
Submitted:
20 November 2024
Posted:
22 November 2024
You are already at the latest version
Abstract
Background: The nuclear encoded enzyme Polymerase gamma (Pol-γ) is crucial for the replication of the mitochondrial genome (mtDNA), which in turn is vital for mitochondria and hence nu-merous metabolic processes and energy production in eukaryotic cells. Variants in the POLG gene, which encodes the catalytic subunit of Pol-γ, can significantly impair Pol-γ enzyme function. Pol-γ-associated disorders are referred to as POLG-spectrum disorders (POLG-SD) and are mainly autosomal-recessively inherited. Clinical manifestations include seizures, muscle weakness and fatigue, severe forms of the disease can lead to premature death in infancy, childhood, and early adulthood, often associated with liver failure or intractable epilepsy. Here, we analyze fibroblasts from a compound heterozygous patient with the established pathogenic variant c.2419C>T; p.(Arg807Cys) and a previously undescribed variant c.678G>C; p.(Gln226His) with a clinical manifestation compatible with POLG-SD, sensory ataxic neuropathy and infantile muscular at-rophy. We conducted a battery of functional studies for Pol-γ and mitochondrial dysfunction on the patient’s fibroblasts, to test whether the novel variant c.678G>C; p.(Gln226His) may be causative for human disease.
Aims/Methods: We analyzed skin-derived fibroblasts in comparison to a first degree relative, an asymptomatic carrier harboring only the established c.2419C>T; p.(Arg807Cys) mutation. As-sessments of mitochondrial function included measurements of mtDNA content, mRNA levels of mitochondrial genes, mitochondrial mass and mitochondrial morphology.
Case Presentation and Results: A 13-year-old male presented with symptoms starting at three years of age, including muscle weakness and atrophy in the lower extremities and facial muscles, which later extended to the upper limbs, voice, and back muscles without further progression. The patient also reported fatigue and muscle pain after physical activity with no sensory deficits. Ex-tensive diagnostic tests such as electromyography, nerve conduction studies, muscle biopsy, and MRI were unremarkable. Exome sequencing revealed that he carries the compound heterozygous variants in POLG c.678G>C; p.(Gln226His) and c.2419C>T; p.(Arg807Cys), but no other potential genetic pathogenic causes. In comparison to a first degree relative who only carried the c.2419C>T; p.(Arg807Cys) pathogenic mutation, in vitro analyses revealed a significant reduction mtDNA content (~50%), and mRNA levels of mtDNA-encoded proteins. Mitochondrial mass was decreased by approximately 20%, and mitochondrial interconnectivity within cells was impaired, as de-termined by (immune-) fluorescence microscopy and mitochondrial stainings.
Conclusion: Our findings suggest that the c.678G>C; p.(Gln226His) mutation, in conjunction with the c.2419C>T; p.(Arg807Cys) mutation, may compromise mtDNA replication and mitochondrial function and could result in clinically significant mitochondriopathy. As this study is based on one patient compared to a first- degree relative, future studies and cases must confirm pathogenicity of c.678G>C; p.(Gln226His), in particular in conjunction with other POLG-variants.
Keywords:
mitochondriopathy
; Alpers-Huttenlocher syndrome
; mtDNA copy number
; mtDNA depletion
; polymerase-gamma
; spinal muscular atrophy
; rare disease
1. Introduction
Mitochondria are essential cellular organelles for eukaryotes. They supply most eukaryotic cells with the majority of adenosine triphosphate (ATP), which is produced as part of oxidative phosphorylation (OXPHOS) by the protein complexes of the electron transport chain (ETC) at the inner mitochondrial membrane [1,2]. In addition, mitochondria are involved in a variety of crucial cellular mechanisms such as divers metabolisms, apoptosis, endoplasmic reticulum stress-response, and generation of reactive oxygen species (ROS) [2].
Mitochondria have their own genome, or their own set of DNA, here referred to as mitochondrial DNA (mtDNA). The double-stranded negatively supercoiled circular genome comprises 16,569 base pairs (bp) and encodes a total of 37 genes (13 proteins essential for the ETC, 22 transfer RNAs (tRNAs), and 2 mitochondrial ribosomal RNAs (rRNAs)) [3,4]. The heterotrimeric enzyme DNA Polymerase-γ (Pol-γ), whose catalytic subunit Pol-γA is encoded by the nuclear gene POLG, is the only polymerase responsible for replication of mtDNA in mammals. Loss or reduced functionality of Pol-γ directly affects mitochondrial DNA synthesis and leads to mtDNA depletion and severe mtDNA deletions [5]. While mtDNA depletion (reduced copy number of mtDNA) often occurs in diseases with an early onset, mtDNA deletions (loss of segments of the mtDNA) occur more often in late onset diseases [6].
Mutations in POLG impair mtDNA replication which causes mitochondrial dysfunction and neurodegenerative disorders [7,8] and are among the most common causes of inherited mitochondriopathies in children and adults, referred to as POLG-spectrum disorders (POLG-SDs) [8,9]. This spectrum of disorders includes motor dysfunction comprising oculomotor dysfunction [10]. More severe defects may manifest as infantile onset epileptiform discharges over occipital brain regions. With disease progression the seizures can generalize and can become resistant to medication. Other clinical features of severe forms of POLG-SDs include liver failure, cortical blindness, myoclonuses and neurodegeneration [11].
