Submitted:
24 July 2024
Posted:
25 July 2024
You are already at the latest version
Abstract
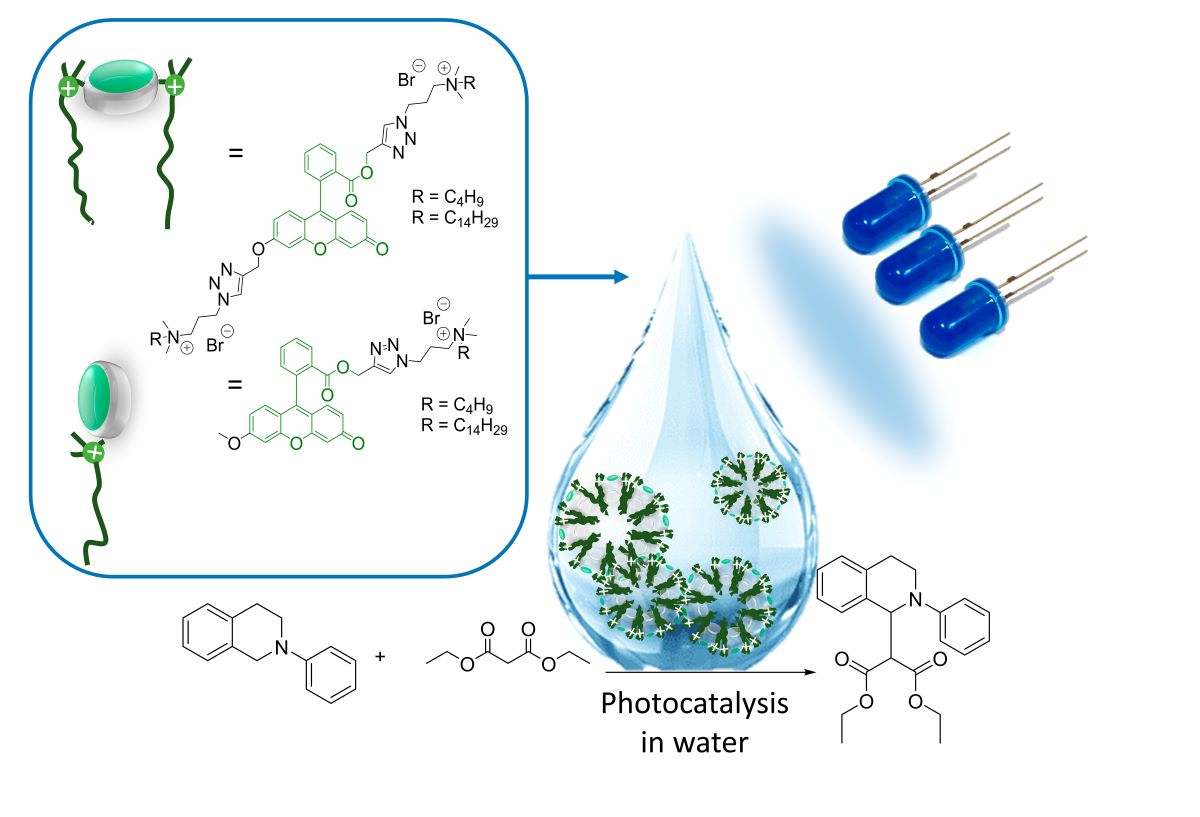
Triazole derivatives of fluorescein containing N,N-dimethylaminopropyl fragments and their ammonium salts were synthesized with yields of 74 - 85%. The resulting compounds exhibit fluorescent properties in the green region of the visible spectrum. The critical aggregation concentration (CAC) was estimated using a py-rene fluorescent probe corresponding to a range of 0.28 - 1.43 mM, and at concentrations above the CAC the compounds form stable aggregates ranging from 165 to 202 nm. A relative quantum yield of 5-24% has been calculated based on fluorescence and UV spectra. The best value is shown by a derivative containing a tetradecyl substituent. When studying the photocatalytic properties of synthesized compounds through the reaction between N-substituted 1,2,3,4-tetrahydroisoquinoline and malonic ester, the mono-tetradecyl deriva-tive demonstrated the best results. According to gas chromatography-mass spectrometry (GC-MS) data, the conversion of the initial heterocycle reached 95%. Therefore, these resulting compounds have the potential to act as an effective photocatalysts.
Keywords:
CuAAC
; photocatalysis
; fluorescein derivatives
; aggregation
1. Introduction
“Green (sustainable) chemistry” is attractive to modern organic synthesis due to a range of benefits relative to classical organic synthesis: milder reaction conditions, absence of toxic solvents or catalysts, short reaction time [1,2]. Therefore, growing attention is being paid to the development of environmental friendly catalysts, the use of aqueous solutions as a medium, the use of microwave, sono- and, in particular, photo-catalysis. Since light, including sunlight, is the source of energy in photocatalytic processes, fluorophore - molecules with fluorescent properties - play an important role [3]. The selection of an appropriate photocatalytic system remains a significant challenge. The variety of reactions that can be accelerated by photocatalysts is extensive, and most research focuses on the photocatalytic treatment of wastewater [4,5,6]. However, there are publications dedicated to the synthesis of various compounds. For example, benzothiophens [7] and carboxylic acids [8] as well as C-C [9,10] or C-N [11,12] cross-coupling reactions were successfully synthesized using photoredox catalysis.
Xanthene dyes, such as fluorescein, are widely used as chemosensors[13,14,15], photocatalysts [16,17,18] and photosensitizers [19,20] due to their high quantum yield [21]. Additionally, fluorescein can undergo modification through various substitutions of the phenolic and carboxylic groups, resulting in a diverse range of derivatives [22]. Combining photoredox catalysis with the reaction in aqueous media will bring us even closer to the concept of Green chemistry and is a very important trend [23]. Among the reactions carried out in water, reactions on water (when water does not solvate the reactants) and in water are distinguished [24]. In the latter case, the reaction can be significantly accelerated by the addition of solubilizing amphiphilic additives. Therefore, the synthesis of amphiphilic compounds from fluorescein derivatives allows for the formation of highly organized structures. These structures, on the one hand, have the ability to solubilize organic molecules in water, and on the other hand, are capable of entering into an excited state upon exposure to quanta of light. This ability enables them to catalyze organic reactions. Thus, it is feasible to integrate micellar and photocatalytic processes within a single catalytic system, thereby enhancing reaction efficiency[25]. However, examples of such systems are still rare [26].
Here, we report on the synthesis of new 1,2,3-triazoles combining fluorescein fragments and dimethylaminopropyl groups using the CuAAC (copper-assisted alkyne-azide cycloaddition) reaction, as well as the production of their ammonium salts. Photophysical properties (quantum yield, fluorescence, UV absorption spectra) and self-assembly (CAC) properties were studied. Based on the collected data, the photocatalytic activity of the synthesized compounds was studied in model condensation reactions between N-phenyl-1,2,3,4-tetrahydroisoquinoline and malonic ester.
2. Materials and Methods
2.1. Synthesis
All reagents were purchased from either Acros, Sigma-Aldrich or Maclin and used without further purification. Solvents were purified by standard methods. Substance purity and the reaction process were monitored by TLC on Merck UV 254 plates and visualized by exposure to UV with a VL-6.LC lamp (Vilber, Marne-la-Vallée, France).
Flash chromatography was done using SepaBeanTM machine 2; detector: DAD variable UV (254 and 280 nm); flow range 5 mL/min; gradient H2O→AcN; column type: Santai Technologies, Inc., SepaFlashTM SW004 Bonded, spherical C18, 20-45 μm, 100 Å, item number: SW-5222-004-SP.
Melting points were measured using the OIptimelt MPA100 melting point apparatus (Stanford Research Systems, Sunnyvale, CA, USA).
1H and 13C NMR spectra were recorded on Bruker Avance 400 Nanobay (Bruker Corporation, Billerica, MA, USA) with signals from residual protons of DMSO-d6 or CDCl3 as internal standard.
ATR-IR spectra and IR spectra in KBr pellets were collected using a Bruker Vector-22 spectrometer (Bruker Corporation, Billerica, MA, USA).
High-resolution mass spectra with electrospray ionization (HRESI MS) were obtained on an Agilent iFunnel 6550 Q-TOF LC/MS (Agilent Technologies, Santa Clara, CA, USA). Carrier gas: nitrogen, temperature 300 °C, carrier flow rate 12.l × min−1, nebulizer pressure 275 kPa, funnel voltage 3500 V, capillary voltage 500 V, total ion current recording mode, 100–3000 m/z mass range, scanning speed 7 spectra × s−1.
Data set for single crystal 5 was collected on a Rigaku XtaLab Synergy S instrument with a HyPix detector and a PhotonJet microfocus X-ray tube using Cu Kα (1.54184 Å) radiation at room temperature. Images were indexed and integrated using the CrysAlisPro data reduction package. Data were corrected for systematic errors and absorption using the ABSPACK module: numerical absorption correction based on Gaussian integration over a multifaceted crystal model and empirical absorption correction based on spherical harmonics according to the point group symmetry using equivalent reflections. The GRAL module was used for analysis of systematic absences and space group determination. The structure was solved by direct methods using SHELXT [27] and refined by the full-matrix least-squares on F2 using SHELXL [28]. Non-hydrogen atoms were refined anisotropically. The hydrogen atoms were inserted at the calculated positions and refined as riding atoms. The figures were generated using Mercury 4.1 [29] program. Crystals were obtained by slow evaporation method.
Prop-2-yn-1-yl 2-(3-oxo-6-(prop-2-yn-1-yloxy)-3H-xanthen-9-yl)benzoate 2 [30], methyl 2-(6-methoxy-3-oxo-3H-xanthen-9-yl)benzoate 3[31], 3’-hydroxy-6’-methoxy-3H-spiro[isobenzofuran-1,9’-xanthen]-3-one 4 [32], 3-azido-N,N-dimethylpropan-1-amine 7 [33] were synthesized according to the literature procedures.
The synthesis of prop-2-yn-1-yl 2-(6-methoxy-3-oxo-3H-xanthen-9-yl)benzoate 5
To a solution of 3’-hydroxy-6’-methoxy-3H-spiro[isobenzofuran-1,9’-xanthen]-3-one 4 (1.5 g, 4.3 mmol, 1 eq.) in 20 ml of dry DMF was added K2CO3 (0.89 g, 6.45 mmol, 1.5 eq.). The mixture was stirred at rt for 30 min, then propargyl bromide (0.62 g, 5.2 mol, 1.2 eq.) was added. The mixture was stirred for 24 hours at rt, and the resultant product was concentrated under vacuum. The residue was suspended in H2O and the formed precipitate was filtered off. After recrystallization in benzene the product 5 was obtained as orange crystals. Yield 1.42 g, 85%. Mp=214 oC.
