Submitted:
10 December 2024
Posted:
11 December 2024
You are already at the latest version
Abstract
This work aimed to analyze pediatric Post-Authorization Studies (PASs) registered in the European Union electronic Register of Post-Authorization Studies (EU PAS Register) from September 2010 until April 2023 to identify trends in terms of timing, age groups and therapeutic areas, and to discuss pediatric specificities and sources of funding for the PASs. A screening process identified PASs conducted exclusively on the pediatric population, and instructions were provided to ensure standardized data collection from the EU PAS Register. A univariate linear regression descriptive analysis was performed to assess trends over time, while a multivariate linear regression analysis helped explore additional characteristics of the studies. Of the 2,574 PASs extracted from the EU PAS Registry, 165 were included in the analysis. The majority of pediatric PASs were observational studies (86%), and most of them utilized secondary data (53%). The annual number of PASs increased significantly between 2010 and 2023. As envisaged, the largest part was funded by pharmaceutical companies (62%). Anti-infectives for systemic uses (25%), medicines for the nervous system (18%), and antineoplastic and immunomodulating agents (15%) resulted the most studied drugs. Our findings show that post-marketing observational research in pediatric populations has increased over time. Nevertheless, industry-academia collaboration should be encouraged, and regulatory guidance is needed to prioritize research in areas of unmet therapeutic need.
Keywords:
Post-authorization studies
; pediatric
; EU PAS register
; descriptive analysis
1. Introduction
The evaluation of drug efficacy and safety is a long-lasting process, also continuing after marketing authorization has been granted. In Europe, post-authorization requirements and commitments may include specific obligations/legally binding measures or recommendations, or additional pharmacovigilance activities detailed in the risk management plan of the medicinal product, which are all aimed at obtaining additional effectiveness and safety data [1]. Moreover, other post-approval studies, including those conducted by industries and independent investigators, should be considered as part of the ongoing, continuous evaluation efforts [2].
Post-Authorization Studies (PASs) play a crucial role in the post-authorization phase of a drug, confirming its benefit-risk profile in real-world settings and helping to detect long-term or rare adverse events that may not have been identified in pre-authorization clinical trials.
In the post-authorization phase, it may be necessary to collect additional data on the safety, especially the long-term effects, and, in certain cases, effectiveness, by investigating how well the treatment works in practice, or quality of authorized medicines to complement the available pre-marketing data [3]. Concerning safety, PASs include those to identify, characterize or quantify a safety hazard, and to confirm the safety profile of the authorized medicinal product or to measure the effectiveness of risk management measures (Post-Authorization Safety Studies, PASS). Concerning efficacy, PASs include those to supplement available efficacy data in the light of well-founded scientific uncertainties on aspects of the proof of benefit that should or can only be addressed after the medicinal product has been approved (Post-Authorization Efficacy Studies, PAES).
Although PASs primarily adopt a non-interventional approach (i.e., observational studies), they may also use interventional study designs (i.e., clinical trials) [4].
PASs are designed with different purposes than pre-marketing studies. Their designs are not systematically submitted to regulatory authorities prior to initiation, because many PASs are conducted by independent investigators and their conduct is less rigorously regulated. In addition, pre-marketing studies are almost exclusively sponsored by manufacturers, whereas PASs may be funded by manufacturers, but also by academic or other types of not-for-profit institutions. Some research suggests that many PASs are frequently carried out with marketing purposes rather than for genuine medical interest [2].
Moreover, PASs should be considered as post-authorization measures that are particularly useful in the context of special populations such as pediatric patients and those affected by rare diseases. The importance of PASs in the context of pediatric medicines lies in the fact that children are typically excluded from pre-authorization clinical trials due to ethical considerations and often respond to medicines differently from adults, highlighting the need for comprehensive investigations into the long-term effects, appropriate dosages, and potential side effects specific to children [5,6]. In addition, part of the PASs conducted in the pediatric population was imposed by regulators based on specific criteria such as the mechanism of action or the potential adverse events of the drug. Concerning rare diseases, where treatment options are limited and adverse effects might have not been detected during pre-marketing studies on small populations, PASs become a lifeline, offering an opportunity to gather real-world evidence and refine therapeutic approaches [6,7].
To strengthen the monitoring of the benefit-risk profile of medicines, the European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP) has been established and is coordinated by the European Medicines Agency (EMA). Its main objective is to facilitate the conduction of high quality, multi-center, independent PASs, with a focus on observational research.
The European Union electronic Post-Authorization Study Register (EU PAS Register) was a publicly available repository developed and supported by the EMA through the ENCePP containing more than 2000 PASs, and it has recently been replaced by the Head of Medicines Agencies (HMA)-EMA Catalogue of real-world data studies [8].
In 2019, the ENCePP Working group 3 (WG3) “Data Sources and Multi-source Studies” carried out a detailed overview of the studies included in the EU PAS Register from its inception to 31st December 2018, with a focus on the multi-database studies [7].
