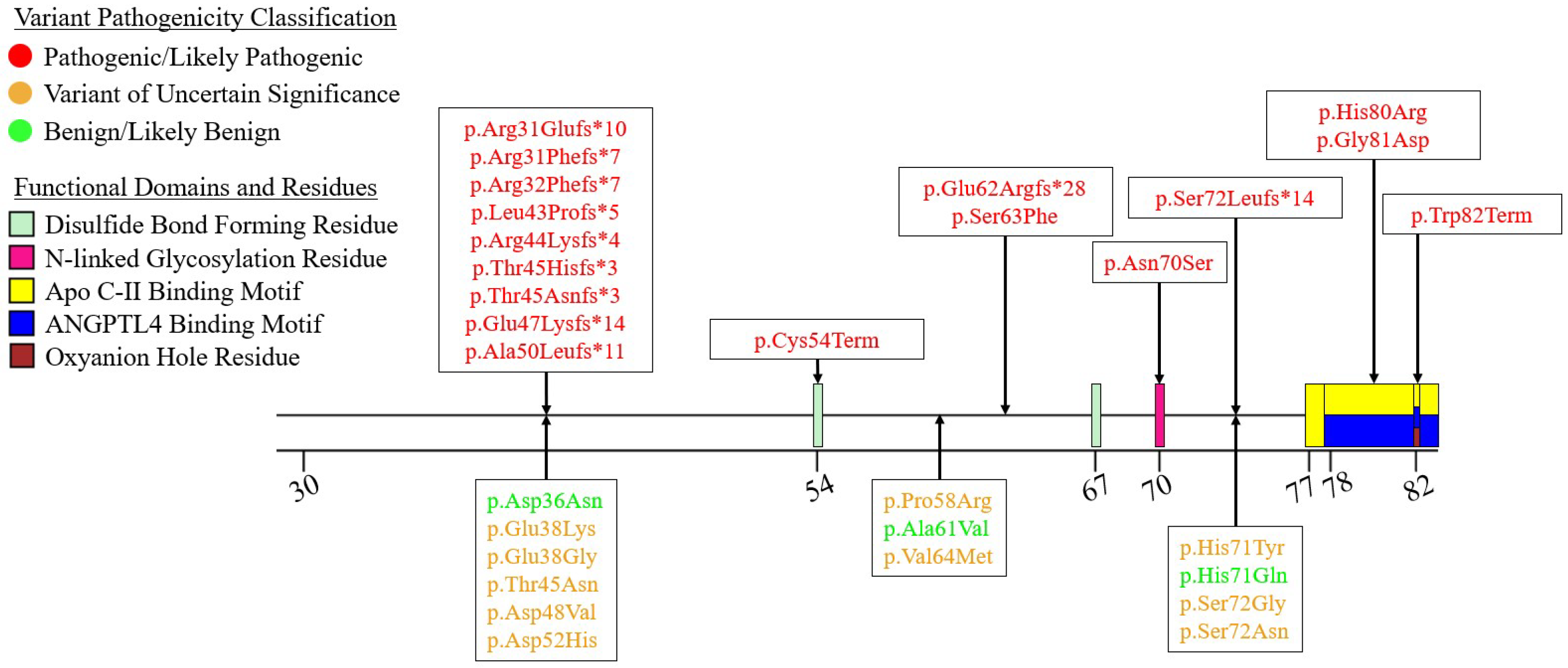
Figure 3.
Protein map of reported LPL coding sequence variants encoded in exon 3. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Yellow boxes represent residues involved in apo C-II binding. Blue boxes represent residues involved in ANGPTL4 binding. Where residues have been found to be involved in multiple functions, multiple boxes are used to represent the overlap. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
Figure 3.