1H NMR (400 MHz, CDCl3, 25 oC) δ, ppm: 2.33 (t, J = 6.9 Hz, 1H, -C≡CH), 3.92 (s, 3H, -OCH3), 4.59 (dABq, 2H, ΔδAB = 0.07, JAB = 12.2 Hz, J = 2.4 Hz, 2H, -OCH2-), 6.45 (s, 1H, ArH), 6.54 (d, J = 9.7, 1H, ArH), 6.74 (d, J = 9.0 Hz, 1H, ArH), 6.80-6.92 (m, 2H, ArH), 6.96 (d, J = 2.4 Hz, 1H, ArH), 7.34 (d, J = 7.2 Hz, 1H, ArH), 7.76 (t, J = 7.4 Hz, 1H, ArH), 7.69 (t, J = 7.6 Hz, 1H, ArH), 8.27 (d, J = 7.8 Hz, 1H, ArH). 13C NMR (101 MHz, CDCl3, 25 oC) δ, ppm: 52.9, 56.1, 56.5, 75.5, 100.5, 105.9, 113.6, 113.6, 114.9, 117.9, 128.9, 129.8, 130.2, 130.3, 130.8, 131.5, 133.2, 134.9, 149.7, 154.5, 159.1, 164.2, 164.6, 185.9. IR (KBr) ν max cm-1: 3294 (C≡CH), 2123, 1726 (C=O), 1643, 1599, 1545, 1518, 1481, 1464, 1417, 1379, 1344, 1280, 1269, 1253, 1211, 1130, 1107, 758. HRESI MS (m/z) [M + H]+: calcd. for C24H17O5+ 385.1076, found: 385.1080. (Fig. S1 in the Supporting Information).
Crystal Data for C24H16O5 (M =384.37 g/mol): monoclinic, space group P21/c (no. 14), a = 14.5495(3) Å, b = 8.7385(2) Å, c = 14.4494(3) Å, β = 99.790(2)°, V = 1810.36(7) Å3, Z = 4, T = 100.0(7) K, μ(Cu Kα) = 0.815 mm-1, Dcalc = 1.410 g/cm3, 12169 reflections measured (6.164° ≤ 2Θ ≤ 151.978°), 3662 unique (Rint = 0.0265, Rsigma = 0.0241) which were used in all calculations. The final R1 was 0.0395 (I > 2σ(I)) and wR2 was 0.1039 (all data). CCDC number 2358771.
General procedure for synthesis of 1,2,3-triazoles 8-9
To a solution of azide 7 (1 mmol, 1 eq. in case of 5 or 2 mmol, 2 eq. in case of 2) in 5 ml of THF was added NEt3 (0.5 ml) following by of alkynes 2 or 5 (1 mmol, 1eq.) and CuI (0.1 mmol, 0.1 eq.). The reaction mixture was stirred at rt in inert atmosphere for 10-14 h. The completion of the reaction was determined by TLC (CHCl3:MeOH=10:1). The reaction mixture was evaporated, diluted with CHCl3 and filtered through amberlite IRA-67®. After evaporation in vacuo the product was obtained as vitreous oil.
(1-(3-(Dimethylamino)propyl)-1H-1,2,3-triazol-4-yl)methyl 2-(6-((1-(3-(dimethylamino)propyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-oxo-3H-xanthen-9-yl)benzoate 8
Yield 0.33 g, 66%. 1H NMR (400 MHz, CDCl3, 25 oC) δ, ppm: 1.98 (p, J = 6.9 Hz, 2H, -CH2-), 2.07 (p, J = 6.9 Hz, 2H, -CH2-), 2.18 (s, 6H, 2*N-CH3), 2.20 (s, 6H, 2*N-CH3), 2.23 – 2.29, (m, 4H, 2* -NCH2-), 4.27 – 4.42 (m, 2H, -NCH2-), 4.28 – 4.40 (m, 2H, -NCH2-), 5.11 (ABq, 2H, ΔδAB = 0.07, JAB = 12.6 Hz, 2H, -OCH2-), 5.32 (s, 2H, -OCH2-), 6.39 (s, 1H, ArH), 6.49 (d, J = 9.7 Hz, 1H, ArH), 6.76 – 6.81 (m, 2H, ArH), 6.81-6.89 (m, 1H, ArH), 7.05 (s, 1H, ArH), 7.25 (s, 1H, TrzH), 7.62 – 7.77 (m, 3H, ArH+TrzH), 8.25 (d, J = 7.8, 1.5 Hz, 1H, ArH). 13C NMR (101 MHz, CDCl3, 25 oC) δ, ppm: 28.1, 28.2, 45.4, 45.4, 48.2, 48.4, 55.8, 55.8, 58.5, 62.7, 76.8, 77.2, 77.5, 101.6, 105.7, 113.8, 117.8, 123.7, 123.9, 129.1, 129.9, 129.9, 130.2, 130.3, 130.6, 131.5, 132.9, 134.4, 142.6, 158.9, 162.7, 165.3, 185.6. IR (KBr) ν max cm-1: 3422, 1718 (C=O), 1641, 1597, 1506, 1381, 1344, 1280, 1253, 1209, 1109, 1003, 852, 760. HRESI MS (m/z) [M + H]+: calcd. for C36H41N8O5+: 665.3200, found: 665.3200. (Fig. S2 in the Supporting Information).
(1-(3-(Dimethylamino)propyl)-1H-1,2,3-triazol-4-yl)methyl 2-(6-methoxy-3-oxo-3H-xanthen-9-yl)benzoate 9
Yield 0.4 g, 78 %. 1H NMR (400 MHz, CDCl3, 25 oC) δ, ppm: 1.98 (t, J = 6.9 Hz, 2H, -CH2-), 2.19 (m, 8H, 2* NCH3, -CH2-), 3.93 (s, 3H, -OCH3), 4.29 – 4.38 (m, 2H, -NCH2-), 5.14 (ABq, 2H, ΔδAB = 0.07, JAB = 12.5 Hz, 2H, -OCH2-), 6.40 (s, 1H, ArH), 6.49 (d, J = 9.8 Hz, 1H, ArH), 6.72 (dd, J = 8.9, 2.5 Hz, 1H, ArH), 6.80 – 6.89 (m, 2H, ArH), 6.84 (dd, J = 18.7, 9.3 Hz, 1H, ArH), 7.30 (d, J = 5.3 Hz, 2H, ArH + TrzH), 7.67 (t, J = 7.1 Hz, 1H, ArH), 7.74 (t, J = 6.8 Hz, 1H, ArH), 8.25 (d, J = 6.8 Hz, 1H, ArH). 13C NMR (101 MHz, CDCl3, 25 oC) δ, ppm: 28.1, 45.4, 48.1, 55.8, 56.1, 58.5, 100.4, 105.6, 113.5, 114.8, 117.5, 123.9, 128.9, 129.8, 130.2, 130.3, 130.5, 131.4, 132.9, 134.4, 141.4, 150.1, 154.3, 158.9, 164.2, 165.3, 185.5. IR (KBr) ν max cm-1: 3422, 2947, 1720 (C=O), 1643, 1599, 1514, 1464, 1381, 1346, 1280, 1267, 1255, 1211, 1107, 1076, 1026, 852, 758. HRESI MS (m/z) [M + H]+: calcd. for C29H29N4O5+: 513.2138, found: 513.2139. (Fig. S3 in the Supporting Information).
General procedure for the synthesis of ammonium salts 10ab, 11ab
To solution of 1,2,3-triazole (0.3 mmol) in 5 ml of fresh distilled MeCN was added alkylbromide (2 eq. in cases of 8 or 1 eq. in case of 9). The reaction mixture was stirred under reflux for 15-30 h and the completion of the reaction was determined by HRESI MS. After evaporation of solvent the product was concentrated under reduced pressure. The residue was purified by flesh chromatography (H2O:MeOH 100:0-0:100) to afford pure product.
N-(3-(4-(((2-(6-((1-(3-(butyldimethylammonio)propyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-oxo-3H-xanthen-9-yl)benzoyl)oxy)methyl)-1H-1,2,3-triazol-1-yl)propyl)-N,N-dimethylbutan-1-aminium bromide 10a
Was synthesized in 20 h. Yield 0.239 g, 85%.1H NMR (400 MHz, CDCl3, 25 oC) δ, ppm: 0.81 – 0.93 (m, 6H, 2*CH3), 1.10-1.17 (m, 2H, CH2), 1.26-1.36 (m, 4H, 2*- CH2), 1.52-1.63 (m, 2H, 2*- CH2), 1.88 (s, 10H, -CH2-), 2.33 – 2.37 (m, 2H, -CH2-), 2.42 – 2.51 (m, 2H, -CH2-), 3.08 (s, 6H, 2*N-CH3), 3.13 (s, 6H, 2*N-CH3), 3.18 – 3.23 (m, 2H, -CH2-), 3.59 (t, J = 5.1 Hz, 2H, -CH2-), 4.27 – 4.45 (m, 2H, -CH2Trz-), 4.49 – 4.55 (m, 2H, -CH2Trz-), 4.96 (ABq, 2H, ΔδAB = 0.07, JAB = 12.5 Hz, 2H, -OCH2-), 5.25 (s, 2H, -OCH2-), 6.23 (s, 1H, ArH), 6.33 (d, J = 9.7 Hz, 1H, ArH), 6.66 (d, J = 8.5 Hz, 1H, ArH), 6.71-6.79 (m, 2H, ArH), 7.03 (d, J = 2.4 Hz, 1H, ArH), 7.21 (d, J = 7.2 Hz, 2H, ArH), 7.35 (s, 1H, TrzH), 7.61 (t, J = 7.1 Hz, 1H, ArH), 7.65 (t, J = 7.8 Hz, 1H, ArH), 8.15 (d, J = 9.4 Hz, 1H, ArH), 8.38 (s, 1H, TrzH). 13C NMR (101 MHz, CDCl3, 25 oC) δ, ppm: 1.4, 1.6, 1.8, 1.9, 2.2, 2.4, 13.9, 14.5, 19.9, 23.0, 24.8, 29.7, 30.0, 32.2, 51.4, 58.4, 62.0, 65.0, 101.6, 105.5, 114.6, 115.3, 116.9, 117.8, 125.1, 129.4, 130.3, 130.7, 130.9, 131.7, 133.3, 134.1, 141.6, 142.8, 151.2, 154.5, 159.4, 163.2, 165.6, 185.7. IR (KBr) ν max cm-1: 3414, 2961, 2928, 1720 (C=O), 1641, 1597, 1502, 1464, 1380, 1346, 1282, 1253, 1211, 1111, 1043, 945. HRESI MS (m/z) [M]2+: calcd. for C44H58N8O52+: 389.2260, found: 389.2261. (Fig. S4 in the Supporting Information).
