Submitted:
18 May 2024
Posted:
21 May 2024
You are already at the latest version
Abstract
Ovarian cancer (OC) represents a significant challenge in the realm of gynecological cancers, characterized by poor survival rates and complex etiology. In this review, we delve into the se-lected genetic, environmental, and hormonal factors underpinning OC development.
We have reviewed scientific databases searching for ovarian cancer genetic and epigenetic factors. We have included studies based on their relevancy. As a result of exploring ovarian carcinogen-esis, this systematic review contains data collected from 126 various works.
The role of prominent genetic players such as BRCA mutations, DNA repair mechanisms, un-derscores the intricate landscape of OC susceptibility. We explore Li-Fraumeni and Lynch syn-drome, which impart a heightened predisposition to OC development. Hormonal factors such as estrogen, progesterone, and androgens are also discussed. Environmental alterations, ranging from lifestyle influences on OC to microbiome dysbiosis and lifestyle factors such as obesity, al-cohol consumption, and physical activity intersect with genetic and epigenetic pathways, shaping the risk landscape for OC.
Through a meticulous examination of current literature, this review provides a nuanced under-standing of the multifactorial nature of OC, emphasizing the need for holistic approaches to prevention, diagnosis, and treatment.
Keywords:
Ovarian cancer
; Genetic factors
; Hormonal factors
; Environmental factors
; BRCA mutations
; DNA repair mechanisms
; Hormone therapy
; Tumor suppressor genes
; Gynecologic cancer
1. Introduction
Ovarian cancer (OC) is one of the most common gynecological cancers, associated with poor survival rates [1,2]. According to the Ovarian Cancer Research Alliance in 2023 there were 19,710 estimated new cases of OC in the United States, which contribute to 1% of all new cancers. OC is responsible for 13,270 deaths in 2023, which is 2.2 % of all cancer deaths. The five-year survival rates for ovarian cancer in the USA are relatively 50,8%. [2]. In 2020 in Central Eastern Europe there were 28 530 new cases and 17 565 deaths. In Northern Europe, the number of new cases was 9457 and the number of deaths was 6530. In Southern Europe, the numbers were 12 779 new cases and 8015 deaths. In Western Europe 12 779 new cases were noted and 8015 deaths [3]. It occurs more often in postmenopausal, white women older than 63 [4] (Figure 1). Ovarian cancer involves numerous factors, complex biological processes, and unpredictable consequences. Non-specific symptoms make early diagnosis more difficult. There is a wide spectrum of factors participating in carcinogenesis, which are worth taking into the deeper analysis. They are divided into genetic and epigenetic factors.
Type I tumors, accounting for 30% of the tumors, are low-grade tumors, characterized by slow growth and large cystic formations with mutations in KRAS, BRAF, PTEN, CTNNB1, PIK3CA, PPP2R1A, and ARID1A genes. Opposite, type II tumors, constituting 70% of all ovarian malignancies, are aggressive, high-grade cancers that almost always present at an advanced stage with high mortality. Type II tumors have alterations in the TP53, BRCA1, and BRCA2 genes and are very genetically unstable [5] (Figure 2). This study is focused mainly on factors related to type II tumors, considering them as a significant medical concern, requiring extensive knowledge and quick action.
Our study aims to provide a comprehensive overview of the main genetic, hormonal, and environmental factors that could be related to the development of ovarian cancer.
As stated above — various genetic factors influence the development of ovarian cancer. We chose to discuss in detail the actions of BRCA1/2, Double Strand Break Repair, Li-Fraumeni and Lynch syndromes, and phosphatase and tensin homolog protein expression. Androgens, estrogens, and progesterone are proven to affect the development of ovarian cancer. That is why we also discuss their mechanism of action. Environmental factors that can be related to the female reproductive system such as late menopause, hormone therapy, polycystic ovary syndrome, as well as pregnancy and breastfeeding. Also, we discuss the effect microbiota and viruses such as HPV, have on the development of ovarian cancer. In this review, the most important factors, the understanding of which is important from a prevention perspective, have been described.
Numerous studies have confirmed the role of epigenetic changes in the development of OC. In ovarian cancer, as in many cancer types, two opposing epigenetic phenomena have been identified: a global decrease in DNA methylation, leading to the demethylation of several oncogenes and repetitive elements; and specific CpG island hypermethylation associated with the promoters of tumor suppressor genes, deactivating these genes [6]. However, a detailed discussion of epigenetic factors falls outside of scope of this review.
2. Materials and Methods
2.1. Overview
Investigation by two autonomous researchers, via a systematic review of scientific literature accessible online, was carried out to uncover valuable publications, analyze the quantity and quality of research, and assess the availability of the materials.
A multistep search through online databases PubMed, Google Scholar, and Science Direct was performed to identify the scientific publications suitable for analysis. Two separate review sessions were conducted using the keywords “ovarian cancer genetic factors” and “ovarian cancer epigenetic factors”.
Articles were chosen in a period since 2000 and then based on the most suitable results, a list of records was organized. All types of scientific papers, which meet the criteria were analyzed. As a result, searching through three databases 89 468 works, in summary, were found after entering the keyword “ovarian cancer genetic factor”. After entering the second keyword “ovarian cancer epigenetic factor” 20 375 publications were found.
2.2. Exclusion and Inclusion Criteria
The research methodology employed a set of exclusion criteria to accurately identify pertinent scientific papers. We have decided to search only for journal articles, published in or after the year 2000, because from this year important works in terms of the described factors began to appear, which contain information confirmed in the subsequent works included.
While searching in PubMed, Embase, and Google Scholar we have utilized the “sort by relevancy” function provided by those databases. We have a priori decided to cut the number of publications screened to the first 200 results in each search in every database.
Papers that were inaccessible or were of any kind of scientific work other than journal articles also were excluded from further review. Only papers in English and Polish were read, as those were the only languages used by the authors. We relied on studies using both human samples and mice.
We have decided to include only papers that directly regard genetic factors connected to ovarian cancer etiology. If a paper was included by one author and excluded by another based on relation to the subject of this review, then it was up for discussion in which all authors could assess the paper’s relevancy until reaching a unanimous decision on the inclusion of that article.
2.3. Records Retrieval
After entering the keyword “ovarian cancer genetic factors” in 3 databases we received 89 468 works. The keyword “ovarian cancer epigenetic factors” produced 20 375 results. The first 200 records from all 3 databases were analyzed by title and abstract. In the next step, 729 works were chosen from the most relevant as sorted by database. After this selection process, the removal of duplicates was carried out to prevent redundancy and uphold data integrity. Duplication exclusion was based on publication identifiers or an analysis of titles, authors, and other metadata, and as a result, 619 publications remain included.
Publications focusing mainly on other than ovarian types of cancers linked to the described genes, as well as articles discussing methods of treatment and causes of cancer resistance for specific chemotherapeutics were excluded.
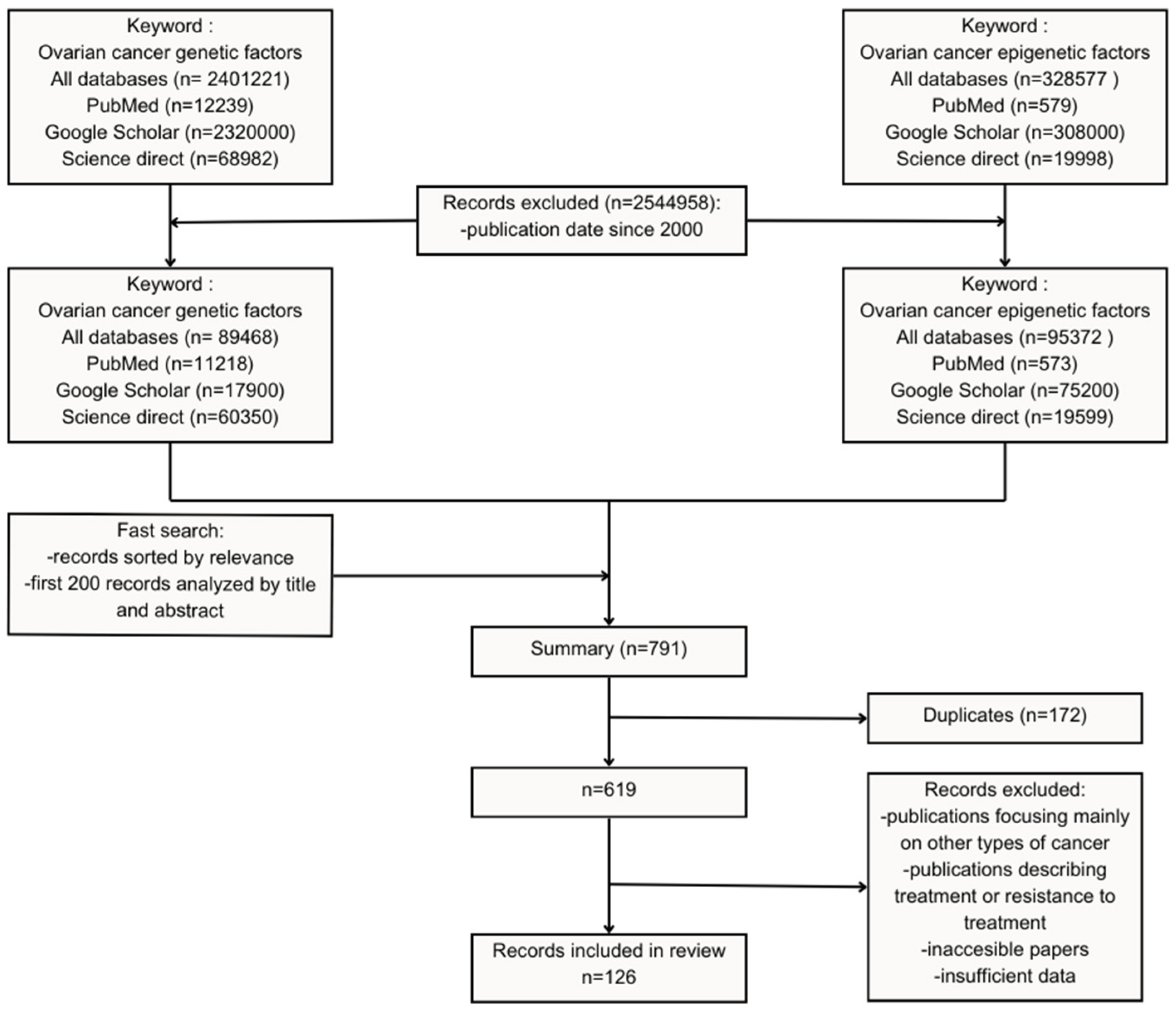
Inaccessible papers and those containing inaccurate data were eliminated to uphold the credibility of the results. 126 papers met the criteria and were included in this work (Figure 10).
Figure 10.
Exclusion criteria and number of works excluded and included in research.
2.4. Data Analysis
All articles were examined by two authors independently. Title, structure, materials, methods, and results were analyzed, including figures and tables. Works unusual or problematic in any way were, as well, fully read.
2.5. Data Compilation
After conducting a comparative examination of the articles (based on discussion and consensus), the researchers established the structure for the classification of papers in specific thematic groups, and the essential qualifiers for each group were defined. Our classification included the most important etiological factors. The factors we deemed most relevant were alterations in BRCA1, BRCA2, and double-strand break repair, TP53 mutations, and genetic syndromes such as Li-Fraumeni and Lynch. The impact of microbiome and HPV infection, obesity, alcohol, and caffeine intake were also included considering them as equal. The paper was, based on this classification, grouped with other papers discussing this distinct etiological factor.
The analysis emphasized commonalities among the papers, comparing various aspects such as the form of the articles, scientific discipline, concepts and types of research, choice of research methods, objectives, and subject matter to identify global tendencies. On this basis, an initial categorization of the papers was carried out. The articles within each category were then analyzed again and compared to ensure alignment with the established criteria, enhancing the method’s sensitivity. A subset of articles was selected from each group for discussion in the review. Our team has experience in systematic reviews in morphological sciences and associated disciplines [124,125,126].
3. Results
3.1. Selected Genetic FACTORS in Ovarian Carcinogenesis
3.1.1. BRCA1/2 and Double Strand Break Repair
BRCA1 and 2 are the tumor suppressor genes, which mutations can lead to breast and ovarian cancer [7]. BRCA1 mutations relate to an increased risk of developing ovarian cancer by the age of 70 from 18% to 54%, and BRCA2 mutations provide an increased risk of developing ovarian cancer by age 70 from 2.4% to 19% [8]. BRCA1 gene mutations were found in 40-50% of families with both breast and ovarian cancer [9].
There are more than 400 mutations detected in BRCA1/2 genes, and presumable numerous of them remain unexplored. At the same time, most of the human mutations are unique, and each family can present specific mutations. Most of them (80%) occur as point mutations or deletion/insertion mutations. Due to this, the p53-dependent DNA breakdown is activated, which may lead to cell cycle arrest and apoptosis [9].
These genetic factors are relevant to such numbers of ovarian cancer cases because of the function that the proteins BRCA, which are products of these genes, perform.
First, BRCA genes through proteins which are their products occur as “caretakers” of chromosome stability [10].
Cells deficient in the murine BRCA2 homolog appear to sustain spontaneous aberrations in chromosome structures, during the cells division. The abnormalities include broken chromosomes and chromatids, triradial and quadradial structures (characteristic for human diseases Bloom’s syndrome and Fanconi’s anemia which can also cause increased susceptibility to cancer), translocations, deletions, and fusions. In BRCA1 deficient mouse cells the structural abnormalities were similar- the abnormality in chromosomal structure occurred, as well as in BRCA1 or BRCA2 deficient human cancer cells [10].
The second important evidence of BRCA’s role in carcinogenesis is that mammalian cells deficient in BRCA have gross chromosomal rearrangement which is a result of inappropriate double-strand break repair (DSB), during the S and G2 phase, when the BRCA proteins are maximally expressed. The main cause of this is the fact that these cells exhibit a lack of homologous recombination (HR), which is the only error-free way to repair DBS. Without HR the altered cells “are doomed to” error-prone DSB repair mechanisms. Nevertheless, in its simplest conception, chromosomal instability in BRCA deficient cells is generated by incorrect directing of DSB processing down inappropriate pathways, rather than the failure of repair [11,12] (Figure 3).
Another role of BRCA2 is that it binds directly with RAD51- a key for DSB repair by HR [9,13]. Studies show that BRCA2 directly regulates the availability and activity of RAD51. This mechanism is responsible for creating nucleoprotein filament which invades and pairs with a homologous DNA duplex, initiating strand exchange between the paired DNA molecules [10,14].
In the presence of BRC peptides, recombinant RAD51 instead of polymerizing spontaneously becomes largely monomeric [10]. Both nonhomologous ends joining (NHEJ) and single-strand annealing (SSA) are processes independent from RAD51 and are error-prone [15].
