
Submitted:
16 December 2024
Posted:
17 December 2024
You are already at the latest version
Abstract
The increasing prevalence of multi-drug-resistant (MDR) bacterial pathogens presents a critical global health threat, highlighting the urgent need for innovative approaches to understanding bacterial patho-genesis and developing effective therapies. This review explores the potential of synthetic biology as a promising tool for investigating host-pathogen interactions and offering alternative therapeutic solu-tions for MDR infections. We first examine the progress of CRISPR-based strategies that have enabled modulation of essential gene expression, providing insights into the molecular mechanisms underly-ing host-pathogen interactions. We then discuss the use of engineered microbial synthetic circuits for rapid pathogen detection, which identify molecular signatures involved in interspecies communica-tion and facilitate swift pathogen elimination. Additionally, we explore the potential of Phage Therapy (PT), which leverages bacteriophages to selectively target and eliminate specific bacterial pathogens, presenting a targeted and promising approach to combat MDR infections. Finally, we review the appli-cation of Organ-On-A-Chip (OOAC) technology, which overcomes the limitations of animal models in predicting human immune responses by using microfluidic devices that simulate organ-level physi-ology and pathophysiology, thereby enabling more accurate disease modeling, drug testing, and the development of personalized medicine.
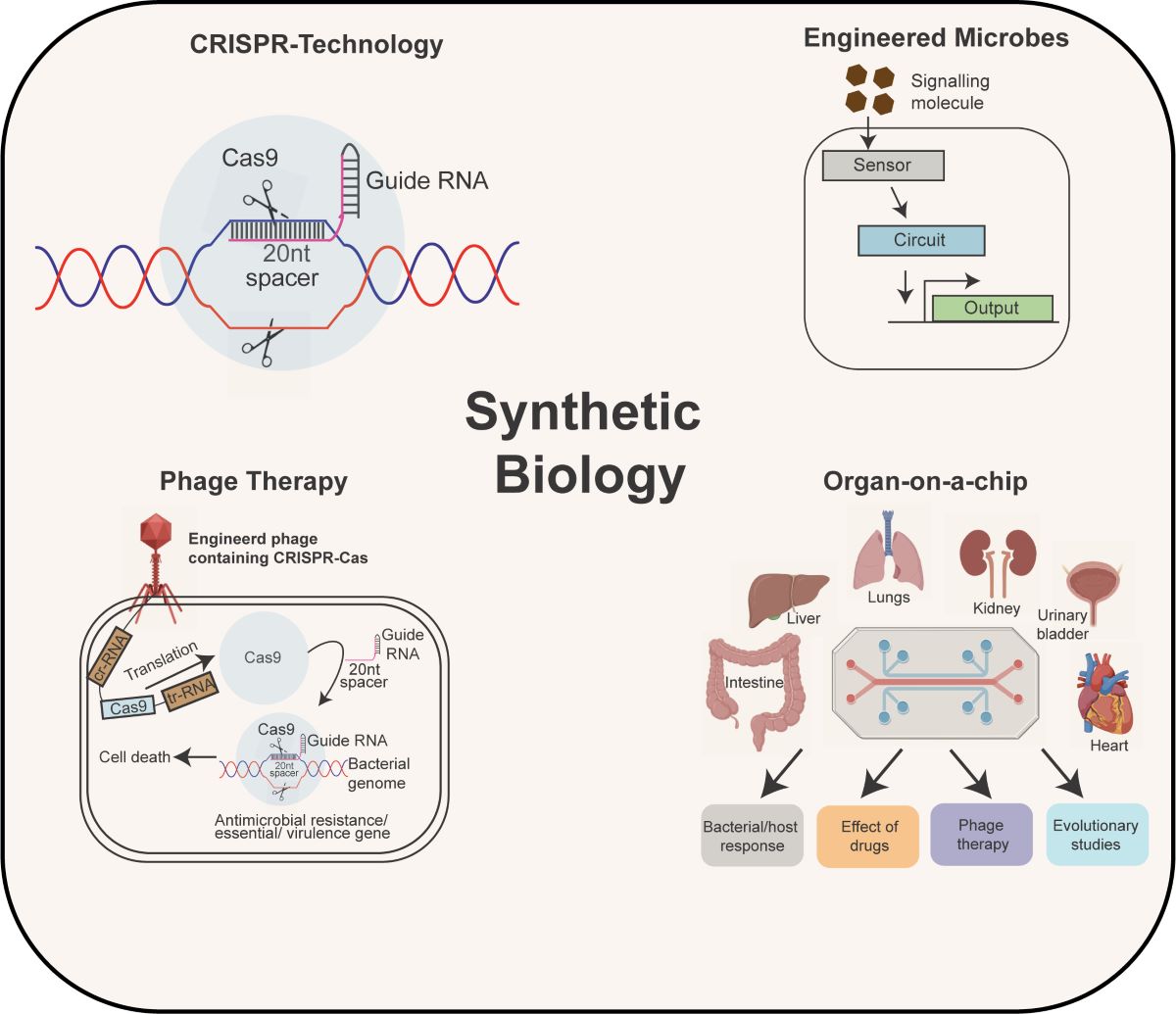
Keywords:
Host-pathogen interactions
; bacterial pathogenesis
; synthetic biology
; CRISPR
; microbial synthetic circuits
; engineered microbes
; phage therapy
; organ-on-a-chip
; multi drug resistance
1. Introduction
Bacterial infections are a worldwide threat that significantly burdens the healthcare systems, with the problem escalating in recent years due to the emergence of multi-drug-resistant bacterial strains [1]. Each year, an estimated 2 million people are infected with Mycobacterium tuberculosis, the etiological agent of tuberculosis, with approximately one-third of the global population serving as carriers of the disease [2]. Similarly, Urinary Tract Infections (UTIs) are becoming increasingly prevalent, with nearly every woman worldwide expected to experience at least one UTI in her lifetime [3]. Alarmingly, reports of multidrug-resistant strains of M. tuberculosis [4] and uropathogenic Escherichia coli, the primary cause of UTIs [5,6] have been on the rise. Recently, Acinetobacter baumannii, a nosocomial acquired pathogen has emerged as a global threat as it exhibits resistance to most commonly used antimicrobial agents [7]. This highlights an urgent need to detect and combat these pathogens [8]. Traditional methods for deciphering the molecular mechanisms of virulence can be labor-intensive, time-consuming, and often rely on murine infection models, the results from these models may not accurately translate to human drug development due to species-specific differences [9]. For example, traditional approaches using l-Red recombination machinery that has been the gold standard for knocking out non-essential genes in model organisms to study their role in virulence [10,11,12] can be time consuming as it involves individually replacing the gene of interest with an antibiotic cassette. Oher drawbacks of this widely used method includes the inability of the method to knock out essential genes and this method was developed only for a few model organisms. Tn-seq was developed to investigate the role of essential genes in determining phenotypes [13,14]. It involves random insertion of a transposable genetic element across the genome. By growing the Tn-seq library under different conditions, essential genes for a particular growth condition can be identified, as these genes will show little or no transposon insertion. Tn-seq has been widely used to identify interaction between essential and non-essential genes [15] as well as genes involved in antibiotic resistance [16,17]. Although Tn-seq has been a powerful tool to identify essential genes, several reports suggest non-random insertions of the transposon across the genome especially for organism with high GC content [18,19,20]. Additionally, the construction of the transposon involves a strong promoter that can lead to overexpression of the genes located downstream of the transposon insertion which can have impact on the phenotype [18]. Furthermore, the role of smaller genes is often missed due the low insertion frequency in them and the method fails to identify the role of overlapping genes (genes which share at least in part the same genomic location) [21]. Finally, when a bacterial pathogen invades a host, it often encounters other bacterial species residing within the host's microbiota. To establish a successful infection, the invading bacteria must engage in molecular communication with these resident species. However, traditional methodologies have limited utility in elucidating the intricate signaling pathways of the resident microbiota that regulate the complex process within the host demanding the need for development of new tools and technologies.
Synthetic biology is an emerging field that combines the concepts and applications of engineering and computational prowess with biological systems to develop tools that has the potential to address important issues related to human health including host-pathogen interactions [22,23,24]. Host-pathogen interactions often involves complex interplay between the defense strategies of the host with the bacterial virulence arsenal. Mutiple genes contribute simutaneously in this intricate interaction between the host and the pathogen which cannnot be deciphered using traditional methods. To achieve this, CRISPR-based approaches were adapted that can knock out or knockdown mutiple genes of interest simultaneously without the involvement of multiple strain construction or antibiotic cassettes [25]. The widely used Type II CRISPR system consists of a chimeric RNA called guide RNA and a single nuclease, Cas9, that can introduce double stranded breaks in the DNA, a process that is lethal for most bacterial species [26]. This principle forms the basis of CRISPR-based technologies (described in detail in the CRISPR section). CRISPR based systems can be inducible, titratable and can be multiplexed to knockout multiple genes in a high-throughput format [27,28,29]. A variant of CRISPR was created to knockdown genes of interest by using a catalytically dead nuclease Cas9 known as CRISPRi [30]. By combining the power of Tn-seq, with CRISPRi, Peters et al created mobile-CRISPRi [31] which enabled researchers to systematically study the role of essential genes and virulence factors across multiple bacterial pathogens including Pseudomonas aeruginosa [32], Acetobacter baumannii [33], and Vibrio sp [34].
One of the key aspects of treating a bacterial infection is early diagnosis of the pathogen within the host. For example, uropathogenic strain of E. coli CFT073 initially infects the urethra. If left untreated, the pathogen migrates upward and infects the urinary bladder causing cystitis [35] followed by infecting the kidney resulting in polynephritis [36]. Currently utilized diagnostic tools exhibit significant limitations, including suboptimal sensitivity, prolonged time requirements for definitive results, and prohibitively high costs, which collectively hinder their clinical efficacy and accessibility [37]. To address this challenge, synthetic biology-based approaches have been adopted to engineer genetic circuits in the bacteria that can act as a sensor for biomarkers for multiple pathogens. Bacterial Whole Cell Biosensors (BWCBs) can easily colonize the human gut, replicate, survive the harsh conditions and establish as a member of the microbiota. These BWCBs are equipped to sense bacterial metabolites or cell surface proteins or lipids and gives detectable output signal (fluorescent proteins, biomarkers or antibodies) which can be used for downstream diagnosis [38]. To achieve this, bacteria are engineered with inducible synthetic circuits utilizing transcription factors whose DNA-binding activity is regulated by a specific ligand. These synthetic circuits have allowed the researchers in deciphering the molecular signatures involved in intra- and inter-species communication [39]. Furthermore, leveraging the natural environmental sensing capabilities of bacteria, researchers have constructed genetic circuits capable of integrating multiple inputs using logic gates, such as AND gates that require all inputs to be present for activation [40], or NOR gates [40] or a combination of diverse array of genetic logic circuits involving OR, NAND, XOR, XNOR gates, broadening the functional applications of BWCBs [40,41].
Another challenge in treating a specific bacterial pathogen stems from its existence within a complex conglomerate of microbiota members. Conventional antimicrobial therapies often employ broad-spectrum antibiotics, which indiscriminately target a wide range of bacteria, including beneficial gut residents, resulting in disruptions to the diversity of the gut microbiome. To overcome this obstacle, synthetic biology based alternative approaches involving bacteriophages have been developed which are collectively referred to as phage therapy [42,43,44,45]. The main advantage of phage therapy (PT) lies in the inherently narrow host range of phages, which helps preserve commensal microflora and results in fewer adverse effects compared to broad-spectrum antibiotic treatments. [44]. In addition, the inherent property of phages to multiply within the host makes them effective at low input doses [44]. Further, phages demonstrate the ability to coevolve with their bacterial hosts during therapeutic applications, enabling effective control of dominant bacterial populations [46]. Although bacterial pathogens may acquire resistance to phages, phage efficacy can be restored or enhanced through mutagenesis [47]. Moreover, bacterial resistance to phages has been shown to come with a trade-off, resulting in a decrease in virulence [48]. The innate ability of phages to infect bacterial cells was exploited by researchers to engineer phages that contains the CRISPR array to target antimicrobial resistance genes of bacterial pathogens [49]. In addition, synthetic biologist have also exploited the property of phages to break open their bacterial host using endolysins. By creating chimeric and conjugated endolysins, researchers were able to target a wide variety of bacterial pathogens [50,51,52,53].
Recent studies have highlighted the critical role of the gut microbiota in human health [54,55,56]. The gut microbiota, consists of approximately 100 trillion bacteria that engages in complex interactions to regulate various gut processes. Disruptions to the gut microbiota, whether through the introduction of pathogenic bacteria or shifts in the abundance of existing species, can have detrimental effects on the host. Traditional methods for studying the immune response to bacterial pathogens have used the murine infection model. Despite the genetic similarities, mice have significant differences in gene expression profiles and mechanism of regulation as compared to humans, leading to inconsistent disease modeling making them less ideal for studying bacterial infection [9]. Furthermore, any drugs that work effectively in mice often fail in human clinical trials due to differences in their pharmacodynamics and pharmacokinetics properties [57,58,59]. To solve this problem, organ-on-a-chip was developed that uses human cell lines in a microfludic device to mimic the host environment more accurately. Using this method, numerous studies have been conducted to unravel the complex interactions between bacterial pathogens, the host immune system, and antimicrobial agents [60,61].
