Submitted:
10 February 2024
Posted:
12 February 2024
You are already at the latest version
Abstract
N-methyl-D-aspartate receptors (NMDARs) are the main class of ionotropic receptor for the ex-citatory neurotransmitter glutamate. It plays a crucial role in the permeability of Ca2+ ions and excitatory neurotransmission in the brain. Being heteromeric receptors, they are composed of sev-eral subunits, including two obligatory GluN1 subunits (eight splice variants) and regulatory GluN2 (GluN2A~D) and/or GluN3 (GluN3A~B) subunits. Widely distributed in the brain, they regulate other neurotransmission systems and are therefore involved in essential functions such as synaptic transmission, learning and memory, plasticity, excitotoxicity. The present review will discuss the physiopathological impacts of NMDAR and particularly GluN2A and GluN2B subu-nits in cognitive processes and neurodegenerative diseases (Alzheimer's disease, Huntington's disease, Parkinson's disease). The pharmacology of different NMDAR antagonists and their po-tentialities will be presented. In particular, a focus will be given on fluoroethylnormemantine (FENM), an investigational drug with very promising developments as a tomography radiotracer for NMDARs, an anxiolytic in post-traumatic stress disorder and a neuroprotective agent in Alzheimer's disease.
Keywords:
N-methyl-D-aspartate receptor
; Alzheimer's disease
; post-traumatic stress disorder
; Fluoroethylnormemantine (FENM)
1. Introduction
Neurotransmission within the central nervous system (CNS) is essentially based on two main neuronal systems that coordinate their excitatory and inhibitory inputs to establish and maintain a constant excitation/inhibition ratio. Glutamate is the most important excitatory neurotransmitter from the CNS, with 50-70% utilization by synapses [1]. Glutamate binds to two types of receptors, namely metabotropic glutamate receptors (mGluRs) which belongs to the G protein-coupled receptor (RCPG) superfamily and ionotropic glutamate receptors, such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), N-methyl-D-aspartate receptor (NMDAR), and kainate receptor. These receptors are present in the entire CNS, with greater density at the cortical and hippocampal level [2,3] and are involved in different neurodevelopmental processes, cognitive functions, learning, memory and synaptic plasticity [4,5,6]. All these receptors are localized in the synaptic and/or extra-synaptic membranes [7], depending on the neuronal maturation stage and receptor function, and with complex inter-relations. The extra-synaptic NMDARs in immature hippocampal neurons represent 75% of NMDARs and are still expressed during adulthood, while, during development, there is an increase in synaptic NMDARs [8,9].
In the CNS, NMDARs are distributed ubiquitously and play a critical role in the synaptic plasticity and learning and memory processes. The glutamate binding leads to an increase in intracellular calcium (Ca2+) inside the neurons which acts as a second messenger to modify synapse necessary for neurotransmission and brain plasticity [10]. Current flow through NMDAR channels is largely blocked by external Mg2+ ions at resting membrane potentials, but can be relieved by depolarization [11]. The receptors are modulated also by different molecules including the co-agonists glycine and D-serine, which bind to a specific site on NMDAR [12]. It is now accepted that the induction of long-term potentiation (LTP) and long-term depression (LTD), in the CAl layer of the hippocampus, is highly dependent on NMDAR activation, permeable to Ca2+ at the postsynaptic synapse [13,14]. NMDAR is a voltage-dependent channel and at resting potential, it is blocked by Mg2+. Astrocytes reuptake about 90% of the glutamate released in the synaptic cleft, through sodium dependent glutamate transporters, named excitatory amino acid transporters (EAATs), and approximately 20% of the synaptic glutamate released fixed on the post-synaptic glutamate receptors, while the rest reached extra-synaptic receptors [15]. Through these transporters, glutamate is metabolized and recycled by glutamine synthetase, which generates glutamine, and redistributed to the pre-synapse to recycle as a neurotransmitter.
In Alzheimer's disease (AD), Huntington's disease (HD), Parkinson's disease (PD), post-traumatic-stress-disorder (PTSD), epilepsy or schizophrenia, NMDARs pathological activation is a main mediator of neuronal injury or dysfunction [16,17,18,19]. In neurodegenerative conditions, an excess of glutamate release resulting from synaptic alterations massively increases intracellular Ca2+ influx. The normal blockade by Mg2+ of the ionophore is removed and abnormally enhanced NMDAR activity. Since the calcium permeability is relatively high, extra-synaptic NMDA receptors are preferentially targeted in these conditions and lead to neuronal excitotoxicity [20].
In the present review, we will describe why and how NMDARs are presently still considered as a potential target for therapeutical treatments, particularly in AD, HD, PD, or PTSD, discussing the therapeutic use of memantine and ketamine as treatments. Finally, we propose a novel drug, fluoroethylnormemantine (FENM) that shows several benefits over memantine in AD models and over ketamine in PTSD models. FENM particularly illustrated that NMDAR dysfunction could still represent an essential target for innovative drugs in neurodegenerative diseases.