BRCA2-deficient cells use NHEJ and SSA to repair site-specific DSBs [10,12]. In BRCA1-deficient cells NHEJ predominates as the mechanism for repair. These examples explain that the role of BRCA proteins in the process of repair relies on directing them to the wrong pathways [10,15]. BRCA2 not only participates in DNA repair but also can regulate the cell cycle (G2/M phase) through its interaction with protein BRAF35 which binds in vitro to the branched DNA structures [8].
What about BRCA1?
BRCA1 can also inhibit the activity of MRE11 – a protein essential for the generation of ssDNA (needed for HR and SSA repair) at sites of DNA breakage and interact with enzymes that alter chromatin and DNA structures. BRCA 1 interacts with SWI/SNF that remodel chromatin, with regulators of histone acetylation/deacetylation, and with DNA helicases [10].
BRCA1 may be responsible for sensing the DNA damage because when the damage in dividing cells, it is done the BRCA1 is rapidly phosphorylated. It has been implicated in a few different checkpoint events- BRCA1 deficient cells fail to arrest scheduled DNA synthesis in S and G2 phases. It can also cohabit in high-molecular-weight complexes with many different proteins that bind to abnormal DNA structures. BRCA1’s precise role here remains to be clarified. It is attractive to posit, however, that it works as a signal “processor” to coordinate DNA damage-sensing mechanisms with appropriate biological responses. On the one hand, BRCA1 participates in protein complexes that have functions intrinsic to the sensing and signaling of different types of DNA lesions. On the other hand, it works as a sequence-specific transcriptional regulator of genes whose expression affects checkpoint enforcement and other downstream biological responses [10].
There is evidence that mutations in p53 are more frequent in cancers with BRCA alterations. The spectrum of mutations is also different [10].
Women who are carriers of BRCA1 and BRCA2 mutations are more susceptible to breast, ovary, colon, stomach, pancreas, and gallbladder cancer. In patients with known BRCA mutations, more frequent monitoring is indicated. The main goal is early detection of malignancy or lesions. A positive BRCA mutation indicates a higher probability of cancer growth but not every mutation has to end that way. Likewise, a negative mutation result is not an exclusion for the development of breast or ovarian cancer in a lifetime [8]
3.1.2. Li-Fraumeni Syndrome
The Li-Fraumeni syndrome (LFS) is a hereditary predisposition to cancers. It is also known as the sarcoma, breast, leukemia, and adrenal gland (SBLA) cancer syndrome [16]. It is related to the development of ovarian cancer. It was first described in 1969 by Dr Frederick Li and Joseph Fraumeni from the National Cancer Institute [17] who observed the high frequency of the same kind of cancer within the young members of one family. We can distinguish three types of the disease.
LFS1, the most common variant, is associated with a mutation in TP53. TP53 is the genetic blueprint for a protein called p53 and most commonly causes this condition by mutations or alterations in that gene. Mutations prevent the gene from functioning properly [18]. In all families with LFS1 sequencing has shown mutations in the p53 DNA binding domain that is caused by germline missense.
These initial findings have been confirmed by several subsequent studies, but mutations in the TP53 gene have also been shown to be distributed throughout the germline Missense mutations account for 74% of germline TP53 mutations, followed by nonsense mutations (∼9%), and splice mutations (∼8%) [18]. Most mutations occur in the highly conserved DNA-binding domain and the six most common “hotspot” mutations are found in codons 175, 245, 248 (two common substitutions), 273, 282, and 337 [18,19]. Mutation can occur in the parent germ cell and be inherited or develop de-novo during embryogenesis. There is a lack of data on the frequency of de novo mutations in LFS patients, although estimates range from 7% to 20% [20] (Figure 4).
Two other less frequent types are LFS 2, and LFS-L. LFS-L are individuals that do not have detectable mutations in P53. LFS2 is associated with a mutation in CHEK2 (checkpoint kinase two), which regulates the activity of P53 [20].
P53 plays a very important part in cancer prevention mechanisms. It controls apoptosis, inhibits angiogenesis, and helps to maintain genomic stability [20]. It participates in fixing DNA by activation of DNA repair proteins. Mutations in LFS lead to the loss of functional P53 protein, which causes the cell’s defense against genetic changes to be decreased.
The most common types of cancer found in families with LFS include osteosarcoma, soft tissue sarcoma, acute leukemia, breast cancer, brain cancer, and adrenal cortical tumors. An increased risk of melanoma, Wilms’ tumor, and cancers of the stomach, colon, pancreas, esophagus, lung, and gonadal germ cells have been reported [5,17]. Although only about 3% of cases of ovarian cancer are associated with LFS, it increases its risk by 47% [5]. Also, they occur much earlier than expected. The median age for patients diagnosed with ovarian cancer is 39,5 years, compared to an average of almost 65 years for those who do not suffer from the Li-Fraumeni syndrome.
In the past, Li-Fraumeni syndrome was diagnosed based on the patient’s and their family’s symptoms and signs. However, with the advent of advanced genetic testing, it’s now possible for patients to determine if they carry a TP53 tumor mutation even before any symptoms of LFS appear [16]. If it’s determined that a patient carries a TP53 mutation, they must be advised about ongoing surveillance and regular screening for cancers associated with LFS.
During the treatment of cancers, radiation therapy on patients with LFS should be avoided due to the increased risk of radiation-induced cancers. Exposure to CT scans or X-rays is not recommended [16]. The treatment remains the same, but to date, cancers developed in LFS have worse survival rates, are more susceptible to malignancy, and are more resistant to chemotherapy.
The probability of individuals with LFS developing any cancer in their lifetime is 75% for men and almost 100% for women [20]. As mentioned previously LFS is associated with only 3 % of all ovarian cancers, but the risk of developing OC is 47% higher for women with LFS compared to ones without it [5].
3.1.3. Lynch Syndrome
Lynch syndrome is a hereditary cancer syndrome that is passed down in an autosomal dominant manner. It may lead to various types of cancer including ovarian cancer. It is caused by pathogenic variants (PVs) in the DNA mismatch repair system [21]. These genes include mutL homolog 1 (MLH1), mutS homolog 2 (MSH2), mutS homolog 6 (MSH6), and PMS1 homolog 2 (PMS2). Deletions in the epithelial cell adhesion molecule (EpCAM) can result in the downstream epigenetic silencing of MSH2. The loss of this gene causes a point or frameshift mutation that can lead to the creation of a non-functioning protein. These mutations accumulate in the areas of microsatellites that can be easily recognized when compared with the microsatellite regions of normal cells [22]. Loss of MSH 6 or MSH 3 alone does not result in cancer formation as the genes share redundant function. If both genes are non-functional then mutations can accumulate with subsequent cancer development [22]. In rarer cases, the inherited inactivation of the MMR system can occur from germline hypermethylation of the promoter region of MLH1 [23]. MLH1 forms a heterodimer with PMS2 to participate in the mismatch repair system, however, its exact function is not known. MLH1 also likely dimerizes with MLH3 [22] (Figure 5, Figure 6, Figure 7 and Figure 8).
MLH1 and MSH2 are two of the most frequently mutated genes in Lynch syndrome tumors. This accounts for approximately 75% of mutations in patients diagnosed with Lynch syndrome [24]. This syndrome is believed to affect 1 in every 278 individuals, but more than 95% of people remain undiagnosed [25]. It increases a lifetime cancer risk by 10-90% [26], which varies based on the specific variant and type of cancer. It exhibits a lack of uniformity. The penetrance level, range of diseases, and the age at which cancer begins can differ based on the specific gene mutation [21] (Figure 9).
Lynch syndrome is the leading cause of inherited colorectal cancer. However, for women, the first sign of Lynch syndrome is more likely to be endometrial cancer.
Additionally, certain variants that cause Lynch syndrome have been shown to increase the risk of ovarian cancer [21]. A lifetime cumulative risk of ovarian cancer for women with Lynch syndrome is about 10% [27], which is especially high for MSH2 - mutation carriers [28]. They constitute 2% of all ovarian cancers [27]. Cancers associated with Lynch syndrome are typically diagnosed at a younger age compared to similar cases in the general population [29]. On average ovarian cancer is diagnosed in individuals with Lynch syndrome between the ages of 43 and 46, compared to the age of 60 in patients without Lynch syndrome (Table 1, Table 2 and Table 3).
Ovarian cancer associated with LS is primarily endometroid and has a better prognosis than the ones linked to BCRA mutations [23]. It is preferable for women diagnosed with Lynch syndrome to consult with a gynecologist around the age of 25 to understand the warning signs of cancer, discuss family planning, and consider strategies to reduce risk [30].
There’s limited evidence on how lifestyle impacts the risk of gynecological cancer in women with Lynch syndrome. The oral contraceptive pill is known to lower the risk of sporadic endometrial and ovarian cancer, as well as ovarian cancer associated with BRCA1/2. Aspirin has been proven to lower the risk of all Lynch syndrome-associated cancers. Also, aspirin has been shown to reduce the incidence of endometrial cancer in women with Lynch syndrome, also in patients with lifestyle-related comorbidities like obesity. This suggests aspirin’s effect on ovarian cancer should be further investigated. Individuals with Lynch syndrome may also be more susceptible to cancer due to lifestyle factors. In people with Lynch syndrome, smoking, alcohol use, and having a higher body mass index increase the risk of colorectal cancer; however, little research has specifically investigated how lifestyle decisions affect the risk of gynecological cancers. Although there isn’t enough proof, it would be wise for women with Lynch syndrome to maintain a healthy weight, exercise frequently, abstain from smoking, and consume alcohol in moderation or not at all [23].
As a risk-reducing measure, the National Comprehensive Cancer Network (NCCN) now advises that MLH1 and MSH2/EPCAM carriers decide whether to have a BSO (bilateral salpingo-oophorectomy) on an individual basis. The date ought to be determined by factors such as family history, specific variants, medical comorbidities, menopausal status, and completion of childbearing. According to NCCN, MSH6 carriers should make their own decisions because there is not enough information to advocate BSO in this case. They offer even more specific advice for PMS2 carriers, noting that they seem to have no increased risk of ovarian cancer and that they can legitimately decide without having an oophorectomy [21].
3.1.4. Phosphatase and Tensin Homolog Protein Expression
It has been demonstrated that there is a link between PTEN (phosphatase and tensin homolog deleted on chromosome 10) protein expression and estrogen receptor expression in EOC (epithelial ovarian cancer). PTEN expression is low in EOC tissues, and estrogen inhibits it through the estrogen receptor 1 (ESR1) in EOC cells [31]. Knocking down PTEN boosted the proliferation and migration of estrogen-driven EOC cells. In addition, estrogen stimulation activated the G protein-coupled receptor 30 (GPR30)-Protein kinase C (PKC) signalling pathway, which phosphorylated PTEN. Inhibiting PTEN phosphorylation inhibited estrogen-induced EOC cell proliferation and migration while lowering AKT and mTOR phosphorylation. These findings revealed that estrogen lowered PTEN expression levels via the ESR1 genomic pathway and phosphorylated PTEN via the GPR30-PKC non-genomic pathway, triggering the PI3K/AKT/mTOR signalling cascade and influencing the fate of EOC cells [43,44].
3.2. Hormonal Factors
3.2.1. Estrogens
The majority of ovarian tumors are thought to emerge in the surface epithelium because of hormonal changes. Prolonged treatment with hormone replacement treatment (HRT) is additionally considered a contributing figure [32]. A 22% increased risk of ovarian cancer was seen in postmenopausal women using unopposed estrogen as HRT [12]. The risk was still significantly increased (by approximately 10%) by the application of a combination of estrogen and progestin [33]. Women who have early menarche, and late menopause, and women who are taking fertility drugs (gonadotropin-releasing hormone antagonists or clomiphene) are at increased risk of developing ovarian cancer [34]. It is caused by a high concentration of estrogen after stimulation of the sex-steroid hormone synthesis in the ovary [35]. Early menarche increases its risk by 4% in Europe and by 5-9% in the USA [36].
3.2.2. Androgens
Androgens are a group of steroid hormones, including testosterone, dihydrotestosterone (DHT), and androstenedione. They play a crucial role in the development and maintenance of male sexual characteristics. Recent research has shed light on the potential role of androgens, a class of male sex hormones, in the development and progression of certain ovarian cancer subtypes [37].
Several studies have reported the expression of androgen receptors (AR) in ovarian cancer cells, particularly in hormone-sensitive subtypes, such as endometrioid and low-grade serous ovarian carcinomas [37]. The presence of AR suggests that these tumors may be responsive to androgen signaling, which could influence their growth and progression. Preclinical studies have demonstrated that androgen stimulation can promote the proliferation and survival of AR-positive ovarian cancer cells in vitro and in vivo models [37,38]. This androgen-driven tumor growth is mediated through the activation of various signaling pathways, including the PI3K/AKT and MAPK pathways, which are known to play crucial roles in cancer cell proliferation and survival [38].
According to global cancer statistics from the World Health Organization (WHO), ovarian cancer accounts for approximately 3% of all new cancer cases and 5% of cancer-related deaths among women worldwide [39] The incidence and mortality rates vary across different regions, with higher rates observed in developed countries compared to developing nations. This variation may be attributed to factors such as differences in risk factors, access to screening and early detection programs, and the availability of effective treatment options [39].
3.2.3. Progesterone
Progesterone, a steroid hormone, has been extensively studied for its potential influence on the development and progression of ovarian cancer. Several recent scientific studies have shed light on the complex relationship between progesterone and ovarian cancer.
Numerous studies have suggested that progesterone may play a protective role against ovarian cancer. A study published in the Journal of the National Cancer Institute in 2013 found that women who used progesterone-only contraceptives had a significantly lower risk of developing ovarian cancer compared to non-users [40]. Similarly, a 2015 study in the International Journal of Cancer reported that higher levels of progesterone were associated with a reduced risk of ovarian cancer [41].
The proposed mechanisms by which progesterone may inhibit ovarian cancer development include its ability to induce apoptosis in ovarian cancer cells, suppress cell proliferation, and inhibit angiogenesis which is crucial for tumor growth. Additionally, progesterone has been shown to modulate the immune system, potentially enhancing the body’s ability to recognize and eliminate cancer cells [42,43,44].
However, the relationship between progesterone and ovarian cancer is not entirely straightforward. Some studies have suggested that progesterone may also have a stimulatory effect on ovarian cancer cells under certain conditions. Another study has found that progesterone could promote the growth and invasion of ovarian cancer cells in the presence of specific genetic alterations [45].
In conclusion, the current scientific evidence suggests that progesterone may play a complex and multifaceted role in the development and progression of ovarian cancer. While many studies have reported a protective effect of progesterone, the specific mechanisms, and the influence of various factors, such as genetic and hormonal profiles, require further investigation to fully understand the relationship between this hormone and ovarian cancer.