This review examines the impact of synthetic biology on the study of host-pathogen interactions through rapid, efficient, and systematic approaches enabled by CRISPR-based technology. It also explores the application of synthetic biology in the early diagnosis of pathogens, utilizing bacterial genetics to develop synthetic circuits capable of sensing pathogens within the host. Additionally, the review discusses alternative treatments for multi-drug-resistant bacteria, focusing on phage therapy, and evaluates the role of organ-on-a-chip technology in advancing the understanding of host-pathogen interactions, thereby facilitating the rapid development of novel therapeutics to combat multi-drug-resistant pathogens
2. CRISPR- A Versatile Tool for Studying Antimicrobial Resistance and Gene Regulation and Bacterial Virulence
Bacteria have by intrinsic restriction systems that can detect and degraded any foreign DNA as part of their adaptive immune defense system [62]. Notably, in 2007, studies on Streptococcus thermophilus during infections with lytic bacteriophages provided the first experimental evidence supporting CRISPR-Cas–mediated adaptive immunity [63]. This discovery inspired the idea of utilizing the natural CRISPR-Cas systems present in bacteria commonly used in the dairy industry for phage immunization, marking the first biotechnological application of CRISPR-Cas [64]. Following this, mature CRISPR RNAs (crRNAs) were identified as guide molecules forming a complex with Cas proteins to inhibit viral replication in E. coli [65] and the DNA-targeting activity of the CRISPR-Cas system was demonstrated in the pathogen Staphylococcus epidermidis [66]. It is estimated that ~40% of bacterial species use CRISPR. There are three types of CRISPR systems, with the Type II system being widely used due to its simplicity. It utilizes a single nuclease, Cas9 and a guide RNA (gRNA) containing a spacer sequence within its first 20 nucleotides that can form a stable RNA-DNA hybrid with the non-template strand of the target DNA. This RNA-DNA hybrid is subsequently recognized by the Cas9 nuclease through a short protospacer adjacent motif (PAM). Cas9 in turn cleaved the target DNA by causing double strand breaks (endonucleolytic cleavage). One of the unique features of the CRISPR system is its inherent flexibility in DNA recognition. By modifying the spacer region of the gRNA, the researchers can achieve precise genome editing (Figure 1A). CRISPR has been widely used for genome editing in bacteria [67], plants [68] and mammals [69,70]. The following section will disscuss three prominent applications of CRISPR namely CRISPR interference (CRISPRi), CRISPR-activation (CRISPRa) and mobile CRISPRi that enabled the study of host-pathogen interactions of both model and non-model pathogens.
2.1. CRISPR-Interference (CRISPRi)
CRISPRi has proven to be a useful tool to study the role of gene regulation in bacteria [25,71,72]. The primary components include a single chimeric RNA, formed by the fusion of crRNA and tracr-RNA, along with a catalytically inactive variant of the Cas9 protein (dCas9) which retains its DNA-binding capability but is unable to induce double-stranded breaks in the DNA. The gRNA-dCas9 complex can inhibit transcription either by occluding the RNA polymerase from binding to its cognate promoter DNA or by sterically hindering the elongation complex through interaction with the non-template DNA strand [25] (Figure 1B). Using this approach, researchers successfully studied the functions of virulence genes across multiple pathogenic bacterial species [72,73,74,75,76]. For example, Wang et al. developed an optimized CRISPRi system for Yersinia pestis that successfully inhibited the expression of genes involved in biofilm formation gene hmsH, cold shock protein cspB and virulence factors yscB and ail. These deletion mutants exhibited decreased virulence in both HeLa cells and mouse models, consistent with previously observed phenotype for yscB and ail knockouts [74]. Similarly, Choudhury et al. applied CRISPRi to target essential genes in the pathogenic Mycobacterium tuberculosis as well as its non-pathogenic counterpart, Mycobacterium smegmatis [73]. In a separate study, a genome-scale CRISPR-KO and CRISPRi libraries were constructed to investigate essential genes for bacterial viability in Mycobacterium tuberculosis (Mtb). Using these libraries, the authors identified genes influencing resistance or susceptibility to the antitubercular drug bedaquiline (BDQ) [77]. The real merit of CRISPRi includes multiplexing and tiltability. By creating a library of gRNAs where each gRNA is specific for a particular gene, the researchers were able to modulate the expression of multiple genes simultaneously. Recently, de Bakker et al. conducted a genome wide CRISPRi screens in Streptococcus pneumoniae in a murine pneumonia model of infection to identify 269 essential genes including genes involved in cell wall biosynthesis, pbp2x, DNA replication, dnaA, cell division, ftsZ and central carbon metabolism, pfkA [75]. In a separate study, Elis et al. developed a novel platform to facilitate the simultaneous knockout of multiple genes in Legionella pneumophila, an intracellular pathogen responsible for legionellosis [76]. This bacterium secretes ~300 effector proteins during infection via the Dot/lcm type IV secretion system. Traditional methods of knocking out individual genes may fail to uncover specific gene contributions to virulence due to functional redundancy. To address this, the authors engineered a CRISPR-based system by designing a plasmid containing dCas9 and an array of 10 distinct spacer sequences. Each spacer was tailored to target a specific gene and was placed under the control of a strong inducible Ptet promoter and a box element. This CRISPR array enabled the simultaneous silencing of 10 genes, allowing the study of their collective impact on cellular behavior in anoxic conditions and during macrophage infection. Notably, the silencing efficiency of each spacer was influenced by its position within the array, with the proximal spacers exhibiting the highest potency, which diminished as the spacers were positioned further distally in the array [76]. Biofilm formation is one of the key defense strategies employed by bacterial pathogens and fungi to limit the accessibility of the antimicrobial agents. To combat biofilm formation, CRISPRi mediated strategies have implemented in E. coli [78], Pseudomonas sp [79] and even in pathogenic fungus Candida albicans [80]. Additionally, a nisin-inducible CRISPRi system was employed in Enterobacter faecalis to investigate the temporal roles of pili in biofilm development. Disruption of pili formation after 24 hours of normal growth using this system led to a significant reduction in biofilm development suggesting that pili play a critical role not only in initial attachment but also in the maintenance of biofilm structure over time [81]. An alternative strategy was adapted to knockdown genes of interest by introducing mismatches within the spacer sequence of the gRNA to reduce its binding affinity to the target DNA. By fine-tuning the degree of mismatches within the gRNA spacer sequence, researchers have precisely modulated target gene expression in bacterial systems [71] and in human cell lines [82]. It is particularly useful when working with essential genes, where complete knockouts can be lethal. Finally, CRISPRi has been used to decipher novel antimicrobial agent targets in various pathogenic bacteria including Acinetobacter bauumanii [83]. Using machine learning algorithms, the authors have identified a compound that targets the lipoprotein trafficking resulting in growth arrest in murine model of infection. A recent study has developed antimicrobial agents targeting the lumazine synthase RibH of Mycobacterium tuberculosis using a CRISPRi based screen [84]. Interstingly, Peters et al have used an essential gene knockout library against antibiotics to dissect out the interaction between genes in response to therapeutics in Bacillus subtilis. The authors found that enzyme undecaprenyl pyrophosphate synthase, UppS, is the primary target of the antimicrobial agent MAC-0170636 [30]. CRISPRi mediated libraries have been also used to determine the genetic hypersensitivity against novel antibiotic agent SCH-79797 against both gram positive and gram-negative bacteria [85].
2.2. CRISPR-Activation
CRISPR activation (CRISPRa) is a robust tool that contrasts with CRISPRi by upregulating the expression of specific target genes. In E. coli, the CRISPRa system comprises a fusion of dCas9 with the ω-subunit of RNA polymerase [71] (Figure 1C). This complex binds to the upstream promoter element, facilitating the recruitment of RNA polymerase. However, a limitation of this approach is this CRISPRa system operates only in a rpoZ-deletion strain. rpoZ encodes the omega subunit of RNA polymerase, and is involved in stringent stress response [86]. Deleting rpoZ can alter metabolic pathways, potentially affecting the outcomes of CRISPRa-mediated gene expression. Additionally, studies have reported the accumulation of suppressor mutants in such systems, which may outcompete knockout strains within the same culture [87]. To address this limitation, Dong et al. have developed a CRISPRa method in which the guide RNA is extended to form scaffold RNA (scRNA) that can bind RNA binding protein, MS2 coat protein, MCP. MCP in turn can recuit transcriptional activators to this complex. To screen for effector proteins that can activate transcription by binding to this CRISPR-MCP complex, a GFP based reporter assay was conducted in E. coli. The authors identified, SoxS, the oxidative stress responsive global regulator in E. coli to be best candidate to act as an activator. Interestingly, the authors observed a clear relationship between the distance of CRISPRa target site to the transcription start site (TSS), and effective gene activation with the most efficient activation was achieved when the gRNA target sites were positioned approximately 60–90 bases upstream of the TSS of a reporter gene [88].
2.3. Mobile CRISPRi
Mobile CRISPRi is a flexible and modular system for gene knockdown or knockout, leveraging CRISPR interference (CRISPRi) technology in conjunction with specific transport mechanisms such as T7 transposases for Gram-negative bacteria and the ICEBs1 conjugative element for Gram-positive bacteria [31,89]. In Gram-negative bacteria, the CRISPR array is delivered to recipient strains through tri-parental conjugation. This process involves a diaminopimelic acid (DAP)-dependent donor strain of Escherichia coli, which transfers the CRISPRi machinery via the RP4 conjugation system, while a second donor provides a helper plasmid carrying Tn7 transposition genes required for integration into the bacterial genome. Once inside the recipient cell, the transposases excise the CRISPRi machinery and facilitate its integration downstream of the glmS genes through recombination. For Gram-positive organisms, the CRISPRi machinery is delivered via biparental conjugation between donor and recipient strains. Upon entry, the CRISPRi machinery is integrated downstream of the trnS-leu2 site with the assistance of an integrase. Importantly, integration of the CRISPRi system downstream of either glnS or trnS-leu2 does not disrupt the function of these genes. Mobile-CRISPRi screens have been used to decipher gene functions in a wide variety of bacterial pathogens [32,49]. In one study, mobile-CRISPRi was used to knockdown essential and non-essential genes in 5 different species of Vibrio [89]. Interestingly, the authors observed a decline in knockdown efficiency as the binding site of the guide RNA shifted from the 5' end to the 3' end of the target gene. In a recent study, using mobile CRISPRi, the authors were able to identify the essential genes of Pseudomonas aeruginosa in murine model of infection. The authors demonstrated that knockdown of exsA, a gene that activates type III secretion system (T3SS) results in attenuated virulence in mice [90]. Interestingly, the authors observed a decline in knockdown efficiency as the binding site of the guide RNA shifted from the 5' end to the 3' end of the target gene. Recently, mobile CRISPRi was used to decipher essential genes in Acinetobacter baumannii [33]. Taken together, CRISPR-based approaches have proven effective in uncovering the roles of key factors involved in host-pathogen interactions (Figure 1D).
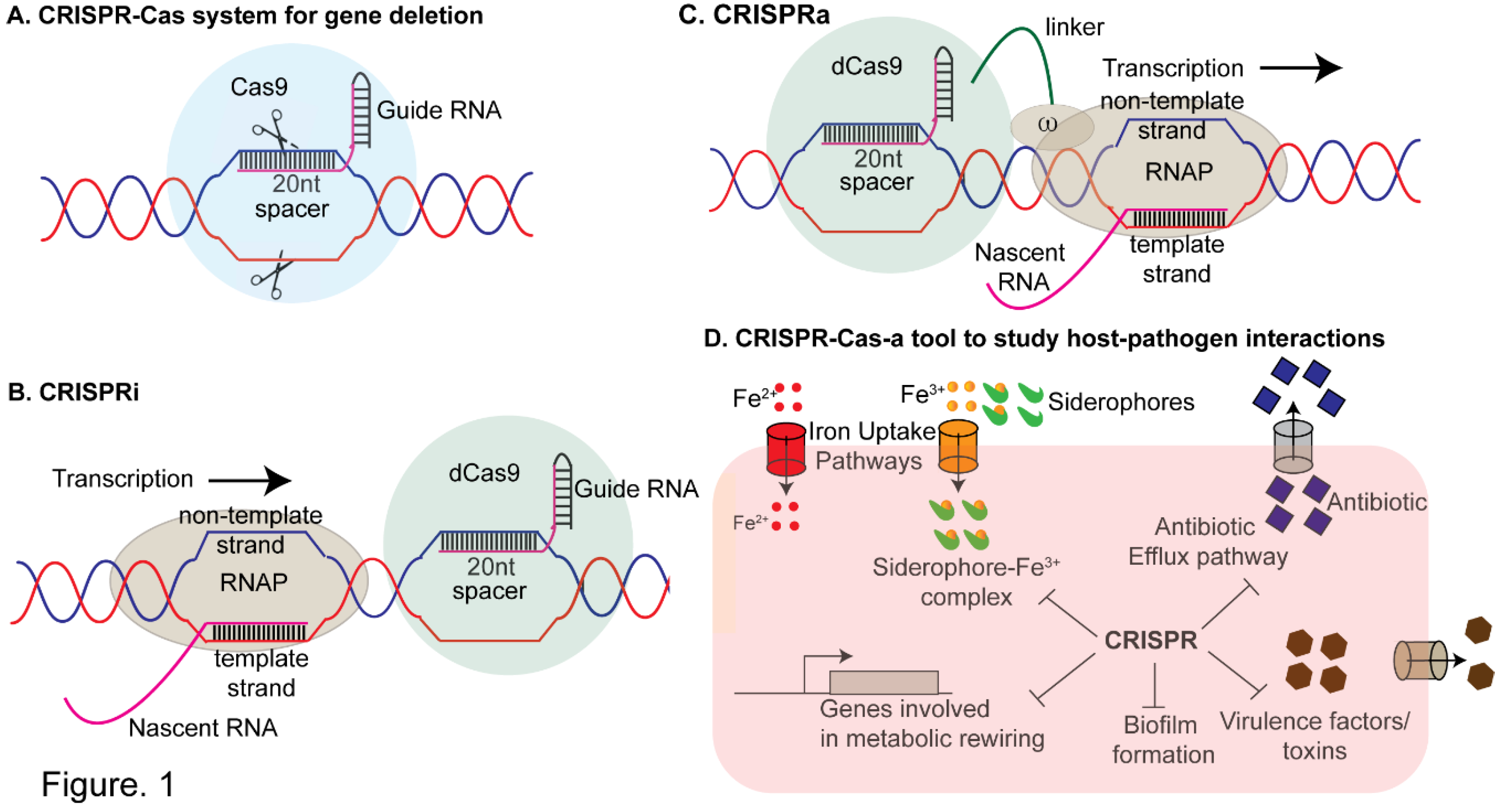
Figure 1.
