Submitted:
11 May 2024
Posted:
13 May 2024
You are already at the latest version
Abstract
The Julia-Kocienski olefination reaction has become over past 30 years since its discovery one of the key C-C connective methods that is used in the late-stage natural product synthesis. The reaction proceeds under mild reaction conditions, with wide substrate scope and functional group tolerance, and with high (E) selectivity. In this review, we discuss the reaction from a mechanistic point of view and disclose key features that play an important role in reaction selectivity. Finally, the mechanistic aspects of the newly developed modification of the Julia-Kocienski reaction, which allows the formation of both (E) and (Z) olefins, from the same reaction partners, are discussed.
Keywords:
Julia-Kocienski reaction
; olefination
; reaction selectivity
; reaction mechanism
1. Introduction
Alkenes belong to a chemical functional group that is omnipresent in literally all natural products. Interestingly, since the early times when organic synthesis slowly became a ‘useful’ scientific discipline, many synthetic strategies have focused on the stereoselective synthesis of these structural motives. Especially, methods that allow for the connective stereoselective introduction of the olefin moiety have become very valuable tools for this achievement. Over the past 100 years, many different connective olefination methods have been developed, but many of them follow the same retrosynthetic pathway [1] - they are based on the reunion of α-negative charge stabilizing reagents 1 with aldehydes or ketones 2 (Table 1).
Since the introduction of the Wittig reaction[7,8] in the late 1950s of the twentieth century, Wittig,[3] Horner-Wadsworth-Emmons,[4] Johnson,[2] Peterson,[5] and Julia olefination[1] established themselves as the most widely used olefination protocols. Each of these methods has, of course, its advantages and drawbacks that change over time because each of the methods went through a long and interesting development since its original disclosure. In this review, we wish to focus on the so-called modified Julia reaction,[1,9,10,11,12,13,14,15,16] also known as the Julia one-pot, Silvestre-Julia, or Julia-Kocienski olefination, and its development in terms of the reaction mechanism and selectivity.
2. Origins and Mechanism of the Julia-Kocienski Olefination Reaction
2.1. Julia-Lythgoe Olefination vs. Julia-Kocienski Olefination: A Comparison
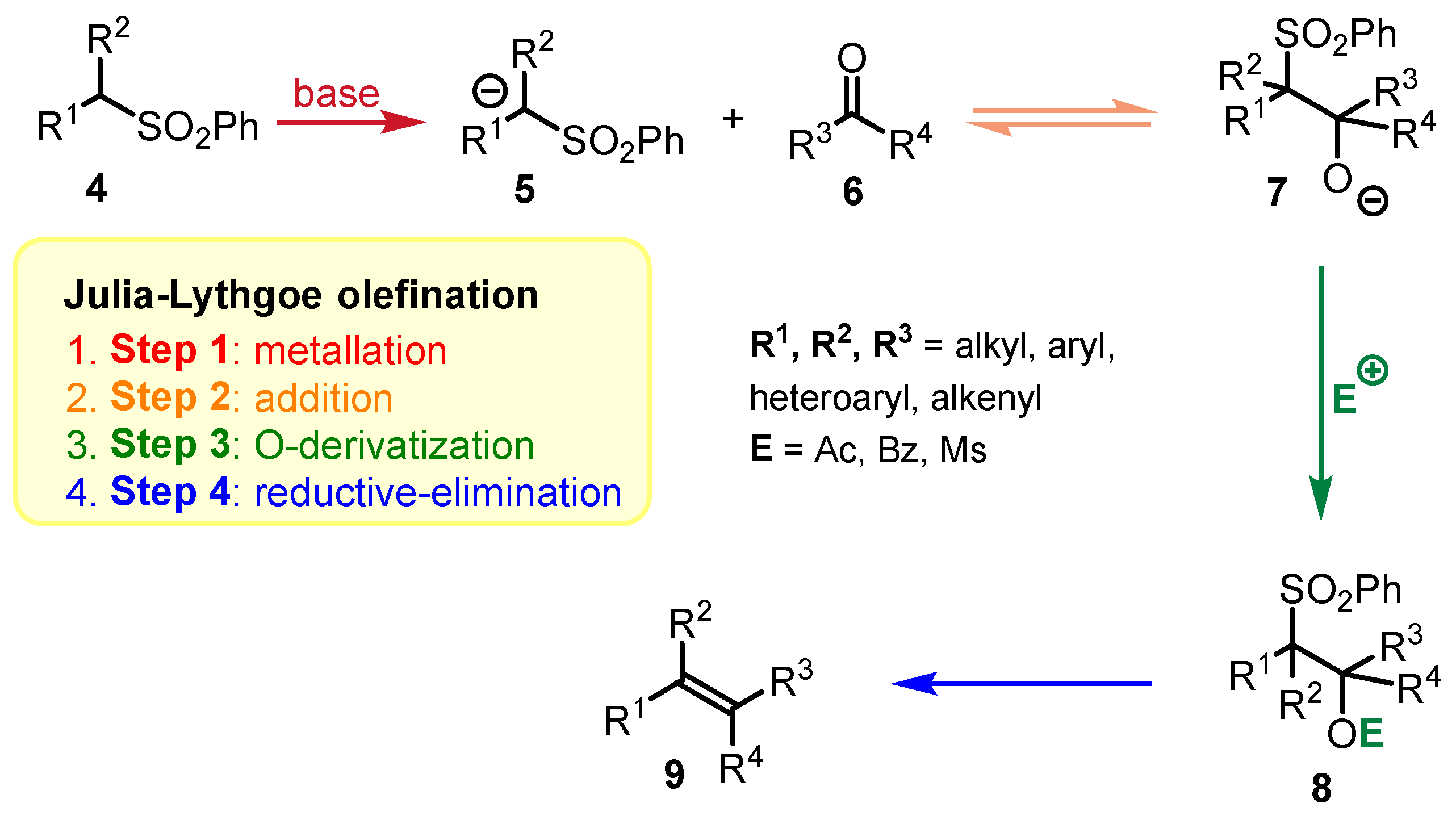
Classical Julia olefination, also known as Julia-Lythgoe olefination, was described for the first time in 1973 by (Mark) Julia and Paris[17] and was later developed by Kocienski and Lythgoe[18]. The original protocol was soon expanded for the beneficial O-derivatization step and thus consisted of four distinct stages carried out commonly in the two-pot protocol (Scheme 1): (1) metalation of an alkylarylsulfone 4, (2) addition of the resulting carbanion species 5 to an aldehyde or ketone 6, (3) O-acylation (sulfonylation) of the adduct 7, and (4) reductive-elimination of the β-acyl (sulfonyl) oxysulfone 8 intermediate. The addition of 5 to 6 typically yields product 7 as a mixture of all possible diastereoisomers; however, this is not of consequence because the stereochemical information encoded in 7 (or 8) is lost during the reductive elimination step. A common feature of Julia-Lythgoe olefination is its high (E)-stereoselectivity[1] – a consequence of the various radical mechanisms that operate in the final stage of reductive elimination[19].
The main drawbacks of Julia-Lythgoe olefination, namely the steric requirement-driven (E/Z)-selectivity, and the two-pot protocol, were in 1993 overcome by Silvestre Julia[20,21] (brother of Mark Julia). Their modification of the standard Julia-Lythgoe olefination protocol was based on the replacement of the phenylsulfonyl group with the benzo[d]thiazol-2-ylsulfonyl (BT) group (Scheme 2). The common feature of the new transformation with Julia-Lythgoe olefination are the first two steps: (1) metalation and (2) addition of metalated sulfone 11 to aldehyde 12. Since in this case the aryl group in the alkyl aryl sulfone is an electron-acceptor, the initially generated β-alkoxy sulfone adduct 13 can undergo to spontaneous Smiles rearrangement (S to O migration of the heteroaryl group) to yield adduct 15. Subsequent β-elimination of SO2 (18) and of an aryloxide anion (17) in 15 directly forms olefin 16.
