
Submitted:
29 October 2024
Posted:
29 October 2024
You are already at the latest version
Abstract
Reactive oxygen species (ROS) encompass various molecular oxygen derivatives naturally produced during aerobic metabolism, including superoxide anion, hydrogen peroxide, and hydroxyl radical. Excessive ROS production leads to oxidative distress, causing cellular damage and contributing to various pathologies, often alongside inflammation. Endogenous sources of ROS include mitochondrial activity and NADPH oxidases. The antioxidant system, comprising enzymes such as superoxide dismutase, peroxiredoxin, and catalase, mitigates ROS-induced damage. This review explores the regulation of ROS by membrane receptors, focusing on B1 and B2 kinin receptors and histamine H2 receptors, which are implicated in vasodilation, angiogenesis, inflammation, and gastric acid secretion. Understanding these interactions provides insights into ROS modulation and its role in disease mechanisms.
Keywords:
Kinin B2 receptor
; Kinin B1 receptor
; Histamine H2 receptor
; oxidative stress
1. Introduction
The term 'reactive oxygen species' (ROS) encompasses a variety of molecular oxygen derivatives that are naturally produced as part of aerobic metabolism [1]. Excessive production of these ROS can lead to molecular damage, a condition referred to as 'oxidative distress'. ROS include superoxide anion, hydrogen peroxide, hydroxyl radical, lipid peroxides, protein peroxides and nucleic acid peroxides [2]. The main endogenous sources of ROS include mitochondrial activity, which generates these species as by-products, and the catalytic actions of NADPH oxidases (NOXs) [3,4].
Free radicals are kept in a critical balance by the body, as they function as molecules with cell signaling and/or second-messenger functions [5,6]. In the event of accumulation, the phenomenon called oxidative stress arises, whereby ROS are capable of causing serious cellular disturbances, participating in the emergence and/or progression of various pathologies, usually in conjunction with some level of inflammation [7,8].
To protect living organisms from oxidative stress, there is a system for removing ROS, called the “antioxidant system”. This system works through the action of a series of enzymes that convert ROS into less toxic species, until they are finally converted into water. The enzymes that are part of this system are: superoxide dismutase (SOD), peroxiredoxin (PRX), glutathione peroxidase (GPx), catalase (CAT), thioredoxin reductase (TRX). NADPH and GSH also participate as electron donors, and these compounds are subsequently reduced again so that they can continue to perform their function [9,10,11].
Because they are intracellular molecules that mediate intermediate cell signaling events, ROS generation may be under the control of membrane receptors. In this review, we intend to relate three receptors from two different systems to this control: the first two, which are part of the same system, are the B1 and B2 kinin receptors (abbreviated throughout the text as B1R and B2R).
These receptors are coupled to the G protein, with some variability in terms of subunit, depending on the tissue. They are effectors of the kallikrein-kinin system (KCS), classically associated with vasodilation, angiogenesis and inflammation [12]. The signaling cascade involves the cleavage of kininogen by the enzyme kallikrein, originating bradykinin (BK) in the circulation or kallidin (KD) in the tissues, which are endogenous B2R ligands. In addition, BK or KD can be cleaved by enzymes called class M and N carboxypeptidases, giving rise to des-arg-9 BK/KD, endogenous ligands of B1R. Another important detail related to this cascade is that the angiotensin-converting enzyme (ACE), an original component of the renin-angiotensin-aldosterone system (RAAS), plays an inhibitory role by degrading BK into an inactive peptide, constituting yet another variable [13]. One differentiation between these receptors is their distribution throughout the tissues: while B2R is expressed in practically all tissues, B1R is found in most only in pathological conditions and generally participates in chronic inflammatory responses [14,15,16].
The relationship with inflammatory responses is also common to the other receptor investigated in our review: histamine receptor 2 (H2R). Histamine is a biogenic monoamine that can act as a neurotransmitter or promote actions at localized sites. The main sources of histamine are mast cells, basophils, histaminergic neurons and ECL gastric cells [17,18,19] and its involvement in various pathologies is well documented [20]. Regarding H2R, this receptor belongs to the class of G-protein-coupled receptors and is characterized as coupled to the g-alpha-s subunit [20,21]. The main known action of H2R is the stimulation of acid secretion by the parietal cells of the stomach, however, this receptor is widely expressed in smooth muscle and endothelial tissues.
In this review we will comment on the possible interactions between these receptors, which have been identified in inflammatory processes, and the modulation of the generation and/or accumulation of reactive oxygen species, as well as identifying points in common between the two systems that may contribute to a general understanding of the regulation of ROS.
2. Methodology
The searches were carried out in the MEDLINE/Pubmed database (https://www.ncbi.nlm.nih.gov/pubmed/) on two dates in April: day 1 and day 30. The reason for carrying out the searches on two different dates was to check for the addition of new articles during this time period. We searched for articles between 1970 and 2024 with the following combinations of keywords, accompanied by the Boolean operator “AND”: "Kinin B2 receptor/ oxidative stress""Kinin B1 receptor/ oxidative stress","Histamine H2 receptor/ oxidative stress"."Kinin B2 receptor antagonist / oxidative stress","Kinin B1 receptor antagonist / oxidative stress" e "Histamine H2 receptor antagonist / oxidative stress". For the articles to be included in this review, they should meet the following inclusion criteria: original communications, article data obtained in mammal species, studies in animal models should have some strategy to ensure that treatment effects would be exclusively resulting from kinin receptors individual signaling and/or H2R signaling, and finally, the articles should be published in English language. The articles were checked according to these criteria at two different times: first by looking at the abstracts when searching and then by reading the article's methodology section. More details about the search process can be found in Figure 1.
Figure 1.
Flow chart. The flowchart illustrates the search strategy applied to find possible interactions between kinin receptors (B1R and B2R) and histamine H2 receptors.
Figure 1.
Flow chart. The flowchart illustrates the search strategy applied to find possible interactions between kinin receptors (B1R and B2R) and histamine H2 receptors.
3. Kinin B2 Receptor on Oxidative Stress
A search for the initial set of keywords, "Kinin B2 receptor/oxidative stress," yielded 54 total results. From this initial number, 25 articles were selected for further analysis. Of these, nine evaluated the effects on the heart, five evaluated the endothelium, five evaluated the kidneys, three evaluated the blood, two evaluated the brain, and another two evaluated the ovaries and monocytes, respectively. The remaining 29 articles were excluded for the following reasons: Eleven articles were deemed to be off-topic, nine were reviews, five could not be directly attributed to B2R, and finally, one article was excluded because it was repeated and another because it had been retracted. It is important to note that in the search carried out using the terms “Kinin B2 receptor antagonist/oxidative stress”, no new results were obtained in relation to the articles listed in the search carried out using the keywords mentioned above. The following section will present the content of the papers that were selected for this review in a qualitative manner.
Delemasure et al [22]assessed the blood parameters of B2R receptor knockout animals by evaluating the ratio between vitamin C content and ascorbyl radical. The authors found a proportional increase in free radicals in the knockout animals, suggesting that the B2R receptor would play a protective role from this perspective.
Given the established links between immunological processes and the generation of reactive oxygen species (ROS), it is natural that researchers are keen to investigate the impact of kinin receptors, particularly B2R, on the behaviour of specific cell populations. The study by Fu C et al. [23] demonstrated an inverse correlation between myeloperoxidase plasma levels and B2R expression in a specific cell population, distinguished by CD34 expression.
Ayres and colleagues studied the activation of the kallikrein kinin system in a model of cisplatin-induced ovarian toxicity. In this study, the authors detected increases in reactive oxygen species, among other markers of cisplatin-induced injury. Specifically, they found increases in superoxide anion, myeloperoxidase (MPO) and N-acetyl-glucosamine (NAG) together with a decrease in reduced glutathione (GSH), suggesting that the antioxidant potential of the tissue was compromised. This was followed by an increase in the expression of the B2R protein [24]. Although this is a descriptive study, we included it due to the well-established association between kinin receptors and the damage caused by cisplatin in other tissues [25,26]. The crucial role of B2R in oxidative effects has also been described in other injury models, such as traumatic brain injury. Ferreira et al [27] showed that B2R activation mediates both the increase in interleukin and TNF-a as well as the activity of nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) and thiobarbituric-acid-reactive species (TBARS).
The combination of B2R and oxidative stress is also frequently investigated in the kidney, with protective or deleterious effects on various physiological parameters, depending on the context. Specifically, in the ischaemia/reperfusion model, B2R has been shown to promote oxidative stress. The authors observed an association between B2R and the local inflammatory response induced by the administration of tissue kallikrein, accompanied by an increase in malonylaldehyde (MDA) and hydrogen peroxide production and a decrease in GSH levels [28]. Bledsoe et al [29] investigated the role of B2R in a gentamicin-induced nephrotoxicity model. In this study, B2R activation was achieved through viral vector-mediated overexpression of human tissue kallikrein. This study showed that B2R activation reduced superoxide anion levels and NADH oxidase (NOX) activity, indicating a protective role for B2R.
Another chronic disease explored as a possible target for renal modulation by B2R is diabetes. Here, B2R activity was stimulated by treatment of mice with an angiotensin-converting enzyme inhibitor (ACEi), whose positive effects on modulating oxidative stress (decreased MDA and increased GPx activity) were mediated by B2R [30]. Investigating diabetes in B2R knockout mice, Jaffa MA et al. [31] found results that corroborate those described in the previous article, since the diabetic knockout animals showed elevated gene expression of the enzyme superoxide dismutase (SOD), which suggests a greater production of free radicals compared to wild type diabetic animals.
Viral vector-mediated overexpression of tissue kallikrein was used again as a strategy to activate B2R and evaluate its potential results in a model of kidney injury caused by gentamicin administration. This therapy brought beneficial effects by decreasing local inflammation and also by correcting decreased NO levels and increases in superoxide production and NADH oxidase activity. These effects were all dependent to some extent on local B2R activation [32]. These results were corroborated in another study, where treatment with kallikrein was infused together with or after gentamicin administration. Again, B2R reduced injury, pro-inflammatory parameters and superoxide generation [33].
The endothelium is a significant tissue of interest for kinin research, particularly in the context of free radical modulation. Studies often explore the intersection of these two topics. For instance, Niewiarowska-Sendo et al. [34] demonstrated using Huvec cells that B2R activation produced a dual effect: it increased ROS and superoxide dismutase levels, while also enhancing catalase (CAT) activity, a well-known scavenger of reactive species.
In another study utilizing human aortic endothelial cells, Yasunari et al. [35] investigated the effects of ACEi treatment on the excessive generation of free radicals induced by high glucose levels. They found that ACEi treatment partially reversed the increase in ROS, with these effects being mediated by B2R activation.
In a thromboangiitis obliterans model, specifically examining the femoral artery of rats with this type of injury, Du et al. [36] demonstrated that basal B2R activity in the tissue served as a protective factor. It mitigated the injury process, reduced apoptosis, and decreased the production of reactive oxygen species following doxorubicin stimulation.
In the study examining the direct impact of B2R activation on hydrogen peroxide-induced damage in a bovine aorta strain, it was demonstrated that receptor activation exerted a protective effect. This effect prevented cell senescence, mitigated the harmful impacts of hydrogen peroxide on cell migration post-injury, and reduced DNA damage. The specificity of this inhibitory effect was confirmed by the lack of impact from B2R activation in the absence of hydrogen peroxide [37]. These findings related to cellular senescence were corroborated in human umbilical cells, further establishing B2R activation as a protective factor against the senescence phenotype induced by hydrogen peroxide [23].
In line with our research strategies, the function of B2R has been most extensively studied in cardiac tissue. One study examined the role of B2R in the basal state by analyzing cardiac tissue from knockout mice. The findings indicated that the absence of B2R in cardiac muscle resulted in an oxidative stress phenotype, evidenced by increased protein expression of the enzyme NADPH oxidase and elevated levels of hydrogen peroxide and superoxide anion [38].
In another model, investigating the effect of tissue kallikrein treatment in a myocardial infarction model, Yao et al. [39] demonstrated that B2R played a crucial role in inhibiting the oxidative stress caused by the injury. This was illustrated by the reduction in superoxide anion levels, NADH oxidase activity and MDA levels.
The link between senescence, oxidative stress and the role of B2R in modulating these processes has been extensively evaluated in cardiac muscle. In one study, 18-month-old mice with B2R knockout at baseline were examined. The results mirrored those observed in younger animals, as discussed above: the absence of B2R led to an increase in oxidative stress, evidenced by elevated levels of ROS, MDA and NADH oxidase activity, along with a decrease in the antioxidant enzymes SOD and catalase. Notably, these markers of oxidative stress in the heart muscle were consistent with those found in the animals' serum [40].
