Submitted:
29 May 2024
Posted:
30 May 2024
You are already at the latest version
Abstract
DFT calculations on the photoisomerization reactions of pyridazine N-oxide derivatives are presented. The irradiation of 3,6-diphenylpyridazine N-oxide allowed to obtain the first excited singlet state that underwent to a ring opening reaction induced to the oxygen migration to the adjacent carbon atom, with the formation of a diazo derivative. This intermediate can give a ring closure reaction to give a pyrazole derivative, or, with a higher transition energy, it can lose nitrogen allowing the formation of 2,5-diphenylfuran. The irradiation of 3-phenylpyridazine N-oxide allowed the formation only of 2-phenylfuran. In this case, calculations allowed to explain this behavior. The first excited singlet state gave a photoisomerization reaction with the formation of the corresponding diazo intermediate. This intermediate could not be converted into the corresponding pyrazole derivative because it was converted into a N-formyl pyrazole never reported in literature. The only possible reaction is the loss of nitrogen and the formation of 2-phenylfuran.
Keywords:
pyridazine N-oxide
; photoisomerization
; DFT calculations
; pyrazole
; furan.
1. Introduction
The photochemical isomerization of heterocyclic compounds has been the object of several studies in the past [1,2,3,4] and the rearrangements of heterocyclic compounds can offer relevant synthetic targets [5]. We can consider, as an example of the type of problems we can find in this field, the photochemical reactivity of pyridine. The photochemical isomerization of pyridine in the excited state to form a Dewar structure has been postulated several years ago (Scheme 1) [6,7,8,9,10,11,12,13,14,15,16]. Transient spectroscopy showed that the decay from S1 and S2 states had a fast component (2.2 ps) attributable to the motion on the ππ* reaction path to form the isomer (an azaprefulvene isomer, see below) [17].
The photochemical reactivity of pyridine is known from the first steps of photochemistry studies. In 1932 Freytag described the reaction of pyridine with water when it is irradiated in aqueous solution [18,19,20]. The same reaction has been reconsidered several years ago [21,22].
Pyridine is able to form a complex in the presence of water and the spectroscopic properties of this complex has been studied [23,24,25,26,27]. Experiments performed in a cryogenic matrix, showed the presence of Dewar pyridine in the reaction of pyridine with water [28]. A very similar reaction has been observed irradiation the pyridine derivative in methanol [29,30].
The 2,4,6-trimethyl derivative gave also a pyrrole derivative [30]. The irradiation of pyridine in cyclohexane gave 2- and 4-cyclohexylpyridine in 10% yield [31]. Furthermore, pentakis(pentafluoroethyl)pyridine gave, if irradiated in perfluoropentane at > 270 nm, the Dewar isomer, that can be converted into the prismane derivative through irradiation at > 200 nm in almost quantitative yields [32].
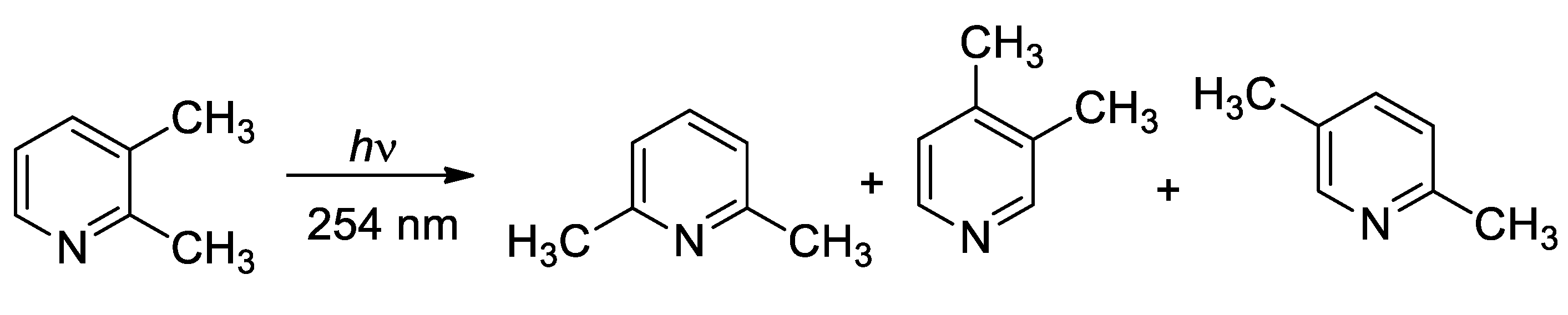
The irradiation of pyridine in the gas phase gave only decomposition products (hydrogen, methane, ethene, acetylene, propylene, allene, methylacetylene, isobutylene) [33]. Gas phase irradiation of 2-picoline gave conflicting results. An article claims as main product of the reaction 4-picoline (together with 2,5-ludidine and pyridine) [34], while another account showed that 2-picoline gave a mixture of 3- and 4-picoline with 5.1 10-4 and 4 10-5, respectively [35]. Another study on gas phase photochemistry of dimethypyridine derivatives proposed a more complex scenario [36]. The irradiation at 254 nm of 2,3-dimethylpyridine (2,3-lutidine) results in the formation of 2,6-dimethylpyridine, 3,4-dimethylpyridine, and 2,5-dimethylpyridine (Scheme 2). However, when the reaction is performed in the presence of nitrogen, while the yields of 2,6-dimethylpyridine and 3,4-dimethylpyridine decreased, the yields of the third product increased. Furthermore, when the irradiation is performed at 290 nm, 2,5-dimethylpyridine was the only reaction product. The authors proposed that these reaction products derived from different pathways. The first two products derived from the S2 state through a mechanism involving electrocyclic ring closure to form an azaprefulvene isomer and sigmatropic shifts.
Furthermore, the formation of 2,5-dimethylpyridine occurred in the triplet state through the formation of a Dewar pyridine derivative followed by a sigmatropic shift of C-2 from C-3 to C-5.
The photochemical reactivity of 3,4,5-trideuteriopyridine is compatible with the formation of an azaprefulvene isomer [37]. When a pyridine derivative is irradiated in methanol some isomerization products have been observed [30]. The mechanism proposed to justify these results involve the formation of Dewar pyridines and prismane.
An aniline derivative was obtained as the main product after the irradiation of an acetonitrile pyridine derivative [38]. Dewar pyridine was supposed to be involved in the reaction together with a [3,3] sigmatropic shift.
The analysis of the literature showed that several hypotheses have been proposed in order to justify the observed reactivity. However, some general observations can be made: the reaction occurs through the excited singlet state. The triplet state is not involved in the reaction. There is a lot of evidence related to the formation of Dewar pyridine. The NMR spectra of Dewar pyridine derivatives obtained performing the reaction at low temperature have been registered [36]. Furthermore, spectroscopic data are in agreement with the formation of an azaprefulvene biradical in the photochemical reaction of pyridine in acetonitrile [17]. CASSCF studies on the formation of Dewar pyridine in the photochemical isomerization of pyridine has been reported [16], while a CASSCF study has been reported also for the formation of the azaprefulvene biradical intermediate [17].
The above reported data clearly showed the presence of an evident confusion of the explanation of the observed photochemical behavior. For this reason, we started a research project devoted to find a lower chaotic description of the reactivity of this type of compounds. However, previous results in this field indicate the presence of a relatively complex frame: in the reaction of pyridinium salts with nucleophiles the observed reaction can be explained admitting the formation of a Dewar isomer [39]; in the case of the reaction with nucleophiles of pyrilium salts, the formation of cyclopentene epoxide cation is favored [40], while in the isomerization of 2,6-dimethylpyrazine, Dewar isomers or benvalene isomers can account for the reaction [41]. Continuing our efforts in this field, we want to describe the results of our calculations on the isomerization of pyridazine N-oxides.
