Submitted:
14 March 2024
Posted:
14 March 2024
You are already at the latest version
Abstract
A Challenging task in routine practice is finding distinction between benign and malignant paragangliomas and pheochromocytomas. The aim of this study is a comparative analysis of angiogenesis by assessing intratumoral microvascular density (MVD) with immunohistochemical (IHC) markers (CD31, CD34, CD105, ERG), and S100 immunoreactivity, Ki67 proliferative index, SDHB expressiveness, tumor size with one the most utilized score Pheochromocytoma of Adrenal Gland Scales Score (PASS), using tissue microarray (TMA) with 115 tumor samples, 61 benign (PASS<4) and 54 potentially malignant (PASS ≥4). We found no notable difference between intratumoral MVD and potentially malignant behavior. Group of potentially malignant tumors are significantly larger in size, have lower intratumoral MVD, and decreased number of S100 labeled sustentacular cells. Both groups have low proliferative activity (mean Ki67 is 1,02 and 1,22 respectively). Most tumors maintains SDHB expression, only 6 cases (5,2%) showed a loss of expression (4 of them in PASS<4 group and 2 in PASS≥4). PASS score is easy available for assessment, and complemented with markers of biological behavior completes risk stratification algorithm. Size is directly related with PASS score and malignancy. Intratumoral MVD is extensively developed but it is not crucial in evaluating the malignant potential.
Keywords:
Angiogenesis
; Pheochomocytoma
; Paraganglioma
; CD105
; ERG
; PASS
; Ki67
; S100
; SDHB
1. Introduction
Paragangliomas are rare non-epithelial neuroendocrine tumors, but potentially life-threatening. Several classifications and terms have been used for these tumors, depending on the place of origin, like the term pheochromocytoma for ones that originate from the medulla of the adrenal gland and paraganglioma for tumors arising in other anatomical places of the autonomic nervous system [1]. Pheochromocytomas are according to the new nomenclature intra-adrenal paragangliomas, and classic sympathetic tumors that produce and release catecholamines: noradrenaline and/or adrenaline [1,2]. Other paragangliomas are extra-adrenal and they can be parasympathetic and sympathetic. Parasympathetic paragangliomas are primarily located in the head and neck (so-called head and neck paraganglioma-HNPGL) and do not release catecholamines, while sympathetic paragangliomas are located in the chest, abdomen, and pelvis, and release large amounts of noradrenaline, so like pheochromocytomas, they are accompanied by symptoms such as hypertension, palpitations, sweating, headaches and diabetes mellitus [2]. These non-epithelial neuroendocrine tumors can occur at any age, with prevalence in the fourth and fifth decade, and they occur equally in men and women [3]. The only known causative factor for paragangliomas is hereditary susceptibility, and for these tumors, genetic contribution is the strongest [3,4,5].
Apart from the clinical symptoms that put at risk the patient, paragangliomas can have a benign clinical course. Ideally, they can be localized, and surgery, as the primary form of therapy, can be a successful and definitive form of therapy [6]. But in the case of malignant behavior and with excessive release of catecholamines, there is no generally effective therapy [7,8]. The prevalence of malignancy is 5% to 26% of cases [9]. It is extremely difficult at the time of diagnosis to predict whether paraganglioma will have a malignant or benign clinical course, especially since metastases can occur much later after the primary tumor [10].
In the Classification of Tumors of Endocrine Organs of the World Health Organization (WHO) from 2017, in the absence of clearly defined malignancy criteria, it is considered that all pheochromocytomas and paragangliomas can potentially metastasize, and instead of classifying them as benign and malignant, it is preferable to stratify the risk of metastasis in these tumors [3]. These recommendations are retained in the latest WHO classification from 2022 [1]. Although it was challenging, a staging system for intra-adrenal and extra-adrenal sympathetic paragangliomas was introduced in 2017 in the 8th Edition AJCC Cancer Staging Manual [3].
Numerous risk factors have been analyzed like localization, and it was observed that 10% of parasympathetic paragangliomas metastasize, up to 25% of pheochromocytoma cases, and 40-70% of sympathetic paraganglioma cases [1,11,12,13,14].
Tumor size and weight showed some correlation with metastatic potential but not as independent variables [11].
Essential histological characteristics that could indicate malignant potential were analyzed and included in the Pheochromocytoma of the Adrenal gland Scaled Score (PASS) by Thompson, who made this scoring system [15]. Although its use remains controversial it is a commonly used scoring system [16,17,18]. PASS scoring system includes twelve histological characteristics: capsular invasion, vascular invasion, extension into the periadrenal adipose tissue, presence of large nests or diffuse growth (in >10% of tumor volume), central tumor or confluent necrosis, high cellularity, tumor cell spindling, cellular monotony, increased mitotic figures (>3/10 high power field), atypical mitotic figures, profound nuclear polymorphism, and nuclear hyperchromasia. The maximum score is 20. The cutoff value is 4, so when the sum is <4 it is considered that the tumor will have a benign course, while a score ≥4, means that the tumor is likely histologically more aggressive and potentially malignant.
Angiogenesis is substantial for tumor growth and metastases so it has been explored in many studies, with different approaches including assessing microvascular density (MVD), vascular patterns, and expression of angiogenic factors but with inconclusive results [19,20].
Ancillary use of many immunohistochemical markers showed some significance and association with the malignant potential of these tumors, such as the proliferative index Ki67, S100 to determine the number of sustentacular cells, and SDHB for screening patients with hereditary paraganglioma pheochromocytoma syndrome (loss of expression of SDHB may indicate malignant biological potential) [1,14,21].
The primary objective of our research is to further explore angiogenesis, determining intratumoral MVD using IHC endothelial markers CD31, CD34, CD105 and nuclear endothelial marker ERG and then look into how intratumoral MVD can contribute to risk stratification by comparing them with histological characteristics using the PASS scoring system. Furthermore the aim is to make a comprehensive analysis of known markers like immunohistochemical expressions of Ki67, S100, and SHDB, along with the tumor's size and weight. The application of TNM staging in a retrospective manner was undertaken to ensure a comprehensive understanding of the clinical context and facilitate future analyses [22].
2. Materials and Methods
We analyzed retrospectively patients who underwent surgery, due to paraganglioma, at the University Clinical Center of Serbia in Belgrade in the period from the end of 2008 to the beginning of 2018. In the study, we included all cases in which there was a consensus on the diagnosis mentioned above, as two pathologists independently performed an examination (revision) of all selected cases. Among 110 consecutive patients who were operated because of tumors, five of them were operated twice during this period (four because of bilateral pheochromocytoma, and one had recurrence). In summary, we gathered 115 tumor samples from 110 consecutive patients. To identify representative areas in the donor paraffin-embedded tissue block, we utilized hematoxylin and eosin-stained (H&E) four-micron sections of each tumor. We manually punched tissue cores from the selected areas and arranged them on a recipient paraffin block. All the patients are represented with at least one representative core tissue cylinder. A tissue microarray was made on which immunohistochemical analysis was performed [23]. There were a total of 107 intra-adrenal paragangliomas with 1 recurrent case and 7 extra-adrenal paragangliomas. We cut 4 µm thick sections from the tissue microarray paraffin blocks using a tissue microtome. These sections were then subjected to deparaffinization and antigen unmasking procedures before being labeled with primary antibodies against CD31, CD105, CD34, ERG, SDHB, S-100, and Ki67. The list of used antibodies and manufacturers is shown in Table 1.
As a control for immunohistochemical staining, external tissue controls were used for all the mentioned antibodies. Immunohistochemical staining was performed manually, following the manufacturer's instructions for each antibody separately [24]. Visualization of the immunohistochemical reaction was performed using the streptavidin-biotin technique (DAKO LSAB+ kit). For further analysis, the IHC stained slide preparations were photographed with a digital camera Olympus DP70 (Olympus Corporation; Tokyo, Japan) through an upright microscope (Olympus BX50, Olympus Corporation) at a magnification of 200x times, creating images at 300x300 resolution, (photo dimensions 4080x3072pixel). The digitalization of all slides was additionally done with the Leica Aperio AT2 slide scanner (Leica Biosystems, Nussloch GmbH, Germany) for analysis and documentation purposes. Virtual slides generated from Leica Aperio AT2 were morphometrically analyzed with a Leica AperioImageScope (version 12.4.6, Leica Biosystems, Nussloch GmbH, Germany) and with FIJI-ImageJ software [25].