Analyses using H2O2-induced senescent cardiomyocytes revealed that B2R activation effectively mitigated ROS generation and NADPH oxidase activity while restoring SOD activity. Importantly, H2O2 induction also resulted in decreased B2R expression, an effect that was reversed by B2R activation, indicating the receptor's protective role in cardiac tissue [41].
In addition, the role of B2R in oxidative stress has been explored in models of hypertension. Using a rat model of hypertension induced by excessive sodium intake, the researchers examined the effects of ACEi administration and its relationship with B2R modulation. The results showed that ACEIs had a positive impact on oxidative stress, reducing NADPH oxidase, both gene and protein expression, and decreasing the expression of the LOX-1 gene, which is associated with oxidative stress. These beneficial effects depended on the activation of B2R [42].
Yoshida et al. [43] investigated the molecular mechanisms underlying the cardioprotective effects of angiotensin II type 1 receptor antagonists (ATRA), in particular valsartan, focusing on their interactions with bradykinin, eNOS and oxidative stress pathways. Using Dahl salt-sensitive hypertensive rats, the study examined various treatment regimens over five weeks. The results indicated that valsartan, at both high and low doses, increased the expression and activity of eNOS and suppressed the expression of NAD(P)H oxidase components and markers of vascular injury. These effects were not observed with the addition of a bradykinin B2 receptor antagonist or with hydralazine alone. The results suggest that the cardioprotective effects of valsartan are partially mediated by the bradykinin-eNOS and oxidative stress pathways.
Cardiac hypertrophy was also assessed in the context of B2R activation. The effect and mechanisms of tissue kallikrein were investigated using hypertrophic animal models in rats and B1 or B2 knockout mice after aortic constriction. They observed that kallikrein had general positive effects on the disease phenotype, which was accompanied by a decrease in the activity of the enzymes NADH/NADPH oxidase and reduced superoxide generation. These adaptations were dependent on B2R activation [44].
Similar results were presented using Angiotensin-converting-enzyme inhibitors (ACEi) as a therapeutic agent, (Lu L et al. 2015). In this study, cardiac hypertrophy was induced in rats through sinoaortic denervation, which caused increases in NADPH oxidase activity, myeloperoxidase and the amount of TBARS, concomitantly with a decrease in the GSH/GSSG ratio. All these alterations were corrected by ACEi treatment, and these effects were mediated by B2R activation [45].
The models of ischemia/reperfusion were evaluated in relation to the role of B2R in the general context of injury and, specifically, in relation to oxidative stress. Evaluating cats, Kumari et al.[46] showed that the basal activation of B2R stimulated the generation of TBARS, which contributed to the oxidative stress promoted by this type of approach. On the other hand, Yin et al. [47] demonstrated that the positive effects of kallikrein therapy after ischemia/reperfusion-induced injury were mediated by B2R, specifically in increasing NO levels and decreasing superoxide generation.
Most studies involving B2R have shown positive results in relation to a protective effect against the exacerbated increase in reactive oxygen species. These findings highlight the critical role of B2R in the homeostasis of various tissues. Among the studies presented above are those in which B2R antagonists were tested exclusively, undertreating the contribution of this constitutive receptor in the KKS.
Interestingly, contrary to the more classical picture of the role of B2R in inflammatory activation and progression, some studies highlight that B2R activation and subsequent generation of reactive oxygen species, can inhibit the activation of inflammatory pathways and mediators such as JNK, TGF-Beta, ERK [29,32,44]. B2R activation followed by eNOS stimulation is a known pathway for the physiological modulation of various processes, such as vasodilation, angiogenesis and modulation of energy metabolism [48,49].
On the other hand, some studies highlight a detrimental effect of B2R activation, resulting in stimulation of ROS generation and consequent amplification of the damaging effects mediated by different agents in different models. One such study used the cisplatin-induced toxicity model [24]. The contribution of kinin receptors to the progression of cisplatin-induced injury has already been described in some studies [25,26]. Ayres et al. [24] highlights inflammation as a central point capable of directing B2R activity in a different way to that presented in most studies. This study points out that the particularities of some models could direct B2R signaling towards the formation of cytokines such as IL-1 Beta and TNF-alpha, leading to the generation of reactive oxygen species, as well as increasing the migration of immune cells to the lesion environment [50].
An interesting question related to the signaling properties of B2R concerns the timing of the pathway's activation. In addition to the aforementioned study on cisplatin, two other studies have reported negative results associated with B2R activity [28,46]. These studies used the ischemia/reperfusion model and exclusively activated B2R during the protocol, while another study that initiated B2R activation before inducing injury produced positive results [47]. These findings, which are consistent with other reports in the literature [51], suggest that B2R function may vary depending on the timing of activation in the context of ischemic injury. Consequently, the moment of activation seems to be a critical factor, possibly explaining the disparate results observed in the same experimental model.
4. Kinin B1 Receptor on Oxidative Stress
The first set of keywords used "Kinin B1 receptor AND oxidative stress" returned 47 articles. Of these, 11 articles were selected, of which 4 evaluated the endothelium, 2 evaluated the kidneys, 2 evaluated the heart, another 2 evaluated the nerves, 2 evaluated the eyes, 2 evaluated the brain/hypothalamus and 1 evaluated the ovary. A total of 36 articles were excluded. The reasons for exclusion were: 22 articles were off topic, 5 articles were repeated in relation to the search for B2R, another 5 articles had non-specific effects and, finally, 4 articles were review articles. The other keyword combination used for B1R "Kinin B1 receptor antagonist AND oxidative stress" did not return any new results. In addition to the 11 articles selected, this section also includes another 5 articles that came from the search for B2R, but which also analyzed the effects of B1R.
Monitoring the basal plasma status of B1R receptor knockout animals with the intention of establishing whether the absence of this receptor had any impact on their oxidative balance was described by Delemasure et al. [22]. Using plasma levels of vitamin C and the ascorbyl radical as parameters, the authors showed that the knockout animals had increased levels of the ascorbyl radical, indicating a greater tendency towards a more oxidative environment [22].
In a study on the effects of cisplatin treatment on mouse ovaries, the authors [24] aimed to determine if the B1R receptor was linked to the injury caused in this tissue. They observed elevated levels of B1R protein, along with increased levels of MPO and superoxide, and decreased levels of GSH.
The kidney is a traditional tissue for studying induced modulations of the CCS. In a study evaluating the impact of an ischemia/reperfusion model, researchers aimed to confirm the effects of kallikrein administration in this context. They found that this treatment intensified inflammation and tissue necrosis, as well as the generation of MDA and superoxide, while also leading to a decrease in GSH levels. However, the overall worsening of the tissue condition was not associated with B1R activation [28].
In a different study focusing on the kidney and its response to salt-induced hypertension, the impact of the B1R receptor by comparing knockout mice lacking this receptor to wild-type mice was examined. The study found that knockout mice showed partial protection from the adverse effects of hypertension on the kidney, attributing this effect to reduced generation of reactive oxygen species (ROS) compared to wild-type mice [52].
In a study examining the factors associated with the production of reactive oxygen species in the heart tissue of animals lacking B1R, it was discovered that these animals had elevated levels of superoxide anion, peroxynitrite, and NO. Additionally, they exhibited reduced SOD activity, indicating the presence of oxidative stress. Moreover, these animals also showed impaired cardiac function, including reduced systolic capacity and increased coronary perfusion pressure [38]. Investigating the heart of B1RKO mice with induced diabetes and comparying the parameters related to oxidative stress the researchers [53] showed that the B1R receptor knockout animals were protected, to a certain extent, against the increase in nitrotyrosine and MPO, which were the two parameters used to assess the effects of diabetes on the animals' hearts.
B1R was also studied in Streptozotocin- induced diabetes model in rats. Systemically administered B1R antagonist to diabetic rats, was able to decrease MDA levels and increase SOD and GSH levels in the animals' sciatic nerve [54].
A study on the optic nerve with a similar experimental design showed that B1R inhibition reversed the increases in MDA and the reductions in GSH [55]. Aside from the optic nerve, these effects were also noticed in the visual cortex and plasma of the animals. A study on the interplay between ROS in diabetes and B1R inhibition revealed that diabetes induction leads to an increase in B1R expression in the retina. Furthermore, the administration of its antagonist was shown to reduce the levels of superoxide anion produced in this tissue in Streptozotocin diabetic rats [56].
Likewise, the inhibition of B1R prevents Angiotensin II-induced neuroinflammation and oxidative stress in primary hypothalamic neurons of mice. The effects caused by Angiotensin II were found to be linked to the activation of B1R. Specifically, B1R was shown to be responsible for increasing the production of reactive oxygen species (ROS) and for increasing the expression of NADPH oxidase [57].
The evaluation of hypothalamic neurons from hypertensive animals showed that treatment with a B1R agonist increased the generation of reactive oxygen species (ROS) in this tissue. This increase was found to be part of the induction of an inflammatory response at the cellular level, as these experiments were carried out ex-vivo [58].
Next, we describe the articles that dealt with the relationship between B1R and reactive oxygen species in the endothelium, where the relationship between glycemic disorders and this receptor is widely explored.
We highlight a study that evaluated the generation of reactive species in the aorta of rats treated with high doses of glucose. The effects of B1R were assessed by administering an inhibitor of the conversion of its agonist (i.e. CPM/CPN enzymes). The authors observed that this treatment reduced the content of superoxide anion, the protein expression of nitrotyrosine and the gene and protein expression of B1R itself [59].
An endothelial cell line treated with high glucose was used to investigate to study the impact of ACE inhibitors on oxidative stress and to determine if B1 receptors might be involved. It was found that ACE inhibitors decreased ROS generation and protected against glucose-induced inhibition of cell proliferation. Interestingly, these effects were not connected to the B1 receptor [60]
Insulin resistance is another model used to study disturbances in glucose homeostasis. Research involving rats in this condition [61] found that a single administration of the B1R antagonist affected the animals' perception of pain but was unable to correct the high levels of superoxide anion in the aorta.
However, a study with a similar design to the previous one yielded different results [62]. The difference lies in the fact that in this study, the B1R antagonist was administered for 7 days. This time, the treatment was able to reduce superoxide anion levels and NADPH oxidase activity.
Most studies involving B1R show that its activation has a negative effect, and therefore promotes oxidative stress, and is related to increased injury and damage induced by free radicals. It is important to note that this receptor, as mentioned in the introduction to this manuscript, is considered to be an inducible protein in various tissues, with basal expression being greatly reduced or absent in situations of homeostasis. The situations in which its expression is usually stimulated involve inflammatory responses, of which this receptor is an intermediary and amplifier, stimulating the secretion of cytokines such as TNF-alpha and IL-1 Beta, which are also involved in the generation of free radicals [63,64].
Another interesting mechanism linking B1R with oxidative stress potentiating effects is its relationship with the generation of reactive oxygen species stimulated by AngII, a peptide from another system that has major connections with the CCS. The direct stimulation via B1R of NADPH oxidase 2 and 4 thus adds another mechanism by which this protein can stimulate the generation of reactive oxygen species and, consequently, the increase in lesions and pathological conditions [57].
However, two studies carried out in the hearts of B1R receptor knockout animals showed opposite results [22,38]. Our first hypotheses to explain these data are related to aspects of indirect regulation due to the model chosen: the absence of B1R may result in an increase in RAAS activity [65], which are closely related to the induction of inflammatory processes and the generation of reactive oxygen species, as demonstrated earlier in this session. Another consequence that could be involved would be an excessive increase in B2R expression, also characteristic of this knockout model [66]. This “excess” could result in an imbalance of B2R activity towards inflammatory activity and therefore stimulating the generation of reactive oxygen species, in a similar way to what [67] demonstrated in relation to negative cardiac adaptations.
A third aspect that can be raised in relation to the possible protective role of the B1R receptor is the tissue in question, since in the heart this receptor is normally expressed in mice and could, in this specific context, play some protective role in a basal situation [38].
5. Histamine H2 Receptor on Oxidative Stress
The first combination of keywords “Histamine H2 receptor AND oxidative stress” resulted in 27 articles. Of this total, 11 articles were selected and the other 16 were rejected. The reasons for rejection were being off topic (13 articles), repeated articles (2 articles), 1 article because it was a review and another because its effects were considered non-specific. The search for the second set of keywords “Histamine H2 receptor antagonist AND oxidative stress” returned 67 total results, of which 30 were selected and 37 were rejected. The reasons for rejection were off topic for 15 articles, 18 repeated articles and language different from those pre-established in the case of one article. Among the articles selected, 21 evaluated the gastric mucosa, 3 evaluated the liver, 2 articles were found for the following tissues: heart, brain, bone marrow and esophagus, while 1 article was found for the following tissues: myeloid cells, neutrophils, testicles, intestine and breast tumor.
