Submitted:
02 April 2024
Posted:
03 April 2024
You are already at the latest version
Abstract
The PAX8/PPARγ rearrangement, producing the PAX8–PPARγ fusion protein (PPFP), is thought to play an essential role in the oncogenesis of thyroid follicular tumors. To identify PPFP-targeted drug candidates and establish an early standard of care for thyroid tumors, we performed ensemble docking-based compound screening. Specifically, we investigated which pocket structure should be adopted to search for a promising ligand compound for the PPFP; the position of the ligand binding pocket on the PPARγ side of the PPFP is common to that of PPARγ; however, the shape is slightly different between them due to environmental factors. We developed a method for selecting a PPFP structure with a relevant pocket and high prediction accuracy for ligand binding. This method was validated using PPARγ, whose structure and activity values are known for many compounds. Then, we performed docking calculations to PPFP for 97 drug or drug-like compounds with a thiazolidine backbone, which is one of the characteristics of ligands that bind well to PPARγ, registered in the DrugBank database. Furthermore, the binding affinities of promising ligand candidates were estimated more reliably using the molecular mechanics-Poisson–Boltzmann surface area method. Thus, we propose promising drug candidates for the PPFP with a thiazolidine backbone.
Keywords:
follicular thyroid neoplasia
; PAX8–PPARγ fusion
; fusion protein
; structure-based drug design
1. Introduction
Thyroid cancer is a malignant tumor that originates in thyroid cells and can lead to abnormalities in hormone production and nodule formation. Most thyroid cancers contain one of several known driver mutations, such as the V600E substitution in BRAF, RAS mutation, RET gene fusion, or PAX8/PPARγ gene fusion. Various drugs targeting these mutants are currently under development and are expected to become new treatment options. For example, if a patient has a BRAF V600E mutation or an ALK fusion gene, they can receive alectinib in an investigator-initiated clinical trial under patient-directed care [1]. Clinical trials on RET inhibitors for RET gene fusion and medullary thyroid cancer with a positive RET gene mutation are ongoing [2,3]. For unresectable thyroid cancer, the multi-targeted kinase inhibitors sorafenib, lenvatinib, and vandetanib are used as molecular-targeted drugs [4,5,6]. For solid tumors, including NTRK fusion gene-positive thyroid cancer, the ROS1/TRK inhibitor enutrectinib has been approved [7]. However, these therapies simultaneously face limitations of side effects and treatment resistance.
The PAX8/PPARγ rearrangement, producing the PAX8–PPARγ fusion protein (PPFP), is rare in follicular adenomas. It has been reported that follicular adenomas with PAX8/PPARγ rearrangement are likely to be follicular carcinomas, as these genetic abnormalities are presumed to be involved in the progression from follicular adenoma to follicular carcinoma [8]. The PAX8/PPARγ fusion gene is a translocation between chromosomal regions 2q13 and 3p25, which contain PAX8 and PPARγ, respectively [9]. Focusing on the PPARγ side of the PPFP, several small molecules have been considered as candidates for PPFP-targeted therapeutics [10,11,12]. The PPFP has been described as an inhibitor of PPARγ activity or a transcription factor with proto-oncogene activity [13]; however, in mice, pioglitazone reportedly binds to the PPFP and induces it to behave like PPARγ, resulting in loss of malignancy as thyroid cancer cells differentiate into adipocyte-like cells [9,10,13]. Furthermore, recent clinical trials have demonstrated that pioglitazone induces a significant decrease in thyroglobulin levels and a mild decrease in tumor size [12]. Therefore, it is possible to design a unique anticancer strategy, namely, suppressing malignant transformation by differentiation into noncancerous cells through enhancing the activity of the aberrant protein PPFP. Despite its importance as an anticancer drug target, there is no effective molecular targeted therapy for the PPFP. The development of small-molecule therapeutics targeting thyroid tumors with PPFP is expected to establish a standard of care for early stages.
The difficulty in drug discovery of fusion proteins, such as PPFP, is that their three-dimensional (3D) structure and complete sequence are not yet known; however, recent attempts have emerged to bridge this critical gap [14,15]. Fusion proteins naturally produced by chromosomal translocations, such as oncogenes, have many residues, and other environmental factors, including neighboring proteins, must also be considered. In our previous study [16], we attempted to construct the entire 3D structure of the PPFP using homology modeling based on expected sequential information [17,18,19] and X-ray crystallographic data of the substructures. The PPFP was assumed to be a 901-residue protein consisting of 396 residues of PAX8 on the N-terminal side and 505 residues of PPARγ on the C-terminal side. When 3D modeling the PPFP, the crystal structure of the PPARγ–rosiglitazone complex was used. This PPARγ sequence was the least deficient. The modeled PPFP ligand-binding sites are common in position but have slightly different shapes. Since the PPFP consists of PAX8 and PPARγ, we also used information on the primary, secondary, and tertiary structures of PPARγ during its modeling. Using the modeled structure, we analyzed the fluctuations using molecular dynamics (MD) simulations to validate the structural stability. Furthermore, the molecular mechanics-Poisson–Boltzmann surface area (MM-PBSA) study of the binding affinities between PPFP/PPARγ and rosiglitazone revealed different binding free energies for the same binding pocket. These results suggest that there may be optimal compounds for the PPFP that are different from PPARγ, indicating the need for drug discovery targeting this fusion protein.
Ensemble docking is an efficient and cost-effective technique for compound screening to identify therapeutic drug candidates and has recently received much attention [20,21]. When preparing multiple protein structures for docking screening, it is important to know in advance which protein structure will likely yield in the most favorable results, that is, the structure with the highest predictive accuracy. If the active compounds are known, structures that correctly predict their active performance can be employed for screening; however, if not, such a policy cannot be followed. Based on the idea that proteins tend to bind to similar molecules even if they are not truly active compounds, Fukunishi et al. [22] proposed a solution to this problem using the following protocol: a relatively small molecular assembly of existing active compounds can be collected as universal active probes (UAPs), and the protein structure with the pocket with the highest sum of docking scores can be adopted as the screening structure. Because there are no known active compounds for the PPFP except for those targeting PPARγ, we can take advantage of the UAP set for docking structure selection.
In this study, we first discuss the selection of the most suitable PPFP pocket for docking simulations from a large number of PPFP structures generated in the MD simulation of the PPFP apo-form and PPFP-rosiglitazone complex structures. We used PPARγ, for which there is already a large amount of data on activity values and ligands, to verify whether the methodology using the UAP set properly performs and, in that case, what conditions are necessary. Verification included the predictive ability of the docking simulation using the Molecular Operating Environment (MOE) tool [23]. To the methodology using the UAP set, we added the condition that the ligand has a thiazolidinedione backbone, which is one of the characteristics of ligands that bind well to PPARγ, and its similar structures. Based on these studies, we selected a PPFP structure with a pocket and high activity prediction potential. The selected PPFP structures were docked with 97 compounds with a thiazolidine (TZD) backbone obtained from DrugBank Online (https://go.drugbank.com/) [24]. The most favorable drug candidate ligands for the PPFP were also investigated. Finally, we propose the most promising candidate ligands for this fusion protein through re-evaluation using a more reliable MM-PBSA method.
2. Results and Discussion
The goal of this study was to predict promising binders for the PPFP using a database of drugs or drug-like compounds through docking calculations. To this end, it was necessary to select PPFP structures suitable for docking from the MD trajectories of apo-PPFP and PPFP–rosiglitazone complexes modeled in our previous study [16]. Although the position of the PPARγ-derived ligand-binding pocket is the same for all of them, their shapes are slightly different. Here, we first established a protocol to select structures suitable for docking using PPARγ, for which there is abundant knowledge on co-crystal structures and binding affinities with active compounds. We then applied this protocol to PPFP, for which there is no prior information on its activity or structure, to propose binders as promising target drug candidates.
2.1. Protocol for Structure Search for Reliable Docking
To establish a protocol for selecting docking structures with good prediction performance using PPARγ, we first prepared 11 PPARγ structures from two origins, the MD trajectories and the protein data bank (PDB) (Table 1). The former is based on the PPARγ–rosiglitazone complex and apo-PPARγ structures modeled in a previous study [16], giving six representative structures (A01–A06) from the equilibrium states in the 300 K MD simulations. The latter provided five representative crystal structures (A07–A11).
We next docked UAPs consisting of 175 small molecules (Table S1) to the aforementioned 11 PPARγ structures to rank their screening performance by the sum of docking scores (Table 1). The results show that structure ID A11 from the PDB had the best UAP with a docking score sum of -1339.0 kcal/mol, and according to a previous study [22], this structure can be judged as the most suitable for docking screening.
