Submitted:
13 August 2024
Posted:
14 August 2024
You are already at the latest version
Abstract
Understanding the structural intricacies of RNA molecules is essential for deciphering numerous biological processes. Traditionally, scientists have relied on experimental methods to gain insights and draw conclusions. However, the recent advent of advanced computational techniques has significantly accelerated and refined the accuracy of research results in several areas. A particularly challenging aspect of RNA analysis is the prediction of its secondary structure, which is crucial for elucidating its functional role in biological systems. This paper deals with the prediction of pseudoknots in RNA, focusing on two types of pseudoknots: K-type and M-type pseudoknots. Pseudoknots are complex RNA formations in which nucleotides in a loop form base pairs with nucleotides outside the loop and thus contribute to essential biological functions. Accurate prediction of these structures is crucial for understanding RNA dynamics and interactions. Building on our previous work in which we developed a framework for the recognition of H- and L-type pseudoknots, an extended grammar-based framework tailored to the prediction of K- and M-type pseudoknots is proposed. This approach uses syntactic pattern recognition techniques and provides a systematic method to identify and characterize these complex RNA structures. Our framework uses context-free grammars (CFGs) to model RNA sequences and predict the occurrence of pseudoknots. By formulating specific grammatical rules for type K and type M pseudoknots, we enable efficient parsing of RNA sequences to recognize potential pseudoknot configurations. This method ensures an exhaustive exploration of possible pseudoknot structures within a reasonable time frame. In addition, the proposed method incorporates essential concepts of biology, such as base pairing optimization and free energy reduction, to improve the accuracy of pseudoknot prediction. These principles are crucial to ensure that the predicted structures are biologically plausible. By embedding these principles into our grammar-based framework, we aim to predict RNA conformations that are both theoretically sound and biologically relevant.
Keywords:
RNA structure prediction
; K-type pseudoknots
; M-type pseudoknots
; syntactic pattern recognition
; context-free grammar
; secondary structure
; computational biology
1. Introduction
RNA molecules play a fundamental role in a considerable number of biological functions. Therefore, understanding the structural intricacies of RNA is crucial to deciphering these processes and gaining insights into cellular functions and disease mechanisms. Among the various RNA structures, pseudoknots are of particular interest due to their complex formation and important biological functions. Pseudoknots are formed when bases in a loop pair with bases outside the loop, resulting in complicated tertiary structures that contribute to the functional repertoire
Traditionally, techniques such as X-ray crystallography, NMR spectroscopy and cryo-electron microscopy have been used to study RNA structures. While these techniques provide detailed insights, they are often time consuming, expensive and limited in their ability to process large RNA datasets. The advent of advanced computational methods has revolutionized the prediction of RNA structures, offering faster and more accurate results. Computational approaches can complement experimental techniques by providing initial structural predictions that guide further experimental investigations.
A key challenge in RNA analysis is the prediction of its secondary structure, which comprises the base-pairing interactions that create a two-dimensional arrangement, including features such as stems, loops, bulges and pseudoknots. Accurate prediction of secondary structure is a crucial step in understanding the functional dynamics and interactions of RNA. Although computational prediction of RNA structures has made considerable progress, accurate prediction of pseudoknots remains a daunting task due to their complexity and variability.
The proposed methodology focuses on the prediction of K-type and M-type pseudoknots in RNA secondary structures. These pseudoknot types are characterized by specific base pairing patterns and structural configurations that are essential for various biological functions. The precise identification and characterization of K- and M-type pseudoknots can provide valuable insights into the behavior of RNA and facilitate the development of RNA-based therapeutics. Building on previous work [1,2,3,4] which developed a framework for the prediction of H- and L-type pseudoknots, an extended grammar-based framework for the prediction of K- and M-type pseudoknots is proposed. Context-free grammars (CFGs) are used to model RNA sequences and predict pseudoknot structures. The definition of specific grammatical rules for K-type and M-type pseudoknots enables efficient parsing of RNA sequences to identify potential pseudoknot configurations. This method ensures a comprehensive coverage of possible pseudoknot structures within a reasonable computational time.
In addition, fundamental biological principles such as base pairing optimization and free energy minimization are used to improve prediction accuracy. These principles are crucial to ensure that the predicted structures are not only theoretically possible but also biologically relevant. Incorporating these principles into a grammar-based framework creates a powerful and accurate tool for predicting RNA structures.
The rest of this paper is organized as follows. Section 2 provides the necessary theoretical background. Section 3 discusses related work in the field of RNA structure prediction and highlights the limitations of existing methods. Section 4 describes the methodology, including the grammar-based framework, the syntactic pattern recognition techniques and the integration of biological principles. Finally, Section 5 summarizes the methodology and outlines future research directions.
2. FTheoretical Background
Essential context regarding RNA, the pseudoknot motif, syntactic pattern recognition, and context-free grammars is provided in this section. This foundational knowledge is crucial for understanding the methodology and significance of the proposed framework for RNA secondary structure prediction.
RNA, or ribonucleic acid, plays an essential role in numerous functions of Biology, such as regulating gene expression and synthesizing proteins. It is made up of ribonucleotides, which include a ribose sugar, a phosphate group, and a nitrogenous base [5]. Unlike DNA, RNA usually exists as a single strand, which enables it to adopt intricate secondary and tertiary structures. These formations result from base-pairing interactions, where adenine (A) pairs with uracil (U), and guanine (G) pairs with cytosine (C). The secondary structure of RNA includes various motifs such as stems, loops, bulges, and pseudoknots, which contribute to its functional diversity.