2. Physiology of NMDA receptors
2.1. Structure, composition, localization
The NMDAR is a transmembrane protein supercomplex with four domains. The extracellular amino-terminal domain (ATD) the extracellular ligand binding domain (LBD), the transmembrane domain (TMD), and the intracellular carboxyl C-terminal domain (CTD) [21]. Structurally, functional NMDARs are heterotetramers (≈ 1.5 KDa) composed of two glycine or D-serine binding GluN1 subunits, two glutamate binding GluN2 subunits, e.g. A, B, C or D, and a glycine binding GluN3 subunit (Figure 1) [22,23]. The difference between each subunit, it’s the length of the GluN CTD region [24]. Non-competitive binding sites are localized within the ion channel pore. First, Mg2+ exerts a voltage-dependent blockade of the ion flux, dependent on an asparagine residue in second transmembrane segment. Depolarization results in Mg2+ ion removal and influx of Ca2+. Second, phencycline (PCP) and PCP-like drugs bind within the pore and exert an uncompetitive antagonism of NMDAR. The PCP site is most accessible when the receptor is in an activated state [25,26,27]. Other antagonists binding to this PCP site include derivatives such as thienylcyclidine or ketamine, by also chemically unrelated molecules like dizocilpine or memantine. Third, Zn2+ inhibits NMDARs current by a high affinity binding site in GluN2A [28], inducing a reduction of channel open probability. The NMDAR subunits are encoded by seven genes. The GluN1 subunit is encoded by a unique gene, GRIN1, but has eight distinct isoforms (a-h, with different splice variants of one single gene). The two GluN1 association with two out of four GRIN2 genes encodes GluN2 subunits are specific on spatiotemporal expression in the brain [21]. And the last is GluN3A/B is encoded by two GRIN3 genes. Compelling evidence indicates that diheteromers and triheteteromers coexist within a single cell or even at a single synapse, adding to the functional diversity of the postsynaptic response [29]. In the adult brain, there is a predominant expression of diheteromeric GluN1/GluN2A or GluN1/GluN2B receptors in hippocampus and cortex. Moreover, triheteromeric GluN1/GluN2A/GluN2B receptors represent, in the hippocampus and cortex, between 15% and 50% of the total receptor population [30,31,32,33]. The distribution is also localization-specific, and there is a predominance of GluN2A subunits in NMDAR at the post-synapse vs. GluN2B subunits at the extra-synaptic membrane [23]. However, NMDARs are mobile on the plasma membrane and GluN2B containing receptors can move through lateral diffusion between synaptic and extra-synaptic spaces.
In the cortex and the hippocampus, GluN2A and GluN2B are the most expressed subunits. Although there is a 70% identical sequence of GluN2A and GluN2B, these subunits play different roles in the physiological or pathological processes [34,35]. GluN3A and GluN3B show a more heterogeneous expression, inducing smaller NMDA currents with reduced calcium permeability in the post-synapse. These subunits therefore act in a dominant-negative manner to block receptor activity. GluN3A is localized in dendritic spines of glutamatergic neurons and influence synaptic stability, receptor assembly and the functional activity in the neuron [36,37]. In particular, GluN1/GluN3A NMDARs are functionally expressed in native neurons, at least in the juvenile brain, and are mediated the excitatory effect of glycine [38]. NMDARs with GluN2C/2D subunits display lower sensitivity to Mg2+ ions and slower deactivation kinetics compared with GluN2A/2B containing NMDARs. The expression of GluN2C/2D has been reported in adult cerebral cortices and more precisely GluN2D subunits were reported to play roles in synaptic transmission of hippocampal neurons [39].
The cellular distribution of NMDARs is also of importance and, in the physiological concept of “tripartite synapse” (Figure 2), based on direct communication between neurons, pre-synaptic terminal, and postsynaptic spine, and peri-synaptic astrocytes, NMDARs are expressed in all cell types with specific roles and impact on the synapse efficiency [40]. NMDARs were also described in oligodendrocytes, which play a major regulatory role on glucose transport and axonal metabolism [41,42]. The existence of functional glial NMDARs shows specificities, notably with an activation at negative membrane potential and differ from most of neuronal NMDARs not only by a weaker Mg2+ block but also by lower calcium permeability. Transcripts for all seven NMDAR subunits have been found in cultured human and rat astrocytes. The permeability of calcium in cortical astrocytes is about 3-4 times lower than in neurons [43]. Astrocytic NMDARs also play an active role in storage, processing of information and neuronal plasticity [44] and astrocytic activation in-creased calcium release pair to post-synaptic depolarization induced LTP [45,46,47]. Moreover, microglia express functional NMDARs in the murine and human CNS system. A microglia treatment with NMDAR antagonist prevented NMDA-induced microglial proliferation and reduced morphological activation and the release of pro-inflammatory cytokines [48].
2.2. NMDARs in the glutamatergic synapse
Glutamate, synthetized from glutamine and sequestered within synaptic vesicles, is released from the pre-synapse into the synaptic cleft and binds to NMDAR in the post-synapse (Figure 1). Only 10% of glutamate present in the synaptic cleft (~1.1 mM) reach glutamate receptors within approximately 1.2 msec. 90% is reuptake by the EAAT-1 transporters in astrocytes and transform back into glutamine by glutamine synthase. Glutamine can be released into the synaptic space by the sodium-coupled neutral amino acid transporter (SNAT3/5). SNAT1/2 transporters on pre-synaptic neurons uptake glutamine and predominantly regulate neuronal glutamine uptake [49,50]. In response to the glutamate synaptic release, NMDARs are activated over hundreds of msec, and activation is maintained long after all glutamate has been removed from the synaptic cleft. In response to glutamate release, AMPARs induced a depolarization in the post-synapse and allowed Na+, K+ and mainly Ca2+ flows through the NMDAR pore. At negative membrane potentials, or near to the resting membrane potential, Mg2+ although reach the NMDAR pore and prevent ion fluxes, but at the positive membrane potentials, Mg2+ is released with the depolarization of NMDAR, allowing an importantly permeability for all ions with a large outward current. Activation of NMDARs lasts thereafter much longer than AMPARs closing rapidly within a few msec. Activation of synaptic NMDARs and large increases in [Ca2+]i are required for the induction of LTP, whereas internalization of synaptic NMDARs, activation of extrasynaptic NMDARs and lower increases in [Ca2+]i are necessary for the induction of LTD. LTP induction promotes recruitment of AMPARs and growth of dendritic spines, whereas LTD induces spine shrinkage and synaptic loss.