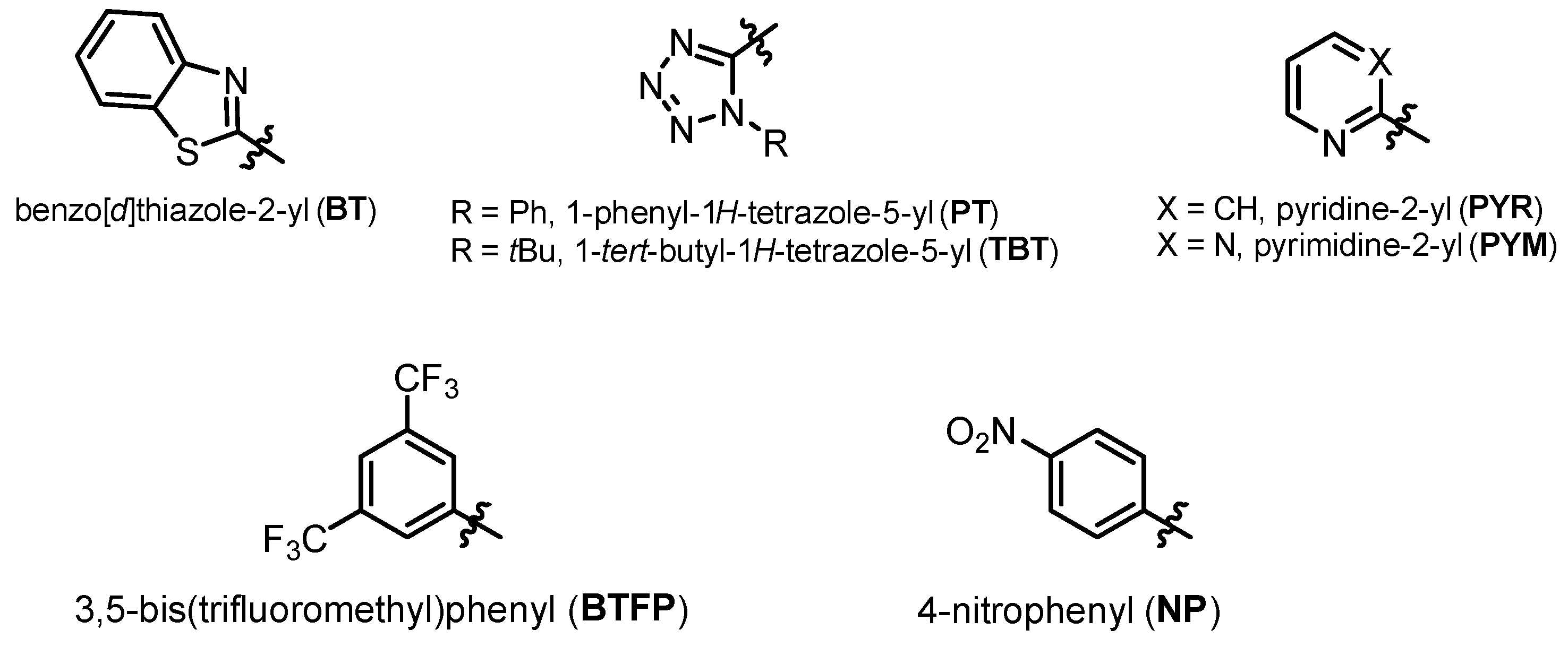
As mentioned above, Silvestre Julia introduced the BT-group as the only electron-acceptor aryl group suitable for the Julia-Kocienski olefination reaction. But this situation did not last long, and many other research groups introduced several different heteroaryl groups such as pyridine-2-yl (PYR),[20,22] 1-phenyl-1H-tetrazole-5-yl (PT),[23] 1-tert-butyl-1H-tetrazole-5-yl (TBT),[24] and 3,5-bis(trifluoromethyl)phenyl (BTFP)[25] and others[20,26]. Interestingly, only the PT group introduced by Kocienski et al.[13,24] possessed sufficiently interesting properties (diminished side reactions such as homocoupling[13], high (E)-selectivity) that remained along with the original BT group as the most widely used heteroaryl acceptor groups explored in olefination reactions.
The generalized scopes and limitations and the achieved (E/Z) selectivities observed for Julia-Lythgoe and Julia-Kocienski olefination are summarized in Table 2. .
2.2. Reaction Mechanism and Its Impact on the Selectivity of Julia-Kocienski Olefination
The Julia-Kocienski reaction mechanism was intensively studied by Silvestre Julia[20,21] and the study was further extended by P. Kocienski and R. Blackmore.[11,12,13,24] Based on these excellent mechanistical works, the reaction mechanism could be established with respect to the stereochemical outcomes of the reaction (Scheme 3). There are three important features of this mechanism that deserve a brief comment.
- (1)
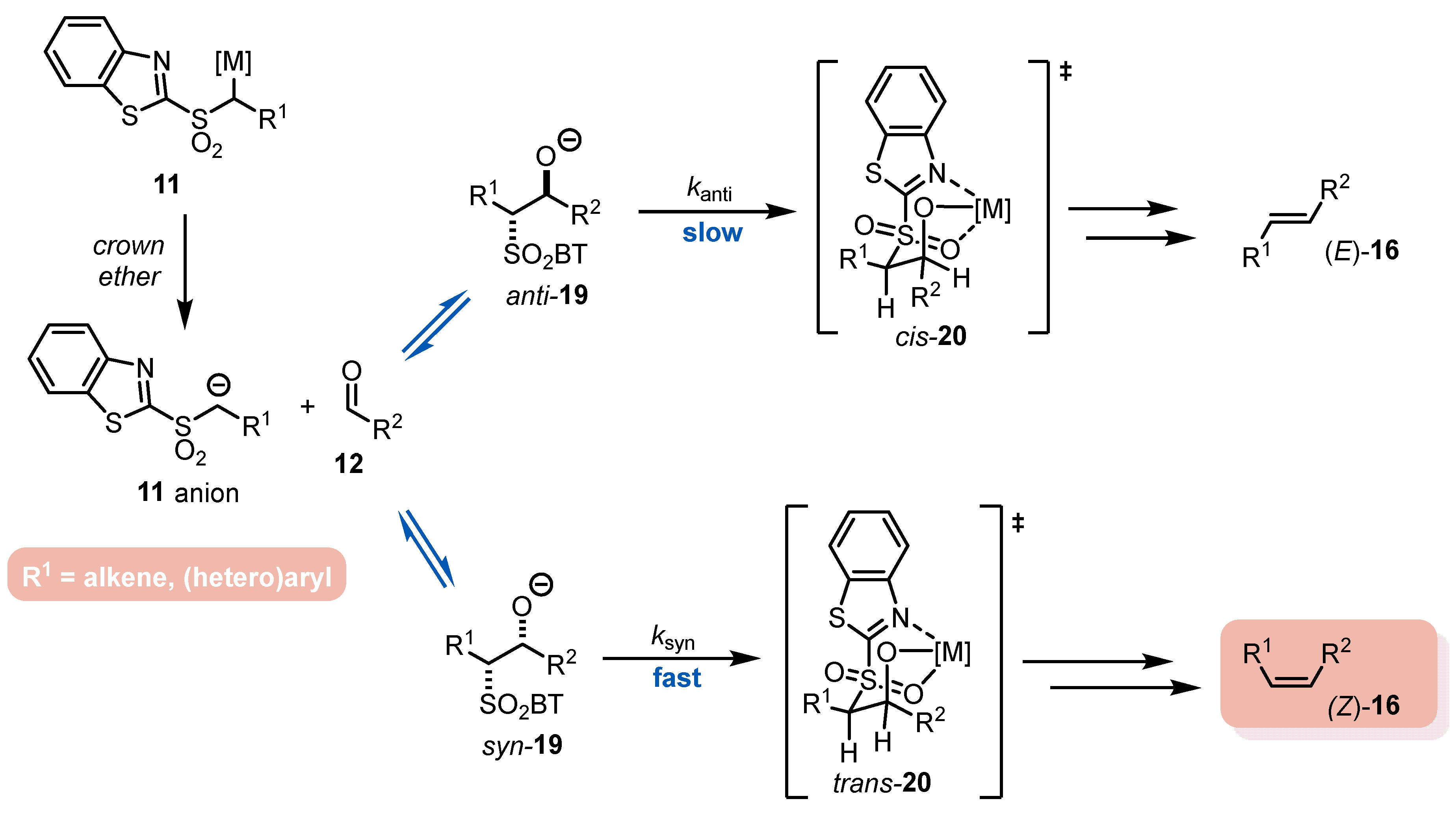
- The addition step of metalated sulfone 11 to aldehyde 12 can provide anti-adduct anti-19 via TS1 or syn-adduct syn-19 via TS2 (Figure 2). The selectivity in this step is extremely important since all subsequent transformations of intermediate 19, Smiles rearrangement and β-elimination, are stereospecific. Thus, the syn/anti-selectivity of the addition step determines the final (E/Z)-olefin ratio. Therefore, in theory, the (E/Z)-selectivity of the reaction could be swapped from (E) to (Z) if proper reaction conditions are applied.
- (2)
- When stabilized metalated sulfonyl anions 11 (R1 = Ph, alkenyl, etc.) are used, the addition step of 11 to 12 becomes reversible (Scheme 3, path A). In this case, the original kinetically driven syn/anti-ratio of adduct 19 becomes less important in comparison with the Smiles rearrangement reaction rates (transformation of 19 to 22). In such cases, the rearrangement of anti-19 adduct leading to (E)-olefin 16 is slower compared to the rearrangement of syn-19 to olefin (Z)-16 due to repulsive 1,2-interactions in the transition state (see cis-20).
- (3)
- For the elimination step, two borderline mechanisms are generally accepted. In the first, which is the most common, the rearranged intermediate 22 undergoes β-elimination. The elimination is stereospecific, and the syn-19 adduct rearranged intermediate syn-22 furnishes (Z)-olefin and the anti-19 adduct rearranged intermediate, compound trans-22 (trans refers to the arrangement of R1 and R2 within the intermediate cycle), yields (E)-olefin. Alternatively, when (hetero)aryl aldehydes 12 (R2 = (hetero)aryl) an alternative elimination pathway (path B) was postulated to occur. In this case, the elimination pathway should proceed through the formation of intermediate carbocation 23. The steric requirements of R1 and R2 then play a crucial role in the final (E/Z)-selectivity of the reaction. Path B was used to explain the unexpected (E)-selectivity of the coupling reactions carried out using (hetero)aryl aldehydes 12 as substrates.
Recently, our group, in collaboration with Robiette’s group, proposed an alternative explanation for the observed (E) selectivity of these reactions. Our explanation is based on a combined experimental and theoretical study that revealed that the key role in the elimination step plays the rearrangement product 22a (Scheme 4).[14] In general, both the anti and syn intermediates 22a, can adopt the cisoid and transoid conformation. Conformational equilibrium is strongly influenced by the steric requirements of the R1 and Ar groups, and in the case of the anti-22a intermediate, the transoid is preferred, while in the case of syn-22a, the cisoid is preferred. Advanced experimental and theoretical study then suggested that in the case of a cisoid conformation, competitive syn elimination can occur,[14] explaining almost exclusive formation of (E)-olefins observed in the case of olefins of general structure 16a.
