Submitted:
08 June 2024
Posted:
11 June 2024
You are already at the latest version
Abstract
The effects of the ferrocenyl moiety to enhance the nucleophilicity of the carbonyl group, situated at its adjacent position, have been explored in a series of α-ferrocenyl ketocarboxylic acids. In the presence of trifluoroacetic anhydride, 3-ferrocenoylpropionic acid and 4-ferrocenoylbutyric acid gave 5-ferrocenyl-4-trifluoroacetyl-2(3H)-furanone and 6-ferrocenyl-5-trifluoroacetyl-3,4-dihydropyran-2-one, respectively. Under similar reaction conditions, 2-ferrocenylcarbonylbenzoic acid, a keto carboxylic acid without a β-hydrogen, gave a dimerized lactone, 3,3’-diferrocenyl-3,3’-diphthalide possibly due to radical coupling. The nucleophilic attack of carbonyl oxygen, activated by the ferrocenyl moiety, to the carboxylic carbon, is assumed to be the crucial mechanistic step in forming these lactones. When the carbonyl group was reduced to an alcohol to break its conjugation with the ferrocenyl moiety, saturated lactones were isolated after the acidic workup. These results indicate that the α-ferrocenyl carbinols readily undergo solvolysis under acidic conditions, giving ferrocenylcarbenium ions, which are attacked by the carboxy oxygen to give lactones.
Keywords:
Ferrocenylcarbenium ion
; nucleophilicity of carbonyl oxygen
; valence tautomerization
; dimerization
; lactone
; ferrocenyl ketocarboxylic acid
; radical coupling
; trifluoroacetic anhydride
1. Introduction
After the discovery of ferrocene in 1951 [1], research has been focused on understanding the role of the ferrocenyl moiety in stabilizing the positive charge at its alpha position. The stability of α-ferrocenyl carbenium ions has been established based on the enhanced rates of solvolysis of ferrocenyl substrates with the SN1 mechanism. For example, ferrocenylmethyl acetate solvolysis in 80% aqueous acetone at 30 °C is 6.7 times faster than triphenylmethyl acetate [2]. Various research groups have studied the existence of α-ferrocenylcarbenium ions using the chemical shift values of protons and carbons at the carbocationic centers in NMR [3,4,5,6,7,8]. The ability of the ferrocenyl moiety to stabilize the positive charge at its α-position has generated further interest in understanding the basic nature of the oxygen atom in acylferrocenes. Protonation of oxygen in acylferrocenes influences their reactivity, particularly in electrophilc aromatic substitution reactions. The electronic effects induced by protonation at the oxygen atom can also significantly alter the electron density of ferrocene moiety, impacting the overall electronic properties of the molecule. Both experimental [9,10,11] and computational [12] studies have shown that carbonyl oxygen is the most basic site in acyl- or formylferrocene. The higher basicity of the carbonyl oxygen in acylferrocenes than that of aryl ketones and aldehydes has been attributed to the cooperative interaction of the proton with both oxygen and the iron center [9].
Understanding the basic nature of the carbonyl oxygen lying alpha to the ferrocenyl moiety, we wished to explore the nucleophilicity of carbonyl oxygen in acylferrocenes, since nucleophilicity correlates with basicity [13,14]. We realized that the α-ferrocenyl ketocarboxylic acid could offer both electrophilic and nucleophilic centers if the carboxy carbon is converted into a better electrophilic center. Moreover, separating ketone and carboxylic acid functions with 2 or 3 carbon atoms can give thermodynamically stable 5- or 6-membered rings by intramolecular nucleophilic attack. We were also interested in studying the effect of breaking conjugation between the carbonyl group and the cyclopentadienyl ring on the reaction outcomes. The study on the nucleophilicity of carbonyl oxygen in acylferrocenes can help understand the competition between cyclopentadienyl carbons and carbonyl oxygen if the molecule contains an electrophilic center. Herein, we report two pathways of lactone formation: 1. intramolecular nucleophilic attack of carbonyl oxygen to the carboxy carbon in the presence of trifluoroacetic anhydride; 2. nucleophilic substitution of the hydroxy group in α-hydroxy carboxylic acids by carboxylic oxygen under acidic conditions.
2. Materials and Methods
2.1. General Procedures
All experiments were performed under nitrogen using standard Schlenk line techniques. All reagents and solvents were purchased from commercial suppliers. Ferrocene, succinic anhydride, magnesium sulfate (Alfa Aesar, Ward Hill, MA), glutaric anhydride (TCI America, Portland, OR), sodium borohydride, phthalic anhydride, aluminum chloride (Acros Organic, Waltham, MA), and trifluoroacetic anhydride (Oakwood Chemicals, SC) were used as received.
1H NMR (400 MHz), 19F NMR (376 MHz), and 13C NMR (100.6 MHz) spectra were recorded on a JEOL-400 ESZ spectrometer at room temperature. The spectra were referenced to the residual protonated solvent (1H) or the solvent signal (13C). 19F NMR spectra were referenced to the external reference of the spectrometer. IR spectra were recorded on a Bruker Alpha-E FTIR spectrometer using a diamond crystal ATR accessory in a range between 400 and 4000 cm−1. ESI-MS were recorded on Agilent 6470 LC/TQ using 1260 infinity II HPLC system. Melting points were taken on Dynalon™ Afon™ DMP100 Melting Point Device and were uncorrected.
X-ray diffraction data were measured at T = 90 K on a Bruker Kappa Apex-II diffractometer equipped with a microfocus CuKα source (λ=1.54184 Å) for 2a' or a sealed-tube MoKα source (λ=0.71073 Å) for 2b and 3b. Structures were solved using SHELXT [15] and refined using SHELXL [16]. Hydrogen atoms were visible in difference maps, placed in idealized positions during refinement, and treated as riding. The crystal of 2a' was an inversion twin, and the BASF parameter refined to 0.481(16). Because the two independent molecules in 2a' are related by an approximate inversion center at 0.484, ½, ¾, restraints were necessary to prevent C atoms from becoming non-positive definite. The crystal and refinement data are presented in Table 1.
2.2. Experimental Procedures
3-(Ferrocenoyl)propionic acid (1a). The compound was synthesized by following the procedure reported in the literature [17].
5-ferrocenyl-4-trifluoroacetyl-2(3H)-furanone (1b). To the stirred solution of 1a (500 mg, 1.74 mmol) in dichloromethane, trifluoroacetic anhydride (0.48 mL, 3.49 mmol) was added. The solution turned purple immediately. The reaction mixture was stirred at room temperature for 30 min. Volatiles were removed in vacuo. The crude product was purified by silica column chromatography using dichloromethane as eluent, and the purple band on the column was collected. Yield (221 mg, 35%). Melting Point: 108–110 °C. IR (ATR, cm-1): 1825.93, 1726.00, 1690.86, 1553.54. 1H NMR (400 MHz, CDCl3, ppm): δ 3.69 (br, 2H), 4.23 (s, 5H) 4.71 (br, 2H), 5.39 (br, 2H) 19F{1 H} NMR (Acetone-d6, ppm): δ −76.5. 13C{1H} NMR (100 MHz, acetone-d6, ppm): δ 33.2, 68.9, 70.0, 70.5, 71.7(cp), 73.2, 103.0, 116.9 (q, 1J = 290.4 Hz, CF3), 171.0, 172.8 (q, 2J = 34.5 Hz, C-O). ESI-MS: m/z 364.1.
(4-ferrocenoyl)butyric acid (2a) and 1,3-diferrocenoylpropane (2a'): To the stirred suspension glutaric anhydride (6.13 g, 53.8 mmol) and anhydrous aluminum chloride (14.3 g, 107 mmol) in 1,2-dichloroethane (80 mL), a solution of ferrocene (12.0 g, 64.6 mmol) in 1,2-dichloroethane (80 mL) was added dropwise. The reaction mixture was stirred at room temperature for 1 hr and poured into ice-cold water. The organic phase was separated, and the aqueous phase was extracted with dichloromethane (3 x 30 mL). The product was extracted in 1.0 M NaOH (3 x 40 mL). The aqueous phase was acidified with conc. HCl until precipitation was complete. The precipitate was separated by filtration, and the residue was dried in air to give 2a (2.34 g, 14.5%) as an orange solid. 1H NMR (400 MHz, CDCl3, ppm) : δ 2.03 (quint, 3J = 7.2 Hz, 2H), 2.50 (t, 3J = 6.8 Hz, 2H), 2.81 (t, 3J = 6.8 Hz, 2H), 4.18 (s, 5H, Cp), 4.49 (br, 2H, Cp), 4.80 (br, 2H, Cp). 13C{1H} NMR (100 MHz, acetone-d6, ppm): δ 19.4, 32.5, 38.1, 68.4, 69.3, 69.6, 79.5, 173.7, 202.3. The melting point and spectroscopic data of the complex were in good agreement with the reported compound [18]. The organic phase was collected, volatiles removed in vacuo, and the products separated by silica column chromatography using a mixture of hexane and ethyl acetate (3:1). The second band of the column, after evaporation of solvents, gave 2a' (1.26 g, 8.3%) as an orange solid. Analytically pure product was obtained by slow solvent evaporation from the diethyl ether and hexane mixture. Melting Point: 131-132.5 °C (Lit.[19] 125 – 127 °C). IR (ATR, cm-1): 1664 (C=O). 1H NMR (400 MHz, CDCl3): δ 2.11 (p, 3J = 7.2 Hz, 2H), 2.83 (t, 3J = 7.6 Hz, 4H), 4.19 (s, 10H, Cp), 4.49 (t, 3J = 2.0 Hz, 4H), 4.82 (t, 3J = 2.0 Hz, 4H). 13C{1H} (100 MHz, CDCl3, ppm): δ 19.5, 38.9, 69.5, 69.9, 72.3, 79.0, 204.3. ESI-MS: m/z 469.0 [M + H]+. The structure of 2a’ was analyzed using single-crystal X-ray analysis.