When penetrating the post-synaptic cell, Ca2+ binds to calmodulin and activates calcium moduling kinase II (CaMKII) which leads to its autophosphorylation at Thr286 [51]. CaMKII is a highly abundant kinase in postsynaptic density, which phosphorylates AMPAR in GluA1 subunit. This phenomenon allows to increase the conductance of AMPAR which bind to postsynaptic density protein 95 (PSD-95), a pivotal scaffolding protein in excitatory neurons also bound to NMDAR and leads to the trans-location of internalized AMPAR to the post-synapse [52,53]. PSD-95 regulates NMDAR activity and in turn neuronal maturation, synapse stabilization, clustering of postsynapse and plasticity [54,55,56]. At the same time, the increase in intracellular Ca2+ activates other kinases such as Ras-Raf, which activates mitogen-activated protein kinases (MAPK family: Map3K, Map2K) that consequently activate extracellular signal-regulated protein kinase 1/2 (ERK1/2). The phosphorylation of ERK1/2 in the nucleus, generates the phosphorylation of cAMP response element binding protein (CREB) principal at Ser133, allowing gene transcription. This pathway is predominantly involved in antioxidant defense, neuroprotection, memory processes, and hippocampal LTP. However, intracellular Ca2+ also activates other second messenger pathways such as adenylate cyclase or guanylate cyclase, leading to increases of 3’,5’-adenosine monophosphate and/or 3’,5’-guanosine monophosphate (cAMP and/or cGMP). Induction of cAMP and/or cGMP activated protein kinase A (PKA), PKG or PKB (AKT). Activations of PKA or AKT, by phosphorylation on Thr197 or Thr308, respectively, also contribute to CREB phosphorylation on Ser133. CREB phosphorylation consequently promotes cellular plasticity by regulating the expression of mRNA levels of CREB targets genes such as c-fos or Nr4a1 different proteins, like tissue-type plasminogen activator (tPA) or brain-derived neurotrophic factor (BDNF). These proteins play indeed a major role in LTP, memory processes and anxiety-related behaviors. tPA, a serine protein, facilitates the conversion of pro-BDNF to mature BDNF a major regulator of brain plasticity [57,58].
Signal diminution or weakening of network connection is also important in the brain. To ensure circuit remodeling, encoding or even erasure of memory, LTD processes make it possible to maintain synaptic efficiency. If in the pre-synapse in the brain, the activity of glutamate released is weak, whereas there is a modest depolarization in the AMPAR and NMDAR post-synapse. The consequence is that there is a weak calcium influx through the NMDAR and activation of PP2B or PP1 phosphatases, and mitochondrial caspase-3. ultimately, it could lead to apoptosis by a mechanism that involves in the dephosphorylation of AMPAR. This phenomenon leads to endocytosis of AMPAR, a degradation by lysosome and long-term depression [59,60,61]. Electrophysiology, LTD could be induced using, for example, a repetition of activation of the presynaptic synapse at low frequencies, without post-synapse activation. In physiological conditions, with neurons at their resting membrane potential and sub-maximal Mg2+ block of NMDAR, calcium influx could induce in response to low frequency synaptic stimulation [61,62]. In rodent models, a stimulation at 1 Hz for 15 min is typically used to induce LTD acutely in hippocampal slices [63].
2.3. Regulation of NMDAR activity
Data suggests that NMDARs can be internalized quickly after physiological triggers [64,65] or that a switch in the NMDAR subunit com-position can happen during early development of neurons [22,66,67]. For instance, in the cortex GluN2B are more expressed than GluN2A in early postnatal brain and there is a shift during the development, and therefore a progressive increase in GluN2A/ GluN2B ratio. This switch could be driven by sensory experience during the brain development [68] and is related to an increase in GluN2A mRNA levels, which are driven by an activation of NMDAR.
In basal conditions, AMPARs (GluA1/2) and NMDARs (GluN2A/2B) are expressed at the post-synapse with the scaffold PSD-95. An important regulation is achieved by the trafficking of NMDARs and AMPARs in synapses during LTP. In the presence of intra-cellular Ca2+, AMPARs are immobilized in post-synapse in a CaMKII-dependent manner and anchored by PSD-95. For NMDARs, GluN2A is tightly anchored at the synapse as it allows an entry of Ca2+ in the developing hippocampus [69], allowing activation of postsynaptic kinase cascades while GluN2B is more diffusible in immature neuron, allowing to diffuse and/or redistribute more intracellular actors such as CaMKII or casein kinase 2 [70,71,72]. Ca2+ increase allows the induction of LTP and concentration of AMPARs in post-synapse. This phenomenon is very important during the initial phase of synaptic potentiation and for the recycling by exocytosis of receptors to the synapse. Moreover, AMPARs diffuse lat-erally in the cell surface when it is not bound to PSD-95. The phosphorylation of the γ2 TARP of PSD-95 auxiliary subunit can separate in C-terminal PSD-95 PDZ domains and allows the diffuse on cell membrane [73,74,75,76]. Some studies highlighted that AMPARs trafficking did not impact NMDAR-mediated LTP, but the reverse is true. It suggested that the expression of NMDAR at the post-synapse could be due to the exocytosis of NMDARs, induced by the phosphorylation of SAP97 by CaMKII at Ser39 in the endoplasmic retic-ulum [53,77,78,79]. Thus, this complex drives GluN2A to the post-synapse compartment in the developing hippocampus.