We began by describing the results of two articles that investigated the interactions between H2 receptor antagonists and free radicals in a tissue-independent manner and, therefore, without binding to the receptors. Although these articles are not directly related to the central theme of the study, we believe it is interesting to report their results in order to contextualize other articles where the intention is to evaluate the direct interaction between receptors and antagonists and their effects.
The first study was carried out in test tubes and sought to assess whether ranitidine, famotidine and cimetidine had any kind of direct interaction with free radicals. They showed that these drugs are capable of sequestering hydroxyl anions, and cimetidine has the additional ability to act as an iron chelator. This is evidence that there is a possibility that these drugs have direct effects independent of their binding to H2R [68]. Another study evaluated the ability of H2R inhibitors to reverse the glycation of bovine protein albumin. Among the drugs studied, ranitidine stood out for its ability to reduce the amount of glycation products, its ability to sequester reactive oxygen species and act as a metal chelator [69].
To begin describing the results found in tissues, we have a study that investigated the effects of the antagonist famotidine in a rat model of testicular ischemia. The antagonist had no effect on GPx and MDA, but restored the decreased levels of SOD and corrected the increases in NO [70].
Another interesting study, which evaluated the modulation of H2R activity in the context of breast tumour development in female mice, showed that cimetidine was able to decrease MDA levels and increase GSH and SOD levels, effects that were enhanced by the addition of vitamin C. This modulation of tumor oxidative stress resulted in lower proliferation and longer survival of the animals [71]
Again evaluating the action of cimetidine, now in the context of a glycerol-induced kidney injury in rats, it was shown that H2R inhibition was able to elevate NO levels, and reduced glutathione levels [72].
Cimetidine, as studied in [73], has shown to have protective effects against oxidative stress caused by a radiation protocol in rats. The analysis of the animals' bone marrow revealed a corrective effect on the increased levels of MDA and reduced levels of GSH and SOD.
In a study involving mice with myeloid cells expressing the KRAS protein, which turned the cells into leukemic cells, researchers found that activating H2R had positive effects on free radical generation in leukemic cells. H2R activation decreased the accumulation of ROS and the momentary generation of superoxide anion. These results were associated with longer survival in animals treated with H2R agonists [74].
Testing the effects of cimetidine on human neutrophils, it was found that H2 receptor inhibition may possess anti-inflammatory properties in certain situations. When neutrophils were stimulated with cytokines and treated with cimetidine, there was a decrease in myeloperoxidase activity. However, the generation of superoxide anion remained unaffected [75].
Cardiac tissue has also been the target of research involving the participation of H2R in adaptations related to the generation of reactive oxygen species. The first article sought to assess whether the H2R receptor had any relationship with the induction of an oxidative response induced by the activation of the Alpha 1 adrenergic receptor in cardiomyoblasts. The authors were able to demonstrate that H2R inhibition attenuated the generation of ROS, lipid peroxidation and decreases in SOD and PRX3 activities caused by alpha 1 activation [76].
Corroborating these data in a model of oxidative stress induction in rats, Kondru et al. [77] show that lipid peroxidation, increased nitrite/nitrate ratio, as well as decreased SOD activity were corrected by administration of the H2R antagonist famotidine.
Below we present two articles that share similarities in the tissue examined (brain) and the agent associated with H2R activity, carnosine. The first study focused on mouse brain endothelial cells treated with rotenone to induce oxidative stress. The authors found that carnosine protected the mitochondria of these cells against loss of membrane potential, which is typically linked to increased generation of reactive species. These protective effects were dependent on H2R activation [78].
In another study [79] that tested the effects of carnosine in a cerebral ischemia/reperfusion protocol in mice, was demonstrated that the corrective effects of carnosine on increased MPO activity, increased TBARS levels, and decreased GSH levels were all mediated by H2R activation.
We will now begin to describe the articles dealing with the gastrointestinal tract. The first article investigated the ability of cimetidine to prevent the damage caused by a radiation protocol in rats. In this study, a protective effect of the inhibition of H2R activity was observed, illustrated by a decrease in MDA levels and an increase in GSH and SOD levels in the intestinal mucosa of the treated animals [80]. The second study found aimed to clarify the potential of quercetin as a mitigating agent for gastric lesions caused by the drug dicofenac. Interestingly, they observed that quercetin was able to decrease the lesion, which involved decreasing oxidative stress levels, and these effects were mediated by H2R activation: increases in MDA were reversed in the presence of quercetin [81].
Another model in which this topic was investigated was that of intestinal damage caused by indomethacin. In this study, a combination of vitamins with ranitidine was used to treat rats, and the results showed that H2R inhibition was able to contribute to decreasing lesions by correcting the increase in MDA and the reductions in GSH and GPx [82].
The relationship between H2R and oxidative stress was also followed in the liver. The activity of the enzyme catalase in mouse liver extract was analyzed upon adding cimetidine. This study demonstrated that cimetidine can directly inhibit catalase activity, suggesting a pro-oxidative role in the liver environment. This inhibition occurred due to cimetidine binding to non-catalytic sites present in the enzyme [83]. However, in the model of chemically induced hepatotoxicity in albino rats, the effects on parameters related to oxidative stress (increases in MDA and decreases in GSH) were partially corrected by the administration of cimetidine [84]. The third liver model discussed here focuses on the hepatic ischemia/reperfusion protocol conducted in rats. In this study, the introduction of histamine was found to have a positive impact on parameters associated with oxidative stress: it led to a decrease in MDA levels and an increase in GSH levels. Interestingly, these effects were not blocked by H2R receptor antagonist drugs, suggesting the involvement of other histamine receptors in the observed outcomes [85].
In the following studies, the involvement of H2R receptors in protecting the esophageal mucosa is discussed. The initial study, conducted on rats, utilized a model of esophagitis and compared the effects of ranitidine with those of "traditional" antioxidants. The findings indicated that only the antioxidant treatment was able to reverse the increase in MDA and the reductions in GSH, as analyzed by the authors [86].
Two additional studies further supported these results by comparing the effects of the antioxidant DA9601 with those of ranitidine in models of chemically-induced esophagitis, Barrett's esophagus, and reflux esophagitis. Once again, H2R inhibition did not have an impact on MDA, GSH, and MPO, while the antioxidant compound successfully reversed the increases in MDA and decreases in GSH [87,88].
The following studies focus on the effects of H2R (histamine H2 receptor) inhibition on gastric mucosal damage. The first study compared the protective effects of famotidine with an extract from the Chinese plant Acanthopanax senticosus on gastric injury induced in rats by a compound called C48/80. The results showed that H2R inhibition reduced the damage caused by C48/80 by correcting the increased levels of MPO, MDA, and XO. The plant extract also showed some protective effects, though to a lesser extent [89].
The second study involved inducing gastric injury in rats by restricting their movement. In this study, histamine and H2R antagonist were administered in the brain to evaluate the central effects of H2R signaling. The researchers found that administering histamine in the brain had positive effects on the gastric lesion by correcting the decreased levels of SOD and the increased levels of MDA. These positive effects were mediated by the activation of H2R in the brain [90].
Also using the movement restriction, but in an aquatic environment, to induce gastric lesions in rats, the protective effects of ranitidine and thymoquinone were compared. Both treatments were found to correct increased TBARs, decreased GSH, and reduced activities of CAT and SOD. However, ranitidine showed superior results in all parameters, with additional benefits observed when the treatments were combined [91].
In a model of gastric injury induced by indomethacin injection, the effects of famotidine and Malus domestica Borkh plant extract were compared. Both treatments showed positive effects in correcting high MDA levels and decreasing GSH content and GPx activity. The plant extract demonstrated superior performance in reducing MDA and increasing GSH [92].
Another study using indomethacin to induce lesions in rats compared the effects of famotidine with felodipine, a calcium channel blocker. Both treatments were equally effective in reducing MDA levels and correcting reduced GSH levels and CAT activity [93].
In a study comparing ranitidine with topiramate using the indomethacin model, both treatments were equally effective in reducing lesions by restoring the activities of SOD, CAT, GPx, GSH levels, and MDA amounts [94].
Further investigations using female rats compared the effects of famotidine with vardenafil, a phosphodiesterase inhibitor, in an indomethacin-induced gastric injury model. The study found that famotidine showed a protective effect by decreasing MDA levels and increasing NO levels, while vardenafil had modest protective effects [95].
Lastly, using the indomethacin model the effects of cimetidine were compared with a Ficus asperifolia plant extract. Both treatments exhibited similar results in correcting reduced SOD and CAT activity, while the plant extract was the only one to decrease MDA levels [96].
Some researchers used the ethanol administration model as a strategy for generating gastric lesions. The first article of the series sought to verify the possible positive effects of famotidine administration in rats submitted to this model. Interestingly, this administration had a dual effect on the amount of TBARs: at 24 hours after treatment, there was a famotidine-induced increase and in subsequent measurements (48 and 72 hours) the effects were positive (reduction of TBARS). On the other hand, free radicals and glutathione levels were corrected by famotidine after 48 hours of treatment [97]
In another ethanol-induced injury protocol, the authors compared the effects of ranitidine with those of the Eremurus persicus plant extract. Both treatments partially restored MDA levels, decreased ROS generation and corrected the reduced GSH content and reduced catalase activity. Ranitidine outperformed in all parameters except MDA [98].
Another comparison between ranitidine and Pulicaria crispa plant extract showed similar actions of the two compounds in reducing the lesions caused by ethanol administration. These effects were mediated by the correction of decreased levels of SOD and GSH [99]
In the last comparison between H2R antagonists and plant extracts in the ethanol gastric injury model, the authors compared ranitidine with Onosma armeniacum root extract. Both treatments had similar effects, which involved correcting the decreased levels of GSH and NO, as well as the increases in MPO [100].
A comparative study explored the effects of famotidine and a standardized antioxidant preparation. Both demonstrated similar protective effects against ethanol-induced lesions, reducing MPO activity and modulating MMP-9 content to inhibit its increase [101].
Another comparison aimed to assess the impact of ranitidine and oleuropein on ethanol-induced injury in rats. Ranitidine effectively regulated elevated TBARs concentrations and the decrease in GPx and GSH. Similarly, oleuropein produced comparable effects [102].
In this final section, we will describe the results of gastric lesions induced by mixed models. The first study compared the effects of ranitidine with those of Elaeagnus conferta Roxb plant extract in rats. Both treatments were equally effective in correcting the decreased levels of GSH and the reduced activities of SOD and CAT, as well as in attenuating the increases in MDA [103].
Continuing to evaluate plant extracts versus H2R antagonists, another study compared the effects of cimetidine with those of rutin. Both agents exhibited protective activity, correcting elevated MDA levels, drops in vitamin C content, and GPx activity. Rutin outperformed in relation to vitamin C, while cimetidine outperformed in relation to GPx [104].
Further comparing ranitidine with the plant extract Ocimum sanctum in different models and animals, it was shown that histamine has a similar effect to that obtained by the injury models used, and both treatments were able to correct the increases in MDA and decreases in SOD [105].
Another comparison with plant extracts, this time involving ranitidine and Garcinia kola, was carried out in the context of gastric injury induced by the combination of HCL and ethanol or indomethacin. Both treatments were able to produce protective effects, correcting the high levels of MDA and the reduced levels of SOD, CAT activity, and the reduced amount of GSH in the two injury protocols [106].
In a comparison between famotidine and pantoprazole (hydrogen pump inhibitor) [107], it was found that only pantoprazole was able to reverse the high levels of MDA caused by the mixed injury model involving acetic acid and ethanol.
Evaluating the effects of these compounds in a model of gastric injury caused by the combination of aspirin and pyloric ligation, they showed that both treatments had significant corrective effects on the increased levels of MDA and on the reduced levels of GSH and the reduced activities of SOD, GPx, and CAT [108].
Finally, we present a study comparing the effects of ranitidine with those obtained by an ARB on the repercussions of gastric injury induced by indomethacin or CRS models. Both treatments showed protective effects, illustrated by an increase in the nitrite/nitrate ratio and a decrease in MDA content [109]. These results are particularly interesting when we consider that the inhibition of AT1 receptor activity can have a direct impact on CRS activation and therefore on B1R and B2R receptor activity.
Although there are important differences in the focus of research involving the three receptors in terms of the tissues investigated, experimental designs and methodologies used, it is possible to draw similarities from the results presented in this review. The first is between the role of the B1R and H2R receptors, which are mostly related to amplifying the generation of reactive oxygen species, often making them good targets for pharmacological inhibition in pathological scenarios. When we look at the results, it is possible to infer that these two receptors may act as redundant players in chronic inflammatory response scenarios and, based on this reasoning, we believe that their joint inhibition would be a good suggestion to improve therapeutic results for gastric diseases
The majority of the studies presented indicate a negative role for H2R activation in the generation of reactive oxygen species, associating its activation with more severe injury. This can be explained by the known inflammatory functions of histamine and the role of H2R activation in promoting local inflammatory responses and recruiting inflammatory cells [110]. However, there is an interesting group of articles suggesting that H2R activation has protective effects against the amplification of oxidative stress. These articles imply a relationship between the role of histamine and the context in which the production of free radicals is evaluated.
