Submitted:
06 June 2024
Posted:
07 June 2024
You are already at the latest version
Abstract
Abstract A multiple number of studies provide evidence that vasopressin (AVP) and steroid hormones are frequently secreted together and closely cooperate in the regulation of blood pressure, metabolism, water-electrolyte balance, and behavior, securing thereby survival and comfort of life. Vasopressin interacts with hormones of the hypothalamo-pituitary-adrenal axis (HPA) at several levels through regulation of release of corticotropin-releasing hormone (CRH), adrenocorticotropic hormone ACTH, and multiple steroid hormones, as well as through interactions with steroids in the target organs. The interactions are facilitated by positive and negative feedbacks occurring between specific components of the HPA axis with engagement of other regulatory molecules. Altogether AVP and HPA cooperate closely as a coordinated functional AVP-HPA system. It has been shown that cooperation between AVP and steroid hormones may be affected by the cellular stress combined with hypoxia, and by metabolic, cardiovascular and respiratory disorders, neurogenic stress and inflammation. Growing evidence indicates that central and peripheral interactions of AVP and steroid hormones are reprogrammed in cardiovascular diseases and that the rearrangements play either beneficial or harmful effects. The present review highlights specific mechanisms of interactions of AVP and steroids at cellular and systemic levels and analyses consequences of inappropriate cooperation between various components of the AVP-HPA system for the regulation of the cardiovascular system and energy balance in cardiovascular and metabolic diseases.
Keywords:
AVP
; CRH
; ACTH
; glucocorticoids
; mineralocorticoids
; androgens
; estrogens
; hypertension
; cardiac failure
; stress
1. Introduction
The first experimental studies showing close functional relationship between vasopressin and steroid hormones were published over 60 years ago. In 1960 Hilton et al., reported that arterial perfusion of the adrenal glands with arginine vasopressin (AVP) or lysine vasopressin (LVP) potently stimulates release of cortisol [1]. Subsequently it has been found that in many instances vasopressin and steroid hormones are secreted together and closely cooperate in regulation of blood pressure, metabolism, water-electrolyte balance and behavior in a manner securing survival and comfort of life. It has been also shown that the cellular stress combined with hypoxia, disturbances of metabolism, cardiovascular and respiratory disorders, neurogenic stress and inflammation may disorganize cooperation between AVP and steroid hormones. Under homeostatic, unstressed conditions both steroid hormones and vasopressin are released in characteristic diurnal rhythms. Expression of AVP mRNA in the suprachiasmatic nucleus shows distinct endogenous circadian rhythm. The diurnal rhythm of secretion is also typical for adrenocorticotropic hormone (ACTH) and glucocorticoids. Interestingly, it appears that the responsiveness of AVP synthesis to steroids also manifests circadian rhythmicity [2]. Vasopressin interacts with the hypothalamo-pituitary-adrenal axis (HPA) at various levels, i.e. in the hypothalamic nuclei, affecting release of corticotropin-releasing hormone (CRH), in the pituitary gland, enhancing ACTH release and in the adrenal glands modulating release or action of multiple steroid hormones. The interactions are facilitated by positive and negative feedbacks occurring between specific components of the HPA system with engagement of other neurotransmittory and neuropeptidergic pathways.
Vasopressin and CRH are synthesized mainly in the hypothalamic nuclei, however both these peptides can be also produced in other regions of the brain. In the hypothalamus vasopressin and CRH are released partly by the same cells of the paraventricular nucleus (PVN). Immunostaining studies showed that virtually all parvocellular CRH neurons in the PVN are stained positively for vasopressin [3] and are co-packaged in neurosecretory vesicles of the hypothalamic-pituitary axons in the median eminence [4]. and the hypophyseal-portal circulation [5,6,7]. There is also evidence that vasopressin may be necessary for appropriate action of glucocorticoids, as it has been shown that the maximal binding capacities for corticosterone and dexamethasone in the hippocampus and the anterior pituitary are significantly lower in homozygous diabetes insipidus (HDI) rats that do not synthesize vasopressin than in their nondiabetic counterparts. Importantly the difference could be eliminated by AVP treatment [8]. On the other hand, glucocorticoids were found to exert negative effect on release and action of vasopressin. For instance corticosterone was found to inhibit release of AVP from the explants containing supraoptic nucleus (SON) and sending projections to the neural lobe of the pituitary [9]. Experiments on Sprague Dawley rats have shown that release of AVP from the hypothalamic slices encompassing PVN and SON neurons could be inhibited by corticosterone, cortisol, testosterone and 17-beta estradiol in a dose-dependent manner, whereas dexamethasone, aldosterone and progesterone were not effective in these experiments [10]. Chronic application of dexamethasone, which is an agonist of corticosterone, significantly decreased plasma corticosterone and ACTH concentrations and elicited differential changes in expressions of CRH, ACTH and AVP in several brain regions, including the cortex, the hippocampus, the hypothalamus and the cerebellum [11]. There is also evidence that vasopressin exerts weak stimulatory effect on secretion of aldosterone from the glomerulosa cells of the adrenal medulla [12].
It is highly probable that some steroid hormones can modulate vasopressin action through actions exerted at the level of AVP receptors. For instance experiments on rats have shown that adrenalectomy, which eliminates the main source of circulating steroid hormones, reduces vasopressin V1a receptors (V1aR) in the hippocampus, the dorsolateral septum and the bed nucleus of the stria terminalis (BNST). Moreover, effects of adrenalectomy on V1aR in the hippocampus, and the BNST can be reduced by treatment with corticosterone and aldosterone [13,14,15]. At the same time it should be noted that glucocorticoids may act in opposite way on expression of V1b vasopressin receptors (V1bR), because administration of dexamethasone was found to increase V1bR mRNA in the pituitary, whereas adrenalectomy reduced V1bR mRNA. The latter effect could be reversed by administration of dexamethasone [16].
It appears that negative feedback between glucocorticoids and vasopressin arises in early life. In the rat applications of dexamethasone and aldosterone to fetal hypothalamic cells cultures have been found to inhibit release of CRH, AVP and oxytocin through mechanisms involving activation of protein kinase A (PKA) and protein kinase C (PKC) [17]. In addition, administration of cortisol to the medium bathing the hypothalamic neurons of the fetal sheep significantly inhibited potassium-induced secretion of AVP [18]. There is also evidence that secretion of several steroid hormones is sex-dependent [19,20].
It is possible that during chronic stress glucocorticoids and mineralocorticoids may act in opposite way on AVP release. Hence, expressions of CRH hnRNA and AVP hnRNA in parvocellular neurons of PVN is significantly elevated in rats exposed to the forced swim stress and the increase is abolished by administration of dexamethasone [21]. On the other hand, application of eplerenone, which is a mineralocorticoid receptor antagonist, alleviated anxiety-like behavior and reduced vasopressin and corticosterone concentrations in the posterior pituitary [22].
Thus far there are still multiple gaps in understanding mutual interactions between vasopressin and steroid hormones in health and cardiovascular diseases. Therefore, the principal aim of the present review is to analyze the cooperation of vasopressin and steroid hormones at the cellular and systematic levels in context of their influence on cardiovascular regulation, tissue metabolism and oxygenation. Specifically we discuss positive and negative consequences of interactions of AVP and steroid hormones in development of hypertension and heart failure.
2. Interactions of the Hypothalamo-Pituitary-Adrenal System with Vasopressin at the Cellular Level
2.1. Genomic and Nongenomic Actions of Steroid Hormones
Multiple studies show that steroid hormones exert rapid as well as delayed cellular effects, that are mediated by specific types of non-genomic and genomic receptors located either in the cellular membrane or intracellularly [23,24,25,26]. Receptors of specific steroids are encoded by different genes and their activation may result either in stimulatory or in inhibitory effects, depending on the cell type. The final action of the specific steroid is determined by dose of the hormone, type of stimulated receptors, number of receptors, and presence of specific enzymatic pathways in the targeted cell. In the cardiovascular system stimulation of steroid receptors causes a wide range of actions that may be either beneficial or detrimental.
In the individual cell steroid ligand cooperates with local transcription factors and other regulatory compounds [27,28]. Essential role in binding to receptors and mobilization of posttranslational modifications play coactivators, which are involved in integration of cellular processes and adjustment of cellular responses to current needs. Especially important role is attributed to Steroid Receptor Coactivators (SRC, namely SRC1, SRC2, SRC3), which are known as Nuclear Receptor Coactivators (NCOAS), and to Coactivator Binding Inhibitors (CBIs) [29,30].
Mineralocorticoid receptors (MR, MCR), glucocorticoid receptors (GR), estrogen receptors (ER, ESR), androgen receptors (AR) and progesterone receptors (PGR) belong to a subfamily 3 of nuclear receptors possessing the ligand binding domain (LBD), the DNA-binding domain (DBD) and the N-terminal domain (NTD). In the inactive state the receptors are present in the cytoplasm in a multiprotein chaperon complex, which contains the ligand-binding cleft identifying and binding the ligand. Binding elicits conformational transformations within the complex that permit subcellular trafficking of the ligand to the target within the cell and its interaction with DNA [31,32].
Glucocorticoid and Mineralocorticoid Receptors. In humans function of glucocorticoid is served by cortisol, whereas in the rat it is attributed to corticosterone, which acts also as mineralocorticoid. Glucocorticoid and mineralocorticoid receptors are members of the nuclear receptor superfamily of transcription factors (TF) that modulate processes of transcription through direct binding to glucocorticoid response element (GRE) or mineralocorticoid response element (MRE) in the DNA. A DNA binding domain is in 96% identical in GR and MR. Initiation of specific transcriptional processes at particular promoter and enhancer regions warrants coordinated cellular responses to steroid hormones. The receptors possess also a C-terminal ligand-binding domain (CT-LBD) and an amino-terminus domain (NTD). GR is a 97 kDA protein encoded by NR3C1/Nr3c1 gene (in humans located in chromosome 5) and cooperates with several co-regulators [32,33,34]. An amino-terminus contains AF-1 and AF-2 regions which interact with CT-LBD and can stimulate transcription in absence of a ligand [35,36,37,38,39].
It has been shown that GR and MR can bind as dimers or tetramers [40,41]. Binding of the ligand by MR or GR initiates a cascade of events enabling translocation of the ligand-bound receptor to the nucleus. In the nucleus the receptors bind to specific DNA sequences which are known as glucocorticoid response elements (GREs) and negative glucocorticoid response elements (nGREs) [42,43]. Regulation of GREs appears to play dominant role in cellular processes of neurons [23,42]. Direct occupancy of nGRE results in repression of the target gene. Steroid receptor coactivators (SRCs) appear to participate in repression of CRH expression by GR in the hypothalamus. In the nucleus MR and GR can also interact with some other active proteins (MAZ- myc-associated zinc finger protein, AP-1 – activator protein 1, NF-kB – nuclear factor κB, and SRC-1/2/3) that operate as ligand-selective co-regulators. They can induce remodeling of gene conformation and may initiate formation of transcription-initiation complexes. GRE-DNA interactions are modulated by chromatin configuration. In the cardiovascular system expression of MR is higher in males than in females [44].
Glucocorticoid receptors have been also identified in mitochondria where they regulate mitochondrial gene transcription. In neuronal mitochondria GRs interact with Bcl-2 protein and form GR/Bcl-2 complexes. Interestingly short time action exerted at low density intensifies formation of theses complexes, whereas in high doses cortisol exert opposite effects [45]. Interaction of Bcl-2 with other regulatory factors determines specificity of actions of glucocorticoids in various organs [37,46,47].
Glucocorticoids modulate also process of transcription indirectly by physical interaction (tethering), which does not require direct contact with DNA but engages activation of transcription factors. In various types of cells these factors may act either as co-activators or as co-repressors. Tissue-cell dependent expression of co-regulators causes specific tissue-cell action of steroid molecules [30,37,48,49,50]. It is likely that the protein-protein interactions mediate rapid effects of steroid hormones and play essential role in trans-repression of genes by glucocorticoids in hypoxia and inflammatory processes. For instance it has been found that they interact with hypoxia induced factors (HIFs) at the level of the promoter region of the inflammatory genes and can either enhance or inhibit activation of the HIF pathway [49,51]. It has been postulated that during the inflammatory processes the co-activating function of steroids determines collagen synthesis, generation of reactive oxygen species and engagement of peroxisome proliferator-receptor gamma co-activator 1-alpha (PGC-1α) as well as activation of p38 mitogen activated protein kinase (MAPK) and nicotinamide adenine dinucleotide phosphate oxidases (NOX) 2 and 4 [52,53]. On the other hand, glucocorticoids can inhibit inflammation through repression of genes engaged in synthesis of pro-inflammatory proteins (AP-1, NFκB) and through enhancement of expression of genes involved in generation of anti-inflammatory compounds [49,54].
There is evidence for reciprocal interactions between glucocorticoid receptors pathways. For instance, it has been shown that GR can induce expression of genes that are promoting or inhibiting p38 MAP kinase pathway (MAPK) [53]. Furthermore, expression of GR and its responsiveness to glucocorticoids is regulated by microRNAs, whereas expression of microRNA is regulated by glucocorticoids [55].
Mineralocorticoid receptor is also known as nuclear receptor of subfamily 3, group C, member 2 (NR3C2). MRs bind mainly aldosterone, but they also show high affinity to cortisol and androgens. Regulatory importance of cortisol and aldosterone in specific cell type largely depends on availability of 11-β-hydroxysteroid dehydrogenase type 2 (11-βHSD2), which converts active cortisol into inactive cortisone. Opposite action is exerted by 11-β HSD1, which transforms cortisone into cortisol. Presence of these isoenzymes significantly determines sensitivity of specific organs and tissues to glucocorticoids and mineralocorticoids. Availability of 11β-HSD2 in several regions of the brain causes that aldosterone has good access to brain MR receptors and can exert potent regulatory effects in spite that its concentration in plasma is hundreds of time lower than the concentration of cortisol [56,57,58]. In the heart cardiomyocytes and macrophages do not express 11-βHSD2 and both cortisol and aldosterone participate in MR stimulation. Moreover, in the heart aldosterone exerts some effects through cross-talk with cardiac G-protein-coupled receptors (GPCRs) [59]. Activated MR can form homodimers or can associate with GR and form heterodimers.
MRs are present in the kidney, heart and vessels where mineralocorticoids participate in the regulation of hypertrophy, fibrosis, inflammation, and apoptosis. Mineralocorticoids can act either directly on NR3C2 receptors or their action can be mediated by formation of other active molecules, such as interleukin-1 (IL-1), tumor necrosis factor α (TNF-α), cardiotrophin-1 (CT-1) and Toll-like receptor 4 (TLR-4). During inflammatory process the inflammatory cytokines (IL-1, IL-6, TNF-α) act synergistically with mineralocorticoids and can act jointly through inhibition of ACTH secretion in the hypothalamic-pituitary-adrenal axis [60]. In the rat mesangial cells aldosterone was found to stimulate NF-κB and glucocorticoid-inducible protein kinase-1 (SGK1) activities. It elevates also promoter activities and protein expressions of intercellular adhesion molecule–1 (ICAM-1) and connective tissue growth factor (CTGF). There is evidence that these factors are involved in aldosterone mediated mesangial fibrosis and inflammation [61].
In cardiomyocytes the genomic action of aldosterone mediated by MR participate in chronotropic and hypertrophic actions. It has been shown that aldosterone enhances expression of mRNA which is coding for the α1H protein and the latter is a constituent of CaV3.2 channel, which is one of the two T channels of the cardiomyocytes [62,63]. Action of aldosterone on T channels is presumably indirect, because the gene CACNA1h, which is coding for the CaV3.2 T type channel does not possess mineralocorticoid response element (MRE). Increased formation of reactive oxygen species (ROS) and their involvement in the regulation of affinity of steroid hormones to MR receptors should be also taken into consideration [63,64,65,66]. In the cardiovascular system the genomic and non-genomic effects of aldosterone are also modified by angiotensin II (Ang II) [35].
In the brain MRs have been identified mainly in the hippocampus, septum and other limbic structures, whereas GRs are expressed in the septum, hippocampus, brain stem and the prefrontal cortex [46,67,68]. Affinity of corticosterone to MR in neurons is 10-fold higher than to GR [68,69]. MRs are associated with cellular membrane and after activation are translocated with help of β-arrestin to the cells where they can exert their actions through non-genomic GPCR processes [25,59].
Androgen, Estrogen and Progesterone. Both in males and females androgens are synthesized in the adrenal glands (mainly in the zona fascicularis and the zona reticularis), in the brain (mainly in the hippocampus), and in the liver [70]. In males testosterone is produced chiefly by Leydig cells of the testis, while in females testosterone and its metabolites are produced primarily in the adrenals and ovaries. Cells of the adrenal cortex synthesize also dehydroepiandrosterone (DHEA), androstenedione, androstendione, androstenediol and 11-β-hydroxyandrostenedione. Testosterone can be converted to dihydrotestosterone by 5α-reductase (aromatase), while deoxycorticosterone is converted into dihydrodeoxycorticosterone. Both DHEA and testosterone are able to stimulate androgen receptors, however DHEA has significantly greater androgenic activity than testosterone. The aromatase producing dihydrotestosterone is also involved in formation of estradiol, which is engaged in stimulation of estrogen receptors. The gene, which is encoding aromatase (CYP19A1) is located on chromatosome 15, and has been identified in lungs, vessels and multiple brain regions [71,72]. The process of aromatization of testosterone to estradiol occurs in several peripheral tissues and in the brain [70].
The main steps of synthesis of progesterone include conversion of cholesterol into pregnenolone by cholesterol side-chain cleavage enzyme (P450scc) belonging to cytochrome p-450 superfamily, and conversion of pregnenolone into progesterone by the 3β-hydoxy-steroid dehydrogenase (3β-HSD). In the brain progesterone is also converted to neuroactive steroids, specifically to 5α-dihydroprogesterone (5-α-DHPROG) and tetrahydroprogesterone (3α-5α-THPROG) [73].
Androgens, estrogens and progesterone interact with receptors in the brain, heart and vessels and participate in the regulation of the cardiovascular system by means of classic genomic and non-classic pathways [74,75,76,77]. Androgen receptors were identified in cells of the reproductive system, bones, vessels and brain [78,79]. In the brain ARs are present in the cortex, midbrain, brain stem and spinal cord. Specifically, high density of AR immunoreactivity was found in the olfactory bulb, the nucleus accumbens, the medial amygdala, the bed nucleus of the lamina terminalis, the medial preoptic area, the septum, the mesencephalic periaqueductal gray (PAG), the dorsal raphe nucleus, the substantia nigra, the area postrema, the dorsal motor vagus nucleus, and in the preganglionic cells of the autonomic nervous system [78,80].
