Submitted:
26 April 2024
Posted:
26 April 2024
You are already at the latest version
Abstract
The direct hydrogenation of greenhouse gas carbon dioxide (CO2) to higher alcohols (C2+OH), as an important research area in C1 chemistry, is one of the most essential approaches to convert CO2 into liquid fuels or other value-added chemicals. The high atom economy and direct pathway make this reaction attractive to convert CO2 to higher alcohols. However, due to the complicated reaction network and the difficulty of C-C coupling, the formation of higher alcohols is more difficult compared with methanol or other compounds containing one carbon. The key issue is development of novel catalysts with high conversion and space time yield. But this is still a huge challenge faced by researchers. In this review, the thermodynamics and reaction pathways of CO2 hydrogenation were firstly clarified. Subsequently, after reviewing single metal-based, bi-and multi-metallic catalysts, the recently progress of tandem catalytic system was emphasized. Finally, we also provided an overview about tandem catalysis for CO2 hydrogenation to higher alcohols.
Keywords:
CO2
; CO2 hydrogenation
; Tandem Catalysts
; Higher Alcohols
1. Introduction
Since the industrial revolution, with the increasing demand for energy by humans, fossil fuels such as coal, oil, and natural gas have been extensively exploited and utilized [1]. Fossil fuels not only provide impetus for the development of human society, but also lead to excessive emissions of greenhouse gases such as CO2, leading to a series of environmental problems such as the greenhouse effect and ocean acidification [2,3]. In order to cope with the rapid increase in CO2 content in the atmosphere, it is necessary not only to reduce emissions, but also to develop the knowledge of carbon capture, utilization and storage (CCUS) [4,5] . Converting CO2 into various high-value chemicals such as alkanes, olefins, alcohols, and aromatics can effectively alleviate environmental problems caused by excessive CO2 emissions and alleviate the excessive reliance on fossil fuels, which is of great significance for the sustainable development of the energy and chemical industries[6,7,8,9,10] . Due to the chemical inertness of CO2 molecular, the conversion of CO2 requires large energy input under harsh reaction conditions, such as high temperature and pressure. Introducing another substance with higher Gibbs free energy as a co-reactant, such as green H2, will make this reaction thermodynamically easier [11]. Moreover, the development of green H2 through water electrolysis from sun and wind electricity make this process promising to realize the goal of carbon neutral. Therefore, the hydrogenation of CO2 to various value-added products is one of the promising methods for utilizing this carbon rich sources [6,12,13,14,15,16,17,18,19].
Higher alcohols generally refer to C2+ alcohols. Due to its high energy density and lower vapor pressure, higher alcohols can be used directly as fuel or fuel additives [20], Higher alcohols are also important raw material or intermediate for the production of other chemical products. It has widespread application in different fields, such as energy, biology, and chemical engineering [21]. At present, higher alcohols is mainly produced through fermentation of crops such as corn and sugarcane, or the hydration of alkenes. However, the former consumes large amount of grain, while the later relies on petroleum resources and has low single-pass conversion[22]. Therefore, the utilization of industrial emissions of CO2 and renewable hydrogen energy to produce higher alcohols is of great significance.
The selective conversion of CO2 hydrogenation to higher alcohol is challenging. The existence of various parallel and sequential reactions makes the reaction network complex, the composition of products are also complicated. The study of CO2 hydrogenation to higher alcohols can be traced back to the 1980s[23], but most catalysts are directly modified from that of syngas to alcohols by hydrogenation. The problems of low activity and limited selectivity hinder its practical application in industry. Recently, with the development of catalyst preparation and characterization technologies, remarkably progress have been made in the area of higher alcohol synthesis (HAS).
The concept of tandem or relay catalysis for the hydrogenation of CO/CO2 to value-added fuels or products, such as low carbon olefin, aromatic, gasoline and alcohol, has drawn the attention of many researchers [24,25]. For example, high aromatic selectivity can be achieved by combining the methanol synthesis catalyst with aromatization zeolite catalyst. The selective synthesis of the target products could cut down the energy consumption for the following separation process, which are crucial for its practical application in industry. Compared with hydrocarbon products, the synthesis of alcohol shows metrics such as higher atom economy, as the oxygen atom can be incorporated into the alcohol molecular. Wang et al.[26] combined catalysts (ZnO-ZrO2│H-MOR│Pt-Sn/SiC) with different functions (methanol synthesis|methanol carbonylation|acetic acid hydrogenation) to give high ethanol selectivity. Thus, the rational design of multifunctional tandem catalysts with controlled reaction channel for the selective synthesis of the target alcohol products.
The hydrogenation of CO/CO2 into high-value C2+ chemicals has drawn the attention of many researchers, which are important to achieve carbon neutrality, but the complicated reaction networks coupled with multiple intermediates and reaction pathways make the selective control difficulty, conventional catalysts still suffer from limited product selectivity. This review paper presents a summary of the research advancements in the catalytic hydrogenation of CO2 to higher alcohols, with particular emphasis on the latest progress of tandem catalysis. The challenges encountered in CO2 hydrogenation to higher alcohols are illustrated, guidance for the subsequent development of tandem catalysts is also provided.
2. Analysis of CO2 Hydrogenation Reaction for Higher Alcohol
2.1. Thermodynamic Analysis
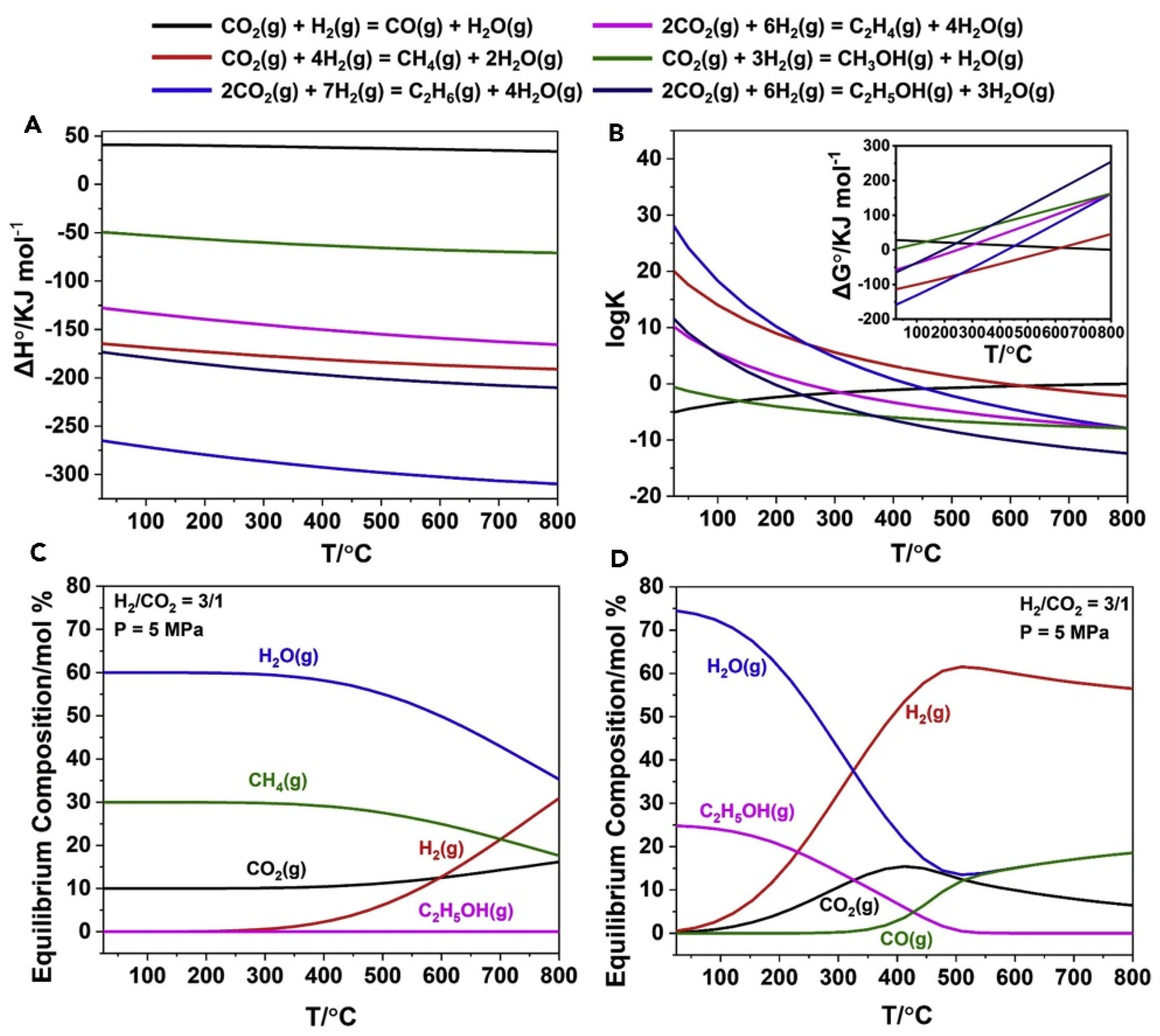
The process of CO2 hydrogenation is intricate, involving numerous parallel and sequential reactions. CO2 hydrogenation encompasses a diverse range of C1 products (e.g., CH3OH, CO, and CH4) as well as C2+ products (e.g., alkanes, alkenes, and higher alcohols) [27,28]. Table 1 [29] provides an overview of the principal reactions involved in the process of CO2 hydrogenation along with their corresponding reaction enthalpies. With the exception of the RWGS reaction, most reactions exhibit exothermic behavior, indicating thermodynamic favorability under low temperatures. Consequently, it is unlikely for the RWGS reaction to occur independently, which may proceed as a parallel side-reaction. Under low temperature, the formation of CO by RWGS reaction can be restrained, but the inertness property of CO2 makes it difficult to be activated. Thus, the reaction conditions must be carefully explored to obtain the target product with high yield.
As depicted in Table 1 and Figure 1 [37], the equilibrium constants are much higher than alcohols under all temperatures, thus alkanes are thermodynamically favored. It’s actually true that methane and other hydrocarbon products occurs as main products in many catalytic systems. The reaction for ethanol formation exhibits lower Gibbs free energy and larger equilibrium constant compared to methanol synthesis, rendering it more favorable thermodynamically. However, due to the shared active sites involved in both methanol synthesis and higher alcohol synthesis reactions for C-O bond hydrogenation, they remain competitive kinetically, resulting in methanol often dominating over ethanol as the primary product [30].
Therefore, based on the aforementioned analysis and given specific reaction conditions, an effective HAS catalyst should possess the ability to impose a high kinetic energy barrier for alkane formation while facilitating alcohol synthesis to achieve superior selectivity towards higher alcohols, and C-C coupling is crucial for the synthesis of high alcohol products.
2.2. Reaction Mechanism
Since the latter part of the 20th century, there has been extensive research conducted on elucidating the intricate reaction mechanisms involved in CO2 hydrogenation. However, the reaction intermediates during the reaction is complicated, such as *CHO、 HCOO*、*CHx、 *CH3CO. Consequently, there remains ongoing debate regarding the precise mechanism and pathway for converting CO2 into higher alcohols. The CO-mediating mechanism and the formate/methoxy-mediating mechanism are widely accepted for HAS.
Arakawa et al. [31] proposed the CO-mediating mechanism, where the initial CO was formed by the reverse water-gas shift reaction(RWGS). Subsequently, the dissociation and activation of CO give *CO species, which react with H2 by non-dissociative adsorption to produce *CH3 species. Finally, higher alcohols are formed by CO insertion into the alkyl intermediates. Xu et al. [32] also revealed the CO-mediating mechanism using in situ DRIFTS analysis over the Cs-CuZnFe catalysts. Their findings revealed that during the course of the reaction, the characteristic peak corresponding to *CO species was firstly observed, followed by adsorption of acetaldehyde and ethoxy intermediates, indicating that formation of higher alcohols on the catalyst follows a CO insertion mechanism (Scheme 1). Methanol steam reforming experiments demonstrated difficulty in breaking C-O bond within methanol, further supporting the preferential formation of alkyl species through CO dissociation.
In their study of the Pt/Co3O4 catalyst, He et al. [33] proposed a mechanism involving formate/methoxy intermediates. They employed isotope labeling technology by adding 13CH3OH prior to the reaction and observed a peak corresponding to 13CH3CH2OH or CH313CH2OH with m/z=47, indicating that the carbon atom in ethanol may originate from methanol. The authors speculated that water could protonate methanol, leading to its decomposition into *CH3 species, which subsequently reacted with CO to form CH3CO*. In the following step, the above CH3CO* intermediate was hydrogenated to form the target ethanol product. The similar formate/methoxy-mediating mechanism was also put forward by Liu et al. [34], the adsorption activation of CO2 can be facilitated over the Cs-modified catalyst, where the energy barrier of HCOO* decomposed into CHO* through formate pathway was lower than that of CO* hydrogenated into CHO* (Scheme 2), thus leading to higher ethanol yield.
The hydrogenation of CO2 to higher alcohols necessitates the RWGS reaction for the generation of CO, followed by subsequent elementary reactions including CO dissociation, C-C coupling, and CO insertion [35,36]. Consequently, the catalyst employed for the hydrogenation of CO2 to low-carbon alcohols must possess dual active sites for both CO dissociation and CO insertion, as well as exhibit excellent reverse water-gas conversion (RWGS) reaction activity while ensuring compatibility between RWGS and CO hydrogenation reactions. Therefore, the design of proper multifunctional catalysts becomes more challenging due to their requirement for multiple active centers.
3. Catalysts
As mentioned above, an effective HAS catalyst should promote CO2 activation, C-C coupling and other elementary reactions. To address this challenge, significant efforts have been made in the development of catalysts, including single metal-based and bimetallic or multi-metallic catalysts. Recently, the reaction coupling/tandem catalytic strategy has drawn the attention of many researchers. The tandem catalysts combine the advantages of catalysts with different function, where the formation or migration of the key intermediates (CO*/CHxO*) can be manipulated, thereby significantly improving the production of higher alcohols. Therefore, after the introduction of single metal-based, bi- and multi-metallic catalysts, this paper focuses on the state of art tandem catalysis.
3.1. Single Metal-Based Catalyst
The initial studies on the formation of higher alcohols via CO2 hydrogenation were conducted in the 1990 s by Kusama et al. [75]. The existing CO2 hydrogenation catalysts can be divided into two types: precious metals (Rh, Pd, Pt, Ir) based catalysts and precious metals (Mo, Co, Cu, Fe) based catalysts (as shown in Table 2) [37,38,39], and many catalyst was derived from alcohol synthesis catalyst using syngas as raw material.
Rh-based catalysts are widely used in ethanol synthesis from syngas, so researchers have begun to use Rh catalysts for CO2 hydrogenation to produce higher alcohols Rh-based catalysts are extensively studied in ethanol synthesis from syngas, prompting researchers to explore the use of Rh catalysts for CO2 hydrogenation to produce higher alcohols [40]. Researchers have found that Rh-TiO2, RhNa-TiO2, and Rh-SiO2 catalysts have good performance for CO2 hydrogenation [31,41,42,43]. Rh has a weak C-C coupling ability, so it is necessary to introduce alkali metals or transition metals onto Rh-based catalysts to promote the formation of C2+ products [44,45]. Liu et al. [46] embedded Rh into Ti-doped CeO2 carrier to construct a Rh1/CeTiOx single-atom catalyst, which exhibited high ethanol selectivity. The oxygen vacancy-Rh Lewis acid-base pairs are conducive to CO adsorption and activation. The C-O bond in CHxOH* and COOH is cleaved into CHx* and CO* substances, and then C-C coupling and hydrogenation to generate ethanol. The strong Rh-O bond generated by Ti-doping-induced crystal reconstruction contributes to the stability of the catalyst. Liu et al. [47] synthesized a dual-atom Pd/CeO2 catalyst with high ethanol selectivity(97.8%). Unfortunately, the diatomic Pd active sites in catalyst were easy to be aggregated into Pd particles. The activity of catalyst declined quickly, and the satability of catalyst was poor. The product selectivity also shift from ethanol to methanol and CO. To solve this problem, the nan-reactor (Pd2Ce@Si16) with hydrophobic shell was prepared, where the diatomic Pd active sites can be stabilized through the microenvironment caused by the in situ enrichment of water. However, the cost of catalysts is one of the key factors for the successful application of catalysts, and the high cost of Rh and other precious metal catalysts limits their industrialization
Mo-based catalysts have been utilized for the synthesis of higher alcohols from syngas, but the reports on their application for CO2 hydrogenation to produce higher alcohols are limited. Tominaga et al. [23] firstly prepared several alkali-modified Mo/SiO2 catalysts for HAS, but the higher alcohol selectivity was low. Thompson et al. [48] synthesized catalysts using Mo2C as the carrier and Cu, Pd, Co and Fe as the active components, and found that the Cu and Pd-modified catalysts showed higher methanol selectivity, and the Co and Fe-modified catalysts had higher C2+ hydrocarbon selectivity.