N-(3-(4-(((2-(6-((1-(3-(dimethyl(tetradecyl)ammonio)propyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-oxo-3H-xanthen-9-yl)benzoyl)oxy)methyl)-1H-1,2,3-triazol-1-yl)propyl)-N,N-dimethyltetradecan-1-aminium bromide 10b
Was synthesized in 30 h. Yield 0.270 g, 74%. 1H NMR (400 MHz, CDCl3, 25 oC) δ, ppm: 0.85 (t, J = 6.7 Hz, 6H, 2*CH3), 1.21 (s, 48H, -CH2-), 1.71 (s, 4H, -CH2-), 2.50 – 2.53 (m, 2H, -CH2-), 2. 58 – 2.69 (m, 2H), 3.30 (s, 6H, 2*N-CH3), 3.35 (s, 6H, 2*N-CH3), 3.39 – 3.49 (m, 4H, 2*-CH2-), 3.83 – 3.92 (m, 4H, 2*-CH2-), 4.49 – 4.62 (m, 2H, -CH2Trz-), 4.72 (t, J = 6.9 Hz, 2H, -CH2Trz-), 4.98 (ABq, 2H, ΔδAB = 0.1, JAB = 12.5 Hz, 2H, -OCH2-), 5.32- 5.35 (m, 2H, -OCH2-), 6.36 (s, 1H, ArH), 6.46 (d, J = 9.9 Hz, 1H, ArH), 6.76 (d, J = 9.0 Hz, 1H, ArH), 6.79-6.86 (m, 2H, ArH), 7.14 – 7.19 (m, 1H, ArH), 7.25 (s, 1H, TrzH), 7.49 (d, 1H, ArH), 7.63 – 7.76 (m, 2H, ArH), 8.23 (d, J = 7.8 Hz, 1H, ArH), 8.59 (s, 1H, TrzH). 13C NMR (101 MHz, CDCl3, 25 oC) δ, ppm: 14.2, 22.8, 22.9, 24.0, 24.1, 26.4, 26.6, 28.8, 29.3, 29.5, 29.6, 29.7, 29.8, 29.9, 32.0, 46.9, 47.2, 50.9, 51.3, 58.2, 61.7, 61.8, 62.5, 65.4, 101.5, 105.1, 114.8, 115.1, 117.5, 124.7, 125.8, 129.3, 130.1, 130.2, 130.3, 130.8, 131.6, 133.7, 141.2, 142.5, 151.6, 154.3, 158.9, 163.2, 165.3, 184.7. IR (KBr) ν max cm-1: 3418, 2924, 2880, 2853, 1720 (C=O), 1643, 1597, 1506, 1468, 1381, 1280, 1253, 1211, 1109, 758. HRESI MS (m/z) [M]2+: calcd. for C64H98N8O52+: 529.3825, found: 529.3882. (Fig. S5 in the Supporting Information).
N-(3-(4-(((2-(6-methoxy-3-oxo-3H-xanthen-9-yl)benzoyl)oxy)methyl)-1H-1,2,3-triazol-1-yl)propyl)-N,N-dimethylbutan-1-aminium bromide 11a
Was synthesized in 15 h. Yield 0.16 g, 79%. 1H NMR (400 MHz, DMSO-d6, 25 oC) δ, ppm: 0.92 (t, J = 7.3 Hz, 3H, CH3), 1.28 (h, J = 7.4 Hz, -СH2-), 1.59 (p, J = 8.3 Hz, 2H, -CH2-), 2.17 – 2.29 (m, 2H, -NCH2-), 3.01 (s, 6H), 3.21 – 3.29 (m, 4H, -CH2-), 3.92 (s, 3H, -OCH3), 4.36 (t, J = 7.1 Hz, 2H, -NCH2-), 5.07 (ABq, 2H, ΔδAB = 0.09, JAB = 12.5 Hz, 2H, -OCH2-), 6.19 (d, J = 1.9 Hz, 1H, ArH), 6.34 (d, J = 9.7Hz, 1H, ArH), 6.74 (d, J = 9.7 Hz, 1H, ArH), 6.82 (d, J = 8.9 Hz, 1H, ArH), 6.79 – 6.86 (m, 2H, ArH), 7.18 (d, J = 2.5 Hz, 1H, ArH), 7.49 (d, J = 7.5 Hz, 1H, ArH), 7.49 (t, J = 7.7 Hz, 1H, ArH), 7.87 (t, J = 7.7 Hz, 1H, ArH), 7.92 (s, 1H, TrzH), 8.18 (d, J = 9.3 Hz, 1H, ArH). 13C NMR (101 MHz, DMSO-d6, 25 oC) δ, ppm: 13.5, 19.1, 22.9, 23.6, 46.4, 50.2, 56.3, 57.9, 60.0, 62.9, 100.5, 104.5, 113.4, 114.3, 116.7, 124.8, 128.9, 129.3, 129.5, 130.1, 130.7, 133.3, 133.6, 140.8, 149.6, 153.6, 158.3, 163.9, 164.7, 183.8. IR (KBr) ν max cm-1: 3418, 2961, 1720 (C=O), 1643, 1599, 1508, 1479, 1383, 1346, 1282, 1255, 1215, 1109, 854, 760. HRESI MS (m/z) [M ]+: calcd. for for C33H37N4O5+: 569.2758, found: 569.2754. (Fig. S6 in the Supporting Information).
N-(3-(4-(((2-(6-methoxy-3-oxo-3H-xanthen-9-yl)benzoyl)oxy)methyl)-1H-1,2,3-triazol-1-yl)propyl)-N,N-dimethyltetradecan-1-aminium bromide 11b
Was synthesized in 30 h. Yield 0.194 g, 82%. 1H NMR (400 MHz, DMSO-d6, 25 oC) δ, ppm: 0.83 (t, J = 6.7 Hz, 3H, CH3), 1.23 (s, 24H, 12*-СH2-), 1.53 – 1.62 (m, 2H, -NCH2-), 2.18 – 2.28 (m, 2H, -NCH2-), 3.00 (s, 6H, -N(CH3)2-), 3.19 – 3.32 (m, 4H, -CH2-), 3.92 (s, 3H, -OCH3), 4.35 (t, J = 7.1 Hz, 2H, -NCH2-), 5.07 (ABq, 2H, ΔδAB = 0.09, JAB = 12.5 Hz, 2H, -OCH2-), 6.18 (d, J = 2.0 Hz, 1H, ArH), 6.33 (d, J = 9.7 Hz, 1H, ArH), 6.74 (d, J = 9.8 Hz, 1H, ArH), 6.79-6.91 (m, 2H, ArH), 7.18 (d, J = 2.4 Hz, 1H, ArH), 7.49 (d, J = 6.5 Hz, 1H, ArH), 7.78 (t, J = 6.5 Hz, 1H, ArH), 7.87 (t, J = 6.5 Hz, 1H, ArH), 7.91 (s, 1H, TrzH), 8.18 (d, J = 7.9 Hz, 1H, ArH). 13C NMR (101 MHz, DMSO-d6, 25 oC): δ, ppm: 13.9, 21.6, 22.1, 25.7, 28.5, 28.7, 28.8, 28.9, 29.0, 31.2, 46.4, 50.2, 56.3, 57.9, 60.0, 63.1, 100.5, 104.5, 113.4, 114.2, 116.7, 124.7, 128.8, 129.2, 129.5, 130.2, 130.6, 130.7, 133.3, 133.6, 140.8, 149.6, 153.6, 158.2, 163.9, 164.7, 183.8. IR (KBr) ν max cm-1: 3422, 2924, 2853, 1720 (C=O), 1643, 1599, 1512, 1466, 1381, 1346, 1280, 1255, 1213, 1107, 1024, 854, 758, 601. HRESI MS (m/z) [M]+: calcd. for C43H57N4O5+: 709.4323, found: 709.4324. (Fig. S7 in the Supporting Information).
2.2. Photophysical and Self-Assembly Investigations
UV-visible spectra were recorded using Shimadzu UV-2600 spectrophotometer equipped with Shimadzu TCC-100 thermostat (Shimadzu Corporation, Kyoto, Japan).
Fluorescence spectra were performed in 10.0 mm quartz cuvettes and recorded on a Fluorolog FL-221 spectrofluorimeter (HORIBA Jobin Yvon, Kyoto, Japan) using exci-tation wavelength 430 nm with 1 nm slit. All studies were conducted at 298 K.
DLS and ELS experiments were carried out on Zetasizer Nano ZS instrument (Malvern Panalytical, Worcestershire, UK) with 4 mW 633 nm He–Ne laser light source and the light scattering angle of 173°. The data were treated with DTS software (Dispersion Technology Software 5.00). The solutions were filtered through 0.8 μM filter before the measurements to remove dust. The experiments were carried out in the disposable plastic cells DTS 0012 (size) or in the disposable folded capillary cells DTS 1070 (zeta potential) (Sigma–Aldrich, USA) at 298K with at least three experiments for each system.
2.3. General Method for Photocatalytic Reaction
A solution of photocatalyst (0.25 μmol, 0.02 eq., 0.05 mL) in acetonitrile was added to N-phenyl-1,2,3,4-tetrahydroisoquinoline (12.5 μmol, 1 eq.) following by malonic ester (62.5 μmol, 5 eq.). Then, 0.2 mL of acetonitrile and 0.25 mL of bidistilled water (or 0,45 ml water) were also added. The exposure time by blue light irradiation was 5 hours. The progress of the reaction was monitored using gas chromatography-mass spectrometry (GC-MS). The reaction mixture was diluted to 1.5 mL with acetonitrile, so the concentration fell within the middle of the calibration curve (8.33 mmol). The final conversion was determined using 1H NMR spectroscopy.
GCMS analysis was performed on a GCMS-QP2010 Ultra gas chromatograph-mass spectrometer (Shimadzu, Kyoto, Japan) equipped with an HP-5MS column (the internal diameter was 0.32 mm and the length was 30 m). The parameters were as follows: Helium 99.995% purity was the carrier gas, the temperature of an injector was 250 °C, the flow rate through the column was 2 mL/min, the thermostat temperature program was a gradient temperature increase from 70 to 250 °C with a step of 10 °C/min. The range of scanned masses was m/z 35 - 400.
3. Results and Discussion
3.1. Synthesis of 1,2,3-Triazoles Containing Fragment of Fluorescein and Its Ammonium Derivatives
The copper-catalyzed cycloaddition of azides and alkynes is a versatile method for the synthesis of 1,4-disubstituted triazoles. For the synthesis of low-molecular-weight 1,2,3-triazoles, containing fragments of fluorescein, several precursors for azide-alkyne cycloaddition were prepared in the first step (Scheme 1). Fluorescein 1 was reacted with excess of propargyl bromide in the presence of K2CO3 to produce dipropargyl ester 2 according to a literature procedure in 98% yield [30]. The alkylation reaction of fluorescein 1 by methyl iodide in the presence of K2CO3 leads to the formation of dimethyl ester 3 with 89% yield [31]. Further hydrolysis using LiOH leads to the formation of monomethyl ester 4 in the form of a lactone form with a yield of 77% [32]. When ester 4 is introduced into reaction with propargyl bromide, the lactone form opens under the action of the base and the formation of ester 5 containing one triple bond occurs.