2. Materials and Methods
Gaussian09 has been used for the discussions about the computed geometries [42]. All the computations were based on the Density Functional Theory (DFT) [43] by using the B3LYP hybrid xc functional [44]. Geometry optimizations from the Gaussian09 program have been obtained at the B3LYP/6-311G+(d,p) level of approximation. Geometry optimizations were performed with default settings on geometry convergence (gradients and displacements), integration grid and electronic density (SCF) convergence. Redundant coordinates were used for the geometry optimization as produced by the Gaussian09 program. Analytical evaluation of the energy second derivative matrix w.r.t. Cartesian coordinates (Hessian matrix) at the B3LYP/6-31G+(d,p) level of approximation confirmed the nature of minima on the energy surface points associated to the optimized structures.
The transition state was determined by using the STQN method for locating transition structures. This method, implemented by H. B. Schlegel and coworkers [45], uses a quadratic synchronous transit approach to get closer to the quadratic region of the transition state and then uses a quasi-Newton or eigenvector-following algorithm to complete the optimization. Like the default algorithm for minimizations, it performs optimizations by default in redundant internal coordinates. This method will converge efficiently when provided with an empirical estimate of the Hessian and suitable starting structures. QST3 option has been used, using dichloromethane as background solvent.
3. Results and Discussion
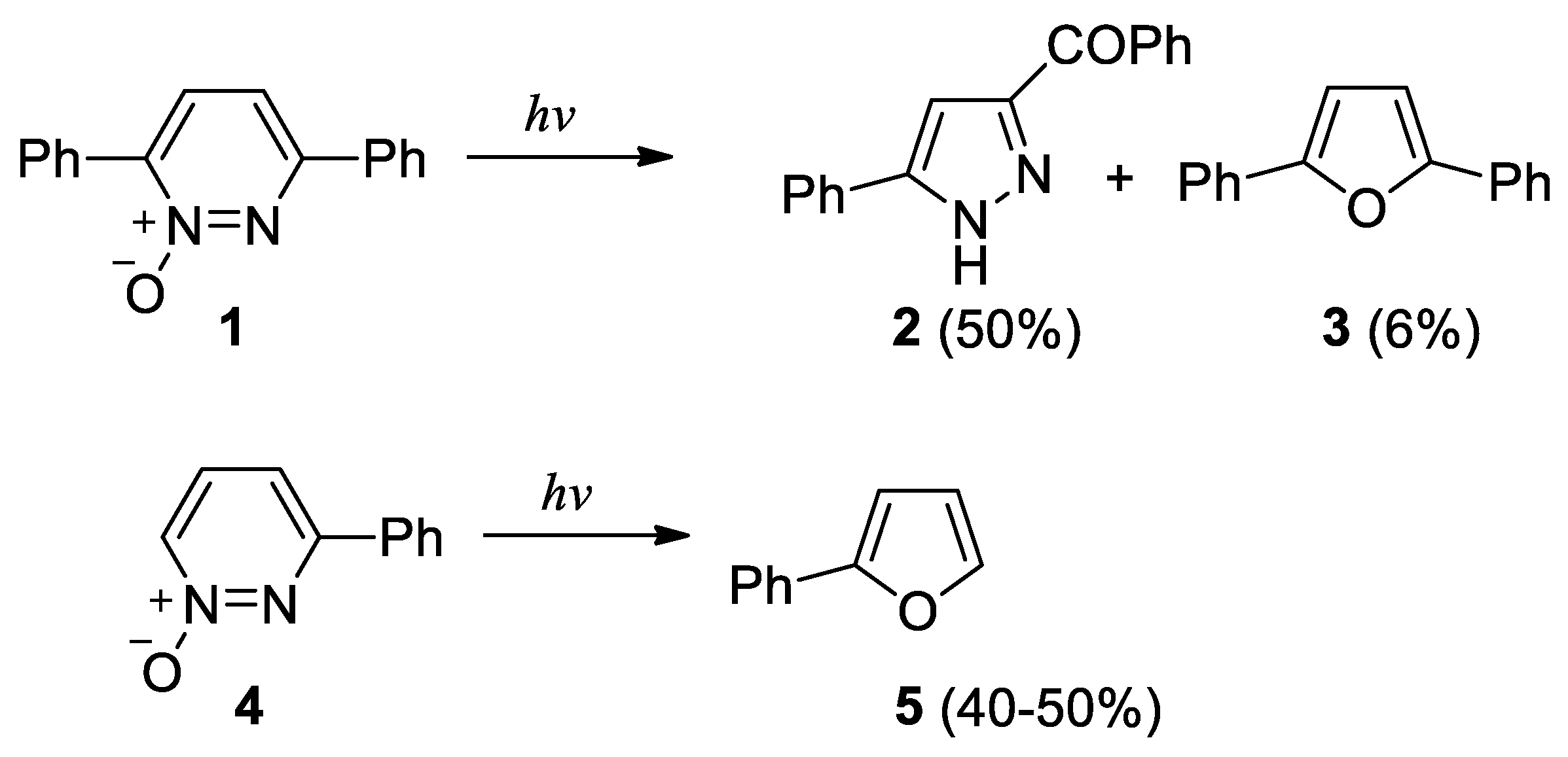
The photochemical isomerization of pyridazine N-oxide derivatives has been the object of some significant research papers [47,48,49,50,51,52,53,54]. The irradiation of 3,6-diphenylpyridazine N-oxide (1) gave 3-benzoyl-5-phenylpyrazole (2) in 50% yield together with 6% of 2,5-diphenylfuran (3) (Scheme 3) [53]. On the other hand, the irradiation of 3-phenylpyridazine N-oxide (4) gave 2-phenylfuran (5) in 40-50% yields (Scheme 3) [52].
The mechanism proposed for these reactions required the formation of the intermediate 6 that allowed a ring opening reaction with the formation of a diazoketone 7. This intermediate had been identified by using flash photolysis experiments. The diazoketone 7 could allow the formation of the pyrazole, or, after the elimination of nitrogen, the formation of the furan derivatives (Scheme 4) [53].
We decided to test this hypothesis with the hope to find a reason for the different reactivity between 3,6-diphenyl and 3-phenylpyridazine N-oxide.
The calculations, performed at DFT/B3LYP/6-311G+(d,p) level of theory on Gaussian 09 allowed to obtain the result depicted in Figure 1 for 3,6-diphenylpyridazine N-oxide.
The first excited singlet state of 3,6-diphenylpyridzine N-oxide was found at 347.59 nm. Attempts to identify the energy of the intermediate 6 failed. The formation of a chemical bond between the oxygen atom and the adjacent carbon atom allowed immediately the cleavage the pyridazine bond with the formation of the diazoketone 7 (Figure 1).
This intermediate can be transformed to give the final products following two different pathways. The attack of nitrogen atom on the caron atom adjacent to the carbonyl group allowed the formation of the ring contraction product 8. This isomerization required the formation of a transition state (TS1) at 1.07 eV (Figure 2). The transition state is an early one, where the nitrogen atom is approaching to the double bond of 7, but the bond between the nitrogen atom and the carbon atom adjacent to the carbonyl group is not formed.