To the best of our knowledge, no studies have identified and described the impact of PASs conducted in the pediatric population. Hence, the aim of this work was to analyze pediatric PASs registered in the ENCePP EU PAS Register to identify trends in terms of timing, age groups and therapeutic areas and to discuss pediatric specificities, dealing with various aspects of study design. The results of this analysis can be used to raise awareness of existing gaps in the post-authorization phase of pediatric medicines and to help industry and not-for-profit organizations find solutions to address unmet needs. By identifying areas in pediatric research that require further attention, this study seeks to guide stakeholders in prioritizing resources effectively, enhancing the quality and impact of pediatric post-marketing surveillance.
2. Materials and Methods
2.1. Data Collection
The data source for this study was a dataset provided by the EMA containing all PASs registered in the EU PAS register from September 2010 to 30th April 2023.
Data collection focused exclusively on PASs related to the pediatric population, excluding all studies involving both adult and pediatric patients. The sample size was determined using information about the age of the study population.
Data collection started in October 2023, and was conducted independently by four researchers (AL, AD, ADE, GSG). To standardize and harmonize the data collection procedure, a case report form and an instruction document were developed.
The publicly accessible EU PAS Register website was searched to retrieve information concerning the availability of study protocol, the summary of study results and the presence of related publications. If a link to the publication(s) was not available on the EU PAS Register website, a specific search was carried out in PubMed. To collect relevant data, priority was given to the information included in the English version of the study protocol uploaded to the ENCePP EU PAS Register, where available. For studies included in a Risk Management Plan (RMP) PASs category 1 (i.e., studies imposed as a condition to the Marketing Authorization) and 2 (i.e., studies imposed as a specific obligation in the context of a Marketing Authorization under exceptional circumstances) and when the study protocol was not available, data was searched on the EMA website. When available, also study reports and/or study synopses were used to retrieve relevant data. In case study protocol, study report, synopsis and/or publication were missing, the information was collected by consulting the EU PAS Register webpage reporting information on each PAS. In case of conflicting information, priority was given to the study protocol.
Data collected for each study is detailed in Table S1. In brief, the information encompassed data concerning study identification (e.g., study title, brief description of the study, status of the study), study scope, countries involved, study type (e.g., observational studies, clinical trials), study design (e.g., descriptive study, cohort studies, cross sectional studies), study objectives (e.g., disease epidemiology, risk assessment), funding sources, age range of the subjects enrolled as well as their classification in pediatric age groups (i.e., preterm newborn infants, term newborn infants, infants and toddlers, children and adolescent), and information on special study populations involved (e.g., lactating mothers). The concerned therapeutic area was assigned to each study based on the Anatomical Therapeutic Chemical (ATC) classification system. The number of drugs/classes of drugs under study was collected; however, it was agreed to limit the collection of information to the most relevant three drugs/drug classes included in the study. Data was gathered for each drug/class of drug, including the name, brand name, ATC code, type, and indications of whether it is a vaccine or an orphan drug. Additionally, the use of primary or secondary data sources for observational studies was reported. The detailed information collected for each study is listed in Table S1.
Moreover, an in-depth look at the different types of funding sources is provided in Table S2.
Once all the data were collected, a two-stage validation phase was carried out by cross-checking information retrieved for a 15% random sample of the identified studies. All cases of ambiguity and/or inconsistency were resolved through discussion, and if consensus was not reached, through the exploration of additional data sources (e.g., literature search) or the involvement of a fifth expert (MF).
2.2. Statistical Analysis
A descriptive analysis was conducted on collected data concerning the identified PASs. Characteristics of study design, eligibility criteria, methodological aspects, study conditions and targets, and source of funding were evaluated stratifying by year of study registration. PASs were grouped by the year of registration. The percentage change in the summarized number of PASs from 2010 to 2023 was determined using the observed annual percentage values. Univariate Linear Regression analysis was used to evaluate trends in PASs registration over time. Linear regression beta coefficients were determined to evaluate potential changes in the number of PASs over time. This statistical analysis approach was also applied to the subsets of data regarding three age categories. To understand more about additional characteristics of the PASs in our data set, we also analyzed information on studies’ source of funding and therapeutic area.
Unless specified otherwise, all statistical tests were two-sided and were performed using a significance (alpha) level of 0.05. Data were analyzed using SPSS package (IBM SPSS Statistics for Windows, version 29.0, IBM Corp., Armonk, N.Y., USA).
3. Results
Out of the 2,574 PASs extracted from the EU PAS Registry from its inception until April 30, 2023, 165 PASs were included in our analysis as they were conducted exclusively on the pediatric population. Out of a total of 165 PASs, 45 were conducted exclusively in countries outside the EU.
At the time of the analysis, 95 (57.6%) PASs were finalized, 49 (29.7%) ongoing and 21 (12.7%) planned (Table 1). Most studies (142; 86.1%) were observational, followed by systematic reviews/meta-analysis (7, 4.2%), and surveys (9, 5.5%). The scope was most frequently risk assessment (82, 49.7%), drug utilization (39, 23.6%), or effectiveness evaluation (35, 21.2%). It is important to note that each study could have more than one study objective.
Moreover, almost three-quarters of the PASs (120; 72.7%) were conducted in at least one European country. Half of the studies (83; 50.3%) were requested by regulatory authorities. Of all studies, 60 (36.4%) were part of a European RMP, while 10 studies (6.1%) were part of a non-EU risk management plan.
Of all 142 observational studies, half (76, 53.2%) used secondary data sources, while a third (48, 33.8%) used primary data collection.