Of particular interest is the brain, where two articles evaluating the effects of histamine activation showed results contrary to the majority of articles. One of these articles, analyzing a model of cerebral ischemia/reperfusion, demonstrated an anti-inflammatory role for histamine, reducing the recruitment of immune cells and oxidative stress through activation of the PKA/AMPc pathway [78,79,111,112]. In another study, brain administration of histamine reduced gastric injury in rats, indicating that the central and peripheral effects of histamine can be different and even antagonistic within the same injury model [90].
Another significant case is that of animals with the Kras mutation in myeloid cells, where treatment with histamine significantly reduced the generation of reactive oxygen species by inhibiting NADPH oxidase [74]. These examples illustrate the need to better understand the variables behind this modulation and how to use this information to prescribe more appropriate treatments for various disorders where the generation of reactive oxygen species plays a key role.
While there are important differences in the focus of research involving the three receptors in terms of the tissues investigated, experimental designs, and methodologies used, it is possible to draw similarities from the results presented in this paper (Table 1). One similarity is between the role of the B1R and H2R receptors, which are mostly associated with amplifying the generation of reactive oxygen species, making them potential targets for pharmacological inhibition in pathological scenarios. Based on the results, it can be inferred that these two receptors may act as redundant players in chronic inflammatory response scenarios, suggesting that their joint inhibition would be beneficial in improving therapeutic outcomes for gastric diseases. The analysis of the three receptors discussed in this review makes it clear that the global context has a significant influence on the potential therapeutic effects of combining these agents (Figure 2). In order to maximize benefits and minimize serious side effects, it is crucial to enhance our understanding of the local and systemic interactions modulated by these receptors. This can be achieved through more mechanistic studies in animal models and ethical human research. Additionally, it is important to continuously improve drug distribution routes to target these agents to specific tissues and avoid off-target effects, which can be detrimental and outweigh the benefits.
Figure 2.
Overview of the role of the B1 and B2 kinin receptors and the H2 histamine receptor in modulating the production of reactive oxide species (ROS) in different organs and tissues. Bradykinin B1 Receptor (B1R), Bradykinin B2 Receptor (B2R), Histamine H2 Receptor (H2R), Reactive Oxygen Species (ROS), Nicotinamide Adenine Dinucleotide Phosphate Oxidase (NADPH oxidase), Superoxide Dismutase (SOD), Glutathione (reduced form) (GSH), Glutathione Disulfide (oxidized form) (GSSG), Catalase (CAT), Malondialdehyde (MDA), Thiobarbituric Acid Reactive Substances (TBARs), Glutathione Peroxidase (GPx), Hydrogen Peroxide (H2O2), Myeloperoxidase (MPO).
Figure 2.
Overview of the role of the B1 and B2 kinin receptors and the H2 histamine receptor in modulating the production of reactive oxide species (ROS) in different organs and tissues. Bradykinin B1 Receptor (B1R), Bradykinin B2 Receptor (B2R), Histamine H2 Receptor (H2R), Reactive Oxygen Species (ROS), Nicotinamide Adenine Dinucleotide Phosphate Oxidase (NADPH oxidase), Superoxide Dismutase (SOD), Glutathione (reduced form) (GSH), Glutathione Disulfide (oxidized form) (GSSG), Catalase (CAT), Malondialdehyde (MDA), Thiobarbituric Acid Reactive Substances (TBARs), Glutathione Peroxidase (GPx), Hydrogen Peroxide (H2O2), Myeloperoxidase (MPO).
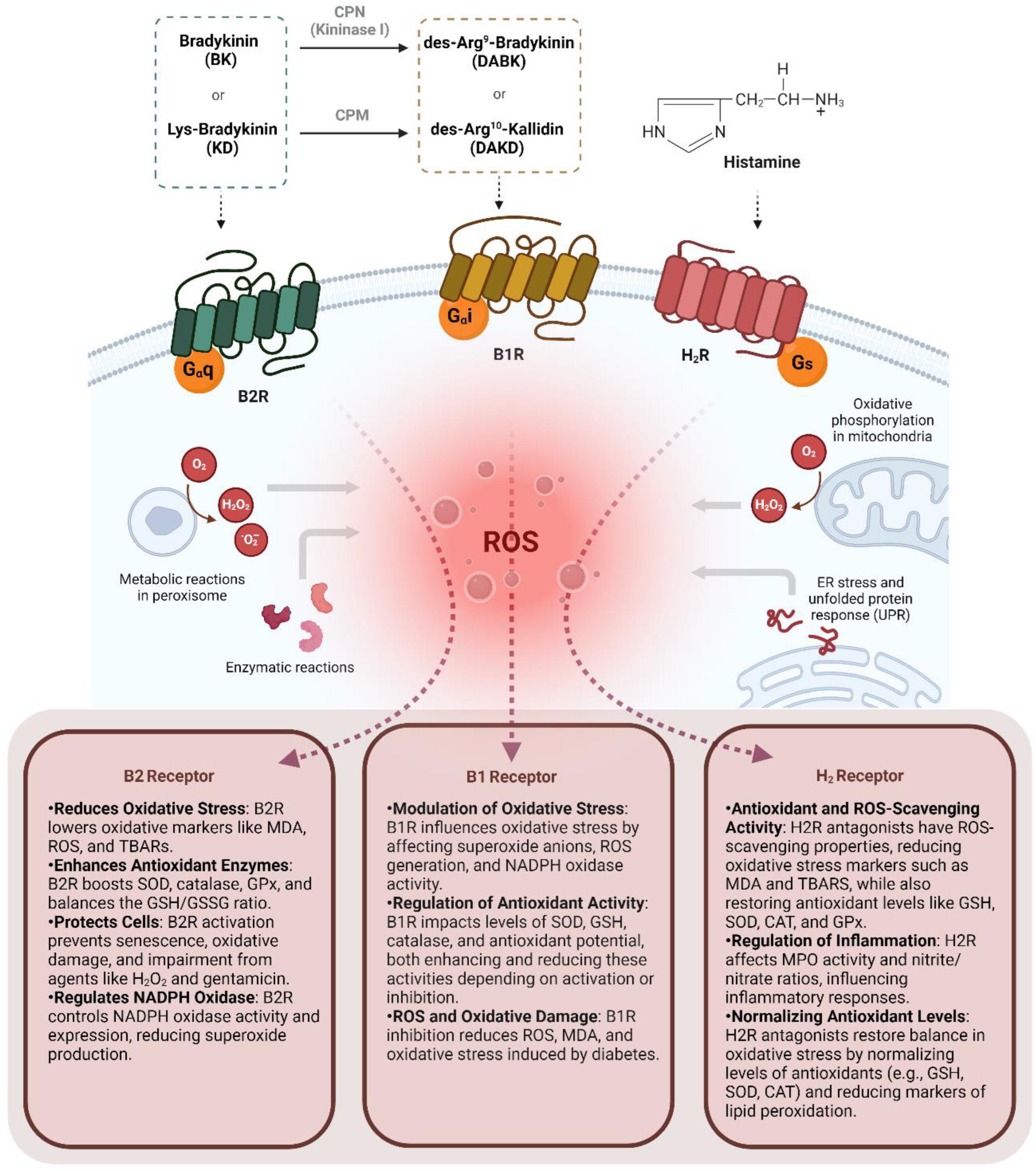

Table 1.
Overview of studies targeting kinin B1, B2, and histamine H2 receptors and their associated oxidative stress endpoints.
Table 1.
Overview of studies targeting kinin B1, B2, and histamine H2 receptors and their associated oxidative stress endpoints.
| Target receptor | Sample | Tissue | Main methods | Main outcomes | Reference |
|---|---|---|---|---|---|
| B1R | Male rats | Thoracic aortic rings | High glucose feeding, B1R antagonist (SSR240612) | B1R inibition did not affected superoxide anions (O2•─) production. | Dias et al., 2007 |
| B1R | C57BL/6 and B1KO mice | Heart | Streptozotocin (STZ)-induced diabetes, B1R antagonist (R-954) | B1R absence partially reversed increased nitrotyrosine and myeloperoxidase levels induced by diabetes. | Westermann et al., 2009 |
| B1R | Male rats | Thoracic aortic rings | High glucose feeding, B1R antagonist (SSR240612), B1R agonist (Sar[D-Phe8]des-Arg9-BK) | B1R activation increased superoxide anions (O2•─) production, increased NADPH oxidase activity, SOD gene expression, and catalase protein expression. | Dias et al., 2010 |
| B1R | Male rats | Ocular tissue (retina) | Streptozotocin (STZ)-induced diabetes, B1R antagonist (LF22-0542) | B1R antagonist normalized the elevated B1R levels and reduced superoxide production. | Pouliot et al., 2012 |
| B1R | Male rats | Sciatic nerve | Streptozotocin (STZ)-induced diabetes, B1R antagonist (R-954) | B1R inhibition reversed diabetes-induced increases in MDA levels and restored the reduced GSH activity, antioxidant potential, and SOD content. | Catanzaro et al., 2013 |
| B1R | Male rats | Optic nerve, visual cortex, plasma | Streptozotocin (STZ)-induced diabetes,B1R antagonist (R-954) | B1R inhibition reversed diabetes-induced increases in MDA levels across all tissues and restored the reduced GSH content in all tissues. | Catanzaro et al., 2017 |
| B1R | Male rats | Thoracic aortic, renal cortex | High glucose infusion, carboxypeptidase M/carboxypeptidase N inhibitor (Mergetpa), iNOS inhibitor (1,400 W) | COM/CPN inhibitor corrected increased aortic superoxide production and increased nitrotyrosine renal cortex protein expression. | Haddad et al., 2017 |
| B1R | Neonatal mice | Hypothalamic neurons | Angiotensin II, B1R antagonist (R715) | B1R activation induces partially ROS generation and NADPH oxidases (Nox2 and 4) gene expression. | Parekh et al., 2020 |
| B1R | Human embryonic kidney (HEK) cells , C57BL/6, and B1KO mice | Kidney | DOCA-salt hypertension, B1R agonist (des-Arg10-kallidin, DAKD), B1R antagonist (R715), B2R antagonist (HOE 140) | B1R absence decreased kidney ROS generation in vivo, B1R increased ROS generation in vitro. | Basuli et al., 2022 |
| B1R | C57BL/6 male mice | Hypothalamic neurons | DOCA-salt hypertension, Lys-[des-Arg9]-Bradykinin (LDABK), B1R antagonist (R715), hydrogen peroxide (H2O2) | B1R activation induces ROS production, with ROS generation partially mediated by TNF, LPS, and H2O2. | Theobald et al., 2023 |
| B1R/ B2R | Human endothelial cells (AECs) | _ | High glucose, ACEi (Temocapril), BK antagonist (Icatibant), B1R antagonist (Lys-(Des-Arg9, Leu8)-Bradykinin) | B2R reversed increased oxidative stress mediated by high glucose treatment. | Yasunari et al., 2003 |
| B1R/ B2R | B1KO and B2KO | Plasma | Reactive Oxygen Species (ROS) Detection | The absence of both B2R and B1R led to an increase in plasma oxidative stress. | Delemasure et al., 2013 |
| B1R/ B2R | C57BL/6, B1KO, and B2KO | Heart | Knockout mice adaptations description | The absence of both B2R and B1R resulted in increased NADPH oxidase protein expression, superoxide anion levels, NO, and peroxynitrite production, while simultaneously decreasing the expression and activity of SOD. | Mesquita et al., 2019 |
| B1R/ B2R | C57BL/6 female mice | Ovary | Cisplatin-induced ovarian toxicity | In the cisplatin-treated group, B1R and B2R regulation resulted in increased levels of superoxide, NAG, and MPO, while GSH levels were reduced. | Ayres et al., 2020 |
| B2R | Female and male cats | Hearts | Ischemimia/reperfusion model, B2R antagonist (HOE 140) | B2R activation led to an increase in thiobarbituric acid reactive substances (TBARS) 60 minutes following reperfusion. | Kumari et al., 2003 |
| B2R | Dahl salt-sensitive hypertensive (DS) rats | Heart (LV) | Hypersodica diet, ACEi (Quinapril), and B2R antagonist (FR172357) | B2R activation increased eNOS, while decreased NADPH oxidase. | Kobayashi et al., 2006 |
| B2R | Rats | Kidney | Isquemia/reperfusion model, kallikrein, B2R antagonist (HOE 140), and B1R antagonist (Lys-(Des-Arg9, Leu8)-Bradykinin) | B2R activation increased ROS, MDA, and oxigen peroxide levels, while decreased GSH. | Chiang et al., 2006 |
| B2R | Male rats | Kidney | Adenovirus carrying the human tissue kallikrein gene, BK antagonist (Icatibant) | B2R partially restored nitrite/nitrate levels reduced by gentamicin and decreased gentamicin-induced NADH oxidase activity and superoxide production. | Bledsoe et al., 2006 |
| B2R | Male rats | Kidney | DOCA-salt hypertension, adenovirus carrying the human tissue kallikrein gene, BK antagonist (Icatibant) | B2R partially corrected the increased NADH oxidase activity and superoxide anion formation. | Bledsoe et al., 2006 |
| B2R | Male rats | Heart | Abdominal aorta contriction, cardiac hipertrophy model | B2R reduced NADH/NADPH oxidase activity, superoxide production, as well as the phosphorylation of MAPKs, ERKs, and AKT. | Li et al., 2007 |
| B2R | Rats | Serum | Streptozotocin (STZ)-induced diabetes, ACEi (Ramipril), B2R antagonist (HOE 140) | B2R increased GPx activity while decreasing MDA content. | Allard et al., 2007 |
| B2R | Rats | Heart (LV) | Hypersodica diet, angiotensin II receptor blocker (Valsartan), B2R antagonist (FR172,357) | B2R reduced both the activity and expression of NADPH oxidase. | Yoshida et al., 2007 |
| B2R | Male rats | Hearts | Coronary ligation infarction model, tissue kallikrein, BK antagonist (Icatibant) | B2R reduced NADH oxidase activity, p22 gene expression, and MDA content, while partially decreasing superoxide production. | Yao et al., 2007 |
| B2R | Male rats | Kidney | Gentamicin, kallikrein infusion, and BK antagonist (Icatibant) | B2R reduced gentamicin-induced superoxide production in the kidney. | Bledsoe et al., 2008 |
| B2R | Male rats | Hearts | Ischemia/reperfusion model, human tissue kallikrein gene, BK antagonist (Icatibant), NG-Nitro- l-Arginine Methyl Ester (L-NAME) | B2R increased heart NO production and normalized superoxide levels. | Yin et al., 2008 |
| B2R | Bovine Aorta Endothelial Cells (BAECs) | _ | ROS-induced senescence, in vitro scratch model, BK, B2R antagonist (HOE 140), NO inhibitor (Nω-Methyl-L-arginine acetate salt) | B2R protected cells from H2O2-induced senescence, DNA damage, and impaired migration. | Oeseburg et al., 2009 |
| B2R | C57BL/6 and B2KO mice | Kidney | Streptozotocin (STZ)-induced diabetes | B2R absence upregulated SOD expression. | Jaffa et al., 2012 |