Theoretical studies also suggested that the syn elimination process should be more favored when the aryl substituent R2 has electron-donating substituents and disfavored when an electron-deficient substituent is present. The postulated prediction was then evaluated using a stereodefined intermediate 24 that was selectively transformed in situ to the corresponding lithiated anion 25 that was allowed to undergo an elimination process. The generated anion cannot undergo the retroaddition process (it is an intermediate after the rearrangement step) and the nucleophile generated in situ (thiolate anion) is not basic enough to trigger the epimerization process of any of the two epimerizable stereogenic centers. Thus, only (Z) olefin (Z)-26 should be produced as the main product of the transformation. If the reaction should proceed through the carbocation-type intermediate of the 23 type (see Scheme 3), an approximately 50:50 ratio of the (E/Z)-isomeric mixture was expected. In all tested cases, the (E)-isomer (E)-26, the product of the synperiplanar elimination process was produced as the main product of the reaction, strongly suggesting that the syn elimination process is the main process that operates during the Julia-Kocienski olefination reaction of alkyl sulfones with aryl aldehydes. The observed stronger preference for electron-donating group containing intermediates to undergo preferentially synperiplanar elimination was also in agreement with the DFT calculation-based prediction. Scheme 5.
2.3. Recent Achievements in the Reaction Selectivity Improvements
Reaction mechanism studies carried out by S. Julia and P. Kocienski, which were later confirmed by our own studies, implies that the reaction selectivity is directly linked with the initial syn/anti-selectivity of the addition step. The adduct ration further directly influences the selectivity (E/Z) of the overall reaction regardless of whether the reaction proceeds through the antiperiplanar elimination (for R1 and R2 = alkyl), or mixed antiperiplanar and synperiplanar (for R1 and/or R2 = (hetero)aryl) elimination in the final step. Not surprisingly, then, most of the methods developed to influence the reaction selectivity in favor of one of the two isomers focus on the key addition step.
2.3.1. Solvent Effect
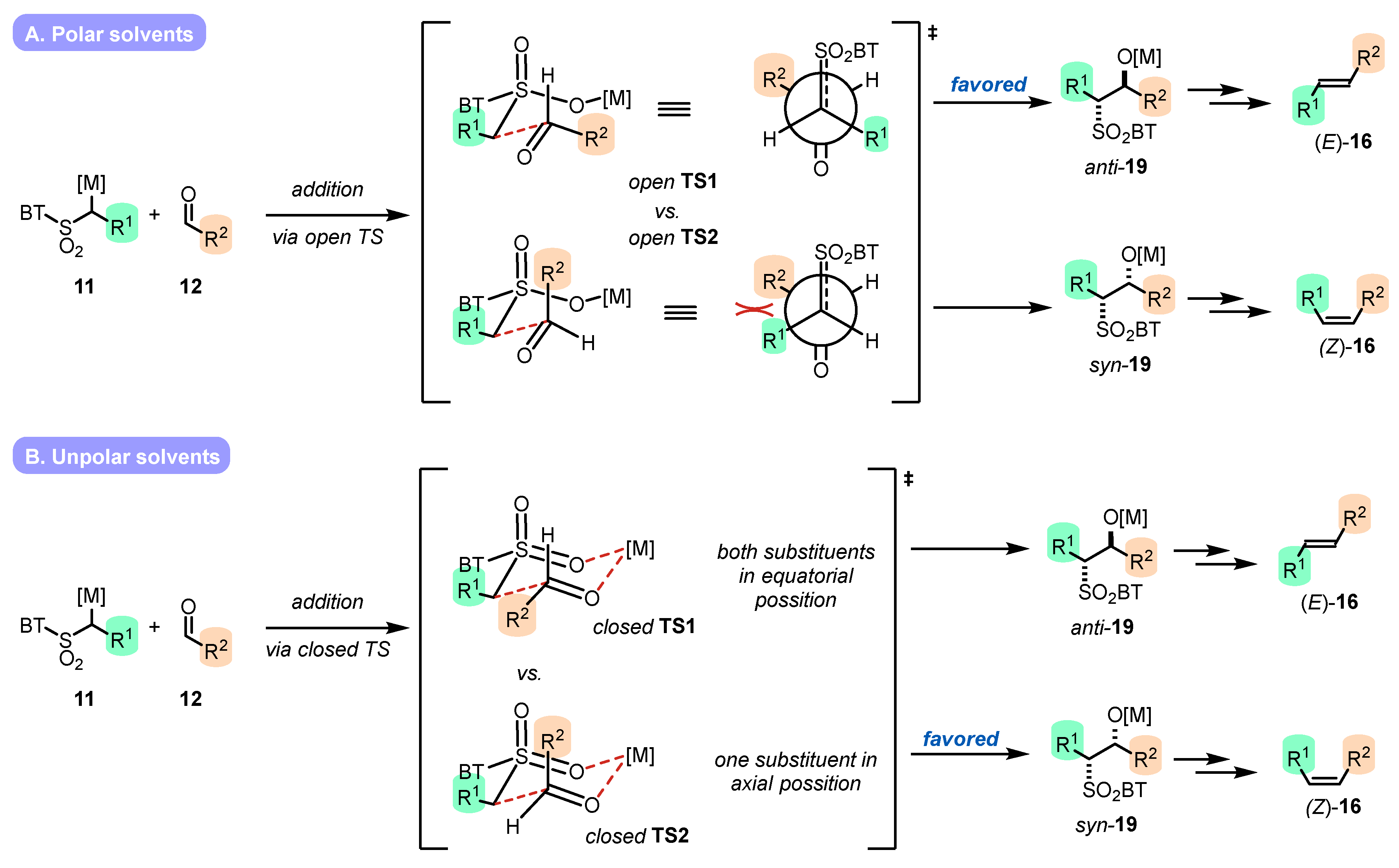
The most important and most straightforward way to influence the syn/anti-selectivity of the addition step is to choose the right solvent for the transformation. When polar solvents such as THF, DME, or DMF are used, anti-adduct anti-19 is the preferred product of the addition due to solvent stabilization (Scheme 6A). On the contrary, when nonpolar solvents such as toluene are used, the reaction proceeds via a closed transition state (Scheme 6B) and syn adduct syn-19 is preferred.
It should be noted that, even though such an approach is generally applicable and correct, the role of the solvent might be altered by several factors that cannot be removed from the reaction mixture and which will be discussed in the following two sub-sections.
- Metal cation
The metal cation, which is always present in the reaction mixture as a ‘residue’ after the deprotonation step, has a key influence on the selectivity of the reaction. In general, cations with the character of a hard Lewis acid such as, e.g., Li+ favor the formation of the (E) olefins. It is assumed that the observed (E) selectivity is caused by a better stabilization of the generated anion 11 that can further add due to its lower reactivity to the aldehyde 12 with a better selectivity that favors the anti-adduct anti-19. On the contrary, when a large cation is used, such as K+, the reaction can proceed preferentially either via closed TS or the solvent can increase the dissociation of the cation from the anion 11 and thus increase the reactivity of it. The first case is typical for nonpolar solvents (e.g., toluene), since the solvent does not provide additional stabilization to the reagents and/or reaction intermediates. In the latter case, dissociation of the cation from reagent 11 increases the reactivity of the anion and leads to the faster production of the kinetic product of the addition step, anti-isomer anti-19. However, it should also be noted that an increase in anion 11 reactivity can also inevitably lead to the undesired self-dimerization of reagent 11 (Scheme 7); thus, a compromise between selectivity and reactivity has to be searched.
- Co-solvents
The addition of the co-solvents to the reaction mixture can also be beneficial when (E)-selectivity is searched. It was observed that the addition of co-solvents such as DMPU or HMPA to the reaction mixtures carried out in the THF or DMF leads to an increase in the (E)-olefin selectivity of the desired product. It is believed that the co-solvent role is in metal cation scavenging with an impact similar to that described in the previous section (increased reactivity that favors the anti-adduct formation).
2.3.2. Additives
Another way to increase the (E/Z) selectivity of the Julia-Kocienski reaction is the addition of additives to the reaction mixture. Over the years, many various additives have been used for such purposes; however, only a few of them have had a significant effect. The relevant ones are listed below.
- Crown ethers
As mentioned in the previous section, the role of (co-)solvent was shown to have a tremendous effect on the reaction yield and selectivity. As a modus operandi, it was postulated that polar solvents increase the reactivity of anion 11 due to a cation/anion separation (reaction kinetic) that leads to the preferential formation of anti-adduct (polar solvents) or syn-adduct (nonpolar solvents). As a disadvantage, self-condensation of metalated sulfone 11 (Scheme 7) was observed. The use of specific cation-chelating co-solvents such as HMPA or DMPU met only with limited success even though in several cases it led to the diminished formation of self-condensation products and an increase in the (E) selectivity.
Based on the same logic, to increase the reactivity of metalated sulfone 11 and thus increase anti-adduct formation (kinetic product), an excess of crown ethers (18-crown-6 for K+; 12-crown-6 for Li+)[27] can be used during the reaction as demonstrated in several recent total syntheses of natural products (e.g., zeaenol,[28] paecilomycins E and F,[29] amphidinolide E,[30] and salarins A and C[31]).
However, it should be noted that if metalated sulfone 11 is used with a group in the lateral chain (R1) that is capable of stabilizing the generated anion, the addition of generated anion 11 to aldehyde 12 is reversible. Consequently, the syn/anti-ratio of adducts 19 is in equilibrium and (Z)-olefin (Z)-16 is formed preferentially due to a faster (kanti < ksyn) Smiles rearrangement step.[32] Scheme 8.