2.1. Pseudoknots in RNA
Pseudoknots are unique RNA motifs where bases in a loop form hydrogen bonds with bases outside the loop, creating interwoven secondary structures. These complex formations play significant roles in many biological functions, including ribosomal frameshifting, viral replication, and RNA catalysis. Pseudoknots are characterized by their intricate base-pairing patterns, making their prediction and identification a challenging task. Understanding pseudoknots is essential for gaining insights into RNA functionality and developing RNA-based therapeutic interventions. Pseudoknots hold significant importance in RNA research and are present across a variety of organisms. These structures feature two helices linked by one or more single-stranded regions, forming loops. The complex nature of pseudoknots makes them difficult to predict, as they involve intersecting base pairs. The pseudoknot motif was initially discovered in the Turnip Yellow Mosaic virus [6]. Various types of pseudoknots exist [7], including H, K, L, and M types (see Figure 1). For example, the H-type pseudoknot [8] consists of two stems and two loops of different lengths, with its formation involving the crossing of two sets of base pairs, known as core stems in our framework. The proposed Grammar-based framework specifically aims to predict K-Type and M-Type pseudoknots in RNA secondary structures.
2.2. Pattern-based Syntax Analysis
Syntactic pattern recognition is a computational approach that analyzes sequences based on their structural patterns and rules. In the context of RNA structure prediction, syntactic pattern recognition involves identifying patterns of base-pairing interactions that conform to specific grammatical rules. This method leverages the formalism of grammars to systematically describe and recognize complex RNA structures, including pseudoknots. By using syntactic pattern recognition, it is possible to efficiently parse RNA sequences and predict their secondary structures based on predefined patterns. This approach employs the concept of defining a formal language [9] through a set of syntactic rules that produce strings within that language. These rules constitute a grammar that defines how sequences of symbols are constructed to form the language. Noam Chomsky’s hierarchy [10] categorizes grammars into four distinct types, with Context-Free Grammars (CFGs) being among the most commonly employed. CFGs are widely applied in areas such as programming language design and other fields [11,12,13,14].
2.2.1. Context Free Grammars
Context-Free Grammars (CFGs) are a formal grammar used to define the syntactic structure of languages. A CFG consists of syntax rules that govern the replacement of symbols with other symbols to generate strings within the defined language. In CFGs, symbols are categorized into terminals and non-terminals.Terminal symbols are the actual symbols appearing in the language strings, while non-terminal symbols act as placeholders that can be expanded into terminals through production rules. For a grammar to be context-free, these rules must be applicable independently of the surrounding symbols, unlike context-sensitive grammars, where the rules’ application depends on the context.
In computational biology, CFGs are utilized to represent the hierarchical arrangement of RNA sequences. A CFG includes syntax rules that define how sequences of symbols—representing nucleotides in this case—can be generated. These rules capture the base-pairing interactions and structural motifs of RNA, enabling the prediction of secondary structures. CFGs are particularly useful for modeling RNA pseudoknots because they can represent nested and recursive structures, which are common in these motifs.
By combining syntactic pattern recognition with context-free grammars, a robust framework for RNA structure prediction can be developed. This approach allows for the systematic identification and characterization of pseudoknots, leveraging the strengths of formal grammars to handle the complexity and diversity of RNA structures. Incorporating biological principles such as optimizing base pairing and reducing free energy further refines the accuracy and biological relevance of predicted RNA structures.
The proposed method for predicting RNA pseudoknots uses syntactic pattern recognition, which involves defining a language with syntax rules that generate relevant symbol sequences. This grammar-based approach specifies how strings of symbols are produced within the language. Various parsing algorithms have been developed for CFGs due to their versatility. Notable examples include the CYK parser [15] and the Earley parser [16], as well as their extensions [17,18,19] and parallel versions [20,21]. For the proposed methodology, the Earley parser is recommended due to its efficiency in handling ambiguous grammars.
3. Related Work
TThe prediction of RNA secondary structures, particularly pseudoknots, is an active area of research due to the complexity associated with accurately modeling these structures. This section provides an overview of the main methods and advances in RNA structure prediction, focusing on both experimental and computational approaches. It also examines the specific challenges and solutions associated with the prediction of pseudoknots.
Experimental techniques have traditionally been the cornerstone of RNA structure analysis. Methods such as X-ray crystallography, NMR spectroscopy and cryo-electron microscopy provide detailed insights into RNA tertiary structures. X-ray crystallography enables high-resolution visualization of RNA molecules, but is often limited by the difficulty of crystallizing RNA. NMR spectroscopy provides information on RNA dynamics in solution, but is limited by the size of the RNA molecule. Cryo-electron microscopy provides high-resolution images of larger RNA complexes, but requires sophisticated equipment and extensive data processing. While powerful, these techniques are typically time-consuming and costly, and they often cannot keep pace with the rapid discovery of new RNA sequences.
In response to the limitations of experimental methods, computational approaches have become increasingly important. Many algorithms for predicting RNA secondary structures rely on dynamic programming to find the configuration with the lowest free energy. For pseudoknots, the challenge is made even greater by the need to account for additional factors such as stability and entropy. Although the problem has been shown to be NP-complete [22], several stochastic and heuristic approaches have been developed to overcome this complexity. For example, Knotty [23] uses a CCJ (Chen-Condon-Jabbari) algorithm with sparsification to efficiently predict pseudoknots. ProbKnot [24] combines base pair probabilities with maximum expected accuracy to predict RNA secondary structures, while IPknot [25] and its extension [26] use integer programming together with the LinearPartition model to improve prediction accuracy.
Machine learning techniques are increasingly used for the prediction of RNA structures. For example, deep learning models such as those in [27] incorporate tertiary constraints to increase accuracy. Another notable approach is the bidirectional LSTM network in combination with IBPMP in [28], which effectively selects the correct base pairs and predicts optimal RNA structures. The 2dRNA model [29] uses a bidirectional LSTM to encode RNA sequences, which are subsequently decoded into dot-bracket structures by a fully connected network. The ATTfold method [30] uses deep learning with an attention mechanism to encode base pairing results, followed by a Convolutional Neural Network (CNN) for decoding. UFold [31] processes RNA sequences as image-like data using Fully Convolutional Networks (FCNs) to make predictions.