CRISPR-Cas System and Its Applications in Host-Pathogen Interaction Studies. A. CRISPR-Cas9-Mediated Gene Knockout: The Type II CRISPR-Cas9 system consists of a single guide RNA (sgRNA) and the Cas9 nuclease. The sgRNA guides Cas9 to its target sequence in the pathogen’s genome, where Cas9 induces double-strand breaks. This disruption results in the elimination or functional inactivation of the target gene. B. CRISPR Interference (CRISPRi): CRISPRi uses a catalytically inactive ("dead") Cas9 (dCas9) that retains DNA-binding ability but lacks nuclease activity. When recruited by an sgRNA to a target sequence, dCas9 sterically blocks transcription by interacting with the non-template DNA strand, leading to gene knockdown. This method can be inducible, allowing the degree of gene knockdown to be modulated by varying inducer concentrations driving CRISPRi expression. C. CRISPR-Associated Gene Activation (CRISPRa): CRISPRa involves a modified Cas9 from the CRISPRi system fused to omega subunit of RNA polymerase. The sgRNA-Cas9 complex binds upstream of the target promoter, facilitating the recruitment of RNA polymerase to enhance gene expression. This approach effectively activates transcription of the target gene. D. Applications in Host-Pathogen Interaction Studies: The CRISPR-Cas system has been widely used in unraveling the mechanisms of pathogenicity in various bacterial pathogens. It has been employed to study the roles of virulence factors, including genes involved in iron acquisition, biofilm formation, toxin production, and antibiotic resistance. Additionally, CRISPR-Cas tools have facilitated the investigation of essential metabolic genes critical for pathogen survival in hostile host environments.
Figure 1.
CRISPR-Cas System and Its Applications in Host-Pathogen Interaction Studies. A. CRISPR-Cas9-Mediated Gene Knockout: The Type II CRISPR-Cas9 system consists of a single guide RNA (sgRNA) and the Cas9 nuclease. The sgRNA guides Cas9 to its target sequence in the pathogen’s genome, where Cas9 induces double-strand breaks. This disruption results in the elimination or functional inactivation of the target gene. B. CRISPR Interference (CRISPRi): CRISPRi uses a catalytically inactive ("dead") Cas9 (dCas9) that retains DNA-binding ability but lacks nuclease activity. When recruited by an sgRNA to a target sequence, dCas9 sterically blocks transcription by interacting with the non-template DNA strand, leading to gene knockdown. This method can be inducible, allowing the degree of gene knockdown to be modulated by varying inducer concentrations driving CRISPRi expression. C. CRISPR-Associated Gene Activation (CRISPRa): CRISPRa involves a modified Cas9 from the CRISPRi system fused to omega subunit of RNA polymerase. The sgRNA-Cas9 complex binds upstream of the target promoter, facilitating the recruitment of RNA polymerase to enhance gene expression. This approach effectively activates transcription of the target gene. D. Applications in Host-Pathogen Interaction Studies: The CRISPR-Cas system has been widely used in unraveling the mechanisms of pathogenicity in various bacterial pathogens. It has been employed to study the roles of virulence factors, including genes involved in iron acquisition, biofilm formation, toxin production, and antibiotic resistance. Additionally, CRISPR-Cas tools have facilitated the investigation of essential metabolic genes critical for pathogen survival in hostile host environments.
3. Engineered Microbes-Modern Biosensors for Pathogen Detection and Elimination
Synthetic biology approaches have also been developed to detect and respond to specific pathogens or pathogen-associated molecular patterns (PAMPs) involved in host-pathogen interactions [24,38,91,92]. By engineering synthetic circuits in bacteria, researchers have created Bacterial Whole Cell Biosensors (BWCBs) that are capable of sensing infection biomarkers and producing easily detectable signals (Figure 2) [93,94,95]. BWCBs offer several advantages over traditional pathogen detection methods. Conventional approaches, such as isolating bacteria from stool samples, are often time-consuming [96]. Moreover, the molecular signatures associated with a pathogen's virulence can be transient, localized to specific gut regions, and absent from stool samples. BWCBs address these limitations by providing a sensitive, rapid, and noninvasive method to identify key molecular markers of pathogenicity at physiologically relevant concentrations [97]. These engineered microbes typically incorporate synthetic circuits composed of three main modules. The first is the "sensor" module, designed to detect specific signals or stimuli, such as pathogen-associated metabolites or secreted toxins. Upon signal detection, the sensor module transmits the information to the second component, the "synthetic circuit," which contains a response regulator. This regulator receives the signal from the sensor and triggers downstream processes to activate the appropriate elements of the final module, the "Output" module. The output module then generates detectable signals, which can include bioluminescence, fluorescence, or colorimetric in nature (Figure 2A). For example, E. coli was engineered to create a genetic memory system capable of sensing anhydrotetracycline (ATC) [98]. This synthetic circuit incorporates a bistable lambda cI/Cro switch, consisting of two components: a “trigger element,” where a tetracycline-inducible promoter regulates the expression of the lambda cro gene, and a “memory element” derived from the cI/Cro region of phage lambda. The circuit is designed to initially reside in the cI state and transition to the Cro state upon induction, with the tetracycline-responsive promoter driving transcription of the cro gene. When this engineered Escherichia coli was administered to mice that was treated with anhydrotetracycline, the recovered bacteria consistently switched to the Cro state, whereas those given to untreated mice retained the cI state. Notably, the engineered E. coli established itself robustly in the murine gut environment suggesting the possibility of using similar genetic circuits to monitor complex, undefined environments and advances the potential for developing living diagnostics and therapeutics [98]. Interestingly, Mimee et al have integrated engineered biological systems with advanced electronics to create a bio-electronic device known as the Ingestible Micro-Bio-Electronic Device (IMBED) for use in diagnostic applications [99]. IMBED combines a probiotic bacterial sensor with ultra-low-power microelectronics to detect biomarkers of the gastrointestinal tract. As a “proof-of-concept”, IMBED was designed to identify gastrointestinal bleeding in a porcine model by sensing heme released from lysed blood cells. Engineered E. coli Nissle 1917 strain responded to increasing blood levels by emitting bioluminescence as a readout, which was detected by phototransistors and subsequently converted to photocurrent signals. Using this device, the authors were able to detect gut infection with 100% specificity at 120 minutes. The IMBED platform has also been adapted to detect other disease-relevant molecules, such as thiosulfate, a biomarker for gut inflammation and N-acyl homoserine lactone (AHL), an indicator of bacterial infections [99]. This technology underscores the potential of ingestible bio-electronic devices for non-invasive and real-time medical diagnostics.
3.1. Engineered Microbes as Biosensors for Gut Inflammation
E. coli Nissle strain was also engineered to develop biosensors capable of detecting markers of gut inflammation in response to Salmonella infection [100]. Previous studies have shown that Salmonella species can exploit the macrophage-mediated inflammatory response to facilitate the establishment of an infection. Reactive oxygen species generated during inflammation react with luminal Sulphur compounds, such as H2S to produce thiosulphate (S₂O₃²⁻) which can be further oxidized to tetrathionate (S4O6²⁻). The ability to utilize tetrathionate as a terminal electron acceptor provides S. Typhimurium with a unique growth advantage over competing species in the inflamed gut lumen. Using synthetic biology tools, Daeffler et al. have modified the widely used probiotic E. coli Nissle strain to sense both the markers of inflammation (thiosulfate and tetrathionate) in a dextran sodium sulfate (DSS) murine model of colitis [100]. To accomplish this, the authors employed a thiosulfate and tetrathionate sensor, along with their respective response regulators, from the marine bacteria Shewanella species as there was no previous report of genetically encoded thiosulfate sensor available, and the only known tetrathionate sensor is the TtrSR two-component system (TCS) from Salmonella typhimurium. In presence of signaling molecule (thiosulfate or tetrathionate), the sensor protein (ThsS or TtrS) phosphorylates its cognate response regulator (ThsR or TtrR) which in turn activates the downstream reductase operon that can utilize thiosulfate or tetrathionate respectively (Figure. 2B and 2C). Although thiosulfate reductase is regulated by the catabolite repressor protein (CRP), the tetrathionate reductase operon is naturally regulated by oxygen and nitrate levels through the global regulator FNR, making it prone to cross-repression in the gut environment. Since oxygen and nitrate levels are elevated during inflammation, these regulatory complexities reduce the reliability of the TtrSR system as a sensor for tetrathionate in this context. To overcome this limitation, the authors engineered the reductase operon under the control of Catabolite Repressor Protein, CRP instead of FNR, thus eliminating the influence of fluctuating O₂ levels in the gut. GFP fluorescence was used as a readout to detect the presence of thiosulfate or tetrathionate in a murine infection model. Using flow cytometry, the authors demonstrated activation of the thiosulfate sensor in response to gut inflammation during the murine infection confirming the validity of the assay [100].
Additionally, recombinase-based circuits have been utilized to engineer the E. coli strain EA3020 to sense nitric oxide, a biomarker of inflammatory bowel disease (IBD), with cyan fluorescent protein (CFP) serving as the output signal. [101]. To achieve this, a bidirectional memory switch was engineered using the canonical E. coli fimbriae (Fim) phase variation system. In the OFF state, the fimS promoter is oriented towards the Inverted Repeat Right (IRR), leading to the constitutive expression of yellow fluorescent protein (YFP). Upon inversion of the switch to the ON state, fimS reorients toward the Inverted Repeat Left (IRL), resulting in the constitutive expression of cyan fluorescent protein (CFP). This inversion is mediated by the DNA recombinase FimE, which specifically binds to the inverted repeat sequences in the promoter region of CFP. By replacing the native promoter of FimE by nitric acid sensor, NorR regulated PnorV, the authors were able to design a synthetic circuit that allowed them to detect the presence of nitric oxide through NorR regulated synthetic circuit in the murine model of infection (Figure. 2D). In a separate study, Vibrio harveyi strain BB170 was engineered to detect Autoinducer-2 (AI-2), a quorum-sensing molecule previously linked to irritable bowel syndrome (IBS), using ex vivo saliva and stool samples from patients. The engineered bacteria sense AI-2 through the periplasmic sensor protein LuxP. LuxP subsequently activates the response regulator LuxQ via phosphorylation, which triggers the downstream luxCDABE cascade to produce bioluminescence as the output signal [102].
3.2. Engineered Microbes as Pathogen Killing Machines
Synthetic biology has facilitated the development of engineered microbes with potential prophylactic and therapeutic applications. For instance, engineered E. coli has been designed to detect Pseudomonas aeruginosa by sensing its quorum-sensing molecule 3OC12HSL [103]. Upon detecting the molecule, sensor LasR of the "detection module" activatesthe expression of GFP fluorescence, confirming the pathogen's presence. The second component, a LasR controlled "destruction module," synthesizes a chimeric bacteriocin called CoPy, which was engineered by replacing the receptor and translocase domains of Colicin E3 with those of Pyocin S3 to specifically target P. aeruginosa. The final component, a "secretion module," utilizes the type IV secretion system FlgM to release CoPy into the environment (Figure. 2E). Using this engineered E. coli strain, the authors demonstrated growth inhibition of P. aeruginosa strain PA01 in co-culture, paving the way for the development of novel therapeutic bacterial strains. In a more recent study, Hwang et al. engineered E. coli Nissle strain to target and kill P. aeruginosa by delivering an antimicrobial peptide and a biofilm-dispersing enzyme [104]. Additionally, a CRISPRi-based synthetic circuit was developed in Bacteroides thetaiotaomicron, a key member of the human gut microbiome, to modulate gene expression and antimicrobial resistance, highlighting the broader potential of synthetic biology in combating pathogens [105].
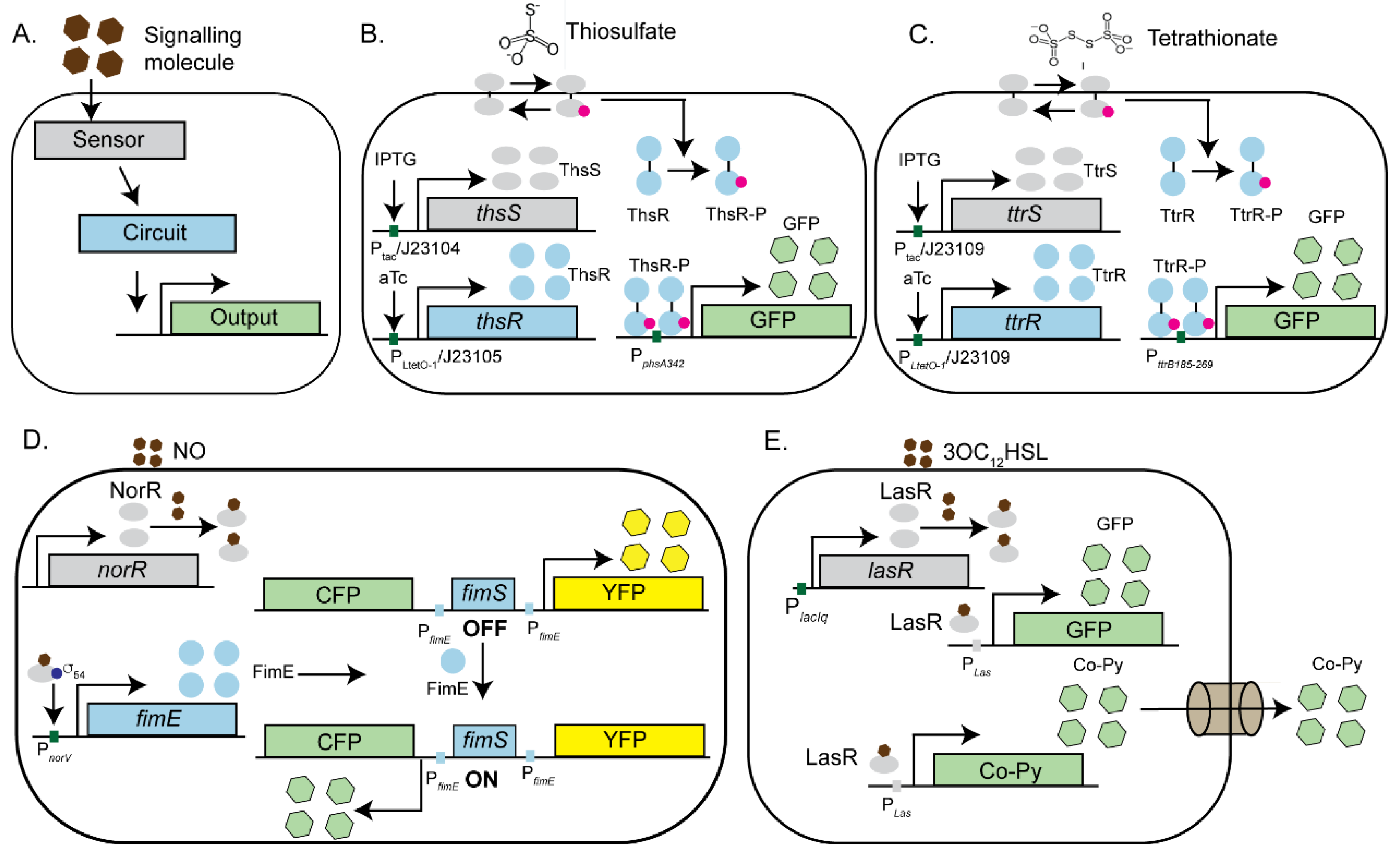
Figure 2.