- Ammonium salts
The use of ammonium salts proved to be also beneficial and in several cases of highly complex molecular scaffolds led to an increase in the observed reaction yield and (E)-selectivity.[33,34] It is believed that the role of ammonium salts is in the activation of aldehyde 12, where, due to its steric requirements, increases the anti-selectivity of the addition step. It should also be noted that the role of counter anion of the ammonium salt is not innocent. The best (E) selectivity was observed when potassium-containing metalated sulfone 11 was reacted in the presence of TBAB (tetra-butylammonium bromide) and lithium-containing metalated sulfone 11 was reacted in the presence of TBAC (tetra-butylammonium chloride). Such observations suggest the beneficial formation of KBr and LiCl salts during the reaction.
- Chelating Metals
Similarly, metal cations as e.g., CeCl3,[35,36] MgCl2,[37] ZnCl2, and LiBr, can be used to activate aldehyde 12 during the reaction. The addition of such salt generally results in an increase in the reaction yield of the transformation. The (E/Z) selectivity of the transformation is influenced only if aldehydes bearing α-alkoxy substituents[37] are used in the presence of an excess of MgCl2 or ZnCl2 (addition via the Cram-chelate transition state).[38]
3. Julia-Kocienski Olefination – Extension to Carboxylic Acid Derivatives
All the above-mentioned olefination methods are based on the reunion of the metalated sulfone 11-type intermediate and a carbonyl-containing intermediate 12 (Scheme 2). The overall transformation can thus be regarded as an addition/rearrangement/elimination sequence, where the final (E/Z)-selectivity of the newly olefinic bond is determined by the addition step. Therefore, the stereoselectivity is dictated by the reaction kinetic of the addition step (kinetic conditions) or by the kinetic of the rearrangement step (the addition step is in equilibrium) (Scheme 3).
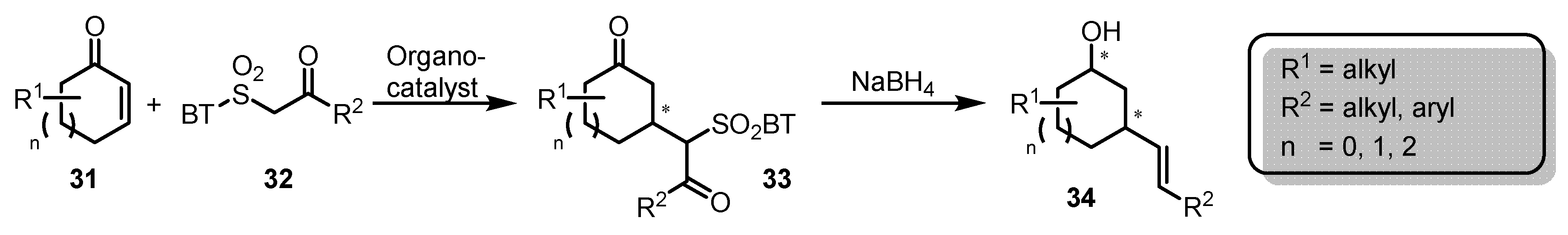
However, recently this paradigm changed since we have introduced ‘reaction work-up driven selectivity’ for the Julia-Kocienski reaction.[39] In analogy to the famous Peterson olefination reaction,[5] we have designed and optimized the new Julia-Kocienski protocol that allows the selective (E) or (Z) olefin formation by a simple change in the reaction work-up procdure. Our protocol is based on the seminal work of Jørgensen et al.[40,41] that demonstrated that β-keto BT sulfones 33 can be successfully transformed into the corresponding olefins 34 in high yields and (E)-stereoselectivity. Scheme 9.
On the basis of these results, we have designed a novel type of the Julia-Kocienski reaction that allows the synthesis of the desired olefins 16 starting from the metalated sulfone 11 and acyl halides 35 (Scheme 10). In this sequence, the reunion of the two reagents (compounds 11 and 35) is carried out using previously described protocol.[42,43] The generated adduct 36 is then quenched in situ with the external source of the proton (protic solvent, e.g., MeOH) and β-keto sulfone 36 is formed. Compound 36 is present in the reaction mixture as a dynamic mixture of its keto and enol derivatives. Compound 36 is present in the reaction mixture as a dynamic mixture of its keto and enol derivatives. When an external mild reducing agent (e.g., NaBH4) is added, the keto-form of keto-36 is selectively reduced, and the nucleophilic hydride approach is directed according to the Felkin-Ahn model[44] (Scheme 11). Carbonyl reduction preferentially generates a syn derivative of β-hydroxy sulfone syn-19, and compound syn-19 is further converted via the Smiles rearrangement/β-elimination sequence of the Julia-Kocienski olefination reaction to olefin (Z)-16. However, if chelating salts such as ZnCl2 are added to the reaction mixture prior to NaBH4, the reduction proceeds through the Cram-chelate model and the anti-β-hydroxy sulfone anti-19 is formed. Consequently, compound anti-19 furnishes after the Smiles rearrangement/β-elimination sequence (E)-olefin (E)-16.
Although only preliminary scope and limitations of the transformation were established (28 examples), the method was successfully applied in the context of the (nitro)fatty acid synthesis.[39]
4. Conclusions
Since its first dissemination in 1993 the reaction sequence that is now referred to as the Julia-Kocienski reaction has become very popular late-stage connective method in natural product synthesis, because it combines highly efficient (reaction yield) and selective (predominantly (E)-selective) connective method that proceeds in one-pot protocol and under mild reaction conditions and with broad substrate and functional group tolerance. The past 30 years of reaction development have also identified key mechanistic properties that allow better control of reaction selectivity. Moreover, we have recently introduced a novel modification of the Julia-Kocienski reaction that not only increases the starting material scope since it allows for the use of previously inaccessible carboxylic acid derivatives as substrates but also allows for the selective (E) or (Z)-olefin formation. In addition, this method allows for the first time in the development of the Julia-Kocienski olefination reaction an independent formation of (E) or (Z) olefins starting from the same starting materials by simple reaction work-up protocol alternation.
Author Contributions
Conceptualization and methodology, C.D. and J.P.; writing: preparation, review and editing of the original draft, C.D. and J.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Internal Grant agency of Palacky University, grant number IGA_PrF_2024_007 and IGA_PrF_2024_028.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Markó, I.E.; Pospíšil, J. Julia, Julia–Kocienski, and Related Sulfur-Based Alkenations. In Science of Synthesis; de Meijere, Ed.; Georg Thieme Verlag: Stuttgart, 2010; Vol. 47a, pp. 105–160. [Google Scholar]
- Johnson, C.R.; Shanklin, J.R.; Kirchhoff, R.A. Olefin Synthesis by Reductive Elimination of.Beta.-Hydroxysulfoximines. Methylenation of Carbonyl Compounds. J Am Chem Soc 1973, 95, 6462–6463. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Reitz, A.B. The Wittig Olefination Reaction and Modifications Involving Phosphoryl-Stabilized Carbanions. Stereochemistry, Mechanism, and Selected Synthetic Aspects. Chem Rev 1989, 89, 863–927. [Google Scholar] [CrossRef]
- Bisceglia, J.A.; Orelli, L.R. Recent Progress in the Horner-Wadsworth-Emmons Reaction. Curr Org Chem 2015, 19, 744–775. [Google Scholar] [CrossRef]
- Staden, L.F. van; Gravestock, D.; Ager, D.J. New Developments in the Peterson Olefination Reaction. Chem Soc Rev 2002, 31, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Coombs, J.R.; Zhang, L.; Morken, J.P. Synthesis of Vinyl Boronates from Aldehydes by a Practical Boron-Wittig Reaction. Org Lett 2015, 17, 1708–1711. [Google Scholar] [CrossRef]
- Wittig, G.; Geissler, G. Zur Reaktionsweise Des Pentaphenyl-phosphors Und Einiger Derivate. Justus Liebigs Ann Chem 1953, 580, 44–57. [Google Scholar] [CrossRef]
- Wittig, G.; Schöllkopf, U. Über Triphenyl-phosphin-methylene Als Olefinbildende Reagenzien (I. Mitteil. Chem Ber 1954, 87, 1318–1330. [Google Scholar] [CrossRef]
- Chatterjee, B.; Bera, S.; Mondal, D. Julia-Kocienski Olefination: A Key Reaction for the Synthesis of Macrolides. Tetrahedron Asymm 2014, 25, 1–55. [Google Scholar] [CrossRef]