6-ferrocenyl-5-trifluoroacetyl-3,4-dihydropyran-2-one (2b). We synthesized this compound by following procedures similar to 1b but with a longer reaction time (12 h). Yield 49%. Melting Point: 86.1–87.9 °C. IR (ATR, cm-1): 1790, 1703 (C=O), 1652 (C=C). 1H NMR (400 MHz, Acetone-d6): δ 2.82 – 2.84 (m, 2H), 2.87 – 2.90 (m, 2H), 4.27 (s, 5H), 4.53 (t, 3J = 1.6 Hz, 2H), 4.65 (t, 3J = 1.6 Hz, 2H). 13C{1H} NMR (100 MHz, Acetone-d6, ppm): δ 20.8, 27.7, 70.1, 70.9, 75.5, 108.1, 116.3 (q, 1J = 291.3, CF3), 163.9, 165.9, 181.5 (q, 2J = 34.5 Hz, CO). 19F NMR (Acetone-d6, ppm): −73.8. The X-ray quality crystals were obtained by cooling its solution in hexane.
2-(Ferrocenoyl)benzoic acid (3a). The compound was synthesized by following the procedure reported in the literature [20].
3,3’-diferrocenyl-3,3’-diphthalide (3b). To the stirred solution of 3a (500 mg, 1.49 mmol) in dichloromethane (10 mL), trifluoroacetic anhydride (1.05 mL, 7.48 mmol) was added. The reaction mixture was stirred at room temperature for 30 min, and then water (20 mL) was added. The product was extracted with dichloromethane (2 x 20 mL). The organic layer was dried with anhydrous MgSO4 and filtered, and the filtrate evaporated to dryness. The crude product was purified by silica gel column chromatography using a mixture of dichloromethane and ethyl acetate (1:1) to give 3b (0.236 g, 48%) as a pale-yellow solid. The 1H NMR analysis of the product showed the mixture of two isomers in a ca. 1:1 ratio. The mixture was suspended in diethyl ether and filtered. The ether-insoluble product was further purified by recrystallization from chloroform by diffusion of ethyl ether. The single-crystal X-ray analysis of the product revealed the formation of a meso isomer. Meso: Melting Point: decomposes above 200 °C. IR (ATR, cm–1): 1767 (C=O). 1H NMR (400 MHz, CDCl3, ppm): δ 3.48 (s, 10H), 3.69 – 3.70 (m, 2H), 3.73 – 3.75 (m, 2H), 4.04 – 4.05 (m, 2H), 4.36 – 4.37 (m, 2H), 7.50 (t, 3J = 7.6 Hz, 2 H), 7.71 – 7.75 (m, 4H, Ar), 7.99 (d, 3J = 7.6 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3, ppm): δ 67.4, 67.8, 68.0, 68.6, 68.9, 84.5, 89.8, 124.0, 125.2, 126.6, 129.7, 133.6, 150.6, 169.6. Racemic: Melting Point: 124 °C. IR (ATR, cm–1): 1728 (C=O). 1H NMR (400 MHz, CDCl3): δ 3.47 (s, 5H, Cp), 3.85 (br, 1H), 3.99 (br, 1H), 4.19 (br, 1H), 4.42 (br, 1H), 7.40 (t, 3J = 7.2 Hz, Ar), 7.56 – 7.63 (m, 2H, Ar), 7.73 (d, 3J = 7.2 Hz, Ar). 13C{1H} NMR (100 MHz, CDCl3, ppm): 67.2, 68.5, 68.6, 68.8, 69.4, 85.5, 89.9, 123.4, 125.5, 126.7, 129.7, 133.8, 150.0, 169.7. Monoferrocenyl dimerized lactone. This product was collected as the first fraction of the column. 1H NMR (400 MHz, CDCl3): δ 2.16 (s, 1H), 3.76 (s, 5H), 4.08 (br, 1H), 4.20 (br, 1H), 4.27 (br, 1H), 4.49 (br, 1H), 4.09 (t, 3J = 2.4 Hz, 1H), 6.37 (dd, 3J = 5.6 Hz, 3J = 2.4 Hz, 1H), 7.03 (dd, 3J = 5.6 Hz, 3J = 1.6 Hz, 1H), 7.35 – 7.37 (m, 1H), 7.50 – 7.59 (m, 4H), 7.69 (t, 3J = 7.2 Hz), 7.84 (d, 3J = 7.6 Hz, 1H), 7.98 (d, 3J = 7.6 Hz, 1H), 7.18 – 7.20 (m, 1H). The reaction was performed by adding two-mole equivalents of ferrocene as 3a. Under similar reaction and workup conditions, the yield of the product was found to be 85%.
5-Ferrocenyldihydro-2(3H)-furanone (4b). To a stirred solution of 1a (1.00 g, 3.49 mmol) in aqueous 1.0 M NaOH (10 mL), NaBH4 (0.661 g, 17.5 mmol) was added. The reaction mixture was refluxed for 1 hr. A brisk gas formation was observed during the reflux, and the compound changed its color from red to orange. The mixture was cooled to room temperature and layered with ethyl acetate. Concentrated HCl was added dropwise with constant stirring until the compound moved from the aqueous to the organic layer. The organic phase was collected, dried with MgSO4, and evaporated to dryness to give a viscous mass. The products were further purified by column chromatography in silica using DCM as eluent, and an orange band was collected. Yield: 0.583 g, 58%. Melting point: 129 -130 °C. IR (ATR cm−1): 1758 (C=O). 1H NMR (400 MHz, CDCl3): δ 2.19 – 2.32 (m, 1H), 2.52 – 2.66 (m, 3H), 4.18 (s, 5H), 4.20 – 4.22 (m, 4H,), 5.31 – 5.34 (m, 1H). 1H NMR (400 MHz, Acetone-d6): δ 2.30 -2.38 (m, 1H), 2.49 - 2.66 (m, 3H), 4.18 (s, 5H), 4.20 (br, 2H), 4.25 (br, 1H), 4.32 (br, 1H), 5.32 – 5.35 (m, 1H). 13C{1H} NMR (100 MHz, CDCl3, ppm): 29.3, 29.7, 67.9, 70.1, 78.8, 78.9, 87.8, 176.9.
6-Ferrocenyl-2H-tetrahydropyran-2-one (5b). We synthesized this compound by following procedures similar to 4b but with a longer reaction time (2 h). Yield: 28%. Melting point: 106 – 107 °C. IR (ATR, cm−1): 1727 (C=O). 1H NMR (400 MHz, CDCl3): δ 1.81 – 2.01 (m, 3H), 2.18 – 2.23 (m, 1H), 2.46 – 2.54 (m, 1H), 2.61 – 2.68 (m, 1H), 4.17 – 4.18 (m, 7H), 4.20 – 4.23 (m, 1H), 4.24 – 4.26 (m, 1H), 5.16 (dd, 3J = 10.4 Hz, 3J = 3.6 Hz). 13C{1H} NMR (100 MHz, CDCl3, ppm): 18.7, 29.0, 29.6, 66.1, 67.3, 68.3, 68.4, 68.9 (Cp), 78.9, 87.1, 171.5.
3. Results and Discussion
Synthesis and Structural Elucidation of Compounds 2a, 2a’, 1b – 5b and Proposed Mechanisms of Their Formation
As shown in Scheme 1, the Friedel-Crafts acylation of ferrocene with succinic anhydride, glutaric anhydride, and phthalic anhydride in the presence of AlCl3 using dichloroethane as solvent gave 3-(ferrocenoyl)propionic acid, 1a, 4-(ferrocenoyl)butyric acid, 2a and 2-(ferrocenoyl)benzoic acid, 3a, respectively. Unlike 1a and 3a, the synthesis of 2a yielded a neutral byproduct that displayed the presence of an unsubstituted cyclopentadienyl ring, two types of methylene protons (in a 1:2 integration ratio), and two sets of substituted cyclopentadienyl protons (in a 1:1 integration ratio) (Figure S1). FTIR (1664 cm−1) and 13C NMR (204 ppm) (Figure S2) indicated the presence of ketone function in the molecule. ESI-MS analysis of the product showed a strong signal at m/z 469 (Figure S3) corresponding to two ferrocenyl moieties, two ketones, and three methylene groups in the molecule. The single crystal X-ray structure of the product unambiguously indicated the formation of 1,3-diferrocenoylpropane, 2a’. Although the compound 2a’ was synthesized from the reaction of ferrocene and glutaryl chloride [21,22], to our knowledge, the formation of such a binuclear product by Friedel-Crafts acylation of ferrocene with glutaric anhydride has not been previously reported.
The compound 2a’ crystallizes in monoclinic space group P21 with two independent molecules in the asymmetric unit. These two molecules are related by an approximate inversion center (Figure 1). The Cp rings display nearly eclipsed conformation as in the closely related crystal structure of 1,2-diferrocenoylethane [23]; the torsional angle C – Cg1 – Cg2 – C in four ferrocenyl moieties ranges from 1.21° to 21.42°. In the crystal, the compound displays weak intermolecular π···π interactions between two Cp rings, C-H···π interactions between Cp-H and the centroid of Cp, and C-H···O interactions between Cp-H and the carbonyl oxygen of two adjacent molecules (Figure S4).