The regulation of NMDARs is also directed by the phosphorylation of its subunits. For example, it was shown that the GluN1 subunit is phosphorylated by PKC on two residues, Ser890 and Ser896. The Ser890 phosphorylation disrupted the clustering of the GluN1 subunit while the Ser896 phosphorylation did not affect clustering. Interestingly, the phosphorylation of Ser896 by PKA appeared necessary to increase NMDAR surface expression at the postsynapse [80,81]. The regulation of the intracellular trafficking of GluN1 is also directed by Ser896 and Ser897 hyperphosphorylation in the endoplasmic reticulum and Golgi apparatus [82]. Among other NMDAR subunits, GluN2A can be phosphorylated on 3 tyrosines, Tyr1325, Tyr1292, and Try1387, resulting in a potentiation of NMDAR currents in the synapse [83]. The phosphorylation of Ser1048 of GluN2A regulated the surface expression of GluN1/GluN2A and the current of NMDAR. It was shown that the dual specificity tyrosine phosphorylation-regulated kinase 1 (DYRK1A)-dependent phosphorylation of GluN2A at Ser1048 blocked GuN1/GluN2A internalization that allowed to express more GluN1/GluN2A receptors at the surface. This phosphorylation resulted in potentiation of NMDAR currents [84]. For GluN2B, the phosphorylation of Ser1303 affected the synapse distribution and activation [85]. GluN2B bears also 3 tyrosines as phosphorylation sites: Tyr1252, Tyr1326, and Tyr1472. These residues are phosphorylated by Fyn, a kinase expressed in the post-synapse. The phosphorylation of Tyr1472 is notably controlled by striatal enriched tyrosine phosphatase, which belongs to a family of protein tyrosine phosphatases in glutamatergic synapse. The action of these phosphatases is to increase endocytosis of NMDAR at the post-synapse. Conversely, the phosphorylation of Tyr1472 stabilizes NMDAR at the post-synapse while phosphorylation of Ser1480 induced a destabilization of NMDAR and involved a decrease of NMDAR expression [86,87]. Numerous studies showed that the phosphorylation of GluN2A or GluN2B subunit reflected NMDAR activity [88,89].
2.4. NMDARs modulators
NMDAR activity can be modulated by small molecules acting through different mechanisms (Table 1). Among competitive NMDAR agonists, the highly selective prototype drug is D-AP5. D-AP5 inhibits excitatory response and blocks plasticity (LTP) in GluN2A subunit-containing NMDAR in rodents, thus having an impact on learning at the behavioral level [92]. Phencyclidine is a dissociative anesthetic with strong addictive properties. PCP induces psychotic and dissociative schizophrenia-like symptoms resulting from the impairment of NMDAR neurotransmission in vivo [93,94]. Its derivative, ketamine also acts as an uncompetitive NMDAR antagonist and is still used as an anesthesic. Ketamine applied in post-synapse induces an inhibition of excitatory pyramidal neuron in extra-synaptic GluN2B subunit. When applied in pre-synapse, the drug induced an inhibition of GluN2D subunit in interneurons and provoked a disinhibition of glutamate release in post-synapse. At the same time, it induced an up-regulation of hippocampal AMPARs (GluA1/GluA2). This phenomenon has a consequence on plasticity and sustains its rapid antidepressant efficacy [95,96]. Among other uncompetitive antagonists acting at the PCP site, dizocilpine (or (+)-MK-801 maleate) is a nanomolar affinity anticonvulsant but amnestic and ataxic drug [97].
Ifenprodil is an uncompetitive antagonist binding specifically on GluN2B (140x preference for GluN2B vs. GluN2A). It was shown that 150 nM of ifenprodil induced 75% inhibition of GluN2B receptor currents in human embryonic kidney (HEK-293) cells [114]. Another blocker on GluN2B subunit receptor, memantine, it was shown that memantine improves cognitive functions and enhance be-havioral disturbance alone as monotherapy or combination with donepezil to moderate at severe form of AD [115]. Amantadine, a demethylated analogue of memantine, was initially assumed to result from effects on dopaminergic systems, before later discovered that it blocks the NMDAR ion channel. The remarkably fast unblocking kinetics of amantadine in comparison of memantine allow it to unblock during the brief depolarization of an action potential at least partly, a characteristic that may decrease deleterious clinical effects [108]. Dextromethorphan is a non-competitive NMAR antagonist which has properties similar to ketamine and phencyclidine. This NMDA receptor antagonism is believed to result in a decreased reuptake of catecholamines. Dextromethorphan also inhibits the reuptake of serotonin [116]. These properties make dextromethorphan have a high abuse and misuse potential. Although the role of dextromethorphan as an NMDAR antagonist appears attractive, its clinical use in the treatment of pain in cancer patients led to controversial results [112,113]. Riluzole has neuroprotective, anticonvulsant, anxiolytic and anesthetic properties. These actions are dominated by its effects on glutamate transmission which are predominately mediated by NMDAR. It decreased glutamatergic transmission probably via inhibition of PKC that regulated presynaptic NMDAR having a facilitatory effect on glutamate release [100]. The anti-cataleptic potential of the drug is the most important finding and is implicate beneficial effects in the treatment of muscle rigidity [117]. Finally, antagonists of the GluN2C and GluN2D subunits also exist, making it possible to determine their localization in the brain, particularly in astrocytes but also in neurons [38].