Androgen signaling engages several molecular pathways. The primary androgen receptor is a nuclear transcription factor that is activated mainly by testosterone and dihydrotestosterone. Non-stimulated ARs are present mainly in cytoplasm and are associated with heat shock proteins (HSPs). Binding of androgen with ARs allows dissociation of the receptor from chaperone proteins and translocation of the complex androgen-AR to the nucleus where it binds to androgen-response element (ARE) and regulates gene transcription. Androgens regulate also rapid non-genomic processes, engaging G-protein coupled receptor family C (GPRC6A), zinc transporter ZIP9 membrane-receptor and oxoeicosanoid receptor (OXER). In consequence through activation of the genomic-dependent and non-genomic dependent signaling pathways androgens initiate transcription processes and activate canonical pathways associated with activation of ionotropic receptors, G-protein coupled receptors activating phosphpholipase C, calcium transporters, and endothelial nitric oxide synthase (eNOS). Testosterone can also stimulate membrane ARs, which are binding to Src and activating MAPK pathway. Transactivation of membrane ARs by other ligands has been also reported [81,82,83,84]. Most likely activation of the rapid non-genomic processes is essential for fast actions of androgens, such as cell migration, mitosis and inflammatory processes [85].
Estrogens easily penetrate through cellular membrane and majority of their molecules is localized within the nucleus [84,86]. In cells they regulate long-lasting processes by means of nuclear receptors and rapid non-genomic processes. Nuclear estrogen receptors ESR1 (ERα) and ESR2 (ERβ) are codified by ESR1 and ESR2 genes. ESR1 is located on chromosome 6 (6q25.1) and ESR2 on chromosome 14 (14q23.2). Both receptors act as transcription factors which mediate transcriptional activity of estrogens with reference to specific genes. In absence of the ligand ESRs are associated with heat shock protein and do not express transcriptional activity. After activation by the ligand ESRs interact with estrogen response element (ERE) and operate either as the monomers or as the dimers (ESR1-ESR1; ESR2-ESR2 or ESR1-ESR2). In the nucleus estrogens enhance transcription of specific target genes [77,87,88] Estrogens can also modulate expression of other genes acting indirectly via activation of PI3K/Akt and MAPK/ERK pathways as well as through inhibition of JNK pathway [89].
Multiple essential actions of estrogens, such as inhibition of ROS production, regulation of mitochondrial ATP level and formation of mitochondrial structural conglomerations occur in mitochondria [90,91]. Estrogens interact also with membrane-associated G-protein-coupled receptor (GPER, named as GPR30), which is encoded on chromosome 7 (7p22.3), and which is found in the endoplasmic nucleus, Golgi apparatus and cellular membrane. GPERs are involved in rapid non-genomic actions of estrogens, that are mediated by extracellularly activated kinase (ERK), cyclic adenosine monophosphate (cAMP) and Ca2. With regard to the cardiovascular system it is essential to note that stimulation of estrogen receptors results in activation of several rapid and long-lasting cellular processes which are essential for function of the heart and vessels. Functionally effective ESRs are present in the cardiac and vascular smooth muscle cells, and modulate function of the perivascular unit [92,93]. GPERs, ERα, ERβ and GPRE1 are widely represented in most cell types of the cardiovascular system and in the adipose tissue [89,94,95]. These processes of stimulation of ERs involve activation of phosphoinoisitide 3-kinase-serin/threonine-specific kinase B (PI3K/Akt /eNOS) and mitogen-activated protein kinase MAPK/e/NOS pathways, that are engaged in production of NO and play essential role in vasodilation. Moreover, it has been shown that in the cardiomyocytes estrogens regulate activity of calcium handling proteins, including L-type Ca2+ channel (LTCC), ryanodine channel (RYR), sarcoplasmic reticulum Ca2+ ATPase and sodium – calcium exchanger. Thus, it has been suggested that complex reciprocal interactions between activation of estrogen receptors and calcium signaling pathways may play essential role in the regulation of cardiomyocytes activities [76,96]. Furthermore, is has been reported that estrogens exert an antioxidant action and regulate cell contractility through effects exerted on calcium-dependent signaling pathways operating in cardiac mitochondria and sarcoplasmic/endoplasmic reticulum, where they modulate Ca 2+-ATPase 2a (SERCA2a) activity and function of calcium ion channels [76,97,98,99,100]. Altogether it is likely that in the heart deficiency of estrogens may result in disturbances of calcium homeostasis. Recently attention has been drawn to essential role of estrogens in the regulation of mitochondrial bioenergetics in human subjects [101]. It has been also reported that interaction of ERα with peroxisome proliferator-activator receptors (PPARs) causes repression of transactivation of the PPAR in the heart and vessels [89].
It is likely that estrogens may also influence function of the cardiovascular system through actions exerted in the brain as their receptors are widely expressed in multiple brain regions involved in the cardiovascular regulation, such as the frontal cortex, and sensorimotor cortex, the thalamus, the hypothalamus, the amygdala, the ventral tegmental area, the hippocampus, the dorsal raphe nucleus and the cerebellum [75,102]. Finally, it should be noted that activation of ERα and GPER1 plays significant role in modulation of immune processes that are mobilized in cardiovascular diseases [77,87,103].
Action of progesterone (P4) is mediated through the genomic signaling engaging two subtypes of nuclear receptors (PGRA, PGRB) and through non-genomic G-protein associated membrane-progestin receptors (mPRs). Some actions of P4 can be also exerted by GRs [104,105,106,107]
Neurosteroids. Some steroids, which are known as neurosteroids, have been identified in the nervous system and act preferably on neuronal membrane receptors. Among them are steroid sulfates, such as DHEAS which is a product of sulfation of DHEA [108]. It is suggested that neurosteroids play significant role in modulation of action of other steroids and classical neurotransmitters. Their action is not a subject of discussion of the present survey [see 109, 110 for further review of this topic] .
2.2. Genomic and Non-Genomic Effects of Vasopressin
Vasopressin, which is a principal vasopressin peptide in mammals, is synthesized mainly in neurons of the hypothalamic supraoptic, paraventricular and suprachiasmatic nuclei. Majority of axons of these neurons reach the posterior pituitary where AVP is released to the blood and can be distributed to peripheral organs. Some of the axons reach the median eminence and release AVP into the hypophyseal portal system and the anterior pituitary, where vasopressin contributes to regulation of ACTH. Vasopressin expressing cells have been also identified in the brain regions engaged in the regulation of blood pressure, metabolism, pain, stress and anxiety. Among them are neurons of the brain cortex, olfactory bulb, BNST, dorsomedial hypothalamic nucleus, nucleus of the diagonal band of Broca (DBB), circumventricular organs (CVO), brain stem, and spinal cord [111,112,113]. In addition AVP mRNA has been detected in the heart and vessels and in the pancreatic tissue [114,115].
Vasopressin gene is located in chromosome 20 and consists of three exons (A, B, and C). The exon A codes for a signal peptide, vasopressin peptide (Cys-Tyr-phe-Gln-Asn-Cys-Pro-Arg-Gly-NH2), a three amino-acid spacer, and the first nine (NH2 terminal) aminoacids of neurophysin (NP). Exon B codes for mid-portion of NP, whereas exon C codes for terminal portion of NP, a cleavage site, and the COOH-terminal glycopeptide (GP, known as copeptin). Copeptin consists of 39 aminoacids and is released from neurons in equimolar quantities with AVP [116,117]. Because copeptin molecule is more stable than vasopressin molecule, measurements of GP concentration are frequently used as a marker of AVP level. Measurements of GP level have been included into ESC guidelines on the myocardial infarction and as a biomarker of inflammation [118,119,120].
Expression of vasopressin gene is influenced by changes of body fluid osmolality and blood volume, as well as by constituents of the hypothalamo-hypophysial-adrenal axis, pain, cytokines and inflammation factors, especially those associated with COVID-19 [116,121,122,123,124,125]. Vasopressin gene expression and AVP mRNA abundance are enhanced by chronic osmotic simulation and are decreased in hypoosmolality. Osmotically-induced increase of AVP mRNA and release of AVP by neurons of the hypothalamo-neurohypophyseal axis are potentiated by administration of lipoplysaccharide and this is associated with enhanced release of IL-1β and IL-6 in the posterior pituitary [126]. Expression of AVP gene in the hypothalamus is potentiated by IL-1 and IL-2. Moreover, IL-1β stimulates release of CRH, AVP, and α-MSH [127,128].
It has been shown that hypoosmolality induces GRs expression and that this is related with corticosterone negative feedback on AVP transcription. The above data support the hypothesis that AVP gene is directly inhibited by glucocorticoids and that the induction of GR sin the hypothalamic cells suppresses AVP expression during prolonged hypoosmolality. [116]. However, it should be noted that during prolonged increase of corticosteroids concentration, such as takes place during autoimmune inflammation, vasopressin neurons can escape from glucocorticoids inhibition, presumably due to increased engagement of inflammatory cytokines [129].
Osmotic stimulation induces rapid upregulation of CRH in vasopressinergic neurons of the hypothalamic magnocellular nuclei [130]. There is evidence for coordinated regulation of AVP and CRH genes by glucocorticoids. It has been shown that adrenalectomy causes enhancement of CRH and AVP immunoreactivity in the hypothalamus and elevated CRH immunoreactivity in the cerebral cortex, the amygdala and the BNST. Since the stimulatory effect of adrenalectomy on expression of AVP and CRH in the hypothalamus could be reduced by administration of dexamethasone, it was concluded that glucocorticoids produced in the adrenal glands play primarily inhibitory role in regulation of AVP and CRH secretion [131]. The PVN neurons express glucocorticoids receptors and glucocorticoids reduce AVP gene expression in parvocellular neurosecretory neurons of the PVN [132]. AVP gene and CRH gene possess cAMP response elements (CRE), that are activated by intracellular cAMP and AP1 and AP2 transcription factors can be repressed by glucocorticoids. In the PVN neurons expression of CRH and AVP genes is regulated by nGRE and serum response element (SRE) [117,133,134]. Vasopressin neurons express also MRs and there is evidence that aldosterone and corticosterone increase ENaC Na+- leak current through an action exerted at the promoter region of the γ ENaC gene [135]. The magnocellular neurosecretory neurons of the PVN and SON, as well as of other brain regions express 11-βHSD2, and this increases their sensitivity for aldosterone [57,136].
It appears that during restraint stress the inhibitory effect of glucocorticoids on CRH and AVP mRNA expressions in the PVN neurons can be modulated by concomitant release of testosterone [137]. During chronic stress glutamergic, gamma-aminobutyric acid (GABA) and noradrenergic terminals exert a number of convergent actions that jointly regulate activity of CRF and AVP neurons of the PVN [138]. Growing evidence indicates that androgens play essential role in the regulation of neurons expressing CRH, AVP, dopamine and serotonin during stress-related behavior [139].
Vasopressin receptors. Arginine vasopressin stimulates two subtypes of V1 receptors (V1R), known as V1aR and V1bR, and one type of V2 receptors (V2R). The AVP receptors belong to a family of G-protein coupled receptors, which includes also receptors for other essential cardiovascular regulators, such as β-agonists, Ang II, endothelin, glucagon-like peptide 1 (GLP-1) and CRH. In high concentrations AVP interacts also with oxytocin receptors [113,140,141,142,143]. AVPR1a gene has been mapped to the 12q14.2 locus and AVPR1b to the 1q32.1 locus. AVP2R gene is located on the long arm of the X-chromosome (Xq28). Mutation of V2R gene is inherited in an X-linked manner and results in congenital nephrogenic diabetes insipidus, which is characterized by strong polydipsia and polyuria [144,145]. It has been shown that vasopressin receptors can act as homodimers and as heterodimers and it is likely that the dimerization influences effectiveness of stimulation of the target cells. Specifically formation of V1aR and OTR, and V1bR and corticotropin-releasing hormone receptor (CRHR) heterodimers has been well documented [146,147]. Expression of vasopressin receptors is regulated by corticosteroids [148]
Vasopressin V1aR mRNA and protein have been detected in multiple organs and tissues, including heart and vessels and their expression is altered in pathological processes [117,142,149]. In the cardiac ventricular sarcolemma vasopressin was found to open KATP channels through action exerted on V1R [150]. AVP was also found to reduce Ca2+ influx through L-type Ca2+ channels, and this effect can be abolished by blockade of V1aR [151]. There is evidence that stimulation of cardiac V1aR decreases cardiac beta receptors responsiveness [152].
In vitro experiments on H9c2 rat ventricle cardiomyocytes exposed to hypoxia revealed that AVP acting on V1aR in a V1aR/GRK2/β- arrestin1/ERK1/2- dependent manner, enhances cell survival. It has been suggested that during heart failure, when levels of circulating AVP are elevated, inhibition of G protein-coupled receptor kinase 2 (GRK2) can potentially exacerbate negative V1aR-mediated effects by preventing receptor desensitization and augmenting Gαq protein-dependent signaling [153,154]. In the heart stimulation of V1aR is also engaged in generation of pro-inflammatory cytokines and in development of inflammation and fibrosis. AVP increases IL-6 mRNA and protein levels in cardiac fibroblasts and this effect requires activation of G protein-coupled receptor kinase 2 (GRK2) and NF-κB [155]. In addition it has been shown that endotoxemia induced by lipopolysaccharide and concomitant increase of IL-1β, TNF-α and interferon gamma causes downregulation of V1aR gene expression in the heart, vessels, liver and lungs, as well as reduction of responsiveness of vascular smooth muscle cells. It is likely that diminished responsiveness of V1aR to vasopressin accounts for their inadequate stimulation during circulatory shock that can occur during endotoxemia [156].
Growing number of studies provide evidence for interaction of AVP with corticosteroids in other organs whose proper action is necessary for appropriate regulation of the cardiovascular functions, such as the brain and the gastrointestinal system. Administration of corticosterone was found to decrease expression of V1aR in the brain lateral septum and the hippocampus [148]. Stimulation of V1aR in cortical astrocytes was found to exert a neuroprotective action and this effect was associated with activation of the nuclear transcription factor cAMP response element-binding protein (CREB) and with prominent decrease of IL-1β and TNF-α gene expressions [150]. Vasopressin plays also essential role in the regulation of the gut microbiome [151]. Enhanced stimulation of V1aR may participate in habituation of release of ACTH, corticosterone and testosterone to repeated stress in Sprague Dawley rats [157].
V1b receptor mRNA is expressed in the corticotropic cells of the brain, and in the anterior pituitary, the adrenal gland, the heart, the kidney, the thymus, the lung, the spleen, the pancreas, the uterus and in the white adipose tissue [158,159,160]. In the brain V1bR mRNA was detected in the olfactory bulb, the cortex, the hippocampus, the hypothalamus, the septum and the cerebellum, although its expression was lower than expression of V1aR mRNA [160]. In the pancreas stimulation of V1bR potentiates secretion of insulin from β cells where vasopressin can act synergistically with CRH [160]. Thus far there is no convincing evidence for presence of V2R in the heart and vessels and for their direct involvement in the regulation of cardiovascular functions by vasopressin. Nevertheless through stimulation of V2R in the kidneys and through regulation of urine concentration and body fluid natremia AVP may modulate effects of stimulation of V1R in the cardiovascular system [157,158,159,160,161].
3. Role of the Hypothalamo-Pituitary- Adrenal System and Vasopressin in Regulation of Energy Balance and Water-Electrolyte Balance at Rest and during Neurogenic Stress
3.1. Regulation of Steroids Secretion
The synthesis of glucocorticoids in the adrenal cortex is regulated mainly by ACTH, which is released from the pituitary gland. Secretion of ACTH is closely regulated by CRH and AVP that are supplied principally by neurons of the PVN [163,164]. Glucocorticoids released from the adrenal cortex and ACTH secreted from the corticotropic cells of the pituitary gland are able to inhibit reciprocally secretion of CRH from the hypothalamic neurones [117,133,165] (Figure 1)
The secretion of glucocorticoids from the adrenal cortex is regulated by the circadian rhythm, however the daily rhythm of glucocorticoid release may be influenced by other factors, such as gender, aging, early life stress, inflammatory diseases, pregnancy and lactation [163,166]. ACTH binds on surface of cells of the fascicular layer of the adrenal cortex with the G protein-coupled melanocortin type 2 receptor (MC2R), which activates the 3′,5′-cyclic AMP signaling cascade, enabling the penetration of cholesterol into the inner mitochondrial membrane. Cholesterol is converted by cytochrome P450 to pregnenolone, which is converted in several steps to progesterone, 11-deoxycortisol, and cortisol (the main glucocorticoid in humans), or corticosterone (the main glucocorticoid in rodents) (Figure 2) [163].
ACTH stimulates also synthesis of aldosterone in the glomerular layer of the adrenal cortex. Aldosterone is synthesized from cholesterol in the inner mitochondrial membrane with the participation of aldosterone synthase (CYP11B2), belonging to the cytochrome P450 (CYP) family. The process of synthesis includes the formation of pregnenolone and conversion of pregnenolone to 11-deoxycorticosterone (DOC) in the smooth endoplasmic reticulum (Figure 2). Further processing to aldosterone takes place again in the mitochondria [167,168]. Synthesis of aldosterone in the adrenal cortex can be also stimulated by the renin-angiotensin system (RAS) and by extracellular potassium concentrations[167].
Androgens, as well as estrogen and progesterone, are released during activation of the HPA axis, which plays crucial role in the regulation of the ovarian cycle. The hypothalamic cells synthesize gonadotropin-releasing-hormone (GnRH), which stimulates the release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) from the anterior pituitary gland. In the woman FSH stimulates follicular maturation and release of estrogen in the follicular phase, while LH triggers ovulation as well as release of progesterone from the corpus luteum (ruptured follicle) in the luteal phase. Estrogens released from the ovaries and acting through reciprocal inhibition are able to slow down the activity of both the pituitary gland (synthesis and secretion of FSH and LH) and the hypothalamic neurons (synthesis and secretion of gonadotropin-releasing hormone), imposing thereby the menstrual cycle [169]. Estrogens are synthesized as a result of action of the aromatase, which is responsible for the aromatization of androgens into estrogens (Figure 2) [170]. The main site of aromatase expression in premenopausal women is located in the ovarian granulosa cells. In pregnant women it is also present in the placental syncytiotrophoblast. Both in men and in postmenopausal women, fibroblasts of adipose tissue and skin contribute to estrogen synthesis [171]. During the reproductive life women show characteristic periodicity of secretion of ovarian hormones, which corresponds to the monthly menstrual cycle. Namely, during the early follicular phase and menstruation, both estradiol (the main circulating estrogen in non-pregnant women of reproductive age) and progesterone levels are low. Estradiol concentration increases before the ovulation in the middle and late follicular phase, and decreases immediately after the ovulation. Subsequently, both estradiol and progesterone levels increase during the luteal phase until menstruation. Apart from the periovulatory phase, progesterone fluctuations correspond to estradiol fluctuations [169].
