
Submitted:
01 April 2024
Posted:
02 April 2024
You are already at the latest version
Abstract
Non-isocyanate polyurethane synthesis by non-Sn catalysis is an essential challenge toward green polyurethane synthesis. Bismuth compounds are attractive candidates due to their low cost, low toxicity, and availability to urethane chemistry. We applied various Bi catalysts to the self-polycondensation of a bishydroxyurethane monomer and found BiCl3 to be an excellent catalyst through optimization. The catalytic activity and price of BiCl3 are comparable to those of Bu2SnO, while its toxicity is significantly low. BiCl3 is, therefore, a promising alternative to Sn-based catalysts in non-isocyanate polyurethane synthesis.
Keywords:
polyurethane
; bismuth
; polycondensation
; non-isocyanate
1. Introduction
Polyurethanes are applied to a wide range of products because of their widely controllable properties and excellent characteristics such as foaming ability, biocompatibility, mechanical strength, and abrasion resistance. The industrial fabrication method for polyurethane is the polyaddition of multifunctional isocyanates and diols, developed by Bayer et al. [1,2,3] and is still used nowadays. Because isocyanates are highly reactive, the reaction proceeds quickly and quantitatively yields polyurethanes. However, isocyanates are not only unstable and highly toxic, and furthermore their raw materials, phosgene derivatives, are even more poisonous and corrosive. In addition, the reaction of amines with phosgenes generates corrosive hydrogen chloride as a byproduct, which requires the plant facilities to be heavily corrosion-resistant. As a substantial solution to these problems, various green polyurethane syntheses have been developed to prevent the use of phosgenes and/or isocyanates [4,5,6,7,8,9,10,11,12,13,14,15,16,17].
Polyaddition of bifunctional five-membered cyclic carbonates obtained by the reaction of diepoxides with carbon dioxide [18,19,20,21] and diamines is a well-explored alternative method [4,5,6,7,8,17]. The resulting polyurethane derivatives bearing hydroxy side chains are called polyhydroxyurethanes, and many studies have been conducted since the patent by Whelan Jr. et al. [4]. However, this method cannot synthesize polyurethanes with the same structure as industrial polyurethanes. Alternatively, polycondensation of dialkylurethanes and bishydroxyurethanes can give such polyurethanes from five-membered cyclic carbonates or linear carbonates and amines without using phosgenes and isocyanates.
Polycondensation of dialkyl urethanes with diols reported by Deepa et al. gives polyurethanes in good yields of 80–90% under solvent-free melting conditions in the presence of titanium-based catalysts such as Ti(OBu)4 [9]. Higher molecular weights are achieved by continuous removal of low-boiling alcohols such as methanol from the polymerization medium under nitrogen purge and subsequent high vacuum. The molecular weight of the resulting polyurethanes ranges from 3,000 to 20,000. However, the four steps of a longer process and the need for reduced pressure are still issues that need to be solved.
Polyurethanes can be obtained by self-polycondensation or transurethane polycondensation with diols of bishydroxyurethanes obtained by the addition reaction of ethylene carbonate (EC) and diamines as monomers, as first reported by Rokicki et al. in 2002 [10]. However, toxic Sn-based catalysts are mainly used. The high boiling point of ethylene glycol, produced as a byproduct, makes sufficient removal difficult, and long reaction times with reduced pressure are required to synthesize high-molecular-weight polymers [10,11]. A safer system has been established for similar polymerization under reduced pressure using less toxic BaO [15] and Ti(OBu)4 [16] as catalysts. Various systems have been explored for these non-isocyanate polyurethane syntheses, and polyurethane with higher molecular weight was developed [14].
For an exploration of new catalysts, we focused on compounds of bismuth, which is an inexpensive heavy element with safety [17]. Many organic and inorganic bismuth compounds have very low toxicity, and some bismuth salts are applied for stomach medicines. Bismuth(III) tricarboxylates such as bismuth 2-ethylhexanoate and bismuth neodecanoate have been developed to replace Sn catalysts for isocyanate-based polyurethane synthesis [23]. The application of BiPh3 as a cocatalyst for a Sn catalyst has also been reported [24]. As related applications, bismuth catalysis has been applied for the depolymerization of polyurethanes catalyzed by bismuth neodecanoate [25], the transurethanization with alcohols catalyzed by bismuth triflate [26], and reprocessing of cross-linked polyurethane through transurethanization [27]. Based on these bismuth-catalyzed urethane chemistries, we investigated bismuth catalysts as a new catalyst for transurethane polymerization. We found that BiCl3 effectively catalyzes the self-polycondensation of a bishydroxyurethane, showing activity comparable to Bu2SnO and higher selectivity.
2. Materials and Methods
2.1. Materials
All the reagents were used as received. EC, dimethylsulfoxide (DMSO) super dehydrated, bismuth subsalicylate, bismuth oxide, and Bu2SnO were purchased from Wako Pure Chemical (Osaka, Japan). Dehydrated xylene, N,N-dimethylformamide (DMF) super dehydrated, N-methyl-2-pyrolidinone (NMP), and bismuth hydroxide nitrate were purchased from Kanto Chemical (Tokyo, Japan). BiCl3, BiPh3, 1,6-hexanediol were purchased from Tokyo Chemical Industry (Tokyo, Japan). BiF3, BiBr3, Bi(OCOCH3)3, and Bi2(SO4)3 were purchased from Sigma Aldrich (St. Louis, MO, USA). Bi(OH)3 was purchased from Nakarai Tesque (Kyoto, Japan). The bishydroxyurethane monomer, bis(2-hydroxyethyl)hexane-1,6-diyldicarbamate (BHU6), was prepared as reported from EC and 1,6-hexanediol [10,11].
2.2. Measurements
1H NMR spectra were measured on a JEOL (Tokyo, Japan) ECX-400 (400 MHz) spectrometer using tetramethylsilane as an internal standard. Fourier-transfer infrared (IR) spectra were measured on a JASCO FT/IR-210 spectrometer equipped with an attenuated total reflectance accessory.
2.3. Polycondensation of BHU6 (Typical Procedure)
BHU6 (292 mg, 1.00 mmol) and BiCl3 (34 mg, 0.11 mmol, 11 mol%) were added to a round-bottom flask equipped with a side-arm distilling adapter, a Liebig condenser, and a receiver. Then, the apparatus was degassed and purged with nitrogen. Dehydrated xylene (5.0 mL) was added. The mixture was stirred at 150 °C for 5 h under a nitrogen atmosphere. The solid produced in the flask was dissolved in NMP at 100 °C, and the insoluble substance (9.2 mg after drying) was removed by centrifugation at 4100–4200 rpm. The supernatant was poured into a mixed solution of acetone (100 mL) and 1 M HCl aq. (0.1 mL). The resulting [6,2]-polyurethane was collected by filtration and drying under reduced pressure at 60 °C (219 g, yield = 95.1%, MnNMR = 5400).