POLG-spectrum disorders summarize a variety of syndromes, including Alpers-Huttenlocher syndrome (AHS), ataxia neuropathy spectrum (ANS), childhood myocerebrohepatopathy spectrum (MCHS), mitochondrial recessive ataxia syndrome (MIRAS), myoclonic epilepsy, myopathy sensory ataxia (MEMSA) and progressive external ophthalmoplegia (PEO) [12]. AHS is one of the most severe conditions among POLG-SDs. While early-onset (neonatal, infancy or childhood) POLG-SDs such as MCHS or AHS are often associated with mtDNA depletion, the mtDNA defects in adolescent or young adult-onset disorders such as ANS, MIRAS or PEO are mostly associated with (accumulating) mtDNA deletions [6]. Patients with POLG-SDs like AHS show decreased mtDNA content and increased mtDNA damage. Loss of mtDNA content (i.e. mtDNA depletion) can be tolerated to a certain extent without causing deleterious effects but is considered pathological when < 70 % [13]. MtDNA content is also correlated with the age onset of POLG-SDs. Infants with POLG-SDs show less mtDNA content with 80% loss whereas adult patients show only 40-50% loss [13].
More than 300 pathological POLG variants have been described so far. The two most common mutations lead to the amino acid substitutions c. 1399G>A; p.(Ala467Thr) and c.2243G>C; p.(Trp748Ser), patients with these mutations show significant residual (~ 5% ) Pol-γ activity [11,14,15]. The c. 1399G>A; p.(Ala467Thr) mutation alone is found in 36% of all alleles associated with POLG-SDs and exists in European populations with the carrier frequency of 0.2% to 1% [16,17]. Yet, there is a range of variants in the POLG gene that are variants of unknown significance, indicating that it is unclear whether the variations have a significant effect on Pol-γ function.
To date, the POLG-variant c.678G>C; p.(Gln226His) is described mostly as a variant of unknown significance, twice as likely benign and as a variant of unclear significance. in Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/variation/206581/). The c.2419C>T; p.(Arg807Cys) mutation is exclusively classified as pathogenic. In some cases, compound heterozygosity can lead to a significant reduction in enzyme activity, causing severe conditions like infant death, especially when the c.2542G>A; p.(Gly848Ser) mutation is located in trans with another disease-causing mutation [12,18]. Since many patients diagnosed with a POLG-SD are compound heterozygous for two variants in POLG [11], the combination of different POLG mutations may strongly influence the phenotype of the patient [6].
2. Materials and Methods
Ethical Permission
The project was approved by the Ethics Commission of the Medical Faculty of the University of Cologne. Tissues were acquired with written informed consent.
Obtaining and Cultivating the Human-Derived Fibroblasts
Skin biopsies were collected from the patient and the carrier and fibroblasts were isolated. Ethical approval was granted from the Ethics Commission of the Medical Faculty of the University of Cologne. Fibroblasts were cultured in medium consisting of DMEM/F-12, GlutaMAX™ (Thermo Fisher Scientific, #10565-018), 10% (v/v) Fetal Bovin Serum (FBS, Biochrom AG) and Antibiotic-Antimycotic (1X Anti/Anti, #15240062, TFS). For maintenance, fibroblasts were cultivated in uncoated T75 cell culture flasks (VWR) in a sterile incubator (Heraeus HeraCell 150, Kendro) at a temperature of 37°C, 95 % air humidity and 5 % CO2 concentration. Culture medium was changed once or twice per week, depending on cell density, which was monitored and images were taken with a brightfield light microscope (DM IL LED, Leica) using objectives with 10x and 20x (air-based) magnification (Leica) and Las X imaging software (Leica). Cells were passaged for further cultivation or seeded for differentiation experiments at 70–80 % confluency, for this cells were washed with phosphate-buffered saline (PBS, TFS), trypsinized (0.05 % Trypsin/EDTA in PBS, TFS) for 10 min, spun down at 1000 g for 5 min and resuspended in culture medium. For passaging, the culture was diluted 1:10 and seeded again in an uncoated T75 flask. For seeding, resuspended fibroblasts were counted with an automatic cell counter (TC20TM, Bio-Rad) and seeded with 3 x 104 cells/cm². For long-term storage, trypsinized and spun down cells were resuspended in FBS with 10 % DMSO, cooled down with -1 °C/min in a cryo-container (Mr. FrostyTM, VWR) and stored at -80°C or in liquid nitrogen.
Whole Exome Sequencing and Annotation
The patient was previously reported in a study published by Keller et al, 2021. Briefly, Exome Sequencing (ES) was conducted for as a parent-child duo. For target enrichment, we used the SureSelect All Exon v7 kit (Agilent) according to the 'SureSelectXT Low Input Automated Target Enrichment for Illumina Paired-End Multiplexed Sequencing' protocol (Version D0, July 2018) provided by Agilent Technologies. Sample processing steps were automated using the NGS Workstation Option B (Agilent), deploying the reagents specified in the protocol. Before target enrichment via hybridization-based capture, an individual molecular-barcoded and indexed library was prepared for each sample. Input samples contained 10–200 ng genomic DNA. After DNA fragmentation to a size of 150–200 base-pair (bp) and DNA end-modification including end-repair, dA-tailing, and ligation of the molecular-barcoded adaptor, adaptor-ligated libraries were amplified via PCR using index primers. Amplified DNA was subsequently purified using AMPure XP beads (Beckman Coulter). After quantity and quality assessment (Agilent tape station), the prepared and aliquoted gDNA library was hybridized to the target-specific SureSelect Capture Library via thermal cycler incubation. The gDNA-Capture Library hybrids were then captured with streptavidin-coated magnetic beads. Postcapture, SureSelect-enriched DNA samples were PCR-amplified and purified. After library validation and quantification (Agilent tape station), equimolar amounts of the library were pooled. Pools were quantified by using the Peqlab KAPA Library Quantification Kit and the Applied Biosystems 7900HT Sequence Detection System and sequenced by using a paired-end 100-nucleotide protocol on the Illumina NovaSeq6000 sequencer, with a resulting average 75× target coverage.