Since the primary goal of this study was to investigate the nucleophilicity of the carbonyl group next to the ferrocenyl moiety, we treated 3-(ferrocenoyl)propionic acid, 1a with trifluoroacetic anhydride (Scheme 2). On adding trifluoroacetic anhydride (TFAA), the compound immediately changed its color from red to deep purple. We separated the product by silica gel column chromatography using DCM as eluent. The 1H NMR analysis of the product shows only four signals with an integration ratio of 2:2:2:5 (Figure S5), indicating the loss of two methylene protons compared to the starting keto-carboxylic acid, 1a. The FTIR showed two strong signals at 1825 cm−1 and 1726 cm−1, indicating the presence of two carbonyl groups in the molecule. A high-energy IR band (1825 cm−1) was consistent with a trifluoroacetyl group in the molecule, which was further confirmed by two quartets at 116.9 (1J = 290.4 Hz) ppm and 172.8 (2J = 34.5 Hz) ppm in 13C NMR and a singlet at −76.5 ppm in 19F NMR (Figures S6, S7). ESI-MS analysis of the product showed a signal at m/z 365.0, but determining the structure of the compound was still a challenge. Finally, the compound yielded dark purple crystals when it was crystallized by slow vapor diffusion of hexane into its solution in dichloromethane under nitrogen. Although a complete structure refinement by X-ray crystallography was not possible due to the tiny size of the crystals and their twinning, the analysis was sufficient to identify the compound. The preliminary crystallographic results showed the formation of an α, β-unsaturated cyclic lactone with a trifluoroacetyl group attached to the β-position of the ring (Figure S8).
To understand the chemistry further, we treated 4-(ferrocenoyl)butyric acid, 2a, with TFAA in DCM (Scheme 2). As expected, the reaction yielded the desired product 2b in 49% isolated yield after chromatographic separation. The spectroscopic data (Figures S9 – S11) of the product were in agreement with the formation of the lactone in a similar way to the formation of 1b. The single crystal X-ray analysis of the product unambiguously confirmed the formation of 6-ferrocenyl-5-trifluoroacetyl-3,4-dihydropyran-2-one (Figure 2). The compound crystallizes in the monoclinic space group P21/n with a single molecule in its asymmetric unit. The bond length between C11 and C12 is 1.3599 (5) Å, confirming a double bond between α and β carbons. The six-membered lactone ring has a half-boat conformation with the Cremer & Pople [24] puckering parameters QT = 0.4736(5) Å, θ = 117.34(6)°, and φ = 30.74(6)°. The two Cp rings in the molecule acquire roughly eclipsed conformation with an average torsional angle C-Cg1–Cg2–C of 11.61°. The Cp rings of the ferrocenyl moiety are almost parallel, the dihedral angle between their planes being 2.73°.
The mechanism of intramolecular cyclization of the keto-carboxylic acid 1a to form an α, β-unsaturated lactone 1b can be proposed as shown in Scheme 3. The reaction between the carboxylic acid and trifluoroacetic anhydride gives a mixed anhydride (I) on which the trifluoroacetate group acts as an excellent leaving group. The carbonyl oxygen of the ketone makes a nucleophilic attack on the carboxy carbon to form a tetrahedral intermediate (II). Such an unusual nucleophilic attack of carbonyl oxygen might be possible due to the electron-donating effects of the ferrocenyl moiety at its alpha position. The tetrahedral intermediate loses trifluoroacetate, forming a resonance-stabilized carbocation (III). The stability of such carbenium ions is attributed to the electron-releasing effects of the ferrocenyl moiety as well as the nearby lone pair of electrons on the oxygen atom. The carbocation loses a proton from the β-position to create a double bond in conjugation with the cyclopentadienyl moiety (IV). The double bond attacks TFAA to give another resonance-stabilized carbocation with a trifluoroacetate group in the ring (V). The inductive effects of trifluoroacetyl group and a positive charge at the adjacent position both facilitate the loss of proton to give the final product (1b).
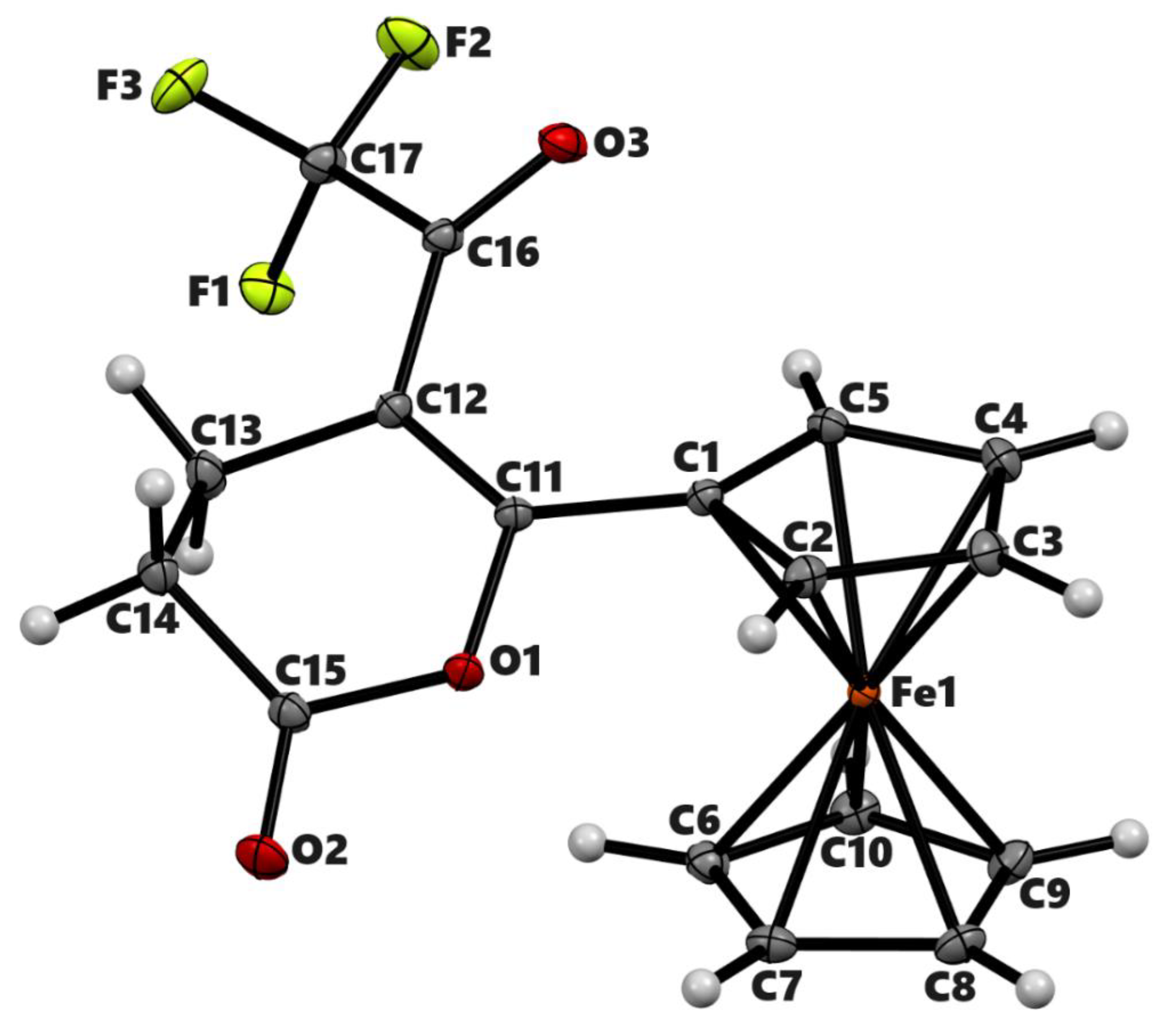
The proposed mechanism involves the formation of 5-ferrocenyl-2(3H)-furanone, IV as an intermediate product from the β-elimination of carbocation III. We were interested in exploring the reaction outcomes in the absence of hydrogen at the β-position of ferrocenyl moiety. For this purpose, we treated 2-(ferrocenoyl)benzoic acid, 3a, with trifluoroacetic anhydride in DCM. On adding TFAA, the compound changed color from dark red to amber at first and then to green. This color change is attributed to the formation of ferrocenium ions [25,26]. During aerobic workup, the reaction product changed to a lighter red color. The 1H NMR analysis of the product indicated the formation of two isomers in a ca. 1:1 ratio. We separated the isomeric mixture by taking advantage of their different solubilities in diethyl ether. The substituted cyclopentadienyl rings of both isomers showed four multiplets, indicating the formation of a chiral center at the alpha position of the ferrocenyl moiety. A single signal in 1H or 13C NMR for the unsubstituted cyclopentadienyl ring for both isomers indicated the presence of either a mirror plane or a rotational axis of symmetry in the molecule (Figures S12 – S15). The ether-insoluble isomer was crystallized from chloroform by vapor diffusion of diethyl ether. The single-crystal X-ray analysis of the product revealed the formation of meso isomer of dimeric lactone, 3,3’-diferrocenyl-3,3’-diphthalide (Figure 3). The compound crystallizes in monoclinic space group P21/c with the molecule lying on an inversion center. The lactone rings bend inward, making a dihedral angle of 48.42° with the cyclopentadienyl ring. The two Cp rings adopt a nearly eclipsed conformation with C-Cp1-Cp2-C dihedral angle of 2.18°. The C-C bond connecting the two monomers is 1.5751(12) Å. The ether-soluble product also contained the same number of signals in NMR as the meso isomer. Therefore, the stereochemistry of the product, by analogy, was assigned to be racemic (R, S). The meso isomer decomposes above 250 °C while the racemic one melts at 124 °C. The isomeric mixture of the complex 3b was reported by Nesmeyanov et al. with limited characterization data [27].