3. Results
3.1. Effect of Catalyst
We screened bismuth catalysts in the self-polycondensation of a bishydroxyurethane obtained from the reaction of 1,6-hexanediol with EC (BHU6) using xylene as a solvent at 145 °C for 3 h under a nitrogen atmosphere (Scheme 1, Table 1). The results were compared with a previously reported reaction using Bu2SnO as a catalyst. The molecular weights of the resulting [6,2]-polyurethane were evaluated by 1H NMR spectroscopic analysis since [6,2]-polyurethane is insoluble in typical solvents for size exclusion chromatography at ambient temperatures. BiCl3 gave polyurethane with lower yield and molecular weight than Bu2SnO while resulting in the highest urethane selectivity among the examined catalysts (Entry 1). BiBr3 gave polyurethane with the highest yield and molecular weight among the examined bismuth catalysts (Entry 2). The urethane selectivity was also high. Bismuth subsalicylate was comparable to BiCl3 and BiBr3 in yield and molecular weight but had a poorer selectivity (Entry 3). Other bismuth compounds resulted in significantly lower yields and selectivity (Entry 4-10). We found BiBr3 and BiCl3 to be the superior catalysts among the bismuth catalysts investigated, by the highest yield and molecular weight polyurethanes of BiBr3 and the highest selectivity and price approximately 1/5 lower than BiBr3 of BiCl3. For further optimization studies, we used BiCl3 because of its better performance under the optimized conditions, as described later.
3.2. Mechanism of Catalysis
We investigated the catalytic mechanism of BiCl3 by IR spectroscopy (Figure 1). The IR spectrum of the 2:1 mixture of BHU6 and BiCl3 showed νN-H and νC=O shifts. The νC=O peak of BHU6 was observed at 1683 cm-1 with a minor shoulder peak at 1651 cm-1. The major and shoulder peaks are assigned to hydrogen-bonded ordered and disordered carbonyl groups, respectively [28]. Absorption assignable to free carbonyl groups above 1700 cm–1 was unobservable. The even number chain between the urethane groups and symmetrical structure are suitable for intermolecular hydrogen bonding. In the spectrum of the mixture of BHU6 and BiCl3, the νC=O absorption clearly shifted to a lower wavenumber with a peak top at 1644 cm-1. This shift indicates the strong coordination of the carbonyl group to BiCl3 more Lewis-acidic than active protons in BHU6. Furthermore, the νN-H peak of BHU6 was observed as a sharp absorption around 3324 cm-1, while that of the mixture of BHU6 and BiCl3 overlapped with a broad νO-H peak at a higher wavenumber (3376 cm-1). This fact also indicates that the N–H proton was released from hydrogen bonding between BHU6 by the interaction between BHU6 and the Lewis acidic BiCl3. The strong activation of the carbonyl group by BiCl3 is a probable driving force of the nucleophilic substitution.
To compare the mechanisms of the Bi- and Sn-catalyzed systems, the IR spectrum of a 2:1 mixture of BHU6 and Bu2SnO was also measured (Figure 1d). In contrast to the BiCl3 catalysis, the wavenumbers of the absorption of the νN–H (3326 cm-1) and νC=O(ordered H-bonded) (1684 cm-1) peaks in the spectrum of the mixture were almost identical to those in the spectrum of BHU6 (3324 and 1683 cm-1, respectively). In addition, the absorption of the νC=O(disordered H-bonded) remained observed at 1651 cm-1. Thus, Bu2SnO negligibly interacted with BHU6. Although the catalytic mechanism for Bu2SnO is unclear, it is likely that the transurethanization mechanisms of the Bi- and Sn- catalysis differ despite the similar reactivity.
3.3. Optimization of Conditions
3.3.1. Effect of Time and Temperature
To optimize the polymerization temperature and time, we performed BiCl3-catalyzed self-polycondensation of BHU6 at different polymerization temperatures (140, 145, 150, 155, and 160 °C) and polymerization times (1, 3, and 5 hours). Before discussing the results of the polycondensation, we describe the aspect of the reaction mixture during the polymerization. In the initial stage, the mixture was in a liquid-liquid separation state due to the insolubility of molten BHU6 in xylene at the polymerization temperature higher than the melting point of BHU6. As the polycondensation progressed, solid polyurethane precipitated. In the late stages of the reaction, most parts of polyurethane precipitated, but fewer parts were dissolved in the supernatant.
We optimized the polymerization temperature and time based on yield, conversion, average molecular weight, and the selectivity of the formation of urethane to urea formed by a side reaction. The conversion is the rate of conversion of the hydroxyethyl group to the repeating unit. The time course of conversion was calculated from the integral ratio of the terminal hydroxyethyl group to the repeating unit in the 1H NMR spectra of the precipitates before purification, which are the main components of this system. The average molecular weight and the ratio of urethane to urea groups were calculated from the 1H NMR spectra of the products obtained by reprecipitation of solutions of both the supernatant and the precipitate in NMP into acetone. The formation of urea groups has been previously shown to occur by the nucleophilic substitution of amines, which is produced by the backbiting reaction of the terminal hydroxy group, on urethanes. Figure 2 shows a typical 1H NMR spectrum of [6,2]-polyurethane obtained by polymerization at 150 °C for 5 h, along with the assignments.
The conversion increased with increasing temperature and time up to 150 °C (Figure 3). On the other hand, at temperatures higher than 150 °C, the conversion rate at 1 hour was almost the same as that at 150 °C, but the conversion rate did not increase as time was extended, and the conversion rate at 5 hours was lower than the rate at 150 °C.
As with the conversion, the yield increased with increasing temperature and time up to 150 °C. Above 155 °C, the yield at 1 h was comparable to that at 150 °C but did not increase with time, resulting in the highest yield of polyurethane at 150 °C for 5 h (Figure 4).
The number-average molecular weight tended to increase with temperature at 1 h (Figure 5). On the other hand, at 3 or 5 h, the molecular weight became highest at 150 °C, and the molecular weights significantly lowered above 155 °C. As a result, the highest molecular weight polyurethane was obtained at 150 °C and 5 h. Yields and molecular weights were correlated with conversions since the conversion of the terminal groups grew polyurethane with the increase of molecular weight.
The ratios of the urea group were suppressed to within 3% at all temperatures at 1 h. Below 150 °C, the urea ratios slightly increased with time but were within 5% (Figure 6). On the other hand, above 155 °C, the urea ratios increased significantly with increasing time. As a result of these optimization experiments, polycondensation at 150 °C for 5 h gave the polyurethane with the highest molecular weight and urethane selectivity in the highest yield.