- Legnani, L.; Porta, A.; Caramella, P.; Toma, L.; Zanoni, G.; Vidari, G. Computational Mechanistic Study of the Julia-Kocieński Reaction. J Org Chem 2015, 80, 3092–3100. [Google Scholar] [CrossRef]
- Aïssa, C. Mechanistic Manifold and New Developments of the Julia-Kocienski Reaction. European J Org Chem 2009, 1831–1844. [Google Scholar] [CrossRef]
- Blakemore, P.R. The Modified Julia Olefination: Alkene Synthesis via the Condensation of Metallated Heteroarylalkylsulfones with Carbonyl Compounds. J Chem Soc Perkin 1 2002, 2, 2563–2585. [Google Scholar] [CrossRef]
- Blakemore, P.R.; Cole, W.J.; Kocieński, P.J.; Morley, A. A Stereoselective Synthesis of Trans-1,2-Disubstituted Alkenes Based on the Condensation of Aldehydes with Metallated 1-Phenyl-1 H -Tetrazol-5-Yl Sulfones. Synlett 1998, 1998, 26–28. [Google Scholar] [CrossRef]
- Robiette, R.; Pospíšil, J. On the Origin of E/Z Selectivity in the Modified Julia Olefination - Importance of the Elimination Step. European J Org Chem 2013, 836–840. [Google Scholar] [CrossRef]
- Baudin, J.B.; Hareau, G.; Julia, S.A.; Ruel, O. A Direct Synthesis of Olefins by Reaction of Carbonyl Compounds with Lithio Derivatives of 2-[Alkyl- or (2′-Alkenyl)- or Benzyl-Sulfonyl]-Benzothiazoles. Tetrahedron Lett 1991, 32, 1175–1178. [Google Scholar] [CrossRef]
- Gueyrard, D. Extension of the Modified Julia Olefination on Carboxylic Acid Derivatives: Scope and Applications. Synlett 2018, 29, 34–45. [Google Scholar] [CrossRef]
- Julia, M.; Paris, J.M. Syntheses a l’aide de Sulfones v(+)- Methode de Synthese Generale de Doubles Liaisons. Tetrahedron Lett 1973, 14, 4833–4836. [Google Scholar] [CrossRef]
- Kocienski, P.J.; Lythgoe, B.; Ruston, S. Scope and Stereochemistry of an Olefin Synthesis from β-Hydroxysulphones. J Chem Soc Perkin 1 1978, 829–834. [Google Scholar] [CrossRef]
- Keck, G.E.; Savin, K.A.; Weglarz, M.A. Use of Samarium Diiodide as an Alternative to Sodium/Mercury Amalgam in the Julia-Lythgoe Olefination. J Org Chem 1995, 60, 3194–3204. [Google Scholar] [CrossRef]
- Baudin, J.B.; Hareau, G.; Julia, S.A.; Lorne, R.; Ruel, O. Stereochemistry of Direct Olefin Formation from Carbonyl Compounds and Lithiated Heterocyclic Sulfones. Bull Soc Chim Fr 1993, 130, 856–878. [Google Scholar]
- Baudin, J.B.; Hareau, G.; Julia, S.A.; Ruel, O. Stereochemistry of the Olefin Formation from Anti and Syn Heterocyclic β-Hydroxy-Sulfones. Bull Soc Chim Fr 1993, 130, 336–357. [Google Scholar]
- Charette, A.B.; Berthelette, C.; St-Martin, D. An Expedient Approach to E, Z-Dienes Using the Julia Olefination. Tetrahedron Lett 2001, 42, 5149–5153. [Google Scholar] [CrossRef]
- Blakemore, P.R.; Cole, W.J.; Kocieński, P.J.; Morley, A. A Stereoselective Synthesis of Trans -1,2-Disubstituted Alkenes Based on the Condensation of Aldehydes with Metallated 1-Phenyl-1 H -Tetrazol-5-Yl Sulfones. Synlett 1998, 1998, 26–28. [Google Scholar] [CrossRef]
- Kocienski, P.J.; Bell, A.; Blakemore, P.R. 1- Tert -Butyl-1 H -Tetrazol-5-Yl Sulfones in the Modified Julia Olefination. Synlett 2000, 2000, 365–366. [Google Scholar] [CrossRef]
- Alonso, D.A.; Fuensanta, M.; Nájera, C.; Varea, M. 3,5-Bis(Trifluoromethyl)Phenyl Sulfones in the Direct Julia−Kocienski Olefination. J Org Chem 2005, 70, 6404–6416. [Google Scholar] [CrossRef] [PubMed]
- Mąkosza, M.; Bujok, R. Synthesis of Benzylidenecyclopropanes from γ-Halopropyl Pentachlorophenyl Sulfones Using a Julia-Kocienski Olefination. Synlett 2008, 2008, 586–588. [Google Scholar] [CrossRef]
- Pospíšil, J. Simple Protocol for Enhanced (E)-Selectivity in Julia–Kocienski Reaction. Tetrahedron Lett 2011, 52, 2348–2352. [Google Scholar] [CrossRef]
- Jana, N.; Nanda, S. Asymmetric Total Syntheses of Cochliomycin A and Zeaenol. European J Org Chem 2012, 4313–4320. [Google Scholar] [CrossRef]
- Mohapatra, D.K.; Reddy, D.S.; Mallampudi, N.A.; Yadav, J.S. Stereoselective Total Syntheses of Paecilomycins e and F through a Protecting Group Directed Diastereoselective Intermolecular Nozaki-Hiyama-Kishi (NHK) Reaction. European J Org Chem 2014, 2014, 5023–5032. [Google Scholar] [CrossRef]
- Sánchez, D.; Andreou, T.; Costa, A.M.; Meyer, K.G.; Williams, D.R.; Barasoain, I.; Díaz, J.F.; Lucena-Agell, D.; Vilarrasa, J. Total Synthesis of Amphidinolide K, a Macrolide That Stabilizes F-Actin. J Org Chem 2016, 80, 8511–8519. [Google Scholar] [CrossRef]
- Wilson, D.M.; Britton, R. Enantioselective Total Synthesis of the Marine Macrolides Salarins A and C. J Am Chem Soc 2024, 146, 8456–8463. [Google Scholar] [CrossRef]
- Billard, F.; Robiette, R.; Pospíšil, J. Julia-Kocienski Reaction-Based 1,3-Diene Synthesis: Aldehyde-Dependent (E, E/E, Z)-Selectivity. J Org Chem 2012, 77, 6358–6364. [Google Scholar] [CrossRef]
- Rehman, M.; Surendran, S.; Siddavatam, N.; Rajendar, G. The Influence of α-Coordinating Groups of Aldehydes on E/Z-Selectivity and the Use of Quaternary Ammonium Counter Ions for Enhanced E-Selectivity in the Julia–Kocienski Reaction. Org Biomol Chem 2022, 20, 329–333. [Google Scholar] [CrossRef]
- Rajendar, G.; Corey, E.J. A Systematic Study of Functionalized Oxiranes as Initiating Groups for Cationic Polycyclization Reactions. J Am Chem Soc 2015, 137, 5837–5844. [Google Scholar] [CrossRef]
- Tsubone, K.; Hashizume, K.; Fuwa, H.; Sasaki, M. Studies toward the Total Synthesis of Gambieric Acids: Convergent Synthesis of the GHIJ-Ring Fragment Having a Side Chain. Tetrahedron Lett 2011, 52, 548–551. [Google Scholar] [CrossRef]
- Tsubone, K.; Hashizume, K.; Fuwa, H.; Sasaki, M. Studies toward the Total Synthesis of Gambieric Acids, Potent Antifungal Polycyclic Ethers: Convergent Synthesis of a Fully Elaborated GHIJ-Ring Fragment. Tetrahedron 2011, 67, 6600–6615. [Google Scholar] [CrossRef]
- Rej, R.K.; Kumar, R.; Nanda, S. Asymmetric Synthesis of Cytospolides C and D through Successful Exploration of Stereoselective Julia-Kocienski Olefination and Suzuki Reaction Followed by Macrolactonization. Tetrahedron 2015, 71, 3185–3194. [Google Scholar] [CrossRef]
- Eliel, E.L.; Frye, S.V.; Hortelano, E.R.; Chen, X.; Bai, X. Asymmetric Synthesis and Cram’s (Chelate) Rule. Pure & App Chem 1991, 63, 1591–1598. [Google Scholar] [CrossRef]
- Bon, D.J. -Y. D.; Chrenko, D.; Kováč, O.; Ferugová, V.; Lasák, P.; Fuksová, M.; Zálešák, F.; Pospíšil, J. Julia-Kocienski-Like Connective C−C and C=C Bond-Forming Reaction. Adv Synth Catal 2024, 366, 480–487. [Google Scholar] [CrossRef]
- Nielsen, M.; Jacobsen, C.B.; Paixão, M.W.; Holub, N.; Jørgensen, K.A. Asymmetric Organocatalytic Formal Alkynylation and Alkenylation of α,β-Unsaturated Aldehydes. J Am Chem Soc 2009, 131, 10581–10586. [Google Scholar] [CrossRef]
- Jacobsen, C.B.; Nielsen, M.; Worgull, D.; Zweifel, T.; Fisker, E.; Jørgensen, K.A. Asymmetric Organocatalytic Monofluorovinylations. J Am Chem Soc 2011, 133, 7398–7404. [Google Scholar] [CrossRef]
- Pospíšil, J.; Sato, H. Practical Synthesis of β-Acyl and β-Alkoxycarbonyl Heterocyclic Sulfones. J Org Chem 2011, 76, 2269–2272. [Google Scholar] [CrossRef] [PubMed]
- Pospíšil, J.; Robiette, R.; Sato, H.; Debrus, K. Practical Synthesis of β-Oxo Benzo[d]Thiazolyl Sulfones: Scope and Limitations. Org Biomol Chem 2012, 10, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Bettens, T.; Alonso, M.; Geerlings, P.; De Proft, F. Mechanochemical Felkin–Anh Model: Achieving Forbidden Reaction Outcomes with Mechanical Force. J Org Chem 2023, 88, 2046–2056. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Julia-Lythgoe olefination protocol.
Scheme 2.
Julia-Kocienski olefination reaction – mechanistic overview.