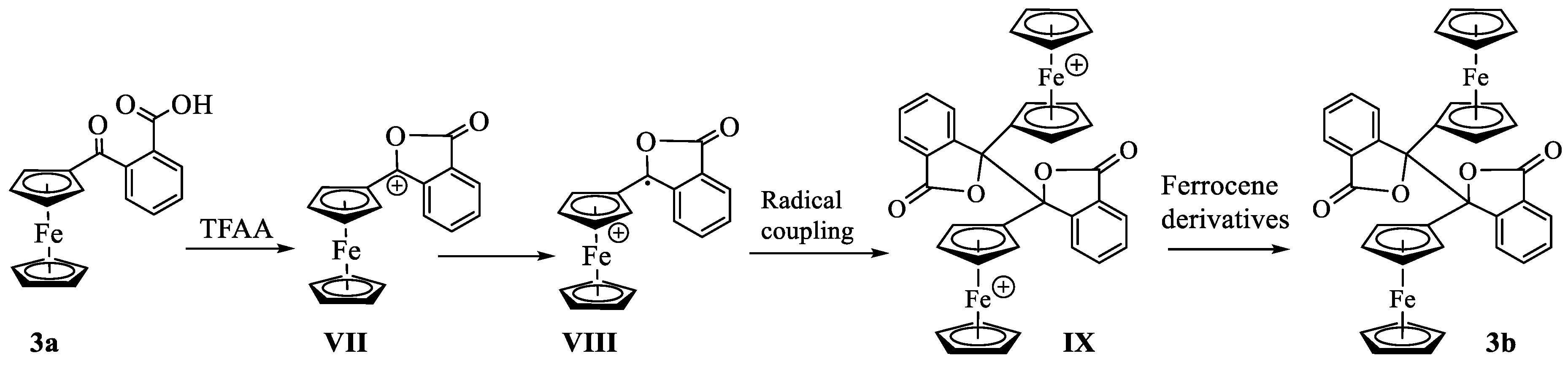
The formation of the dimerized lactone from 3a in the presence of TFAA further supports our hypothesis of intramolecular nucleophilic attack of carbonyl oxygen on the carboxylic carbon. As shown in Scheme 4, the nucleophilic attack of the carbonyl oxygen on the carboxy carbon, followed by a loss of the leaving group (vide supra), forms the α-ferrocenylcarbenium ion (VII). The carbenium ion undergoes internal oxidation-reduction (valence tautomerization), whereby the non-bonding electron from the d2g set of iron is transferred to the positively charged carbon atom, giving a radical cation (VIII) [28]. The two radical cations undergo coupling to give a dication dimer (IX). The intermolecular oxidation-reduction between the dimer and ferrocene derivative in the solution produces the neutral dimer 3b. Similar reaction mechanisms have been proposed earlier by Cais and others [25,29,30,31,32,33] to explain the formation of dimerized products obtained from the solvolysis of α-ferrocenylcarbinols under acidic conditions.
We attempted to isolate and characterize all the reaction products and their byproducts to validate the proposed mechanism. We could not isolate more than 48% of the dimeric products in our multiple synthetic efforts. We observed the formation of a black intractable residue, possibly due to the decomposition of cationic intermediates caused by the sacrificial oxidation of ferrocene derivatives. Careful separation of the reaction products by column chromatography indicated the product with one Fe(Cp) loss from dimer 3a, as evidenced by 1H NMR. The compound contains only one unsubstituted cyclopentadienyl group and 17 other chemically quique protons, indicating the loss of CpFe moiety from the dimer (Figure S16). We added two equivalents of ferrocene in the reaction mixture to prevent the loss of the starting compound during reductive dimerization of the radical cation VIII. Under these conditions, the yield of 3b increased to 85%, and no decomposition product was detected. We assume that ferrocene preferentially oxidizes to provide the electron to the carbenium ion.
To get a better insight into the participation of the ferrocenyl moiety in activating the carbonyl oxygen for a nucleophilic attack, we decided to reduce the ketone to secondary alcohol and treat the hydroxycarboxylic acid with TFAA. The compound changed from red to yellow when compounds 1a and 2a were treated with NaBH4 in NaOH(aq). The product remained soluble in NaOH, indicating the presence of the carboxylic acid group in the molecule. However, when the solutions were acidified using HCl, both compounds gave orange precipitates no longer soluble in aqueous NaOH (Scheme 5). The neutral product showed a strong signal at 1758 cm−1 (4b) and 1727 cm−1 (5b) in IR and a doublet of doublet corresponding to one proton at 5.31 ppm (4b) and 5.16 (5b) in 1H NMR indicating the formation of lactones (Figures S17 – S20). The lactone 5b was crystallized from a diethyl ether and hexane mixture by slow evaporation. A single-crystal X-ray analysis of the product showed the formation of the desired product (Figure S21); however, its complete refinement was difficult due to weak scattering and crystal twinning. Formation of lactone 4b under similar reaction conditions has been reported in the literature with limited characterization data [34,35].
These results indicate that isolating hydroxycarboxylic acids from reducing ferrocenyl keto-carboxylic acid is difficult after acid workup. The instability of such hydroxycarboxylic acid in the presence of acid might be due to the facile solvolysis of the 2° alcohol to give a stable α-ferrocenyl carbenium ion. The carbenium ion is intramolecularly attacked by the oxygen atom of the carboxylic acid, giving a lactone as shown in Scheme 6.
4. Conclusions
We have studied the uncommon lactonization in α-ferrocenyl ketocarboxylic acids in the presence of trifluoroacetic anhydride. We believe the important mechanistic step of lactone formation is the nucleophilic attack of ketone oxygen to carboxy carbon. The electron-donating effect of ferrocenyl moiety increases the electron density of carbonyl oxygen to facilitate the attack. These reactions yielded 5-ferrocenyl-4-trifluoroacetyl-2(3H)-furanone from 3-(ferrocenoyl)propionic acid and 6-ferrocenyl-5-trifluoroacetyl-3,4-dihydropyran-2-one from 4-(ferrocenoyl)butyric acid with structurally rare but biologically and pharmacologically important furanone [36] and pyranone [37] motifs. In these reactions, trifluoroacetic anhydride behaves as an electrophile, and its fragmentation product, trifluoroacetate, acts as a base. The 2-(ferrocenoyl)benzoic acid, on the other hand, gave a dimerized lactone, 3,3'-diferrocenyl-3,3'-diphthalide caused by the valence tautomerization of α-ferrocenylcarbenium ion followed by dimerization of two ferrocenylphthalide units. The current work explains the necessity of reducing ketone to methylene before nucleophilic ring-closing in ferrocene-bound ketocarboxylic acids to synthesize homoannular ferrocene derivatives [17,38,39]. Our attempts to synthesize α-ferrocenyl hydroxycarboxylic acids by reducing the ketone function in 3-(ferrocenoyl)propionic acid and 4-(ferrocenoyl)butyric acid were unsuccessful. We believe that the incipient hydroxycarboxylic acid cyclizes by an intramolecular SN1-type mechanism. We are currently exploring the effects of acids on the redox potentials of the iron centers in acylferrocenes by electrochemical methods.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: 1H NMR of 2a’; Figure S2: 13C NMR of 2a’; Figure S3: ESI-MS of 2a’; Figure S4: Crystal packing of 2a’ along the crystallographic a axis; Figure S5: 1H NMR of 1b; Figure S6: 13C NMR of 1b; Figure S7: 19F NMR of 1b; Figure S8: Molecular structure of 1b; Figure S9: 1H NMR of 2b; Figure S10: 13C NMR of 2b; Figure S11: 19F NMR of 2b; Figure S12: 1H NMR of 3b (meso); Figure S13: 13C NMR of 3b (meso); Figure S14: 1H NMR of 3b (racemic); Figure S15: 13C NMR of 3b (racemic); Figure S16: 1H NMR of demetallated product from lactone 3b; Figure S17: 1H NMR of 4b; Figure S18: 13C NMR of 4b; Figure S19: 1H NMR of 5b; Figure S20: 13C NMR of 5b; Figure S20: Molecular Structure of 5b.
Author Contributions
Conceptualization, U.R.P.; synthesis and spectroscopic characterization, A.M.A., B.J.C. and B.P.L.; X-ray crystallography, F.R.F.; writing—original draft preparation, U.R.P.; writing—review and editing, U.R.P., A.M.A., B.J.C., B.P.L., and F.R.F.; visualization, U.R.P.; supervision, U.R.P.; project administration, U.R.P.; funding acquisition, U.R.P. All authors have read and agreed to the published version of the manuscript.
Funding
The work was funded by the Louisiana Board of Regents. Contract Number: LEQSF (2017-18)-RD-A-28 and Supervised Undergraduate Research Experience (SURE) from the Louisiana Board of Regents.
Data Availability Statement
Original contributions included in the study are included in the article and supplementary material; further inquiries can be directed to the corresponding authors. CIFs have been deposited at the Cambridge Crystallographic Data Centre under the CCDC deposition numbers given in Table 1.