As already mentioned, androgens are synthesized mainly in the adult female ovaries and in the male testes. In addition, their synthesis is stimulated by ACTH in the reticular zone of the adrenal glands (mainly dehydroepiandrosterone sulfate, DHEA/-S; 4-androstenedione, A4 and 11β-hydroxyandrostenedione). Androgens are metabolized in the liver and subsequently excreted by the kidneys [172]. In the man the dominant androgen is testosterone; its concentration is 15 times higher than in the woman [173,174] Testosterone synthesis in men takes place in Leydig cells of the testes (Figure 1). Activation of the Leydig cell LH receptors (LHR, G protein-coupled receptor) by LH initiates an intracellular signaling cascade, which involves stimulation of adenylate cyclase, increased production of cAMP, and the activation of adenosine-dependent kinase [175]. As a consequence of activation of cAMP-dependent intracellular signal transduction, cholesterol can be transferred to the mitochondria by the steroidogenic acute regulatory protein (STAR), the translocator protein (18 kDa; TSPO), and other transducosome proteins [176]. In the mitochondria of Leydig cells, cholesterol is converted to pregnenolone by the cytochrome P450 enzyme located on the matrix side of the inner mitochondrial membrane, which cleaves the C27 cholesterol side chain (CYP11A1). Subsequently, after transfer to the smooth endoplasmic reticulum, pregnenolone is metabolized to testosterone by 3β-hydroxysteroid dehydrogenase (3b-HSD; HSD3B), 17α-hydroxylase/17,20 lyase (CYP17A1), and 17β-hydroxysteroid dehydrogenase type 3 (17b-HSD3, HSD17B) (Figure 2) [177,178,179].
3.2. Interaction of Steroids and AVP in Regulation of Energy Balance
Multiple studies indicate that both steroid hormones and vasopressin play an important role in regulation of energy balance. Glucocorticoids promote glucose production in the liver and reduce peripheral glucose uptake by muscle and adipose tissue. They also increase the breakdown of constituents of fat and muscle, providing thereby additional substrates for glucose and free fatty acids production [180].
Vasopressin is involved in regulation of food intake and energy balance via a wide range of central and peripheral actions exerted on cellular growth and proliferation, protein turnover, lipid metabolism, and glucose homeostasis [181]. For instance, endogenous nucleobindin-2 (NUCB2)/nesfatin-1 has been shown to regulate AVP and oxytocin in the PVN of male mice, and this was associated with resetting of fluid balance and food intake, and with an increase of body weight without the change of energy expenditure [182]. Experiments on mice showed that administration of AVP analogue (Ac3IV) twice a day for 22 days not only reduced energy consumption, body weight and fat content, but also decreased blood glucose concentration, increased insulin sensitivity and significantly improved glucose tolerance and glucose-induced insulin secretion. Moreover, it lowered total cholesterol, low density lipoprotein (LDL) cholesterol and triglycerides, and increased high density lipoprotein (HDL) cholesterol [183]. Several hormones affecting energy balance play significant role in regulation of AVP secretion. Ghrelin, an orexigenic hormone, has been shown to stimulate vasopressinergic neurons in the hypothalamic paraventricular nucleus and AVP secretion in a nutritional status-dependent manner in in vivo experiments. It also activated excitatory GABAergic synaptic input via a retrograde neuron-glial circuit in the hypothalamic slices obtained from fasted rats [184]. On the other hand, i.c.v. administration of adiponectin significantly reduced basal plasma AVP concentrations in conscious rats in a dose-dependent manner, with the maximum effect achieved 10 min after administration [185].
Additionally, it appears that AVP may exert thermogenic functions, as exposure of brown adipose tissue (BAT) adipocytes to AVP was found to increase uncoupling protein-1 (UCP-1) protein expression, to induce a time- and dose-dependent increase in p38 MAP kinase phosphorylation, as well as to increase monocyte chemoattractant protein-1 (MCP-1) mRNA and IL-6 expressions. In contrast adiponectin mRNA expression was reduced [186]. Former studies have shown that acting in the adipose tissue on V1aR AVP may exert both lipolytic and antilipolytic effects [187,188]. Its action on lipid metabolism through V1bR is different. It has been shown that V1bR deficient mice have lower body weight and greater epididymal adipose tissue mass in comparison to wild-type mice. It is believed that AVP acting on V1bR receptors is able to influence lipid metabolism through modulation of insulin signaling. Currently it appears that stimulation of V1aRs results in the impairment of glucose tolerance and in the lipolytic action, whereas activation of V1bRs improves glucose tolerance and exerts an antilipolytic effect [189,190]
Elevated plasma copeptin (AVP substitute) concentrations have been demonstrated in patients with type 2 diabetes and with type 1 diabetes [191]. Moreover, recent studies indicate that higher concentrations of AVP or copeptin predispose to development of type 2 diabetes and metabolic syndrome and that consumption of larger amounts of water by people with high plasma copeptin concentrations results in reduction of fasting glucose or glucagon levels [192]. Studies on human populations revealed an association of polymorphisms of the gene encoding AVP with metabolic disorders. A significant relationship was found between high copeptin level and reduced insulin sensitivity as well as between AVP gene tagSNPs (CC genotype rs6084264, TT genotype rs2282018, C allele rs2770381 and CC genotype rs1410713) and the incidence of hyperglycemia [193]. Similarly, specific polymorphism of V1aR gene (T allele of rs1042615) has been associated with an increased incidence of type 2 diabetes in people who consume large amounts of fat or are overweight [194]. In another study the major A allele of rs35810727, a tagSNP of the V1bR, has been associated with an increased body mass index (BMI), and type 2 diabetes [195].
3.3. Interaction of AVP and Steroids in Regulation of Water-Electrolyte Balance
It has been well established that the vasopressin system plays pivotal role in the regulation of water and electrolyte balance and that lack of AVP results in excretion of large amounts of free water in the urine (polyuria) and in subsequent polydipsia [112,196,197]. Release of AVP is regulated by a variety of internal and external cooperating factors, acting as anticipatory or consecutive signals [192,198,199]. Regulation of water-electrolyte balance via AVP is a complex process in which the osmolality of the extracellular fluid (ECF), including plasma and cerebrospinal fluid, plays a key role. ECF tonicity is sensed by osmoreceptive neurons located mainly in the subfornical organ (SFO) and the organum vasculosum of the lamina terminalis (OVLT) which are able to sense the osmolarity using aquaporin (AQP) receptor, and have direct connections with the PVN and SON [200,201]. Osmotically-induced shrinkage of osmoreceptive cells during dehydration causes activation of TRPV1 delta-N channels (a family of transient receptor potential cation channels) that sense mechanical stretch and allow the influx of cations into the cell [200,202]. In addition activity of the brain osmoreceptors may be regulated by anticipatory signals generated in receptors of the oropharyngeal region, esophagus or stomach, which participate in the regulation of fluid intake. Specifically, the information about the taste of water is transmitted centripetally by the tympanic cord belonging to the facial nerve, the information about the dry mouth is received via the trigeminal nerve and the information about the stretching of the esophagus and stomach and volume of water ingested are transmitted by the glossopharyngeal nerve and by the vagus nerve [203].
In the kidneys AVP regulates the activity of the aquaporin-2 (AQP2) water channel in the collecting duct and takes part in the process of urine concentration [204]. Moreover, it stimulates sodium reabsorption acting on the luminal sodium channel ENaC in the cortical and outer medullary parts of the collecting tubules, and activates urea transporters UT-A1 and UT-A3 in the inner terminal medullary increasing urea reabsorption in the collecting tubules [205,206]. Additionally, at higher concentrations, AVP increases also sodium reabsorption through activation of Na-K-2Cl cotransporters (NKCC2) in the thick ascending limb of the nephron loop (Henle’s loop) [207].
There is strong evidence that the regulation of water-electrolyte balance by vasopressin is significantly influenced by steroid hormones. Experimental studies have shown that glucocorticoids deficiency facilitates activation of V2 receptors in the renal collecting tubule [208].It has been also shown that application of dexamethasone (a cortisol analogue) improves cardiac functions in rats with congestive heart failure elicited by coronary artery ligation, and this effect is associated with reduced expression of V2R and AQP2 and AQP3, and with reduced number of ENaC and Na-K-2Cl cotransporters 3 in the renal collecting tubule [209]. The effects of dexamethasone were abolished by the use of the glucocorticoid receptor inhibitor RU486 [209]. However, the clinical studies could not confirm significant involvement of glucocorticoids applied alone in fluid retention in critically ill patients and it has been postulated that effects of steroid hormones on water-electrolyte balance are mediated mainly by activation of mineralocorticoid receptors [210]. It is wells known that mineralocorticoids have a significant impact on electrolyte balance, especially on sodium and potassium turnover as well as on clearance of free water [211,212]. Moreover, components of the renin-angiotensin-aldosterone system (RAAS), acting in the hypothalamus are able to stimulate vasopressin secretion, thirst, and sodium appetite [213].
Currently it appears that also gonadal steroids have a significant effect on the vasopressinergic system [214]. Experimental studies revealed that the antidiuretic function of AVP is more effective in male rats than in female rats [215]. Furthermore, its effect is more potent in females during the estrus when the level of circulating estrogen is lowest [215]. In addition, ovariectomy was found to increase the antidiuretic response to AVP to the level comparable to that observed in males, whereas estradiol substitution reduced the antidiuretic effect of AVP to the level observed in non-estrus females [215]. It appears that estrogens may also influence the hypothalamic vasopressinergic neurons acting both through ERα and ERβ receptors. In this context presence of ERα on osmoreceptive neurons of the SFO and OVLT, which send projections to the SON has been demonstrated [216,217,218]. Moreover, both ERα and ERβ have been detected in the kidney, where the expression of ERα is prevailing [219]. Interestingly, GPER is a newly discovered aldosterone receptor that is believed to mediate nongenomic aldosterone pathways. GPER activation by aldosterone mediates water and sodium retention in the body and contributes to vasoconstriction [220]. Recently it has been shown that production of aldosterone may be influenced by progesterone [221].
3.4. Interaction of AVP and Steroids in Neurogenic Stress
A number of studies provide evidence that neurogenically mediated stress causes joint stimulation of the vasopressin secreting neurons and CRH secreting neurons which suggests that a coordinated regulation of AVP and CRH release and action may play an important role in modulation of neurogenic stress. Moreover, it has been found that in chronic stress and depression the effects of CRH on ACTH release are strongly enhanced by vasopressin, which is produced in increasing amounts when the hypothalamic PVN and SON neurons are chronically activated [223,224]. It has been found that AVP is engaged in stress-induced tachycardia and baroreceptor reflex (BRR) desensitization [225]. Clinical studies have shown that healthy adults exposed to a social stress test respond with an increase in serum copeptin concentration [223]. Experiments on mice showed that chronic unpredictable stress (UCS and ovariectomy influence AVP immunoreactivity in the lateral magnocellular and the medial magnocellular subdivisions of the PVN [226]. More recently it has been shown that during the chronic stress, hypothalamic activation of pituitary changes from the dominant CRH phenotype to the dominant AVP phenotype, however cortisol level remains elevated because its metabolism is reduced [227]. The role of vasopressin in the response to stress appears to differ in female and male C57BL/6 mice. Namely, a six-week chronic variable stress (CVS) paradigm has been found to increase sociability in female mice and to decrease AVP mRNA in the PVN, whereas in male mice, CVS had no effect on social behavior or AVP expression [228]. Studies conducted on male Sprague Dawley rats subjected to a 4-week unpredictable chronic mild stress (UCMS) showed that injections of small interfering RNA (siRNA) directed against V1aR into the PVN prevent the increase in blood pressure and the elevation of renal sympathetic nerve activity (RSNA), whereas administration of scrambled RNA (scrRNA) into the PVN elicit an increase in blood pressure. The results suggested that V1aR signaling in the PVN contribute to generation of neuro-cardiovascular responses to stress [229]. In addition recent studies provided evidence that the rs10877969 polymorphism of V1aR gene is associated with elevation of symptoms of stress and acute pain [230]. Research performed by Komnenov et al. (2021) showed that rats subjected to UCMS have higher plasma AVP levels and greater abundance of V1aR and V1bR transcripts in the PVN. The rats manifested also higher resting MAP, heart rate, and RSNA and these effects could be eliminated by combined inhibition of V1aR and V1bR [231]
4. Altered Interactions of Vasopressin with the Hypothalamo-Pituitary-Adrenal System in Cardiovascular and Metabolic Diseases
4.1. Cardiovascular Diseases
Growing evidence indicates that central and peripheral interactions of steroid hormones and vasopressin are reprogrammed in hypertension and that the occurring modifications may initiate and/or potentiate cardiovascular complications. Pietranera et al. (2004) revealed that s.c. injections of deoxycorticosterone (DOC) acetate cause significantly greater increases of AVP and V1aR mRNA in the magnocellular divisions of the PVN in the spontaneously hypertensive (SHR) rats than in the control SHR rats receiving oil vehicle [232]. It is possible that AVP contributes to elevation of blood pressure in DOCA-induced hypertension through augmentation of the neurogenic component of vascular resistance. Studies conducted on DOC- treated and saline-treated rats revealed a significant increase in vascular resistance that was associated with amplification of the central sympathetic tone only in DOC-salt rats. These effects were intensified by vasopressin. Moreover, a reduction in vascular resistance was observed in DOC-salt rats treated with AVP antagonist (I-deaminopenicillamine, 4-valine, 8-D-arginine vasopressin, dPVDAVP) and in those who underwent lumbar sympathectomy [233]. Experiments on dogs maintained on normal salt diet showed that subcutaneous administration of DOC to dogs elicited an increase of blood pressure, that was accompanied by an increase of cardiac output, hypernatremia, and elevation of vasopressin concentration in plasma and in the cerebrospinal fluid (CSF) at early stage of hypertension [234].
Some evidence suggests that inappropriate interaction of steroid hormones and vasopressin may play a role in development of congestive heart failure. For instance, it has been reported that treatment of the sheep with paced-induced heart failure with urocortin 2 (Ucn2; group of peptides sharing structural similarities with CRH combined with canrenoic acid, which is an antagonist of mineralocorticoid receptor improved better hemodynamics than application of canrenoic acid alone. The combined treatment contributed also to reductions of PRA, Ang II and AVP concentrations, as well as to improvement of the kidneys functions [235].
4.2. Metabolic Diseases
Growing number of studies address a question whether interactions of AVP and steroids are altered in metabolic diseases. Clinical data, based on measurements of blood copeptin concentration, suggest that AVP may play a role in pathogenesis of the metabolic syndrome (MetS). In this line, obese men with elevated fat free mass and total fat mass show higher fasting glucose and insulin concentrations as well as enhanced pituitary response to combined CRH/AVP stimulation. The results suggest that joint stimulation of CRH and AVP, which is associated with excessive stimulation of ACTH release may promote development of body overweight, adiposity and hyperinsulinemia [236]. There is also evidence that obesity causes significant rearrangement of activation of the HPA by the noradrenergic system. For instance noradrenergic transporter activity (NAT) assessed in positron emission tomography (PET) was found to correlate differently in obese and non-obese patients. In the non-obese patients, a positive correlation was found between orbitofronal NAT and HPA axis activity, whereas in the obese patients the correlation was negative. The study suggested that in the obese patients regulation of HPA axis by the noradrenergic system activates mainly the hypothalamic neurons whereas in the non-obese subjects it engages more intensively the prefrontal-limbic cortex [237]. It has been also postulated that altered association of copeptin and insulin resistance is related to elevation of hepatic glycogenolysis, increased insulin, glucagon and ACTH secretions, and enhanced activation of 11β-HSD2 [238]. Links between specific steroid hormones and vasopressin in hypoglycemia are less evident. It has been reported that insulin-induced hypoglycemia increases secretion of cortisol, but it does not affect plasma AVP level [239]. In another study more drastic hypoglycemia (1.6 mmol/l), probably producing hypoglycemic stress, caused a rapid significant increase of serum copeptin concentration, which was positively correlated with ACTH and cortisol concentrations. Interestingly, these effects were observed only in women. In the man, there was no correlation between copeptin and blood ACTH level during hypoglycemia and only poor correlation was found between copeptin and blood cortisol level during hypoglycemia [240]. Studies conducted on AVP-deficient Brattleboro rats that were also suffering from diabetes mellitus showed that AVP does not play a significant role in the regulation of release of the HPA axis hormones during acute stress [241].
Balapattabi et al. (2021) examined associations of the liver failure with AVP secretion and hyponatremia in relevance to sex differences. They showed that male rats with a bile duct ligation (BDL) were hyponatremic, manifested significantly higher concentrations of plasma copeptin, and higher FosB expression in the supraoptic AVP neurons than the sham males, whereas similar associations were not observed in female BDL rats [242].The study conducted in women with polycystic ovary syndrome, revealed that AVP peak responses to hypoglycemia were negatively correlated with testosterone, androstenedione, and endogenous insulin levels, however, there was no correlation between AVP and basal and hypoglycemia-induced peak cortisol concentrations [243].
4.3. Neurogenic Stress with Relevance to Sex Differences
It is well known that there are sex differences in tolerance of stress, and in predisposition to anxiety, and depression, and that they may at least partly result from different regulation of the HPA axis [244,245]. Experimental studies demonstrated that the female rats respond with larger increases of ACTH to neurogenic stress than the male rats and suggested that during the neurogenic stress the gonadal steroids involved in the regulation of sexual behavior and reproduction may have a potential impact on the pituitary secretion of ACTH [246]. Subsequently it has been shown that secretion of ACTH is regulated by testosterone-dependent effect on the synthesis of AVP and by corticosterone-dependent effect on the synthesis of CRH in the PVN [247]. It is likely that one of the reasons of differences in sex-related susceptibility to stress results from functional heterogeneity of GR and MR in the brains of females and males, because higher expression of MR was found in the brain of the male mice than in the brain of female mice manifesting depressive behavior [248]. Research performed by Woodward et al. (2023) on female and male mice, subjected to the UCMS procedure showed that female mice show anxiety and depressive behavior associated with FosB activation in neurons of the medial prefrontal cortex (mPFC) expressing parvalbumin after 4 weeks of exposure, while in male mice the behavioral and biochemical alterations is observed not earlier than after 8 weeks of UCMS. Furthermore, the use of patch-clamp electrophysiology allowed to demonstrate that there were time-corresponding sex-specific differences in altered neuronal excitability of the mPFC cells after 4 and 8 weeks of the exposure [245]. Rosinger et al. (2019) using a corticotropin-releasing factor 1 receptor (CRFR1) reporter mouse line, demonstrated that male mice showed a significantly higher distribution of cells expressing CRFR1 in the PVN compared to females, however it should be noted that this relationship was age-related, because it was observed only in older (20-24-month) mice, and not in mice during the early post-natal life. Gonadectomy in adult six-week-old mice resulted in a significant decrease in the number of CRFR1-immunoreactive cells in the PVN in males but not in females. In addition CRFR1 cells showed moderate co-expression with estrogen receptor alpha and high co-expression with androgen receptor. The use of restraint stress resulted in greater activation of CRFR1 cells in the PVN of male than in the PVN of female mice [249]. Cox et al. (2015) examined the association of X chromosome genes with neurobehavioral disorders in an experimental model (fragile X syndrome, autism) using a mouse model with an atypical sex chromosome configuration resembling Turner (45, XO) and Klinefelter (47, XXY) syndromes. The researchers showed higher AVP expression in the amygdala of female mice with one copy of X-chromosome gene. Reduced level of plasma AVP was found in girls with Turner syndrome [250].
