Submitted:
06 December 2024
Posted:
09 December 2024
You are already at the latest version
Abstract
The mRNA technology can replace the expensive recombinant technology for every type of protein, making biological drugs more affordable. It can also expedite the entry of new biological drugs, and the copies of approved mRNA products can be treated as generic or biosimilar products due to their chemical nature. Hundreds of new protein drugs have been blocked due to the high cost of recombinant development. The low CAPEX and OPEX associated with mRNA technology bring it within the reach of developing countries currently deprived of lifesaving biological drugs. In this paper, we advise developers to introduce novel proteins, switch recombinant manufacturing to mRNA delivery, and further advise the regulatory authorities to allow approval of copies of mRNA products with less testing. We anticipate that mRNA technology will make protein drugs like natural and engineered proteins, monoclonal antibodies, and vaccines accessible to billions of patients worldwide.
Keywords:
1. Introduction
2. Protein Drugs
3. Recombinant Technology
4. mRNA Technology
4.1. Developing mRNA Protein Delivery
4.2. mRNA Purification and Quality Control
4.3. Storage and Cold Chain Management
4.4. Lipid Compositions and Ligand Targeting
5. Recombinant vs Ribosomal
- Eliminating Protein Production: Traditional recombinant protein production requires creating, optimizing, and scaling up cell lines to produce the protein. mRNA eliminates this process, as mRNA directly encodes the protein of interest, which is then made within the patient's cells. This reduces both preclinical and manufacturing timelines.
- Simplified Manufacturing: mRNA synthesis is more straightforward and faster than protein production, as it involves in vitro transcription, which can be completed within weeks. In contrast, recombinant proteins require complex bioreactors, extensive purification, and scaling-up processes, which can take months or even years.
- Rapid Design and Iteration: mRNA sequences can be quickly modified if adjustments are needed, such as changes in dose or specific protein regions. This adaptability is especially valuable in early development, allowing researchers to iterate quickly without lengthy cell line adjustments.
- Streamlined Preclinical Testing: mRNA drugs, mainly vaccines, may bypass some traditional preclinical studies, as they have a different safety profile than recombinant proteins. The risk of toxicity is generally lower, as mRNA does not integrate into the genome and is rapidly degraded after the protein is expressed.
- Potential for Faster Clinical Trials: mRNA's faster production enables quicker scale-up for clinical trials, reducing the wait time between phases. mRNA therapies also tend to elicit a robust immune response, which may shorten dose-ranging and efficacy assessment stages, especially in vaccines.
- Regulatory Pathways and Accelerated Approval: The success of COVID-19 mRNA vaccines has led to new regulatory insights and expedited review pathways, potentially offering shorter approval timelines for future mRNA-based therapeutics.
- Estimated Time Reduction: MRNA technology can potentially reduce the typical drug development timeline from around 10–15 years to as short as 5–8 years in some cases, depending on the disease and the regulatory pathway chosen.
- Lower Manufacturing Costs: mRNA production uses more straightforward and more scalable processes. Traditional recombinant proteins require costly mammalian or microbial cell culture systems, which are resource-intensive and involve complex purification steps. In contrast, mRNA synthesis can be done rapidly in vitro, cutting manufacturing costs by approximately 30-40%.
- Reduced Infrastructure and Facility Requirements: Recombinant protein production often demands specialized bioreactors, sterile environments, and stringent quality controls. In contrast, mRNA manufacturing can use more straightforward equipment and smaller facilities, reducing fixed costs and the capital investment needed for facilities.
- Streamlined Preclinical and Clinical Phases: mRNA's flexibility reduces the need for extended preclinical testing and enables faster transition through clinical trials. This can lower costs associated with maintaining and managing trials, reducing expenses for staffing, data monitoring, and patient recruitment.
- Less Batch Variability and Simplified Quality Control: Each batch is less variable with mRNA since it's made through a predictable in vitro transcription process. This contrasts with protein products, which vary depending on cell line behavior and production conditions. Reduced variability translates to less waste and fewer quality control expenditures.
- Reduced R&D Costs Through Rapid Iteration: Because mRNA sequences are easily adjustable, early-stage development costs drop as researchers can adjust sequences without creating new cell lines. This makes R&D less expensive, especially in preclinical phases, as modifications to dose or protein regions don’t involve extensive re-optimization.
- Faster Development Timeline: Producing mAbs typically involves generating stable cell lines, optimizing them, and scaling production. This process can take years. With mRNA, the sequence coding for the mAb can be synthesized and delivered directly to the body’s cells, which then produce the antibody. This approach skips cell line development, reducing early-stage development by up to 50%. Additionally, mRNA-encoding mAbs can be optimized and modified more rapidly than protein-based mAbs, allowing faster adjustments and fewer delays if dose modifications or structural changes are needed.
- Lower Production Costs: Traditional mAb production is complex, requiring costly cell culture facilities, extensive purification processes, and quality control. mRNA bypasses these since manufacturing involves producing mRNA, which is more straightforward and can be done in smaller facilities. mRNA is highly scalable, allowing manufacturers to adjust production quickly to meet demand without needing major infrastructure expansion, which is costly in conventional mAb production. This reduces batch-production costs significantly. Producing mRNA with consistent quality is often simpler than ensuring consistency in mAb production across batches, especially in complex systems like CHO cells.
- Reduced R&D Costs: The mRNA platform is highly adaptable, allowing rapid iteration with lower costs if changes are required. For mAbs, adjustments to the mRNA sequence do not require creating new cell lines, thus eliminating associated time and expense. Since mRNA allows in vivo expression of mAbs, early testing can begin faster and at potentially lower costs.
- Speed of Regulatory Approval: The FDA has become increasingly familiar with mRNA technology thanks to recent approvals and accelerated pathways. For mAb-based treatments, this familiarity could result in quicker regulatory processes if safety profiles are favorable, though this is still a developing area. For novel mAbs, the use of mRNA can cut development costs and timelines even more than typical mRNA-based proteins. Costs could be reduced by over half compared to conventional mAb production methods, with timelines potentially shortened from 8–12 years to as little as 4–7 years for specific applications, especially if regulatory bodies offer expedited paths for mRNA-based mAbs.
6. New Protein Drugs
- Adipocyte Fatty Acid-Binding Protein (AFABP) transports fatty acids within adipose tissue. It may help regulate fat metabolism, with targeting AFABP proposed as a potential strategy to reduce fat storage and enhance fat mobilization [56].
- AMP-Activated Protein Kinase (AMPK) is an energy-sensing enzyme that promotes autophagy, reduces inflammation, and prevents age-related diseases; it can be activated through diet, exercise, or compounds like metformin [57].
- Amyloid Precursor Protein (APP) is essential for neuron health, though improper processing can lead to Alzheimer’s; maintaining normal APP levels while preventing harmful byproducts is a research focus [58].
- Brain-derived neurotrophic Factor (BDNF) is crucial for synaptic plasticity, and its deficiency is linked to learning deficits and cognitive decline, with therapeutic approaches aimed at increasing BDNF levels showing promise. Nerve Growth Factor (NGF) supports neuron survival, with NGF gene therapy being explored for Alzheimer’s disease [59].
- Calcium/Calmodulin-Dependent Protein Kinase II (CaMKII) is essential for memory processes, and targeting CaMKII signaling through therapy may support cognition. CREB (cAMP Response Element-Binding Protein) regulates memory consolidation, and therapies enhancing CREB may benefit learning in neurodegenerative conditions [60].