All tissue samples are then divided into two categories according to the PASS score (PASS <4 and PASS ≥4).
We counted sustentacular cells labeled with S 100 antibody on a two-dimensional section in a created network over the image. Cells whose positive nuclei are located in cubes, and cell extensions and nuclei that cross the upper and right border, are included and those that cross the left and lower border are excluded [26,27]. The average number of sustentacular cells per 1 mm2 was calculated according to the measured tumor surface.
We analyzed endothelial cytoplasmic and/or membrane markers of angiogenesis CD31, CD34, CD105 and counted blood vessels according to the authors Weidner and Tanigawa, where each individual or group of endothelial cells that are clearly separated from adjacent microvessels, tumor cells, or other connective tissue elements is counted as a single blood microvessel. Vessel lumen, although often present, is not necessary to define a structure as a microvessel. Every 40 µm of microvessel length was counted as a new microvessel [28,29,30].
In addition to cytoplasmic/or membrane endothelial markers, we also counted blood vessels using the nuclear marker ERG. In the photo of each cylinder, using the Cell Counter option, we counted blood vessels and displayed intratumoral MVD as an average number per 1mm2.
Proliferation index Ki67 was determined on each tissue cylinder by the percentage of positive tumor nuclei which are counted using the Cell Counter option. We noticed granular cytoplasmic staining in tumor cells as positive for SDHB, and the absence of staining in tumor cells with positive endothelial cells, as negative and loss of immunohistochemical SDHB expression.
We employed both parametric (t-test) and non-parametric (Chi-square test, Mann-Whitney U test) difference tests, along with Spearman's correlation analysis as a correlation test. The selected significance level, which represents the probability of the first type of error, is 0.05.
Clinical data (blood pressure, catecholamines, and/or their metabolites in plasma or urine, radiological imaging, genetic testing), macroscopic findings (tumor size and weight) were obtained from medical records, and paraffin blocks from the archives, with the approval of Ethical committees.
3. Results
3.1. Clinical and macroscopic findings
The research was done on 115 tumor tissues, 75 (65.2%) females, and 40 (34.8%) males. All cases underwent preoperative radiological imaging with a CT scan, in some it was supplemented with magnetic resonance imaging. Elevated arterial pressure and/or paroxysmal pressure spikes were recorded in 91 (79.8%) patients. Among the total of 109 patients (accounting for 94.8%), pertinent information regarding the analysis of catecholamines and/or their metabolites in plasma or urine was available, and of these, 93 patients (representing 85.3%) exhibited elevated values. Amongst the cohort of 52 patients (constituting 45.2%), a supplementary diagnostic MIBG scintigraphy was conducted, revealing an augmented accumulation in the tumor region for 39 of the cases (equating to 75%). For 47 tissue samples (40,83%) information about genetic testing was available. Seventeen were confirmed to have Multiple Endocrine Neoplasia Type 2a (MEN2a), 10 patients had Von Hippel-Lindau Disease (VHL), and four patients had Von Recklinghausen Disease (Neurofibromatosis Type 1). In this group, only one patient was confirmed to have an SDHB mutation. At the time of diagnosis and surgery, 64 (55,7%) were over 50 years old, and 51 (44,3%) were under 50 years old. Clinicopathological data according to the PASS score, are shown in Table 2.
In 60 (52.2%) cases, tumors were ≥50 mm, and in 55 (47.8%) cases, they were <50 mm. The mass of the tumor was greater than 60 grams in 34 (29.6%) cases and less than 60 grams in 81 (70.4%) cases. Comparing tumor size (in mm) and PASS values our research has shown that there is a significant association (p=0,00) with medium strong correlation (r=0,431).
3.2. Microscopic and immunohistochemical findings
Intratumoral MVD was examined as previously described for the endothelial nuclear marker (ERG) and for endothelial cytoplasmic markers (CD34, CD105, CD31) and compared with group PASS <4 benign and PASS ≥4 malignant (Figure 1). Staining for each endogenous marker is shown in Figure 2. No significant statistical difference in average intratumoral MVD in group PASS <4 benign and PASS ≥4 malignant was found, (ERG p=0.071, CD34 p=0.077, CD105 p=0.088, and CD31 p=0.337). Average intratumoral MVD was lower in the malignant group, for all four antibodies. Comparing average intratumoral MVD and PASS score, there was no correlation between average intratumoral MVD and potential malignant behavior (ERG p=0.590, CD34 p=0.213, CD105 p=0.139, and CD31 p=0.849). Intratumoral MVD, labeled with ERG, also has no considerable correlation with tumor size (mm) and tumor weight (g), p=0,752 and p=0,786 respectively.
Sustentacular cells were labeled with S100 antibody and shown in Figure 3. In our study, we counted the total number of sustentacular cells and expressed it as an average number per mm2 of tumor tissue surface (Figure 4). The average number of sustentacular cells per mm2 is 203.43, and we found no statistically notable correlation (p=0,080). In the potentially benign (61 cases) group, the average number of sustentacular cells is 243, and in the potentially malignant (54 cases) group, that number was lower, 158.61. The number of sustentacular cells is decreased in the group of PASS≥4 tumors but without statistically meaningful difference (p=0.062).
The proliferative index Ki67 was determined as described and the values shown in relation to PASS (PASS<4 and PASS≥4) (Figure 5). In our study, the PASS≥4 group, the potentially malignant had a mean Ki67 of 1.22% and the potentially benign group (PASS<4) had a mean Ki67 of 1.02%, with no statistically significant difference in these two groups (p=0,598). Examining the relationship between values of Ki67 and PASS scores no significant correlation was found (p=0,114). Detailed analysis by different values of Ki67 showed among 54 cases in PASS≥4 group, 10 (18.5%) had a Ki67 value of ≥1%, 6 (11.1%) had a Ki67 value of ≥2% and 4 (7.4%) had a Ki67 value of >3%. The remaining 34 (62.96%) cases in group PASS≥4 had a Ki67 value <1%. Among the 61 cases in the PASS <4 group, 39 (69.9%) had a Ki67 value of <1%, and 53 (86.9%) had a Ki67 value of <2%. However, 7 (11.5%) cases had a Ki67 value of ≥2%, 14 (22.9%) had a Ki67 value of ≥1%, and 1 case had a Ki67 value of >3% (1.64%) with the same conclusion, changing the cutoff value of Ki67 does not change differences in these two groups (PASS<4 and PASS≥4).
We examined the SDHB immunohistochemical expression, in tumor cells, some cases were shown in Figure 6 and Figure 7. In our study 109 cases (94,8%) were positive and 6 cases (5,2%), lost immunoreactivity. In the PASS <4 group 4 cases (6,6%) were SDHB negative and 57 cases (93,4%) were SDHB positive, while in PASS≥4 group 2 cases (3,7%) were negative and 52 (96,3%) were positive. There was no notable difference in losing SDHB immunoreactivity among group PASS <4 and PASS≥4 (p=0,683).
Some patients underwent genetic testing, so data were available for 47 tissue samples (40,87%), with 32 (27,83%) of them having a confirmed mutation, data are summarized in Table 3.
The mean number of ERG-labeled blood vessels in the group of patients with a confirmed mutation was 277.15, whereas in the group without the mutation, it was 266.65. Our analysis showed that the difference in intratumoral MVD was not statistically significant between these two groups (p=0.583).