Engineered microbes as tools to study host-pathogen interactions. A. Schematic representation of an engineered microbial system designed to detect specific pathogens or their metabolites using synthetic circuits. The system comprises a sensor that responds to a target signal, an inducible, titratable, gated or bistable synthetic circuit, and a downstream reporter gene. Upon sensing the signal, the sensor activates a response regulator, which triggers a gene cascade to produce a detectable output signal (colorimetric, fluorescent, or bioluminescent). B-C. The E. coli Nissle strain was engineered to act as a biosensor for gut inflammation by detecting thiosulfate (B) and tetrathionate (C), biomarkers of Salmonella-induced inflammation. Detection involves specific sensors (ThsS for thiosulfate and TtrS for tetrathionate) that phosphorylate their respective response regulators (ThsR or TtrR) in presence of the signal. The phosphorylated response regulators activate reductase operons that can utilize these metabolites. Sensors and response regulators were driven by either inducible promoters (e.g., IPTG or ATC) in vitro or constitutive promoters (J23104, J23105, and J23109) in murine infection models. D. The E. coli strain EA3020 was engineered to sense nitric oxide (NO), a biomarker of inflammatory bowel disease (IBD), with cyan fluorescent protein (CFP) as the output. In the presence of NO, the NorR sensor activates the recombinase FimE, which switches the FimS invertible promoter element from an OFF to an ON state, driving CFP expression. E. E. coli was designed to detect Pseudomonas aeruginosa by sensing its quorum-sensing molecule, 3OC12HSL. The LasR sensor activates the detection module, resulting in GFP fluorescence, confirming pathogen presence. Additionally, LasR activates the expression of chimeric bacteriocin Co-Py, which is secreted through the FlgM-mediated Type IV secretion system to eliminate the target pathogen.
Figure 2.
Engineered microbes as tools to study host-pathogen interactions. A. Schematic representation of an engineered microbial system designed to detect specific pathogens or their metabolites using synthetic circuits. The system comprises a sensor that responds to a target signal, an inducible, titratable, gated or bistable synthetic circuit, and a downstream reporter gene. Upon sensing the signal, the sensor activates a response regulator, which triggers a gene cascade to produce a detectable output signal (colorimetric, fluorescent, or bioluminescent). B-C. The E. coli Nissle strain was engineered to act as a biosensor for gut inflammation by detecting thiosulfate (B) and tetrathionate (C), biomarkers of Salmonella-induced inflammation. Detection involves specific sensors (ThsS for thiosulfate and TtrS for tetrathionate) that phosphorylate their respective response regulators (ThsR or TtrR) in presence of the signal. The phosphorylated response regulators activate reductase operons that can utilize these metabolites. Sensors and response regulators were driven by either inducible promoters (e.g., IPTG or ATC) in vitro or constitutive promoters (J23104, J23105, and J23109) in murine infection models. D. The E. coli strain EA3020 was engineered to sense nitric oxide (NO), a biomarker of inflammatory bowel disease (IBD), with cyan fluorescent protein (CFP) as the output. In the presence of NO, the NorR sensor activates the recombinase FimE, which switches the FimS invertible promoter element from an OFF to an ON state, driving CFP expression. E. E. coli was designed to detect Pseudomonas aeruginosa by sensing its quorum-sensing molecule, 3OC12HSL. The LasR sensor activates the detection module, resulting in GFP fluorescence, confirming pathogen presence. Additionally, LasR activates the expression of chimeric bacteriocin Co-Py, which is secreted through the FlgM-mediated Type IV secretion system to eliminate the target pathogen.
4. Phage Therapy-Based Approaches to Decipher Host-Pathogen Interactions
Bacteriophages have long been employed as valuable tools for generating gene knockouts across various bacterial species [106]. With advances in synthetic biology, phages have been further repurposed for the detection and elimination of multidrug-resistant bacterial strains [42,106,107]. This has been achieved through CRISPR interference (CRISPRi)-based approaches [107,108] or by inducing bacterial cell lysis using endolysins [45,109].
4.1. Application of CRISPRi Based Methods on Phage Therapy
CRISPRi-based technology has enabled the creation of engineered bacteriophages that can be precisely tailored to target specific DNA sequences of the desired pathogens in the microbiome. This method offers a significant advantage over antibiotics, the later often cause the loss of beneficial bacteria in the microbiome due to their broad-spectrum effects [108]. For example, Citorik et al. developed a CRISPRi-based-PT system in which RNA-guided nucleases (RGNs) were delivered into E. coli strain EMG2 via a phagemid to target antibiotic resistance genes blaNDM-1 and blaSHV-18, conferring β-lactam resistance, or a mutated DNA gyrase gene (gyrAD87G) associated with quinolone resistance [109]. Once inside the pathogen, the cr-RNA directed nucleases to the target DNA, inducing double-stranded breaks that led to plasmid loss and bacterial death (Figure. 3A). Importantly, this method demonstrated specificity, as bacteria lacking the targeted sequences remained unaffected. The authors further tested their CRISPRi-based method in vivo using Galleria mellonella larvae infected with Enterohemorrhagic E. coli O157:H7 (EHEC). By targeting the eae gene, which encodes a virulence factor critical for the pathogen's attachment to the host epithelial cells, the authors demonstrated enhanced survival of Galleria mellonella larvae treated with the engineered phage compared to the control. This in vivo study highlighted the potential of CRISPRi-based phage therapy in improving host survival by specifically targeting bacterial virulence factors. Similarly, Bikard et al. successfully treated Staphylococcus aureus infections in a murine skin model by targeting key virulence factors, including the aminoglycoside phosphotransferase gene (aph), the methicillin resistance gene (mecA), and the enterotoxin gene (sek) using CRISPR-Cas based approach that was delivered by a bacteriophage (Figure. 3A). This study showcases the potential of CRISPR-based phage therapy as a precise antimicrobial strategy [110].
4.2. Phage Based Antimicrobials-Endolysins
One promising approach to combating pathogenic bacteria involves modifying the native endolysins of bacteriophages [111]. Endolysins are enzymes produced by bacteriophages during the lytic phase. These enzymes degrade the peptidoglycan layer of the bacterial host cell wall from within, leading to cell lysis and the subsequent release of progeny virions (Figure. 3B). Endolysins exhibit structural variability depending on its target. Endolysin that target Gram-positive bacteria typically have a modular structure, consisting of a catalytic domain responsible for enzymatic activity and a cell wall-binding domain (CBD) that ensures the enzyme binds effectively to the bacterial cell wall. In contrast, endolysins targeting Gram-negative bacteria are generally smaller, single-domain proteins without a distinct CBD. The outer membrane of Gram-negative bacteria restricts access to the peptidoglycan layer, necessitating different structural adaptations in the endolysins [111]. Previous studies have demonstrated that recombinantly purified endolysins are effective in killing both Gram-positive and Gram-negative bacteria [112,113]. Building on this, researchers engineered a chimeric endolysin, Cpl-7-11, by combining the catalytic, linker, and cell wall-binding domains of two separate endolysins, Cpl1 and Cpl7S. This chimeric endolysin was shown to be 50% more effective against Streptococcus pneumoniae in a murine infection model compared to the parental Cpl1 alone [114]. Similar studies have been conducted with chimeric endolysins to treat Streptococcus sp [115,116], Staphylococcus sp [117] and Listeria sp [118]. While chimeric endolysins have shown success in treating Gram-positive bacteria, their application to Gram-negative bacteria has been limited by the presence of the outer membrane. To overcome this challenge, researchers have used permeabilizing agents such as EDTA, citric acid, polymyxin B, and poly-L-lysine. A combination of EDTA, citric acid, and malic acid was found to be particularly effective against Pseudomonas aeruginosa strain PA01 and Pseudomonas fluorescens when treated with the Salmonella phage endolysin Lys68 [119]. Another innovative approach involved fusing endolysins with lipopolysaccharide (LPS)-destabilizing peptides, leading to the creation of "Artilysins." These engineered enzymes combine the antimicrobial activity of endolysins with the ability to disrupt the outer membrane of Gram-negative bacteria, enhancing their effectiveness in targeting a broader range of pathogens. In one study, polycationic nonapeptides (PCNP) were fused with the endolysin OBPgp279, isolated from the P. fluorescens phage OBP, to create an Artilysin that was found to be effective against Pseudomonas aeruginosa [120]. Similarly, Lood et al. discovered the endolysin PlyF307, which was effective against Acinetobacter baumannii in a murine infection model [121]. This strategy of combining endolysins with LPS-destabilizing peptides shows promise in enhancing the efficacy of phage-derived therapies, especially against difficult-to-treat Gram-negative pathogens.
4.3. Application of Engineered Phages for Bacterial Detection and Diagnostics
Recent developments in phage-based pathogen detection have addressed some limitations of traditional methods, which often fail to identify pathogens present in low numbers due to the enrichment step [122]. To overcome this, researchers have created luminescence-based assays by fusing a luciferase gene with a phage protein through homologous recombination. Upon infection, the engineered phage produces light, which can be detected using luminescence assays. For example, Sarkis et al. engineered a phage by cloning the luciferase gene into the tRNA region of the L5 mycobacteriophage, which successfully detected live Mycobacterium smegmatis [123]. Similarly, an assay for detecting Listeria monocytogenes was developed by fusing the major capsid protein gene of Listeria phage A511 with the luxA and luxB genes from Vibrio harveyi. This system emitted a luminescent signal when encountering the pathogen, enabling detection of Listeria at concentrations as low as CFU <1/g in contaminated food [124].
Additionally, GFP-based phages were engineered to detect enterohemorrhagic strain of E. coli O157:H7. In this engineered phage, the gene encoding green fluorescent protein (GFP) was recombined into the genome of a T4 phage mutant that lacks lytic activity. Upon encountering the target pathogen, GFP fluorescence is expressed on the small outer capsid (soc), with the fluorescence intensity directly proportional to the infection time [125]. In a different approach, Piuri et al. engineered the mycobacteriophage TM4 to emit GFP or ZsYellow in presence of Mycobacterium tuberculosis, enabling detection using flow cytometry. This assay was further used to detect multi-drug-resistant strains of M. tuberculosis. The addition of Rifampicin decreased fluorescence intensity in susceptible bacterial populations, while any remaining fluorescence after Rifampicin treatment indicated the presence of drug-resistant bacteria, providing a rapid detection method for drug-resistant M. tuberculosis [126]. Finally, a T7 coliphage was engineered to display a biotinylated peptide on its major capsid protein. These biotinylated phages were conjugated to streptavidin-coated quantum dots (QDs), which emit fluorescence only after infection. In the absence of the host, the biotinylated phages were not produced, offering a novel method for pathogen detection [127].
Inspite of its versatility, phage therapy have several limitations that hinder their widespread use as therapeutic agents. Their narrow cleavage spectrum restricts them to targeting only specific bacteria, which makes them less effective against infections caused by multiple bacterial species. Additionally, lysogenic bacteriophages can transfer toxins and antibiotic resistance genes, complicating their therapeutic use. There is also a lack of standardized policies and regulations for bacteriophage therapy (PT), as well as insufficient guidance on phage isolation, purification, and clinical application. Importanntly, bacteria can evolve resistance to bacteriophages through various mechanisms, such as the CRISPR-Cas system, which complicates their long-term effectiveness. Furthermore, the lack of pharmacokinetic data, the difficulty of maintaining effective phage concentrations in the body, and potential interactions with the immune system or release of bacterial toxins pose significant challenges for PT's clinical application [128].