Protein map of reported LPL coding sequence variants encoded in exon 3. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Yellow boxes represent residues involved in apo C-II binding. Blue boxes represent residues involved in ANGPTL4 binding. Where residues have been found to be involved in multiple functions, multiple boxes are used to represent the overlap. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
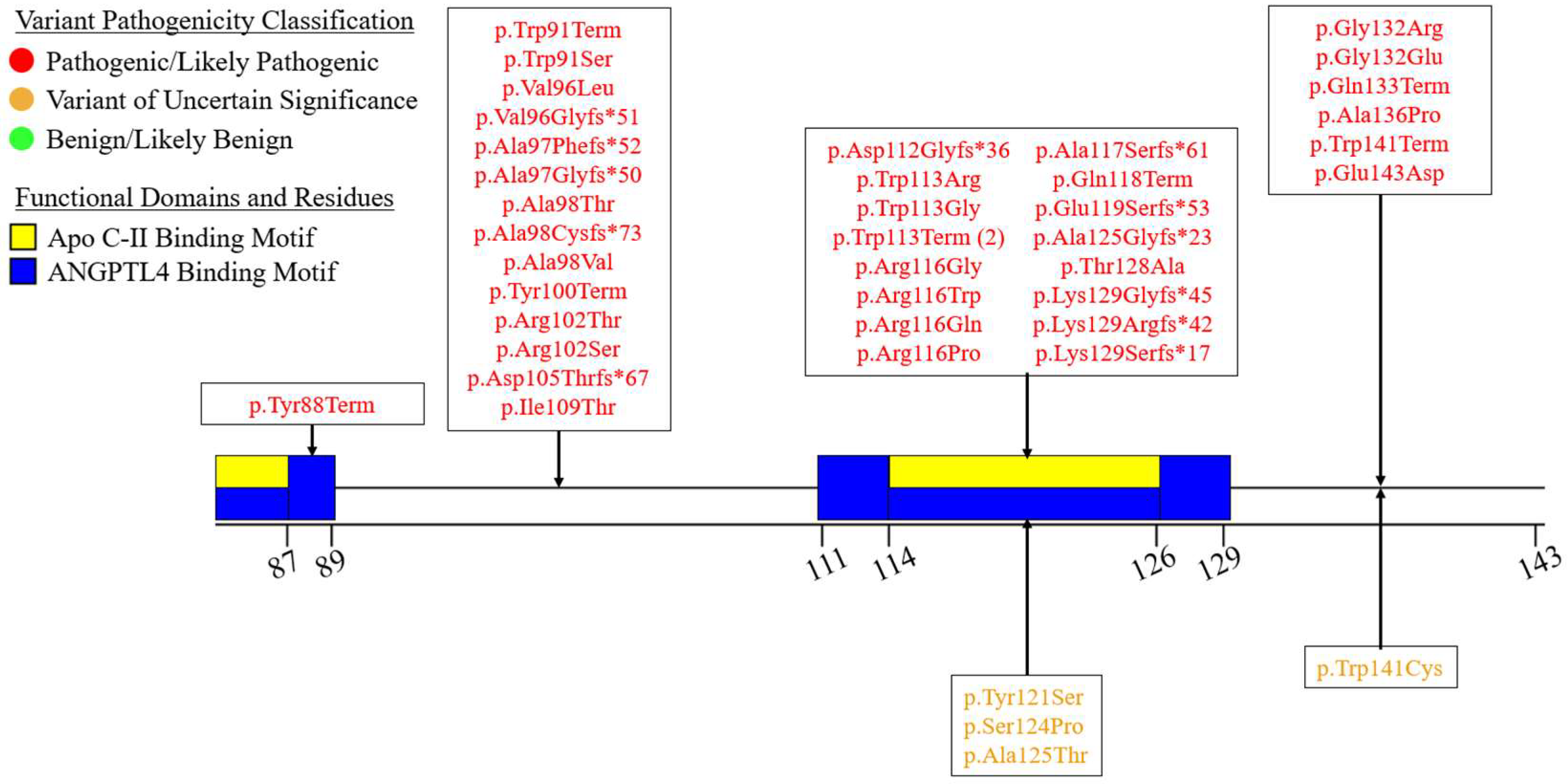
Figure 4.
Protein map of reported LPL coding sequence variants encoded in exon 4. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Green boxes represent residues forming the catalytic triad of the LPL hydrolase domain. Brown box represents residues involved in the formation of an oxyanion hole in mature protein. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
Figure 4.
Protein map of reported LPL coding sequence variants encoded in exon 4. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Green boxes represent residues forming the catalytic triad of the LPL hydrolase domain. Brown box represents residues involved in the formation of an oxyanion hole in mature protein. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.

Figure 5.
Protein map of reported LPL coding sequence variants encoded in exon 5. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Green boxes represent residues forming the catalytic triad of the LPL hydrolase domain. Purple boxes represent residues involved in the coordination of Ca2+ into the mature LPL protein. Yellow boxes represent residues involved in apo C-II binding. Light-green box indicates cysteine residues involved in the formation of intramolecular disulfide bonds. Red boxes represent residues forming the lid region involved in regulating substrate access to active site of mature LPL. Blue boxes represent residues involved in ANGPTL4 binding. Where residues have been found to be involved in multiple functions, multiple boxes are used to represent the overlap. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
Figure 5.