To verify the performance of the UAP protocol on the PPARγ target, we prepared a compound set consisting of 41 PPARγ binders with co-crystal structures registered with PPARγ in the PDB and at least one of the activity values of IC50, Kd, or Ki reported in ChEMBL. Then, the sums of the docking scores for the compound set with the 11 PPARγ structures were compared to those of the UAP set. Figure 1 shows the correlation between the sum of docking scores for the 11 structures in the UAP- and PDB-derived compound sets. The high correlation coefficient, R2=0.84, suggests that the UAP protocol would be applicable to PPARγ for the selection of good pockets for screening with high probability.
To examine the predictive ability of docking for a wide range of PPARγ known active compounds, we performed docking for 11 PPARγ structures of 1696 compounds with activity values (IC50, Kd, Ki) for PPARγ registered in ChEMBL. Table 2 shows the correlation coefficient between docking scores and activity values for the corresponding compounds for each activity type and for each PPARγ structure (denoted as “All” in Table 2). Almost no correlation was observed for IC50, and weak correlations were observed for Kd and Ki. We then limited our analysis to compounds with TZD and phenylacetic acid (PA) backbones that play important roles in binding to PPARγ; an existing marketed drug, pioglitazone, shares the TZD backbone. Even with these restrictions, we found little improvement in the correlation with IC50; however, we obtained noteworthy correlation coefficients (R2) with Ki: for the TZD compounds, 0.482 with the structure ID A03, 0.481 with A05, and 0.491 with A06, and for the PA compounds, greater than 0.5 with many structures. As hypothesized, a binding mode similar to that of TZD, including PA, is expected to improve the prediction performance of Ki.
Since the PPARγ pocket is softer, the chemical space of possible binders is wider. The PPARγ compound group registered in ChEMBL has a wide variation. The selection criteria for the pocket that is expected to give the best docking results are as follows: It can be expected that good results will be obtained if the condition is focused on PPARγ. Among the PPARγ ligands, it seems best to focus on the TZD skeleton characteristic of pioglitazone, which is already a marketed drug. In the case of the PPFP, it is not possible to know the activity value in advance; therefore, it would be beneficial to select based on some molecular characteristics. In terms of the position of the pocket, PPARγ and PPFP have a pocket in the same position. Therefore, the basic idea of drug discovery is to change the side chain based on the mother skeleton, which is expected to have some activity. First, we focused our discussion on the analogous structure of pioglitazone. Therefore, we propose that the best approach is to have the TZD backbone characteristic of pioglitazone, which is already known to bind well among PPARγ ligands, in addition to previously used methods. Following only the methods of previous studies, the structure selected for PPARγ was PDB-derived A11. The correlation between the docking score and activity value showed that the R2 value was not necessarily good. Based on the proposed method, the best structure was A06, which was derived from MD simulations. It had sufficient Ki data and a larger R2 value than A11. Thus, an even better docking score prediction was expected.
2.2. Application to the PPFP
Based on the PPARγ considerations above, a screening protocol that provided the highest predictive accuracy was applied to the PPFP. As in the case of PPARγ, but without any crystal structures, 19 representative structures with structural IDs B01–B19 were extracted from the MD trajectories for the rosiglitazone complex and apo structure in our previous study (Table 3). After evaluation of binding capacities by the UAP set, structure ID B14, showing the highest value of the sum of docking scores (-1303.2 kcal/mol) was selected as the best pocket structure for screening the PPFP (Figure 2).
In this study, we attempted to identify PPFP target drug candidates from the perspective of drug repositioning. The compounds to be screened were obtained from DrugBank [24], an online database of drug and drug candidate compounds and their targets, which includes 9213 compounds available as of November 2022. Based on the scope of the proposed protocol, 97 compounds with TZD backbones were identified (Table S2). Because we focused on comparing the drug candidates with existing drugs, such as pioglitazone and rosiglitazone, candidates with a PA backbone were not considered in this study. The 97 compounds were docked to the PPFP structure with an ID of B14. Table 4 shows the top 10 scoring compounds, along with pioglitazone and rosiglitazone. Pioglitazone and rosiglitazone are widely known PPARγ drugs; however, their binding scores with PPFP were not very high in this study.
For a more reliable binding-affinity evaluation, multi-sampling MM-PBSA calculations were performed to rescore the 12 docked compounds, including pioglitazone and rosiglitazone. Table 5 shows that teneligliptin (L72), a DDP-4 inhibitor, was the most promising binder for the PPFP. Rosiglitazone (L4) and pioglitazone (L18) had high binding affinities. For ligand candidate L13, the docking score was good; however, the binding free energy was -11.98 kcal/mol, excluding it from the candidate list. Although pioglitazone and rosiglitazone are naturally good candidates, we also determined better binders, such as teneligliptin (see Figure 3), using our computational protocol.
3. Materials and Methods
3.1. Protein Structures for Docking
3.1.1. PPARγ Structures
We prepared 11 PPARγ structures categorized into two structure types. Numerous structures were generated by MD simulations during modeling the PPARγ apo-body and the PPARγ-rosiglitazone complex structures in our previous study [16]. These were clustered based on the shape of the ligand-binding sites using the density-based spatial clustering of applications with noise (DBSCAN) method of the Amber tool in the MD analysis program. Finally, six structures were selected. On the other hand, the Site Finder application in MOE software was used to detect pocket shapes in 256 PPARγ-ligand complex crystal structures downloaded from the PDB. The features of the pocket shapes were quantified using Sterimol parameters [20,21]. The numerical values were used to cluster the molecules using the k-means method; the Sterimol parameters are a set of vectors describing the steric occupancy of a molecule. They have been extensively used in quantitative structure-activity relationship (QSAR) studies for drug discovery [22,23]. To this end, five structures were selected PDB ID: 1I7I, 2ZK1, 3ADS, 3H0A, and 3VJI. Structures downloaded from the PDB were preprocessed using MOE for docking simulations [23]. Hydrogen atoms were added using Protonate3D. AMBER10:EHT was applied to the force field. The hydrogen atoms were structurally optimized.
3.1.2. PPFP Structures
We created 19 PPFP structures. In our previous study [16], we proposed a homology modeling construction of the entire 3D structure of the PPFP, which is still unresolved, based on X-ray crystal structure data for PPARγ and PAX8. To validate these model structures, we analyzed the fluctuations using MD simulations and predicted the physical properties based on the structures and sequences. Many trajectories extracted from these MD simulations were clustered using the DBSCAN method of the Amber tool in the MD analysis program [25]. The parameter radius distance was set to ε=0.9 and the threshold with neighborhood density was set to minPts=25. The clustering criterion was the root-mean-square deviation (RMSD) of the residues around the PPFP pocket. The sampled temporal range was between 10 and 100 ns, which was determined to be the equilibrium state, and 18,000 frames were read every five frames. The top clusters, which accounted for more than 1% of all frames, were extracted, and their centroid was used as the representative structure in the equilibrium state. Numerous PPFP-rosiglitazone complex and PPFP apo-body structures, which were previously modeled [16], were generated using MD simulations. These were also clustered based on the shape of the ligand-binding sites. Because the crystal structure of the PPFP was not available, only the MD simulation-derived structures were used. The same procedure as for PPARγ was performed for the 19 prepared PPFP structures.
3.2. Compound Sets for Docking
3.2.1. UAPs
Fukunishi et al. [22] proposed the UAP set, which is a collection of relatively small molecules among active compounds against various targets, to evaluate the screening ability of the pocket. This evaluation is based on the idea that similar molecules tend to bind to a pocket, even if it is not the actual active compound of the pocket. Therefore, the pocket with the highest score obtained by docking UAPs to multiple pockets is considered the binding pocket suitable for screening active compounds. In this study, the UAP set consisting of 175 small drug-like compounds registered in the PDB was obtained from MyPresto version 5 (https://www.mypresto5.jp).
3.2.2. PPARγ Ligands Registered in ChEMBL
One hundred and ninety five (195) PPARγ–ligand complex crystal structures were available from the PDB. One hundred and forty eight (148) PPARγ ligand activity values (IC50, Kd, Ki) were in ChEMBL. Among them, 63 PPARγ ligands had both crystal structures and activity values. Excluding duplicates, 41 PPARγ ligands were obtained. These 41 PPARγ ligands were docked against 11 structures of PPARγ.
