Submitted:
11 April 2024
Posted:
15 April 2024
You are already at the latest version
Abstract
Systemic inflammation and coronary microvascular endothelial dysfunction are essential pathophysiological factors in heart failure with preserved ejection fraction (HFpEF) that support the use of statins. The pleiotropic properties of statins, such as anti-inflammatory, antihypertrophic, antifibrotic, and antioxidant effects, are generally accepted and may be beneficial in HF, especially in HFpEF. Numerous observational clinical trials have consistently shown a beneficial prognostic effect of statins in patients with HFpEF, while the results of two larger trials in patients with HFrEF have been controversial. Such differences may be related to a more pronounced impact of the pleiotropic properties of statins on the pathophysiology of HFpEF and pro-inflammatory comorbidities (arterial hypertension, diabetes mellitus, obesity, chronic kidney disease) that are more common in HFpEF. This review discusses the potential mechanisms of statin action that may be beneficial for patients with HFpEF, as well as clinical trials that have evaluated the statin effects on left ventricular diastolic function and clinical outcomes in patients with HFpEF.
Keywords:
statins
; heart failure with preserved ejection fraction
; inflammation
; diastolic dysfunction
; prognosis
1. Introduction
Approximately half of patients with heart failure have normal (preserved) ejection fraction (HFpEF) [1]. HFpEF is becoming one of the key problems of modern healthcare. The prevalence of HFpEF is steadily increasing due to aging population and high incidence of arterial hypertension, obesity, and diabetes mellitus. The prognosis is almost as unsatisfactory as in HF with reduced ejection fraction (HFrEF), while effective treatments for HFpEF are still limited [1]. Most of therapies that improve outcomes in HFrEF have not been as effective in HFpEF, which may be explained by the difference in the initial pathophysiological process in these two major HF phenotypes – cardiomyocyte death in HFrEF and microvascular myocardial inflammation in HFpEF [2].
The results of multiple clinical trials with anti-inflammatory/immunomodulatory therapy for HFrEF (with corticosteroids, methotrexate, immunoglobulin, or tumor necrosis factor [TNF] inhibitors) have been inconclusive and contradictory [3]. The lack of success of these trials is partly due to the fact that myocardial inflammation in HFrEF is detected only at advanced stages and is a result of reactive response to severe LV systolic dysfunction, while at earlier stages left ventricular (LV) remodeling is controlled primarily through cardiomyocyte loss [2]. In contrast, in HFpEF, remodeling is initially governed by chronic microvascular inflammation, which is supported by compelling preclinical and clinical evidence [4].
The pro-inflammatory paradigm of HFpEF was proposed in 2013 by Paulus W.J. and Tschöpe C. [2]. Most patients with HFpEF have multiple extracardiac pro-inflammatory comorbidities such as obesity, hypertension, atrial fibrillation, type 2 diabetes mellitus, coronary artery disease, chronic kidney disease, chronic obstructive pulmonary disease, anemia [5]. These comorbidities along with advanced unhealthy ageing have been postulated to trigger a sustained low-grade systemic inflammatory response with systemic endothelial dysfunction, including microvascular coronary dysfunction. Activated endothelial cells begin to express adhesion molecules on their surface [6] with subsequent myocardial infiltration with activated leukocytes, the transformation of fibroblasts into myofibroblasts and the development of cardiac interstitial fibrosis [2,7]. Endothelial microvascular inflammation also triggers oxidative stress, reduces nitric oxide (NO) bioavailability and impairs the NO - cyclic guanosine monophosphate (cGMP) - protein kinase G (PKG) signaling pathway, resulting in cardiomyocyte hypertrophy, altered myofilament protein phosphorylation and increased cardiomyocyte stiffness [8]. In turn, interstitial fibrosis and stiff cardiomyocytes contribute to increased LV stiffness and LV filling pressure with the development of HFpEF.
Thus, chronic low-intensity myocardial inflammation may play a key role in the pathogenesis of HFpEF, supporting the use of anti-inflammatory therapy. In these circumstances, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (or statins) hold considerable promise [2]. In addition to their hypolipidaemic effects and a high safety profile, statins stimulate nitric oxide production in endothelial cells and exhibit anti-inflammatory, antioxidant, antihypertrophic and antifibrotic activities [9]. In this review, we consider the potential mechanisms of statin action explored in experimental studies that may be useful in HFpEF, as well as clinical trials that have evaluated the effects of statins on LV diastolic function and clinical outcomes in patients with HFpEF.
2. Statins: General Presentation
Statins act as competitive and reversible inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme of cholesterol biosynthesis, and thereby significantly reduce de novo cholesterol synthesis in the liver. Statins reduce the blood levels of atherogenic lipoproteins and thereby reduce the risk of coronary artery disease and its complications, regardless of blood cholesterol levels [10]. Statins stabilise existing atheromatous plaques and prevent the formation of new plaques, thereby reducing the overall coronary burden [11], which may be of great importance for HFrEF of ischaemic aetiology.
In addition to their hypolipidaemic effects, statins exert a plethora of pleiotropic effects without a clear relationship to the low-density lipoprotein cholesterol (LDL-C) levels [12]. The pleiotropic properties of statins, such as an improvement of endothelial function and increase in the bioavailability of nitric oxide [13], anti-inflammatory [14], immunomodulatory and antioxidant effects [15], regression of cardiac fibrosis and hypertrophy [16,17] are generally accepted and may be beneficial in HF population (Figure 1). The spectrum and magnitude of pleiotropic activity of statins depends on their chemical and pharmacokinetic properties as well as on the sensitivity of cells to these drugs [18]. The pleiotropic effects of statins are thought to be mainly due to a decrease in the production of mevalonate and its numerous metabolites, such as the isoprenoid intermediates farnesylpyrophosphate (FPP) and geranyl-geranylpyrophosphate (GGPP) [12]. FPP and GGPP are involved in posttranslational modification (prenylation) of a wide range of biological molecules, primarily small GTPases, leading to an increase in their hydrophobicity and anchoring in the plasma membrane and, thereby, their activation. Small GTPases prenylated by FPPs include members of the Ras family (K-Ras, H-Ras, N-Ras, RhoE, Rheb, and RhoB); other GTPases are prenylated by GGPP (Rac1, Rac2, Ra1A, Rap1B, RhoA, RhoB, RhoC, Cdc42, Rab family) [19]. Small GTPases are involved in signaling pathways controlling many important cellular processes, such as cell proliferation, differentiation, apoptosis, adhesion, migration, cytokine production, and function of cytoskeleton [20].
One of the key substrates for activated Rho GTPases are serine/threonine Rho-associated coiled-coil-containing protein kinases (ROCKs). ROCKs are represented by two isoforms (ROCK1 and ROCK2) that have different subcellular distribution in different tissues (vascular smooth muscle cells, endothelium, fibroblasts, inflammatory cells, cardiomyocytes), but perform similar functions, regulating many cellular processes including proliferation, motility, and cell viability [21]. Activation of ROCKs may suppress the pro-survival phosphatidylinositol 3-kinase (PI3K)/Akt (protein kinase B) pathway and induce cardiac fibrosis (via activation of profibrotic gene expression and transdifferentiation of fibroblasts into myofibroblasts) and myocardial hypertrophy in pathologic conditions [9]. ROCKs activity has been shown to be significantly increased in leukocytes from patients with cardiovascular diseases such as metabolic syndrome, arterial hypertension and coronary heart disease [22,23,24]. Hence, statins, by preventing the geranylgeranylation of Rho and its activation of ROCKs, may inhibit their downstream effects [9].
Statins can also realize their pleiotropic effects through epigenetic modifications, binding to some proteins and modulating their activity. Among mevalonate-independent targets of statins are histone deacetylase 2 (HDAC2) [9]. Inhibition of HDAC2 activity promotes the accumulation of acetylated histone H3 and increased expression of the cyclin-dependent protein kinase inhibitor protein p21 [25].
3. Microcirculatory Endothelial Dysfunction in HFpEF and Endothelial Effects of Statins
The pivotal event in the pathogenesis of HFpEF is the development of endothelial microvascular endothelial dysfunction. A hallmark of endothelial dysfunction is a decrease in NO production and its diffusion to the underlying smooth muscle myocytes/cardiomyocytes (decreased NO bioavailability) as a result of decreased expression/activation of endothelial NO synthase (eNOS) and/or eNOS uncoupling. The decrease in NO production in HFpEF is largely due to the activation of free-radical oxidation processes within endothelial cells via nicotinamide adenine dinucleotide phosphate (NADH)-oxidase [6,26], resulting in NO binding to superoxide (O2–) with formation of a cytotoxic reactive oxidant peroxynitrite (ONOO–) [26]. In addition, reactive oxygen species (ROS) contribute to reduced bioavailability of tetrahydrobiopterin (BH4), a cofactor of eNOS, leading to subsequent substrate uncoupling of eNOS with production of O2– instead of NO (Figure 2). To date, oxidative stress is a highly evident component of the pathophysiology of HFpEF [27].
Statins induce eNOS upregulation (increase eNOS mRNA stability) and thereby increase NO bioavailability (Figure 2) [28]. This effect may be achieved through inhibition of the Rho/ROCK pathway [29] and activation of PI3K/protein kinase Akt pathway [29]. In endothelial cells, ROCKs inhibit the pro-survival PI3K/protein kinase Akt pathway, resulting in decreased eNOS expression and cGMP levels [30]. ROCKs also induce NADPH-oxidase, leading to the formation of ROS. In clinical trials, statins reduced ROCK activity in patients with different cardiac diseases such as atherosclerosis, heart failure and hypertension [31,32,33] regardless of the reduction in LDL-C blood levels [33]. The inhibition of the Rho/ROCK pathway by statins also results in decreased ex-expression of miRNA-155, which suppresses eNOS expression [34].
Other mechanisms of statins to increase NO bioavailability have also been suggested, such as a downregulation in membrane protein caveolin-1 that directly binds to and inhibits eNOS [35], an induction of kruppel-like factor-2 mRNA that are required for eNOS expression [36], an increase in the concentration of a cofactor for eNOS function BH4 [37], or increased mobilisation of endothelial progenitor cells [38]. The increase in NO bioavailability with statins has been associated with an enhancement of myocardial blood flow under hypoxia [39], as well as an increase in capillary density and improvement in myocardial perfusion [40].
4. Microcirculatory Inflammation in HFpEF and Anti-Inflammatory Effects of Statins
Patients with HFpEF usually have elevated blood levels of pro-inflammatory cytokines, that trigger systemic endothelial dysfunction and endothelial activation [4]. Activated endothelial cells express adhesion molecules such as E-selectin, P-selectin, intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), which interact reversibly with the corresponding ligands on the surface of circulating monocytes (leukocyte function-associated antigen-1 [LFA-1] and integrin dimer CD11b [αM-subunit β2 integrin] Mac-1) [41], the expression and affinity of which, in turn, is enhanced under the action of endothelial chemokines. As a result of this interaction, the movement of monocytes in coronary capillaries slows down up to a complete stop. High expression of adhesion molecules (ICAM-1 and E-selectin) was detected in the coronary microcirculation of patients with HFpEF [6]. The adhesion of monocytes to endothelial cells is a prerequisite for a key step in the whole chain of the inflammatory process – the migration of monocytes from the bloodstream into the subendothelial space. This process occurs according to a concentration gradient of chemoattractants such as monocyte chemoattractant protein-1 (MCP-1) and interleukin (IL)-8 released from the stressed myocardium. Once migrated into the tissue monocytes quickly transform into macrophages and start producing the main cytokine of fibrosis – transforming growth factor-β (TGF-β) (Figure 2) [6]. TGF-β triggers transformation of fibroblasts into myofibroblasts. The last ones produce collagen intensively, resulted in progression of fibrosis and LV diastolic dysfunction (DD). In experimental models with pressure overload, 1) features of inflammation are always found in hypertrophied myocardium together with fibrosis; 2) the areas of fibrosis and inflammation usually coincide; 3) inflammation always occurs before fibrosis, and 4) suppression of inflammation allows to prevent fibrosis as well [42]. In biopsy studies in patients with HFpEF, a significant accumulation of activated macrophages producing TGF-β and other fibroblast triggers such as galectin-3 and osteopontin was found in the myocardium, which was associated with fibroblast activation and excessive collagen deposition [7,43,44].
One of the key pleiotropic properties of statins is their well-documented anti-inflammatory effects [12]. Statins have been shown to be effective in a number of anti-inflammatory diseases: atherosclerosis [45], myocarditis [46], diabetic nephropathy [47], Alzheimer's disease [48], bone diseases [49], and autoimmune diseases [50]. In addition, the anti-inflammatory effects of statins can be observed at all stages of the inflammation in hypertensive heart disease [51].
Statins reduce monocyte adhesion and migration through inhibition of expression/activity of endothelial adhesion molecules (VCAM-1, E-selectin), leukocyte integrins (Mac-1, LFA-1), chemokines (MCP-1) and their receptors, and some of these effects were independent of HMG-CoA reductase inhibition (Figure 2) [52,53,54]. One potential mechanism for the anti-inflammatory action of statins is an inhibition of pro-inflammatory cytokine production with elimination of their activating effects on nuclear factor κ-light-chain-enhancer of activated B cells (NF-kB). NF-kB is a ubiquitous transcription factor that influences the production of pro-inflammatory cytokines such as TNF-α [12] and is involved in cell proliferation, differentiation and immune defense [55]. According to one hypothesis, the ability of statins to scavenge free oxygen radicals and stimulate nitric oxide production may contribute to the stabilisation of IkBα protein, a natural inhibitor of NF-kB [56]. The anti-inflammatory effect of statins through inhibition of NF-kB has been convincingly demonstrated in numerous experimental and clinical studies [57,58,59,60].
Statins may exert anti-inflammatory impact through the regulation of the Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasomes – cytosolic protein oligomeric complexes within the immune cells. NLRP3 inflammasomes detect bacterial and viral pathogens as well as by-products of tissue damage or cellular stress, and trigger an inflammatory response via proteolytic activation of the inflammatory cytokines, interleukin-1β and -18 by caspase-1 [61]. The NLRP3 inflammasome assembly is triggered by the NF-κB pathway, which is activated by damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), and endogenous cytokines. The activation signal is provided by a variety of stimuli including extracellular ATP, mitochondria-generated ROS, and particulate matter including cholesterol crystals [62]. Statins may inhibit the activation of NLRP3 in response to atherogenic stimuli by activating the pregnane X receptor [63]. Statins are also able to reduce the expression of toll-like receptors 2 and 4 on the surface of immune cells and thereby prevent lipopolysaccharide-induced activation of monocytes and endothelial cells [12]. Reduced expression of toll-like receptors leads to reduced activation of the NF-κB [64], reduced NLRP3 activation [65] and ultimately cause a switch of the immune response to anti-inflammatory responses [12]. Statins also may suppress transcription of protein components of the inflammasome through inhibition of NF-kB [66]. Nevertheless, the cumulative effect of statins on NLRP3 inflammasome is still a matter of debate. Lipophilic statins are thought to have a stronger effect on NLRP3 inflammasome complexes than hydrophilic statins [12]. Further studies are needed to clarify the effect of statins on inflammasomes.
One of the most sensitive markers of inflammation is C-reactive protein (CRP). CRP is an acute phase reactant and is produced in hepatocytes under stimulation with IL-6, IL-1 and TNF-α [67]; however, it can be produced locally by macrophages in foci of inflammation [68]. The relationship between CRP levels and cardiovascular risk is well established. In patients with HFpEF, CRP is associated with greater comorbidity burden [69], and poor outcome [70]. Numerous studies have shown the ability of statins to reduce CRP blood levels (by about 15-30%) [71,72,73], while meta-analysis has shown that lipophilic statins were more effective in decreasing hsCRP and IL-6 in patients with HF [74]. Statin suppression of CRP effects, including effects on monocyte MCP-1, MIP-1α and MIP-1β cytokine synthesis and cell motility, is mediated by L-mevalonate/FPP-dependent inhibition of ERK1/2 MAP kinases [75]. In the Korean Acute Heart Failure registry, CRP was a clear prognostic marker for any form of heart failure (HFrEF, HF with mildly reduced EF, or HFpEF), but only in subgroup of patients with EF >40% and upper tertiles of CRP (>1.14 mg/dL) who were on statins showed a better survival trend than those who were not on statins [76].
