Submitted:
07 November 2024
Posted:
08 November 2024
You are already at the latest version
Abstract
Despite great progress in the treatment of atrial fibrillation (AF), especially with the development of increasingly effective invasive techniques, many questions remain unanswered regarding the pathogenic mechanism of the arrhythmia and its prevention methods. The development of AF is based on anatomical and functional changes in the myocardium that result from disturbed ion flows and altered electrophysiology of the cardiomyocyte. Electrical instability and electrical remodeling underlying arrhythmia may result from oxidative stress, caused by mitochondrial dysfunction. The role of mitochondrial dysfunction in the pathogenesis of AF is not yet completely clarified; however, it is emphasized by the reduction of AF burden after therapeutic interventions that improve mitochondrial well-being. This selected review aims to summarize the mechanisms of mitochondrial dysfunction related to AF and current pharmacological treatment options tar-geting mitochondria to prevent or improve the outcome of AF.
Keywords:
atrial fibrillation
; mitochondrial dysfunction
; arrhythmias
; oxidative stress
1. Introduction
Mitochondria have critical role in the cellular homeostasis. They represent the “fuel” of the cell and so in case of mitochondrial disfunction there would be significant and severe alterations. In addition to specific mitochondrial syndromes, mitochondrial alterations can determine the onset of diseases with multifactorial pathogenesis, in which mitochondria can play a role. Interestingly mitochondria could also contribute to the development of arrythmias. This selected review aims to verify whether there is a connection between mitochondrial dysfunction and the development of atrial fibrillation (AF) and whether mitochondria can become a therapeutic target of AF.
2. Role of the Mitochondria
The mitochondria are essential organelles present in nucleated cells, involved in adenosine triphosphate (ATP) production. The oxidative phosphorylation system includes five protein complexes and two factors (coenzyme Q10 and cytochrome c) which produce ATP through electron transfer along inner membrane of mitochondria. As a byproduct of phosphorylation, mitochondria also represent the main source of radical of oxygen species (ROS), which in great amount could lead to mitochondrial and cell dysfunction. Mitochondrial disease can be linked by cardiovascular risk factors or include complex and massive genetic disorders caused by dysfunction of mitochondria [1] Several cardiovascular risk factors, such as hyperglycemia can cause augmented ROS (Table 1). In addition, mitochondrial deoxyribonucleic acid (DNA) covers only a little number of mitochondrial proteins; about 1500 proteins essential to the respiratory chain are encoded by nuclear DNA. So mitochondrial disorders are mainly caused by mutation in nuclear DNA, and they are inherited according to Mendelian Model (including autosomal recessive, dominant and X-linked pattern with the autosomal recessive being the most common modality of transmission) [2]. The genetic basis of Mitochondriopathies is extremely various, since some diseases are caused by a single gene mutation and others are secondary to multiple gene mutation, such as in Leigh syndrome in which 75 genes can be involved [3]. Moreover, concerning the mitochondrial DNA, another source of genetic heterogeneity is caused by heteroplasmy. Heteroplasmy refers to a combination of mutated and wild type mitochondrial DNA molecules inside human cells. The pathological phenotype could occur only in case of more than 60% of mutated DNA molecules (with a kind of threshold effect) [1,2]. In addition to these general mechanisms of AF pathogenesis, there are studies demonstrating a primary role of mitochondrial DNA dysfunction in the pathogenesis of AF. Yamazoe et al. aimed to evaluate the level of cell-free DNA (cfDNA) in AF patients and AF-mimicking models and to clarify its impact on inflammation. Nuclear and mitochondrial DNA were extracted separately and fragmented to mimic nuclear cfDNA (n-cfDNA) and mitochondrial cfDNA (mt-cfDNA). The AF group showed a higher cfDNA concentration than the non-AF group (p < 0.001). The copy numbers of n-cfDNA and mt-cfDNA were higher in the AF groups than in the non-AF groups; the difference particularly of mt-cfDNA was evident (p = 0.011 and p < 0.001, respectively). Administration of total cfDNA and mt-cfDNA to macrophages significantly promoted the expression of IL-1β and IL-6 via TLR9, whereas n-cfDNA did not. The induction of cytokine expression by methylated mt-cfDNA was lower than that by unmethylated mt-cfDNA. Collectively, AF was associated with an increased level of cfDNA, particularly mt-cfDNA. Poorly methylated mt-cfDNA released from cardiomyocytes may be involved in the sterile systemic inflammation accompanied by AF [3].
4. Acquired Mitochondrial Dysfunction
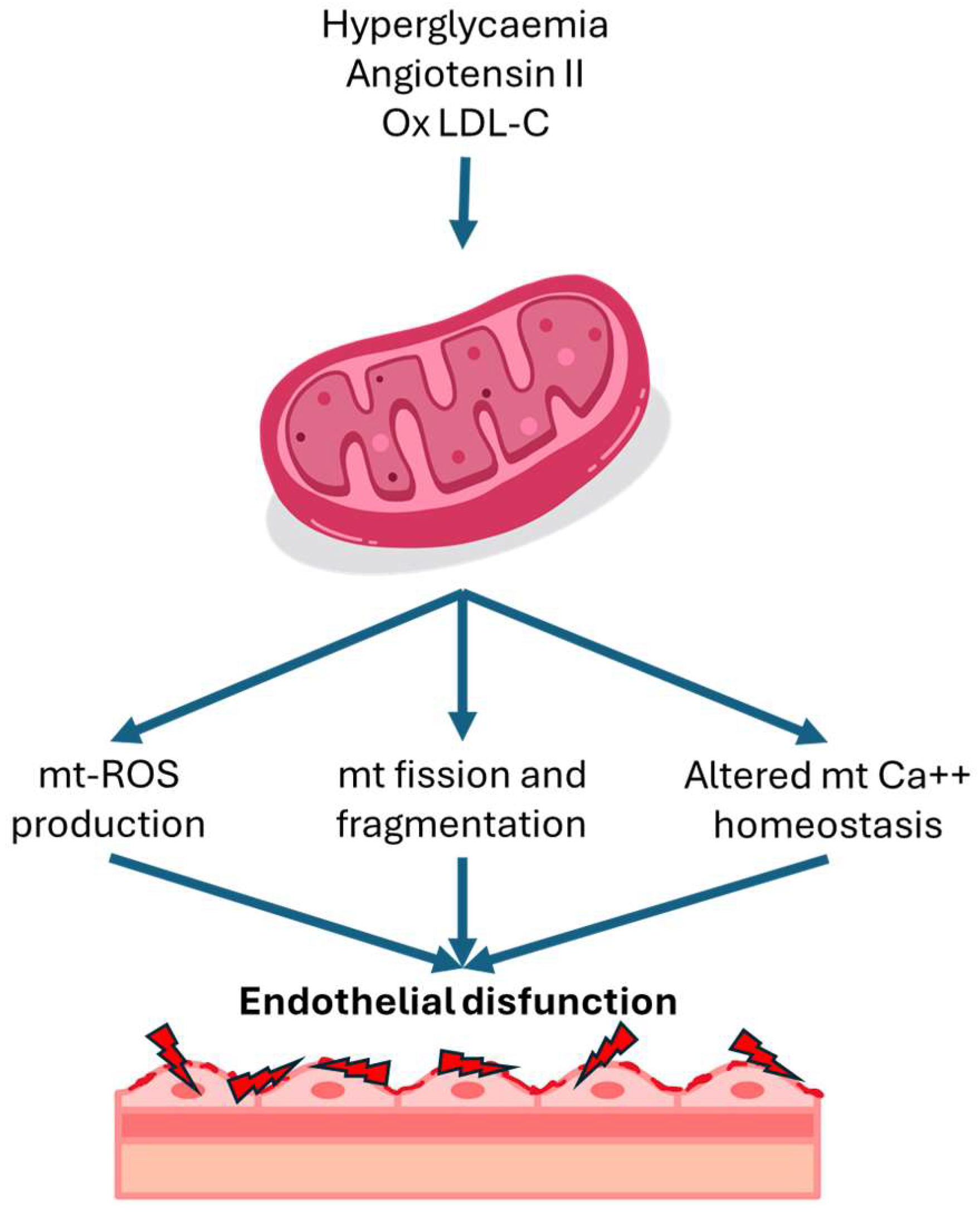
In addition of genetic disorders of mitochondria, several cardiovascular risk factors can cause augmented ROS production in endothelial cells. The main proven risk factors are hyperglycemia, increased angiotensin II and oxidized LDL. Hyperglycaemia has been shown to trigger an overproduction of mitochondrial ROS [11]. Under normal circumstances, protons are shuttled into the intermembrane space of mitochondria during the electron transport chain, establishing a proton gradient that fuels ATP synthesis. However, in high intracellular glucose levels, additional electron donors like nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide hydride (FADH)-2 enter the electron transport chain, boosting oxidation in the tricarboxylic acid cycle. This culminates in an increased voltage gradient across the mitochondrial membrane until it reaches a critical threshold. When this threshold is surpassed, electron transfer within complex III of the electron transport chain is blocked, causing electrons to revert to Coenzyme Q and subsequently donate electrons to oxygen molecules, thereby generating superoxide [4]. Angiotensin II induces mitochondrial dysfunction through a protein kinase C dependent pathway by activating endothelial nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2). In this process, mitochondrial protein kinase C (PKC) epsilon is a downstream target of NOX2, and it activates mitochondrial ATP-sensitive potassium channels, leading to mitochondrial reverse electron transfer and subsequent generation of superoxide [12]. In addition to the factors mentioned above, several other cardiovascular risk factors, including oxidized low-density lipoprotein (LDL) and turbulent flow has been associated to an increase in mitochondrial ROS levels in endothelial cells [6].
Figure 1.
Mitochondrial alterations in endothelial disfunction. LDL-C: lipoprotein low density-concentration; Mt: mitochondrial; ROS: reactive oxygen species
Figure 1.
Mitochondrial alterations in endothelial disfunction. LDL-C: lipoprotein low density-concentration; Mt: mitochondrial; ROS: reactive oxygen species
5. Cellular Alterations in the Heart and Electrogenesis of Atrial Fibrillation
The development of an arrhythmia is related to several mechanisms i.e., augmented or reduced automaticity, triggered activity, and re-entrant circuit [13]. The underlying mechanisms initiating and promoting AF are incompletely resolved. However, calcium (Ca2+) handling abnormalities and oxidative stress are thought to play central roles in the pathophysiology of AF. Mitochondria are the main producers of cellular adenosine triphosphate (ATP) in cardiac myocytes, and both Ca2+ and adenosine diphosphate (ADP) are key regulators of respiratory flux to match energy supply to the constantly varying demands in the heart [14].