The molecular weights and urea ratios were correlated. In the polymerizations for 1 h, the molecular weight did not decrease even at 160 °C, and the urea ratio stayed lower than 3%. On the other hand, in the polymerizations for 3 and 5 h, molecular weights lowered above 150 °C, and the urea ratios increased significantly from 3–5% at lower temperatures to 7–18%. This relationship suggests that the formation of urea may inhibit the increase in molecular weight.
3.3.2. Discussion on the Mechanism
We discuss the results described above based on the mechanism of this polycondensation (Figure 7). The primary reaction in this system is transurethanization [29], in which the terminal hydroxy group of a monomer or polymer nucleophilically attacks the carbonyl carbon of another monomer or a polymer chain end, and this transurethanization reaction extends the polymer chain. On the other hand, ureation occurs as a side reaction [10,11,16]. This ureation is initiated by a backbiting reaction, in which the terminal hydroxy group intramolecularly attacks the urethane carbonyl group, forming a polymer with an amine terminus that is more reactive than the hydroxy terminus and EC. A nucleophilic attack of the amine terminus on a urethane group in another polymer chain results in chain extension or disproportionation, accompanied by the formation of polymers with urea groups. However, since the average molecular weight remains almost identical after disproportionation, the intermolecular ureation of the amine and urethane hardly interferes with the increase in molecular weight. On the other hand, if the amine terminus generated by backbiting of the hydroxy terminus intramolecularly attacks a urethane carbonyl group and further backbites, this reaction splits the chain into a cyclic oligomer with urethane or urea groups and a linear oligomer with terminal amine or terminal alcohol groups. As a result, the polymer chain shortens. Therefore, intramolecular backbiting accompanied by ureation can be assumed to cause the inhibition of molecular weight increase.
Next, we discuss the reason why ureation occurred in the later stages of polymerization at high temperatures. As mentioned above, the solvent and monomer are in a liquid-liquid separation state in the initial stage of polymerization. As the polycondensation proceeds to form polymers with shorter chains, their polarity becomes lower than that of the monomer, and their solubility improves. However, as the polymerization proceeds to form longer polymers, the polymers become insoluble and precipitate out due to entropic disadvantages and increased crystallinity. Since the melting point of the polymer is 150–160 °C, the polymerization temperature below 150 °C, which is lower than the melting point, results in precipitation of the crystallized polymers. Both transurethanization extending polymer chain and ureation shortening polymer chain are suppressed for the precipitated polymers. This crystallization will be an essential factor in inhibiting ureation at low temperatures. In contrast, when the polymerization temperature is above 155 °C, the precipitated polymer is in a molten state, and as a result, the extension of the molecular chain continues. However, in the late stage, the probability of reaction between mostly consumed hydroxy termini becomes significantly lower, and the hydroxy termini backbites to produce amine termini, which causes the scission of polymer chains via ureation. In other words, polycondensation at low temperatures stops the change of molecular weights by precipitation of polymers with sufficient length for crystallinity. In contrast, polycondensation at high temperatures causes a decrease in molecular weight due to continuous reactions of molten terminal groups, including ureation-induced chain scission.
3.3.3. Effect of Solvent
We investigated the effect of solvents on the self-polycondensation of BHU6 using DMF and DMSO, solvents used in isocyanate-based polyurethane synthesis, and anisole, an aromatic ether with a similar boiling point to xylene (Table 2). Polymerization did not proceed in DMF or DMSO (Entry 1 and 2) despite the good solubilities of BHU6 and [6,2]-polyurethane in these polar solvents. A probable reason is the loss of catalytic activity of these highly polar solvents tightly coordinating with Lewis-acidic BiCl3. Another possible factor is the high reaction temperature degrading DMF and DMSO to give dimethylamine and disproportionation products of DMSO, which also coordinate with BiCl3 to deteriorate its catalytic activity.
Polycondensation in anisole proceeded through a liquid-liquid separation state in the early stages and produced [6,2]-polyurethane as a precipitate in the late stages (Run 3), as did the polycondensation in xylene. However, the yield, molecular weight, and selectivity of the resulting polyurethane were lower than those of xylene. The polarity of anisole higher than xylene, while not as high as those of DMF and DMSO, probably resulted in coordination with BiCl3, which competes with the activation of BHU6 with BiCl3. Therefore, polar solvents are unsuitable for this polycondensation due to the deactivation of BiCl3 by coordination, and xylene was the best azeotropic solvent among the solvents examined.
We optimized the amount of xylene, which proved the best azeotropic solvent (Table 3). The appearance of polyurethane obtained at the end of polymerization differed with the solvent amount. Polyurethane obtained using 3 L/mol of xylene was molten (Entry 5), but those obtained using higher amounts of xylene were solid (Entry 1–4). This difference was attributed to the lower molecular weight and urethane selectivity of the polyurethane obtained in Entry 5, which was less crystallizable than the others. This unsuccessful result may be attributed to the insufficient azeotropic removal of ethylene glycol due to the insufficient amount of xylene, which inhibited polycondensation despite the advantages in the reaction progression in the molten state. This cessation of transurethanization led to the ureation, so that the highest urea ratio in the polyurethane obtained using 3 L/mol of xylene was probably responsible for the synergistic reduction of the melting point.
The increase in solvent volume increased yield, molecular weight, and reaction selectivity. This improvement is attributable to the accelerated azeotropic removal of ethylene glycol, which enhanced the polycondensation. However, further improvement was not achieved by increasing the amount above 5 L/mol. This limitation may originate from the precipitation of the product after a certain degree of growth, which inactive both the polycondensation and side reactions. We concluded the optimal solvent volume to 5 L/mol.
3.3.4. Effect of Catalyst Amount
Finally, the amount of catalyst was optimized (Table 4). Increasing the catalyst amount from 5 mol% to 10 mol% improved yield and molecular weight without deteriorating the selectivity. However, a further increase in catalyst amount to 15 mol% resulted in a slight decrease in yield and molecular weight. Considering the higher urea ratio, we attributed this decrease to the promotion of ureation, which shortens the polymer chain.
3.3.5. Polycondensation under Optimized Conditions Using BiCl3, BiBr3, and Bu2SnO
We performed polycondensation of BHU6 under conditions optimized for the BiCl3 catalyst system using BiBr3 and Bu2SnO (Table 5), which gave good results in the catalyst screening described above (Table 1). BiBr3 resulted in a lower urethane selectivity than BiCl3, as per the conditions mentioned above (Entry 1). The formation of the urea group probably reduced the yield and molecular weight. Bu2SnO also had a lower urethane selectivity (Entry 2), although the molecular weight and yield of the polymer were almost comparable to those of BiCl3. Since the upper limit of molecular weight attainable in this polycondensation is the molecular weight at which sufficient crystallization occurs, catalysts with adequate activity and selectivity will give comparable results. Thus, BiCl3, a Bi catalyst with high selectivity and activity, showed comparable ability to the previously reported Bu2SnO.