| B2R | Rat cardiomyocytes cell line (H9C2) | _ | ROS-induced senescence, eNOS inhibitor (Nω-methyl-L-arginine acetate salt), BK, and B2R antagonist (HOE 140) | B2R inhibited H2O2-induced effects: reduced B2R expression, increased ROS, decreased SOD levels and activity, and elevated NADPH oxidase expression and activity. | Dong et al., 2013 |
| B2R | Swiss mice | Ipsilateral cortex | Traumatic brain injury model | B2R partially reduced NADPH oxidase activity and TBARs. | Ferreira et al., 2014 |
| B2R | Humans DM patients mononuclear cells | _ | Plasmatic measurements, BK or B2R antagonist (HOE 140) treatments | B2R increased RB mRNA, AKT phosphorylation, and cyclin D1; decreased ROS and cellular senescence; inversely correlated with plasma MPO levels. | Fu et al., 2015 |
| B2R | Male rats | Hearts | Sinoaortic denervation, ACEi (Ramipril), B2R antagonist (HOE 140), AT1R antagonist (Losartan) | B2R normalized TBARS, GSH/GSSG ratio, and NADPH oxidase activity. | Lu et al., 2015 |
| B2R | C57BL/6 and B2KO mice | Heart, Serum | Reactive Oxygen Species (ROS) Detection | B2R absence increased ROS, serum/heart MDA, and NADPH oxidase expression; decreased heart/serum SOD activity, heart SOD protein, and catalase expression. | Feng et al., 2016 |
| B2R | Human umbilical vein endothelial cells (HUVECs) | Endothelium | BK and B2R antagonist (HOE 140) treatments | B2R increased ROS, SOD, and catalase. | Niewiarowska-Sendo et al., 2018 |
| B2R | Female rats | Lower extremity veins | Thromboangitis obliterans model, B2R antagonist (HOE 140) treatment | B2R blockade increased reactive species and caspase-3 activity, and decreased Pi3k expression. | Du et al., 2019 |
| H2R | Cardiomyoblasts lineage (H9C2) | _ | Phenylephrine (alfa 1 agonist), H2 antagonist (Famotidine) | Famotidine reduced ROS and lipid peroxidation and restored SOD and PRX levels after phenylephrine treatment. | Potnuri et al., 2020 |
| H2R | Bovine serum albumin (BSA) | Bovine serum albumin (BSA) | BSA glycation, H2R antagonist (Famotidine, Ranitdine, Cimetidine) | Ranitidine showed the strongest anti-glycation and ROS-scavenging effects. | Biedrzycki et al., 2023 |
| H2R | Human blood and rats | Plasma and gastric lumen | H2R antagonist (Famotidine, Ranitdine, Cimetidine) reaction test | H2 antagonists scavenge OH radicals; cimetidine also chelates iron. | Lapenna et al., 1994 |
| H2R | Male rats | Gastric mucosa | Chronic ethanol-induced mucosal injury, H2R antagonist (Famotidine) | H2 inhibition raised TBARS at 24-48h, lowered it at 72h, reduced SROS at 48h, and increased glutathione at 48-72h. | Hernández-Muñoz et atl., 2000 |
| H2R | Male rats | Esophageal mucosa | Reflux esophagitis model, antioxidant (DA9601), H2R antagonist (Ranitidine) | H2 antagonist have antioxidant properties, scavenging ROS and offering protection against oxidative stress. | Oh et al., 2001 |
| H2R | Male rats | Esophageal mucosa | Reflux esophagitis model, antioxidant (DA9601), H2R antagonist (Ranitidine) | H2 antagonist did not affect MDA, GSH, or MPO levels. | Lee et al., 2001 |
| H2R | Male rats | Gastric mucosa | Reflux esophagitis model, antioxidant (DA9601), H2R antagonist (Ranitidine) | H2 antagonist had no impact on MDA, GSH, or MPO activity. | Oh et al., 2001 |
| H2R | Male humans | Neutrophils | Opzonized zymosan (OZ), Acetate phorbol (PMA), calcium ionophore, Rebamipide, H2R antagonist (Cimetidine) | H2 antagonist reduced MPO activity but not superoxide generation. | Shimoyama et al., 2003 |
| H2R | Rats, guinea pigs (both genders) | Blood | Ethanol gastric injutry model, piloric ligation gastric model, histamine, H2R antagonist (Ranitidine), plant extract (ocimum sanctum) | H2 antagonist partially reduced histamine-induced MDA increase and SOD decrease. | Kath et al., 2006 |
| H2R | Male rats | Gastric mucosa | HCl/ethanol gastric lesion model, indomethacin gastric lesion model, plant extract (kolaviron), H2R antagonist (Ranitidine) | H2 antagonist partially restored GSH, SOD, CAT, and reduced MDA. | Olaleye et al., 2006 |
| H2R | Male rats | Gastric mucosa | Ethanol gastric injury model, H2R antagonist (Ranitidine, Famotidine) | H2 antagonist corrected MPO activity raised by gastric ulcers. | Singh et al., 2007 |
| H2R | Rats | Gastric mucosa | Ethanol gastric injury model, plant extract (Onosma armeniacum), H2R antagonist (Ranitidine) | H2 antagonist partially restored GSH and NO and reduced MPO and MDA, but not SOD. | Cadirci et al., 2007 |
| H2R | Male rats | Gastric mucosa | Acetic acid gastric injury model, proton-pump inhibitor (Pantoprazole), H2R antagonist (Famotidine), Indomethacin | H2 antagonist did not affect ulcer-induced MDA increase. | Fornai et al., 2009 |
| H2R | Female rats | Gastric mucosa | Indomethacin gastric ulcer model, H2R antagonist (Famotidine), PDE inhibitor (Vardenafil) | H2 antagonist lowered MDA and restored NO levels. | Karakaya et al., 2009 |
| H2R | Rats | Gastric mucosa | Indomethacin gastric ulcer model, CRS ulcer model, angiotensin II receptor antagonist (Telmisartan, Candesartan), H2R antagonist (Ranitidine) | H2 antagonist normalized MDA and nitrite/nitrate ratio. | Morsy et al., 2009 |
| H2R | Male rats | Hepatic tissue | DDC hepatic toxicity model, ascorbic acid, H2R antagonist (Cimetidine), calcium channel antagonist (Nifedipine) | H2 antagonist partially corrected MDA and GSH; SOD was unaffected. | Gaafa et al., 2011 |
| H2R | Male rats | Gastric mucosa | Indomethacin gastric ulcer model, plant extract (Ficus asperifolia bark), H2R antagonist (Cimetidine) | H2 antagonist restored SOD and CAT activities; MDA was unaffected. | Raji et al., 2011 |
| H2R | Mouse brain-derived endothelial cells | _ | Rotenone, Carnosine, H2R antagonist (Cimetidine e Zolantidine) | H2 inhibition reversed carnosine's mitochondrial protective effects. | Zhang et al., 2012 |
| H2R | Male rats | Gastric mucosa | Ethanol gastric injury model, oleuropein (OLE), H2R antagonist (Ranitidine) | H2 antagonist partially corrected GSH, GPx, and TBARS but not SOD and CAT. | Alirezaei et al., 2012 |
| H2R | Male rats | Hepatic tissue | Ischemia/reperfusion model, histamine, H2R antagonist (Ranitidine) | Positive histamine effects on MDA and GSH were H2-independent. | El-Mahdy et al., 2013 |
| H2R | Male rats | Gastric mucosa | Ulceral models, rutin, H2R antagonist (Cimetidine) | H2 antagonist reduced MDA, restored vitamin C, and increased GPx. | Olaleye et al., 2013 |
| H2R | Female and male rats | Gastric mucosa | Pylorus ligation gastric ulcer model, acetylsalicylic acid (ASA), gallic acid, H2R antagonist (Famotidine) | H2 antagonist partially restored SOD, GSH, CAT, GPx, and reduced MDA. | Asokkumar et al., 2014 |
| H2R | Male rats | Gastric mucosa | Brain microinjections of histamine, H1R antagonist (Tripolidine), H2R antagonist (Ranitidine) | H2 activation reduced MDA and restored SOD activity. | Qiao et al., 2015 |
| H2R | Male rats | Kidney | Glycerol kidney injury model, L-carnitine, H2R antagonist (Cimetidine) | H2 antagonist normalized NO and glutathione, reduced cytochrome p450. | Estaphan et al., 2015 |
| H2R | BALB/c mice | Liver extracts | Catalase activity, H2O2, H2R antagonist (Cimetidine) | H2 antagonist inhibited catalase and lowered optimal temperature. | Jahangirvand et al., 2016 |
| H2R | Male rats | Heart | Oxidative stress inductor (Doxorubicin), ACE inhibitor (Captopril), H2R antagonist (Famotidine) | H2 inhibition corrected lipid peroxidation and nitrite/nitrate ratio, partially restored SOD. | Kondru et al., 2018 |
| H2R | Male rats | Gastrintestinal tract | Dicofenac enterophaty induced model, adenosine receptor antagonist (Quercetin), H2R antagonist (Ranitidine) | H2 antagonist worsened diclofenac-induced MDA increase. | Singh et al., 2017 |
| H2R | Male rats | Gastric mucosa | Water- immersion restraint stress ulcer model, Thymoquinone, H2R antagonist (Ranitidine) | H2 antagonist corrected TBARS, GSH, SOD, and CAT in ulcers. | Ahmad et al., 2017 |
| H2R | Transgenic mice Kras/NoxKO | Myeloid cells | H2R agonist (N-methylhistamine) and H2R antagonist (Ranitidine) | H2 activation inhibited superoxide and reduced ROS in Kras mice. | Aydin et al., 2019 |
| H2R | Male rats | Gastric mucosa | Ethanol gastric injutry model, plant extract (Pulicaria crispa), H2R antagonist (Ranitidine) | H2 antagonist partially restored GSH and SOD in ulcers. | Fahmi et al., 2019 |
| H2R | Male rats | Small intestine | Indomethacin small intestine lesion model, Vitamin C, Vitamin E, β-Carotene, sodium selene, H2R antagonist (Ranitidine) | H2 antagonist partially restored GSH, CAT, GPx, and increased SOD; no MDA effect. | Turkyilmaz et al., 2019 |
| H2R | Male rats | Bone marrow and intestinal tissue | Irradiation, H2R antagonist (Cimetidine) | H2 antagonist corrected MDA, GSH, and SOD levels. | Estaphan et al., 2020 |
| H2R | Female and male mice | Brain | Brain ischemia/reperfusion model, L-carnosine, H2R antagonist (Ranitidine) | H2 antagonist blocked L-carnosine effects on TBARS, GSH, and MPO in brain ischemia. | Virdi et al., 2020 |
| H2R | Male rats | Testis | Testicular ischemia model, H2R antagonist (Famotidine) | H2 antagonist normalized NO and SOD; MDA and GPx were unaffected. | Tanriverdi et al., 2021 |
| H2R | Male rats | Gastric tissue | Indomethacin gastric ulcer model, water immersion stress model, plant extract (Elaeagnus conferta Roxb.), H2R antagonist (Ranitidine) | H2 antagonist partially restored CAT, GSH, SOD, and reduced MDA. | Gupta et al., 2021 |
| H2R | Male rats | Gastric mucosa | Ethanol gastric injutry model, plant extract (E. persicus), H2R antagonist (Ranitidine) | H2 antagonist restored CAT, GSH, and reduced ROS and MDA. | Beiranvand et al., 2021 |
| H2R | Female swiss mice | Tumor tissue | Breast tumor model, vitamin C, H2R antagonist (Cimetidine) | H2 antagonist partially restored tumor GSH, SOD, and reduced MDA. | Ibrahim et al., 2022 |
| H2R | Male rats | Gastric mucosa | Indomethacin gastric lesion model, Topiramate, H2R antagonist (Ranitidine) | H2 antagonist restored SOD, CAT, GPx activities, GSH, and reduced MDA. | Jafari et al., 2022 |
| H2R | Male rats | Gastric mucosa | Indomethacin gastric ulcer model, plant extract (Malus domestica Borkh), H2R antagonist (Famotidine) | H2 antagonist partially restored GSH, GPx, and reduced MDA. | Mahmoud et al., 2023 |
| H2R | Rats | Gastric tissue | Indomethacin gastric ulcer model, Felodipine, H2R antagonist (Famotidine) | H2 antagonist corrected MDA, GSH, and catalase levels. | Akbaş et al., 2023 |
6. Conclusion
The review emphasizes that while Reactive Oxygen Species (ROS) are linked to various human diseases, there have been limited clinical applications for ROS modulation. However, recent developments are concentrating on selectively targeting ROS-generating enzymes, especially NADPH oxidases, to maintain normal bodily functions and enhance treatment effectiveness. This indicates a move towards precision medicine within the field of ROS pharmacology.