5.1. ATP Homeostasis, ROS Production and Atrial Fibrillation
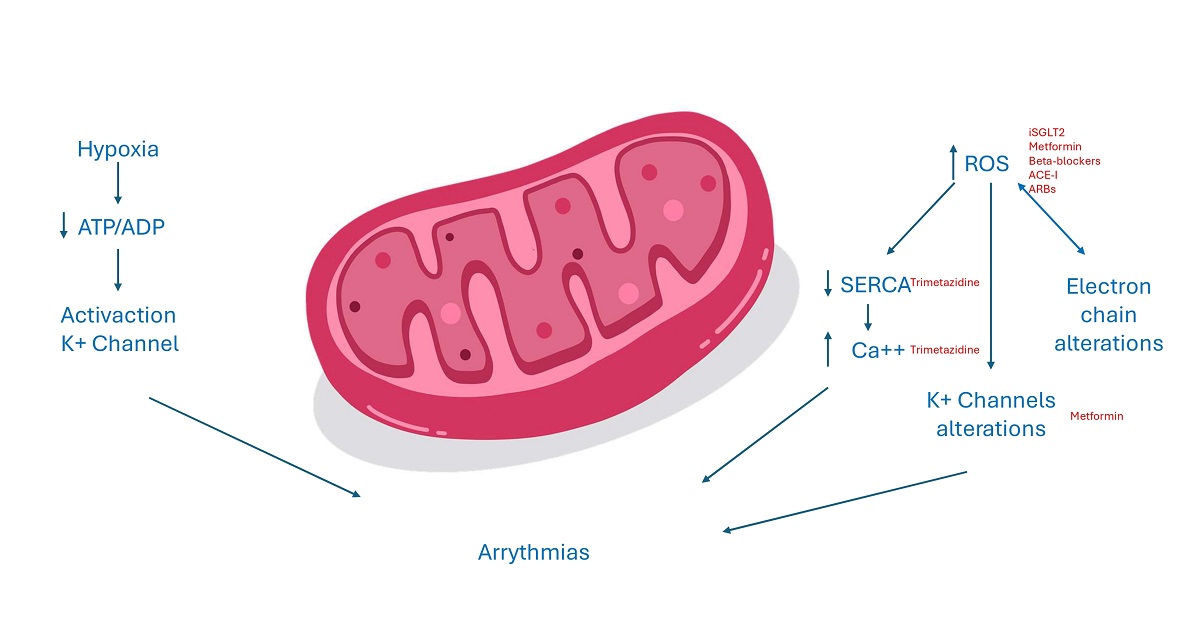
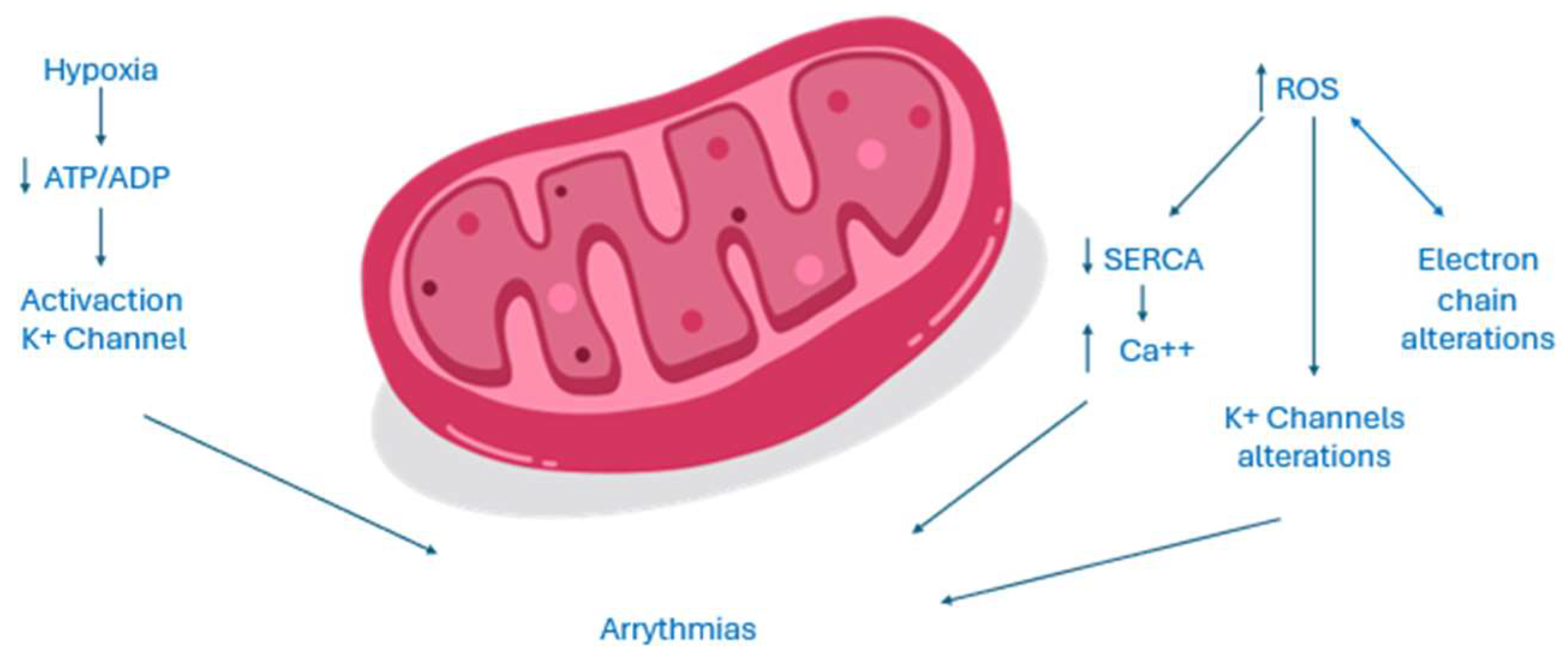
The dysfunction of mitochondria has a negative impact since it could reduce ATP production and at same time increase ROS production. The reduction of ATP source alters all enzymes and ion transporters which are ATP dependent and so the normal cell excitability. An increased ADP/ATP ratio as an acidosis status, or variation of magnesium concentration, all conditions present in case of myocardium ischemia, could in fact activated sarcKATP channels. The inwardly potassium current causes a reduced action potential duration and could be cytoprotective since it reduces calcium overload. However, this could be detrimental too, increasing risk of arrhythmias since an open status of these channels shortens duration of action potential and the duration of effective refractory period and creates an electrical dispersion which increases the risk of re-entrant arrhythmias. Moreover, the increase of ROS, could activate a maladaptive response in the long term and could alter normal gene expression, inducing cell death [13]. ROS could increase the release of calcium interfering with ryanodine receptor 2 (RyR2), thus inhibiting the activity of Sarco-Endoplasmic Reticulum Calcium ATPase (SERCA). SERCA is the main channel involved in diastolic relaxation of myocardium, absorbing calcium into reticulum. Moreover, ROS induces a kind of vicious circle, in which the damage of all the components of transfer electron chain and ion channels leads to an increased production of ROS too. The final effect is an intracellular calcium overload, which represents a proarrhythmic stimulus [15]. Furthermore, oxidative stress increases arrhythmic burden activating late Sodium current, which leads to early after depolarizations, prolonging duration of action potential. ROS could also interfere with potassium balance in different ways. They could in fact modify gene expression of potassium currents but also could alter potassium channels functioning, acting on kinase or phosphatase proteins which regulate the phosphorylation status of these channels. The cell integrity and the normal function of myocardial syncytium is essential, especially to lead to transmission of cardiac impulse. The linkage between cardiomyocytes is regulated by connexins, present in three different isoforms in myocardial tissue. An increased stimulus to ROS production that may be secondary to different pathological scenarios associated to activation of RAS system causes a downregulation of connexin 43. The dysregulation of gap junctions could impair electrical conduction, increasing arrhythmic events through an increased electrical dispersion [13,15]. Recent publications have explored the correlation between vascular function and AF, suggesting the involvement of endothelial dysfunction in AF development [15,16,17,18]. Endothelial dysfunction has been demonstrated to contribute to the initiation and perpetuation of the atrial arrhythmic substrate regulating immune cell infiltration and inflammation within the cardiac tissue, augmenting the fibrous burden of the atria [19,20] and promoting oxidative stress through the overproduction of reactive oxygen species with known arrhythmogenic effects [19].
Figure 2.
Electrogenesis of AF in mitochondrial dysfunction.
6. Drugs with Mitochondrial Effects
Current medications and nutraceutical products utilized in cardiovascular medicine target these issues, with existing in vitro and animal studies indicating positive impacts on mitochondrial function and cellular metabolism. Nevertheless, to broaden their clinical applications in this setting, rigorous randomized double-blind placebo-controlled trials specifically within the context of AF are imperative for medications that influence mitochondrial function. These trials are essential to validate and establish the efficacy of such interventions, paving the way for wider adoption in clinical practice [21]. Therapeutic strategies and their respective main mechanism of influence on mitochondrial function and AF are resumed in Table 3.
6.1. Oral Hypoglycaemic Agents
Oxidative stress and mitochondrial disfunction due to chronic hyperglycaemia are a crucial element in the pathogenesis and progression of diabetes and its complications [22]. Though it is not by chance that many studies about the effects of oral hypoglycaemic agents on oxidative damage were conducted.
SGLT2 inhibitors, targeting the sodium-glucose co-transporter 2, enhance glycaemic control, induce natriuresis and uricosuria. Beyond diabetes management, these inhibitors demonstrate favourable cardiovascular outcomes, especially in heart failure (HF) and chronic kidney disease (CKD). Subgroup analyses indicate benefits in preventing arrhythmias, including AF, suggesting potential pleiotropic effects in cellular metabolism and mitochondrial function. Li et al. demonstrated that in diabetic mice the use of Empagliflozin suppressed oxidative stress and myocardial fibrosis by inhibiting the transforming growth factor- beta (TGF-beta/Smad) pathway and activating the nuclear factor erythroid 2–related factor 2/antioxidant responsive element (Nrf2/ARE) signalling [23]. In Shao et al.‘s study, SGLT2 inhibition with empagliflozin restores mitochondrial membrane potential enhances the respiratory rate and promotes mitochondrial biogenesis through increased expression of Peroxisome proliferator-activated receptor-gamma coactivator 1 (PGC-1), Nuclear respiratory factor-1 (NRF-1), and Mitofusin-1 (Mfn-1). This improvement correlates with reduced reactive oxidative species (ROS) synthesis, systemic inflammation, atrial fibrosis, and cardiomyocyte hypertrophy, resulting in a significant 36.8% reduction in tachypacing-induced AF susceptibility [24]. Analog results were found by Koizumi et al. that the treatment lead to a reduction in inflammation, atrial fibrosis, atrial tachyarrhythmia inducibility, atrial tachyarrhythmia duration and normalized the interatrial conduction time, in parallel with a decrease in mitochondrial ROS generation, increase in superoxide dismutase (SOD) activity and increase in mitochondrial content through adenosine monophosphate-activated protein kinase (AMPK) signalling pathways [25]. Recently Zhao et al. analysed the effects of Dapagliflozin in rats with a sepsis-like condition and demonstrated significant improvement in myocardial injury and susceptibility to AF correlated with the activation of the Nrf2 signalling pathway, leading to a reduction in oxidative stress [26].