The target-associated bioactive values for PPARγ were extracted from CHEMBL29 (https://www.ebi.ac.uk/chembl/) with the target ID CHEMBL235. This resulted in 1696 compounds with SMILES (Table S3).
The computational analysis procedure was the same even when a different type of ligand was docked to PPARγ. The median and logP values for the activity were used in the calculation and analysis; Ki had 20 small activity values with logP 2, which were excluded as outliers (Figure S1). First, when all IC50 values were used (All), there was little correlation; the 1696 PPARγ ligands obtained from ChEMBL included those with large molecular weights. Lipinski’s rule of five [26,27] summarizes the chemical properties of compounds that are likely to become oral drugs and suggests that one of the properties of the drug is a molecular weight of less than 500.
TZDs are a class of heterocyclic compounds with thioether and amine groups attached at the first and third positions of the saturated five-membered ring, respectively (Figure 4). The TZD skeleton is characteristic of pioglitazone. Pioglitazone is a ligand for PPARγ, and is already a marketed drug; a two-dimensional view of the ligand binding sites of PPARγ and pioglitazone is displayed in Figure 4. Positional relationships and interactions of the peripheral residues are seen, where hydrogen bonds, ionic bonds, π-π interactions, CH-π interactions, cation-π interactions, halogen bonds, and coordination bonds are anticipated. The TZD backbone plays an important role in the bonding; thus, we extracted 101 compounds with this backbone from the aforementioned 1696 bioactive compounds (Table S3).
PA is another common skeleton of PPARγ binding compounds with a similar binding mode to TZD (Figure 4). To investigate the predicted activity performance of compounds with such similar binding modes, we extracted 31 compounds with a PA backbone from the aforementioned 1696 bioactive compounds of PPARγ (Table S3). Using these experimental values (IC50, Kd, and Ki), we confirmed that they could be reproduced in the order of increasing activity and correlation. This method was applied to the PPFP. In the case of PPARγ, we established a procedure to predict the superiority of activity values from docking calculations, determining the ligand that binds well to the PPFP. All activity values for PPARγ were used for calculation and analysis and were obtained by evaluating median and logP values. For Ki, 366 original values were obtained, of which 20 extremely low activity values were excluded as outliers.
3.2.3. TZD-Backbone Compounds Registered in the DrugBank Database
As of November 2022, there were 9213 ligands registered in the DrugBank database [24]. The TZD backbone portion is a characteristic structure of pioglitazone and plays an important role in the binding of pioglitazone to the ligand binding site of PPARγ. Similarly, the TZD backbone is thought to play an important role in the binding to the ligand-binding site of the PPFP. A total of 97 ligands with the TZD skeleton were identified. In this study, we did not consider the case of PA because we wanted to compare the candidates with existing commercial drugs, pioglitazone and rosiglitazone.
3.3. Computational Methods
3.3.1. Docking
Docking simulations were performed using MOE software [23]. The final binding free energies and docking scores were evaluated using the London dG and GBVI/WSA dG scoring functions.
3.3.2. MM-PBSA Method
The MM-PBSA approach is an efficient method for evaluating the binding free energy () of protein-ligand complexes [28]. We used the MMPBSA.py module of AmberTools 14 to calculate the binding free energies of 10 ligands considered promising candidates for PPFP with PPFP, pioglitazone with PPFP, and rosiglitazone with PPFP, and investigated their binding affinities [29]. First, MD simulations were run for 10 ns to investigate the conformational fluctuations at equilibrium in solution. The protocol for this MD simulation was the same as that previously published [16]. Consequently, 100 structures were extracted from the MD trajectories of the production runs. The scores were reevaluated using the MM-PBSA method [30]. The binding free energies were calculated using the MM-PBSA method to determine the binding affinity of the receptor (PPFP) and ligand (several small molecules of the candidate drugs). The difference between the free energy of the complex and those of the ligand and receptor is the binding free energy (Equation 1):
The can also be decomposed into ligand–residue interactions [29]. In this study, the dielectric constant of the solute (protein, internal dielectric constant) was 4.0, while that of the solvent (water, external dielectric constant) was 80.0.
4. Conclusions
A method for selecting protein structures with pockets suitable for ligand docking from numerous protein structures was developed, and correlations between docking scores of PPARγ and PPARγ ligands and their activity values were examined. Correlations were checked to determine whether they could be reproduced in the order of the experimental activity. The method used in a previous study using UAP is promising. To this method, we added the condition that the PPARγ ligands have a TZD backbone characteristic of the existing drugs, pioglitazone and rosiglitazone. In this way, the correlations improved. We applied this method to the PPFP and identified small molecules with a TZD moiety as promising drug compounds. In addition to PPARγ ligands included in existing drugs, compounds with structures similar to TZDs, such as PA, may also be candidates. This suggests the need to search for drug candidates within the PPFP pocket rather than the PPARγ pocket. The top ten compounds reported herein are expected to be potential candidate drugs for the PPFP, for which the ligands are promising.
Supplementary Materials
The following supporting information can be downloaded at website of this paper posted on Preprints.org, Table S1: SMILES of UAP; Table S2: Small molecule SMILES with a thiazolidinedione skeleton registered in DrugBank; Table S3: SMILES of ligands of PPARγ with activity values registered in ChEMBL and their respective conditional classification.; Figure S1: (a) Outliers in Ki in docking to structure A03 to show correlation (b).
Author Contributions
Conceptualization, K.S., Y.O. and S.T.; methodology, K.S., Y.O. and S.T.; software, K.S., Y.O. and S.T.; validation, K.S., Y.O. and S.T.; formal analysis, K.S., Y.O. and S.T.; investigation, K.S., Y.O. and S.T.; resources, S.T.; data curation, K.S. and Y.O.; writing—original draft preparation, K.S.; writing—review and editing, Y.O. and S.T.; visualization, K.S. and Y.O.; supervision, S.T.; project administration, S.T.; funding acquisition, S.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Grants-in-Aid for Scientific Research (No. 21K06098) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and Japan Society for the Promotion of Science (JSPS), and by MEXT Quantum Leap Flagship Program (Grant No. JPMXS0120330644).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tahara, M. Genomic Medicine in Thyroid Cancer toward Precision Medicine. Folia Endocrinol. Jpn. 2020, 37, 110–114. [Google Scholar] [CrossRef]
- Subbiah, V.; Velcheti, V.; Tuch, B.B.; Ebata, K.; Busaidy, N.L.; Cabanillas, M.E.; Wirth, L.J.; Stock, S.; Smith, S.; Lauriault, V.; et al. Selective RET Kinase Inhibition for Patients with RET-Altered Cancers. Ann. Oncol. 2018, 29, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W.; Cao, Q.; Sheets, M.P.; Wilson, D.; et al. Precision Targeted Therapy with BLU-667 for RET -Driven Cancers. Cancer Discov. 2018, 8, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in Patients with Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in Radioactive Iodine-Refractory, Locally Advanced or Metastatic Differentiated Thyroid Cancer: A Randomised, Double-Blind, Phase 3 Trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in Patients with Advanced or Metastatic NTRK Fusion-Positive Solid Tumours: Integrated Analysis of Three Phase 1–2 Trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Klemke, M.; Drieschner, N.; Laabs, A.; Rippe, V.; Belge, G.; Bullerdiek, J.; Sendt, W. On the Prevalence of the PAX8-PPARG Fusion Resulting from the Chromosomal Translocation t(2;3)(Q13;P25) in Adenomas of the Thyroid. Cancer Genet. 2011, 204, 334–339. [Google Scholar] [CrossRef]
- Kroll, T.G. PAX8-PPARgamma 1 Fusion in Oncogene Human Thyroid Carcinoma. Science (80-. ). 2000, 289, 1357–1360. [Google Scholar] [CrossRef]
- Dobson, M.E.; Diallo-Krou, E.; Grachtchouk, V.; Yu, J.; Colby, L.A.; Wilkinson, J.E.; Giordano, T.J.; Koenig, R.J. Pioglitazone Induces a Proadipogenic Antitumor Response in Mice with PAX8-PPARγ Fusion Protein Thyroid Carcinoma. Endocrinology 2011, 152, 4455–4465. [Google Scholar] [CrossRef]
- Xu, B.; O’Donnell, M.; O’Donnell, J.; Yu, J.; Zhang, Y.; Sartor, M.A.; Koenig, R.J. Adipogenic Differentiation of Thyroid Cancer Cells through the Pax8-PPARγ Fusion Protein Is Regulated by Thyroid Transcription Factor 1 (TTF-1). J. Biol. Chem. 2016, 291, 19274–19286. [Google Scholar] [CrossRef] [PubMed]
- Giordano, T.J.; Haugen, B.R.; Sherman, S.I.; Shah, M.H.; Caoili, E.M.; Koenig, R.J. Pioglitazone Therapy of PAX8-PPARγ Fusion Protein Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2018, 103, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Giordano, T.J. Delineation, Functional Validation, and Bioinformatic Evaluation of Gene Expression in Thyroid Follicular Carcinomas with the Pax8-Pparg Translocation. Clin. Cancer Res. 2006, 12, 1983–1993. [Google Scholar] [CrossRef]
- Kumar, H.; Tang, L.; Yang, C.; Kim, P. F UsionPDB : A Kno Wledg Ebase of Human Fusion Prot Eins. 2023, 1–16.