3.2.4. Progesterone Receptor
The type of progesterone receptor (PR) and its genetic variations have been implicated in the development of ovarian cancer. One significant factor is the loss of heterozygosity (LOH) at the 11q23.3-24.3 locus, where the PR gene is located [46].
LOH at 11q23.3-24.3 has been frequently observed in ovarian cancer cases, suggesting that this region harbors tumor suppressor genes crucial for ovarian carcinogenesis [47,48]. The loss of one functional copy of the PR gene in this region may contribute to the dysregulation of progesterone signaling, potentially promoting ovarian cancer development.
Regarding specific PR gene polymorphisms, scientists have identified four variants: +44C/T, +331G/A, G393G, and V660L [47,48]. After extensive research, it was found that the +44C/T, +331G/A, and G393G polymorphisms did not appear to be associated with an increased risk of ovarian cancer [47,48]. However, an inverse relationship was observed between the V660L variant and ovarian cancer risk (odds ratio = 0.70, 95% confidence interval: 0.57, 0.85) [47,48]. This suggests that individuals carrying the V660L variant may have a reduced risk of developing ovarian cancer compared to those without this genetic variation.
The V660L polymorphism is thought to potentially alter the biological functions of the progesterone receptor, which could influence the development and progression of ovarian cancer [47,48,49]. However, the specific mechanisms by which this variant confers a protective effect are not fully understood and require further investigation. The association between PR gene variations and ovarian cancer risk may be influenced by various factors, such as ovarian cancer histology, reproductive history, and other risk factors [50]. Further research is needed to fully elucidate the complex interplay between progesterone receptor genetics, hormone signaling pathways, and the development of ovarian cancer.
3.3. Environmental Factors
3.3.1. PCOS and Age of Menopause
The direct correlation between polycystic ovary syndrome (PCOS) and ovarian malignancies is not yet confirmed but it has been shown in many studies that they share the same risk factors, such as obesity and hormonal imbalances [51]. Because of that it is now believed that PCOS may lead to ovarian cancer. Endometriosis also is one of the risk factors.
The proof for age at menopause as a risk factor is conflicting [6]. In the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort, age above 52 was associated with an increased risk of ovarian cancer by 5.7% (HR = 1.57, 95% CI 1.16-2,13) compared with age of menopause under 45. However, when excluding women who were diagnosed with OC within their first 2 years of follow-up, the risk decreased slightly and amounted to 4% (HR = 1.40, 95% CI 0.98–2.00) [52].
3.3.2. Pregnancy and Breastfeeding
Pregnancy has a great impact on decreasing the risk of OC. It has been proven that women who had given birth had 30-40% lower chances of developing ovarian cancer [53,57,58]. Some other studies calculated the risk of OC decreasing by 8% with every pregnancy and another one calculated a decline of 18%, 26%, 33%, and 42% for the first, second, third, and fourth pregnancy, respectively [54,55]. In Finland, scientists have proven that the odds ratio (OR) between parity and developing OC for serous cancer was 0.65 (95% confidence interval 0.56-0.77), for mucinous cancer 0.66 (0.52-0.83), for endometrioid cancer 0.52 (0.40-0.68), for clear-cell cancer 0.30 (0.19-0.46) and other types 0.59 (0.43-0.80). In women aged 55 or older, the respective (ORs) were 0.86 (0.75-0.99), 0.78 (0.57-1.07), 0.61 (0.47-0.79), 0.44 (0.29-0.66) and 0.74 (0.57- 0.95), adjusted for hormone therapy [56].
The number of childbirths was associated with a trend toward reduction of risk, especially in serous and clear-cell cancers. Higher age at first birth was associated with a higher risk of clear-cell cancer but otherwise, age at first or last birth did not have an impact on the incidence of cancer subtypes [56]. Breastfeeding decreases the risk of OC, especially with long-term duration [57], but some studies have proven that prolactin may induce carcinogenesis by regulating gene expression or by activating signaling pathways associated with proliferation and inhibition of apoptosis [57].
3.3.3. Combined Oral Contraceptive Pills
Combined oral contraceptive pills (COCPs) are unquestionably the strongest protective factor and play an important role in preventing ovarian cancer. Substantial reduction in epithelial ovarian cancer risk was observed among women who used COCPs for < 1 year if they were recent users (time since first or last COCP use within 20 years), each year of COCP use provided an average 5% reduction in the odds ratio (OR 0.95; CI 0.92–0.98) [58]. The greatest reduction in risk was [58] use before age 20 years and stopped after age 30 years [47,59].
3.3.4. Role of Microbiome
Our microbiome can impact a lot of different parts of our body. More and more scientific papers are being published about the higher risk of developing cancer caused by dysbiosis [60,61,62,63]. There is evidence that dysbiosis, which can be called oncobiosis, may also contribute to the carcinogenesis of ovarian malignancies [64,65,66]. Many different factors can cause the transformation of the microbiome. The most common are lifestyle choices such as smoking, obesity, type of diet, changes to the diurnal rhythm, aging, underlying diseases, exercise, and antibiotic and probiotic use [67].
Women who have vaginal colonies poor in Lactobacillus spp. are proven to be carriers of BRCA 1 mutations. Deficiency of Lactobacillus is also observed in women with ovarian cancer. This relation is more observable in the group of patients who are less than 40 years old [68]. Nevertheless, scoliosis is not only caused by a decrease but also an increased number of some other species. The tumor tissue is enriched in Gram-negative bacteria such as Proteobacteria and Fusobacteria [67]. Potentially pathogenic microorganisms such as Brucella, Mycoplasma, and Chlamydia spp. were found in 60–76% of ovarian tumors, as well as HSV virus, cytomegalovirus, or C. trachomatis [69].
Presumably changes in the microbiome affect carcinogenesis by inducting inflammation and regulating immune responses. Bacterial metabolites and components play a major role in these processes. Specifically, lipopolysaccharides, lysophosphatides, tryptophan metabolites, short-chain fatty acids, secondary bile acids, and polyamines are shown to participate in ovarian cancer pathogenesis [67].
First, lipopolysaccharides (LPS), which are the components of gram-negative bacteria’s outer membrane, can activate cancer cells and tumor-associated macrophages, moreover, the tumor tissue is more reactive to the LPS than normal tissue. LPS also drives the inflammation in cancer cells, by activating TLR4 receptors [70,71].
Lysophosphatides can also induce cell proliferation, migration, and invasion of cancer cells and increase the expression of the elements essential for cancer angiogenesis [67]. Other bacterial metabolites were shown to engage in carcinogenesis in general, however their impact on ovarian cancer specifically is still ambiguous.
3.3.5. Role of Viruses
Another factor, associated with dysbiosis and ovarian cancer development is a viral infection. The analysis found that potentially tumorigenic viruses were present in more than 50% of the analyzed tumor tissues. In this group, the Herpesviruses and Human Papillomaviruses have a great share [72].
Human herpesvirus-6a (HHV6a) is present in 50% of tumor samples and may exhibit two oncogenic mechanisms. First is its ability to block insulin growth factor binding protein, which causes a great share of free and active growth factors with potentially mitogenic consequences. The second mechanism may consist of activation of some oncogenic genes like SH3RF2 [73].
Viruses of the human papillomavirus group are one of the most common sexually transmitted viruses in the world. HPV16, 18, 31, and 45 have a high oncogenic potential and these types cause intraepithelial neoplasia, which can lead to invasive cancers.
The early region of HPV genomes encodes six proteins participating in viral replication- E1, E2, E3-E4. E6 and E7 act as oncogenes by promoting tumor growth and malignant transformation. These proteins target several negative regulators of the cell cycle, including p53, which can be an important factor in ovarian carcinogenesis. The HR-HPV E6 oncoprotein supports proteasomal degradation of p53, removes the trophic sentinel response for viral DNA synthesis, and increases telomerase activity to elude cell senescence [74]. Some reports have confirmed the presence of HPV in malignant ovarian cancer [75,76,77]. However, the frequency of occurrence varies significantly by geographical region with a prevalence of zero in most studies from Western Europe and North America and an HPV prevalence reaching almost 19% in Eastern Europe and 67% in Asia. This may be the result of environmental and genetic factors or differences in lifestyle factors such as smoking [78].
3.3.6. Obesity, Physical Activity, and Metabolic Basis of Ovarian Cancer
It is indisputable that obesity and physical activity are important factors in carcinogenesis in general. There are studies describing the correlation between these aspects and the risk of OC [79,80,81,82]. Reserved evidence associates a small amount of physical activity with a higher risk of ovarian cancer [83]. Obesity is a risk factor for 13 different cancers including OC. According to the Sook Bae meta-analysis presence of obesity 5 years before the diagnosis of ovarian cancer and at a young age is related to a poor prognosis [83].
The main biological mechanism whereby these components are related to cancer incidence is the fact that a high concentration of adipocytes in the human organism can lead to adipose tissue impairment, which affects immune and hormonal alternations in the microenvironment which is an important part of carcinogenesis [84]. The other participating factors may be altered adipokine expression, increased levels of circulating growth factors, and chronic inflammation [85]. Obesity and lack of physical activity relate to pathways related to oxidative stress, DNA methylation, telomere length, immune function, and gut microbiome [83].
Adipose tissue produces several interleukins IL such as Il-6, Il-8 as well as leptin, C reactive protein, IFNs, monocyte chemotactic protein 1 (MCP1), and tumor necrosis factor α (TNF-α). It is proven that IL-6 is increased in ovarian patient’s serum, and it is related to poor outcomes and chemotherapy resistance [86]. IL-6 activates the JAK-STAT3 pathway and enables ovarian cancer cell invasion and metastasis. Moreover, IL-6 induces Mcl-1 antiapoptotic protein expression which happens to be overexpressed in OC. Higher levels of IL-8, TNF-α, and CRP are also related to increased risk of ovarian carcinogenesis.
Adipose tissue also produces leptin, which is related to the estradiol secretion from the ovaries. Furthermore, in ovarian cancer patients reduced serum leptin levels have been observed. On the other hand, overexpression of leptin receptors in ovarian cancer tissue has indicated aggressive disease [87]. Besides, leptin is involved in the proinflammatory process, which as we mentioned earlier is an important component of OC. Leptin contributes to the metastatic advancement of epithelial ovarian cancer by participating in tissue invasion and cell migration by binding Ob-Rb mediated via JAK/STAT3, MAPK, AKT, mTOR, RhoA/ROCK, and MYPT1 signaling pathways. Leptin also inhibits some of the apoptosis pathway elements such as TNF receptor 1, caspase-6, caspase-3, and Bad, which are a part of the carcinogenic mechanism.
There is some evidence adiponectin is also a factor in ovarian carcinogenesis, it is known that adiponectin has antiangiogenic, anti-inflammatory, and anti-neoplastic properties and its levels are decreased in obesity. Increased levels of Il-6, leptin, and VEGF (vascular endothelial growth factor) and decreased levels of adiponectin have been observed to be caused by hypoxia [85]. Hypoxia itself is closely connected with higher fatty mass [88]. IGF-1 (insulin growth factor), which is also related to obesity can activate HIF 1 (hypoxia-inducible factor) in conditions of lower oxygen availability. Increased level of IGF-1 is inversely linked with the survival of epithelial ovarian cancer [85].
Metabolic derangements can also contribute to the progress of OC by influencing levels of sex hormones important in ovarian carcinogenesis. As mentioned, there are a lot of potential processes connected with obesity that may increase ovarian cancer risk. A cohort study of 461,646 women (≤49 years of age) registered in the Danish Medical Birth Registry found a correlation that the risk of premenopausal ovarian cancer increased by 23 % per 5 kg/m2 increase in BMI [89].
3.3.7. Alcohol
There are increasing numbers of evidence showing that alcohol consumption may induct epigenetic changes such as suppression of DNAm mechanism [90]. DNAm alterations are the early step in ovarian carcinogenesis [91], which is why there is a possible association between drinking alcohol and the development of epithelial ovarian cancer. Despite all, these days, there are more studies describing no such relation [90,92,93]. Moreover, there is some evidence showing a decreasing risk of ovarian cancer connected with higher red wine consumption [93].
3.3.8. Caffeine
Although coffee is proven to have a few antioxidant and anti-carcinogenic compounds, we also know that it correlates with a higher level of sex hormones (testosterone, estradiol), which is associated with an enhanced risk of ovarian cancer [94]. Coffee contains acrylamide which can also affect carcinogenesis [95]. Previous studies have shown an increased risk of ovarian cancer associated with caffeine intake in premenopausal women and no or slight association in postmenopausal women [96].
4. Discussion
Evaluation of available global literature regarding genetic and epigenetic factors of ovarian cancer revealed significant aspects influencing the development of this cancer, which remain unclear. Our article focused solely on factors of highest importance to the etiology of ovarian cancer. There are still many other significant factors, both genetic and epigenetic — such as polymorphisms of matrix metalloproteinases [97,98], non-coding RNAs [99], TIPARP gene [100], XRCC3 18067 C>T (Thr241Met) and XRCC2 rs3218536 [101,102], as well as, RAD21 amplification [103], transcription factor KLF5 [104] or vitamin D [105,106,107].
But crucial ones are BRCA1/2 genes variations which are already proven as a major etiologic factor of various cancers. Said variations play an important factor in the development of not only ovarian but also breast and endometrial cancer. Research regarding BRCA1/2 shall be therefore number one priority for researchers in modern gynecologic oncology. Numerous articles about BRCA mutations are being written every year, although it can still be assumed that not all mutations have been identified. Knowledge about processes related to these mutations is essential for discovering new, effective treatments. An example of that is therapy by poly-(ADP-ribose) polymerase (PARP) inhibitors (PARPis), such as olaparib, niraparib, and rucaparib, which with the new diagnostic possibilities, provide the greatest clinical benefit in patients with BRCA mutations or who test positive for homologous recombination deficiency [108,109].
Furthermore, we have found cancer-related syndromes, such as Li-Fraumeni syndrome and Lynch syndrome to be serious factors in the development of ovarian cancer. From 5000 to 20000 people are diagnosed with Li-Fraumeni syndrome [110]. Lynch syndrome is estimated to affect 1 in 280 to 1 in 2000 people in the general population [111]. This creates a considerable group of patients who require better prophylaxis, diagnostics, and treatment options. Hereditary cancer syndromes such as Li-Fraumeni syndrome and Lynch syndrome are important areas of research regarding the genetic causes of ovarian cancer. Both syndromes increase susceptibility to various types of cancer, including ovarian cancer. However, there is limited data characterizing ovarian cancer in these specific inherited cancer syndromes. In the case of Li-Fraumeni syndrome, the association between mutations in the tumor suppressor gene p53 and the occurrence of ovarian cancer is well-documented, yet the detailed mechanisms underlying this association remain to be investigated. Similarly, in Lynch syndrome, mutations in DNA repair genes are associated with an increased risk of developing ovarian cancer, but further research is needed to comprehensively understand these relationships. Precisely identifying specific gene mutations in these inherited cancer syndromes and their impact on ovarian cancer development is crucial for effective clinical research, genetic diagnostics, and the development of personalized treatment and prevention strategies.
