
Submitted:
29 April 2024
Posted:
30 April 2024
You are already at the latest version
Abstract
This study aimed to identify whether gyrate atrophy of the choroid and retina (GACR) heterozygous individuals have possible clinical manifestations and to explore the potential pathogenic mechanism. In this retrospective study, we surveyed a two-generation pedigree of an individual diagnosed with GACR. Two family members underwent ophthalmological, hematologic, and genetic tests. An arginine-restricted diet with vitamin B6 supplementation was implemented; clinical assessments were repeated every 3 months during follow-up. Relative OAT mRNA expression was determined using real-time quantitative polymerase chain reaction. The 19-year-old compound heterozygous daughter (OAT: c.1186C>T; c.748C>T), had bilateral high myopia, posterior staphyloma, chorioretinal atrophy, macular abnormalities, and elevated hematologic ornithine. The 54-year-old heterozygous mother (OAT: c.1186C>T), presented with bilateral severe myopia, asymmetric posterior staphyloma, retina and choroidal capillary layer atrophy, retinal pigment epithelium abnormalities, and mildly elevated hematologic ornithine. Compared to normal individuals, the daughter and mother had 29% and 46% relative OAT mRNA expression, respectively (P<0.001). We believe that this is the first report of a carrier of one OAT variant allele with mild phenotypes, suggesting that family members should be aware of the possible involvement of autosomal recessive conditions. Additional data suggests nonsense mediated decay-initiated mRNA degradation may cause GACR.
Keywords:
gyrate atrophy of the choroid and retina (GACR)
; OAT gene
; carrier
; allele
; autosomal recessive
1. Introduction
Gyrate atrophy of the choroid and retina (GACR; OMIM#258870) is a rare autosomal recessive metabolic disease [1], with an estimated worldwide prevalence of approximately 1 per 2,770,000 to 1 per 1,500,000 except in the Finnish population [2]. Since it was first described by Fuchs in 1896, only three patients from two families have been reported in China [3].
The OAT (OMIM#613349) gene on chromosome 10q26, which encodes ornithine-δ-aminotransferase, is responsible for the conversion of ornithine (Orn) to glutamate, and is the causative gene of GACR. Variants in OAT can result in OAT deficiency and excessive accumulation of Orn [4]. The elevated serum Orn levels are toxic to the retina and choroid, causing chorioretinal degeneration and atrophy [5]. As a severe vision-threatening disease, nyctalopia and decreased visual acuity early in childhood are common symptoms in patients with GACR. Eventually, the disease leads to severe visual impairment and functional blindness at 40–50 years of age. Reduced electroretinogram responses and macular abnormalities are also frequently observed in GACR [6,7,8,9]. In addition, extraocular manifestations in the muscle and nervous systems have also been reported, but not consistently. Currently, a protein-restricted diet focusing on lowering plasma Orn levels or supplementation with pyridoxine (vitamin B6) is experimentally administered to patients with GACR to prevent vision loss [10,11,12].
In autosomal recessive diseases, individuals with a heterozygous variant can maintain normal biological conditions and have a normal phenotype according to Mendel’s Law of Dominance [13]. However, in some autosomal recessive genetic metabolic disorders, including phenylketonuria, cystic fibrosis, and Usher syndrome, few carriers display mild symptoms [14,15,16,17]. Although this is uncommon, it indicates that necessary clinical examinations should be suggested for this population group.
To date, no reports have identified carriers with GACR symptoms. Here, we report a GACR family within two generations: a daughter with compound heterozygous variants and mother with a heterozygous variant in OAT, both of whom exhibited manifestations of GACR despite OAT heterozygosity. The potential pathogenic mechanisms of OAT nonsense variants and the relationship between variants and clinical manifestations were analysed to improve understanding of GACR pathogenesis.
2. Materials and Methods
2.1. Patients
A two-generation pedigree proband diagnosed with GACR at our clinic in August 2023 was enrolled in this retrospective study. Approval for data collection and analysis was granted by the Institutional Review Board of the Eye and ENT Hospital of Fudan University, Shanghai, China. Written informed consent, consistent with the Declaration of Helsinki, was acquired from the participants before performing all examinations and genetic testing.
2.2. Clinical Examinations, Biochemical Analysis, and Treatments
Two participants from one family underwent standard ophthalmological examinations including slit-lamp biomicroscopy (Keeler, Windsor, UK), best corrected visual acuity (BCVA, Snellen Chart), intraocular pressure (NCT, Canon TX-20, Japan), ultra-widefield fundus and fundus autofluorescence imaging (Optos UWF™, Scotland, UK), spectral domain optical coherence tomography (OCT; SD-OCT, Heidelberg Engineering, Heidelberg, Germany), optical coherence tomography angiography (VG200S, Svision Imaging, Ltd. Luoyang, China), and full-field electroretinography (RetiMINERTM, IRC, Chongqing, China). Axial length was measured using IOL Master (IOLMaster 700, Carl ZEISS, Germany). Refraction was assessed by both autorefraction and subjective refraction, and spherical equivalence was calculated as spherical dioptre (D) + ½ cylindrical dioptric power. Posterior staphyloma was classified according to Curtin’s and Kyoko’s systems [18,19]: the contour of the outermost border of the posterior staphyloma was analyzed using ultra-widefield fundus, fundus autofluorescence, and B-ultrasound. Of these, Type I corresponds to wide macular staphyloma, and Type V refers to inferior staphyloma. Peripheral blood metabolic tests were performed using a GCMS-QP2010 (Shimadzu, Kyoto, Japan) and an ACQUITY TQ-D instrument (Waters Corp., USA). Therapy with an arginine-restricted diet and vitamin B6 supplementation was conducted as previously described, and clinical examinations were repeated every 3 months during follow-up from August 2023 [20].
2.3. Genetic Testing and Pathogenicity Assessment
Exome sequencing, genome sequencing, and Sanger sequencing
Peripheral blood samples from both participants were collected in EDTA-anticoagulant tubes and then outsourced to Shanghai WeHealth Biomedical Technology Co., Ltd. Genomic DNA was subjected to exome sequencing, comprising more than 23,000 genes, as previously reported [21]. All reads were aligned to the National Centre for Biotechnology Information (https://www.ncbi.nlm.nih.gov/) human reference genome GRCh38/hg19). The amplified genomic sequences were compared with the OAT reference sequence NM_000274.4.
Genome sequencing was conducted by Shanghai WeHealth Biomedical Technology Co., Ltd., as previously reported [22]. The TWIST Library Preparation EF Kit 2.0 (104207) was used for DNA fragmentation and genome library construction. The detected variants were further validated by Sanger sequencing as previously reported [21]. Polymerase chain reaction (PCR) primers were designed using Primer3Plus (http://www.primer3plus.com/cgi-bin/dev/primer3plus.cgi).