- Collagen: Collagen is a structural protein in connective tissues. Its extensive post-translational modifications, such as hydroxylation and glycosylation, are challenging to replicate in recombinant systems. While recombinant collagen-like proteins are produced, they may not fully mimic the properties of native collagen [61].
- Elastin: Elastin provides elasticity to tissues like skin and blood vessels. Recombinant production is complicated by its repetitive sequences and cross-linking requirements. Some elastin-like polypeptides are synthesized recombinantly but may not fully replicate native elastin's properties [62].
- Fibroblast Growth Factor (FGF) supports tissue repair, blood vessel formation, and cell survival in heart tissue [63].
- Forkhead Box O (FOXO) proteins, such as FOXO3, regulate cell survival, stress resistance, and metabolism, enhancing antioxidant defenses and DNA repair, with FOXO3 associated with longer lifespans [64].
- Fragile X Mental Retardation Protein (FMRP) is essential for cognitive development, with therapies targeting its affected pathways in intellectual disabilities [65].
- Heat Shock Proteins (HSPs), particularly HSP70, protect other proteins from damage by refolding misfolded proteins, preventing aggregation associated with Alzheimer’s [66].
- Irisin, a protein produced by muscles during exercise, is believed to help convert white fat, which stores energy, into brown fat, which burns energy in a process called “browning,” potentially increasing energy expenditure and reducing fat stores [67].
- Keratin is a structural protein in hair, nails, and skin. Its insolubility and tendency to form strong disulfide bonds make recombinant production challenging. Recombinant keratin-like proteins have been developed, but they may not fully replicate the characteristics of native keratin [68].
- Klotho, a protein associated with lifespan extension, regulates calcium and phosphate metabolism, supports kidney and cardiovascular health, and reduces inflammation, with higher levels linked to improved brain function [69].
- Leptin is a hormone that regulates energy balance by signaling the brain to reduce appetite when fat stores are adequate, potentially promoting fat burning in individuals with leptin sensitivity [70].
- Lipoprotein Lipase (LPL) breaks down triglycerides in the bloodstream into free fatty acids, which can be used for energy or stored in fat cells. [71].
- Myosin: Myosin is a motor protein complex essential for muscle contraction. Its large size and complex assembly make recombinant production challenging. While some subunits or fragments are available, full-length functional myosin is less commonly produced recombinantly [72].
- NAD+ is used for cellular energy production and repair, with NAD+ levels declining with age [73].
- Neuregulin-1, essential for brain development, supports cognitive health, particularly schizophrenia [74].
- Oxytocin receptors influence social learning, and oxytocin treatments are explored for autism [75].
- p53, known as the “guardian of the genome,” prevents DNA damage by regulating cell cycle and apoptosis, reducing cancer risk and supporting longevity [76].
- Platelet-derived growth Factor (PDGF), which recruits fibroblasts and smooth muscle cells for wound healing and scar formation [77];
- Reelin, vital for brain development, is linked to autism and schizophrenia, with signaling modulation as a potential treatment [78].
- SIRT1 activates pathways for stress protection, DNA repair, and antioxidant defenses, while SIRT3, located in mitochondria, regulates energy production and protects against oxidative stress [79].
- Synaptic proteins like PSD-95 and Synapsin are fundamental to memory, while GABA and glutamate receptor imbalances relate to intellectual disabilities and neurological disorders [80].
- Tubulin: Tubulin forms microtubules, crucial for cell structure and division. Recombinant expression of tubulin is complex due to its tendency to form aggregates and the need for specific chaperones for proper folding. Native tubulin is often purified from natural sources for research purposes [81].
- Vascular endothelial growth factor (VEGF) promotes nutrient and oxygen supply, which is crucial for healing, especially in muscle, bone, or skin injuries [82].
Regulatory Guidelines
7. Recombinant Switchover
- Designing the mRNA Sequence for the Biosimilar Protein: Sequence Fidelity: The mRNA sequence should encode the exact amino acid sequence of the approved biosimilar protein. This requires designing the mRNA with codon optimization for efficient translation in the target cells (e.g., human cells), which helps achieve high expression levels [85].
- 5’ and 3’ Untranslated Regions (UTRs): Optimized UTRs can enhance mRNA stability and translational efficiency. Choosing UTRs that increase translation in specific tissues or cells can help achieve targeted protein delivery. Codon-optimized mRNA constructs have significantly improved translation rates for proteins like erythropoietin, commonly delivered as a recombinant protein.
- Modified Nucleosides: Incorporating modified nucleosides such as pseudouridine and 1-methyl pseudouridine into the mRNA can reduce immune recognition while increasing stability and translational efficiency [2].
- Poly(A) Tail and Cap Structure: A cap structure (e.g., Cap 1) and a poly(A) tail increase mRNA stability and translation, enhancing the duration and consistency of protein production.
- LNP Formulation: Encapsulating mRNA in lipid nanoparticles (LNPs) protects it from degradation in the bloodstream, facilitates cellular uptake, and promotes endosomal escape, enabling the mRNA to reach the cytoplasm, which can be translated into protein [86].
- Demonstrating Biosimilarity: To meet biosimilar standards, the mRNA-produced protein must demonstrate similarity in structure, function, and therapeutic efficacy to the original recombinant protein. This often involves comparative testing, including in vitro and in vivo assays for pharmacokinetics, pharmacodynamics, immunogenicity, and stability.
- FDA and EMA Requirements: Regulatory agencies require evidence that the mRNA-delivered protein matches the reference product regarding potency and safety, which may include animal studies and clinical trials.
- Applications: mRNA delivery is under investigation for therapeutic proteins that traditionally require regular injections, such as monoclonal antibodies, clotting factors, and enzymes for genetic diseases. This approach aims to enable endogenous protein production, potentially reducing dosing frequency.
- Clinical Example: mRNA-based delivery of Factor VIII for hemophilia is being studied as a long-lasting alternative to recombinant protein infusions [87,88]. This approach to delivering biosimilar proteins via mRNA is in its early stages, but it holds promise for making therapeutic protein delivery more effective and patient-friendly. The development process includes complex regulatory and manufacturing considerations to ensure that the therapeutic effect and safety of the mRNA-delivered protein match those of the approved recombinant versions.
Regulatory Guideline
- Animal toxicology studies will be required to establish the product's safety due to its LNP formulation; if the developer uses a composition that the FDA has approved, the extent of nonclinical studies will be reduced but not removed as proof of safety is required. Regardless of whether only the subcutaneous route administers the product, intravenous research will also be necessary. Table 2 lists a proposed animal toxicology plan.
- Protein yield calculations will be made from animal expression studies that can be satisfied with rodent species since their expression is higher than in humans. Notably, a single mRNA molecule can yield hundreds or even thousands of protein molecules depending on mRNA stability and functionality. Based on animal conversion data, an early phase 0 study in humans will be required to confirm the mRNA dose-protein yield profile.
- A short phase II study with 4-6 subjects will establish a dose-response relationship and the pharmacokinetic disposition profile. These profiles are expected to not match well due to the inevitable difference in the absorption half-life and yield appearance. However, the FDA will at least require a comparison of the total AUC to relate it to the total dose reaching the body. Notably, the developer requests the same indications, requiring this equivalence to be established.
- A phase III or comparative efficacy study will likely not be required, but this waiver will require the developer to present a scientific argument to the FDA.