These ambiguities make us unable to apply full ovarian cancer treatment and prevention, which can result in a greater number of new cases.
Furthermore, epigenetic factors related to the reproductive system play an important role in the field of ovarian cancer. Many relationships have been established, such as the effects of contraception, childbirth, breastfeeding, hormonal factors, and even coffee and alcohol l however there is still much research to be done and many relationships remain uncertain [112,113,114,115,116,117,118,119,120].
For example, polycystic ovary syndrome (PCOS) represents a complex scenario. While it is now believed that there is a potential association between PCOS and an increased risk of ovarian cancer, the data remains inconclusive and varies across different studies [121,122]. Further research is needed to clarify the relationship between PCOS and ovarian cancer development. In Lynch syndrome, the data on gynecological surveillance is of low quality, with most studies being single-center and retrospective. The results are inconsistent, with some studies showing benefits and others not. The United Kingdom Familial Ovarian Cancer Screening Study (UKFOCS) found that a combination of serum CA125 testing and transvaginal ultrasound scanning was sensitive and resulted in a shift in disease stage in women with a lifetime risk of ovarian cancer greater than 10% [123].
A significant number of works concerning the role of microbiome in carcinogenesis remain ambiguous and require further research. The great quantity of articles explains potentially important processes, connected with dysbiosis and the presence of specific pathogens and ovarian carcinogenesis, however many speculations need to be confirmed.
Human papillomavirus (HPV) infection, consists of, as well, the subject of more and more publications including it as a possible OC factor, however, the results are different in various geographic regions. Regrettably, there is no substantial evidence to verify its impact. HPV infection is a significant issue on a broad scale, and that is why more works regarding this problem are needed.
5. Conclusions
Our review shows that ovarian cancer is a multifactor disease. We attempted to summarize our actual knowledge about the most important genetic and epigenetic factors in ovarian carcinogenesis. This condition can occur alone, but it also maintains a part of complex disorders like Lynch and Li-Fraumeni syndrome. Although BRCA-1 and BRCA-2 seem to be the most common genetic causes of OC, the variety of genes and mutations are numerous.
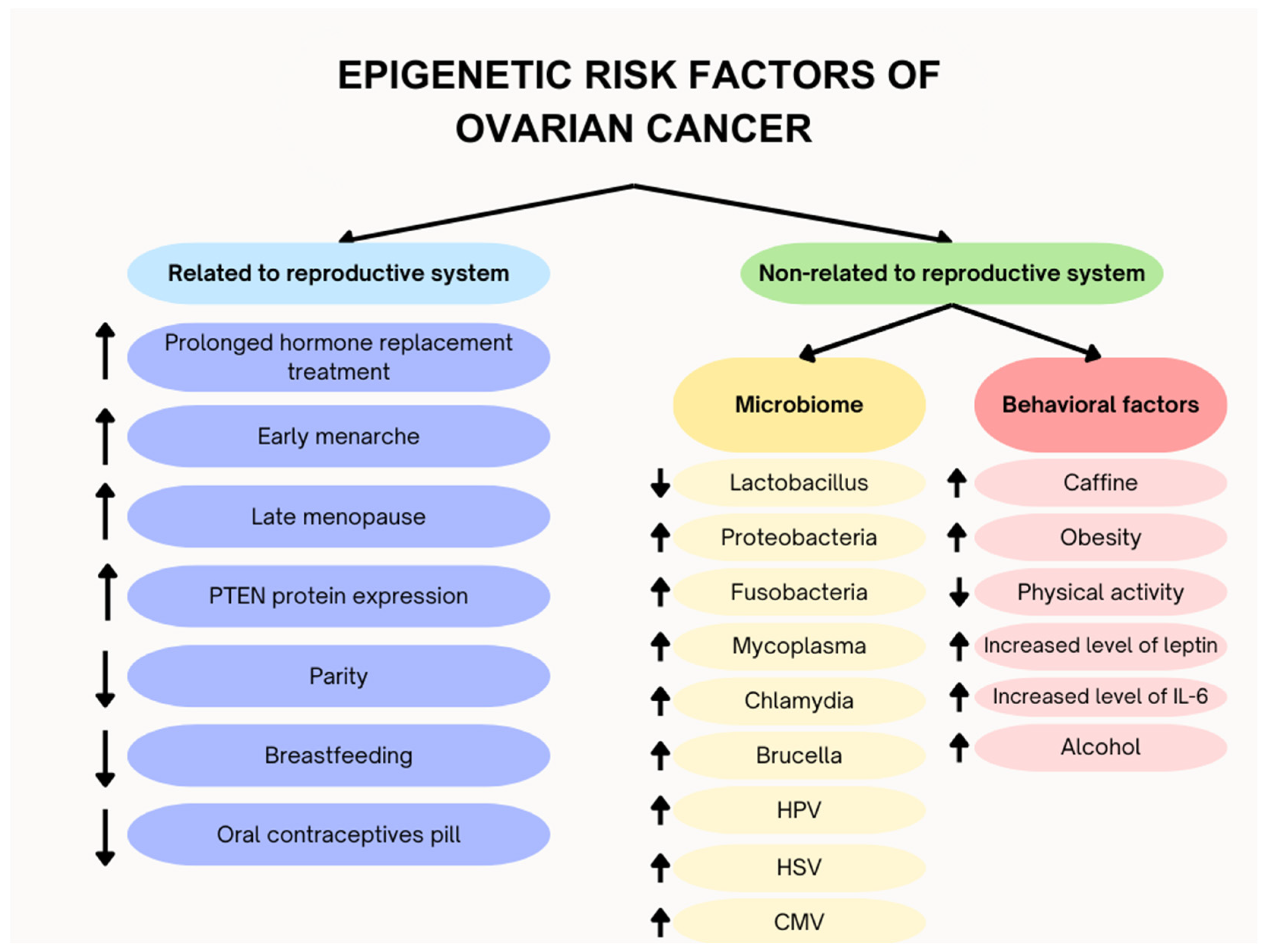
Knowledge about described genes is crucial to early diagnosis and treatment of OC, which, as mentioned earlier, is one of the most common gynecologic cancers with the worst prognosis. While exploring the available articles about genetics and epigenetics of ovarian cancer we also came across materials regarding some substances’ influence and lifestyle on mutation development, thereby development of ovarian cancer. Although still a lot of them remain ambiguous, awareness of their potential role in ovarian carcinogenesis can also be an important element of OC prevention (Figure 11).
Figure 11.
Differentation of epigentic factos into related and non-related to reproductive system.
Please, as a reader keep in mind our research discusses only the most important factors only shedding some light on this subject, which still is shrouded in darkness. In search of answers to one of the most important questions of today’s oncologic gynecology, we hope to aid frontline researchers who are constantly searching in laboratories and clinics for solutions to this page-one problem.
Author Contributions
Alicja Florczak was responsible for the investigation, data curation, formal analysis, writing the original draft, and visual preparation of the manuscript. She was a leading contributor to materials and methods and preparation of the final manuscript. Aleksandra Królikowska was responsible for the investigation, data curation, formal analysis, and writing the original draft. She was also a leading contributor to materials and methods and preparation of the final manuscript preparation. Mateusz Mazurek was responsible for the visual preparation of the manuscript; he was a supporting contributor in materials and methods and final manuscript preparation. He was also a supporting contributor in the supervision of work. Sławomir Woźniak MD, PhD was responsible for the conceptualization of the study and the support in the supervision of the work. Zygmunt Domagała MD, PhD was the leading supervisor and supporting contributor in the final manuscript preparation.
Funding
The study did not require any additional funding. Databases and articles were retrieved using our Institution’s accesses.
Institutional Review Board Statement
Bioethics committee approval was not required for the review study.
Acknowledgments
The authors would like to thank Małgorzata Smazek Ryndak for the language correction and Piotr Baron for help with creation of tables and figures.
Conflicts of Interest
The authors declare no conflict of interest.
References
- S. Gupta et al., “Prevalence of BRCA1 and BRCA2 Mutations Among Patients With Ovarian, Primary Peritoneal, and Fallopian Tube Cancer in India: A Multicenter Cross-Sectional Study,” JCO Glob Oncol, vol. 7, no. 7, pp. 849–861, Dec. 2021. [CrossRef]
- “Ovarian Cancer By The Numbers | OCRA.” Accessed: Mar. 03, 2024. [Online]. Available online: https://ocrahope.org/get-the-facts/statistics/.
- C. J. Cabasag et al., “Ovarian cancer today and tomorrow: A global assessment by world region and Human Development Index using GLOBOCAN 2020,” Int J Cancer, vol. 151, no. 9, pp. 1535–1541, Nov. 2022. [CrossRef]
- “Ovarian Cancer Statistics | How Common is Ovarian Cancer | American Cancer Society.” Accessed: Feb. 28, 2024. [Online]. Available online: https://www.cancer.org/cancer/types/ovarian-cancer/about/key-statistics.html.
- A. Toss et al., “Hereditary ovarian cancer: Not only BRCA 1 and 2 Genes,” Biomed Res Int, vol. 2015, 2015. [CrossRef]
- A. S. Avramenko and J. M. Flanagan, “An epigenetic hypothesis for ovarian cancer prevention by oral contraceptive pill use,” Clin Epigenetics, vol. 15, no. 1, Dec. 2023. [CrossRef]
- K. Xu, S. Yang, and Y. Zhao, “Prognostic significance of BRCA mutations in ovarian cancer: an updated systematic review with meta-analysis,” Oncotarget, vol. 8, no. 1, p. 285, Jan. 2017. [CrossRef]
- J. T. Casaubon, S. Kashyap, and J.-P. Regan, “BRCA1 and BRCA2 Mutations,” StatPearls, Jul. 2023, Accessed: Feb. 28, 2024. [Online]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK470239/.
- A. Alacacioglu et al., “BRCA genes: BRCA 1 and BRCA 2,” JBUON, vol. 23, no. 4, pp. 862–866, 2018.
- A. R. Venkitaraman, “Cancer susceptibility and the functions of BRCA1 and BRCA2,” Cell, vol. 108, no. 2, pp. 171–182, Jan. 2002. [CrossRef]
- I. Vergote et al., “European experts consensus: BRCA/homologous recombination deficiency testing in first-line ovarian cancer,” Annals of Oncology, vol. 33, no. 3, pp. 276–287, Mar. 2022. [CrossRef]
- A. N. J. Tutt et al., “Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer,” Cold Spring Harb Symp Quant Biol, vol. 70, pp. 139–148, 2005. [CrossRef]
- X. Li and W. D. Heyer, “Homologous recombination in DNA repair and DNA damage tolerance,” Cell Research 2008 18:1, vol. 18, no. 1, pp. 99–113, Jan. 2008. [CrossRef]
- J. Baselga et al., “A positive defect,” Nature Reviews Cancer 2005 5:5, vol. 5, no. 5, pp. 333–333, Apr. 2005. [CrossRef]
- K. D. Doig, A. P. Fellowes, and S. B. Fox, “Homologous Recombination Repair Deficiency: An Overview for Pathologists,” Modern Pathology, vol. 36, no. 3, p. 100049, Mar. 2023. [CrossRef]
- A. Villani, T. Frebourg, and D. Malkin, “Li-Fraumeni Syndrome,” The Hereditary Basis of Childhood Cancer, pp. 1–21, Aug. 2023. [CrossRef]
- “Li-Fraumeni Syndrome | Cancer.Net.” Accessed: May 06, 2024. [Online]. Available online: https://www.cancer.net/cancer-types/li-fraumeni-syndrome.
- T. Guha and D. Malkin, “Inherited TP53 Mutations and the Li-Fraumeni Syndrome,” Cold Spring Harb Perspect Med, vol. 7, no. 4, Apr. 2017. [CrossRef]
- A. D. Sorrell, C. R. Espenschied, J. O. Culver, and J. N. Weitzel, “TP53 Testing and Li-Fraumeni Syndrome: Current Status of Clinical Applications and Future Directions,” Mol Diagn Ther, vol. 17, no. 1, p. 31, Feb. 2013. [CrossRef]
- T. Kumamoto et al., “Medical guidelines for Li–Fraumeni syndrome 2019, version 1.1,” International Journal of Clinical Oncology 2021 26:12, vol. 26, no. 12, pp. 2161–2178, Oct. 2021. [CrossRef]
- K. A. Underkofler and K. L. Ring, “Updates in gynecologic care for individuals with lynch syndrome,” Front Oncol, vol. 13, 2023. [CrossRef]
- B. J. Bansidhar and J. Silinsky, “History and Pathogenesis of Lynch Syndrome,” Clin Colon Rectal Surg, vol. 25, no. 2, p. 63, 2012. [CrossRef]
- N. A. Ryan, R. F. McMahon, N. C. Ramchander, M. W. Seif, D. G. Evans, and E. J. Crosbie, “Lynch syndrome for the gynaecologist,” The Obstetrician & Gynaecologist, vol. 23, no. 1, p. 9, Jan. 2021. [CrossRef]
- B. Liu et al., “Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients,” Nat Med, vol. 2, no. 2, pp. 169–174, 1996. [CrossRef]
- F. E. McRonald et al., “Identification of people with Lynch syndrome from those presenting with colorectal cancer in England: baseline analysis of the diagnostic pathway,” European Journal of Human Genetics 2024 32:5, vol. 32, no. 5, pp. 529–538, Feb. 2024. [CrossRef]
- H. Hampel and A. De La Chapelle, “The search for unaffected individuals with Lynch syndrome: do the ends justify the means?,” Cancer Prev Res (Phila), vol. 4, no. 1, pp. 1–5, Jan. 2011. [CrossRef]
- E. M. Grindedal et al., “Survival in women with MMR mutations and ovarian cancer: a multicentre study in Lynch syndrome kindreds,” J Med Genet, vol. 47, no. 2, pp. 99–102, Feb. 2010. [CrossRef]
- R. R. Broaddus et al., “Pathologic features of endometrial carcinoma associated with HNPCC: a comparison with sporadic endometrial carcinoma,” Cancer, vol. 106, no. 1, pp. 87–94, Jan. 2006. [CrossRef]
- C. A. Christianson, K. P. Powell, S. E. Hahn, S. H. Blanton, J. Bogacik, and V. C. Henrich, “The use of a family history risk assessment tool within a community health care system: views of primary care providers,” J Genet Couns, vol. 21, no. 5, pp. 652–661, 2012. [CrossRef]
- E. J. Crosbie et al., “The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome,” Genet Med, vol. 21, no. 10, pp. 2390–2400, Oct. 2019. [CrossRef]
- X. Li et al., “Estrogen promotes Epithelial ovarian cancer cells proliferation via down-regulating expression and activating phosphorylation of PTEN,” Arch Biochem Biophys, vol. 743, p. 109662, Jul. 2023. [CrossRef]
- S. Sarwar, A. Alamro, F. Huq, and A. Alghamdi, “Insights Into the Role of Epigenetic Factors Determining the Estrogen Response in Estrogen-Positive Ovarian Cancer and Prospects of Combining Epi-Drugs With Endocrine Therapy,” Front Genet, vol. 13, Jul. 2022. [CrossRef]
- B. Zhou et al., “Hormone replacement therapy and ovarian cancer risk: A meta-analysis,” Gynecol Oncol, vol. 108, no. 3, pp. 641–651, Mar. 2008. [CrossRef]
- F. Mungenast and T. Thalhammer, “Estrogen Biosynthesis and Action in Ovarian Cancer,” Front Endocrinol (Lausanne), vol. 5, no. NOV, 2014. [CrossRef]
- J. H. Choi, A. S. T. Wong, H. F. Huang, and P. C. K. Leung, “Gonadotropins and Ovarian Cancer,” Endocr Rev, vol. 28, no. 4, pp. 440–461, Jun. 2007. [CrossRef]
- H. Yang et al., “Age at menarche and epithelial ovarian cancer risk: A meta-analysis and Mendelian randomization study,” Cancer Med, vol. 8, no. 8, p. 4012, Jul. 2019. [CrossRef]
- N.-M. Ewa et al., “Ovarian cancer – modern approach to its origin and histogenesis,” Ginekol Pol, vol. 83, no. 6, pp. 454–457, 2012, Accessed: Apr. 28, 2024. [Online]. Available online: https://journals.viamedica.pl/ginekologia_polska/article/view/46183.