Furthermore, despite the differences in research focus on the three receptors, there are similarities. Both B1R and H2R receptors amplify the generation of reactive oxygen species, making them suitable targets for pharmacological intervention in chronic inflammatory responses. Combined inhibition of these receptors may improve treatment outcomes for conditions such as gastric diseases. Understanding the local and systemic interactions regulated by these receptors through mechanistic studies, as well as improving drug delivery methods to target specific tissues, is crucial for maximizing benefits and minimizing adverse effects.
B1 bradykinin receptor (B1R), B2 bradykinin receptor (B2R); histamine H2 receptor (H2R), bradykinin (BK), angiotensin-converting enzyme (ACE) superoxide anions ( O2•─), superoxide dismutase (SOD) malondialdehyde (MDA), glutathione (GSH), Reactive Oxygen Species (ROS), Tumor Necrosis Factor (TNF), Lipopolysaccharide (LPS), Hydrogen Peroxide (H2O2), Nitric Oxide (NO), N-acetyl-β-D-glucosaminidase (NAG), Myeloperoxidase (MPO), Endothelial Nitric Oxide Synthase (eNOS), Mitogen-Activated Protein Kinases (MAPKs), Extracellular Signal-Regulated Kinases (ERKs), Protein Kinase B (AKT), Glutathione Peroxidase (GPx), Catalase (CAT), Thiobarbituric Acid Reactive Substances (TBARs).
Acknowledgments
We regret that due to space limitations, we couldn't acknowledge all scientists who contributed to the investigation of kinin and histamine interactions on oxidative stress. COST Action CA18133 is acknowledged (W.A.F). The figure was create with BioRender.
Data availability: No data was used for the research described in the manuscript.
CRediT authorship contribution statement: Conceptualization: M.F.G; L.M; W.A.F; investigation: M.F.G; L.M; W.A.F; visualization: M.F.G; L.M; W.A.F; writing—original draft: M.F.G; L.M; writing—review and editing: M.F.G; L.M; W.A.F; designing and preparing figures: L.M.; The authors read and approved the final manuscript.
Declaration of competing interest: The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: L.M. Mast cells as effectors in hemorrhage and acute cardiovascular diseases (MASTer Consortium) approved by the European Granting Authority as part of the ERA4Health Joint Transnational Call “Research targeting development of innovative therapeutic strategies in cardiovascular disease” (CARDINNOV/2023) program. ERA4Health EU Network (ERA-NET), Funded by the European Union under the Horizon Europe Framework Programme (GA N°101095426 Horizon_EU) #CardInnov_E4H, Co-fund by The Research Foundation – Flanders (FWO).
References
- Sies H, Jones DP. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat Rev Mol Cell Biol. 2020 Jul;21(7):363-383. [CrossRef] [PubMed]
- Lushchak, VI. Free radicals, Reactive Oxygen Species, Oxidative Stress and its Classification. Chem Biol Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Xue C, Li X, Liu G, Liu W. Evaluation of Mitochondrial Respiratory Chain on the Generation of Reactive Oxygen Species and Cytotoxicity in HaCaT Cells Induced by Nanosized Titanium Dioxide Under UVA Irradiation. Int J Toxicol. 2016, 35, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Iatsenko I, Boquete JP, Lemaitre B. Microbiota-Derived Lactate Activates Production of Reactive Oxygen Species by the Intestinal NADPH Oxidase Nox and Shortens Drosophila Lifespan. Immunity, 49. [CrossRef] [PubMed]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial Reactive Oxygen Species (ROS) and ROS-induced ROS Release. Physiol Rev, 94. [CrossRef] [PubMed]
- Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, Dong W. ROS and ROS-Mediated Cellular Signaling. Oxid Med Cell Longev, 4350. [CrossRef] [PubMed]
- Scialò F, Fernández-Ayala DJ, Sanz A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front Physiol. 2017, 8, 428. [CrossRef] [PubMed]
- El-Kenawi A, Ruffell B. Inflammation, ROS, and Mutagenesis. Cancer Cell, 32. [CrossRef] [PubMed]
- Nguyen NH, Tran GB, Nguyen CT. Anti-Oxidative Effects of Superoxide Dismutase 3 on inflammatory Diseases. J Mol Med (Berl), 98. [CrossRef] [PubMed]
- Rhee SG, Woo HA, Kil IS, Bae SH. Peroxiredoxin Functions as a Peroxidase and a regulator and sensor of local peroxides. J Biol Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Couto N, Wood J, Barber J. The Role of Glutathione Reductase and Related Enzymes on Cellular Redox Homoeostasis Network. Free Radic Biol Med. 2016, 95, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Regoli D, Marceau F, Barabé J. De Novo Formation of Vascular Receptors for Bradykinin. Can J Physiol Pharmacol. 1978, 56, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Leeb-Lundberg LM, Marceau F, Müller-Esterl W, Pettibone DJ, Zuraw BL. International Union of Pharmacology. XLV. Classification of the Kinin Receptor Family: from Molecular Mechanisms to Pathophysiological Consequences. Pharmacol Rev, 57. [CrossRef] [PubMed]
- Ewald DA, Pang IH, Sternweis PC, Miller RJ. Differential G Protein-Mediated Coupling of Neurotransmitter Receptors to Ca2+ Channels in Rat Dorsal Root Ganglion Neurons In Vitro. Neuron. 1989, 2, 1185-1193. [CrossRef] [PubMed]
- Austin CE, Faussner A, Robinson HE, Chakravarty S, Kyle DJ, Bathon JM, Proud D. Stable Expression of the Human Kinin B1 Receptor in Chinese Hamster Ovary cCells. Characterization of Ligand Binding and Effector Pathways. J Biol Chem. 1997, 272, 11420–11425. [Google Scholar] [CrossRef] [PubMed]
- Ignjatovic T, Stanisavljevic S, Brovkovych V, Skidgel RA, Erdös EG. Kinin B1 ReceptorsStimulate Nitric Oxide Production in Endothelial Cells: Signaling Pathways Activated by Angiotensin I-Converting Enzyme Inhibitors and Peptide Lgands. Mol Pharmacol, 1310; 66. [CrossRef] [PubMed]
- Thangam EB, Jemima EA, Singh H, Baig MS, Khan M, Mathias CB, Church MK, Saluja R. The Role of Histamine and Histamine Receptors in Mast Cell-Mediated Allergy and Inflammation: The Hunt for New Therapeutic Targets. Front Immunol. 1873. [CrossRef] [PubMed]
- Branco ACCC, Yoshikawa FSY, Pietrobon AJ, Sato MN. Role of Histamine in Modulating the Immune Response and Inflammation. Mediators Inflamm, 9: 2018, 2018. [CrossRef] [PubMed]
- Borriello F, Iannone R, Marone G. Histamine Release from Mast Cells and Basophils. Handb Exp Pharmacol. 2017, 241, 121–139. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P. The Basics of Histamine Biology. Ann Allergy Asthma Immunol, S: Suppl). [CrossRef] [PubMed]
- Parsons ME, Ganellin CR. Histamine and its Receptors. Br J Pharmacol. 2006, 147 (Suppl 1), S127–5135. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Delemasure S, Blaes N, Richard C, Couture R, Bader M, Dutartre P, Girolami JP, Connat JL, Rochette L. Antioxidant/Oxidant Status and Cardiac Function in Bradykinin B(1)- and B(2)-Receptor Null Mice. Physiol Res, 62. [CrossRef] [PubMed]
- Fu C, Li B, Sun Y, Ma G, Yao Y. Bradykinin Inhibits Oxidative Stress-Induced Senescence of Endothelial Progenitor Cells through the B2R/AKT/RB and B2R/EGFR/RB Signal Pathways. Oncotarget. 2467. [CrossRef] [PubMed]
- Ayres LS, Berger M, Durli ICLO, Kuhl CP, Terraciano PB, Garcez TNA, Dos Santos BG, Guimarães JA, Passos EP, Cirne-Lima EO. Kallikrein-Kinin System and Oxidative Stress in Cisplatin-Induced Ovarian Toxicity. Reprod Toxicol, 93. [CrossRef] [PubMed]
- Estrela GR, Wasinski F, Bacurau RF, Malheiros DM, Câmara NO, Araújo RC. Kinin B2 Receptor Deletion and Blockage Ameliorates Cisplatin-Induced Acute Renal Injury. Int Immunopharmacol. [CrossRef] [PubMed]
- Estrela GR, Wasinski F, Gregnani MF, Freitas-Lima LC, Arruda AC, Morais RL, Malheiros DM, Camara NOS, Pesquero JB, Bader M, Barros CC, Araújo RC. Angiotensin-Converting Enzyme Inhibitor Protects Against Cisplatin Nephrotoxicity by Modulating Kinin B1 Receptor Expression and Aminopeptidase P Activity in Mice. Front Mol Biosci, 7. [CrossRef] [PubMed]
- Ferreira APO, Rodrigues FS, Della-Pace ID, Mota BC, Oliveira SM, de Campos Velho Gewehr C, Bobinski F, de Oliveira CV, Brum JS, Oliveira MS, Furian AF, de Barros CS, dos Santos AR, Ferreira J, Fighera MR, Royes LF. HOE-140, an Antagonist of B2 Receptor, Protects Against Memory Deficits and Brain Damage Induced by Moderate Lateral Fuid Percussion Injury in Mice. Psychopharmacology (Berl), 1935. [CrossRef] [PubMed]
- Chiang WC, Chien CT, Lin WW, Lin SL, Chen YM, Lai CF, Wu KD, Chao J, Tsai TJ. Early Activation of Bradykinin B2 Receptor Aggravates Reactive Oxygen Species Generation and Renal Damage in Ischemia/Reperfusion Injury. 2006; 41. [CrossRef] [PubMed]
- Bledsoe G, Crickman S, Mao J, Xia CF, Murakami H, Chao L, Chao J. Kallikrein/Kinin Protects Against Gentamicin-Induced Nephrotoxicity by Inhibition of Inflammation and Apoptosis. Nephrol Dial Transplant, 21. [CrossRef] [PubMed]
- Allard J, Buléon M, Cellier E, Renaud I, Pecher C, Praddaude F, Conti M, Tack I, Girolami JP. ACE Inhibitor Reduces Growth Factor Receptor Expression and Signaling but also Albuminuria through B2-Kinin Glomerular Receptor Activation in Diabetic Rats. Am J Physiol Renal Physiol, 1083. [CrossRef] [PubMed]
- Jaffa MA, Kobeissy F, Al Hariri M, Chalhoub H, Eid A, Ziyadeh FN, Jaffa AA. Global Renal Gene Expression Profiling Analysis in B2-Kinin Receptor Null Mice: Impact of Diabetes. PLoS One, 4471; 7. [CrossRef] [PubMed]