One of the key modulators of cell metabolism, survival and proliferation is PI3K/Akt pathway, and the maintenance of myocardial inflammation may be mediated through enhanced activity of this pathway in immune cells, in particular, macrophages [50]. The data regarding statins effect on PI3K/Akt signaling pathway are ambiguous. Reduced ROCK activity may stimulate the intracellular PI3K/Akt signaling pathway and, thus, enhance immune cell activation. On the other hand, there are some data demonstrating the inhibitory effects of statins on PI3K/Akt activation; the anticipated effect depends on cell type, experiment conditions and applied concentrations [77]. While reducing ROCK and downstream molecules activation statins may reduce immune cell migration to the myocardium and thereby suppress inflammation, which may ultimately benefit cardiac remodeling processes [78]. Statins decrease the expression of interferon (INF)-γ-inducible major histocompatibility complex molecules class II (MHC II) on the surface of antigen-presenting cells, thereby reducing antigen presentation to CD4+ T lymphocytes [79]. In different modes of induction of dendritic cell maturation, statins inhibited the expression of MHC II involved in antigen presentation and of the T-cell costimulatory molecules CD80/CD86 (B7-1/2), CD40 and CD83 [80,81]. Dendritic cells derived in culture from blood monocytes in the presence of statins not only failed to provide full antigen presentation to T cells, but also promoted the expansion of T cells with suppressor activity [80]. Dose-dependent inhibition of CD4+ T-cells proliferation accompanied by accumulation of regulatory Foxp3+ cells by statins was detected in vitro. Statins impaired spontaneous and chemokine-induced lymphocyte migration apparently by inhibiting cellular motility [82]. Statins inhibit monocyte differentiation to macrophages and cytokine synthesis by activated macrophages at the posttranscriptional level [83].
Other anti-inflammatory mechanisms of statins include decreased TGF-β signaling in T cells, impaired T cell activation and induction of T regulatory cells [81]. Ex vivo, atorvastatin suppressed the expression of neutrophil chemoattractant IL-8 produced by endothelial cells [84], and simvastatin decreased chemokine production and chemokine receptor expression in human endothelial cells and macrophages [85]. Statins may directly block LFA-1 integrin [53], which is involved in leukocyte adhesion and migration and co-stimulatory signal transduction to T cells during antigen presentation. Activation of integrins, including LFA-1, is accompanied by conformational changes leading to an increase in the affinity for their substrates. Statins may bind to LFA-1 and prevent such conformational changes, which can partly explain the anti-inflammatory activity of statins [53].
5. Low NO-cGMP-PKG Pathway Activity Contributes to Myocardial Dysfunction. Role of Statins
The main consequence of reduced NO bioavailability in HFpEF is impaired signaling through the intracellular NO-cGMP-PKG pathway (Figure 2) [86]. NO diffusion into the underlying vascular smooth muscle cells leads to microvessel dilatation. NO also inhibits the expression of adhesion molecules and reduces microvascular inflammation, regulates platelet function, and promotes the mobilisation of stem cells to maintain vascular integrity. NO also penetrates into adjacent cardiomyocytes, where it binds to its intracellular receptor soluble guanylate cyclase (sGC) and activates it with subsequent formation of the secondary intracellular messenger cGMP. cGMP exerts its physiological effects mainly through the activation of cGMP-dependent protein kinase type I (PKGI). PKGI directly phosphorylates a variety of proteins, exerting numerous beneficial cardiovascular effects [87].
When NO bioavailability is low, cGMP formation and PKGI activity in cardiomyocytes are reduced. Suppression of the NO-cGMP-PKG signaling network has been clearly demonstrated in animal models of HFpEF [6,88,89]. In LV biopsies, cGMP content and PKG activity were significantly lower in patients with HFpEF compared with patients with HFrEF or asymptomatic aortic stenosis [6,8], that was associated with low NO bioavailability and high nitrosative/oxidative stress [6]. ROS can directly oxidize PKGI reducing its activity. In patients with HFpEF, higher myocardial oxidative stress-dependent activation of eNOS leading to PKGIα oxidation was revealed [90].
NO-cGMP-PKG pathway suppress prohypertrophic signaling cascades, including Gq-coupled pathway via phosphorylation of the regulator of G protein signaling (RGS) 2/4 [91], and the calcineurin/NFAT (nuclear factor of activated T-cells) pathway via phosphorylation of the canonical transient receptor potential calcium ions (Ca2+) channel (TRPC) 6 [92]. At low NO-cGMP-PKG signaling pathway activity, this inhibitory effect on prohypertrophic signaling pathways may be significantly attenuated. The cGMP-PKG pathway also prevents fibrosis, presumably by suppressing TGF-β and preventing the conversion of fibroblasts to active myofibroblasts [93]. TGF-β1 is synthesized in many myocardial cells and transmits a signal to the cell nucleus via the suppressor of mothers against decapentaplegic (SMAD) 2/3 pathway [94]. A decreased activity of the cGMP-PKG pathway in fibroblasts is associated with stimulation of collagen synthesis and its excessive myocardial deposition [2,86]. On the contrary, increased activity of the NO-cGMP-PKG pathway attenuates signaling through the SMAD2/3 pathway and, preventing the development of myocardial fibrosis [95]. In addition, the transdifferentiation of fibroblasts into myofibroblasts depends on TRPC6 activity, that is inhibited by PKGI [93].
Activated NO-cGMP-PKG pathway participates in the intracellular Ca2+ handling and contributes to the normal LV relaxation. When PKG activity is low, the phosphorylation of proteins involved in active relaxation is impaired, namely 1) phospholamban, decreasing the rate of Ca2+ removal from the cytosol and 2) troponin I, decreasing the actin and myosin bridges detachment rate, resulting in an increase in passive tension of cardiomyocytes (Fpassive). In LV biopsies, Fpassive was significantly higher in patients with HFpEF compared to patients with HFrEF (Figure 2) [8].
PKGI is involved in maintaining high myocardial compliance through phosphorylation of spring elements of titin molecules. At low PKG activity, molecules of the N2B titin isoform are hypophosphorylated, that leads to an increase in Fpassive and, accordingly, a steeper rise in LV filling pressure [96]. It has been shown that titin hypophosphorylation is more pronounced in HFpEF than in HFrEF [97]. High stiffness of cardiomyocytes together with myocardial fibrosis plays the most important role in the myocardial stiffening and increased LV filling pressure [98]. Thus, enhancement of the NO-cGMP-PKG pathway may be a promising target in HFpEF. In preclinical studies, therapeutic manipulations that target this pathway have improved LV compliance, and reduced cardiac hypertrophy and fibrosis [99,100,101,102]. However, clinical trials with therapeutic interventions targeting the NO-cGMP-PKG pathway by angiotensin receptor neprilysin inhibitors, phosphodiesterase-5 inhibitors, sGC stimulators, NO-inducing drugs including β3 adrenergic receptor agonists, inorganic nitrates, and inorganic nitrates/nitrites showed neutral outcomes in patients with HFpEF [86]. One can assumed that more "natural" activation of this signaling axis and, consequently, improvement of myocardial function can be achieved via reducing inflammation, oxidative stress and endothelial dysfunction, i.e., via eliminating the main contributors to low NO bioavailability (Figure 2). This approach opens great perspectives for statins, as they would be expected not only to activate the NO-cGMP-PKG pathway, but also to reduce diverse pro-inflammatory and oxidative signaling pathways involved in the pathogenesis of HFpEF.
Statins rapidly improve endothelial redox balance and increase bioavailability of endothelium-derived NO to neighboring cardiomyocytes, macrophages, fibroblasts etc., leading to prevention of cardiac hypertrophy and fibrosis and amelioration of LVDD [16,103,104]. In a retrospective analysis of biopsy specimens from the study of van Heerebeek et al. [8], patients with HFpEF taking statins had higher protein kinase G activity along with lower cardiomyocyte hypertrophy and lower nitrotyrosine levels (a marker of oxidative processes activity) and lower Fpassive compared to patients not taking statins [2].
6. Effect of Statins on Myocardial Hypertrophy and Fibrosis
LV hypertrophy (LVH) is a common structural cardiac HFpEF alteration [105]. LVH is a compensatory adaptative mechanism to increased demands on LV function, including pressure overload. Subsequently, after a series of poorly characterized events ("transition to failure"), the LV predominantly develops progressive LVDD and elevated filling pressures with preserved EF (HFpEF-phenotype) [106]. LVH is invariably accompanied by LVDD due to delayed relaxation, hypertrophy, increased stiffness of cardiomyocytes, and myocardial fibrosis [51]. Myocardial fibrosis, as well as LVH, at initial stages apparently represents a compensatory response, providing temporal and spatial coordination of force efforts of both hypertrophied muscle fibers and the whole LV under pathological conditions (high afterload). Thereafter, myocardial fibrosis contributes to increased LV stiffness and LV filling pressures.
The GTP-binding proteins Ras, Rho and Rac play an important role in LVH [9], and statins by inhibiting their activity may reduce myocyte hypertrophy [107,108,109]. LVH may be mediated by increased NADPH-oxidase activity and myocardial oxidative stress, while Rac1 (Ras-related C3 botulinum toxin substrate) is able to directly activate NADPH-oxidase. Statins, via inhibition of isoprenylation, can suppress Rac1 activation and thereby prevent cardiomyocyte hypertrophy. Mice with a cardiac-specific deletion of Rac1 exhibited reduced NADPH oxidase activity and myocardial oxidative stress, which was accompanied with decreased LVH [110]. In vitro, statins increased levels of the small GTP-binding protein GDP dissociation stimulator (SmgGDS) and decreased levels of Rac-1 [111]. In wild-type mice, statins reduced Rac1 expression, myocardial hypertrophy and fibrosis, but not in mice lacking SmgGDS [112]. In clinical study, perioperative administration of atorvastatin 40 mg daily to patients undergoing cardiac surgery was accompanied by Rac1-mediated inhibition of NADPH oxidase activity in specimens of the right atrial appendage [104]. In a clinical study of Lee et al. (2002), pravastatin therapy in patients with hypercholesterolemia was accompanied by a decrease in LV mass, presumably through suppression of free-radical oxidation processes, since in multivariate analysis, a regression of LV mass was correlated only with a change in the level of oxidative damage marker 8-iso-prostaglandin F [109]. In a clinical study by Su et al. (2000), the addition of pravastatin to antihypertensive therapy in hypertensive patients with hypercholesterolemia was associated with an additional decrease in LV mass index regardless of the effect on cholesterol levels [113].
The effect of statins on the myocardium may also be mediated through inhibition of the Rho/ROCK pathway [21]. The suppression of ROCK activation can lead to numerous beneficial effects including reduced cell proliferation, myocardial hypertrophy and fibrosis [9]. Mice with a genetic deletion of ROCK1 were characterized by a lower degree of ischemia-induced myocardial fibrosis compared to wild-type mice [114]. In an experiment with angiotensin II infusion and transverse aortic constriction, mice without ROCK2 had less severe LVH and fibrosis compared with wild-type mice [115]. In another experimental study, mice with deletion of both ROCK1 and ROCK2 exhibited less severe fibrosis and increased autophagy compared to mice with single deletion [116]. Patients with hypertensive LVH had several times higher levels of ROCKs in leukocytes than patients with arterial hypertension but without LVH [117].
Statins may also reduce myocardial fibrosis, presumably via inhibition of Rho proteins and increased NO bioavailability [118]. Statins were superior to other drugs blocking the activity of the mevalonate pathway (alendronate and fasudil) in their effects on the proliferation of myocardial fibroblasts and inhibited collagen synthesis in vitro [119]. In vitro studies have shown the ability of statins to prevent the differentiation of fibroblasts into myofibroblasts [120] and to induce reverse differentiation of profibrotic myofibroblasts through inhibition of GGPP [121], that abrogates a key process in myocardial fibrosis. Simvastatin in vivo suppressed the differentiation of fibroblasts into myofibroblasts and inhibited the angiotensin II-induced cardiac fibrosis by regulating exosome-mediated cell-cell communication within the myocardium [122].
In rat and human cardiac fibroblast cell cultures, atorvastatin significantly decreased collagen synthesis, α(I)-procollagen mRNA expression, and connective tissue growth factor gene expression [123]. In a rat model of LV hypertrophy and HFpEF (Dahl salt-sensitive rats fed with high-salt), treatment with pitavastatin resulted in decreased expression of hypertrophic and profibrotic genes, which was accompanied by a significant reduction of myocardial fibrosis [124]. In another rat model of HFpEF (transgenic rats overexpressing the renin gene with elevated cardiac levels of angiotensin II), rosuvastatin significantly reduced Rac1 expression, cardiac hypertrophy and perivascular fibrosis, and improved LV contractility [125]. In a mouse model of HFpEF (C57BL/6 mice with a high-fat diet and N-arginine methyl ester treatment), simvastatin reduced LVDD through a reduction of myocardial fibrosis, which was associated with prevention of phosphorylation of Smad (Smad2 and Smad3) and Ras/Raf/MAPK (mitogen-activated protein kinase) profibrotic signaling pathways downstream of the TGF-β receptor in cardiac tissue [126]. In another mouse model of HF (with transverse aortic constriction), simvastatin reduced myocardial fibrosis and hypertrophy by suppressing fibroblast proliferation and myofibroblast formation through modulation of endothelial Krüppel-like factor 2 (Klf2)-TGFβ1 or KLF2-forkhead box P1 (Foxp1, endothelial transcription factor)-TGFβ1 signaling pathways [127]. Lipophilic but not hydrophilic statins increased the expression of cardioprotective interleukin-33 in human cardiomyocytes and cardiac fibroblasts in vitro, presumably through inhibition of GGPP, which may also contribute to the antihypertrophic and antifibrotic effects of statins [128].
7. Effect of Statins on Diastolic Dysfunction and Epicardial Adipose Tissue
Ultimately, the above-mentioned pleiotropic effects of statins may lead to an improvement in LVDD, which has been shown in multiple preclinical studies [16,129,130,131]. Clinically, the ability of statins to improve LVDD has also been shown in patients with a variety of cardiovascular conditions, such as ischaemic heart disease, hyperlipidaemia, hypertensive heart disease, as well as in patients on peritoneal dialysis. The retrospective cohort study of Okura et al. (2007) enrolled 430 patients with ischemic heart disease but without HF [132]. Statin treatment (31% of participants) was associated with significant decrease in the ratio of the early mitral flow velocity to early diastolic velocity of the mitral annulus (E/e’ ratio, as a measure of LV filling pressures), and higher free survival rate (cardiac death and congestive heart failure) compared with patients without statins. In the prospective study of Yagi S. et al. (2011), a low-dose pitavastatin (1.0 mg/day) significantly decreased E/e’ ratio and albuminuria in patients with hyperlipidemia, which was associated with suppression of oxidative stress [133]. In a retrospective cohort study by Ovchinnikov et al. (2022), involving 223 asymptomatic patients with hypertensive concentric LV hypertrophy and ejection fraction ≥ 50% statin treatment significantly prevented the transition to HFpEF and LVDD deterioration during a follow-up of 8 years [134]. In the study of Wu et. al. (2017) with 213 patients undergoing peritoneal dialysis and without hyperlipidemia but with elevated hsCRP, atorvastatin significantly improved the tissue Doppler imaging or global strain imaging diastolic parameters [135].