- K. A. Kujawa and K. M. Lisowska, “Rak jajnika – od biologii do kliniki,” Postepy Hig Med Dosw, vol. 69, pp. 1275–1290, Dec. 2015. [CrossRef]
- Maciej Serda et al., “Utrata heterozygotyczności loci sprzężonych z genami BRCA1 i BRCA2 w rakach jajnika,” Uniwersytet śląski, vol. 7, no. 1, pp. 343–354, 2006.
- P. G. Moorman et al., “Oral contraceptives and risk of ovarian cancer and breast cancer among high-risk women: a systematic review and meta-analysis,” J Clin Oncol, vol. 31, no. 33, pp. 4188–4198, Nov. 2013. [CrossRef]
- M. A. Merritt et al., “Reproductive factors and risk of mortality in the European Prospective Investigation into Cancer and Nutrition; a cohort study,” BMC Med, vol. 13, no. 1, pp. 1–15, Oct. 2015. [CrossRef]
- “Expression of Gonadotropin Receptor and Growth Responses to Key Reproductive Hormones in Normal and Malignant Human Ovarian Surface Epithelial Cells1 | Cancer Research | American Association for Cancer Research.” Accessed: Apr. 16, 2024. [Online]. Available online: https://aacrjournals.org/cancerres/article/61/18/6768/507837/Expression-of-Gonadotropin-Receptor-and-Growth.
- V. Syed, G. Ulinski, S. C. Mok, and S. M. Ho, “Reproductive Hormone-Induced, STAT3-Mediated Interleukin 6 Action in Normal and Malignant Human Ovarian Surface Epithelial Cells,” JNCI: Journal of the National Cancer Institute, vol. 94, no. 8, pp. 617–629, Apr. 2002. [CrossRef]
- V. Syed and S. M. Ho, “Progesterone-induced apoptosis in immortalized normal and malignant human ovarian surface epithelial cells involves enhanced expression of FasL,” Oncogene 2003 22:44, vol. 22, no. 44, pp. 6883–6890, Oct. 2003. [CrossRef]
- Q. Ye, W. Cai, Y. Zheng, B. M. Evers, and Q. B. She, “ERK and AKT signaling cooperate to translationally regulate survivin expression for metastatic progression of colorectal cancer,” Oncogene, vol. 33, no. 14, pp. 1828–1839, Apr. 2014. [CrossRef]
- F. Modugno, “Ovarian Cancer and Polymorphisms in the Androgen and Progesterone Receptor Genes: A HuGE Review,” Am J Epidemiol, vol. 159, no. 4, pp. 319–335, Feb. 2004. [CrossRef]
- C. L. Pearce et al., “Progesterone receptor variation and risk of ovarian cancer is limited to the invasive endometrioid subtype: results from the ovarian cancer association consortium pooled analysis,” Br J Cancer, vol. 98, no. 2, p. 282, Jan. 2008. [CrossRef]
- K. L. Terry, I. De Vivo, L. Titus-Ernstoff, P. M. Sluss, and D. W. Cramer, “Genetic Variation in the Progesterone Receptor Gene and Ovarian Cancer Risk,” Am J Epidemiol, vol. 161, no. 5, pp. 442–451, Mar. 2005. [CrossRef]
- D. B. Leite et al., “Progesterone receptor (PROGINS) polymorphism and the risk of ovarian cancer,” Steroids, vol. 73, no. 6, pp. 676–680, Jul. 2008. [CrossRef]
- I. B. Runnebaum et al., “Progesterone receptor variant increases ovarian cancer risk in BRCA1 and BRCA2 mutation carriers who were never exposed to oral contraceptives,” Pharmacogenetics, vol. 11, no. 7, pp. 635–638, 2001. [CrossRef]
- H. P. K. Throwba et al., “The epigenetic correlation among ovarian cancer, endometriosis and PCOS: A review,” Crit Rev Oncol Hematol, vol. 180, p. 103852, Dec. 2022. [CrossRef]
- B. M. Reid, J. B. Permuth, and T. A. Sellers, “Epidemiology of ovarian cancer: a review,” Cancer Biol Med, vol. 14, no. 1, p. 9, Mar. 2017. [CrossRef]
- R. Troisi et al., “The Role of Pregnancy, Perinatal Factors, and Hormones in Maternal Cancer Risk: A review of the evidence,” J Intern Med, vol. 283, no. 5, p. 430, May 2018. [CrossRef]
- H. O. Adami et al., “Parity, age at first childbirth, and risk of ovarian cancer,” The Lancet, vol. 344, no. 8932, pp. 1250–1254, Nov. 1994. [CrossRef]
- K. K. Tsilidis et al., “Oral contraceptive use and reproductive factors and risk of ovarian cancer in the European Prospective Investigation into Cancer and Nutrition,” British Journal of Cancer 2011 105:9, vol. 105, no. 9, pp. 1436–1442, Sep. 2011. [CrossRef]
- V. Toufakis, S. Katuwal, E. Pukkala, and J. S. Tapanainen, “Impact of parity on the incidence of ovarian cancer subtypes: a population-based case-control study,” Acta Oncol, vol. 60, no. 7, pp. 850–855, 2021. [CrossRef]
- A. Ramírez-de-Arellano, J. C. Villegas-Pineda, C. D. Hernández-Silva, and A. L. Pereira-Suárez, “The Relevant Participation of Prolactin in the Genesis and Progression of Gynecological Cancers,” Front Endocrinol (Lausanne), vol. 12, Oct. 2021. [CrossRef]
- G. Lurie et al., “Combined oral contraceptive use and epithelial ovarian cancer risk: time-related effects,” Epidemiology, vol. 19, no. 2, pp. 237–243, Mar. 2008. [CrossRef]
- J. S. Ferris, M. B. Daly, S. S. Buys, J. M. Genkinger, Y. Liao, and M. B. Terry, “Oral contraceptive and reproductive risk factors for ovarian cancer within sisters in the breast cancer family registry,” Br J Cancer, vol. 110, no. 4, p. 1074, Feb. 2014. [CrossRef]
- A. A. Samkari et al., “Body Microbiota and Its Relationship With Benign and Malignant Breast Tumors: A Systematic Review,” Cureus, vol. 14, no. 5, May 2022. [CrossRef]
- E. Reitano et al., “Oral Bacterial Microbiota in Digestive Cancer Patients: A Systematic Review,” Microorganisms, vol. 9, no. 12, Dec. 2021. [CrossRef]
- X. Yu et al., “Microbial dysbiosis in oral squamous cell carcinoma: A systematic review and meta-analysis,” Heliyon, vol. 9, no. 2, Feb. 2023. [CrossRef]
- L. Su Mun et al., “Association of Microbiome with Oral Squamous Cell Carcinoma: A Systematic Review of the Metagenomic Studies,” Int J Environ Res Public Health, vol. 18, no. 14, Jul. 2021. [CrossRef]
- S. Banerjee et al., “The ovarian cancer oncobiome,” Oncotarget, vol. 8, no. 22, p. 36225, May 2017. [CrossRef]
- X. Hu et al., “Gut microbiota dysbiosis promotes the development of epithelial ovarian cancer via regulating Hedgehog signaling pathway,” Gut Microbes, vol. 15, no. 1, 2023. [CrossRef]
- R. Siddiqui, Z. Makhlouf, A. M. Alharbi, H. Alfahemi, and N. A. Khan, “The Gut Microbiome and Female Health,” Biology (Basel), vol. 11, no. 11, p. 1683, Nov. 2022. [CrossRef]
- A. Sipos et al., “The role of the microbiome in ovarian cancer: mechanistic insights into oncobiosis and to bacterial metabolite signaling,” Molecular Medicine, vol. 27, no. 1, p. 33, Dec. 2021. [CrossRef]
- N. R. Nené et al., “Association between the cervicovaginal microbiome, BRCA1 mutation status, and risk of ovarian cancer: a case-control study,” Lancet Oncol, vol. 20, no. 8, pp. 1171–1182, Aug. 2019. [CrossRef]
- P. Łaniewski, Z. E. Ilhan, and M. M. Herbst-Kralovetz, “The microbiome and gynaecological cancer development, prevention and therapy,” Nat Rev Urol, vol. 17, no. 4, p. 232, 2020. [CrossRef]
- Y. C. Lu, W. C. Yeh, and P. S. Ohashi, “LPS/TLR4 signal transduction pathway,” Cytokine, vol. 42, no. 2, pp. 145–151, May 2008. [CrossRef]
- B. Bertani and N. Ruiz, “Function and Biogenesis of Lipopolysaccharides,” EcoSal Plus, vol. 8, no. 1, Dec. 2018. [CrossRef]
- M. Cazzaniga, M. Cardinali, F. Di Pierro, and A. Bertuccioli, “Ovarian Microbiota, Ovarian Cancer and the Underestimated Role of HPV,” International Journal of Molecular Sciences 2022, Vol. 23, Page 16019, vol. 23, no. 24, p. 16019, Dec. 2022. [CrossRef]
- N. Gulve and T. Rudel, “Chlamydia trachomatis and human herpesvirus 6 infections in ovarian cancer—Casual or causal?,” PLoS Pathog, vol. 15, no. 11, 2019. [CrossRef]
- S. Pathak, J. R. Wilczyński, and E. Paradowska, “Factors in Oncogenesis: Viral Infections in Ovarian Cancer,” Cancers 2020, Vol. 12, Page 561, vol. 12, no. 3, p. 561, Feb. 2020. [CrossRef]
- E. Paradowska, A. Jabłońska, M. Studzińska, M. Wilczyński, and J. R. Wilczyński, “Detection and genotyping of CMV and HPV in tumors and fallopian tubes from epithelial ovarian cancer patients,” Sci Rep, vol. 9, no. 1, Dec. 2019. [CrossRef]
- E. Malisic, R. Jankovic, and K. Jakovljevic, “Detection and genotyping of human papillomaviruses and their role in the development of ovarian carcinomas,” Arch Gynecol Obstet, vol. 286, no. 3, pp. 723–728, Sep. 2012. [CrossRef]
- S. Banerjee et al., “The ovarian cancer oncobiome,” Oncotarget, vol. 8, no. 22, pp. 36225–36245, May 2017. [CrossRef]
- M. F. Svahn, M. T. Faber, J. Christensen, B. Norrild, and S. K. Kjaer, “Prevalence of human papillomavirus in epithelial ovarian cancer tissue. A meta-analysis of observational studies,” Acta Obstet Gynecol Scand, vol. 93, no. 1, pp. 6–19, Jan. 2014. [CrossRef]
- C. M. Olsen, A. C. Green, D. C. Whiteman, S. Sadeghi, F. Kolahdooz, and P. M. Webb, “Obesity and the risk of epithelial ovarian cancer: A systematic review and meta-analysis,” Eur J Cancer, vol. 43, no. 4, pp. 690–709, Mar. 2007. [CrossRef]
- A. Urbute, K. Frederiksen, and S. K. Kjaer, “Early adulthood overweight and obesity and risk of premenopausal ovarian cancer, and premenopausal breast cancer including receptor status: prospective cohort study of nearly 500,000 Danish women,” Ann Epidemiol, vol. 70, pp. 61–67, Jun. 2022. [CrossRef]
- Z. Y. Liu et al., “Dietary inflammatory index and risk of gynecological cancers: a systematic review and meta-analysis of observational studies,” J Gynecol Oncol, vol. 30, no. 3, May 2019. [CrossRef]
- N. Ding, J. Zhan, Y. Shi, T. Qiao, P. Li, and T. Zhang, “Obesity in children and adolescents and the risk of ovarian cancer: A systematic review and dose–response meta-analysis,” PLoS One, vol. 17, no. 12, Dec. 2022. [CrossRef]
- C. M. Friedenreich, C. Ryder-Burbidge, and J. McNeil, “Physical activity, obesity and sedentary behavior in cancer etiology: epidemiologic evidence and biologic mechanisms,” Mol Oncol, vol. 15, no. 3, pp. 790–800, Mar. 2021. [CrossRef]
- M. Baczewska, K. Bojczuk, A. Kołakowski, J. Dobroch, P. Guzik, and P. Knapp, “Obesity and Energy Substrate Transporters in Ovarian Cancer—Review,” Molecules, vol. 26, no. 6, Mar. 2021. [CrossRef]
- N. Khanlarkhani et al., “Metabolic risk factors of ovarian cancer: a review,” JBRA Assist Reprod, vol. 26, no. 2, p. 335, 2022. [CrossRef]
- N. Kolomeyevskaya et al., “Cytokine profiling of ascites at primary surgery identifies an interaction of tumor necrosis factor-α and interleukin-6 in predicting reduced progression-free survival in epithelial ovarian cancer,” Gynecol Oncol, vol. 138, no. 2, pp. 352–357, Aug. 2015. [CrossRef]
- A. Ray, J. Fornsaglio, S. Dogan, S. Hedau, S. D. Naik, and A. De, “Gynaecological cancers and leptin: A focus on the endometrium and ovary,” Facts Views Vis Obgyn, vol. 10, no. 1, p. 5, Mar. 2018, Accessed: Mar. 04, 2024. [Online]. Available: /pmc/articles/PMC6260667/.