Glucagon-like peptide-1 (GLP1)-RA acts as analogues of the hormone GLP1, which stimulates glucose-dependent insulin secretion. Nuamnaichati et al. demonstrated that GLP-1 receptor activation has antioxidant and antiapoptotic effects, reducing intracellular and mitochondrial ROS production in cardiomyoblasts. This improvement is mediated through the Phosphoinositide 3-kinases/Protein kinase B (PI3K/PKB) signalling pathway [27]. In addition, GLP1- ameliorated Interleukin-1 beta (IL-1β)-induced ROS production, reduced nicotinamide adenine dinucleotide phosphate oxidase-4 (NOX-4) expression and increased the expression of SOD-1 and glutathione peroxidase [28,29]. In diabetic animal models, the amelioration of mitochondrial function due to GLP1-RA administration led to a significant reduction in collagen deposition, improving cardiac remodelling, diastolic function, and the incidence of AF [28] However, a meta-analysis showed no significant reduction of the risk of major arrhythmias in more than 60,000 diabetic patient treated with GLP1-RA, including AF [relative risk (RR)=0.96, 95% confidential interval (CI) (0.86, 1.07), p =0.43] [29] and in a French cohort study showed an increased risk of AF in patients treated with GLP1-RA [30]
The Dipeptidyl peptidase-4 (DPP-4) inhibitors belong to a novel class of oral antidiabetic medications that target the incretin system. DPP-4 is an enzyme responsible for deactivating gastric inhibitory polypeptide (GIP) and GLP-1. The nationwide cohort study taking place in Taiwan and including over 90.000 diabetics patients already treated with metformin showed that the use of DDP4i as a second-line drug (mostly Sitagliptin) lowered the risk of new-onset AF compared with non-DPP4i users [hazard ratio (HR): 0.65; 95% confidential interval (CI) 0.56–0.76; P < 0.0001] [31]. In vitro studies demonstrated that the expression of DDP-4 is augmented in cells exposed to hypoxic conditions leading to the generation of ROS and inner mitochondrial membrane potential reduction. Therefore, inhibition of this enzyme could have cardioprotective effects mostly due to attenuation of oxidative stress and amelioration of mitochondrial function [31]. Zhang et al. found that in rabbits with alloxan-induced diabetes mellitus (DM), the DDP-4 inhibitor Alogliptin demonstrated significant protective effects on mitochondria, reducing the production rate of mitochondrial reactive oxygen species, preventing membrane depolarization, and alleviating mitochondrial swelling. Additionally, Alogliptin improved mitochondrial biogenesis through the adiponectin/AMP-activated protein kinase pathway, specifically activating PGC-1/NRF1 signalling. These positive mitochondrial modifications contributed to a reduction in AF inducibility [32]. Positive effects were observed also for Linagliptin in a canine AF model in which the treatment determined a suppression of the inducibility of AF and atrial fibrosis, alongside with a suppression of the ROS expression [33]. However, in the recent meta-analysis conducted by Patoulias et al. involving 52520 patients from 6 trials comparing DPP-4i with placebo, the treatment did not seem to confer any significant cardiovascular benefits and did not affect the risk for AF (RR = 0.95, 95% CI: 0.78-1.17, I 2 = 0%), while they were associated with a significant increase in the risk for atrial flutter, equal to 52% (RR = 1.52, 95%CI: 1.03-2.24, I2 = 0%) [34].
In the 2022 Chan et al. published a meta-analysis including 2,826,059 patients in which SGLT2i treatment was associated with lower risk of new-onset AF in participants with type 2 DM compared with either DPP4i [hazard ratio (HR):0.90; 95% CI 0.84–0.96; P = 0.0028] or GLP-1RA [HR 0.74; 95% CI 0.63–0.88; P = 0.0007], with no statistically significant difference between GLP-1RA and DDP4i [HR 1.01; 95% CI 0.86–1.19; p= 0.8980,35].
Metformin primarily exerts its metabolic effects in the liver, where it reduces glucose and lipid synthesis through the phosphorylation and activation of 5′ AMP-activated protein kinase (AMPK). Furthermore, AMPK activation leads to glucose transporter-4 mediated glucose uptake, contributing to higher systemic insulin sensitivity. The impact of metformin on atrial remodelling has been investigated, as evidenced by a study analysing data from 645,710 patients with DM2 over a 13-year follow-up period from the Taiwan National Health Insurance Research Database. The findings revealed a 19% decrease in the incidence of AF with metformin use [HR 0.81, 95% CI 0.76-0.86, p < 0.001)] [36]. Experimental studies involving pacing-induced AF offer insights into the possible molecular mechanism of this evidence. Metformin activated AMPK, Src kinase, and normalized connexin expression, reducing pacing-induced AF effects and preventing atrial remodelling by activating the AMPK/PGC-1/peroxisome proliferator-activated receptor(PPAR)-pathway. Metformin’s electronegativity concentrates it in mitochondria, influencing cellular energy metabolism, and preserving mitochondrial function, with improved oxygen consumption and augmented activity of complexes I, II and IV and upregulation of PGC-1alfa [19,37].
Thiazolidinediones (TZDs) constitute a medication group in DM treatment, impacting mitochondrial function by acting as PPAR- agonists to reduce insulin resistance. Studies about their effect in AF showed conflicting results. Clinical evidence from Danish nationwide registries published by Pallisgaard et al., with over 100,000 diabetic patients, revealed that TZDs reduced AF incidence by 24%, adjusted for age, sex, and comorbidities, compared to other second-line antidiabetic treatments [HR 0.76, 95% CI 0.57–1.00, p = 0.047) [38]. A meta-analysis combining three randomized clinical trials and four observational studies supported the preventive potential of TZDs against AF showing an overall reduction of 30% [odds ratio (OR): 0.73, 95% CI 0.62-0.87, p = 0.0003], with a 23% reduction in new-onset AF and a 59% risk reduction in AF recurrence [38]. Animal models such as alloxan-induced DM in rabbits, demonstrated that TZDs attenuated atrial remodelling, reduced AF inducibility, and improved ion channel function (ICa and INa) [39]. Xu et al. found that pioglitazone pretreatment decreased AF duration and age-related atrial remodelling, suggesting protective mechanisms involving upregulation of antioxidant pathways and inhibition of mitochondrial apoptotic signalling [40]. However, in patients with coronary artery disease undergoing TZD or other second-line medications during a median follow-up of 4.2 years, TZDs did not affect AF prevalence [41]. Moreover, in a small randomized prospective study, TZDs did not affect AF recurrence after electrical cardioversion [42].
6.2. Hypolipidemic Drugs
Statins act as 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors and are the cornerstone of treatment in dyslipidaemias. Beyond the effect on lipidic metabolism, Statins also exert a pleiotropic cardioprotective effect. Statins, theoretically, can offer protection against AF not only by diminishing the burden of vascular disease but also by addressing atrial remodelling thanks to anti-inflammatory, anti-oxidative, anti-proliferative, and antithrombotic actions. Additionally, statins may enhance endothelial function and neurohormonal regulation [43]. According to the Taiwan National Health Insurance research database, statins demonstrated a 13% decrease in the risk of new-onset AF [HR: 0.935; 95% CI: 0.877–0.998; p =0.0427] [44]. However, a Spanish registry study by Cabratosa-Alves et al. reported only minimal protective effects of statins against new-onset AF in cases of lone hypertension without vascular ischemic disease [45]. A meta-analysis conducted by Fang et al. revealed an overall significant reduction in the risk of AF incidence/recurrence [odds ratio (OR)=0.49, 95% CI 0.37-0.65; p < 0.00001]; this effect appeared to be more pronounced in secondary prevention [OR=0.34, 95% CI 0.18-0.64; p < 0.0001] than in primary prevention [OR=0.54, 95% CI 0.40-0.74; p < 0.0001] [46]. Inflammation and abnormal oxidative stress are identified as key pathophysiological features linked to atrial remodeling and heightened myocardial tissue inflammation, leading to AF onset, recurrence, and persistence [43]. The mechanisms by which Statins exert their antioxidant action are suppression of the activity of Rhodopsin/Rho-associated protein kinase (Rho/ROCK) pathways, activation of PI(3)K/PKB pathway, inhibition of GTPase Ras-related C3 botulinum toxin substrate 1 (Rac1) required for nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) activity, and of the expression of mRNA expression of NADPH oxidase subunits (Nox1, p22phox) and indirectly by reduction of pro-inflammatory cytokines production [43].
Recent evidence suggests a potential association between Proprotein Convertase Subtilisin/Kexin type 9 (PCSK9) and oxidative stress, particularly about oxidized LDL-induced endothelial cell apoptosis and regulation of PCSK9 expression by ROS. Furthermore, mitochondrial ROS appears to influence the interaction between lectin-type oxidized LDL receptor 1 (LOX-1) and PCSK9, key players in atherosclerosis. Evolocumab, a PCSK9 inhibitor, has demonstrated significant reductions in oxidative stress-related cytotoxicity and atherosclerotic progression in both cellular and animal models. Safeian et al. found that Evolocumab led to a significant reduction in cytotoxicity induced by H2O2 in human umbilical vein endothelial cells [47]. In a mice model Evolocumab lowered atherosclerotic progression reducing oxidative stress through promoting macrophage autophagy [48]. It was proposed that the antioxidant activity of PCSK9i may be at least in part mediated by NAD-dependent deacetylase sirtuin-3 (SIRT3), which counteracts mitochondrial ROS accumulation [49]. A significant reduction in leucocytes ROS production (H2O2) was observed in a sample of 18 hypercholesterolemic patient after 2 weeks of treatment with Evolocumab (p-value = 0.004) [50]. However, the antioxidant effects of PCSK9 inhibitors have not yet been investigated in the context of AF.
Fibrates, acting as PPAR-alfa agonists, are commonly utilized to address hypertriglyceridemia, decrease hepatic apolipoprotein C-III (apoC-III) levels, and enhance lipoprotein lipase-mediated lipolysis. By operating on PPAR/PGC-1 pathway these medications influence mitochondrial function. In experimental AF models, fenofibrate contributes to mitigating metabolic remodelling by regulating the PPAR-/sirtuin route 1/PGC-1, effectively reversing the shortening of the atrial refractory period [51].