4. Discussion
The primary difficulty particularly with these non-epithelial neuroendocrine tumors is that there are no established standards for determining their malignancy, and patients must undergo long-term monitoring and face uncertainty since metastases can develop even many years after the initial tumor [10,31,32]. The primary objective during histological examination following surgery is to accurately diagnose a malignant tumor that has not yet spread to other parts of the body. Numerous studies have been conducted, and efforts are being made to develop an algorithmic model based on known factors that indicate malignancy. In many studies, the PASS score is a reliable prognostic factor, even though the evaluation of certain histological parameters such as hyperchromasia, nuclear pleomorphism, and cellularity are subjective. Tumors with a PASS score less than 4 did not develop metastases, indicating that only patients with a PASS score of 4 or higher would require follow-up, including annual radiological imaging and biochemical measurements of metanephrines [18]. In recent assessments of the Pheochromocytoma of the Adrenal Gland Scaled Score (PASS), as well as other scoring systems that have been introduced since its development, the World Health Organization (WHO) Classification of Endocrine Tumors recognizes that while these systems have their advantages and disadvantages, their use should not be discouraged in further research, but instead should be optimized in conjunction with molecular analyses [1]. Considering this, we employed the PASS score as a suitable standard for our diagnostic algorithm. In our study, we categorized our cases based on the value of the PASS score and analyzed additional prognostic factors. In numerous studies, angiogenesis has been recognized as a critical factor for the proliferation of tumors and the development of metastases [19]. Given the highly vascular nature of paragangliomas as a solid tumor, there has been interest in assessing the significance of angiogenesis in its pathogenesis, and investigations were prompted. Gao X. et al investigated CD31 as an angiogenic marker, by counting the number of CD31 positive vessels within the highest expressed tumor area (MVD), and the number was significantly higher in pheochromocytomas with PASS<4 than in those with PASS≥4. Gao X. et al. also detected that intratumoral hemorrhage was significantly higher in pheochromocytomas with PASS≥4, than PASS<4, which could also result in relatively low CD31 status in histologically low-grade tumors [16]. H. Ohji et al counted CD34 labeled blood vessels (MVD) and found no statistical association between the MVD and malignancy, while Q. Liu et al, which assessed MVD by staining endothelial cells with antibody to factor VIII/von Willebrand factor antigen, found a highly significant difference between the groups of benign and malignant [20,21]. In the former study the cases were divided into benign and malignant according to the presence of metastatic disease and in the latter group, besides the presence of metastasis, tumors with evidence of capsular or vascular invasion were included, which are histological criteria included in PASS scoring scale. Q. Lui also noticed that areas of highest vascularization were usually along the periphery of the tumor [20]. M. Bialas et al studied angiogenesis status in pheochromocytomas, including MVD and vascular architecture, after immunostaining endothelial cells with antibodies CD31 and CD105 [33]. Vascular architecture patterns were highly heterogeneous within pheochromocytomas, both benign and malignant. The MVD in the central areas of the tumor was higher than in the subcapsular areas. Secondary changes in tumors, like hemorrhage and cystic degeneration, influenced counting MVD and assessing vascular architecture [33]. Favier et al used immunostaining for CD34 and α-actin smooth muscle cells to define vascular architecture, and they reported differences, noticing two different patterns, mainly benign pheochromocytomas having more regular patterns, with short and straight vascular segments, while malignant presented with an irregular pattern, longer vascular segments, of irregular length [34]. The density of vascular segments was lower in irregular patterns, but blood vessel counting was not considered appropriate for these tumors [34]. L. Oudiijk et al found that the mean sensitivity and specificity of vascular architecture as a predictor of potential malignant behavior of paragangliomas was 59,7% and 72,9%, with a significant agreement between the observers [35]. To assess tumor angiogenesis and to minimize the potential influence of marker choice on our results we chose to evaluate intratumoral MVD using several IHC markers at the same time. By making TMA we tried to select areas of the tumor with a high density of blood vessels without hemorrhages and cystic degeneration. While most studies, as was mentioned, have utilized CD31 and CD34, which are pan-endothelial cell markers that react with both proliferating and pre-existing vessels in tumors, CD105 (endoglin) is an endothelial antibody that primarily binds to activated endothelial cells in angiogenesis. Thus, it may be a more specific marker for tumor angiogenesis, as indicated by prior research [36,37,38,39]. In addition to the established antibodies used to measure intratumoral MVD, we examined ERG, a transcription factor belonging to the ETS family that is expressed by endothelial cells involved in angiogenesis. This approach has not been previously explored, to the best of our knowledge, on paragangliomas [40]. All four antibodies used for intratumoral MVD indicated that these were well-vascularized tumors. There was no statistically significant difference in intratumoral MVD with regard to PASS potentially malignant behavior. However, all four endothelial antibodies showed lower intratumoral MVD in the group of malignant tumors, suggesting that cells may be able to overcome hypoxia. Hypoxia has been mentioned as essential for angiogenesis in paraganliomas and due to the pseudohypoxia signaling pathway, they are well known for increased angiogenesis [41,42]. Although neoplastic angiogenesis allows tumor spread, it does not imply that these well-vascularized tumors will metastasize. Additional knowledge is needed to understand the crucial features that drive metastasis of paraganglioma and the role of angiogenesis in this process. In light of the high replicative potential observed in many malignant tumors, several studies have focused on the proliferative Ki67 index [43,44,45,46]. However, it has been found that this index has low sensitivity when it comes to intra-adrenal and extra-adrenal paraganglioma despite its high specificity. Paragangliomas are slow-growing tumors, and the most show very low Ki-67 labeling [18]. Proliferative index Ki67 have high specificity which implies poor sensitivity, with only 50% of malignant tumors have a score greater than 2-3%, so the cut-off value for malignancy is low [18,47]. Our study confirmed the low sensitivity of Ki67 in this context. Nevertheless, Ki67 is an immunohistochemical marker that should be measured, as high Ki67 values may warrant attention for other prognostic factors, such as genetic predisposition and careful consideration of significant histologic predictors of malignancy, such as tumor necrosis [18]. Paragangliomas consist of chief cells that are visible on H&E staining, as well as sustentacular cells, that often necessitate a specific immunohistochemical antibody. While their precise role in metastasis development is not yet clear, several studies, including our own, have demonstrated a decrease in the number of sustentacular cells in malignant tumors [43,48,49,50]. In some cases in our study these cells were entirely absent. These tumors have the strongest genetic contribution with nearly 40% of them associated with mutations in one of the known susceptibility genes, including NF1, VHL, RET, EPAS1, EGLN1, SDHA, SDHB, SDHC, SDHD, SDHAF2, FH, TMEM127, MAX, MDH2, GOT2, SLC25A11, DLST, H3F3A, DNMT3A, MET, MERTK, and KIF1B. There are different types of mutations including germline only, germline and somatic, somatic and somatic mosaicism [3,4,5,51]. Germline mutations involving genes coding for succinate dehydrogenases (SDHx) are the most common genetic cause of paraganglioma, occurring in up to 25% of cases [52,53]. They are followed by genes VHL (4-10%), RET (1-5%), and NF1 (1-5%) [54]. Testing for the germline SDHB mutation in patients with paraganglioma is recommended by Clinical Practice Guidelines [6,55]. Immunohistochemically SDHB negative staining in tumors carries a high risk for the presence of SDHx mutations, whether in SDHB, SDHC, SDHD or SDHA [14]. Loss of SDHB immunoreactivity in tumor cells with SDHx mutations is reported with 100% sensitivity and 84% specificity, with a positive predictive value of 92% and a negative predictive value of 100% [56]. In our study the majority of cases retained immunopositivity to SDHB, and only one had a confirmed mutation. Since genetic testing was not available in all cases, and unfortunately still is not routinely done, immunohistochemistry helped to potentially rule out an SDHx mutation. It was found that the malignant paraganliomas were larger than the benign ones with a median tumor size of 61mm and 44mm, respectively. However, the precise cutoff value that could precisely predict malignant behavior is not established. Since they are all potentially malignant, a staging system is introduced, with size 5cm as cut off value for T1 and T2 [22]. Tumor size and weight are routinely reported, which makes them available for risk stratification, but they are not considered an independent parameter. Invasive features, like an invasion of periadrenal fatty tissue despite being more common in malignant tumors, were not significantly correlated regarding potential malignant behavior in some studies, but it is a criterion for T3 [15,18,22]. The prognosis of malignant paragangliomas remains poor with overall 5-year survival less than 50% [57]. Management of malignant tumor includes surgery, radiation therapy or chemotherapy and several targeted therapies have been investigated, but currently, there are no curative therapy options [58].
Limitations: In our study, we had a large number of consecutive cases, but the majority were adrenal tumors and a small number were extra-adrenal tumors. Intra-adrenal paragangliomas usually do not have the SDHB mutation [14,59]. Some investigations indicated that extra-adrenal paragangliomas also have a higher and more heterogeneous proliferative index Ki67 than intra-adrenal [60]. Genetic testing was not performed on all patients, so the angiogenesis was not correlated with the tumor’s background completely.