2.4. In Silico Analysis and Protein Structure Modelling
We reviewed publications in the 1000 Genome database, ExAC, gnomAD, PubMed (https://pubmed.ncbi.nlm.nih.gov/), and Exome Sequencing Project (https://evs.gs.washington.edu/EVS/) to search for population frequencies and determine if the variants had been previously reported. The pathogenicity of the identified OAT variants was predicted using various in silico prediction tools, including LRT, FATHMM-MKL, ClinPred, CADD, DANN, GERP, Mutation Taster, and SPIDEX, as previously described [21]. All variants were subsequently evaluated according to the American College of Medical Genetics and Genomics (ACMG) guidelines [23].
Three-dimensional structural modelling of the OAT protein was performed using PyMOL (https://pymol.org/2/) based on a known structure (PDB ID: 7T9Z). To analyse variant conservation, the amino acid sequence of OAT in different species was obtained from UniProt (https://www.uniprot.org/), and multiple sequence alignment analysis of the variant sites in several species was performed using DNAMAN software (https://www.lynnon.com/download/).
2.5. OAT mRNA Analysis
Whether nonsense variants impact gene transcription was confirmed by detecting the relative gene expression levels of OAT cDNA utilizing real-time fluorescence quantitative PCR (RT-qPCR). Total RNA was extracted from patients and healthy controls using a RNA extraction kit (TIANGEN, Beijing, China). Normal control samples were obtained from healthy volunteers without pathogenic OAT variants. RNA was then reverse transcribed into cDNA using the Hifair®-II-1st-Strand cDNA-Synthesis Kit (Yeasen, Beijing, China). Targeted RNA was amplified by qPCR using the SYBR Green PCR master mix (Novoprotein, Shanghai, China) and then analysed on an RT-qPCR System (Roche, Shanghai, China). The primers used are listed in Table S1. Relative OAT mRNA expression was calculated and normalized against an internal reference gene GAPDH based on the 2-ΔΔCt method [24]. All experiments were performed in triplicate.
2.6. Statistical Analyses
Statistical analysis of relative OAT mRNA expression was performed using one-way ANOVA, followed by Bonferroni's multiple-comparison correction, using GraphPad Prism 9 (Boston, MA, USA) (https://www.graphpad.com/). P<0.05 was considered statistically significant.
3. Results
3.1. Ocular Characteristics
The proband (F-II1), a 19-year-old daughter, visited our clinic complaining of significant bilateral vision loss. In addition to a history of high myopia and regular visits to optometry doctors since childhood, the patient had no known systemic or ocular disorders. BCVA was 20/66 in both eyes, with a spherical equivalence refraction error of −17.00 D and −18.25 D in the right and left eye, respectively. Ophthalmological examination revealed punctate posterior subcapsular cataracts in the right eye (Figure S1). Both eyes presented with wide, macular posterior staphyloma and were classified as Type 1 bilaterally. Multiple, sharply demarcated, scallop-shaped chorioretinal atrophy was found in the mid-peripheral and peripheral regions of the fundus in both eyes (Figure 1A-B). Enhanced-depth OCT showed increased central macular thickness with multiple intraretinal cystic spaces and bilateral chorioretinal atrophy (Figure 1C1-C2). Extensive loss of the outer layers and significant thinning of the retina are indicated by yellow arrows (Figure 1C2). OCT imaging of the left eye revealed hyperreflective deposits below the retinal pigment epithelium (RPE) (Figure 1C2b, white arrows), whereas the corresponding region on the fundus image appeared relatively normal. Wide optical coherence tomography angiography images showed significant bilateral vascular loss (Figure 1D1-D2). An electroretinography examination revealed a nearly extinguished bilateral rod and cone response (Figure 1E).
As the mother (F-I2) presented with unusual unilateral high myopia and fluttering dark shadows in her right eye, an ophthalmological examination was also conducted. She had no history of systemic or ocular disorders other than myopia. BCVA was 20/28 in both eyes with a spherical equivalence refraction error of −9.00 D and −2.75 D in the right and left eye, respectively. Her right eye presented with a wide, macular posterior staphyloma (Type 1) (Figure 2A1, Dc’, yellow dotted square), and the axial length was 26.45 mm. Furthermore, focal circular areas of retinal atrophy were super-temporally observed in her right eye, and fundus autofluorescence imaging revealed the corresponding area was hypoautofluorescent surrounded by hyperautofluorescent areas (Figure 2A). An OCT B-scan demonstrated atrophy of the retina and choroidal capillary layers in the parapapillary and supratemporal retina (Figure 2Dc-f,yellow arrow). Pigment epithelial detachment (PED) and an area of disorganized RPE layer were detected at the edge of one cilioretinal atrophy lesion, close to the superior vascular arch (Figure 2Df, white arrow). However, the left eye presented with mild myopia and an axial length of 24.52-mm. An inferior posterior staphyloma (Type 5) (Figure 2B1, Ca-b, yellow dotted square) with regional retinoschisis (Figure 2Cb) was found on fundus imaging, B-scan, and OCT examination. More detailed ocular examination results are displayed in Table 1. Unfortunately, the daughter’s father was not available for ocular examination or genetic testing.
3.2. Hematologic Biochemical Findings
Based on clinical findings, serum amino acid levels were tested for both members (Table 1). Consistent with our expectations, the daughter’s (F-II2) Orn levels showed a marked elevation (257.92 µM, reference range: 10–100 µM) and the creatine level was slightly decreased to 94.83 µM (reference range: 95–1000 µM). Surprisingly, the mother exhibited a slight increase in Orn of 102.08 µM.
3.3. Genetic Findings
Exome sequencing and Sanger sequencing identified a compound heterozygous OAT in F-II1 (M1: c.1186C>T, p.R396*; M2: c.748C>T, p.R250*) and a heterozygous OAT in F-I2 (M1: c.1186C>T, p.R396*) (Figure 3A, C). Because exome sequencing may miss deep intron variants or structural variants, which can influence the phenotype, genome sequencing was performed on F-I2. No other pathogenic OAT variants were identified. The pathogenicity analysis of the identified variants is shown in Figure 3B. The amino acid residues of the identified variants were highly conserved among the species (Figure 3D). The three-dimensional structures of the wild-type and variant OAT proteins are shown in Figure 4A; both variant proteins were predicted to be shorter than the wild-type protein (493 amino acids). The catalytic sites for substrate specificities in the OAT protein are shown in Figure 4B in stick mode, which shows that the nonsense variant of M2 at position 250 was deleterious owing to the loss of the catalytic sites due to early termination of protein translation.