4. Conclusions
We applied various Bi catalysts to the self-polycondensation of BHU6 and found BiCl3 to be an excellent catalyst through optimization. The catalytic activity and price of BiCl3 are comparable to those of Bu2SnO, while its toxicity is significantly low. BiCl3 is, therefore, a promising alternative to Sn-based catalysts. The effectiveness of the BiCl3 catalyst will contribute to the development of green non-isocyanate polyurethane synthesis by expansion to polycondensation with other hydroxyurethanes and diols.
Author Contributions
Conceptualization, B.O.; methodology, B.O. and Y.K.; validation, B.O. and Y.K.; formal analysis, B.O. and Y.K.; investigation, Y.K.; resources, B.O. and Y.K.; data curation, Y.K.; writing—original draft preparation, B.O. and Y.K.; writing—review and editing, B.O.; visualization, B.O. and Y.K.; supervision, B.O.; project administration, B.O.; funding acquisition, B.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bayer, O. Das Di-Isocyanat-Polyadditionsverfahren (Polyurethane). Angew. Chem., 1947, 59, 257–272. [Google Scholar] [CrossRef]
- Bayer, O.; Müller, E.; Petersen, S.; Piepenbrink, H.-F.; Windemuth, E. Über neuartige hochelastische Stoffe „Vulcollan”. Angew. Chem., 1950, 62, 57–66. [Google Scholar] [CrossRef]
- Müller, E.; Bayer, O.; Petersen, S.; Piepenbrink, H.-F.; Schmidt, F.; Weinbrenner, E. Über neuartige hochelastische Stoffe „Vulcollan”︁. II. Teil 9. Mitteilung über Polyurethane. Angew. Chem., 1952, 64, 523–531. [Google Scholar] [CrossRef]
- Whelan, J.M., Jr.; Cotter, R. J. Multiple cyclic carbonate polymers US3072613, 1963.
- Mikheev, V.V.; Svetlaov, N. V.; Sysoev, V. A.; Brus’ko, N. V. Synthesis and some properties of linear hydroxyl containing polyurethanes. Deposited Doc.
- Rokicki, G.; Łaziński, R. Polyamines containing β-hydroxyurethane linkages as curing agents for epoxy resin. Angew. Makromol. Chem., 1989, 170, 211–225. [Google Scholar] [CrossRef]
- Rokicki, G.; Gzajkowaska, J. Badania nad syntezq polihydroksyuretanów z wykorzystaniem diepoksydów, dwutlenku węgla i diamin. Polimery 1989, 34, 141–147. [Google Scholar] [CrossRef]
- Kihara, N.; Endo, T. Synthesis and properties of poly(hydroxyurethane)s J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2765–2773. [Google Scholar] [CrossRef]
- Deepa, P.; Jayakannan, M. Solvent-free and nonisocyanate melt transurethane reaction for aliphatic polyurethanes and mechanistic aspects. J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2445–2458. [Google Scholar] [CrossRef]
- Rokicki, G.; Piotrowska, A. A new route to polyurethanes from ethylene carbonate, diamines and diols. Polymer, 2002, 43, 2927–2935. [Google Scholar] [CrossRef]
- Ochiai, B.; Utsuno, T. Non-isocyanate synthesis and application of telechelic polyurethanes via polycondensation of diurethanes obtained from ethylene carbonate and diamines. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 525–533. [Google Scholar] [CrossRef]
- Deng, Y.; Li, S.; Zhao, J.; Zhang, Z.; Yang, W. Crystallizable and tough aliphatic thermoplastic poly(ether urethane)s synthesized through a non-isocyanate route. RSC Adv., 2014, 4, 43406–43414. [Google Scholar] [CrossRef]
- Ochiai, B.; Endo, T. Carbon dioxide and carbon disulfide as resources for functional polymers. Prog. Polym. Sci., 2005, 30, 183–215. [Google Scholar] [CrossRef]
- Maisonneuve, L.; Lamarzelle, O.; Rix, E.; Grau, E.; Cramail, H. Isocyanate-free routes to polyurethanes and poly(hydroxy urethane)s. Chem. Rev. 2015, 115, 12407–12439. [Google Scholar] [CrossRef] [PubMed]
- Dyer, E.; Scott, H. The preparation of polymeric and cyclic urethans and ureas from ethylene carbonate and amines J. Am. Chem. Soc., 1957, 79, 672–675. [Google Scholar] [CrossRef]
- Wołosz, D.; Parzuchowski, P.; Rolińska, K. Environmentally friendly synthesis of urea-free poly(carbonate-urethane) elastomers. Macromolecules, 2022, 55, 4995–5008. [Google Scholar] [CrossRef]
- Bourguignon, M.; Grignard, B.; Detrembleur, C. Water-induced self-blown non-isocyanate polyurethane foams. Angew. Chem. Int. Ed. 2022, 61, e202213422. [Google Scholar] [CrossRef]
- Peppel, W. J. Preparation and properties of the alkylene carbonates Ind. Eng. Chem., 1958, 50, 767–770. [Google Scholar]
- Brindoepke, G.; Marten, M. Verfahren zur herstellung von 2-oxo-1,3-dioxolanen. DE3529263, 1987. Chem. Abstr., 1987, 107, 156951z. [Google Scholar]
- Kihara, N.; Hara, M.; Endo, T. Catalytic activity of various salts in the reaction of 2,3-epoxypropyl phenyl ether and carbon dioxide under atmospheric pressure J. Org. Chem., 1993, 58, 6198–6202. [Google Scholar] [CrossRef]
- Rollin, P.; Soares, L.K.; Barcellos, A.M.; Araujo, D.R.; Lenardão, E.J.; Jacob, R.G.; Perin, G. Five-membered cyclic carbonates: versatility for applications in organic synthesis, pharmaceutical, and materials sciences. Appl. Sci. 2021, 11, 5024. [Google Scholar] [CrossRef]
- Mohan, R. Green bismuth. Nat. Chem, 2010, 2, 336. [Google Scholar] [CrossRef]
- Leckart, A.R.; Hansen, H. V. Bismuth catalyst system for preparing polyurethane elastomers. US458 4362A, 1985.