5. Conclusions
In the end, it seems that it all comes down to genetics and molecular testing, which is not surprising when these tumors have so strong genetic contribution, but it’s discouraging because routine genetic testing in all health centers is not yet available. Eventually, genetic testing will complete our knowledge and clarify some doubts, but in everyday practice, it is possible to assess stratification risk with greater accuracy using accessible data like tumor size, histological parameters (included in PASS score), and additional immunohistochemical markers (proliferative index Ki67, S100, SDHB). Angiogenesis in these highly vascularized tumors has been studied and showed different conclusions regarding MVD and vascular architecture. Although angiogenesis cannot predict malignant behavior like in other solid tumors, assessment of intratumoral MVD is important, and on readily available scanned images it is quick, so furthermore may help understand tumor biology and malignant transformation, in order to recognize genes which would be targets for therapy.
Author Contributions
Conceptualization, M.M. and D.D.; methodology, M.M. and D.D.; software, S.I.; validation, D.D., T.S. and B.V.; formal analysis, M.M. and D.D.; investigation, M.M.; resources, Z.V., Z.M, and B.V.; data curation, M.M. and Z.M.; writing—original draft preparation, M.M.; writing—review and editing, D.D.; visualization, M.M.; supervision, D.D.; project administration, M.M. and D.D.; funding acquisition, D.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by Ethics Committee of University Clinical Center of Serbia (1322/VII-18 07.07.2022.).
Informed Consent Statement
The written informed consent from patients was not obtained, because of the rule of Ethical Committee of University Clinical Center of Serbia, for the retrospective studies, which analyzes the data from period preceding the beginning of the research, research can be approved in accordance with the special approvals of the director of the clinics, and with appropriate statements.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors want to thank for support of Immunohistochemical Laboratory of Institute of Pathology, for antibodies and digital scanning.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Mete, O.; Asa, S.L.; Gill, A.J.; Kimura, N.; de Krijger, R.R.; Tischler, A. Overview of the 2022 WHO Classification of Paragangliomas and Pheochromocytomas. Endocr Pathol 2022, 33, 90–114. [Google Scholar] [CrossRef]
- Lam, A.K-Y. Update on Adrenal Tumors in 2017 World Health Organisation (WHO) of Endocrine Tumours. Endocr Pathol 2017, 28, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.V.; Osamura, R.Y.; Kloppel, G.; Rosai, J. Tumors of the adrenal medulla and extra-adrenal paraganglia. In WHO classification of tumours of endocrine organs: pathology and genetics of tumours of endocrine organ, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloppel, G., Rosai, J., Eds.; IARC Press: Lyon, France, 2017; pp. 179–192. [Google Scholar]
- Fishbein, L.; Merrill, S.; Fraker, D.L.; Cohen, D.L.; Nathanson, K.L. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 2013, 20, 1444–1450. [Google Scholar] [CrossRef]
- Dahia, P.L.M. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer 2014, 14, 108–19. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.M.; Duh, Q-Y. ; Eisenhofer, G.; Gimenez-Roqueplo, A-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young Jr, W.F. Pheochromocytoma and paraganglioma: an endocrine society clinical and practice guideline. J Clin Endocrinol Metab 2014, 99, 1915–42. [Google Scholar] [CrossRef] [PubMed]
- Grogan, R.H.; Mitmaker, E.J.; Duh, Q-Y. Changing paradigms in the treatment of malignant pheochromocytoma. Cancer control 2011, 18, 104–12. [Google Scholar] [CrossRef]
- Druce, M.R.; Kaltsas, G.A.; Fraenkel, M.; Gross, D.J.; Grossman, A.B. Novel and evolving therapies in the treatment of malignant pheochromocytoma: experience with the mTOR inhibitor everolimus (RAD001). Horm Metab Res 2009, 41, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Edstrom Elder, E.; Hjelm Skog, A.-L.; Hoog, A.; Hamberger, B. The management of benign and malignant pheochromocytoma and abdominal paraganglioma. Eur J Surg Oncol 2003, 29, 278–83. [Google Scholar] [CrossRef]
- Tischler, A.S. Pheochromocytoma and extra-adrenal paraganlioma: updates. Arch Pathol Lab Med 2008, 132, 1272–84. [Google Scholar] [CrossRef]
- Ayala-Ramirez, M.; Feng, L.; Johnson, M.M.; Ejaz, S.; Habra, M.A.; Rich, T.; Busaidy, N.; Cote, G.J.; Perrier, N.; Phan, A.; Patel, S.; Waguespack, S.; Jimenez, C. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas; primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab 2011, 96, 717–25. [Google Scholar] [CrossRef]
- Hamidi, O.; Young Jr, W.F.; Iniguez-Ariza, N.M.; Kittah, N.E.; Gruber, L.; Bancos, C.; Tamhane, S.; Bancos, I. Malignant Pheochromocytoma and Paraganglioma: 272 Patients Over 55 Years. J Clin Endocrinol Metab 2017, 102, 3296–3305. [Google Scholar] [CrossRef] [PubMed]
- Jasim, S.; Jimenez, C. Metastatic pheochromocytoma and paraganglioma: Management of endocrine manifestations, surgery and ablative procedures, and systemic therapies. Best Prac Res Clin Endocrinol Metab 2020, 34, 101354. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Tischler, A.S.; de Krijger, R.R. Diagnostic tests and biomarkers for pheochromocytoma and extra-adrenal paraganglioma: from routine laboratory methods to disease stratification. Endocr Pathol 2012, 23, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.D.R. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Path 2002, 26, 551–66. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yamazaki, Y.; Pecori, A.; Tezuka, Y.; Ono, Y.; Omata, K.; Morimoto, R.; Nakamura, Y.; Satoh, F.; Sasano, H. Histopatological Analysis of Tumor Microinviroment and Angiogenesis in Pheochromocytoma. Front Endocrinol 2020, 10, 587779. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Tischler, A.S.; Lloyd, R.V.; DeLellis, R.A.; de Krijger, R.; van Nederveen, F.; Nose, V. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol 2009, 33, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Strong, V.E.; Kennedy, T.; Al-Ahmadie, H.; Tang, L.; Coleman, J.; Fong, Y.; Brennan, M.; Ghossein, R.A. Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic and cell cycle/apoptosis gene expression analysis. Surgery 2008, 143, 759–68. [Google Scholar] [CrossRef]
- Folkman, J.; Shing, Y. Angiogenesis. J Biol Chem 1992, 267, 10931–4. [Google Scholar] [CrossRef]
- Liu, Q.; Djuricin, G.; Staren, E.D.; Gattuso, P.; Gould, V.E.; Shen, J.; Saclarides, T.; Rubin, D.B.; Prinz, R.A. Tumor angiogenesis in pheochromocytomas and paragangliomas. Surgery 1996, 120, 938–42, discussion 942-3. [Google Scholar] [CrossRef]
- Ohji, H.; Sasagawa, I.; Iciyanagi, O.; Suzuki, Y.; Nakada, T. Tumour angiogenesis and Ki67 expression in phaeochromocytoma. BJU Int 2001, 87, 381–5. [Google Scholar] [CrossRef]
- Jimenez, C.; Junsheng, M.; Gonzalez, A.R.; Varghese, J.; Zhang, M.; Perrier, N.; Habra, M.A.; Graham, P.; Waguespack, S.G. TNM Staging and Overall Survival in Patients With Pheochromocytoma and Sympathetic Paraganglioma. J Clin Endocrinol Metab 2023, 108, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Skacel, M.; Skilton, B.; Pettay, J.D.; Tubbs, R.R. Tissue microarrays: a powerful tool for high-throughput analysis of clinical specimens: a review of the method with validation data. Appl Immunohistochem Mol Morphol 2002, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.R.; Rudbeck, L. The Staining Proscess. In Immunohistochemical Staining Methods, 6th ed.; Taylor, C.R., Rudbeck, L., Eds.; Dako Denmark A/S: Glostrup, Denmark, 2013; pp. 20–91. [Google Scholar]
- Schnidein, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; Tinevez, J-Y.; White, D.J.; Hartenstein, V.; Eliceiri, K.; Tomancak, P.; Cardona, A. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012, 9, 676–82. [Google Scholar]
- https://www.studocu.com/en-gb/document/the-open-university/cell-biology/cell-counts-histology-guidance-sxhl288/6880909.
- https://pdf4pro.com/view/exercise-3-biology-105-estimating-the-size-of-cells-4aac9.html. Exercise 3.