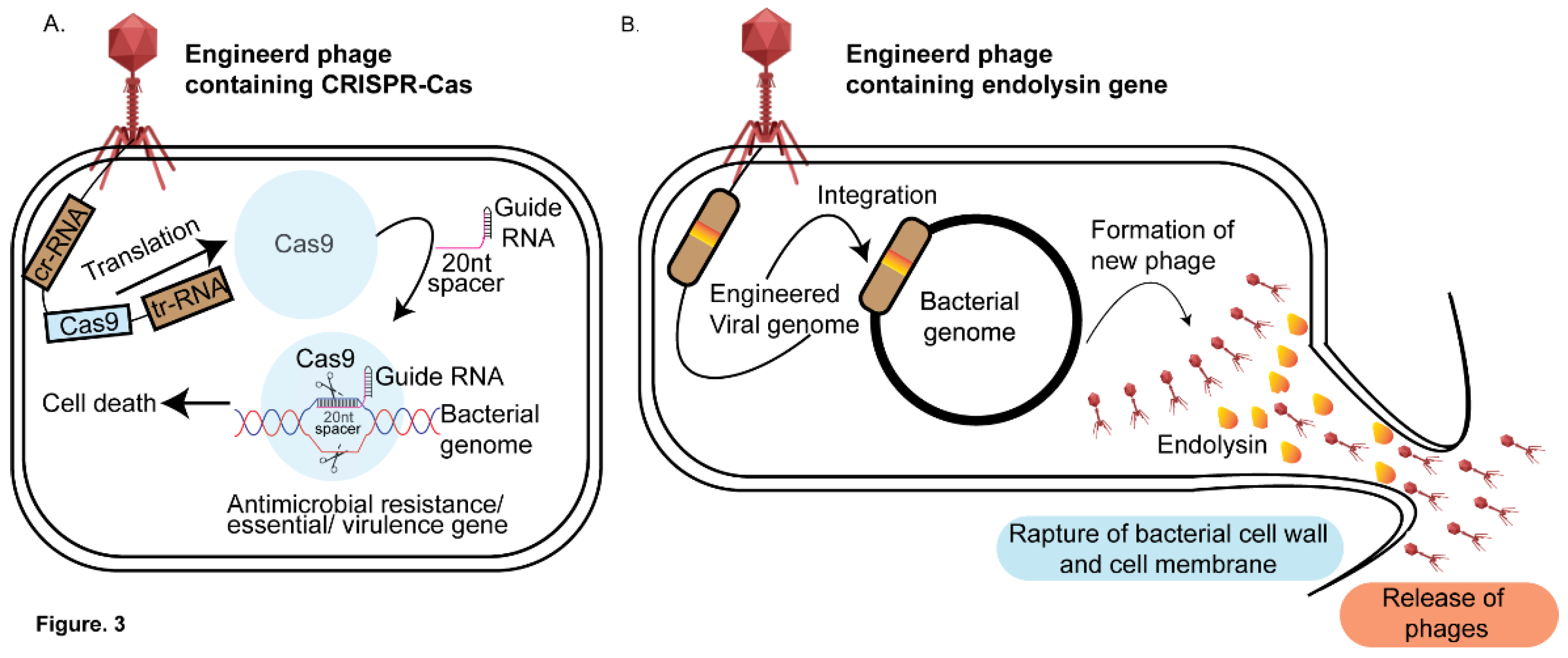
Figure 3.
Phage therapy-based approaches for targeted pathogen elimination. A. CRISPR-Cas phage engineering: The bacteriophage genome is modified to include CRISPR-Cas genes. Upon infection, the phage transfers its genome into the bacterial host where the Cas9 gene gets translated by the host translational machinery and guided by a specific gRNA to target sequences that can be an antimicrobial resistance gene or a virulence factor, or an essential gene. Cas9-induced double-stranded breaks in the target DNA result in bacterial cell death.B. Endolysin phage engineering: The bacteriophage genome is engineered to carry endolysin genes. During the lytic phase, these genes are expressed by the bacterial host, producing endolysins that degrade the peptidoglycan layer of the cell wall. This leads to bacterial lysis, pathogen death, and the release of new phage progeny.
Figure 3.
Phage therapy-based approaches for targeted pathogen elimination. A. CRISPR-Cas phage engineering: The bacteriophage genome is modified to include CRISPR-Cas genes. Upon infection, the phage transfers its genome into the bacterial host where the Cas9 gene gets translated by the host translational machinery and guided by a specific gRNA to target sequences that can be an antimicrobial resistance gene or a virulence factor, or an essential gene. Cas9-induced double-stranded breaks in the target DNA result in bacterial cell death.B. Endolysin phage engineering: The bacteriophage genome is engineered to carry endolysin genes. During the lytic phase, these genes are expressed by the bacterial host, producing endolysins that degrade the peptidoglycan layer of the cell wall. This leads to bacterial lysis, pathogen death, and the release of new phage progeny.
5. Organ-on-a-Chip-an Emerging Platform for Investigating Host-Pathogen Interactions
Traditional approaches to study host-pathogen interactions are limited by the inaccessibility of human organs, ethical concerns, and the lack of translatability of murine models to human systems [9]. Additionally, fecal microbiota analysis fails to capture real-time microbial dynamics under physiological or disease conditions [129]. To address these challenges, organ-on-a-chip technology was developed to replicate human organ-level functions for accurate drug testing and disease modeling while reducing dependence on animal models [60,130]. By integrating shear stress conditions that replicate the in vivo environment, this microfluidics-based approach offers a unique platform for investigating pathogen interactions with various human organ tissues, including the gut, lungs, liver, intestine, and kidney [60,61,131,132,133,134,135] (Figure 4). This methodology enables the generation of physiologically relevant insights as discussed below.
5.1. Organ-on-a-Chip as a Tool to Study Interaction of Pathogens with the Host
Organ-on-a-chip technology has been widely applied to explore the adaptative strategies employed by the pathogens to counteract the host immune defense (Figure 4A) [60,61,136]. Uropathogenic Escherichia coli (UPEC) evade host immune defenses and antibiotic treament by forming intracellular bacterial communities (IBCs) within the umbrella cells of the urinary bladder [137]. Commonly used antibiotics fail to completely eliminate IBCs, allowing the persistent population to reactivate within the host and cause recurrent UTIs. Understanding the molecular mechanisms of IBC formation could pave the way for developing novel therapeutics to effectively treat recurrent UTIs. The inaccessibility of the urinary bladder presents significant challenges for studying the IBC formation in real time within the host environment. To address this problem, Sharma et al. developed a chip to investigate bacterial persistence by uropathogenic E. coli [137]. The chip, which cocultured umbrella cells and bladder microvascular epithelial cells in the presence of either urine or nutrient-rich media, mimicked the human urinary bladder environment. By incorporating linear strain to simulate bladder filling and voiding, the authors observed that E. coli was able to form IBCs with the umbrella cells. Interestingly, upon antibiotic treatment, the umbrella cells exfoliated, leading to the formation of new IBCs. This study highlighted the pivotal role of IBCs as reservoirs for pathogens, providing valuable insights into the molecular mechanisms of virulence of urinary tract infections. (Figure 4B).
Organ-on-a-chip models have proven valuable in investigating the the host immune response to Mycobacterium tuberculosis (Mtb). Pulmonary surfactant, which comprises dipalmitoylphosphatidylcholine (a unique phospholipid) and four surfactant-associated proteins—SP-A, SP-B, SP-C, and SP-D—plays a crucial role in host defense. It lines the alveolar wall and facilitates the binding and clearance of various pathogens, including Mycobacterium tuberculosis [138]. Previous studies have indicated that mice deficient in pulmonary surfactant production exhibit increased susceptibility to Mycobacterium tuberculosis [139], highlighting the importance of pulmonary surfactant in mitigating tuberculosis. To explore the molecular mechanisms behind these interactions, Thacker et al. recently developed a lung-on-a-chip model incorporating alveolar cells and macrophages to investigate the early dynamics of Mycobacterium tuberculosis (Mtb) infection at an air-liquid interface [140]. The chip was engineered to circulate air, blood, and surfactant, closely replicating the lung’s in vivo environment. Through time-lapse microscopy, the study revealed a crucial role of pulmonary surfactant in controlling tuberculosis by mitigating bacterial growth within the macrophages. In another study, Pseudomonas aeruginosa exhihibited favourable growth in the lung airway chips designed to model the cystic fibrosis (CF) airway, reflecting key features of the disease. These chips incorporate primary epithelium isolated from CF patients and effectively replicate the CF airway's characteristics, such as increased cilia density, mucus accumulation, ciliary beating frequency, and IL-8 secretion. As a result, there is greater adhesion of circulating immune cells to the endothelium and their transmigration into the airway compartment, compared to healthy airway chips [141]. Organ-on-a-chip technology has applications in studying secondary bacterial infections, such as Staphylococcus aureus infections following influenza. Pneumonia, often exacerbated by bacterial co-infections, significantly increases morbidity and mortality during influenza epidemics. A microfluidic chip system was developed to model these interactions by coculturing alveolar epithelial cells (NCI-H441) and monocyte-derived macrophages in the upper chamber under air-phase conditions for 14 days, while human umbilical vein endothelial cells (HUVECs) were cultured in the lower chamber with medium perfused via a peristaltic pump. The authors observed significant damage to the endothelial layer following Staphylococcus aureus infection. This platform allowed spatiotemporal monitoring of S. aureus spread and host immune responses, offering an advanced tool for studying pneumonia pathophysiology and exploring potential therapeutic targets. [142].
Furthermore, by designing a chip that replicates the human intestinal environment, incorporating shear stress from intestinal flow and tensile stress from peristaltic movement, the authors demonstrated that Shigella flexneri entry through the apical side of enterocytes results in a loss of barrier integrity [143]. This effect was further aggravated by physical deformities caused by peristaltic movement-induced stress on the cells. In a separate study, the importance of tensile stress was highlighted using enterotoxigenic E. coli, which increased the secretion of cGMP by human jejunal enteroids in response to enterotoxins released by the pathogen. This study provided new insights into the mechanisms underlying enterotoxin-induced diarrhea [144].
5.2. Organ Chips as a Tool to Decipher Host-Microbiota Interactions
There is growing evidence about the significant role of gut microbiota in human health [54,55], with disruptions linked to conditions such as, inflammatory bowel disease (IBD) [145], celiac disease [146] and gastrointestinal malignancies [147]. Understanding the crosstalk between individual microbes, gut epithelial cells, and immune cells under standard and disease conditions could open new avenues for drug development. Recent studies have demonstrated the potential of organ-on-a-chip models to uncover these interactions. For example, Kim et al. conducted a “proof-of-concept” study where the authors incubated eight probiotic bacterial strains with intestinal epithelial cells in an organ-on-a-chip model [148]. The authors found that peristaltic motion is crucial in regulating microbiota overgrowth in the gut. In addition, the authors also found that the exogenous addition of lipopolysaccharide (LPS) endotoxin induced the production of proinflammatory cytokines (IL-8, IL-6, IL-1B, and TNF-α), leading to villus restructuring and impaired barrier function. Interestingly, this damage could be reversed by adding probiotic bacteria.
Recently, significant progress has been made in replicating the human gut microbiome in vitro. Using an advanced two-channel intestine-on-a-chip model, the authors successfully co-cultured a microbiome containing over 200 bacterial species, closely mimicking the diversity and complexity of a human stool microbiome, in direct contact with human intestinal epithelium and its naturally secreted mucus layer for five days [149]. These models were lined with either Caco-2 cells or primary human organoid-derived epithelium, interfaced with primary intestinal microvascular endothelium. This was accomplished by creating a hypoxia gradient across the epithelial–endothelial interface, replicating the in vivo conditions, which was confirmed through the use of integrated on-chip oxygen sensors. The presence of living bacteria in the system either maintained or improved intestinal barrier function, underscoring the microbiome's role in supporting intestinal health within this model. This setup provides a sophisticated platform for investigating host-microbiome interactions in a controlled, physiologically relevant environment In a separate study, Shin et al, deciphered DSS-induced oxidative stress caused changes in villus microarchitecture and barrier function in a gut-inflammation-on-a-chip model, further underscoring the importance of gut homeostasis in disease. [150]. Addition of non-pathogenic E. coli or lipopolysaccharide (LPS) results in the production of proinflammatory cytokines and the recruitment of immune cells. Additionally, the inclusion of probiotic bacteria was also shown to reduce oxidative stress, further highlighting the importance of gut microbiota dynamics on human health. Finally, Tovaglieri et al. investigated the relationship between host pathophysiology and the gut microbiome using a human colonic epithelium gut-chip model to simulate enterohemorrhagic E. coli (EHEC) infection [151]. The authors found that human microbiome-derived metabolites caused greater epithelial cell damage compared to those from mice and identified four metabolites that triggered the expression of flagellin, a bacterial protein essential for motility. These findings suggest that specific metabolites in the human gut microbiome may influence the host’s immune response, offering insights into host-microbiome interactions that could enhance the translational relevance of microbial research for human health.
The organ-on-chip platform, like any emerging technology, faces several challenges that need to be addressed for broader adoption and effective application. Notably, organ chips can exhibit significant variability across different production batches, laboratories, and users [152]. Interestingly, Polydimethylsiloxane (PDMS), the materials used in the fabrication of these chips despite being favored for its high biocompatibility, transparency, and oxygen permeability, has been shown in multiple studies to adsorb proteins onto its surface and can interfere with the interaction of drugs or stimulatory substances with the cells within the chip [153]. Additionally, organ-on-a-chip devices often require specialized equipment adding complexity and cost to their setup.
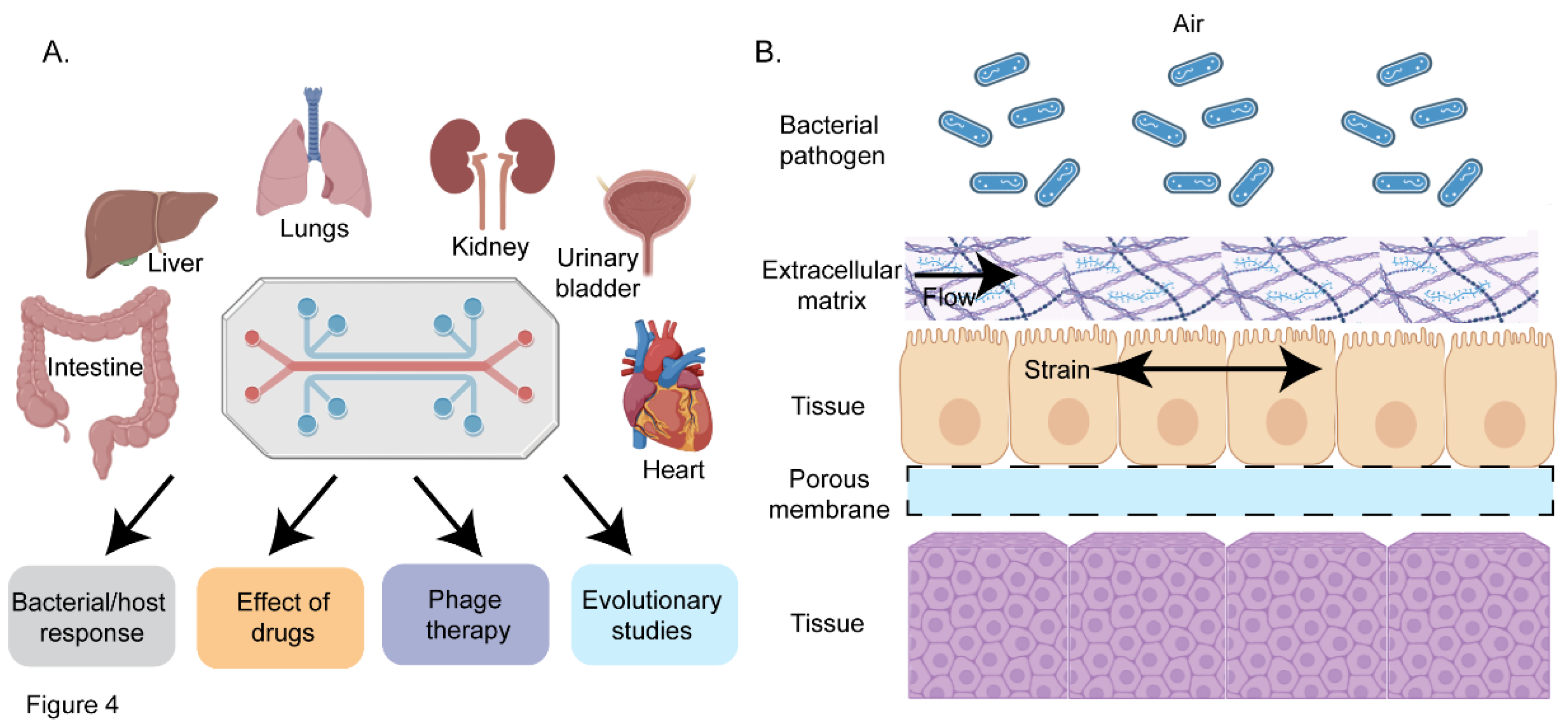
Figure 4.