As classical FTS catalyst, Co is commonly employed in CO hydrogenation to produce higher alcohols. However, Co based catalyst possesses strong hydrogenation capability, which lead to methane as the dominant product in CO2 hydrogenation, Co based catalyst exhibits low activity for the RWGS. Arakawa et al. [49] first reported the Co-based catalyst for CO2 hydrogenation to ethanol, with ethanol selectivity of 7.9%. Davis et al. [50] prepared Na-promoted cobalt oxide catalysts with SiO2 as the carrier (1% Na-20% Co/SiO2). Co carbide produced by carburizing has higher activity and selectivity for HAS, mostly ethanol as product. Li et al.[51] synthesized mesoporous structured Co3O4 (Co3O-m) catalysts using mesoporous silica (KIT-6) as template. It’s deemed that Co3O4 was the main active site, and the promoted formation of long chain hydrocarbons and alcohols were ascribed to the ordered mesoporous structure. The formation of the undesirable CH4 is alao hindered.
Cu is commonly utilized for methanol synthesis, and it is widely acknowledged that Cu is responsible for the non-dissociative of CO to form *CO intermediates, resulting methanol or CO as main product. However, the coupling to C-C is difficult over Cu catalyst. To address this problem, other elements can be incorporated in to Cu catalyst for the CHx species formation and the following CHx-C(Hx)O coupling to produce higher alcohols [36,52]. Ding et al. [53] obtained high density of Cu active sites by embedding Cu nanoparticles into Na-β zeolite to achieve high C2+OH STY(17.1mmol-g cat-1 h-1), and the formation of other byproducts was also restricted by the ordered zeolite porous structure. Dimitra et al. [54] used MFI and BEA zeolite as carriers to recrystallize and encapsulate Cu nanoparticles to produce mesoporous zeolite catalysts. The introduction of mesopores into zeolite improves the conversion and selectivity. Mesoporous Cu-containing Beta zeolite catalysts are more favorable for ethanol production, while non-mesoporous catalysts produce more CO.
Fe has emerged as a promising catalyst for CO2 conversion owing to its high activity for RWGS and Fischer-Tropsch synthesis. Fe is recognized as an active component for the dissociation of carbon monoxide, thereby contributing to the formation of alkyl intermediates and resulting hydrocarbon products [55,56,57,58]. To elevate the synthesis of alcohols, it is usually necessary to combine Fe with Pd, Rh or Cu to enhance the CO insertion to the alkyl species. Sun et al. [59] modified Fe catalyst both with Na and S, where Na modification contributes to CO dissociation, while sulfate facilitates non-dissociated CO adsorption. The promoted formation of alcohols was due to the reduction of the energy barrier, while allowing Fe sites to be in a different electronic environment, the matching between dissociated and non-dissociated CO activation are provide for HAS. Wu et al. [60] prepared carbon-based electron buffer layer on the ternary catalytic component ZnOx·Fe5C2·Fe3O4 to adjust the catalytic system, and the electron transfer pathway (ZnOx→Fe species or carbon layer) ensures appropriate CO* adsorption strength at the catalytic interface. In the ethanol synthesis process, it promotes the C-C coupling between CHx* and CO*, and finally achieves an ultra-high ethanol yield of 366.6 g EtOH kg cat-1 h-1.

Table 2.
Catalytic Performance of HAS from CO2 Hydrogenation over Single component catalyst.
| Enter | Catalysts | T (℃) |
P (MPa) |
GHSV/ L.g-1h-1 |
H2/ CO2 |
XCO2 (%) |
SCO (%) |
SHC (%) |
SMeOH (%) |
SHA (%) |
STYHA (mmolgcat-1h-1) |
Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Rh-TiO2 | 300 | 10 | 6 | 3 | / | / | 69.8 | 21.1 | 9.3 | 0.7 | [40] | |
| 2 | RhLi-SiO2 | 240 | 5 | 6 | 3 | 7.0 | 15.7 | 63.5 | 5.2 | 15.5 | 0.36 | [42] | |
| 4 | 1Pd2Ce@Si16 | 250 | 3 | 3 | / | 5.9 | / | / | / | 98.7 | 11.6 | [47] | |
| 3 | Mo/SiO2 | 500 | 1.6 | / | 1 | / | / | 46.6 | 22.1 | 2.8 | / | [23] | |
| 4 | Cu/Mo2C | 200 | 4 | / | 3 | 4.6 | 8.6 | 15.7 | 63 | 14 | / | [48] | |
| 5 | Co/Mo2C | 200 | 4 | / | 3 | 4.8 | 9.5 | 17.1 | 46 | 25 | / | [48] | |
| 6 | Fe/Mo2C | 200 | 4 | / | 3 | 3.9 | 6.8 | 18.6 | 58 | 16 | / | [48] | |
| 7 | Na-Co/SiO2 | 220 | 1.9 | 3 | 3 | 15.7 | / | 99.3 | / | 0.7 | / | [50] | |
| 8 | Co3O4 | 200 | 2 | 6 | 3 | 28.9 | 0 | 53.6 | / | 19.2 | 1.6 | [51] | |
| 9 | Cu@Na-Beta | 300 | 1.3 | 12 | 1 | 7.9 | 30.5 | / | / | ~100 | 17.1 | [53] | |
| 10 | FeNaS-0.6 | 320 | 3 | 8 | / | 32.0 | 20.7 | / | / | 12.8 | 78.5a | [59] | |
| 11 | Na-ZnFe@C | 320 | 5 | 0.9 | 3 | 38.4 | 7.6 | 43.2 | 1.8 | 20.3 | 158.1a | [60] | |
a mg gcat-1h-1
3.2. Bi- and Multi-Metallic Catalysts
Since an excellent HAS catalyst usually needs to balance CO dissociation activation (alkylation) and CO non-dissociative activation (alcohol formation), however, single component catalyst is difficult to solve all problems in CO2 hydrogenation to higher alcohols, so researchers began to consider introducing other active components, and the catalytic performance is shown in Table 3. CO2 is prone to produce C1 products such as CH4, CO and CH3OH on multi-component catalysts, so developing efficient multi-component catalysts to combine C=O activation and C-C coupling for HAS is still a huge challenge.
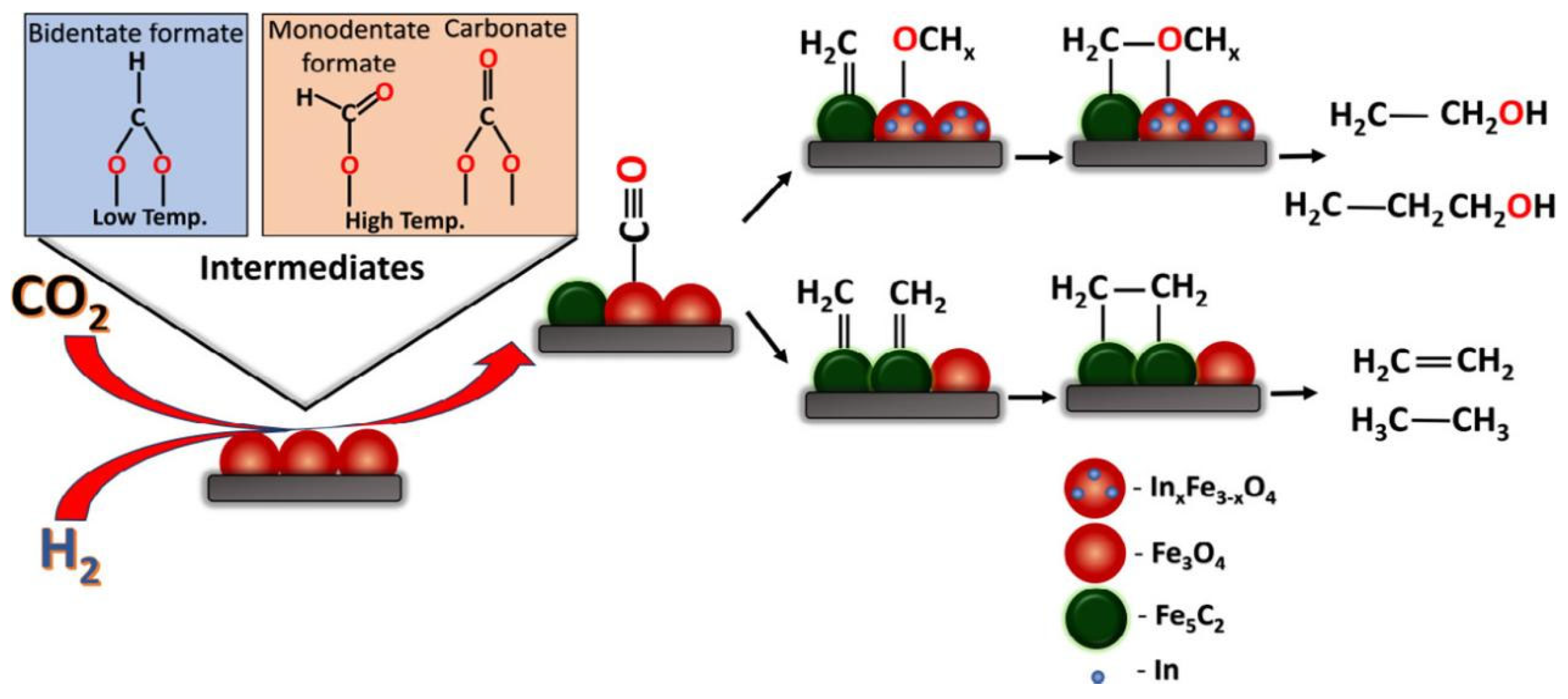
In2O3 exhibits high activity in the hydrogenation of CO2 to methanol [61], prompting researchers to explore its potential for CO2 hydrogenation to low-carbon alcohols and further enhance its catalytic function based on the excellent CO2 activation ability of In2O3 catalyst [62,63]. Witoon et al. [64] developed a K-Co catalyst supported on In2O3, which created multi-active sites (K-Co-In) that significantly reduced weak H2 adsorption and strengthened the interaction between adsorbed H and the catalyst surface. This improved the insertion capability of CO into adsorbed CxHy prior to hydrocarbon hydroforming, resulting in enhanced total selectivity and STY of higher alcohols. The catalyst achieved high STY of 169.6 mg gcat-1 h-1 with an 87.4% proportion of C2+OH among all alcohols at 4 MPa and 320°C. Peter et al. [65] introduced In into a conventional iron-based catalyst along with K as promoter, positing that In modification altered the reaction pathway of traditional Fe3O4 catalyst (Fig. 2). Initially, CO2 is converted into CO on Fe3O4 via RWGS reaction followed by hydrocarbon formation on FexCy phase. The introduction of induced strain generation facilitated the formation of OCHx intermediates, which subsequently combined with CHx intermediate to form higher alcohols species (hydrocarbons). Addition of K enhanced CO2 adsorption and higher alcohols selectivity. Huang et al. [66] prepared a single-atom catalyst Ir1-In2O3, which not only facilitates the adsorption and activation of CO2 into CO*, but also provides better opportunity for C−C coupling between CH3O*−Ov and Irδ+−CO* due to the bifunctional nature of Ir1−In2O3 (consisting of partially reduced In2O3 and Lewis acid base pair between a single atom of Ir and adjacent oxygen vacancy (Ov)).
Figure 2.
Brief schematic of the mechanism of formation of HAs and other hydrocarbons based on the intermediates observed during the in situ DRIFTS measurement [65].
Figure 2.
Brief schematic of the mechanism of formation of HAs and other hydrocarbons based on the intermediates observed during the in situ DRIFTS measurement [65].
An et al. [67] obtained Co/La4Ga2O9 catalyst by the reduction LaCo0.5Ga0.5O3 perovskite. Unlike other La-based catalysts, strong interactions between Co nanoparticles and La4Ga2O9 were generated on this catalyst. RWGS reactions were carried out on La4Ga2O9, converting CO2 into CO, and then CO migrated to Co0-Co2+ on CoNPs, and then hydrogenated to generate ethanol. Sun et al. [68] adjusted the interaction force between Na and Co2C by controlling the content of Na. The interaction force between Na and Co2C was adjusted by Sun et al. [68] through the control of Na content. The strong interaction caused by the generation of Na-Co bond formed stable Na-Co2C active sites in the reaction process. The strong interaction resulting from the formation of Na-Co bonds led to the creation of stable Na-Co2C active sites during the reaction process. The increase of interaction significantly improved the dispersity of Co2C and reduced the particle size, thus improving the RWGS formation rate and ethanol STY. The enhanced interaction significantly improved the dispersion of Co2C and reduced the particle size, thereby enhancing the RWGS formation rate and ethanol STY. At the same time, with the increase of interaction, the adsorption of CO2 and non-deionized adsorption of CO on Na-Co2C active sites increased, and then adjusted the surface CO/CHx ratio, promoting the insertion of CO to form ethanol. Concurrently, as the interaction increased, the adsorption of CO2 and non-deionized adsorption of CO on Na-Co2C active sites also increased. This adjustment led to a modified surface CO/CHx ratio, thereby promoting the insertion of CO to form ethanol. However, too strong interaction is conducive to CO desorption, resulting in high CO selectivity, while moderate interaction under 2 wt% Na obtains the best alcohol selectivity and ethanol STY. Nevertheless, excessive interaction promotes CO desorption, resulting in high CO selectivity. Conversely, moderate interaction at 2 wt% Na achieves the highest alcohol selectivity and ethanol STY.
Wang et al. [69] prepared Mo-Co-K sulfidation catalysts with activated carbon as carrier. The performance of the catalysts was further optimized by adjusting the Co/Mo and K/Mo molar ratios to improve the selectivity of C2+OH. Finally, the Mo1Co0.3K0.9/AC catalyst with Mo/activated carbon weight percentage of 12wt% achieved a CO2 conversion rate of 23.7% and a C2+OH selectivity of 10.0% under the conditions of 320°C, 5.0MPa and 3000 ml gcat-1h-1. Wu et al. [70] prepared a series of CoMoCx catalysts with ionic liquid as precursors, and optimized the efficiency of CO2 hydrogenation on ethanol by changing the carbonization temperature to adjust the catalyst proportion and electronic performance. The synergistic effect of Co, Mo2C and Co6Mo6C2 in CoMoCx-800 promoted the activation of H2 and CO2, which was beneficial to the C-C coupling of methanol and formyl substances produced by HCOO* and DMF substances, and finally showed good activity and high ethanol selectivity.
Li et al. [71] introduced Ga species in CuCo catalyst, and the mutual electronic effect between Ga and Co in spinel structure improved the reduction of Co3+, resulting in more defective CoGaOx species. Scheme 3 illustrate the proposed mechanism of CO2 hydrogenation to form ethanol. Firstly, the raw CO2 molecules are adsorbed at the oxygen vacancies of catalyst. Subsequently, carbonate species are hydrogenated to produce formate and methoxy species, which are further hydrogenated to form formate and methoxy species. Afterward, the adsorbed methoxy species were converted into CHx species. Meanwhile, CO was inserted into the adjacent CHx species to form C2H5O* species. Eventually, ethoxy species were hydrogenated to produce ethanol. Under the reaction conditions of 220°C and 3MPa, the selectivity of ethanol was 23.8% and the STY of EtOH was 1.35mmol gcat-1h-1.