- Luo, S.-G.; Tan, H.-M.; Zhang, J.-G.; Wu, Y.-J.; Pei, F.-K.; Meng, X.-H. Catalytic mechanisms of triphenyl bismuth, dibutyltin dilaurate, and their combination in polyurethane-forming reaction. J. Appl. Polym. Sci., 1997, 65, 1217–1225. [Google Scholar] [CrossRef]
- Vanbergen, T.; Verlent, I.; De Geeter, J.; Haelterman, B.; Claes, L.; De Vos, D. Recycling of flexible polyurethane foam by split-phase alcoholysis: identification of additives and alcoholyzing agents to reach higher efficiencies. ChemSusChem, 2020, 13, 3835–3843. [Google Scholar] [CrossRef] [PubMed]
- Jousseaume, B.; Laporte, C.; Toupance, T.; Bernard, J. M. Efficient bismuth catalysts for transcarbamoylation. Tetrahedron Lett., 2002, 43, 6305–6307. [Google Scholar] [CrossRef]
- Fortman, D.J.; Sheppard, D. T.; Dichtel, W. R. Reprocessing cross-linked polyurethanes by catalyzing carbamate exchange. Macromolecules 2019, 52, 6330–6335. [Google Scholar] [CrossRef]
- Guo, R.; Zhang, Q.; Wu, Y.; Chen, H.; Liu, Y.; Wang, J.; Duan, X.; Chen, Q.; Ge, Z.; Zhang, Y. Extremely strong and tough biodegradable poly(urethane) elastomers with unprecedented crack tolerance via hierarchical hydrogen-bonding interactions. Adv. Mater. 2023, 35, 2212130. [Google Scholar] [CrossRef]
- Bakkali-Hassani, C.; Berne, D.; Ladmiral, V.; Caillol, S. Transcarbamoylation in polyurethanes: underestimated exchange reactions? Macromolecules 2022, 55, 7974–7991. [Google Scholar] [CrossRef]
Scheme 1.
Bi-catalyzed self-polycondensation of BHU6.
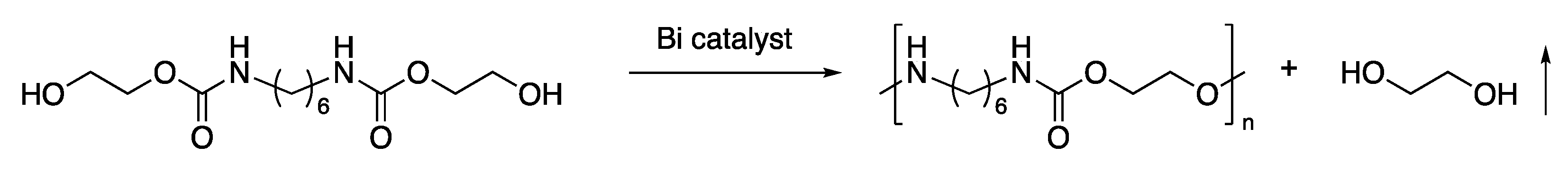
Figure 1.
(a) Wide-range and (b) magnified IR spectra of BHU6, BiCl3, Bu2SnO, 2:1 mixture of BHU6 and BiCl3, and 2:1 mixture of BHU6 and Bu2SnO.
Figure 1.
(a) Wide-range and (b) magnified IR spectra of BHU6, BiCl3, Bu2SnO, 2:1 mixture of BHU6 and BiCl3, and 2:1 mixture of BHU6 and Bu2SnO.
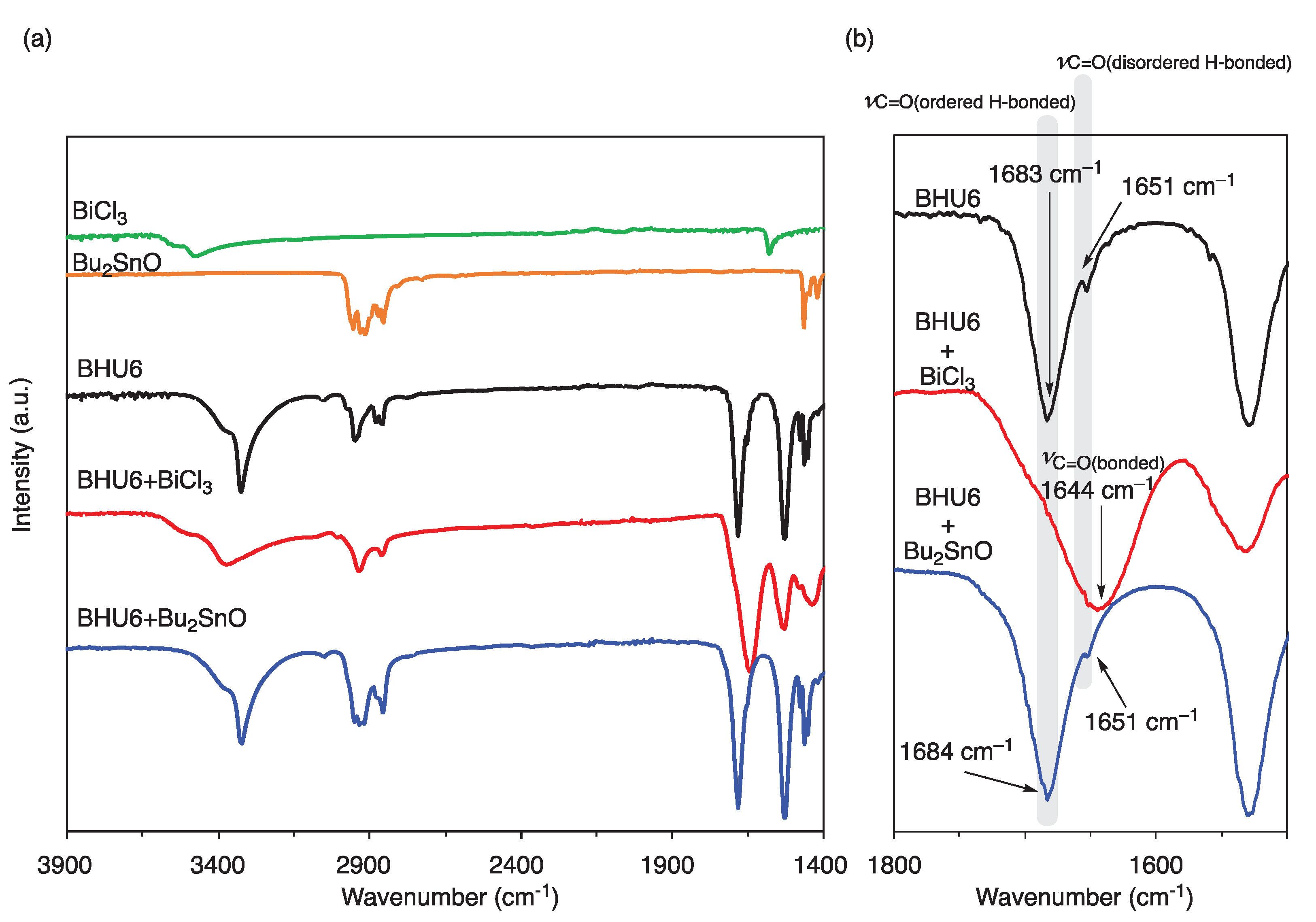
Figure 2.
1H NMR spectrum of [6,2]-polyurethane obtained by self-condensation of BHU6 at 150 °C for 5 h in xylene using BiCl3 (10 mol%).
Figure 2.
1H NMR spectrum of [6,2]-polyurethane obtained by self-condensation of BHU6 at 150 °C for 5 h in xylene using BiCl3 (10 mol%).