The compound 8 was not stable and a hydrogen shift allowed its transformation in the main product of the reaction, the pyrazole derivative 9 (Figure 1). This isomerization can occur through the transition state TS2, showing an energy of 1.03 eV (Figure 3). The early transition state showed the hydrogen atom while it is migrating from the carbon atom to the nitrogen atom.
The second path showed by 7 to give the reaction product is the lost of nitrogen atom. The authors supposed the formation of an unstable intermediate that underwent the attack of the oxygen atom to give the final product, 2,5-diphenylfuran (Figure 1). The calculations did not support this mechanism (Figure 2). In fact, the intermediate 7 was converted directly into the furan derivative 10, through the formation of a transition state TS3 showing an energy of 1.34 eV. The energy difference of 0.27 eV can account for the observed yields of the reaction products between 9 and 10. In fact, the proposed intermediate was the transition state TS3 (Figure 4).
The above reported results accounts to justify the observed reactivity, allowing to give a simple framework of the observed reactions. To confirm this result, this delineated scheme has to justify also the reactivity of 3-phenylpyridazine N-oxide that was able to give only 2-phenylfuran. In this case, the reaction could be described, on the basis of the above reported calculations, considering the Scheme 5.
The results of the calculations on the photoisomerization of 3-phenylpyridazine N-oxide is summarized in the Figure 5.
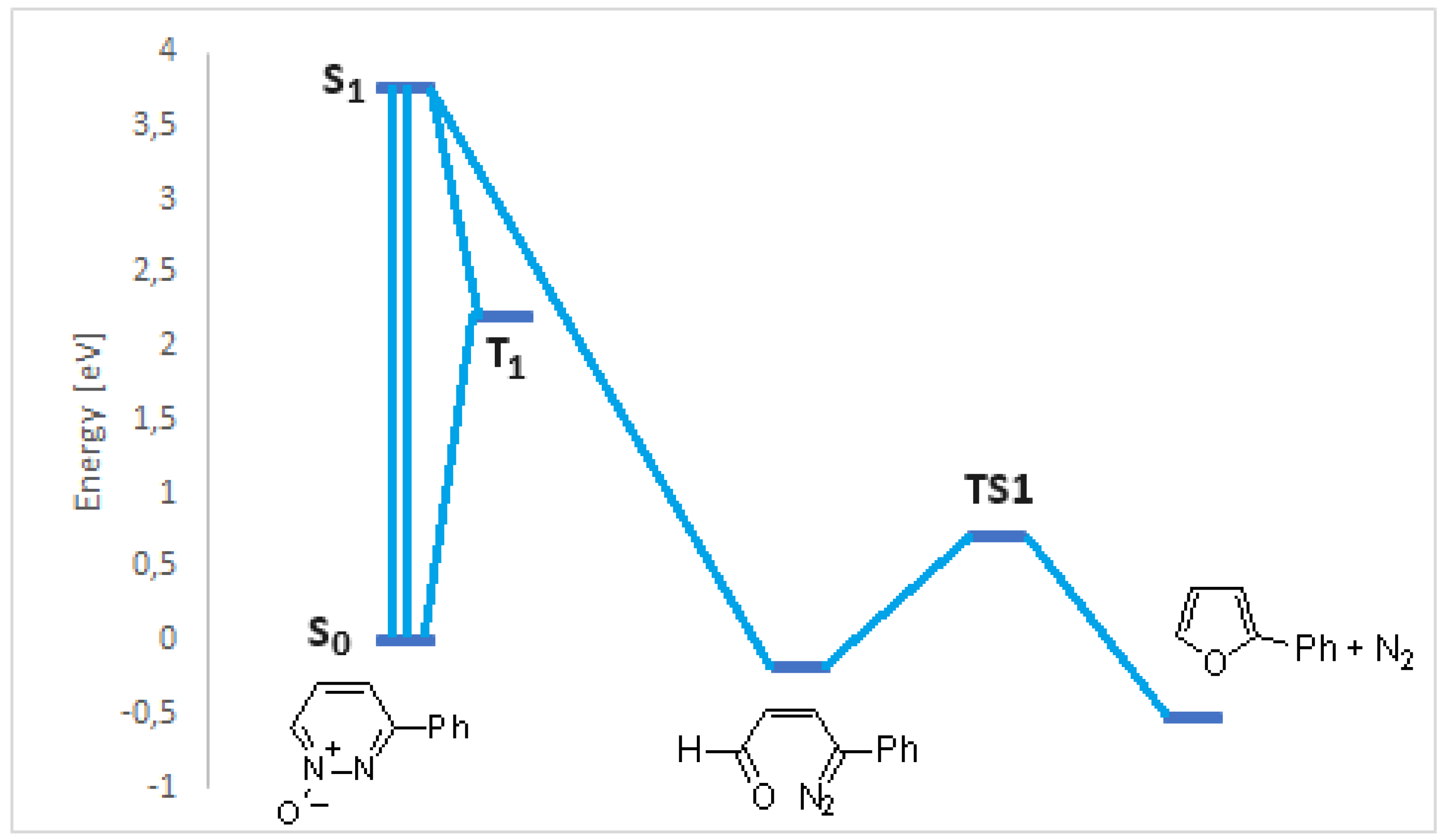
3-Phenylpyridazine N-oxide showed an excited singlet state at 330 nm. The first excited singlet state can give the first isomerization reaction to give the diazo derivative 11. The isomerization reaction of 11 to give 12 did not occur. In fact, the calculations allowed to obtain a different reaction product, the compound 14 (Scheme 6).
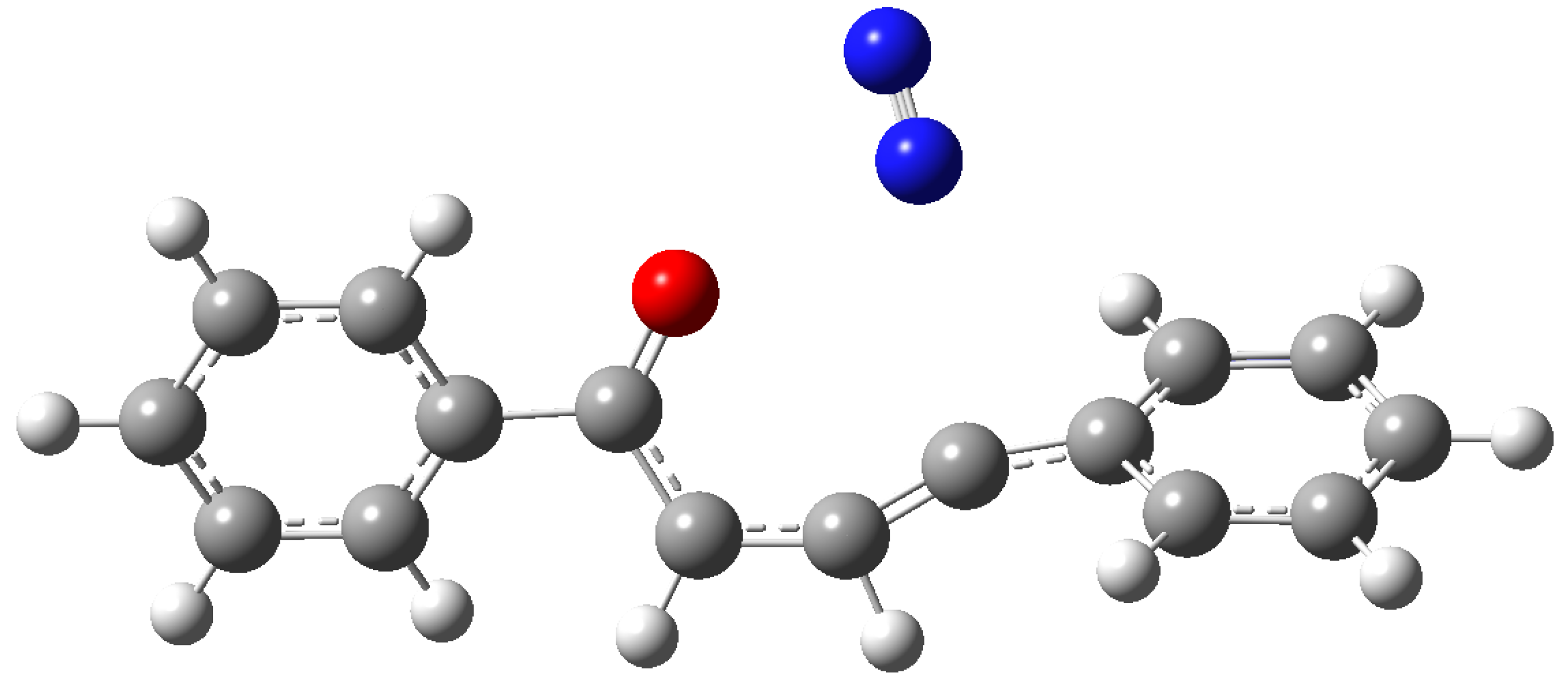
Compound 14 has not been reported in literature and, probably, decomposed. The only possible reaction was the loss of nitrogen from 11 to give 2-phenylfuran 13. The reaction occurred through the formation of the transition state TS1, showing an energy of 0.87 eV (Figure 6).