3.4. OAT mRNA Detection
Figure 4C shows that the relative OAT mRNA expression levels in F-II1 and F-I2 were approximately only 29% and 46%, respectively, of those in normal individuals. Statistically significant differences were found between F-II1 and normal controls (n=3, P<0.001), F-I2 and normal controls (n=3, P<0.001), and F-II1 and F-I2 (n=3, P=0.003).
3.5. Treatment and Follow-Up
An arginine-restricted diet and vitamin B6 supplementation were administered to both individuals. Serum Orn levels of F-II1 and F-I2 decreased to 209.68 µM and 92.60 µM, respectively, after a 3-month follow-up, while fundus lesions, including the macular structure of F-II1, remained bilaterally stable.
4. Discussion
In this study, we identified a Chinese family in which the proband was diagnosed with GACR; two previously reported OAT variants (c.1186C>T and c.748C>T) were found. Interestingly, the girl’s mother, who only had one OAT variant (c.1186C>T) following genome sequencing analysis, displayed some ocular manifestations (severe myopia, atrophy of retina and choroidal capillary layer, RPE abnormalities of the right eye, and bilateral posterior staphyloma) and a slightly elevated serum Orn level, indicating that she might be an “affected” carrier. Additionally, the relative mRNA expression levels of OAT were decreased in both the mother and daughter. Accordingly, this report is the first to report a carrier presenting with a mild phenotype of GACR.
GACR is a rare progressive autosomal recessive disorder. With an error in Orn metabolism attributed to OAT deficiency, ocular manifestations mainly manifest as the aggravation of gyrate chorioretinal atrophy with age, which significantly threatens vision and frequently leads to blindness. In this study, the proband (F-II1) manifested typical multiple sharply demarcated, scallop-shaped chorioretinal atrophy, high myopia, and bilateral posterior staphyloma. In particular, hyperreflective deposits below the RPE layer occurred in areas where the retina and choroid had not yet atrophied, consistent with previous theories showing that Orn cytotoxicity in ornithine-δ-aminotransferase-deficient human RPE cells could be the main pathogenic mechanism of GACR in an in vitro model [25,26]. The participant’s accumulation of serum Orn levels was higher than the reference value. Previous studies have demonstrated the effectiveness of an arginine-restricted diet and vitamin B6 supplementation for GACR [20]. Similarly, after the definitive diagnosis and initiation of treatment, serum Orn levels began to decrease, and the macular abnormalities were stable. Notably, the daughter had a history of long-term follow-up at an optometry clinic. However, her fundus manifestations were ignored for a long time until macular abnormalities appeared. A thorough examination and follow-up of the fundus, including the peripheral fundus, is suggested for children with uncommon rapid myopia progression and poor BCVA. Fundus diseases include not only GACR, but also familial exudative vitreoretinopathy [27], vitreoretinal degeneration diseases (such as Stickler Syndrome [28] and Wagner syndrome [29]), and some other inherited choroidal retinal diseases, which should be considered in these children. The diagnosis of GACR should be confirmed, not only by thorough multimodal fundus imaging, but also by the presence of hyperornithinaemia and OAT variants. In summary, accurate identification of genetic causes, timely diagnosis, and early treatment may protect the vision of patients with GACR.
The mother was heterozygous for OAT, as confirmed using genome sequencing. As the patient complained of ocular discomfort, detailed ophthalmological examinations were performed, which revealed unilateral severe myopia and bilateral posterior staphyloma, as well as atrophy of the retina and choroidal capillary layer and PED with an area of disorganized RPE layer in her right eye, which led us to suspect a mild phenotype of GACR. Subsequent serum metabolic tests also showed mild elevation of Orn. As mentioned, RPE was the most vulnerable in GACR; moreover, we also detected PED and an area of disorganized RPE layer adjacent to the boundary of one cilioretinal atrophy lesion. Further, the patient’s right eye showed high myopia, which is a common manifestation of GACR. Her left eye had an axial length of only 24.52 mm, but also showed posterior staphyloma. Previous studies have reported that damaged RPE cells may be associated with posterior staphyloma formation. On one hand, damaged RPE cells would allow passive spreading of the remaining cells, stretching the retina in the posterior pole and compressing the choroid between the expanding Bruch’s membrane and the sclera [30,31]; on the other hand, reduction of oxygen demand by RPE cells and photoreceptors might lead to vascular constriction and thinning at the level of the choroid, stimulating posterior staphyloma development [32]. These might be the reasons for the phenotypes found in the proband’s mother.
This phenomenon of “affected” carriers in autosomal recessive conditions is not rare, especially in inherited metabolic abnormalities diseases. For instance, carriers for phenylketonuria only have 7.3–10% of enzymatic activity compared with the normal controls not carrying any pathogenic variants of PAH genes (100% enzymatic activity) in liver biopsy studies [14,15]. Further, for cystic fibrosis, some studies indicate that carriers are at increased risk for some conditions associated with cystic fibrosis, including pancreatitis, male infertility, bronchiectasis, and cholelithiasis [16,33]. Contrary to the carriers who are frequently described as “unaffected,” the concept of allele-dose responses indicate that they might actually present with an intermediate phenotype or be “moderately affected” [34]. However, there are no prior reports of carriers presenting with a clinical phenotype of GACR. We hypothesize that this might be due to the lack of serious complaints of vision loss, thus these carriers may neglect undergoing detailed ophthalmologic examinations. Therefore, we recommend that individuals heterozygous for recessive genetic disorders should be evaluated and screened in future research to determine possible clinical implications that could reduce the risk of serious complications and prevent damage from genetic diseases.
To further investigate the association between the mother’s phenotype and the OAT variant, the relative mRNA expression levels of OAT in both the mother and daughter were analysed. We previously demonstrated that triggering cystic fibrosis transmembrane conductance regulator (CFTR) mRNA degradation via nonsense mediated decay (NMD) may be a pathogenic cause of GACR. NMD is a transcriptional regulatory mechanism that detects mRNA with premature termination codons and triggers truncated protein degradation to prevent potentially harmful effects [35]. Compared with that in the normal control, the relative OAT mRNA expression levels of both mother and daughter were decreased, indicating that transcription of OAT was impaired. Differences between the mother and daughter may suggest an association with phenotypic severity. Given the difficulty of detecting ornithine-δ-aminotransferase activity within target tissues, including the eyes, muscles, and nerves in patients with GACR, relative OAT mRNA expression of peripheral blood may not only identify the pathogenicity, but also indicate the processes and prognosis, although this will require further study and multivariate analyses.
There are some limitations to the present study. Owing to the rarity of the disease, we did not observe enough cases of carriers with GACR, and it would be preferable to measure OAT enzyme activity. Additionally, information on this family was still partly missing because the father was not available for examination.