- The FDA will approve the plasmid design and construction, and its tests may include, as shown in Table 3)

| Animal Species | Mouse |
|---|---|
| Duration in-life | 28 days to 3 months |
| Administration | Repeated, subcutaneous |
| Dosage level | 2 |
| Groups | 2: saline control, dose group |
| Group size | 20 |
| Total Animals | 40 |
| Monitoring | Mortality, body weight, clinical observation, food consumption, local reaction, body temperature, FOB (mod. Irwin), hematology, clinical chemistry, urinalysis, bone marrow smear, blood coagulation parameters, cytokines TNF-alpha, IFN-gamma, IL-10, etc. |
| Post mortem | Necropsy and weight of selected organs; histopathological evaluation, RNA biodistribution |
| Duration | 3 months |
| Test | Method | Specification |
|---|---|---|
| Identity | Restriction Enzyme Analysis with Agarose Gel Electrophoresis | Consistent with the reference standard |
| Plasmid Identity | Sanger Sequencing | Matches reference standard |
| Purity | A260/A280 | 1.8 – 2.0 |
| Concentration | A260 | Default 1 mg/mL |
| Residual Host Cell Protein | HCP ELISA | < 1% |
| Residual Host Genomic DNA | Quantitative PCR | < 5% |
| Residual Host RNA | Agarose Gel Electrophoresis | Non-detectable at 200 ng |
| Endotoxin | Quantitative LAL assay | < 0.01 EU/µg |
| Bioburden | USP <61> | No growth after 48 hours |
8. “Biosimilar” mRNA
- Comirnaty (BNT162b2), developed by Pfizer-BioNTech, became the first mRNA vaccine approved for COVID-19 prevention in August 2021, marking a significant milestone in mRNA vaccine technology with FDA approval. Shortly after, Spikevax (mRNA-1273) by Moderna was approved in December 2020 as the second mRNA vaccine for COVID-19 prevention, further solidifying the role of mRNA vaccines in the pandemic response. In May 2024, Moderna also introduced mRESVIA (mRNA-1345), the first mRNA vaccine approved specifically for preventing Respiratory Syncytial Virus (RSV) in adults aged 60 and over, expanding mRNA applications beyond COVID-19 [93].
- Moderna, Inc. has a robust pipeline of mRNA-based vaccines and therapeutics across multiple areas. For infectious diseases, it includes the approved COVID-19 Vaccine (mRNA-1273), marketed as Spikevax®, a next-generation COVID-19 Vaccine (mRNA-1283) designed for more accessible storage and administration, and seasonal influenza vaccines (mRNA-1010, mRNA-1020, mRNA-1030) targeting multiple influenza strains. Moderna is also developing an RSV Vaccine (mRNA-1345) for older adults and combination vaccines, such as the flu and COVID-19 vaccine (mRNA-1083) and a triple vaccine targeting flu, COVID-19, and RSV (mRNA-1230). In oncology, Moderna collaborates with Merck on Individualized Neoantigen Therapy (INT) (mRNA-4157), a personalized cancer vaccine targeting melanoma and other cancers. Additionally, for rare diseases, Moderna is developing therapies for Propionic Acidemia (mRNA-3927) and Methylmalonic Acidemia (mRNA-3705) (wwww.moderna.com).
- BioNTech SE, widely recognized for its COVID-19 vaccine developed with Pfizer, is advancing its pipeline with mRNA therapies for infectious diseases, such as the influenza vaccine BNT161 and a shingles vaccine (BNT163), also developed with Pfizer. BioNTech's oncology portfolio includes FixVac (BNT111), an mRNA vaccine targeting advanced melanoma, and the Individualized Neoantigen Specific Immunotherapy (iNeST) (BNT122), developed in collaboration with Genentech (www.biointech.com).
- CureVac N.V. is developing mRNA-based vaccines and therapeutics, including a second-generation COVID-19 vaccine (CVnCoV) and a rabies vaccine (CV7202) for infectious diseases. It also explores mRNA-based cancer vaccines targeting various tumors (www.curevac.com).
- Translate Bio, now part of Sanofi, focuses on mRNA therapeutics, including influenza and COVID-19 vaccines and an inhaled mRNA therapy for cystic fibrosis (MRT5005) (www.translatebio.com).
- Arcturus Therapeutics is developing self-amplifying mRNA vaccines, such as a COVID-19 vaccine (ARCT-021) and an influenza vaccine, as well as mRNA therapy for Ornithine Transcarbamylase (OTC) Deficiency (ARCT-810), a urea cycle disorder (https://arcturusrx.com).
- Gritstone bio is working on mRNA-based cancer immunotherapies, including Granite (SLATE), personalized immunotherapy for solid tumors, and the CORAL program, which develops mRNA vaccines for infectious diseases, including COVID-19 (https://gritstonebio.com)
- eTheRNA Immunotherapies focuses on mRNA-based immunotherapies, specifically TriMix-Based Cancer Vaccines designed to stimulate immune responses against tumors (https://www.etherna.be).
- Imperial College London is advancing a self-amplifying RNA (saRNA) vaccine platform, including a saRNA COVID-19 vaccine targeting SARS-CoV-2 https://www.imperial.ac.uk).
- Chimeron Bio is developing mRNA therapies with OncoRNA to target solid tumors and various infectious disease vaccines. Providence Therapeutics is advancing mRNA vaccines, including PTX-COVID19-B, an mRNA COVID-19 vaccine candidate targeting SARS-CoV-2, and personalized cancer vaccines (https://www.chimeron.com).
8.1. Scenario 1
8.2. Scenario 2
8.3. Scenario 3
- Dose Adjustment Based on AUC: If the AUC differs, adjusting the dose to match the original product’s AUC can help achieve comparable exposure over time, potentially leading to similar clinical efficacy.
- Peak and Duration Adjustments: Differences in C_max or half-life may require adjusting the frequency or amount of dosing. For instance, if the modified mRNA degrades faster, more frequent dosing or higher doses might be necessary to maintain therapeutic levels.
- Fine-tuning with Therapeutic Drug Monitoring: If achieving an exact match is challenging, therapeutic drug monitoring (TDM) could be employed to individualize dosing in response to observed PK variability, ensuring patients reach the intended therapeutic range.
8.4. Scenario 4
8.5. Toxicology Comparison with Identical LNP Formulation
- In Vitro Assays: Start with in vitro toxicity assays to assess cellular toxicity and immune responses. Cytotoxicity, cellular uptake, and inflammatory marker assays (e.g., cytokine release profiles) can highlight any subtle differences in toxicity due to minor sequence variations.
- Acute and Chronic Toxicity Studies: Conduct acute and chronic toxicity studies in animal models to observe any short-term or long-term toxicological impacts. Monitor biomarkers, organ weights, and histopathology to identify potential differences.
- Biodistribution and Target Organ Toxicity: With identical LNPs, differences in biodistribution are less likely; however, conducting biodistribution studies using labeled mRNA or LNPs can confirm this. Focus mainly on target organ toxicity (such as liver and spleen, where LNPs tend to accumulate) to assess any subtle toxicological effects.
- Comparative Immune Response: After administration, assess immune activation markers (e.g., interferons, cytokines). Even with identical LNPs, slight mRNA sequence differences can alter immune response intensity or duration.