- Bledsoe G, Shen B, Yao Y, Zhang JJ, Chao L, Chao J. Reversal of Renal Fibrosis, Inflammation, and Glomerular Hypertrophy by Kallikrein Gene Delivery. Hum Gene Ther, 17. [CrossRef] [PubMed]
- Bledsoe G, Shen B, Yao YY, Hagiwara M, Mizell B, Teuton M, Grass D, Chao L, Chao J. Role of Tissue Kallikrein in Prevention and Recovery of Gentamicin-Induced Renal injury. Toxicol Sci. [CrossRef] [PubMed]
- Niewiarowska-Sendo A, Kozik A, Guevara-Lora I. Influence of Bradykinin B2 Receptor and Dopamine D2 Receptor on the Oxidative Stress, Inflammatory Response, and Apoptotic Process in Human Endothelial Cells. PLoS One, 0206; 13. [CrossRef] [PubMed]
- Yasunari K, Maeda K, Watanabe T, Nakamura M, Asada A, Yoshikawa J. Converting Enzyme Inhibitor Temocaprilat Prevents High Glucose-Mediated Suppression of Human Aortic Endothelial Cell Proliferation. J Cardiovasc Pharmacol. [CrossRef] [PubMed]
- Du YM, Du BH, Yang J, Zang S, Wang XP, Mao X, Zhang W, Jiang LP. Effect of Bradykinin on Rats with Thromboangiitis Obliterans through PI3K/Akt Signaling Pathway. Eur Rev Med Pharmacol Sci. 1016. [CrossRef] [PubMed]
- Oeseburg H, Iusuf D, van der Harst P, van Gilst WH, Henning RH, Roks AJ. Bradykinin Protects Against Oxidative Stress-Induced Endothelial Cell Senescence. Hypertension, 53. [CrossRef] [PubMed]
- Mesquita TRR, Miguel-Dos-Santos R, Jesus ICG, de Almeida GKM, Fernandes VA, Gomes AAL, Guatimosim S, Martins-Silva L, Ferreira AJ, Capettini LDSA, Pesquero JL, Lauton-Santos S. Ablation of B1- and B2-Kinin Receptors Causes Cardiac Dysfunction through Redox-Nitroso Unbalance. Life Sci. [CrossRef] [PubMed]
- Yao YY, Yin H, Shen B, Chao L, Chao J. Tissue Kallikrein Infusion Prevents Cardiomyocyte Apoptosis, Inflammation and Ventricular Remodeling after Myocardial Infarction. Regul Pept. [CrossRef] [PubMed]
- Feng W, Xu X, Zhao G, Zhao J, Dong R, Ma B, Zhang Y, Long G, Wang DW, Tu L. Increased Age-Related Cardiac Dysfunction in Bradykinin B2 Receptor-Deficient Mice. J Gerontol A Biol Sci Med Sci, 71. [CrossRef] [PubMed]
- Dong R, Xu X, Li G, Feng W, Zhao G, Zhao J, Wang DW, Tu L. Bradykinin Inhibits Oxidative Stress-Induced Cardiomyocytes Senescence via Regulating Redox State. PLoS One, 7703; 8. [CrossRef] [PubMed]
- Kobayashi N, Honda T, Yoshida K, Nakano S, Ohno T, Tsubokou Y, Matsuoka H. Critical Role of Bradykinin-eNOS and Oxidative Stress-LOX-1 Pathway in Cardiovascular Remodeling Under Chronic Angiotensin-Converting Enzyme Inhibition. Atherosclerosis. 2006, 187, 92-100. [CrossRef] [PubMed]
- Yoshida K, Kobayashi N, Ohno T, Fukushima H, Matsuoka H. Cardioprotective Effect of Angiotensin II Type 1 Receptor Antagonist Associated with Bradykinin-Endothelial Nitric Oxide Synthase and Oxidative Stress in Dahl Salt-Sensitive Hypertensive Rats. J Hypertens, 1633; 25. [CrossRef] [PubMed]
- Li HJ, Yin H, Yao YY, Shen B, Bader M, Chao L, Chao J. Tissue Kallikrein Protects Against Pressure Overload-Induced Cardiac Hypertrophy through Kinin B2 Receptor and Glycogen Synthase Kinase-3beta Activation. Cardiovasc Res. 2007, 73, 130-142. [CrossRef] [PubMed]
- Lu L, Guo L, Xie TT, Xin HL. Angiotensin Converting Enzyme is Involved in the Cardiac Hypertrophy Induced by Sinoaortic Denervation in Rats. Cardiovasc Pathol. 2015, 24, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Kumari R, Maulik M, Manchanda SC, Maulik SK. Protective Effect of Bradykinin Antagonist Hoe-140 During In Vivo Myocardial Ischemic-Reperfusion Injury in the Cat. Regul Pept. [CrossRef] [PubMed]
- Yin H, Chao L, Chao J. Nitric Oxide Mediates Cardiac Protection of Tissue Kallikrein by Reducing Inflammation and Ventricular Remodeling After Myocardial Ischemia/Reperfusion. Life Sci, 82. [CrossRef] [PubMed]
- Cargnoni A, Comini L, Bernocchi P, Bachetti T, Ceconi C, Curello S, Ferrari R. Role of Bradykinin and eNOS in the Anti-Ischaemic Effect of Trandolapril. Br J Pharmacol. [CrossRef] [PubMed]
- Wu Y, Fu C, Li B, Liu C, He Z, Li XE, Wang A, Ma G, Yao Y. Bradykinin Protects Human Endothelial Progenitor Cells from High-Glucose-Induced Senescence through B2 Receptor-Mediated Activation of the Akt/eNOS Signalling Pathway. J Diabetes Res, 2021. [CrossRef] [PubMed]
- Akash MSH, Rehman K, Liaqat A. Tumor Necrosis Factor-Alpha: Role in Development of Insulin Resistance and Pathogenesis of Type 2 Diabetes Mellitus. J Cell Biochem. 2018, 119, 105-110. [CrossRef] [PubMed]
- Souza DG, Pinho V, Pesquero JL, Lomez ES, Poole S, Juliano L, Correa A Jr, de A Castro MS, Teixeira MM. Role of the Bradykinin B2 Receptor for the Local and Systemic Inflammatory Response that Follows Severe Reperfusion Injury. Br J Pharmacol. 2003, 139, 129–139. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Basuli D, Parekh RU, White A, Thayyil A, Sriramula S. Kinin B1 Receptor Mediates Renal Injury and Remodeling in Hypertension. Front Med (Lausanne). 2022, 8, 780834. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Westermann D, Walther T, Savvatis K, Escher F, Sobirey M, Riad A, Bader M, Schultheiss HP, Tschöpe C. Gene Deletion of the Kinin Receptor B1 Attenuates Cardiac Inflammation and Fibrosis During the Development of Experimental Diabetic Cardiomyopathy. Diabetes. 2009, 58, 1373–1381. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Catanzaro O, Capponi JA, Michieli J, Labal E, Di Martino I, Sirois P. Bradykinin B₁ antagonism inhibits oxidative stress and restores Na+K+ ATPase activity in diabetic rat peripheral nervous system. Peptides, 44. [CrossRef] [PubMed]
- Catanzaro OL, Capponi JA, Di Martino I, Labal ES, Sirois P. Oxidative Stress in the Optic Nerve and Cortical Visual Area of Steptozotocin-Induced Diabetic Wistar Rats: Blockade with a Selective Bradykinin B1 Receptor Antagonist. Neuropeptides. [CrossRef] [PubMed]
- Pouliot M, Talbot S, Sénécal J, Dotigny F, Vaucher E, Couture R. Ocular Application of the Kinin B1 Receptor Antagonist LF22-0542 Inhibits Retinal Inflammation and Oxidative Stress in Streptozotocin-Diabetic Rats. PLoS One, 3386; 7. [CrossRef] [PubMed]
- Parekh RU, Robidoux J, Sriramula S. Kinin B1 Receptor Blockade Prevents Angiotensin II-induced Neuroinflammation and Oxidative Stress in Primary Hypothalamic Neurons. Cell Mol Neurobiol, 40. [CrossRef] [PubMed]
- Theobald D, Sriramula S. Kinin B1 Receptor Mediates Bidirectional Interaction between Neuroinflammation and Oxidative Stress. Antioxidants (Basel). 2023, 2023 12, 150. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Haddad Y, Couture R. Kininase 1 As a Preclinical Therapeutic Target for Kinin B1 Receptor in Insulin Resistance. Front Pharmacol. 2017, 8, 509. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yasunari K, Maeda K, Watanabe T, Nakamura M, Asada A, Yoshikawa J. Converting Enzyme Inhibitor Temocaprilat Prevents High Glucose-Mediated Suppression of Human Aortic Endothelial Cell Proliferation. J Cardiovasc Pharmacol. 2003, 42 (Suppl 1):S55-560. [CrossRef] [PubMed]
- Dias JP, Ismael MA, Pilon M, de Champlain J, Ferrari B, Carayon P, Couture R. The Kinin B1 Receptor Antagonist SSR240612 Reverses Tactile and Cold allodynia in an Experimental Rat Model of Insulin Resistance. Br J Pharmacol. 2007, 152, 280–287. [CrossRef] [PubMed] [PubMed Central]
- Dias JP, Talbot S, Sénécal J, Carayon P, Couture R. Kinin B1 Receptor Enhances the Oxidative Stress in a Rat Model of Insulin Resistance: Outcome in Hypertension, Allodynia and Metabolic Complications. PLoS One, 1262; 5. [CrossRef] [PubMed]
- Marceau F, Sabourin T, Houle S, Fortin JP, Petitclerc E, Molinaro G, Adam A. Kinin Receptors: Functional Aspects. Int Immunopharmacol. 2002, 214, 1729-1739. [CrossRef] [PubMed]
- Csiszar A, Wang M, Lakatta EG, Ungvari Z. Inflammation and Endothelial Dysfunction During Aging: Role of NF-kappaB. J Appl Physiol (1985). 1333. [CrossRef] [PubMed]
- Schmaier, AH. The Kallikrein-Kinin and the Renin-Angiotensin Systems Have a Multilayered iInteraction. Am J Physiol Regul Integr Comp Physiol. 2003, 285, R1–13. [Google Scholar] [CrossRef] [PubMed]
- Duka A, Kintsurashvili E, Duka I, Ona D, Hopkins TA, Bader M, Gavras I, Gavras H. Angiotensin-Converting Enzyme Inhibition After Experimental Myocardial Infarct: Role of the Kinin B1 and B2 Receptors. Hypertension, 1352; 51. [CrossRef] [PubMed]
- Ribuot C, Godin D, Couture R, Regoli D, Nadeau R. In Vivo B2-Receptor-Mediated Negative Chronotropic Effect of Bradykinin in Canine Sinus Node. Am J Physiol. [CrossRef] [PubMed]
- Lapenna D, De Gioia S, Mezzetti A, Grossi L, Festi D, Marzio L, Cuccurullo F. H2-Receptor Antagonists are Scavengers of Oxygen Radicals. Eur J Clin Invest, 24. [CrossRef] [PubMed]
- Biedrzycki G, Wolszczak-Biedrzycka B, Dorf J, Michalak D, Żendzian-Piotrowska M, Zalewska A, Maciejczyk M. Antioxidant and Anti-Glycation Potential of H2 Receptor Antagonists-In Vitro Studies and a Systematic Literature Review. Pharmaceuticals (Basel), 16. [CrossRef] [PubMed]
- Tanriverdi HI, Şenel U, Gevrek F, Akbaş A. Protective Effect of Famotidine on Ischemia-Reperfusion Injury Following Testicular Torsion in Rats. J Pediatr Urol, 17. [CrossRef] [PubMed]
- Ibrahim SSA, El-Aal SAA, Reda AM, Achy SE, Shahine Y. Anti-Neoplastic Action of Cimetidine/Vitamin C on Histamine and the PI3K/AKT/mTOR Pathway in Ehrlich Breast Cancer. Sci Rep, 1151. [CrossRef] [PubMed]
- Estaphan S, Eissa H, Elattar S, Rashed L, Farouk M. A Study on the Effect of Cimetidine and L-Carnitine on Myoglobinuric Acute Kidney Injury in Male Rats. Injury, 1223; 46. [CrossRef] [PubMed]