Figure 3.
Conversion of terminal group into repeating unit in precipitate before purification.
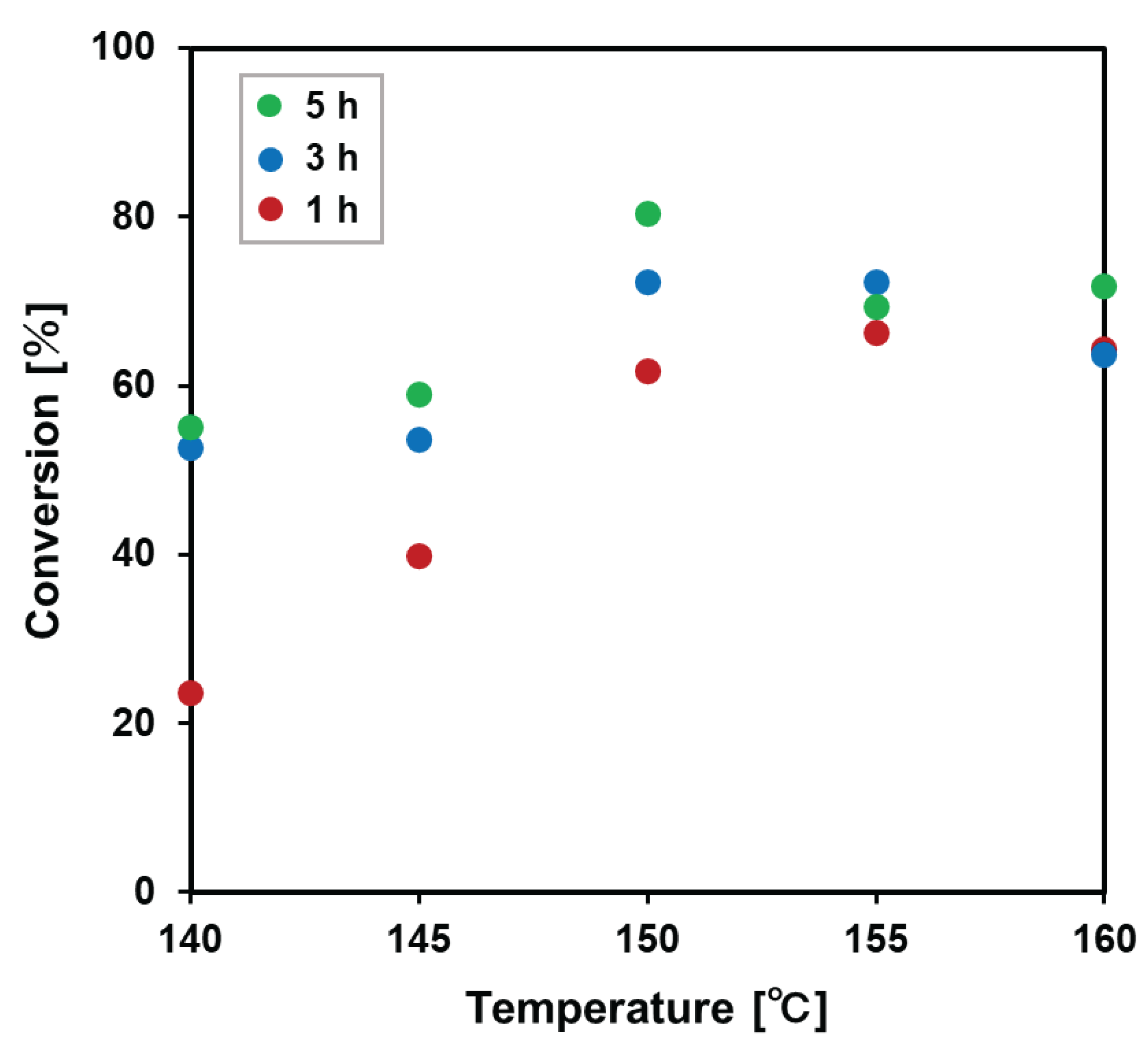
Figure 4.
Yield of [6,2]-polyurethane.
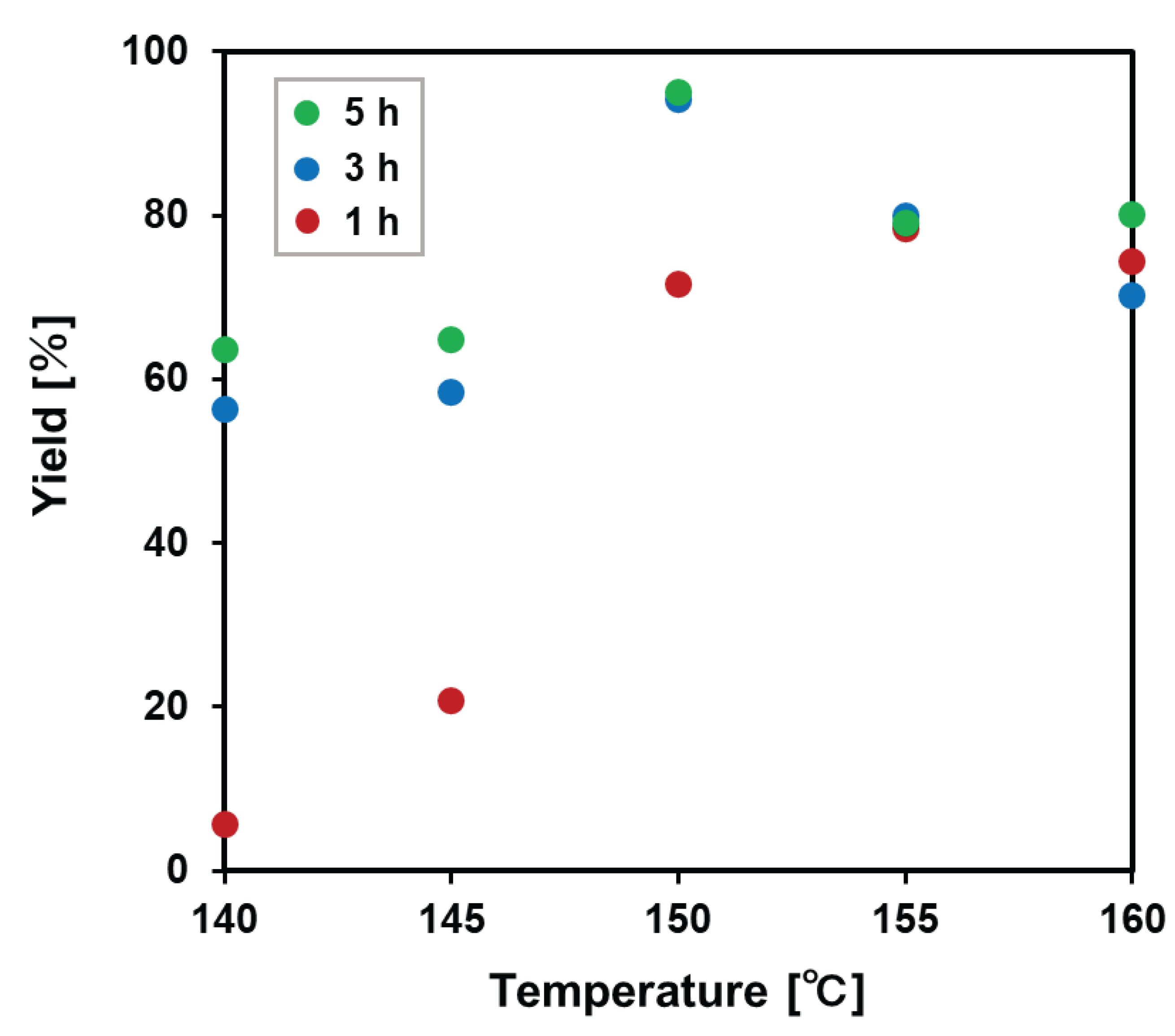
Figure 5.