Recent clinical studies of Cohen et al., (2023) have revealed that gender differences in reaction to stress occur already in young people aged 18-25, and they may be related to different activation of the prefrontal cortex. Using GABA in magnetic resonance spectroscopy the authors were able to demonstrate significant differences in neuronal activity in responses to stress among the sexes. It should be noted that the differences were especially evident in the ventromedial prefrontal cortex, which plays an essential role in the regulation of the hypothalamic-pituitary, hypothalamic-pituitary adrenal and hypothalamic-pituitary-gonadal axes [251].
5. Impact of Therapeutic Interventions on Interactions of Vasopressin with the Hypothalamo-Pituitary-Adrenal System in Health and Cardiovascular and Metabolic Diseases
As discussed in former parts of this review there are several interconnections between steroids and vasopressin that are significantly affected in cardiovascular, metabolic and psychogenic disorders [157,168,237]. Thus it is justified to assume that various therapeutic interventions, having impact on vasopressinergic system and the hypothalamo-pituitary-adrenal axis. may potently influence effectiveness of the AVP-HPA interactions.
Systemically applied corticosteroids are widely used in therapy of inflammatory and autoimmune disorders [163,236,252]. Their influence on the HPA axis and vasopressin secretion resembles the role of endogenous corticosteroids in negative feedback loop. A post-mortem study of Erkut et al.(1998) on 9 patients treated with corticosteroid and on 8 control subjects demonstrated significant decrease of CRH and AVP releasing neurons in the hypothalamus. The study showed that the corticosteroid-treated patients retained only 3.3% of CRH releasing cells and 33% of AVP-releasing cells in comparison to the control group [253]. Thus far, clinical implications of these changes have not been fully recognized. A study on premature infants with bronchopulmonary dysplasia revealed that treatment with dexamethasone improves pulmonary functions, but does not modify AVP levels [254]. On the other hand, administration of corticosteroid in supraphysiological doses in the clinical trial on children with acute lymphoblastic leukaemia (ALL) resulted in suppression of the HPA axis for a few weeks in majority of patients. [255]. The decrease in AVP mRNA was also found in the SChN of patients treated with corticosteroid (2-fold lower level in the SChN of patients exposed to corticosteroid in comparison to the control subjects). Accordingly, it has been proposed that corticosteroid-induced changes in AVP synthesis may account for circadian rhythms abnormalities and sleep disturbances in steroid-treated patients [256].
It has been postulated that vasopressin synthesized in magnocellular cells of the hypothalamus participates in a phenomenon known as „corticosteroid escape”. According to this hypothesis inflammatory mediators (for instance IL-6) increase AVP release from the hypothalamic magnocellular PVN cells above the physiological concentrations, which results in strong activation of adenylyl cyclases (AC2 and AC7) and elevation of cAMP to a level, at which it can inhibit the negative feedback mediated by endogenous corticosteroids in the HPA axis. Most likely, rearrangement of mutual relations between CRH, AVP, interleukins and corticosteroids accounts for the “corticosteroid escape” that is observed during activation of the host immune system and that maintains corticosteroid release in spite of high dose of exogenous corticosteroid imposed by the therapy (Figure 3) [257]. However, the role of the „corticosteroid escape” in achievement of optimum results of systemic corticosteroid therapy is not yet fully determined.
5.1. Steroids and Vasopressin Treatment in Cardiovascular Diseases
It is likely that resetting of vasopressin-HPA system by corticosteroids plays beneficial role in treatment of severe cardiovascular disorders such as myocardial infarction, shock and cardiac arrest. A study on the rat model of myocardial infarction revealed that hydrocortisone administration during early reperfusion period results in decreased infarct size and in reduced oxidative stress [258]. The potentially beneficial role of corticosteroids may result from their anti-inflammatory activity, thogh there are some concerns regarding the use of corticosteroids in acute myocardial infarction because of their negative effects on wound healing and scar formation. The meta-analysis performed by Giugliano et al [259] indicated slight reduction in cardiovascular mortality in patients with acute myocardial infarction treated with corticosteroids. However, it should be noted that this survey was based mainly on studies including small number of patients (less than 100) and did not equivocally indicated benefit of treatment with corticosteroids [259]. In another study the best survival rate (91%) was found in patients with low baseline cortisol level and with appropriate adrenal response to ACTH analogue (difference between minimum and maximum cortisol levels higher than 9 μg/dl) [260]. A study on a cohort of 159 patients with septic shock revealed that survival rate was significantly greater when vasopressin was used together with hydrocortisone [261]. This finding suggested a potential benefit of joint application of corticosteroids and vasopressin in critically ill patients. More recently, randomized clinical trial on 512 patients of 10 hospitals in Denmark showed significant increase in probability of return of spontaneous circulation (ROSC) in patients receiving combined treatment of methylprednisolone and vasopressin in comparison to patients receiving placebo. However, there was no difference in 30-days mortality and neurological outcome of the patients [262].
5.2. Impact of Anti-Depressive and Neuroleptic Treatments on Vasopressin-HPA Interactions
As it was mentioned above, the vasopressin-HPA system is controlled by monoamine neurotransmitters, and administration of anti-depressive or other neuroleptic compounds, which are interfering with monoaminergic transmission, significantly influences AVP-HPA interactions (see 3.3). Moreover, it has been reported that activation of the HPA system during stress seems to be altered in patients with depression, and that vasopressin plays more important role than CRH in regulation of ACTH release in this group of patients [263,264]. Altered action of vasopressin in patients with depressive and stress-related disorders opens a discussion on the usefulness of selective AVP antagonists as anti-depressive or anxiolytic drugs [264,265,266]. Undoubtedly, further studies are needed to explain whether down-regulation of the vasopressinergic pathways would play a beneficial role in these disorders.
The study upon animal model of depression (olfactory bulbectomy in mice, OB mice) revealed that OB mice demonstrated higher vasopressin and ACTH plasma levels. Moreover , it was possible to revers theses elevations by chronic treatment with anti-depressive drugs, namely fluoxetine (SSRI) and venlafaxine (SNRI) [267]. Another study on healthy rats showed that antidepressant compounds, such as SSRI and desipramine (tricyclic antidepressant) influence function of HPA axis by means of vasopressin V1bRs [268]. Studies on Wistar rats have shown that AVP release from magnocellular and parvocellular cells may be affected by neuroleptic drugs and that clozapine and olanzapine are more effective than haloperidol [269].
A study upon six patients with atrial fibrillation qualified for electrical cardioversion and 6 patients with depression qualified for electroconvulsive therapy revealed significantly enhanced activation of HPA after these interventions. In particular, AVP concentration increased 7 times after electrical cardioversion and 2 times after electroconvulsive therapy, whereas plasma ACTH levels were elevated 3 times and 4 times, respectively [270].
6. Summary and Conclusions
The present review analyses complex mechanisms underlying cooperation of vasopressin with hormones of the hypothalamo-pituitary-adrenal axis. As shown in Figure 4 AVP cooperates with specific components of the HPA axis through interactions occurring both in the central nervous system and in the peripheral organs.
The cooperation plays significant role in the regulation of blood circulation, metabolism, water-electrolyte balance and behavioral adaptation to stress challenges. Vasopressin and components of the HPA axis closely interact and can be considered as the functionally united AVP-HPA system. Growing evidence shows that cardiovascular and metabolic diseases as well as inflammation and stress are associated with an inappropriate function of the AVP-HPA system and this question should be taken into consideration when the pharmacological treatment is planned.
7. Conclusions
- Vasopressin (AVP) and steroid hormones are frequently released together and closely cooperate in the regulation of blood pressure, metabolism, water-electrolyte balance and behavior.
- Vasopressin interacts with specific components of the hypothalamo-pituitary-adrenal axis in the brain and in several peripheral organs and tissues, including the heart, vessels, kidney and adipose tissue.
- Appropriate interactions of AVP with the HPA axis is essential for efficient regulation of the water-electrolyte balance, blood pressure, and energy balance, and it is justified to consider vasopressin and the hypothalamo-pituitary axis as the highly coordinated functional AVP-HPA system.
- Interactions between AVP and HPA are significantly altered in the cardiovascular, respiratory and metabolic diseases and during inflammation and the neurogenic stress.
- Inappropriate interactions of AVP with steroid hormones may initiate or potentiate cardiovascular complications and metabolic diseases.
- The interplay of vasopressin and steroid hormones is not yet fully recognized and further studies are needed to determine potentially beneficial or harmful consequences of interference with these factors in treatment of specific pathological states.
Author Contributions
E.S.-S proposed the design and the final version of the review; E.S.-S., K.C., W.B.-R., and M.K. searched the literature; K.C. and M.K. designed figures. All authors wrote the first draft of the manuscript. All authors have read and accepted the final version of the manuscript.
Funding
The study was supported by the Medical University of Warsaw Scientific Projects 1MA/N. The work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors wish to thank to Mr Marcin Kumosa from the Department of Experimental and Clinical Physiology of the Medical University of Warsaw for the technical preparation of figures.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
ACE1: ACE2 - angiotensin converting enzymes, Ach – acetylcholine, ACTH – adrenocorticotropic hormone , AGT – angiotensinogen, AP – area postrema, AT1R, AT2R - angiotensin receptors, AVP – arginine vasopressin, BDL – bile duct ligation, cAMP – cyclic adenosine monophosphate, CBF – coronary blood flow, CBI – Coactivator Binding Inhibitor, CES – coronary endothelial cell, COVID - 19 – coronavirus disease 2019, CRF – cortictropin releasing factor, CRH – corticotropin-releasing hormone, dDAVP – desmopressin, an analog of AVP, dVNc – dorsal nucleus of vagus nerve, ERK – extracellular signal-regulated kinase, EAT – epicardial adipose tissue, EDR – endothelium dependent relaxation, ET – endothelin, GABA – gamma aminobutyric acid GLUT – glucose transporter, GRK2 - G protein-coupled receptor kinase 2, HDL – high density lipoprotein, HOMA – homeostatic model assessment, HIF – hypoxia inducible factor, ICV – intracerebroventricular , IL – interleukin, IRAP – insulin regulated aminopeptidase, IRS –insulin receptor substrate, ISI insulin sensitivity index, JNK – Jun N-terminal kinase, LDL – low density lipoprotein, L-NAME - N(ω)-nitro-L-arginine methyl ester, LV – left ventricle, MAPK - mitogen-activated protein kinase, MasR - MAS receptor for angiotensin-(1-7), MCP-1 - monocyte chemoattractant protein-1, MPTP - 1-methyl-4phenyl-1,2,3,6-tetrahydropyridine, mTOR – mammalian target of rapamycine, NADPH - dinicotinamide adenine dinucleotide phosphate, NAT - noradrenergic transporter activity, NcAmb – nucleus ambiguous, NCOAS – nuclear receptor co-activator, NFκB – nuclear factor kappa B, NO – nitric oxide, NTS – nucleus of the solitary tract, OVLT – organum vasculosum of the lamina terminalis, PET - positron emission tomography, PLA2 - phospholipase A2, PLC- phospholipase C, PVN – parventricular nucleus, RAS – renin angiotensin system, RAAS – renin-angiotensin-aldosterone system, ROS – reactive oxygen species, ROSC- return of spontaneous circulation, RVLM – rostral ventrolateral medulla, SFO – subfornical organ, SHR – spontaneously hypertensive rat, SON – supraoptic nucleus, SNP - single nucleotide polymorphism, SRC – steroid receptor co-activator, TNF-α- tumor necrosis factor alpha, T1DM1 – diabetes mellitus of type 1, T1DM2 – diabetes mellitus of type 2, UCMS - unpredictable chronic mild stress UCP – uncoupling protein, UT – urea transporter, VSMC – vascular smooth muscle cell, V1aR – vasopressin receptor of type V1a, V1bR – vasopressin receptor of type V1b, V2R – V2 vasopressin receptor of type 2, VS – vasopressin system, WKY – Wistar Kyoto rat, ZDF – Zucker diabetic fat.
References
- Hilton JG, Scian LF, Westermann SD, Nakano J, Kruesi OR. Vasopressin stimulation of the isolated adrenal glands: nature and mechanism of hydrocortisone secretion. Endocrinology. 1960;67:298-310. [CrossRef]
- Larsen PJ, Vrang N, Møller M, Jessop DS, Lightman SL, Chowdrey HS, Mikkelsen JD. The diurnal expression of genes encoding vasopressin and vasoactive intestinal peptide within the rat suprachiasmatic nucleus is influenced by circulating glucocorticoids. Brain Res Mol Brain Res. 1994;27(2):342-346. [CrossRef]
- Mouri T, Itoi K, Takahashi K, Suda T, Murakami O, Yoshinaga K, Andoh N, Ohtani H, Masuda T, Sasano N. Colocalization of corticotropin-releasing factor and vasopressin in the paraventricular nucleus of the human hypothalamus. Neuroendocrinology. 1993;57(1):34-39.
- Otubo A,Kawakami N, Maejima S, Ueda Y, Morris JF, Sakamoto T, Sakamoto H. Vasopressin gene products are colocalised with corticotrophin-releasing factor within neurosecretory vesicles in the external zone of the median eminence of the Japanese macaque monkey (Macaca fuscata). J Neuroendocrinol. 2020;32(8):e12875. [CrossRef]
- Engler D, Pham T, Fullerton MJ, Ooi G, Funder JW, Clarke IJ. Studies of the secretion of corticotropin-releasing factor and arginine vasopressin into the hypophysial-portal circulation of the conscious sheep. I. Effect of an audiovisual stimulus and insulin-induced hypoglycemia. Neuroendocrinology. 1989;49(4):367-381. [CrossRef]
- Familari M, Smith AI, Smith R, Funder JW. Arginine vasopressin is a much more potent stimulus to ACTH release from ovine anterior pituitary cells than ovine corticotropin-releasing factor. 1. In vitro studies. Neuroendocrinology. 1989;50(2):152-157. [CrossRef]
- Labrie F, Giguere V, Proulx L, Lefevre G. Interactions between CRF, epinephrine, vasopressin and glucocorticoids in the control of ACTH secretion. J Steroid Biochem. 1984;20(1):153-160. [CrossRef]
- Veldhuis HD, de Kloet ER. Vasopressin-related peptides increase the hippocampal corticosterone receptor capacity of diabetes insipidus (Brattleboro) rat. Endocrinology. 1982;110(1):153-157. [CrossRef]
- Papanek PE, Sladek CD, Raff H. Corticosterone inhibition of osmotically stimulated vasopressin from hypothalamic-neurohypophysial explants. Am J Physiol. 1997;272(1 Pt 2):R158-162. [CrossRef]
- Liu X, Wang CA, Chen YZ. Nongenomic effect of glucocorticoid on the release of arginine vasopressin from hypothalamic slices in rats. Neuroendocrinology. 1995;62(6):628-633. [CrossRef]
- Calogero AE, Liapi C, Chrousos GP. Hypothalamic and suprahypothalamic effects of prolonged treatment with dexamethasone in the rat. J Endocrinol Invest. 1991;14(4):277-286. [CrossRef]
- Woodcock EA, Mcleod JK, Johnston CI. Vasopressin stimulates phosphatidylinositol turnover and aldosterone synthesis in rat adrenal glomerulosa cells: comparison with angiotensin II. Endocrinology. 1986;118(6):2432-2436. [CrossRef]
- Saito R, Ishiharada N, Ban Y, Honda K, Takano Y, Kamiya H. Vasopressin V1 receptor in rat hippocampus is regulated by adrenocortical functions. Brain Res. 1994;646(1):170-174. [CrossRef]
- Watters JJ, Poulin P, Dorsa DM. Steroid hormone regulation of vasopressinergic neurotransmission in the central nervous system. Prog Brain Res. 1998;119:247-261. [CrossRef]
- Watters JJ, Wilkinson CW, Dorsa DM. Glucocorticoid regulation of vasopressin V1a receptors in rat forebrain. Brain Res Mol Brain Res. 1996;38(2):276-284. [CrossRef]
- Rabadan-Diehl C, Makara G, Kiss A, Lolait S, Zelena D, Ochedalski T, Aguilera G. Regulation of pituitary V1b vasopressin receptor messenger ribonucleic acid by adrenalectomy and glucocorticoid administration. Endocrinology. 1997;138(12):5189-5194. [CrossRef]
- Hu SB,Tannahill LA, Biswas S, Lightman SL. Release of corticotrophin-releasing factor-41, arginine vasopressin and oxytocin from rat fetal hypothalamic cells in culture: response to activation of intracellular second messengers and to corticosteroids. J Endocrinol. 1992;132(1):57-65. [CrossRef]
- Currie IS, Gillies G, Brooks AN. Modulation of arginine vasopressin secretion from cultured ovine hypothalamic cells by glucocorticoids and opioid peptides. Neuroendocrinology. 1994;60(4):360-367. [CrossRef]
- Kokras N, Hodes GE, Bangasser DA, Dalla Sex differences in the hypothalamic-pituitary-adrenal axis: An obstacle to antidepressant drug development? C.Br J Pharmacol. 2019;176(21):4090-4106. [CrossRef]
- Orshal JM, Khalil RA. Gender, sex hormones, and vascular tone. Am J Physiol Regul Integr Comp Physiol. 2004;286(2):R233-R249. [CrossRef]
- Jiang YQ, Kawashima H,Iwasaki Y,Uchida K, Sugimoto K, Itoi K. Differential effects of forced swim-stress on the corticotropin-releasing hormone and vasopressin gene transcription in the parvocellular division of the paraventricular nucleus of rat hypothalamus. Neurosci Lett. 2004;358(3):201-204. [CrossRef]