In addition to thermodynamic models and machine learning methods, stochastic context-free grammars (SCFGs) are also used to predict RNA structures. SCFGs, as implemented in Pfold [32,33], predict secondary structures based on RNA alignments. Extensions such as multi-threading in PPfold [34] improve execution time. RNA decoder [35] applies an SCFG, taking into account the context of protein-coding RNAs. Other SCFG-based approaches include Contrafold [36], Evfold [37], Infernal [38], Oxfold [39] and Stemloc [40]. The research has also demonstrated the adaptation of Zuker’s thermodynamic model to an SCFG by calculating production probabilities from thermodynamic constants [41]. The diversity of approaches, from dynamic programming to machine learning and SCFGs, illustrates the complexity of RNA structure prediction and the need for effective integration of these methods. The wealth of research in this area emphasizes the need to combine these techniques to improve prediction accuracy. To this end, the proposed grammar-centered framework aims to improve the prediction of K- and M-type pseudoknots by extending the existing tools for RNA secondary structure analysis and addressing the current limitations in pseudoknot prediction. Recently, approaches using CFGs [1,2,3,4] have shown promising results.
4. Overview of Our Approach
This section describes the proposed methodology, which builds on and extends the Knotify and Knotify+ systems described in [1,2]. The updated platform extends Knotify’s capabilities to predict K-type and M-type pseudoknots in RNA sequences. The original Knotify system comprises three main steps: First, a CFG parser examines the RNA sequence to generate all potential trees that contain a pseudoknot pattern. Then, these trees are analyzed to determine the core strains of the pseudoknot and to identify possible base pairs in the surrounding regions. Finally, the best tree is selected based on two key criteria: the highest number of base pairs and the lowest free energy of the sequence. In this paper, we introduce two new CFGs that can be integrated into the original Knotify platform task and improve its ability to predict K and M type pseudoknots.
4.1. Grammar Definition for the Detection of K-type Pseudoknots
The proposed approach to identify type K pseudoknots in RNA sequences uses syntactic pattern recognition and a CFG parser. A major focus is on the careful selection of primitive patterns, which is essential for accurate recognition. In RNA sequences, the nitrogenous bases adenine (A), cytosine (C), guanine (G) and uracil (U) serve as fundamental components. Therefore, the proposed grammar uses these four terminal symbols to represent RNA sequences linguistically, such as "ACUGACCGCAGCU".
To identify a specific pattern, the method uses a pattern grammar to analyse the linguistic representation of the RNA sequence. The design of this grammar is crucial for accurate pattern recognition and significantly influences the result. Therefore, the creation of an effective CFG to describe pseudoknots is essential. CFGs are particularly effective in representing structural features, and for this purpose the grammar , detailed in Table 1, is used to predict pseudoknots of type K.
In the grammar, which is listed in Table 1, the syntactic rules are described in the column "Syntactic rules". This grammar contains four non-terminal symbols: NT = {R, X, D, Y}, where R serves as the root-start symbol. The rules in which R appears on the left-hand side (rules 0 to 63) are used to recognize potential pseudoknots of type K within the RNA sequence. A type K pseudoknot is characterized by at least three core stems, as shown in Figure 1c. To syntactically identify a type K pseudoknot, a suitable pattern grammar is used to analyze the linguistic representation of the RNA sequence. The CFG was developed to effectively describe the syntax of a type K pseudoknot where the core stems form inserted base pairs. For example, the rule R → "A" X "G" X "U" D "C" L "C" L "G" represents a pseudoknot structure of the form A..G..U..C..C..G, where the base pairs A–U, G–C and C–G are intercalated, as indicated by the colors. These intercalated base pairs are referred to as core strains in this study. There are 64 () possible syntax rules with R on the left, given the four base pairs (A–U, U–A, C–G, G–C) and three possible intercalations. Rules 16 to 59 are omitted as they are simple. Figure 2 illustrates how these core stems are nested to form a pseudo node of type K in the given example.
The proposed method for identifying K-type pseudoknots in RNA sequences uses syntactic pattern recognition together with a context-free grammar (CFG) parser. RNA sequences are represented by the four nucleotide bases — adenine (A), cytosine (C), guanine (G) and uracil (U) — with each base represented by a corresponding character. Thus, each RNA sequence can be represented as a chain of these symbols. The grammar is constructed with these four terminal symbols and contains four non-terminal symbols in the set NT.
The syntactic rules of the grammar, which are listed in the second column of the Table 1, aim to identify potential pseudo nodes in the input string. All rules with the start symbol R on the left-hand side are used for this purpose. A pseudo node of type K is defined by the presence of at least three base pairs that form inserted core stems. The non-terminal symbol X generates sequences for the four inner loops of the pseudoknot, while the non-terminal symbol D generates base sequences that are located between the two intersecting main base pairs.
The CFG parser can identify pseudo nodes in strings where the start and end symbols are part of the main stems and can handle substrings using a sliding windows technique. This method parses all substrings of the RNA sequence, starting with the shortest and gradually increasing the length by one symbol until the entire sequence is processed. Parsing is stopped if the length of the substring falls below a certain threshold value, which corresponds to the minimum length for a
To handle grammatical ambiguity, the YAEP [47] parser, which uses the Earley algorithm, is recommended for CFG parsing. Section 4.2.1 describes how these substrings are integrated into the original RNA sequence and how additional base pairs are integrated into the pseudoknot. The process for selecting the optimal parse tree from the generated options is described in detail in Section 4.3.
4.2. Grammar Definition for the Detection of K-type Pseudoknots
Using the approach described above, the grammar , which is described in detail in Table 2, has been tailored to identify M-type pseudoknots. This grammar uses the same four non-terminal symbols and four terminal symbols as . All syntactic rules with R on the left (i.e., rules 0 to 255) are designed to recognize potential M-type pseudoknots within the input RNA sequence. A type M pseudoknot is defined by having at least four core stems, as shown in Figure 1d. The CFG is able to capture the syntax of type M pseudoknots where the four core stems form nested base pairs.