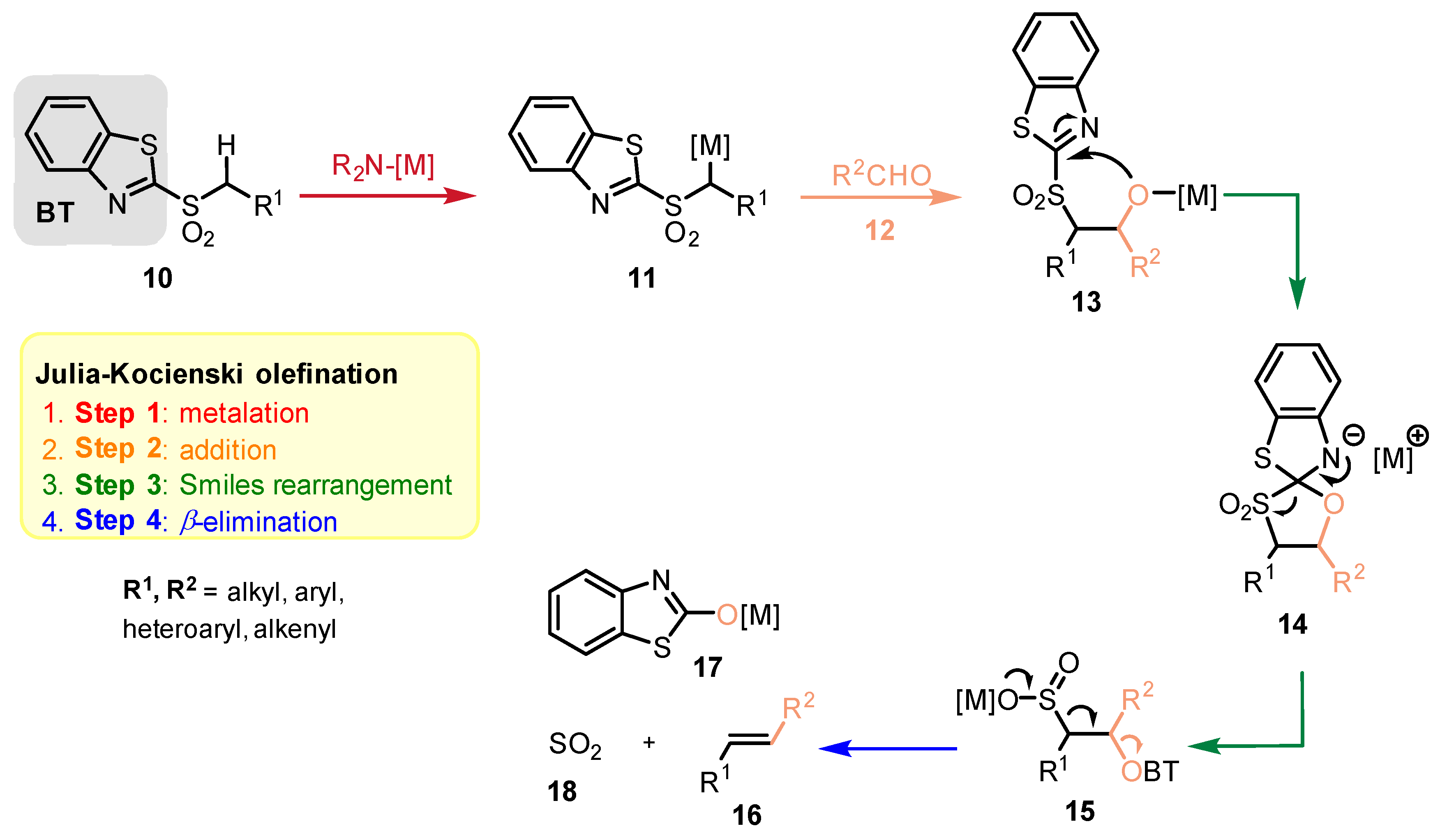
Figure 1.
Most commonly used activators in Julia-Kocienski olefination.
Scheme 3.
Detailed reaction mechanism of the Julia-Kocienski reaction.
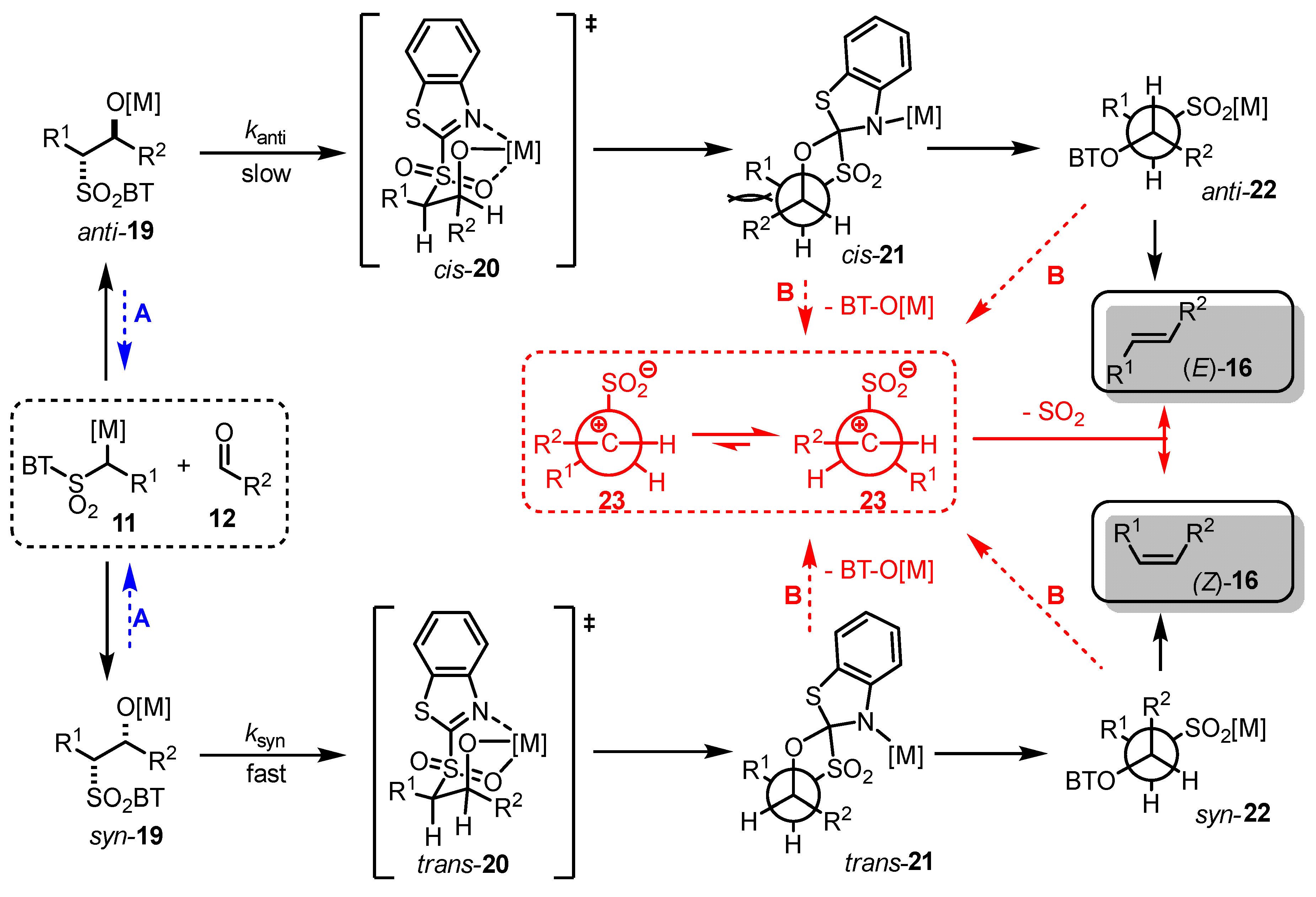
Figure 2.
Addition of the metalated sulfone 11 to aldehyde 12. Mechanistic rationale.
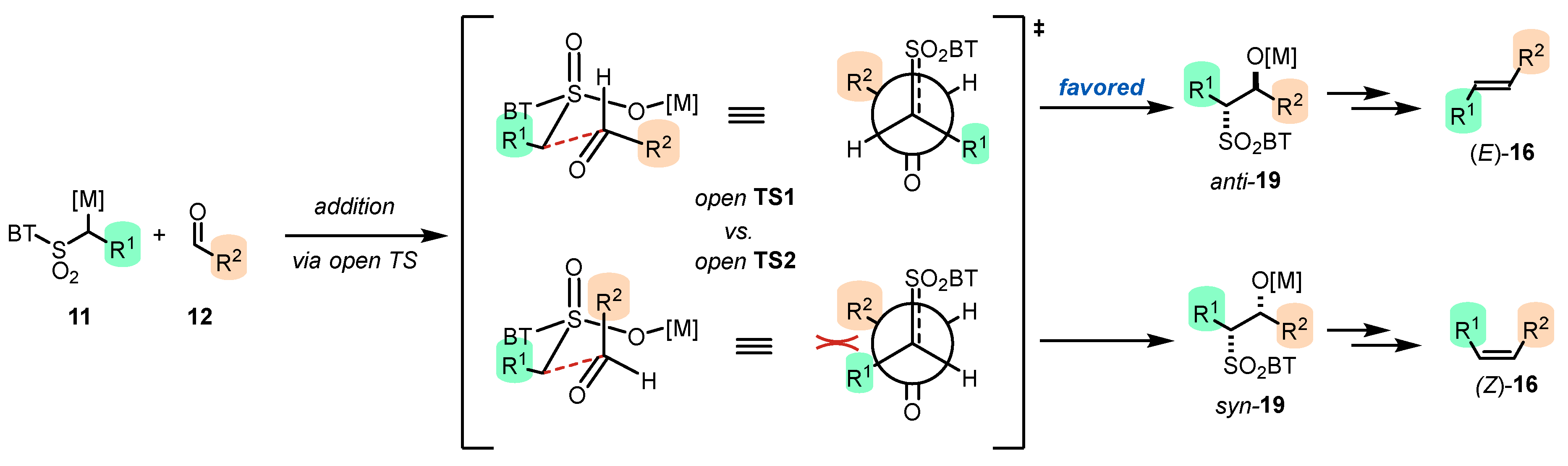
Scheme 4.
The rationale for the observed high (E)-selectivity in the Julia-Kocienski olefination of aromatic aldehydes.
Scheme 4.
The rationale for the observed high (E)-selectivity in the Julia-Kocienski olefination of aromatic aldehydes.