Omega-3 fatty acids are used in the treatment of hypertriglyceridemia and exhibit potent anti-inflammatory effects by replacing arachidonic acid in cell membranes especially eicosapentaenoic acid and docosahexaenoic acid. The direct impact of omega-3 fatty acids on ionic channels, coupled with their modulation of the cell membrane properties, could influence the occurrence of AF. Supplementation with these fatty acids affects ion channel function, influencing cardiac action potentials, stabilizing electrical activity, and prolonging the refractory period of cardiomyocyte thanks to the ability to reduce oxidative stress in heart cells and to preserve cells integrity. However, the efficacy of omega-3 fatty acids in preventing AF may vary based on individual conditions and clinical backgrounds [51]
6.3. Others
Trimetazidine (TMZ), an approved anti-anginal drug for treating ischemic heart disease, exerts beneficial effects by enhancing cellular energetic balance. It achieves this by inhibiting long-chain 3-ketoacyl Coenzyme A thiolase, a key player in mitochondrial fatty acid oxidation, redirecting mitochondrial substrate utilization toward glucose, thereby improving ATP synthesis. However, it seems to influence many aspects of mitochondrial function [21]. In ischemic conditions, Trimetazidine (TMZ) directly activates complex I in the respiratory chain, enhancing electron transport chain (ETC) function with a significant reduction in the production of ROS; Additionally, in ischemic conditions, TMZ normalizes the expression of factors regulating mitochondrial biogenesis, such as PPAR and PGC-1α, and adjusts the expression of Mfn-1, Dystrophin Related Protein-1 (Drp1), and Mitochondrial dynamin like GTPase (Opa-1), impacting mitochondrial fusion/fission dynamics [52]. In addition, preclinical studies suggested an antiarrhythmic activity. Using a dog model of HF Li et al. suggest that TMZ may prevent tachycardia-induced atrial ultrastructural remodelling, decrease AF inducibility, and shorten AF duration [53]. However, it remains unclear if TMZ’s protective effects in non-ischemic conditions are related to improvements in mitochondrial function [21].
Ranolazine functions mainly as a late sodium channel influx inhibitor during repolarization, leading to decreased intracellular sodium and calcium concentrations, and so to reduced oxygen consumption. Additionally, ranolazine is thought to act as a partial fatty acid oxidation inhibitor, contributing to the attenuation of oxidative stress. By the action on sodium and potassium channel Ranolazine showed antiarrhythmic proprieties, particularly at the atrial level. In a meta-analysis the combination of ranolazine to amiodarone significantly increased the sinus rhythm restoration rate compared to amiodarone alone (RR=2.87, 95% CI 2.48–3.32, p<0.05), in patients with LV systolic dysfunction [54]. Ranolazine’s suggested mechanism of action involves extending post-repolarization refractoriness and slowing conduction velocity. Beyond these electrophysiological effects, studies have demonstrated that ranolazine enhances mitochondrial function, mitigates oxidative stress, and inhibits apoptosis [21]
Carvedilol exhibits a mild β1-blocking selectivity that transitions to non-selective at elevated doses. Additionally, it possesses α1-blocking and antioxidant attributes, influencing diverse ion channels and currents. Notably, it outperforms selective β1 blockers like metoprolol and atenolol in the suppression of postoperative AF. The proposition that carvedilol excels over other beta-blockers in AF treatment is partially elucidated by its antioxidative effects [51].
In atrial tissues of individuals AF increased levels of angiotensin II (Ang II), Ang II receptor, aldosterone, and an augmented activity of angiotensin-converting enzyme (ACE) was found [43]. As already mentioned in section 4, the expression of ATII could led to ROS production. Thereby inhibiting Ang II production aids in reducing oxidative stress (OS) in vascular structures [51]. Moreover the ATII-induced production of H2O2 triggers the binding action of nuclear factor kappa B (NF-κB) to the sodium channel protein type 5 subunit alpha (SCN5A), reducing the inward current of sodium (INa). The downregulation of SCN5A and INa altered the cellular electrical propriety promoting arrhythmias. This can be prevented by renine-angiotensine system (RAS) inhibition with Angiotensin converting enzyme inhibitors (ACE-I) or Angiotensin II receptor type 1 (AT1R) blocker [43]. Recently Zhao et al. highlight the favorable effects of aliskiren (ALS) in attenuating atrial remodeling and diminishing susceptibility to AF by reducing inflammation and oxidative stress via PI3K/PKB, in dogs subjected to tachycardic pacing [55].
Xanthine oxidase (XO) plays an instrumental role in redox signalling across various cardiovascular disorders, particularly emphasizing its involvement in AF. Though the effects of drugs that act on XO was thought to be beneficial [51]. In their investigation, Xu et al. explored the impact of Febuxostat and allopurinol, XO inhibitors, on AF susceptibility. Both Febuxostat and Allopurinol demonstrated significant suppression of atrial remodelling associated with hypertension and the perpetuation of AF in a murine model. This potential effect was attributed to the inhibition of the ox-Ca2+-calmodulin-dependent protein-kinase type-II (CaMKII) signalling pathway, a regulator of heart contraction. Moreover, Febuxostat exhibited antioxidant effects by directly combating reactive oxygen species (ROS) [56].
Inflammation and oxidative stress are strictly related. Recent clinical trials have revealed that corticosteroids and colchicine may prevent AF development and support the fundamental impact of inflammatory pathways in managing AF. A meta-analysis by Liu et al. including fourteen studies with 13,803 patients showed that corticosteroid significantly decreased the risk of post operative AF (relative risk [RR], 0.7; 95% CI 0.55–0.89; P = 0.003) [59]. In a murine model Colchicine taken up by leucocytes inhibits cytokine and interleukin expression and modulates leucocyte superoxide production [57]. Table 3 summarized mentioned drugs [21,51,58,59,60,61,62,63,64,65,66,67].
7. Nutraceutical with Mitochondrial Effects
Ubiquinone, or coenzyme Q10 (CoQ10) is a coenzyme crucial for mitochondrial complexes engaged in ATP production, CoQ10 stands out as one of the most widely used nutritional supplements. Its significance in cellular bioenergetics has been extensively researched in both animal and human studies. The proposed application of CoQ10 in patients with cardiovascular disease primarily focuses on two clinical entities: statin-associated muscle symptoms (SAMS) and HF [58,59]. However, emerging evidence hints at its potential in AF prevention. In HF patients, Zhao et al. demonstrated that CoQ10 treatment significantly reduced AF incidence compared to placebo over a 12-month period (6.3% vs 22.2%; P = 0.02) [60]. While a small randomized controlled trial suggested a potential reduction in postoperative AF with short-term CoQ10 treatment, these findings lacked consistent support in subsequent studies, as outlined in a meta-analysis. However, given its good tolerability and favorable safety profile, CoQ10 could be considered as an adjunctive therapy to mitigate AF risk in specific scenarios [21].
Treatment with vitamin C has been shown to decrease the incidence of post-surgical AF, as well as arrhythmia recurrence after electrical cardioversion in a prospective study involving 44 patients (p=0.024). N-acetyl cysteine was shown to reduce the risk of AF in dogs undergoing tachycardic pacing by increasing the density of L-type calcium current [61].
L-glutamine is increasingly recognized as a potential nutraceutical for AF treatment thanks to its ability to reduce ROS production and stabilize the microtubule network through heightened heat shock protein (HSP) expression [62]. Costunolide, a sesquiterpene lactone renowned for its anti-inflammatory and anti-fibrotic properties, effectively mitigates inflammation and fibrosis induced by Ang II in mice. Notably, costunolide has demonstrated the ability to preserve mitochondrial function and reduce oxidative stress [51]. Andrographolide, an active ingredient in the medicinal plant Andrographis paniculata, has numerous pharmacological properties and has proven beneficial in AF by reducing heart cell apoptosis, improving mitochondrial function, demonstrating antioxidant properties, and regulating inflammation and calcium homeostasis genes. It also activates the transcription pathways involved in the antioxidant response, such as factor-2-related erythroid nuclear [51]. Table 4 summarizes mentioned nutraceutical drugs.
8. Experimental Drugs with Mitochondrial Effects
Elamipretide, also known as Bendavia, MTP-131, or SS-31, is a pioneering mitochondria-targeted drug under clinical investigation for primary mitochondrial myopathy [63,64] and in HF treatment. It enhances mitochondrial energetics and reduces reactive oxygen species, potentially by stabilizing the mitochondrial membrane and cytochrome c [65]. Despite initial positive studies in animals and human with HF, showing potential benefits, a recent phase 2 clinical trial with repeated elamipretide administration over 28 days in a small cohort of heart failure patients did not demonstrate improvement in left ventricular ejection fraction [66]. KL1333, by elevating NAD+ levels and activating AMPK/PGC-1 signaling, enhances mitochondrial function and reduces oxidative stress in fibroblasts derived from patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. Conversely, the medication KH176 demonstrates efficacy in lowering cellular ROS levels. This protective mechanism involves interaction with the thioredoxin system/peroxiredoxin enzyme pathway. Idebenone, an analog of synthetic coenzyme Q10, exhibits potential for treating various conditions associated with impaired mitochondrial function. While it demonstrates cardioprotective effects in an animal model of ischemia/reperfusion, further exploration is necessary to understand its broader impact on heart diseases [67].
In the realm of potential therapies, gene therapy represents an experimental avenue, albeit in its early stages. Genetic constructs, typically delivered through adenoviral vectors, can be transported to the myocardium via direct intramyocardial injection, epicardial gene painting, or intracoronary infusion. These gene-based approaches, tested in animal models of AF have shown success in restoring sinus rhythm or improving ventricular rate control. However, their introduction into clinical practice is still premature at this stage [21]
9. Effect of Anticoagulant Drugs on Mitochondrial Function
Oral anticoagulants are essential for preventing stroke and systemic embolisms in AF patients. Direct oral anticoagulants (DOACs) are now preferred over vitamin K antagonists (VKAs) due to their similar efficacy and better safety profile. Consequently, DOACs have received a class I recommendation in AF management guidelines, while VKAs are reserved for specific conditions like mechanical prosthetic valves, severe mitral stenosis, or antiphospholipid syndrome (APS) [72,73]. These guidelines are based on data from randomized clinical trials (RCTs) and real-world studies of underrepresented patient subgroups [74,75]. DOACs work by inhibiting activated factor X (e.g., Rivaroxaban, Edoxaban, Apixaban) or thrombin (e.g., Dabigatran). Furthermore, FXa and FIIa have “pleiotropic” effects, including influence on mitochondrial function and regulation of oxidative stress, through the activation of the protease-activated receptors (PARs) family [76]. Thrombin primarily activates PAR-1 but also PAR-3 and PAR-4, while FXa activates PAR-1 and PAR-2. Activation of PAR receptors triggers pro-inflammatory and pro-fibrotic effects in various cell types, mediating conditions such as atherosclerosis, atrial remodeling, cardiac hypertrophy, and chronic inflammatory pulmonary disorders, all of which may contribute to the incidence of AF [77,78]. Mitochondrial dysfunction, oxidative stress, inflammation, and coagulation are closely intertwined. ROS operate at multiple levels in the coagulation cascade, regulating endothelial functions, platelet activation, and the production of coagulation factors. This creates a vicious cycle, elevating thrombotic risk in conditions associated with oxidative stress [79]. Consequently, interest has been piqued regarding the potential effects of antithrombotic therapies on mitochondrial function and REDOX homeostasis. Even if studies conducted specifically on AF in mitochondrial disease are lacking, both in vitro and in vivo studies have suggested that direct oral anticoagulants possess antioxidant properties acting on mitochondrial function that may contribute to their beneficial effects on clinical outcomes [75,80,81]. These effects could, at least in part, contribute to the advantageous effects of DOACs compared to VKA. Indeed, Warfarin was found to cause mitochondrial damage in lymphocytes and reduce the cellular ATP levels in hepatocytes, leading to compromised viability [82].