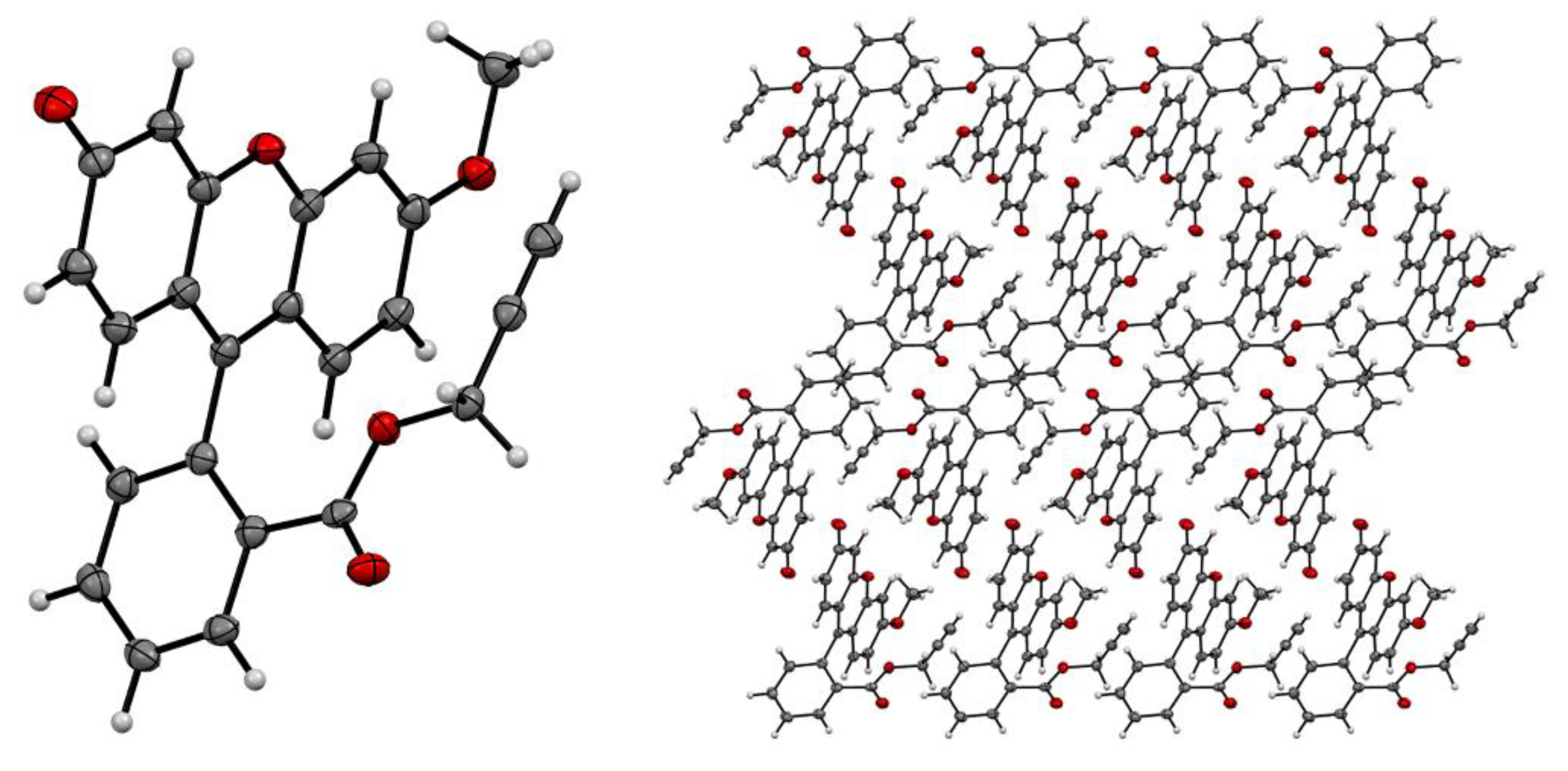
A mono-crystal was grown for compound 5, and the structure of the synthesized compound was confirmed by X-Ray crystallography data (Figure 1). Compound 5 was also characterized using a full range of physicochemical analysis methods, including NMR 1H, 13C, IR and high-resolution electrospray ionization mass spectrometry (HRESI MS).
The terminal proton of the triple bond resonates as a triplet at 2.33 ppm. As is the case with the dipropargyl derivative 2, the methylene protons in the ester group are diastereotopic and appear as an AB quadruplet at 4.59 ppm. This characteristic specificity allows for additional monitoring of the direction of alkylation reaction. Aromatic protons from the benzoate group appear in the weak magnetic field region as doublets at 8.27 ppm (J=7.8 Hz) and 7.34 ppm (J = 7.2 Hz) and two triplets at 7.76 ppm and 7.68 ppm with J = 7.4 and 7.6 Hz, respectively. The xanthene protons resonate as doublets at 6.96 ppm (J =2.4 Hz), 6.74 ppm (J =9.0 Hz), and 6.54 ppm (J =9.7 Hz), a singlet at 6.45 ppm, as well as two overlapping proton signals as a multiplet at the range 6.80 – 6.92 ppm. The composition of the compound 5 is also defined by HRESI MS: mono-protonated quasimolecular ion [M+H]+ was found with m/z 385.1080 (calcd. for C24H17O5+ 385.1076). There is an absorption band in IR spectrum corresponding to valence vibrations of C≡CH bond at 3294 cm⁻¹ (Fig.S1).
3-Сhloro-N,N-dimethylpropan-1-amine hydrochloride 6 was used as a precursor for the synthesis of azides. It’s heating in the presence of NaN3 led to the nucleophilic substitution of chlorine with the azide functional group. After adjusting the pH of the reaction mixture to 10 with KOH and extracting the product, 3-azido-N,N-dimethyl propane-1 amine 7 was obtained in a yield of 85% [33]. The presence of a tertiary amino group in the structure of azide 7 allows further obtaining quaternized derivatives that are more soluble in water.
The next step was the CuAAC reaction of acetylenes 2 or 5 with azide 7 in the presence of catalytic amounts of CuI in THF and triethylamine (NEt3) as a base (Scheme 2). After the filtration through Amberlite IRA-67 and evaporation, new 1,2,3-triazoles 8 and 9 were isolated as vitreous oils with good yields. In the 1H NMR spectrum of compound 8 there is the absence of signals of terminal acetylene protons at 2.33 ppm and 2.62 ppm. Also signals, related to N,N-dimethylaminopropyl groups are observed: two singlets of methyl protons at 2.18 and 2.20 ppm and methylene protons at 1.98, 2.07 and 2.23 – 2.29 ppm). Signals of methylene protons attached to nitrogen appear as a multiplet at 4.35 ppm and a triplet at 4.46 ppm with J = 7 Hz. Methylene linkers associated with oxygen are observed as a singlet at 5.32 ppm and AB-quadruplet from diastereotopic protons at 5.11 ppm. Triazoles protons appear as singlets at 7.25 and 7.72 ppm (Fig.S2).
According to the HRESI MS data, there is the presence of [M+H]+ quasimolecular ion (calcd. for C36H41N8O5+: 665.3200, found: 665.3200). The 1H NMR spectrum of compound 9 shows proton signals of the N,N-dimethylpropyl fragment and a proton signal of the new triazole as a singlet at 7.30 ppm. The diastereotopic protons of the methylene bridge between the fluorescein fragment and the triazole appear as an AB-quadruplet at 5.14. The methylene protons of the linker between the fluoresceins and the triazoles fragments appears as a multiplet at the range of 4.29 – 4.38 ppm. The formation of triazole 9 is also terminated by the presence of the [M+H]+ quasimolecular ion with m/z = 513.2139, which corresponds to the calculated for C29H29N4O5+ m/z= 513.2132 (Fig S3).
At the next step obtained 1,2,3-triazole derivates 8 and 9 were introduced into the quaternization reaction with 1 eq. of C4H9Br or C14H29Br per 1 amino group. The competiton of reaction was monitored by HRESI MS. The reaction took approximately 15–20 hours for monosubstituted and 20–30 hours for disubstituted fluorescein derivates. After the evaporation of the solvent and dispersion in hexane compounds 10ab and 11ab were obtained as viscous oil with yields from 74% to 85%. The ammonium salts 11ab were additionally purified using flash chromatography. On the 1H NMR spectrum of dibutyl fluoresceine derivate 10a appears signals, related to alkyl group as triplets of methyl protons at 0.87 ppm and singlet of methylens protons at 1.88 ppm. Methylene protons, attached to quaternary nitrogen, resonates as the broad signals at 3.28 and 3.59 ppm. The composition of 10a was well-defined by HRESI MS: the double charged [M - 2Br]2+ quasimolecular ion with m/z 389.2261 (calcd. for C44H58N8O52+: 389.2260) was found (Fig. S4). 1H NMR spectrum of 10b contains new signals of the alkyl substitute as a triplet at 0.86 ppm, a singlet at 1.21 ppm and the multiplets at 3.28 and 3.59 ppm. According to the HRESI MS data, there is a presence of quasimolecular ion [M - 2Br]2+ m/z 529.3882 (calcd. for C64H98N8O52+ m/z 529.3825) (Fig. S5). According to the 1H NMR spectra of monoquaternized fluorescein derivatives (11ab), the protons of the methyl moiety are evident at 0.92 0.83 ppm, the methylene protons resonate at 1.28, 1.59 ppm for (11a), at 1.23 ppm for (11b), respectively. In the mass-spectrum of compound 11a quasimolecular ion [M-Br]+ with m/z 569.2754 (calcd. for C33H37N4O5+ m/z 569.2758) was found; and for 11b - [M-Br]+ with m/z 709.4324 (calcd. for C43H57N4O5+ m/z 709.4323) (Fig. S6-S7).
3.2. Photophysical and Self Assembly Properties
The next step was the study of some photophysical properties of the synthesized compounds. It was found that fluorescein derivates 10-11, which contain one or two alkyl groups, exhibited fluorescent properties in the green range of the visible light spectrum. The change of the fluorescence intensity with respect to concentration was studied. The spectra of all compounds exhibited a similar profile with a maximum at λmax =513 nm and a shoulder at λ=554 nm. In the case of compound 11b with two long hydrophobic groups the maximum was red-shifted to λmax =525 nm (Figure 2A) in regard to fluorescein. And vice versa, in the absorption spectra of 10-11, the absorption maximum shifts bathochromically in the regard to fluorescein and two maxima were observed at λmax =458 and 486 nm, with a shoulder that presents at λ=428 nm (Figure 2B). In the case of compound 10b, as well as in the emission spectra, a bathochromic shift at 4 nm is observed. Such a bachochromic shift indicates a change in the polarity of the surroundings and is consistent with the solvatochromic behavior of fluorescein upon transfer to nonpolar solvents[34], indirectly indicating the formation of aggregates.
One of the most significant photophysical properties of fluorophore molecules is the fluorescent quantum yield. There are two methods for determining the quantum yield: absolute and relative[35]. In contrast to the absolute quantum yield, which requires the use of an integrating sphere, to determine relative yields only emission and absorption spectra are used. So the next step was the calculating the relative quantum yield for compounds 10ab-11ab. We used fluorescein in a 0.1 M NaOH solution for comparison purposes with the known quantum yield is 95%. [36]. For all obtained compounds 10ab-11ab in the concentration range of 0.01-0.05 mM, the Lambert-Bouguer-Beer law applies and there is a linear correlation between absorption intensity and concentration. Relative quantum yield was calculated using emission and absorption spectra of fluorescein derivates 10ab-11ab and formula (1).
where Qs is the fluorescence quantum yield of an unknown sample, and Qr is the fluorescence quantum yield of a known standard, ms and mr are the gradients of the integrated fluorescence intensity graphs depending on absorption, ns and nr are the refractive indexes of solvents used for an unknown sample and a reference, respectively[37].

Table 1.