4. Conclusions
DFT calculations allowed to explain the apparently complex behavior of aryl substituted pyridazine N-oxide derivatives. In the case of 3,6-diphenyl derivative, the first excited singlet state can allow directly the ring opening reaction with formation of a diazo derivative. The diazo compound can allow a ring closure reaction to give a pyrazole derivative or, with a higher transition energy, a loss of nitrogen with the formation of a furan derivatives. In the case of 3-phenylpyridazine N-oxide, the reaction followed the same scheme, allowing the formation of the diazo derivative 11. This compound cannot give a pyrazole derivative 12 allowing the formation of only the furan derivative 13.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Structural information on all the compounds studied in this article.
Author Contributions
Conceptualization, M.D.A. and L.E.; investigation, M.D.A.; data curation, L. E.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Braslavsky, S.; Heicklen, J. The gas-phase thermal and photochemical decomposition of heterocyclic compounds containing nitrogen, oxygen, or sulfur. Chem. Rev. 1977, 77, 473–511. [Google Scholar] [CrossRef]
- Lablache-Combier, A. Photorearrangments of nitrogen-containing arenes. In CRC Handbook of Photochemistry and Photobiology; Horspool, W.M., Song, P.-S., Eds.; CRC Press: Boca Raton, NJ, USA, 1995; pp. 1063–1120. [Google Scholar]
- Pavlik, J. WPhotoisomerization of some nitrogen-containing heteroaromatic compounds. In CRC Handbook of Organic Photochemistry and Photobiology; Horspool, W., Lenci, F., Eds.; CRC Press: Boca Raton, NJ, USA, 2004; pp. 97–1. [Google Scholar]
- Pavlik, J. W. Photochemistry of thiazoles, isothiazoles, and 1,2,4-thiadiazoles. In CRC Handbook of organic photochemistry and photobiology, Horspool, W.; Lenci, F., Eds. CRC Press: Boca Raton. 2004, pp. 98–1,98-14.
- Lefebvre, C.; Fortier, L.; Hoffmann, N. Photochemical rearrangements in heterocyclic chemistry. Eur. J. Org. Chem. 2020, 1393–1404. [Google Scholar] [CrossRef]
- Wilzbach, K. E.; Rausch D., J. Photochemistry of nitrogen heterocycles. Dewar pyridine and its intermediacy in photoreduction and photohydration of pyridine. J. Am. Chem. Soc. 1970, 92, 2178–2179. [Google Scholar] [CrossRef]
- Chapman, O. L.; McIntosh, C. L.; Pacansky, J. Photochemical transformations. XLVIII. Cyclobutadiene. J. Am. Chem. Soc. 1973, 95, 614–617. [Google Scholar] [CrossRef]
- Ratajczak, E.; Sztuba, B. The gas phase photolysis of pentafluoropyridine. J. Photochem. 1980, 13, 233–242. [Google Scholar] [CrossRef]
- Yamazaki, I.; Murao, T.; Yamanaka, T.; Yoshihara, K. Intramolecular electronic relaxation and photoisomerization processes in the isolated azabenzene molecules pyridine, pyrazine and pyrimidine. Farad. Discuss. Chem. Soc. 1983, 75, 395–405. [Google Scholar] [CrossRef]
- Vysotskii, Y. S.; Sivyakova, L. N. Quantum-chemical interpretation of recyclization reactions. 10. Photoisomerization of six-membered heterocycles. Khim. Geterosikl. Soedin. 1986, 22, 357–363. [Google Scholar] [CrossRef]
- Johnstone, D. E.; Sodeau, J. R. Matrix-controlled photochemistry of benzene and pyridine. J. Phys. Chem. 1991, 95, 165–169. [Google Scholar] [CrossRef]
- Sobolewski, A. L.; Domcke, W. Photophysically relevant potential energy functions of low-lying singlet states of benzene, pyridine and pyrazine: an ab initio study. Chem. Phys. Lett. 1991, 180, 381–386. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, X.; Pulay, P. Geometries, force fields, and vibrational assignments of Dewar benzene and Dewar pyridine. J. Phys. Chem. 1992, 96, 3669–3674. [Google Scholar] [CrossRef]
- Kudoth, S.; Takayanagi, M.; Nakata, M. Dewar pyridine studied by matrix isolation infrared spectroscopy and DFT calculation. J. Photochem. Photobiol. A: Chem. 1999, 123, 25–30. [Google Scholar] [CrossRef]
- Wang, B.; Liu, B.; Wang, Y.; Wang, L. Ultrafast dynamics of pyridine in “channel three” region. Int. J. Mass Spectrom. 2010, 289, 92–97. [Google Scholar] [CrossRef]
- Su, M. -D. Model Study on the Pyridine−Dewar pyridine and some related photoisomerization reactions. J. Phys. Chem. A 2007, 111, 971–975. [Google Scholar] [CrossRef]
- Chachisvilis, M.; Zewail, A. H. Femtosecond dynamics of pyridine in the condensed phase: valence isomerization by conical intersections. J. Phys. Chem. A 1999, 103, 7408–7418. [Google Scholar] [CrossRef]
- Freytag, H.; Neudert, W. Einwirkung ultravioletter Strahlen auf Pyridin. (I. Mitteilung). Ein neuer Nachweis einiger primärer aromatischer Amine und des Pyridins. J. Prakt. Chem. 1932, 135, 15–35. [Google Scholar] [CrossRef]
- Freytag, H.; Hlučka, F. Einwirkung ultravioletter Strahlen auf Pyridin. III. Mitteilung: Über Photopyridinbildung im Spektrum. J. Prakt. Chem. 1933, 136, 288–292. [Google Scholar] [CrossRef]
- Freytag, H. Einwirkung ultravioletter Strahlen auf Pyridin. V. Mitteilung: Über den qualitativen Nachweis weiterer primärer aromatischer Amine, über das Verhalten von Pyridinderivaten im UV-Licht und über die Natur des „Photopyridins”︁. J. Prakt. Chem. 1934, 139, 44–62. [Google Scholar] [CrossRef]
- Joussot-Dubien, J.; Houdard, J. Reversible photolysis of pyridine in aqueous solution. Tetrahedron Lett. 1967, 4389–4391. [Google Scholar] [CrossRef]
- Joussot-Dubien, J.; Houdard-Pereyre, J. Photolyse de la pyridine en solution aqueuse. Bull. Soc. Chim. Fr. 1969, 2619–2623. [Google Scholar]