Study results and publications could be gathered for 70% of the studies (116/165) and were considered primary sources of information together with the study protocol, when available.
An overview of all 165 pediatric PASs is also provided below using descriptive statistics.
The annual number of registered PASs increased significantly between 2010 and 2023, with a peak in 2014 (Table 2).
In the linear regression on relationship between the number of registered PASs (dependent variable) and the year of registration (independent variables X), the regression coefficient of 0.47 informed that the number of registered PASs increased by 0.47 with each additional year (Figure 1). A correlation coefficient (R) of 0.251 showed a positive linear relationship between number of registered PASs and the year of registration, however the relationship was non-significant (Regression p-value = 0.386).
Most of the pediatric PASs (103, 62.4%) were funded by pharmaceutical companies, followed by those funded by government bodies (25, 4.1%) (Table 3).
When the study registration year was described by funding source, no clear time trends were identified by funding source, apart from the increase in PASs from 2011 to 2013, in PASs funded both by pharmaceutical companies and government agencies (Table 4).
With reference to the therapeutic area, anti-infective drugs for systemic uses (41, 24.8%), medicines for the nervous system (29, 17.6%), and the antineoplastic and immunomodulating agents (24, 14.5%) were the most studied drugs (Table 5). Moreover, it is relevant to note that the number of study drugs under investigation vary widely among the PASs and that sometimes PASs focus on classes of drugs (e.g., a class of vaccines or antibiotics) rather than on specific drug(s). Most, 67.9% (112/165) of the PASs focused on one study drug, the 3% (5/165) on two, the 4.8% (8/165) on more than three while the 13.3% (22/165) on an entire class of medicines. For the 10.9% (18/165), no specific study drug was considered since the focus of the PAS was on the disease.
With reference to the age groups, preterm newborns were involved in 43 studies (26%), term newborns (from 0 to 27 days) were involved in 71 (43%), infants and toddlers (from 28 days to 23 months) in 102 (61.8%), children (from 2 to 11 years) in 121 (73.3%) and adolescents (from 12 to 16/18 years, dependent on region) were included in 94 (57%) of the analyzed PASs.
The results of the linear regression model, considering the age categories (i.e., preterm, term newborns and infants; children; adolescents), indicated an increasing trend in the number of registered PASs over time in all age groups (Figure 2; all the regression coefficients were positive: preterm/term newborn and infants 0.44 with R2 = 0.144; children with a regression coefficient 0.49 with R2 = 0.118; adolescents 0.33 with R2 = 0.073).
The greatest increase in the number of registered PASs over time (0.49/year) is observed in children, a slightly smaller increase (0.44/year) is observed in preterm/term newborns, while a smaller increase (0.33/year) is shown in the adolescent group followed by a decrease after 2019.
4. Discussion
An analysis of the pediatric PASs, as retrieved from the EU PAS register from its inception till April 2023, was performed using descriptive statistics to identify trends in terms of timing, age groups and therapeutic areas and to discuss pediatric specificities. Sources of funding for the PASs were investigated as well.
During the collection phase, a systematic approach was followed to retrieve information from the main data sources and store it in a harmonized and standardized way to facilitate the analysis. Some challenges were encountered during this phase, mainly related to the unavailability of study protocols for some studies, leading to incomplete data retrieval or difficulties in interpreting the information available on the EU PAS Register website for those studies without the disposal of additional protocol information. Nevertheless, these challenges were overcome by consulting the available study documents (e.g., the summary of the study results) or by looking at the available publications related to the studies. Moreover, all cases of inconsistency in data collection were addressed through discussion, exploration of additional data sources and reaching consensus among the involved researchers.
The results of our study provide valuable insights into the evolving landscape of pediatric pharmacovigilance and pharmacoepidemiology. In particular, the increase in the number of pediatric PASs registered over the years underlines the growing attention that the healthcare community, regulators and pharmaceutical industry paid to the pediatric population and the efforts made to investigate and ensure the safety and effectiveness of medicines developed for this vulnerable population. The increase in the number of registered PASs reflects a concerted effort to address the regulatory and ethical imperatives surrounding the development and use of medicines in children, encouraged by the entry into force of the Pediatric Regulation in January 2007 [9]. The utilization of linear regression to explore the temporal trend in the identified PASs further elucidates this progression, revealing a statistically significant annual increase of 0.47 PASs per year. This finding not only highlights the proactive approach taken by regulatory authorities and pharmaceutical companies but also emphasizes the ongoing commitment to pediatric pharmacovigilance, thereby fostering safer medication practices for children. Moreover, the increase in the number of these pediatric studies, together with the proportion of observational studies, indicates that observational studies are increasingly considered to complement the available evidence from pediatric clinical trials. This represents an indicator of the direction in which clinical research is going on, thus increasing the observational and post-marketing research to complement trial evidence.
Interestingly, out of a total of 165 PASs, 45 were conducted exclusively in countries outside the EU. At first glance, this may seem confusing, as the EU PAS Register is an EU register, thus only studies conducted in EU Member States could have been registered. However, it is important to clarify and emphasize that the EU PAS Register welcomed the registration of all pharmacovigilance and pharmacoepidemiology studies, regardless of the countries in which they were conducted. This inclusive approach underlines the Register’s commitment to providing a comprehensive overview of research efforts in these areas, ensuring that valuable data from a wide range of geographical locations are available to relevant stakeholders for analysis and decision-making. This is something that the EU will continue with the new HMA-EMA Catalogue, which is open for registration to all studies regardless of the country(ies) in which they are conducted [10].