Relative quantum yield for compounds 10ab-11ab against fluorescein disodium salt.1.
| System |
, % (water with 1% DMF) |
|---|---|
| 10a | 16 |
| 10b | 5 |
| 11a | 18 |
| 11b | 24 |
1Emission and absorption spectra were recorded in 10 mm quartz cell in the range of 0.01-0.1mM.
For the fluorescein derivatives 10ab-11ab, was found a significant decrease in relative quantum yield compared to disodium salt of fluorescein. It has been shown that the highest quantum yield is observed for monosubstituted derivative with single tetradecyl fragment 11b. Presence of two hydrophobic substituents leads to decrease in quantum yield due to aggregation in solution. The lowest quantum yield has compound 10a, which is also characterized by a small bathochromic shift in absorption and emission spectra. The correlation between fluorescence intensity and concentration of obtained derivates 10ab, 11ab was also studied. It is worth noting that, as the concentration increases, a significant increase in the fluorescence intensity was observed. This may be due to the formation of associates, which is accompanied by a decrease in polarity. Furthermore, further decreases in fluorescence intensity could be associated with quenching at high concentrations, where the decreased and quenched luminescence is associated with an increased number of collisions between particles in solution. (Figure 3). Interestingly, for all four compounds, the maximum emission intensity occurred at a CI max = 0.05 mM. The highest relative emission intensity was typically observed for the monosubstituted derivative 11b with one tetradecyl group.
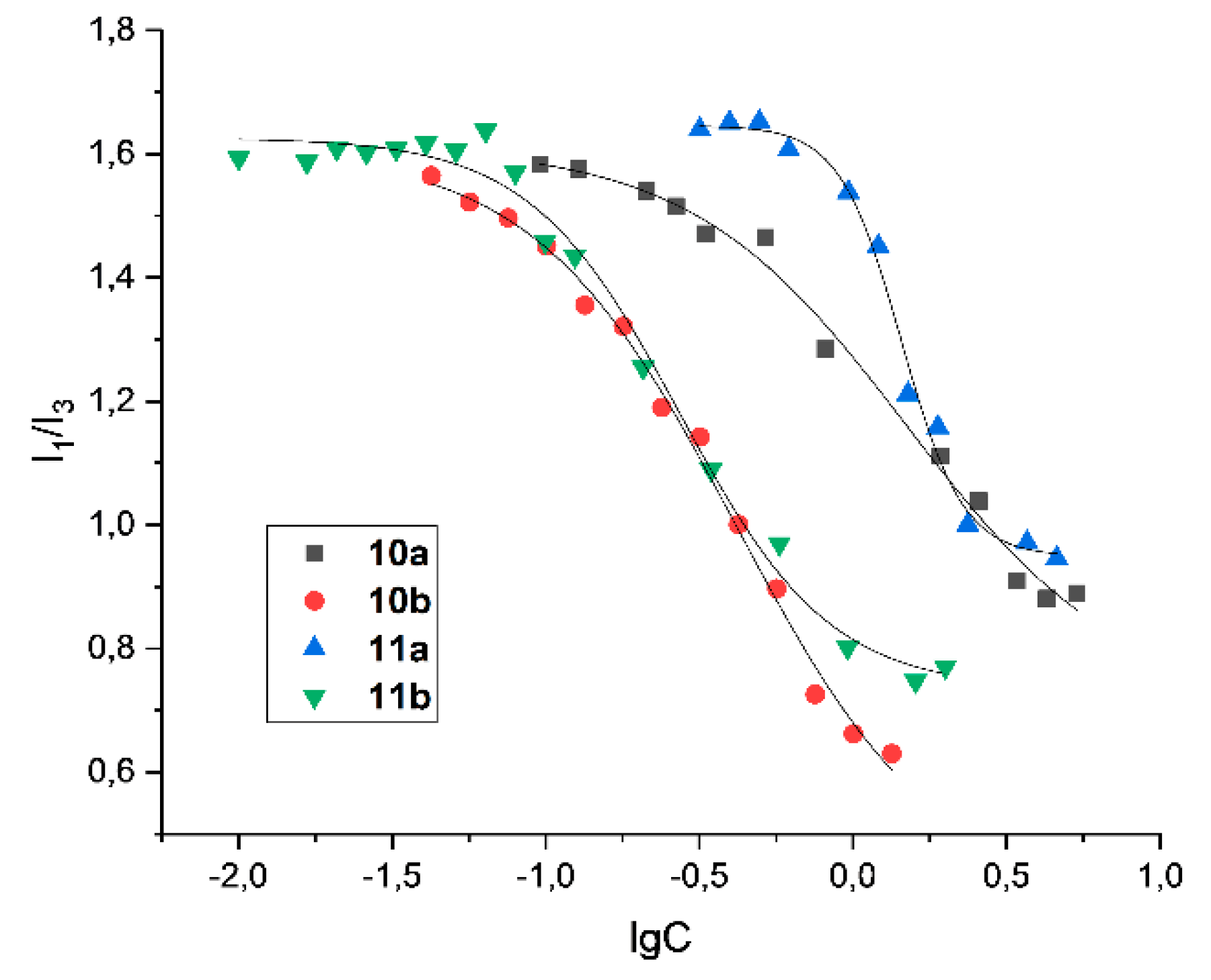
The presence of a positively charged ammonium fragment and a long hydrophobic alkyl substituent in the structure of compounds 10ab, 11ab creates the prerequisites for aggregation of particles in solution. To confirm the formation of aggregates, it was necessary to determine the critical aggregation concentration (CAC) values using the pyrene fluorescent probe [38]. CAC was determined as the value at which a change in slope was observed in the corresponding graphs of the intensity ratio between the first (373 nm) and third (383 nm) peaks in the pyrene emission spectrum (Figure 4). The polarity of pyrene decreased because of its solubilization in the hydrophobic part of the aggregates. Less lipophilic derivatives 10a and 11a with one/two butyl fragments showed higher CAC values (1.43 and 0.82 mM) than the corresponding tetradecyl-containing compounds 10b and 11b (0.43 and 0.28 mM). Surprisingly, in contrast to classical hemini surfactants, which are characterized by a decrease in CAC compared to their single-charged analogs [39], in this case the introduction of two lipophilic fragments leads to an increase in CAC. Such an increase can be attributed to the high rigidity of the fluorescein spacer, which prevents effective hydrophobic interactions between the alkyl moieties.
On the next step was performed an experiment on dynamic light scattering to confirm the formation of aggregates. At concentrations below CAC, aqueous solutions of compounds 10-11 formed disordered aggregates with a high polydispersity index (PDI). Above the critical concentration, all systems formed sub-micron aggregates (Table 2). The lower PDI was found for 10a. The largest size and polydispersity index were found for 10b with two tetradecyl substituents.
3.3. Photophysical and Self Assembly Properties
Photocatalysis has become a versatile tool for C-H activation reactions of sp3-hybridized carbon atoms, allowing direct and selective C-C bond formation [40,41]. The use of photocatalysis allows the easy generation of highly reactive iminium cations, which has been used previously in the coupling reactions of N-phenyl-1,2,3,4-tetrahydroisoquinoline (THI) with various carbon-containing nucleophiles [42]. Due to their unique photophysical characteristics, fluorescein and its derivatives have also become widely used as photoredox catalysts [43,44], including for reactions involving iminium ions [45]. In our study, the photocatalytical activity of amphiphilic 1,2,3-triazole derivatives of fluorescein in the model reaction of malonic ether to THI was studied (Scheme 3). The photocatalytic activity of fluorescein derivatives was carried out in pure acetonitrile and an acetonitrile-water mixtures. The reaction mixture was irradiated for 5 hours at room temperature using 24w blue led photoreactor (Fig. S8).
The conversion of the initial THI was evaluated by gas chromatography–mass spectrometry (GC-MS), using an absolute calibration method (Fig. S9). The method of reaction estimation by the product calibration line was not used because two peaks were recorded in the GC-MS spectra: the product peak (m/z = 367) and the peak corresponding to the product decomposition (m /z = 208). The ratio of these peaks varied, which can be explained by the fact that the product decomposed during evaporation in the injector. In addition to GC-MS, product yield was estimated from the integrated intensity of the TGI (product) CH proton signal at 5.57 ppm relative to the TGI (reagent) CH2 proton signal at 4.27 ppm (Fig. S10).
Table 3.
THI conversion (GCMS) or product yield (1H-NMR) in the reaction of THI in with malonic ester in the presence of 0.02 equiv. of 10ab, 11ab.
Table 3.
THI conversion (GCMS) or product yield (1H-NMR) in the reaction of THI in with malonic ester in the presence of 0.02 equiv. of 10ab, 11ab.
| 10a | 10b | 11a | 11b | |||||
|---|---|---|---|---|---|---|---|---|
| GCMS, % | 1H-NMR % | GCMS, % | 1H-NMR % | GCMS, % | 1H-NMR % | GCMS, % | 1H-NMR % | |
| MeCN | 87 | 94 | 86 | 93 | 86 | 91 | 84 | 87 |
| MeCN:Water, 50:50 | 87 | 95 | 86 | 94 | 87 | 91 | 87 | 89 |
| MeCN:Water, 10:90 | 92 | 94 | 84 | 90 | 66 | 71 | 95 | 100 |
*12.5 µmol THI, 0.25 µmol of catalyst, 62.5 µmol malonic ester, blue LED.
In all cases, the product yield by 1H NMR was higher than the conversion by GC-MS on average by 5%, which is due to the lower sensitivity of the NMR method. When the reaction was carried out in pure acetonitrile, the GC-MS conversion did not reach 90%. Similar data were obtained at 50% water content. At transition to 90% water content photocatalysts began to behave differently - the lowest conversion was demonstrated by 11a, and its analog 11b having tetradecyl fragment, on the contrary, was the best, demonstrating 95% conversion by GC-MS and 100% product formation on 1H NMR. The data obtained are consistent with the fact that 11b has the highest quantum yield in the series. Also 11b has the lowest CAC value in the series, a positive contribution of micellar catalysis [46] in this case is highly probable.