Protein map of reported LPL coding sequence variants encoded in exon 5. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Green boxes represent residues forming the catalytic triad of the LPL hydrolase domain. Purple boxes represent residues involved in the coordination of Ca2+ into the mature LPL protein. Yellow boxes represent residues involved in apo C-II binding. Light-green box indicates cysteine residues involved in the formation of intramolecular disulfide bonds. Red boxes represent residues forming the lid region involved in regulating substrate access to active site of mature LPL. Blue boxes represent residues involved in ANGPTL4 binding. Where residues have been found to be involved in multiple functions, multiple boxes are used to represent the overlap. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
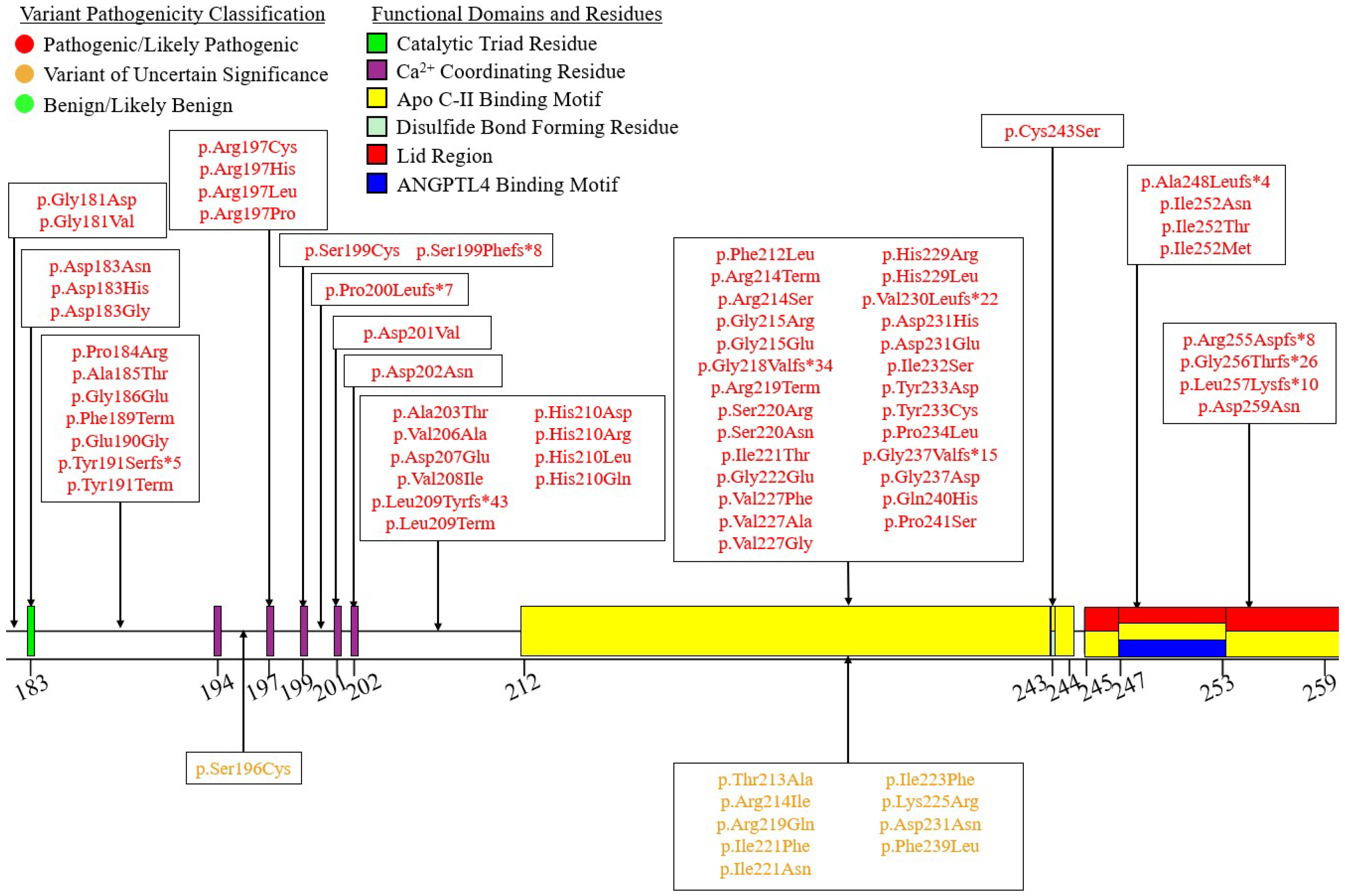
Figure 6.