Of relevance is also the classification of the 165 pediatric PASs according to their source of funding (public or private) that provides valuable insights into the financial dynamics shaping pediatric pharmacovigilance efforts. As expected, the majority of PASs (62.4%) were funded by pharmaceutical companies, highlighting the key role of industry in driving research in this area. Conversely, the comparatively lower proportion of PASs funded by not-for-profit organizations (e.g. universities) and public funds (28.5%) highlights a potential gap in public sector support for pediatric pharmacovigilance initiatives. The same trend is confirmed when looking at funding over time, while PASs funded by pharmaceutical companies show an increase, a similar trend is not observed for studies funded by other institutions. This discrepancy may be potentially driven by regulatory requirements reflecting different priorities and funding dynamics between the private and public sectors and underlines the need for joint efforts to ensure comprehensive and balanced pediatric drug safety research. Industry-academia collaborations should be encouraged especially in areas of unmet therapeutic need to speed up scientific research. Regulatory guidance is deemed necessary to enhance collaboration in the pediatric field.
Moreover, while the EU PAS register is comprehensive in its coverage of study data on the source of funding, there is a notable lack of information on the study sponsorship, especially regarding whether the research is conducted by profit or non-profit organizations. This omission could pose a significant challenge in assessing the purpose of the study, as it becomes difficult to determine whether the intention behind the research is for profit or not-for-profit. This shows that without transparency and a clear understanding of the aims and objectives of a study, it is difficult to make well-informed conclusions about PASs in pediatrics. To overcome this limitation, and considering the evolving landscape for the PASs, currently included in the new HMA-EMA Catalogue of real-world data studies [8], we strongly recommend that this information is also mentioned for each study. In this way, stakeholders can gain a clearer understanding of the aims and objectives behind each study, thereby increasing transparency and facilitating informed decision-making within the scientific community.
With regard to the results concerning the distribution of PASs by therapeutic area, the majority of study drugs/classes of drug under investigation belonged to the following three: anti-infectives, antineoplastic and immunomodulating agents and drugs for the nervous system. This is partially in line with the main pediatric medicines studied in pre-marketing studies, namely anti-infectives, antineoplastic and immunomodulators. In fact, a systematic review by Deejesh Subramanian et al., 2022 found that while oncology drugs are widely represented in early phase clinical trials, the most studied drugs in phase I-III pediatric trials were antivirals (27% of all trials), dominated by HIV/AIDS drugs, followed by blood products and modifiers (10%), drugs for genetic or enzyme protein disorders (8%), immunological agents (7%), and antibacterials (6%) [11]. The anti-infectives for systemic use representing the group with the highest ratio (21%) of the total number of pediatric authorized medicines are also confirmed by an analysis performed by Toma M and colleagues in 2021 [12].
This outcome suggests that it would be necessary to increase the percentage of PASs in certain therapeutic areas that are less represented, but it could be worth considering that this result may contain a source of bias, as some diseases do not affect the pediatric population, and therefore some therapeutic areas may be less represented.
Finally, looking at the pediatric age groups and discussing pediatric specificities, our analysis shows a difference in the number of PASs reported over time. Specifically, the number of studies reported for children (aged 2-11 years) shows an increasing trend over time, which is higher than the slightly increasing trend for adolescents (aged 12-16/18 years). This may give rise to different considerations, mainly related to the differences that exist between Member States with regard to the adolescent group. Firstly, the age of legal capacity varies between Member States, allowing, for example, the inclusion of subjects over 16 years of age in adult clinical studies and thus excluding them from pediatric studies and therefore a less need for additional post-authorization data. Secondly, it may be easier to conduct PASs than pre-marketing studies in children, which raise more ethical and regulatory concerns and more recruitment difficulties. It may also be due to the fact that the adolescent population is often studied with the adult population and our analysis, which focuses on studies conducted exclusively in the pediatric population, may not cover this sample. Moreover, as highlighted in the paper by Toma M and colleagues [11], most of the medicines were tested in the adolescent age group in the pre-marketing setting.
Of relevance is to highlight that the regulatory landscape related to the medicinal products is currently evolving. On March 19, 2024, the Committee on the Environment, Public Health and Food Safety (ENVI) at the European Parliament, in charge for the procedure, adopted its position on the new Directive and Regulation on medicinal products for human use and the proposed Directive also includes specific mention of pharmacovigilance to monitor long-term post-authorization safety and efficacy studies in children, including relevant data from off-label use of the product. The dossier will be taken up by the new Parliament after the European elections on 6-9 June, 2024 [13,14].
5. Conclusions
In conclusion, our results show that PASs, including observational studies, are increasing over time, to complement results of pre-marketing pediatric clinical trials. Moreover, greater availability of study documents for each study, including study protocols and results, is needed to have a better understanding and level of detail of the information available on each study and to improve the transparency of pediatric studies. This could be implemented in the new HMA-EMA Catalogue of real-world data studies. Finally, collaborations between industry and academia should be promoted as well as regulatory guidance is needed to prioritize research where there is a major unmet therapeutic need in the pediatric research in terms of pediatric populations and therapeutic areas less studied in the post-authorization phase.