Systemic inflammation, being an important pathologic mechanism of HFpEF, is closely related to the expansion and inflammation of epicardial adipose tissue (EAT). In inflamed EAT, adipocytes and activated macrophages produce proinflammatory cytokines and chemokines, that via endocrine and paracrine mechanisms cause local inflammation, oxidative stress, microvascular endothelial dysfunction, and interstitial matrix deposition, thereby leading to slower relaxation, increased cardiomyocyte stiffness, and myocardial fibrosis – the main contributors of LVDD [136]. EAT expansion imposes the pericardial restrain and impedes ventricular filling. A meta-analysis of 11 studies showed that increased EAT is independently associated with LVDD [137]. EAT expansion correlates with a poor prognosis in patients with HFpEF [138]. Therefore, the inhibition of EAT expansion and inflammation may be a promising therapeutic strategy for HFpEF.
Statins as anti-inflammatory drugs have some therapeutic potential to attenuate EAT expansion. In patients with aortic stenosis, statin therapy was accompanied by a significant reduction in EAT thickness and levels of EAT-secreted inflammatory mediators [139]. In patients who underwent percutaneous coronary intervention, atorvastatin reduced EAT thickness more significantly than simvastatin in combination with ezetimibe [140]. In patients who underwent pulmonary vein isolation for atrial fibrillation, intensive therapy with atorvastatin (80 mg/day) was accompanied by a significant reduction in EAT thickness [141]. In postmenopausal women with hyperlipidemia, intensive therapy with atorvastatin (80 mg/day) resulted in a greater reduction in EAT compared to intensive therapy with pravastatin (40 mg/day) [142].
8. Statins in Heart Failure
Given the numerous pleiotropic effects of statins, it was once hypothesized that statins might be effective in HF [143]. Data on the use of statins in patients with HFrEF are controversial [144,145,146,147]. In two large randomized, double-blind, placebo-controlled trials (GISSI-HF and CORONA), statins did not improve the prognosis of patients with HFrEF [144,145]. Both studies used hydrophilic rosuvastatin with less pleiotropic potential and low myocardial penetration [148] and at a low starting dose of 10 mg, which may have limited its prognostic impact. More recent observational prospective cohort studies and meta-analyses have shown a reduction in mortality on statin treatment in patients with HFrEF [149,150]. Nevertheless, the challenge associated with the use of statins exists. Current guidelines do not recommend routine use of statins in most patients with HFrEF unless otherwise indicated (hypercholesterolemia, coronary artery disease), but they can be continued in patients who have already been receiving treatment [151].
In contrast, benefits of statins were consistently observed in HFpEF (see below). The differences may be related to the different pathophysiological drivers for LV remodeling [2,152] suggesting different targets for statins. In an observational study, patients with HFpEF treated with statins were less likely to experience atrial fibrillation [153]. In the prospective study, involving 59 statin-naïve patients with HFpEF, statin therapy consistently reduced blood levels of inflammatory biomarkers (hsCRP and MCP-1), and a biomarker of LV wall stress N-terminal pro-B-type natriuretic peptide (NT-proBNP (NT-proBNP), that was associated with improvement in LVDD and reduction in LV filling pressures [154]. One of the most frequent complications of HFpEF is a pulmonary hypertension, occurring in the majority of patients with HFpEF and associated with a poor prognosis [155]. In a retrospective study involving 762 patients with severe pulmonary hypertension (pulmonary artery systolic pressure ≥ 60 mm Hg) and preserved EF (≥ 50%), administration of statins was associated with a 58% reduction in overall mortality (P < 0.001) and was independent of the presence of chronic obstructive pulmonary disease [156].
Although statin therapy for HFpEF has not been evaluated in randomised trials, many observational studies have consistently reported a positive effect of statins on mortality in patients with HFpEF of different ethnicity (Table 1) [157,158,159,160,161,162,163,164,165,166,167,168], including HFpEF patients without coronary artery disease [169]. Several meta-analyses have confirmed beneficial prognostic effect of statins in HFpEF, where statins reduced all-cause mortality in patients with HFpEF by 25% to 40% [150,170,171]. But in any case, the results of all these observational studies should be validated in specially designed large randomised clinical trials with sufficient statistical power.
The effects of statins extend beyond HFpEF patients. A higher prevalence of non-cardiac pro-inflammatory and metabolic comorbidities (obesity, hypertension, diabetes, chronic kidney disease, chronic obstructive pulmonary disease, liver disease) in HFpEF may also be relevant [172]. These comorbidities may be involved in the pathophysiology of HFpEF [2]. The association between these comorbidities and myocardial dysfunction may to be mediated by systemic inflammation [173], and statins have been shown to be beneficial in these conditions as well [174,175,176].
9. Which Statins are Better for HFpEF?
The ability of statins to pass through the cell membrane depends on their chemical properties and the expression of the appropriate membrane transporters in the tissues [18]. Statins can be divided into hydrophilic (rosuvastatin and pravastatin) and lipophilic (simvastatin, pitavastatin, atorvastatin), depending on their ability to dissolve in water or lipid-containing media, respectively [148]. Hydrophilic statins may exhibit greater hepatoselectivity than lipophilic statins, whereas lipophilic statins may undergo passive diffusion through both hepatic and non-hepatic tissues [177]. Active transport of statins into cells is mainly performed by proteins of the OATP (organic-anion-transporting polypeptide) and NTCP (Na+ tauro-cholate co-transporting polypeptide) families [177]. Hepatocytes express all types of transporters from these protein families; some members of the OATP family are also found in the membranes of enterocytes, endothelial and skeletal muscle cells [18]. In contrast, all other cells including myocardial cells – cardiomyocytes, fibroblasts, and macrophages – lack these transporters, so lipophilic but not hydrophilic statins can enter these cells by passive diffusion through cell membranes [177,178]. Hydrophilic statins have low uptake by cardiac muscles (approximately 1% of liver uptake), whereas lipophilic statins have a higher uptake (up to 80% of liver uptake) [178]. Thus, hydrophilic statins may not exhibit pleotropic effects to the same extent as lipophilic statins, which penetrate extrahepatic tissues such as hypertrophic myocardium.
A meta-analysis of 15 randomised controlled trials showed that lipophilic statins have more pronounced pleiotropic effects compared with hydrophilic statins [179]. Several meta-analyses have shown that lipophilic statins may be more effective in improving cardiac function and reducing inflammation [74,180], reducing all-cause mortality and hospitalisations due to worsening of heart failure in patients with HFpEF [150,180]. In the Stat-LVDF study, treatment with lipophilic pitavastatin but not hydrophilic rosuvastatin delayed an increase in BNP in patients with hypercholesterolemia and asymptomatic LVDD [181]. Equivalent doses of lipophilic atorvastatin resulted in greater reductions in lipid oxidation markers compared to hydrophilic pravastatin in subjects with hyperlipidaemia and metabolic syndrome [182]. In a prospective clinical study by Ovchinnikov et al. (2019), administration of lipophilic atorvastatin was associated with a more pronounced increase in exercise tolerance, haemodynamic improvement and decrease in fibrosis biomarkers compared with hydrophilic rosuvastatin [154].
10. Potential Adverse Effects of Statins in HFpEF
Despite the numerous beneficial effects of statins in LVDD/HFpEF, there are known concerns about adverse effects of statins that may limit their use in HFpEF, particularly muscle myopathy and new-onset type 2 diabetes mellitus (DM) [183].
Myopathy is a frequent dose-dependent side effect of statins, and its occurrence in patients with HFpEF may lead to even greater exercise limitation. Statin-associated muscle symptoms may affect 5% to 10% of patients. Risk factors for statin-induced myopathy include advanced age, female sex, and physical disability [184], making this a highly relevant issue in HFpEF. In most cases, myopathy manifestations resolve after discontinuation or reduction of the dose of statins; however, in some patients this does not happen, that may indicate necrotizing autoimmune myopathy induced by statins [184]. Statins through prenylation inhibition can reduce the expression of ubiquinone (coenzyme Q10), a cofactor in the mitochondrial electron transport chain, that can lead to impaired oxidative phosphorylation and energy cell supply, damage and apoptosis of skeletal muscle cells [183]. In several studies, adding coenzyme Q10 reduced muscle symptoms associated with statins [185]. Statins also potentiate uncoupling of the stabilizing protein FKBP12 from ryanodine receptor 1, leading to leakage of Ca2+ from the sarcoplasmic reticulum, excessive muscle contraction, thereby provoking myopathy [186]. Impaired activation of small GTPases and inhibition of the PI3K/Akt pathway may also be important, inducing muscle atrophy via activation of the forkhead box class O (FOXO) protein group followed by increased expression of atrogenic proteins such as atrogin-1 and muscle-specific ring finger (MuRF)-1 [184].
Statins may increase the risk of DM in a dose-dependent manner by 9% [187], which may be relevant in HFpEF, as DM plays an important role in the pathogenesis of HFpEF. It is believed that these effects of statins are realized through dysfunction of pancreatic β-cells and insulin resistance of peripheral tissues [188]. Statins can inhibit the expression of the GLUT-2 transporter and inhibit pancreatic ß-cell calcium channels, thereby impairing insulin release [189]. In addition, intensive therapy with statins may induce ß-cell apoptosis [190]. Statins may also reduce peripheral tissue insulin sensitivity via decreased translocation of the GLUT4 transporter and inhibition of insulin receptor substrate (IRS-1) and its downstream signaling pathways [191]. Of note, pitavastatin and pravastatin do not affect glucose levels [188].
11. Conclusions and Future Perspectives
Currently, the therapeutic treatment approaches for HFpEF are very limited, and the search for effective treatment is of great need. One potential treatment option is statins, which have shown multiple pleiotropic effects into the different cell types that may be extremely beneficial in HFpEF (Figure 1). Various hypotheses have been proposed to explain the beneficial effects of statins in HFpEF; however, the exact mechanisms remain incompletely understood and should be confirmed and more rigorously investigated in additional specially designed studies. To date, the ability of statins to improve outcomes in HFpEF has been consistently shown in multiple observational studies, where statins reduced total mortality by 20% to 80%. Of note, these impressive results were obtained in observational studies, where adherence to therapy is usually significantly lower than in controlled large-scale randomized trials. These findings should be tested in adequately powered randomized clinical trials. However, the implementation of such studies is severely limited by the fact that statins are indicated for most patients with HFpEF due to high or very high risk of cardiovascular complications.
Lipid-lowering therapy continues to be a challenge in the treatment of cardiovascular diseases, including elderly patients with HFpEF, as statins are the group of drugs that are most commonly underused and underdosed [192]. In recent large randomized clinical trials (IMPERIOR-PRESERVED, PARAGON-HF, TOP-CAT), 31-47% of participants did not receive lipid-lowering medications [193]. One of the best ways to improve adherence to statin therapy is counselling and education of patients, combined with frequent and comprehensive monitoring of patients' adherence.
Since subclinical microvascular inflammation is a common pathogenetic mechanism of HFpEF regardless of the predominant HFpEF phenotype and comorbidities [4], various anti-inflammatory targeting strategies other than statins are being actively tested. An anti-interleukin-1 strategy has held great promise, but clinical trials with a recombinant IL-1 receptor antagonist anakinra in HFpEF have yielded conflicting results [194,195]. To date, it is not clear whether canakinumab, which blocks the effects of IL-1β and has been shown to be highly effective in postinfarction patients [196], can also improve prognosis in HFpEF. Other emerging anti-inflammatory strategies with potential benefit in HFpEF include administration of colchicine (ongoing COLpEF trial [NCT04857931]), immunomodulation by intracoronary injection of cardiosphere-derived cells (ongoing REGRESS-HFpEF trial [NCT02941705]), myeloperoxidase inhibition (ongoing ENDEAVOR trial [NCT04986202]), and others [4]. A deeper understanding and detailed characterization of the inflammatory mechanisms responsible for the onset and progression of HFpEF is needed to enable more targeted treatment and prognostic improvement in HFpEF.