8.6. Toxicology Comparison with Different LNP Formulations
- Comparative PK and PD Studies: Begin with PK and pharmacodynamic (PD) comparisons to understand how differences in LNP formulations affect mRNA biodistribution and protein expression. Different LNP formulations may alter tissue targeting, clearance rates, and protein exposure levels.
- Immune Response Profiling: Different LNP formulations will likely elicit varied immune responses. If applicable, use immune profiling assays to measure systemic immune markers, such as cytokines (IL-6, TNF-α) and adaptive immune responses. This will help assess any LNP-induced immunogenicity.
- In Vivo Toxicology Studies with Emphasis on LNP-Sensitive Organs: In vivo studies should assess organs where LNPs commonly accumulate (e.g., liver, spleen, lymph nodes). Detailed histopathological analysis and liver enzyme assays (e.g., ALT, AST) are critical to detect potential hepatotoxicity or immune cell infiltration in these organs.
- Comparative Inflammatory and Complement Activation Studies: Different LNP formulations can varying degrees, activate the complement system or inflammatory pathways. In vivo and ex vivo assays, such as the hemolysis assay for complement activation and inflammatory biomarker panels, will help detect LNP-specific immune and inflammatory responses.
- Repeat-Dose Toxicity Studies: Conduct repeat-dose toxicity studies in relevant animal models to evaluate cumulative toxic effects, which may be more pronounced with different LNP formulations. Monitoring clinical signs, body weight, hematology, and organ function over time will help identify chronic toxicity risks.
8.7. Manufacturing Quality and Consistency
8.8. Structural Characterization
8.9. Stability Testing
8.10. Functional Testing
8.11. Immunogenicity Assessment
8.12. Comparative Analytical Testing and Documentation
9. Proposed Guideline
9.1. Quality by Design (QbD) and Manufacturing Controls
- Process Development and Control: Following a QbD approach, ensure the manufacturing process is consistent and capable of producing high-quality mRNA with controlled sequence, structure, and modifications. Key aspects include:
- Template DNA Quality: DNA template purity is essential as it determines mRNA sequence fidelity.
- In Vitro Transcription (IVT) Consistency: Controls on enzyme concentrations, nucleotide substrates, and buffer systems to achieve consistent transcription.
- Modified Nucleotides: Validation is required to confirm their precise incorporation if modified nucleotides (e.g., pseudouridine) are used.
- Good Manufacturing Practices (GMP): Ensure GMP compliance, including aseptic processing for sterility and safety. This encompasses monitoring environmental conditions to protect RNA from degradation.
- Lipid Nanoparticle (LNP) Encapsulation: If using LNPs, specify particle size, encapsulation efficiency, and homogeneity.
9.2. Structural Characterization
- Sequence Identity: Use high-throughput RNA sequencing to verify that the mRNA sequence matches the reference sequence.
- Secondary Structure Verification: Secondary structures like hairpins can influence translation efficiency and stability. Techniques such as Selective 2’-Hydroxyl Acylation analyzed by Primer Extension (SHAPE) or cryo-electron microscopy can validate these structures.
- Modified Nucleotide Analysis: Confirm the presence, location, and ratio of modified nucleotides using mass spectrometry or high-performance liquid chromatography (HPLC) to match the reference profile.
9.3. Stability Testing
- Accelerated and Real-Time Stability: Conduct stability studies under accelerated (e.g., 40°C, 75% RH) and real-time storage conditions to define shelf life. Real-time studies are essential to simulate actual storage conditions (e.g., -20°C).
- Thermal Cycling Studies: Test stability under repeated freezing and thawing cycles to mimic standard handling practices.
- Forced Degradation Studies: Subject the mRNA to stress conditions such as extreme pH, oxidative stress, and light exposure to identify degradation pathways and establish degradation products.
- LNP Stability: For mRNA in LNPs, monitor particle size and zeta potential over time. Use dynamic light scattering (DLS) or transmission electron microscopy (TEM) for LNP integrity analysis.
9.4. Functional Testing
- In Vitro Translation Efficiency: Conduct in vitro translation assays to ensure the mRNA product consistently translates into the desired protein with expected yield and activity.
- In Vivo Protein Expression and PK Studies: Perform animal studies to compare in vivo protein expression levels and pharmacokinetics (PK) with the reference mRNA product. This should include protein quantification and assessment of distribution, metabolism, and excretion.
- Bioactivity of Expressed Protein: Confirm that the protein expressed from the mRNA has comparable bioactivity to the reference product, using cell-based or biochemical assays to match therapeutic efficacy.
9.5. Immunogenicity Assessment
- Innate Immune Activation: Assess immune activation potential, especially if using unmodified mRNA. Testing can involve cytokine release assays or toll-like receptor (TLR) assays to evaluate the activation of the innate immune response.
- Adaptive Immune Response: Evaluate the immunogenicity of the expressed protein, primarily if the therapeutic mRNA is intended for repeated administration. Animal studies and in vitro assays can provide preliminary data, though clinical testing is ultimately required.
- Anti-mRNA Antibody Formation: If patients receive multiple doses, assess the risk of anti-drug antibodies (ADA) against the mRNA or the lipid nanoparticles, as these could impact product efficacy or safety.
9.6. Comparative Analytical Testing (Drawing from Biosimilar Guidelines)
- Comparability Protocol: Create a side-by-side analytical comparison with the reference product, covering:
- Purity and Impurities: Use capillary electrophoresis, HPLC, and other analytical methods to quantify impurities, such as truncated RNA and double-stranded RNA by-products.
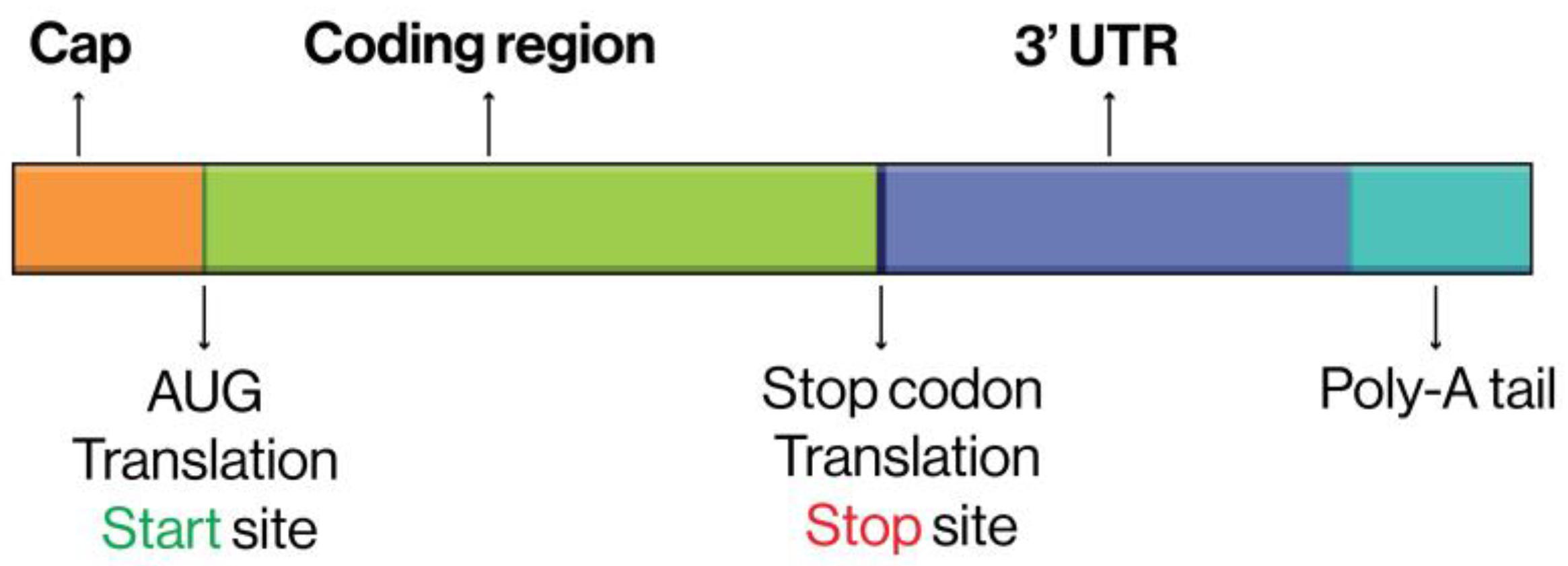
- 5’ Cap and 3’ Poly(A) Tail Length: Confirm that the mRNA’s 5’ cap structure and poly(A) tail length are consistent with the reference, as these impact translation initiation and stability.