6. Patents
Not applicable.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Information collected for each study, Table S2: Information collected for funding source.
Author Contributions
Conceptualization, A.C., M.F. and G.T.; methodology, A.L., A.C., G.R., D.B., F.B.; validation, S.C., G.T. and M.F.; formal analysis, A.L., A.D., G.R., A.DE., G.S.G.; investigation, A.L., A.D., A.DE., G.S.G..; data curation, A.L., A.D., G.R., A.DE., G.S.G.; writing—original draft preparation, A.L., A.D., G.R., A.DE.; writing—review and editing, AL., A.DE., G.R., A.C., D.B., F.B., S.C., G.T., F.K., M.F., K.M.H., F.B.A., A.U, K.G., A.U.; visualization, G.R.; supervision, M.F. AL and GR also contributed as first authors to the drafting of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable for this study, as it was not necessary to directly create or manage databases in which identifiable or identified patient data could be present; therefore, according to local regulations, it was not necessary to obtain the approval of an ethics committee to carry out this study.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data used were collected from the EU PAS register, which contains public data, although the link is currently no longer active as the EU PAS register has been replaced by the Head of Medicines Agencies (HMA)-EMA Catalogue of real-world data studies [8]. - Available online: https://catalogues.ema.europa.eu/catalogue-rwd-studies.
Acknowledgments
The extracted file from the EU PAS register provided by colleagues working at the European Medicines Agency.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Post-authorisation measures: questions and answers. European Medicines Agency post-authorisation procedural advice for users of the centralised procedure. Available online: https://www.ema.europa.eu/en/pharmacovigilance-post-authorisation/post-authorisation-measures-questions-and-answers (accessed on 23 September 2024).
- Zeitoun, J.D.; Ross, J.S.; Atal, I.; Vivot, A.; Downing, N. S.; Baron, G.; Ravaud, P. Postmarketing studies for novel drugs approved by both the FDA and EMA between 2005 and 2010: a cross-sectional study. BMJ Open 2017, 7(12), e018587. [Google Scholar] [CrossRef] [PubMed]
- Guideline on good pharmacovigilance practices (GVP). Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-module-viii-post-authorisation-safety-studies-rev-3_en.pdf. (accessed on 23 September 2024).
- Carroll, R.; Ramagopalan, S.V.; Cid-Ruzafa, J.; Lambrelli, D.; McDonald, L. An analysis of characteristics of post-authorisation studies registered on the ENCePP EU PAS Register. F1000Res. 2017, 6, 1447. [Google Scholar] [CrossRef]
- ICH E11(R1) guideline on clinical investigation of medicinal products in the pediatric population - Scientific guideline. Available online: https://www.ema.europa.eu/en/ich-e11r1-guideline-clinical-investigation-medicinal-products-pediatric-population-scientific-guideline (accessed on 8 November 2024).
- Ethical considerations for clinical trials on medicinal products conducted with minors. Recommendations of the expert group on clinical trials for the implementation of Regulation (EU) No 536/2014 on clinical trials on medicinal products for human use. Revision 1. Available online: https://health.ec.europa.eu/system/files/2018-02/2017_09_18_ethical_consid_ct_with_minors_0.pdf. (accessed on 8 November 2024).
- Sultana, J.; Crisafulli, S.; Almas, M.; Ippazio, C.A.; Esme, B.; et al. Overview of the European post-authorisation study register post-authorization studies performed in Europe from September 2010 to December 2018. Pharmacoepidemiol Drug Saf 2022, 31(6), 689–705. [Google Scholar] [CrossRef] [PubMed]
- Maier, W.C.; Christensen, R.A.; Anderson, P. Post-approval Studies for Rare Disease Treatments and Orphan Drugs. In Rare Diseases Epidemiology: Update and Overview, 2nd ed.; Posada De La Paz, M., Taruscio, D., Groft, S.C., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Volume 1031, pp. 197–205. [Google Scholar] [CrossRef]
- Catalogue of RWD studies. Available online: https://catalogues.ema.europa.eu/catalogue-rwd-studies (accessed on 23 September 2024).
- REGULATION (EC) No 1901/2006 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 12 December 2006 on medicinal products for pediatric use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32006R1901 (accessed on 23 September 2024).
- FAQs on the RWD Catalogues, the old EU PAS Register & ENCePP Resource Database. Question 47. Can I register a study that is not conducted in the EU? Available online: https://catalogues.ema.europa.eu/support (accessed on 6 November 2024).
- Subramanian, D.; Cruz, C.V.; Garcia-Bournissen, F. Systematic Review of Early Phase Pediatric Clinical Pharmacology Trials. J Pediatr Pharmacol Ther 2022, 27(7), 609–617. [Google Scholar] [CrossRef] [PubMed]
- Toma, M.; Felisi, M.; Bonifazi, D.; Bonifazi, F.; Giannuzzi, V.; et al. Pediatric Medicines in Europe: The Pediatric Regulation—Is It Time for Reform? Front Med 2021, 8, 593281. [Google Scholar] [CrossRef] [PubMed]
- ENVI POSITION FOR THE NEW PHARMACEUTICAL LEGISLATION: WHAT’S NEW FOR PEDIATRICS? Accessed May 20, 2024 Available online: https://eptri.eu/news/envi-position-for-the-new-pharmaceutical-legislation-whats-new-for-pediatrics (accessed on 23 September 2024). Parliament adopts its position on EU pharmaceutical reform. European Parliament. Available online: https://www.europarl.europa.eu/news/en/press-room/20240408IPR20308/parliament-adopts-its-position-on-eu-pharmaceutical-reform (accessed on 23 September 2024).