3. The impact of NMDARs in neurodegenerative diseases
3.1. Alzheimer's disease
According to World Health Organization, there are currently more than 55 million peoples with dementia in worldwide. Every year, 10 million of new cases are detected. This dementia result from different injuries and pathology in the brain. More 60-70% of the total cases are people who were affected by AD. It is an aging-related neurodegenerative disorder characterized by slow deterioration of cognitive functions, such as autonomy, declarative memory, and recognition memory. AD is a progressive disorder, asymptomatic in its early stage but with symptoms evolving from mild cognitive impairment to severe dementia. Histopathologically, AD is characterized by extracellular accumulation of aggregated amyloid-β (Aβ) proteins in the hippocampus and cortex and intraneuronal fibrillary tangles composed of hyperphosphorylated tau protein. Brain neuroinflammation is also massive and typified by a reactive gliosis (microgliosis and astrogliosis), oxidative stress, loss of synapses and neurons in layers III/IV of the neocortex, hippocampus, and cortex [118,119,120,121]. Pathologically elevated oligomeric Aβ may indirectly cause a partial block of NMDARs and shift the activation of NMDAR-dependent signaling cascades toward pathways involved in the induction of LTD and synaptic loss. The consequence is a progressive cognitive decline, due to a deficient cholinergic transmission and excitotoxicity in glutamatergic synapses. Indeed, in the AD brain and human cortical neurons, excitatory synapses containing the GluN2B subunit of the NMDARs appear to be the main sites of Aβ oligomers accumulation inducing a high level of glutamate in the synaptic cleft (Figure 2). Moreover, oligomeric Aβ block glutamate recapture by the EAATs in astrocytes and activate mGluR in the post-synapse [122], resulting in oxidative stress and apoptosis. The mGluR over-activation led to AMPARs internalization, desensitization of NMDARs and an en-docytosis of GluN2B in post-synapse resulting in LTP alteration and increased LTD, and, finally, more neuroinflammation [123,124,125]. In the extra-synapse region, the binding of glutamate to ionotropic glutamatergic receptors, with a GluN2B subunit, generates a massive extra-synaptic Ca2+ entry and activates deleterious signaling pathways involving MAPK, GSK-3β, or JNK. Activations of these pathways result in an increase in apoptosis and hyperphosphorylation of the tau protein [21,126]. Moreover, Aβ deposits increase the Src kinase Fyn, which phosphorylates GluN2B in Try1472. This GluN2B phosphorylation increases the subunit association with its scaffold PSD-95 and causes a disruption of synaptic plasticity and LTP in the brain leading to death of glutamatergic neuron [86,127]. The massive entry of Ca2+ also concours to inhibit signaling pathways beneficial to LTP, involving CAMKII, calcineurin, pCREB, and ERK, to reduce cellular survival factors, including BNDF and CREB, contributing to LTP/LTD alterations [128]. The activation of extra-synaptic NMDARs leads to an increase in the production of Aβ [129]. In post-mortem AD human brain, the levels of GluN1, GluN2A, and GluN2B were reported to be de-creased in the hippocampus and entorhinal cortex [130,131,132,133,134]. GluN2C or GluN2D mRNA level did not change in AD patient brain [133]. Clinical trials showed that a significant efficacy for memantine over placebo in moderate to severe forms of AD with an improvement of cognitive functions and activities of daily living [135,136]. Memantine is well tolerated by the patient but shows side effects like dizziness, headache, or constipations [137]. The drug was approved by FDA in 2003.
3.2. Huntington's disease
In the world, the prevalence of Huntington’s disease (HD) is approximately 5 cases per 100,000 individuals [138]. HD is an autosomal dominant neurodegenerative disorder characterized by the presence of motor symptoms due to the brain atrophy, degeneration of striatal neurons (caudate nucleus and putamen) and cerebral cortex with specific loss of efferent medium spiny neurons [139]. Patients are affected around 35-50 years of age and the symptoms include movement disorders, dementia, cognitive impairment and behavioral or psychiatric manifestations [140]. HD is caused by a pathological expansion of cytosine-adenine-guanine (CAG) repeats, between 10 to 35, in the exon 1 of the huntingtin gene, on chromosome 4p16.3. This gene encodes the huntingtin (Htt) protein that shows, in HD, an elongation of polyQ, gener-ating mutant huntingtin (mHtt) [141]. There is a strong correlation between length of the polyQ repeats and the age of the disease onset [142]. Htt protein is necessary for the formation of cortical and striatal excitatory synapses, embryonic shaping of the nervous system, protein trafficking, post-synaptic signaling, vesicle transport, transcription factor regulation and regulation of cell death [143,144]. Htt is localized in the cytoplasm and associated with vesicle membranes by the scaffold PSD95 [145]. The mHtt aggregates in the brain in the nucleus and cytoplasm and this accumulation results in alterations of gene transcription, neurotransmitter metabolism, and alterations of BDNF and its TrkB receptor expressions and distributions. Post-mortem studies showed a loss of striatal NMDARs in early symptomatic and pre-symptomatic stages of the disease [146,147]. In the R6/2 transgenic mouse model of HD, the expression of GluN2A and GluN2B, but not GluN1, decreased in the striatal region [148,149]. In YAC128 mice, no change in NR1, NR2A, or NR2B in the striatum of mice with symptoms was reported but a decrease in phosphorylation of NR1 at Ser897, previously reported to decrease NMDAR current [150].
The pathological context resulted in an imbalance between glutamate and dopamine in the striatum with an increase in dopamine in the post-synapse and an alteration of glutamate reuptake by the glial glutamate transporter in corticostriatal synapses. GLT mRNA was reduced in rodent models in HD in relation with motor dysfunction [151]. Glutamatergic alterations therefore included: (i) an accumulation of glutamate in the synaptic cleft, (ii) a decrease of post-synaptic NMDARs, and (iii) an increase of GluN2B extra-synaptic expression in striatal spiny neurons that could be responsible for cell death when GluN2A is decreased [152,153]. Glutamatergic NMDAR neurotransmission promotes the expression of an-ti-apoptotic factor Bcl-2, antioxidant, and of the pro-survival trophic factor BDNF. In HD condition, BDNF release and TrkB activation are decreased in the post-synapse, together with increases in pro-apoptotic factor Bax, pro-death genes such as FOXO or FBS, dysregulation of mitochondrial calcium and CREB-inactivating dephosphorylation signals [154,155]. mHtt indeed contributed to suppress CREB gene by sequestering its coactivator CBP [156]. Current therapy developments focus on gene therapies and antisense oligos. Alternatively, symptomatic treatments rely on neuroleptics, dopaminergic inhibitors, such as tetrabenazine, and antidepressants adapted to reduce motor and psychiatric signs, and sleep disorders [157].
3.3. Parkinson's disease
Parkinson’s disease (PD) is the second most frequent neurodegenerative disorder after AD and represents a large health burden to society. Approximately 1% of the population over 60 years of age is affected. PD is a progressive neurodegenerative disorder resulting from the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc) and the accumulation of intracytoplasmic inclusions, known as Lewy bodies [158]. A series of alterations in the circuitry of basal ganglia nuclei leads to severe motor control impairments such as tremor, muscular rigidity, and bradykinesia [159]. Pharmacological approaches to PD predominantly target the dopaminergic system, with dopamine replacement by its precursor, L-3,4-dihydroxyphenylalanine (L-Dopa) [160]. However, a prolonged treatment leads to the development of motor complications, known as L-Dopa-induced dyskinesia, involving choreic movements, dystonia, and ballism [161].