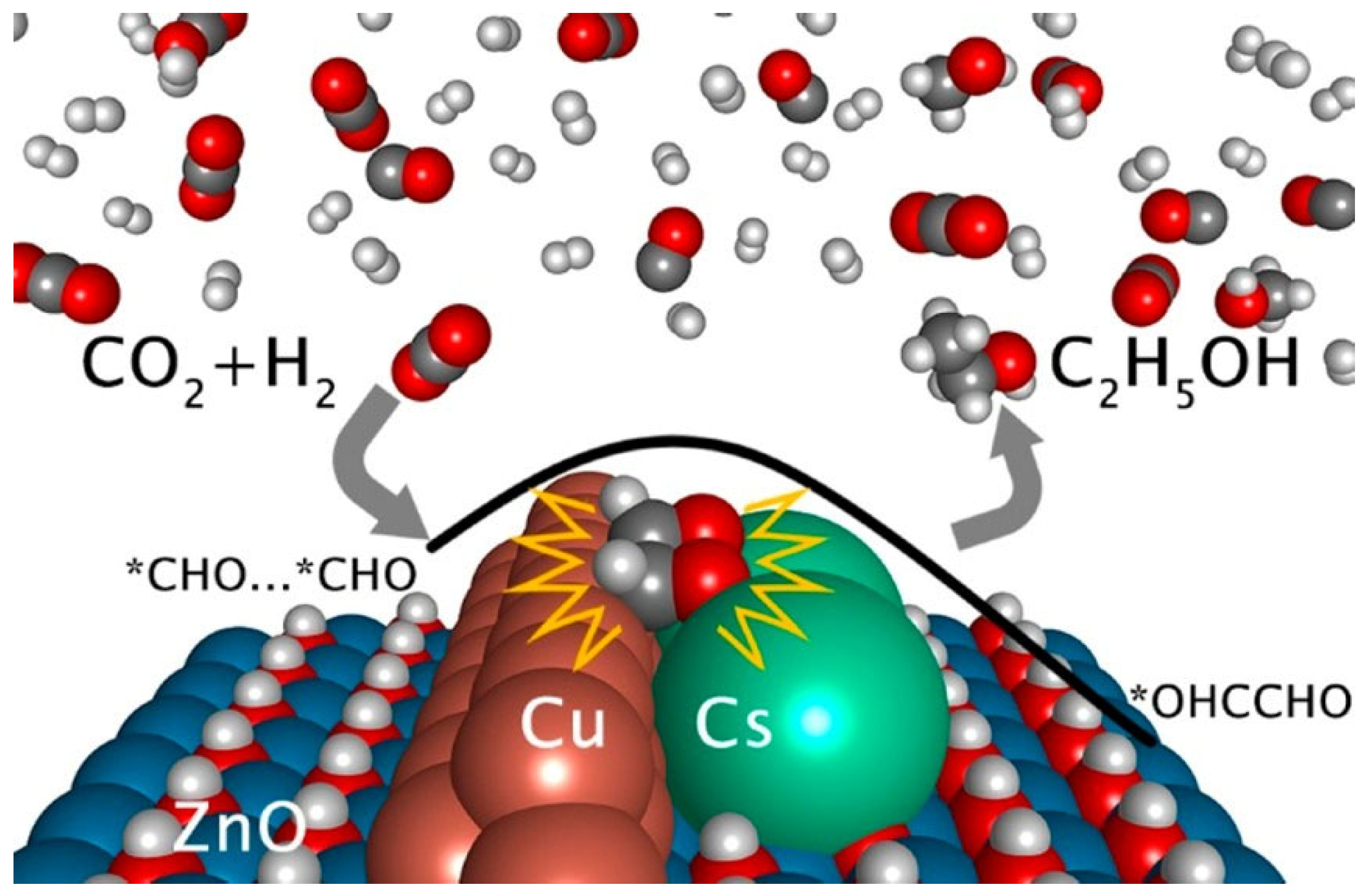
Cs/Cu/ZnO catalyst was prepared by Wang et al. [34] through the co-deposition of Cs and Cu on a ZnO support. The deposition of Cs introduced multifunctional sites with a unique structure at the Cu-Cs-ZnO interface, facilitating enhanced interaction with CO2 and promoting methanol synthesis predominantly via the formate pathway. The introduction of Cs deposition created multifunctional sites with a unique structure at the Cu-Cs-ZnO interface, which facilitated enhanced interaction with CO2 and predominantly promoted methanol synthesis via the formate pathway. Importantly, this regulation of CHO binding strength enabled efficient decomposition of HCOOH into CHO through the formate pathway while allowing for subsequent hydrogenation into methanol. Furthermore, fine-tuning CHO binding facilitated close arrangement of CHO pairs to promote C−C coupling and ultimately enhance ethanol synthesis. Jaehoon et al. [72] designed Na-promoted bimetallic CuCo catalyst (Na-CuCo-y), which can directly hydrogenate CO2 to produce C3+OH. The metal Co sites were responsible for the dissociation of CO and FTS, and Cu produced alcohols by inserting CO into the growth chain generated near it.
Liu et al. [32] prepared Cs-modified CuFeZn catalyst with synergistic active sites. High STY of 73.4 mg g cat-1 h-1 and the C2+OH/ROH ratio of 93.8% were achieved due to the interface synergy of Cu-ZnO and Cu-Fe carbides and the regulation of the hydrogenation ability of the catalyst by Cs. In this multi-component catalyst, ferric carbide is responsible for the dissociation of CO (forming alkyl intermediates), Cu is responsible for the non-dissociative activation of CO, and the balance of the two components (Cu/Fe=0.8) produces high and higher alcohol selectivity. Fan et al. [73] prepared a Cr-modified CuFe catalyst by sol-gel method. higher alcohols were formed on the CuFe catalyst by the CO intermediate route. Firstly, RWGS occurs on the Cu active site to produce CO, and part of CO dissociate and hydrogenate to CHx intermediates on FeCx. Then CHx intermediate and CO form higher alcohols by C-C coupling at the Cu-FeCx interface. The addition of Cr enhances the interaction between Cu and Fe species, promoting the formation of more Cu-FeCx interfaces. Sun et al. [74] obtained a ternary catalyst by sputtering highly active nano-Cu particles onto a Na-modified Fe3O4 carrier through a physical sputtering device. The sputtered Cu on NaFe provides a highly active Cu-Na-Fe coordination, leading to the metal Cu around the Fe5C2 active site in the CO2 hydrogenation atmosphere. resulting in the presence of metal Cu around the Fe5C2 active site during CO2 hydrogenation. The sputtered Cu enhances the adsorption of CO2 and promotes the reduction of Fe, and the subsequent carbonization in CO2 hydrogenation, forming more active Fe5C2 sites. The sputtered Cu enhances CO2 adsorption, facilitates Fe reduction, and subsequent carbonization during CO2 hydrogenation, leading to the formation of more active Fe5C2 sites. The unique structure allows for a perfect match between C-O activation, C-C coupling and C-O insertion. High-value olefins and ethanol can be produced under mild conditions with a high STY.
3.3. Tandem Catalyst
Due to the complex reaction network, the production of higher alcohols by CO2 hydrogenation using conventional catalysts becomes challenging. It’s hard to simultaneously consider both CO2 activation and C-C coupling over conversional catalyst. Therefore, the reaction-coupling strategy/tandem catalysis emerge as a practical choice for HAS, where the formation, adsorption/ desorption or migration of intermediates (CHxO* or CO*) can be manipulated. This reaction-coupling strategy inspired researchers to combine two or more functional catalyst to regulate the reaction network (as depicted in Table 4).
In 1998, Yamaguchi et al. [75] firstly combined Cu-based catalyst CuZnAlK with Fe-based FeCuAlK by , which was responsible for methanol synthesis and Fischer-Tropsch synthesis, respectively. Compared with single catalyst, and the yield of ethanol over the combined catalysts with multifunction, such as C-C bond growth, CO2 to CO reduction and OH insertion, was enhanced.
Chen et al. [76] combined RWGS catalyst(K/Cu–Zn) and Fischer-Tropsch synthesis catalyst (Cu25Fe22Co3K3) in two-stage bed model for HAS. When the reactant gas was fed in firstly downstream through CFCK catalyst and then through CZK catalyst, the STY of higher alcohols was much higher than the inverse model. It’s deemed that generated CO from RWGS catalyst was used as the feedstock for subsequent CO hydrogenation over Cu25Fe22Co3K3 catalyst, thus promoting the production of higher alcohols and other C2+ hydrocarbons or/and oxygenates. It’s also observed that the yield of C2+ hydrocarbons and oxygenates were increased, while the selectivity of CO was decreased.
Yang et al. [77] prepared Na-Fe@C catalyst by pyrolyzing the Fe-based MOFs under inert atmosphere, the selectivity of alkene reached 54.9% for 3%Na-Fe@C, while the ethanol selectivity was only 14.0%. Because the alkali Na metal can donate electron to the Fe-based catalyst, the hydrogenation of alkene to alkane can be hindered by the unfavorable H2 adsorption. After combination with CuZnAl catalyst, the selectivity of ethanol increased significantly from 14.0% to 35.0%, while the alkene selectivity decreased to 33.0%. It’s reasonable to conclude that the HAS follows the RWGS+FTS pathway.
CuZnAlZr, ZnZr and ZnCrAl are typical methanol synthesis catalysts, Liu et al. [78] combined them with the KCuFeZn (KCFZ) catalyst, considering CHxO*/CO* intermediates can be supplied to regulate the reaction network of HAS to C2+OH products. It’s found that the CuZnAlZr with high rWGSR activity and the resulting CO products are most favorable among the methanol synthesis catalyst, with the STY of higher alcohols increased to 84.0 mg g cat -1 h-1. While the promoting effect of ZnZr and ZnCrAl on HA selectivity is limited. It’s interesting to find that a linear correlation between higher alcohols STY and CH3OH+CO yield, confirming the importance of CHxO*/CO* intermediates in HAS.
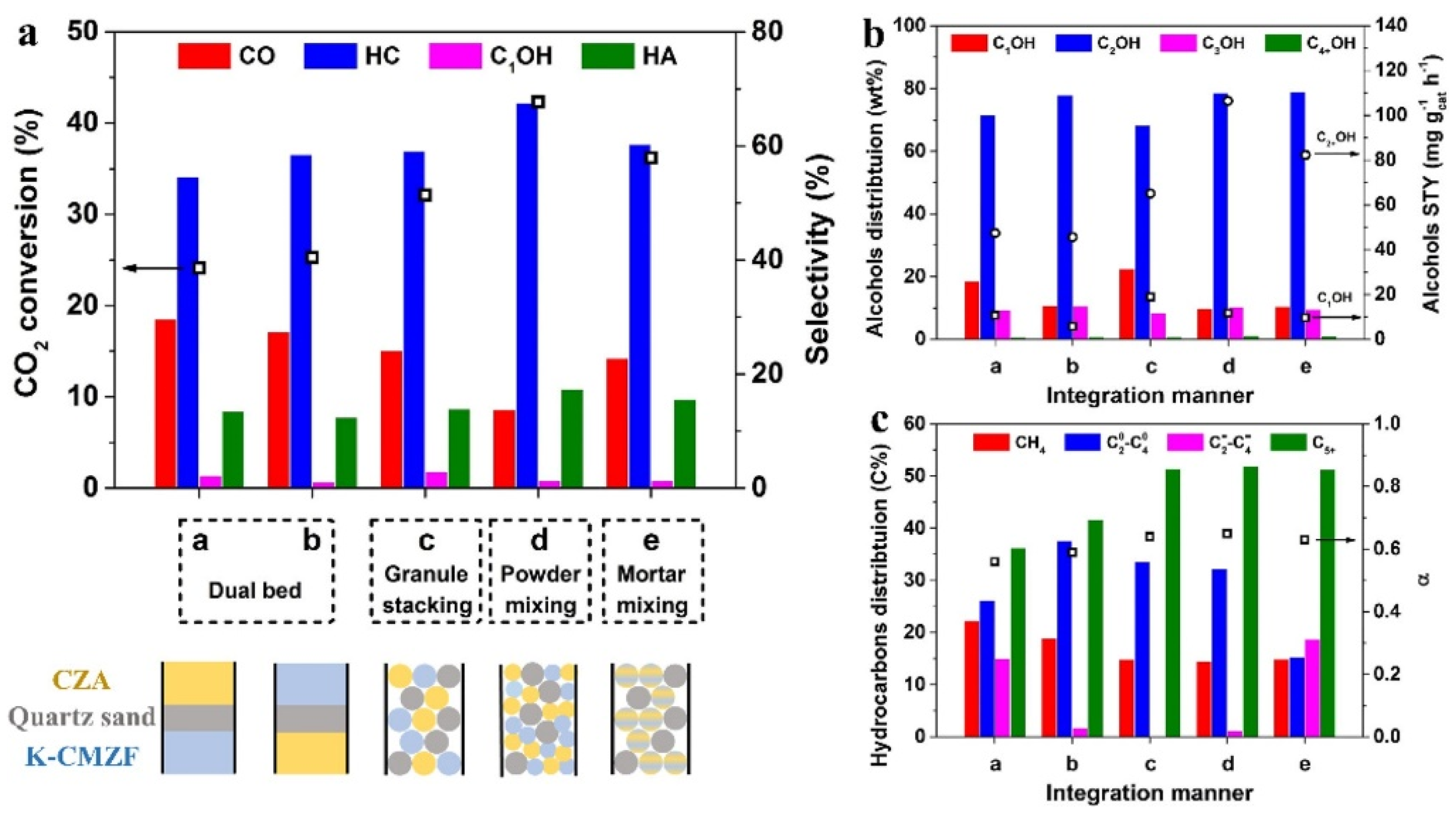
Liu et al. [78] combined CuZnAl and K-CuMgZnFe oxides into a new type of multifunctional catalyst. the CuZnAl is responsible for the produced *CO species, which migrate to the surface of K-CuMgZnFe for HAS. The five catalyst configuration methods were carefully compared to better under the reaction network, including dual bed, granule stacking, powder mixing, and mortar mixing. It’s demonstrated the the proximity between the different components are crucial to the migration of chemisorbed *CO, the highest STY of higher alcohols was observed when the above to components was powder mixing.
Guo et al. [79] also coupled Mn-Cu-K modified FeC catalyst(MnCuK-Fe5C2) with CuZnAlZr catalyst to produce higher alcohols. The best performance was obtained by powder mixing mode, and the resulting higher alcohols selectivity reached 15.5%, which was much higher than other integration modes. It’s noticeable that the selectivity towards propanol and butanol with more carbon number reaches 40% among the total higher alcohols products, which was ascribed to the co-modification of ferric carbide with transition metal Mn, Cu and alkaline metal K. The synergistic effect of the multifunctional active sites resulted to the enhanced formation of *C2H5O and the final higher alcohols products. Liu et al. [80] also explored the relationship between the components in the reaction coupling strategy, and finally combined CuZnAlZr and 4.7KCFZ into a multifunctional catalyst, achieving the selectivity from 17.4% to 33.0%, and the STY from 42.1 mg gcat-1h-1 to 84.0 mg g cat-1 h-1, which was nearly doubled.
Considering the RWGS and C−C coupling capability of the Co2C nanoprisms, Sun et al. [81] combined Co2C nanoprisms with CuZnAl. After investigating different tandem modes, the STY of higher alcohols reached 2.2 mmol g−1 h−1 for the dual bed Co2C||CuZnAl tandem catalyst, where Co2C and CuZnAl are configured on upstream and downstream layer, respectively. The fraction of C2+OH in total alchol products was 85.5%. Compared with the single Co2C catalyst, the yield of higher alchols increases approximately 3 times for the tandem catalyst. It’s deemed that the olefins formed on the upstream Co2C layer diffused downstream to the CuZnAl layer. The hydrogenated R-CHx intermediates coupled with the CHxO intermediates (produced by CuZnAl) to form higher alcohols.