Molecular weight of polyurethane after purification calculated by 1H NMR spectroscopy.
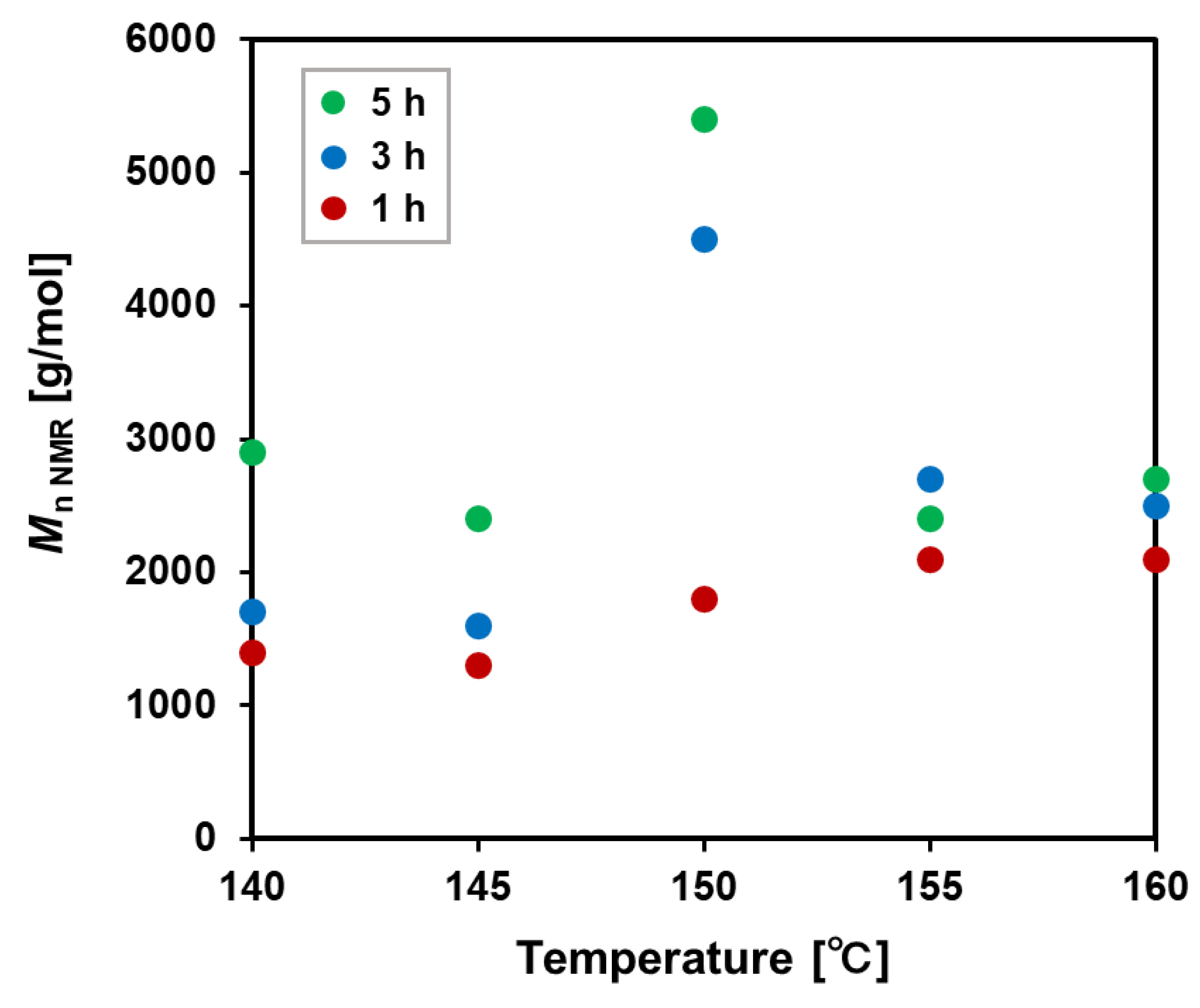
Figure 6.
Urea group ratio of [6,2]-polyurethane obtained after purification determined by 1H NMR spectroscopy.
Figure 6.
Urea group ratio of [6,2]-polyurethane obtained after purification determined by 1H NMR spectroscopy.
Figure 7.
Mechanism of BiCl3-catalyzed polycondensation of BHU6.