Reads were aligned to human reference genome hg38/GRCh38 (Genome Reference Consortium Human GRCh38). For read improvement, PCR duplicates were removed using Picard tools, and local realignment and base quality score recalibration (BQSR) were performed using Genome Analysis Toolkit (GATK). Single nucleotide polymorphisms (SNPs) and short insertions/deletions (INDELs) were called by Platypus, Haplotype Caller, and Mpileup programs and subsequently filtered via variant quality score calibration (VQSR) using GATK. For CNV detection based on coverage analysis XHMM, Conifer, and ExomeDepth algorithms were applied. ALLEGRO program was used for ROH detection based on multipoint linkage analysis. Data combining and functional variant annotation were enabled by the COMBINE and FUNC modules developed by Cologne Center for Genomics (CCG).
Real-Time Quantitative PCR
DNA was extracted using a QIAGEN Mini DNA Kit (QIAGEN) according to the manufacturer’s protocol. Quantification of mtDNA copy number was performed using RT-qPCR. The MT-CYTB, MT-3319R gene and the household genes APP and 18S were amplified from all the fibroblasts using the specific primers (see next section). RNA was extracted using NucleoSpin RNA XS kit (MACHEREY-NAGEL) and cDNA was produced using ProtoScript® II First Strand cDNA Synthesis Kit (NEB). Quantification of mitochondrial encoded mRNA levels was performed using RT-qPCR. Genes which were investigated were MT-CYTB, MT-ATP6 and MT-ND1. Specific primers were used flanking the named genes. RT-qPCR was performed with an initial denaturation step of 95°C for 10 minutes sec, then 95°C denaturation 15 seconds, followed by primer and probe hybridization and DNA synthesis at 60°C for 60 seconds; the last two steps were repeated for 40 cycles. The reaction was measured using SYBR® Green Reagent (Thermo Fisher) and the Applied Biosystems StepOnePlus (Thermo Fisher). The readouts were analysed with the ΔΔCT-methode.
Primer used for RT-qPCR:
| APP Fw: | TTTTTGTGTGCTCTCCCAGGTCT |
| APP Rev: | TGGTCACTGGTTGGTTGGC |
| 18S rRNA Fw: | TAGAGGGACAAGTGGCGTTC |
| 18S rRNA Rev: | CGCTGAGCCAGTCAGTGT |
| MT-CYTB Fw: | GCCTGCCTGATCCTCCAAAT |
| MT-CYTB Rev: | AAGGTAGCGGATGATTCAGCC |
| MT-R3319 Fw: | CACCCAAGAACAGGGTTTGT |
| MT-R3319 Rev: | TGGCCATGGGTATGTTG |
| MT-ATP6 RT-qPCR Fw: | CCCACTTCTTACCACAAGGC |
| MT-ATP6 RT-qPCR Rev: | TGGCGCTTCCAATTAGGTGC |
| MT-ND1 RT-qPCR Fw: | GCTTCAACATCGAATACGCCGC |
| MT-ND1 RT-qPCR Rev: | TAGAGTTCAGGGGAGAGTGCG |
Immunofluorescence and Microscopy
Cells were cultivated as stated above in 24-well plates on glass coverslips (VWR), previously coated for 3 hours at 37°C with 20 µg/ml Poly-D-lysine (PDL, AppliChem), fixed for 30 min at room temperature in 3.7 % formaldehyde and 4 % sucrose (in PBS) and permeabilized + blocked with 5 % bovine serum albumin (BSA, Sigma-Aldrich) and 0.2 % Triton X-100 (AppliChem) in PBS for 10 min. Before fixation cells were incubated with MitoTracker™ (Diulution 1:10 000) for 30 min at 37 °C and AlexaFluor™ Phalloidin (dilution 1:40) for 45 min at room temperature also before fixation. Nuclei were stained with 1 drop/ml NucBlueTM (Hochest 33342, TFS) for 20-30 min and samples were mounted on objective slides (Bio-Rad) with aqueous PolyMountTM (Polysciences), dried for 24 at room temperature in the dark and long-term stored at 4°C in the dark.
Immunostained cells were imaged with a widefield fluorescence microscope (Axioscope 5, Zeiss), using an LED excitation lamp (Colibri 7, Zeiss), a fluorescence camera device (Axiocam 705 mono, Zeiss), objective 40x (oil-based) magnification (Zeiss), ApoTome.2 (Zeiss) and Zen imaging software (Zen 3.6 blue edition, Zeiss). For the analysis of protein expression levels, all images were taken with identical settings (laser intensity and exposure time) to ensure statistical comparability.
Images were anaylzed with ImageJ 1.54f using a macro which is based on “Image J Mitophagy Macro” [19,20]. The macro calculated relative area using the mitochondrial area (determined with MitoTracker™ Deep Red FM (Thermo Fisher)) and dividing it by the Alexa Fluor™ 488 Phalloidin (Thermo Fisher) staining as a reference for the cell size. Interconnectivity was measured by dividing the mean area of the mitochondria by the perimeter of the mitochondria in the cell.
3. Results
Case Presention
The patient, a 13-year-old male, first exhibited symptoms at 3 years of age. Initial symptoms were weakness and atrophy in the proximal muscles of the lower extremities and facial muscles. Over the years, the condition has involved the upper proximal extremities, voice, and back muscles, but has not shown progression afterwards. Physical examination revealed muscle atrophy, with more severe involvement of the lower than the upper limbs. The patient displayed dysarthria, ptosis, scapular winging, pes cavus, and pes planovalgus. Muscle tone was notably decreased, and deep tendon reflexes were weakly elicitable on both sides. The patient reported significant fatigue and muscle pain following physical activity, although no sensory deficits were observed. Diagnostic tests including electromyography revealed no abnormalities, and nerve conduction velocity was within normal limits. Muscle biopsy did not show any pathological changes. Imaging studies, including MRI of the brain and spine, were normal, and serum creatine kinase levels were within normal range.