- Mass and Charge Profile: Analyze the molecular mass and charge distribution to detect any variations in mRNA composition.
9.7. Additional Clinical Studies for Safety and Efficacy
- Pharmacodynamics (PD) and PK Matching: Conduct clinical PK and PD studies in human volunteers to confirm that the mRNA product has bioavailability and therapeutic efficacy similar to the reference.
- Comparative Safety Assessment: Include safety assessments for single-dose and repeated administrations, especially monitoring for differences in immune response.
9.8. Documentation and Regulatory Compliance
- Detailed Documentation: Compile all testing data, including batch records, stability reports, and clinical trial data, by regulatory requirements.
- Risk Management: Follow risk assessment protocols to identify and mitigate potential risks unique to mRNAs, such as enzymatic degradation or immune activation.
| Testing Category | mRNA-Specific Considerations | Biosimilar Guideline Parallels |
|---|---|---|
| Sequence Identity | Complete sequence and modified nucleotides | Amino acid sequence identity |
| Secondary Structure | SHAPE and cryo-EM for folding patterns | Protein folding and glycosylation |
| Stability Testing | Thermal cycling, forced degradation | Real-time, accelerated, forced degradation |
| Functional Testing | In vitro translation, in vivo protein expression | Cell-based assays for protein activity |
| Immunogenicity | Innate and adaptive response assessments | Anti-drug antibody and immune response testing |
| PK/PD Clinical Testing | Protein expression PK and therapeutic bioactivity | PK/PD comparison in clinical studies |
10. Naming
11. Intellectual Property
- Patisiran (Onpattro) Approval Year: 2018. Treatment of polyneuropathy caused by hereditary transthyretin-mediated amyloidosis (hATTR). Utilizes LNPs to deliver small interfering RNA (siRNA) targeting the transthyretin (TTR) gene, reducing the production of the TTR protein.
- Pfizer-BioNTech's Comirnaty and Moderna's Spikevax: Approval Year: 2020. Indication: Prevention of COVID-19. Employ LNPs to encapsulate messenger RNA (mRNA) encoding the SARS-CoV-2 spike protein, facilitating cellular uptake and subsequent immune response.
- Moderna's RSV Vaccine (mRESVIA): Approval Year: 202. Prevention of respiratory syncytial virus (RSV) in adults aged 60 and older. Uses LNPs to deliver mRNA encoding RSV antigens, stimulating an immune response against the virus.
- Lipid nanoparticle (LNP) formulations are widely used for delivering therapeutic agents, particularly nucleic acids like mRNA and siRNA. While patents protect many LNP technologies, some formulations are no longer under patent protection and are considered patent-free. These include early-generation LNPs and particular naturally occurring lipid-based delivery systems.
-
Early LNP formulations, developed in the 1990s and early 2000s, often utilized cationic lipids such as DOTMA (N-[1-(2,3-dioleyloxy)propyl]-N, N,N-trimethylammonium chloride) and DOPE (dioleoylphosphatidylethanolamine). Many of these early patents have expired, rendering these formulations patent-free.Certain lipid-based delivery systems, like those using naturally occurring lipids without proprietary modifications, may not be covered by active patents. However, their efficacy and stability might be limited compared to more advanced, patented LNP technologies. This lower efficiency is significant for vaccines but not as much when used to express functional proteins. Other formulations with expired patents utilized in FDA-approved products make a great choice. For example, the original lipid nanoparticle (LNP) formulation used by Onpattro (patisiran) during its development and early stages involved a specific combination of lipids optimized to deliver small interfering RNA (siRNA) efficiently. The primary components included:
- Ionizable Lipid: DLin-MC3-DMA: DLin-MC3-DMA was the critical ionizable lipid in Onpattro's formulation, specifically designed to become positively charged in the acidic environment of endosomes, which aids in the release of the siRNA into the cell's cytoplasm.
- Phospholipid: DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine). DSPC is a phospholipid that provides structural stability to the lipid nanoparticle and supports its interaction with cell membranes.
- Cholesterol: Cholesterol enhances the fluidity and stability of the LNP, improving its integrity and ability to circulate in the bloodstream.
- Polyethylene Glycol (PEG)-Lipid: DMG-PEG2000 (1,2-dimyristoyl-rac-glycero-3-methoxy polyethylene glycol-2000)
12. Conclusions
Funding
Conflicts of Interest
References
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhong, Y.; Wang, X.; Shen, J.; An, W. Advances in Circular RNA and Its Applications. Int J Med Sci. 2022, 19, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Glass, Z.; Huang, M.; Chen, Z.Y.; Xu, Q. Ex vivo cell-based CRISPR/Cas9 genome editing for therapeutic applications. Biomaterials 2020, 234, 119711. [Google Scholar] [CrossRef]
- Government, U. Clinical Trials with term “mRNA” https://clinicaltrials.gov/search?intr=mRNA2024 [.
- Requena, J.R. The protean prion protein. PLOS Biology 2020, 18, e3000754. [Google Scholar] [CrossRef]
- Sanger, F. The Arrangement of Amino Acids in Proteins. In: Anson, M.L.; Bailey, K.; Edsall, J.T.; editors. Advances in Protein Chemistry. 7: Academic Press; 1952. p. 1-67.