Protein map of reported LPL coding sequence variants encoded in exon 6. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Red boxes represent residues forming the lid region involved in regulating substrate access to active site of mature LPL. Light-green box indicates cysteine residues involved in the formation of intramolecular disulfide bonds. Green boxes represent residues forming the catalytic triad of the LPL hydrolase domain. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
Figure 6.
Protein map of reported LPL coding sequence variants encoded in exon 6. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Red boxes represent residues forming the lid region involved in regulating substrate access to active site of mature LPL. Light-green box indicates cysteine residues involved in the formation of intramolecular disulfide bonds. Green boxes represent residues forming the catalytic triad of the LPL hydrolase domain. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
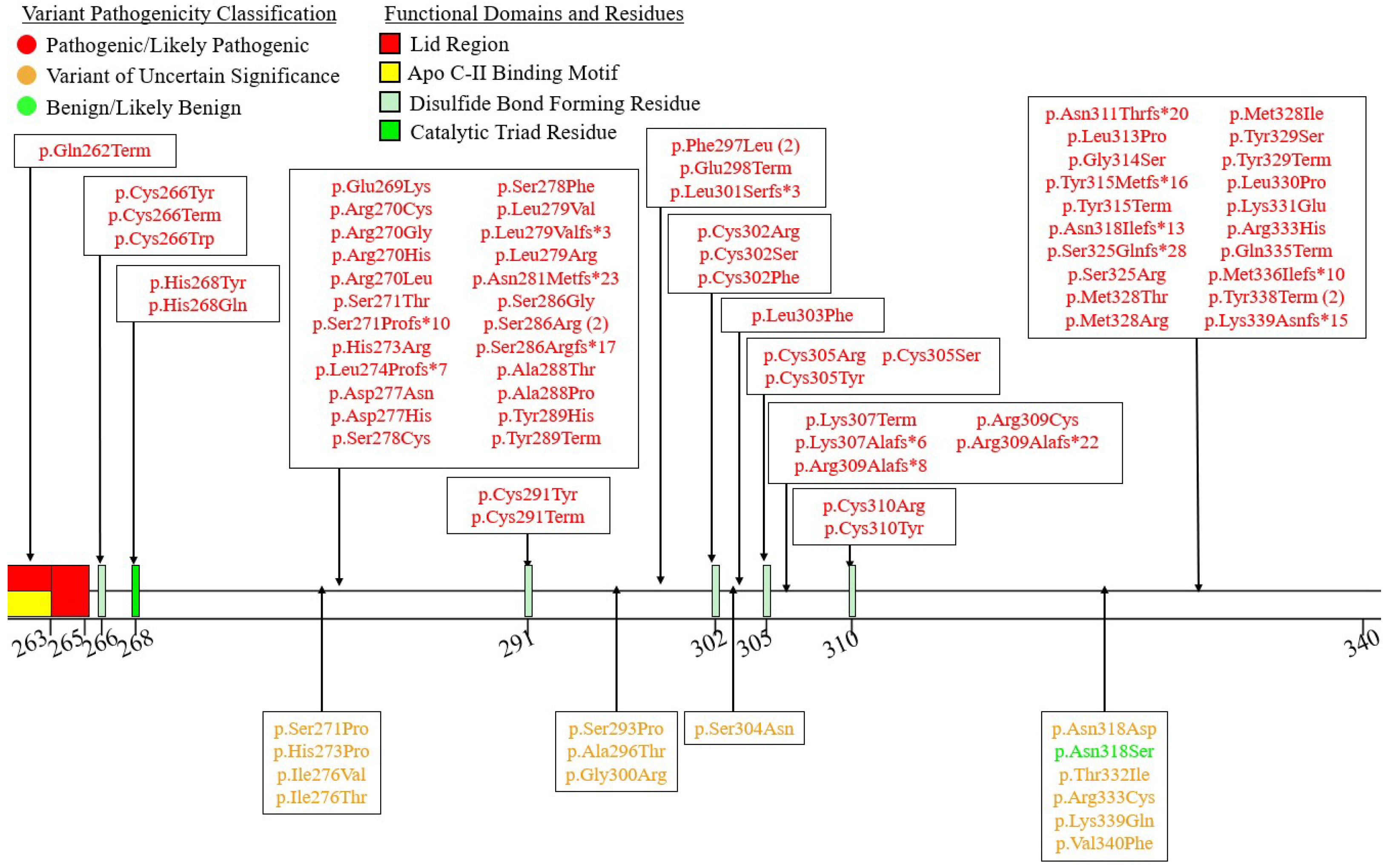
Figure 7.