Author Contributions
conceptualization, A.O., A.P., T.A. and E.B.; literature search; writing—original draft preparation, A.O. A.P., A.F. and E.B.; writing—review and editing, T.A. and F.A.; supervision, A.O and F.A.; funding acquisition, A.O. and F.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Ministry of Health of the Russian Federation (registration number 124020200111-6).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Acknowledgments
The authors are grateful to Olga Filatova, for her help in English editing of manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vasan, R.S.; Xanthakis, V.; Lyass, A.; Andersson, C.; Tsao, C.; Cheng, S.; Aragam, J.; Benjamin, E.J.; Larson, M.G. Epidemiology of Left Ventricular Systolic Dysfunction and Heart Failure in the Framingham Study: An Echocardiographic Study Over 3 Decades. JACC Cardiovasc. Imaging. 2018, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Panahi, M.; Papanikolaou, A.; Torabi, A.; Zhang, J.G.; Khan, H.; Vazir, A.; Hasham, M.G.; Cleland, J.G.F.; Rosenthal, N.A.; Harding, S.E.; Sattler, S. Immunomodulatory interventions in myocardial infarction and heart failure: a systematic review of clinical trials and meta-analysis of IL-1 inhibition. Cardiovasc. Res. 2018, 114, 1445–1461. [Google Scholar] [CrossRef]
- Pugliese, N.R.; Pellicori, P.; Filidei, F.; De Biase, N.; Maffia, P.; Guzik, T.J.; Masi, S.; Taddei, S.; Cleland, J.G.F. Inflammatory pathways in heart failure with preserved left ventricular ejection fraction: implications for future interventions. Cardiovasc. Res. 2023, 118, 3536–3555. [Google Scholar] [CrossRef] [PubMed]
- Dunlay, S.M.; Roger, V.L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2017, 14, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; Paulus, W.J.; Hamdani, N. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; Poller, W.; Schultheiss, H.P.; Tschöpe, C. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef] [PubMed]
- van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A.; Verheugt, F.W.; Niessen, H.W.; Paulus, W.J. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012, 126, 830–839. [Google Scholar] [CrossRef]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef]
- Sacks, F.M.; Pfeffer, M.A.; Moye, L.A.; Rouleau, J.L.; Rutherford, J.D.; Cole, T.G.; Brown, L.; Warnica, J.W.; Arnold, J.M.; Wun, C.C.; Davis, B.R.; Braunwald, E. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N. Engl. J. Med. 1996, 335, 1001–1009. [Google Scholar] [CrossRef]
- Lee, M.S.; Duan, L.; Clare, R.; Hekimian, A.; Spencer, H.; Chen, W. Comparison of Effects of Statin Use on Mortality in Patients With Heart Failure and Preserved Versus Reduced Left Ventricular Ejection Fraction. Am. J. Cardiol. 2018, 122, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory Action of Statins in Cardiovascular Disease: the Role of Inflammasome and Toll-Like Receptor Pathways. Clin. Rev. Allergy Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Bakogiannis, C.; Leeson, P.; Guzik, T.J.; Zhang, M.H.; Tousoulis, D.; Antonopoulos, A.S.; Demosthenous, M.; Marinou, K.; Hale, A.; Paschalis, A.; Psarros, C.; Triantafyllou, C.; Bendall, J.; Casadei, B.; Stefanadis, C.; Channon, K.M. Rapid, direct effects of statin treatment on arterial redox state and nitric oxide bioavailability in human atherosclerosis via tetrahydrobiopterin-mediated endothelial nitric oxide synthase coupling. Circulation. 2011, 124, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Sola, S.; Mir, M.Q.; Lerakis, S.; Tandon, N.; Khan, B.V. Atorvastatin improves left ventricular systolic function and serum markers of inflammation in nonischemic heart failure. J. Am. Coll. Cardiol. 2006, 47, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Jiang, H.; Sun, A.; Wang, Y.; Zou, Y.; Ge, J.; Chen, H. Effects of statin therapy on inflammatory markers in chronic heart failure: a meta-analysis of randomized controlled trials. Arch. Med. Res. 2010, 41, 464–471. [Google Scholar] [CrossRef]
- Akahori, H.; Tsujino, T.; Naito, Y.; Matsumoto, M.; Sasaki, N.; Iwasaku, T.; Eguchi, A.; Sawada, H.; Hirotani, S.; Masuyama, T. Atorvastatin ameliorates cardiac fibrosis and improves left ventricular diastolic function in hypertensive diastolic heart failure model rats. J. Hypertens. 2014, 32, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Garre, D.; Gonzalez-Rubio, M.L.; Munoz-Pacheco, P.; Caro-Vadillo, A.; Aragoncillo, P.; Fernandez-Cruz, A. Rosuvastatin added to standard heart failure therapy improves cardiac remodelling in heart failure rats with preserved ejection fraction. Eur. J. Heart Fail. 2010, 12, 903–912. [Google Scholar] [CrossRef]
- Arefieva, T.I.; Filatova, A.Y.; Potekhina, A.V.; Shchinova, A.M. Immunotropic Effects and Proposed Mechanism of Action for 3-Hydroxy-3-methylglutaryl-coenzyme A Reductase Inhibitors (Statins). Biochemistry (Mosc). 2018, 83, 874–889. [Google Scholar] [CrossRef]
- Xu, N.; Shen, N.; Wang, X.; Jiang, S.; Xue, B.; Li, C. Protein prenylation and human diseases: a balance of protein farnesylation and geranylgeranylation. Sci. China Life Sci. 2015, 58, 328–335. [Google Scholar] [CrossRef]
- Soh, J.E.C.; Shimizu, A.; Sato, A.; Ogita, H. Novel cardiovascular protective effects of RhoA signaling and its therapeutic implications. Biochem. Pharmacol. 2023, 218, 115899. [Google Scholar] [CrossRef]
- Seccia, T.M.; Rigato, M.; Ravarotto, V.; Calò, L.A. ROCK (RhoA/Rho Kinase) in Cardiovascular-Renal Pathophysiology: A Review of New Advancements. J. Clin. Med. 2020, 9, 1328. [Google Scholar] [CrossRef]
- Liu, P.Y.; Chen, J.H.; Lin, L.J.; Liao, J.K. Increased Rho kinase activity in a Taiwanese population with metabolic syndrome. J. Am. Coll. Cardiol. 2007, 49, 1619–1624. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Soga, J.; Hidaka, T.; Idei, N.; Fujii, Y.; Fujimura, N.; Mikami, S.; Maruhashi, T.; Kihara, Y.; Chayama, K.; Kato, H.; Noma, K.; Liao, J.K.; Higashi, Y.; ROCK Study Group. Calcium channel blocker and Rho-associated kinase activity in patients with hypertension. J. Hypertens. 2011, 29, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Nohria, A.; Grunert, M.E.; Rikitake, Y.; Noma, K.; Prsic, A.; Ganz, P.; Liao, J.K.; Creager, M.A. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ. Res. 2006, 99, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lin, J.H.; Chou, C.W.; Chang, Y.F.; Yeh, S.H.; Chen, C.C. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer Res. 2008, 68, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; Hill, T.M.; Mammen, P.P.A.; Huang, J.; Lee, D.I.; Hahn, V.S.; Sharma, K.; Kass, D.A; Lavandero, S.; Gillette, T.G.; Hill, J.A. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Breitkreuz, M.; Hamdani, N. A change of heart: oxidative stress in governing muscle function? Biophys Rev. 2015, 7, 321–341. [Google Scholar] [CrossRef] [PubMed]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. J. Clin. Med. 2019, 8, 2051. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Liu, X.B.; Bi, S.J.; Lu, Q.H.; Zhang, J. Inhibition of Rho-kinase is involved in the therapeutic effects of atorvastatin in heart ischemia/reperfusion. Exp. Ther. Med. 2020, 20, 3147–3153. [Google Scholar] [CrossRef]
- Dimmeler, S.; Assmus, B.; Hermann, C.; Haendeler, J.; Zeiher, A.M. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ. Res. 1998, 83, 334–341. [Google Scholar] [CrossRef]
- Liu, P.Y.; Liu, Y.W.; Lin, L.J.; Chen, J.H.; Liao, J.K. Evidence for statin pleiotropy in humans: differential effects of statins and ezetimibe on rho-associated coiled-coil containing protein kinase activity, endothelial function, and inflammation. Circulation. 2009, 119, 131–138. [Google Scholar] [CrossRef]
- Nohria, A.; Grunert, M.E.; Rikitake, Y.; Noma, K.; Prsic, A.; Ganz, P.; Liao, J.K.; Creager, M.A. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ. Res. 2006, 99, 1426–1432. [Google Scholar] [CrossRef]
- Rawlings, R.; Nohria, A.; Liu, P.Y.; Donnelly, J.; Creager, M.A.; Ganz, P.; Selwyn, A.; Liao, J.K. Comparison of effects of rosuvastatin (10 mg) versus atorvastatin (40 mg) on rho kinase activity in caucasian men with a previous atherosclerotic event. Am. J. Cardiol. 2009, 103, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Margaritis, M.; Channon, K.M.; Antoniades, C. Statins as regulators of redox state in the vascular endothelium: beyond lipid lowering. Antioxid. Redox Signal. 2014, 20, 1198–215. [Google Scholar] [CrossRef]
- Pelat, M.; Dessy, C.; Massion, P.; Desager, J.P.; Feron, O.; Balligand, J.L. Rosuvastatin decreases caveolin-1 and improves nitric oxide-dependent heart rate and blood pressure variability in apolipoprotein E-/- mice in vivo. Circulation. 2003, 107, 2480–2486. [Google Scholar] [CrossRef]
- Rossi, J.; Rouleau, L.; Tardif, J.C.; Leask, R.L. Effect of simvastatin on Kruppel-like factor2, endothelial nitric oxide synthase and thrombomodulin expression in endothelial cells under shear stress. Life Sci. 2010, 87, 92–99. [Google Scholar] [CrossRef]
- Bleda, S.; De Haro, J.; Florez, A.; Varela, C.; Esparza, L.; Acin, F. Long-term pleiotropic effect of statins upon nitric oxide and C-reactive protein levels in patients with peripheral arterial disease. Heart Asia. 2011, 3, 130–134. [Google Scholar] [PubMed]
- Vasa, M.; Fichtlscherer, S.; Adler, K.; Aicher, A.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001, 103, 2885–2890. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, P.; Taccardi, A.A.; Grilli, A.; De Lutiis, M.A.; Barsotti, A.; Felaco, M.; De Caterina, R. Chronic treatment with rosuvastatin modulates nitric oxide synthase expression and reduces ischemia-reperfusion injury in rat hearts. Cardiovasc. Res. 2005, 66, 462–471. [Google Scholar] [CrossRef] [PubMed]
- von Haehling, S.; Anker, S.D.; Bassenge, E. Statins and the role of nitric oxide in chronic heart failure. Heart Fail. Rev. 2003, 8, 99–106. [Google Scholar] [CrossRef]
- Leick, M.; Azcutia, V.; Newton, G.; Luscinskas, F.W. Leukocyte recruitment in inflammation: basic concepts and new mechanistic insights based on new models and microscopic imaging technologies. Cell Tissue Res. 2014, 355, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Glezeva, N.; Baugh, J.A. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart Fail. Rev. 2014, 19, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Muñoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; Osborne, M.T.; Hung, J.; Vinegoni, C.; Naxerova, K.; Sosnovik, D.E.; Zile, M.R.; Bradshaw, A.D.; Liao, R.; Tawakol, A.; Weissleder, R.; Rosenzweig, A.; Swirski, F.K.; Sam, F.; Nahrendorf, M. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef] [PubMed]
- González, G.E.; Rhaleb, N.E.; D'Ambrosio, M.A.; Nakagawa, P.; Liao, T.D.; Peterson, E.L.; Leung, P.; Dai, X.; Janic, B.; Liu, Y.H.; Yang, X.P.; Carretero, O.A. Cardiac-deleterious role of galectin-3 in chronic angiotensin II-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1287–H1296. [Google Scholar] [CrossRef] [PubMed]
- Schönbeck, U.; Libby, P. Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation. 2004, 109, II18–II26. [Google Scholar] [CrossRef] [PubMed]
- Parsamanesh, N.; Karami-Zarandi, M.; Banach, M.; Penson, P.E.; Sahebkar, A. Effects of statins on myocarditis: A review of underlying molecular mechanisms. Prog. Cardiovasc. Dis. 2021, 67, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yuan, Y.; Sun, Z.L. Cholesterol Contributes to Diabetic Nephropathy through SCAP-SREBP-2 Pathway. Int. J. Endocrinol. 2013, 2013, 592576. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.C.; Resende, R.; Oliveira, C.R.; Pereira, C.M. Cholesterol and statins in Alzheimer's disease: current controversies. Exp. Neurol. 2010, 223, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Chamani, S.; Liberale, L.; Mobasheri, L.; Montecucco, F.; Al-Rasadi, K.; Jamialahmadi, T.; Sahebkar, A. The role of statins in the differentiation and function of bone cells. Eur. J. Clin. Invest. 2021, 51, e13534. [Google Scholar] [CrossRef]
- Lee, Y.G.; Lee, J.; Byeon, S.E.; Yoo, D.S.; Kim, M.H.; Lee, S.Y.; Cho, J.Y. Functional role of Akt in macrophage-mediated innate immunity. Front. Biosci. (Landmark Ed). 2011, 16, 517–530. [Google Scholar] [CrossRef]
- Ovchinnikov, A.G.; Arefieva, T.I.; Potekhina, A.V.; Filatova, A.Y.; Ageev, F.T.; Boytsov, S.A. The Molecular and Cellular Mechanisms Associated with a Microvascular Inflammation in the Pathogenesis of Heart Failure with Preserved Ejection Fraction. Acta Nat. 2020, 12, 40–51. [Google Scholar] [CrossRef]
- Romano, M.; Diomede, L.; Sironi, M.; Massimiliano, L.; Sottocorno, M.; Polentarutti, N.; Guglielmotti, A.; Albani, D.; Bruno, A.; Fruscella, P.; et al. Inhibition of monocyte chemotactic protein-1 synthesis by statins. Lab. Investig. 2000, 80, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Weitz-Schmidt, G.; Welzenbach, K.; Brinkmann, V.; Kamata, T.; Kallen, J.; Bruns, C.; Cottens, S.; Takada, Y.; Hommel, U. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med. 2001, 7, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Veillard, N.R.; Braunersreuther, V.; Arnaud, C.; Burger, F.; Pelli, G.; Steffens, S.; Mach, F. Simvastatin modulates chemokine and chemokine receptor expression by geranylgeranyl isoprenoid pathway in human endothelial cells and macrophages. Atherosclerosis. 2006, 188, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.Z.; Xin, S.L.; Chen, C.; Liu, T. Effect of atorvastatin on expression of TLR4 and NF-κB p65 in atherosclerotic rabbits. Asian Pac. J. Trop. Med. 2013, 6, 493–496. [Google Scholar] [CrossRef]