Organ-on-a-Chip to study host-pathogen interactions. A. Overview of various organs modeled using organ-on-a-chip technology to investigate the molecular mechanisms of infectious diseases and the potential applications of this approach. Organ on a chip has been used to decipher bacterial and host response, to study the effect of drugs, antimicrobials and engineered bacteriophages (phage therapy) on bacterial pathogens, and to study genetic and epigenetic changes in response to the environment. B. Simplified diagram illustrating the layered structure of the chip. The top layer contains the pathogenic bacteria of interest, followed by the extracellular matrix. Nutrients, growth factors, and drugs flow through this layer to replicate the host environment (e.g., gut). Beneath this, the first host cell layer is represented, such as umbrella cells in a urinary bladder model. The chip design includes mechanical strain to simulate physiological conditions, such as peristaltic motion in a gut model or the stress bladder epithelial cells undergo during filling and voiding in a urinary bladder model. A porous membrane is located below the host cell layer, enabling efficient exchange of chemicals with the underlying tissue layer.
Figure 4.
Organ-on-a-Chip to study host-pathogen interactions. A. Overview of various organs modeled using organ-on-a-chip technology to investigate the molecular mechanisms of infectious diseases and the potential applications of this approach. Organ on a chip has been used to decipher bacterial and host response, to study the effect of drugs, antimicrobials and engineered bacteriophages (phage therapy) on bacterial pathogens, and to study genetic and epigenetic changes in response to the environment. B. Simplified diagram illustrating the layered structure of the chip. The top layer contains the pathogenic bacteria of interest, followed by the extracellular matrix. Nutrients, growth factors, and drugs flow through this layer to replicate the host environment (e.g., gut). Beneath this, the first host cell layer is represented, such as umbrella cells in a urinary bladder model. The chip design includes mechanical strain to simulate physiological conditions, such as peristaltic motion in a gut model or the stress bladder epithelial cells undergo during filling and voiding in a urinary bladder model. A porous membrane is located below the host cell layer, enabling efficient exchange of chemicals with the underlying tissue layer.
6. Conclusions and Future Perspective
In the past decade, the emergence of Multi-Drug-Resistant (MDR) bacterial strains has become a significant global health challenge, driven by factors such as de novo mutations, horizontal gene transfer, and the inappropriate use of antibiotics in both human medicine and livestock farming [154,155]. These factors foster conditions that promote bacterial resistance, enabling them to survive in sublethal concentrations of antibiotics. Furthermore, to counteract host immune responses, bacteria have evolved a range of virulence factors, sensors, and response regulators that activate downstream stress response pathways to counteract the host mediated nutritional immunity [156,157]. For instance, the Ferric Uptake Regulator (Fur) plays a crucial role in equipping uropathogenic E. coli with the necessary adaptations to survive the nutrient-limited urinary tract [157]. Similarly, the global Fe-S cluster regulator, IscR, has been shown to be pivotal in the survival and virulence of Yersinia pestis [158]. Additionally, Mycobacterium tuberculosis has developed several adaptive strategies to evade antimicrobial treatments, including its ability to enter a dormant state and reactivate under immunocompromised conditions, establishing chronic infections. Central to this adaptation is the DevR/DosR response regulator, which is crucial for the virulence, dormancy adaptation, and antibiotic tolerance of M. tuberculosis. By modulating the dormancy regulon, DevR/DosR facilitates the persistence of M. tuberculosis in the host, enabling it to endure environmental stresses that promote dormancy [159]. Moreover, PolyPhosphate Kinase 1 (PPK1) plays a key role during phosphate starvation, likely through a bistable signaling pathway [160], while the transcription factor MtrA regulates dormancy and reactivation, promoting long-term survival of M. tuberculosis [161]. We have previously developed recombinant tools to study the role of transcription factors in M. tuberculosis under in vitro [162] and in vivo conditions [163]. Using these methods, we identified the role of the novel transcription factor Rv1222 in M. tuberculosis [164]. Additionally, these tools have proven valuable in discovering a novel anti-tuberculosis molecule, D-AAP1 to treat drug resistant M. tuberculosis [165]. With the growing emergence of resistant strains worldwide, there is an urgent need to develop new tools to better understand the roles of these factors in host-pathogen interactions.
Synthetic biology-based strategies have greatly enhanced our ability to dissect the roles of virulence factors and essential genes in bacterial pathogens [24,38,91,92]. Many pathogenic bacteria contain horizontally acquired genes located within their genomic pathogenicity islands. These islands frequently encode genes critical for bacterial survival and virulence, enabling pathogens to counteract host immune response. For instance, the uropathogenic Escherichia coli strain CFT073 harbors 13 pathogenicity islands, which include genes involved in iron acquisition, toxin production and transport, biofilm formation, and antimicrobial resistance [157,166]. The CRISPR-Cas systems have emerged as powerful tools in bacterial research due to their versatility in targeting bacterial defense mechanisms. These methods can be utilized to elucidate the roles of pathogenicity island genes with unknown functions, thereby revealing novel adaptive strategies underlying host-pathogen interactions. Mobile-CRISPRi has emerged as new player to study host-pathogen interactions. Currently, this method can be only utilized in a limited number of Gram-positive pathogens. Further development of mobile-CRISPRi-based platforms will unlock the untapped potential to study a greater range of pathogens. Furthermore, CRISPR-based functional genomics techniques, such as CRISPR interference (CRISPRi), combined with sequencing technologies like CRISPRi-seq, have proven effective in identifying key gene subsets and disrupting essential processes involved in bacterial host-pathogen interaction [75]. Although the application of CRISPRi-seq is still limited, its integration with other techniques, such as Tn-seq, has the potential to reveal niche-specific metabolic processes and identify synergistic antibiotic targets that may be concealed by genetic redundancy. Finally, CRISPR-mediated transposition (CAST) [167] and CRISPR activation (CRISPRa) systems provide innovative methods for large-scale bacterial genome analysis, offering promising solutions to the limitations of current methods.
The development of engineered microbes to detect microbial pathogens and pathogen associated metabolites in vivo has opened new avenues for understanding the molecular signatures associated with disease progression, offering valuable insights for drug development. BWCBs have proven to be an effective method for identifying both intra- and inter-species communication signals (quorum sensing molecules) within the gut microbiota. These reporter gene-based synthetic circuits allow for bacterial imaging, but over time, the signal intensity may diminish, often due to the depletion of essential exogenous substrates required for bioluminescent bacteria [168]. Furthermore, fluorescence imaging is hindered by background autofluorescence, which can compromise sensitivity [169]. To address these limitations, strategies to correct for autofluorescence can be employed, or alternative optical imaging techniques, such as bioluminescence or photoacoustic imaging, can be explored to improve accuracy and reliability.
Phage therapy (PT) has emerged as a promising alternative to traditional antibiotics, utilizing bacteriophages to target and eliminate antibiotic-resistant pathogens [42,106,107]. While it holds significant promise, its widespread adoption is hindered by challenges such as the need for extensive clinical research, regulatory approvals, and overcoming misconceptions regarding the potential for phages to cause infections. PT is especially promising in personalized medicine, where phage biobanks are used to match specific phages to bacterial strains [128]. Despite these hurdles, PT could serve as a valuable adjunct to conventional antibiotics, particularly in treating infections resistant to standard therapies. Additionally, advancements in phage therapy, driven by clinical trials and biotechnological innovations, provide a foundation for developing alternative anti-infectives. However, challenges such as the longevity of phages, their interactions with antibiotics, pharmacokinetics, and safety concerns regarding endotoxins and protein toxins remain. Efforts to develop large phage banks and improve isolation and purification methods are crucial to ensuring the safety and efficacy of PT, while genetic engineering and sequencing technologies offer hope for the future, provided that logistical and regulatory hurdles are addressed.
Finally, organ-on-a-chip models offer significant advantages for studying conditions that are challenging to replicate in humans or animals, including genetic disorders and effects of severe radiation exposure. These models provide a human-relevant alternative to genetically modified mice, facilitating more accurate therapeutic development. Organ chips also present a safer alternative for investigating pregnancy-related drug effects on the placental barrier [130]. Recent advancements have led to the development of multi-organ-on-a-chip systems designed to explore how various organs respond under normal and diseased conditions [170]. These systems allow for more accurate modeling of inter-organ interactions and disease progression. By simulating complex drug metabolism and pharmacokinetics, they will provide a more relevant approach to preclinical drug testing. The potential of multi-organ chips to transform personalized medicine is immense, providing insights into individual responses to therapeutics. Looking forward, the developmment of bacterial platforms for antimicrobial therapy and the combination of innovative strategies such as phage therapy, CRISPR-based genomics, and synthetic microbial communities offer promising directions to address antibiotic resistance and enhance therapeutic approaches. These emerging technologies have the potential to reshape treatments for infectious diseases, heralding a pivotal shift in the fight against antimicrobial resistance.
Author Contributions
“Conceptualization, writing—original draft preparation. writing—review and editing, visualization, supervision-Rajdeep Banerjee.
Funding
The author has not received any funding for writing this review.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Pulingam, T., et al., Antimicrobial resistance: Prevalence, economic burden, mechanisms of resistance and strategies to overcome. Eur J Pharm Sci, 2022. 170: p. 106103. [CrossRef]
- Organization, W.H., Global Tuberculosis Report. 2024.
- Rahn, D.D., Urinary tract infections: contemporary management. Urol Nurs, 2008. 28(5): p. 333-41; quiz 342.
- Seung, K.J., S. Keshavjee, and M.L. Rich, Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harb Perspect Med, 2015. 5(9): p. a017863. [CrossRef]
- Khan, M.I., et al., Assessment of multidrug resistance in bacterial isolates from urinary tract-infected patients. Journal of Radiation Research and Applied Sciences, 2020. 13(1): p. 267-275. [CrossRef]
- Brumbaugh, A.R. and H.L. Mobley, Preventing urinary tract infection: progress toward an effective Escherichia coli vaccine. Expert Rev Vaccines, 2012. 11(6): p. 663-76.
- Clark, N.M., G.G. Zhanel, and J.P. Lynch, 3rd, Emergence of antimicrobial resistance among Acinetobacter species: a global threat. Curr Opin Crit Care, 2016. 22(5): p. 491-9. [CrossRef]
- Chua, H.C., et al., Combatting the Rising Tide of Antimicrobial Resistance: Pharmacokinetic/Pharmacodynamic Dosing Strategies for Maximal Precision. Int J Antimicrob Agents, 2021. 57(3): p. 106269. [CrossRef]
- Seok, J., et al., Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A, 2013. 110(9): p. 3507-12. [CrossRef]
- Baba, T., et al., Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol, 2006. 2: p. 2006.0008.
- Winzeler, E.A., et al., Functional Characterization of the S. cerevisiae Genome by Gene Deletion and Parallel Analysis. Science, 1999. 285(5429): p. 901-906.
- Koo, B.-M., et al., Construction and Analysis of Two Genome-Scale Deletion Libraries for Bacillus subtilis. Cell Systems, 2017. 4(3): p. 291-305.e7. [CrossRef]
- van Opijnen, T., K.L. Bodi, and A. Camilli, Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nature Methods, 2009. 6(10): p. 767-772.
- Cain, A.K., et al., A decade of advances in transposon-insertion sequencing. Nature Reviews Genetics, 2020. 21(9): p. 526-540. [CrossRef]
- Jana, B., et al., CRISPRi–TnSeq maps genome-wide interactions between essential and non-essential genes in bacteria. Nature Microbiology, 2024. 9(9): p. 2395-2409. [CrossRef]
- Leshchiner, D., et al., A genome-wide atlas of antibiotic susceptibility targets and pathways to tolerance. Nature Communications, 2022. 13(1): p. 3165. [CrossRef]
- Gallagher Larry, A., J. Shendure, and C. Manoil, Genome-Scale Identification of Resistance Functions in Pseudomonas aeruginosa Using Tn-seq. mBio, 2011. 2(1). [CrossRef]
- DeJesus Michael, A., et al., Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. mBio, 2017. 8(1). [CrossRef]
- Green, B., et al., Insertion site preference of Mu, Tn5, and Tn7 transposons. Mobile DNA, 2012. 3(1): p. 3. [CrossRef]
- Javaid, N. and S. Choi, CRISPR/Cas System and Factors Affecting Its Precision and Efficiency. Front Cell Dev Biol, 2021. 9: p. 761709. [CrossRef]
- Fonseca, M.M., D.J. Harris, and D. Posada, Origin and Length Distribution of Unidirectional Prokaryotic Overlapping Genes. G3 Genes|Genomes|Genetics, 2014. 4(1): p. 19-27. [CrossRef]
- Mishra, D., et al., A load driver device for engineering modularity in biological networks. Nat Biotechnol, 2014. 32(12): p. 1268-75. [CrossRef]
- Benner, S.A. and A.M. Sismour, Synthetic biology. Nature Reviews Genetics, 2005. 6(7): p. 533-543.