NMDARs largely regulated by dopaminergic afferents are very abundant in the basal ganglia and control the release of the neurotransmitters γ-aminobutyric acid (GABA) and acetylcholine [162]. NMDAR antagonists could be used to decrease L-Dopa dosage and diminish any potential oxidative damage as oxidative products of dopaminergic neurons play a role in cell death [163]. In addition, expression and activity of glutamate receptors are also affected during L-Dopa-induced dyskinesia [164]. NMDAR antagonists, such as dextromethorphan, have been reported to suppress dyskinesia in PD patients. Adverse effects at high doses limit however this treatment strategy [165]. Alternatively, amantadine, a non-selective NMDAR antagonist approved by FDA in 2003, remains an interesting therapeutic option in the management of L-Dopa-induced dyskinesias [166]. Amantadine was associated with an increased lifespan in patients with PD, suggesting that it may have neuroprotective properties [167]. Memantine has also been tested in PD patients with a moderate success, as it did not appear to share the anti-dyskinetic activity of amantadine [168]. A second generation of adamantane-based drugs are being designed in the hope of improving its clinical efficacy [169].
4. Fluoroethylnormemantine (FENM): a new generation NMDAR uncompetitive antagonist
4.1. 18F-FENM as a PET NMDAR radiotracer
Memantine and several derivatives have been fluorinated with a positon-emitting isotope 18F and tested as radiotracers of positron emission tomography (PET) for the in vivo labeling of NMDARs [170,171,172]. 18F-memantine was homogeneously distributed in the cortex and basal ganglia regions, as well as the cerebellum. However, the observed pattern of 18F-memantine uptake in the whole brain was not consistent with autoradiographic studies performed on postmortem human brain using 3H- thienylcyclidine [173] and did not reflect the regional NMDAR distribution [170,171,172]. Moreover, the radioligand binding was also partly displaced by haloperidol, suggesting some binding at sigma-1 receptors. Therefore, the radiotracer did not appear suitable for the PET imaging of the NMDA receptors. Among derivatives, 18F-fluoroethylnormemantine (18F-FENM) showed an excellent selectivity and specificity for NMDAR in preclinical models in vivo In particular, 18F-FENM injected intravenously has been shown to effectively cross the blood brain barrier and to bind to the grey matter, cerebellar cortex and central grey nuclei [174,175]. Its specific distribution matched the one of NMDAR GluN1 mandatory subunit, and a low non-specific binding level was seen after pre-injection with ketamine at anesthetic doses. Moreover, FENM competed in vitro with 3H-thienylcyclidine in rat brain membranes with a Ki of 3.5 µM [174,175]. As observed for memantine, FENM is poorly metabolized in vivo with good stability in plasma and plasma protein binding, but with a low effective dosimetric dose as compared to other PET radiotracers [175]. Moreover, the tracer was used recently in a rat preclinical model of excitotoxicity [176]. In vivo brain lesions were performed using stereotaxic quinolinic acid injections in the left motor area of Sprague Dawley rats. PET detected a significant increase in 18F-FENM uptake 24 and 72 h after excitotoxic lesions compared to the control group. So, although FENM showed only a moderate affinity for the NMDAR PCP site, the tracer appeared suitable to track activation of NMDARs in neurological diseases.
4.2. FENM as an anxiolytic agent in PTSD
NMDAR antagonists have shown efficacy against a variety of neuropsychiatric disorders. Ketamine is widely known for its rapid-acting long-lasting antidepressant effect and its efficacy in treatment-resistant depression [101,177,178]. Ketamine also prevented stress-induced behavioral despair and, when administered prior to stress, attenuated learned fear [179,180,181]. Available PTSD treatments, including medications and therapy, effectively treat only a fraction of people who try them for adequate dose and duration. This indicates a need to develop treatments targeting durable remission of PTSD. Agencies (FDA and EMA) have approved only two pharmacotherapies for PTSD, the selective serotonin reuptake inhibitors paroxetine and sertraline [182,183]. FENM therefore appeared as a putative alternative and preclinical studies examined its efficacy in rodent models of PTSD, compared to the reference NMDAR antagonists ketamine and memantine [184,185]. FENM or memantine were administered in rats submitted to a battery of behavioral assays, including paired-pulse inhibition, open field, light dark test, forced swim test, and cued fear conditioning [184]. When administered at a variety of timepoints prior to both cued fear conditioning and extinction training, FENM exerted long-lasting reductions in fear behavior in a dose-specific manner. FENM was also effective in attenuating learned fear when administered acutely prior to fear conditioning, indicating that it may also be leveraged as a resilience-enhancing prophylactic. Importantly, FENM did not alter sensorimotor gating during paired-pulse inhibition or locomotion in the open field acutely after injection. These results are in direct contrast with data obtained using memantine, as the drug significantly reduced startle response and locomotion 30 min after injection. This data suggests that FENM is devoid of non-specific side effects [184]. In a PTSD model, the drug was tested as a prophylactic or antidepressant against stress-induced maladaptive behavior, contextual fear conditioning in mice, and compared to ketamine [185]. Given after stress, FENM decreased behavioral despair and reduced perseverative behavior. When administered after re-exposure, FENM facilitated extinction learning. As a prophylactic, FENM attenuated learned fear and decreased stress-induced behavioral despair [185]. Interestingly, ketamine but not FENM increased expression of c-fos in CA3 ventral hippocampal area while both ketamine and FENM attenuated large-amplitude AMPA receptor-mediated bursts in the same area suggesting at least some common neurobiological mechanism [185]. These data outlined the potentialities of FENM as a NMDAR antagonism-acting anxiolytic drug and seeded a future development in PTSD.