4. Conclusions
Thus, new 1,2,3-triazole derivatives of fluorescein containing one or two fragments of tertiary amines and their ammonium salts were synthesized for the first time. The structures of these compounds were confirmed by a wide range of physical and chemical methods. The relative quantum yields were calculated using emission and ultraviolet absorption spectra data. Values of the critical aggregation concentrations were obtained for ammonium salts. Formation of aggregates was also confirmed by dynamic light scattering experiments. It has been shown that the presence of one or two hydrophobic tetradecyl fragments in the structure of a fluorescein derivative leads to an increase in the size of aggregates. The smallest size of aggregates and polydispersity index were recorded for a derivative with two butyl substituents in the structure. The study of photocatalytic activity using the model coupling reaction of malonic ester with N-phenyl 1,2,3,4-tetrahydroisoquinoline under the blue LED light, showed good efficiency for all derivatives. However, the conversion of initial THI reached 100% when compound 11b, containing a tetradecyl substituent, was used. These data confirmed the calculation of quantum yield, which gave the best value for compound 11b.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: NMR 1H (a), 13C (b), FT IR (c) and HRESI MS (d) spectra of prop-2-yn-1-yl 2-(6-methoxy-3-oxo-3H-xanthen-9-yl)benzoate (5); Figure S2: NMR 1H (a), 13C (b), FT IR (c) and HRESI MS (d) spectra of (1-(3-(dimethylamino)propyl)-1H-1,2,3-triazol-4-yl)methyl 2-(6-((1-(3-(dimethylamino)propyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-oxo-3H-xanthen-9-yl)benzoate (8); Figure S3: NMR 1H (a), 13C (b), FT IR (c) and HRESI MS (d) spectra of (1-(3-(dimethylamino)propyl)-1H-1,2,3-triazol-4-yl)methyl 2-(6-methoxy-3-oxo-3H-xanthen-9-yl)benzoate (9); Figure S4: NMR 1H (a), 13C (b), FT IR (c) and HRESI MS (d) spectra of N-(3-(4-(((2-(6-((1-(3-(butyldimethylammonio)propyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-oxo-3H-xanthen-9-yl)benzoyl)oxy)methyl)-1H-1,2,3-triazol-1-yl)propyl)-N,N-dimethylbutan-1-aminium bromide (10a); Figure S5: NMR 1H (a), 13C (b), FT IR (c) and HRESI MS (d) spectra of N-(3-(4-(((2-(6-((1-(3-(dimethyl(tetradecyl)ammonio)propyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-oxo-3H-xanthen-9-yl)benzoyl)oxy)methyl)-1H-1,2,3-triazol-1-yl)propyl)-N,N-dimethyltetradecan-1-aminium bromide (10b); Figure S6: NMR 1H (a), 13C (b), FT IR (c) and HRESI MS (d) spectra of N-(3-(4-(((2-(6-methoxy-3-oxo-3H-xanthen-9-yl)benzoyl)oxy)methyl)-1H-1,2,3-triazol-1-yl)propyl)-N,N-dimethylbutan-1-aminium bromide (11a); Figure S7: NMR 1H (a), 13C (b), FT IR (c) and HRESI MS (d) spectra of N-(3-(4-(((2-(6-methoxy-3-oxo-3H-xanthen-9-yl)benzoyl)oxy)methyl)-1H-1,2,3-triazol-1-yl)propyl)-N,N-dimethyltetradecan-1-aminium bromide (11b); Figure S8: Photo of a 24W LED air-cooled photoreactor; Figure S9: Plot of the GC-MS peak area of N-phenyl-1,2,3,4-tetrahydroisoquinoline vs its concentration; Figure S9: 1H NMR spectrum of the reaction of THI with malonic ester in the presence of 11b (CDCl3).
Author Contributions
Conceptualization, V.B., I.A. and S.E., methodology, V.B. and E.D.; investigation, A.A., A.A., E.B., D.I., K.U., D.M.; resources, I.A.; data curation, , V.B., D.I. and E.D; writing—original draft preparation, A.A., A.A.; writing—review and editing, V.B.; visualization, V.B.; supervision, V.B.; project administration, V.B.; funding acquisition, V.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, grant number 21-73-10062”.
Data Availability Statement
Data is contained within the article or supplementary material.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ardila-Fierro, K.J.; Hernández, J.G. Sustainability Assessment of Mechanochemistry by Using the Twelve Principles of Green Chemistry. ChemSusChem 2021, 14, 2145–2162. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.N.; Zhang, D.; Miller, S.J.; Rossen, K.; Chirik, P.J.; Kozlowski, M.C.; Zimmerman, J.B.; Brooks, B.W.; Savage, P.E.; Allen, D.T.; et al. Green Chemistry: A Framework for a Sustainable Future. Org. Process Res. Dev. 2021, 25, 1455–1459. [Google Scholar] [CrossRef]
- Bobo, M.V.; Kuchta, J.J.; Vannucci, A.K. Recent advancements in the development of molecular organic photocatalysts. Org. Biomol. Chem. 2021, 19, 4816–4834. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shi, W. Recent developments in visible-light photocatalytic degradation of antibiotics. Cuihua Xuebao/Chinese J. Catal. 2016, 37, 792–799. [Google Scholar] [CrossRef]
- Koe, W.S.; Lee, J.W.; Chong, W.C.; Pang, Y.L.; Sim, L.C. An overview of photocatalytic degradation: photocatalysts, mechanisms, and development of photocatalytic membrane. Environ. Sci. Pollut. Res. 2020, 27, 2522–2565. [Google Scholar] [CrossRef]
- Kumari, H.; Sonia; Suman; Ranga, R.; Chahal, S.; Devi, S.; Sharma, S.; Kumar, S.; Kumar, P.; Kumar, S.; et al. A Review on Photocatalysis Used For Wastewater Treatment: Dye Degradation; Springer International Publishing, 2023; Vol. 234; ISBN 0123456789. [CrossRef]
- Hari, D.P.; Hering, T.; König, B. Visible light photocatalytic synthesis of benzothiophenes. Org. Lett. 2012, 14, 5334–5337. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Wang, W.; Yue, J.-P.; Jiang, Y.-X.; Wei, M.-K.; Zhang, H.-P.; Yan, S.-S.; Liao, L.-L.; Yu, D. Visible-light photocatalytic di- and hydro-carboxylation of unactivated alkenes with CO2. Nat. Catal. 2022, 5, 1–7. [Google Scholar] [CrossRef]
- Dissanayake, K.C.; Ebukuyo, P.O.; Dhahir, Y.J.; Wheeler, K.; He, H. A BODIPY-functionalized PdII photoredox catalyst for Sonogashira C-C cross-coupling reactions. Chem. Commun. 2019, 55, 4973–4976. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Yang, J.; Yang, B.; Han, Y.; Zhu, L.; Xue, X.S.; Zhu, F. Photoredox Nickel-Catalysed Stille Cross-Coupling Reactions. Angew. Chemie - Int. Ed. 2023, 62. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Lei, T.; Wei, X.Z.; Ye, C.; Liu, Z.; Chen, B.; Tung, C.H.; Wu, L.Z. Metal-free, redox-neutral, site-selective access to heteroarylamine via direct radical−radical cross-coupling powered by visible light photocatalysis. J. Am. Chem. Soc. 2020, 142, 16805–16813. [Google Scholar] [CrossRef] [PubMed]
- Jati, A.; Dey, K.; Nurhuda, M.; Addicoat, M.A.; Banerjee, R.; Maji, B. Dual Metalation in a Two-Dimensional Covalent Organic Framework for Photocatalytic C-N Cross-Coupling Reactions. J. Am. Chem. Soc. 2022, 144, 7822–7833. [Google Scholar] [CrossRef] [PubMed]
- Zavalishin, M.N.; Gamov, G.A.; Kiselev, A.N.; Nikitin, G.A. A fluorescein conjugate as colorimetric and red-emissive fluorescence chemosensor for selective recognition Cu2+ ions. Opt. Mater. (Amst). 2024, 153, 115580–10. [Google Scholar] [CrossRef]
- Wagay, S.A.; Alam, M.; Ali, R. Synthesis of two novel fluorescein appended dipyromethanes (DPMs): Naked-eye chemosensors for fluoride, acetate and phosphate anions. J. Mol. Struct. 2023, 1291, 135982–10. [Google Scholar] [CrossRef]
- Keerthana, S.; Sam, B.; George, L.; Sudhakar, Y.N.; Varghese, A. Fluorescein Based Fluorescence Sensors for the Selective Sensing of Various Analytes. J. Fluoresc. 2021, 31, 1251–1276. [Google Scholar]
- Li, Z.; Song, H.; Guo, R.; Zuo, M.; Hou, C.; Sun, S.; He, X.; Sun, Z.; Chu, W. Visible-light-induced condensation cyclization to synthesize benzimidazoles using fluorescein as a photocatalyst. Green Chem. 2019, 21, 3602–3605. [Google Scholar] [CrossRef]
- Tu, X.P.; Wei, L.L.; Zhang, K.X.; Chen, Y.; Zhou, M.D. Synthesis of fluorescein-containing polymeric heterogeneous photocatalyst and its applications. Tetrahedron 2024, 160, 134028–10. [Google Scholar] [CrossRef]
- Sun, W.; Chen, H.; Wang, K.; Wang, X.; Lei, M.; Liu, C.; Zhong, Q. Synthesis of benzothiazoles using fluorescein as an efficient photocatalyst under visible light. Mol. Catal. 2021, 510, 111693–10. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Zhou, Z.; Zhou, R.; Sun, Q.; Wu, P. Halo-fluorescein for photodynamic bacteria inactivation in extremely acidic conditions. Nat. Commun. 2021, 12, 526–10. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.; Llanos, A.; Gomila, R.M.; Frontera, A.; Rodríguez, L. Ligand and Gold(I) Fluorescein-AIEgens as Photosensitizers in Solution and Doped Polymers. Inorg. Chem. 2023, 62, 7131–7140. [Google Scholar] [CrossRef] [PubMed]
- Gaigalas, A. Measurement of the Fluorescence Quantum Yield Using a Spectrometer With an Integrating Sphere Detector. J. Res. Natl. Inst. Stand. Technol. 2008, 113. [Google Scholar] [CrossRef] [PubMed]
- Rajasekar, M. Recent development in fluorescein derivatives. J. Mol. Struct. 2021, 1224, 129085–10. [Google Scholar] [CrossRef]
- Russo, C.; Brunelli, F.; Tron, G.C.; Giustiniano, M. Visible-Light Photoredox Catalysis in Water. J. Org. Chem. 2023, 88, 6284–6293. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Clerget, M.; Yu, J.; Kincaid, J.R.A.; Walde, P.; Gallou, F.; Lipshutz, B.H. Water as the reaction medium in organic chemistry: From our worst enemy to our best friend. Chem. Sci. 2021, 12, 4237–4266. [Google Scholar] [CrossRef] [PubMed]
- Pallini, F.; Sangalli, E.; Sassi, M.; Roth, P.M.C.; Mattiello, S.; Beverina, L. Selective photoredox direct arylations of aryl bromides in water in a microfluidic reactor. Org. Biomol. Chem. 2021, 19, 3016–3023. [Google Scholar] [CrossRef] [PubMed]
- Artemenko, A.A.; Burilov, V.A.; Solov’eva, S.E.; Antipin, I.S. Covalent and Supramolecular Conjugates of Calixarenes with Some Fluorescent Dyes of the Xanthene Series. Colloid J. 2022, 84, 563–580. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A, Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. A Short History of ShelX. Acta Crystallogr. A. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.; Edgington, P.; McCabe, P.; Pidcock, E.; Shields, G.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. - J APPL CRYST 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Berscheid, R.; Nieger, M.; Vögtle, F. Konkave Farbstoffmoleküle vom Triphenylmethan-Typ. Chem. Ber. 1992, 125, 2539–2552. [Google Scholar] [CrossRef]
- Xiang, Y.; He, B.; Li, X.; Zhu, Q. The design and synthesis of novel “turn-on” fluorescent probes to visualize monoamine oxidase-B in living cells. RSC Adv. 2013, 3, 4876–4879. [Google Scholar] [CrossRef]
- Du, L.; Risinger, A.L.; Yee, S.S.; Ola, A.R.B.; Zammiello, C.L.; Cichewicz, R.H.; Mooberry, S.L. Identification of C-6 as a New Site for Linker Conjugation to the Taccalonolide Microtubule Stabilizers. J. Nat. Prod. 2019, 82, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Carboni, B.; Benalil, A.; Vaultier, M. Aliphatic amino azides as key building blocks for efficient polyamine syntheses. J. Org. Chem. 1993, 58, 3736–3741. [Google Scholar] [CrossRef]
- Cheptea, C.; Zara, A.; Ambrosi, E.; Morosanu, A.C.; Diaconu, M.; Miron, M.; Dorohoi, D.O.; Dimitriu, D.G. On the Solvatochromism of Fluorescein Sodium. Symmetry (Basel). 2024, 16, 673–10. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of fluorescence spectroscopy, 3rd Principles of fluorescence spectroscopy, Springer, New York, USA, 3rd edn, 2006.; 2006; ISBN 0387312781. 10.1007/978-0-387-46312-4.