Experimental Results
To confirm a phenotype with mitochondrial impairment in-vitro, we generated fibroblasts from skin biopsies derived from the patient and the mother, as the only available first degree relative. Both fibroblast lines were expandable, yet the patient’s cells grew slower that the cells of the carrier. Fibroblasts of the carrier show a more linear shape and appeared less branched (Figure 1), with the patient’s fibroblast sparser, less linear, and more diverse cellular morphologies, implying relative growth defects in an in-vitro environment providing optimal conditions for fast cell proliferation.
c.678G>C; p.(Gln226His) Fibroblasts Have a Reduced mtDNA Content.
Next, we used RT-qPCR to determine mtDNA content. We investigated the mitochondrial genes MT-CYTB and MT-3319R. For normalization, we used the household genes APP and 18S. We found that the patient shows decreased mtDNA levels compared to the carrier (reduction of ~ 50 %) , indicating a severe loss of mtDNA replication due to impaired function of Pol-γ in fibroblasts (Figure 2E).
c.678G>C; p.(Gln226His) Fibroblast mRNA Levels of mtDNA Encoded Genes Are Reduced
Next, we investigated the effect of diminished mtDNA on mtDNA-encoded gene expression, using RT-qPCR. To ensure that the whole circular mtDNA molecule is represented and to detect signs of premature termination of transcription, we chose three genes located close to the transcription initiation of the heavy strand promotor 2 (HSP2) (MT-ND1), in the middle of it (MT-ATP6) and in the end of the process (MT-CYTB) (Figure 2A). Readouts show that mRNA levels are affected by the malfunction of Pol-γ (Figure 2B-D), suggesting a reduction in mitochondrial function and fitness in these cells.
Mitochondrial Mass and Interconnectivity Are Negatively Affected by the Malfunction of Pol-γ
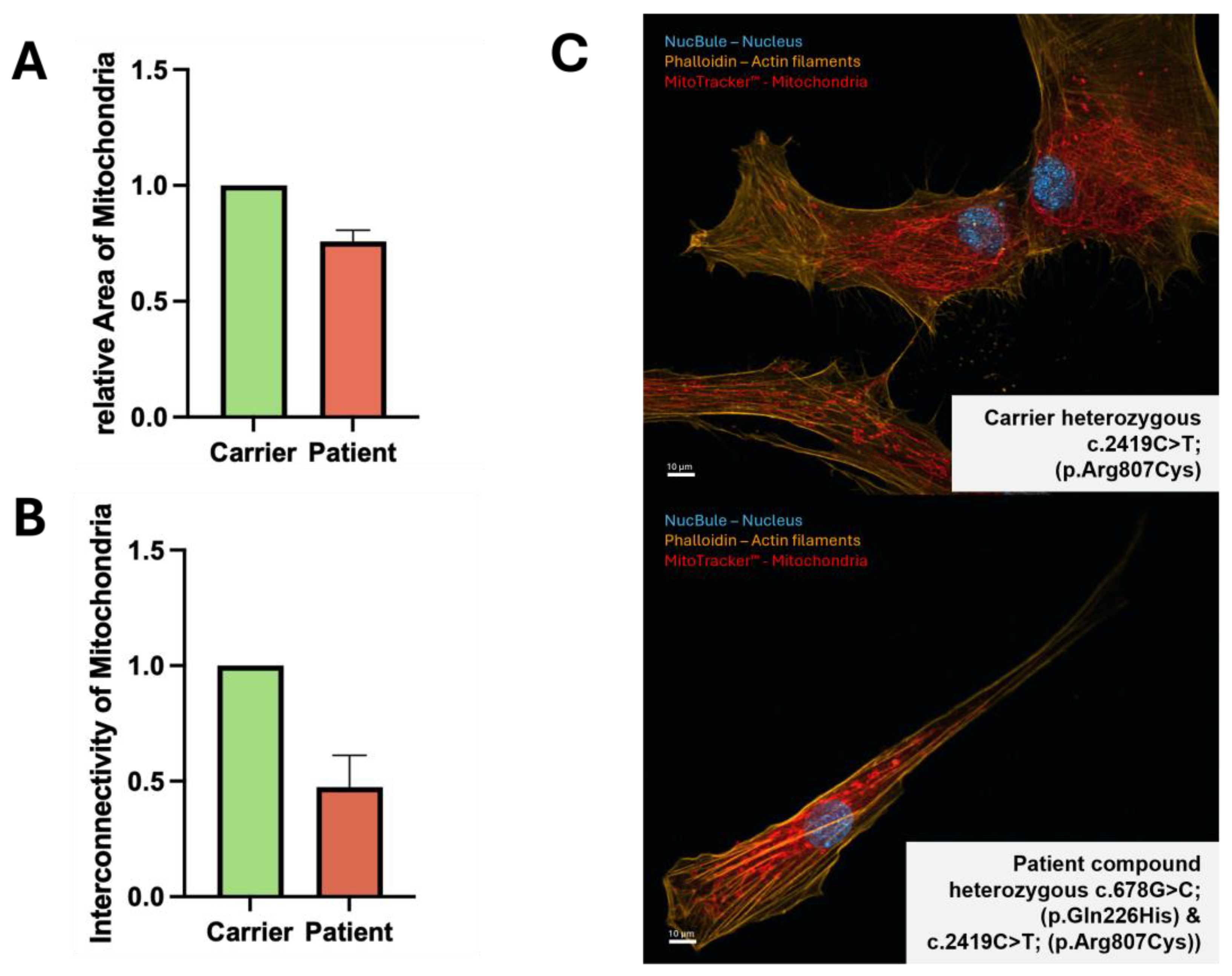
To investigate changes in mitochondrial morphology, we stained mitochondria in fibroblasts with MitoTracker™. We visualized the relative area of mitochondria (total area of mitochondria divided by total area of cell) in individual cells, and additionally calculate interconnectivity (mean area divided by mean perimeter) (Figure 3). Relative area was decreased by ~ 20 % (Figure 3B) and interconnectivity by ~ 50 % (Figure 3C) in patient cells.