Protein map of reported LPL coding sequence variants encoded in exon 7. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Orange boxes represent residues involved in interacting with finger 3 of the LU domain of GPIHBP1. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
Figure 7.
Protein map of reported LPL coding sequence variants encoded in exon 7. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Orange boxes represent residues involved in interacting with finger 3 of the LU domain of GPIHBP1. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
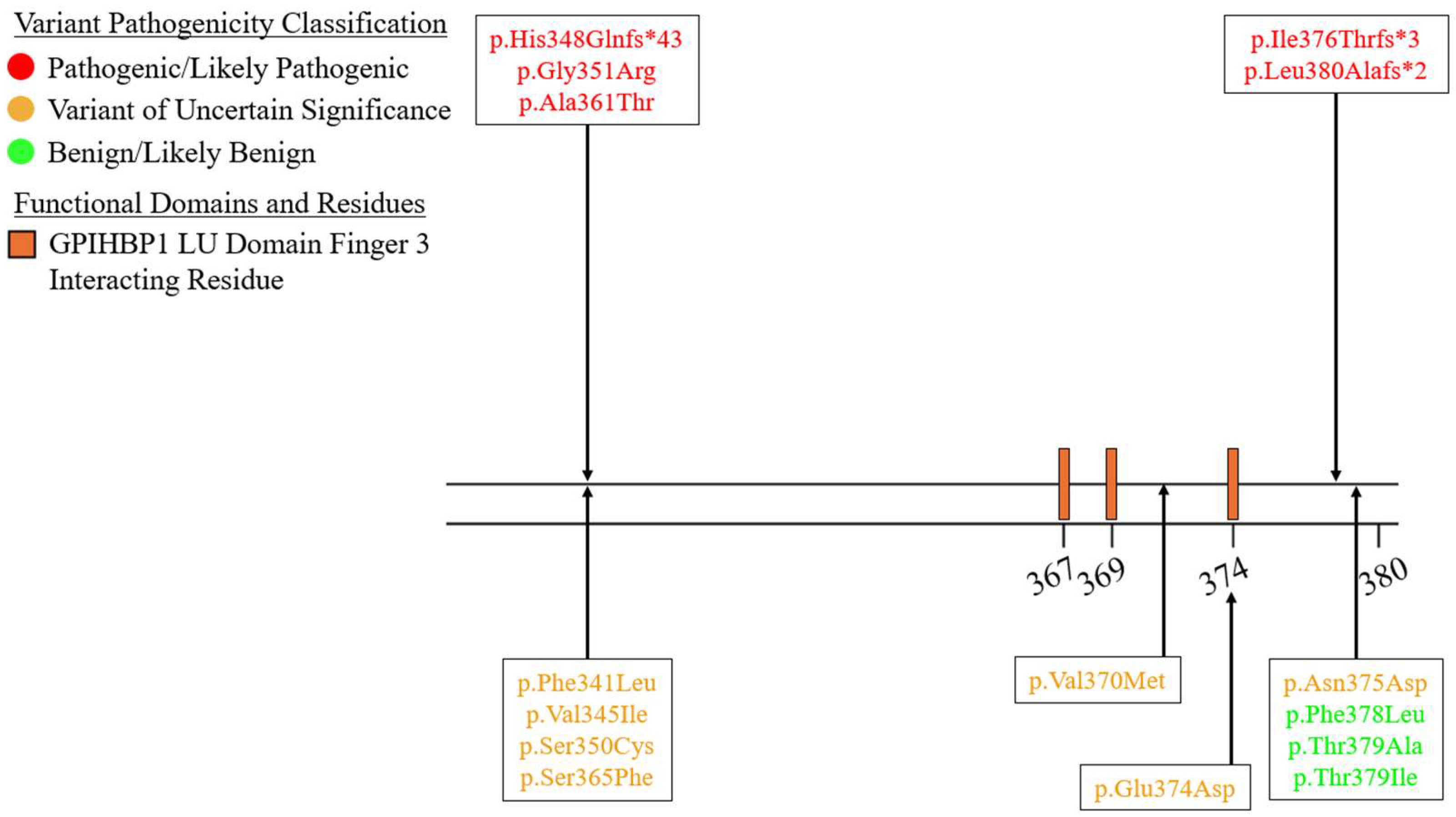
Figure 8.