- Shamriz, S.; Ofoghi, H. Design, Structure Prediction and Molecular Dynamics Simulation of a Fusion Construct Containing Malaria Pre-Erythrocytic Vaccine Candidate, PfCelTOS, and Human Interleukin 2 as Adjuvant. BMC Bioinf 2016, 17, 71. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Okiyama, Y.; Tanaka, S. In Silico Modeling of PAX8–PPARγ Fusion Protein in Thyroid Carcinoma: Influence of Structural Perturbation by Fusion on Ligand-Binding Affinity. J. Comput. Aided. Mol. Des. 2021, 35, 629–642. [Google Scholar] [CrossRef]
- Raman, P.; Koenig, R.J. Pax-8-PPAR-γ Fusion Protein in Thyroid Carcinoma. Nat. Rev. Endocrinol. 2014, 10, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Vuttariello, E.; Biffali, E.; Pannone, R.; Capiluongo, A.; Monaco, M.; Sica, V.; Aiello, C.; Matuozzo, M.; Chiofalo, M.G.; Botti, G.; et al. Rapid Methods to Create a Positive Control and Identify the PAX8/PPARγ Rearrangement in FNA Thyroid Samples by Molecular Biology. Oncotarget 2018, 9, 19255–19262. [Google Scholar] [CrossRef]
- Pasca di Magliano, M.; Di Lauro, R.; Zannini, M. Pax8 Has a Key Role in Thyroid Cell Differentiation. Proc. Natl. Acad. Sci. 2000, 97, 13144–13149. [Google Scholar] [CrossRef]
- Tian, S.; Sun, H.; Pan, P.; Li, D.; Zhen, X.; Li, Y.; Hou, T. Assessing an Ensemble Docking-Based Virtual Screening Strategy for Kinase Targets by Considering Protein Flexibility. J. Chem. Inf. Model. 2014, 54, 2664–2679. [Google Scholar] [CrossRef]
- Uehara, S.; Tanaka, S. Cosolvent-Based Molecular Dynamics for Ensemble Docking: Practical Method for Generating Druggable Protein Conformations. J. Chem. Inf. Model. 2017, 57, 742–756. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Ohno, K.; Orita, M.; Nakamura, H. Selection of In Silico Drug Screening Results by Using Universal Active Probes (UAPs). J. Chem. Inf. Model. 2010, 50, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- ULC, C.C.G. Molecular Operating Environment (MOE), 2020.09. Chemical Computing Group ULC, McGill University; Montreal, QC, Canada: 2020. 2020, 2020. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Proceedings of the Proceedings of the 2nd International Conference on Knowledge Discovery and Data Mining; p. 1996.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings 1PII of Original Article: S0169-409X(96)00423-1. The Article Was Originally Published in Advanced Drug Delivery Reviews 23 (1997). Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Miller, B.R.; Mcgee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. Py : An E Ffi Cient Program for End-State Free Energy Calculations. 2012.
- Sahakyan, H. Improving Virtual Screening Results with MM/GBSA and MM/PBSA Rescoring. J. Comput. Aided. Mol. Des. 2021, 35, 731–736. [Google Scholar] [CrossRef]
Figure 1.
Correlation between the sum of scores obtained by docking the UAP and PDB PPARγ ligands to the 11 PPARγ structures. The UAP is a drug-like small molecule population consisting of 175 compounds. The PDB PPARγ ligands are 41 small molecules with crystal structures and active values. Units are in kcal/mol.
Figure 1.
Correlation between the sum of scores obtained by docking the UAP and PDB PPARγ ligands to the 11 PPARγ structures. The UAP is a drug-like small molecule population consisting of 175 compounds. The PDB PPARγ ligands are 41 small molecules with crystal structures and active values. Units are in kcal/mol.
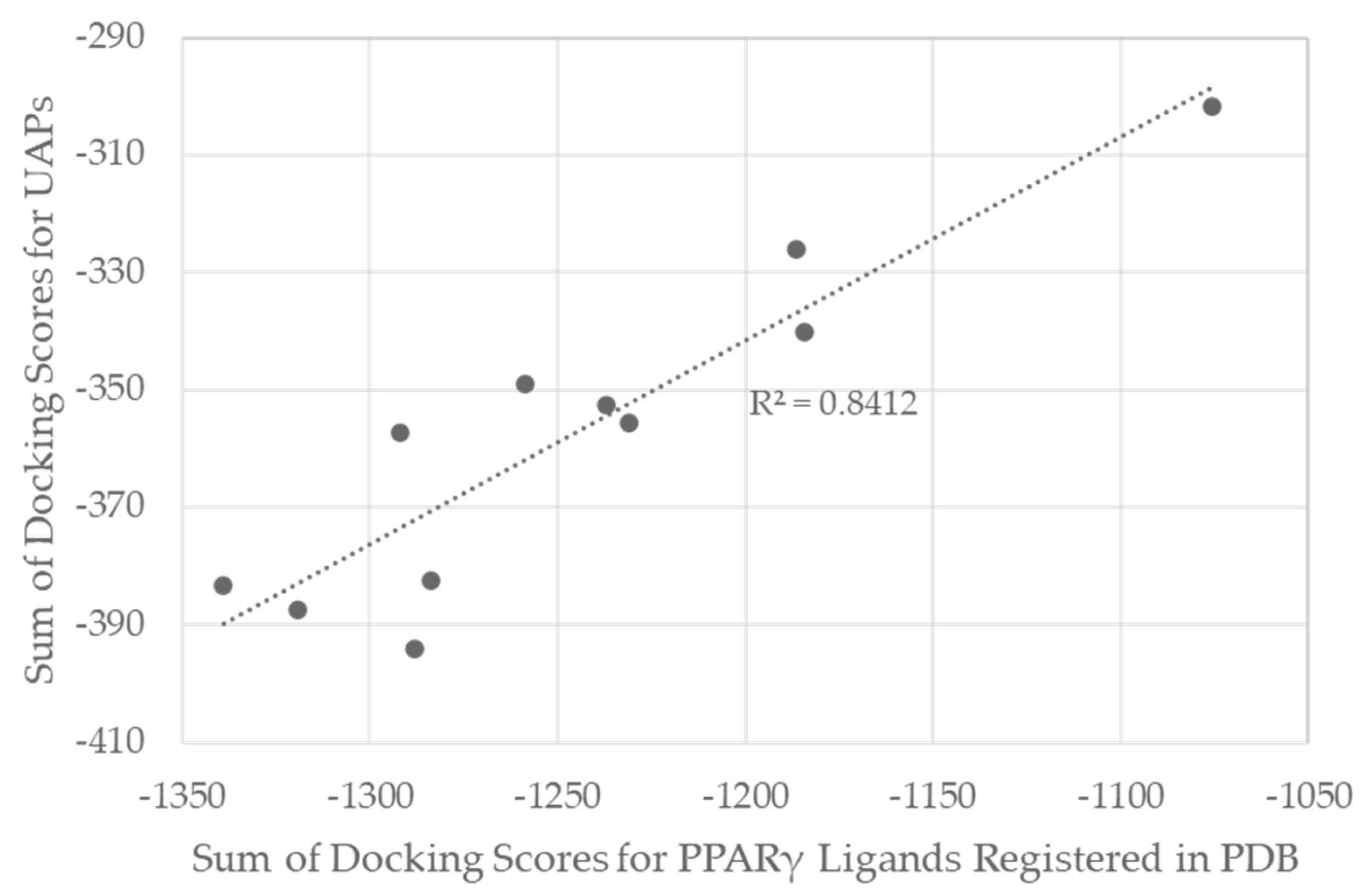
Figure 2.