5. Conclusions
In conclusion, to the best of our knowledge, this study reports for the first time, a carrier with one variant allele of OAT who exhibited mild GACR phenotypes; this suggests that family members should be aware of the possible involvement of heterozygous variants in autosomal recessive conditions. Furthermore, triggering of CFTR mRNA degradation by NMD may be the causative mechanism of GACR. The present study deepens understanding of this disease, while emphasizing the importance of fundus screening for high myopia in children.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Anterior segment photographs of F-II1 (right eye); Table S1: Sequences of qRT-PCR primers.
Author Contributions
Conceptualization, Yuqiao Ju, Qing Chang, Yuan Zong and Xin Huang; Funding acquisition, Fengjuan Gao, Yuan Zong and Xin Huang; Investigation, Yuqiao Ju and Xiao Li; Project administration, Yuan Zong and Xin Huang; Resources, Yuqiao Ju and Xiao Li; Validation, Yuqiao Ju and Xiao Li; Writing – original draft, Yuqiao Ju and Xiao Li; Writing – review & editing, Fengjuan Gao, Yuan Zong and Xin Huang. All authors will be informed about each step of manuscript processing including submission, revision, revision reminder, etc. via emails from our system or assigned Assistant Editor.
Funding
This study was supported by the National Natural Science Foundation of China (grant nos. 82070975, 82201204, and 82101149) and Shanghai Science and Technology Commission (grant no. 21ZR1411400).
Institutional Review Board Statement
Approval for data collection and analysis was granted by the Institutional Review Board of the Eye and ENT Hospital of Fudan University, Shanghai, China on November 26, 2020. The ethical approval number was [2020]2020119.
Informed Consent Statement
Written informed consent, consistent with the Declaration of Helsinki, was acquired from the participants before all examinations and genetic testing.
Data Availability Statement
Data are available from the authors upon reasonable request.
Acknowledgments
The authors would like to thank all the members of this project as well as all the volunteers who participated in this project.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Inborn errors of metabolism: Gyrate atrophy. Adv Exp Med Biol 2018, 1085, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Montioli, R.; Bellezza, I.; Desbats, M.A.; Borri Voltattorni, C.; Salviati, L.; Cellini, B. Deficit of human ornithine aminotransferase in gyrate atrophy: Molecular, cellular, and clinical aspects. Biochim Biophys Acta Proteins Proteom 2021, 1869, 140555. [Google Scholar] [CrossRef]
- Ramesh, V.; Benoit, L.A.; Crawford, P.; Harvey, P.T.; Shows, T.B.; Shih, V.E.; Gusella, J.F. The ornithine aminotransferase (OAT) locus: Analysis of RFLPs in gyrate atrophy. Am J Hum Genet 1988, 42, 365–372. [Google Scholar] [PubMed]
- Kaiser-Kupfer, M.I.; Valle, D.; Del Valle, L.A. A specific enzyme defect in gyrate atrophy. Am J Ophthalmol 1978, 85, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Hayasaka, S.; Kodama, T.; Ohira, A. Retinal risks of high-dose ornithine supplements: A review. Br J Nutr 2011, 106, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Peltola, K.E.; Näntö-Salonen, K.; Heinonen, O.J.; Jääskeläinen, S.; Heinänen, K.; Simell, O.; Nikoskelainen, E. Ophthalmologic heterogeneity in subjects with gyrate atrophy of choroid and retina harboring the L402P mutation of ornithine aminotransferase. Ophthalmology 2001, 108, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Gekka, T.; Hayashi, T.; Ida, H.; Ohashi, T.; Eto, Y.; Tsuneoka, H. OAT mutations and clinical features in two Japanese brothers with gyrate atrophy of the choroid and retina. Doc Ophthalmol 2014, 128, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.M.; Elnahry, A.G.; Tripathy, K.; Foster, R.E.; Mehanna, C.J.; Vishal, R.; Çavdarlı, C.; Arrigo, A.; Parodi, M.B. Analysis of optical coherence angiography in cystoid macular oedema associated with gyrate atrophy. Eye (Lond) 2021, 35, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, K.; Chawla, R.; Sharma, Y.R.; Gogia, V. Ultrawide field fluorescein angiogram in a family with gyrate atrophy and foveoschisis. Oman J Ophthalmol 2016, 9, 104–106. [Google Scholar] [CrossRef]
- Kaiser-Kupfer, M.I.; de Monasterio, F.; Valle, D.; Walser, M.; Brusilow, S. Visual results of a long-term trial of a low-arginine diet in gyrate atrophy of choroid and retina. Ophthalmology 1981, 88, 307–310. [Google Scholar] [CrossRef]
- McInnes, R.R.; Arshinoff, S.A.; Bell, L.; McCulloch, C. Treatment of gyrate atrophy of the choroid and retina with low arginine diet. Trans Am Ophthalmol Soc 1980, 78, 226–242. [Google Scholar] [PubMed]
- Valle, D.; Walser, M.; Brusilow, S.W.; Kaiser-Kupfer, M. Gyrate atrophy of the choroid and retina: Amino acid metabolism and correction of hyperornithinemia with an arginine-deficient diet. J Clin Invest 1980, 65, 371–378. [Google Scholar] [CrossRef]
- Hames, A.; Khan, S.; Gilliland, C.; Goldman, L.; Lo, H.W.; Magda, K.; Keathley, J. Carriers of autosomal recessive conditions: Are they really “unaffected? ” J Med Genet 2023, 61, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hillert, A.; Anikster, Y.; Belanger-Quintana, A.; Burlina, A.; Burton, B.K.; Carducci, C.; Chiesa, A.E.; Christodoulou, J.; Đorđević, M.; Desviat, L.R.; et al. The genetic landscape and epidemiology of phenylketonuria. Am J Hum Genet 2020, 107, 234–250. [Google Scholar] [CrossRef] [PubMed]