3.4. The Regulation Mechanism of CO2 Hydrogenation to Higher Alcohols on Tandem Catalysts
The catalytic performance in CO2 hydrogenation to higher alcohols is determined by the synergetic effect between the multifunctional components of tandem catalyst. Liu et al. [78] fixed the mass ratio of CZA and K-CMZF catalysts at 1:1 and investigated the effects of the five integration methods on the catalysts, as shown in Figure 3.
When the catalysts were integrated by dual bed manner, the CO2 conversion is low (24−26%). The higher alcohols selectivity and STY are also limited (Figure 3b). It’s noticeable that the fraction of light olefins decreases significantly from 15.0% to 1.7% when K-CMZF is loaded above CZA instead of CZA loaded above K-CMZF (Figure 6c), indicating that the alkenes produced from K-CMZF are hydrogenated to alkanes by CZA. When shortening the distance of the two components by granule mixing, both CO2 conversion and higher alcohols STY increase. Thus, closer proximity between the two catalytic components can facilitate the conversion of CO2 to higher alcohols through multi-step reaction channel. For the catalyst with powder mixing, the highest CO2 conversion (42.3%), higher alcohols selectivity (17.4%), and STY (106.5 mg g cat−1 h−1) with a higher alcohols fraction of 90.2% are obtained. This implies that this catalyst configuration is the most conducive to HAS and C−C coupling reactions. Consequently, short distance between tandem catalysts facilitates the migration of the CO intermediate. Actually, adsorbed *CO species rather than gaseous CO are the key intermediates for the formation of high alcohols in this tandem catalysis. However, further shortening the distance with mortar mixing leads to decline of catalytic activity, which was due to the migration or ion exchange of an active metallic ion (e.g., K+), which could weaken the hydrogenation ability. It’s further found find that the CZA component can be poisoned by K from K-CMZF when overly close proximity occurs, hence reducing its hydrogenation ability. Consequently, proper proximity with certain distance between active sites is most favorable for CO2 hydrogenation to higher alcohols, this explained the better performance of powder mixing catalyst rather than mortar mixing. It is also reasonable that a suitable ratio of the two components can reach a rate matching of the key intermediates (*CO, *CHx) for HAS.
Based on the above research, the reaction pathway was proposed in Scheme 4. The CZA component is in favor of CO species (*CO) formation, while the K-CMZF component is in charge of the formation of *CHx by non-dissociation adsorption of *CO. The transformation of COx to higher alcohols via the CO-mediating mechanism, which was introduced previously in Part 2.2. The synergy between the two components with proper mass ratio guarantee the continuous and rapid transformations of CO2 to *CO and *CO to higher alcohols, the rate match of above two reactions is important for HAS. CO/*CO is the key intermediate in HAS and it’s of great importance to achieve high activity. However, this reaction is considered to be restricted by kinetics rather than thermodynamics, which was due to the low CO concentration in gas phase, and the coverage of chemisorbed CO (*CO) on catalyst was also limited. Thus, the proximity between the two components determines the migration of chemisorbed *CO and the sequential synthesis of higher alcohols.
Yang et al. [77] also found that the granule-mixed Na-Fe@C/K-CuZnAl catalyst with short distance achieved higher ethanol selectivity and CO2 conversion than that of physical-mixing (Figure 4). Enlarged spatial distance between the different components of catalyst led to the decrease in ethanol selectivity, which can be attributed to the difficult C-C coupling by CO insertion to synthesis higher alcohols. This point of view was supported by the increased CO selectivity with enlarged spatial distance. The ethanol selectivity was further declined under the dual-bed mode, where the intermediates produced from the two catalysts bed were difficult to combine directly due to the enlarged distance of the reaction active sites. By reversing the catalyst arrangement (K-CuZnAl||Na-Fe@C), the ethanol selectivity was lowest, which was even lower than that of the sole Na-Fe@C catalyst. Although this configuration mode of the multifunctional catalysts could elevate the partial pressure of CO in the HAS system. The methanol produced from the upper CuZnAl catalyst downstream to cover the surface of the NaFe@C catalyst, which are not beneficial to ethanol production.
Based on the results of catalytic performances and in situ characterization, four plausible reaction pathways were proposed for the synthesis of ethanol by tandem catalysis, as shown in Figure 5. In route I and route II, CO* species from CuZnAl catalyst diffuse onto the surface of the Fe-based catalyst (Fe5C2 as mainly active site), where the dissociation and hydrogenation of CO* species for CHm* formation. Finally, ethanol is formed by different C−C coupling processes.
Furthermore, CuZnAl is also a highly efficient catalyst for methanol synthesis with CHxO* or formate as the reaction intermediates. Thus, the formate or CHxO* intermediates desorbed from CuZnAl catalyst can diffuse onto the interface of Fe-based catalyst for C−C coupling. In route III, the CHxO* species from CuZnAl catalyst couple with CHm* species from Fe-based catalyst to produce aldehyde species, and ethanol is formed by the following hydrogenation; Different from route III, the species couple with CHm* species from CuZnAl catalyst are formate species in route IV, the resulting CHmCOO* species are hydrogenated to ethanol with aldehyde as the intermediate (2H* + CHmCOO*→CHmCHO*+ OH*).
Guo et al. [79] also investigated the effects of integration modes of the multifunctional catalysts. The mass ratio of MnCuK-FeC and CuZnAlZr was fixed at 1: 1. For the dual-bed configuration, the conversion of CO2 was 38.5% and 38.9%, and the selectivity of HAs was 13.0% and 10.5%, respectively. Powder mixing-filling method can further improve the selectivity of higher alcohols from 13.5% (MnCuK-FeC alone) to 15.5%. In-situ DRIFT results show tandem catalyst promotes the formation of *C2H5O. Thus, shorter distance between the two components facilitates the formation of long-chain oxygenated intermediates and the following production of higher alcohols. Therefore, a shorter distance between the two components facilitates the formation of long-chain oxygenated intermediates and subsequently enhances the production of higher alcohols.
Sun et al. [81] found the sole Co2C nanoprisms displayed low C2+OH and CO selectivities of 5.3 and 12.5%, respectively. The selectivity of hydrocarbon reached 82.1%, in which the olefin selectivity reached 11.0%. For CuZnAl catalyst, the selectivity of CH3OH and CO are 66.1% and 33.9%, respectively. The CO2 conversion reached 22.0%, which supply plentiful CHxO intermediates. The due bed Co2C||CuZnAl catalysts show 3-fold increase in C2+OH selectivity to 16.4%, and the C2+OH/ROH fraction reaches 85.5%. The corresponding STY reached 1.6 mmol g−1 h−1(Figure 6b). It should be noticed that the produced olefins Co2C decrease from 11.0 to 0.7%, which is almost in line with the increase in C2+OH (from 5.3 to 16.4%). It’s reasonable to conclude that the multifunctional tandem catalysts promoted the C−C coupling and the synergy of elementary reactions. However, the CuZnAl||Co2C catalysts with inverse sequence show negligible C2+OH selectivity (0.2%), and CH4 is the dominate product (88.3%). For Co-CuZnAl and Co2C/CuZnAl catalysts, CH4 and C2+ alkanes were also the predominant products, the formation of higher alcohols is also difficult. Obviously, the HAS can be regulated by combining multifunctional catalysts with different configuration mode.
The C−C coupling mechanism can be further illustrated with the help of in situ chemical transient kinetics (CTK) experiments. Olefins generated by the upstream Co2C catalyst diffuse to the surface of the downstream CuZnAl catalyst, where they were hydrogenated to form R-CHx intermediates. Then, R-CHx couple with the CHxO intermediates produced from CuZnAl to form higher alcohols. When switching from H2/CO2 to H2/CO, the response intensity of C2H5OH/C3H7OH is still lower than that when switching to H2/CO/C2H4 (Figure 7b). This indicate that the formation of R-CHx intermediates via CO hydrogenation is more difficult than the formation of R-CHx intermediates via olefins hydrogenation or dissociation, confirming that the insertion of CO into CHx has higher energy barrier than the CHx−CHxO coupling.
As described in Part 2, the CO2 hydrogenation involved many parallel and sequential reactions. Thus, the influence of reaction conditions, such as temperature, pressure, and residence time on the tandem reaction is also complicated. Liu et al. [82] discovered that high temperature benefits CO2 conversion and formation of higher alcohols and hydrocarbons, as shown in Figure 8. It’s reasonable to conclude that high pressure facilitates HAS and both formation of alcohols and hydrocarbons, according to Le Chatelier’s principle. Besides, the increased H2 partial pressure facilitates hydrogenation ability, which is demonstrated by the decrease of olefins. The increased coverage of adsorbed *CO species facilitates the formation of higher alcohols, hence leading to the increase of higher alcohols yield.
Sun et al. [81] found the formation of methanol was more preferred under low temperature, while the C−C coupling needed more energy input under high temperature. However, the hydrogenation ability and undesirable CH4 production are prone to happen when the temperature was higher than 260 °C). Different from the results of Liu et al[82], the effects of reaction pressure on the selectivity of C2+OH was not prominent, implying that the HAS may not obey the CO insertion mechanism, because the insertion ability should be favored under higher CO partial pressure.
Under the present reaction conditions and CO2 conversion, the HAS was still under kinetic control. The conversion of *CO intermediates can be facilitated by prolonged residence time. Moreover, long residence time also improved C-C coupling ability, thus higher yield of higher alcohols and C5+ hydrocarbons was obtained. However, Sun et al. observed the contradictory trend, the selectivity of hydrocarbon dropped considerably with the decrease of residence time, yet the selectivity of CO and higher alcohols gradually increased, which was ascribed to the avoid of excessive hydrogenation.
Liu et al. [80] found that the CO and alkane selectivity showed an opposite trend with the alkene selectivity, which is ascribed to the high hydrogenation ability of CuZnAlZr catalysts. The yield of higher alcohols increases firstly and then drop, peaking at 42.0 mg g cat −1 h−1 at mass ratio of 1/1 under 300 °C. Sun et al. also studied the effects of the mass ratio on the HAS using Co2C||CuZnAl tandem catalyst. However, with the declining of the mass ratio, the CO selectivity was remarkably improved, and the formation of methanol gradually exceeded ethanol, which indicated that the mass ratio could regulate the cover of intermediates and the following C−C coupling ability. The yield of higher alcohols peaked at 2.2 mmol g−1 h−1 at a ratio of 2/1. The above results also infer that the C−C coupling may be manipulated to the desired reaction channel by bonding between HCOO, CHx, or CHxO on the tandem catalyst.
Different reaction conditions exert distinct effects on different multi-component catalysts; thus, it is not possible to draw an unanimous conclusion. The matching the of multifunctional catalysts is crucial to the HAS performance. Due to the complexity of HAS reaction network, further investigation is warranted to ascertain the optimal catalytic system and reaction conditions.
4. Conclusions
The hydrogenation of CO2 to higher alcohols is of significant importance for the conversion of CO2 into value-added chemicals. This route also hold the potential to addresses the issue of greenhouse effect by excessive CO2 emissions. The hydrogenation of CO2 to higher alcohols remains a major challenge due to the complexity of the reaction and existence of many by-products, such as CO, CH4 and other C2+ hydrocarbons. In this review, we summarize the latest progress in CO2 hydrogenation to higher alcohols, emphasizing the stat of art work strategy of tandem catalysis.
Tandem catalysis demonstrates considerable promise for the synthesis of higher alcohols, while further work is still required to give better selectivity control. First, considering the thermodynamics and kinetics of the existing reactions in HAS, the matching of different kind of components for the tandem catalysis is crucial, thus the rational design of the active sites of the tandem catalyst should be highlighted. The functional matching between catalysts with different functions can be achieved through genetic algorithm and artificial intelligence screening. DFT calculation and molecular dynamic simulation can be applied to better understand the reaction mechanism, which is important for the design of catalyst composition and structure in HAS. Secondly, due to the existence of various possible reaction pathway, the reaction mechanism of tandem catalysis is still far from clear. Key intimidates and real active sites during HAS should be identified by in situ characterization techniques, such as IR, XPS, XRD, TEM. The catalytic structure-performance relationship should be further revealed.
Thirdly, to control the transport of intermediates or products and thus the reaction sequence, the appropriate spatial arrangement of different components holds potential to avoid byproduct formation. The kinetic matching of cascade reaction steps, such as C-C coupling and CO insertion, are the key issues guiding the selection of each catalyst component and the construction of an efficient catalysis system. For this purpose, the rational arrangement of different functional components in mesoscale with controllable proximity deserves more research endeavors, where the transportation of intermediates/products in sequence can be manipulated.
Lastly, novel reactors, such as nano-reactors or membrane reactors, can contribute to the reaction optimization via adjusting the surroundings or kinetics of HAS. Cu@Na-Beta nano-reactor reached high STY space-time yield of 398 mg gcat-1 h-1, as the HAS was promoted by the confinement surrounding of reactive centers in zeolitic frameworks [53]. Water enriched nano-reactor with dual Pd active sites exhibited high ethanol selectivity (98.7%) by breaking the restriction of HAS [83]. Membrane reactors can remove the by-products (e.g. water) in situ through selective separation to effectively enhance higher alcohols yield. Novel reactors hold great possibility to overcome the thermodynamic equilibrium limitation.
Tandem catalysis is expected to play more important roles in the direct synthesis of value-added products in C1 chemistry, by enabling the development of new reaction channels with precise control. Future advances are expected for HAS. We anticipate that this review will provide a solid foundation for the ongoing development of tandem catalysis, and we look forward to the emergence of innovative research efforts aimed at addressing the challenges associated with CO2 hydrogenation to alcohols.
Author Contributions
Y.C.; Design, Writing and editing original draft, J Z.L.; validation, X Y.C.; Writing and Investigate, S Y.G.; Investigate, Y. B.W.; Revise and Funding acquisition, L.W.; writing − review & editing, H.W.; validation, resources, project administration, G.F.G.; resources, project administration, funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (22078159 and U19B2001) and the Ningxia Key Research & Development Program (2023BDE03001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data will be made available on request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zhang, Z.; Pan, S.-Y.; Li, H.; Cai, J.; Olabi, A.G.; Anthony, E.J.; Manovic, V. Recent advances in carbon dioxide utilization. Renew. Sustain. Energy Rev. 2020, 125, 109799. [CrossRef]
- Lacis, A. A.; Schmidt, G. A.; Rind, D.; Ruedy, R. A., Atmospheric CO2: Principal Control Knob Governing Earth's Temperature. Science 2010, 330, (6002), 356-359.
- Solomon, S.; Plattner, G.-K.; Knutti, R.; Friedlingstein, P. Irreversible climate change due to carbon dioxide emissions. Proc. Natl. Acad. Sci. USA 2009, 106, 1704–1709. [CrossRef]
- Zhang, Z.; Wang, T.; Blunt, M.J.; Anthony, E.J.; Park, A.-H.A.; Hughes, R.W.; Webley, P.A.; Yan, J. Advances in carbon capture, utilization and storage. Appl. Energy 2020, 278, 115627. [CrossRef]
- Corma, A. Preface to Special Issue of ChemSusChem on Green Carbon Science: CO2 Capture and Conversion. ChemSusChem 2020, 13, 6054–6055. [CrossRef]
- Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J.G. Recent Advances in Carbon Dioxide Hydrogenation to Methanol via Heterogeneous Catalysis. Chem. Rev. 2020, 120, 7984–8034. [CrossRef]
- Hepburn, C.; Adlen, E.; Beddington, J.; Carter, E.A.; Fuss, S.; Mac Dowell, N.; Minx, J.C.; Smith, P.; Williams, C.K. The technological and economic prospects for CO2 utilization and removal. Nature 2019, 575, 87–97. [CrossRef]
- Tapia, J. F. D.; Lee, J. Y.; Ooi, R. E. H.; Foo, D. C. Y.; Tan, R. R., A review of optimization and decision-making models for the planning of CO2 capture, utilization and storage (CCUS) systems. Sustain. Prod. Consump. 2018, 13, 1-15.