Ribbon drawing of PPFP/RXRα on DNA. Red denotes the PPFP and blue RXRα. The PPARγ used for modeling PPFP is known to form a dimer with RXRα. Zinc ions are shown as balls. The ligand binding pocket in PPFP is shown in green.
Figure 2.
Ribbon drawing of PPFP/RXRα on DNA. Red denotes the PPFP and blue RXRα. The PPARγ used for modeling PPFP is known to form a dimer with RXRα. Zinc ions are shown as balls. The ligand binding pocket in PPFP is shown in green.
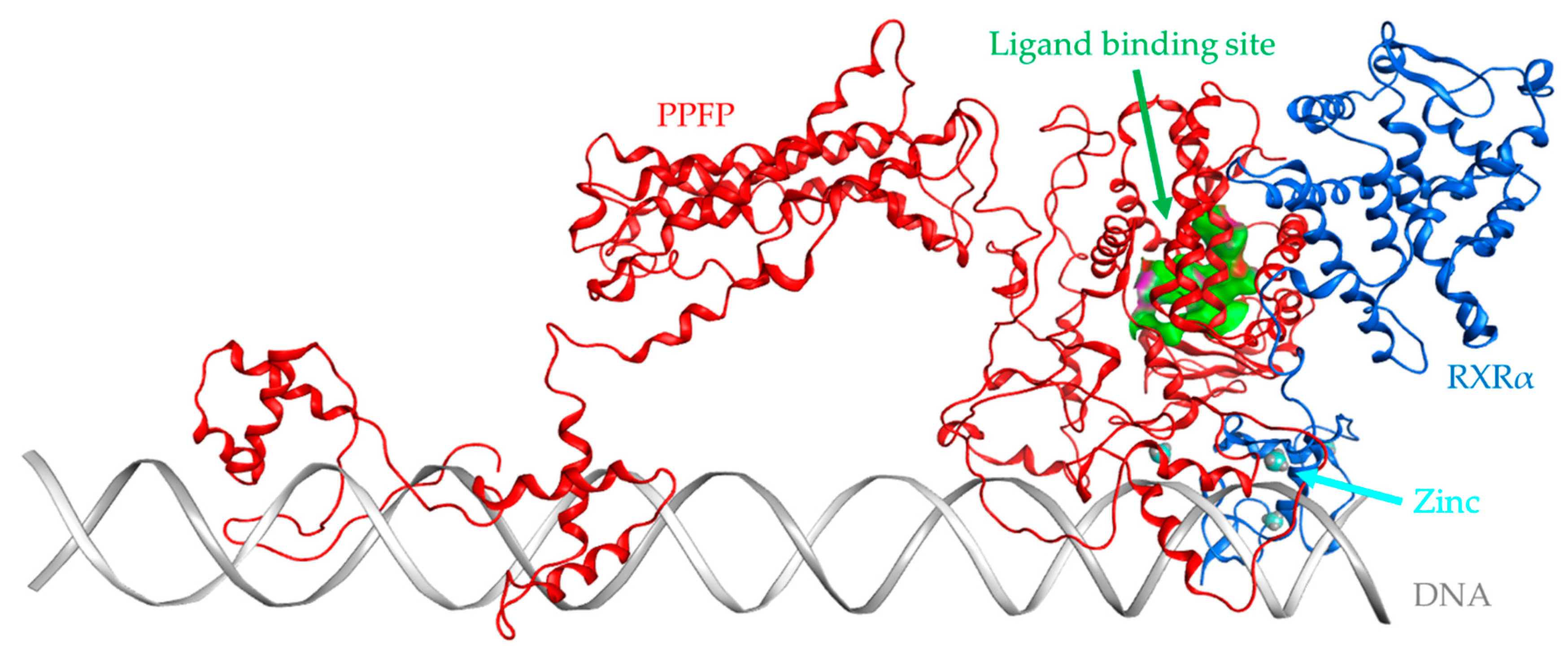
Figure 3.
Binding pose of tenegliptin (L72) in PPFP. (a) Three-dimensional view, where L72 is represented by stick. (b) Two-dimensional view. (c) Superposition of L72 (stick) to pioglitazone (cyan) in binding pocket. (d) Superposition of L72 (stick) to rosiglitazone (magenta) in binding pocket.
Figure 3.
Binding pose of tenegliptin (L72) in PPFP. (a) Three-dimensional view, where L72 is represented by stick. (b) Two-dimensional view. (c) Superposition of L72 (stick) to pioglitazone (cyan) in binding pocket. (d) Superposition of L72 (stick) to rosiglitazone (magenta) in binding pocket.
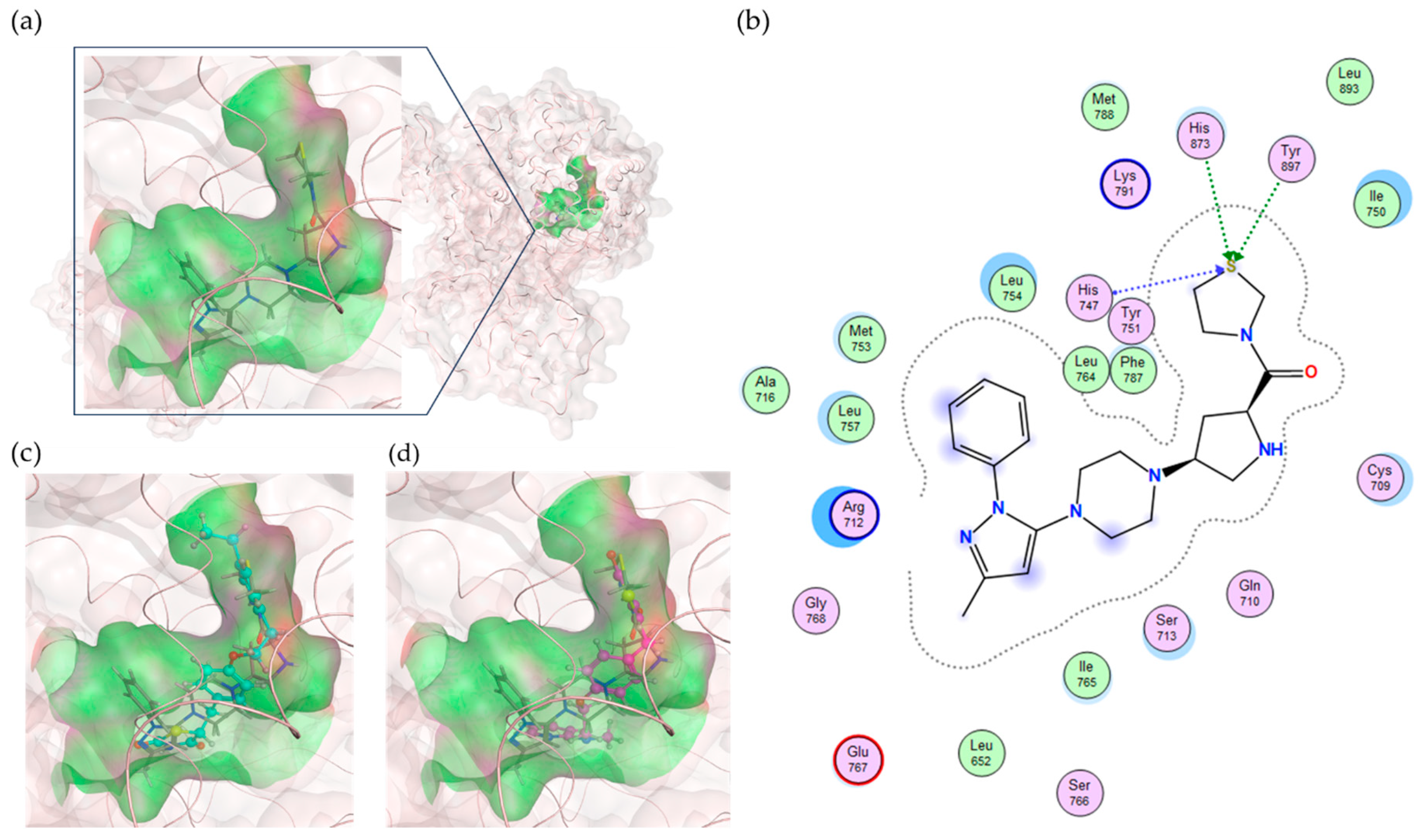
Figure 4.
(a) Two-dimensional chemical structure of TZD. (b) Two-dimensional view of the characterization of the ligand binding sites of PPARγ and pioglitazone. Dotted green arrows indicate interactions between TZD and Gln640, Tyr827, and His677. (c) Two-dimensional chemical structure of PA.