- Destexhe, A.; Smets, J.; Adamowicz, L.; Maes, G. Matrix isolation FT-IR studies and ab initio calculations of hydrogen-bonded complexes of molecules modeling cytosine or isocytosine tautomers. 1. Pyridine and pyrimidine complexes with water in argon matrixes. J. Phys. Chem. 1994, 98, 1506–1514. [Google Scholar] [CrossRef]
- Dkhissi, A.; Adamowicz, L.; Maes, G. Density Functional Theory Study of the Hydrogen-Bonded Pyridine−H2O Complex: A Comparison with RHF and MP2 Methods and with Experimental Data. J. Phys. Chem. A 2000, 104, 2112–2119. [Google Scholar] [CrossRef]
- Reimers, J. R.; Cai, Z. -L. Hydrogen bonding and reactivity of water to azines in their S1 (n,π*) electronic excited states in the gas phase and in solution. Phys. Chem. Chem. Phys. 2012, 14, 8791–8802. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sobolewski, A. L.; Borrelli, R.; Domcke, W. Computational investigation of the photoinduced homolytic dissociation of water in the pyridine–water complex. Phys. Chem. Chem. Phys. 2013, 15, 5957–5966. [Google Scholar] [CrossRef] [PubMed]
- Esteves-López, N.; Coussan, S.; Dedonder-Lardeux, C.; Jouvet, C. Photoinduced water splitting in pyridine water clusters. Phys. Chem. Chem. Phys. 2016, 18, 25637–25644. [Google Scholar] [CrossRef] [PubMed]
- Esteves-López, N.; Coussan, S. UV photochemistry of pyridine-water and pyridine-ammonia complexes trapped in cryogenic matrices. J. Mol. Struct. 2018, 1172, 65–73. [Google Scholar] [CrossRef]
- Kellogg, R. M.; van Bergen, T. J.; Wynberg, H. Photochemical ring contraction, reduction, and solvent addition in pyridines. Tetrahedron Lett. 1969, 5211–5214. [Google Scholar] [CrossRef]
- Van Bergen, T. J.; Kellogg, R. M. Photochemistry of 3,5-dicarboalkoxypyridines. Reduction and rearrangement. J. Am. Chem. Soc. 1972, 94, 8451–8471. [Google Scholar] [CrossRef]
- Caplain, S.; Catteau, J. P.; Lablache-Combier, A. Liquid-phase photochemistry of pyridine and 2- and 4-picoline. J. Chem. Soc. D, Chem. Commun, 1475. [Google Scholar] [CrossRef]
- Barlow, M. G.; Dingwall, J. G.; Haszeldine, R. N. Valence-bond isomers of heterocyclic compounds: isomers of pentakis(pentafluoroethyl)pyridine. J. Chem. Soc. D, Chem. Commun. [CrossRef]
- Mathias, E.; Heicklen, J. The gas phase photolysis of pyridine. Mol. Photochem. 1972, 4, 483–500. [Google Scholar]
- Caplain, S.; Lablache-Combier, A. Gas-phase photochemistry of picolines and lutidines. J. Chem. Soc. D, Chem. Commun. 1248. [Google Scholar] [CrossRef]
- Roebke, W. Gas-phase photolysis of 2-picoline. J. Phys. Chem. 1970, 74, 4198–4203. [Google Scholar] [CrossRef]
- Pavlik J., W.; Kebede, N.; Thompson, M.; Colin Day, A.; Barltrop, J. A. Vapor-phase photochemistry of dimethylpyridines. J. Am. Chem. Soc. 1999, 121, 5666–5673. [Google Scholar] [CrossRef]
- Pavlik, J. W.; Laohhasurayotin, S. The photochemistry of 3,4,5-trideuteriopyridine. Tetrahedron Lett. 2003, 44, 8109–8111. [Google Scholar] [CrossRef]
- Ogata, Y.; Takagi, K. Photoisomerization of 2-pyridylacetonitrile to anthranilonitrile. J. Am. Chem. Soc. 1974, 96, 5933–5934. [Google Scholar] [CrossRef]
- D’Auria, M. On the photochemical reaction of pyridinium salts with nucleophiles, Photochem. Photobiol. Sci. 2021, 20, 923–926. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, M. The photoreaction of pyrilium cation with water: a DFT study. Lett. Org. Chem. 2022, 19, 739–742. [Google Scholar] [CrossRef]
- D’Auria, M. A theoretical study on the photochemical isomerization of 2,6-dimethylpyrazine. Organics 2022, 3, 95–101. [Google Scholar] [CrossRef]
- Gaussian 09, Revision A.1, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- Parr, R. G.; Yang, W. Density Functional Theory of Atoms and Molecules, Oxford University Press: Oxford, UK, 1989.
- Becke, A. D. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Peng, C.; Schlegel, H. B. Combining Synchronous Transit and Quasi-Newton Methods to Find Transition State. Israel J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P. Y.; Schlegel, H. B.; Frisch, M. J. Using Redundant Internal Coordinates to Optimize Equilibrium Geometries and Transition States. J. Comp. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Kumler, P. L.; Buchardt, O. The photolysis of 3,6-diphenylpyridazine N-oxide. Detection of a transient diazo compound. J. Am. Chem. Soc. 1968, 90, 3640–3641. [Google Scholar] [CrossRef]
- Buchardt, O. The photolysis of 1,4-diphenylphthalazine N-oxide to 1,3-diphenylisobenzofuran. Tetrahedron Lett. 1968, 1911–1912. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Arai, H.; Igeta, H. Photochemistry. III. The photolysis of 3,4,5,6-tetraphenylpyridazine N.oxides. Tetrahedron Lett. 2582. [Google Scholar]
- Tsuchiya, T.; Arai, H.; Igeta, H. Photolysis of pyridazine N-oxides; formation of cyclopropenyl ketones. J. Chem. Soc. Chem. Commun. 1972, 550–551. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Arai, H.; Tonami, T.; Igeta, H. Photochemistry. VII. Photolyses of polyphenyl pyridazine N-oxides. Chem. Pharm. Bull. 1972, 20, 300–303. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Arai, H.; Igeta, H. Photochemistry – IX. Formation of cyclopropenyl ketones and furans from pyridazine N-oxides by irradiation. Tetrahedron 1973, 29, 2747–2751. [Google Scholar] [CrossRef]
- Tomer, K. B.; Harrit, N.; Rosenthal, I.; Buchardt, O.; Kumler, P. L.; Creed, D. Photochemical behavior of aromatic 1,2-diazine N-oxides. J. Am. Chem. Soc. 1973, 95, 7402–7406. [Google Scholar] [CrossRef]
- Portillo, M.; Maxwell, M. A.; Frederich J., H. Synthesis of nitrogen heterocycles via photochemical ring opening of pyridazine N-oxides. Org. Lett. 2016, 18, 5142–5145. [Google Scholar] [CrossRef]
Scheme 1.