- Ni, Y.; Chen, Z.; Fu, Y.; Liu, Y.; Zhu, W.; Liu, Z. Selective conversion of CO2 and H2 into aromatics. Nat. Commun. 2018, 9, 1–7. [CrossRef]
- Gao, P.; Li, S.; Bu, X.; Dang, S.; Liu, Z.; Wang, H.; Zhong, L.; Qiu, M.; Yang, C.; Cai, J.; et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017, 9, 1019–1024. [CrossRef]
- Song, C. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing Catal. Today 2006, 115, 2–32. [CrossRef]
- Sancho-Sanz, I.; Korili, S.; Gil, A. Catalytic valorization of CO2 by hydrogenation: current status and future trends. Catal. Rev. 2021, 65, 698–772. [CrossRef]
- Mandal, S.C.; Das, A.; Roy, D.; Das, S.; Nair, A.S.; Pathak, B. Developments of the heterogeneous and homogeneous CO2 hydrogenation to value-added C2+-based hydrocarbons and oxygenated products. Co-ord. Chem. Rev. 2022, 471, 214737. [CrossRef]
- Ateka, A.; Rodriguez-Vega, P.; Ereña, J.; Aguayo, A.; Bilbao, J. A review on the valorization of CO2. Focusing on the thermodynamics and catalyst design studies of the direct synthesis of dimethyl ether. Fuel Process. Technol. 2022, 233. [CrossRef]
- Wang, L. X.; Wang, L.; Xiao, F. S., Tuning product selectivity in CO2 hydrogenation over metal-based catalysts. Chem Sci 2021, 12, (44), 14660-14673.
- Atsbha, T.A.; Yoon, T.; Seongho, P.; Lee, C.-J. A review on the catalytic conversion of CO2 using H2 for synthesis of CO, methanol, and hydrocarbons. J. CO2 Util. 2020, 44, 101413. [CrossRef]
- Chen, G. B.; Waterhouse, G. I. N.; Shi, R.; Zhao, J. Q.; Li, Z. H.; Wu, L. Z.; Tung, C. H.; Zhang, T. R., From Solar Energy to Fuels: Recent Advances in Light-Driven C1 Chemistry. Angew. Chem.-Int. Edit. 2019, 58, (49), 17528-17551.
- Dalena, F.; Senatore, A.; Basile, M.; Knani, S.; Basile, A.; Iulianelli, A. Advances in Methanol Production and Utilization, with Particular Emphasis toward Hydrogen Generation via Membrane Reactor Technology. Membranes 2018, 8, 98. [CrossRef]
- Wang, W.; Wang, S. P.; Ma, X. B.; Gong, J. L., Recent advances in catalytic hydrogenation of carbon dioxide. Chem Soc Rev 2011, 40, (7), 3703-3727.
- Christensen, E.; Yanowitz, J.; Ratcliff, M.; McCormick, R.L. Renewable Oxygenate Blending Effects on Gasoline Properties. Energy Fuels 2011, 25, 4723–4733. [CrossRef]
- Angelici, C.; Weckhuysen, B.M.; Bruijnincx, P.C.A. Chemocatalytic Conversion of Ethanol into Butadiene and Other Bulk Chemicals. ChemSusChem 2013, 6, 1595–1614. [CrossRef]
- Bai, F.; Anderson, W.; Moo-Young, M. Ethanol fermentation technologies from sugar and starch feedstocks. Biotechnol. Adv. 2008, 26, 89–105. [CrossRef]
- Tatsumi, T.; Muramatsu, A.; Tominaga, H. O., Alcohol synthesis from CO2/H2 on silica-supported molybdenum catalysts. Chem Let 1985, (5), 593-594.
- Li, Z. Qu, Y. Wang, J. Liu, H. Li, M. Miao, S. Li, C., Highly selective conversion of carbon dioxide to aromatics over tandem catalysts. Joule 2019, (3), 570−583.
- Wei, J. Yao, R. Han, Y. Ge, Q. Sun, J., Towards the development of the emerging process of CO2 heterogenous hydrogenation into high-value unsaturated heavy hydrocarbons. Chem SocvRev 2021, 50, (19), 10764-10805.
- Kang, J.; He, S.; Zhou, W.; Shen, Z.; Li, Y.; Chen, M.; Zhang, Q.; Wang, Y. Single-pass transformation of syngas into ethanol with high selectivity by triple tandem catalysis. Nat. Commun. 2020, 11, 1–11. [CrossRef]
- Nie, X. W.; Li, W. H.; Jiang, X.; Guo, X. W.; Song, C. S., Recent advances in catalytic CO2 hydrogenation to alcohols and hydrocarbons. Adv Catal 2019, 65, 121-233.
- Yi, Q.; Li, W. Y.; Feng, J.; Xie, K. C., Carbon cycle in advanced coal chemical engineering. Chem Soc Rev 2015, 44, (15), 5409-5445.
- Yi, X. H., CO2 Hydrogenation for Ethanol Production: A Thermodynamic Analysis. Science PG 2017, 5, (6), 145-152.
- Jia, C.; Gao, J.; Dai, Y.; Zhang, J.; Yang, Y. The thermodynamics analysis and experimental validation for complicated systems in CO 2 hydrogenation process. J. Energy Chem. 2016, 25, 1027–1037. [CrossRef]
- Hitoshi, K.; Kiyomi, O.; Kazuhiro, S.; Hironori, A., CO2 hydrogenation to ethanol over promoted Rh/SiO2 catalysts. Catal Today. 1996, 28, 261-266.
- Xu, D.; Ding, M.; Hong, X.; Liu, G.; Tsang, S. C. E., Selective C2+ Alcohol Synthesis from Direct CO2 Hydrogenation over a Cs-Promoted Cu-Fe-Zn Catalyst. Acs Catal 2020, 10, (9), 5250-5260.
- He, Z.; Qian, Q.; Ma, J.; Meng, Q.; Zhou, H.; Song, J.; Liu, Z.; Han, B., Water-Enhanced Synthesis of Higher Alcohols from CO2 Hydrogenation over a Pt/Co3O4 Catalyst under Milder Conditions. Angew. Chem.-Int. Edit. 2016, 55, (2), 737-741.
- Wang, X.; Ramirez, P. J.; Liao, W.; Rodriguez, J. A.; Liu, P., Cesium-Induced Active Sites for C-C Coupling and Ethanol Synthesis from CO2 Hydrogenation on Cu/ZnO(000(1)over-bar) Surfaces. J Am Chem Soc 2021, 143, (33), 13103-13112.
- Aresta, M.; Dibenedetto, A.; Angelini, A., Catalysis for the Valorization of Exhaust Carbon: from CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, (3), 1709-1742.
- Gupta, M.; Smith, M.L.; Spivey, J.J. Heterogeneous Catalytic Conversion of Dry Syngas to Ethanol and Higher Alcohols on Cu-Based Catalysts. ACS Catal. 2011, 1, 641–656. [CrossRef]
- Xu, D.; Wang, Y.; Ding, M.; Hong, X.; Liu, G.; Tsang, S.C.E. Advances in higher alcohol synthesis from CO2 hydrogenation. Chem 2021, 7, 849–881. [CrossRef]
- Zeng, F.; Mebrahtu, C.; Xi, X.; Liao, L.; Ren, J.; Xie, J.; Heeres, H.J.; Palkovits, R. Catalysts design for higher alcohols synthesis by CO2 hydrogenation: Trends and future perspectives. Appl. Catal. B Environ. 2021, 291, 120073. [CrossRef]
- Ye, R.-P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 1–15. [CrossRef]
- Sheerin, E.; Reddy, G. K.; Smirniotis, P., Evaluation of Rh/CexTi1-xO2 catalysts for synthesis of oxygenates from syngas using XPS and TPR techniques. Catal Today 2016, 263, 75-83.
- Inoue, T.; Iizuka, T.; Tanabe, K. Hydrogenation of carbon dioxide and carbon monoxide over supported rhodium catalysts under 10 bar pressure. Appl. Catal. 1989, 46, 1–9. [CrossRef]
- Bando, K.K.; Soga, K.; Kunimori, K.; Arakawa, H. Effect of Li additive on CO2 hydrogenation reactivity of zeolite supported Rh catalysts. Appl. Catal. A: Gen. 1998, 175, 67–81. [CrossRef]
- Kusama, H.; Okabe, K.; Sayama, K.; Arakawa, H., Ethanol synthesis by catalytic hydrogenation of CO2 over Rh-Fe/SiO2 catalysts. Energy 1997, 22, (2-3), 343-348.
- Wang, G.; Luo, R.; Yang, C.; Song, J.; Xiong, C.; Tian, H.; Zhao, Z.-J.; Mu, R.; Gong, J. Active sites in CO2 hydrogenation over confined VOx-Rh catalysts. Sci. China Chem. 2019, 62, 1710–1719. [CrossRef]
- Yang, C.; Mu, R.; Wang, G.; Song, J.; Tian, H.; Zhao, Z.-J.; Gong, J. Hydroxyl-mediated ethanol selectivity of CO2 hydrogenation. Chem. Sci. 2019, 10, 3161–3167. [CrossRef]
- Zheng, K.; Li, Y.; Liu, B.; Jiang, F.; Xu, Y.; Liu, X., Ti-doped CeO2 Stabilized Single-Atom Rhodium Catalyst for Selective and Stable CO2 Hydrogenation to Ethanol. Angew. Chem.-Int. Edit. 2022, 61, (44).
- Chen, J.; Zha, Y.; Liu, B.; Li, Y.; Xu, Y.; Liu, X. Rationally Designed Water Enriched Nano Reactor for Stable CO2 Hydrogenation with Near 100% Ethanol Selectivity over Diatomic Palladium Active Sites. ACS Catal. 2023, 13, 7110–7121. [CrossRef]
- Chen, Y.; Choi, S.; Thompson, L.T. Low temperature CO2 hydrogenation to alcohols and hydrocarbons over Mo2C supported metal catalysts. J. Catal. 2016, 343, 147–156. [CrossRef]
- Okabe, K.; Yamada, H.; Hanaoka, T.; Matsuzaki, T.; Arakawa, H.; Abe, Y., CO2 hydrogenation to alcohols over highly dispersed Co/SiO2 catalysts derived from acetate. Chem Lett 2001, (9), 904-905.
- Gnanamani, M.K.; Jacobs, G.; Keogh, R.A.; Shafer, W.D.; Sparks, D.E.; Hopps, S.D.; Thomas, G.A.; Davis, B.H. Fischer-Tropsch synthesis: Effect of pretreatment conditions of cobalt on activity and selectivity for hydrogenation of carbon dioxide. Appl. Catal. A: Gen. 2015, 499, 39–46. [CrossRef]
- Liu, B.; Ouyang, B.; Zhang, Y.; Lv, K.; Li, Q.; Ding, Y.; Li, J. Effects of mesoporous structure and Pt promoter on the activity of Co-based catalysts in low-temperature CO2 hydrogenation for higher alcohol synthesis. J. Catal. 2018, 366, 91–97. [CrossRef]
- Luk, H.T.; Mondelli, C.; Ferré, D.C.; Stewart, J.A.; Pérez-Ramírez, J. Status and prospects in higher alcohols synthesis from syngas. Chem. Soc. Rev. 2017, 46, 1358–1426. [CrossRef]
- Ding, L.; Shi, T.; Gu, J.; Cui, Y.; Zhang, Z.; Yang, C.; Chen, T.; Lin, M.; Wang, P.; Xue, N.; et al. CO2 Hydrogenation to Ethanol over Cu@Na-Beta. Chem 2020, 6, 2673–2689. [CrossRef]
- Iltsiou, D.; Mielby, J.; Kegnaes, S., Direct Conversion of CO2 into Alcohols Using Cu-Based Zeolite Catalysts. ChemPlusChem 2023, 89, (1).
- Aitbekova, A.; Goodman, E. D.; Wu, L.; Boubnov, A.; Hoffman, A. S.; Genc, A.; Cheng, H.; Casalena, L.; Bare, S. R.; Cargnello, M., Engineering of Ruthenium-Iron Oxide Colloidal Heterostructures: Improved Yields in CO2 Hydrogenation to Hydrocarbons. Angew. Chem.-Int. Edit. 2019, 58, (48), 17451-17457.
- Albrecht, M.; Rodemerck, U.; Schneider, M.; Bröring, M.; Baabe, D.; Kondratenko, E.V. Unexpectedly efficient CO2 hydrogenation to higher hydrocarbons over non-doped Fe2O3. Appl. Catal. B: Environ. 2017, 204, 119–126. [CrossRef]
- Jiang, J.; Wen, C.; Tian, Z.; Wang, Y.; Zhai, Y.; Chen, L.; Li, Y.; Liu, Q.; Wang, C.; Ma, L., Manganese-Promoted Fe3O4 Microsphere for Efficient Conversion of CO2 to Light Olefins. Ind Eng Chem Res 2020, 59, (5), 2155-2162.
- Kasipandi, S.; Bae, J.W. Recent Advances in Direct Synthesis of Value-Added Aromatic Chemicals from Syngas by Cascade Reactions over Bifunctional Catalysts. Adv. Mater. 2019, 31, e1803390. [CrossRef]
- Yao, R.; Wei, J.; Ge, Q.; Xu, J.; Han, Y.; Ma, Q.; Xu, H.; Sun, J. Monometallic iron catalysts with synergistic Na and S for higher alcohols synthesis via CO2 hydrogenation. Appl. Catal. B: Environ. 2021, 298, 120556. [CrossRef]
- Wang, Y.; Wang, W.; He, R.; Li, M.; Zhang, J.; Cao, F.; Liu, J.; Lin, S.; Gao, X.; Yang, G.; Wang, M.; Xing, T.; Liu, T.; Liu, Q.; Hu, H.; Tsubaki, N.; Wu, M., Carbon-Based Electron Buffer Layer on ZnOx-Fe5C2-Fe3O4 Boosts Ethanol Synthesis from CO2 Hydrogenation. Angew. Chem.-Int. Edit. 2023, 62, (46).
- Wang, J.; Zhang, G.; Zhu, J.; Zhang, X.; Ding, F.; Zhang, A.; Guo, X.; Song, C., CO2 Hydrogenation to Methanol over In2O3-Based Catalysts: From Mechanism to Catalyst Development. Acs Catal 2021, 11, (3), 1406-1423.
- Umegaki, T.; Kuratani, K.; Yamada, Y.; Ueda, A.; Kuriyama, N.; Kobayashi, T.; Xu, Q. Hydrogen production via steam reforming of ethyl alcohol over nano-structured indium oxide catalysts. J. Power Sources 2008, 179, 566–570. [CrossRef]
- Lorenz, H.; Jochum, W.; Klötzer, B.; Stöger-Pollach, M.; Schwarz, S.; Pfaller, K.; Penner, S. Novel methanol steam reforming activity and selectivity of pure In2O3. Appl. Catal. A: Gen. 2008, 347, 34–42. [CrossRef]
- Witoon, T.; Numpilai, T.; Nijpanich, S.; Chanlek, N.; Kidkhunthod, P.; Cheng, C.K.; Ng, K.H.; Vo, D.-V.N.; Ittisanronnachai, S.; Wattanakit, C.; et al. Enhanced CO2 hydrogenation to higher alcohols over K-Co promoted In2O3 catalysts. Chem. Eng. J. 2022, 431. [CrossRef]
- Goud, D.; Churipard, S.R.; Bagchi, D.; Singh, A.K.; Riyaz, M.; Vinod, C.P.; Peter, S.C. Strain-Enhanced Phase Transformation of Iron Oxide for Higher Alcohol Production from CO2. ACS Catal. 2022, 12, 11118–11128. [CrossRef]
- Ye, X.; Yang, C.; Pan, X.; Ma, J.; Zhang, Y.; Ren, Y.; Liu, X.; Li, L.; Huang, Y., Highly Selective Hydrogenation of CO2 to Ethanol via Designed Bifunctional Ir1-In2O3 Single-Atom Catalyst. J Am Chem Soc 2020, 142, (45), 19001-19005.