- Hlavacova N, Bakos J, Jezova D. Eplerenone, a selective mineralocorticoid receptor blocker, exerts anxiolytic effects accompanied by changes in stress hormone release. J Psychopharmacol. 2010;24(5):779-786. [CrossRef]
- de Kloet ER, Meijer OC, de Nicola AF, de Rijk RH, Joëls M. Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front Neuroendocrinol. 2018;49:124-145. [CrossRef]
- Karst H, Berger S, Turiault M, Tronche F, Schütz G, Joëls M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci U S A. 2005;102(52):19204-19207. [CrossRef]
- Karst H, den Boon FS, Vervoort N, Adrian M, Kapitein LC, Joëls M. Non-genomic steroid signaling through the mineralocorticoid receptor: Involvement of a membrane-associated receptor? Mol Cell Endocrinol. 2022;541:111501. [CrossRef]
- Vasudevan N, Pfaff DW. Non-genomic actions of estrogens and their interaction with genomic actions in the brain. Front Neuroendocrinol. 2008;29(2):238-257. [CrossRef]
- Lachize S, Apostolakis EM, van der Laan S, Tijssen AM, Xu J, de Kloet ER, Meijer OC. Steroid receptor coactivator-1 is necessary for regulation of corticotropin-releasing hormone by chronic stress and glucocorticoids. Proc Natl Acad Sci U S A. 2009;106(19):8038-8042. [CrossRef]
- van Weert LTCM, Buurstede JC, Mahfouz A, Braakhuis PSM, Polman JAE, Sips HCM, Roozendaal B, Balog J, de Kloet ER, Datson NA, et al. NeuroD Factors Discriminate Mineralocorticoid From Glucocorticoid Receptor DNA Binding in the Male Rat Brain. Endocrinology. 2017;158(5):1511-1522. [CrossRef]
- Skowron KJ, Booker K, Cheng C, Creed S, David BP, Lazzara PR, Lian A, Siddiqui Z, Speltz TE, Moore TW. Steroid receptor/coactivator binding inhibitors: An update. Mol Cell Endocrinol. 2019 ;493:110471. [CrossRef]
- Stashi E, York B, O’Malley BW. Steroid receptor coactivators: servants and masters for control of systems metabolism. Trends Endocrinol Metab. 2014;25(7):337-347. [CrossRef]
- Grossmann C, Almeida-Prieto B, Nolze A, Alvarez de la Rosa D. Structural and molecular determinants of mineralocorticoid receptor signalling.Br J Pharmacol. 2022;179(13):3103-3118. [CrossRef]
- Vanderhaeghen T, Beyaert R, Libert C. Bidirectional Crosstalk Between Hypoxia Inducible Factors and Glucocorticoid Signalling in Health and Disease. Front Immunol. 2021 ;12:684085. [CrossRef]
- Fadel L, Dacic M, Fonda V, Sokolsky BA, Quagliarini F, Rogatsky I, Uhlenhaut NH. Modulating glucocorticoid receptor actions in physiology and pathology: Insights from coregulators Pharmacol Ther. 2023;251:108531. [CrossRef]
- Vandevyver S, Dejager L, Libert C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr Rev. 2014 ;35(4):671-693. [CrossRef]
- Clayton SA, Jones SW, Kurowska-Stolarska M, Clark AR. The role of microRNAs in glucocorticoid action. J Biol Chem. 2018;293(6):1865-1874. [CrossRef]
- Knutti D, Kaul A, Kralli A. A tissue-specific coactivator of steroid receptors, identified in a functional genetic screen. Mol Cell Biol. 2000;20(7):2411-2422. [CrossRef]
- Meijer OC, Buurstede JC, Schaaf MJM. Corticosteroid Receptors in the Brain: Transcriptional Mechanisms for Specificity and Context-Dependent Effects. Cell Mol Neurobiol. 2019;39(4):539-549. [CrossRef]
- Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. 2013;132(5):1033-1044. [CrossRef]
- Oakley RH, Cruz-Topete D, He B, Foley JF, Myers PH, Xu X, Gomez-Sanchez CE, Chambon P, Willis MS, Cidlowski JA. Cardiomyocyte glucocorticoid and mineralocorticoid receptors directly and antagonistically regulate heart disease in mice. Sci Signal. 2019;12(577):eaau9685. [CrossRef]
- Nelson CC, Hendy SC, Shukin RJ, Cheng H, Bruchovsky N, Koop BF, Rennie PS. Determinants of DNA sequence specificity of the androgen, progesterone, and glucocorticoid receptors: evidence for differential steroid receptor response elements. Mol Endocrinol. 1999;13(12):2090-2107. [CrossRef]
- Presman DM, Hager GL. More than meets the dimer: What is the quaternary structure of the glucocorticoid receptor?Transcription. 2017;8(1):32-39. [CrossRef]
- Koning ACAM, Buurstede JC, van Weert LTCM, Meijer OC. Glucocorticoid and Mineralocorticoid Receptors in the Brain: A Transcriptional Perspective. J Endocr Soc. 2019;3(10):1917-1930. [CrossRef]
- Sacta MA, Chinenov Y, Rogatsky I. Glucocorticoid Signaling: An Update from a Genomic Perspective. Annu Rev Physiol. 2016;78:155-180. [CrossRef]
- Faulkner JL,Belin de Chantemèle EJ. Mineralocorticoid Receptor and Endothelial Dysfunction in Hypertension. Curr Hypertens Rep. 2019;21(10):78. [CrossRef]
- Kokkinopoulou I, Moutsatsou P. Mitochondrial Glucocorticoid Receptors and Their Actions. Int J Mol Sci. 2021;22(11):6054. [CrossRef]
- Viho EMG, Buurstede JC, Mahfouz A, Koorneef LL, van Weert LTCM, Houtman R, Hunt HJ, Kroon J, Meijer OC. Corticosteroid Action in the Brain: The Potential of Selective Receptor Modulation. Neuroendocrinology. 2019;109(3):266-276. [CrossRef]
- Winnay JN, Xu J, O’Malley BW, Hammer GD. Steroid receptor coactivator-1-deficient mice exhibit altered hypothalamic-pituitary-adrenal axis function. Endocrinology. 2006;147(3):1322-1332. [CrossRef]
- Yi P, Yu X, Wang Z, O’Malley BW. Steroid receptor-coregulator transcriptional complexes: new insights from CryoEM. Essays Biochem. 2021;65(6):857-866. [CrossRef]
- Marchi D, van Eeden FJM. Homeostatic Regulation of Glucocorticoid Receptor Activity by Hypoxia-Inducible Factor 1: From Physiology to Clinic. Cells. 2021 Dec 7;10(12):3441. [CrossRef]
- Syed AP, Greulich F, Ansari SA, Uhlenhaut NH. Anti-inflammatory glucocorticoid action: genomic insights and emerging concepts. Curr Opin Pharmacol. 2020;53:35-44. [CrossRef]
- Kodama T, Shimizu N,Yoshikawa N, Makino Y, Ouchida R, Okamoto K, Hisada T, Nakamura H, Morimoto C, Tanaka H. Role of the glucocorticoid receptor for regulation of hypoxia-dependent gene expression. J Biol Chem. 2003;278(35):33384-33391. [CrossRef]
- Callera GE, Touyz RM, Tostes RC, Yogi A, He Y, Malkinson S, Schiffrin EL. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension. 2005; 45(4):773-779. [CrossRef]
- Zeyen L, Seternes OM, Mikkola I. Crosstalk between p38 MAPK and GR Signaling. Int J Mol Sci. 2022;23(6):3322. [CrossRef]
- Quatrini L, Ugolini S. New insights into the cell-and tissue-specificity of glucocorticoid actions. Cell Mol Immunol. 2021;18(2):269-278. [CrossRef]
- Davel AP, Anwar IJ, Jaffe IZ. The endothelial mineralocorticoid receptor: mediator of the switch from vascular health to disease. Curr Opin Nephrol Hypertens. 2017;26(2):97-104. [CrossRef]
- Geerling JC, Loewy AD. Aldosterone in the brain. Am J Physiol Renal Physiol. 2009 Sep;297(3):F559-F576. [CrossRef]
- Haque M, Wilson R, Sharma K, Mills NJ, Teruyama R. Localisation of 11β-Hydroxysteroid Dehydrogenase Type 2 in Mineralocorticoid Receptor Expressing Magnocellular Neurosecretory Neurones of the Rat Supraoptic and Paraventricular Nuclei. J Neuroendocrinol.27(11):835-849. [CrossRef]
- Vassiliou AG, Athanasiou N, Vassiliadi DA, Jahaj E, Keskinidou C, Kotanidou A, Dimopoulou I. Glucocorticoid and mineralocorticoid receptor expression in critical illness: A narrative review. World J Crit Care Med. 2021;10(4):102-111. [CrossRef]
- Parker BM, Wertz SL, Pollard CM, Desimine VL, Maning J, McCrink KA, Lymperopoulos A. Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease. Int J Mol Sci. 2018;19(12):3764. [CrossRef]
- Perlstein RS, Whitnall MH, Abrams JS, Mougey EH, Neta R. Synergistic roles of interleukin-6, interleukin-1, and tumor necrosis factor in the adrenocorticotropin response to bacterial lipopolysaccharide in vivo. Endocrinology. 1993;132(3):946-952. [CrossRef]
- Terada Y, Ueda S, Hamada K, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Kagawa T, Horino T, Takao T. Aldosterone stimulates nuclear factor-kappa B activity and transcription of intercellular adhesion molecule-1 and connective tissue growth factor in rat mesangial cells via serum- and glucocorticoid-inducible protein kinase-1. Clin Exp Nephrol. 2012;16(1):81-88. [CrossRef]
- Maturana A, Lenglet S, Python M, Kuroda S, Rossier MF. Role of the T-type calcium channel CaV3.2 in the chronotropic action of corticosteroids in isolated rat ventricular myocytes. Endocrinology. 2009;150(8):3726-3734. [CrossRef]
- Rossier MF. The Cardiac Mineralocorticoid Receptor (MR): A Therapeutic Target Against Ventricular Arrhythmias. Front Endocrinol (Lausanne). 2021;12:694758. [CrossRef]
- Funder JW. Aldosterone and Mineralocorticoid Receptors-Physiology and Pathophysiology. Int J Mol Sci. 2017;18(5):1032. [CrossRef]
- Igbekele AE, Jia G, Hill MA, Sowers JR, Jia G. Mineralocorticoid Receptor Activation in Vascular Insulin Resistance and Dysfunction. Int J Mol Sci. 2022;23(16):8954. [CrossRef]
- Vanderhaeghen T, Timmermans S, Watts D, Paakinaho V, Eggermont M, Vandewalle J, Wallaeys C, Van Wyngene L, Van Looveren K, Nuyttens L, et al., Reprogramming of glucocorticoid receptor function by hypoxia. EMBO Rep. 2022;23(1):e53083. [CrossRef]
- McEown K, Treit D. Mineralocorticoid receptors in the medial prefrontal cortex and hippocampus mediate rats’ unconditioned fear behaviour. Horm Behav. 2011;60(5):581-588. [CrossRef]
- Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117(6):2505-2511. [CrossRef]
- Reul JM, de Kloet ER, van Sluijs FJ, Rijnberk A, Rothuizen J. Binding characteristics of mineralocorticoid and glucocorticoid receptors in dog brain and pituitary. Endocrinology. 1990;127(2):907-915. [CrossRef]
- Ahmadpour D, Grange-Messent V. Involvement of Testosterone Signaling in the Integrity of the Neurovascular Unit in the Male: Review of Evidence, Contradictions, and Hypothesis. Neuroendocrinology. 2021;111(5):403-420. [CrossRef]
- Castelli MP, Casti A, Casu A, Frau R, Bortolato M, Spiga S, Ennas MG. Regional distribution of 5α-reductase type 2 in the adult rat brain: an immunohistochemical analysis. Psychoneuroendocrinology. 2013;38(2):281-293. [CrossRef]
- Takahashi K, Hosoya T, Onoe K, Takashima T, Tanaka M, Ishii A, Nakatomi Y, Tazawa S, Takahashi K, Doi H, et al., Association between aromatase in human brains and personality traits. Sci Rep. 2018;8(1):16841. [CrossRef]
- Guennoun R. Progesterone in the Brain: Hormone, Neurosteroid and Neuroprotectant. Int J Mol Sci. 2020 ;21(15):5271. [CrossRef]
- Ghoumari AM, Abi Ghanem C, Asbelaoui N, Schumacher M, Hussain R Roles of Progesterone, Testosterone and Their Nuclear Receptors in Central Nervous System Myelination and Remyelination. Int J Mol Sci. 2020;21(9):3163. [CrossRef]
- Hwang WJ, Lee TY, Kim NS, Kwon JS. The Role of Estrogen Receptors and Their Signaling across Psychiatric Disorders. Int J Mol Sci. 2020 Dec 31;22(1):373. [CrossRef]
- Jiao L, Machuki JO, Wu Q, Shi M, Fu L, Adekunle AO, Tao X, Xu C, Hu X, Yin Z, Sun H. Estrogen and calcium handling proteins: new discoveries and mechanisms in cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2020;318(4):H820-H829. [CrossRef]
- Trenti A, Tedesco S, Boscaro C, Trevisi L, Bolego C, Cignarella A. Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. Int J Mol Sci. 2018;19(3):859. [CrossRef]
- Coolen RL, Cambier JC, Spantidea PI, van Asselt E, Blok BFM. Androgen receptors in areas of the spinal cord and brainstem: A study in adult male cats. J Anat. 2021 Jul;239(1):125-135. [CrossRef]
- Orshal JM, Khalil RA. Gender, sex hormones, and vascular tone. Am J Physiol Regul Integr Comp Physiol. 2004;286(2):R233-R249. [CrossRef]
- Cunningham RL, Lumia AR, McGinnis MY. Androgen receptors, sex behavior, and aggression. Neuroendocrinology. 2012;96(2):131-140. [CrossRef]
- Cheng J, Watkins SC, Walker WH. Testosterone activates mitogen-activated protein kinase via Src kinase and the epidermal growth factor receptor in sertoli cells. Endocrinology. 2007;148(5):2066-2074. [CrossRef]
- Davey RA, Grossmann M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin Biochem Rev. 2016;37(1):3-15.
- Thomas P. Membrane Androgen Receptors Unrelated to Nuclear Steroid Receptors. Endocrinology. 2019;160(4):772-781. [CrossRef]
- Venkatesh VS, Grossmann M, Zajac JD, Davey RA. The role of the androgen receptor in the pathogenesis of obesity and its utility as a target for obesity treatments. Obes Rev. 2022;23(6):e13429. [CrossRef]
- Lucas-Herald AK, Touyz RM. Androgens and Androgen Receptors as Determinants of Vascular Sex Differences Across the Lifespan. Can J Cardiol. 2022;38(12):1854-1864. [CrossRef]
- Arterburn JB, Prossnitz ER. G Protein-Coupled Estrogen Receptor GPER: Molecular Pharmacology and Therapeutic Applications. Annu Rev Pharmacol Toxicol. 2023;63:295-320. [CrossRef]
- Chakraborty B, Byemerwa J, Krebs T, Lim F, Chang CY, McDonnell DP. Estrogen Receptor Signaling in the Immune System. Endocr Rev. 2023 ;44(1):117-141. [CrossRef]
- Gregorio KCR, Laurindo CP, Machado UF. Estrogen and Glycemic Homeostasis: The Fundamental Role of Nuclear Estrogen Receptors ESR1/ESR2 in Glucose Transporter GLUT4 Regulation. Cells. 2021 Jan 7;10(1):99. [CrossRef]
- Rzemieniec J, Castiglioni L, Gelosa P, Muluhie M, Mercuriali B, Sironi L. Nuclear Receptors in Myocardial and Cerebral Ischemia-Mechanisms of Action and Therapeutic Strategies. Int J Mol Sci. 2021;22(22):12326. [CrossRef]
- Guajardo-Correa E, Silva-Agüero JF, Calle X, Chiong M, Henríquez M, García-Rivas G, Latorre M, Parra V. Estrogen signaling as a bridge between the nucleus and mitochondria in cardiovascular diseases. Front Cell Dev Biol. 2022 ;10:968373. [CrossRef]
- Wang H, Zhao Z, Lin M, Groban L Activation of GPR30 inhibits cardiac fibroblast proliferation. Mol Cell Biochem. 2015;405(1-2):135-148. [CrossRef]
- Kurmann L, Okoniewski M, Dubey RK. Estradiol Inhibits Human Brain Vascular Pericyte Migration Activity: A Functional and Transcriptomic Analysis. Cells. 2021;10(9):2314. [CrossRef]
- Machuki JO, Zhang HY, Harding SE, Sun H. Molecular pathways of oestrogen receptors and β-adrenergic receptors in cardiac cells: Recognition of their similarities, interactions and therapeutic value. Acta Physiol (Oxf). 2018;222(2):e12978. [CrossRef]
- da Silva JS, Montagnoli TL, Rocha BS, Tacco MLCA, Marinho SCP, Zapata-Sudo G. Estrogen Receptors: Therapeutic Perspectives for the Treatment of Cardiac Dysfunction after Myocardial Infarction. Int J Mol Sci. 2021 Jan 7;22(2):525. [CrossRef]
- Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. 2019;116:135-170. [CrossRef]
- Tran QK Reciprocality Between Estrogen Biology and Calcium Signaling in the Cardiovascular System. Front Endocrinol (Lausanne). 2020;11:568203. [CrossRef]
- Menazza S, Murphy E. The Expanding Complexity of Estrogen Receptor Signaling in the Cardiovascular System. Circ Res. 2016;118(6):994-1007. [CrossRef]
- Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81(4):1535-1565. [CrossRef]
- Sun J, Aponte AM,Menazza S, Gucek M, Steenbergen C, Murphy E. Additive cardioprotection by pharmacological postconditioning with hydrogen sulfide and nitric oxide donors in mouse heart: S-sulfhydration vs. S-nitrosylation. Cardiovasc Res. 2016;110(1):96-106. [CrossRef]
- Zimmerman MA, Budish RA, Kashyap S, Lindsey SH. GPER-novel membrane oestrogen receptor. Clin Sci (Lond). 2016;130(12):1005-1016. [CrossRef]
- Klinge CM. Estrogenic control of mitochondrial function. Redox Biol. 2020 Apr;31:101435. [CrossRef]
- Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol. 1997;388(4):507-525. [CrossRef]
- Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;7(12):715-726. [CrossRef]
- Aickareth J, Hawwar M, Sanchez N, Gnanasekaran R, Zhang J. Membrane Progesterone Receptors (mPRs/PAQRs) Are Going beyond Its Initial Definitions. Membranes (Basel). 2023 Feb 22;13(3):260. [CrossRef]
- Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Oprea TI, Prossnitz ER, Dun NJ. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J Endocrinol. 2007;193(2):311-321. [CrossRef]
- Shah NM, Imami N, Johnson MR. Progesterone Modulation of Pregnancy-Related Immune Responses. Front Immunol. 2018;9:1293. [CrossRef]
- Singh M, Su C, Ng S. Non-genomic mechanisms of progesteroneaction in the brain. Front Neurosci. 2013 Sep 19;7:159. [CrossRef]
- Vítků J, Hampl R. Steroid Conjugates and Their Physiological Role. Physiol Res. 2023 ;72(S4):S317-S322. [CrossRef]
- Pinna G. Allopregnanolone (1938-2019): A trajectory of 80 years of outstanding scientific achievements. Neurobiol Stress. 2020;13:100246. [CrossRef]
- Brann DW, Lu Y, Wang J, Zhang Q, Thakkar R, Sareddy GR, Pratap UP, Tekmal RR, Vadlamudi RK. Brain-derived estrogen and neural function. Neurosci Biobehav Rev. 2022;132:793-817. [CrossRef]
- Buijs RM, Hermes MH, Kalsbeek A. The suprachiasmatic nucleus-paraventricular nucleus interactions: a bridge to the neuroendocrine and autonomic nervous system. Prog Brain Res. 1998;119:365-382. [CrossRef]
- Szczepanska-Sadowska E, Czarzasta K, Cudnoch-Jedrzejewska A. Dysregulation of the Renin-Angiotensin System and the Vasopressinergic System Interactions in Cardiovascular Disorders. Curr Hypertens Rep. 2018;20(3):19. [CrossRef]
- Szczepanska-Sadowska E, Zera T, Sosnowski P, Cudnoch-Jedrzejewska A, Puszko A, Misicka A. Vasopressin and Related Peptides; Potential Value in Diagnosis, Prognosis and Treatment of Clinical Disorders. Curr Drug Metab. 2017;18(4):306-345. [CrossRef]
- Amico JA, Finn FM, Haldar J. Oxytocin and vasopressin are present in human and rat pancreas. Am J Med Sci. 1988;296(5):303-307. [CrossRef]
- Hupf H, Grimm D, Riegger GA, Schunkert H. Evidence for a vasopressin system in the rat heart. Circ Res. 1999;84(3):365-370. [CrossRef]
- Burbach JP, Luckman SM, Murphy D, Gainer H. Gene regulation in the magnocellular hypothalamo-neurohypophysial system. Physiol Rev. 2001;81(3):1197-1267. [CrossRef]
- Sparapani S, Millet-Boureima C, Oliver J, Mu K, Hadavi P, Kalostian T, Ali N, Avelar CM, Bardies M, Barrow B, et al., The Biology of Vasopressin. Biomedicines. 2021;9(1):89. [CrossRef]
- Morgenthaler NG, Struck J, Alonso C, Bergmann A. Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin Chem. 2006;52(1):112-119. [CrossRef]
- Rechlin T, Hochholzer W, Stelzi C, Laule K, Freidank H, Morgenthaler NG, et al. Incremental value of Copeptin for rapid rule out of acute myocardial infarction. J Am Coll Cardiol.2009;54:60–68. [CrossRef]
- Roffi M, Patrono C. CardioPulse: ‘Ten Commandments’ of 2015 European Society of Cardiology Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation (NSTE-ACS). Eur Heart J. 2016;37(3):208.