Figure 4.
(a) Two-dimensional chemical structure of TZD. (b) Two-dimensional view of the characterization of the ligand binding sites of PPARγ and pioglitazone. Dotted green arrows indicate interactions between TZD and Gln640, Tyr827, and His677. (c) Two-dimensional chemical structure of PA.
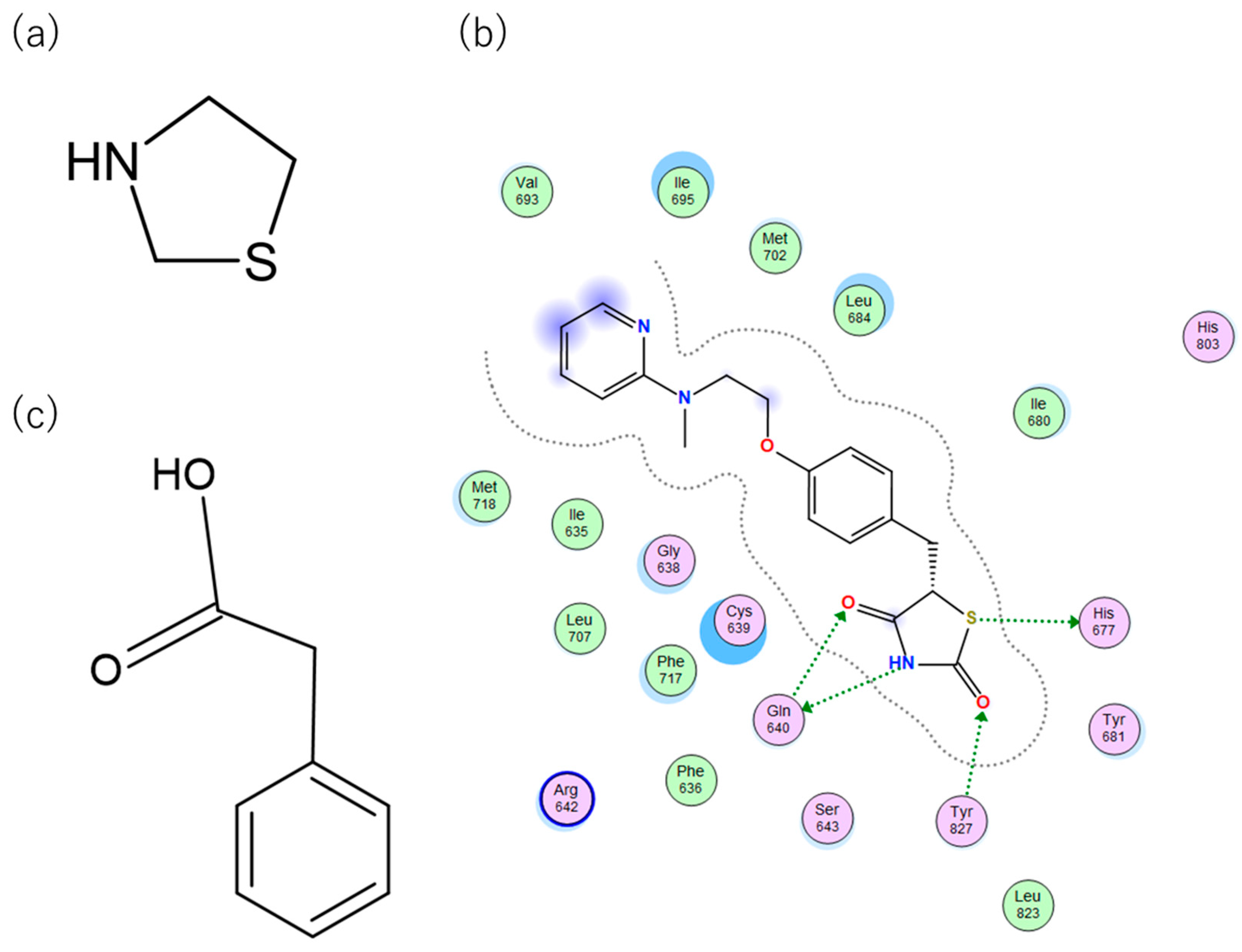

Table 1.
Score and ranking of UAP docking to prepared PPARγ structures. The scores are in kcal/mol and UAP is a small-molecule population consisting of 175 drug-like molecules. The sum of the docking scores was obtained by adding all scores obtained for a single structure. Preparation of PPARγ structures and the origin and ID name of each structure.
Table 1.
Score and ranking of UAP docking to prepared PPARγ structures. The scores are in kcal/mol and UAP is a small-molecule population consisting of 175 drug-like molecules. The sum of the docking scores was obtained by adding all scores obtained for a single structure. Preparation of PPARγ structures and the origin and ID name of each structure.
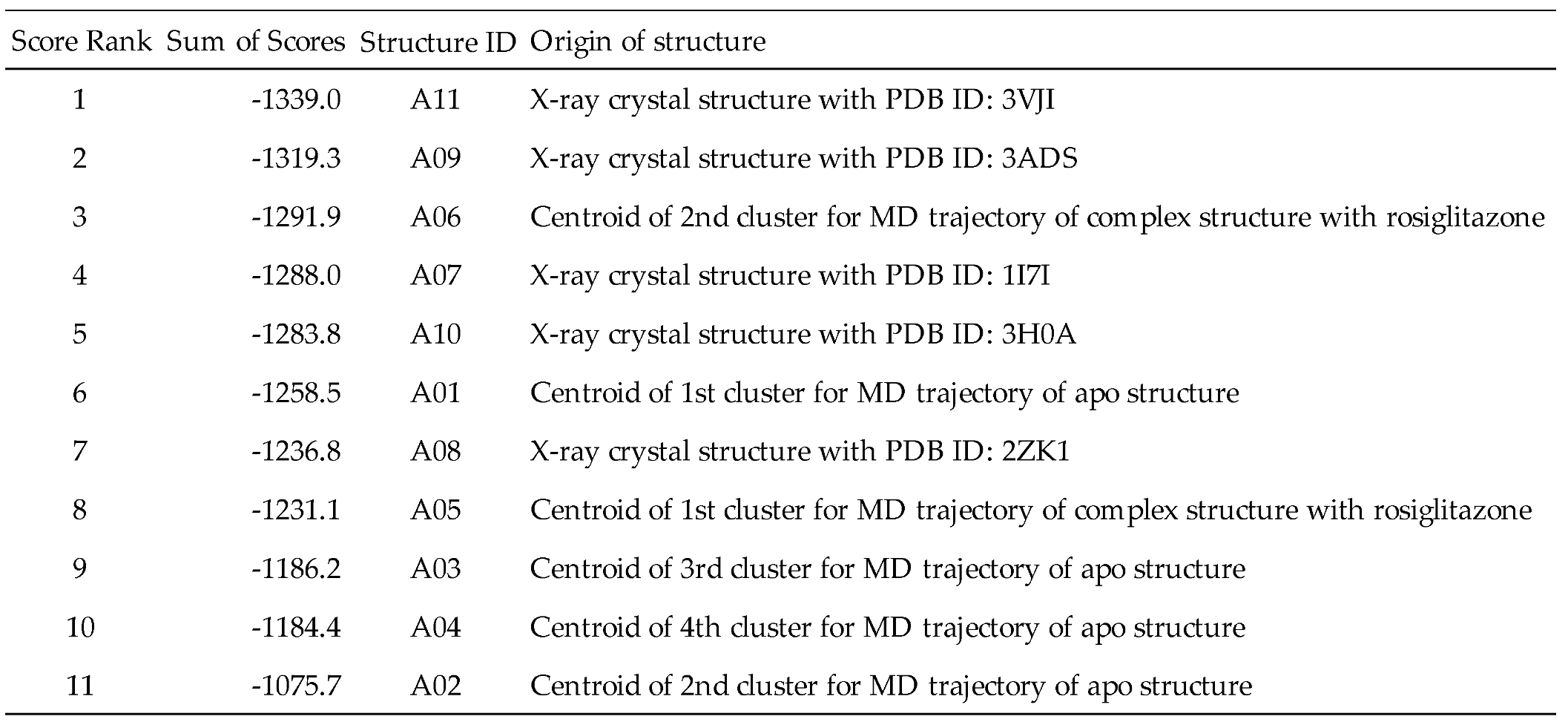 |
Table 2.