For example, the rule R → "U" X "G" X "C" X "A" D "A" X "C" X "G" X "U" represents a pseudo node with the structure U..G..C...A...A.C..G...U, where the base pairs U–A, G–C, C–G and A–U are intercalated, with the colors indicating the respective pairs. These intercalated base pairs, known as core stems, are crucial for recognizing pseudoknots of type M. Considering the four possible base pairs (A–U, U–A, C–G, G–C) and the four possible intercalations, there are 256 () syntactic rules with R on the left, with rules 16 to 251 omitted for brevity. Figure 3 illustrates how these core stems merge in the given example to form the pseudoknot of type M.
4.2.1. Core Stems Decoration
After the parse trees have been generated as described above, the next step is to add additional base pairs to the pseudonode by analyzing all generated trees. To increase the efficiency of the CFG parser, it focuses exclusively on identifying the key stems of the pseudoknot. Although this approach simplifies the CFG by reducing the number of syntactic rules and increases performance, it requires a thorough examination of all parse trees to determine the base pairs surrounding these essential stems.
The parser carefully evaluates each base within the pseudo node loops to check if it can be paired with a matching base outside the loops. The decoration process for the example in Section 4.1 is shown in Table 3. After identifying the core stems at positions 2-8, 5-15 and 12-18 (highlighted in bold), which correspond to base pairs A–U, G–C and C–G (colored for clarity), the parser examines the bases within the loops at positions 9-11 for possible pairings with the bases at positions 0-1 and 19-21 respectively. Then the bases in the internal loops at positions 3-4 and 16-17 are examined for a possible base pairing. In summary, the base pairs are determined as follows: positions 1–9 in phase 1, 11–18 in phase 2 and positions 4–16 and 3–17 in phase 3.
4.3. Optimal Pseudoknot Selection
According to the methodology described by our research team in [1,2,3], the final phase of our approach consists in selecting the most accurate pseudoknot. Several strategies for predicting RNA base pairing have been proposed in the literature, including
- The Minimum Free Energy (MFE) method [42], which determines the RNA structure with the lowest free energy. Although this approach is based on the second law of thermodynamics, the predicted structure does not always correspond to natural conditions.
- The maximum pairing principle [43] emphasizes the counting of base pairs around the critical stems of the pseudoknot. In dot-bracket notation, the configurations with the highest number of base pairs around the pseudoknot generally correspond to the structures with minimum free energy.
- The partition function method [44] assumes that the true base pairs are those that are most likely to lie within the minimum free energy distribution and improves accuracy by including the free energy of neighboring pairs at a given temperature.
- Comparative Sequence Analysis [45] investigates substitution patterns in pairwise alignments of homologous sequences.
- Physical Experiments [46] includes laboratory techniques to validate predictions.
For the selection of the optimal tree, a hybrid model is proposed that combines aspects of the two most widely used methods: Maximum Pairing and MFE. This combined approach aims to predict RNA secondary structures, including complex motifs such as K-type and M-type pseudoknots, with greater precision and efficiency. First, the trees are ranked based on the number of base pairs surrounding the pseudoknot, and the MFE method is applied exclusively to the trees with the highest base pair numbers. This heuristic approach shows superior performance compared to the traditional MFE method. This hybrid model has succesfully been used in all Knotify versions [1,2,3,4], which follow a similar architecture, and is therefore also proposed for the presented method.
5. Conclusion and Future Work
In this work, a comprehensive methodology for the detection and optimal selection of K- and M-type pseudoknots in RNA sequences was presented. By utilising the power of context-free grammars (CFGs) and applying a hybrid model that integrates the principles of the minimum free energy (MFE) method with the maximum pairing principle, our approach efficiently identifies and predicts complex RNA secondary structures, including those with intricate pseudoknot motifs. The proposed CFGs are carefully designed to capture the essential features of K- and M-type pseudoknots and ensure both accuracy and computational efficiency. By focusing on the core stems of the pseudoknot, we reduce the complexity of the parsing process, which is further improved by selectively applying the MFE method to the most promising parse trees. This combination of techniques enables more accurate prediction of RNA structures while maintaining computational feasibility, making it a valuable tool for researchers in the field of RNA bioinformatics. Furthermore, our approach to parse tree decoration ensures that additional base pairs, especially those flanking the essential stems, are accurately identified, resulting in a more complete representation of the RNA pseudoknot structure. The next step in this research is the implementation of the proposed methodology using the YAEP [47] parser. By implementing our approach within the YAEP framework, we expect further improvements in parsing speed and accuracy, especially when dealing with large and complex RNA sequences. Furthermore, we plan to integrate this improved methodology into the Knotify+ [2] platform, a versatile and user-friendly tool for RNA secondary structure prediction. By integrating our advanced pseudoknot detection and selection algorithm into Knotify+, we aim to provide researchers with a powerful and easily accessible platform to study RNA pseudoknots and other secondary structures. This integration will not only streamline the prediction process, but also expand the capabilities of the platform, making it a comprehensive resource for RNA bioinformatics research.
In summary, our methodology represents a significant advance in the field of RNA structure prediction. With the planned implementation in YAEP and integration into Knotify, we are confident that this work will contribute to more accurate and efficient RNA structure analysis, which will ultimately support the understanding of the role of RNA in biological processes and the development of RNA-based therapeutics.
Author Contributions
Conceptualization, C.P.; methodology, C.P.; validation, C.P.; formal analysis, C.P.; writing—original draft preparation, C.P.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Andrikos, C., Makris, E., Kolaitis, A., Rassias, G., Pavlatos, C. & Tsanakas, P. Knotify: An Efficient Parallel Platform for RNA Pseudoknot Prediction Using Syntactic Pattern Recognition. Methods Protoc. 2022, 5, 14.