Photochamical isomerization of pyridine.
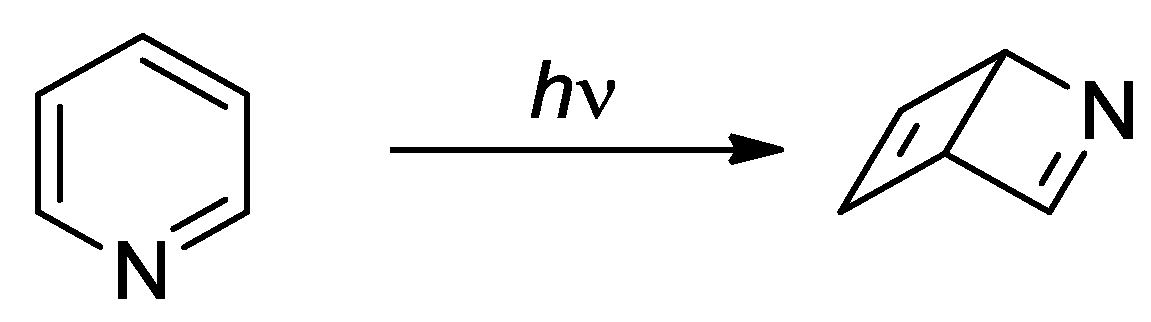
Scheme 2.
Photochemical isomerization of 2,3-dimethylpyridine.
Scheme 3.
Photochemical isomerization of 3,6-diphenyl- and 3-phenylpyridazine N-oxide.
Scheme 4.
Proposed mechanism for the formation of pyrazoles and furans in the photoisomerization of pyridazine N-oxide.
Scheme 4.
Proposed mechanism for the formation of pyrazoles and furans in the photoisomerization of pyridazine N-oxide.
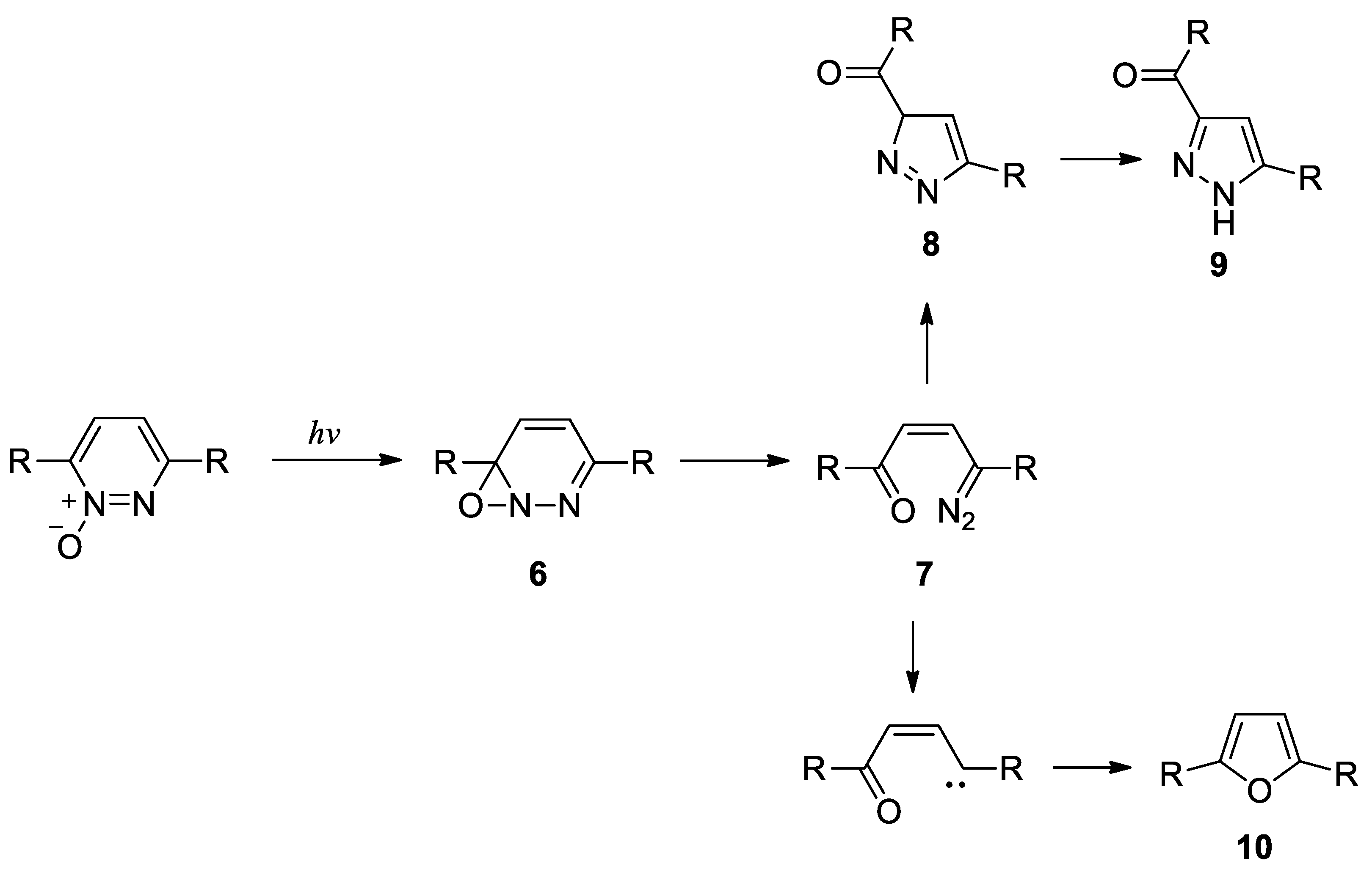
Figure 1.
Photochemical isomerization of 3,6-diphenylpyridazine N-oxide.