- Yao, Y.; Pan, Y.; Chen, J.; Sun, X.; Qiu, Y.; Ding, Y. Endoglin (CD105) expression in angiogenesis of primary hepatocellular carcinomas: analysis using tissue microarrays and comparisons with CD34 and VEGF. Ann Clin Lab Sci 2007, 37, 39–48. [Google Scholar] [PubMed]
- Tanigawa, N.; Lu, C.; Mitsui, T.; Miura, S. Quantitation of sinusoid-like vessels in hepatocellular carcinoma: its clinical and prognostic significance. Hepatology 1997, 26, 1216–23. [Google Scholar] [PubMed]
- Weidner, N. Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast Cancer Res Treat 1995, 36, 169–80. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Gicquel, C.; Lumbroso, J.; Tenenbaum, F.; Comoy, E.; Bosq, J.; Foncesa, E.; Ghillani, P.P.; Aubert, B.; Travagli, J.P.; Gardet, P.; Parmentier, C. Malignant pheochromocytoma: clinical, biological, histologic and therapeutic data in a series of 20 patients with distant metastases. J Endocrinol Invest 1992, 15, 631–42. [Google Scholar] [CrossRef]
- Pattarino, F.; Bouloux, P.M. The diagnosis of malignancy in phaeochromocytoma. Clin Endocrinol (Oxf) 1996, 44, 239–41. [Google Scholar] [CrossRef]
- Bialas, M.; Dyduch, G.; Dudala, J.; Bereza-Buziak, M.; Hubalewska-Dydejczyk, A.; Budzynski, A.; Okon, K. Study of microvessel density and the expression of vascular endothelial growth factors in adrenal gland pheochromocytomas. Int J Endocrinol 2014, 2014, 104129. [Google Scholar] [CrossRef]
- Favier, J.; Plouin, P-F.; Corvol, P.; Gasc, J-M. Angiogenesis and vascular architecture in pheochromocytomas: distinctive traits in malignant tumors. Am J Pathol 2002, 161, 1235–46. [Google Scholar] [CrossRef]
- Oudijk, L.; van Nederveen, F.; Badoual, C.; Tissier, F.; Tischler, A.S.; Smid, M.; Gaal, J.; Lepoutre-Lussey, C.; Gimenez-Roqueplo, A-P.; Dinjens, W.N.M.; Korpershoek, E.; de Krijger, R.; Favier, J. Vascular pattern analysis for the prediction of clinical behaviour in pheochromocytomas and paragangliomas. PLoS One 2015, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Seon, B.K.; Matsuno, F.; Haruta, Y.; Kondo, M.; Barcos, M. Long-lasting complete inhibition of human solid tumors in SCID mice by targeting endothelial cells of tumor vasculature with antihuman endoglin immunotoxin. Clin Cancer Res 1997, 3, 1031–44. [Google Scholar] [PubMed]
- Westphal, J.R.; Willems, H.W.; Schalkwijk, C.J.; Ruiter, D.J.; de Waal, R.M. Characteristics and possible function of endoglin, a TGF-beta binding protein. Behring Inst Mitt 1993, (92), 15–22. [Google Scholar]
- Saad, R.S.; Jasnosz, K.M.; Tung, M.Y.; Silverman, J.F. Endoglin (CD105) expression in endometrial carcinoma. Int J Gynecol Pathol 2003, 22, 248–53. [Google Scholar] [CrossRef] [PubMed]
- Behrem, S.; Zarkovic, K.; Eskinja, N.; Jonjic, N. Endoglin is a better marker than CD31 in evaluation of angiogenesis in glioblastoma. Croat Med J. 2005, 46, 417–22. [Google Scholar] [PubMed]
- Birdsey, G.M.; Dryden, N.H.; Amsellem, V.; Gebhardt, F.; Sahnan, K.; Haskard, D.O.; Dejana, E.; Mason, J.C.; Randi, A.M. Transcription factor Erg regulates angiogenesis and endothelial apoptosis through VE-cadherin. Blood. 2008, 111, 3498–506. [Google Scholar] [CrossRef] [PubMed]
- Favier, J.; Gimenez-Roqueplo, A-P. Pheochromocytomas: the (pseudo)-hypoxia hypothesis. Best Pract Res Clin Endocrinol Metab 2010, 24, 957–68. [Google Scholar] [CrossRef] [PubMed]
- Amorim-Pires, D.; Peixoto, J.; Lima, J. Hypoxia Pathway Mutations in Pheochromocytomas and Paragangliomas. Cytogenet Genome Res 2016, 150, 227–41. [Google Scholar] [CrossRef]
- De Wailly, P.; Oragano, L.; Rade, F.; Beaulieu, A.; Arnault, V.; Levillain, P.; Kraimps, J.L. Malignant pheochromocytoma: new malignancy criteria. Langenbecks Arch Surg 2012, 397, 239–246. [Google Scholar] [CrossRef]
- Nagura, S.; Katoh, R.; Kawaoi, A.; Kobayashi, M.; Obara, T.; Omata, K. Immunohistohemical estimation of growth activity to predict biological behavior of pheochromocytomas. Mod Pathol 1999, 12, 1107–11. [Google Scholar]
- August, C.; August, K.; Schroeder, S.; Bahn, H.; Hinze, R.; Baba, H.A.; Kersting, C.; Buerger, H. CGH and CD44/MIB-1 immunohistochemisty are helpful to distinguish metastasized from nonmetastasized sporadic pheochromocytomas. Mod Pathol 2004, 17, 1119–28. [Google Scholar] [CrossRef] [PubMed]
- Elder, E.E.; Xu, D.; Hoog, A.; Enberg, U.; Hou, M.; Pisa, P.; Gruber, A.; Larsson, C.; Backdahl, M. KI67 AND hTERT expression can aid in distinction between malignant and benign pheochromocytoma and paraganglioma. Mod Pathl 2003, 16, 246–55. [Google Scholar] [CrossRef] [PubMed]
- Tavangar, S.M.; Shojaee, A.; Tabriz, M.H.; Haghpanah, V.; Larijani, B.; Heshmat, R.; Lashkari, A.; Azimi, S. Immunohistochemical expression of Ki-67, c-erbB-2, and c-kit antigens in benign and malignant pheochromocytoma. Pathol Res Pract 2010, 206, 305–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, T-H.; Chen, Y-J.; Wu, S-F.; Gao, J.; Jiang, W-J.; Lu, Z-H.; Guan, J.; Wei, S-Z.; Luo, Y-F.; Cao, J-L.; Wan, J-W. Distinction between benign and malignant pheochromocytomas. Zhonghua Bing Li Xue Za Zhi 2004, 33, 198–202. [Google Scholar] [PubMed]
- Unger, P.; Hoffman, K.; Pertsemlidis, D.; Thung, S.; Wolfe, D.; Kaneko, M. S100 protein-positive sustentacular cells in malignant and locally aggressive adrenal pheochromocytomas. Arch Path Lab Med 1991, 115, 484–7. [Google Scholar] [PubMed]
- Schlumberg, M.; Gicquel, C.; Lumbroso, J.; Tenenbaum, F.; Comoy, E.; Bosq, J. Fonseca, E.; Ghillani, P.P.; Aubert, B.; Travagli, J.P.; Gardet, P.; Parmentier, C. Malignant pheochromocytoma: clinical, biological, histologic and therapeutic data in a series of 20 patients with distant metastases. J Endocinol Invest 1992, 15, 631–42. [Google Scholar] [CrossRef] [PubMed]
- Buffet, A.; Burnichon, N.; Favier, J.; Gimenez-Roqueplo, A-P. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab 2020, 34, 101416. [Google Scholar] [CrossRef]
- Baysal, B.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Drovdlic, C.M.; Savul, S.A.; McLeod, D.R.; Yee, H.A.; Brackmann, D.E.; Slattery 3rd, W.H; Myers, E.N.; Farrell, R.E.; Rubinsterin, W.S. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet 2002, 39, 178–83. [Google Scholar] [CrossRef]
- Buffet, A.; Venisse, A.; Nau, V.; Roncellin, I.; Boccio, V.; Le Pottier, N.; Boussion, M.; Travers, C.; Simian, C.; Burnichon, N.; Abermil, N.; Favier, J.; Jeunemaitre, X.; Gimenez-Roqueplo, A-P. A decade (2001–2010) of genetic testing for pheochromocytoma and paraganglioma. Horm Metab Res 2012, 44, 359–66. [Google Scholar] [CrossRef]
- Benn, D.E.; Robinson, B.G.; Clifton-Bligh, R.J. 15 YEARS OF PARAGANGLIOMA: Clinical manifestations of paraganglioma syndromes types 1-5. Endocr Relat Cancer 2015, 22, T91–T103. [Google Scholar] [CrossRef]
- Plouin, P.F.; Amar, L.; Dekkers, O.M.; Fassnacht, M.; Gimenez-Roqueplo, A-P.; Lenders, J.W.M.; Lussey-Lepoutre, C.; Steichen, O. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 2016, 174, G1–G10. [Google Scholar] [CrossRef]
Figure 1.