- Makris E, Kolaitis A, Andrikos C, Moulos V, Tsanakas P, Pavlatos C. Knotify+: Toward the Prediction of RNA H-Type Pseudoknots, Including Bulges and Internal Loops. Biomolecules. 2023; 13(2):308. [CrossRef]
- Koroulis C, Makris E, Kolaitis A, Tsanakas P, Pavlatos Syntactic Pattern Recognition for the Prediction of L-Type Pseudoknots in RNA Applied Sciences 13 (8), 5168, 2023. [CrossRef]
- Makris, E., Kolaitis, A., Andrikos, C., Moulos, V., Tsanakas, P., Pavlatos, C. An intelligent grammar-based platform for RNA H-type pseudoknot prediction. Artificial Intelligence Applications and Innovations. AIAI 2022 IFIP WG 12.5 International Workshops. IFIP Advances in Information and Communication Technology 2022, vol 652, Springer.
- Watson, J.; Crick, F. Molecular Structure Of Nucleic Acids. Am. J. Psychiatry 2003, 160, 623–624. [Google Scholar] [CrossRef]
- Rietveld, K.; Van Poelgeest, R.; Pleij, C.W.; Van Boom, J.; Bosch, L. The tRNA-Uke structure at the 3’ terminus of turnip yellow mosaic virus RNA. Differences and similarities with canonical tRNA. Nucleic Acids Res. 1982, 10, 1929–1946. [Google Scholar] [CrossRef] [PubMed]
- Kucharík, M.; Hofacker, I.L.; Stadler, P.F.; Qin, J. Pseudoknots in RNA folding landscapes. Bioinformatics 2016, 32, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Pseudoknots: RNA structures with diverse functions.
- Hopcroft, J.E.; Ullman, J.D. Formal languages and their relation to automata; Addison-Wesley Longman Publishing Co., Inc.: Boston, MA, USA, 1969. [Google Scholar]
- Chomsky, N. Three models for the description of language. IRE Trans. Inf. Theory 1956, 2, 113–124. [Google Scholar] [CrossRef]
- Pavlatos, C., Vita, V., Ekonomou, L. Syntactic pattern recognition of power system signals In Proceedings of the 19th WSEAS international conference on systems (part of CSCC’15), Zakynthos Island, Greece (pp. 16-20).
- Panagopoulos, I., Pavlatos C., Papakonstantinou G. An Embedded System for Artificial Intelligence Applications International Journal of Computational Intelligence, vol.1 no1-4,( 2004).
- Pavlatos, C., Panagopoulos, I., Papakonstantinou, G. A programmable pipelined coprocessor for parsing applications In Workshop on application specific processors (WASP) CODES, Stockholm (Vol. 294).
- Pavlatos, C., Dimopoulos, A., Papakonstantinou, G. An intelligent embedded system for control applications In Workshop on modeling and control of complex systems, Cyprus (2005).
- Younger, D.H. Recognition and parsing of context-free languages in n3. Inf. Control. 1967, 10, 189–208. [Google Scholar] [CrossRef]
- Earley, J. An efficient context-free parsing algorithm. Commun. ACM 1970, 13, 94–102. [Google Scholar] [CrossRef]
- Graham, S.L.; Harrison, M.A.; Ruzzo, W.L. An improved context-free recognizer. ACM Trans. Program. Lang. Syst. 1980, 2, 415–462. [Google Scholar] [CrossRef]
- Ruzzo, W.L. General context-free language recognition. PhD Thesis, University of California, Berkeley, CA, USA, 1978. [Google Scholar]
- Geng, T.; Xu, F.; Mei, H.; Meng, W.; Chen, Z.; Lai, C. A practical GLR parser generator for software reverse engineering. JNW, 2014, 9(3), 769–776.
- Pavlatos, C.; Dimopoulos, A.C.; Koulouris, A.; Andronikos, T.; Panagopoulos, I.; Papakonstantinou, G. Efficient reconfigurable embedded parsers. Comput. Lang. Syst. Struct. 2009, 35, 196–215. [Google Scholar] [CrossRef]
- Chiang, Y.; Fu, K. Parallel parsing algorithms and VLSI implementations for syntactic pattern recognition. IEEE Trans. Pattern Anal. Mach. Intell. 1984, 6, 302–314. [Google Scholar] [CrossRef]
- Akutsu, T. Dynamic programming algorithms for RNA secondary structure prediction with pseudoknots. Discret. Appl. Math. 2000, 104, 45–62. [Google Scholar] [CrossRef]
- Jabbari, H.; Wark, I.; Montemagno, C.; Will, S. Knotty: efficient and accurate prediction of complex RNA pseudoknot structures. Bioinformatics 2018, 34, 3849–3856. [Google Scholar] [CrossRef] [PubMed]
- Bellaousov, S.; Mathews, D.H. ProbKnot: fast prediction of RNA secondary structure including pseudoknots. RNA 2010, 16, 1870–80. [Google Scholar] [CrossRef]
- Sato, K.; Kato, Y.; Hamada, M.; Akutsu, T.; Asai, K. IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics 2011, 27, 85–93. [Google Scholar] [CrossRef]
- Sato, K. ; Kato, Y. Prediction of RNA secondary structure including pseudoknots for long sequences. Briefings In Bioinformatics 2021 23.