Rivaroxaban has been extensively investigated for its potential antioxidant properties in experimental and animal models. Preclinical studies using human umbilical vein endothelial cells (HUVECs) have demonstrated a dose-dependent reduction in ROS production and other oxidative stress biomarkers with Rivaroxaban treatment [83,84,85]. Similar findings were observed in human atrial cells [86]. Preclinic studies by Zekri-Nechar et al. have demonstrated that Rivaroxaban, when used in combination with cardioaspirin, improved mitochondrial functionality in human coronary artery endothelial cells (HCAECs) exposed to high glucose. This improvement was attributed to increased mitophagy promotion, enhancement of mitochondrial membrane potential, and reduction in ROS production [87]. Animal models showed that rivaroxaban may safeguard mitochondria by modifying the expression levels of various genes linked to mitochondrial function in angiotensin II infused KKAy mice. Furthermore, it may alleviate the angiotensin II-induced decrease in cardiac ROS levels and ATP production [5]. In rat kidney mitochondria, rivaroxaban’s effects were found to be dose-dependent: at low concentrations, it induced mitochondrial dysfunction and oxidative stress by reducing the activity of mitochondrial succinate dehydrogenase and the mitochondrial membrane potential, while increasing ROS production, mitochondrial swelling, and cytochrome c release. Conversely, at high concentrations, these effects were averted [88]. In a human ex vivo study focusing on abdominal aortic aneurysmal sites with intraluminal mural thrombus, Rivaroxaban improved the enzymatic activity of citrate synthase and cytochrome C oxidase, biomarkers of mitochondrial density and respiration, respectively, in the same model [89].
Numerous studies have highlighted the pleiotropic effects of edoxaban, encompassing not only its anti-inflammatory and antioxidant properties but also its antifibrotic and antiremodeling effects [46,90]. Regarding the effects on mithocondria, Bukowska et al. demonstrated that exposure to factor Xa in the human lung carcinoma cell line A549 edoxaban prevented activated clotting factor X-induced mitochondrial impairment by augmenting mitochondrial oxygen consumption during maximal oxidative phosphorylation and, consequently, mitochondrial ATP production [91]. Moreover, edoxaban showed beneficial impact on AF induction and duration. In a canine model of congestive HF, edoxaban treatment attenuated atrial fibrosis and reduced the duration of AF episodes induced by ventricular tachypacing (VTP). It also suppressed PAR-2 and fibronectin upregulation, indicating inhibition of AF progression and structural remodeling. Additionally, in a murine model, edoxaban mitigated vulnerability to AF episodes induced by AngII, potentially through anti-oxidant mechanisms [77].
Information regarding consequences of apixaban use on mitochondrial functionality are lacking. Limited data existed about its antioxidant effects. A preclinical in vitro study conducted by Torramade-Moix et al. shed light on this aspect. In the study, HUVECs and human dermal microvascular endothelial cells (HMECs-1) exposed to uremic plasma showed normalized ROS levels following pretreatment with apixaban [92]. Additionally, Durmaz et al. demonstrated in a sample of 35 Wistar albino rats that administration of direct oral anticoagulants, including apixaban, alongside rivaroxaban and dabigatran, resulted in increased total antioxidant capacity and decreased total oxidant status [93].
Dabigatran, a direct inhibitor of thrombin, has also shown potential antioxidative activity. In a HUVECs model, both Dabigatran and rivaroxaban reduced ROS levels and total oxidative stress (TOS). Additionally, dabigatran mitigated oxidative damage of pyrimidines induced by oxysterol to levels comparable to control cells [93,94,95,96,97,98,99]. This antioxidative effect could be at least in part due to improvement in mitochondrial function. However, in a study using rat gastric epithelial cell line, dabigatran induced cytotoxic effects mediated through increased ROS generation, decreased mitochondrial membrane potential, and elevated lipid peroxidation [100]
10. Conclusions
Mitochondropathy and cardiac arrhythmias have a close etiopathogenetic relationship. In addition to mitochondrial diseases, mitochondrial dysfunction can also cause the development of cardiac arrhythmias, including AF. Several risk factors such as hyperglicemia and dyslipidemia can be cause of mitochondrial disease. Several drugs and nutraceuticals act indirectly at the mitochondrial level and can represent a therapeutic approach for the treatment of cardiac arrhythmias. Our narrative review data were extrapolated from several subanalyses, so randomized clinical trials with specific hypotheses are needed to validate these findings. Based on the data from our narrative review, metformin, iSGLT2, statins, trimetazidine, beta-blockers, ACE-I, ARBs and AT1R antagonist appear to be the most promising drugs.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, A.D.A., V.R., A.M.; methodology, V.R..; validation, V.R., A.M.; formal analysis, A.M.;—original draft preparation A.C., G.E.D.V.,R.M.,A.M.; writing—review and editing, A.C.,G.E.D.V.,R.M.,A.M.; visualization, A.M.; supervision, V.R.. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- G. S. Gorman et al., “Mitochondrial diseases,” Nat Rev Dis Primers, vol. 2, no. 1, p. 16080, Oct. 2016. [CrossRef]
- T. Klopstock, C. Priglinger, A. Yilmaz, C. Kornblum, F. Distelmaier, and H. Prokisch, “Mitochondrial Disorders.,” Dtsch Arztebl Int, vol. 118, no. 44, pp. 741–748, Nov. 2021. [CrossRef]
- M. Yamazoe et al., “Sparsely methylated mitochondrial cell free DNA released from cardiomyocytes contributes to systemic inflammatory response accompanied by atrial fibrillation,” Sci Rep, vol. 11, no. 1, p. 5837, Mar. 2021. [CrossRef]
- T. Fiorentino, A. Prioletta, P. Zuo, and F. Folli, “Hyperglycemia-induced Oxidative Stress and its Role in Diabetes Mellitus Related Cardiovascular Diseases,” Curr Pharm Des, vol. 19, no. 32, pp. 5695–5703, Aug. 2013. [CrossRef]
- Y. Rao, J. Chen, Y. Guo, T. Ji, and P. Xie, “Rivaroxaban ameliorates angiotensin II-induced cardiac remodeling by attenuating TXNIP/Trx2 interaction in KKAy mice,” Thromb Res, vol. 193, pp. 45–52, Sep. 2020. [CrossRef]
- K. Qu et al., “Mitochondrial dysfunction in vascular endothelial cells and its role in atherosclerosis,” Front Physiol, vol. 13, Dec. 2022. [CrossRef]
- T.-A. Popoiu, J. Dudek, C. Maack, and E. Bertero, “Cardiac Involvement in Mitochondrial Disorders,” Curr Heart Fail Rep, vol. 20, no. 1, pp. 76–87, Feb. 2023. [CrossRef]
- S. L. Stenton and H. Prokisch, “Genetics of mitochondrial diseases: Identifying mutations to help diagnosis.,” EBioMedicine, vol. 56, p. 102784, Jun. 2020. [CrossRef]
- Y. S. Ng and D. M. Turnbull, “Mitochondrial disease: genetics and management,” J Neurol, vol. 263, no. 1, pp. 179–191, Jan. 2016. [CrossRef]
- R. L. Davis, C. Liang, and C. M. Sue, “Mitochondrial diseases,” 2018, pp. 125–141. [CrossRef]
- T. Yu, J. L. Robotham, and Y. Yoon, “Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology,” Proceedings of the National Academy of Sciences, vol. 103, no. 8, pp. 2653–2658, Feb. 2006. [CrossRef]
- S. I. Dikalov et al., “Nox2-Induced Production of Mitochondrial Superoxide in Angiotensin II-Mediated Endothelial Oxidative Stress and Hypertension,” Antioxid Redox Signal, vol. 20, no. 2, pp. 281–294, Jan. 2014. [CrossRef]