- van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat Rev Dis Primers 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Polgreen, P.M.; Comellas, A.P. Clinical phenotypes of cystic fibrosis carriers. Annu Rev Med 2022, 73, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Wagenaar, M.; ter Rahe, B.; van Aarem, A.; Huygen, P.; Admiraal, R.; Bleeker-Wagemakers, E.; Pinckers, A.; Kimberling, W.; Cremers, C. Clinical findings in obligate carriers of type I Usher syndrome. Am J Med Genet 1995, 59, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Matsui, K. Proposed classification of posterior staphylomas based on analyses of eye shape by three-dimensional magnetic resonance imaging and wide-field fundus imaging. Ophthalmology 2014, 121, 1798–1809. [Google Scholar] [CrossRef] [PubMed]
- Curtin, B.J. The posterior staphyloma of pathologic myopia. Trans Am Ophthalmol Soc 1977, 75, 67–86. [Google Scholar]
- Bergen, A.A.; Buijs, M.J.; Ten Asbroek, A.L.; Balfoort, B.M.; Boon, C.J.; Dutch GACR “Bird’s Eye View” Consortium; Brands, M.M.; Wanders, R.J.; van Karnebeek, C.D.; Houtkooper, R.H. Vision on gyrate atrophy: Why treat the eye? EMBO Mol Med 2024, 16, 4–7. [Google Scholar] [CrossRef]
- Ju, Y.; Zhang, L.; Gao, F.; Zong, Y.; Chen, T.; Ruan, L.; Chang, Q.; Zhang, T.; Huang, X. Genetic characteristics and clinical manifestations of foveal hypoplasia in familial exudative vitreoretinopathy. Am J Ophthalmol 2024, 262, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Liao, K.; Zhao, Y.; Long, Z.; Zhu, S.; Wu, J.; Xiao, M.; Zhou, J.; Zhang, S.; Li, L.; et al. A novel system for the detection of spontaneous abortion-causing aneuploidy and its erroneous chromosome origins through the combination of low-pass copy number variation sequencing and NGS-based STR tests. J Clin Med 2023, 12, 1809. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ohnaka, M.; Okuda-Ashitaka, E.; Kaneko, S.; Ando, A.; Maeda, M.; Furuta, K.; Suzuki, M.; Takahashi, K.; Ito, S. Induction of arginase II mRNA by nitric oxide using an in vitro model of gyrate atrophy of choroid and retina. Invest Ophthalmol Vis Sci 2011, 52, 1493–1500. [Google Scholar] [CrossRef]
- Sergouniotis, P.I.; Davidson, A.E.; Lenassi, E.; Devery, S.R.; Moore, A.T.; Webster, A.R. Retinal structure, function, and molecular pathologic features in gyrate atrophy. Ophthalmology 2012, 119, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Tauqeer, Z.; Yonekawa, Y. Familial exudative vitreoretinopathy: Pathophysiology, diagnosis, and management. Asia Pac J Ophthalmol (Phila) 2018, 7, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Snead, M.P.; Yates, J.R. Clinical and molecular genetics of Stickler syndrome. J Med Genet 1999, 36, 353–359. [Google Scholar] [CrossRef]
- Araújo, J.R.; Tavares-Ferreira, J.; Estrela-Silva, S.; Rocha, P.; Brandão, E.; Faria, P.A.; Falcão-Reis, F.; Rocha-Sousa, A. Wagner syndrome: Anatomic, functional and genetic characterization of a Portuguese family. Graefes Arch Clin Exp Ophthalmol 2018, 256, 163–171. [Google Scholar] [CrossRef]
- Ohno-Matsui, K.; Jonas, J.B. Posterior staphyloma in pathologic myopia. Prog Retin Eye Res 2019, 70, 99–109. [Google Scholar] [CrossRef]
- Komori, S.; Ueno, S.; Ito, Y.; Sayo, A.; Meinert, M.; Kominami, T.; Inooka, D.; Kitagawa, M.; Nishida, K.; Takahashi, K.; et al. Steeper macular curvature in eyes with non-highly myopic retinitis pigmentosa. Invest Ophthalmol Vis Sci 2019, 60, 3135–3141. [Google Scholar] [CrossRef] [PubMed]
- El Matri, L.; Falfoul, Y.; El Matri, K.; El Euch, I.; Ghali, H.; Habibi, I.; Hassairi, A.; Chaker, N.; Schorderet, D.; Chebil, A. Posterior staphylomas in non-highly myopic eyes with retinitis pigmentosa. Int Ophthalmol 2020, 40, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.C.; Comellas, A.P.; Hornick, D.B.; Stoltz, D.A.; Cavanaugh, J.E.; Gerke, A.K.; Welsh, M.J.; Zabner, J.; Polgreen, P.M. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc Natl Acad Sci U S A 2020, 117, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Keathley, J.; Garneau, V.; Zavala-Mora, D.; Heister, R.R.; Gauthier, E.; Morin-Bernier, J.; Green, R.; Vohl, M.C. A systematic review and recommendations around frameworks for evaluating scientific validity in nutritional genomics. Front Nutr 2021, 8, 789215. [Google Scholar] [CrossRef]
- Karousis, E.D.; Mühlemann, O. Nonsense-mediated mRNA decay begins where translation ends. Cold Spring Harb Perspect Biol 2019, 11, a032862. [Google Scholar] [CrossRef]
Figure 1.
Ocular characteristics of F-II1. A-B: Ultra-widefield fundus images, including colour fundus and autofluorescence imaging. C: Optical coherence tomography (OCT) reveals macular abnormalities (C1a and C2a) as well as retinal and choriocapillaris atrophy (C1b and C2b) in both eyes. An OCT B-scan of the left eye reveals deposits below the retinal pigment epithelium cells, and choroidal atrophy (C2b,). D: Wide angio-OCT (OCTA) images of F-II1 in the right (D1) and left eye (D2). E: Electroretinography of F-II1 cells and normal controls. Yellow arrows indicate retinal and choriocapillaris atrophy. White arrows indicate deposits below the retinal pigment epithelium cells.
Figure 1.
Ocular characteristics of F-II1. A-B: Ultra-widefield fundus images, including colour fundus and autofluorescence imaging. C: Optical coherence tomography (OCT) reveals macular abnormalities (C1a and C2a) as well as retinal and choriocapillaris atrophy (C1b and C2b) in both eyes. An OCT B-scan of the left eye reveals deposits below the retinal pigment epithelium cells, and choroidal atrophy (C2b,). D: Wide angio-OCT (OCTA) images of F-II1 in the right (D1) and left eye (D2). E: Electroretinography of F-II1 cells and normal controls. Yellow arrows indicate retinal and choriocapillaris atrophy. White arrows indicate deposits below the retinal pigment epithelium cells.