Box plot: shows the number of blood vessels per mm2 (marked with antibodies CD31, CD105, ERG, CD34) in relation to the PASS score (PASS<4 and PASS≥4).
Figure 1.
Box plot: shows the number of blood vessels per mm2 (marked with antibodies CD31, CD105, ERG, CD34) in relation to the PASS score (PASS<4 and PASS≥4).
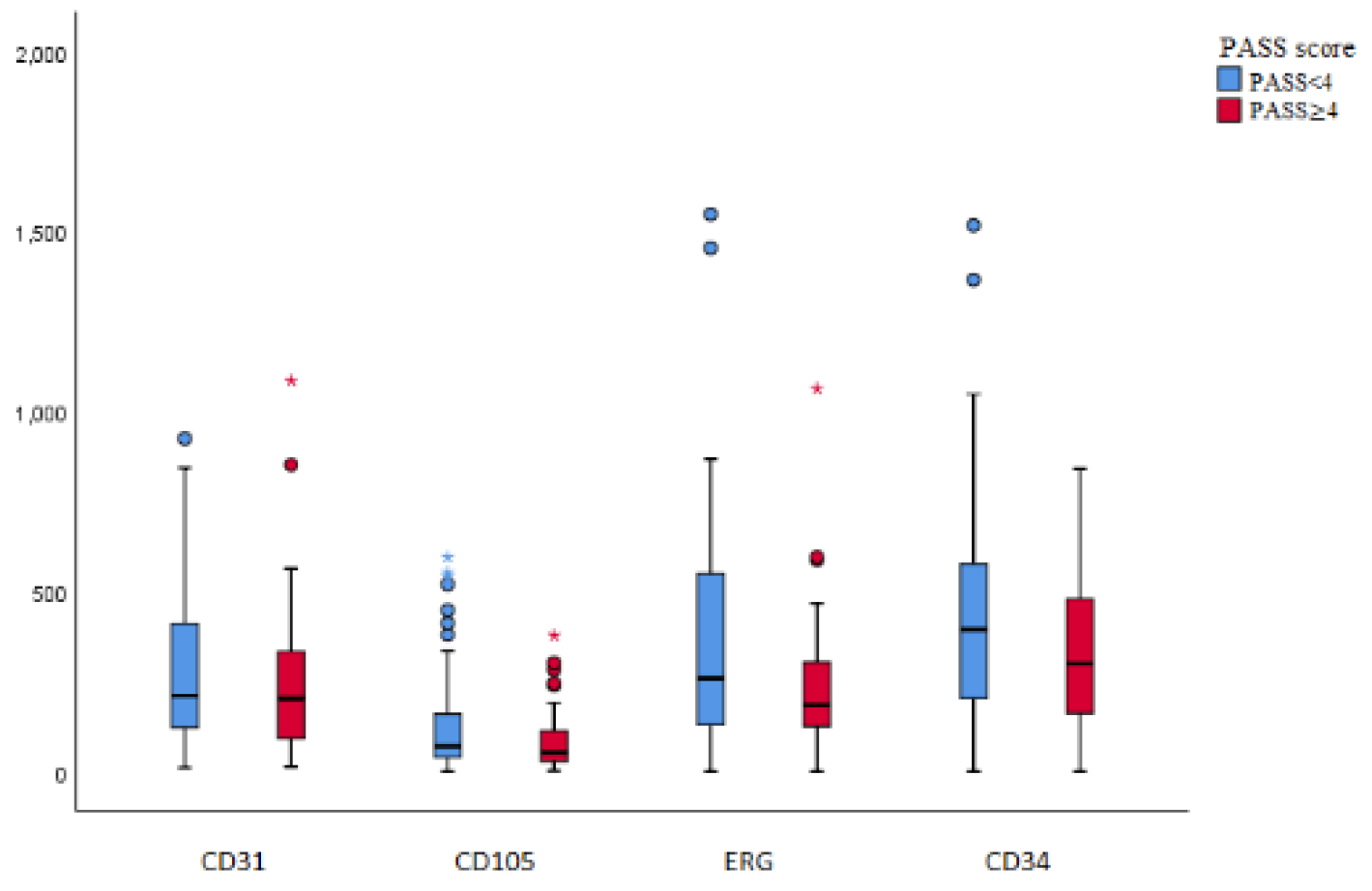
Figure 2.
Intratumoral MVD immunostaining of blood vessels, in case of PASS<4, labeled with ERG (A), CD31(C), CD 105 (E), CD 34 (G) and PASS ≥4 labeled with ERG (B), CD31(D), CD105 (F), CD34 (H). All images are at the same magnification, scale bar of 200μm.
Figure 2.
Intratumoral MVD immunostaining of blood vessels, in case of PASS<4, labeled with ERG (A), CD31(C), CD 105 (E), CD 34 (G) and PASS ≥4 labeled with ERG (B), CD31(D), CD105 (F), CD34 (H). All images are at the same magnification, scale bar of 200μm.
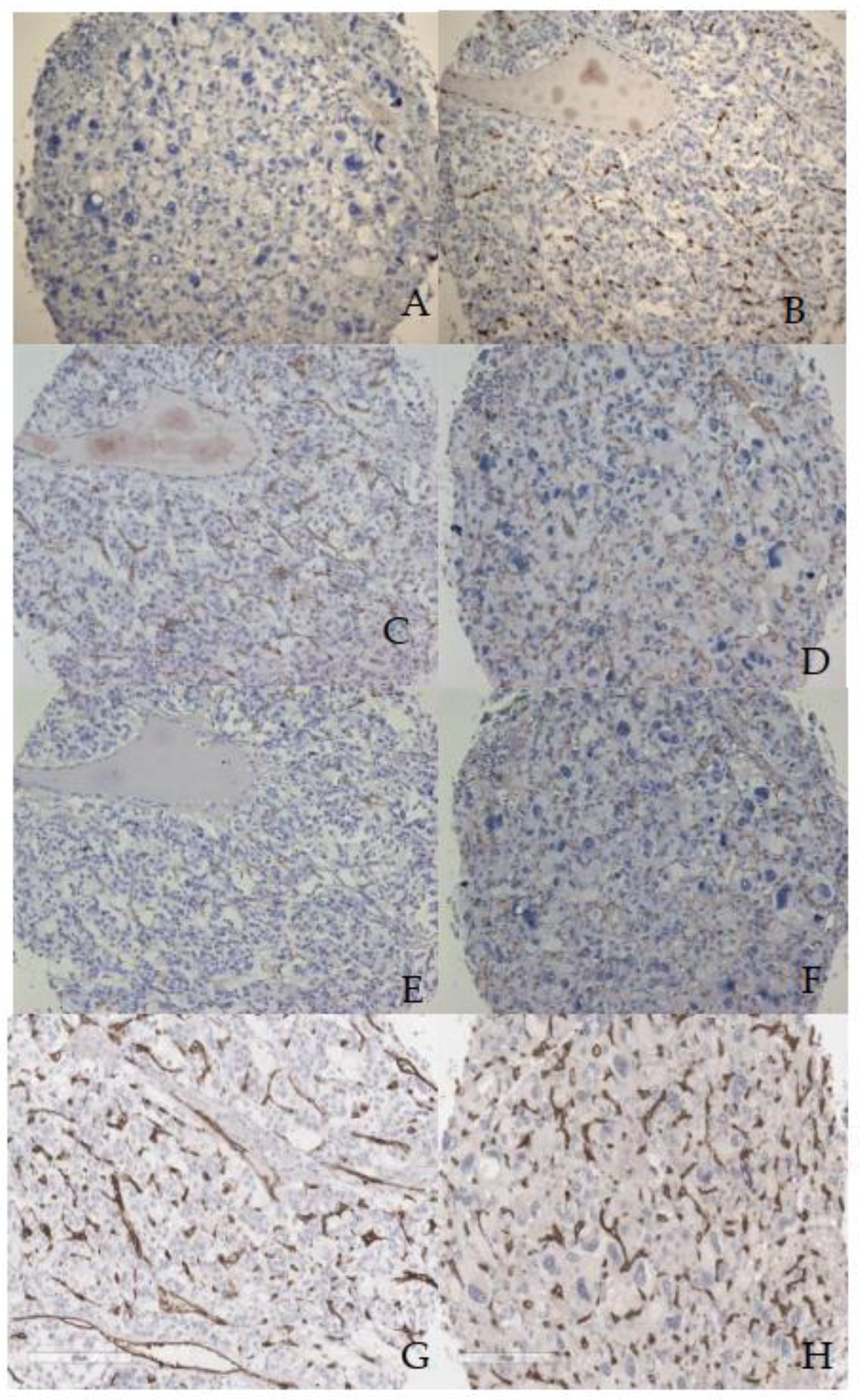
Figure 3.