4.3. FENM as a neuroprotective agent in AD
FENM was examined, in comparison with memantine, as a neuroprotectant in AD models. Two models were used in parallel, a pharmacological model induced in mice by intracerebroventricular injection of pre-aggregated/oligomeric amyloid-β25-35 (Aβ25-35) peptide [186,187] and the APPswe/PSEN1∂E9 transgenic line [[188,189]; Carles et al., submitted]. Both memantine and FENM showed symptomatic anti-amnesic effects at a low mg/kg dose-range, in Aβ25-35-treated mice submitted, one week after the peptide injection, to a battery of behavioral tests: spontaneous alternation, passive avoidance, object recognition, place learning in the water-maze, and topographic memory in the Hamlet. Interestingly, FENM was not amnesic when tested alone at a high dose, 10 mg/kg, contrarily to memantine. When drugs were injected in the same low mg/kg dose-range but once-a-day during one week, they prevented Aβ25-35-induced memory deficits, oxidative stress (lipid peroxidation, cytochrome c release), inflammation (IL-6, TNFα increases; GFAP and Iba1 immunoreactivity in the hippocampus and cortex), and apoptosis and cell loss (Bax/Bcl-2 ratio; cell loss in the hippocampus CA1 area) [187]. FENM effects, notably on neuroinflammation, were more robust than observed with memantine. A second study examined FENM efficacy when the drug was subcutaneously infused using an osmotic minipump during one week after the Aβ25–35 injection in mice. Deficits in spontaneous alternation and object recognition were prevented by infused FENM [Carles et al., submitted]. Similar effects were observed with daily intraperitoneal FENM or memantine treatments. Animals infused at 0.1 mg/kg/day showed prevention of Aβ25–35-induced neuroinflammation, oxidative stress and apoptosis. Aβ25–35 provoked, in hippocampal homogenates or synaptosomes, a decrease in PSD95 level, an increase in GluN2A subunit of the NMDAR with a decreased phosphorylation level, and no change in GluN2B but an increase in its phosphorylation. The FENM infusion attenuated Aβ25–35-induced alteration in PSD95, GluN2A and P-GluN2B levels but not P-GluN2A, showing a direct regulation of NMDAR in AD mice. GluN2D levels were unchanged whatever the treatment. In 10-month-old APPswe/PSEN1∂E9 and wildtype control mice, FENM was administered for four weeks, either by daily intraperitoneal injections at 0.3 mg/kg or chronic subcutaneous infusion at 0.1 mg/kg/day using osmotic minipump. Animals were then examined for spatial working memory, neuroinflammation, apoptosis, amyloid load markers, and synaptic LTP in hippocampal slices [Carles et al., submitted]. Both infused and repeated intraperitoneal FENM treatments attenuated the behavioral deficits, microglial activation, resulting increases in cytokines TNFα and IL-6, and increase in Bax levels in the mouse hippocampus. Both soluble and insoluble Aβ1-40 and soluble Aβ1-42 cortical levels were attenuated by the treatments. Alteration of long-term potentiation maintenance in the dentate gyrus of APPswe/PSEN1∂E9 mice was also improved by the treatments. These data confirmed that a post-symptomatic treatment with FENM allowed a significant prevention of AD pathology in a transgenic mouse model. Moreover, under a pre-symptomic long-term treatment, 1-5 mg/kg/day in drinking water for 9 months starting at the age of 3-month-old, APPswe/PSEN1∂E9 mice showed a prevention of the memory deficits in the same battery of behavioral tests, spontaneous alternation, passive avoidance, object recognition, and place learning in the water-maze. Post-mortem, the FENM treatment decreased neuroinflammation (GFAP and Iba1 immunoreactivity) in the hippocampus, and insoluble Aβ1-40 Aβ1-42 levels. This decrease of amyloid load was associated to a decrease of amyloid plaques in hippocampus [unpublished data].
Recombinant NMDARs were expressed in a canonical electrophysiological model [unpublished data]. A dose-response curve of inhibition of currents elicited by application of agonists by FENM was performed, with memantine as a benchmark and internal control, and in presence of physiologically relevant level of magnesium and at resting potential of -60 mV, the IC50 values of FENM on the different NMDAR subtypes were: 168 µM for GluN1/GluN2A, 127.3 µM for GluN1/GluN2B, 13.9 µM for GluN1/GluN2C, and 11.5 µM for GluN1/GluN2D receptors. FENM therefore appeared a direct NMDAR antagonist, with a significant subtype selectivity for GluN2C and GluN2D subunit-containing receptors. The potentialities as a neuroprotective and anxiolytic NMDAR antagonist of FENM could be link to this selectivity. Further studies will be essential to clearly identify how its mode of action at NMDAR is translated into its neuroprotective action, and to determine whether biochemical or regional specifications sustain its pharmacological profile.
5. Conclusions
NMDARs generated tremendous research efforts in neuropharmacology since the mid 80's, not only for their prominent role in the balance between excitation and inhibition in the brain, their role in major physiological responses, such as neurological, cognitive and motor functions, but also due to their direct responsibilities in numerous pathological conditions. Among them, neurodegenerative pathologies are due or amplified by a chronic excitotoxic status and involve specific alterations, as observed with Aβ affecting the excitatory synapse in AD. NMDAR antagonists, and mainly uncompetitive antagonists targeting the PCP site within the ionophore, have led to major clinical breakthroughs, including memantine in AD, ketamine in anesthesia or major depression, amantadine in PD. Novel drugs are still in development and we particularly develop FENM, a memantine derivative that present superior neuroprotective activity in AD, as compared to memantine, and anxiolytic properties in PTSD, as compared with ketamine. The drug is expected to enter clinical trials for the second semester of 2024.
Author Contributions
Writing—original draft preparation, A.C. and T.M.; writing—review and editing, A.C., A.F., F.P.D., G.R. and T.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been funded by ReST Therapeutics and the SATT AXLR (maturation project #0682). A.C. acknowledges a PhD grant from ReST Therapeutics.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
A.C. received PhD grant from ReST Therapeutics; A.F. is employee and co-inventor on patents from ReST Therapeutics; F.P.D. is co-founder and employee of ReST Therapeutics; GR is founder and co-inventor on patents from ReST Therapeutics. T.M. is co-inventor on patents from ReST Therapeutics.