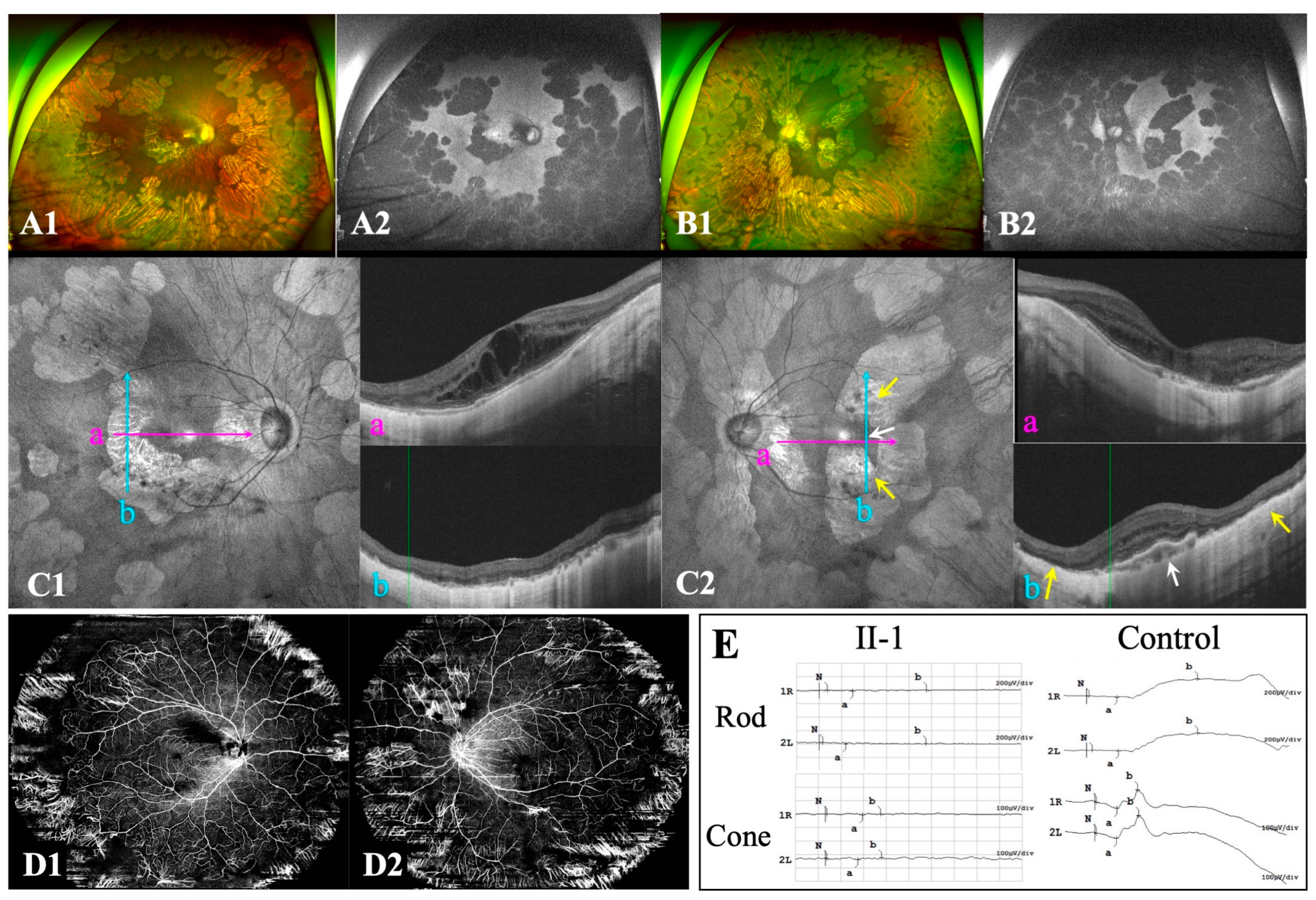
Figure 2.
Ocular characteristics of F-I2. A-B: Ultra-widefield fundus images include both colour fundus and autofluorescent images. C: Optical coherence tomography (OCT) B-scan (Ca, b) and B-ultrasound (Ca’) of the left eye reveal inferior posterior staphyloma (PS) with retinoschisis. D: OCT B-scan (Dc, f) and B-ultrasound (Dc’) of the right eye reveal a wide macular PS. OCT B-scan of the right eye reveals focal circular areas of retinal atrophy and choriocapillaris atrophy located in the region adjacent to the disc (Dc, d) and the supertemporal vascular arcade (De, f), with pigment epithelial detachment (PED) close to the retinal atrophy lesion (Df). Yellow arrows refer to focal circular areas of retinal atrophy and choriocapillaris atrophy. Yellow dotted squares indicate the site of PS. White arrows indicate PED.
Figure 2.
Ocular characteristics of F-I2. A-B: Ultra-widefield fundus images include both colour fundus and autofluorescent images. C: Optical coherence tomography (OCT) B-scan (Ca, b) and B-ultrasound (Ca’) of the left eye reveal inferior posterior staphyloma (PS) with retinoschisis. D: OCT B-scan (Dc, f) and B-ultrasound (Dc’) of the right eye reveal a wide macular PS. OCT B-scan of the right eye reveals focal circular areas of retinal atrophy and choriocapillaris atrophy located in the region adjacent to the disc (Dc, d) and the supertemporal vascular arcade (De, f), with pigment epithelial detachment (PED) close to the retinal atrophy lesion (Df). Yellow arrows refer to focal circular areas of retinal atrophy and choriocapillaris atrophy. Yellow dotted squares indicate the site of PS. White arrows indicate PED.
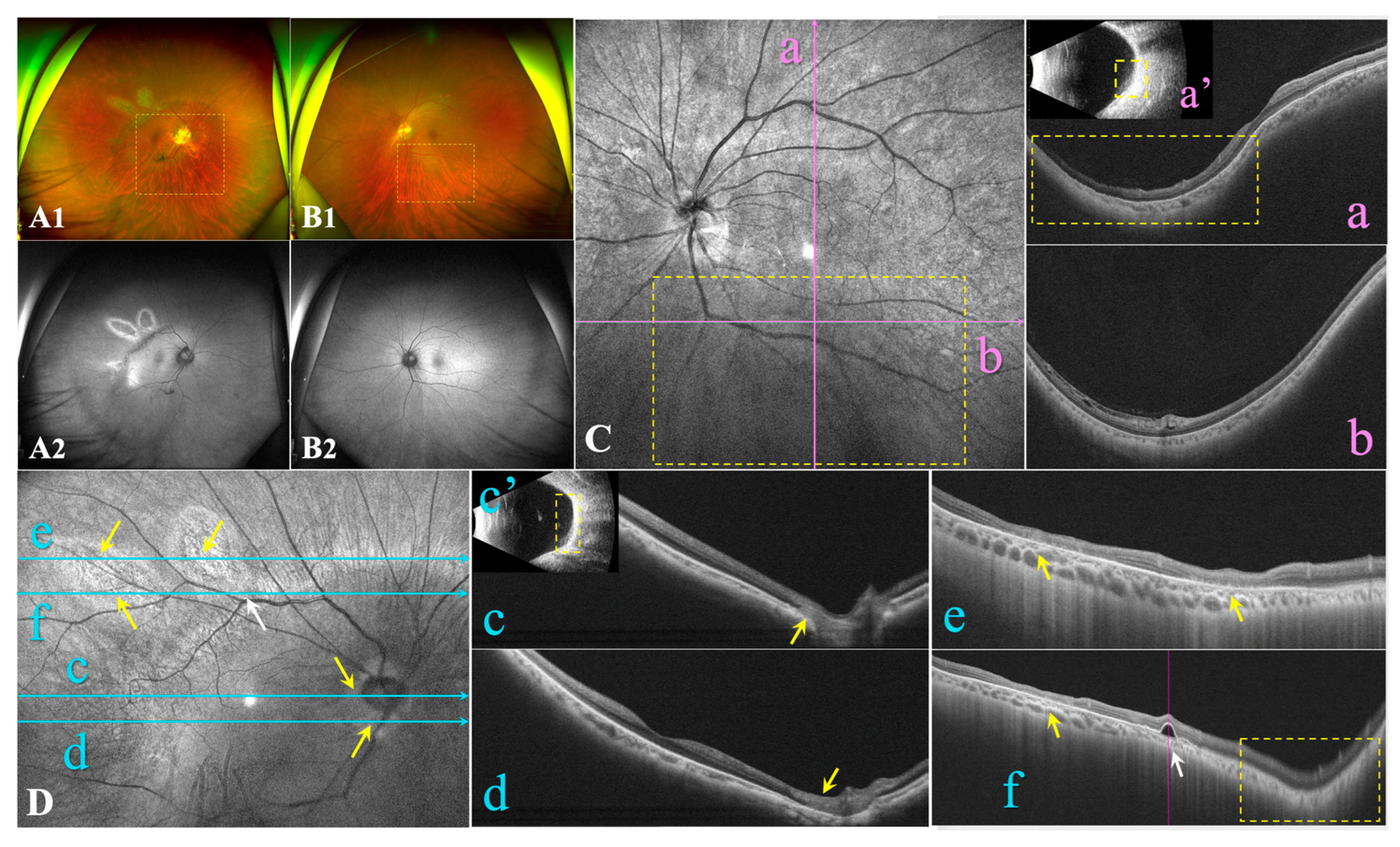
Figure 3.