Protein map of reported LPL coding sequence variants encoded in exon 8. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Turquoise boxes represent residues involved in the formation of a stabilizing hydrogen bond between LPL and GPIHBP1. Pink box represents asparagine residues at which N-linked glycosylation has been found to occur. Orange boxes represent residues involved in interacting with finger 3 of the LU domain of GPIHBP1. Light pink-purple boxes represent the Trp-rich lipid binding domain. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
Figure 8.
Protein map of reported LPL coding sequence variants encoded in exon 8. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Turquoise boxes represent residues involved in the formation of a stabilizing hydrogen bond between LPL and GPIHBP1. Pink box represents asparagine residues at which N-linked glycosylation has been found to occur. Orange boxes represent residues involved in interacting with finger 3 of the LU domain of GPIHBP1. Light pink-purple boxes represent the Trp-rich lipid binding domain. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
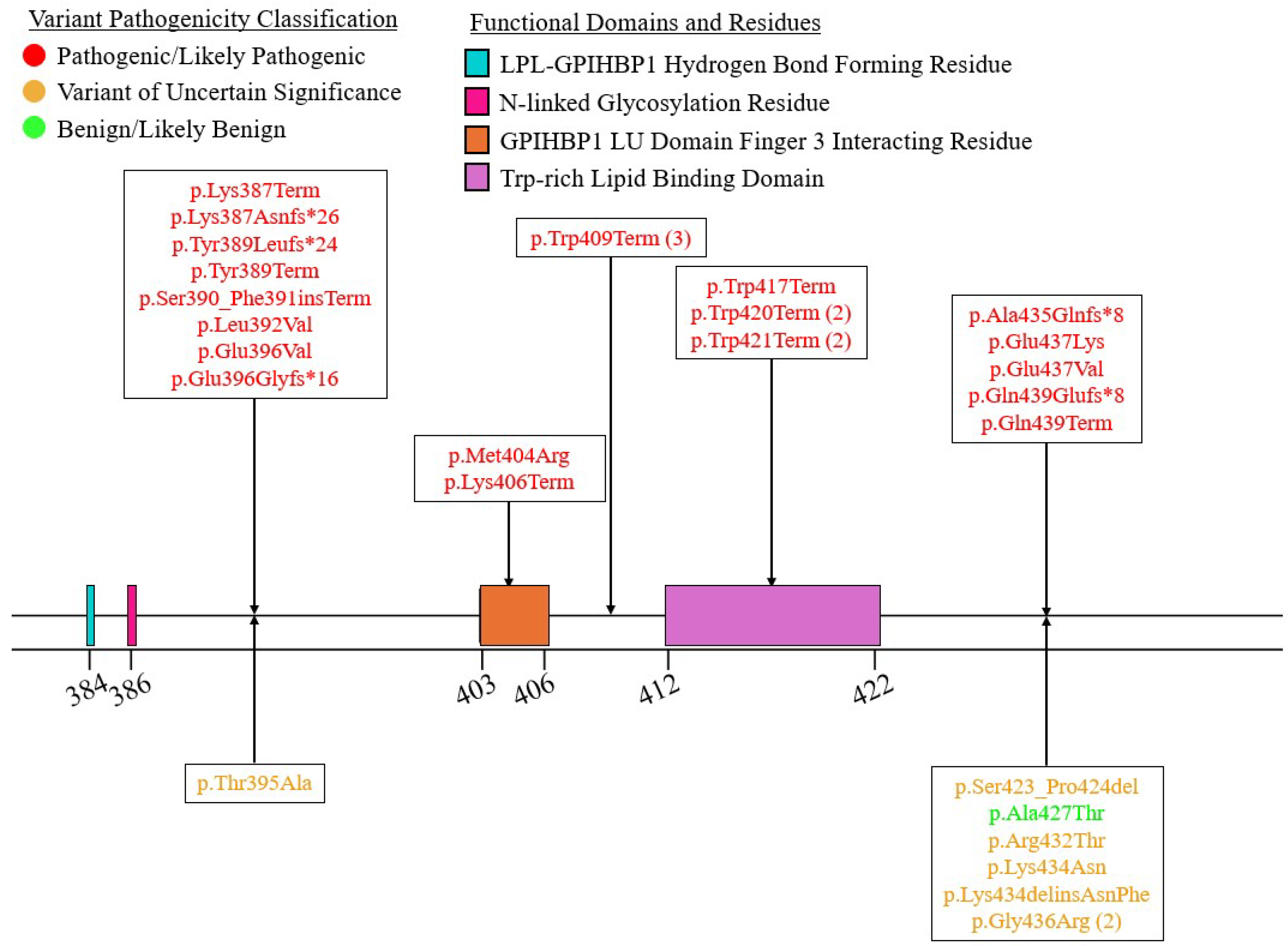
Figure 9.