- Yamamoto, A.; Hoshi, K.; Ichihara, K. Fluvastatin, an inhibitor of 3-hydroxy-3-methylglutaryl-CoA reductase, scavenges free radicals and inhibits lipid peroxidation in rat liver microsomes. Eur. J. Pharmacol. 1998, 361, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Presa, M.A.; Martín-Ventura, J.L.; Ortego, M.; Gómez-Hernández, A.; Tuñón, J.; Hernández-Vargas, P.; Blanco-Colio, L.M.; Mas, S.; Aparicio, C.; Ortega, L.; et al. Atorvastatin reduces the expression of cyclooxygenase-2 in a rabbit model of atherosclerosis and in cultured vascular smooth muscle cells. Atherosclerosis. 2002, 160, 49–58. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Mummidi, S.; Mahimainathan, L.; Patel, D.N.; Bailey, S.R.; Imam, S.Z.; Greene, W.C.; Valente, A.J. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-κB- and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J. Biol. Chem. 2006, 281, 15099–15109. [Google Scholar] [CrossRef]
- Ozbek, E.; Cekmen, M.; Ilbey, Y.O.; Simsek, A.; Polat, E.C.; Somay, A. Atorvastatin prevents gentamicin-induced renal damage in rats through the inhibition of p38-MAPK and NF-κB pathways. Ren. Fail. 2009, 31, 382–392. [Google Scholar] [CrossRef]
- Sheridan, A.; Wheeler-Jones, C.P.D.; Gage, M.C. The Immunomodulatory Effects of Statins on Macrophages. Immuno. 2022, 2, 317–343. [Google Scholar] [CrossRef]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging. Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef]
- Lim, K.H.; Staudt, L.M. Toll-like receptor signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a011247. [Google Scholar]
- Wang, S.; Xie, X.; Lei, T.; Zhang, K.; Lai, B.; Zhang, Z.; Guan, Y.; Mao, G.; Xiao, L.; Wang, N. Statins Attenuate Activation of the NLRP3 Inflammasome by Oxidized LDL or TNFα in Vascular Endothelial Cells through a PXR-Dependent Mechanism. Mol. Pharmacol. 2017, 92, 256–264. [Google Scholar] [CrossRef]
- Bahrami, A.; Parsamanesh, N.; Atkin, S.L.; Banach, M.; Sahebkar, A. Effect of statins on toll-like receptors: a new insight to pleiotropic effects. Pharmacol. Res. 2018, 135, 230–238. [Google Scholar] [CrossRef]
- Moutzouri, E.; Tellis, C.C.; Rousouli, K.; Liberopoulos, E.N.; Milionis, H.J.; Elisaf, M.S.; Tselepis, A.D. Effect of simvastatin or its combination with ezetimibe on Toll-like receptor expression and lipopolysaccharide-induced cytokine production in monocytes of hypercholesterolemic patients. Atherosclerosis. 2012, 225, 381–387. [Google Scholar] [CrossRef]
- Dichtl, W.; Dulak, J.; Frick, M.; Alber, H.F.; Schwarzacher, S.P.; Ares, M.P.; Nilsson, J.; Pachinger, O.; Weidinger, F. HMG-CoA reductase inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 58–63. [Google Scholar] [CrossRef]
- Arévalo-Lorido, J.C. Clinical relevance for lowering C-reactive protein with statins. Ann. Med. 2016, 48, 516–524. [Google Scholar] [CrossRef]
- Calabró, P.; Willerson, J.T.; Yeh, E.T. Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation. 2003, 108, 1930–1932. [Google Scholar] [CrossRef]
- DuBrock, H.M.; AbouEzzeddine, O.F.; Redfield, M.M. High-sensitivity C-reactive protein in heart failure with preserved ejection fraction. PLoS One. 2018, 13, e0201836. [Google Scholar] [CrossRef]
- Lakhani, I.; Wong, M.V.; Hung, J.K.F.; Gong, M.; Waleed, K.B.; Xia, Y.; Lee, S.; Roever, L.; Liu, T.; Tse, G.; Leung, K.S.K.; Li, K.H.C. Diagnostic and prognostic value of serum C-reactive protein in heart failure with preserved ejection fraction: a systematic review and meta-analysis. Heart Fail. Rev. 2021, 26, 1141–1150. [Google Scholar] [CrossRef]
- Puri, R.; Nissen, S.E.; Libby, P.; Shao, M.; Ballantyne, C.M.; Barter, P.J.; Chapman, M.J.; Erbel, R.; Raichlen, J.S.; Uno, K.; Kataoka, Y.; Nicholls, S.J. C-reactive protein, but not low-density lipoprotein cholesterol levels, associate with coronary atheroma regression and cardiovascular events after maximally intensive statin therapy. Circulation. 2013, 128, 2395–2403. [Google Scholar] [CrossRef]
- Komukai, K.; Kubo, T.; Kitabata, H.; Matsuo, Y.; Ozaki, Y.; Takarada, S.; Okumoto, Y.; Shiono, Y.; Orii, M.; Shimamura, K.; et al. Effect of atorvastatin therapy on fibrous cap thickness in coronary atherosclerotic plaque as assessed by optical coherence tomography: The EASY-FIT study. J. Am. Coll. Cardiol. 2014, 64, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Tanaka, A.; Kawasaki, T.; Goto, Y.; Morita, Y.; Asaumi, Y.; Nakao, K.; Fujiwara, R.; Nishimura, K.; Miyamoto, Y.; Ishihara, M.; Ogawa, H.; Koga, N.; Narula, J.; Yasuda, S. Effect of Intensive Statin Therapy on Coronary High-Intensity Plaques Detected by Noncontrast T1-Weighted Imaging: The AQUAMARINE Pilot Study. J. Am. Coll. Cardiol. 2015, 66, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Bonsu, K.O.; Reidpath, D.D.; Kadirvelu, A. Effects of Statin Treatment on Inflammation and Cardiac Function in Heart Failure: An Adjusted Indirect Comparison Meta-Analysis of Randomized Trials. Cardiovasc. Ther. 2015, 33, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Burger, F.; Pelli, G.; Poku, N.K.; Berlier, C.; Steffens, S.; Mach, F. Statins inhibit C-reactive protein-induced chemokine secretion, ICAM-1 upregulation and chemotaxis in adherent human monocytes. Rheumatology (Oxford). 2009, 48, 233–242. [Google Scholar] [CrossRef]
- Park, J.J.; Yoon, M.; Cho, H.W.; Cho, H.J.; Kim, K.H.; Yang, D.H.; Yoo, B.S.; Kang, S.M.; Baek, S.H.; Jeon, E.S.; Kim, J.J.; Cho, M.C.; Chae, S.C.; Oh, B.H.; Choi, D.J. C-reactive protein and statins in heart failure with reduced and preserved ejection fraction. Front. Cardiovasc. Med. 2022, 9, 1064967. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, Y. , Fard J.K.; Ghafoor D.; Eid A.H.; Sahebkar A. Paradoxical effects of statins on endothelial and cancer cells: the impact of concentrations. Cancer Cell Int. 2023, 23, 43. [Google Scholar] [CrossRef] [PubMed]
- Walkowski, B.; Kleibert, M.; Majka, M.; Wojciechowska, M. Insight into the Role of the PI3K/Akt Pathway in Ischemic Injury and Post-Infarct Left Ventricular Remodeling in Normal and Diabetic Heart. Cells. 2022, 11, 1553. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Qin, H.; Benveniste, E.N. The IFN-γ-Induced Transcriptional Program of the CIITA Gene Is Inhibited by Statins. Eur. J. Immunol. 2008, 38, 2325. [Google Scholar] [CrossRef]
- Leuenberger, T.; Pfueller, C.F.; Luessi, F.; Bendix, I.; Paterka, M.; Prozorovski, T.; Treue, D.; Luenstedt, S.; Herz, J.; Siffrin, V.; Infante-Duarte, C.; Zipp, F.; Waiczies, S. Modulation of dendritic cell immunobiology via inhibition of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase. PLoS One. 2014, 9, e100871. [Google Scholar] [CrossRef]
- Frostegård, J.; Zhang, Y.; Sun, J.; Yan, K.; Liu, A. Oxidized Low-Density Lipoprotein (OxLDL)-Treated Dendritic Cells Promote Activation of T Cells in Human Atherosclerotic Plaque and Blood, Which Is Repressed by Statins: MicroRNA let-7c Is Integral to the Effect. J. Am. Heart Assoc. 2016, 5, e003976. [Google Scholar] [CrossRef] [PubMed]
- Radyukhina, N.V.; Ruleva, N.Yu.; Filatova, A.Yu.; Arefieva, T.I. Inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase (statins) suppress human CD4+ T lymphocytes proliferation and motility in vitro. Bull. Exp. Biol. Med. 2021, 172, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Ruleva, N.Yu.; Radyukhina, N.V.; Zubkova, E.S.; Filatova, A.Yu.; Arefieva, T.I. Inhibitors of 3-Hydroxy-3-Methylglutaryl Coenzyme a Reductase (Statins) Suppress Differentiation and Reduce LPS IFNγ-Induced Cytokine Production in Human Monocyte Macrophage Culture. Bull. Exp. Biol. Med. 2020, 170, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Ghittoni, R.; Napolitani, G.; Benati, D.; Ulivieri, C.; Patrussi, L.; Laghi Pasini, F.; Lanzavecchia, A.; Baldari, C.T. Simvastatin inhibits the MHC class II pathway of antigen presentation by impairing Ras superfamily GTPases. Eur. J. Immunol. 2006, 36, 2885–2893. [Google Scholar] [CrossRef] [PubMed]
- Weitz-Schmidt, G. Statins as anti-inflammatory agents. Trends Pharmacol. Sci. 2002, 23, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Wu, C.; Xu, Y.; Cai, J.; Zhao, M.; Zu, L. The NO-cGMP-PKG Axis in HFpEF: From Pathological Mechanisms to Potential Therapies. Aging Dis. 2023, 14, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C.; Meyer, A.; Charloux, A.; Andrès, E.; Gény, B.; Talha, S. The Endocrine Function of the Heart: Physiology and Involvements of Natriuretic Peptides and Cyclic Nucleotide Phosphodiesterases in Heart Failure. J. Clin. Med. 2019, 8, 1746. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, K.; Liu, M.; Su, J.; Qin, X.; Wang, X.; Zhang, J.; Li, S.; Fan, G. An herbal preparation ameliorates heart failure with preserved ejection fraction by alleviating microvascular endothelial inflammation and activating NO-cGMP-PKG pathway. Phytomedicine. 2021, 91, 153633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Feng, B.; Ma, X.; Sun, K.; Xu, G.; Zhou, Y. Dapagliflozin improves left ventricular remodeling and aorta sympathetic tone in a pig model of heart failure with preserved ejection fraction. Cardiovasc. Diabetol. 2019, 18, 107. [Google Scholar] [CrossRef]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lódi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovács, Á.; Fülöp, G.Á.; Falcão-Pires, I.; Reusch, P.H.; Linthout, S.V.; Papp, Z.; van Heerebeek, L.; Vecchione, C.; Maier, L.S.; Ciccarelli, M.; Tschöpe, C.; Mügge, A.; Bagi, Z.; Sossalla, S.; Hamdani, N. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc. Res. 2021, 117, 495–507. [Google Scholar] [CrossRef]
- Takimoto, E.; Koitabashi, N.; Hsu, S.; Ketner, E.A.; Zhang, M.; Nagayama, T.; Bedja, D.; Gabrielson, K.L.; Blanton, R.; Siderovski, D.P.; Mendelsohn, M.E.; Kass, D.A. Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. J. Clin. Invest. 2009, 119, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.; Rainer, P.P.; Lee, D.I.; Hao, S.; Bedja, D.; Birnbaumer, L.; Cingolani, O.H.; Kass, D.A. Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cGMP-protein kinase G modulation. Circ. Res. 2014, 114, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, F. A concise discussion of the regulatory role of cGMP kinase I in cardiac physiology and pathology. Basic.Res. Cardiol. 2018, 113, 31. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Venugopal, H.; Frangogiannis, N.G. Smad-dependent pathways in the infarcted and failing heart. Curr. Opin. Pharmacol. 2022, 64, 102207. [Google Scholar] [CrossRef] [PubMed]
- Buxton, I.L.; Duan, D. Cyclic GMP/protein kinase G phosphorylation of Smad3 blocks transforming growth factor-beta-induced nuclear Smad translocation: a key antifibrogenic mechanism of atrial natriuretic peptide. Circ. Res. 2008, 102, 151–153. [Google Scholar] [CrossRef] [PubMed]
- LeWinter, M.M; Granzier, HL. Cardiac titin and heart disease. J. Cardiovasc. Pharm. 2014, 63, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–441. [Google Scholar] [CrossRef] [PubMed]
- Zile, M.R.; Baicu, C.F.; Ikonomidis, J.S.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; Redfield, M.M.; Bull, D.A.; Granzier, H.L.; LeWinter, M.M. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015, 131, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P.; Stasch, J.P. Anti-fibrotic effects of soluble guanylate cyclase stimulators and activators: a review of the preclinical evidence. Resp. Med. 2017, 122, S1–S9. [Google Scholar] [CrossRef]
- Wilck, N.; Markó, L.; Balogh, A.; Kräker, K.; Herse, F.; Bartolomaeus, H.; Szijártó, I.A.; Gollasch, M.; Reichhart, N.; Strauss, O.; Heuser, A.; Brockschnieder, D.; Kretschmer, A.; Lesche, R.; Sohler, F.; Stasch, J.P.; Sandner, P.; Luft, F.C.; Müller, D.N.; Dechend, R.; Haase, N. Nitric oxide-sensitive guanylyl cyclase stimulation improves experimental heart failure with preserved ejection fraction. JCI Insight. 2018, 3, e96006. [Google Scholar] [CrossRef]
- Burke, R.M.; Lighthouse, J.K.; Mickelsen, D.M.; Small, E.M. Sacubitril/Valsartan Decreases Cardiac Fibrosis in Left Ventricle Pressure Overload by Restoring PKG Signaling in Cardiac Fibroblasts. Circ. Heart Fail. 2019, 12, e005565. [Google Scholar] [CrossRef] [PubMed]
- Numata, G.; Takimoto, E. Cyclic GMP and PKG Signaling in Heart Failure. Front. Pharmacol. 2022, 13, 792798. [Google Scholar] [CrossRef]
- Ramasubbu, K.; Estep, J.; White, D.L.; Deswal, A.; Mann, D.L. Experimental and clinical basis for the use of statins in patients with ischemic and nonischemic cardiomyopathy. J. Am. Coll. Cardiol. 2008, 51, 415–426. [Google Scholar] [CrossRef]
- Antoniades, C.; Demosthenous, M.; Reilly, S.; Margaritis, M.; Zhang, M.H.; Antonopoulos, A.; Marinou, K.; Nahar, K.; Jayaram, R.; Tousoulis, D.; Bakogiannis, C.; Sayeed, R.; Triantafyllou, C.; Koumallos, N.; Psarros, C.; Miliou, A.; Stefanadis, C.; Channon, K.M.; Casadei, B. Myocardial redox state predicts in-hospital clinical outcome after cardiac surgery effects of short-term pre-operative statin treatment. J. Am. Coll. Cardiol. 2012, 59, 60–70. [Google Scholar] [CrossRef]
- Hahn, V.S.; Yanek, L.R.; Vaishnav, J.; Ying, W.; Vaidya, D.; Lee, Y.Z.J.; Riley, S.J.; Subramanya, V.; Brown, E.E.; Hopkins, C.D.; Ononogbu, S.; Perzel Mandell, K.; Halushka, M.K.; Steenbergen, C. Jr.; Rosenberg, A.Z.; Tedford, R.J.; Judge, D.P.; Shah, S.J.; Russell, S.D.; Kass, D.A.; Sharma, K. Endomyocardial Biopsy Characterization of Heart Failure With Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020, 8, 712–724. [Google Scholar] [CrossRef]
- Frohlich, E.D.; Apstein, C.; Chobanian, A.V.; Devereux, R.B.; Dustan, H.P.; Dzau, V.; Fauad-Tarazi, F.; Horan, M.J.; Marcus, M.; Massie, B.; et al. The heart in hypertension. N. Engl. J. Med. 1992, 327, 998–1008. [Google Scholar] [CrossRef]