Identification of OAT variants in the family. A: Pedigree of the family with gyrate atrophy with choroid and retina (GACR). Question marks indicate that the data were unavailable. B: Prediction of pathogenicity of the identified variants. C: Sanger sequencing of identified variants. Variant or wild-type nucleotides are indicated by red arrows. D: Conservative analysis. Abbreviations: LRT: Likelihood Ratio Test; FATHMM: Functional Analysis Through Hidden Markov Models; CADD: Combined Annotation; GERP: Genomic Evolutionary Rate Profiling; Dependent Depletion SPIDEX: Pre-computed Index of Splicing Variants; ACMG: American College of Medical Genetics. M1: c.1186C>T, p.R396*; M2: c.748C>T, p.R250*; WT: wild-type.
Figure 3.
Identification of OAT variants in the family. A: Pedigree of the family with gyrate atrophy with choroid and retina (GACR). Question marks indicate that the data were unavailable. B: Prediction of pathogenicity of the identified variants. C: Sanger sequencing of identified variants. Variant or wild-type nucleotides are indicated by red arrows. D: Conservative analysis. Abbreviations: LRT: Likelihood Ratio Test; FATHMM: Functional Analysis Through Hidden Markov Models; CADD: Combined Annotation; GERP: Genomic Evolutionary Rate Profiling; Dependent Depletion SPIDEX: Pre-computed Index of Splicing Variants; ACMG: American College of Medical Genetics. M1: c.1186C>T, p.R396*; M2: c.748C>T, p.R250*; WT: wild-type.
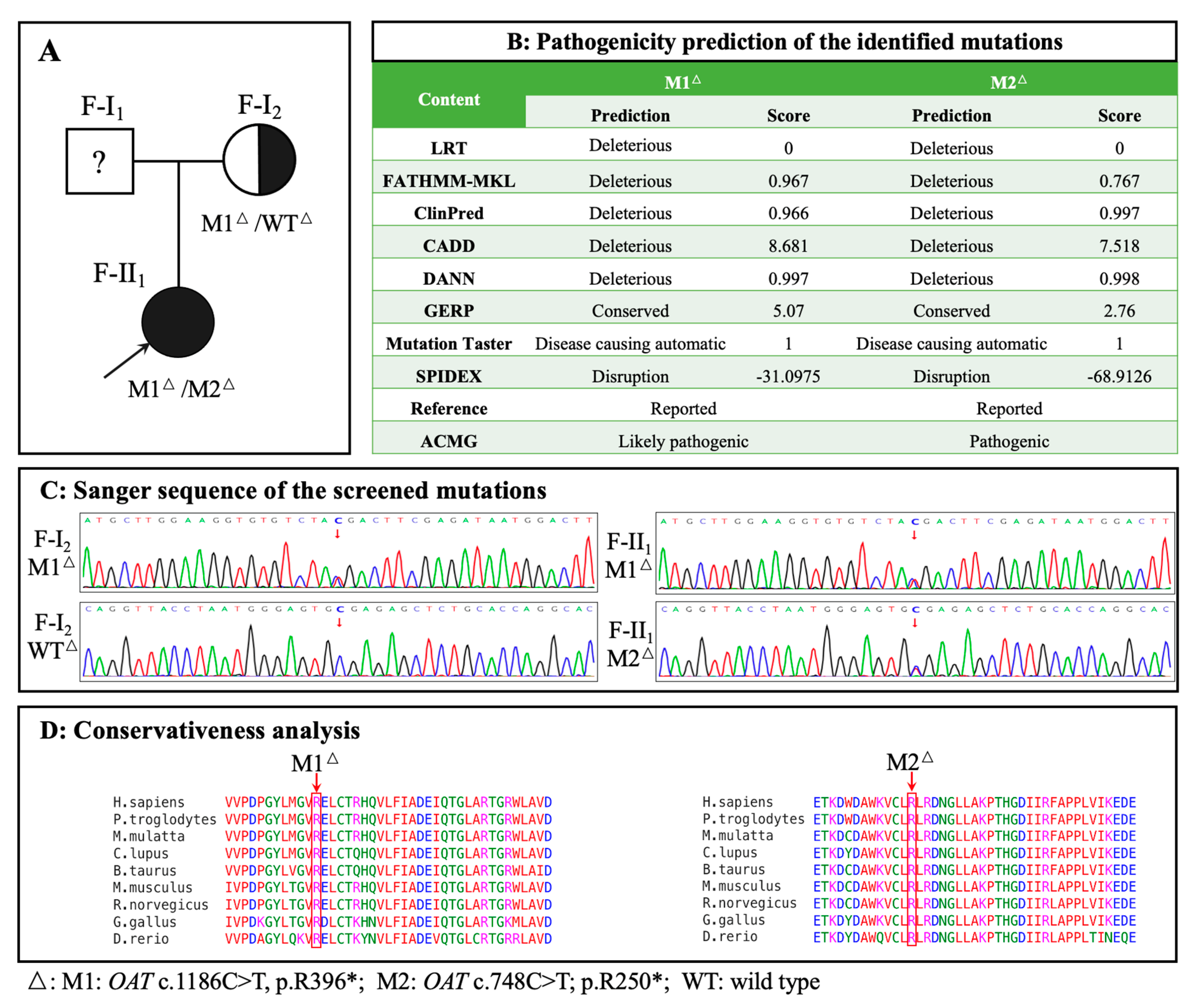
Figure 4.