- K.-C. Yang, M. G. Bonini, and S. C. Dudley, “Mitochondria and arrhythmias,” Free Radic Biol Med, vol. 71, pp. 351–361, Jun. 2014. [CrossRef]
- F. E. Mason, J. R. D. Pronto, K. Alhussini, C. Maack, and N. Voigt, “Cellular and mitochondrial mechanisms of atrial fibrillation,” Basic Res Cardiol, vol. 115, no. 6, p. 72, Dec. 2020. [CrossRef]
- J. Deng, Y. Jiang, Z. B. Chen, J.-W. Rhee, Y. Deng, and Z. V. Wang, “Mitochondrial Dysfunction in Cardiac Arrhythmias,” Cells, vol. 12, no. 5, p. 679, Feb. 2023. [CrossRef]
- N. Black, F. Mohammad, K. Saraf, and G. Morris, “Endothelial function and atrial fibrillation: A missing piece of the puzzle?,” J Cardiovasc Electrophysiol, vol. 33, no. 1, pp. 109–116, Jan. 2022. [CrossRef]
- A. Khan, G. N. Thomas, G. Y. H. Lip, and A. Shantsila, “Endothelial function in patients with atrial fibrillation,” Ann Med, vol. 52, no. 1–2, pp. 1–11, Feb. 2020. [CrossRef]
- S. Qin, M. Boidin, B. J. R. Buckley, G. Y. H. Lip, and D. H. J. Thijssen, “Endothelial dysfunction and vascular maladaptation in atrial fibrillation,” Eur J Clin Invest, vol. 51, no. 5, May 2021. [CrossRef]
- M. Guazzi and R. Arena, “Endothelial dysfunction and pathophysiological correlates in atrial fibrillation,” Heart, vol. 95, no. 2, pp. 102–106, Jan. 2009. [CrossRef]
- B. Balint, V. Jaremek, V. Thorburn, S. N. Whitehead, and L. A. Sposato, “Left atrial microvascular endothelial dysfunction, myocardial inflammation and fibrosis after selective insular cortex ischemic stroke,” Int J Cardiol, vol. 292, pp. 148–155, Oct. 2019. [CrossRef]
- P. Muszyński and T. A. Bonda, “Mitochondrial Dysfunction in Atrial Fibrillation—Mechanisms and Pharmacological Interventions,” J Clin Med, vol. 10, no. 11, p. 2385, May 2021. [CrossRef]
- Caturano et al., “Oxidative Stress in Type 2 Diabetes: Impacts from Pathogenesis to Lifestyle Modifications,” Curr Issues Mol Biol, vol. 45, no. 8, pp. 6651–6666, Aug. 2023. [CrossRef]
- Li et al., “SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart,” Cardiovasc Diabetol, vol. 18, no. 1, p. 15, Dec. 2019. [CrossRef]
- Q. Shao et al., “Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats,” Cardiovasc Diabetol, vol. 18, no. 1, p. 165, Dec. 2019. [CrossRef]
- T. Koizumi et al., “Empagliflozin suppresses mitochondrial reactive oxygen species generation and mitigates the inducibility of atrial fibrillation in diabetic rats,” Front Cardiovasc Med, vol. 10, Feb. 2023. [CrossRef]
- X. Zhao et al., “Dapagliflozin attenuates the vulnerability to atrial fibrillation in rats with lipopolysaccharide-induced myocardial injury,” Int Immunopharmacol, vol. 125, p. 111038, Dec. 2023. [CrossRef]
- N. Nuamnaichati, S. Mangmool, N. Chattipakorn, and W. Parichatikanond, “Stimulation of GLP-1 Receptor Inhibits Methylglyoxal-Induced Mitochondrial Dysfunctions in H9c2 Cardiomyoblasts: Potential Role of Epac/PI3K/Akt Pathway,” Front Pharmacol, vol. 11, May 2020. [CrossRef]
- L. Zhang, J. Tian, S. Diao, G. Zhang, M. Xiao, and D. Chang, “GLP-1 receptor agonist liraglutide protects cardiomyocytes from IL-1β-induced metabolic disturbance and mitochondrial dysfunction,” Chem Biol Interact, vol. 332, p. 109252, Dec. 2020. [CrossRef]
- V. A. Myasoedova et al., “Anti-Inflammation and Anti-Oxidation: The Key to Unlocking the Cardiovascular Potential of SGLT2 Inhibitors and GLP1 Receptor Agonists,” Antioxidants, vol. 13, no. 1, p. 16, Dec. 2023. [CrossRef]
- G. Fauchier et al., “Glucose-lowering drug use and new-onset atrial fibrillation in patients with diabetes mellitus,” Diabetologia, vol. 64, no. 11, pp. 2602–2605, Nov. 2021. [CrossRef]
- Y. Ma et al., “DPP-4 inhibitor anagliptin protects against hypoxia-induced cytotoxicity in cardiac H9C2 cells,” Artif Cells Nanomed Biotechnol, vol. 47, no. 1, pp. 3823–3831, Dec. 2019. [CrossRef]
- X. Zhang et al., “Alogliptin, a Dipeptidyl Peptidase-4 Inhibitor, Alleviates Atrial Remodeling and Improves Mitochondrial Function and Biogenesis in Diabetic Rabbits,” J Am Heart Assoc, vol. 6, no. 5, May 2017. [CrossRef]
- T. Igarashi et al., “Linagliptin prevents atrial electrical and structural remodeling in a canine model of atrial fibrillation,” Heart Vessels, vol. 33, no. 10, pp. 1258–1265, Oct. 2018. [CrossRef]
- D. I. Patoulias et al., “Cardiovascular efficacy and safety of dipeptidyl peptidase-4 inhibitors: A meta-analysis of cardiovascular outcome trials,” World J Cardiol, vol. 13, no. 10, pp. 585–592, Oct. 2021. [CrossRef]
- Y.-H. Chan et al., “The risk of incident atrial fibrillation in patients with type 2 diabetes treated with sodium glucose cotransporter-2 inhibitors, glucagon-like peptide-1 receptor agonists, and dipeptidyl peptidase-4 inhibitors: a nationwide cohort study,” Cardiovasc Diabetol, vol. 21, no. 1, p. 118, Dec. 2022. [CrossRef]
- S.-H. Chang et al., “Association of metformin with lower atrial fibrillation risk among patients with type 2 diabetes mellitus: a population-based dynamic cohort and in vitro studies,” Cardiovasc Diabetol, vol. 13, no. 1, p. 123, Dec. 2014. [CrossRef]
- D. Sun and F. Yang, “Metformin improves cardiac function in mice with heart failure after myocardial infarction by regulating mitochondrial energy metabolism,” Biochem Biophys Res Commun, vol. 486, no. 2, pp. 329–335, Apr. 2017. [CrossRef]
- Z. Zhang et al., “Thiazolidinedione use and atrial fibrillation in diabetic patients: a meta-analysis,” BMC Cardiovasc Disord, vol. 17, no. 1, p. 96, Dec. 2017. [CrossRef]
- C. Liu et al., “Pioglitazone attenuates atrial remodeling and vulnerability to atrial fibrillation in alloxan-induced diabetic rabbits,” Cardiovasc Ther, vol. 35, no. 5, Oct. 2017. [CrossRef]
- D. XU et al., “PPAR-γ Activator Pioglitazone Prevents Age-Related Atrial Fibrillation Susceptibility by Improving Antioxidant Capacity and Reducing Apoptosis in a Rat Model,” J Cardiovasc Electrophysiol, vol. 23, no. 2, pp. 209–217, Feb. 2012. [CrossRef]
- J. L. Pallisgaard, M. M. Brooks, B. R. Chaitman, D. B. Boothroyd, M. Perez, and M. A. Hlatky, “Thiazolidinediones and Risk of Atrial Fibrillation Among Patients with Diabetes and Coronary Disease,” Am J Med, vol. 131, no. 7, pp. 805–812, Jul. 2018. [CrossRef]
- J. Gu, W. Hu, Z. Song, X. Liu, and D. Zhang, “PPARγ agonist use and recurrence of atrial fibrillation after successful electrical cardioversion,” Hellenic Journal of Cardiology, vol. 58, no. 5, pp. 387–390, Sep. 2017. [CrossRef]
- E. A. Williams, V. Russo, S. Ceraso, D. Gupta, and R. Barrett-Jolley, “Anti-arrhythmic properties of non-antiarrhythmic medications.,” Pharmacol Res, vol. 156, p. 104762, Jun. 2020. [CrossRef]
- C.-H. Tseng et al., “Statins reduce new-onset atrial fibrillation after acute myocardial infarction: A nationwide study.,” Medicine, vol. 99, no. 2, p. e18517, Jan. 2020. [CrossRef]
- L. Alves-Cabratosa et al., “Statins and new-onset atrial fibrillation in a cohort of patients with hypertension. Analysis of electronic health records, 2006–2015,” PLoS One, vol. 12, no. 10, p. e0186972, Oct. 2017. [CrossRef]
- W. Fang, H. Li, H. Zhang, and S. Jiang, “The role of statin therapy in the prevention of atrial fibrillation: a meta-analysis of randomized controlled trials,” Br J Clin Pharmacol, vol. 74, no. 5, pp. 744–756, Nov. 2012. [CrossRef]
- L. Safaeian, M. Mirian, and S. Bahrizadeh, “Evolocumab, a PCSK9 inhibitor, protects human endothelial cells against H 2 O 2 -induced oxidative stress,” Arch Physiol Biochem, vol. 128, no. 6, pp. 1681–1686, Nov. 2022. [CrossRef]
- J. Yang et al., “PCSK9 inhibitors suppress oxidative stress and inflammation in atherosclerotic development by promoting macrophage autophagy.,” Am J Transl Res, vol. 15, no. 8, pp. 5129–5144, 2023.