All in all, in our patient derived fibroblasts in comparison to a first degree relative, we found that Pol-γ is likely impaired as we found reduced mtDNA, lower mitochondrial mRNA and protein levels, all of which likely disrupts mitochondrial biogenesis, leading decreased mitochondrial mass and interconnectivity.
4. Discussion
The present study aimed to investigate the impact of a specific variants in the POLG-gene c.678G>C; p.(Gln226His) on mitochondrial health by assessing mtDNA content, mRNA levels of mitochondrial genes, and mitochondrial mass and morphology, in patient-derived fibroblasts of a patient carrying an additional and established heterozygous pathogenic variant in trans and showing clinical symptoms typical of mitochondriopathy. Our findings demonstrate a clear detrimental effect of the mutation on mitochondrial function and biogenesis in the studied cells in comparison to cells derived from a first degree relative and are in agreement with the clinical manifestation as mitochondriopathy.
Experimental results show that compound heterozygous genetic variants in POLG c.678G>C; p.(Gln226His) and c.2419C>T; p.(Arg807Cys) result in impaired mitochondrial function. We found reduced mtDNA, mRNA levels of mtDNA encoded genes, and mitochondrial area and interconnectivity. Our results show a significant reduction in mtDNA content in the mutant samples (i.e. the patient-derived fibroblast with the Compound heterozygous genetic variants in POLG c.678G>C; p.(Gln226His) and c.2419C>T; p.(Arg807Cys) compared to the carrier control. This suggests that the heterozygous mutation c.678G>C; p.(Gln226His) in the POLG-gene impairs mitochondrial DNA replication when in-trans with the established c.2419C>T; p.(Arg807Cys) mutation, in line with Pol-γ being the only mammalian enzyme responsible for mitochondrial DNA replication, and with the patient showing a mitochondriopathy-typical phenotype. The observed decrease in mtDNA leads to reduced expression of essential mitochondrial proteins (e.g. for OXPHOS), further compromising mitochondrial activity, such as energy production in forms of ATP in eukaryotic cells. Also clinically, the patient phenotype would be in agreement with reduction of mtDNA, with symptoms that can be caused by mitochondrial dysfunction (s. below).
The mRNA levels of mitochondrial genes, including those involved in oxidative phosphorylation, were significantly lower in patient cells. This reduction in mRNA levels indicates a disruption in the transcriptional regulation of mitochondrial genes, which could stem from the decreased mtDNA content naturally resulting in at least relatively decreased net transcription in vitro in conditions of optimal cellular growth. The decreased transcription or premature termination of mtDNA of these genes alone is likely to impair the production of proteins necessary for mitochondrial respiratory function, leading to diminished ATP production and increased oxidative stress.
In the same way mtDNA content and mRNA levels of mitochondrial encoded genes were reduced, our measurements of mitochondrial mass revealed a reduction in the patient samples. This finding indicates that the mutation c.678G>C; p.(Gln226His) in conjunction with another mutation in trans negatively affects not only the genetic and transcriptional components of mitochondria, but also their overall abundance and structural integrity. This decrease in mitochondrial mass suggests a reduction in mitochondrial biogenesis and/or an increase in mitochondrial degradation. Moreover, the loss of mitochondrial mass will certainly lead to impaired cellular energy production, as fewer mitochondria are available to meet the metabolic demands of the cell. Additionally, mitochondrial interconnectivity was significantly reduced. Mitochondrial interconnectivity refers to the dynamic network formed by mitochondria within a cell. This network facilitates efficient energy distribution, communication, and the exchange of mitochondrial DNA and proteins, ensuring optimal cellular function and adaptation to metabolic demands. Mitochondrial interconnectivity is thus crucial for maintaining mitochondrial function and cellular homeostasis.
Clinical View and Conclusion
From a clinical perspective, patients with pathogenic variants in the POLG-gene affecting mitochondrial health may present a wide range of clinical symptoms, ranging from mild muscle weakness to stroke-like episodes and liver failure. The symptoms of our patient (e.g. muscle weakness and atrophy in different parts of the body, fatigue and muscle pain following exercise) are compatible with a mitochondriopathy. However, mitochondriopathy, in particular those caused by variants in POLG, can display a great variety of symptoms, it thus difficult to argue that the patient presented here shows typical symptoms of a certain POLG-spectrum disorder, but symptoms are in principle in agreement with a mitochondriopathy. Nonetheless, our study provides first cellular evidence that the mutation c.678G>C; p.(Gln226His) when present in trans with an known pathogenic POLG variant may well significantly impair mitochondrial health and function by reducing mtDNA content, mRNA levels of mitochondrial genes, and mitochondrial mass. These findings help explain the clinical symptoms observed in patient with a mitochondrial disease. The donor of the control fibroblasts, being directly related to the patient, shares a 50% identical genetic background and also the second mutation present in the patient, which allowed us to investigate the c.678G>C; p.(Gln226His) mutation independently of the other mutation present in the patient.