Protein structure modelling and transcription analysis. A: Three-dimensional structural model of the OAT protein. The truncated portion of the protein is coloured orange, and the mutants are shown as M1-b and M2-b. WT: wild-type. B: Catalytic sites for substrate specificity in the OAT protein. Capital letters correspond to abbreviations of different amino acid names and numbers correspond to amino acid orders. C: Relative OAT mRNA expression levels in peripheral blood of the two family members and normal controls. **: P<0.01; ***P<0.001. P<0.05 was considered statistically significant.
Figure 4.
Protein structure modelling and transcription analysis. A: Three-dimensional structural model of the OAT protein. The truncated portion of the protein is coloured orange, and the mutants are shown as M1-b and M2-b. WT: wild-type. B: Catalytic sites for substrate specificity in the OAT protein. Capital letters correspond to abbreviations of different amino acid names and numbers correspond to amino acid orders. C: Relative OAT mRNA expression levels in peripheral blood of the two family members and normal controls. **: P<0.01; ***P<0.001. P<0.05 was considered statistically significant.
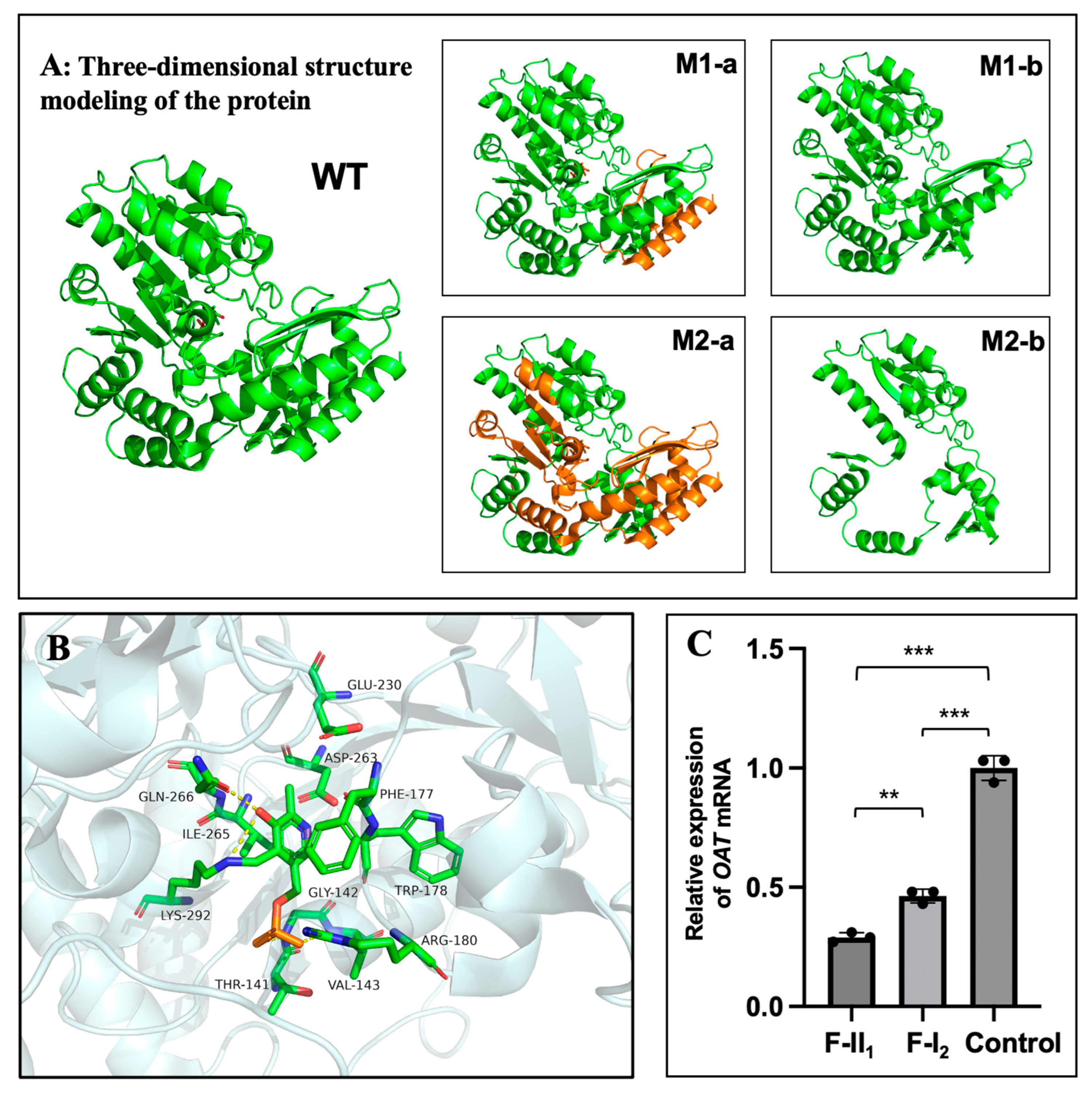

Table 1.
Ocular manifestation and results of haematological test.
| II-1 | I-2 | ||||
| Age (years) | 19 | 54 | |||
| Chief complaint | Decreased vision bilaterally | High myopia and fluttering dark shadows in the right eye | |||
| Ophthalmic parameters |
OD | OS | OD | OS | |
| IOP (mmHg) | 12.6 | 12.4 | 14.5 | 13.6 | |
| AL (mm) | 29.40 | 29.40 | 26.45 | 24.52 | |
| SE→BCVA | -17.00D→20/66 | -18.25D→20/66 | -9.00D→20/28 | -2.75D→20/28 | |
| Ocular manifestations | Punctate posterior subcapsular cataract | / | Nuclear cataract | Nuclear cataract | |
| Bilateral high myopia Bilateral PS (Type 1) Bilateral sharply demarcated circular areas of peripheral chorioretinal atrophy Bilateral macular atrophy |
High myopia PS (Type 1) Focal circular areas of retinal atrophy / |
Mild myopia Inferior PS (Type 5) / / |
|||
| Haematological testing (μM) | Serum value | Serum value | Reference | ||
| Ornithine | 257.92↑ | 102.08↑ | 10-100 | ||
| Creatine | 94.83↓ | 121.75 | 95-1000 | ||
Abbreviations: IOP: intraocular pressure; AL: Axial length; SE: spherical equivalent, = sphere + cylinder/2 D; BCVA: best corrected visual acuity; PS: posterior staphyloma; OD: the right eye; OS: the left eye. Type 1: Wide, macular PS, Type 5: Inferior PS. “↑” indicate an elevated value, while “↓” indicate a decreased value.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