- Hattori, T.; Shimokawa, H.; Higashi, M.; Hiroki, J.; Mukai, Y.; Tsutsui, H.; Kaibuchi, K.; Takeshita, A. Long-term inhibition of Rho-kinase suppresses left ventricular remodeling after myocardial infarction in mice. Circulation. 2004, 109, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Horinaka, S.; Mita, S.; Nakano, S.; Honda, T.; Yoshida, K.; Kobayashi, T.; Matsuoka, H. Critical role of Rho-kinase pathway for cardiac performance and remodeling in failing rat hearts. Cardiovasc. Res. 2002, 55, 757–777. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Chou, T.F.; Tsai, C.H. Association of pravastatin and left ventricular mass in hypercholesterolemic patients: role of 8-iso-prostaglandin f2alpha formation. J. Cardiovasc. Pharmacol. 2002, 40, 868–874. [Google Scholar] [CrossRef]
- Satoh, M.; Ogita, H.; Takeshita, K.; Mukai, Y.; Kwiatkowski, D.J.; Liao, J.K. Requirement of Rac1 in the development of cardiac hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 7432–7437. [Google Scholar] [CrossRef]
- Tanaka, S.; Fukumoto, Y.; Nochioka, K.; Minami, T.; Kudo, S.; Shiba, N.; Takai, Y.; Williams, C.L.; Liao, J.K.; Shimokawa, H. Statins exert the pleiotropic effects through small GTP-binding protein dissociation stimulator upregulation with a resultant Rac1 degradation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1591–600. [Google Scholar] [CrossRef] [PubMed]
- Kudo, S.; Satoh, K.; Nogi, M.; Suzuki, K.; Sunamura, S.; Omura, J.; Kikuchi, N.; Kurosawa, R.; Satoh, T.; Minami, T.; Ikeda, S.; Miyata, S.; Shimokawa, H. SmgGDS as a Crucial Mediator of the Inhibitory Effects of Statins on Cardiac Hypertrophy and Fibrosis: Novel Mechanism of the Pleiotropic Effects of Statins. Hypertension. 2016, 67, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Su, S.F.; Hsiao, C.L.; Chu, C.W.; Lee, B.C.; Lee, T.M. Effects of pravastatin on left ventricular mass in patients with hyperlipidemia and essential hypertension. Am. J. Cardiol. 2000, 86, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Haudek, S.B.; Gupta, D.; Dewald, O.; Schwartz, R.J.; Wei, L.; Trial, J.; Entman, M.L. Rho kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc. Res. 2009, 83, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, R.; Li, Y.; Noma, K.; Hiroi, Y.; Liu, P.Y.; Taniguchi, M.; Ito, M.; Liao, J.K. FHL2 prevents cardiac hypertrophy in mice with cardiac-specific deletion of ROCK2. FASEB J. 2013, 27, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Surma, M.; Yang, Y.; Wei, L. Disruption of both ROCK1 and ROCK2 genes in cardiomyocytes promotes autophagy and reduces cardiac fibrosis during aging. FASEB J. 2019, 33, 7348–7362. [Google Scholar] [CrossRef]
- Gabrielli, L.; Winter, J.L.; Godoy, I.; McNab, P.; Padilla, I.; Cordova, S.; Rigotti, P.; Novoa, U.; Mora, I.; García, L.; Ocaranza, M.P.; Jalil, J.E. Increased rho-kinase activity in hypertensive patients with left ventricular hypertrophy. Am. J. Hypertens. 2014, 27, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Somers, T.; Siddiqi, S.; Morshuis, W.J.; Russel, F.G.M.; Schirris, T.J.J. Statins and Cardiomyocyte Metabolism, Friend or Foe? J. Cardiovasc. Dev. Dis. 2023, 10, 417. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Shen, Y.; Liang, C.; Wang, H.; Huang, J.; Xue, P.; Luo, M. Inhibition of the mevalonate pathway improves myocardial fibrosis. Exp. Ther. Med. 2021, 21, 224. [Google Scholar] [CrossRef]
- Rizvi, F.; Siddiqui, R.; DeFranco, A.; Homar, P.; Emelyanova, L.; Holmuhamedov, E.; Ross, G.; Tajik, A.J.; Jahangir, A. Simvastatin reduces TGF-β1-induced SMAD2/3-dependent human ventricular fibroblasts differentiation: Role of protein phosphatase activation. Int. J. Cardiol. 2018, 270, 228–236. [Google Scholar] [CrossRef]
- Emelyanova, L.; Sra, A.; Schmuck, E.G.; Raval, A.N.; Downey, F.X.; Jahangir, A.; Rizvi, F.; Ross, G.R. Impact of statins on cellular respiration and de-differentiation of myofibroblasts in human failing hearts. ESC Heart. Fail. 2019, 6, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.F.; Hsieh, C.C.; Wang, S.C.; Chang, C.Y.; Hung, C.H.; Kuo, P.L.; Liu, Y.R.; Li, C.Y.; Liu, P.L. Simvastatin Attenuates Cardiac Fibrosis via Regulation of Cardiomyocyte-Derived Exosome Secretion. J. Clin. Med. 2019, 8, 794. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Denver, R.; Bailey, M.; Krum, H. In vitro inhibitory effects of atorvastatin on cardiac fibroblasts: implications for ventricular remodelling. Clin. Exp. Pharmacol. Physiol. 2005, 32, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Saka, M.; Obata, K.; Ichihara, S.; Cheng, X.W.; Kimata, H.; Nishizawa, T.; Noda, A.; Izawa, H.; Nagata, K.; Murohara, T.; Yokota, M. Pitavastatin improves cardiac function and survival in association with suppression of the myocardial endothelin system in a rat model of hypertensive heart failure. J. Cardiovasc. Pharmacol. 2006, 47, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Habibi, J.; Whaley-Connell, A.; Qazi, M.A.; Hayden, M.R.; Cooper, S.A.; Tramontano, A.; Thyfault, J.; Stump, C.; Ferrario, C.; Muniyappa, R.; Sowers, J.R. Rosuvastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, decreases cardiac oxidative stress and remodeling in Ren2 transgenic rats. Endocrinology. 2007, 148, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Marunouchi, T.; Matsumura, K.; Fuji, E.; Iwamoto, A.; Tanonaka, K. Simvastatin Attenuates Cardiac Fibrosis under Pathophysiological Conditions of Heart Failure with Preserved Left Ventricular Ejection Fraction by Inhibiting TGF-β Signaling. Pharmacology. 2024, 109, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Liu, J.; Chen, X.; Duan, Y.; Wang, X.; Shen, Y.; Kuang, Y.; Zhuang, T.; Tomlinson, B.; Chan, P.; Yu, Z.; Cheng, Y.; Zhang, L.; Liu, Z.; Zhang, Y.; Zhao, Z.; Zhang, Q.; Liu, J. Endothelial Klf2-Foxp1-TGFβ signal mediates the inhibitory effects of simvastatin on maladaptive cardiac remodeling. Theranostics. 2021, 11, 1609–1625. [Google Scholar] [CrossRef] [PubMed]
- Pentz, R.; Kaun, C.; Thaler, B.; Stojkovic, S.; Lenz, M.; Krychtiuk, K.A.; Zuckermann, A.; Huber, K.; Wojta, J.; Hohensinner, P.J.; Demyanets, S. Cardioprotective cytokine interleukin-33 is up-regulated by statins in human cardiac tissue. J. Cell Mol. Med. 2018, 22, 6122–6133. [Google Scholar] [CrossRef] [PubMed]
- Mannheim, D.; Herrmann, J.; Bonetti, P.O.; Lavi, R.; Lerman, L.O.; Lerman, A. Simvastatin preserves diastolic function in experimental hypercholesterolemia independently of its lipid lowering effect. Atherosclerosis. 2011, 216, 283–291. [Google Scholar] [CrossRef]
- Lu, J.; Liu, F.; Chen, F.; Jin, Y.; Chen, H.; Liu, D.; Cui, W. Amlodipine and atorvastatin improve ventricular hypertrophy and diastolic function via inhibiting TNF-alpha, IL-1beta and NF-kappaB inflammatory cytokine networks in elderly spontaneously hypertensive rats. Biomed. Pharmacother. 2016, 83, 330–339. [Google Scholar] [CrossRef]
- Yamada, Y.; Takeuchi, S.; Yoneda, M.; Ito, S.; Sano, Y.; Nagasawa, K.; Matsuura, N.; Uchinaka, A.; Murohara, T.; Nagata, K. Atorvastatin reduces cardiac and adipose tissue inflammation in rats with metabolic syndrome. Int. J. Cardiol. 2017, 240, 332–338. [Google Scholar] [CrossRef]
- Okura, H.; Asawa, K.; Kubo, T.; Taguchi, H.; Toda, I.; Yoshiyama, M.; Yoshikawa, J.; Yoshida, K. Impact of statin therapy on systemic inflammation, left ventricular systolic and diastolic function and prognosis in low risk ischemic heart disease patients without history of congestive heart failure. Intern. Med. 2007, 46, 1337–1343. [Google Scholar] [CrossRef]
- Yagi, S.; Akaike, M.; Aihara, K.-I.; Iwase, T.; Ishikawa, K.; Yoshida, S.; Sumitomo-Ueda, Y.; Kusunose, K.; Niki, T.; Yamaguchi, K.; Koshiba, K.; Taketani, Y.; Tomita, N.; Yamada, H.; Soeki, T.; Wakatsuki, T.; Matsumoto, T.; Sata, M. Effect of low-dose (1 mg/day) pitavastatin on left ventricular diastolic function and albuminuria in patients with hyperlipidemia. Am. J. Cardiol. 2011, 107, 1644–1649. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, A.; Belyavskiy, E.; Potekhina, A.; Ageev, F. Asymptomatic Left Ventricular Hypertrophy Is a Potent Risk Factor for the Development of HFpEF but Not HFrEF: Results of a Retrospective Cohort Study. J. Clin. Med. 2022, 11, 3885. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.K.; Yeh, C.F.; Chiang, J.Y.; Lin, T.T.; Wu, Y.F.; Chiang, C.K.; Kao, T.W.; Hung, K.Y.; Huang, J.W. Effects of atorvastatin treatment on left ventricular diastolic function in peritoneal dialysis patients - The ALEVENT clinical trial. J. Clin. Lipidol. 2017, 11, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G. Epicardial adipose tissue in contemporary cardiology. Nat. Rev. Cardiol. 2022, 19, 593–606. [Google Scholar] [CrossRef]
- Nerlekar, N.; Muthalaly, R.G.; Wong, N.; Thakur, U.; Wong, D.T.L.; Brown, A.J.; Marwick, T.H. Association of Volumetric Epicardial Adipose Tissue Quantification and Cardiac Structure and Function. J. Am. Heart Assoc. 2018, 7, e009975. [Google Scholar] [CrossRef] [PubMed]
- van Woerden, G.; van Veldhuisen, D.J.; Manintveld, O.C.; van Empel, V.P.M.; Willems, T.P.; de Boer, R.A.; Rienstra, M.; Westenbrink, B.D.; Gorter, T.M. Epicardial Adipose Tissue and Outcome in Heart Failure With Mid-Range and Preserved Ejection Fraction. Circ. Heart Fail. 2022, 15, e009238. [Google Scholar] [CrossRef]
- Parisi, V.; Petraglia, L.; D'Esposito, V.; Cabaro, S.; Rengo, G.; Caruso, A.; Grimaldi, M.G.; Baldascino, F.; De Bellis, A.; Vitale, D.; Formisano, R.; Ferro, A.; Paolillo, S.; Davin, L.; Lancellotti, P.; Formisano, P.; Perrone Filardi, P.; Ferrara, N.; Leosco, D. Statin therapy modulates thickness and inflammatory profile of human epicardial adipose tissue. Int. J. Cardiol. 2019, 274, 326–330. [Google Scholar] [CrossRef]
- Park, J.H.; Park, Y.S.; Kim, Y.J.; Lee, I.S.; Kim, J.H.; Lee, J.H.; Choi, S.W.; Jeong, J.O.; Seong, I.W. Effects of statins on the epicardial fat thickness in patients with coronary artery stenosis underwent percutaneous coronary intervention: comparison of atorvastatin with simvastatin/ezetimibe. J. Cardiovasc. Ultrasound. 2010, 18, 121–126. [Google Scholar] [CrossRef]
- Soucek, F.; Covassin, N.; Singh, P.; Ruzek, L.; Kara, T.; Suleiman, M.; Lerman, A.; Koestler, C.; Friedman, P.A.; Lopez-Jimenez, F.; Somers, V.K. Effects of Atorvastatin (80 mg) Therapy on Quantity of Epicardial Adipose Tissue in Patients Undergoing Pulmonary Vein Isolation for Atrial Fibrillation. Am. J. Cardiol. 2015, 116, 1443–1446. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, N.; Melek, B.H.; Arepalli, C.D.; Hartlage, G.R.; Chen, Z.; Kim, S.; Stillman, A.E.; Raggi, P. Effect of intensive versus moderate lipid-lowering therapy on epicardial adipose tissue in hyperlipidemic post-menopausal women: a substudy of the BELLES trial (Beyond Endorsed Lipid Lowering with EBT Scanning). J. Am. Coll. Cardiol. 2013, 61, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
- Bonsu, K.O.; Kadirvelu, A.; Reidpath, D.D. Statins in heart failure: do we need another trial? Vasc. Health Risk Manag. 2013, 9, 303–319. [Google Scholar] [PubMed]
- Tavazzi, L.; Maggioni, A.P.; Marchioli, R.; Barlera, S.; Franzosi, M.G.; Latini, R.; Lucci, D.; Nicolosi, G.L.; Porcu, M.; Tognoni, G.; Gissi-HF Investigators. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008, 372, 1231–1239. [Google Scholar] [PubMed]
- Kjekshus, J.; Apetrei, E.; Barrios, V.; Böhm, M.; Cleland, J.G.; Cornel, J.H.; Dunselman, P.; Fonseca, C.; Goudev, A.; Grande, P.; Gullestad, L.; Hjalmarson, A.; Hradec, J.; Jánosi, A.; Kamenský, G.; Komajda, M.; Korewicki, J.; Kuusi, T.; Mach, F.; Mareev, V.; McMurray, J.J.; Ranjith, N.; Schaufelberger, M.; Vanhaecke, J.; van Veldhuisen, D.J.; Waagstein, F.; Wedel, H.; Wikstrand, J.; CORONA Group. Rosuvastatin in older patients with systolic heart failure. N. Engl. J. Med. 2007, 357, 2248–2261. [Google Scholar] [CrossRef] [PubMed]
- Gastelurrutia, P.; Lupón, J.; de Antonio, M.; Urrutia, A.; Díez, C.; Coll, R.; Altimir, S.; Bayes-Genis, A. Statins in heart failure: the paradox between large randomized clinical trials and real life. Mayo Clin. Proc. 2012, 87, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Maison, P.; Desamericq, G.; Hemery, F.; Elie, N.; Del'volgo, A.; Dubois-Randé, J.L.; Hittinger, L.; Macquin-Mavier, I. Relationship between recommended chronic heart failure treatments and mortality over 8 years in real-world conditions: a pharmacoepidemiological study. Eur. J. Clin. Pharmacol. 2013, 69, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Climent, E.; Benaiges, D.; Pedro-Botet, J. Hydrophilic or Lipophilic Statins? Front Cardiovasc Med. 2021, 8, 687585. [Google Scholar] [CrossRef]
- Alehagen, U.; Benson, L.; Edner, M.; Dahlström, U.; Lund, L.H. Association between use of statins and outcomes in heart failure with reduced ejection fraction: prospective propensity score matched cohort study of 21 864 patients in the Swedish Heart Failure Registry. Circ. Heart Fail. 2015, 8, 252–260. [Google Scholar] [CrossRef]
- Bielecka-Dabrowa, A.; Ibadete Bytyç, I.; Von Haehling, S.; Anker, S.; Jozwiak, J.; Rysz, J.; Hernandez, A.V.; Bajraktari, G.; Mikhalidis, D.M.; Banach, M. Association of statin use and clinical outcomes in heart failure patients: a systematic review and meta-analysis. Lipids Health Dis. 2019, 18, 188. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; Graham, I.M.; Halliday, A.; Landmesser, U.; Mihaylova, B.; Pedersen, T.R.; Riccardi, G.; Richter, D.J.; Sabatine, M.S.; Taskinen, M.R.; Tokgozoglu, L.; Wiklund, O.; ESC Scientific Document Group. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Ohte, N.; Little, W.C. Statins beneficial for heart failure with preserved ejection fraction but not heart failure with reduced ejection fraction? Circ. J. 2015, 79, 508–509. [Google Scholar] [CrossRef]