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proceedings of the National Academy of Sciences 1951, 37, 205–211. [Google Scholar] [CrossRef]
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M.; Parrish, R.G.; Wyckoff, H.; Phillips, D.C. A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef] [PubMed]
- CHAFFEY, N. Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K. and Walter, P. Molecular biology of the cell. 4th edn. Annals of Botany 2003, 91, 401. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; et al. Therapeutic peptides: current applications and future directions. Signal Transduction and Targeted Therapy 2022, 7, 48. [Google Scholar] [CrossRef]
- Liu, W.; Li, Y.; Patrinos, G.P.; Xu, S.; Thong, M.K.; Chen, Z.; et al. The 1% gift to humanity: The Human Genome Project II. Cell Res. 2024, 34, 747–750. [Google Scholar] [CrossRef]
- Omenn, G.S.; Lane, L.; Overall, C.M.; Lindskog, C.; Pineau, C.; Packer, N.H.; et al. The 2023 Report on the Proteome from the HUPO Human Proteome Project. J Proteome Res. 2024, 23, 532–549. [Google Scholar] [CrossRef]
- Radoux, C.J.; Vianello, F.; McGreig, J.; Desai, N.; Bradley, A.R. The druggable genome: Twenty years later. Frontiers in Bioinformatics. 2022, 2. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, I.; Tornali, C.; Bragazzi, N.L.; Martini, M. The Discovery of Insulin: An Important Milestone in the History of Medicine. Front Endocrinol (Lausanne). 2018, 9, 613. [Google Scholar] [CrossRef] [PubMed]
- Ayyar, V.S. History of growth hormone therapy. Indian J Endocrinol Metab. 2011, 15 (Suppl. 3), S162–S165. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, P.M.; Tuddenham, E.G. The hemophilias--from royal genes to gene therapy. N Engl J Med. 2001, 344, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Greenwalt, T.J. A short history of transfusion medicine. Transfusion. 1997, 37, 550–563. [Google Scholar] [CrossRef]
- Bergman, S.J.; Ferguson, M.C.; Santanello, C. Interferons as therapeutic agents for infectious diseases. Infect Dis Clin North Am. 2011, 25, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Leão Rde, B.; Esteves, S.C. Gonadotropin therapy in assisted reproduction: an evolutionary perspective from biologics to biotech. Clinics (Sao Paulo). 2014, 69, 279–293. [Google Scholar] [CrossRef]
- Szmuness, W.; Stevens, C.E.; Harley, E.J.; Zang, E.A.; Oleszko, W.R.; William, D.C.; et al. Hepatitis B vaccine: demonstration of efficacy in a controlled clinical trial in a high-risk population in the United States. N Engl J Med. 1980, 303, 833–841. [Google Scholar] [CrossRef]
- Xu, S.; Ye, X.-T.; Zhang, D.; Dong, P.; Wu, Y.-H.; Pan, C.-W. Predicting clinical outcomes in chronic hepatitis B patients receiving nucleoside analogues and pegylated interferon alpha: a hematochemical and clinical analysis. BMC Infectious Diseases. 2024, 24, 1149. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yang, Y.; Wang, G.; Liu, M. Current landscape and future directions of bispecific antibodies in cancer immunotherapy. Frontiers in Immunology. 2022, 13. [Google Scholar] [CrossRef]
- Palanques-Pastor, T.; Megías-Vericat, J.E.; Bosó Ribelles, V.; Gómez Seguí, I.; Poveda Andrés, J.L. Effectiveness of Caplacizumab Nanobody in Acquired Thrombotic Thrombocytopenic Purpura Refractory to Conventional Treatment. Acta Haematologica. 2021, 145, 72–77. [Google Scholar] [CrossRef]
- Fox, G.E. Origins and Early Evolution of the Ribosome. In: Hernández G, Jagus R, editors. Evolution of the Protein Synthesis Machinery and Its Regulation. Cham: Springer International Publishing; 2016. p. 31-60.
- Palade, G.E. A small particulate component of the cytoplasm. J Biophys Biochem Cytol. 1955, 1, 59–68. [Google Scholar] [CrossRef]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef] [PubMed]
- Meselson, M.; Stahl, F.W. THE REPLICATION OF DNA IN ESCHERICHIA COLI. Proc Natl Acad Sci U S A. 1958, 44, 671–682. [Google Scholar] [CrossRef]
- Nirenberg, M.W.; Matthaei, J.H. The dependence of cell-free protein synthesis in <i>E. coli</i> upon naturally occurring or synthetic polyribonucleotides. Proceedings of the National Academy of Sciences. 1961, 47, 1588–1602. [Google Scholar]
- Zorca, S.M.; Zorca, C.E. The legacy of a founding father of modern cell biology: George Emil Palade (1912-2008). Yale J Biol Med. 2011, 84, 113–116. [Google Scholar]
- Schmidt, C.; Haefner, E.; Gerbeth, J.; Beissert, T.; Sahin, U.; Perkovic, M.; et al. A taRNA vaccine candidate induces a specific immune response that protects mice against Chikungunya virus infections. Molecular Therapy Nucleic Acids. 2022, 28, 743–754. [Google Scholar] [CrossRef]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; et al. Reversible methylation of m(6)A(m) in the 5' cap controls mRNA stability. Nature. 2017, 541, 371–375. [Google Scholar] [CrossRef]
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Synthesis and properties of mRNAs containing the novel "anti-reverse" cap analogs 7-methyl(3'-O-methyl)GpppG and 7-methyl (3'-deoxy)GpppG. Rna. 2001, 7, 1486–1495. [Google Scholar] [PubMed]
- Henderson, J.M.; Ujita, A.; Hill, E.; Yousif-Rosales, S.; Smith, C.; Ko, N.; et al. Cap 1 Messenger RNA Synthesis with Co-transcriptional CleanCap(®) Analog by In Vitro Transcription. Curr Protoc. 2021, 1, e39. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; et al. The challenge and prospect of mRNA therapeutics landscape. Biotechnol Adv. 2020, 40, 107534. [Google Scholar] [CrossRef]
- Kim, S.C.; Sekhon, S.S.; Shin, W.R.; Ahn, G.; Cho, B.K.; Ahn, J.Y.; et al. Modifications of mRNA vaccine structural elements for improving mRNA stability and translation efficiency. Mol Cell Toxicol. 2022, 18, 1–8. [Google Scholar] [CrossRef]
- Sander, I.M.; Chaney, J.L.; Clark, P.L. Expanding Anfinsen's principle: contributions of synonymous codon selection to rational protein design. J Am Chem Soc. 2014, 136, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Wethmar, K. The regulatory potential of upstream open reading frames in eukaryotic gene expression. Wiley Interdiscip Rev RNA. 2014, 5, 765–778. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted LVW, Ebbesen, K. K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Yang, L.; Wilusz, J.E.; Chen, L.L. Biogenesis and Regulatory Roles of Circular RNAs. Annu Rev Cell Dev Biol. 2022, 38, 263–289. [Google Scholar] [CrossRef]
- McKenna, S.A.; Kim, I.; Puglisi, E.V.; Lindhout, D.A.; Aitken, C.E.; Marshall, R.A.; et al. Purification and characterization of transcribed RNAs using gel filtration chromatography. Nat Protoc. 2007, 2, 3270–3277. [Google Scholar] [CrossRef]
- Xiao, F.; Chen, Z.; Wei, Z.; Tian, L. Hydrophobic Interaction: A Promising Driving Force for the Biomedical Applications of Nucleic Acids. Advanced Science. 2020, 7, 2001048. [Google Scholar] [CrossRef]
- Nwokeoji, A.O.; Kung, A.W.; Kilby, P.M.; Portwood, D.E.; Dickman, M.J. Purification and characterisation of dsRNA using ion pair reverse phase chromatography and mass spectrometry. J Chromatogr, A. 2017, 1484, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Whitley, J.; Zwolinski, C.; Denis, C.; Maughan, M.; Hayles, L.; Clarke, D.; et al. Development of mRNA manufacturing for vaccines and therapeutics: mRNA platform requirements and development of a scalable production process to support early phase clinical trials. Transl Res. 2022, 242, 38–55. [Google Scholar] [CrossRef]