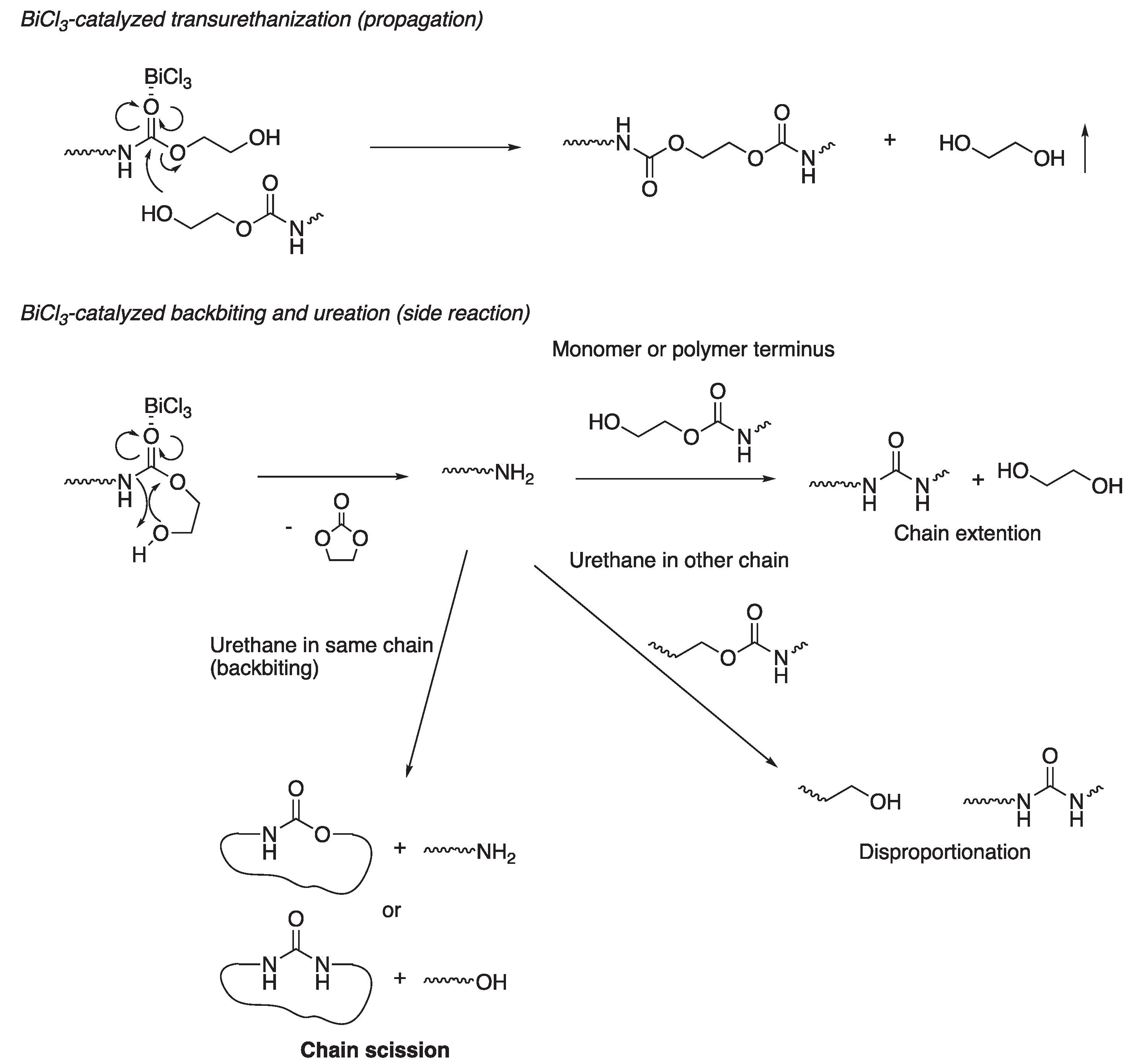

Table 1.
Effect of catalyst in self-polycondensation of BHU6.
| entry | catalyst | Yield (%)a | MnNMRb | Urethane/Ureab |
|---|---|---|---|---|
| 1 * | BiCl3 | 59 | 1,600 | 97/3 |
| 2 | BiBr3 | 74 | 2,100 | 95/5 |
| 3 | Bismuth subsalicylate | 66 | 2,000 | 87/13 |
| 4 | Bi(OH)3 | 51 | 1,400 | 92/8 |
| 5 | BiF3 | 45 | 1,400 | 88/12 |
| 6 | Bi2O3 | 23 | 1,400 | 91/9 |
| 7 | Bi(OCOCH3)3 | 25 | 1,300 | 88/12 |
| 8 | Bi2(SO4)3 | 20 | 1,300 | 99/1 |
| 9 | Bismuth hydroxide nitrate | 16 | 1,300 | 97/3 |
| 10 | BiPh3 | 1 | 860 | 39/61 |
| c.f. | Bu2SnO | 82 | 2,400 | 86/14 |
Conditions: BHU6, 1 mmol; catalyst, 10 mol%; xylene, 5 mL; 145 °C; 3 h; N2. a Isolated yield after precipitation into acetone. b Determined by 1H NMR spectroscopy (400 MHz, d6-DMSO).
Table 2.
Effect of solvents on self-polycondensation of BHU6 catalyzed by BiCl3.
| Entry | Solvent | Yield (%)a | MnNMRb | Urethane/Ureab |
|---|---|---|---|---|
| 1 | DMF | No polymerization | ||
| 2 | DMSO | No polymerization | ||
| 3 | Anisol | 48 | 1,800 | 89/11 |
| c.f. | Xylene | 95 | 5,400 | 96/4 |
Conditions: BHU6, 1 mmol; BiCl3, 10 mol%; solvent, 5 mL; 150 °C; 5 h; N2. a Isolated yield after precipitation into acetone. b Determined by 1H NMR spectroscopy (400 MHz, d6-DMSO).
Table 3.
Effect of amount of xylene on self-polycondensation of BHU6 catalyzed by BiCl3.
| Entry | Xylene/BHU6 (L/mol) | Yield (%)a | MnNMRb | Urethane/Ureab |
|---|---|---|---|---|
| 1 | 15 | 91 | 4,700 | 97/3 |
| 2 | 10 | 91 | 4,600 | 90/10 |
| 3 | 8 | 89 | 3,400 | 93/7 |
| 4 | 5 | 95 | 5,400 | 96/4 |
| 5 | 3 | 67 | 2,300 | 87/13 |
Conditions: BHU6, 1 mmol; BiCl3, 10 mol%; 150 °C; 5 h; N2. a Isolated yield after precipitation into acetone. b Determined by 1H NMR spectroscopy (400 MHz, d6-DMSO).
Table 4.
Effect of amount of BiCl3 on self-polycondensation of BHU6 catalyzed by BiCl3.
| Entry | BiCl3 (mol%) | Yield (%)a | MnNMRb | Urethane/Ureab |
|---|---|---|---|---|
| 1 | 5 | 91 | 3,700 | 96/4 |
| 2 | 10 | 95 | 5,400 | 96/4 |
| 3 | 15 | 90 | 5,100 | 94/6 |
Conditions: BHU6, 1 mmol; xylene, 5 mL; 150 °C; 5 h; N2. a Isolated yield after precipitation into acetone. b Determined by 1H NMR spectroscopy (400 MHz, d6-DMSO).
Table 5.
Effect of catalyst on self-polycondensation of BHU6 catalyzed by BiCl3 under conditions optimized for BiCl3.
Table 5.
Effect of catalyst on self-polycondensation of BHU6 catalyzed by BiCl3 under conditions optimized for BiCl3.
| Entry | Catalyst | Yield (%)a | MnNMRb | Urethane/Ureab |
|---|---|---|---|---|
| 1 | BiBr3 | 90 | 4,000 | 93/7 |
| 2 | Bu2SnO | 95 | 4,900 | 91/9 |
| c.f. | BiCl3 | 95 | 5,400 | 96/4 |
Conditions: BHU6, 1 mmol; catalyst, 10 mol%; xylene, 5 mL; 150 °C; 5 h; N2. a Isolated yield after precipitation into acetone. b Determined by 1H NMR spectroscopy (400 MHz, d6-DMSO).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