Protein map of reported LPL coding sequence variants encoded in exon 9. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Dark grey boxes represent residues involved in interacting with finger 1 of the LU domain of GPIHBP1. Yellow-Green boxes represent residues involved in interacting with figure 2 of the LU domain of GPIHBP1. Orange boxes represent residues involved in interacting with finger 3 of the LU domain of GPIHBP1. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
Figure 9.
Protein map of reported LPL coding sequence variants encoded in exon 9. Number line indicates amino acid residue numbering in primary structure of newly synthesized LPL peptide, with the specific numbers indicating the beginning and/or end of the functional domain they border. Dark grey boxes represent residues involved in interacting with finger 1 of the LU domain of GPIHBP1. Yellow-Green boxes represent residues involved in interacting with figure 2 of the LU domain of GPIHBP1. Orange boxes represent residues involved in interacting with finger 3 of the LU domain of GPIHBP1. All variants are color-coded according to their ACMG pathogenicity classification: red indicates pathogenic or likely pathogenic, yellow orange indicates a variant of uncertain significance (VUS), and green indicates benign or likely benign. Pathogenic/likely pathogenic variants are shown above the linear map, while VUS, and benign/likely benign variants are shown below.
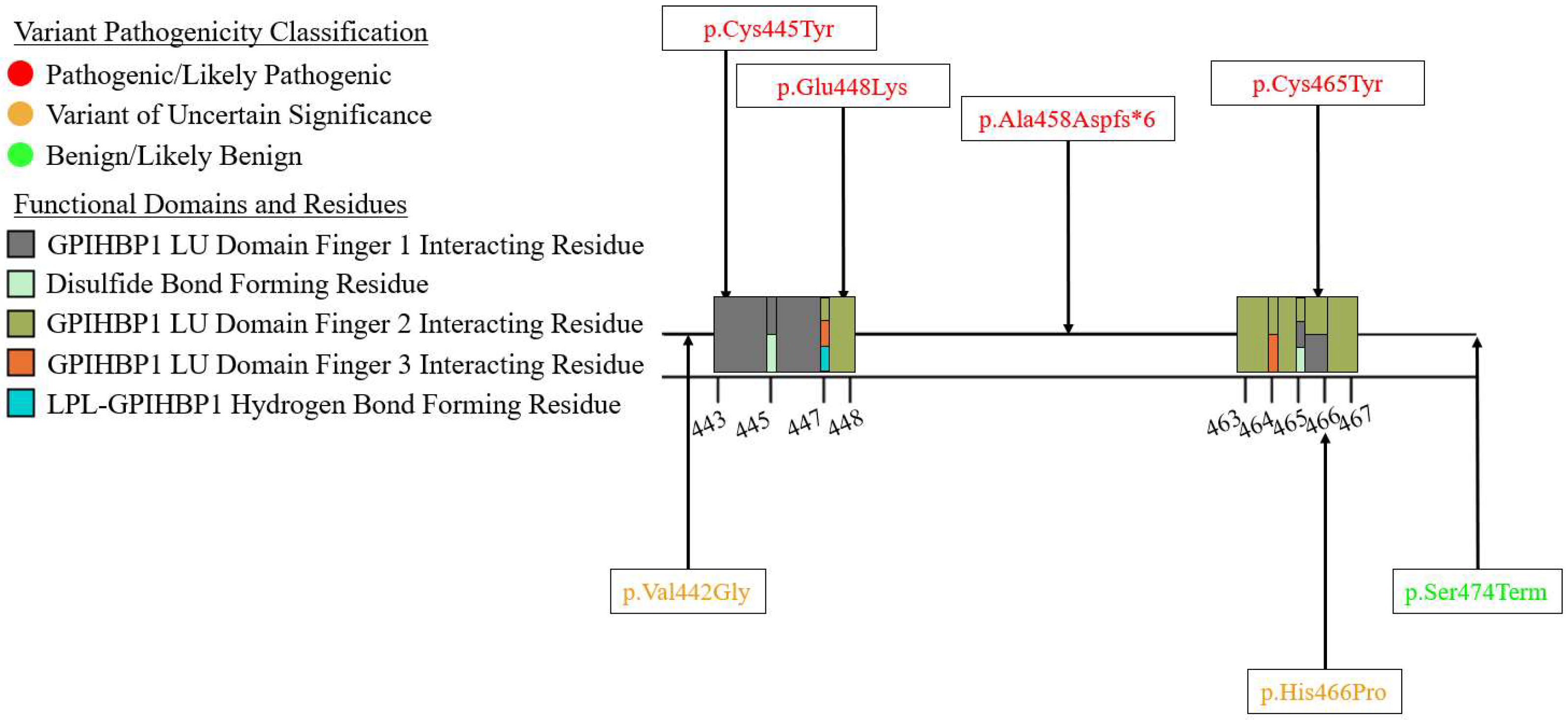

Table 1.