- Shen, J.; Snook, R.D. Thermal lens measurement of absolute quantum yields using quenched fluorescent samples as references. Chem. Phys. Lett. 1989, 155, 583–586. [Google Scholar] [CrossRef]
- Kandi, D.; Mansingh, S.; Behera, A.; Parida, K. Calculation of relative fluorescence quantum yield and Urbach energy of colloidal CdS QDs in various easily accessible solvents. J. Lumin. 2021, 231, 117792. [Google Scholar] [CrossRef]
- Piñeiro, L.; Novo, M.; Al-Soufi, W. Fluorescence emission of pyrene in surfactant solutions. Adv. Colloid Interface Sci. 2015, 215, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vasileva, L.; Gaynanova, G.; Valeeva, F.; Romanova, E.; Pavlov, R.; Kuznetsov, D.; Belyaev, G.; Zueva, I.; Lyubina, A.; Voloshina, A.; et al. Synthesis, Properties, and Biomedical Application of Dicationic Gemini Surfactants with Dodecane Spacer and Carbamate Fragments. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Dumur, F.; Lalevée, J. Recent Advances in Photoredox Catalysts. Catalysts 2024, 14, 1–5. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Franz, J.F.; Kraus, W.B.; Zeitler, K. No photocatalyst required-versatile, visible light mediated transformations with polyhalomethanes. Chem. Commun. 2015, 51, 8280–8283. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.O.; Jung, Y.J.; Kim, M.; Kim, H.; Li, J.; Ko, H.; Lee, H.I.; Lee, H.J.; Lee, J.K. Substituent Effects of Fluorescein on Photoredox Initiating Performance under Visible Light. ACS Omega 2023, 8, 40277–40286. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Yadav, R.K.; Kim, T.W.; Kumar, A.; Dwivedi, D.K. Chitosan-based fluorescein isothiocyanate film as a highly efficient metal-free photocatalyst for solar-light-mediated direct C-H arylation. Int. J. Energy Res. 2021, 45, 5964–5973. [Google Scholar] [CrossRef]
- Hari, D.P.; König, B. Eosin Y Catalyzed Visible Light Oxidative C–C and C–P bond Formation. Org. Lett. 2011, 13, 3852–3855. [Google Scholar] [CrossRef] [PubMed]
- Acharjee, A.; Rakshit, A.; Chowdhury, S.; Saha, B. Micelle catalysed conversion of ‘on water’ reactions into ‘in water’ one. J. Mol. Liq. 2021, 321, 114897–10. [Google Scholar] [CrossRef]
Scheme 1.
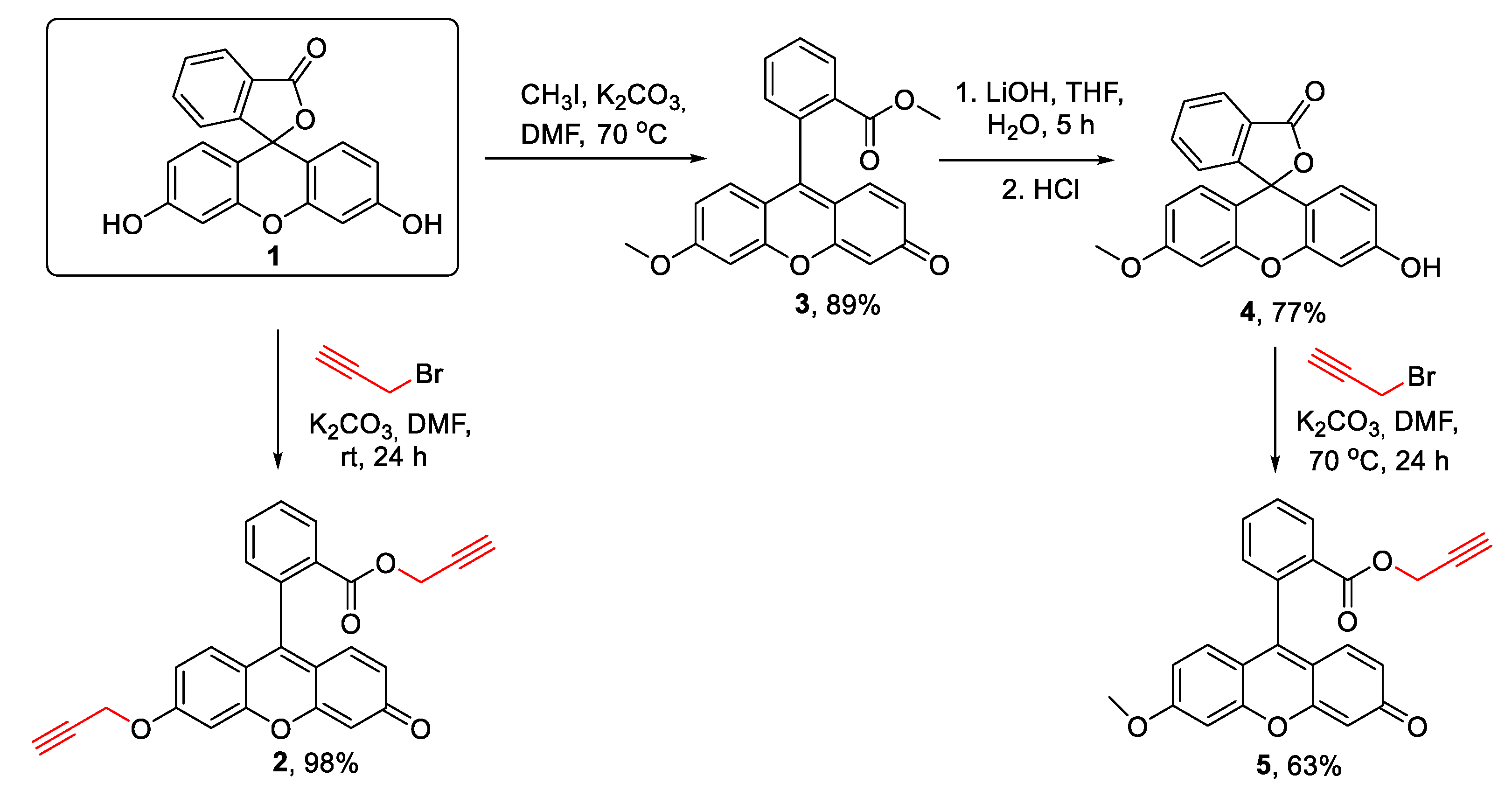
Figure 1.
ORTEP representation of 5 showing 50% probability thermal ellipsoids and crystal packing of 5, view along b axes. C atoms – grey, O atoms – red.
Figure 1.
ORTEP representation of 5 showing 50% probability thermal ellipsoids and crystal packing of 5, view along b axes. C atoms – grey, O atoms – red.
Scheme 2.
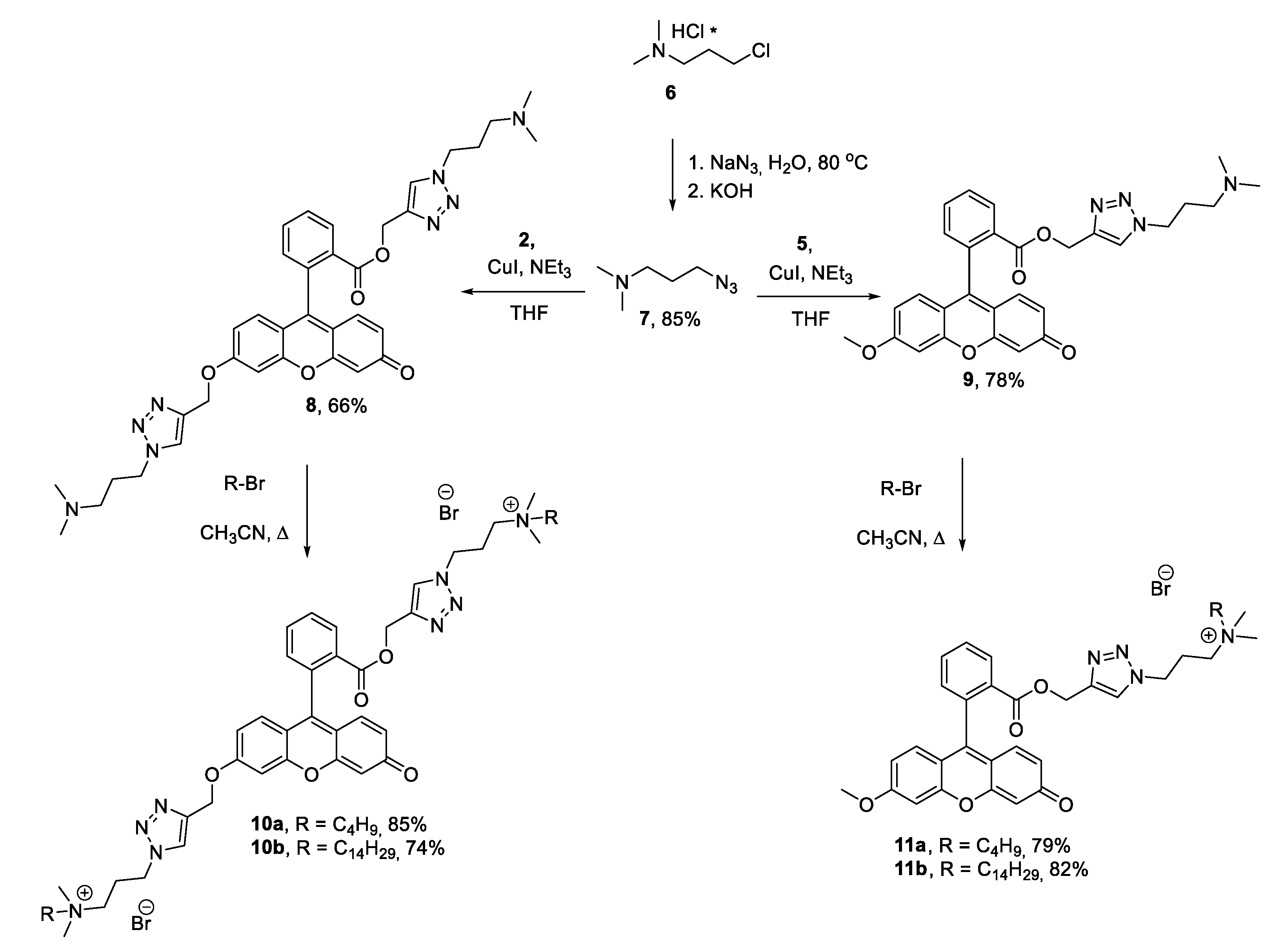
Figure 2.