ICH staining showing S100 labeled sustentacular cells in case evaluated PASS score 10 (A), PASS 6 (B), and PASS 3 (C) scale bar 90µm.
Figure 3.
ICH staining showing S100 labeled sustentacular cells in case evaluated PASS score 10 (A), PASS 6 (B), and PASS 3 (C) scale bar 90µm.
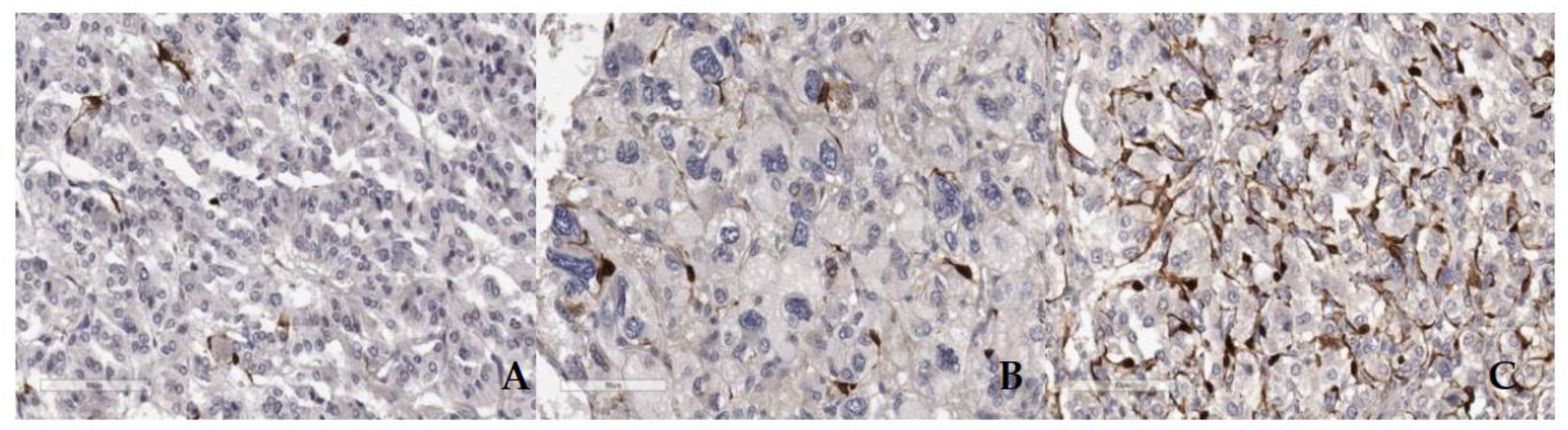
Figure 4.
Box plot S10 labeled cells in groups PASS<4 and PASS≥4.
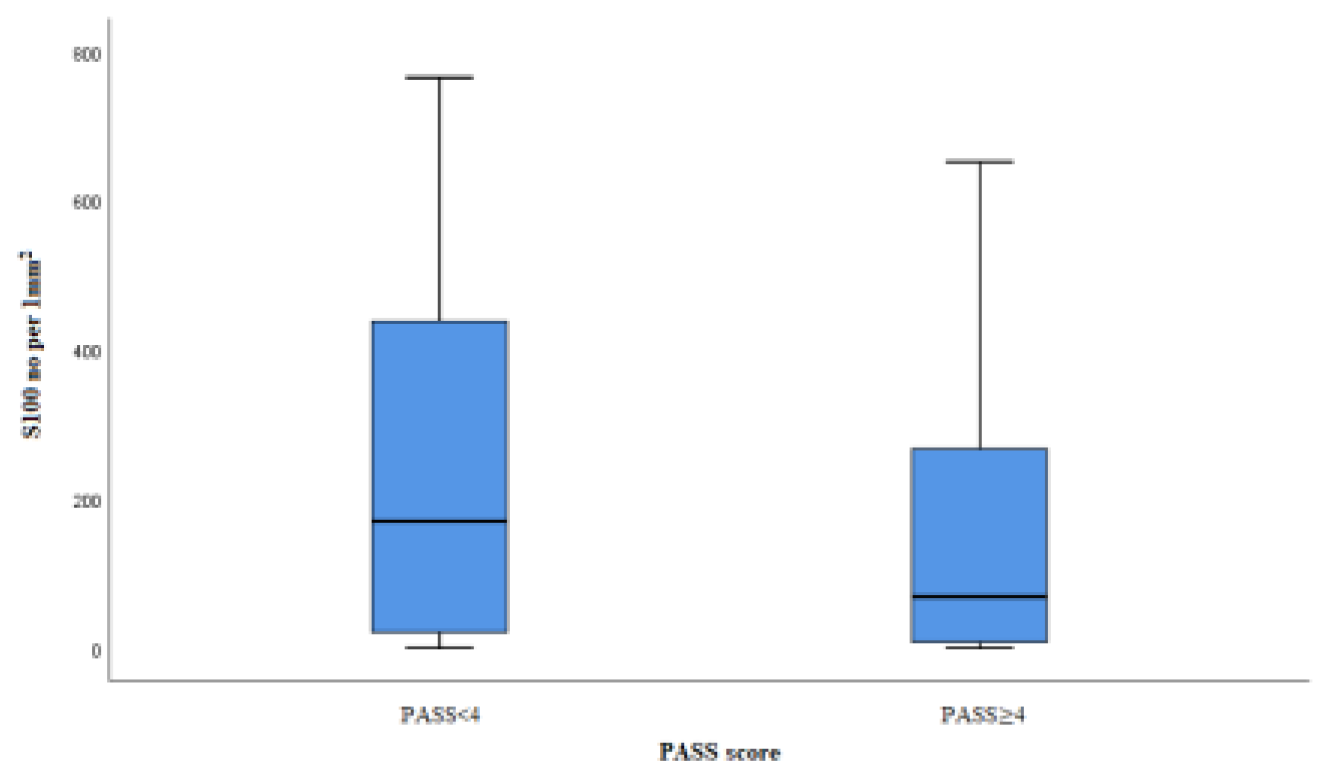
Figure 6.
IHC SDHB lost expresion in cases PASS score 1 (A), PASS score 4 (B) and PASS score 8 (C).
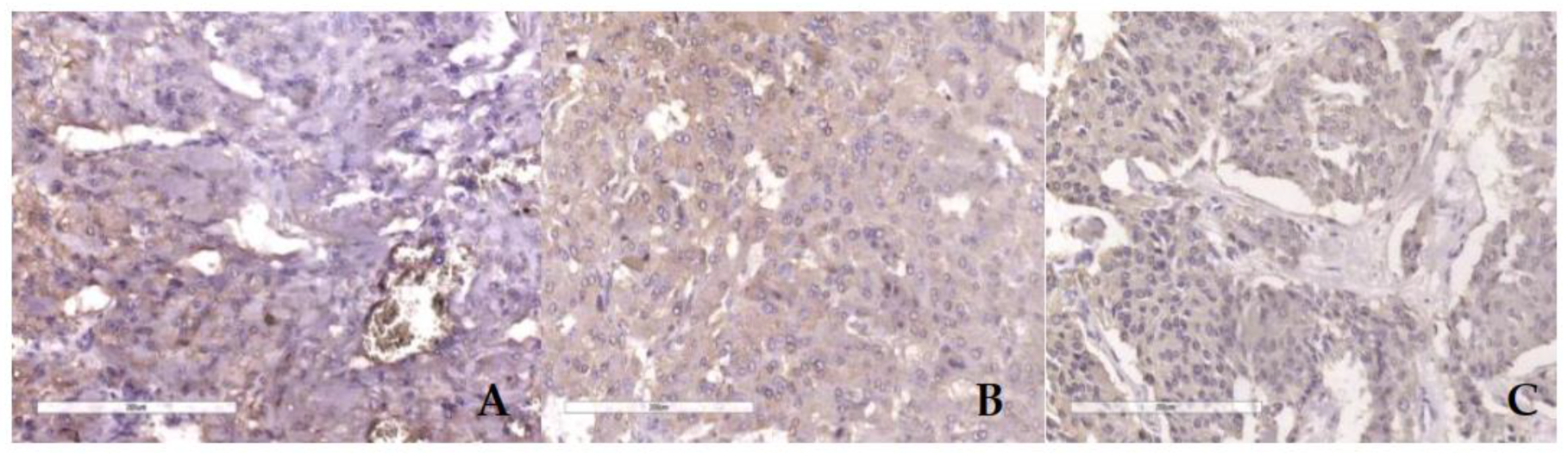
Figure 7.