Figure 1.
Number of registered PASs by year, among all 165 included PASs. The red line represents the regression line estimated by the model.
Figure 1.
Number of registered PASs by year, among all 165 included PASs. The red line represents the regression line estimated by the model.
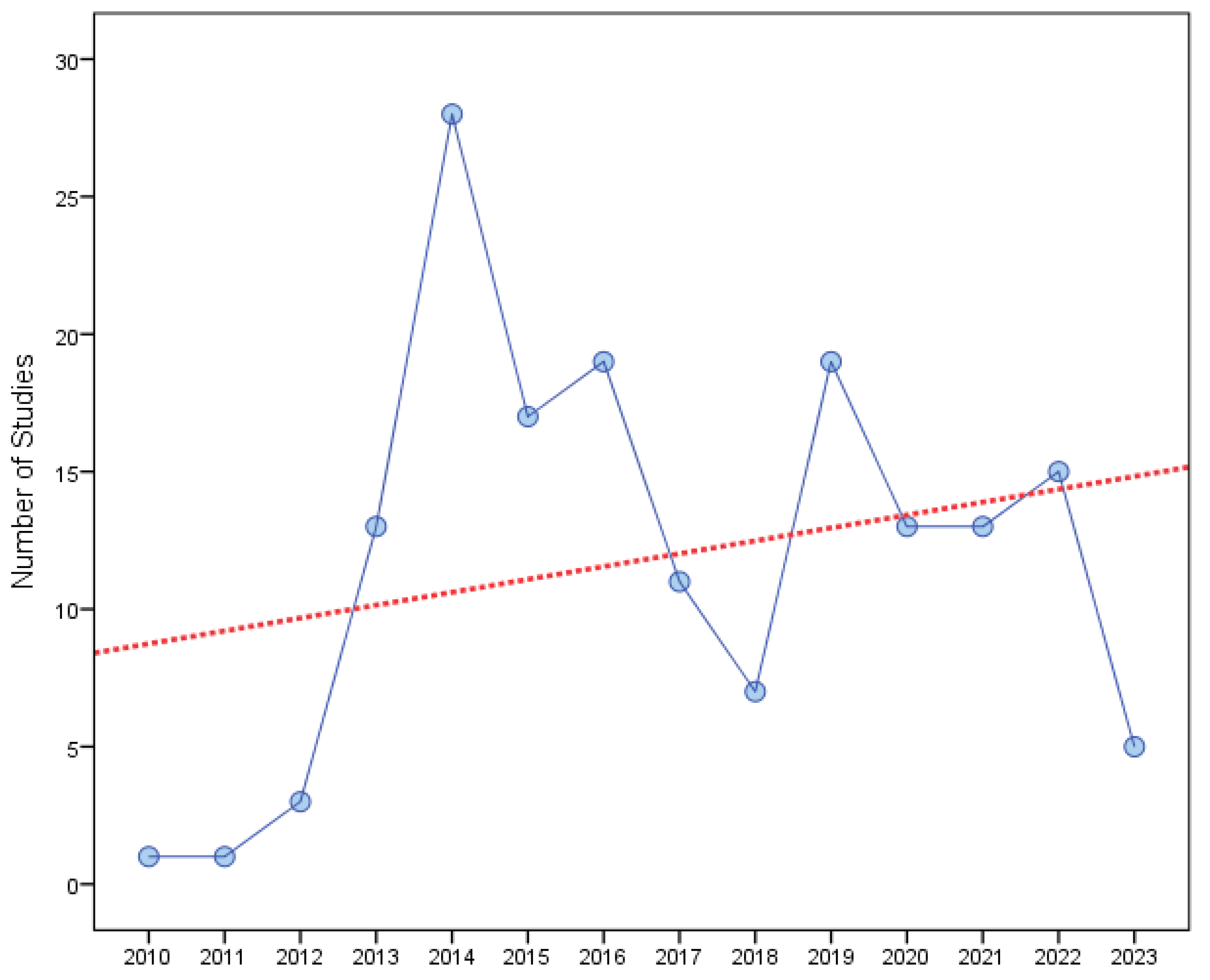
Figure 2.
Number of registered PASs by year and age category, among all 165 included PASs. The colored lines represent the regression lines of the three groups estimated by the model.
Figure 2.
Number of registered PASs by year and age category, among all 165 included PASs. The colored lines represent the regression lines of the three groups estimated by the model.