- An, K.; Zhang, S.; Wang, J.; Liu, Q.; Zhang, Z.; Liu, Y. A highly selective catalyst of Co/La4Ga2O9 for CO2 hydrogenation to ethanol. J. Energy Chem. 2020, 56, 486–495. [CrossRef]
- Zhang, S.; Wu, Z.; Liu, X.; Shao, Z.; Xia, L.; Zhong, L.; Wang, H.; Sun, Y. Tuning the interaction between Na and Co2C to promote selective CO2 hydrogenation to ethanol. Appl. Catal. B: Environ. 2021, 293, 120207. [CrossRef]
- Liu, S.; Zhou, H.; Zhang, L.; Ma, Z.; Wang, Y., Activated Carbon-Supported Mo-Co-K Sulfide Catalysts for Synthesizing Higher Alcohols from CO2. Chem Eng Technol 2019, 42, (5), 962-970.
- Zhang, H.; Han, H.; Xiao, L.; Wu, W., Highly Selective Synthesis of Ethanol via CO2 Hydrogenation over CoMoCx Catalysts. Chemcatchem 2021, 13, (14), 3333-3339.
- Zhang, G.; Fan, G.; Zheng, L.; Li, F., Ga-Promoted CuCo-Based Catalysts for Efficient CO2 Hydrogenation to Ethanol: The Key Synergistic Role of Cu-CoGaOx Interfacial Sites. Acs Appl Mater Interfaces 2022, 14, (31), 35569-35580.
- Irshad, M.; Chun, H.-J.; Khan, M.K.; Jo, H.; Kim, S.K.; Kim, J. Synthesis of n-butanol-rich C3+ alcohols by direct CO2 hydrogenation over a stable Cu–Co tandem catalyst. Appl. Catal. B: Environ. 2024, 340. [CrossRef]
- Zhang, Q.; Wang, S.; Geng, R.; Wang, P.; Dong, M.; Wang, J.; Fan, W. Hydrogenation of CO2 to higher alcohols on an efficient Cr-modified CuFe catalyst. Appl. Catal. B: Environ. 2023, 337. [CrossRef]
- Si, Z.; Wang, L.; Han, Y.; Yu, J.; Ge, Q.; Zeng, C.; Sun, J. Synthesis of Alkene and Ethanol in CO2 Hydrogenation on a Highly Active Sputtering CuNaFe Catalyst. ACS Sustain. Chem. Eng. 2022, 10, 14972–14979. [CrossRef]
- Yamamoto, T.; Inui, T., Highly effective synthesis of ethanol from CO2 on Fe, Cu-based novel catalysts. In Advances in Chemical Conversions for Mitigating Carbon Dioxide, Inui, T.; Anpo, M.; Izui, K.; Yanagida, S.; Yamaguchi, T., Eds 1998, 114, 513-516.
- Guo, H.; Li, S.; Peng, F.; Zhang, H.; Xiong, L.; Huang, C.; Wang, C.; Chen, X. Roles Investigation of Promoters in K/Cu–Zn Catalyst and Higher Alcohols Synthesis from CO2 Hydrogenation over a Novel Two-Stage Bed Catalyst Combination System. Catal. Lett. 2014, 145, 620–630. [CrossRef]
- Wang, Y.; Wang, K.; Zhang, B.; Peng, X.; Gao, X.; Yang, G.; Hu, H.; Wu, M.; Tsubaki, N. Direct Conversion of CO2 to Ethanol Boosted by Intimacy-Sensitive Multifunctional Catalysts. ACS Catal. 2021, 11, 11742–11753. [CrossRef]
- Xu, D.; Yang, H.; Hong, X.; Liu, G.; Tsang, S.C.E. Tandem Catalysis of Direct CO2 Hydrogenation to Higher Alcohols. ACS Catal. 2021, 11, 8978–8984. [CrossRef]
- Huang, J.; Zhang, G.; Wang, M.; Zhu, J.; Ding, F.; Song, C.; Guo, X. The synthesis of higher alcohols from CO2 hydrogenation over Mn-Cu-K modified Fe5C2 and CuZnAlZr tandem catalysts. Front. Energy Res. 2023, 10. [CrossRef]
- Wang, Y.; Xu, D.; Zhang, X.; Hong, X.; Liu, G., Selective C2+ alcohol synthesis by CO2 hydrogenation via a reaction-coupling strategy. Catalysis Science & Technology 2022, 12, (5), 1539-1550.
- Zhang, S.; Huang, C.; Shao, Z.; Zhou, H.; Chen, J.; Li, L.; Lu, J.; Liu, X.; Luo, H.; Xia, L.; Wang, H.; Sun, Y., Revealing and Regulating the Complex Reaction Mechanism of CO2 Hydrogenation to Higher Alcohols on Multifunctional Tandem Catalysts. Acs Catal 2023, 13, (5), 3055-3065.
- Liu, T.; Xu, D.; Song, M.; Hong, X.; Liu, G., K-ZrO2 Interfaces Boost CO2 Hydrogenation to Higher Alcohols. Acs Cata 2023, 13, 4667-4674.
- Chen, J.; Zha, Y.; Liu, B.; Li, Y.; Xu, Y.; Liu, X. Rationally Designed Water Enriched Nano Reactor for Stable CO2 Hydrogenation with Near 100% Ethanol Selectivity over Diatomic Palladium Active Sites. ACS Catal. 2023, 13, 7110–7121. [CrossRef]
Figure 1.
Thermodynamic analyses of CO2 hydrogenation reactions (A and B) Diagram of (A) standard enthalpy variation (ΔH°) and (B) equilibrium constant (K) and standard Gibbs free energy change (ΔG°) of involving reactions along with temperature.(C and D) Equilibrium composition of the hydrogenation of CO2 to (C) methane and ethanol, and (D) CO and ethanol at 5 MPa (H2/CO2 = 3/1). All the thermodynamics were calculated with HSC Chemistry 9 software [37].
Figure 1.
Thermodynamic analyses of CO2 hydrogenation reactions (A and B) Diagram of (A) standard enthalpy variation (ΔH°) and (B) equilibrium constant (K) and standard Gibbs free energy change (ΔG°) of involving reactions along with temperature.(C and D) Equilibrium composition of the hydrogenation of CO2 to (C) methane and ethanol, and (D) CO and ethanol at 5 MPa (H2/CO2 = 3/1). All the thermodynamics were calculated with HSC Chemistry 9 software [37].
Scheme 1.
Illustration for the Reaction Pathways of CO2 Hydrogenation over the Cs-CuFeZn Catalysts [32].
Scheme 1.
Illustration for the Reaction Pathways of CO2 Hydrogenation over the Cs-CuFeZn Catalysts [32].
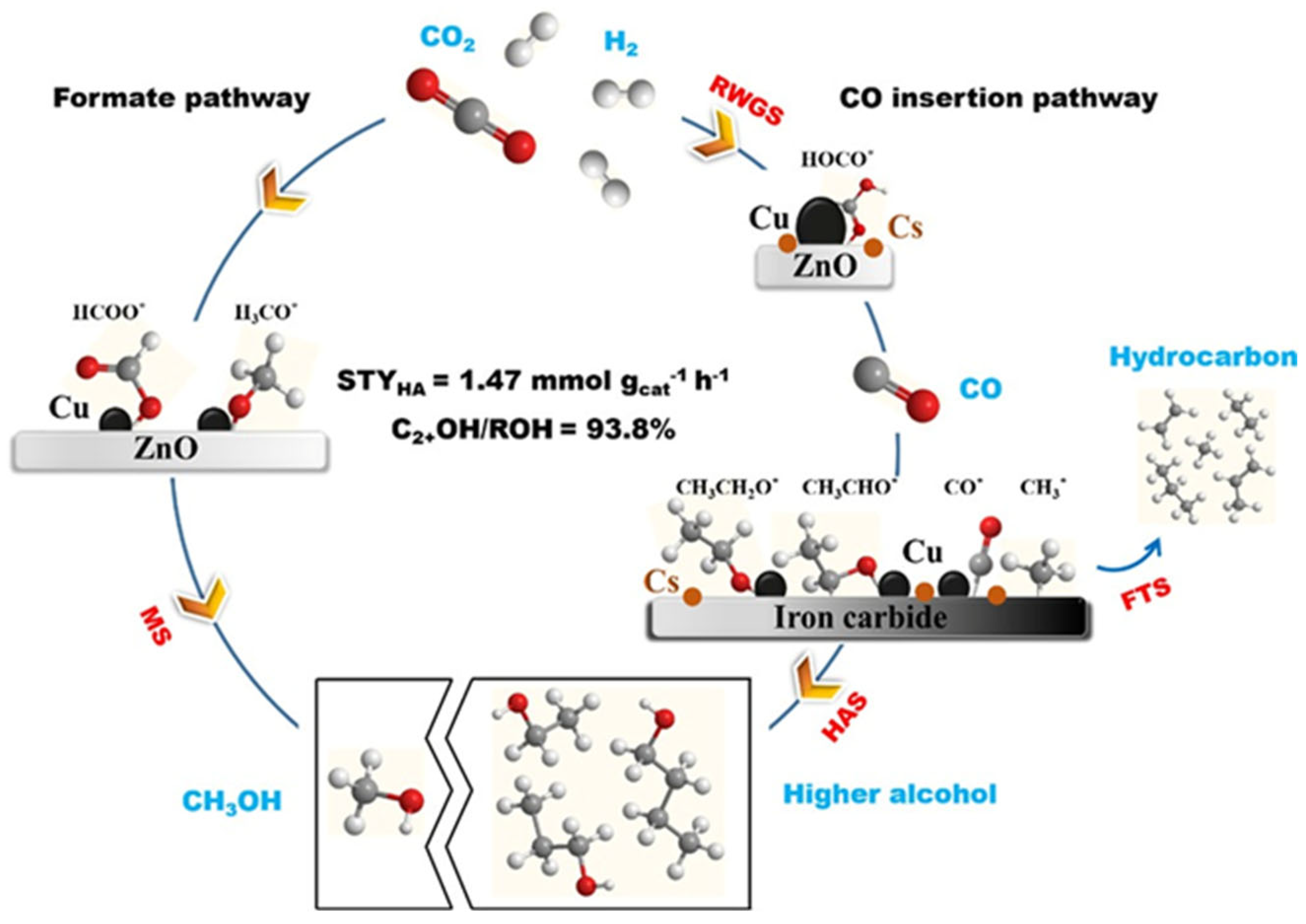
Scheme 2.
Illustration for the Reaction Pathways of CO2 Hydrogenation over the Cs/Cu/ZnO Catalyst [34].
Scheme 2.
Illustration for the Reaction Pathways of CO2 Hydrogenation over the Cs/Cu/ZnO Catalyst [34].
Scheme 3.
Proposed Mechanism for the CO2 Hydrogenation to Produce Ethanol over Ga-Promoted CuCo-Based Catalysts [71].
Scheme 3.
Proposed Mechanism for the CO2 Hydrogenation to Produce Ethanol over Ga-Promoted CuCo-Based Catalysts [71].
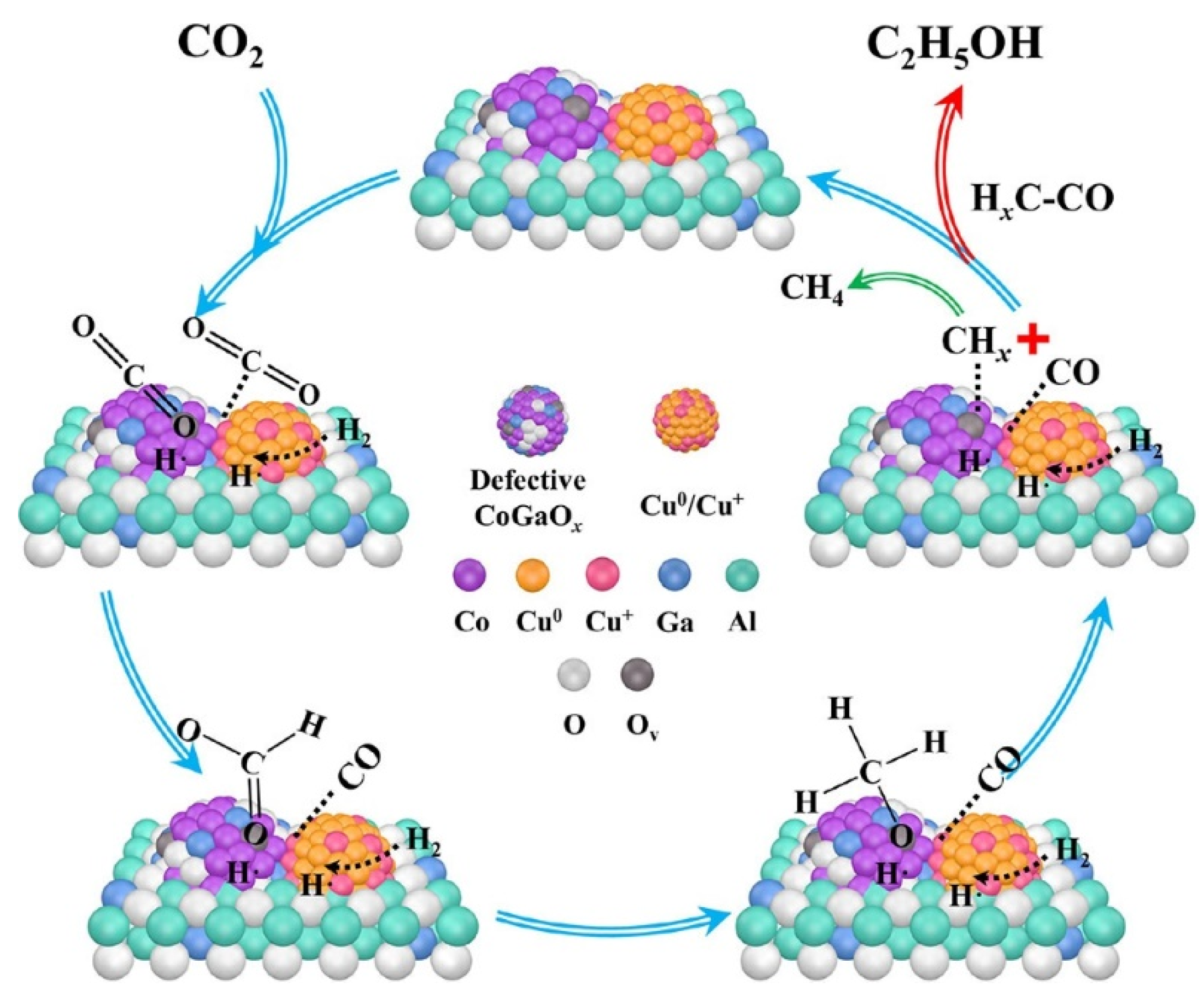
Figure 3.
Results of CO2 hydrogenation over the catalysts packed in different manners. (a) CO2 conversion and products selectivity, (b) alcohol distribution and STY, and (c) HC distribution and α over a series of CZA/K-CMZF multifunctional catalysts (CZA/K-CMZF mass ratio of 1) with different integration manners. Reaction conditions: 5 MPa, CO2/H2 = 1/3, 6 L gcat−1 h−1, and 320 °C [78].
Figure 3.
Results of CO2 hydrogenation over the catalysts packed in different manners. (a) CO2 conversion and products selectivity, (b) alcohol distribution and STY, and (c) HC distribution and α over a series of CZA/K-CMZF multifunctional catalysts (CZA/K-CMZF mass ratio of 1) with different integration manners. Reaction conditions: 5 MPa, CO2/H2 = 1/3, 6 L gcat−1 h−1, and 320 °C [78].