Acknowledgments
The authors extend their appreciation to the Department of Chemistry & Physical Sciences, Nicholls State University, for providing funds for purchasing chemicals, and the Department of Chemistry, Louisiana State University, for providing X-ray crystallography services free of charge.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kealy, T.J.; Pauson, P.L. A New Type of Organo-Iron Compound. Nature 1951, 168, 1039–1040. [Google Scholar] [CrossRef]
- Richards, J.H.; Hill, A.A. α-Metallocenyl carbonium ions. J. Am. Chem. Soc. 1959, 81, 3484. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Buchholz, H.; Reddy, V.P.; De Meijere, A.; Olah, G.A. Stable carbocations. 285. 1-Ferrocenyl-1-cyclopropyl cation: the first long-lived cyclopropyl cation. J. Am. Chem. Soc. 1992, 114, 1097. [Google Scholar] [CrossRef]
- Cully, N.; Watts, W.E. Stable carbocations. XX. A kinetic study of the SN1 hydrolysis of aryl(ferrocenyl)methyl acetates. J. Organomet. Chem. 1979, 182, 99–103. [Google Scholar] [CrossRef]
- Abram, T.S.; Watts, W.E. Stable carbocations. Part 12. Generation, observation, and properties of ferrocenyl-stabilized vinyl cations. J. Chem. Soc., Perkin Trans. 1. [CrossRef]
- Cerichelli, G.; Floris, B.; Ortaggi, G. Ferrocenyl carbocations. Ionization of ferrocenyl alcohols in aqueous sulfuric acid. J. Organomet. Chem. 1974, 78, 241. [Google Scholar] [CrossRef]
- Natsume, S.; Kurihara, H.; Yamaguchi, T.; Erabi, T.; Wada, M. Stability and reactivity of ferrocenyl(2,4,6-trimethoxyphenyl)carbenium salts. J. Organomet. Chem. 1999, 574, 86–93. [Google Scholar] [CrossRef]
- Korb, M.; Mahrholdt, J.; Liu, X.; Lang, H. Reactivity of Planar-Chiral α-Ferrocenyl Carbocations towards Electron-Rich Aromatics. Eur. J. Inorg. Chem. 2019, 2019, 973–987. [Google Scholar] [CrossRef]
- Rubalcava, H.E.; Thomson, J.B. A spectroscopic study of the protonation of acylferrocenes. Spectrochim. Acta 1962, 18, 449–459. [Google Scholar] [CrossRef]
- Olah, G.A.; Mo, Y.K. Organometallic chemistry: V. Protonation of acylferrocenes under stable ion conditions in fso3h-so2c1f solution. J. Organomet. Chem. 1973, 60, 311–321. [Google Scholar] [CrossRef]
- Roberts, R.M.G.; Silver, J.; Wells, A.S. Mössbauer and NMR studies of protonated acyl diphosphaferrocenes. Inorganica Chim. Acta. 1986, 119, 171–176. [Google Scholar] [CrossRef]
- Saric, A.; Vrcek, V.; Buehl, M. Density functional study of protonated formylmetallocenes. Organometallics 2008, 27, 394–401. [Google Scholar] [CrossRef]
- Bunnett, J.F. Nucleophilic Reactivity. Annu. Rev. Phys. Chem. 1963, 14, 271–290. [Google Scholar] [CrossRef]
- Jaramillo, P.; Pérez, P.; Fuentealba, P. Relationship between basicity and nucleophilicity. Journal of Physical Organic Chemistry 2007, 20, 1050–1057. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr., Sect A: Foundations and Advances 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Structural Chemistry 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, U.R.; Daigle, D.P.; Naquin, S.D.; Engeron, G.S.; Lo, M.A.; Fronczek, F.R. Synthesis, Crystal Structure, and Electrochemistry of Mono- and Bis-Homoannular Ferrocene Derivatives. Crystals 2024, 14, 141. [Google Scholar] [CrossRef]
- Wieczorek, A.; Blauz, A.; Makal, A.; Rychlik, B.; Plazuk, D. Synthesis and evaluation of biological properties of ferrocenyl-podophyllotoxin conjugates. Dalton Trans. 2017, 46, 10847–10858. [Google Scholar] [CrossRef]
- Woltersdorf, M.; Kranich, R.; Schmalz, H.-G. Enantioselective synthesis of new C2-symmetric ferrocenylalkylamines via sonochemical amination of 1-ferrocenylalkyl acetates. Tetrahedron 1997, 53, 7219–7230. [Google Scholar] [CrossRef]
- Pokharel, U.R.; Bergeron, J.T.; Fronczek, F.R. Synthesis and crystal structures of 2-(ferrocenylcarbonyl)benzoic acid and 3-ferrocenylphthalide. Acta Crystallogr., Sect. E 2020, 76, 1163–1167. [Google Scholar] [CrossRef]
- Goldberg, S.I.; Breland, J.G. Ferrocene studies. XIX. Synthesis of 1,2-terferrocene. J. Org. Chem. 1971, 36, 1499–1503. [Google Scholar] [CrossRef]
- Tombul, M.; Gemici, S.; Bulut, A. Alkyl Lewis Acid Catalyzed Syntheses of Dicarbonyl Ferrocenes. Asian J. Chem. 2010, 22, 7070. [Google Scholar]
- Tombul, M.; Bulut, A.; Guven, K.; Buyukgungor, O. 1,4-Diferrocenylbutane-1,4-dione. Acta Crystallogr., Sect. E 2008, 64, m444–m445. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.t.; Pople, J. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Cais, M.; Modiano, A.; Raveh, A. Organometallic Studies. XVII.1 A Novel Approach to the Synthesis of the Benzopentalene System2. J. Am. Chem. Soc. 1965, 87, 5607–5614. [Google Scholar] [CrossRef]
- Casper, L.A.; Ebel, V.; Linseis, M.; Winter, R.F. Five shades of green: substituent influence on the (spectro-) electrochemical properties of diferrocenyl(phenyl)methylium dyes. Dalton Trans. 2021, 50, 15336–15351. [Google Scholar] [CrossRef] [PubMed]
- Nesmeyanov, A.N.; Vil'chevskaya, V.D.; Kochetkova, N.S. Reactions of ο-carboxybenzoylferrocene. Dokl. Akad. Nauk SSSR 1965, 165, 835–837. [Google Scholar]
- Gleiter, R.; Seeger, R. The Structure of the Ferrocenyl-Methyl Cation. Preliminary communication. Helv. Chim. Acta 1971, 54, 1217–1220. [Google Scholar] [CrossRef]
- Cais, M.; Eisenstadt, A. Organometallic Studies. X.1a Reductive Dimerization of α-Metallocenylcarbonium Ions. I1b. J. Org. Chem. 1965, 30, 1148–1154. [Google Scholar] [CrossRef]
- Fedin, E.I.; Blumenfeld, A.L.; Petrovskii, P.V.; Kreindlin, A.Z.; Fadeeva, S.S.; Rybinskaya, M.I. Conversion of the diamagnetic nonamethylferrocenylcarbenium salts into the paramagnetic salts of bis(nonamethylferroceniumyl)ethane. J. Organomet. Chem. 1985, 292, 257–268. [Google Scholar] [CrossRef]
- Banide, E.V.; Ortin, Y.; Chamiot, B.; Cassidy, A.; Niehaus, J.; Moore, A.; Seward, C.M.; Müller-Bunz, H.; McGlinchey, M.J. Syntheses, Structures, and Dimerizations of Ferrocenyl- and Fluorenylideneallenes: Push−Pull Multiple Bonds? Organometallics 2008, 27, 4173–4182. [Google Scholar] [CrossRef]
- Casper, L.A.; Deuter, K.L.; Rehse, A.; Winter, R.F. Dimerization of 9-Phenyl-ferroceno [2,3]indenylmethyl Radicals: Electrochemical and Spectroelectrochemical Studies. ACS Org. & Inorg. Au. [CrossRef]
- Casper, L.A.; Linseis, M.; Demeshko, S.; Azarkh, M.; Drescher, M.; Winter, R.F. Tailoring Valence Tautomerism by Using Redox Potentials: Studies on Ferrocene-Based Triarylmethylium Dyes with Electron-Poor Fluorenylium and Thioxanthylium Acceptors. Chem. Eur. J. 2021, 27, 10854–10868. [Google Scholar] [CrossRef] [PubMed]
- Huffman, J.W.; Rabb, D.J. 1,2-(α-Oxotetramethylene)ferrocene. J. Org. Chem. 1961, 26, 3588. [Google Scholar] [CrossRef]
- Sugiyama, N.; Suzuki, H.; Shioura, Y.; Teitei, T. Reaction of ferrocene with acyl chlorides. Bull. Chem. Soc. Jpn. 1962, 35, 767. [Google Scholar] [CrossRef]
- Husain, A.; Khan, S.A.; Iram, F.; Iqbal, M.A.; Asif, M. Insights into the chemistry and therapeutic potential of furanones: A versatile pharmacophore. Eur. J. Med. Chem. 2019, 171, 66–92. [Google Scholar] [CrossRef] [PubMed]
- Bhat, Z.S.; Rather, M.A.; Maqbool, M.; Lah, H.U.; Yousuf, S.K.; Ahmad, Z. α-pyrones: Small molecules with versatile structural diversity reflected in multiple pharmacological activities-an update. Biomedicine & Pharmacotherapy 2017, 91, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, K.L., Jr.; Curby, R.J., Jr. Ferrocene bridging and homoannular cyclizations. J. Am. Chem. Soc. 1957, 79, 3290. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Curby, R.J., Jr.; Gustafson, D.H.; Harrison, K.G.; Bozak, R.E.; Bublitz, D.E. Organic chemistry of ferrocene. V. Cyclization of ω-ferrocenylaliphatic acids. J. Am. Chem. Soc. 1962, 84, 3263. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of ferrocenyl keto-carboxylic acids.