By comparing the heterozygous carrier (who shows no clinical symptoms) with the compound heterozygous patient (who exhibits clear and phenotypical symptoms), it is possible that the c.678G>C; p.(Gln226His) mutation is responsible for the patient's condition. The observed detrimental effects, including reduced mtDNA content and mitochondrial mass, point to compromised mitochondrial function. Future research should focus on unravelling the molecular mechanisms by which this mutation disrupts mtDNA replication, maintenance, gene expression, and mitochondrial biogenesis. It will also be important to determine whether the observed reduction in mitochondrial mass and potential disruption of interconnectivity is a general response to mitochondrial dysfunction or specific to certain mutations, cell types, or conditions.
Author Contributions
Conceptualization, I.M., F.L., C.C., HZ; methodology, I.M., F.L., C.C.; software, I.M.; validation, H.Z. and M.K.; formal analysis, I.M.; investigation, I.M.; writing—original draft preparation, I.M.; writing—review and editing, H.Z, M.K.; visualization, I.M.; supervision, H.Z.; project administration, H.Z.; funding acquisition, I.M., H.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by a scholarship for doctoral candidates from the Faculty of Medicine at Otto von Guericke University Magdeburg and the Jürgen Manchot Foundation.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data will be provided upon reasonable request by the corresponding author.
Acknowledgments
We thank Professor Martin Zenker for co-supervising this project. We would also like to thank Nur Cengiz Winter and Andrea Delle Vedove for their assistance in the analysis of the microscopy images.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sullivan, E.D.; Longley, M.J.; Copeland, W.C. Polymerase γ Efficiently Replicates through Many Natural Template Barriers but Stalls at the HSP1 Quadruplex. Journal of Biological Chemistry 2020, 295, 17802–17815. [Google Scholar] [CrossRef] [PubMed]
- van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef] [PubMed]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the Mitochondrial DNA Replication Machinery in Mitochondrial DNA Mutagenesis, Aging and Age-Related Diseases. Ageing Res Rev 2017, 33, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Young, M.J.; Copeland, W.C. Human Mitochondrial DNA Replication Machinery and Disease. Curr Opin Genet Dev 2016, 38, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Hance, N.; Ekstrand, M.I.; Trifunovic, A. Mitochondrial DNA Polymerase Gamma Is Essential for Mammalian Embryogenesis. Hum Mol Genet 2005, 14, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Farnum, G.A.; Nurminen, A.; Kaguni, L.S. Mapping 136 Pathogenic Mutations into Functional Modules in Human DNA Polymerase γ Establishes Predictive Genotype–Phenotype Correlations for the Complete Spectrum of POLG Syndromes. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2014, 1837, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Fadic, R.; Johns, D. Clinical Spectrum of Mitochondrial Diseases. Semin Neurol 1996, 16, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Mitochondriopathies. Eur J Neurol 2004, 11, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.-J.C.; Naviaux, R.K.; Brunetti-Pierri, N.; Zhang, Q.; Schmitt, E.S.; Truong, C.; Milone, M.; Cohen, B.H.; Wical, B.; Ganesh, J.; et al. Molecular and Clinical Genetics of Mitochondrial Diseases Due to POLG Mutations. Hum Mutat 2008, 29, E150–E172. [Google Scholar] [CrossRef] [PubMed]
- Fuke, S.; Kametani, M.; Yamada, K.; Kasahara, T.; Kubota-Sakashita, M.; Kujoth, G.C.; Prolla, T.A.; Hitoshi, S.; Kato, T. Heterozygous Polg Mutation Causes Motor Dysfunction Due to Mt <scp>DNA</Scp> Deletions. Ann Clin Transl Neurol 2014, 1, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Hans, Z.; Bernard, S.; Natja, H. SM Gr up SM Journal of Neurology and Neuroscience Treatment Avenues for the Juvenile and Adult Onset Mitochondriopathies Alpers Syndrome, Ataxia Neuropathy Spectrum, MEMSA and PEO Caused by Polymerase-Gamma Mutations. SM Journal of Neurology and Neuroscience 2017, 3, 2–7. [Google Scholar]
- Stumpf, J.D.; Saneto, R.P.; Copeland, W.C. Clinical and Molecular Features of POLG-Related Mitochondrial Disease. Cold Spring Harb Perspect Biol 2013, 5, a011395–a011395. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, C.; Tran, G.T.; Coxhead, J.; Bertelsen, B.; Lilleng, P.K.; Balafkan, N.; Payne, B.; Miletic, H.; Chinnery, P.F.; Bindoff, L.A. Molecular Pathogenesis of Polymerase Gamma–Related Neurodegeneration. Ann Neurol 2014, 76, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.S.L.; Longley, M.J.; Copeland, W.C. The Common A467T Mutation in the Human Mitochondrial DNA Polymerase (POLG) Compromises Catalytic Efficiency and Interaction with the Accessory Subunit. Journal of Biological Chemistry 2005, 280, 31341–31346. [Google Scholar] [CrossRef] [PubMed]
- Hakonen, A.H.; Heiskanen, S.; Juvonen, V.; Lappalainen, I.; Luoma, P.T.; Rantamäki, M.; Goethem, G. Van; Löfgren, A.; Hackman, P.; Paetau, A.; et al. Mitochondrial DNA Polymerase W748S Mutation: A Common Cause of Autosomal Recessive Ataxia with Ancient European Origin. The American Journal of Human Genetics 2005, 77, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Winterthun, S.; Ferrari, G.; He, L.; Taylor, R.W.; Zeviani, M.; Turnbull, D.M.; Engelsen, B.A.; Moen, G.; Bindoff, L.A. Autosomal Recessive Mitochondrial Ataxic Syndrome Due to Mitochondrial Polymerase Mutations. Neurology 2005, 64, 1204–1208. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R. Phenotypic Spectrum Associated with Mutations of the Mitochondrial Polymerase Gene. Brain 2006, 129, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, M.-E.; Ng, Y.S.; Taylor, R.W.; McFarland, R. Epilepsy Due to Mutations in the Mitochondrial Polymerase Gamma (POLG) Gene: A Clinical and Molecular Genetic Review. Epilepsia 2016, 57, 1531–1545. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Zhu, J.; Kulich, S.M.; Chu, C.T. Mitochondrially Localized ERK2 Regulates Mitophagy and Autophagic Cell Stress. Autophagy 2008, 4, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Dagda, R.K.; Chu, C.T. Monitoring Mitophagy in Neuronal Cell Cultures. In; 2011; pp. 325–339.