Emission (A) and absorbtion (B) spectra of fluorescein (0.1 M NaOH) and derivates 10ab, 11ab in water, C=0.01 mM.
Figure 2.
Emission (A) and absorbtion (B) spectra of fluorescein (0.1 M NaOH) and derivates 10ab, 11ab in water, C=0.01 mM.
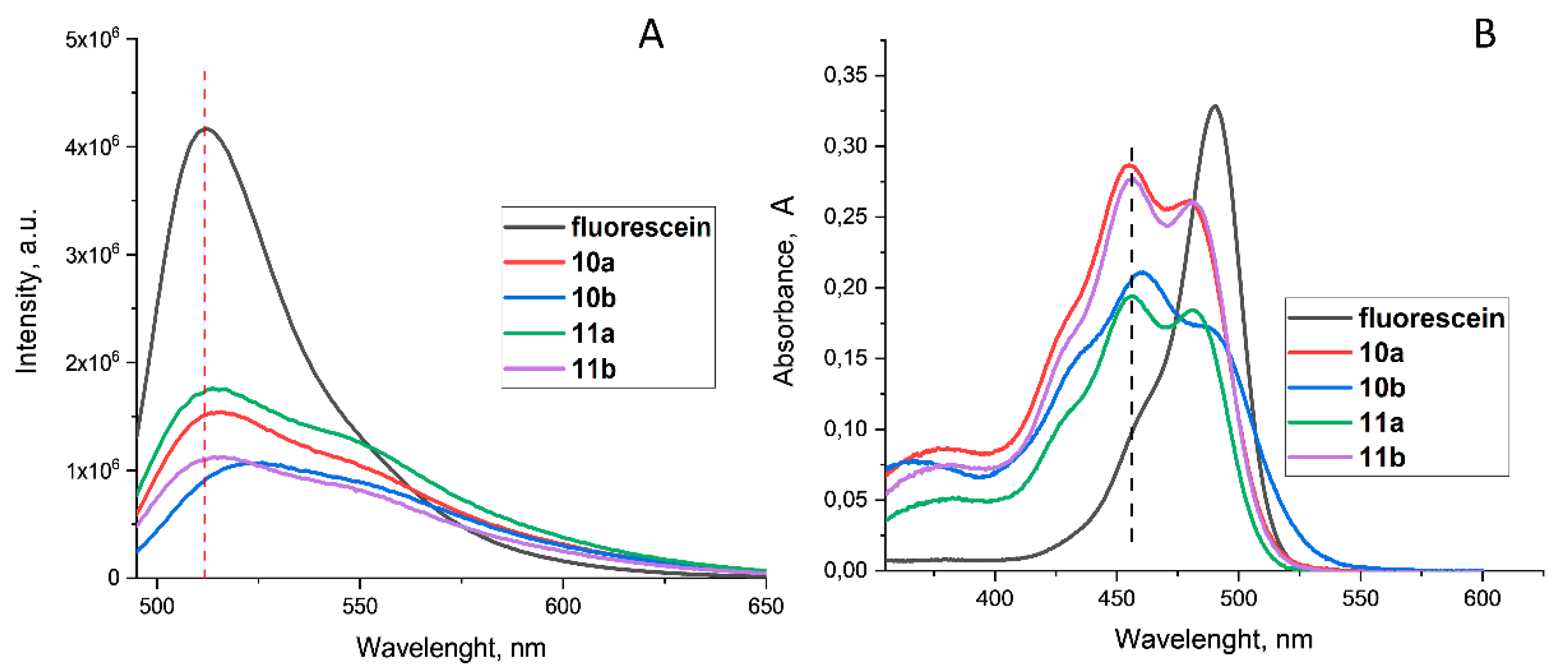
Figure 3.
The dependence of the fluorescence intensity of compounds (10ab-11ab) on the concentration at λ= 518 nm. Concentration range: 0.08 – 1 mM, λex = 430 nm, H2O, 25 oC.
Figure 3.
The dependence of the fluorescence intensity of compounds (10ab-11ab) on the concentration at λ= 518 nm. Concentration range: 0.08 – 1 mM, λex = 430 nm, H2O, 25 oC.
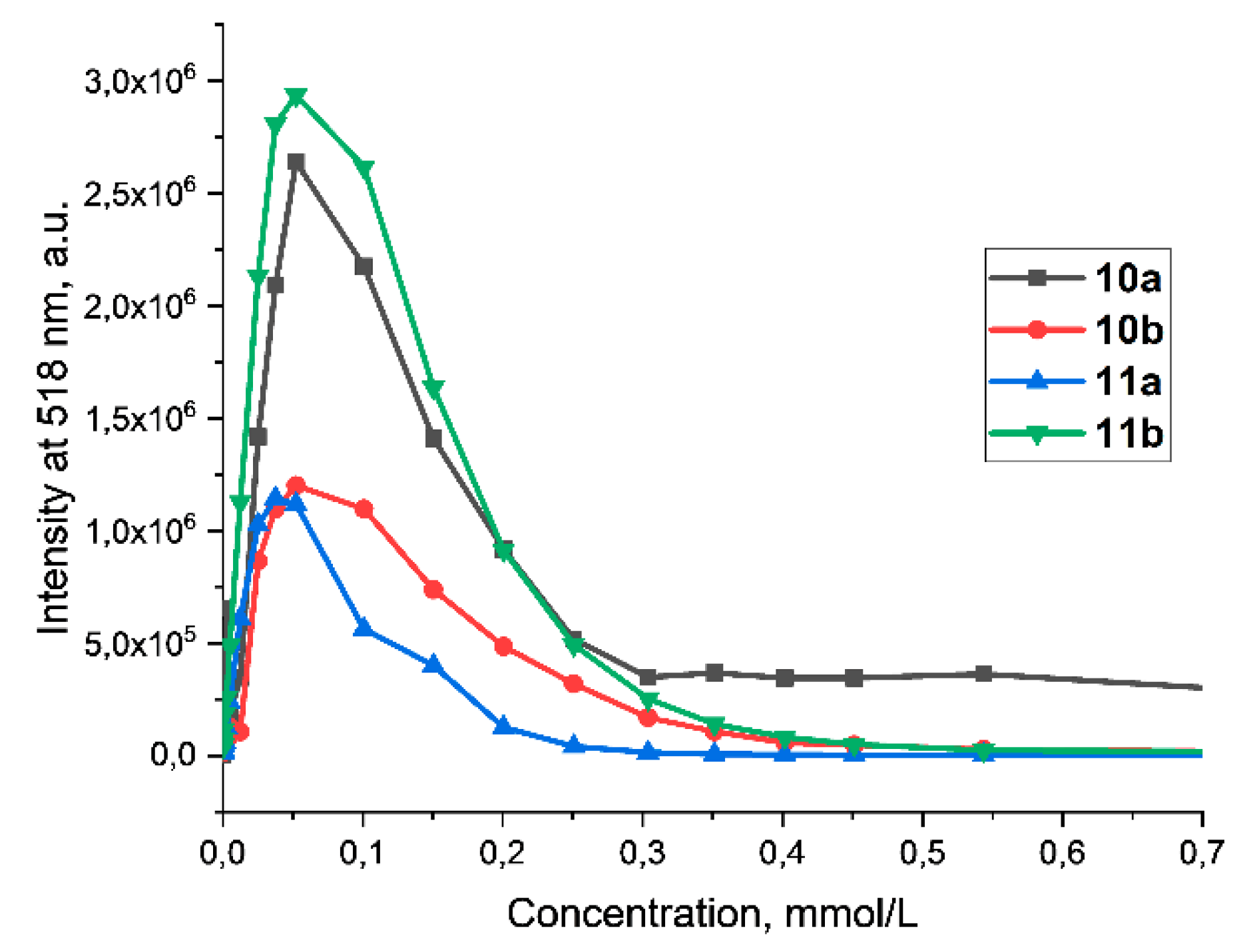
Figure 4.
Dependence of the ratio of the fluorescence intensities of the first (373 nm) and third (384 nm) vibrational peaks of pyrene on the concentration of 10-11 in binary systems 10/11 - pyrene. C(Pyrene)=0.002mM, C(fluorescein derivatives) = 0.005 - 5 mM.
Figure 4.
Dependence of the ratio of the fluorescence intensities of the first (373 nm) and third (384 nm) vibrational peaks of pyrene on the concentration of 10-11 in binary systems 10/11 - pyrene. C(Pyrene)=0.002mM, C(fluorescein derivatives) = 0.005 - 5 mM.
Scheme 3.
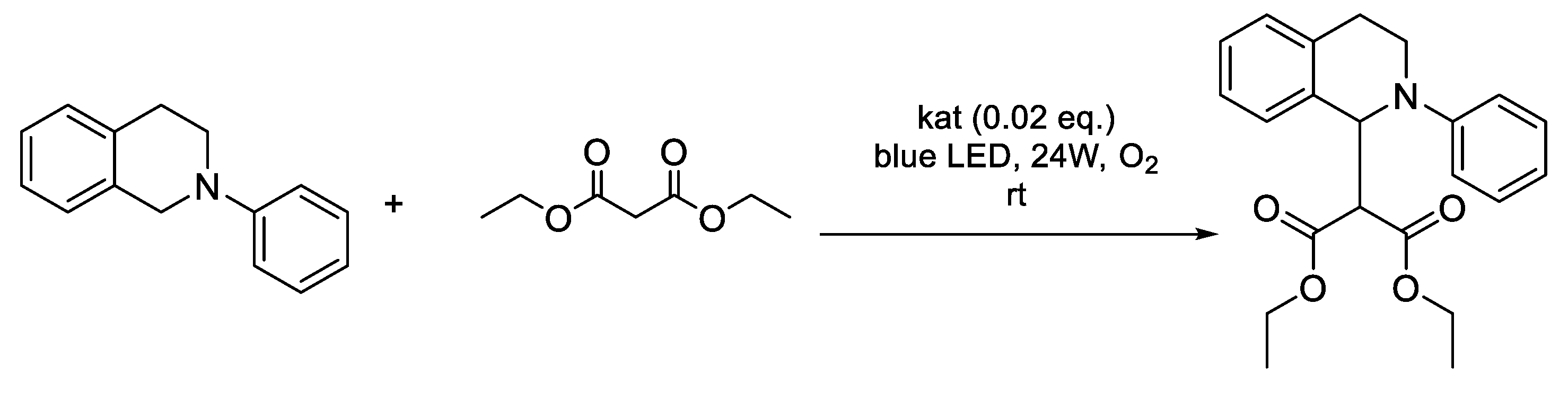
Table 2.
CAC values, average hydrodynamic diameters d and PDI of compounds 10ab-11ab, H2O, 25 oC.1.
| System | 10a | 10b | 11a | 11b |
|---|---|---|---|---|
| CAC, mM | 1.43 | 0.43 | 0.82 | 0.28 |
| d, nm | 165±1 | 267±25 | 171±2 | 202±12 |
| PDI | 0.244±0.018 | 0.433±0.029 | 0.429±0.029 | 0.310±0.052 |
1C (10, 11) = 2 mM.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