- Singh, J.; Hanson, J.; Paliwal, K.; Zhou, Y. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Zhong, X.; Liu, H.; Lu, C.; Li, C.; Zhang, H. DMfold: A novel method to predict RNA secondary structure with pseudoknots based on deep learning and improved base pair Maximization Principle. Front. Genet. 2019, 10, 143. [Google Scholar] [CrossRef]
- Kangkun, M.; Jun, W.; Yi, X. Prediction of RNA secondary structure with pseudoknots using coupled deep neural networks. Biophys. Rep. 2020, 6, 146–154. [Google Scholar]
- Wang, Y.; Liu, Y.; Wang, S.; Liu, Z.; Gao, Y.; Zhang, H.; Dong, L. ATTfold: RNA secondary structure prediction with pseudoknots based on attention mechanism. Front. Genet. 2020, 11, 1564. [Google Scholar] [CrossRef]
- Fu, L., Cao, Y., Wu, J., Peng, Q., Nie, Q. & Xie, X. UFold: fast and accurate RNA secondary structure prediction with deep learning. Nucleic Acids Research 2021 50, e14.
- Knudsen, B.; Hein, J. RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics 1999, 15, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, B.; Hein, J. Pfold: RNA Secondary Structure Prediction Using Stochastic Context-Free Grammars. Nucleic Acids Res. 2003, 31, 3423–3428. [Google Scholar] [CrossRef] [PubMed]
- Sukosd, Z.; Knudsen, B.; Vaerum, M.; Kjems, J.; Andersen, E.S. Multithreaded comparative RNA secondary structure prediction using stochastic context-free grammars. BMC Bioinform. 2011, 12, 103. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Meyer, I.M.; Forsberg, R.; Simmonds, P.; Hein, J. A comparative method for finding and folding RNA secondary structures within protein-coding regions. Nucleic Acids Res. 2004, 32, 4925–4936. [Google Scholar] [CrossRef]
- Do, C.B.; Woods, D.A.; Batzoglou, S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics 2006, 22, e90–e98. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.S.; Bejerano, G.; Siepel, A.; Rosenbloom, K.; Lindblad-Toh, K.; Lander, E.S.; Kent, J.; Miller, W.; Haussler, D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput. Biol. 2006, 2, e33. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.W.; Haas, P.A.; Mathieson, L.A.; Volynkin, V.; Lyngsø, R.; Tataru, P.; Hein, J. Oxfold: kinetic folding of RNA using stochastic context-free grammars and evolutionary information. Bioinformatics 2013, 29, 704–710. [Google Scholar] [CrossRef]
- Bradley, R.K.; Pachter, L.; Holmes, I. Specific alignment of structured RNA: stochastic grammars and sequence annealing. Bioinformatics 2008, 24, 2677–2683. [Google Scholar] [CrossRef]
- Isambert, H.; Siggia, E.D. Modeling RNA folding paths with pseudoknots: application to hepatitis delta virus ribozyme. Proc. Natl. Acad. Sci. USA 2000, 97, 6515–6520. [Google Scholar] [CrossRef] [PubMed]
- Trotta, E. On the normalization of the minimum free energy of RNAs by sequence length. PLoS ONE 2014, 9, e113380. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Jacobson, A.B. Fast algorithm for predicting the secondary structure of single-stranded RNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6309–6313. [Google Scholar] [CrossRef]
- Mathews, D.H. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA 2004, 10, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Rivas, E.; Eddy, S.R. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinform. 2001, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; R.Corey, D. RNA Sequencing: Platform Selection, Experimental Design, and Data Interpretation. Nucleic Acid Ther. 2012, 22, 271–274.
- Available online: https://github.com/vnmakarov/yaep (accessed on 25 March 2020).
Figure 1.
Different types of RNA pseudoknot
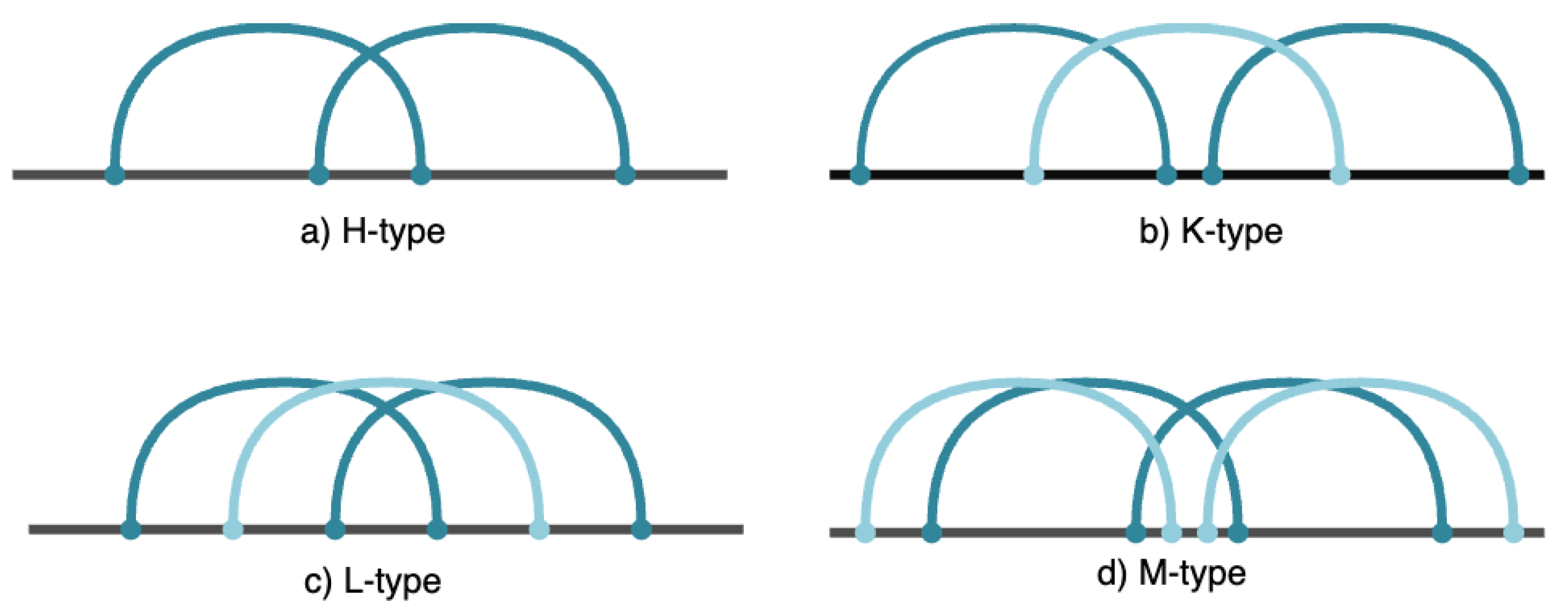
Figure 2.