- N. D’Onofrio et al., “SIRT3 mediates the effects of PCSK9 inhibitors on inflammation, autophagy, and oxidative stress in endothelial cells,” Theranostics, vol. 13, no. 2, pp. 2023. [CrossRef]
- Silla et al., “Treatment with PCSK9 Inhibitor Evolocumab Improves Vascular Oxidative Stress and Arterial Stiffness in Hypercholesterolemic Patients with High Cardiovascular Risk,” Antioxidants, vol. 12, no. 3, p. 578, Feb. 2023. [CrossRef]
- da S. Menezes Júnior, A. L. G. de França-e-Silva, J. M. de Oliveira, and D. M. da Silva, “Developing Pharmacological Therapies for Atrial Fibrillation Targeting Mitochondrial Dysfunction and Oxidative Stress: A Scoping Review,” Int J Mol Sci, vol. 25, no. 1, p. 535, Dec. 2023. [CrossRef]
- W. Shi, “Effects of trimetazidine on mitochondrial respiratory function, biosynthesis, and fission/fusion in rats with acute myocardial ischemia,” The Anatolian Journal of Cardiology, 2017. [CrossRef]
- Z. Li et al., “GW28-e0789 Trimetazidine decreases inducibility and duration of atrial fibrillation in a dog model of congestive heart failure,” J Am Coll Cardiol, vol. 70, no. 16, p. C29, Oct. 2017. [CrossRef]
- Ratte, F. Wiedmann, M. Kraft, H. A. Katus, and C. Schmidt, “Antiarrhythmic Properties of Ranolazine: Inhibition of Atrial Fibrillation Associated TASK-1 Potassium Channels,” Front Pharmacol, vol. 10, Nov. 2019. [Google Scholar] [CrossRef]
- Z. Zhao et al., “Attenuation of atrial remodeling by aliskiren via affecting oxidative stress, inflammation and PI3K/Akt signaling pathway,” Cardiovasc Drugs Ther, vol. 35, no. 3, pp. 587–598, Jun. 2021. [CrossRef]
- D. Xu et al., “Xanthine oxidase inhibitor febuxostat reduces atrial fibrillation susceptibility by inhibition of oxidized CaMKII in Dahl salt-sensitive rats,” Clin Sci, vol. 135, no. 20, pp. 2409–2422, Oct. 2021. [CrossRef]
- K. Andelova, B. S. Bacova, M. Sykora, P. Hlivak, M. Barancik, and N. Tribulova, “Mechanisms Underlying Antiarrhythmic Properties of Cardioprotective Agents Impacting Inflammation and Oxidative Stress,” Int J Mol Sci, vol. 23, no. 3, p. 1416, Jan. 2022. [CrossRef]
- E. Raizner and M. A. Quiñones, “Coenzyme Q10 for Patients With Cardiovascular Disease,” J Am Coll Cardiol, vol. 77, no. 5, pp. 609–619, Feb. 2021. [CrossRef]
- Martelli, L. Testai, A. Colletti, and A. F. G. Cicero, “Coenzyme Q10: Clinical Applications in Cardiovascular Diseases,” Antioxidants, vol. 9, no. 4, p. 341, Apr. 2020. [Google Scholar] [CrossRef]
- Q. Zhao, A. H. Kebbati, Y. Zhang, Y. Tang, E. Okello, and C. Huang, “Effect of Coenzyme Q10 on the Incidence of Atrial Fibrillation in Patients with Heart Failure,” Journal of Investigative Medicine, vol. 63, no. 5, pp. 735–739, Jun. 2015. [CrossRef]
- I. Balan, V. B. Halațiu, and A. Scridon, “Oxidative Stress, Inflammation, and Mitochondrial Dysfunction: A Link between Obesity and Atrial Fibrillation,” Antioxidants, vol. 13, no. 1, p. 117, Jan. 2024. [CrossRef]
- L. Pool, L. F. J. M. Wijdeveld, N. M. S. de Groot, and B. J. J. M. Brundel, “The Role of Mitochondrial Dysfunction in Atrial Fibrillation: Translation to Druggable Target and Biomarker Discovery,” Int J Mol Sci, vol. 22, no. 16, p. 8463, Aug. 2021. [CrossRef]
- Karaa, R. Haas, A. Goldstein, J. Vockley, W. D. Weaver, and B. H. Cohen, “Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy,” Neurology, vol. 90, no. 14, Apr. 2018. [Google Scholar] [CrossRef]
- Karaa et al., “Efficacy and Safety of Elamipretide in Individuals With Primary Mitochondrial Myopathy,” Neurology, vol. 101, no. 3, Jul. 2023. [CrossRef]
- K. C. Chatfield et al., “Elamipretide Improves Mitochondrial Function in the Failing Human Heart,” JACC Basic Transl Sci, vol. 4, no. 2, pp. 147–157, Apr. 2019. [CrossRef]
- J. Butler et al., “Effects of Elamipretide on Left Ventricular Function in Patients With Heart Failure With Reduced Ejection Fraction: The PROGRESS-HF Phase 2 Trial,” J Card Fail, vol. 26, no. 5, pp. 429–437, May 2020. [CrossRef]
- K.-S. Seo et al., “KL1333, a Novel NAD+ Modulator, Improves Energy Metabolism and Mitochondrial Dysfunction in MELAS Fibroblasts.,” Front Neurol, vol. 9, p. 552, 2018. [CrossRef]
- D. Wang, L. Jiang, B. Feng, N. He, Y. Zhang, and H. Ye, “Protective effects of glucagon-like peptide-1 on cardiac remodeling by inhibiting oxidative stress through mammalian target of rapamycin complex 1/p70 ribosomal protein S6 kinase pathway in diabetes mellitus,” J Diabetes Investig, vol. 11, no. 1, pp. 39–51, Jan. 2020. [CrossRef]
- J. Wei, R. Wang, H. Ye, Y. Wang, L. Wang, and X. Zhang, “Effects of GLP-1 receptor agonists on arrhythmias and its subtypes in patients with type 2 diabetes: A systematic review and meta-analysis.,” Front Endocrinol (Lausanne), vol. 13, p. 910256, 2022. [CrossRef]
- C.-Y. Chang et al., “Dipeptidyl peptidase-4 inhibitor decreases the risk of atrial fibrillation in patients with type 2 diabetes: a nationwide cohort study in Taiwan,” Cardiovasc Diabetol, vol. 16, no. 1, p. 159, Dec. 2017. [CrossRef]
- P. Korantzopoulos, K. P. Letsas, and T. Liu, “Xanthine Oxidase and Uric Acid in Atrial Fibrillation,” Front Physiol, vol. 3, 2012. [CrossRef]
- G. Hindricks et al., “2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS),” Eur Heart J, vol. 42, no. 5, pp. 373–498, Feb. 2021. [CrossRef]
- D. O. Kleindorfer et al., “2021 Guideline for the Prevention of Stroke in Patients With Stroke and Transient Ischemic Attack: A Guideline From the American Heart Association/American Stroke Association,” Stroke, vol. 52, no. 7, Jul. 2021. [CrossRef]
- T. Ruff et al., “Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials,” The Lancet, vol. 383, no. 9921, pp. 955–962, Mar. 2014. [CrossRef]
- L. Falco et al., “Antioxidant Properties of Oral Antithrombotic Therapies in Atherosclerotic Disease and Atrial Fibrillation,” Antioxidants (Basel), vol. 12, no. 6, Jun. 2023. [CrossRef]
- V. Russo and D. Fabiani, “Put out the fire: The pleiotropic anti-inflammatory action of non-vitamin K oral anticoagulants,” Pharmacol Res, vol. 182, Aug. 2022. [CrossRef]
- A. Goette, M. Mollenhauer, V. Rudolph, M. Lamparter, M. Meier, and M. Böhm, “Pleiotropic effects of NOACs with focus on edoxaban: scientific findings and potential clinical implications,” Herzschrittmachertherapie + Elektrophysiologie, vol. 34, no. 2, pp. 142–152, Jun. 2023. [CrossRef]
- H. ten Cate, T. J. Guzik, J. Eikelboom, and H. M. H. Spronk, “Pleiotropic actions of factor Xa inhibition in cardiovascular prevention: mechanistic insights and implications for anti-thrombotic treatment,” Cardiovasc Res, vol. 117, no. 9, pp. 2030–2044, Jul. 2021. [CrossRef]
- E. Masselli et al., “ROS in Platelet Biology: Functional Aspects and Methodological Insights,” Int J Mol Sci, vol. 21, no. 14, p. 4866, Jul. 2020. [CrossRef]
- M. Gori et al., “Pleiotropic effects of anti-thrombotic therapies: have direct oral anticoagulants any anti-inflammatory effect?,” Bleeding, Thrombosis, and Vascular Biology, vol. 1, no. 3, Dec. 2022. [CrossRef]
- V. Russo et al., “Dual Pathway Inhibition with Rivaroxaban and Aspirin Reduces Inflammatory Biomarkers in Atherosclerosis,” J Cardiovasc Pharmacol, vol. 81, no. 2, pp. 129–133, Feb. 2023. [CrossRef]
- M. Bețiu et al., “Mitochondrial Effects of Common Cardiovascular Medications: The Good, the Bad and the Mixed,” Int J Mol Sci, vol. 23, no. 21, p. 13653, Nov. 2022. [CrossRef]
- M. Maeda, T. Tsuboi, and T. Hayashi, “An Inhibitor of Activated Blood Coagulation Factor X Shows Anti-Endothelial Senescence and Anti-Atherosclerotic Effects,” J Vasc Res, vol. 56, no. 4, pp. 2019. [CrossRef]
- P. Ellinghaus et al., “Expression of pro-inflammatory genes in human endothelial cells: Comparison of rivaroxaban and dabigatran,” Thromb Res, vol. 142, pp. 44–51, Jun. 2016. [CrossRef]
- Y. Ishibashi, T. Matsui, S. Ueda, K. Fukami, and S. Yamagishi, “Advanced glycation end products potentiate citrated plasma-evoked oxidative and inflammatory reactions in endothelial cells by up-regulating protease-activated receptor-1 expression,” Cardiovasc Diabetol, vol. 13, no. 1, p. 60, Dec. 2014. [CrossRef]
- Bukowska et al., “Coagulation factor Xa induces an inflammatory signalling by activation of protease-activated receptors in human atrial tissue,” Eur J Pharmacol, vol. 718, no. 1–3, pp. 114–123, Oct. 2013. [CrossRef]
- K. Zekri-Nechar et al., “Mitochondrial mitophagy protection combining rivaroxaban and aspirin in high glucose-exposed human coronary artery endothelial cell. An in vitro study,” Diab Vasc Dis Res, vol. 19, no. 5, p. 147916412211298, Sep. 2022. [CrossRef]
- F. Samiei, H. Sajjadi, A. Jamshidzadeh, E. Seydi, and J. Pourahmad, “Contrasting Role of Concentration in Rivaroxaban Induced Toxicity and Oxidative Stress in Isolated Kidney Mitochondria,” Drug Res, vol. 69, no. 10, pp. 523–527, Oct. 2019. [CrossRef]
- J. J. Zamorano-Leon et al., “Factor Xa Inhibition by Rivaroxaban Modified Mitochondrial-Associated Proteins in Human Abdominal Aortic Aneurysms,” Ann Vasc Surg, vol. 67, pp. 482–489, Aug. 2020. [CrossRef]
- Y. Narita et al., “Edoxaban Exerts Antioxidant Effects Through FXa Inhibition and Direct Radical-Scavenging Activity,” Int J Mol Sci, vol. 20, no. 17, p. 4140, Aug. 2019. [CrossRef]
- Bukowska et al., “Activated clotting factor X mediates mitochondrial alterations and inflammatory responses via protease-activated receptor signaling in alveolar epithelial cells,” Eur J Pharmacol, vol. 869, p. 172875, Feb. 2020. [CrossRef]
- S. Torramade-Moix et al., “Apixaban Downregulates Endothelial Inflammatory and Prothrombotic Phenotype in an In Vitro Model of Endothelial Dysfunction in Uremia,” Cardiovasc Drugs Ther, vol. 35, no. 3, pp. 521–532, Jun. 2021. [CrossRef]
- S. Durmaz et al., “Direct oral anticoagulant agents attenuate temporary aortic occlusion-induced renal oxidative and inflammatory responses in rats,” Turkish Journal of Thoracic and Cardiovascular Surgery, vol. 30, no. 2, pp. 184–191, Apr. 2022. [CrossRef]
- S. L. Johnson, J. Iannucci, N. P. Seeram, and P. Grammas, “Inhibiting thrombin improves motor function and decreases oxidative stress in the LRRK2 transgenic Drosophila melanogaster model of Parkinson’s disease,” Biochem Biophys Res Commun, vol. 527, no. 2, pp. 532–538, Jun. 2020. [CrossRef]
- J. Iannucci et al., “Short-term treatment with dabigatran alters protein expression patterns in a late-stage tau-based Alzheimer’s disease mouse model,” Biochem Biophys Rep, vol. 24, p. 100862, Dec. 2020. [CrossRef]
- Sanchez, X. Yin, J. Luo, J. Martinez, and P. Grammas, “Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia,” Front Aging Neurosci, vol. 5. 2013. [CrossRef]
- S. Pingel, V. Tiyerili, J. Mueller, N. Werner, G. Nickenig, and C. Mueller, “Experimental research Thrombin inhibition by dabigatran attenuates atherosclerosis in ApoE deficient mice,” Archives of Medical Science, vol. 1, pp. 2014. [CrossRef]
- N. P. E. Kadoglou et al., “The Beneficial Effects of a Direct Thrombin Inhibitor, Dabigatran Etexilate, on the Development and Stability of Atherosclerotic Lesions in Apolipoprotein E-deficient Mice,” Cardiovasc Drugs Ther, vol. 26, no. 5, pp. 367–374, Oct. 2012. [CrossRef]
- E. Woźniak, M. Broncel, B. Bukowska, and P. Gorzelak-Pabiś, “The Protective Effect of Dabigatran and Rivaroxaban on DNA Oxidative Changes in a Model of Vascular Endothelial Damage with Oxidized Cholesterol,” Int J Mol Sci, vol. 21, no. 6, p. 1953, Mar. 2020. [Google Scholar] [CrossRef]
- H. Kurokawa, A. Taninaka, H. Shigekawa, and H. Matsui, “Dabigatran Etexilate Induces Cytotoxicity in Rat Gastric Epithelial Cell Line via Mitochondrial Reactive Oxygen Species Production,” Cells, vol. 10, no. 10, p. 2508, Sep. 2021. [CrossRef]
- G. E. Raskob et al., “Edoxaban for the Treatment of Cancer-Associated Venous Thromboembolism,” New England Journal of Medicine, vol. 378, no. 7, pp. 615–624, Feb. 2018. [CrossRef]
- V. Russo et al., “Anti-Inflammatory and Anticancer Effects of Anticoagulant Therapy in Patients with Malignancy,” Life 2023, Vol. 13, Page 1888, vol. 13, no. 9, p. 1888, Sep. 2023. [CrossRef]

Table 1.