Figure 1.
Brightfield microscopy picture from fibroblasts (passage 4) from the patient and the relative show reduced growth in the patient cell line. The fibroblasts of the patient (A) are less confluent and have a polyclonal shape. The fibroblasts of the carrier (B) grow faster, are more confluent and have more of a linear shape. Scale bar: 50 μm.
Figure 1.
Brightfield microscopy picture from fibroblasts (passage 4) from the patient and the relative show reduced growth in the patient cell line. The fibroblasts of the patient (A) are less confluent and have a polyclonal shape. The fibroblasts of the carrier (B) grow faster, are more confluent and have more of a linear shape. Scale bar: 50 μm.
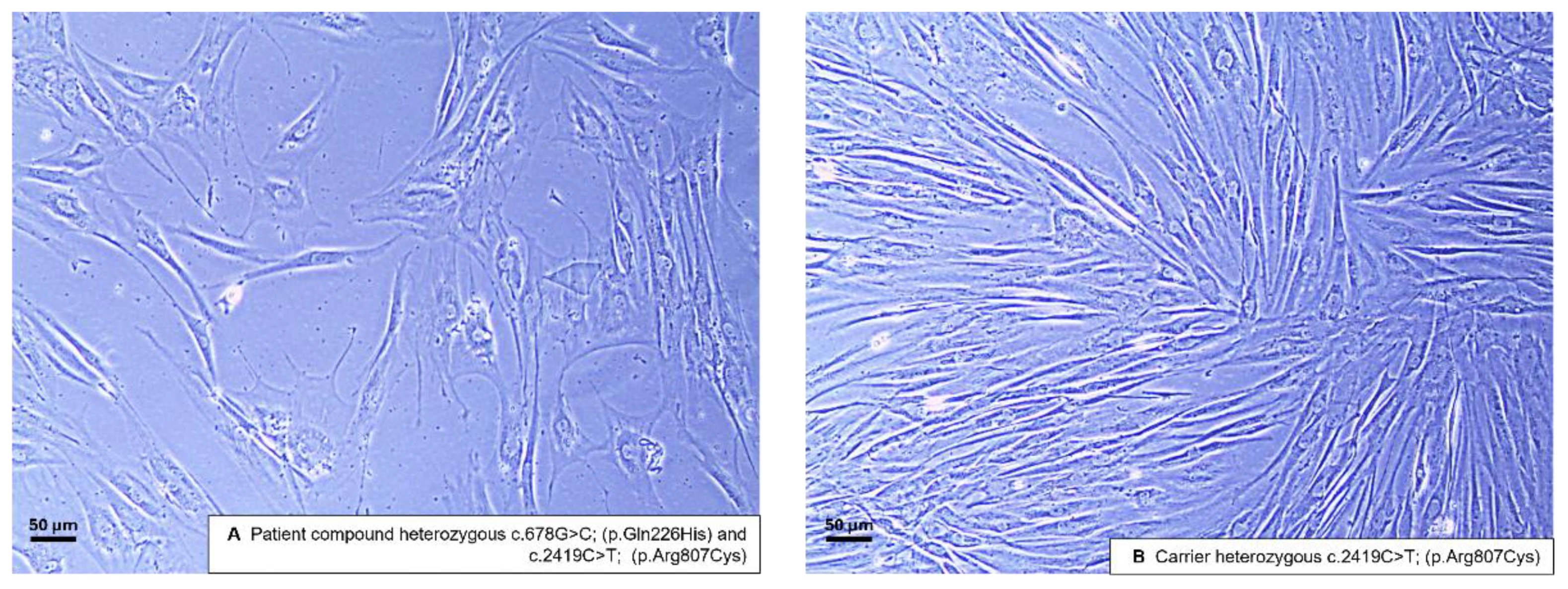
Figure 2.
c.678G>C; p.(Gln226His) fibroblasts show reduced mtDNA levels and mtDNA encoded mRNA levels as determined by RT-qPCR. Fibroblast of the patient and the relative were lysed and DNA/ RNA was isolated to perform RT-qPCR analysis. E shows the relative mtDNA content of the patient (red) and the carrier (green): Reduction of mtDNA content ~ 50 %. B-D show mRNA levels of the two cell lines of the mtDNA encoded genes. B displays MT-CYTB: mRNA levels are reduced by ~ 75 %. C displays MT-ATP6: mRNA levels are reduced by ~ 70 %. D displays MT-ND1: mRNA levels are reduced by ~ 45 %.
Figure 2.
c.678G>C; p.(Gln226His) fibroblasts show reduced mtDNA levels and mtDNA encoded mRNA levels as determined by RT-qPCR. Fibroblast of the patient and the relative were lysed and DNA/ RNA was isolated to perform RT-qPCR analysis. E shows the relative mtDNA content of the patient (red) and the carrier (green): Reduction of mtDNA content ~ 50 %. B-D show mRNA levels of the two cell lines of the mtDNA encoded genes. B displays MT-CYTB: mRNA levels are reduced by ~ 75 %. C displays MT-ATP6: mRNA levels are reduced by ~ 70 %. D displays MT-ND1: mRNA levels are reduced by ~ 45 %.