ICH SDHB positivity in cases with PASS score 3 (A) and PASS score 9 (B).
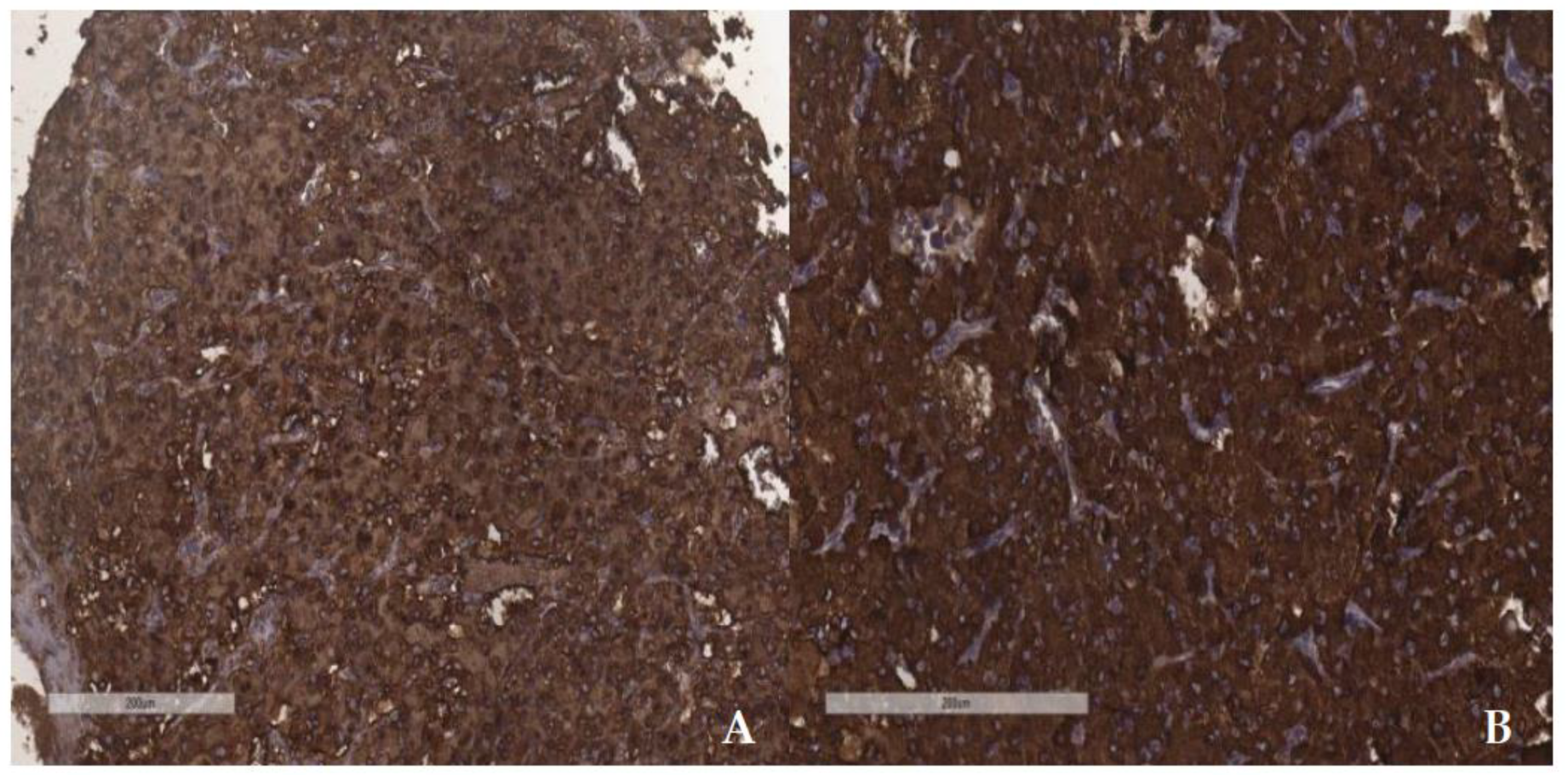

Table 1.
List of used antibodies and their manufacturers.
| Antibody | Dilution | Source |
|---|---|---|
| CD31 | 1:100 | BIO SB |
| CD34 | 1:200 | NOVOCASTRA |
| CD105 | 1:100 | Termo Fisher |
| ERG | 1:100 | ABCAM |
| SDHB | 1:50 | ABCAM |
| S-100 | 1:100 | NOVOCASTRA |
| Ki67 | 1:200 | DAKO |
Table 2.
Clinicopathological data in groups PASS<4 and PASS≥4.
| Feature | PASS<4 | PASS≥4 |
|---|---|---|
| Total number (%)a (m,f) b | 61 (53%) (17m, 44f) | 54 (47%) (23m, 31f) |
| Age at resection mean/range | 48,4 (22-78) | 48,8 (19-73) |
| Site of tumor | ||
| Intraadrenal total/percentages | 55 (47,83%) | 52 (45,22%) |
| extraadrenal total/percentages | 6 (5,22%) | 2 (1,74%) |
| Tumor weight in grams mean/range | 31,7 (3-159) | 89,7 (2-361) |
| Tumor size in mm mean/range | 44 (4-110) | 61 (20-130) |
| Catecholamines/metabolites | ||
| elevated total/percentages | 47 (40,87%) | 46 (40%) |
| normal total/percentages | 9 (7,83%) | 7 (6,08%) |
| unknown data total/percentages | 5 (4,35%) | 1 (0,87%) |
| Blood pressure/spikes | ||
| elevated total/percentages | 47 (40,87%) | 44 (38,26%) |
| normal total/percentages | 13 (11,3%) | 10 (8,7%) |
| unknown data total/percentages | 1 (0,87%) | |
| MIBI scintigraphy | ||
| increased accumulation | 24 (20,87%) | 16 (13,91%) |
| normal distribution | 8 (6,96%) | 4 (3,49%) |
| unknown data | 29 (25,22%) | 34 (29,56%) |
| Genetic testing | ||
| mutation present | 16 (13,91%) | 16 (13,91%) |
| mutation excluded | 8 (6,96%) | 8 (6,09%) |
| unknown data | 37 (32,17%) | 31 (26,96%) |
| TNM | ||
| T1 tumor < 5cmc | 37 | 13 |
| T2 tumor ≥ 5cmd | 22 | 24 |
| T3 tumor of any size with invasion | 2 | 16 |
apercentages; bmale; f, female, cwithout invasion into surrounding tissues, d without invasion or sympathetic PG of any size.
Table 3.
Results of genetic testing, age of patients and PASS score values.
| Syndrome | Number (m,f)x | Age mean/range | PASS mean/range |
|---|---|---|---|
| VHL | 10 (6m, 4f) | 37,7 (19-55) | 4,2 (1-12) |
| MEN2a | 17 (6m; 11f) | 42,6 (22-64) | 3,1 (1-8) |
| NF1 | 4 (1m; 3f) | 44,3 (34-53) | 6,3 (4-9) |
| SDHB | 1(1f) | 51 | 9 |
| Without mutation | 15 (3m; 11f) | 45 (29-64) | 5,1 (1-10) |
| Not available | 68 (24m; 44f) | 52,5 (23-78) | 3,7 (1-10) |
x m, male; f, female.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