Scheme 4.
Proposed Reaction Pathway and Proximity Effect for CO2 Hydrogenation to higher alcohols over the CZA/K-CMZF Multifunctional Catalyst [78].
Scheme 4.
Proposed Reaction Pathway and Proximity Effect for CO2 Hydrogenation to higher alcohols over the CZA/K-CMZF Multifunctional Catalyst [78].
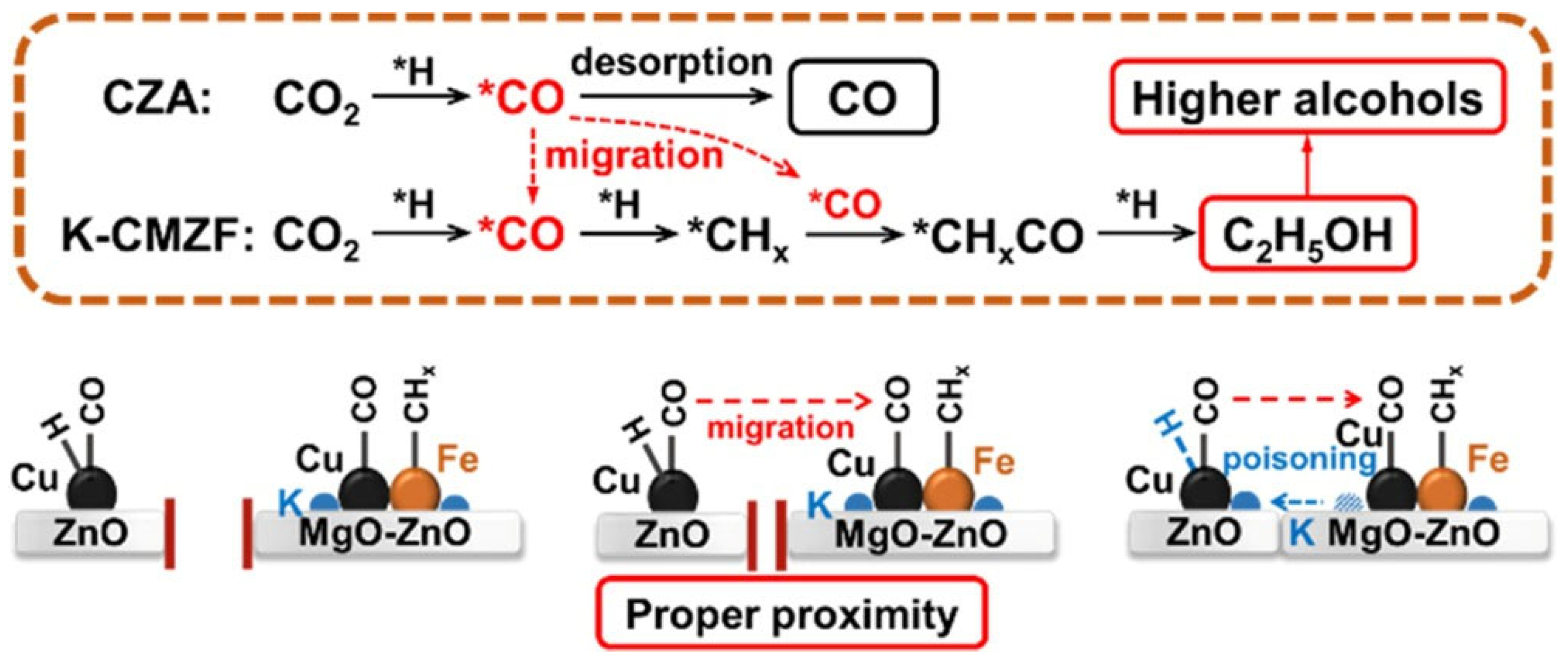
Figure 4.
Effect of intimacy modes on the catalytic performance. (a)Physical-mixing of the catalyst powder, 2% Na-Fe@C/5%-K-CuZnAl. (b) Granule-mixing, 2% Na-Fe@C/5% K-CuZnAl with different amounts of quartz sand (0, 1, and 2 g). (c) Dual-bed with 2% Na-Fe@C loaded above 5% K-CuZnAl and 2% Na-Fe@C||5% K-CuZnAl.(d) Dual-bed with 2% Na-Fe@C loaded below 5% K-CuZnAl and 5%K-CuZnAl||2% Na-Fe@C. Reaction conditions: 320 °C, 5 MPa(25.6% CO2, 71.36% H2, and 3.04% Ar), 15 mL min−1, and time on stream (TOS) = 8 h. Catalyst weight: 0.1 g of Na-Fe@C, 0.1 g of 5%K-CuZnAl, and 1 g of quartz sand [77].
Figure 4.
Effect of intimacy modes on the catalytic performance. (a)Physical-mixing of the catalyst powder, 2% Na-Fe@C/5%-K-CuZnAl. (b) Granule-mixing, 2% Na-Fe@C/5% K-CuZnAl with different amounts of quartz sand (0, 1, and 2 g). (c) Dual-bed with 2% Na-Fe@C loaded above 5% K-CuZnAl and 2% Na-Fe@C||5% K-CuZnAl.(d) Dual-bed with 2% Na-Fe@C loaded below 5% K-CuZnAl and 5%K-CuZnAl||2% Na-Fe@C. Reaction conditions: 320 °C, 5 MPa(25.6% CO2, 71.36% H2, and 3.04% Ar), 15 mL min−1, and time on stream (TOS) = 8 h. Catalyst weight: 0.1 g of Na-Fe@C, 0.1 g of 5%K-CuZnAl, and 1 g of quartz sand [77].
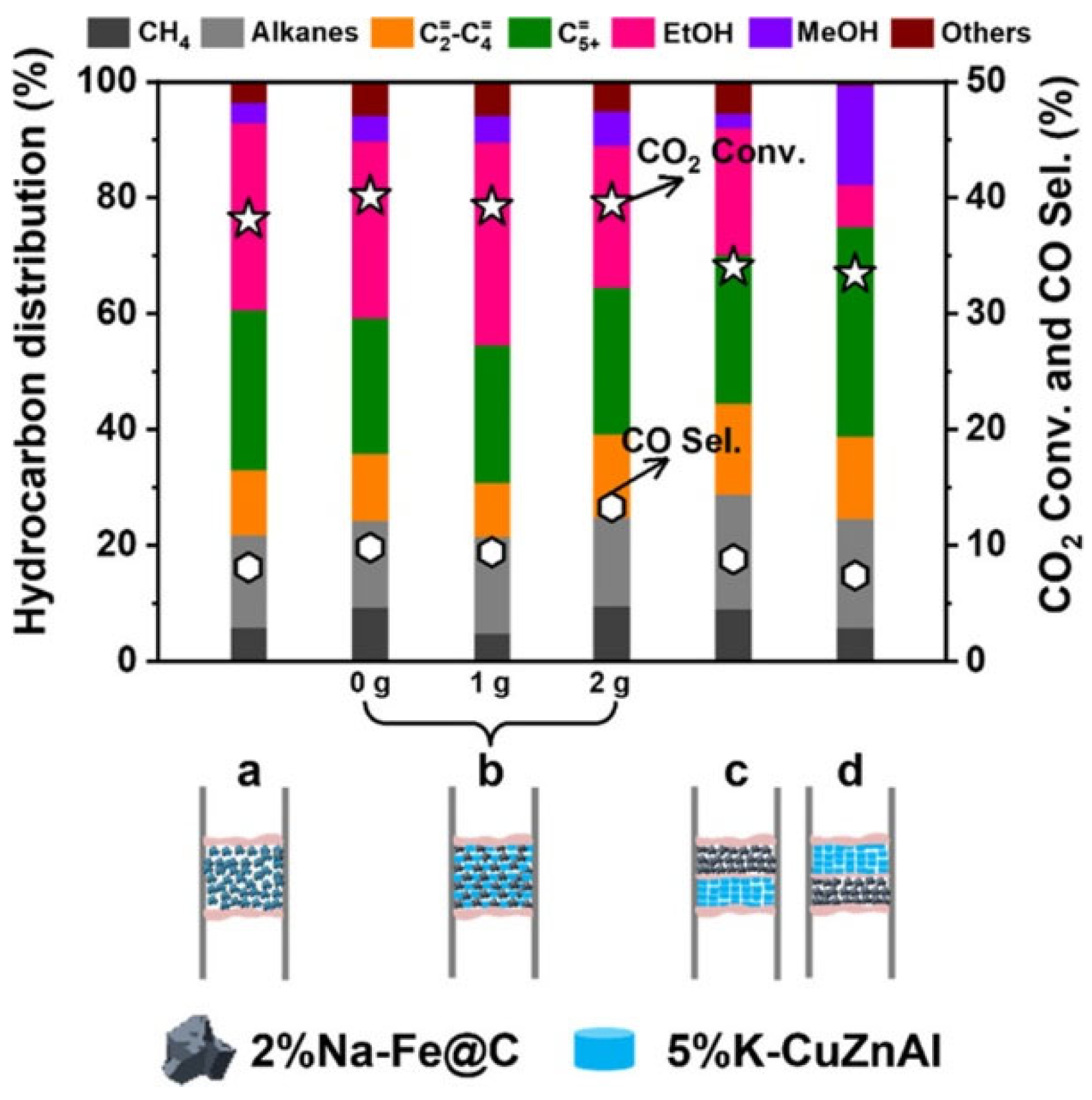
Figure 5.
Reaction network for ethanol synthesis from CO2 hydrogenation via the Na-Fe@C/K-CuZnAl multifunctional catalyst [77].
Figure 5.
Reaction network for ethanol synthesis from CO2 hydrogenation via the Na-Fe@C/K-CuZnAl multifunctional catalyst [77].
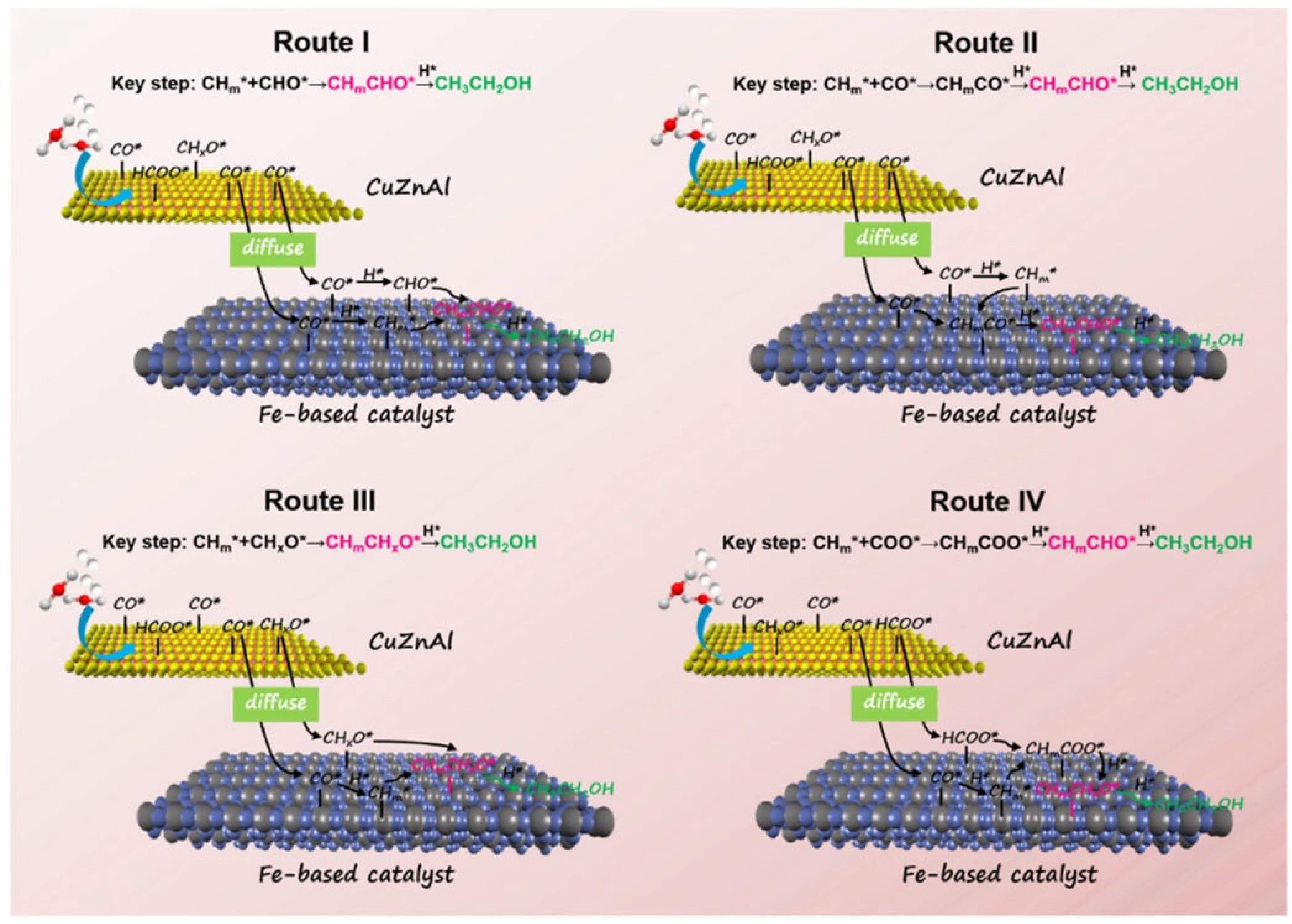
Figure 6.
Catalytic performance of different multifunctional catalysts in CO2 hydrogenation. (a) CO2 conversion and product selectivity. (b)Alcohol distribution and the STY. (c) Effects of space velocity on the Co2C||CuZnAl catalyst. (d) Effects of mass ratio on the Co2C||CuZnAl catalyst [81].
Figure 6.
Catalytic performance of different multifunctional catalysts in CO2 hydrogenation. (a) CO2 conversion and product selectivity. (b)Alcohol distribution and the STY. (c) Effects of space velocity on the Co2C||CuZnAl catalyst. (d) Effects of mass ratio on the Co2C||CuZnAl catalyst [81].