- Zakeri, R.; Chamberlain, A.M.; Roger, V.L.; Redfield, M.M. Temporal relationship and prognostic significance of atrial fibrillation in heart failure patients with preserved ejection fraction: a community-based study. Circulation. 2013, 128, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, A.G.; Dreeva, Z.V.; Potekhina, A.V.; Arefieva, T.I.; Masenko, V.P.; Ageev, F.T. Statins improves functional capacity and restores LV diastolic reserve in patients with heart failure with preserved left ventricular ejection fraction. Eur. J. Heart Fail. 2019, 21 (Suppl 1), 418. [Google Scholar]
- Vanderpool, R.R.; Saul, M.; Nouraie, M.; Gladwin, M.T.; Simon, M.A. Association between hemodynamic markers of pulmonary hypertension and outcomes in heart failure with preserved ejection fraction. JAMA Cardiol. 2018, 3, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Holzhauser, L.; Hovnanians, N.; Eshtehardi, P.; Mojadidi, M.K.; Deng, Y.; Goodman-Meza, D.; Msaouel, P.; Ko, Y.A.; Zolty, R. Statin therapy improves survival in patients with severe pulmonary hypertension: a propensity score matching study. Heart Vessels. 2017, 32, 969–976. [Google Scholar] [CrossRef]
- Fukuta, H.; Sane, D.C.; Brucks, S.; Little, W.C. Statin therapy may be associated with lower mortality in patients with diastolic heart failure: a preliminary report. Circulation. 2005, 112, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Roik, M.; Starczewska, M.H.; Huczek, Z.; Kochanowski, J.; Opolski, G. Statin therapy and mortality among patients hospitalized with heart failure and preserved left ventricular function--a preliminary report. Acta Cardiol. 2008, 63, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Wang, Y.; Foody, J.M. Effect of statins, angiotensin-converting enzyme inhibitors, and beta blockers on survival in patients >or=65 years of age with heart failure and preserved left ventricular systolic function. Am. J. Cardiol. 2008, 101, 217–222. [Google Scholar] [CrossRef]
- Tehrani, F.; Morrissey, R.; Phan, A.; Chien, C.; Schwarz, E.R. Statin therapy in patients with diastolic heart failure. Clin Cardiol. 2010, 33, E1–5. [Google Scholar] [CrossRef]
- Gomez-Soto, F.M.; Romero, S.P.; Bernal, J.A.; Escobar, M.A.; Puerto, J.L.; Andrey, J.L.; Ruiz, P.; Gomez, F. Mortality and morbidity of newly diagnosed heart failure treated with statins: a propensity-adjusted cohort study. Int. J. Cardiol. 2010, 140, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Quirós López, R.; García Alegría, J.; Martín Escalante, M.D.; Trujillo Santos, J.; Villena Ruiz, M.Á.; Perea Milla, E. Prognostic factors and long-term survival after initial diagnosis of heart failure. Med. Clin. (Barc). 2012, 138, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Suzuki, S.; Yajima, J.; Oikawa, Y.; Sagara, K.; Otsuka, T.; Matsuno, S.; Kano, H.; Uejima, T.; Koike, A.; Nagashima, K.; Kirigaya, H.; Sawada, H.; Aizawa, T.; Yamashita, T. Clinical characteristics and long-term clinical outcomes of Japanese heart failure patients with preserved versus reduced left ventricular ejection fraction: a prospective cohort of Shinken Database 2004-2011. J. Cardiol. 2013, 62, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Alehagen, U.; Benson, L.; Edner, M.; Dahlström, U.; Lund, L.H. Association Between Use of Statins and Mortality in Patients With Heart Failure and Ejection Fraction of ≥50. Circ. Heart. Fail. 2015, 8, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Duan, L.; Clare, R.; Hekimian, A.; Spencer, H.; Chen, W. Comparison of Effects of Statin Use on Mortality in Patients With Heart Failure and Preserved Versus Reduced Left Ventricular Ejection Fraction. Am. J. Cardiol. 2018, 122, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Nochioka, K.; Sakata, Y.; Miyata, S.; Miura, M.; Takada, T.; Tadaki, S.; Ushigome, R.; Yamauchi, T.; Takahashi, J.; Shimokawa, H.; CHART-2 Investigators. Prognostic impact of statin use in patients with heart failure and preserved ejection fraction. Circ. J. 2015, 79, 574–582. [Google Scholar] [CrossRef]
- Yap, J.; Sim, D.; Lim, C.P.; Chia, S.Y.; Go, Y.Y.; Jaufeerally, F.R.; Sim, L.L.; Liew, R.; Ching, C.K. Predictors of two-year mortality in Asian patients with heart failure and preserved ejection fraction. Int J Cardiol. 2015, 183, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, T.; Kajio, H. Favorable effects of statins in the treatment of heart failure with preserved ejection fraction in patients without ischemic heart disease. Int. J. Cardiol. 2018, 255, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Marume, K.; Takashio, S.; Nagai, T.; Tsujita, K.; Saito, Y.; Yoshikawa, T.; Anzai, T. Effect of Statins on Mortality in Heart Failure With Preserved Ejection Fraction Without Coronary Artery Disease- Report From the JASPER Study. Circ. J. 2019, 83, 357–367. [Google Scholar] [CrossRef]
- Liu, G.; Zheng, X.-X.; Xu, Y.-L.; Ru, J.; Hui, R.T.; Huang, X.H. Meta-analysis of the effect of statins on mortality in patients with preserved ejection fraction. Am. J. Cardiol. 2014, 113, 1198–1204. [Google Scholar] [CrossRef]
- Fukuta, H.; Goto, T.; Wakami, K.; Ohte, N. The effect of statins on mortality in heart failure with preserved ejection fraction: a meta-analysis of propensity score analyses. Int. J. Cardiol. 2016, 214, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Ergatoudes, C.; Schaufelberger, M.; Andersson, B.; Pivodic, A.; Dahlström, U.; Fu, M. Non-cardiac comorbidities and mortality in patients with heart failure with reduced vs. preserved ejection fraction: a study using the Swedish Heart Failure Registry. Clin. Res. Cardiol. 2019, 108, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Sanders-van Wijk, S.; Tromp, J.; Beussink-Nelson, L.; Hage, C.; Svedlund, S.; Saraste, A.; Swat, S.A.; Sanchez, C.; Njoroge, J.; Tan, R.S.; Fermer, M.L.; Gan, L.M.; Lund, L.H.; Lam, C.S.P.; Shah, S.J. Proteomic Evaluation of the Comorbidity-Inflammation Paradigm in Heart Failure With Preserved Ejection Fraction: Results From the PROMIS-HFpEF Study. Circulation. 2020, 142, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Armitage, J.; Parish, S.; Sleigh, P.; Peto, R.; Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003, 361, 2005–2016. [Google Scholar] [PubMed]
- Tonelli, M.; Moyé, L.; Sacks, F.M.; Kiberd, B.; Curhan, G.; Cholesterol and Recurrent Events (CARE) Trial Investigators. Pravastatin for secondary prevention of cardiovascular events in persons with mild chronic renal insufficiency. Ann. Intern. Med. 2003, 138, 98–104. [Google Scholar] [CrossRef]
- Raymakers, A.J.N.; Sadatsafavi, M.; Sin, D.D.; De Vera, M.A.; Lynd, L.D. The Impact of Statin Drug Use on All-Cause Mortality in Patients With COPD: A Population-Based Cohort Study. Chest. 2017, 152, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef]
- Nezasa, K.; Higaki, K.; Matsumura, T.; Inazawa, K.; Hasegawa, H.; Nakano, M.; Koike, M. Liver-specific distribution of rosuvastatin in rats: comparison with pravastatin and simvastatin. Drug Metab. Dispos. 2002, 30, 1158–1163. [Google Scholar] [CrossRef]
- Sahebkar, A.; Kotani, K.; Serban, C.; Ursoniu, S.; Mikhailidis, D.P.; Jones, S.R.; Ray, K.K.; Blaha, M.J.; Rysz, J.; Toth, P.P.; Muntner, P.; Lip, G.Y.; Banach, M.; Lipid and Blood Pressure Meta-analysis Collaboration (LBPMC) Group. Statin therapy reduces plasma endothelin-1 concentrations: A meta-analysis of 15 randomized controlled trials. Atherosclerosis. 2015, 241, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.J.; Cauthen, C.A.; Biondi-Zoccai, G.G.; Abbate, A.; Vrtovec, B.; Khan, B.V.; Vetrovec, G.W. Meta-analysis of randomized controlled trials of statins versus placebo in patients with heart failure. Am. J. Cardiol. 2009, 104, 1708–1716. [Google Scholar] [CrossRef]
- Morimoto, T.; Katanasaka, Y.; Sunagawa, Y.; Hirano, S.; Miyazaki, Y.; Funamoto, M.; Hojo, Y.; Suzuki, H.; Morimoto, E.; Ueno, M.; Shimatsu, A.; Satoh-Asahara, N.; Yamakage, H.; Wada, H.; Hasegawa, K. Effects of Statins on Left Ventricular Diastolic Function in Patients with Dyslipidemia and Diastolic Dysfunction (Stat-LVDF Study). Biol. Pharm. Bull. 2015, 38, 1404–1409. [Google Scholar] [CrossRef]
- Murrow, J.R.; Sher, S.; Ali, S.; Uphoff, I.; Patel, R.; Porkert, M.; Le, N.A.; Jones, D.; Quyyumi, A.A. The differential effect of statins on oxidative stress and endothelial function: atorvastatin versus pravastatin. J. Clin. Lipidol. 2012, 6, 42–49. [Google Scholar] [CrossRef]
- Patel, K.K.; Sehgal, V.S.; Kashfi, K. Molecular targets of statins and their potential side effects: Not all the glitter is gold. Eur. J. Pharmacol. 2022, 922, 174906. [Google Scholar] [CrossRef]
- Thompson, P.D.; Panza, G.; Zaleski, A.; Taylor, B. Statin-Associated Side Effects. J. Am. Coll. Cardiol. 2016, 67, 2395–2410. [Google Scholar] [CrossRef]
- Qu, H.; Guo, M.; Chai, H.; Wang, W.T.; Gao, Z.Y.; Shi, D.Z. Effects of Coenzyme Q10 on Statin-Induced Myopathy: An Updated Meta-Analysis of Randomized Controlled Trials. J. Am. Heart Assoc. 2018, 7, e009835. [Google Scholar] [CrossRef]
- Turner, R.M.; Pirmohamed, M. Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components. J. Clin. Med. 2019, 9, 22. [Google Scholar] [CrossRef]
- Sattar, N.; Preiss, D.; Murray, H.M.; Welsh, P.; Buckley, B.M.; de Craen, A.J.; Seshasai, S.R.; McMurray, J.J.; Freeman, D.J.; Jukema, J.W.; Macfarlane, P.W.; Packard, C.J.; Stott, D.J.; Westendorp, R.G.; Shepherd, J.; Davis, B.R.; Pressel, S.L.; Marchioli, R.; Marfisi, R.M.; Maggioni, A.P.; Tavazzi, L.; Tognoni, G.; Kjekshus, J.; Pedersen, T.R.; Cook, T.J.; Gotto, A.M.; Clearfield, M.B.; Downs, J.R.; Nakamura, H.; Ohashi, Y.; Mizuno, K.; Ray, K.K.; Ford, I. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010, 375, 735–742. [Google Scholar] [CrossRef]
- Mach, F.; Ray, K.K.; Wiklund, O.; Corsini, A.; Catapano, A.L.; Bruckert, E.; De Backer, G.; Hegele, R.A.; Hovingh, G.K.; Jacobson, T.A.; Krauss, R.M.; Laufs, U.; Leiter, L.A.; März, W.; Nordestgaard, B.G.; Raal, F.J.; Roden, M.; Santos, R.D.; Stein, E.A.; Stroes, E.S.; Thompson, P.D.; Tokgözoglu, L.; Vladutiu, G.D.; Gencer, B.; Stock, J.K.; Ginsberg, H.N.; Chapman, M.J.; European Atherosclerosis Society Consensus Panel. Adverse effects of statin therapy: perception vs. the evidence - focus on glucose homeostasis, cognitive, renal and hepatic function, haemorrhagic stroke and cataract. Eur. Heart J. 2018, 39, 2526–2539. [Google Scholar] [CrossRef]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, S.P. Different effects of statins on induction of diabetes mellitus: an experimental study. Drug Des. Devel. Ther. 2015, 9, 6211–6223. [Google Scholar] [CrossRef]
- Robinson, J.G. Statins and diabetes risk: how real is it and what are the mechanisms? Curr. Opin. Lipidol. 2015, 26, 228–235. [Google Scholar] [CrossRef]
- Drexel, H.; Coats, A.J.S.; Spoletini, I.; Bilato, C.; Mollace, V.; Perrone Filardi, P.; Rosano, G.M.C. An expert opinion paper on statin adherence and implementation of new lipid-lowering medications by the ESC Working Group on Cardiovascular Pharmacotherapy: Barriers to be overcome. Eur. Heart J. Cardiovasc. Pharmacother. 2020, 6, 115–121. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Shahzeb Khan, M.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure, E.; Giannetti, N.; Gomez-Mesa, J.E.; Janssens, S.; Januzzi, J.L.; Gonzalez-Juanatey, J.R.; Merkely, B.; Nicholls, S.J.; Perrone, S.V.; Piña, I.L.; Ponikowski, P.; Senni, M.; Seronde, M.F.; Sim, D.; Spinar, J.; Squire, I.; Taddei, S.; Tsutsui, H.; Verma, S.; Vinereanu, D.; Zhang, J.; Jamal, W.; Schnaidt, S.; Schnee, J.M.; Brueckmann, M.; Pocock, S.J.; Zannad, F.; Packer, M.; EMPEROR-Preserved Trial Committees and Investigators. Baseline characteristics of patients with heart failure with preserved ejection fraction in the EMPEROR-Preserved trial. Eur. J. Heart Fail. 2020, 22, 2383–2392. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Arena, R.; Biondi-Zoccai, G.; Canada, J.M.; Oddi, C.; Abouzaki, N.A.; Jahangiri, A.; Falcao, R.A.; Kontos, M.C.; Shah, K.B.; Voelkel, N.F.; Dinarello, C.A.; Abbate, A. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am. J. Cardiol. 2014, 113, 321–327. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Trankle, C.R.; Canada, J.M.; Carbone, S.; Buckley, L.; Kadariya, D.; Del Buono, M.G.; Billingsley, H.; Wohlford, G.; Viscusi, M.; Oddi-Erdle, C.; Abouzaki, N.A.; Dixon, D.; Biondi-Zoccai, G.; Arena, R.; Abbate, A. IL-1 Blockade in Patients With Heart Failure With Preserved Ejection Fraction. Circ. Heart Fail. 2018, 11, e005036. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; Kastelein, J.J.P.; Cornel, J.H.; Pais, P.; Pella, D.; Genest, J.; Cifkova, R.; Lorenzatti, A.; Forster, T.; Kobalava, Z.; Vida-Simiti, L.; Flather, M.; Shimokawa, H.; Ogawa, H.; Dellborg, M.; Rossi, P.R.F.; Troquay, R.P.T.; Libby, P.; Glynn, R.J.; CANTOS Trial Group. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
Figure 1.
The beneficial myocardial effects of statins in heart failure with preserved ejection fraction (HFpEF) may be due to their impact on different cell types. HDAC indicates histone deacetylase; LFA-1, leukocyte function-associated antigen-1; NO, nitic oxide; ROS reactive oxygen species. The upward pointing arrows denote stimulatory and downward pointing arrows inhibitory effects.
Figure 1.
The beneficial myocardial effects of statins in heart failure with preserved ejection fraction (HFpEF) may be due to their impact on different cell types. HDAC indicates histone deacetylase; LFA-1, leukocyte function-associated antigen-1; NO, nitic oxide; ROS reactive oxygen species. The upward pointing arrows denote stimulatory and downward pointing arrows inhibitory effects.
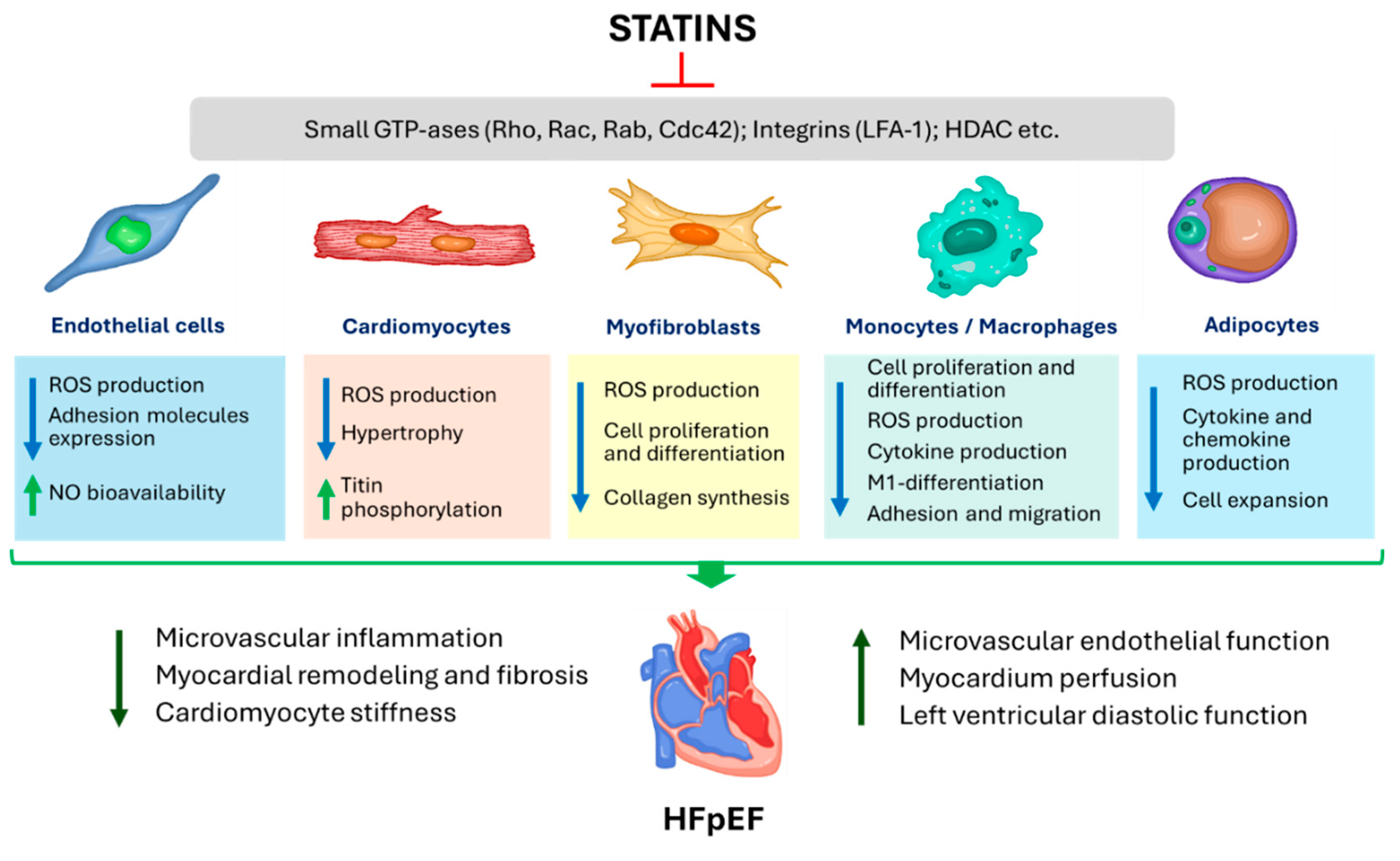
Figure 2.