Correlation coefficient R2 between docking scores of 11 prepared PPARγ structures with PPARγ ligands obtained from ChEMBL and their activity values. “All” represents all of the respective activity value data; “TZD” represents compounds with a thiazolidine backbone; “PA” represents compounds with a phenylacetic acid backbone.
Table 2.
Correlation coefficient R2 between docking scores of 11 prepared PPARγ structures with PPARγ ligands obtained from ChEMBL and their activity values. “All” represents all of the respective activity value data; “TZD” represents compounds with a thiazolidine backbone; “PA” represents compounds with a phenylacetic acid backbone.
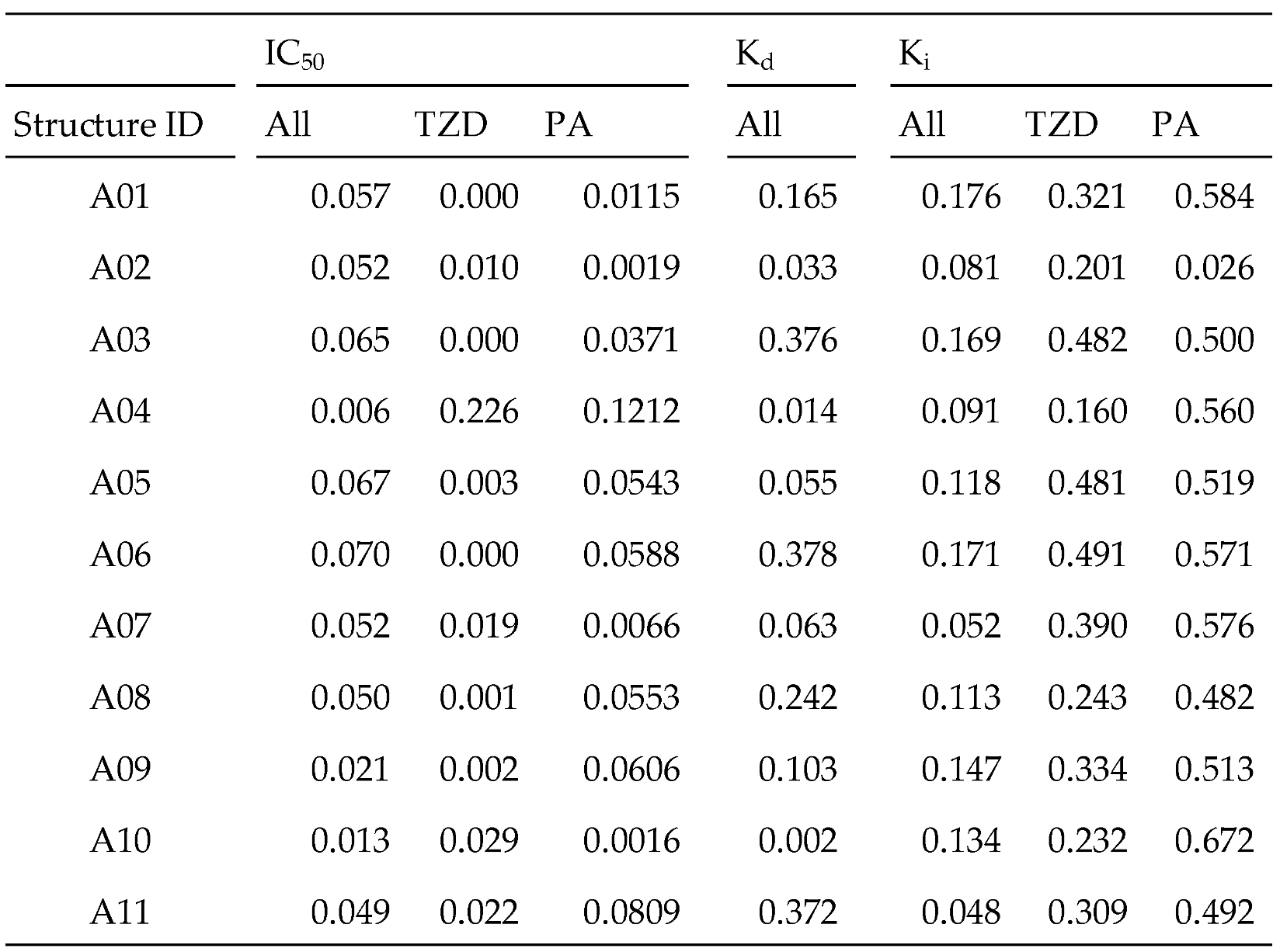 |
Table 3.
Docking scores and ranking of UAP docking to prepared PPFP structures. The scores are in kcal/mol and UAP is a small-molecule population consisting of 175 drug-like molecules. The sum of the docking scores was obtained by adding all scores obtained for a single structure. Preparation of PPFP structures and the origin and ID name of each structure.
Table 3.
Docking scores and ranking of UAP docking to prepared PPFP structures. The scores are in kcal/mol and UAP is a small-molecule population consisting of 175 drug-like molecules. The sum of the docking scores was obtained by adding all scores obtained for a single structure. Preparation of PPFP structures and the origin and ID name of each structure.
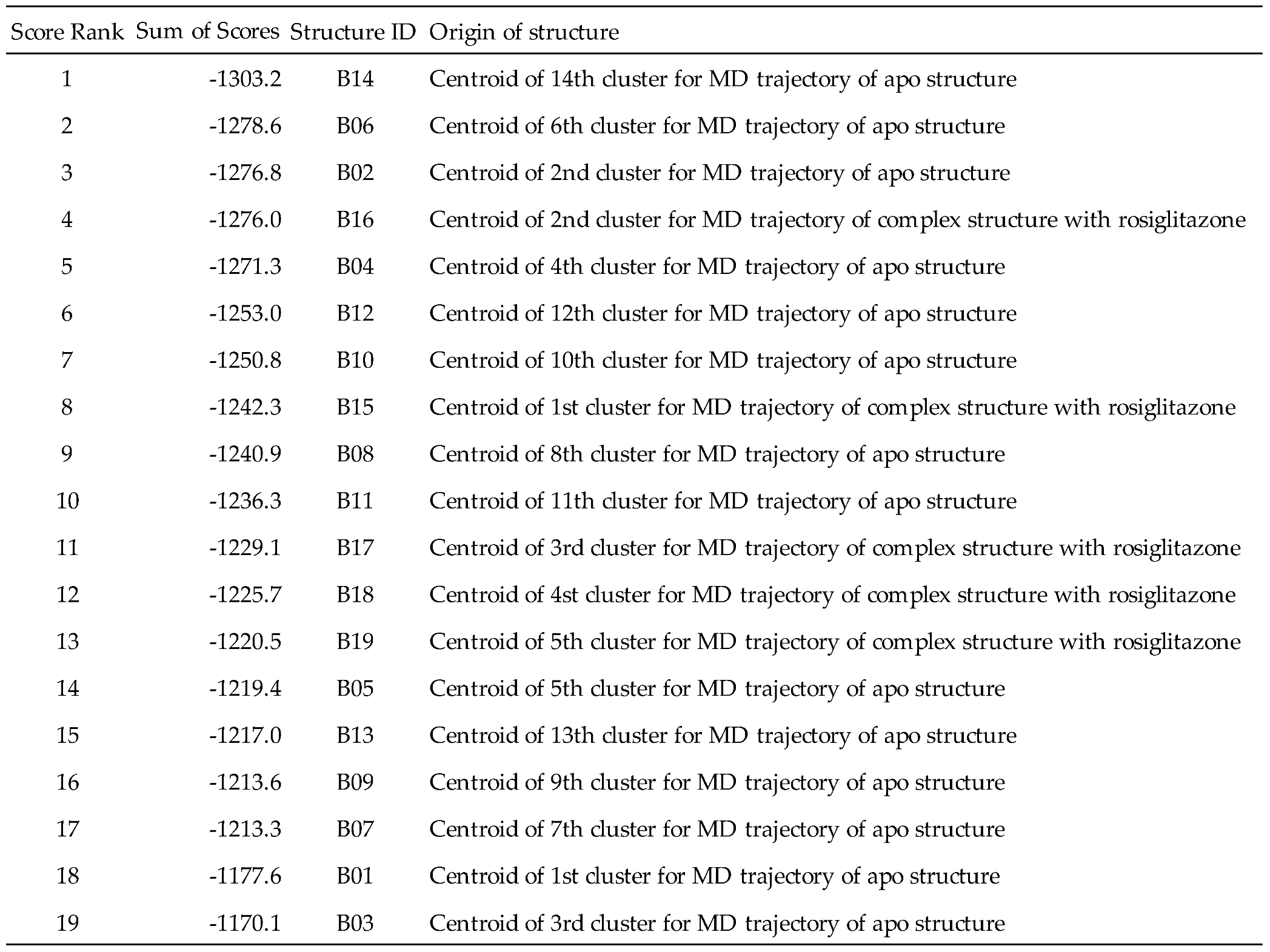 |
Table 4.