- Aikins AO, Nguyen DH, Paundralingga O, Farmer GE, Shimoura CG, Brock C, Cunningham JT. Cardiovascular Neuroendocrinology: Emerging Role for Neurohypophyseal Hormones in Pathophysiology. Endocrinology. 202;162(8):bqab082. [CrossRef]
- Costello HM, Krilis G, Grenier C, Severs D, Czopek A, Ivy JR, Nixon M, Holmes MC, Livingstone DEW, Hoorn EJ et al. High salt intake activates the hypothalamic-pituitary-adrenal axis, amplifies the stress response, and alters tissue glucocorticoid exposure in mice. Cardiovasc Res. 2023;119(8):1740-1750. [CrossRef]
- Goldstein DS. The extended autonomic system, dyshomeostasis, and COVID-19. Clin Auton Res. 2020;30(4):299-315. [CrossRef]
- Iwasaki Y, Oiso Y, Saito H, Majzoub JA. Positive and negative regulation of the rat vasopressin gene promoter. Endocrinology. 1997;138(12):5266-5274. [CrossRef]
- Lee S, Rivier C. Hypophysiotropic role and hypothalamic gene expression of corticotropin-releasing factor and vasopressin in rats injected with interleukin-1 beta systemically or into the brain ventricles. J Neuroendocrinol. 1994 ;6(2):217-224. [CrossRef]
- Grinevich V, Ma XM, Jirikowski G, Verbalis J, Aguilera G. Lipopolysaccharide endotoxin potentiates the effect of osmotic stimulation on vasopressin synthesis and secretion in the rat hypothalamus. J Neuroendocrinol. 2003;15(2):141-149. [CrossRef]
- Pardy K, Murphy D, Carter D, Hui KM. The influence of interleukin-2 on vasopressin and oxytocin gene expression in the rodent hypothalamus. J Neuroimmunol. 1993;42(2):131-138. [CrossRef]
- Zelazowski P, Patchev VK, Zelazowska EB, Chrousos GP, Gold PW, Sternberg EM. Release of hypothalamic corticotropin-releasing hormone and arginine-vasopressin by interleukin 1 beta and alpha MSH: studies in rats with different susceptibility to inflammatory disease. Brain Res. 1993;631(1):22-26. [CrossRef]
- Antoni FA. Magnocellular Vasopressin and the Mechanism of “Glucocorticoid Escape”.Front Endocrinol (Lausanne). 2019 Jun 26;10:422. [CrossRef]
- Luo X, Kiss A, Makara G, Lolait SJ, Aguilera G Stress-specific regulation of corticotropin releasing hormone receptor expression in the paraventricular and supraoptic nuclei of the hypothalamus in the rat. J Neuroendocrinol. 1994;6(6):689-696. [CrossRef]
- Sawchenko PE. Adrenalectomy-induced enhancement of CRF and vasopressin immunoreactivity in parvocellular neurosecretory neurons: anatomic, peptide, and steroid specificity. J Neurosci. 1987r;7(4):1093-1106. [CrossRef]
- Kovács KJ, Földes A, Sawchenko PE. Glucocorticoid negative feedback selectively targets vasopressin transcription in parvocellular neurosecretory neurons. J Neurosci. 2000;20(10):3843-3852. [CrossRef]
- Kageyama K, Suda T. Regulatory mechanisms underlying corticotropin-releasing factor gene expression in the hypothalamus. Endocr J. 2009;56(3):335-344. [CrossRef]
- Yoshida M. Gene regulation system of vasopressin and corticotropin-releasing hormone.Gene Regul Syst Bio. 2008;2:71-88. [CrossRef]
- Mills NJ, Sharma K, Haque M, Moore M, Teruyama R. Aldosterone Mediated Regulation of Epithelial Sodium Channel (ENaC) Subunits in the Rat Hypothalamus. Neuroscience. 2018;390:278-292. [CrossRef]
- Geerling JC, Loewy AD. Aldosterone in the brain. Am J Physiol Renal Physiol. 2009; 297(3):F559-F576. [CrossRef]
- Viau V, Chu A, Soriano L, Dallman MF. Independent and overlapping effects of corticosterone and testosterone on corticotropin-releasing hormone and arginine vasopressin mRNA expression in the paraventricular nucleus of the hypothalamus and stress-induced adrenocorticotropic hormone release. J Neurosci. 1999;19(15):6684-6693. [CrossRef]
- Herman JP,Tasker JG. Paraventricular Hypothalamic Mechanisms of Chronic Stress Adaptation. Front Endocrinol (Lausanne). 2016;7:137. [CrossRef]
- Sheng JA, Tan SML, Hale TM, Handa RJ. Androgens and Their Role in Regulating Sex Differences in the Hypothalamic/Pituitary/Adrenal Axis Stress Response and Stress-Related Behaviors. Androg Clin Res Ther. 2021;2(1):261-274. [CrossRef]
- Aguilera G,Liu Y. The molecular physiology of CRH neurons. Front Neuroendocrinol. 2012 ;33(1):67-84. [CrossRef]
- Bous J, Fouillen A, Orcel H, Granier S, Bron P, Mouillac B. Structures of the arginine-vasopressin and oxytocin receptor signaling complexes. Vitam Horm. 2023;123:67-107. [CrossRef]
- Holmes CL, Landry DW, Granton JT. Science Review: Vasopressin and the cardiovascular system part 2 - clinical physiology. Crit Care. 2004;8(1):15-23. [CrossRef]
- Thai BS, Chia LY, Nguyen ATN, Qin C, Ritchie RH, Hutchinson DS, Kompa A, White PJ, May LT. Targeting G protein-coupled receptors for heart failure treatment. Br J Pharmacol. 2023. Online ahead of print. [CrossRef]
- Morello JP,Bichet DG. Nephrogenic diabetes insipidus. Annu Rev Physiol. 2001;63:607-630. [CrossRef]
- Zang L, Gong Y, Li Y, Dou J, Lyu Z, Su X, Zhang Y, Mu Y. A Novel Missense Mutation of Arginine Vasopressin Receptor 2 in a Chinese Family with Congenital Nephrogenic Diabetes Insipidus: X-Chromosome Inactivation in Female CNDI Patients with Heterozygote 814A>G Mutation. Biomed Res Int. 2022;2022:7073158. [CrossRef]
- Dekan Z, Kremsmayr T, Keov P, Godin M, Teakle N, Dürrauer L, Xiang H, Gharib D, Bergmayr C, Hellinger R et al. Nature-inspired dimerization as a strategy to modulate neuropeptide pharmacology exemplified with vasopressin and oxytocin. Chem Sci. 2021;12(11):4057-4062. [CrossRef]
- Murat B, Devost D, Andrés M, Mion J, Boulay V, Corbani M, Zingg HH, Guillon G. V1b and CRHR1 receptor heterodimerization mediates synergistic biological actions of vasopressin and CRH. Mol Endocrinol. 2012;26(3):502-520. [CrossRef]
- Patchev VK, Almeida OF. Corticosteroid regulation of gene expression and binding characteristics of vasopressin receptors in the rat brain. Eur J Neurosci. 1995;7(7):1579-1583. [CrossRef]
- Wasilewski MA, Grisanti LA, Song J, Carter RL, Repas AA, Myers VD, Gao E, Koch WJ, Cheung JY, Feldman AM et al., Vasopressin type 1A receptor deletion enhances cardiac contractility, β-adrenergic receptor sensitivity and acute cardiac injury-induced dysfunction. Clin Sci (Lond). 2016 ;130(22):2017-2027. [CrossRef]
- Tsuchiya M, Tsuchiya K, Maruyama R, Takemura G, Minatoguchi S, Fujiwara H. Vasopressin inhibits sarcolemmal ATP-sensitive K+ channels via V1 receptors activation in the guinea pig heart. Circ J. 2002;66(3):277-282. [CrossRef]
- Hantash BM, Thomas AP, Reeves JP. Regulation of the cardiac L-type calcium channel in L6 cells by arginine-vasopressin. Biochem J. 2006;400(3):411-419. [CrossRef]
- Tilley DG, Zhu W, Myers VD, Barr LA, Gao E, Li X, Song J, Carter RL, Makarewich CA, Yu D et al., β-adrenergic receptor-mediated cardiac contractility is inhibited via vasopressin type 1A-receptor-dependent signaling. Circulation. 2014;130(20):1800-1811. [CrossRef]
- Zhu W, Tilley DG, Myers VD, Coleman RC, Feldman AM. Arginine vasopressin enhances cell survival via a G protein-coupled receptor kinase 2/β-arrestin1/extracellular-regulated kinase 1/2-dependent pathway in H9c2 cells. Mol Pharmacol. 2013:227-235. [CrossRef]
- Zhu W, Tilley DG, Myers VD, Tsai EJ, Feldman AM. Increased vasopressin 1A receptor expression in failing human hearts. J Am Coll Cardiol. 2014;63(4):375-376. [CrossRef]
- Xu F, Sun S, Wang X, Ni E, Zhao L, Zhu W. GRK2 Mediates Arginine Vasopressin-Induced Interleukin-6 Production via Nuclear Factor-kappaB Signaling Neonatal Rat Cardiac Fibroblast.Mol Pharmacol. 2017;92(3):278-284. [CrossRef]
- Bucher M, Hobbhahn J, Taeger K, Kurtz A. Cytokine-mediated downregulation of vasopressin V(1A) receptors during acute endotoxemia in rats. Am J Physiol Regul Integr Comp Physiol. 2002;282(4):R979-984. [CrossRef]
- Gray M, Innala L, Viau V. Central vasopressin V1A receptor blockade impedes hypothalamic-pituitary-adrenal habituation to repeated restraint stress exposure in adult male rats. Neuropsychopharmacology. 2012;37(12):2712-2719. [CrossRef]
- Roper J, O’Carroll AM, Young W 3rd, Lolait S. The vasopressin Avpr1b receptor: molecular and pharmacological studies. Stress. 2011:98-115. [CrossRef]
- Lolait SJ, O’Carroll AM, Mahan LC, Felder CC, Button DC, Young WS 3rd, Mezey E, Brownstein MJ. Extrapituitary expression of the rat V1b vasopressin receptor gene. Proc Natl Acad Sci U S A. 1995;92(15):6783-6787. [CrossRef]
- O’Carroll AM, Howell GM, Roberts EM, Lolait SJ. Vasopressin potentiates corticotropin-releasing hormone-induced insulin release from mouse pancreatic beta-cells. J Endocrinol. 2008;197(2):231-239. [CrossRef]
- Khan S, Raghuram V, Chen L, Chou CL, Yang CR, Khundmiri SJ, Knepper MA. Vasopressin V2 receptor, tolvaptan, and ERK1/2 phosphorylation in the renal collecting duct. Am J Physiol Renal Physiol. 2024;326(1):F57-F68. [CrossRef]
- Schrier RW. Molecular mechanisms of clinical concentrating and diluting disorders. Prog Brain Res. 2008;170:539-550. [CrossRef]
- Lightman SL, Birnie MT, Conway-Campbell BL. Dynamics of ACTH and Cortisol Secretion and Implications for Disease. Endocr Rev. 2020 Jun 1;41(3):bnaa002. [CrossRef]
- Yates FE, Russell SM, Dallman MF, Hodge GA, McCann SM, Dhariwal AP. Potentiation by vasopressin of corticotropin release induced by corticotropin-releasing factor. Endocrinology. 1971;88(1):3-15. [CrossRef]
- Joseph DN, Whirledge S. Stress and the HPA Axis: Balancing Homeostasis and Fertility. Int J Mol Sci. 2017 Oct 24;18(10):2224. [CrossRef]
- Robertson-Dixon I, Murphy MJ, Crewther SG, Riddell N. The Influence of Light Wavelength on Human HPA Axis Rhythms: A Systematic Review. Life (Basel). 2023 Sep 26;13(10):1968. [CrossRef]
- Otsuka H, Abe M, Kobayashi H. The Effect of Aldosterone on Cardiorenal and Metabolic Systems. Int J Mol Sci. 2023 Mar 11;24(6):5370. [CrossRef]
- Sztechman D, Czarzasta K, Cudnoch-Jedrzejewska A, Szczepanska-Sadowska E, Zera T. Aldosterone and mineralocorticoid receptors in regulation of the cardiovascular system and pathological remodelling of the heart and arteries. J Physiol Pharmacol. 2018 Dec;69(6). [CrossRef]
- Albert KM, Newhouse PA. Estrogen, Stress, and Depression: Cognitive and Biological Interactions. Annu Rev Clin Psychol. 2019;15:399-423. [CrossRef]
- Patel S, Homaei A, Raju AB, Meher BR. Estrogen: The necessary evil for human health, and ways to tame it. Biomed Pharmacother. 2018;102:403-411. [CrossRef]
- Nelson LR, Bulun SE. Estrogen production and action. J Am Acad Dermatol. 2001 Sep;45(3 Suppl):S116-24. [CrossRef]
- Naamneh Elzenaty R, du Toit T, Flück CE. Basics of androgen synthesis and action. Best Pract Res Clin Endocrinol Metab. 2022 Jul;36(4):101665. [CrossRef]
- Fanelli F, Baronio F, Ortolano R, Mezzullo M, Cassio A, Pagotto U, Balsamo A. Normative Basal Values of Hormones and Proteins of Gonadal and Adrenal Functions from Birth to Adulthood. Sex Dev. 2018;12(1-3):50-94. [CrossRef]
- Kulle AE, Riepe FG, Melchior D, Hiort O, Holterhus PM. A novel ultrapressure liquid chromatography tandem mass spectrometry method for the simultaneous determination of androstenedione, testosterone, and dihydrotestosterone in pediatric blood samples: age- and sex-specific reference data. J Clin Endocrinol Metab. 2010;95(5):2399-2409. [CrossRef]
- Zirkin BR, Papadopoulos V. Leydig cells: formation, function, and regulation. Biol Reprod. 2018;99(1):101-111. [CrossRef]
- Midzak A, Akula N, Lecanu L, Papadopoulos V. Novel androstenetriol interacts with the mitochondrial translocator protein and controls steroidogenesis. J Biol Chem. 2011;286(11):9875-9887. [CrossRef]
- Beattie MC, Adekola L, Papadopoulos V, Chen H, Zirkin BR. Leydig cell aging and hypogonadism. Exp Gerontol. 2015;68:87-91. [CrossRef]
- Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev. 2004;25(6):947-970. [CrossRef]
- Wang Y, Chen F, Ye L, Zirkin B, Chen H. Steroidogenesis in Leydig cells: effects of aging and environmental factors. Reproduction. 2017;154(4):R111-R122. [CrossRef]
- Kuo T, McQueen A, Chen TC, Wang JC. Regulation of Glucose Homeostasis by Glucocorticoids. Adv Exp Med Biol. 2015;872:99-126. [CrossRef]
- Yoshimura M, Conway-Campbell B, Ueta Y. Arginine vasopressin: Direct and indirect action on metabolism. Peptides. 2021 Aug;142:170555. [CrossRef]
- Nakata M, Gantulga D, Santoso P, Zhang B, Masuda C, Mori M, Okada T, Yada T. Paraventricular NUCB2/Nesfatin-1 Supports Oxytocin and Vasopressin Neurons to Control Feeding Behavior and Fluid Balance in Male Mice. Endocrinology. 2016;157(6):2322-2332. [CrossRef]
- Mohan S, Flatt PR, Irwin N, Moffett RC. Weight-reducing, lipid-lowering and antidiabetic activities of a novel arginine vasopressin analogue acting at the V1a and V1b receptors in high-fat-fed mice. Diabetes Obes Metab. 2021;23(10):2215-2225. [CrossRef]
- Haam J, Halmos KC, Di S, Tasker JG. Nutritional state-dependent ghrelin activation of vasopressin neurons via retrograde trans-neuronal-glial stimulation of excitatory GABA circuits. J Neurosci. 2014;34(18):6201-6213. [CrossRef]
- Iwama S, Sugimura Y, Murase T, Hiroi M, Goto M, Hayashi M, Arima H, Oiso Y. Central adiponectin functions to inhibit arginine vasopressin release in conscious rats. J Neuroendocrinol. 2009;21(9):753-759. [CrossRef]
- Küchler S, Perwitz N, Schick RR, Klein J, Westphal S. Arginine-vasopressin directly promotes a thermogenic and pro-inflammatory adipokine expression profile in brown adipocytes. Regul Pept. 2010;164(2-3):126-132. [CrossRef]
- Rofe AM, Williamson DH. Metabolic effects of vasopressin infusion in the starved rat. Reversal of ketonaemia. Biochem J. 1983;212(1):231-239. [CrossRef]
- VAUGHAN M. EFFECT OF PITRESSIN ON LIPOLYSIS AND ON PHOSPHORYLASE ACTIVITY IN RAT ADIPOSE TISSUE. Am J Physiol. 1964;207:1166-1168. [CrossRef]
- Hiroyama M, Aoyagi T, Fujiwara Y, Oshikawa S, Sanbe A, Endo F, Tanoue A. Hyperammonaemia in V1a vasopressin receptor knockout mice caused by the promoted proteolysis and reduced intrahepatic blood volume. J Physiol. 2007;581(Pt 3):1183-1192. [CrossRef]
- Hiroyama M, Fujiwara Y, Nakamura K, Aoyagi T, Mizutani R, Sanbe A, Tasaki R, Tanoue A. Altered lipid metabolism in vasopressin V1B receptor-deficient mice. Eur J Pharmacol. 2009;602(2-3):455-461. [CrossRef]
- Velho G, El Boustany R, Lefèvre G, Mohammedi K, Fumeron F, Potier L, Bankir L, Bouby N, Hadjadj S, Marre M, Roussel R. Plasma Copeptin, Kidney Outcomes, Ischemic Heart Disease, and All-Cause Mortality in People With Long-standing Type 1 Diabetes. Diabetes Care. 2016;39(12):2288-2295. [CrossRef]
- Vanhaecke T, Perrier ET, Melander O. A Journey through the Early Evidence Linking Hydration to Metabolic Health. Ann Nutr Metab. 2020;76 Suppl 1:4-9. [CrossRef]
- Roussel R, El Boustany R, Bouby N, Potier L, Fumeron F, Mohammedi K, Balkau B, Tichet J, Bankir L, Marre M, Velho G. Plasma Copeptin, AVP Gene Variants, and Incidence of Type 2 Diabetes in a Cohort From the Community. J Clin Endocrinol Metab. 2016 Jun;101(6):2432-9. Enhörning S, Leosdottir M, Wallström P, Gullberg B, Berglund G, Wirfält E, Melander O. Relation between human vasopressin 1a gene variance, fat intake, and diabetes. Am J Clin Nutr. 2009;89(1):400-406. [CrossRef]
- Enhörning S, Leosdottir M, Wallström P, Gullberg B, Berglund G, Wirfält E, Melander O. Relation between human vasopressin 1a gene variance, fat intake, and diabetes. Am J Clin Nutr. 2009;89(1):400-406. [CrossRef]
- Enhörning S, Sjögren M, Hedblad B, Nilsson PM, Struck J, Melander O. Genetic vasopressin 1b receptor variance in overweight and diabetes mellitus. Eur J Endocrinol. 2016 ;174(1):69-75. [CrossRef]