Summary of Coding Region Pathogenic/Likely Pathogenic and VUS variants in LPL.
| Variant Type (P-LP1/VUS2) | ||||||
|---|---|---|---|---|---|---|
| Genomic Location |
Missense | Nonsense | Small Deletion |
Small Insertion |
Small Indel |
Total |
| Exon 1 | 4/1 | 2/0 | 3*/0 | 2/0 | 1/0 | 12/1 |
| Exon 2 | 4/10 | 2/0 | 6/0 | 5/0 | 0/0 | 17/10 |
| Exon 3 | 18/4 | 7/0 | 6/0 | 4/0 | 3/0 | 38/4 |
| Exon 4 | 11/2 | 1/0 | 4/0 | 0/0 | 0/0 | 16/2 |
| Exon 5 | 51/10 | 4/0 | 11/0 | 1/0 | 0/0 | 67/10 |
| Exon 6 | 46/14 | 10/0 | 12/0 | 4/0 | 0/0 | 72/14 |
| Exon 7 | 2/7 | 0/0 | 3/0 | 0/0 | 0/0 | 5/7 |
| Exon 8 | 5/5 | 10/0 | 5/1 | 3/0 | 0/1 | 23/7 |
| Exon 9 | 3/2 | 0/0 | 1/0 | 0/0 | 0/0 | 4/2 |
| Total | 144/55 | 36/0 | 51/1 | 19/0 | 4/1 | 254/57 |
1P-LP = Pathogenic/Likely Pathogenic; 2VUS = Variant of Uncertain Significance; *One of these small deletion variants also affects the first nucleotide of intron 1.
Table 2.
Summary of Non-Coding Region Pathogenic/Likely Pathogenic and VUS variants in LPL.
| Variant Type (P-LP1/VUS2) | |||||
|---|---|---|---|---|---|
| Genomic Location |
Splicing | Small Deletion |
Small Insertion |
Other | Total |
|
Regulatory (Promoter, 5’ UTR, and 3’ UTR) |
N/A3 | 0/0 | 0/0 | 1/1 | 1/1 |
| Intron 1 | 7/0 | 1/0 | 1/0 | N/A | 9/0 |
| Intron 2 | 5*/0 | 0/0 | 1/0 | N/A | 6/0 |
| Intron 3 | 1/0 | 0/0 | 0/0 | N/A | 1/0 |
| Intron 4 | 2/0 | 0/0 | 0/0 | N/A | 2/0 |
| Intron 5 | 1/0 | 0/0 | 0/0 | N/A | 1/0 |
| Intron 6 | 5/0 | 0/0 | 0/0 | N/A | 5/0 |
| Intron 7 | 2/0 | 0/0 | 0/0 | N/A | 2/0 |
| Intron 8 | 5/0 | 0/0 | 0/0 | N/A | 5/0 |
| Intron 9 | 1/0 | 0/0 | 0/0 | N/A | 1/0 |
| Total | 29/0 | 1/0 | 2/0 | 1/0 | 33/1 |
1P-LP = Pathogenic/Likely Pathogenic; 2VUS = Variant of Uncertain Significance; 3N/A = Not Applicable; *One of these small deletion variants also affects the first nucleotide of intron 1.
Table 3.
Summary of Pathogenic/Likely Pathogenic and VUS Large-Scale Variants in LPL.
| Variant Type (P-LP1/VUS2) | ||
|---|---|---|
| Gross Deletions | Gross Insertions | Complex Rearrangements |
| 10/0 | 2/0 | 1/0 |
1P-LP = Pathogenic/Likely Pathogenic; 2VUS = Variant of Uncertain Significance.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