- Ruder, W.C., T. Lu, and J.J. Collins, Synthetic Biology Moving into the Clinic. Science, 2011. 333(6047): p. 1248-1252. [CrossRef]
- Qi, Lei S., et al., Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell, 2013. 152(5): p. 1173-1183. [CrossRef]
- Sharda, M., A. Badrinarayanan, and A.S.N. Seshasayee, Evolutionary and Comparative Analysis of Bacterial Nonhomologous End Joining Repair. Genome Biology and Evolution, 2020. 12(12): p. 2450-2466. [CrossRef]
- Doudna, J.A. and E. Charpentier, The new frontier of genome engineering with CRISPR-Cas9. Science, 2014. 346(6213): p. 1258096. [CrossRef]
- McNeil, M.B., et al., CRISPR interference identifies vulnerable cellular pathways with bactericidal phenotypes in Mycobacterium tuberculosis. Mol Microbiol, 2021. 116(4): p. 1033-1043. [CrossRef]
- Shalem, O., N.E. Sanjana, and F. Zhang, High-throughput functional genomics using CRISPR–Cas9. Nature Reviews Genetics, 2015. 16(5): p. 299-311. [CrossRef]
- Peters, J.M., et al., A Comprehensive, CRISPR-based Functional Analysis of Essential Genes in Bacteria. Cell, 2016. 165(6): p. 1493-1506. [CrossRef]
- Peters, J.M., et al., Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. Nature Microbiology, 2019. 4(2): p. 244-250.
- Qu, J., et al., Modulating Pathogenesis with Mobile-CRISPRi. Journal of Bacteriology, 2019. 201(22): p. 10.1128/jb.00304-19. [CrossRef]
- Ward, R.D., et al., Essential gene knockdowns reveal genetic vulnerabilities and antibiotic sensitivities in Acinetobacter baumannii. mBio, 2024. 15(2): p. e02051-23. [CrossRef]
- Geyman, L.J., et al., Mobile-CRISPRi as a powerful tool for modulating Vibrio gene expression. Applied and Environmental Microbiology, 2024. 90(6): p. e00065-24. [CrossRef]
- Flores-Mireles, A.L., et al., Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol, 2015. 13(5): p. 269-84. [CrossRef]
- Wiles, T.J., R.R. Kulesus, and M.A. Mulvey, Origins and virulence mechanisms of uropathogenic Escherichia coli. Exp Mol Pathol, 2008. 85(1): p. 11-9. [CrossRef]
- Fu, E., et al., Perspective on diagnostics for global health. IEEE Pulse, 2011. 2(6): p. 40-50. [CrossRef]
- Rogers, J.K., N.D. Taylor, and G.M. Church, Biosensor-based engineering of biosynthetic pathways. Current Opinion in Biotechnology, 2016. 42: p. 84-91. [CrossRef]
- Kumari, A., et al., Biosensing systems for the detection of bacterial quorum signaling molecules. Anal Chem, 2006. 78(22): p. 7603-9. [CrossRef]
- Bonnet, J., et al., Amplifying genetic logic gates. Science, 2013. 340(6132): p. 599-603. [CrossRef]
- Wang, B., et al., Engineering modular and orthogonal genetic logic gates for robust digital-like synthetic biology. Nature Communications, 2011. 2(1): p. 508. [CrossRef]
- Strathdee, S.A., et al., Phage therapy: From biological mechanisms to future directions. Cell, 2023. 186(1): p. 17-31. [CrossRef]
- Lammens, E.-M., P.I. Nikel, and R. Lavigne, Exploring the synthetic biology potential of bacteriophages for engineering non-model bacteria. Nature Communications, 2020. 11(1): p. 5294. [CrossRef]
- Loc-Carrillo, C. and S.T. Abedon, Pros and cons of phage therapy. Bacteriophage, 2011. 1(2): p. 111-114. [CrossRef]
- Samson, J.E., et al., Revenge of the phages: defeating bacterial defences. Nature Reviews Microbiology, 2013. 11(10): p. 675-687. [CrossRef]
- Koskella, B. and M.A. Brockhurst, Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol Rev, 2014. 38(5): p. 916-31. [CrossRef]
- Weigle, J.J., Induction of Mutations in a Bacterial Virus*. Proceedings of the National Academy of Sciences, 1953. 39(7): p. 628-636.
- León, M. and R. Bastías, Virulence reduction in bacteriophage resistant bacteria. Front Microbiol, 2015. 6: p. 343. [CrossRef]
- Enright, A.L., et al., CRISPRi functional genomics in bacteria and its application to medical and industrial research. Microbiol Mol Biol Rev, 2024. 88(2): p. e0017022. [CrossRef]
- Rahman, M.U., et al., Endolysin, a Promising Solution against Antimicrobial Resistance. Antibiotics (Basel), 2021. 10(11). [CrossRef]
- Belete, M.A., et al., Phage endolysins as new therapeutic options for multidrug resistant Staphylococcus aureus: an emerging antibiotic-free way to combat drug resistant infections. Frontiers in Cellular and Infection Microbiology, 2024. 14. [CrossRef]
- Abdelrahman, F., et al., Phage-Encoded Endolysins. Antibiotics (Basel), 2021. 10(2). [CrossRef]
- Hermoso, J.A., J.L. García, and P. García, Taking aim on bacterial pathogens: from phage therapy to enzybiotics. Current Opinion in Microbiology, 2007. 10(5): p. 461-472. [CrossRef]
- Fan, Y. and O. Pedersen, Gut microbiota in human metabolic health and disease. Nature Reviews Microbiology, 2021. 19(1): p. 55-71. [CrossRef]
- Hou, K., et al., Microbiota in health and diseases. Signal Transduction and Targeted Therapy, 2022. 7(1): p. 135.
- Jandhyala, S.M., et al., Role of the normal gut microbiota. World J Gastroenterol, 2015. 21(29): p. 8787-803. [CrossRef]
- Van Norman, G.A., Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic Transl Sci, 2019. 4(7): p. 845-854.
- Andes, D. and W.A. Craig, Animal model pharmacokinetics and pharmacodynamics: a critical review. International Journal of Antimicrobial Agents, 2002. 19(4): p. 261-268. [CrossRef]
- Moore, J.N., et al., Animal pharmacokinetics/pharmacodynamics (PK/PD) infection models for clinical development of antibacterial drugs: lessons from selected cases. Journal of Antimicrobial Chemotherapy, 2023. 78(6): p. 1337-1343. [CrossRef]
- Baddal, B. and P. Marrazzo, Refining Host-Pathogen Interactions: Organ-on-Chip Side of the Coin. Pathogens, 2021. 10(2). [CrossRef]
- Leung, C.M., et al., A guide to the organ-on-a-chip. Nature Reviews Methods Primers, 2022. 2(1): p. 33. [CrossRef]
- Tock, M.R. and D.T.F. Dryden, The biology of restriction and anti-restriction. Current Opinion in Microbiology, 2005. 8(4): p. 466-472. [CrossRef]
- Barrangou, R., et al., CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science, 2007. 315(5819): p. 1709-1712. [CrossRef]
- Barrangou, R. and P. Horvath, CRISPR: New Horizons in Phage Resistance and Strain Identification. Annual Review of Food Science and Technology, 2012. 3(Volume 3, 2012): p. 143-162. [CrossRef]
- Brouns, S.J.J., et al., Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science, 2008. 321(5891): p. 960-964. [CrossRef]
- Marraffini, L.A. and E.J. Sontheimer, CRISPR Interference Limits Horizontal Gene Transfer in Staphylococci by Targeting DNA. Science, 2008. 322(5909): p. 1843-1845. [CrossRef]
- Mojica, F.J., et al., Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol, 2000. 36(1): p. 244-6. [CrossRef]
- Jaganathan, D., et al., CRISPR for Crop Improvement: An Update Review. Front Plant Sci, 2018. 9: p. 985. [CrossRef]
- Xu, X., et al., Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Molecular Cell, 2021. 81(20): p. 4333-4345.e4. [CrossRef]
- Adli, M., The CRISPR tool kit for genome editing and beyond. Nature Communications, 2018. 9(1): p. 1911. [CrossRef]
- Bikard, D., et al., Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Research, 2013. 41(15): p. 7429-7437. [CrossRef]
- Liu, X., et al., High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol Syst Biol, 2017. 13(5): p. 931. [CrossRef]
- Choudhary, E., et al., Gene silencing by CRISPR interference in mycobacteria. Nat Commun, 2015. 6: p. 6267. [CrossRef]
- Wang, T., et al., Reversible Gene Expression Control in Yersinia pestis by Using an Optimized CRISPR Interference System. Appl Environ Microbiol, 2019. 85(12). [CrossRef]
- de Bakker, V., et al., CRISPRi-seq for genome-wide fitness quantification in bacteria. Nature Protocols, 2022. 17(2): p. 252-281.
- Ellis, N.A., et al., A multiplex CRISPR interference tool for virulence gene interrogation in Legionella pneumophila. Communications Biology, 2021. 4(1): p. 157. [CrossRef]
- Yan, M.-Y., et al., Application of combined CRISPR screening for genetic and chemical-genetic interaction profiling in Mycobacterium tuberculosis. Science Advances, 2022. 8(47): p. eadd5907. [CrossRef]
- Wan, X., et al., Engineering a CRISPR interference system targeting AcrAB-TolC efflux pump to prevent multidrug resistance development in Escherichia coli. Journal of Antimicrobial Chemotherapy, 2022. 77(8): p. 2158-2166.
- Noirot-Gros, M.-F., et al., CRISPR interference to interrogate genes that control biofilm formation in Pseudomonas fluorescens. Scientific Reports, 2019. 9(1): p. 15954. [CrossRef]
- Gervais, N.C., et al., Development and applications of a CRISPR activation system for facile genetic overexpression in Candida albicans. G3 (Bethesda), 2023. 13(2). [CrossRef]
- Afonina, I., et al., Multiplex CRISPRi System Enables the Study of Stage-Specific Biofilm Genetic Requirements in Enterococcus faecalis. mBio, 2020. 11(5): p. 10.1128/mbio.01101-20. [CrossRef]
- Jost, M., et al., Titrating gene expression using libraries of systematically attenuated CRISPR guide RNAs. Nature Biotechnology, 2020. 38(3): p. 355-364. [CrossRef]
- Liu, G., et al., Deep learning-guided discovery of an antibiotic targeting Acinetobacter baumannii. Nature Chemical Biology, 2023. 19(11): p. 1342-1350. [CrossRef]
- Singh, M., et al., Discovery of potent antimycobacterial agents targeting lumazine synthase (RibH) of Mycobacterium tuberculosis. Scientific Reports, 2024. 14(1): p. 12170. [CrossRef]
- Martin, J.K., et al., A Dual-Mechanism Antibiotic Kills Gram-Negative Bacteria and Avoids Drug Resistance. Cell, 2020. 181(7): p. 1518-1532.e14. [CrossRef]
- Mathew, R. and D. Chatterji, The evolving story of the omega subunit of bacterial RNA polymerase. Trends in Microbiology, 2006. 14(10): p. 450-455. [CrossRef]
- Patel, U.R., S. Gautam, and D. Chatterji, Validation of Omega Subunit of RNA Polymerase as a Functional Entity. Biomolecules, 2020. 10(11). [CrossRef]
- Dong, C., et al., Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nature Communications, 2018. 9(1): p. 2489. [CrossRef]
- Geyman, L.J., et al., Mobile-CRISPRi as a powerful tool for modulating Vibrio gene expression. Appl Environ Microbiol, 2024. 90(6): p. e0006524. [CrossRef]
- Qu, J., et al., Modulating Pathogenesis with Mobile-CRISPRi. J Bacteriol, 2019. 201(22). [CrossRef]
- Mukherji, S. and A. van Oudenaarden, Synthetic biology: understanding biological design from synthetic circuits. Nat Rev Genet, 2009. 10(12): p. 859-71.
- Khalil, A.S. and J.J. Collins, Synthetic biology: applications come of age. Nature Reviews Genetics, 2010. 11(5): p. 367-379. [CrossRef]
- Ahmed, A., et al., Biosensors for whole-cell bacterial detection. Clin Microbiol Rev, 2014. 27(3): p. 631-46. [CrossRef]
- Naydich, A.D., et al., Synthetic Gene Circuits Enable Systems-Level Biosensor Trigger Discovery at the Host-Microbe Interface. mSystems, 2019. 4(4). [CrossRef]
- Hernández-Sancho, J.M., et al., A versatile microbial platform as a tunable whole-cell chemical sensor. Nature Communications, 2024. 15(1): p. 8316. [CrossRef]
- Croxen, M.A., et al., Recent advances in understanding enteric pathogenic Escherichia coli. Clin Microbiol Rev, 2013. 26(4): p. 822-80. [CrossRef]
- Park, M., S.L. Tsai, and W. Chen, Microbial biosensors: engineered microorganisms as the sensing machinery. Sensors (Basel), 2013. 13(5): p. 5777-95. [CrossRef]
- Kotula, J.W., et al., Programmable bacteria detect and record an environmental signal in the mammalian gut. Proc Natl Acad Sci U S A, 2014. 111(13): p. 4838-43. [CrossRef]
- Mimee, M., et al., An ingestible bacterial-electronic system to monitor gastrointestinal health. Science, 2018. 360(6391): p. 915-918. [CrossRef]
- Daeffler, K.N.M., et al., Engineering bacterial thiosulfate and tetrathionate sensors for detecting gut inflammation. Molecular Systems Biology, 2017. 13(4): p. 923. [CrossRef]
- Archer, E.J., A.B. Robinson, and G.M. Süel, Engineered E. coli that detect and respond to gut inflammation through nitric oxide sensing. ACS Synth Biol, 2012. 1(10): p. 451-7.