- S. C. Larsson, N. Spyrou, and C. S. Mantzoros, “Body fatness associations with cancer: evidence from recent epidemiological studies and future directions,” Metabolism, vol. 137, Dec. 2022. [CrossRef]
- A. Urbute, K. Frederiksen, and S. K. Kjaer, “Early adulthood overweight and obesity and risk of premenopausal ovarian cancer, and premenopausal breast cancer including receptor status: prospective cohort study of nearly 500,000 Danish women,” Ann Epidemiol, vol. 70, pp. 61–67, Jun. 2022. [CrossRef]
- D. Wu et al., “Mediation analysis of alcohol consumption, DNA methylation, and epithelial ovarian cancer,” J Hum Genet, vol. 63, no. 3, p. 339, Mar. 2018. [CrossRef]
- D. Cvetkovic, “Early events in ovarian oncogenesis,” Reprod Biol Endocrinol, vol. 1, no. 1, p. 68, 2003. [CrossRef]
- S. Liu, S. Feng, F. Du, K. Zhang, and Y. Shen, “Association of smoking, alcohol, and coffee consumption with the risk of ovarian cancer and prognosis: a mendelian randomization study,” BMC Cancer, vol. 23, no. 1, Dec. 2023. [CrossRef]
- L. S. Cook et al., “Adult lifetime alcohol consumption and invasive epithelial ovarian cancer risk in a population-based case-control study,” Gynecol Oncol, vol. 140, no. 2, pp. 277–284, Feb. 2016. [CrossRef]
- K. Tanha et al., “Investigation on factors associated with ovarian cancer: an umbrella review of systematic review and meta-analyses,” J Ovarian Res, vol. 14, no. 1, p. 153, Dec. 2021. [CrossRef]
- 95. C. Pelucchi, C. Bosetti, C. Galeone, and C. La Vecchia, “Dietary acrylamide and cancer risk: An updated meta-analysis,” Int J Cancer, vol. 136, no. 12, pp. 2912–2922, Jun. 2015. [CrossRef]
- J. Kotsopoulos et al., “Coffee Intake, Variants in Genes Involved in Caffeine Metabolism and the Risk of Epithelial Ovarian Cancer,” Cancer Causes Control, vol. 20, no. 3, p. 335, Apr. 2009. [CrossRef]
- M. C. Vos, A. A. M. van der Wurff, T. H. van Kuppevelt, and L. F. A. G. Massuger, “The role of MMP-14 in ovarian cancer: a systematic review,” J Ovarian Res, vol. 14, no. 1, p. 101, Dec. 2021. [CrossRef]
- X. M. Zhu and W. F. Sun, “Association between matrix metalloproteinases polymorphisms and ovarian cancer risk: A meta-analysis and systematic review,” PLoS One, vol. 12, no. 9, Sep. 2017. [CrossRef]
- H. Liu, L. Sun, X. Liu, R. Wang, and Q. Luo, “Associations between non-coding RNAs genetic polymorphisms with ovarian cancer risk: A systematic review and meta-analysis update with trial sequential analysis,” Medicine, vol. 102, no. 39, p. E35257, Sep. 2023. [CrossRef]
- M. vahidi, M. Houshmand, M. Banoei, and F. Heidari, “The association between TIPARP gene polymorphisms rs2665390 and ovarian cancer susceptibility,” Gynecol Oncol Rep, vol. 47, p. 101175, Jun. 2023. [CrossRef]
- M. Kamali et al., “Association of XRCC2 rs3218536 Polymorphism with Susceptibility of Breast and Ovarian Cancer: A Systematic Review and Meta-Analysis,” Asian Pac J Cancer Prev, vol. 18, no. 7, p. 1743, Jul. 2017. [CrossRef]
- M. Karimi-Zarchi et al., “Association of XRCC3 18067 C>T (Thr241Met) polymorphism with risk of cervical and ovarian cancers: A systematic review and meta-analysis,” Interv Med Appl Sci, vol. 11, no. 3, p. 172, Sep. 2019. [CrossRef]
- E. Mikó et al., “Microbiome—Microbial Metabolome—Cancer Cell Interactions in Breast Cancer—Familiar, but Unexplored,” Cells, vol. 8, no. 4, Apr. 2019. [CrossRef]
- Y. Wu et al., “KLF5 Promotes Tumor Progression and Parp Inhibitor Resistance in Ovarian Cancer,” Advanced Science, vol. 10, no. 31, Nov. 2023. [CrossRef]
- A. Dovnik and N. F. Dovnik, “Vitamin D and Ovarian Cancer: Systematic Review of the Literature with a Focus on Molecular Mechanisms,” Cells, vol. 9, no. 2, Feb. 2020. [CrossRef]
- E. Deuster, U. Jeschke, Y. Ye, S. Mahner, and B. Czogalla, “Vitamin D and VDR in Gynecological Cancers—A Systematic Review,” Int J Mol Sci, vol. 18, no. 11, Nov. 2017. [CrossRef]
- T. Lawler and S. Warren Andersen, “Serum 25-Hydroxyvitamin D and Cancer Risk: A Systematic Review of Mendelian Randomization Studies,” Nutrients, vol. 15, no. 2, Jan. 2023. [CrossRef]
- X. Jiang, X. Li, W. Li, H. Bai, and Z. Zhang, “PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms,” J Cell Mol Med, vol. 23, no. 4, pp. 2303–2313, Apr. 2019. [CrossRef]
- D. M. O’Malley, T. C. Krivak, N. Kabil, J. Munley, and K. N. Moore, “PARP Inhibitors in Ovarian Cancer: A Review,” Target Oncol, vol. 18, no. 4, p. 471, Jul. 2023. [CrossRef]
- T. Kumamoto et al., “Medical guidelines for Li–Fraumeni syndrome 2019, version 1.1,” International Journal of Clinical Oncology 2021 26:12, vol. 26, no. 12, pp. 2161–2178, Oct. 2021. [CrossRef]
- J. Y. Pan et al., “Worldwide Practice Patterns in Lynch Syndrome Diagnosis and Management, Based on Data From the International Mismatch Repair Consortium.,” Clin Gastroenterol Hepatol, vol. 16, no. 12, pp. 1901-1910.e11, Dec. 2018. [CrossRef]
- D. P. Li et al., “Breastfeeding and ovarian cancer risk: a systematic review and meta-analysis of 40 epidemiological studies,” Asian Pac J Cancer Prev, vol. 15, no. 12, pp. 4829–4837, 2014. [CrossRef]
- A. Babic et al., “Association Between Breastfeeding and Ovarian Cancer Risk,” JAMA Oncol, vol. 6, no. 6, Jun. 2020. [CrossRef]
- J. M. Genkinger et al., “Alcohol intake and ovarian cancer risk: a pooled analysis of 10 cohort studies,” Br J Cancer, vol. 94, no. 5, pp. 757–762, Mar. 2006. [CrossRef]
- H. Yan-Hong, L. Jing, L. Hong, H. Shan-Shan, L. Yan, and L. Ju, “Association between alcohol consumption and the risk of ovarian cancer: a meta-analysis of prospective observational studies,” BMC Public Health, vol. 15, no. 1, Mar. 2015. [CrossRef]
- D. Huber, S. Seitz, K. Kast, G. Emons, and O. Ortmann, “Use of oral contraceptives in BRCA mutation carriers and risk for ovarian and breast cancer: a systematic review,” Arch Gynecol Obstet, vol. 301, no. 4, pp. 875–884, Apr. 2020. [CrossRef]
- Y. Y. Xia and J. Kotsopoulos, “Beyond the pill: contraception and the prevention of hereditary ovarian cancer,” Hered Cancer Clin Pract, vol. 20, no. 1, Dec. 2022. [CrossRef]
- J. Steevens, L. J. Schouten, B. A. J. Verhage, R. A. Goldbohm, and P. A. Van Den Brandt, “Tea and coffee drinking and ovarian cancer risk: results from the Netherlands Cohort Study and a meta-analysis,” Br J Cancer, vol. 97, no. 9, p. 1291, Nov. 2007. [CrossRef]
- J. M. Genkinger et al., “Alcohol intake and ovarian cancer risk: a pooled analysis of 10 cohort studies,” Br J Cancer, vol. 94, no. 5, pp. 757–762, Mar. 2006. [CrossRef]
- H. Yan-Hong, L. Jing, L. Hong, H. Shan-Shan, L. Yan, and L. Ju, “Association between alcohol consumption and the risk of ovarian cancer: a meta-analysis of prospective observational studies,” BMC Public Health, vol. 15, no. 1, Mar. 2015. [CrossRef]
- H. R. Harris and K. L. Terry, “Polycystic ovary syndrome and risk of endometrial, ovarian, and breast cancer: a systematic review,” Fertil Res Pract, vol. 2, no. 1, Dec. 2016. [CrossRef]
- H. P. K. Throwba et al., “The epigenetic correlation among ovarian cancer, endometriosis and PCOS: A review,” Crit Rev Oncol Hematol, vol. 180, p. 103852, Dec. 2022. [CrossRef]
- A. N. Rosenthal et al., “Evidence of Stage Shift in Women Diagnosed With Ovarian Cancer During Phase II of the United Kingdom Familial Ovarian Cancer Screening Study,” J Clin Oncol, vol. 35, no. 13, pp. 1411–1420, May 2017. [CrossRef]
- J. Domański, Z. Domagala, J. E. Simmons, and M. Wanat, “Terra Incognita in anatomical museology – A literature review from the perspective of evidence-based care,” Annals of Anatomy - Anatomischer Anzeiger, vol. 245, p. 152013, Jan. 2023. [CrossRef]
- M. Mateusz et al., “Decoding the Mysteries of the Obturator Nerve,” J Anat Soc India, vol. 73, no. 1, pp. 64–69, 2024. [CrossRef]
- D. Domagała et al., “Cellular, Molecular and Clinical Aspects of Aortic Aneurysm—Vascular Physiology and Pathophysiology,” Cells 2024, Vol. 13, Page 274, vol. 13, no. 3, p. 274, Feb. 2024. [CrossRef]
Figure 1.
Percent of new cases of ovarian cancer in particular age groups.
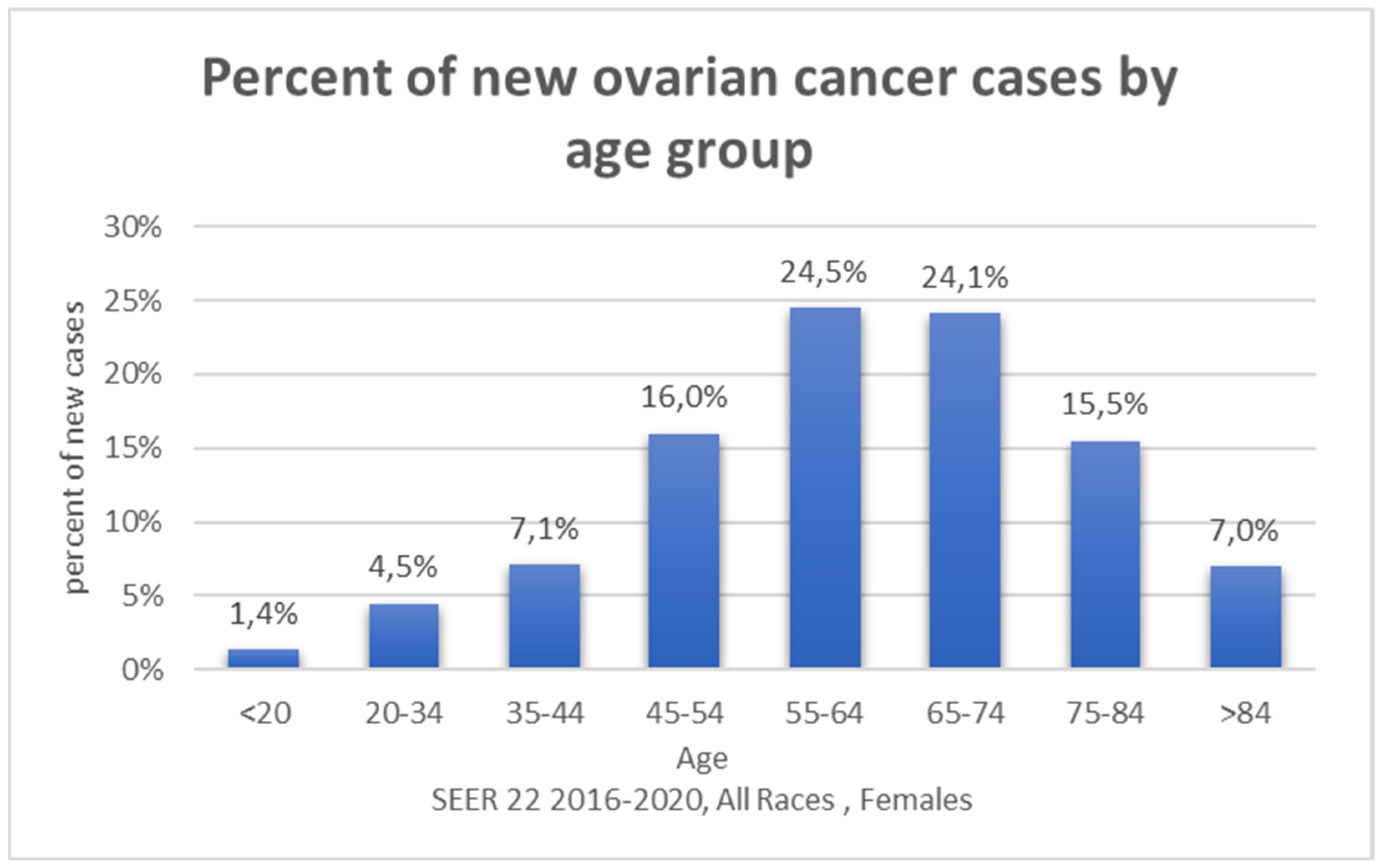
Figure 2.
Frequency of mutations of particular genes in hereditary ovarian cancer.