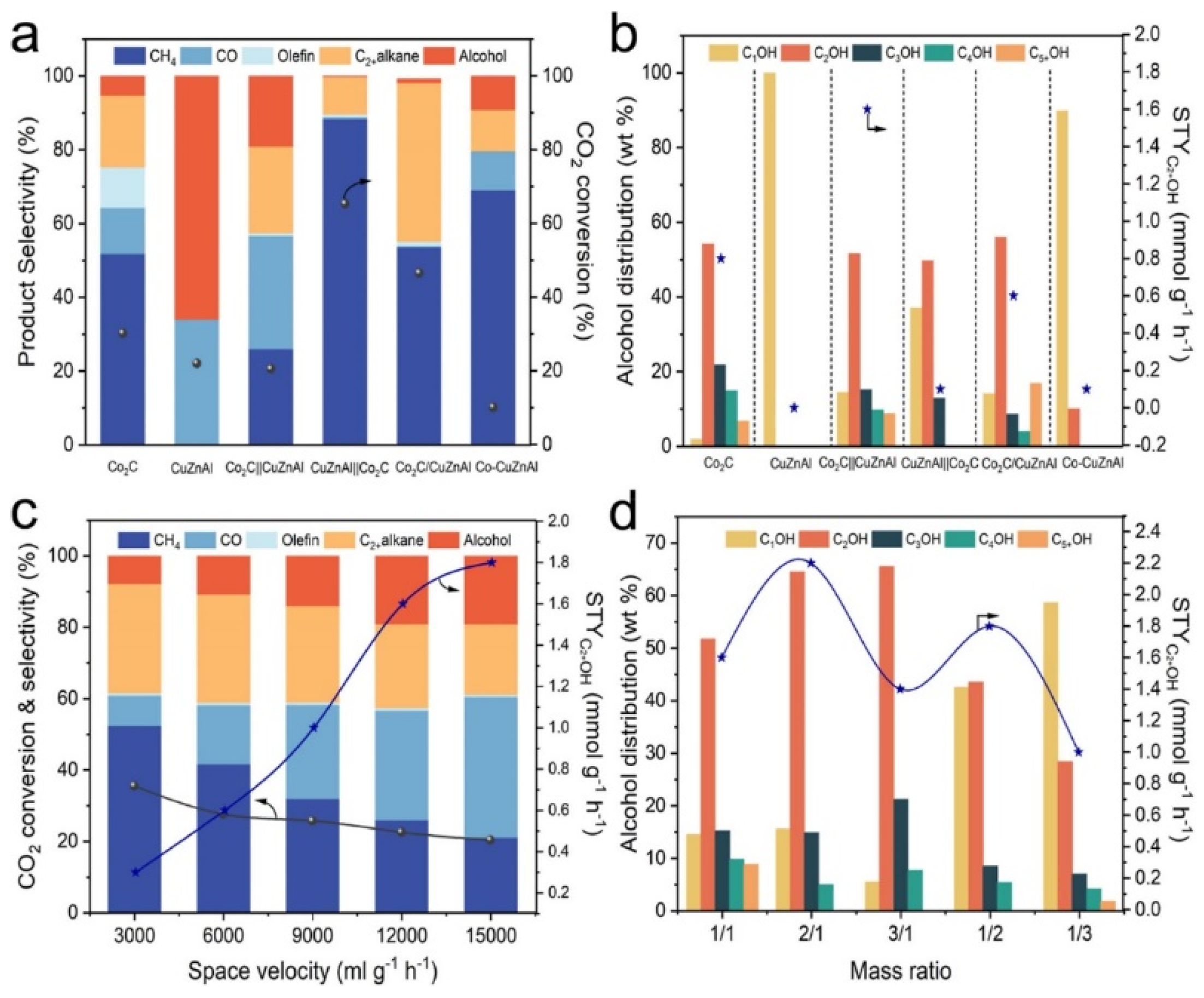
Figure 7.
Reaction simulations of different products on CuZnAl in CTK tests. (a) Response of alcohols in switching H2/CO2 to CO, CH4, C2H4, and C3H6. (b) Response of alcohols in switching H2/CO2 to H2/CO, H2/CO/C2H4, and CH3OH/Ar [81].
Figure 7.
Reaction simulations of different products on CuZnAl in CTK tests. (a) Response of alcohols in switching H2/CO2 to CO, CH4, C2H4, and C3H6. (b) Response of alcohols in switching H2/CO2 to H2/CO, H2/CO/C2H4, and CH3OH/Ar [81].
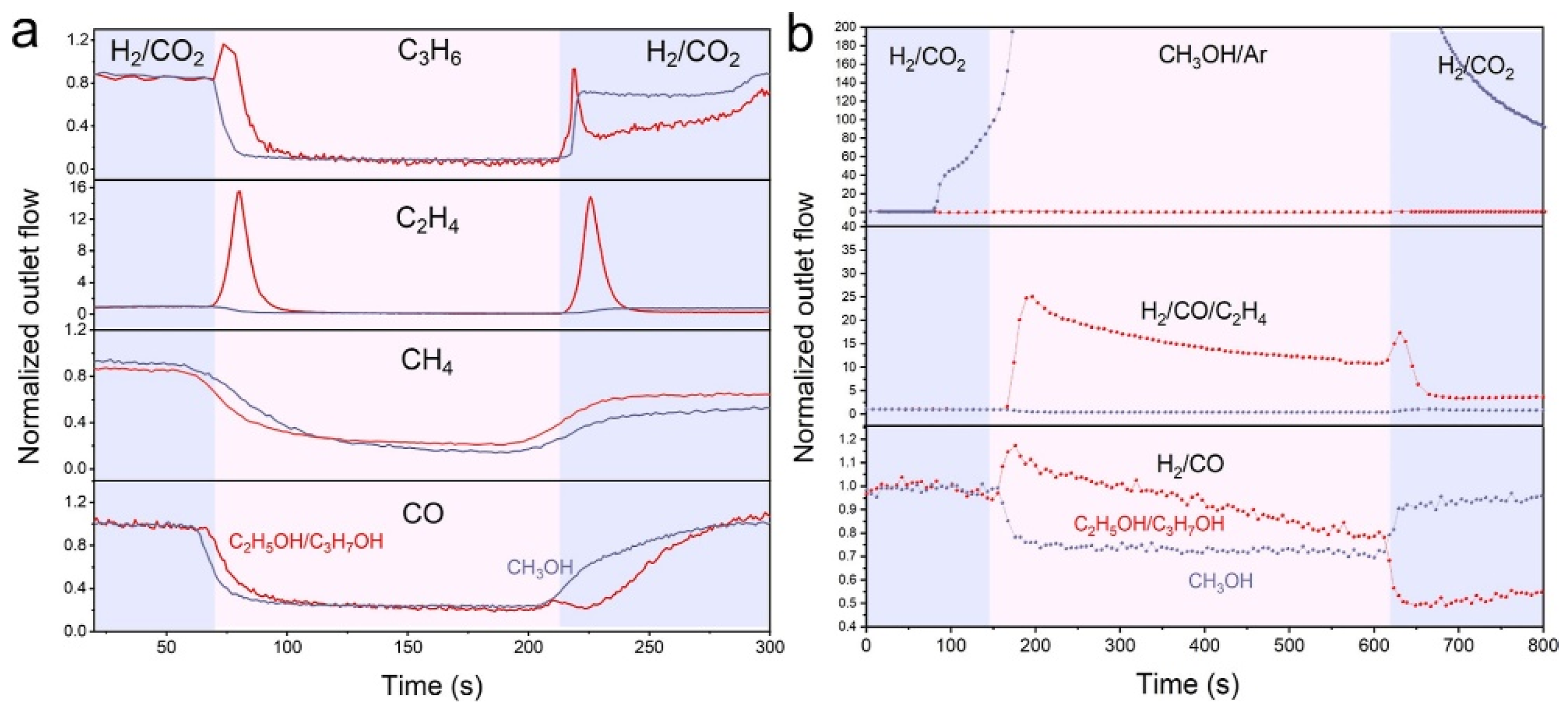
Figure 8.
CO2 conversion and product selectivity over CZA/K-CMZF multifunctional catalyst (CZA/K-CMZF mass ratio of 1, powder mixing) at different (a) reaction temperatures, (b) reaction pressures, and (c) WHSV. (d) Correlation between CO2 conversion and CO selectivity under different reaction conditions. Standard reaction conditions: 5 MPa, CO2/H2 = 1/3, 6 L gcat−1 h−1, and 320 °C [81].
Figure 8.
CO2 conversion and product selectivity over CZA/K-CMZF multifunctional catalyst (CZA/K-CMZF mass ratio of 1, powder mixing) at different (a) reaction temperatures, (b) reaction pressures, and (c) WHSV. (d) Correlation between CO2 conversion and CO selectivity under different reaction conditions. Standard reaction conditions: 5 MPa, CO2/H2 = 1/3, 6 L gcat−1 h−1, and 320 °C [81].
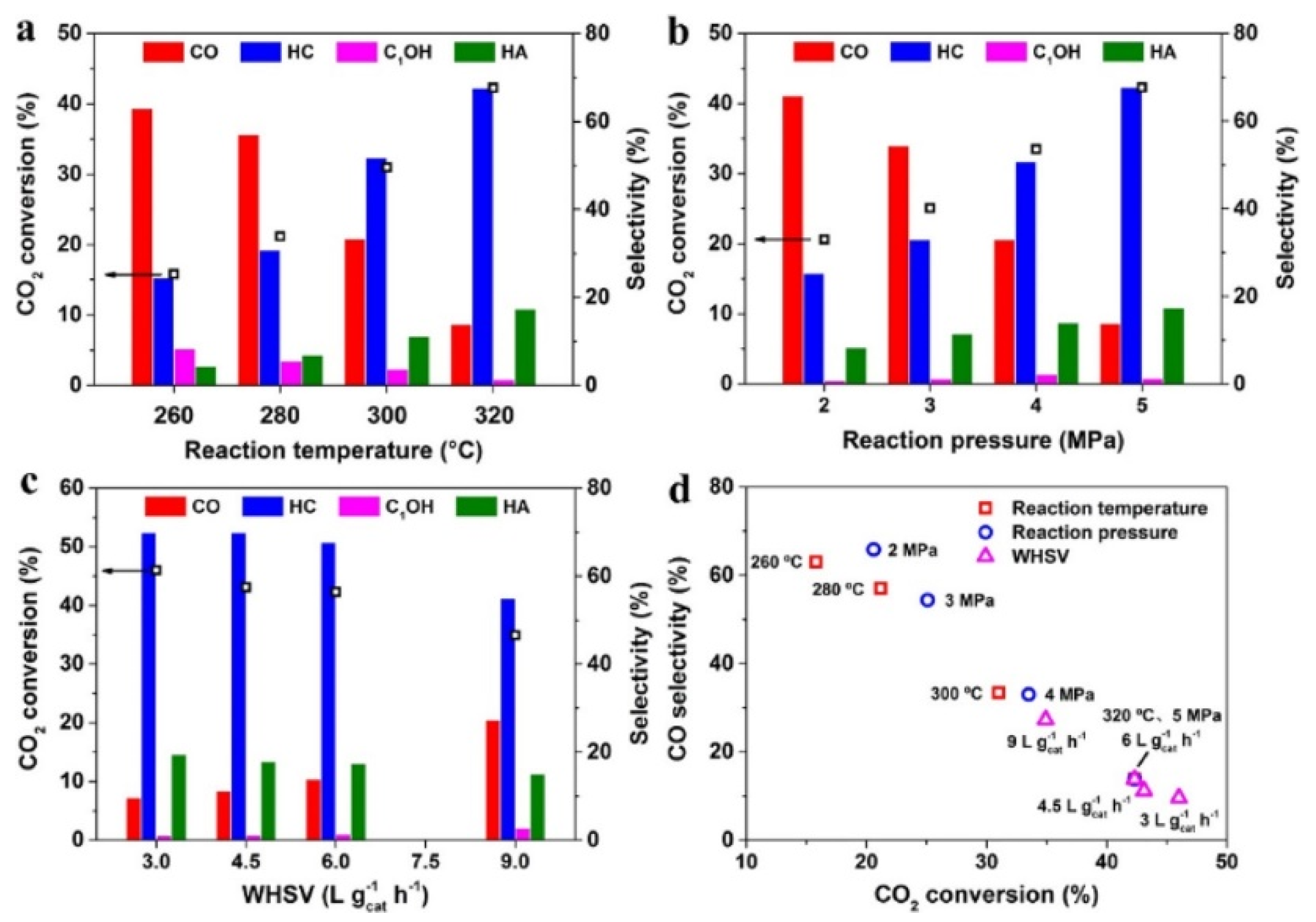
Table 1.
The main-side reactions and △G298K, △H298K,K298K of CO2 hydrogenation system [29].
Table 1.
The main-side reactions and △G298K, △H298K,K298K of CO2 hydrogenation system [29].
| Reaction Equation | △G298 K (kJ/mol) |
△H298 K (kJ/mol) |
K298K |
|---|---|---|---|
| CO2 + H2 ↔ CO + H2O | 28.6 | 41.1 | 9.67×10-6 |
| CO2 + 4H2 ↔ CH4 + 2H2O | -113.5 | -165.0 | 7.79×1019 |
| n CO2 + (3n+1) H2 ↔ CnH2n+2 + 2n H2O | |||
| n CO2 + 3n H2 ↔ CnH2n + 2n H2O | |||
| CO2 +3H2 ↔ CH3OH + H2O | 3.5 | -49.3 | 2.45×10-1 |
| 2CO2 + 6H2 ↔ C2H5OH +3H2O | -32.4 | -86.7 | 4.70×105 |
| n CO2 + 3n H2 ↔ CnH2n+1OH + (2n-1) H2O |
Table 3.
Catalytic Performance of HAS from CO2 Hydrogenation over Multi component catalyst.
| Enter | Catalysts | T (℃) |
P (MPa) |
GHSV/ L.g-1h-1 |
H2/ CO2 |
XCO2 (%) |
SCO (%) |
SHC (%) |
SMeOH (%) |
SHA (%) |
STYHA (mmolgcat-1h-1) |
Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | In2O3 | 200 | 5 | / | 4 | 0.1 | / | / | 100 | / | / | [61] |
| 2 | 2.5K5Co-In2O3 | 380 | 4 | / | 3 | 36.6 | 80.8 | 33.9 | 8.2 | 57.9 | 3.73 | [64] |
| 3 | In2Fe/K-Al2O3 | 300 | 2 | 4 | 3 | 36.7 | 7.4 | 37.9 | 2.3 | 42 | / | [65] |
| 4 | Ir1-In2O3 | 200 | 6 | / | 5 | / | / | / | / | 99.7 | 0.99 | [66] |
| 5 | Co/La4Ga2O9 | 280 | 3 | 3 | 3 | 9.6 | 10.8 | 52.2 | 13.7 | 23.3 | / | [67] |
| 6 | Na-Co/SiO2 | 250 | 5 | 6 | 3 | 21.5 | 26.3 | 61.2 | 1.7 | 10.8 | 0.47 | [68] |
| 7 | Mo-Co-K | 320 | 5 | 3 | 3 | 23.5 | / | 21 | 70 | 8.9 | / | [29] |
| 8 | CoMoCx-800 | 180 | 2 | / | 3 | / | / | / | / | / | 0.53 | [69] |
| 9 10 11 |
Cu-CoGa-0.4 Na-CuCo-9 Cs-C0.8F1.0Z1.0 |
220 330 330 |
3 4 5 |
6 5 4 |
3 1 3 |
17.8 20.1 36.6 |
2.3 26.5 / |
43.5 53.8 / |
27.5 0.5 1.2 |
23.8 26.3 18.6 |
1.35 1.09a 1.47 |
[70] [71] [72] |
| 12 | Cr-CuFe | 320 | 4 | 6 | 3 | 38.4 | 14.8 | / | / | 29.2 | 104.1b | [73] |
| 13 | sp-CuNaFe | 310 | 3 | 28.8 | / | 32.2 | / | / | / | 10 | 153b | [74] |
a C3+ alcohol space time yield b mg gcat-1h-1
Table 4.
Catalytic Performance of HAS from CO2 Hydrogenation over Tandem catalyst.
| Enter | Catalysts | T (℃) |
P (MPa) |
GHSV/ L.g-1h-1 |
H2/ CO2 |
XCO2 (%) |
SCO (%) |
SHC (%) |
SMeOH (%) |
SHA (%) |
STYHA (mmolgcat-1h-1) |
Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CuZnAlK/FeCuAlK | 330 | 8 | 20 | 3 | 54.5 | 9.7 | 64.5 | 5.2 | 17.0 | / | [75] | |
| 2 | CZK||CFCK | 350 | 6 | 5 | 3 | 32.4 | 45.3 | 42.9 | 5.3 | 6.5 | 1.37 | [76] | |
| 3 | Na-Fe@C/KCuZnAl | 320 | 5 | / | 2.8 | 39.2 | 9.4 | 54.5 | 4.6 | 35 | / | [77] | |
| 4 | MnCuK-FeC/CuZnAlZr | 300 | 3 | 6 | 3 | 42.1 | 22.7 | 60.6 | 1.2 | 15.5 | / | [79] | |
| 5 | CuZnAl/K-CuMgZnFe | 320 | 5 | 6 | 3 | 42.3 | 13.8 | 67.6 | 1.3 | 17.4 | 2.24 | [78] | |
| 6 | 4.7KCuFeZn/CuZnAlZr | 300 | 5 | 3 | 3 | 29.4 | 26.1 | 66.7 | 2.1 | 31.2 | / | [80] | |
| 7 | Co2C||CuZnAl | 250 | 5 | 12 | 3 | 21.2 | / | / | / | 18.3 | 2.20 | [81] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