Risk Factor of Mitochondrial Disease through increasing ROS.
| Risk Factors of Mitochondrial Disease |
| Smoke [3] |
| Hyperglicemia [4] |
| Fatty foods [3] |
| Sedentariety [3] |
| Alcohol [3] |
| Angiotensin II [5] |
| Use of drugs [3] |
| Dyslipidemia [6] |
ROS: radical oxidative species.
Table 2.
Main mitochondrial pathologies with cardiac involvement.
| Syndrome | Causative Genes | Inheritance Pattern | Clinical manifestations | Onset |
|---|---|---|---|---|
| Leigh Syndrome | More than 80 genes in Mitochondrial (MtDNA) and nuclear DNA (nDNA) including SURF1 | AR (mainly) |
Seizures, encephalopathy, failure to thrive, dysphagia, cardiac involvement (HCM or DCM; valvular disease, arrhythmia, conduction defect) | Childhood |
| Sengers Syndrome | acylglycerol kinase AGK (nDNA) | AR | cataracts, HCM, skeletal myopathy, and lactic acidosis | Childhood/adulthood |
| Kearns–Sayre syndrome (KSS) | MtDNA deletion | Maternal inheritance pattern | Neurological involvement (ataxia,dementia), Diabetes mellitus, cardiac conduction disorders (possible onset with sudden death), pigmentary retinopathy | Adulthood |
| Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) syndrome | MtDNA mutations (m.3243A>Gin MT-TL1, and other pathogenetic variants in MT-TL1) or MT-TV and MT-TQ | Maternal inheritance pattern | Ataxia, seizures, stroke like episodes, myopathy, lactis acidosis, HCM, LVnon compactation, pre-excitation and atrioventricular block deafness. | Adulthood |
| Leber hereditary optic neuropathy (LHON) | mutations in Mt-DNA m.11778G>A (MT-ND4), m.14484T>C (MT-ND6) and m.3460G>A (MT-ND1) |
AR | Visual loss, cardiac involvement (pre-excitation) | Adulthood |
AR, autosomal recessive; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; MtDNA, mitochondrial Deoxyribonucleic Acid; nDNA, nuclear Deoxyribonucleic Acid.
Table 3.
Therapeutic strategies (drugs) and their respective main mechanism of influence on mitochondrial function and AF.
Table 3.
Therapeutic strategies (drugs) and their respective main mechanism of influence on mitochondrial function and AF.
| Medication | Main effect on mithocondrial function and AF |
|---|---|
| SGLT-2 inhibitors [22,23,24,25,26,29] | ↓ ROS production; restoration of mitochondrial membrane potential; ↑ mitochondrial biogenesis acting on PGC-1, NRF-1, Mfn-1 and AMPK; regulation of intracellulare electrolyte balance; ↓ in myocardial remodeling and fibrosis acting on TGF-beta/smad and NRF2/ARE; ↓ AF inducibility and in AF incidence |
| GLP1R antagonists [27,28,29,68,69] | ↓ ROS production and ↑ in ROS scavengers’ mechanisms; antiapoptotic effects acting on cAMP/Epac/PI3K/Akt pathway; ↓ in myocardial remodeling and fibrosis; ↓ AF inducibility in animal models, contrasting data on humans. |
| DDP-4 inhibitors [30,31,32,33,34,35,70] | ↓ mitochondrial ROS production; ↓ mitochondrial membrane depolarization; ↑ mitochondrial biogenesis acting on PGC-1 /NRF1/Tfam; ↓ AF inducibility in animal models, contrasting data on humans. |
| Metformin [36,37] | ↑ mitochondrial oxygen consumption and activity of complexes I, II and IV; ↓ atrial remodeling by activating the AMPK/PGC-1/PPAR; ↓ of AF incidence by 19% |
| Thiazolidinediones [38,39,41,42] | ↓ oxidative stress; ↓ mitochondrial apoptotic signaling acting on PPAR; ↓ atrial remodeling ↑ ion channel function (ICa and INa); ↓ AF inducibility in animal models, contrasting data on humans. |
| Statins [44,45,46] | ↓ oxidative stress through ↓ Rho/ROCK pathways, ↑ PI(3)K/Akt pathway, and ↓ NAD(P)H oxidase activity; ↓incidence of AF by 19% |
| Fibrates [51] | ↑ mitochondrial function acting on PPAR/PGC-1; ↓ atrial remodeling and inducibility of AF prolonging atrial refractory period |
| Omega 3 fatty acids [51] | ↓ ROS production; Regulation of ion channels and cardiac electrical activity |
| Trimetazidine [52,53] | ↓ miotochondrial ROS production by activate complex I and ETC ↑ in mitochondrial biogenesis acting on PPAR/PGC-1α; Improvement on mitochondrial fusion/fission dynamics acting on Mfn-1/Drp1/Opa-1; ↓ atrial remodeling; ↓ AF inducibility and duration in ischemic conditions |
| Ranolazine [54] | ↓ mitochondrial ROS production due to inhibition of fatty acid oxidation; Antiarrhythmic proprieties due to action on sodium and potassium channels. |
| Carvedilol [51] | Block on alfa1 and beta1 adrenergic receptors; Antioxidative proprieties |
| ACE-I, ARB and AT1R blocker [51] | ↓ROS production by XO and NADPH oxidase, induced by AngII; Stabilization of cellular electrical proprieties blocking of the NF-κB action on SCN5A |
| Febuxostat and Allopurinol [56,71] | ↓of oxidative stress inhibiting XO; ↓of AF susceptibility inhibiting ox-Ca2+-calmodulin-dependent protein-kinase type-II (CaMKII) |
| Ubiquinone (CoQ10) [58,60] | Cofactor involved in electron transport within the respiratory chain. Anti-inflammatory and anti-oxidant activity. |
ACE-I: Angiotensin converter enzyme inhibitors; AF: atrial fibrillation; AMPK: Adenosine monophosphate kinase; AngII: Angiotensin II; ARB: Angiotensin receptors blockers; AT1R: Angiotensin II receptor 1; cAMP: Cyclic adenosin monophosphate; CoQ10: Coenzyme Q 10; DPP-4i: Dipeptidyl peptidase-4 inhibitors; Drp: Dystrophin Related Protein; Epac: Rap guanine nucleotide exchange factor 3; GLP1RA: Glucagon-like peptide receptor antagonists; Mfn: Mitofusin; NADPH: Nicotinamide adenine dinucleotide phosphate; NF-kB: Nuclear transcription factor-B; NRF: Nuclear respiratory factor; Opa1: Mitochondrial dynamin like GTPase; PGC: Peroxisome proliferator-activated receptor-gamma coactivator; PI3K: Phosphoinositide 3-kinase; PPAR: Peroxisome proliferator–activated receptors; ROS: Reactive oxygen species; Rho/ROCK: Rhodopsin/Rho-associated protein kinase; SCN5A: Sodium channel protein type 5 subunit alpha; SGLT-2: Sodium/glucose cotransporter 2; TGF-beta: Transforming growth factor-beta; Tfam: Transcription factor A mitochondrial; akt: Serine/threonine kinase 1; XO: Xanthine oxidase.
Table 4.
Therapeutic strategies (nutraceuticals) and their respective main mechanism of influence on mitochondrial function and AF.
Table 4.
Therapeutic strategies (nutraceuticals) and their respective main mechanism of influence on mitochondrial function and AF.
| Ubiquinone (CoQ10) [58,60] | Cofactor involved in electron transport within the respiratory chain. Anti-inflammatory and anti-oxidant activity. |
| Vitamin C and E [61] | Anti-inflammatory and anti-oxidant activity; ↓ post-surgical AF and AF recurrence after electrical cardioversion. |
| N-acetyl cysteine [61] | ↓ risk of AF by ↑ the density of L-type calcium current |
| L-glutamine [62] | ↓ ROS production and stabilize the microtubule network through heightened heat shock protein (HSP) expression. |
| Costunolide [51] | ↑ mitochondrial function and ↓ in ROS production, anti-inflammatory and anti-fibrotic properties |
| Andrographolide [51] | ↑ mitochondrial function and ↓ in ROS production, anti-inflammatory proprieties through regulation of calcium homeostasis genes |
AF: atrial fibrillation; ROS: reactive oxidative species.
Table 5.
Antioxidant proprieties and effects on mitochondrial function of direct oral anticoagulation (DOAC).
Table 5.
Antioxidant proprieties and effects on mitochondrial function of direct oral anticoagulation (DOAC).
| Medication | Mechanism | Effect on mitochondrial function |
|---|---|---|
| Rivaroxaban [5,81,84,87,88,89,99] | Factor Xa inhibitor | ↓ ROS production; restoration of mitochondrial membrane potential; ↑ mitophagy; ↑ citrate synthase; ↑ cytochrome C oxidase |
| Edoxaban [77,90,101] | Factor Xa inhibitor | ↓ ROS production; ↑ mitochondrial oxigen consumption; ↑ ATP production; ↓ atrial remodeling |
| Apixaban [92,102] | Factor Xa inhibitor | ↓ ROS production; |
| Dabigatran [100] | Thrombin inhibitor | ↓ ROS production; ↓ ROS-induced DNA strand breakage; ↓ SOD |
ATP: adenosine triphosphate; DNA: Deoxyribonucleic acid; ROS: Reactive oxygen species; SOD: superoxide dismutase.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