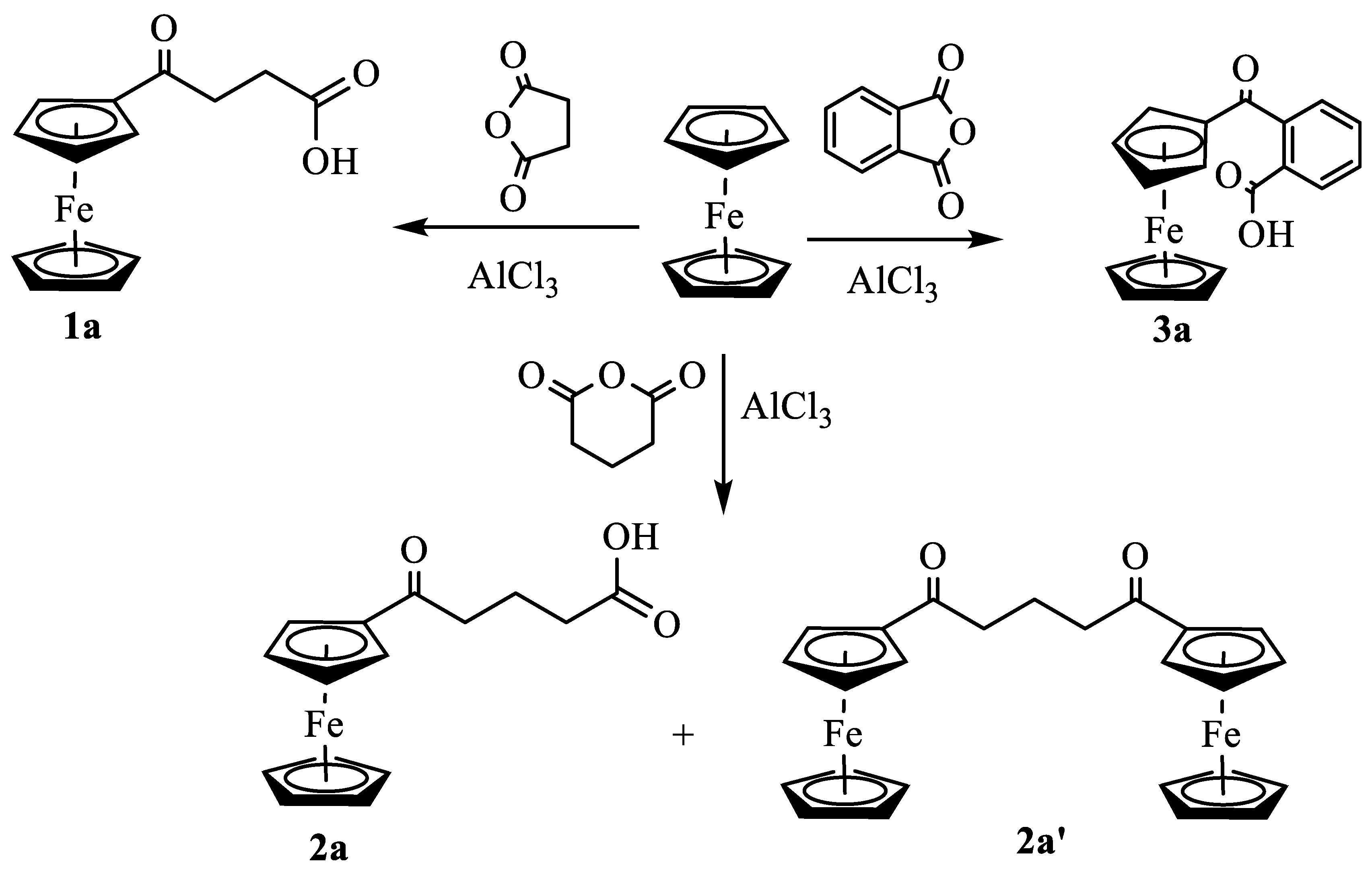
Figure 1.
ORTEP diagram of solid-state structure showing the atom-numbering scheme of compound 2a'. Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complex: Fe1–C1 2.035(16), Fe1–C2 2.022(16), Fe1–C3 2.037(17), Fe1–C4 2.055(17), Fe1–C5 2.023(16), Fe1–C16 2.017(17), Fe1–C17 2.054(17), Fe1–C18 2.016(18), Fe1–C19 2.027(18), Fe1–C20 2.029(18), C1–C6 1.48(2), O1–C6 1.218(18), C6–C7 1.52(2), C7–C8 1.54(2).
Figure 1.
ORTEP diagram of solid-state structure showing the atom-numbering scheme of compound 2a'. Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complex: Fe1–C1 2.035(16), Fe1–C2 2.022(16), Fe1–C3 2.037(17), Fe1–C4 2.055(17), Fe1–C5 2.023(16), Fe1–C16 2.017(17), Fe1–C17 2.054(17), Fe1–C18 2.016(18), Fe1–C19 2.027(18), Fe1–C20 2.029(18), C1–C6 1.48(2), O1–C6 1.218(18), C6–C7 1.52(2), C7–C8 1.54(2).
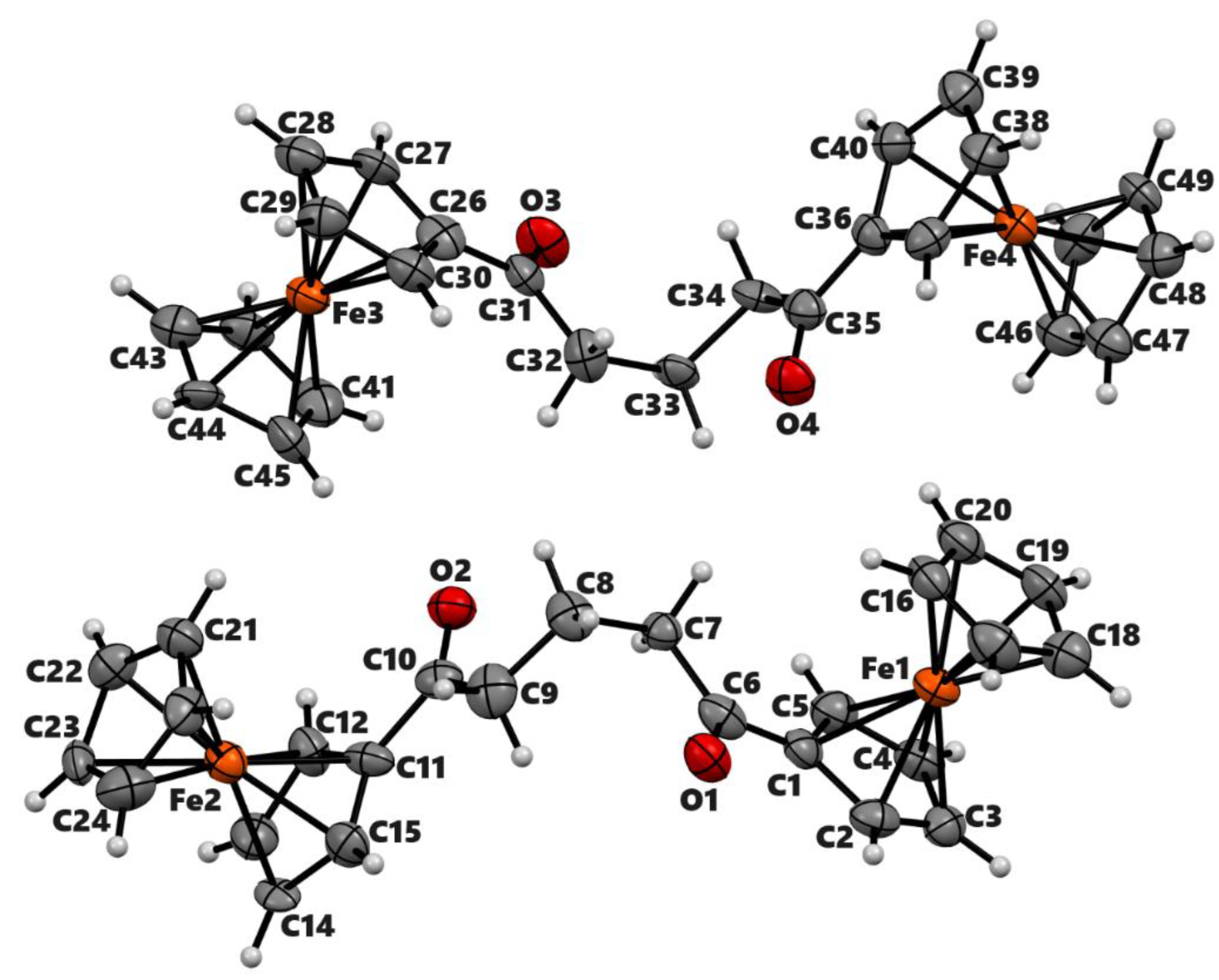
Scheme 2.
Lactonization of ketocarboxylic acids in the presence of TFAA.
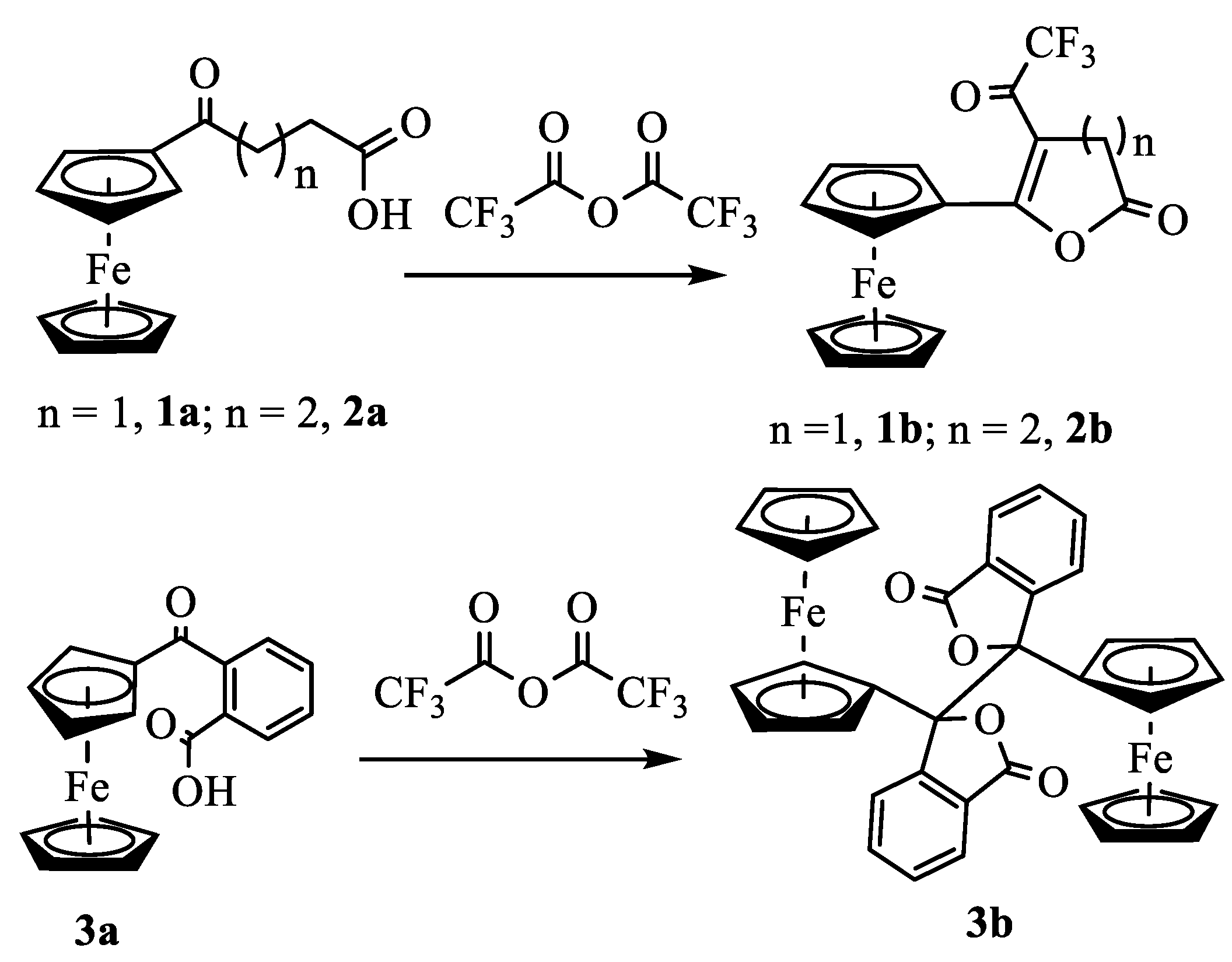
Figure 2.