The rule R → "A" X "G" X "U" D "C" X "C" X "G" that detects an K-type pseudoknot
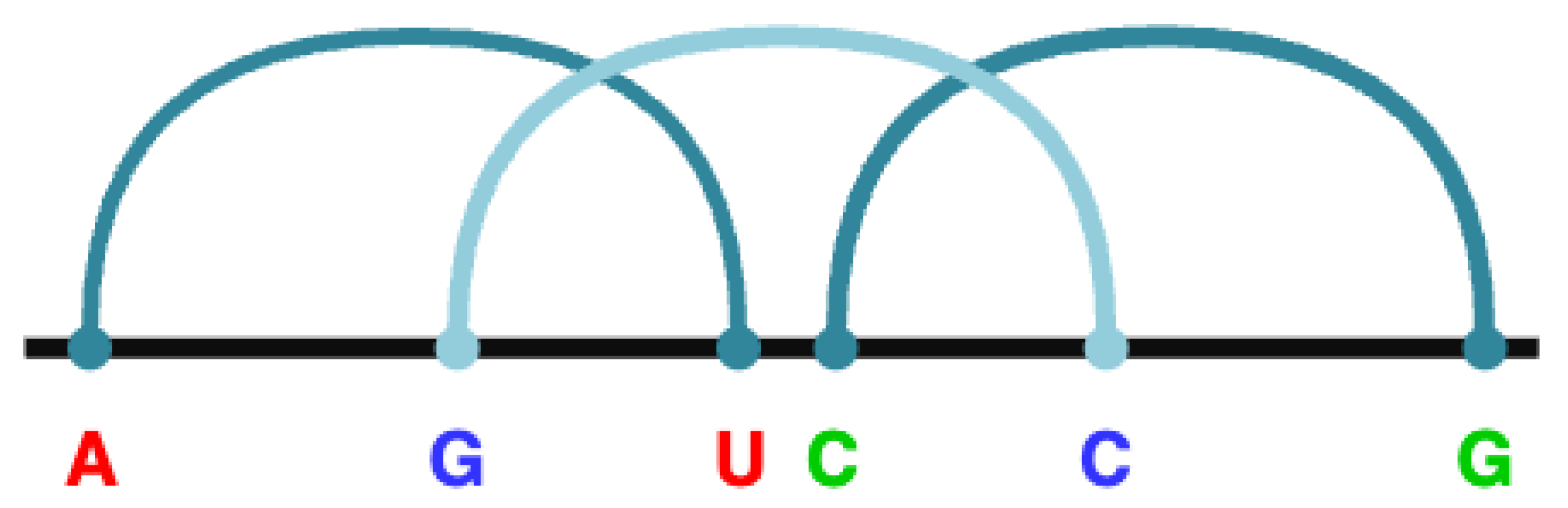
Figure 3.
The rule R → "U" X "G" X "C" X "A" D "A" X "C" X "G" X "U" that detects an M-type pseudoknot
Figure 3.
The rule R → "U" X "G" X "C" X "A" D "A" X "C" X "G" X "U" that detects an M-type pseudoknot
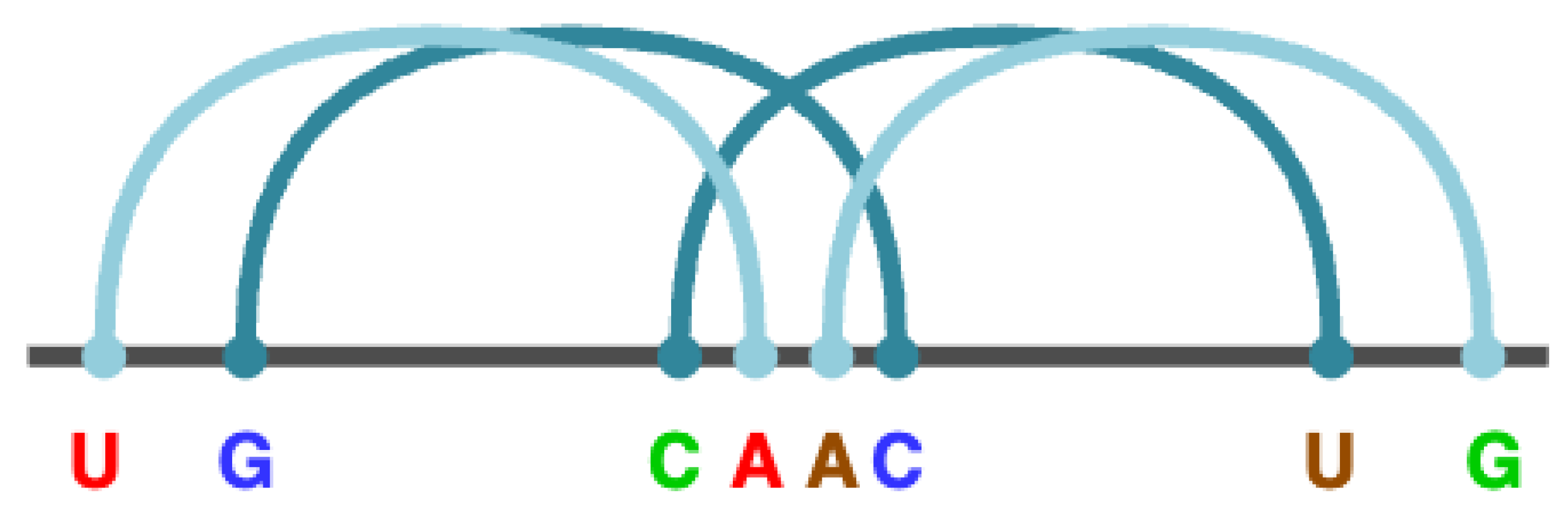

Table 1.