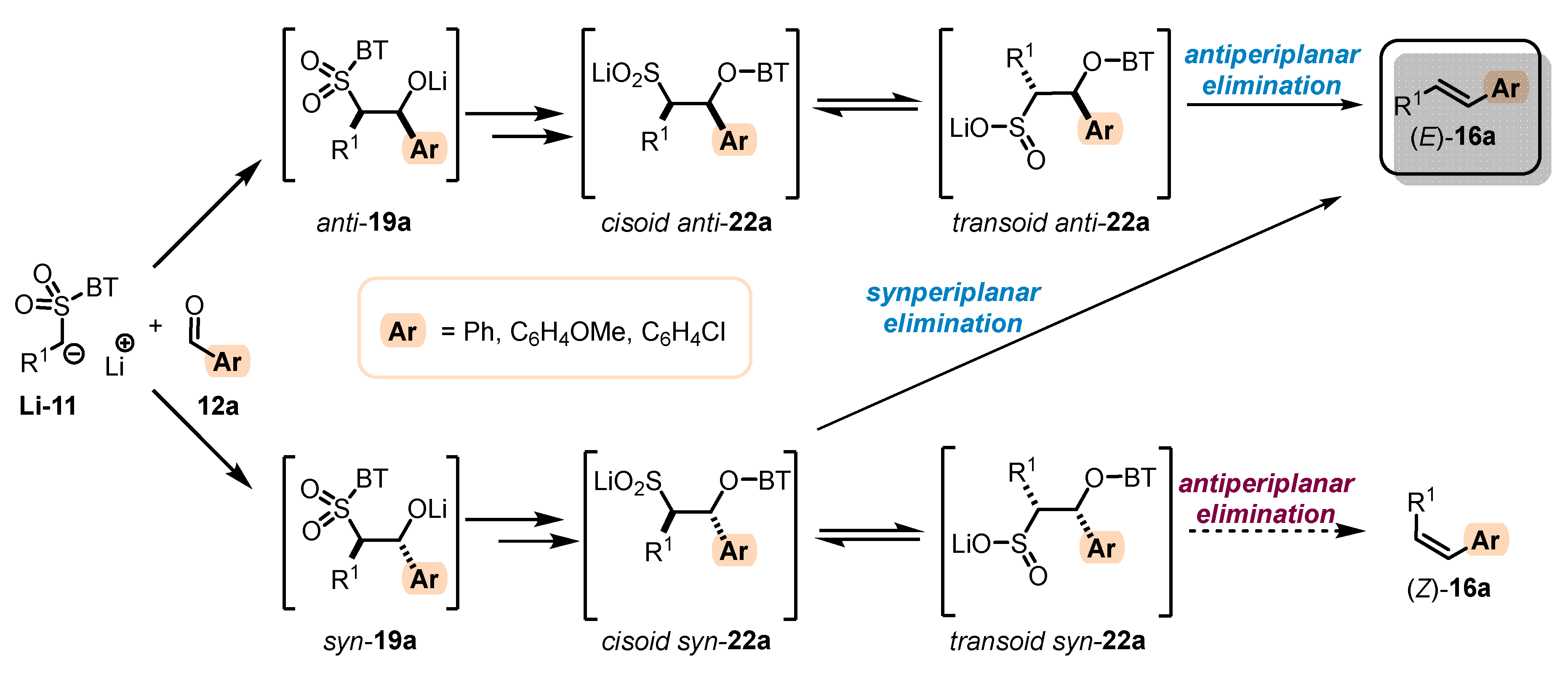
Scheme 5.
Stereoselectivity in the elimination step: a competition between the synperiplanar and antiperiplanar elimination processes.
Scheme 5.
Stereoselectivity in the elimination step: a competition between the synperiplanar and antiperiplanar elimination processes.
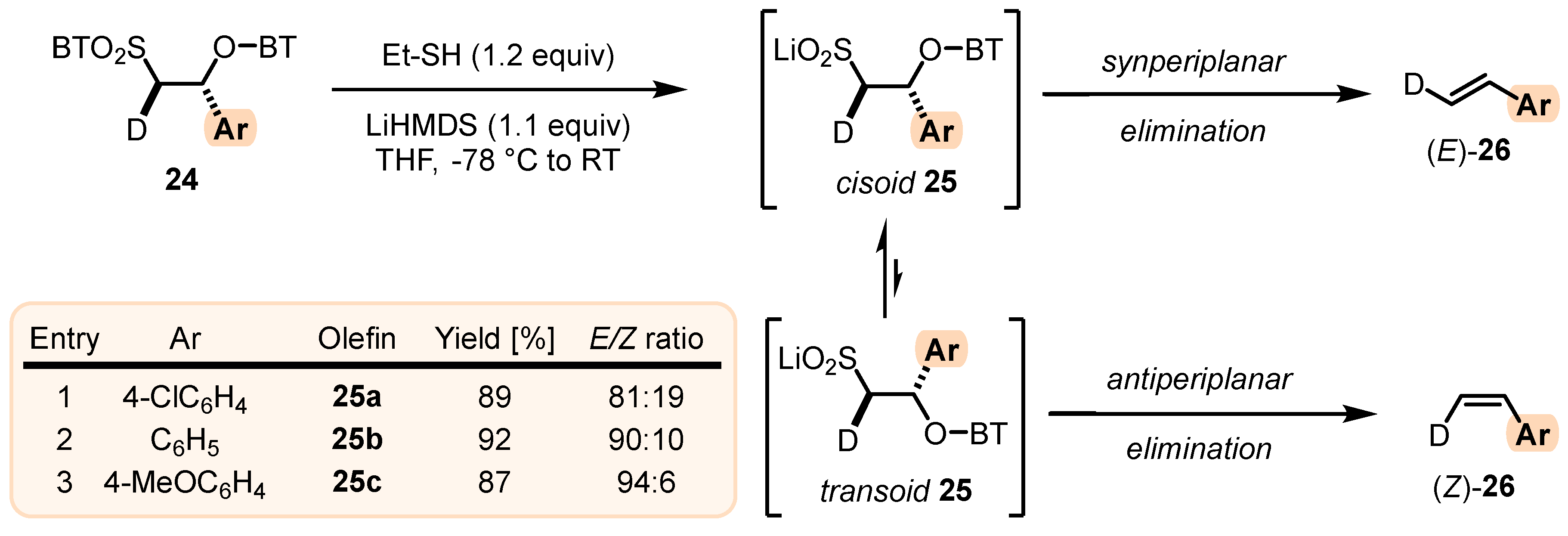
Scheme 6.
The impact of the solvent polarity on the stereochemical outcome of the Julia-Kocienki reaction.
Scheme 6.
The impact of the solvent polarity on the stereochemical outcome of the Julia-Kocienki reaction.
Scheme 7.
Self-condensation reaction that accompanies the reaction of anion 11.
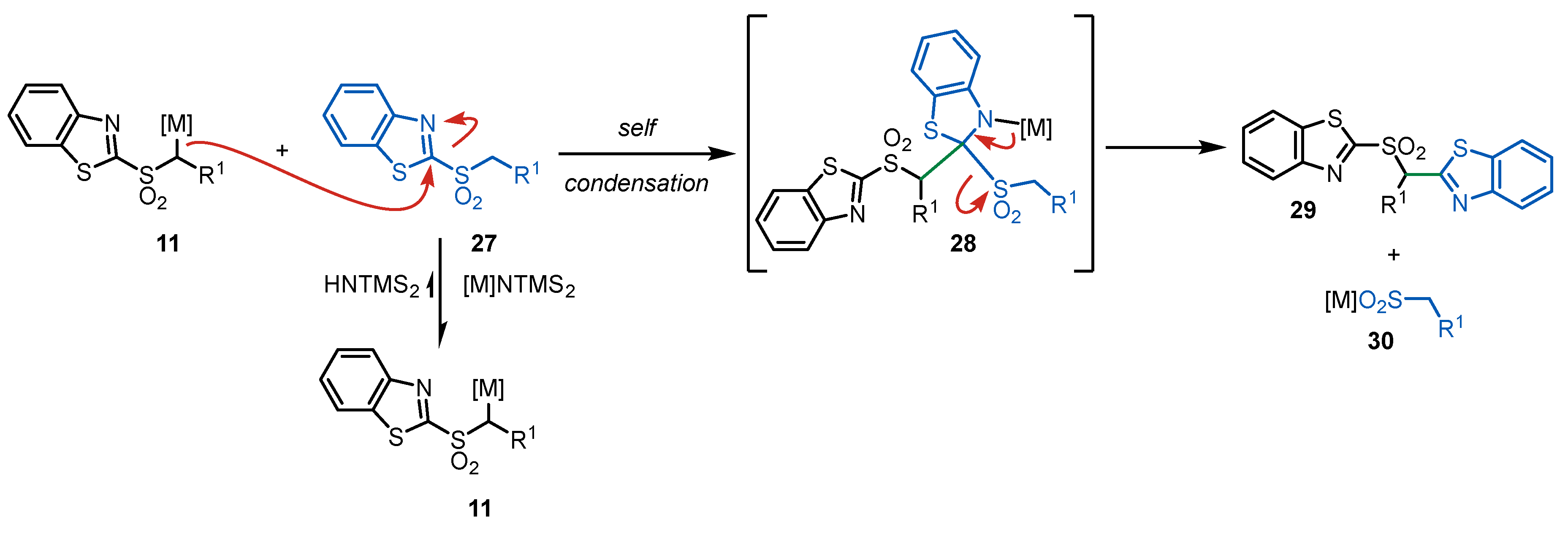
Scheme 8.
Role of crown ethers in the Julia-Kocienski reaction. High (Z)-selectivity in the case of stabilized metalated sulfones.