- Loan Young, T.; Chang Wang, K.; James Varley, A.; Li, B. Clinical delivery of circular RNA: Lessons learned from RNA drug development. Adv Drug Deliv Rev. 2023, 197, 114826. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Song, H.; Tropea, J.E.; Needle, D.; Waugh, D.S.; Gu, S.; et al. The molecular mechanism of dsRNA processing by a bacterial Dicer. Nucleic Acids Res. 2019, 47, 4707–4720. [Google Scholar] [CrossRef] [PubMed]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; et al. A Facile Method for the Removal of dsRNA Contaminant from <em>In Vitro</em>-Transcribed mRNA. Molecular Therapy Nucleic Acids. 2019, 15, 26–35. [Google Scholar]
- Childs-Disney, J.L.; Yang, X.; Gibaut QMR, Tong, Y. ; Batey, R.T.; Disney, M.D. Targeting RNA structures with small molecules. Nat Rev Drug Discov. 2022, 21, 736–762. [Google Scholar] [CrossRef] [PubMed]
- Kaukinen, U.; Lyytikäinen, S.; Mikkola, S.; Lönnberg, H. The reactivity of phosphodiester bonds within linear single-stranded oligoribonucleotides is strongly dependent on the base sequence. Nucleic Acids Res. 2002, 30, 468–474. [Google Scholar] [CrossRef]
- Abdelwahed, W.; Degobert, G.; Stainmesse, S.; Fessi, H. Freeze-drying of nanoparticles: formulation, process and storage considerations. Adv Drug Deliv Rev. 2006, 58, 1688–1713. [Google Scholar] [CrossRef]
- Thorn, C.R.; Sharma, D.; Combs, R.; Bhujbal, S.; Romine, J.; Zheng, X.; et al. The journey of a lifetime - development of Pfizer's COVID-19 vaccine. Curr Opin Biotechnol. 2022, 78, 102803. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, Z.; Jin, P.; Jiang, K.; Xu, Y.; Pan, F.; et al. Effects of PEG antibodies on in vivo performance of LNP-mRNA vaccines. Int J Pharm. 2024, 650, 123695. [Google Scholar] [CrossRef]
- Qin, S.; Tang, X.; Chen, Y.; Chen, K.; Fan, N.; Xiao, W.; et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal Transduct Target Ther. 2022, 7, 166. [Google Scholar] [CrossRef]
- Hsu, F.F.; Liang, K.H.; Kumari, M.; Chen, W.Y.; Lin, H.T.; Cheng, C.M.; et al. An efficient approach for SARS-CoV-2 monoclonal antibody production via modified mRNA-LNP immunization. Int J Pharm. 2022, 627, 122256. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, F.B.; Shen, W.-J. Hormone-sensitive lipase. Journal of Lipid Research. 2002, 43, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Chen, J.; Zhang, Z.; Shi, H.; Sun, W.; Yi, Q. The role of AMPK in macrophage metabolism, function and polarisation. Journal of Translational Medicine. 2023, 21, 892. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Zhang, Y.; Wang, J.-Z. Updates in Alzheimer's disease: from basic research to diagnosis and therapies. Translational Neurodegeneration. 2024, 13, 45. [Google Scholar] [CrossRef]
- Rajamanickam, G.; Lee ATH, Liao, P. Role of Brain Derived Neurotrophic Factor and Related Therapeutic Strategies in Central Post-Stroke Pain. Neurochemical Research. 2024, 49, 2303–2318. [Google Scholar] [CrossRef]
- Jeong, S.; Jeong Hwan, L.; CHOWDHURY MAR. Multi-faceted regulation of CREB family transcription factors. Frontiers in Molecular Neuroscience. 2024, 17. [Google Scholar]
- Sorushanova, A.; Delgado, L.M.; Wu, Z.; Shologu, N.; Kshirsagar, A.; Raghunath, R.; et al. The Collagen Suprafamily: From Biosynthesis to Advanced Biomaterial Development. Adv Mater. 2019, 31, e1801651. [Google Scholar] [CrossRef] [PubMed]
- Vindin, H.; Mithieux, S.M.; Weiss, A.S. Elastin architecture. Matrix Biol. 2019, 84, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, D.H.; Senyo, S.E. mRNA therapy for myocardial infarction: A review of targets and delivery vehicles. Frontiers in Bioengineering and Biotechnology. 2022, 10. [Google Scholar] [CrossRef]
- Omorou, M.; Huang, Y.; Gao, M.; Mu, C.; Xu, W.; Han, Y.; et al. The forkhead box O3 (FOXO3): a key player in the regulation of ischemia and reperfusion injury. Cellular and Molecular Life Sciences. 2023, 80, 102. [Google Scholar] [CrossRef]
- Sandoval, S.O.; Méndez-Albelo, N.M.; Xu, Z.; Zhao, X. From wings to whiskers to stem cells: why every model matters in fragile X syndrome research. Journal of Neurodevelopmental Disorders. 2024, 16, 30. [Google Scholar] [CrossRef]
- Zuo, W.-F.; Pang, Q.; Zhu, X.; Yang, Q.-Q.; Zhao, Q.; He, G.; et al. Heat shock proteins as hallmarks of cancer: insights from molecular mechanisms to therapeutic strategies. Journal of Hematology & Oncology. 2024, 17, 81. [Google Scholar]
- Zhang, P.; He, Y.; Wu, S.; Li, X.; Lin, X.; Gan, M.; et al. Factors Associated with White Fat Browning: New Regulators of Lipid Metabolism. International Journal of Molecular Sciences. 2022, 23, 7641. [Google Scholar] [CrossRef]
- Zhang, W.; Fan, Y. Structure of Keratin. Methods Mol Biol. 2021, 2347, 41–53. [Google Scholar] [PubMed]
- Prud’homme, G.J.; Kurt, M.; Wang, Q. Pathobiology of the Klotho Antiaging Protein and Therapeutic Considerations. Frontiers in Aging. 2022, 3. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; et al. Leptin and Obesity: Role and Clinical Implication. Front Endocrinol (Lausanne). 2021, 12, 585887. [Google Scholar] [CrossRef] [PubMed]
- Mead, J.R.; Irvine, S.A.; Ramji, D.P. Lipoprotein lipase: structure, function, regulation, and role in disease. Journal of Molecular Medicine. 2002, 80, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Guhathakurta, P.; Prochniewicz, E.; Thomas, D.D. Actin-Myosin Interaction: Structure, Function and Drug Discovery. Int J Mol Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Chen, W.; Hou, P.; Liu, Z.; Zuo, M.; Liu, S.; et al. NAD+ metabolism-based immunoregulation and therapeutic potential. Cell & Bioscience. 2023, 13, 81. [Google Scholar]
- Zhang, Z.; Huang, J.; Shen, Y.; Li, R. BACE1-Dependent Neuregulin-1 Signaling: An Implication for Schizophrenia. Frontiers in Molecular Neuroscience. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zhang, H.; Wang, P.; Cui, W.; Xu, K.; Chen, D.; et al. Oxytocin and serotonin in the modulation of neural function: Neurobiological underpinnings of autism-related behavior. Frontiers in Neuroscience. 2022, 16. [Google Scholar] [CrossRef]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; et al. p53 signaling in cancer progression and therapy. Cancer Cell International. 2021, 21, 703. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Tang, L.; Zhang, D. Molecular mechanisms of uterine incision healing and scar formation. European Journal of Medical Research. 2023, 28, 496. [Google Scholar] [CrossRef]
- Scala, M.; Grasso, E.A.; Di Cara, G.; Riva, A.; Striano, P.; Verrotti, A. The Pathophysiological Link Between Reelin and Autism: Overview and New Insights. Frontiers in Genetics. 2022, 13. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. Epigenetic Changes in Aging: The Contribution of SIRT1 to Longevity. In: Bueno, V.; editor. Cellular and Molecular Aspects of Ageing. Cham: Springer Nature Switzerland; 2024. p. 33-49.