- Hew-Butler T, Smith-Hale V, Pollard-McGrandy A, VanSumeren M. Of Mice and Men-The Physiology, Psychology, and Pathology of Overhydration. Nutrients. 2019 Jul 7;11(7):1539. [CrossRef]
- Szczepanska-Sadowska E, Wsol A, Cudnoch-Jedrzejewska A, Żera T. Complementary Role of Oxytocin and Vasopressin in Cardiovascular Regulation. Int J Mol Sci. 2021 Oct 24;22(21):11465. [CrossRef]
- Bichet DG. Regulation of Thirst and Vasopressin Release. Annu Rev Physiol. 2019; 81:359-373. [CrossRef]
- Szczepanska-Sadowska E. Neuromodulation of Cardiac Ischemic Pain: Role of the Autonomic Nervous System and Vasopressin. J Integr Neurosci. 2024;23(3):49. [CrossRef]
- Danziger J, Zeidel ML. Osmotic homeostasis. Clin J Am Soc Nephrol. 2015;10(5):852-862. [CrossRef]
- Zimmerman CA, Lin YC, Leib DE, Guo L, Huey EL, Daly GE, Chen Y, Knight ZA. Thirst neurons anticipate the homeostatic consequences of eating and drinking. Nature. 2016;537(7622):680-684. [CrossRef]
- Zaelzer C, Hua P, Prager-Khoutorsky M, Ciura S, Voisin DL, Liedtke W, Bourque CW. ΔN-TRPV1: A Molecular Co-detector of Body Temperature and Osmotic Stress. Cell Rep. 2015;13(1):23-30. [CrossRef]
- Saker P, Farrell MJ, Adib FR, Egan GF, McKinley MJ, Denton DA. Regional brain responses associated with drinking water during thirst and after its satiation. Proc Natl Acad Sci U S A. 2014;111(14):5379-5384. [CrossRef]
- Cheung PW, Bouley R, Brown D. Targeting the Trafficking of Kidney Water Channels for Therapeutic Benefit. Annu Rev Pharmacol Toxicol. 2020;60:175-194. [CrossRef]
- Bankir L, Bichet DG, Bouby N. Vasopressin V2 receptors, ENaC, and sodium reabsorption: a risk factor for hypertension? Am J Physiol Renal Physiol. 2010;299(5):F917-F928. [CrossRef]
- Fenton RA. Essential role of vasopressin-regulated urea transport processes in the mammalian kidney. Pflugers Arch. 2009;458(1):169-177. [CrossRef]
- Bankir L. Antidiuretic action of vasopressin: quantitative aspects and interaction between V1a and V2 receptor-mediated effects. Cardiovasc Res. 2001;51(3):372-390. [CrossRef]
- Wang W, Li C, Summer SN, Falk S, Cadnapaphornchai MA, Chen YC, Schrier RW. Molecular analysis of impaired urinary diluting capacity in glucocorticoid deficiency. Am J Physiol Renal Physiol. 2006;290(5):F1135-1142. [CrossRef]
- Zhu X, Huang Y, Li S, Ge N, Li T, Wang Y, Liu K, Liu C. Glucocorticoids Reverse Diluted Hyponatremia Through Inhibiting Arginine Vasopressin Pathway in Heart Failure Rats. J Am Heart Assoc. 2020;9(10):e014950. [CrossRef]
- Frenkel A, Abuhasira R, Bichovsky Y, Bukhin A, Novack V, Brotfain E, Zlotnik A, Klein M. Examination of the association of steroids with fluid accumulation in critically ill patients, considering the possibility of biases. Sci Rep. 2021;11(1):5557. [CrossRef]
- Kenyon CJ, Saccoccio NA, Morris DJ. Aldosterone effects on water and electrolyte metabolism. J Endocrinol. 1984 Jan;100(1):93-100. [CrossRef]
- Johnston JG, Welch AK, Cain BD, Sayeski PP, Gumz ML, Wingo CS. Aldosterone: Renal Action and Physiological Effects. Compr Physiol. 2023;13(2):4409-4491. [CrossRef]
- Patel S, Rauf A, Khan H, Abu-Izneid T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed Pharmacother. 2017;94:317-325. [CrossRef]
- Sladek CD, Somponpun SJ. Estrogen receptors: their roles in regulation of vasopressin release for maintenance of fluid and electrolyte homeostasis. Front Neuroendocrinol. 2008;29(1):114-127. [CrossRef]
- Wang YX, Crofton JT, Liu H, Sato K, Brooks DP, Share L. Estradiol attenuates the antidiuretic action of vasopressin in ovariectomized rats. Am J Physiol. 1995;268(4 Pt 2):R951-R957. [CrossRef]
- Somponpun SJ, Johnson AK, Beltz T, Sladek CD. Estrogen receptor-alpha expression in osmosensitive elements of the lamina terminalis: regulation by hypertonicity. Am J Physiol Regul Integr Comp Physiol. 2004;287(3):R661-R669. [CrossRef]
- Somponpun SJ. Neuroendocrine regulation of fluid and electrolyte balance by ovarian steroids: contributions from central oestrogen receptors. J Neuroendocrinol. 2007;19(10):809-818. [CrossRef]
- Voisin DL, Simonian SX, Herbison AE. Identification of estrogen receptor-containing neurons projecting to the rat supraoptic nucleus. Neuroscience. 1997;78(1):215-228. [CrossRef]
- Kuiper GG, Carlsson B, Grandien K, Enmark E, Häggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138(3):863-870. [CrossRef]
- Li X, Kuang W, Qiu Z, Zhou Z. G protein-coupled estrogen receptor: a promising therapeutic target for aldosterone-induced hypertension. Front Endocrinol (Lausanne). 2023 Aug 17;14:1226458. [CrossRef]
- Rodriguez-Giustiniani P, Rodriguez-Sanchez N, Galloway SDR. Fluid and electrolyte balance considerations for female athletes. Eur J Sport Sci. 2022;22(5):697-708. [CrossRef]
- Swenson KL, Sladek CD. Gonadal steroid modulation of vasopressin secretion in response to osmotic stimulation. Endocrinology. 1997;138(5):2089-2097. [CrossRef]
- Siegenthaler J, Walti C, Urwyler SA, Schuetz P, Christ-Crain M. Copeptin concentrations during psychological stress: the PsyCo study. Eur J Endocrinol. 2014;171(6):737-742. [CrossRef]
- Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Aging Res Rev.2005; 4; 141-194. [CrossRef]
- Milutinović-Smiljanić S, Šarenac O, Lozić-Djurić M, Murphy D, Japundžić-Žigon N. Evidence for involvement of central vasopressin V1b and V2 receptors in stress-induced baroreflex desensitization. Br J Pharmacol. 2013;169(4):900-908. [CrossRef]
- Grassi D, Lagunas N, Calmarza-Font I, Diz-Chaves Y, Garcia-Segura LM, Panzica GC. Chronic unpredictable stress and long-term ovariectomy affect arginine-vasopressin expression in the paraventricular nucleus of adult female mice. Brain Res. 2014 Nov 7;1588:55-62. [CrossRef]
- Russell G, Lightman S. The human stress response. Nat Rev Endocrinol. 2019;15(9):525-534. [CrossRef]
- Borrow AP, Bales NJ, Stover SA, Handa RJ. Chronic Variable Stress Induces Sex-Specific Alterations in Social Behavior and Neuropeptide Expression in the Mouse. Endocrinology. 2018;159(7):2803-2814. [CrossRef]
- Das S, Komnenov D, Newhouse L, Rishi AK, Rossi NF. Paraventricular Nucleus V1a Receptor Knockdown Blunts Neurocardiovascular Responses to Acute Stress in Male Rats after Chronic Mild Unpredictable Stress. Physiol Behav. 2022 Sep 1;253:113867. [CrossRef]
- Powell-Roach KL, Yao Y, Jhun EH, He Y, Suarez ML, Ezenwa MO, Molokie RE, Wang ZJ, Wilkie DJ. Vasopressin SNP pain factors and stress in sickle cell disease PLoS One. 2019 Nov 11;14(11):e0224886. [CrossRef]
- Komnenov D, Quaal H, Rossi NF. V1a and V1b vasopressin receptors within the paraventricular nucleus contribute to hypertension in male rats exposed to chronic mild unpredictable stress. Am J Physiol Regul Integr Comp Physiol. 2021;320(3):R213-R225. [CrossRef]
- Pietranera L, Saravia F, Roig P, Lima A, De Nicola AF. Mineralocorticoid treatment upregulates the hypothalamic vasopressinergic system of spontaneously hypertensive rats. Neuroendocrinology. 2004;80(2):100-110. [CrossRef]
- Matsuguchi H, Schmid PG. Acute interaction of vasopressin and neurogenic mechanisms in DOC-salt hypertension. Am J Physiol. 1982;242(1):H37-H43. [CrossRef]
- Ferrario CM, Mohara O, Ueno Y, Brosnihan KB. Hemodynamic and neurohormonal changes in the development of DOC hypertension in the dog. Am J Med Sci. 1988;295(4):352-359. [CrossRef]
- Rademaker MT, Charles CJ, Nicholls MG, Richards AM. Interactions of enhanced urocortin 2 and mineralocorticoid receptor antagonism in experimental heart failure. Circ Heart Fail. 2013;6(4):825-832. [CrossRef]
- Pasquali R, Gagliardi L, Vicennati V, Gambineri A, Colitta D, Ceroni L, Casimirri F. ACTH and cortisol response to combined corticotropin releasing hormone-arginine vasopressin stimulation in obese males and its relationship to body weight, fat distribution and parameters of the metabolic syndrome. Int J Obes Relat Metab Disord. 1999;23(4):419-424. [CrossRef]
- Schinke C, Hesse S, Rullmann M, Becker GA, Luthardt J, Zientek F, Patt M, Stoppe M, Schmidt E, Meyer K, Meyer PM, Orthgieß J, Blüher M, Kratzsch J, Ding YS, Then Bergh F, Sabri O. Central noradrenaline transporter availability is linked with HPA axis responsiveness and copeptin in human obesity and non-obese controls. Stress. 2019;22(1):93-102.
- Canivell S, Mohaupt M, Ackermann D, Pruijm M, Guessous I, Ehret G, Escher G, Pechère-Bertschi A, Vogt B, Devuyst O, Burnier M, Martin PY, Ponte B, Bochud M. Copeptin and insulin resistance: effect modification by age and 11 β-HSD2 activity in a population-based study. J Endocrinol Invest. 2018;41(7):799-808. [CrossRef]
- Nye EJ, Bornstein SR, Grice JE, Tauchnitz R, Hockings GI, Strakosch CR, Jackson RV, Torpy DJ. Interactions between the stimulated hypothalamic-pituitary-adrenal axis and leptin in humans. J Neuroendocrinol. 2000;12(2):141-145. [CrossRef]
- Kacheva S, Kolk K, Morgenthaler NG, Brabant G, Karges W. Gender-specific co-activation of arginine vasopressin and the hypothalamic-pituitary-adrenal axis during stress. Clin Endocrinol (Oxf). 2015;82(4):570-576. [CrossRef]
- Zelena D, Mergl Z, Makara GB. The role of vasopressin in diabetes mellitus-induced hypothalamo-pituitary-adrenal axis activation: studies in Brattleboro rats. Brain Res Bull. 2006;69(1):48-56. [CrossRef]
- Balapattabi K, Little JT, Bachelor ME, Cunningham RL, Cunningham JT. Sex Differences in the Regulation of Vasopressin and Oxytocin Secretion in Bile Duct-Ligated Rats. Neuroendocrinology. 2021;111(3):237-248. [CrossRef]
- Coiro V, Volpi R, Capretti L, Bacchi-Modena A, Cigarini C, Bianconi L, Rossi G, Gramellini D, Chiodera P. Arginine vasopressin secretion in non-obese women with polycystic ovary syndrome. Acta Endocrinol (Copenh). 1989;121(6):784-790. [CrossRef]
- Pavlidi P, Kokras N, Dalla C. Sex Differences in Depression and Anxiety. Curr Top Behav Neurosci. 2023;62:103-132. [CrossRef]
- Woodward E, Rangel-Barajas C, Ringland A, Logrip ML, Coutellier L. Sex-Specific Timelines for Adaptations of Prefrontal Parvalbumin Neurons in Response to Stress and Changes in Anxiety- and Depressive-Like Behaviors. eNeuro. 2023 Mar 2;10(3):ENEURO.0300-22.2023. [CrossRef]
- Rivier C. Gender, sex steroids, corticotropin-releasing factor, nitric oxide, and the HPA response to stress. Pharmacol Biochem Behav. 1999;64(4):739-751. [CrossRef]
- Viau V. Functional cross-talk between the hypothalamic-pituitary-gonadal and -adrenal axes. J Neuroendocrinol. 2002;14(6):506-513. [CrossRef]
- Teo CH, Wong ACH, Sivakumaran RN, Parhar I, Soga T. Gender Differences in Cortisol and Cortisol Receptors in Depression: A Narrative Review. Int J Mol Sci. 2023 Apr 12;24(8):7129. [CrossRef]
- Rosinger ZJ, Jacobskind JS, De Guzman RM, Justice NJ, Zuloaga DG. A sexually dimorphic distribution of corticotropin-releasing factor receptor 1 in the paraventricular hypothalamus. Neuroscience. 2019;409:195-203. [CrossRef]
- Cox KH, Quinnies KM, Eschendroeder A, Didrick PM, Eugster EA, Rissman EF. Number of X-chromosome genes influences social behavior and vasopressin gene expression in mice. Psychoneuroendocrinology. 2015;51:271-81. [CrossRef]
- Cohen JE, Holsen LM, Ironside M, Moser AD, Duda JM, Null KE, Perlo S, Richards CE, Nascimento NF, Du F, Zuo C, Misra M, Pizzagalli DA, Goldstein JM. Neural response to stress differs by sex in young adulthood. Psychiatry Res Neuroimaging. 2023 Jul;332:111646. [CrossRef]
- Zoorob RJ, Cender D. A different look at corticosteroids. Am Fam Physician. 1998;58(2):443-450.
- Erkut ZA, Pool C, Swaab DF. Glucocorticoids suppress corticotropin-releasing hormone and vasopressin expression in human hypothalamic neurons. J Clin Endocrinol Metab. 1998;83(6):2066-2073. [CrossRef]
- Zanardo V, Golin R, Chiozza ML, Faggian D. Dexamethasone does not affect vasopressin release in bronchopulmonary dysplasia. Pediatr Nephrol. 2000;15(3-4):241-244. [CrossRef]
- Gordijn MS, Gemke RJ, van Dalen EC, Rotteveel J, Kaspers GJ. Hypothalamic-pituitary-adrenal (HPA) axis suppression after treatment with glucocorticoid therapy for childhood acute lymphoblastic leukaemia. Cochrane Database Syst Rev. 2012 May 16;(5):CD008727. Update in: Cochrane Database Syst Rev. 2015;(8):CD008727. [CrossRef]
- Liu RY, Unmehopa UA, Zhou JN, Swaab DF. Glucocorticoids suppress vasopressin gene expression in human suprachiasmatic nucleus. J Steroid Biochem Mol Biol. 2006;98(4-5):248-253. [CrossRef]
- Antoni FA. Magnocellular Vasopressin and the Mechanism of “Glucocorticoid Escape”. Front Endocrinol (Lausanne). 2019 Jun 26;10:422. [CrossRef]
- Escudero DS, Fantinelli JC, Martínez VR, González Arbeláez LF, Amarillo ME, Pérez NG, Díaz RG. Hydrocortisone cardioprotection in ischaemia/reperfusion injury involves antioxidant mechanisms. Eur J Clin Invest. 2024 Jan 31:e14172. Epub ahead of print. [CrossRef]
- Giugliano GR, Giugliano RP, Gibson CM, Kuntz RE. Meta-analysis of corticosteroid treatment in acute myocardial infarction. Am J Cardiol. 2003;91(9):1055-1059. [CrossRef]
- Tol MM, Shekar K, Barnett AG, McGree J, McWhinney BC, Ziegenfuss M, Ungerer JP, Fraser JF. A preliminary investigation into adrenal responsiveness and outcomes in patients with cardiogenic shock after acute myocardial infarction. J Crit Care. 2014 Jun;29(3):470.e1-6. [CrossRef]
- Torgersen C, Luckner G, Schröder DC, Schmittinger CA, Rex C, Ulmer H, Dünser MW. Concomitant arginine-vasopressin and hydrocortisone therapy in severe septic shock: association with mortality. Intensive Care Med. 2011;37(9):1432-1437. [CrossRef]
- Penn J, Douglas W, Curran J, Chaudhuri D, Dionne JC, Fernando SM, Granton D, Mathew R, Rochwerg B. Efficacy and safety of corticosteroids in cardiac arrest: a systematic review, meta-analysis and trial sequential analysis of randomized control trials. Crit Care. 2023 Jan 11;27(1):12. [CrossRef]
- Andersen LW, Isbye D, Kjærgaard J, Kristensen CM, Darling S, Zwisler ST, Fisker S, Schmidt JC, Kirkegaard H, Grejs AM, et al. Effect of Vasopressin and Methylprednisolone vs Placebo on Return of Spontaneous Circulation in Patients With In-Hospital Cardiac Arrest: A Randomized Clinical Trial. JAMA. 2021;326(16):1586-1594. [CrossRef]
- Scott LV, Dinan TG. Vasopressin and the regulation of hypothalamic-pituitary-adrenal axis function: implications for the pathophysiology of depression. Life Sci1998;62(22):1985-1998;. [CrossRef]
- Scott LV, Dinan TG. Vasopressin as a target for antidepressant development: an assessment of the available evidence. J Affect Disord. 2002;72(2):113-124. [CrossRef]
- Simon NG, Guillon C, Fabio K, Heindel ND, Lu SF, Miller M, Ferris CF, Brownstein MJ, Garripa C, Koppel GA. Vasopressin antagonists as anxiolytics and antidepressants: recent developments. Recent Pat CNS Drug Discov. 2008;3(2):77-93. [CrossRef]
- Poretti MB, Sawant RS, Rask-Andersen M, de Cuneo MF, Schiöth HB, Perez MF, Carlini VP. Reduced vasopressin receptors activation mediates the anti-depressant effects of fluoxetine and venlafaxine in bulbectomy model of depression. Psychopharmacology (Berl). 2016 ;233(6):1077-1086. [CrossRef]
- Stewart LQ, Roper JA, Young WS 3rd, O’Carroll AM, Lolait SJ. The role of the arginine vasopressin Avp1b receptor in the acute neuroendocrine action of antidepressants. Psychoneuroendocrinology. 2008;33(4):405-415. [CrossRef]
- Kiss A, Bundzikova J, Pirnik Z, Mikkelsen JD. Different antipsychotics elicit different effects on magnocellular oxytocinergic and vasopressinergic neurons as revealed by Fos immunohistochemistry. J Neurosci Res. 2010;88(3):677-685.