The inflammation paradigm in heart failure with preserved ejection fraction (HFpEF) and potential effects of statins. Unhealthy aging and common pro-inflammatory comorbidities via elevated blood levels of pro-inflammatory cytokines drive chronic systemic low-intensity inflammation leading to coronary microvascular endothelial dysfunction/activation and nitrosative/oxidative stress. Activated/dysfunctional endothelial cells express adhesion molecules (ICAM-1 and VCAM-1) and secrete chemokines (MCP-1), leading to the migration of monocytes from the bloodstream into the myocardium, where they transform into macrophages and release transforming growth factor-β (TGF-β), which promotes the conversion of fibroblasts to myofibroblasts with subsequent deposition of collagen, resulted in progression of fibrosis and diastolic dysfunction. The formation of reactive oxygen species (ROS) within endothelial cells leads to decreased nitric oxide (NO) bioavailability with subsequent suppression of NO - cyclic guanyl monophosphate (cGMP) - protein kinase G (PKG) signaling and cardiac structural and functional adverse changes. Statins have numerous pleiotropic effects and may be useful at all stages of this inflammation paradigm. Statins reduce the production of proinflammatory cytokines and decrease the expression of adhesion molecules and chemokines. In endothelial cells, statins induce endothelial NO synthase (eNOS) and reduce ROS levels through direct ROS scavenging and suppression of NADPH-oxidase activity. This prevents eNOS uncoupling by suppressing the oxidation of a cofactor of eNOS tetrahydrobiopterin (BH4) via peroxynitrite (OONO–), which is formed by the interaction of superoxide anions (O2–) and NO. Ultimately, all these effects increase NO bioavailability and restore the NO-cGMP-PKG signaling network with the numerous resultant beneficial effects. PKG mediates phosphorylation (P) of phospholamban (PLB) to activate sarcoplasmic reticulum (SR) calcium ions (Ca2+)-ATPase pump and increase Ca2+ uptake into the SR, as well as the phosphorylation of troponin I (Tn I) to reduce myofilament Ca2+ sensitivity and thereby increase lusitropy. The PKG-mediated phosphorylation of titin reduces cardiomyocyte stiffness. PKG suppresses prohypertrophic signaling cascades, including Gq-coupled pathway via phosphorylation of the regulator of G protein signaling (RGS) 2/4, and the calcineurin/ nuclear factor of activated T-cells (NFAT) pathway via phosphorylation of the canonical transient receptor potential Ca2+ channel (TRPC) 6 signaling. Statins may also reduce myocardial fibrosis presumably via increased NO bioavailability, prevention of fibroblasts differentiation, inhibition of profibrotic signaling pathways downstream of the TGF-β receptors, and collagen synthesis. AT indicates angiotensin; CKD, chronic kidney disease; ET, endothelin; GDF-15, growth differentiation factor-15; GPCR, G-protein controlled receptor; GTP indicates guanosine triphosphate; hsCRP, high-sensitivity C-reactive protein; ICAM-1, intercellular adhesion molecule 1; IL, interleukin; IP3, inositol triphosphate; MCP-1, monocyte chemoattractant protein-1; NADPH, nicotinamide adenine dinucleotide phosphate; PDE5, phosphodiesterase-5; RyR, ryanodine receptor; TNF-α, tumor necrosis factor-α, VCAM 1, vascular cell adhesion molecule 1; PLC, phospholipase C; sGC, soluble cyclases; ST2, soluble suppression of tumorigenicity 2; Tm, tropomyosin. The arrow lines denote stimulatory and ꟶ lines inhibitory effects.
Figure 2.
The inflammation paradigm in heart failure with preserved ejection fraction (HFpEF) and potential effects of statins. Unhealthy aging and common pro-inflammatory comorbidities via elevated blood levels of pro-inflammatory cytokines drive chronic systemic low-intensity inflammation leading to coronary microvascular endothelial dysfunction/activation and nitrosative/oxidative stress. Activated/dysfunctional endothelial cells express adhesion molecules (ICAM-1 and VCAM-1) and secrete chemokines (MCP-1), leading to the migration of monocytes from the bloodstream into the myocardium, where they transform into macrophages and release transforming growth factor-β (TGF-β), which promotes the conversion of fibroblasts to myofibroblasts with subsequent deposition of collagen, resulted in progression of fibrosis and diastolic dysfunction. The formation of reactive oxygen species (ROS) within endothelial cells leads to decreased nitric oxide (NO) bioavailability with subsequent suppression of NO - cyclic guanyl monophosphate (cGMP) - protein kinase G (PKG) signaling and cardiac structural and functional adverse changes. Statins have numerous pleiotropic effects and may be useful at all stages of this inflammation paradigm. Statins reduce the production of proinflammatory cytokines and decrease the expression of adhesion molecules and chemokines. In endothelial cells, statins induce endothelial NO synthase (eNOS) and reduce ROS levels through direct ROS scavenging and suppression of NADPH-oxidase activity. This prevents eNOS uncoupling by suppressing the oxidation of a cofactor of eNOS tetrahydrobiopterin (BH4) via peroxynitrite (OONO–), which is formed by the interaction of superoxide anions (O2–) and NO. Ultimately, all these effects increase NO bioavailability and restore the NO-cGMP-PKG signaling network with the numerous resultant beneficial effects. PKG mediates phosphorylation (P) of phospholamban (PLB) to activate sarcoplasmic reticulum (SR) calcium ions (Ca2+)-ATPase pump and increase Ca2+ uptake into the SR, as well as the phosphorylation of troponin I (Tn I) to reduce myofilament Ca2+ sensitivity and thereby increase lusitropy. The PKG-mediated phosphorylation of titin reduces cardiomyocyte stiffness. PKG suppresses prohypertrophic signaling cascades, including Gq-coupled pathway via phosphorylation of the regulator of G protein signaling (RGS) 2/4, and the calcineurin/ nuclear factor of activated T-cells (NFAT) pathway via phosphorylation of the canonical transient receptor potential Ca2+ channel (TRPC) 6 signaling. Statins may also reduce myocardial fibrosis presumably via increased NO bioavailability, prevention of fibroblasts differentiation, inhibition of profibrotic signaling pathways downstream of the TGF-β receptors, and collagen synthesis. AT indicates angiotensin; CKD, chronic kidney disease; ET, endothelin; GDF-15, growth differentiation factor-15; GPCR, G-protein controlled receptor; GTP indicates guanosine triphosphate; hsCRP, high-sensitivity C-reactive protein; ICAM-1, intercellular adhesion molecule 1; IL, interleukin; IP3, inositol triphosphate; MCP-1, monocyte chemoattractant protein-1; NADPH, nicotinamide adenine dinucleotide phosphate; PDE5, phosphodiesterase-5; RyR, ryanodine receptor; TNF-α, tumor necrosis factor-α, VCAM 1, vascular cell adhesion molecule 1; PLC, phospholipase C; sGC, soluble cyclases; ST2, soluble suppression of tumorigenicity 2; Tm, tropomyosin. The arrow lines denote stimulatory and ꟶ lines inhibitory effects.
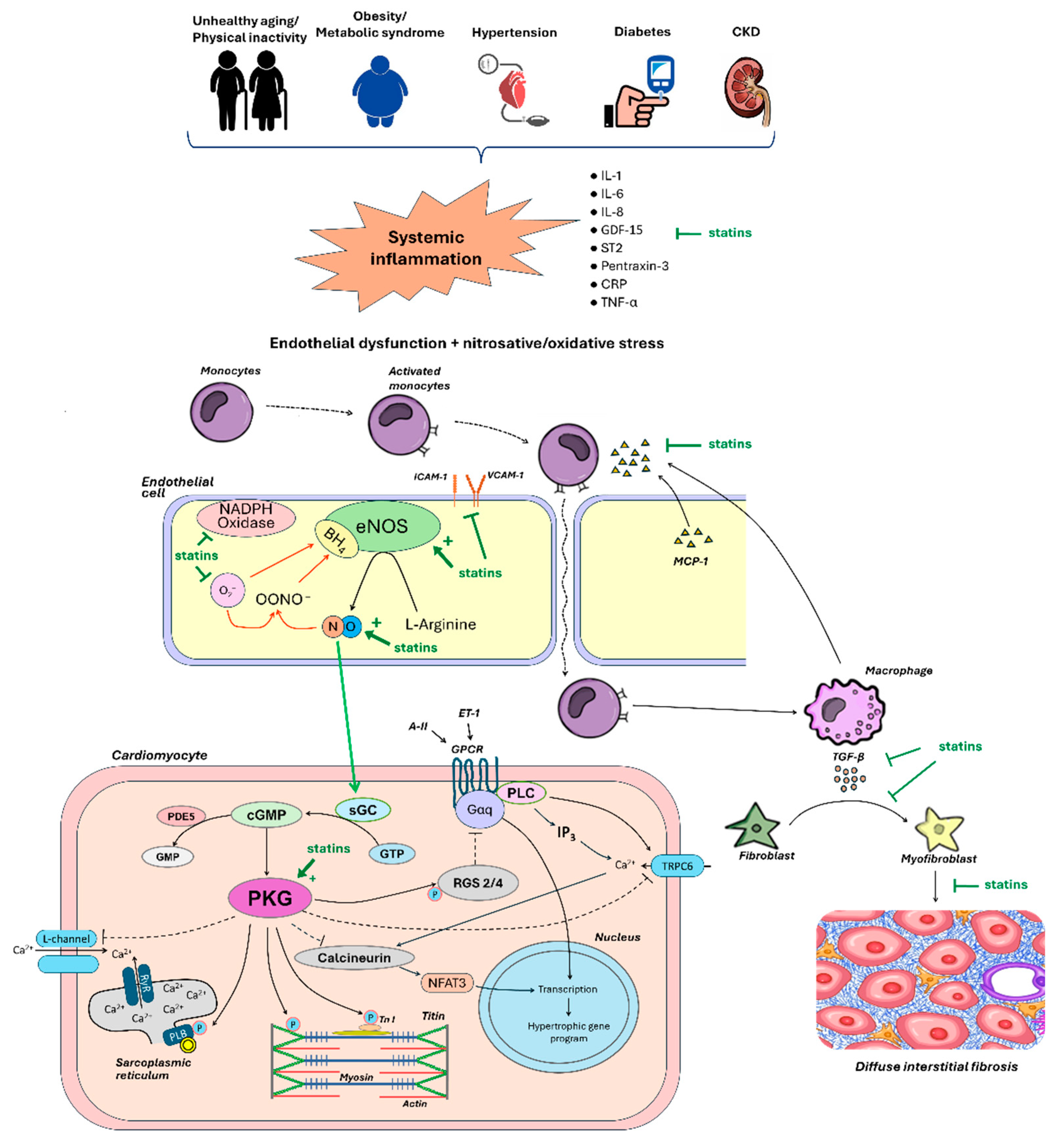

Table 1.
Summary of clinical studies evaluating the effect of statins on prognosis in patients with heart failure with preserved ejection fraction.
Table 1.
Summary of clinical studies evaluating the effect of statins on prognosis in patients with heart failure with preserved ejection fraction.
| Study, Year [References] | Study Design |
Country | Patients, n |
Proportion on Statins, % | Type of Statin |
Follow-Up, Years | Main Results |
|---|---|---|---|---|---|---|---|
| Fukuta H. et al., 2005 [157] | Prospective | United States | 137 | 50 | Atorvastatin, simvastatin, pravastatin | 2 | ↓All-cause mortality (HR 0.20; 95% CI 0.06 to 0.62; P < 0.01) |
| Roik M. et al., 2008 [158] | Prospective | Poland | 146 | 71 | Simvastatin, atorvastatin, lovastatin | 1 | ↓ All-cause mortality (HR 0.24, 95% CI 0.07 to 0.90; P < 0.05) ↓ CV admission (HR 0.55, 95% CI 0.33 to 0.92; P< 0.05) |
| Shah R. et al., 2008 [159] | Retrospective | United States | 13533 | 17 | N/A | 3 | ↓All-cause mortality (HR 0.73, 95% CI 0.68 to 0.79, P < 0.001) |
| Tehrani F. et al., 2010 [160] | Retrospective | United States | 270 | 30 | N/A | 5 | ↓ All-cause mortality (HR 0.65; 95% CI 0.45 to 0.95; P = 0.029) No significant reduction in CV admission |
| Gomez-Soto F.M. et al., 2010 [161] | Prospective | Spain | 1120 | 50 | Simvastatin, lovastatin, pravastatin | 5 | ↓ All-cause mortality (HR 0.34, 95% CI 0.21 to 0.47, P < 0.001) ↓ HF admission (13.9 vs. 19.7 per 100 persons-year, P < 0.001) |
| Quirós López R. et al., 2012 [162] | Retrospective | Spain | 231 | 21 | N/A | 10 | ↓ All-cause mortality (HR 0.27, 95% CI 0.15 to 0.50, P = 0.01) |
| Kaneko H. et al., 2013 [163] | Prospective | Japan | 1121 | 55 | N/A | 3 | ↓ All-cause mortality (HR 0.26, 95% CI 0.140 to 0.479, P < 0.001) |
| Nochioka K. et al., 2015 [164] | Prospective | Japan | 3124 | 37 | N/A | 3 | ↓All-cause mortality (HR 0.71; 95% CI 0.62 to 0.82; P < 0.001.) No significant reduction in HF admission |
| Alehagen U. et al., 2015 [165] | Prospective | Sweden | 9140 | 38 | N/A | 1 | ↓ All-cause mortality (HR 0.80, 95% CI 0.72 to 0.89, P < 0.001) ↓ All-cause mortality + CV admission (HR 0.89, 95% CI 0.82 to 0.96; P < 0.01) |
| Yap J. et al., 2015 [166] | Prospective | Singapore | 751 | 61 | N/A | 2 | ↓ All-cause mortality (HR 0.59, 95% CI 0.44 to 0.79; P < 0.001) ↓ CV mortality (HR 0.58, 95% CI 0.37-0.89; P = 0.012) |
| Lee M.S.et al., 2018 [167] | Retrospective | United States | 7563 | 72 | N/A | 6.7 | ↓ All-cause mortality (HR 0.73, 95% CI 0.66 to 0.81; P < 0.001) |
| Tsujimoto T. et al., 2018 [168] | Prospective (data of TOPCAT trial) | United States, Canada, Brazil, Argentina, Russia, Georgia | 3378 | 52 | N/A | 3.3 | ↓ All-cause mortality (HR 0.79, 95% CI 0.63 to 0.99; P = 0.04) |
| Marume K. et al., 2019 [169] | Prospective | Japan | 414 | 20 | N/A | 2 | ↓ All-cause mortality (HR 0.21, 95% CI 0.06 to 0.72; P < 0.02) |
CI, confidence interval; CV, cardiovascular; HR, hazard ratio; ↓, decrease.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