Scheme 8.
Role of crown ethers in the Julia-Kocienski reaction. High (Z)-selectivity in the case of stabilized metalated sulfones.
Scheme 9.
A seminal work by Jørgensen et al. [40,41] that demonstrated the possibility of stereoselective transformation of β-keto sulfones into the corresponding (E)-olefins 34.
Scheme 10.
Proposed reaction sequence for the modified Julia-Kocienski olefination reaction, where the stereoselectivity of the generated olefin is not determined in the addition step.
Scheme 10.
Proposed reaction sequence for the modified Julia-Kocienski olefination reaction, where the stereoselectivity of the generated olefin is not determined in the addition step.
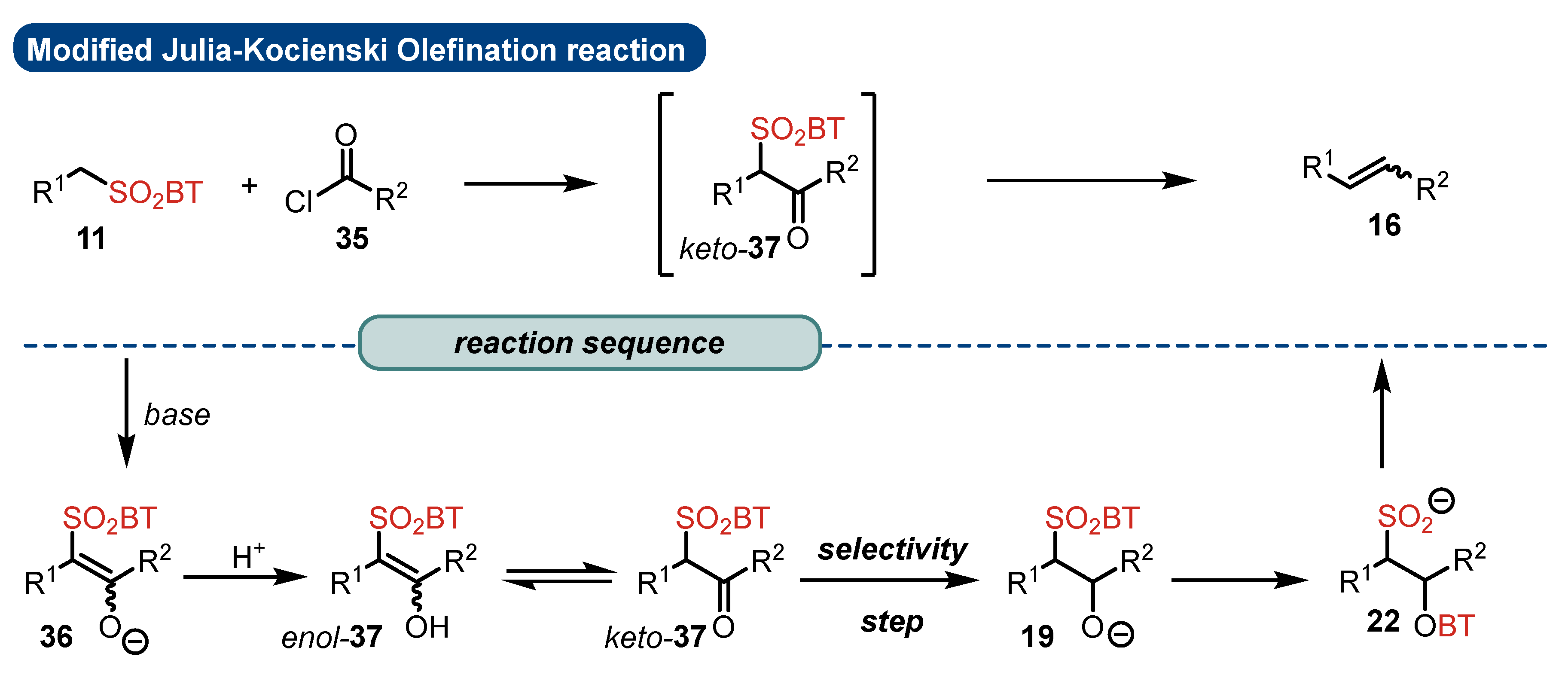
Scheme 11.
The rational design behind the stereoselective modified Julia-Kocienski olefination reaction.
Scheme 11.
The rational design behind the stereoselective modified Julia-Kocienski olefination reaction.
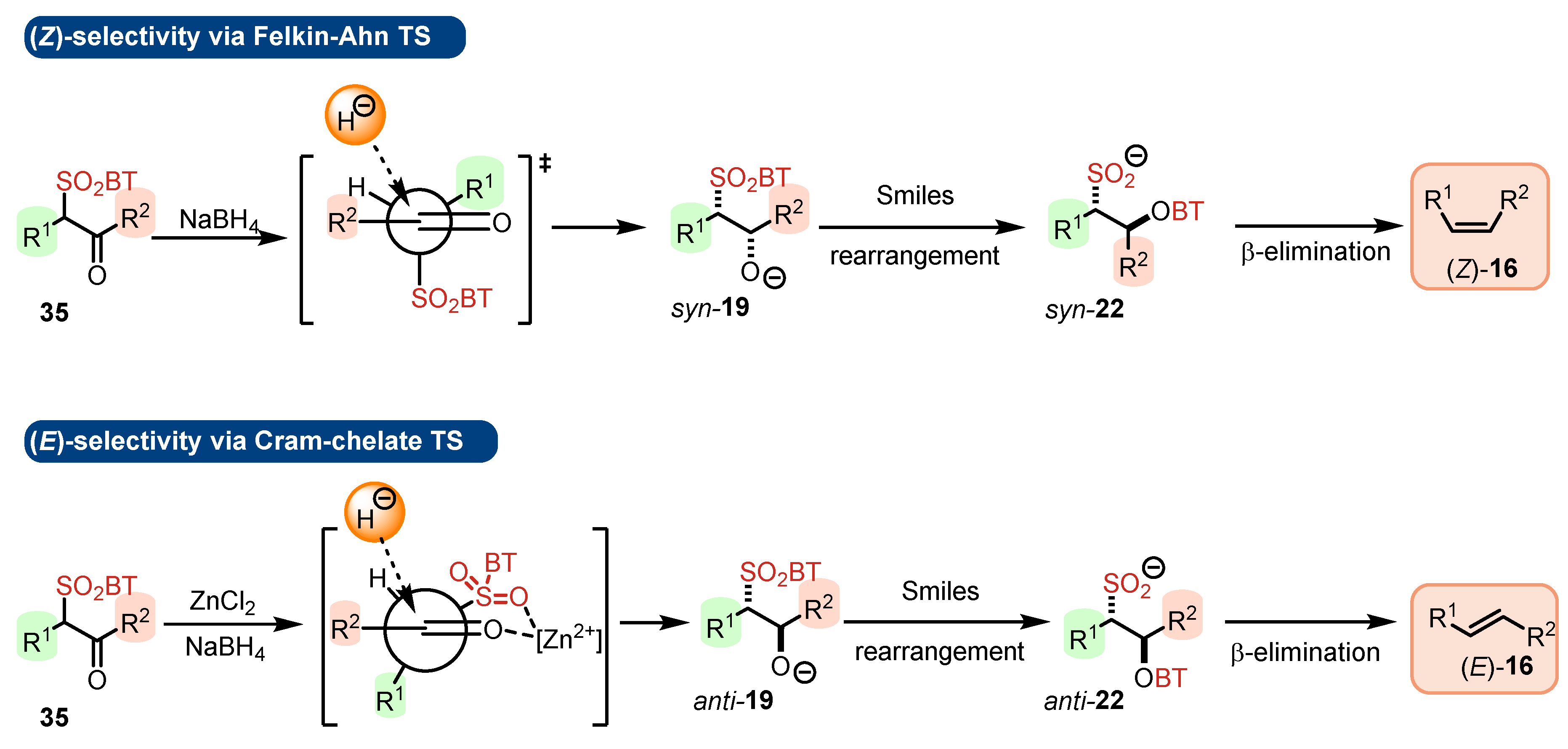

Table 1.
Common carbonyl-based olefination methods used in organic synthesis.
 | ||
| Activating unit X | Olefination Method | Litt. reference |
| PhSO2 | Julia-Lythgoe | Ref.[1] |
| ActSO2 | Julia-Kocienski | Ref.[1] |
| PhSO(NMe) | Johnson | Ref.[2] |
| R3P+ | Wittig | Ref.[3] |
| R2P(=O) | Wittig-Horner | Ref.[3] |
| (RO)2P(=O) | Horner-Wodsworth-Emmons (HWE) | Ref.[4] |
| R3Si | Peterson | Ref.[5] |
| R2B | Boron-Wittig | Ref.[6] |
Table 2.
Comparison of the Julia-Lythgoe and Julia-Kocienski olefination: General features.
| Key Features | Julia-Lythgoe | Julia-Kocienski |
| Practical Difference | Two-pot protocol | One-pot protocol |
| Origin of Stereoselectivity | Reductive Elimination Step | The addition step |
| Scope of olefin formation | ||
| Terminal |  |
 |
| 1,2-disubstituted |  |
 |
| Trisubstituted |  |
 |
| Tetrasubstituted |  |
 |
| Scope of(E)-Stereoselectivity | ||
| 1,2-disubstituted |  |
 |
| Trisubstituted |  |
 |
| Tetrasubstituted |  |
 |
| Scope of(Z)-Stereoselectivity | ||
| 1,2-disubstituted |  |
 if theTBT-activatinggroup is used; if theTBT-activatinggroup is used;
|
| Trisubstituted |  |
 |
| Tetrasubstituted |  |
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