ORTEP diagram of solid-state structure showing the atom-numbering scheme of compound 2b. Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complex: Fe1–C1 2.0338(4), Fe1–C2 2.0310(4), Fe1–C3 2.0510(4), Fe1–C4 2.0607(4), Fe1–C5 2.0495(4), Fe1–C6 2.0515(4), Fe1–C7 2.0431(4), Fe1–C8 2.0528(4), Fe1–C9 2.0597(4), Fe1–C10 2.0621(4), C1–C11 1.4593(5), C11–C12 1.3599(5), O1–C11 1.3837(5), O1–C15 1.3823(5), O2–C15 1.1996(5), C13–C14 1.5262(6), C12–C16 1.4630(5), O3–C16 1.2149(5), C16–C17 1.5579(5), F1–C17 1.3377(5), F2–C17 1.3284(5), F3–C17 1.3458(5) Fe–Cp (centroid, substituted) 1.643, Fe–Cp (centroid, unsubstituted) 1.657.
Figure 2.
ORTEP diagram of solid-state structure showing the atom-numbering scheme of compound 2b. Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complex: Fe1–C1 2.0338(4), Fe1–C2 2.0310(4), Fe1–C3 2.0510(4), Fe1–C4 2.0607(4), Fe1–C5 2.0495(4), Fe1–C6 2.0515(4), Fe1–C7 2.0431(4), Fe1–C8 2.0528(4), Fe1–C9 2.0597(4), Fe1–C10 2.0621(4), C1–C11 1.4593(5), C11–C12 1.3599(5), O1–C11 1.3837(5), O1–C15 1.3823(5), O2–C15 1.1996(5), C13–C14 1.5262(6), C12–C16 1.4630(5), O3–C16 1.2149(5), C16–C17 1.5579(5), F1–C17 1.3377(5), F2–C17 1.3284(5), F3–C17 1.3458(5) Fe–Cp (centroid, substituted) 1.643, Fe–Cp (centroid, unsubstituted) 1.657.
Scheme 3.
Proposed mechanism of the formation of the lactone.
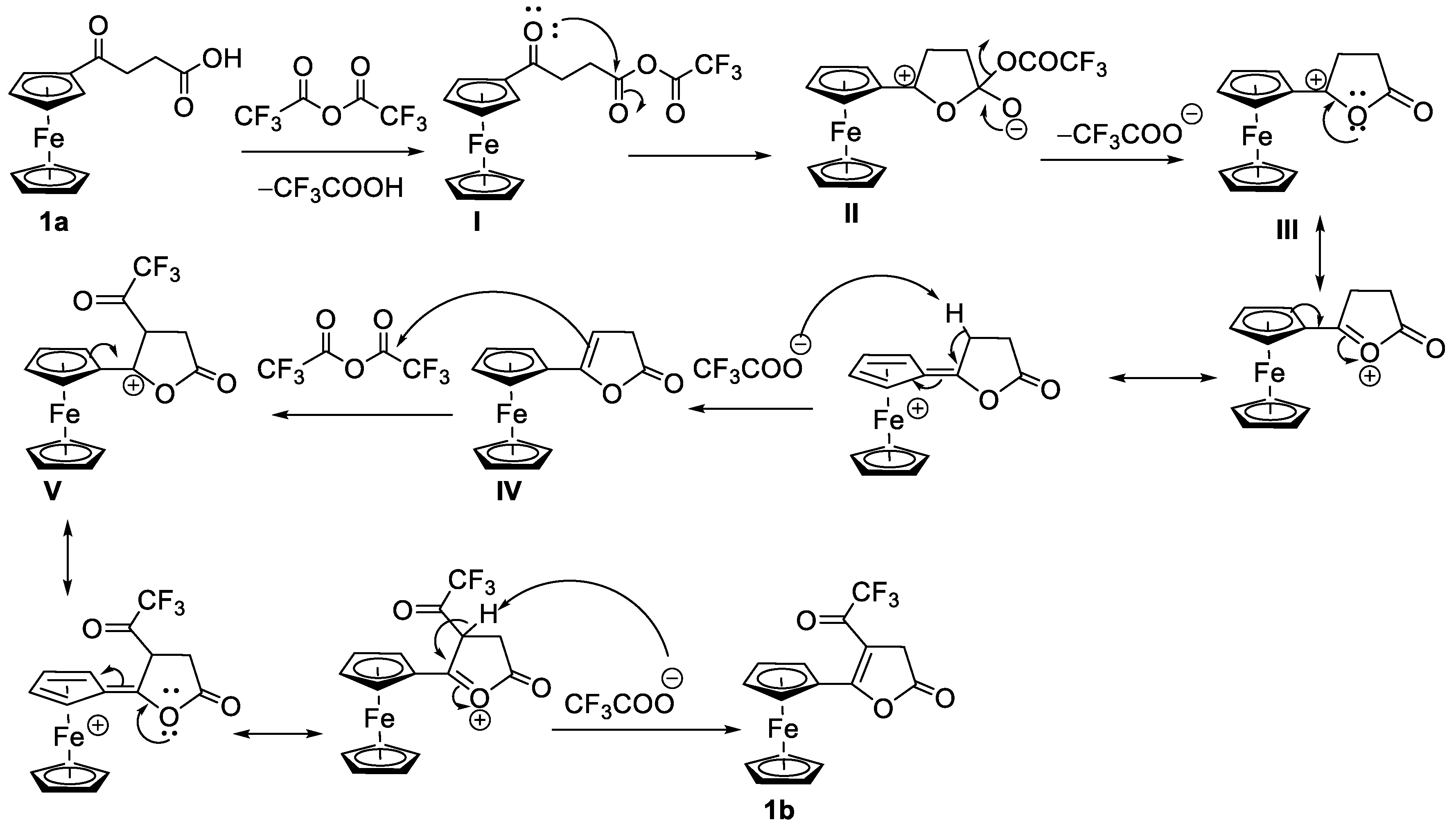
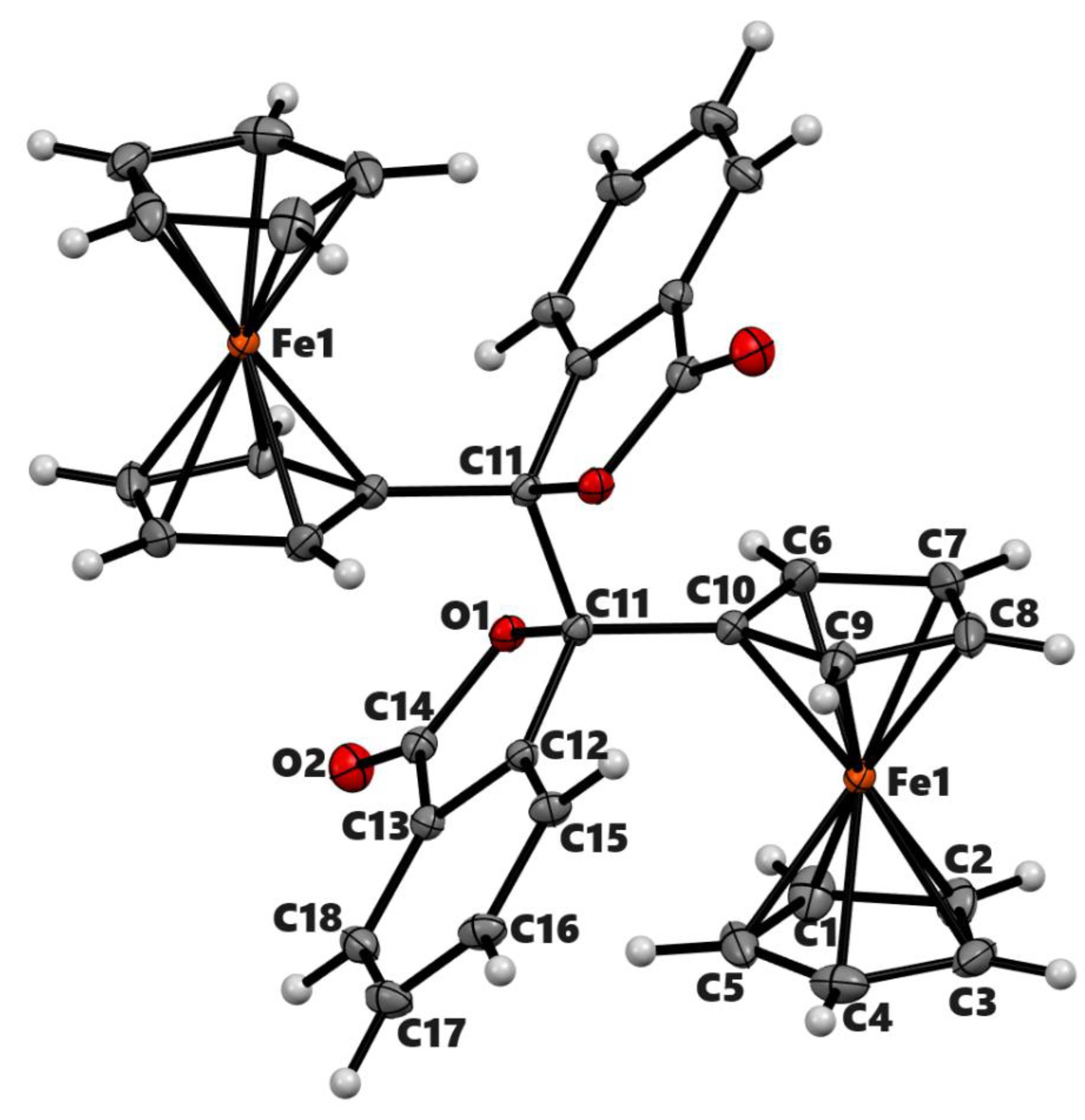
Figure 3.