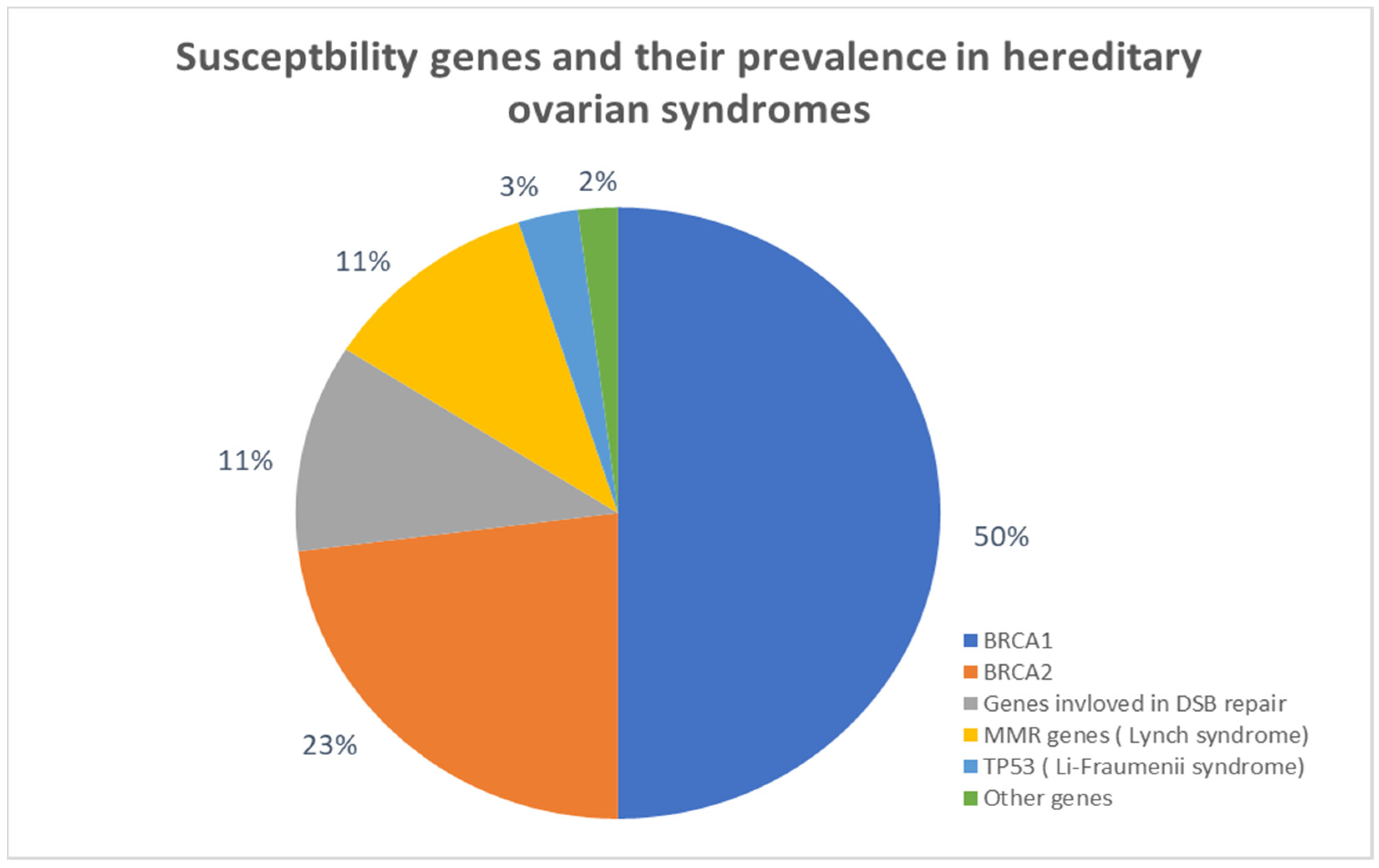
Figure 3.
Preferred ways of Double Strand Break repair in normal cells and in BRCA deficient cells.
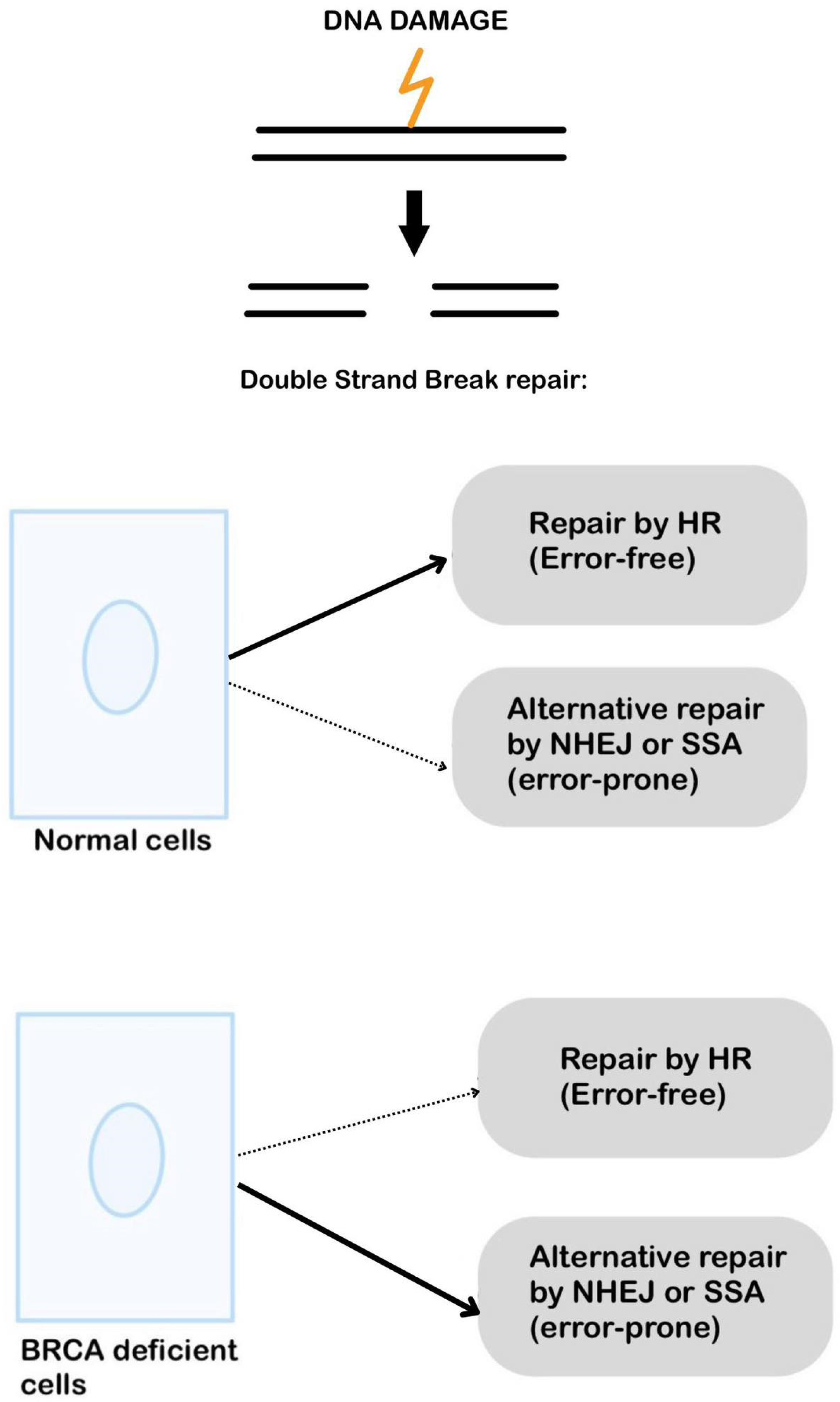
Figure 4.
The distribution of point variants in the TP53 gene at specific codon numbers.
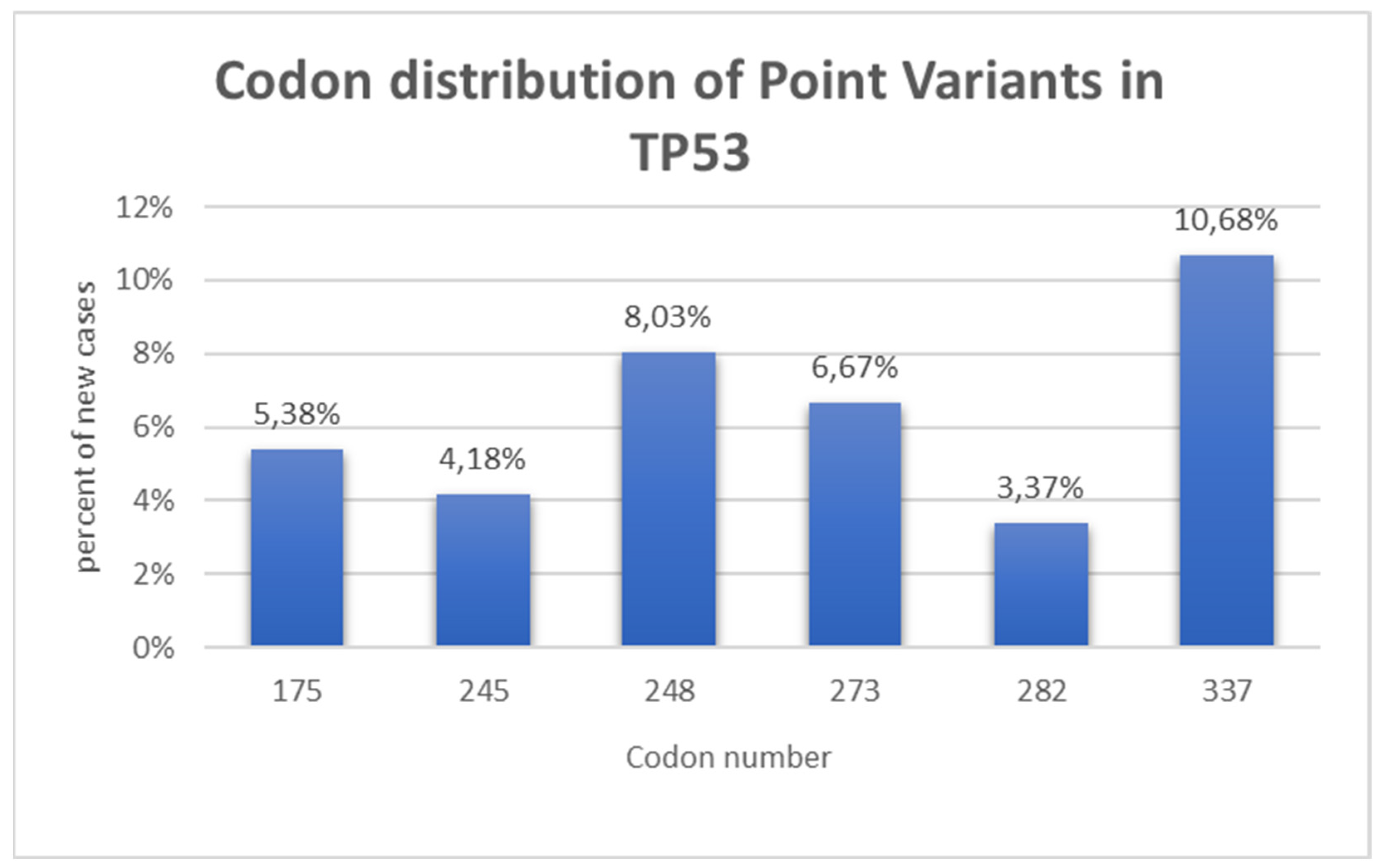
Figure 5.
The distribution of types of germline variants across MLH1 gene. Each chart represents different types of variants: Missense, Silent, In frame, Intronic, and Nonsense.
Figure 5.
The distribution of types of germline variants across MLH1 gene. Each chart represents different types of variants: Missense, Silent, In frame, Intronic, and Nonsense.

Figure 6.
The distribution of types of germline variants across MSH2 gene. Each chart represents different types of variants: Missense, Silent, In frame, Intronic, and Nonsense.
Figure 6.
The distribution of types of germline variants across MSH2 gene. Each chart represents different types of variants: Missense, Silent, In frame, Intronic, and Nonsense.
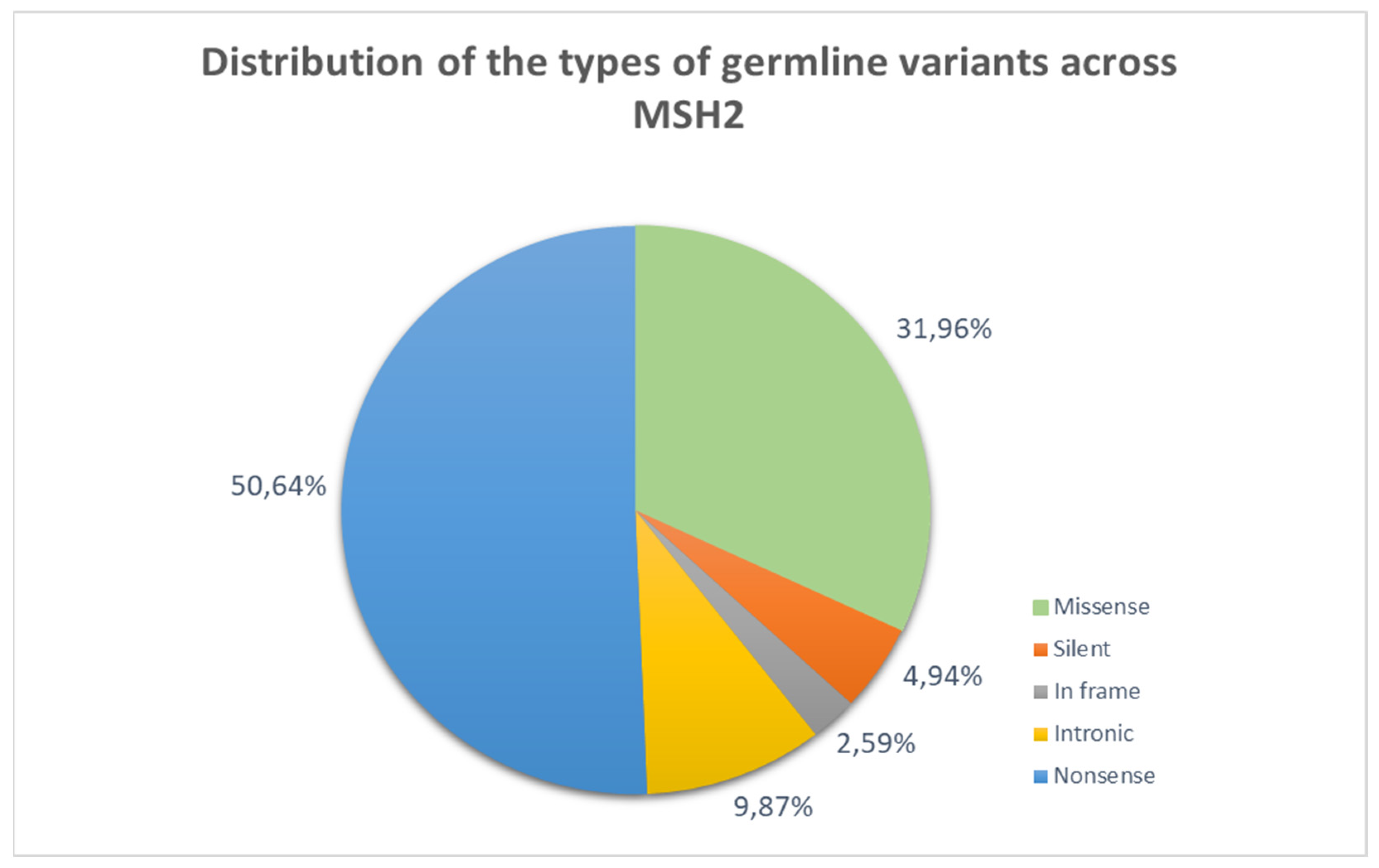
Figure 7.