- Florkowski CM, Crozier IG, Nightingale S, Evans MJ, Ellis MJ, Joyce P, Donald RA. Plasma cortisol, PRL, ACTH, AVP and corticotrophin releasing hormone responses to direct current cardioversion and electroconvulsive therapy. Clin Endocrinol (Oxf). 1996;44(2):163-168. [CrossRef]
Figure 1.
Regulation of secretion of hormones of the hypothalamo-pituitary-adrenal axis. ACTH – adrenocorticotropic hormone, AVP – arginine vasopressin, CRH – corticotropine-releasing hormone, FSH - follicle-stimulating hormone GnRH – gonadotropine-releasing hormone, LH - luteinizing hormone. Other explanations in the text.
Figure 1.
Regulation of secretion of hormones of the hypothalamo-pituitary-adrenal axis. ACTH – adrenocorticotropic hormone, AVP – arginine vasopressin, CRH – corticotropine-releasing hormone, FSH - follicle-stimulating hormone GnRH – gonadotropine-releasing hormone, LH - luteinizing hormone. Other explanations in the text.
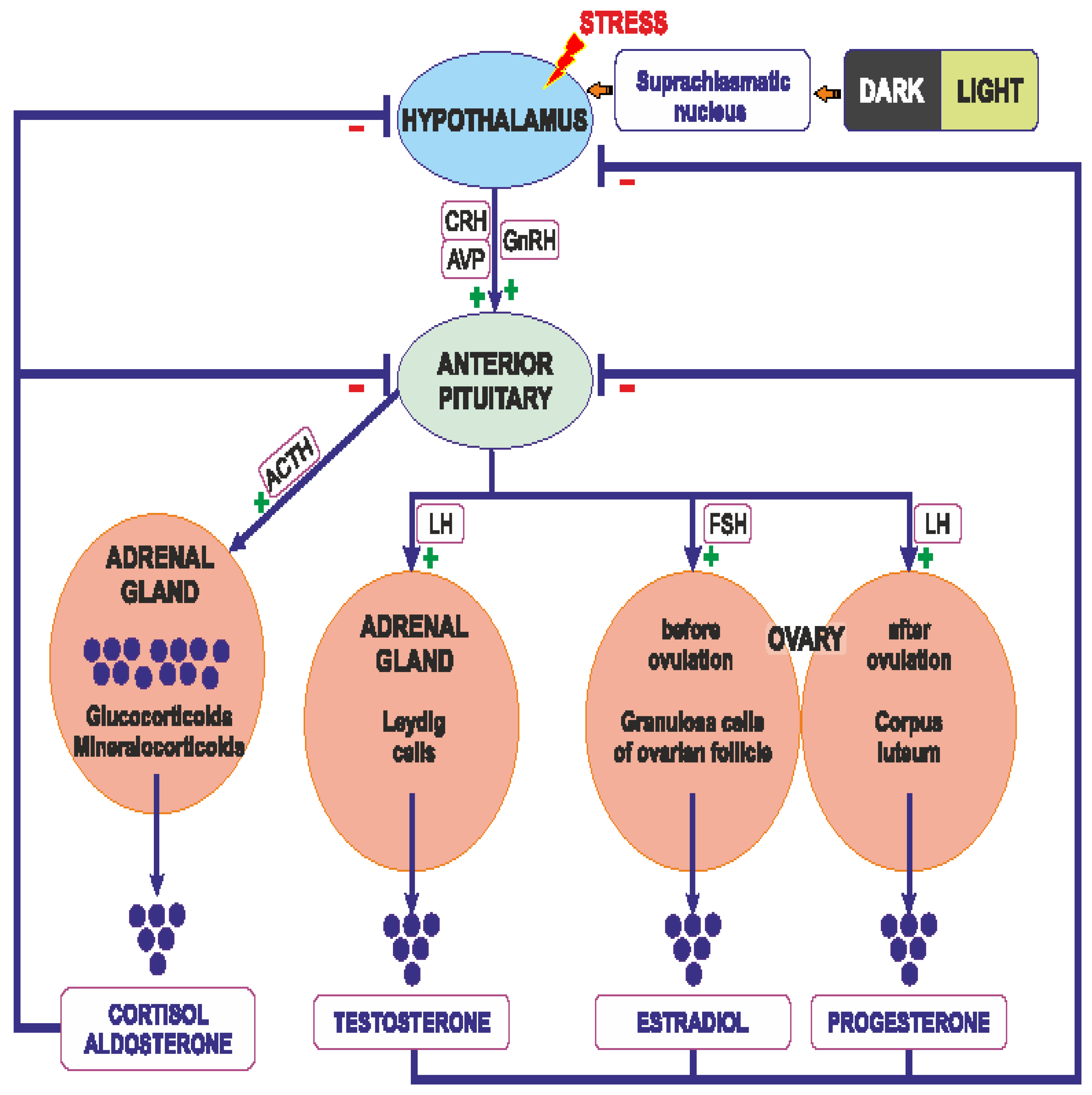
Figure 2.
Steps of synthesis of steroid hormones in the adrenal glands and gonads. Other explanations in the text.
Figure 2.
Steps of synthesis of steroid hormones in the adrenal glands and gonads. Other explanations in the text.
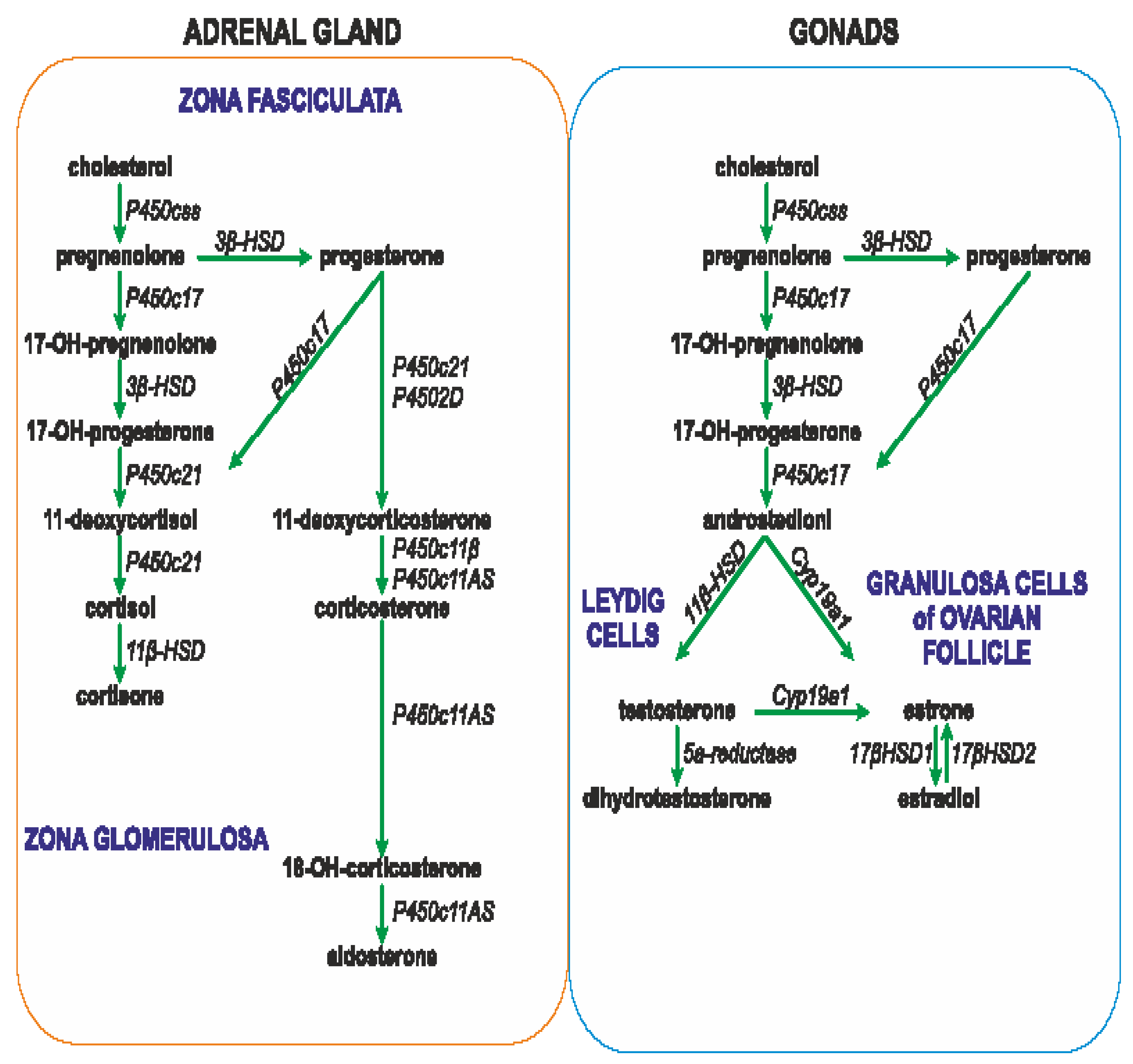
Figure 3.
A phenomenon of „corticosteroid escape”. A - under normal conditions corticosteroids inhibit release of corticotrpine-releasing hormone (CRH), B – during inflammatory state and activation of autoimmune processes inhibition of CRH secretion by negative feedback is impaired and activation of the HPA axis is maintained in spite of high concentration of corticosteroids. ACTH – adrenocorticotropic hormone, IL-6 – interleukin 6.
Figure 3.
A phenomenon of „corticosteroid escape”. A - under normal conditions corticosteroids inhibit release of corticotrpine-releasing hormone (CRH), B – during inflammatory state and activation of autoimmune processes inhibition of CRH secretion by negative feedback is impaired and activation of the HPA axis is maintained in spite of high concentration of corticosteroids. ACTH – adrenocorticotropic hormone, IL-6 – interleukin 6.
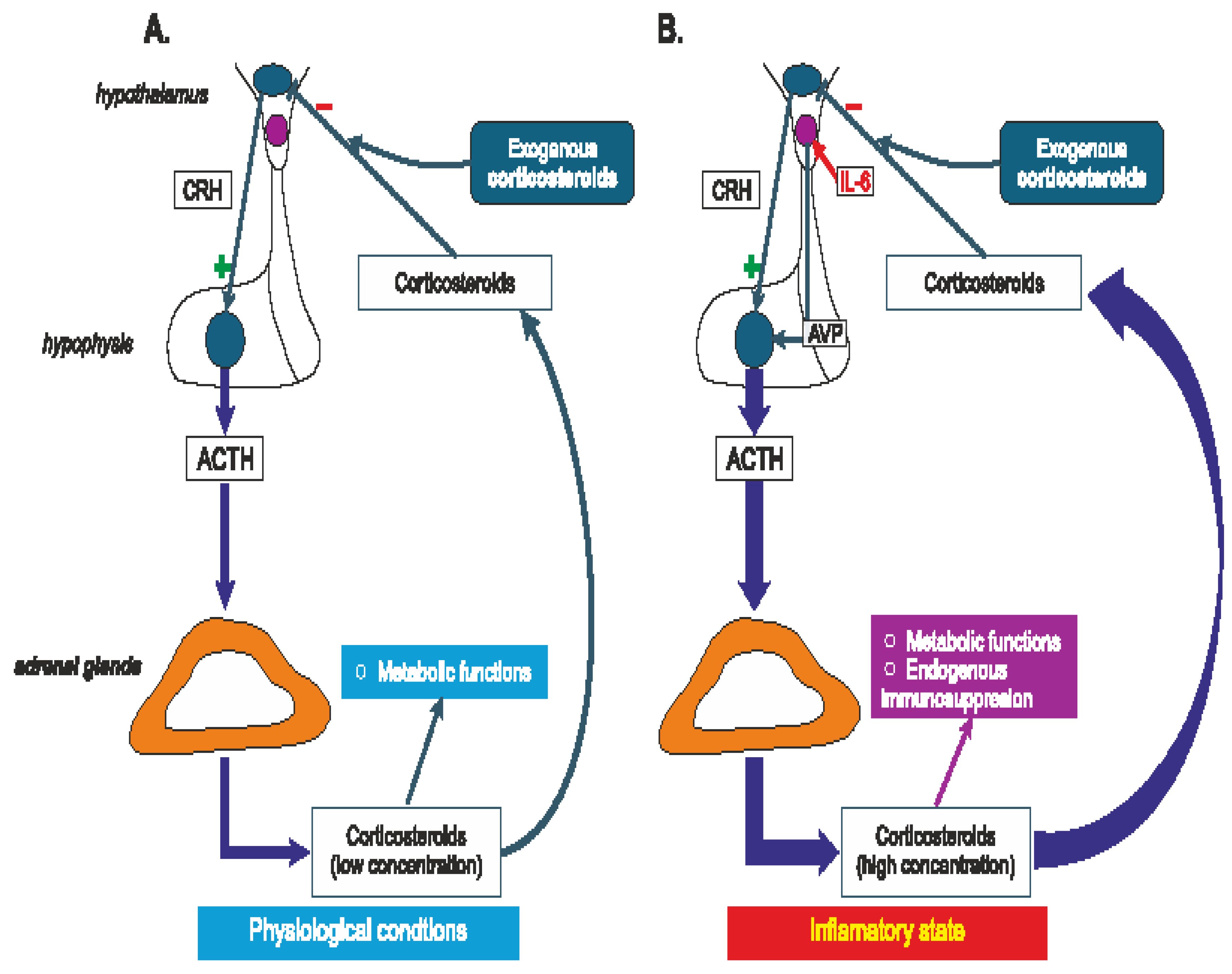
Figure 4.
Stimulatory and inhibitory interactions between vasopressin and hormones secreted by the hypothalamo-pituitary-adrenal system that play essential role in the regulation of energy balance, water-electrolyte balance and blood pressure. ANS – autonomic nervous system, AVP – arginine vasopressin, GR – glucocorticoid receptor, MR – mineralocorticois receptor, V1R, V1aR, V1bR – vasopressin receptors.
Figure 4.
Stimulatory and inhibitory interactions between vasopressin and hormones secreted by the hypothalamo-pituitary-adrenal system that play essential role in the regulation of energy balance, water-electrolyte balance and blood pressure. ANS – autonomic nervous system, AVP – arginine vasopressin, GR – glucocorticoid receptor, MR – mineralocorticois receptor, V1R, V1aR, V1bR – vasopressin receptors.
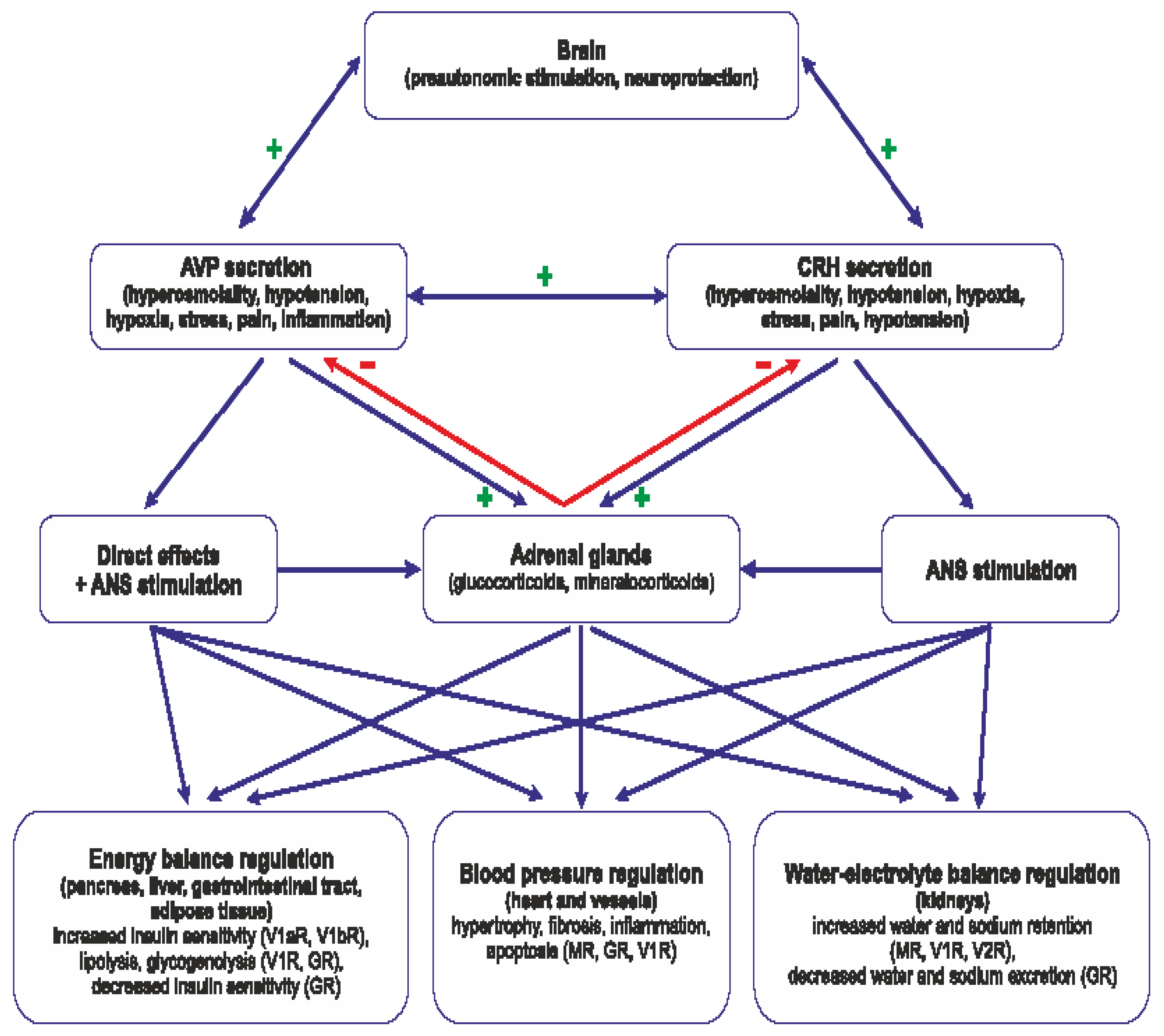
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