ORTEP diagram of solid-state structure showing the atom-numbering scheme of compound 3b. Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complex: Fe1–C1 2.0494(8), Fe1–C2 2.0576(8), Fe1–C3 2.0567(8), Fe1–C4 2.0500(9), Fe1–C5 2.0491(8), Fe1–C6 2.0502(8), Fe1–C7 2.0587(8), Fe1–C8 2.0542(8), Fe1–C9 2.0475(7) Fe1–C10 2.0405(7), O1–C14 1.3711(9), O1–C11 1.4578(8), O1–C14 1.3711(9), O2–C14 1.2084(9), Fe–Cp (centroid, substituted) 1.650, Fe–Cp (centroid, unsubstituted) 1.658.
Figure 3.
ORTEP diagram of solid-state structure showing the atom-numbering scheme of compound 3b. Displacement ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) for the complex: Fe1–C1 2.0494(8), Fe1–C2 2.0576(8), Fe1–C3 2.0567(8), Fe1–C4 2.0500(9), Fe1–C5 2.0491(8), Fe1–C6 2.0502(8), Fe1–C7 2.0587(8), Fe1–C8 2.0542(8), Fe1–C9 2.0475(7) Fe1–C10 2.0405(7), O1–C14 1.3711(9), O1–C11 1.4578(8), O1–C14 1.3711(9), O2–C14 1.2084(9), Fe–Cp (centroid, substituted) 1.650, Fe–Cp (centroid, unsubstituted) 1.658.
Scheme 4.
Proposed mechanism of the formation of dimerized lactone.
Scheme 5.
Reduction of ketone in keto-carboxylic acids and isolation of lactones after acidic workup.
Scheme 5.
Reduction of ketone in keto-carboxylic acids and isolation of lactones after acidic workup.
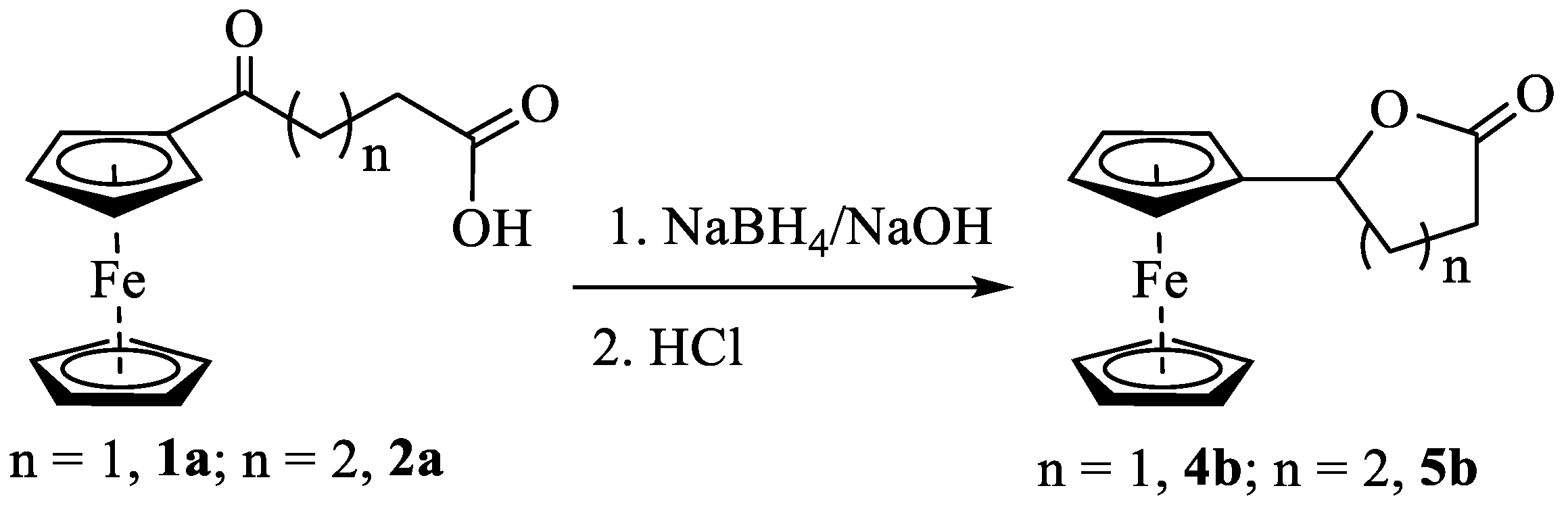
Scheme 6.
Mechanism of the formation of lactone from α-ferrocenylhydroxycarboxylic acids.
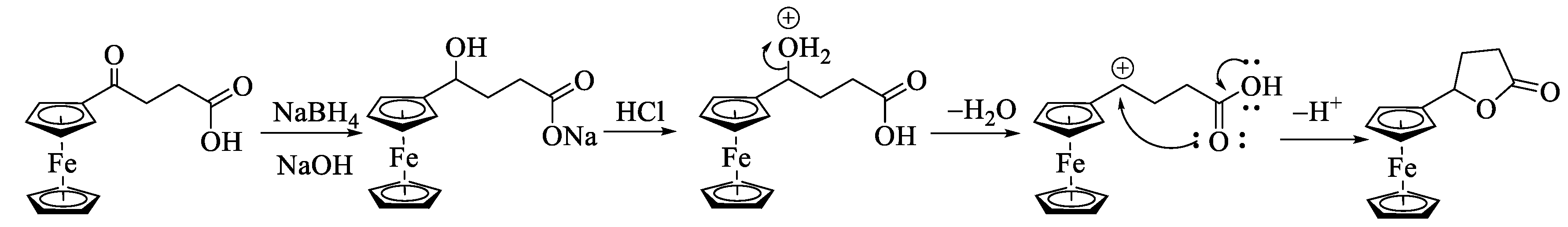

Table 1.
Crystal data and refinement parameters.
| 2a’ | 2b | 3b | |
| Chemical formula | C25H24Fe2O2 | C17H13F3FeO3 | C36H26Fe2O4 |
| Mr | 468.14 | 378.12 | 634.27 |
| Deposition number | CCDC 2355187 | CCDC 2355188 | CCDC 2355189 |
| Crystal system, space group | Monoclinic, P21 | Monoclinic, P21/n | Monoclinic, P21/c |
| Temperature (K) | 90 | 90 | 90 |
| a, b, c (Å) | 5.7838 (5), 11.8770 (9), 28.071 (2) | 10.6270 (2), 9.7968 (2), 14.9960 (3) | 9.8279 (8), 13.3787 (10), 10.8256 (8) |
| β (°) | 93.773 (6) | 110.1183 (9) | 111.064 (4) |
| V (Å3) | 1924.2 (3) | 1465.98 (5) | 1328.29 (18) |
| Z | 4 | 4 | 2 |
| Radiation type | Cu Kα | Mo Kα | Mo Kα |
| µ (mm−1) | 12.24 | 1.08 | 1.14 |
| Crystal size (mm) | 0.15 × 0.04 × 0.01 | 0.38 × 0.30 × 0.13 | 0.22 × 0.15 × 0.14 |
| Diffractometer | Bruker Kappa APEX-II DUO | Bruker Kappa APEX-II DUO | Bruker Kappa APEX-II DUO |
| Absorption correction | Multi-scan | Multi-scan | Multi-scan |
| Tmin, Tmax | 0.564, 0.887 | 0.764, 0.873 | 0.811, 0.857 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 12160, 5225, 3851 | 74201, 13090, 12014 | 65909, 9349, 7963 |
| Rint | 0.074 | 0.019 | 0.031 |
| (sin θ/λ)max (Å−1) | 0.568 | 1.086 | 0.945 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.068, 0.178, 1.04 | 0.021, 0.059, 1.05 | 0.031, 0.086, 1.05 |
| No. of reflections | 5225 | 13090 | 9349 |
| No. of parameters | 524 | 244 | 190 |
| Δρmax, Δρmin (e Å−3) | 1.01, −0.55 | 0.88, −0.48 | 1.02, −0.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