The distribution of types of germline variants across MSH6 gene. Each chart represents different types of variants: Missense, Silent, In frame, Intronic, and Nonsense.
Figure 7.
The distribution of types of germline variants across MSH6 gene. Each chart represents different types of variants: Missense, Silent, In frame, Intronic, and Nonsense.
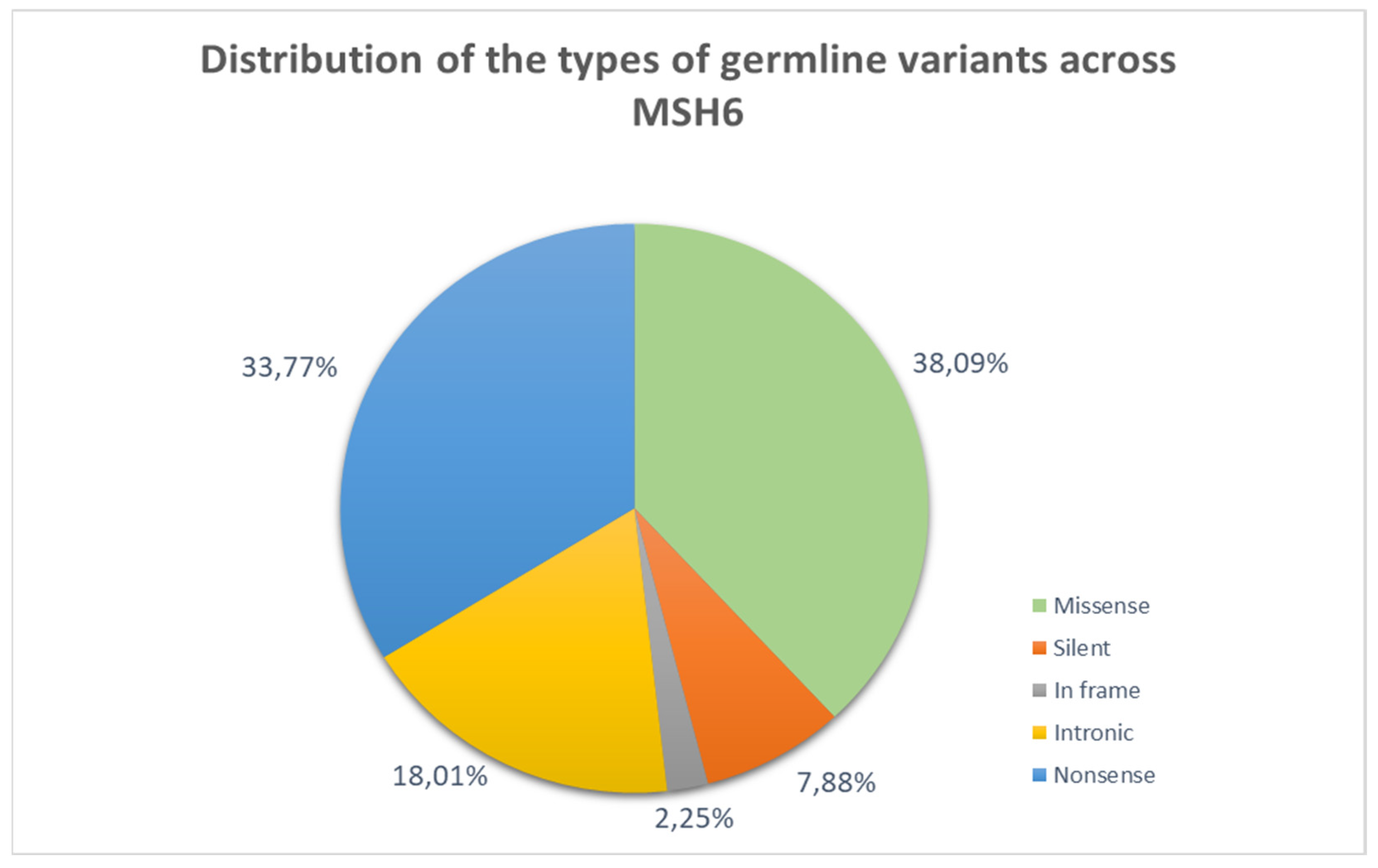
Figure 8.
The distribution of types of germline variants across PMS2 gene. Each chart represents different types of variants: Missense, Silent, In frame, Intronic, and Nonsense.
Figure 8.
The distribution of types of germline variants across PMS2 gene. Each chart represents different types of variants: Missense, Silent, In frame, Intronic, and Nonsense.
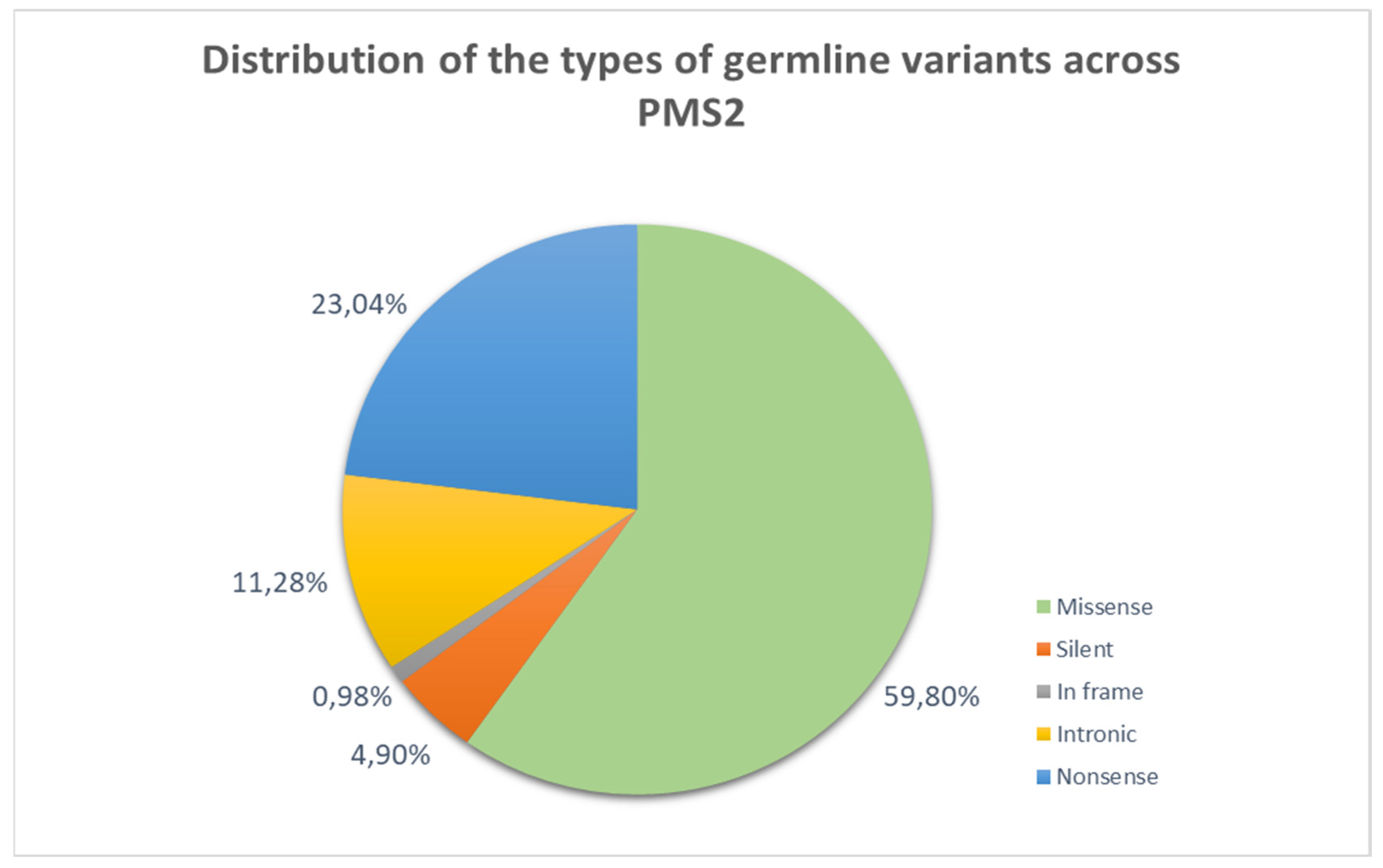
Figure 9.
An overview of the mutation frequency of individual genes associated with Lynch syndrome.
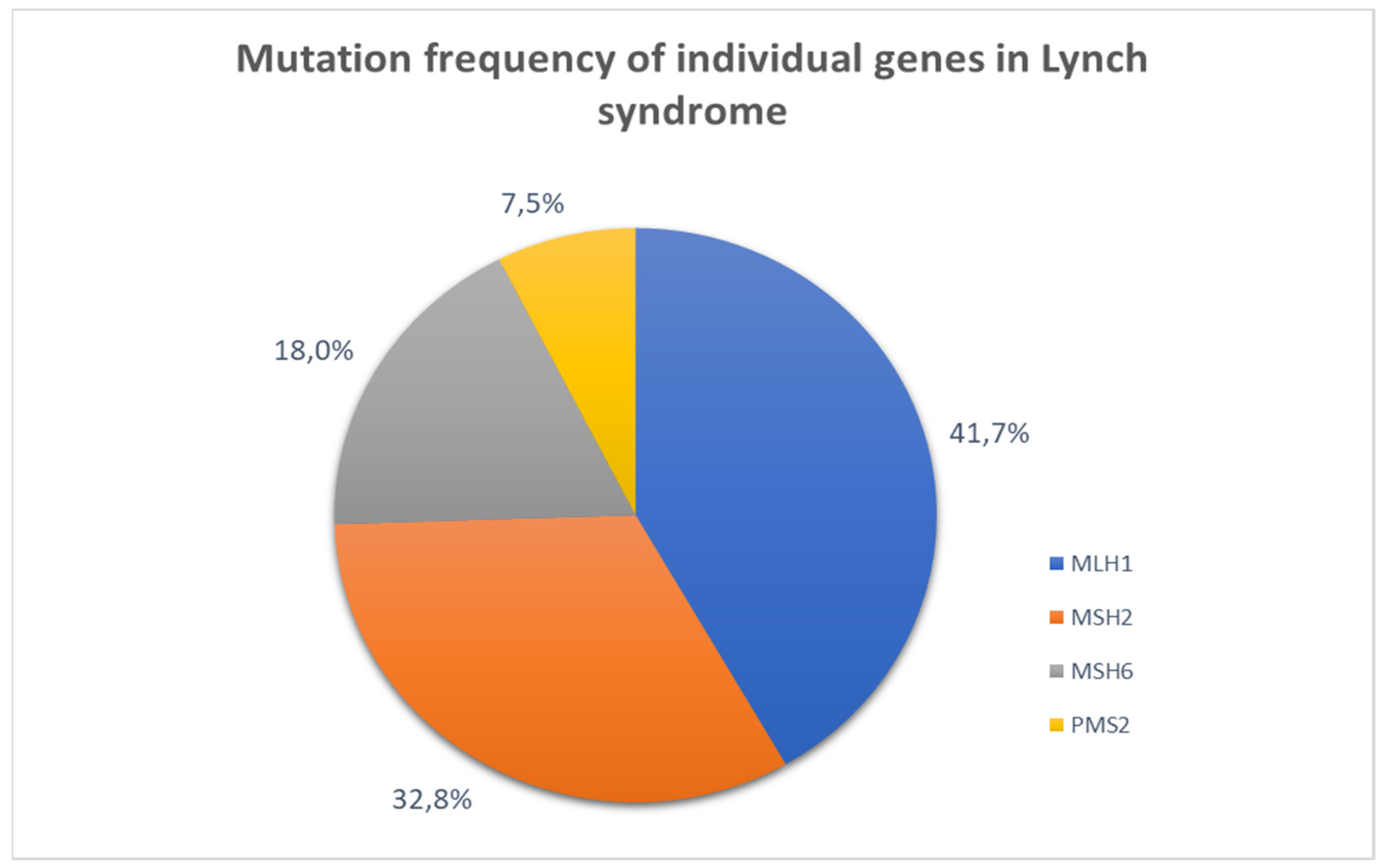

Table 1.
A comparative analysis of the lifetime risk and average age of diagnosis for ovarian, colorectal and endometrial cancers in the general population versus individuals with MLH1 mutation.
Table 1.
A comparative analysis of the lifetime risk and average age of diagnosis for ovarian, colorectal and endometrial cancers in the general population versus individuals with MLH1 mutation.
| Cancer Type | Lifetime Risk in general population | Lifetime Risk with MLH1 mutation | Average age of diagnosis in general population | Average age of diagnosis with MLH1 mutation |
|---|---|---|---|---|
| Ovarian | 4-20% | 1,30% | 46 | 63 |
| Colorectal | 46-61% | 4,20% | 44 | 68-72 |
| Endometrial | 34-54% | 3,10% | 49 | 60 |
Table 2.
A comparative analysis of the lifetime risk and average age of diagnosis for ovarian, colorectal and endometrial cancers in the general population versus individuals with MSH2 mutation.
Table 2.
A comparative analysis of the lifetime risk and average age of diagnosis for ovarian, colorectal and endometrial cancers in the general population versus individuals with MSH2 mutation.
| Cancer Type | Lifetime Risk in general population | Lifetime Risk with MSH2 mutation | Average age of diagnosis in general population | Average age of diagnosis with MSH2 mutation |
|---|---|---|---|---|
| Ovarian | 8-38% | 1,30% | 43 | 63 |
| Colorectal | 33-52% | 4,20% | 44 | 68-72 |
| Endometrial | 21-57% | 3,10% | 47-48 | 60 |
Table 3.
A comparative analysis of the lifetime risk and average age of diagnosis for ovarian, colorectal and endometrial cancers in the general population versus individuals with MSH6 mutation.
Table 3.
A comparative analysis of the lifetime risk and average age of diagnosis for ovarian, colorectal and endometrial cancers in the general population versus individuals with MSH6 mutation.
| Cancer Type | Lifetime Risk in general population | Lifetime Risk with MSH6 mutation | Average age of diagnosis in general population | Average age of diagnosis with MSH6 mutation |
|---|---|---|---|---|
| Ovarian | <1-13% | 1,30% | 46 | 63 |
| Colorectal | 10-44% | 4,20% | 42-69 | 68-72 |
| Endometrial | 16-49% | 3,10% | 53-55 | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