- Mirza, F.J.; Zahid, S. The Role of Synapsins in Neurological Disorders. Neuroscience Bulletin. 2018, 34, 349–358. [Google Scholar] [CrossRef]
- Abbaali, I.; Truong, D.; Day, S.D.; Mushayeed, F.; Ganesh, B.; Haro-Ramirez, N.; et al. The tubulin database: Linking mutations, modifications, ligands and local interactions. PLoS One. 2023, 18, e0295279. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yuan, Z.; Sun, W.; Shafiq, M.; Zhu, J.; Chen, J.; et al. Vascular Endothelial Growth Factor-Recruiting Nanofiber Bandages Promote Multifunctional Skin Regeneration via Improved Angiogenesis and Immunomodulation. Advanced Fiber Materials. 2023, 5, 327–348. [Google Scholar] [CrossRef]
- Banoun, H. mRNA: Vaccine or Gene Therapy? The Safety Regulatory Issues. Int J Mol Sci. 2023, 24. [Google Scholar] [CrossRef]
- Rohner, E.; Yang, R.; Foo, K.S.; Goedel, A.; Chien, K.R. Unlocking the promise of mRNA therapeutics. Nature Biotechnology. 2022, 40, 1586–1600. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nature Reviews Materials. 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Youssef, M.; Hitti, C.; Puppin Chaves Fulber, J.; Kamen, A.A. Enabling mRNA Therapeutics: Current Landscape and Challenges in Manufacturing. Biomolecules. 2023, 13, 1497. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, S.; Fu, R.; Zhang, L.; Huang, K.; Peng, H.; et al. Therapeutic Prospects of mRNA-Based Gene Therapy for Glioblastoma. Front Oncol. 2019, 9, 1208. [Google Scholar] [CrossRef] [PubMed]
- University, P. Clinical Drug Experience Knowledgebase: FDA Approvals | Recombinant protein https://cdek.pharmacy.purdue.edu/fda-approvals/Recombinant%20protein/2024 [.
- Markets, R. Recombinant Proteins Market https://www.researchandmarkets.com/report/protein-drugs#tag-pos-12024 [.
- Niazi, S.K. Biosimilars Adoption: Recognizing and Removing the RoadBlocks. Clinicoecon Outcomes Res. 2023, 15, 281–294. [Google Scholar] [CrossRef]
- Niazi, S.K. The FDA's New Guideline "Generally Accepted Scientific Knowledge" (GASK): An Opportunity to Expedite the Approval of Biosimilars. Pharmaceuticals (Basel). 2023, 16. [Google Scholar] [CrossRef] [PubMed]
- FDA. mRNA https://www.fda.gov/search?s=mRNA2024 [.
- Morais, P.; Adachi, H.; Yu, Y.T. The Critical Contribution of Pseudouridine to mRNA COVID-19 Vaccines. Front Cell Dev Biol. 2021, 9, 789427. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, R.E.; Minnell, J.J.; Ganley, M.; Painter, G.F.; Draper, S.L. mRNA Synthesis and Encapsulation in Ionizable Lipid Nanoparticles. Current Protocols. 2023, 3, e898. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.E.; Sahin, E. Chemical and physical instabilities in manufacturing and storage of therapeutic proteins. Curr Opin Biotechnol. 2019, 60, 159–167. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hsu, H.-E.; Perez, S.J.L.P.; Kumari, M.; Chen, G.-H.; Hong, M.-H.; et al. Current landscape of mRNA technologies and delivery systems for new modality therapeutics. Journal of Biomedical Science. 2024, 31, 89. [Google Scholar] [CrossRef] [PubMed]
| mRNA Type | Applications | Properties | Regulatory Status |
| Conventional mRNA [1] | Vaccines | Basic structure with 5' cap, UTRs, poly(A) tail | Approved for COVID-19 vaccines |
| Modified mRNA [2] | Protein replacement, cancer vaccines | Chemically modified nucleosides | Clinical trials for genetic diseases, COVID-19 vaccines |
| Self-amplifying mRNA (saRNA) [3] | Vaccines, gene therapies | Self-replicating sequences for dose-sparing | Early clinical trials |
| Circular RNA (circRNA) [4] | Long-term protein replacement therapies | Circular structure for enhanced stability | Preclinical and early clinical stages |
| Therapeutic Guide RNA [5] | Gene editing for genetic disorders | Guide sequence directs gene editing | Clinical trials under ATMP designation |
| Quality | Attribute | Method (s) |
|---|---|---|
| Identity | mRNA sequence identity confirmation | Sanger sequencing |
| Content | RNA concentration | Ultraviolet Spectroscopy (UV) |
| Integrity | mRNA intactness | Capillary electrophoresis |
| Purity | 5’ capping efficiency | Reverse-phase liquid chromatography-mass spectroscopy (RP-LC-MS-MS) |
| 3’ poly (A) tail length | Capillary Electrophoresis | |
| Product-related impurities - dsRNA | Slot-blot | |
| Product-related impurities - aggregate quantitation | Size exclusion-high-performance liquid chromatography (SE-HPLC) | |
| Process-related impurities - residual DNA template | Quantitative PCR (qPCR) | |
| Process-related impurities-quantitation of free/nonincorporated nucleosides | Reverse-phase liquid chromatography-mass spectroscopy (RP-LC-MS-MS) | |
| Process-related impurities - residual T7 RNA polymerase content | Enzyme-linked immunosorbent assay (ELISA) | |
| Potency | Expression of target protein | Cell-based assay |
| Safety | Endotoxin | USP<85> |
| Bioburden | USP <61> | |
| Other | Appearance | USP <790> |
| Residual solvents | USP <467> | |
| pH | USP <791> |
| Quality | Attribute | Method (s) |
|---|---|---|
| Identity | RNA identification | Sanger sequencing |
| Identity of lipids | Reversed-phase high-performance liquid chromatography with charged aerosol detector (RP-HPLC-CAD) | |
| Content | RNA concentration/RNA encapsulation efficiency | Ribogreen Assay |
| Lipid content | Reversed-phase high-performance liquid chromatography with charged aerosol detector (RP-HPLC-CAD) | |
| Integrity | LNP size and polydispersity | Dynamic light scattering (DLS) |
| RNA size and integrity | Capillary electrophoresis (CE) | |
| Potency | Expression of target protein | Cell-based assay |
| Purity | Product-related impurities - aggregate quantitation | Size exclusion-high-performance liquid chromatography (SE-HPLC) |
| Product-related impurities – the percentage of fragment mRNA | Ion pair reversed-phase high-performance liquid chromatography (IP-RP-HPLC) | |
| Safety | Endotoxin | USP<85> |
| Sterility | USP <71> | |
| Other | Appearance | USP <790> |
| pH | USP <791)> | |
| Subvisible particles | USP <787)> | |
| Osmolality | USP <785)> | |
| Residual solvents | USP <467> | |
| Extractable volume | USP <1>, USP <698> | |
| Container closure integrity | USP <1207> |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