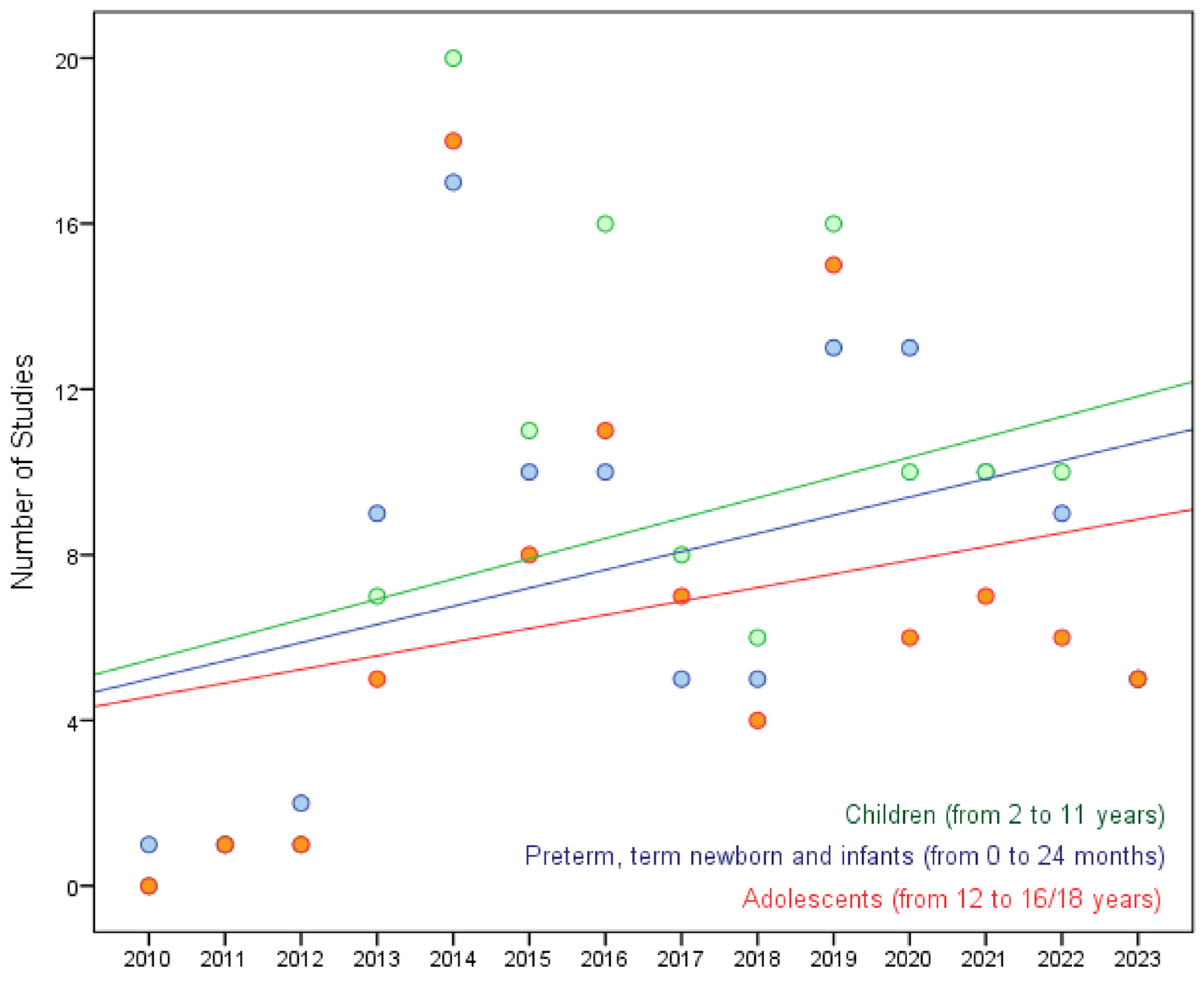

Table 1.
Characteristics of the 165 PASs on pediatric populations registered in the EU PAS Register from its inception to 30th April 2023.
Table 1.
Characteristics of the 165 PASs on pediatric populations registered in the EU PAS Register from its inception to 30th April 2023.
| Studies on pediatric populations registered in the EU PAS Register N= 165 (%) |
|
|---|---|
| Study type | |
| Observational study | 142 (86.1) |
| Survey | 9 (5.5) |
| Review or meta-analysis | 7 (4.2) |
| Clinical trial | 4 (2.4) |
| Other | 3 (1.8) |
| Study design | |
| Descriptive study | 18 (10.9) |
| Cohort studies | 103 (62.4) |
| Cross sectional studies | 8 (4.8) |
| Case control studies | 7 (4.2) |
| Other | 14 (8.5) |
| More than 1 | 13 (8.0) |
| Unknown | 2 (1.2) |
| Status of the Study | |
| Finalized | 95 (57.6) |
| Ongoing | 49 (29.7) |
| Planned | 21 (12.7) |
| Funding Details | |
| Private | 109 (66.1) |
| Public | 45 (27.3) |
| Mixed | 9 (5.5) |
| Unknown | 1 (0.6) |
| Scope of the study* | |
| Risk Assessment | 82 (49.7) |
| Drug Utilization | 39 (23.6) |
| Effectiveness Evaluation | 35 (21.2) |
| Disease Epidemiology | 27 (16.4) |
| Other Scope | 22 (13.3) |
| ENCePP Seal | |
| Yes | 7 (4.2) |
| No | 158 (95.8) |
| Requested by a regulator | |
| Yes | 83 (50.3) |
| No | 80 (48.5) |
| Unknown | 2 (1.2) |
| Country | |
| EU | 120 (72.7) |
| Non-EU | 45 (27.3) |
| RMP status | |
| EU RMP 1 | 12 (7.4) |
| EU RMP 2 | 4 (2.4) |
| EU RMP 3 | 44 (26.6) |
| Non-EU RMP only | 10 (6.1) |
| Unknown | 13 (7.8) |
| Not applicable | 82 (49.7) |
| Data collection** | |
| Primary data | 48 (33.8) |
| Secondary data | 76 (53.2) |
| Mixed | 18 (12.7) |
| Use of reference drug for formal comparison | |
| Yes | 36 (21.8) |
| No | 129 (78.2) |
| Subjects age range*** | |
| Preterm newborn infants | 43 (26.1) |
| Term newborn infants (0 to 27 days) | 71 (43.0) |
| Infants and toddlers (28 days to 23 months) | 102 (61.8) |
| Children (2 to 11 years) | 121 (73.3) |
| Adolescents 12 to 16/18 ₸ | 94 (57.0) |
* Each study could have more than one scope category; hence, the scope categories were not mutually exclusive. ** The data collection variable was calculated only with reference to the 142 observational studies. *** Each study could have more than one category of age range. ₸ The adolescent upper age limit depends on the region.