Data on top 10 docking scores, ligand ID names, generic names, and 2D structures.
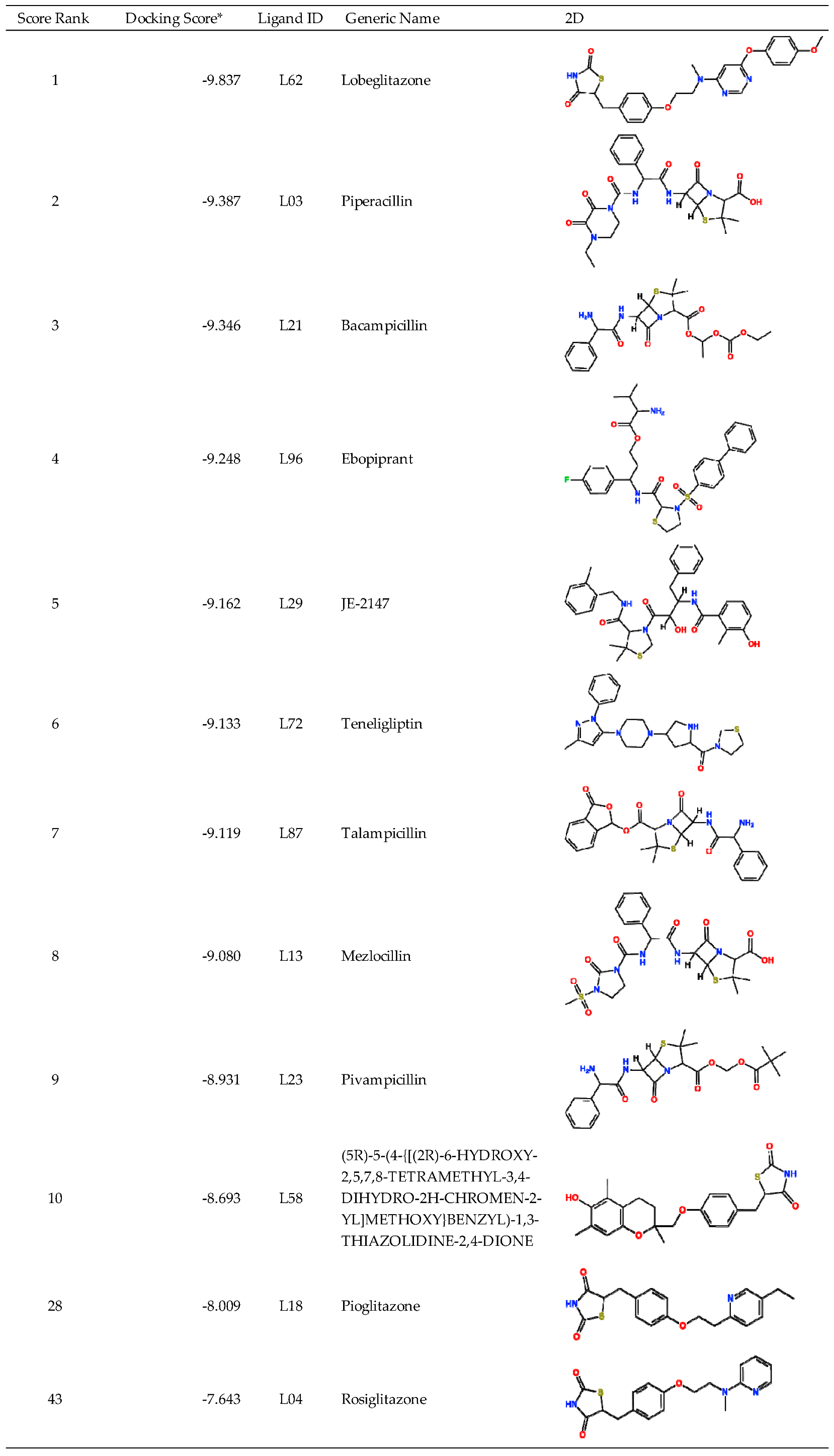 |
*Score values are in kcal/mol.
Table 5.
Calculation of MM/PBSA binding free energies for PPFP candidates.
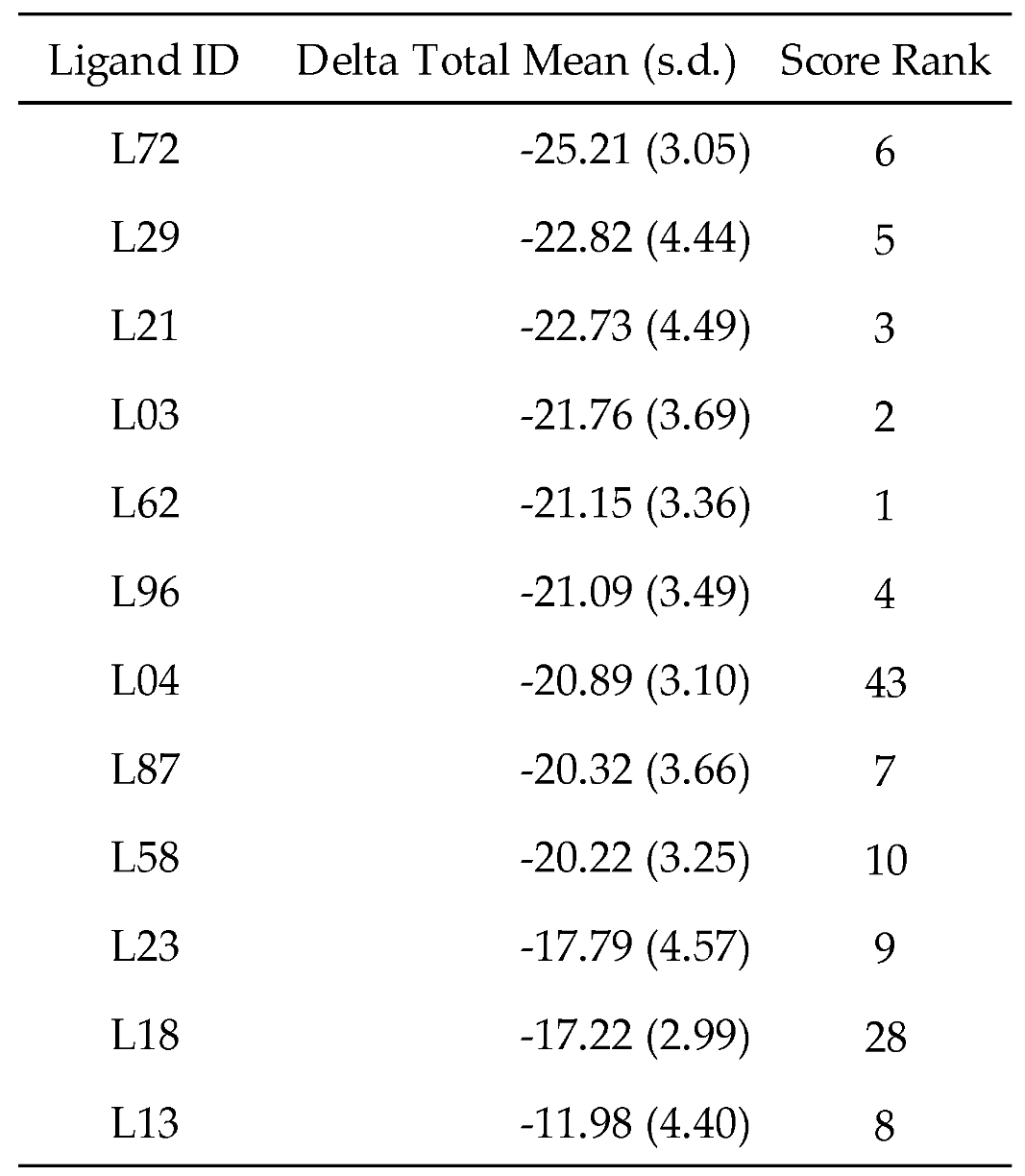 |
The unit of binding free energy (Delta Total) is kcal/mol. “s.d.” means the standard deviation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