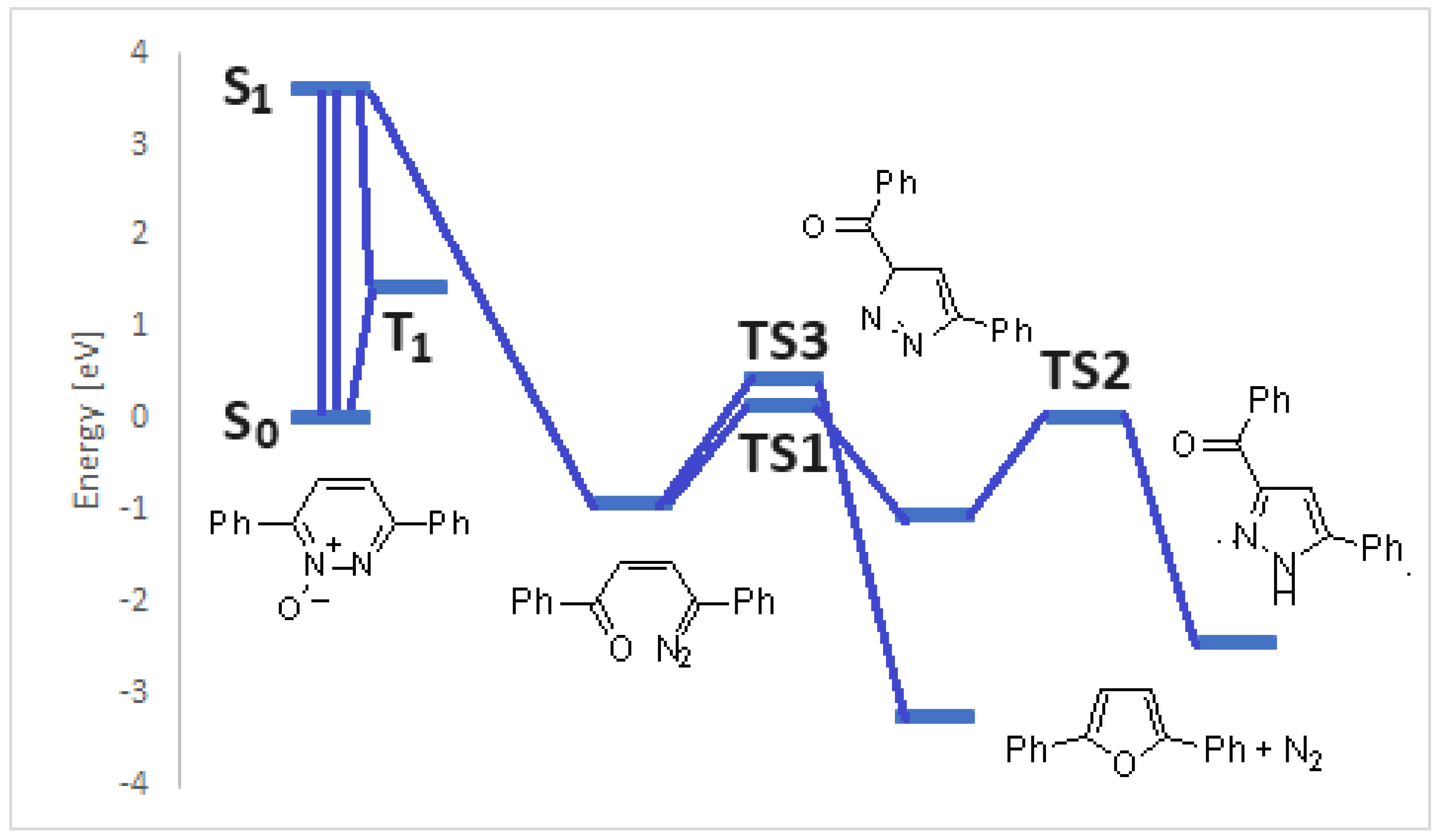
Figure 2.
Transition state in the conversion 7 → 8, TS1.
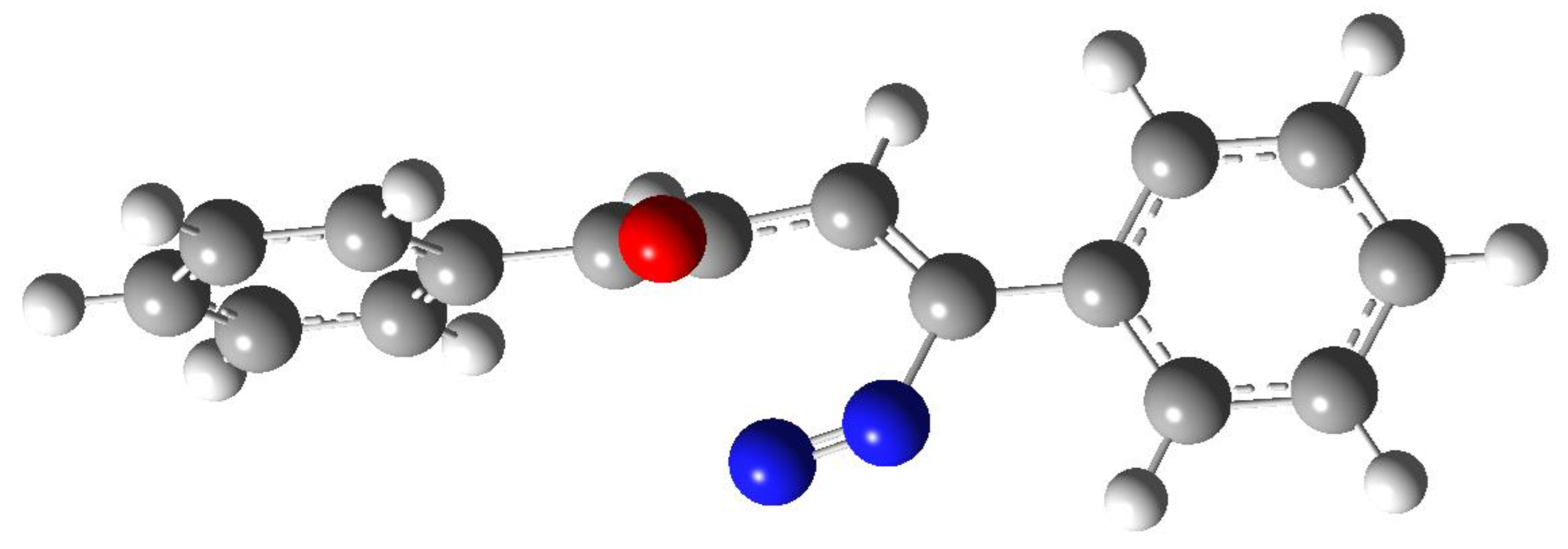
Figure 3.
Transition state in the conversion 8 → 9, TS2.
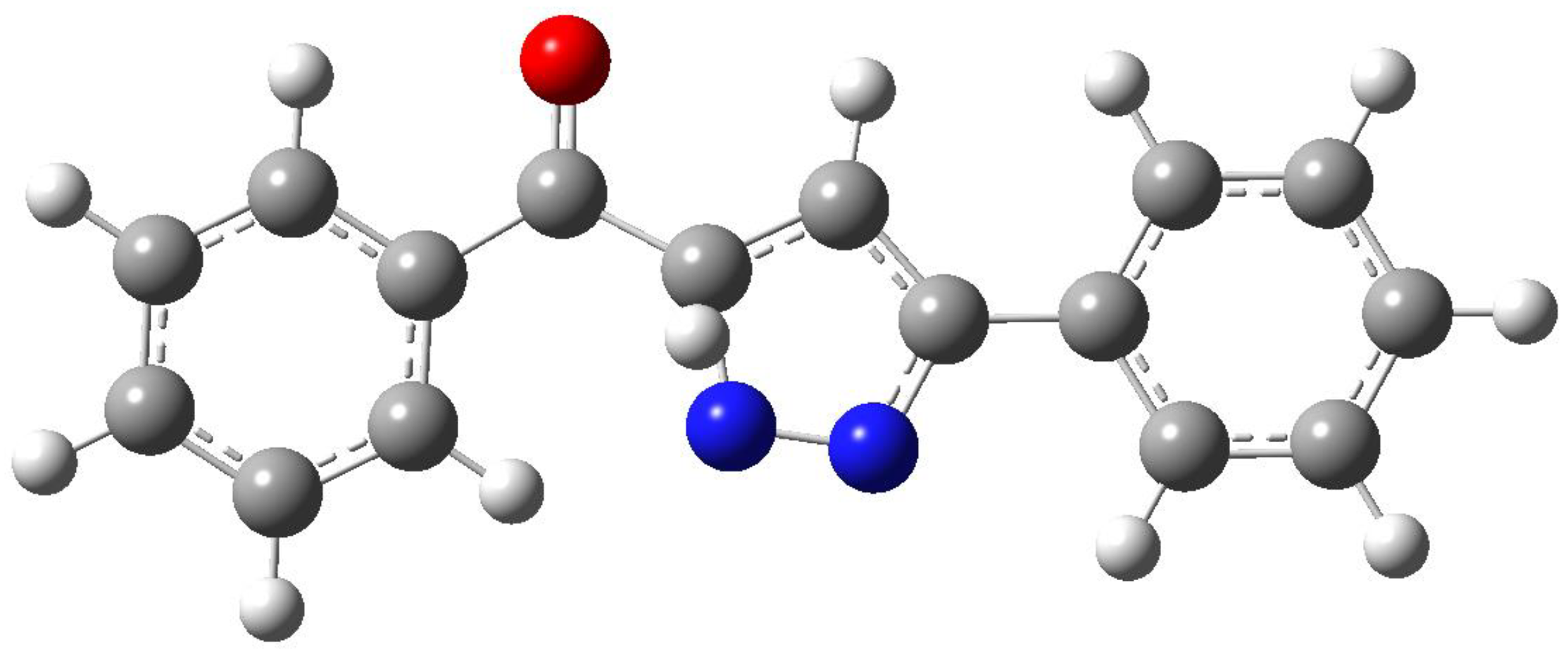
Figure 4.