- Estaphan S, Abdel-Malek R, Rashed L, Mohamed EA. Cimetidine a Promising Radio-Protective Agent Through Modulating Bax/Bcl2 Ratio: An In Vivo Study in Male Rats. J Cell Physiol, 8495. [CrossRef] [PubMed]
- Aydin E, Hallner A, Grauers Wiktorin H, Staffas A, Hellstrand K, Martner A. NOX2 Inhibition Reduces Oxidative Stress and Prolongs Survival in Murine KRAS-Induced Myeloproliferative Disease. Oncogene, 1534; 38. [CrossRef] [PubMed]
- Shimoyama T, Fukuda S, Liu Q, Fukuda Y, Nakaji S, Sugawara K. Characteristics of Attenuating Effects of Rebamipide, an Anti-Ulcer Agent, on Oxidative Burst of Human Neutrophils. J Pharmacol Sci, 91. [CrossRef] [PubMed]
- Potnuri AG, Allakonda L, Saheera S. Involvement of Histamine 2 Receptor in Alpha 1 Adrenoceptor Mediated Cardiac Hypertrophy and Oxidative Stress in H9c2 Cardio Myoblasts. J Cardiovasc Transl Res. 2021, 14, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Kondru SK, Potnuri AG, Allakonda L, Konduri P. Histamine 2 Receptor Antagonism Elicits Protection Against Doxorubicin-Induced Cardiotoxicity in Rodent Model. Mol Cell Biochem. [CrossRef] [PubMed]
- Zhang L, Yao K, Fan Y, He P, Wang X, Hu W, Chen Z. Carnosine Protects Brain Microvascular Endothelial Cells Against Rotenone-Induced Oxidative Stress Injury Through Histamine H₁ and H₂ Receptors In Vitro. Clin Exp Pharmacol Physiol. 1019. [CrossRef] [PubMed]
- Virdi JK, Bhanot A, Jaggi AS, Agarwal N. Investigation on Beneficial Role of L-Carnosine in Neuroprotective Mechanism of Ischemic Postconditioning in Mice: Possible Role of Histidine Histamine pathway. Int J Neurosci. 2020, 130, 983-998. [CrossRef] [PubMed]
- Estaphan S, Abdel-Malek R, Rashed L, Mohamed EA. Cimetidine a Promising Radio-Protective Agent Through Modulating Bax/Bcl2 Ratio: An In Vivo Study in Male Rats. J Cell Physiol, 8495. [CrossRef] [PubMed]
- Singh DP, Borse SP, Nivsarkar M. Overcoming the Exacerbating Effects of Ranitidine on NSAID-Induced Small Intestinal Toxicity With Quercetin: Providing a Complete GI Solution. Chem Biol Interact. [CrossRef] [PubMed]
- Turkyilmaz IB, Arda Pirincci P, Bolkent S, Yanardag R. The Effects of Vitamins and Selenium Mixture or Ranitidine Against Small Intestinal Injury Induced by Indomethacin in Adult Rats. J Food Biochem. 2019, 43, e12808. [CrossRef] [PubMed]
- Jahangirvand M, Minai-Tehrani D, Yazdi F, Minai-Tehrani A, Razmi N. Binding of Cimetidine to Balb/C Mouse Liver Catalase; Kinetics and Conformational Studies. Curr Clin Pharmacol, 11. [CrossRef] [PubMed]
- El-Mahdy NA, El-Sisi AE, Dewidar BI, El-Desouky KI. Histamine Protects Against the Acute Phase of Experimentally-Induced Hepatic Ischemia/Reperfusion. J Immunotoxico, ,10. [CrossRef] [PubMed]
- Gaafa KM, Badawy MM, Hamza AA. The Protective Effects of Ascorbic Acid, Cimetidine, and Nifedipine on Diethyldithiocarbamate-Induced Hepatic Toxicity in Albino Rats. Drug Chem Toxicol, 34. [CrossRef] [PubMed]
- Oh TY, Lee JS, Ahn BO, Cho H, Kim WB, Kim YB, Surh YJ, Cho SW, Hahm KB. Oxidative Damages are Critical in Pathogenesis of Reflux Esophagitis: Implication of in its Treatment. Free Radic Biol Med, 30. [CrossRef] [PubMed]
- Lee JS, Oh TY, Ahn BO, Cho H, Kim WB, Kim YB, Surh YJ, Kim HJ, Hahm KB. Involvement of Oxidative Stress in Experimentally Induced Reflux Esophagitis and Barrett's Esophagus: Clue for the Chemoprevention of Esophageal Carcinoma by Antioxidants. Mutat Res. [CrossRef] [PubMed]
- Oh TY, Lee JS, Ahn BO, Cho H, Kim WB, Kim YB, Surh YJ, Cho SW, Lee KM, Hahm KB. Oxidative Stress is More Important Than Acid in the Pathogenesis of Reflux Oesophagitis in Rats. Gut. 2001, 49, 364–371. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim HK, Kim MG, Leem KH. Extrusion Process of Acanthopanax Senticosus Leaves Enhances the Gastroprotective Effect of Compound 48/80 on Acute Gastric Mucosal Lesion in Rats. J Tradit Chin Med, 36. [CrossRef] [PubMed]
- Qiao X, Tang X, Zhang J, Chen K, Zhang Y, Wang C, Fei S, Zhu J, Zhu S, Liu Z, Li T, Lv S, Liang Y. Protective Effect of Histamine Microinjected Into the Cerebellar Fastigial Nucleus on Stress-Induced Gastric Mucosal Damage in Rats. Am J Transl Res, 2015; 2, 1648–1659.
- Ahmad SS, Najmi AK, Kaundal M, Akhtar M. Gastroprotective Effect of Thymoquinone on Water Immersion Restraint Stress Induced Ulceration in Rats. Drug Res (Stuttg). 2017, 67, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud MF, Abdo W, Nabil M, Drissi B, El-Shazly AM, Abdelfattah MAO, Sobeh M. Apple (Malus domestica Borkh) lLeaves Attenuate Indomethacin-Induced Gastric Ulcer in Rats. Biomed Pharmacother, 1143. [CrossRef] [PubMed]
- Akbaş N, Süleyman B, Mammadov R, Gülaboğlu M, Akbaş EM, Süleyman H. Effect of Felodipine on Indomethacin-Induced Gastric Ulcers in Rats. Exp Anim, 72. [CrossRef] [PubMed]
- Jafari A, Andishfar N, Esmaeilzadeh Z, Khezri MR, Ghasemnejad-Berenji M. Gastroprotective Effect of Topiramate on Indomethacin-Induced Peptic Ulcer in Rats: Biochemical and Histological Analyses. Basic Clin Pharmacol Toxicol. [CrossRef] [PubMed]
- Karakaya K, Hanci V, Bektas S, Can M, Ucan HB, Emre AU, Tascilar O, Ozkocak Turan I, Comert M, Irkorucu O, Karadeniz Cakmak G. Mitigation of Indomethacin-Induced Gastric Mucosal Lesions by a Potent Specific Type V Phosphodiesterase Inhibitor. World J Gastroenterol, 5091; 15. [CrossRef] [PubMed]
- Raji Y, Oyeyemi WA, Shittu ST, Bolarinwa AF. Gastro-Protective Effect of Methanol Extract of Ficus Asperifolia Bark on Indomethacin-Induced Gastric Ulcer in Rats. Niger J Physiol Sci, 26. [PubMed]
- Hernández-Muñoz R, Montiel-Ruíz C, Vázquez-Martínez O. Gastric Mucosal Cell Proliferation in Ethanol-Induced Chronic Mucosal Injury is Related to Oxidative Stress and Lipid Peroxidation in Rats. Lab Invest, 1161; 80. [CrossRef] [PubMed]
- Beiranvand M, Bahramikia S, Dezfoulian O. Evaluation of Antioxidant and Anti-Ulcerogenic Effects of Eremurus Persicus (Jaub & Spach) Boiss Leaf Hydroalcoholic Extract on Ethanol-Induced Gastric Ulcer in Rats. Inflammopharmacology, 1503. [CrossRef] [PubMed]
- Fahmi AA, Abdur-Rahman M, Aboul Naser AF, Hamed MA, Abd-Alla HI, Nasr MI. Pulicaria Crispa Mitigates Gastric Ulcer Induced by Ethanol in Rats: Role of Treatment and Auto Healing. Biomarkers. [CrossRef] [PubMed]
- Cadirci E, Suleyman H, Aksoy H, Halici Z, Ozgen U, Koc A, Ozturk N. Effects of Onosma Armeniacum Rroot Extract on Ethanol-Induced Oxidative Stress in Stomach Tissue of Rats. Chem Biol Interact. [CrossRef] [PubMed]
- Pradeepkumar Singh L, Kundu P, Ganguly K, Mishra A, Swarnakar S. Novel Role of Famotidine in Downregulation of Matrix Metalloproteinase-9 During Protection of Ethanol-Induced Acute Gastric Ulcer. Free Radic Biol Med, 43. [CrossRef] [PubMed]
- Alirezaei M, Dezfoulian O, Neamati S, Rashidipour M, Tanideh N, Kheradmand A. Oleuropein Prevents Eethanol-Induced Gastric Ulcers Via Elevation of Antioxidant Enzyme Activities in Rats. J Physiol Biochem, 68. [CrossRef] [PubMed]
- Gupta M, Gulati M, Kapoor B, Kumar B, Kumar R, Kumar R, Khurana N, Gupta R, Singh N. Anti-Ulcerogenic Effect of Methanolic Extract of Elaeagnus Conferta Roxb. Seeds in Wistar Rats. J Ethnopharmacol, 1141. [CrossRef] [PubMed]
- Olaleye MT, Akinmoladun AC. Comparative Gastroprotective Effect of Post-Treatment with Low Doses of Rutin and Cimetidine in Rats. Fundam Clin Pharmacol, 27. [CrossRef] [PubMed]
- Kath RK, Gupta RK. Antioxidant Activity of Hydroalcoholic Leaf Extract of Ocimum Sanctum in Animal Models of Peptic Ulcer. Indian J Physiol Pharmacol. 2006, 50, 391–396. [Google Scholar] [PubMed]
- Olaleye SB, Farombi EO. Attenuation of Indomethacin- and HCl/Ethanol-Induced Oxidative Gastric Mucosa Damage in Rats by Kolaviron, a Natural Biflavonoid of Garcinia Kola Seed. Phytother Res, 20. [CrossRef] [PubMed]
- Fornai M, Colucci R, Antonioli L, Ghisu N, Tuccori M, Blandizzi C, Del Tacca M. Effects of Pantoprazole on Ulcer Healing Delay Associated with NSAID Treatment. Naunyn Schmiedebergs Arch Pharmacol. 2009, 379, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Asokkumar K, Sen S, Umamaheswari M, Sivashanmugam AT, Subhadradevi V. Synergistic Effect of the Combination of Gallic acid and Famotidine in Protection of Rat Gastric Mucosa. Pharmacol Rep. 2014, 66, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Morsy M, Ashour O, Amin E, Rofaeil R. Gastroprotective Effects of Telmisartan on Experimentally-Induced Gastric Ulcers in Rats. Pharmazie, 1: 64, 590-594. PMID, 1982; 64.
- Panula, P. Histamine Receptors, Agonists, and Antagonists in Health and Disease. Handb Clin Neurol. 2021, 180, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Nogueira-Machado JA, Lima e Silva FC, Cunha EP, Calsolari MR, Costa DC, Perilo CS, Horta BC, Ferreira IC, Chaves MM. Modulation of the Production of Reactive Oxygen Species (ROS) by cAMP-Elevating Agents in Granulocytes from Diabetic Patients: an Akt/PKB-Dependent Phenomenon. Diabetes Metab, 32. [CrossRef] [PubMed]
- Torres-Román AL, Rodríguez-Flores KL, Hernández-Mora VM, Ruiz-García E, Prospero-García O, Guijosa A, Molina A, Morales-Mulia M, Aschner M, Santamaría A, Ortega-Gómez A. Examining the Role of Histaminergic, Orexinergic, and Cannabinergic Systems in Redox Regulation in Gastric Adenocarcinoma. Mini Rev Med Chem, 1806. [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