Syntactic rules .
| Rule Number | Syntactic Rules |
|---|---|
| 0 | R → "A" X "A" X "U" D "A" X "U" X "U" |
| 1 | R → "A" X "A" X "U" D "U" X "U" X "A" |
| 2 | R → "A" X "A" X "U" D "C" X "U" X "G" |
| 3 | R → "A" X "A" X "U" D "G" X "U" X "C" |
| 4 | R → "A" X "U" X "U" D "A" X "A" X "U" |
| 5 | R → "A" X "U" X "U" D "U" X "A" X "A" |
| 6 | R → "A" X "U" X "U" D "C" X "A" X "G" |
| 7 | R → "A" X "U" X "U" D "G" X "A" X "C" |
| 8 | R → "A" X "G" X "U" D "A" X "C" X "U" |
| 9 | R → "A" X "G" X "U" D "U" X "C" X "A" |
| 10 | R → "A" X "G" X "U" D "C" X "C" X "G" |
| 11 | R → "A" X "G" X "U" D "G" X "C" X "C" |
| 12 | R → "A" X "C" X "U" D "A" X "G" X "U" |
| 13 | R → "A" X "C" X "U" D "U" X "G" X "A" |
| 14 | R → "A" X "C" X "U" D "C" X "G" X "G" |
| 15 | R → "A" X "C" X "U" D "G" X "G" X "C" |
| . | |
| . | |
| . | |
| 60 | R → "C" X "C" X "G" D "A" X "G" X "U" |
| 61 | R → "C" X "C" X "G" D "U" X "G" X "A" |
| 62 | R → "C" X "C" X "G" D "G" X "G" X "C" |
| 63 | R → "C" X "C" X "G" D "C" X "G" X "G" |
| 64 | X → "A" X |
| 65 | X → "U" X |
| 66 | X → "C" X |
| 67 | X → "G" X |
| 68 | X → "A" |
| 69 | X → "U" |
| 70 | X → "C" |
| 71 | X → "G" |
| 72 | D → Y Y |
| 73 | Y → "A" |
| 74 | Y → "U" |
| 75 | Y → "C" |
| 76 | Y → "G" |
| 77 | Y |
Table 2.
Syntactic rules .
| Rule Number | Syntactic Rules |
|---|---|
| 0 | R → "A" X "A" X "A" X "U" D "A" X "U" X "U" X "U" |
| 1 | R → "A" X "A" X "A" X "U" D "U" X "U" X "U" X "A" |
| 2 | R → "A" X "A" X "A" X "U" D "G" X "U" X "U" X "C" |
| 3 | R → "A" X "A" X "A" X "U" D "C" X "U" X "U" X "G" |
| 4 | R → "A" X "A" X "U" X "U" D "A" X "U" X "A" X "U" |
| 5 | R → "A" X "A" X "U" X "U" D "U" X "U" X "A" X "A" |
| 6 | R → "A" X "A" X "U" X "U" D "G" X "U" X "A" X "C" |
| 7 | R → "A" X "A" X "U" X "U" D "C" X "U" X "A" X "G" |
| 8 | R → "A" X "A" X "G" X "U" D "A" X "U" X "C" X "U" |
| 9 | R → "A" X "A" X "G" X "U" D "U" X "U" X "C" X "A" |
| 10 | R → "A" X "A" X "G" X "U" D "G" X "U" X "C" X "C" |
| 11 | R → "A" X "A" X "G" X "U" D "C" X "U" X "C" X "G" |
| 12 | R → "A" X "A" X "C" X "U" D "A" X "U" X "G" X "U" |
| 13 | R → "A" X "A" X "C" X "U" D "U" X "U" X "G" X "A" |
| 14 | R → "A" X "A" X "C" X "U" D "G" X "U" X "G" X "C" |
| 15 | R → "A" X "A" X "C" X "U" D "C" X "U" X "G" X "G" |
| . | |
| . | |
| . | |
| 252 | R → "C" X "C" X "C" X "G" D "A" X "G" X "G" X "U" |
| 253 | R → "A" X "A" X "A" X "U" D "U" X "U" X "U" X "A" |
| 254 | R → "A" X "A" X "A" X "U" D "G" X "U" X "U" X "C" |
| 255 | R → "A" X "A" X "A" X "U" D "C" X "U" X "U" X "G" |
| 256 | X → "A" X |
| 257 | X → "U" X |
| 258 | X → "C" X |
| 259 | X → "G" X |
| 260 | X → "A" |
| 261 | X → "U" |
| 262 | X → "C" |
| 263 | X → "G" |
| 264 | D → Y Y |
| 265 | Y → "A" |
| 266 | Y → "U" |
| 267 | Y → "C" |
| 268 | Y → "G" |
| 269 | Y |
Table 3.
Decorating the core stems
| Index | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RNA | C | U | A | C | C | G | C | C | U | A | C | U | C | A | A | C | G | G | G | A | C | C |
| Parser output: | . | . | ( | . | . | [ | . | . | ) | . | . | . | { | . | . | ] | . | . | } | . | . | . |
| Phase 1 | . | ( | ( | . | . | [ | . | . | ) | ) | . | . | { | . | . | ] | . | . | } | . | . | . |
| Phase 2 | . | ( | ( | . | . | [ | . | . | ) | ) | . | { | { | . | . | ] | . | . | } | } | . | . |
| Phase 3 | . | ( | ( | [ | [ | [ | . | . | ) | ) | . | { | { | . | . | ] | ] | ] | } | } | . | . |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