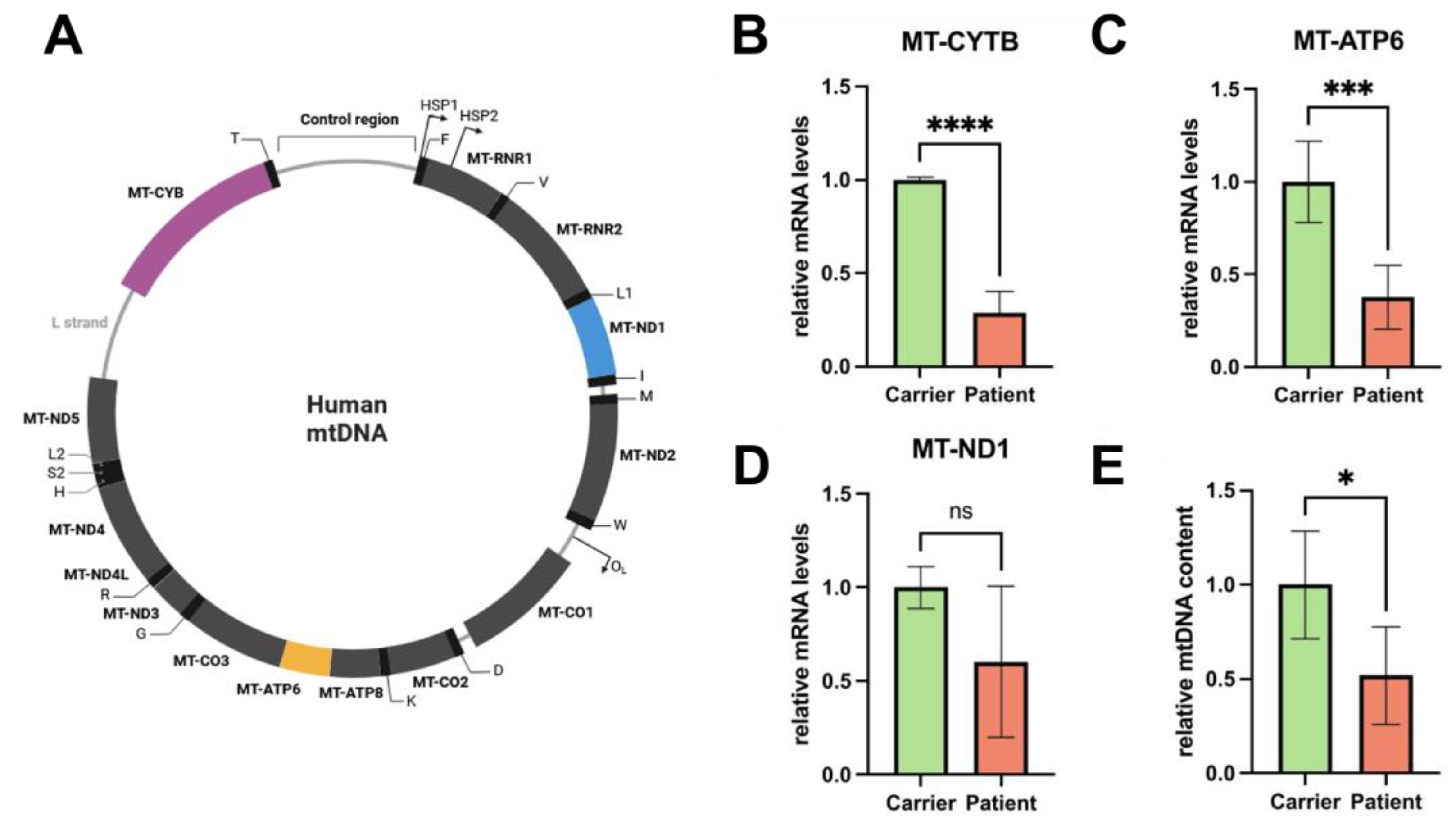
Figure 3.
Immunofluorescent Microscopy of fibroblasts visualizing mitochondria with MitoTracker™ showing reduced mitochondrial mass and interconnectivity of mitochondria. The fibroblasts were fixed and stained with MitoTracker™ (red colour) to display mitochondria and Phalloidin (yellow colour) to recognize the cell size. A Representative image of the patient fibroblast (upper panels) and the carrier (lower panels). B The relative area (also referred to as mitochondrial mass) is measured by calculating the area of mitochondria divided by the cell area (stained with CellTracker Phalloidin™): the relative area is reduced by ~ 20 %. C The interconnectivity is calculated by dividing the mean area of the single mitochondria by the mean perimeter of one mitochondria: the interconnectivity is reduced by ~ 50 %. Scale bar: 10 μm.
Figure 3.
Immunofluorescent Microscopy of fibroblasts visualizing mitochondria with MitoTracker™ showing reduced mitochondrial mass and interconnectivity of mitochondria. The fibroblasts were fixed and stained with MitoTracker™ (red colour) to display mitochondria and Phalloidin (yellow colour) to recognize the cell size. A Representative image of the patient fibroblast (upper panels) and the carrier (lower panels). B The relative area (also referred to as mitochondrial mass) is measured by calculating the area of mitochondria divided by the cell area (stained with CellTracker Phalloidin™): the relative area is reduced by ~ 20 %. C The interconnectivity is calculated by dividing the mean area of the single mitochondria by the mean perimeter of one mitochondria: the interconnectivity is reduced by ~ 50 %. Scale bar: 10 μm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