Table 2.
Number and percentage of registered PASs by year, among all 165 included PASs.
| Year of study registration | Frequency | Percent | |
|---|---|---|---|
| 2010 | 1 | .6 | |
| 2011 | 1 | .6 | |
| 2012 | 3 | 1.8 | |
| 2013 | 13 | 7.9 | |
| 2014 | 28 | 17.0 | |
| 2015 | 17 | 10.3 | |
| 2016 | 19 | 11.5 | |
| 2017 | 11 | 6.7 | |
| 2018 | 7 | 4.2 | |
| 2019 | 19 | 11.5 | |
| 2020 | 13 | 7.9 | |
| 2021 | 13 | 7.9 | |
| 2022 | 15 | 9.1 | |
| 2023 | 5 | 3.0 | |
| Total | 165 | 100.0 | |
Table 3.
Number and percentage of registered PASs by funding source, among all 165 included PASs.
| Funding Source | Frequency | Percent | |
|---|---|---|---|
| Pharmaceutical companies | 103 | 62.4 | |
| Government body | 25 | 15.2 | |
| EU funding scheme | 5 | 3.0 | |
| Research councils | 1 | .6 | |
| Other* | 16 | 9.7 | |
| More than one | 14 | 8.5 | |
| Unknown | 1 | .6 | |
| Total | 165 | 100.0 | |
* The ’other’ category included mainly universities, hospitals and national grants as sources of funding.
Table 4.
Number and percentage of registered PASs by funding source and year, among 126 PASs funded by pharmaceutical companies or government body.
Table 4.
Number and percentage of registered PASs by funding source and year, among 126 PASs funded by pharmaceutical companies or government body.
| Year of study registration | Pharmaceutical Companies |
Government Body |
||
|---|---|---|---|---|
| Count | % | Count | % | |
| 2011 | 0 | 0.0 | 0 | 0.0 |
| 2012 | 0 | 0.0 | 0 | 0.0 |
| 2013 | 3 | 27.3 | 3 | 27.3 |
| 2014 | 24 | 85.7 | 2 | 7.1 |
| 2015 | 9 | 52.9 | 4 | 23.5 |
| 2016 | 12 | 66.7 | 3 | 16.7 |
| 2017 | 7 | 63.6 | 3 | 27.3 |
| 2018 | 4 | 66.7 | 0 | 0.0 |
| 2019 | 12 | 80.0 | 3 | 20.0 |
| 2020 | 9 | 81.8 | 1 | 9.1 |
| 2021 | 9 | 69.2 | 1 | 7.7 |
| 2022 | 9 | 64.3 | 3 | 21.4 |
| 2023 | 5 | 100.0 | 0 | 0.0 |
| Total | 103 | 68.2 | 23 | 15.2 |
Table 5.
Number and percentage of registered PASs by therapeutic area, among all 165 included PASs.
| Therapeutic area | Frequency | Percent | |
|---|---|---|---|
| A: Alimentary tract and metabolism | 6 | 3.6 | |
| B: Blood and blood forming organs | 8 | 4.8 | |
| C: Cardiovascular system | 3 | 1.8 | |
| D: Dermatologicals | 1 | .6 | |
| H: Systemic hormonal preparation, excluding sex hormones and insulin | 9 | 5.5 | |
| J: Anti-infective for systemic uses | 41 | 24.8 | |
| L: Antineoplastic and immunomodulating agents | 24 | 14.5 | |
| M: Muscolo-skeletal system | 2 | 1.2 | |
| N: Nervous system | 29 | 17.6 | |
| P: Antiparasitic products, insecticides and repellents | 1 | .6 | |
| R: Respiratory system | 9 | 5.5 | |
| S: Sensory organs | 1 | .6 | |
| V: Various | 6 | 3.6 | |
| Others* | 25 | 15.2 | |
| Total | 165 | 100.0 | |
*The ’others’ category mainly included the subcategories “more than one therapeutic area,” “unknown therapeutic area,” and “not applicable”.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