Transition state in the conversion 7 → 10, TS3.
Scheme 5.
Possible photoisomerization intermediates and products of 3-phenylpyridazine N-oxide.
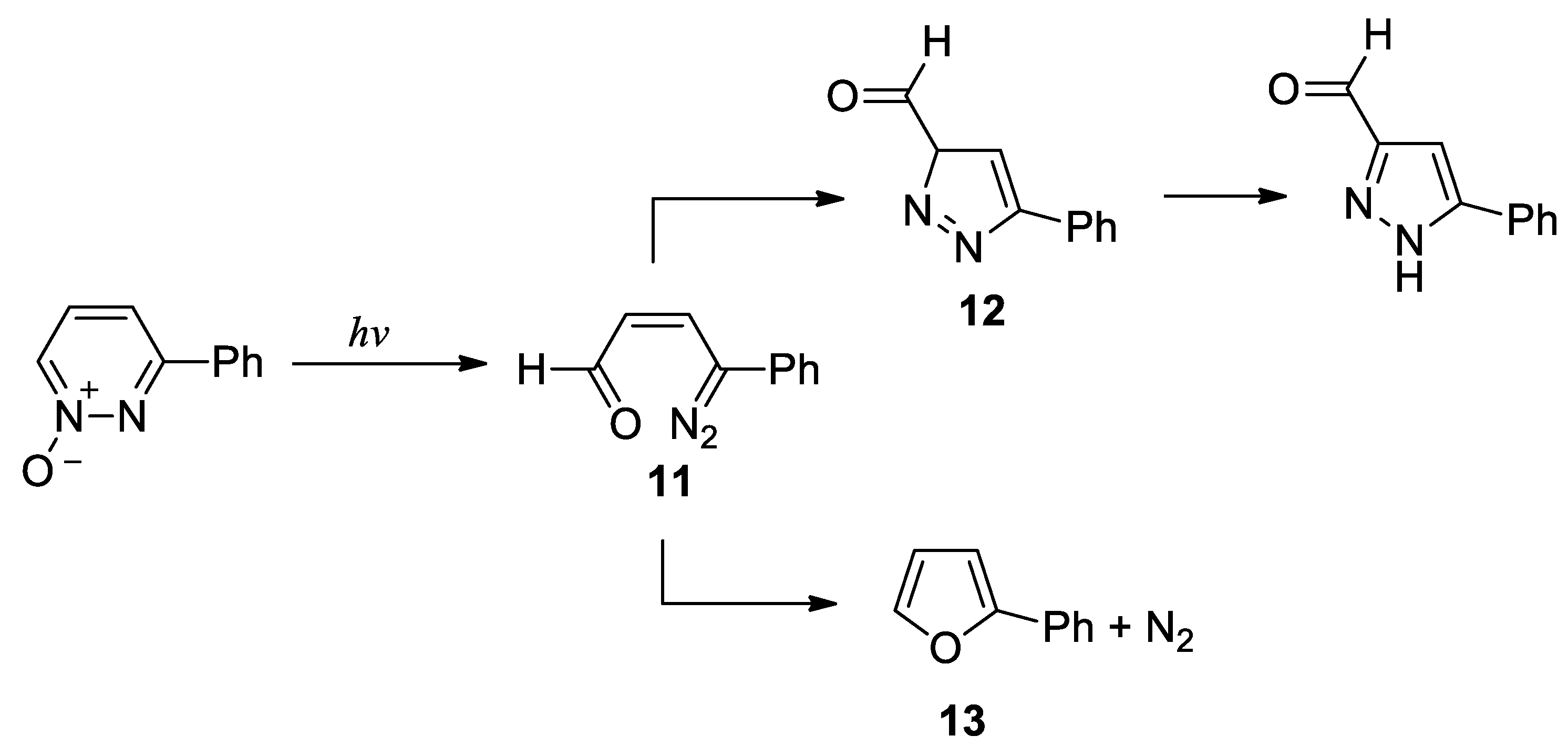
Figure 5.
Photochemical isomerization of 3-phenylpyridazine N-oxide.
Scheme 6.
Possible isomerization of diazo derivative 11.
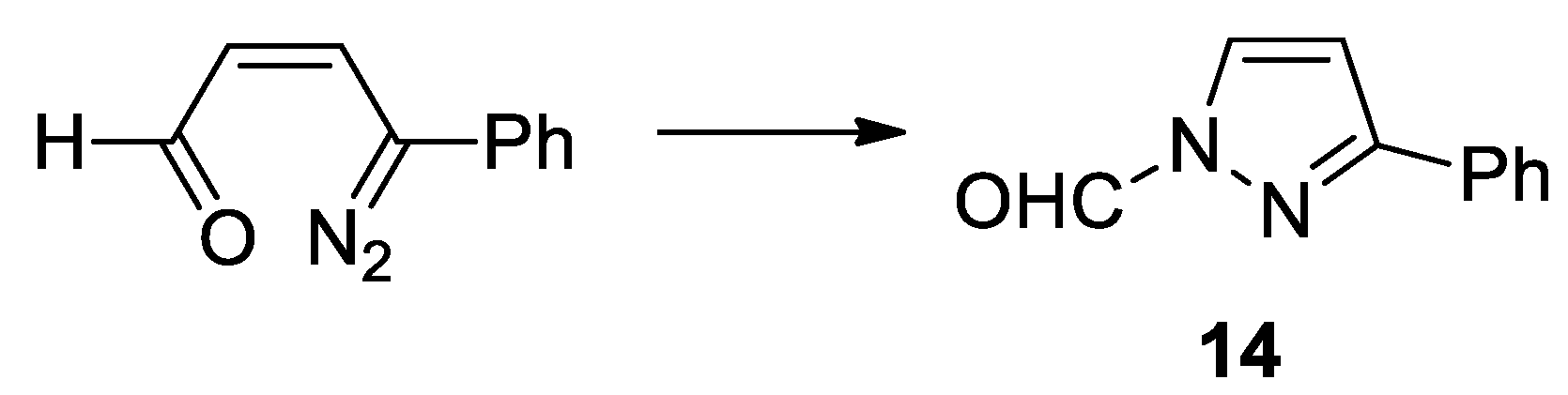
Figure 6.
Transition state in the reaction of 11 to give 13.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