- Raut, N., P. Pasini, and S. Daunert, Deciphering bacterial universal language by detecting the quorum sensing signal, autoinducer-2, with a whole-cell sensing system. Anal Chem, 2013. 85(20): p. 9604-9. [CrossRef]
- Gupta, S., E.E. Bram, and R. Weiss, Genetically programmable pathogen sense and destroy. ACS Synth Biol, 2013. 2(12): p. 715-23.
- Hwang, I.Y., et al., Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nature Communications, 2017. 8(1): p. 15028. [CrossRef]
- Zheng, L., et al., CRISPR/Cas-Based Genome Editing for Human Gut Commensal Bacteroides Species. ACS Synthetic Biology, 2022. 11(1): p. 464-472. [CrossRef]
- Zalewska-Piątek, B., Phage Therapy-Challenges, Opportunities and Future Prospects. Pharmaceuticals (Basel), 2023. 16(12). [CrossRef]
- Lin, D.M., B. Koskella, and H.C. Lin, Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J Gastrointest Pharmacol Ther, 2017. 8(3): p. 162-173. [CrossRef]
- Dörner, P.J., et al., Clinically used broad-spectrum antibiotics compromise inflammatory monocyte-dependent antibacterial defense in the lung. Nature Communications, 2024. 15(1): p. 2788. [CrossRef]
- Citorik, R.J., M. Mimee, and T.K. Lu, Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nature Biotechnology, 2014. 32(11): p. 1141-1145. [CrossRef]
- Bikard, D., et al., Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol, 2014. 32(11): p. 1146-50. [CrossRef]
- Schmelcher, M., D.M. Donovan, and M.J. Loessner, Bacteriophage endolysins as novel antimicrobials. Future Microbiol, 2012. 7(10): p. 1147-71. [CrossRef]
- O'Flaherty, S., et al., The Recombinant Phage Lysin LysK Has a Broad Spectrum of Lytic Activity against Clinically Relevant Staphylococci, Including Methicillin-Resistant Staphylococcus aureus. Journal of Bacteriology, 2005. 187(20): p. 7161-7164. [CrossRef]
- Zhang, S., et al., Characterization of Salmonella endolysin XFII produced by recombinant Escherichia coli and its application combined with chitosan in lysing Gram-negative bacteria. Microbial Cell Factories, 2022. 21(1): p. 171. [CrossRef]
- Díez-Martínez, R., et al., A novel chimeric phage lysin with high in vitro and in vivo bactericidal activity against Streptococcus pneumoniae. Journal of Antimicrobial Chemotherapy, 2015. 70(6): p. 1763-1773. [CrossRef]
- Becker, S.C., et al., Differentially conserved staphylococcal SH3b_5 cell wall binding domains confer increased staphylolytic and streptolytic activity to a streptococcal prophage endolysin domain. Gene, 2009. 443(1): p. 32-41. [CrossRef]
- Dong, Q., et al., Construction of a chimeric lysin Ply187N-V12C with extended lytic activity against staphylococci and streptococci. Microb Biotechnol, 2015. 8(2): p. 210-20. [CrossRef]
- Yang, H., et al., Novel Chimeric Lysin with High-Level Antimicrobial Activity against Methicillin-Resistant Staphylococcus aureus In Vitro and In Vivo. Antimicrobial Agents and Chemotherapy, 2014. 58(1): p. 536-542. [CrossRef]
- Schmelcher, M., V.S. Tchang, and M.J. Loessner, Domain shuffling and module engineering of Listeria phage endolysins for enhanced lytic activity and binding affinity. Microb Biotechnol, 2011. 4(5): p. 651-62. [CrossRef]
- Oliveira, H., et al., A thermostable Salmonella phage endolysin, Lys68, with broad bactericidal properties against gram-negative pathogens in presence of weak acids. PLoS One, 2014. 9(10): p. e108376. [CrossRef]
- Briers, Y., et al., Engineered Endolysin-Based “Artilysins” To Combat Multidrug-Resistant Gram-Negative Pathogens. mBio, 2014. 5(4): p. 10.1128/mbio.01379-14. [CrossRef]
- Lood, R., et al., Novel phage lysin capable of killing the multidrug-resistant gram-negative bacterium Acinetobacter baumannii in a mouse bacteremia model. Antimicrob Agents Chemother, 2015. 59(4): p. 1983-91. [CrossRef]
- Lazcka, O., F.J.D. Campo, and F.X. Muñoz, Pathogen detection: A perspective of traditional methods and biosensors. Biosensors and Bioelectronics, 2007. 22(7): p. 1205-1217. [CrossRef]
- Sarkis, G.J., W.R. Jacobs, Jr., and G.F. Hatfull, L5 luciferase reporter mycobacteriophages: a sensitive tool for the detection and assay of live mycobacteria. Mol Microbiol, 1995. 15(6): p. 1055-67.
- Loessner, M.J., et al., Construction of luciferase reporter bacteriophage A511::luxAB for rapid and sensitive detection of viable Listeria cells. Appl Environ Microbiol, 1996. 62(4): p. 1133-40. [CrossRef]
- Oda, M., et al., Rapid detection of Escherichia coli O157:H7 by using green fluorescent protein-labeled PP01 bacteriophage. Appl Environ Microbiol, 2004. 70(1): p. 527-34. [CrossRef]
- Piuri, M., W.R. Jacobs, Jr., and G.F. Hatfull, Fluoromycobacteriophages for Rapid, Specific, and Sensitive Antibiotic Susceptibility Testing of Mycobacterium tuberculosis. PLOS ONE, 2009. 4(3): p. e4870.
- Edgar, R., et al., High-sensitivity bacterial detection using biotin-tagged phage and quantum-dot nanocomplexes. Proceedings of the National Academy of Sciences, 2006. 103(13): p. 4841-4845. [CrossRef]
- Lin, J., et al., Limitations of Phage Therapy and Corresponding Optimization Strategies: A Review. Molecules, 2022. 27(6). [CrossRef]
- Ahn, J.-S., et al., Fecal Microbiome Does Not Represent Whole Gut Microbiome. Cellular Microbiology, 2023. 2023(1): p. 6868417. [CrossRef]
- Ingber, D.E., Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat Rev Genet, 2022. 23(8): p. 467-491. [CrossRef]
- Zamprogno, P., et al., Second-generation lung-on-a-chip with an array of stretchable alveoli made with a biological membrane. Communications Biology, 2021. 4(1): p. 168. [CrossRef]
- Kim, H.J. and D.E. Ingber, Gut-on-a-Chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr Biol (Camb), 2013. 5(9): p. 1130-40. [CrossRef]
- Feaugas, T. and N. Sauvonnet, Organ-on-chip to investigate host-pathogens interactions. Cellular Microbiology, 2021. 23(7): p. e13336.
- Wang, J., et al., A virus-induced kidney disease model based on organ-on-a-chip: Pathogenesis exploration of virus-related renal dysfunctions. Biomaterials, 2019. 219: p. 119367. [CrossRef]
- Ashammakhi, N., et al., Gut-on-a-chip: Current progress and future opportunities. Biomaterials, 2020. 255: p. 120196. [CrossRef]
- Yokoi, F., S. Deguchi, and K. Takayama, Organ-on-a-chip models for elucidating the cellular biology of infectious diseases. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 2023. 1870(6): p. 119504. [CrossRef]
- Sharma, K., et al., Dynamic persistence of UPEC intracellular bacterial communities in a human bladder-chip model of urinary tract infection. Elife, 2021. 10. [CrossRef]
- Han, S. and R.K. Mallampalli, The Role of Surfactant in Lung Disease and Host Defense against Pulmonary Infections. Ann Am Thorac Soc, 2015. 12(5): p. 765-74. [CrossRef]
- Chroneos, Z.C., et al., Pulmonary surfactant and tuberculosis. Tuberculosis (Edinb), 2009. 89 Suppl 1: p. S10-4. [CrossRef]
- Thacker, V.V., et al., A lung-on-chip model of early Mycobacterium tuberculosis infection reveals an essential role for alveolar epithelial cells in controlling bacterial growth. eLife, 2020. 9: p. e59961. [CrossRef]
- Plebani, R., et al., Modeling pulmonary cystic fibrosis in a human lung airway-on-a-chip. Journal of Cystic Fibrosis, 2022. 21(4): p. 606-615. [CrossRef]
- Deinhardt-Emmer, S., et al., Co-infection with Staphylococcus aureus after primary influenza virus infection leads to damage of the endothelium in a human alveolus-on-a-chip model. Biofabrication, 2020. 12(2): p. 025012. [CrossRef]
- Grassart, A., et al., Bioengineered Human Organ-on-Chip Reveals Intestinal Microenvironment and Mechanical Forces Impacting Shigella Infection. Cell Host & Microbe, 2019. 26(4): p. 565. [CrossRef]
- Sunuwar, L., et al., Mechanical Stimuli Affect Escherichia coli Heat-Stable Enterotoxin-Cyclic GMP Signaling in a Human Enteroid Intestine-Chip Model. Infection and Immunity, 2020. 88(3): p. 10.1128/iai.00866-19. [CrossRef]
- Ni, J., et al., Gut microbiota and IBD: causation or correlation? Nature Reviews Gastroenterology & Hepatology, 2017. 14(10): p. 573-584.
- Valitutti, F., S. Cucchiara, and A. Fasano, Celiac Disease and the Microbiome. Nutrients, 2019. 11(10). [CrossRef]
- Ağagündüz, D., et al., Understanding the role of the gut microbiome in gastrointestinal cancer: A review. Front Pharmacol, 2023. 14: p. 1130562. [CrossRef]
- Kim, H.J., et al., Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proceedings of the National Academy of Sciences, 2016. 113(1): p. E7-E15. [CrossRef]
- Jalili-Firoozinezhad, S., et al., A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nature Biomedical Engineering, 2019. 3(7): p. 520-531. [CrossRef]
- Shin, W. and H.J. Kim, Intestinal barrier dysfunction orchestrates the onset of inflammatory host–microbiome cross-talk in a human gut inflammation-on-a-chip. Proceedings of the National Academy of Sciences, 2018. 115(45): p. E10539-E10547. [CrossRef]
- Tovaglieri, A., et al., Species-specific enhancement of enterohemorrhagic E. coli pathogenesis mediated by microbiome metabolites. Microbiome, 2019. 7(1): p. 43. [CrossRef]
- Ingber, D.E., Developmentally inspired human ‘organs on chips’. Development, 2018. 145(16).
- Wong, I. and C.M. Ho, Surface molecular property modifications for poly(dimethylsiloxane) (PDMS) based microfluidic devices. Microfluid Nanofluidics, 2009. 7(3): p. 291-306. [CrossRef]
- Nikaido, H., Multidrug resistance in bacteria. Annu Rev Biochem, 2009. 78: p. 119-46.
- Uddin, T.M., et al., Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. Journal of Infection and Public Health, 2021. 14(12): p. 1750-1766. [CrossRef]
- Banerjee, R., et al., Iron-sulfur Rrf2 transcription factors: an emerging versatile platform for sensing stress. Curr Opin Microbiol, 2024. 82: p. 102543. [CrossRef]
- Banerjee, R., et al., Tailoring a Global Iron Regulon to a Uropathogen. mBio, 2020. 11(2). [CrossRef]
- Balderas, D., et al., Genome Scale Analysis Reveals IscR Directly and Indirectly Regulates Virulence Factor Genes in Pathogenic Yersinia. mBio, 2021. 12(3): p. e0063321. [CrossRef]
- Sharma, S., et al., Functional insights into Mycobacterium tuberculosis DevR-dependent transcriptional machinery utilizing Escherichia coli. Biochem J, 2021. 478(16): p. 3079-3098.
- Sanyal, S., et al., Polyphosphate kinase 1, a central node in the stress response network of Mycobacterium tuberculosis, connects the two-component systems MprAB and SenX3-RegX3 and the extracytoplasmic function sigma factor, sigma E. Microbiology (Reading), 2013. 159(Pt 10): p. 2074-2086.
- Sharma, A.K., et al., MtrA, an essential response regulator of the MtrAB two-component system, regulates the transcription of resuscitation-promoting factor B of Mycobacterium tuberculosis. Microbiology (Reading), 2015. 161(6): p. 1271-81. [CrossRef]
- Banerjee, R., et al., Optimization of recombinant Mycobacterium tuberculosis RNA polymerase expression and purification. Tuberculosis (Edinb), 2014. 94(4): p. 397-404. [CrossRef]
- Banerjee, R., et al., Recombinant reporter assay using transcriptional machinery of Mycobacterium tuberculosis. J Bacteriol, 2015. 197(3): p. 646-53. [CrossRef]
- Rudra, P., et al., Novel mechanism of gene regulation: the protein Rv1222 of Mycobacterium tuberculosis inhibits transcription by anchoring the RNA polymerase onto DNA. Nucleic Acids Res, 2015. 43(12): p. 5855-67.
- Lin, W., et al., Structural Basis of Mycobacterium tuberculosis Transcription and Transcription Inhibition. Mol Cell, 2017. 66(2): p. 169-179.e8. [CrossRef]
- Lloyd, A.L., et al., Genomic islands of uropathogenic Escherichia coli contribute to virulence. J Bacteriol, 2009. 191(11): p. 3469-81. [CrossRef]
- Klompe, S.E., et al., Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature, 2019. 571(7764): p. 219-225. [CrossRef]
- James, M.L. and S.S. Gambhir, A molecular imaging primer: modalities, imaging agents, and applications. Physiol Rev, 2012. 92(2): p. 897-965. [CrossRef]
- Surre, J., et al., Strong increase in the autofluorescence of cells signals struggle for survival. Scientific Reports, 2018. 8(1): p. 12088. [CrossRef]
- Picollet-D’hahan, N., et al., Multiorgan-on-a-Chip: A Systemic Approach To Model and Decipher Inter-Organ Communication. Trends in Biotechnology, 2021. 39(8): p. 788-810. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instruc |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