Abbreviations
| ABD | agonist binding domain |
| AD | Alzheimer's disease |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| Aβ | amyloid-β |
| BDNF | brain-derived neurotrophic factor |
| CaMKII | calcium moduling kinase II |
| cAMP | 3’,5’-adenosine monophosphate |
| cGMP | 3’,5’-guanosine monophosphate |
| CNS | central nervous system |
| CREB | cAMP response element binding protein |
| CTD | carboxyl C-terminal domain |
| DYRK1A | dual specificity tyrosine-phosphorylation-regulated kinase 1 |
| EAAT | excitatory amino acid transporter |
| ERK1/2 | extracellular signal-regulated protein kinase 1/2 |
| FENM | fluoroethylnormemantine |
| GABA | γ-aminobutyric acid |
| HD | Huntington's disease |
| HEK-293 | human embryonic kidney 293 cells |
| L-Dopa | L-3,4-dihydroxyphenylalanine |
| LTD | long-term depression |
| LTP | long-term potentiation |
| MAPK | mitogen-activated protein kinases |
| NMDAR | N-methyl-D-aspartate receptor |
| PCP | phencycline |
| PD | Parkinson's disease |
| PET | positron emission tomography |
| PKA | protein kinase A |
| PSD-95 | postsynaptic density protein 95 |
| PTSD | post-traumatic-stress-disorder |
| RCPG | G protein-coupled receptor |
| SANT | sodium-coupled neutral amino acid transporter |
| tPA | tissue-type plasminogen activator |
References
- Greenamyre, J.T.; Maragos, W.F.; Albin, R.L.; Penney, J.B.; Young, A.B. Glutamate transmission and toxicity in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatr 1988, 12, 421–30. [Google Scholar] [CrossRef] [PubMed]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol Rev 1999, 51, 7–61. [Google Scholar] [PubMed]
- Volianskis, A.; France, G.; Jensen, M.S.; Bortolotto, Z.A.; Jane, D.E.; Collingridge, G.L. Long-term potentiation and the role of N-methyl-D-aspartate receptors. Brain Res 2015, 1621, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Lynch, G. , Baudry, M. The biochemistry of memory: a new and specific hypothesis. Science 1984, 8, 1057–63. [Google Scholar] [CrossRef]
- Wang, H.; Peng, R.-Y. Basic roles of key molecules connected with NMDAR signaling pathway on regulating learning and memory and synaptic plasticity. Mil Med Res 2016, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E. NMDA receptor C-terminal signaling in development, plasticity, and disease. F1000Res 2019, 8, F1000. [Google Scholar] [CrossRef] [PubMed]
- Pol, A.N.V.D.; Hermans-Borgmeyer, I.; Hofer, M.; Ghosh, P.; Heinemann, S. Ionotropic glutamate-receptor gene expression in hypothalamus: Localization of AMPA, kainate, and NMDA receptor RNA with in situ hybrid-ization. J Comp Neurol 1994, 343, 428–44. [Google Scholar] [CrossRef] [PubMed]
- Tovar, K.R.; Westbrook, G.L. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci 1999, 19, 4180–8. [Google Scholar] [CrossRef]
- Cottrell, J.R.; Dubé, G.R.; Egles, C.; Liu, G. Distribution, density, and clustering of functional glutamate receptors before and after synaptogenesis in hippocampal neurons. J Neurophysiol 2000, 84, 1573–87. [Google Scholar] [CrossRef]
- Usherwood, P.N.; Machili, P.; Leaf, G. L-Glutamate at insect excitatory nerve-muscle synapses. Nature 1968, 219, 1169–72. [Google Scholar] [CrossRef]
- Nowak, L.; Bregestovski, P.; Ascher, P.; Herbet, A.; Prochiantz, A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature 1984, 307, 462–5. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.W.; Ascher, P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 1987, 325, 529–31. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Manabe, T.; Takahashi, T. Presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapse. Science 1996, 273, 648–50. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Kauer, J.A.; Perkel, D.J.; Nicoll, R.A. The impact of postsynaptic calcium on synaptic trans-mission--its role in long-term potentiation. Trends Neurosci 1989, 12, 444–50. [Google Scholar] [CrossRef] [PubMed]
- Marin, P.; Delumeau, J.C.; Tence, M.; Cordier, J.; Glowinski, J.; Premont, J. Somatostatin potentiates the alpha 1-adrenergic activation of phospholipase C in striatal astrocytes through a mechanism involving arachidonic acid and glutamate. Proc Natl Acad Sci USA. 1991, 88, 9016–20. [Google Scholar] [CrossRef]
- Geddes, J.W.; Cotman, C.W. Plasticity in hippocampal excitatory amino acid receptors in Alzheimer's disease. Neurosci Res 1986, 3, 672–8. [Google Scholar] [CrossRef]
- Weiss, J.; Goldberg, M.P.; Choi, D.W. Ketamine protects cultured neocortical neurons from hypoxic injury. Brain Res 1986, 380, 186–90. [Google Scholar] [CrossRef]
- Etienne, P.; Baudry, M. Calcium dependent aspects of synaptic plasticity, excitatory amino acid neurotransmission, brain aging and schizophrenia: a unifying hypothesis. Neurobiol Aging 1987, 8, 362–6. [Google Scholar] [CrossRef]
- Adamec, R. Transmitter systems involved in neural plasticity underlying increased anxiety and defense-implications for understanding anxiety following traumatic stress. Neurosci Biobehav Rev 1997, 21, 755–65. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Nicklas, W.J. Evidence that the loss of the voltage-dependent Mg2+ block at the N-methyl-D-aspartate receptor underlies receptor activation during inhibition of neuronal metabolism. J Neurochem 1992, 59, 1211–20. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 2012, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Chen, H.S.; Zhang, D.; Lipton, S.A. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci 2010, 30, 11246–50. [Google Scholar] [CrossRef]
- Stroebel, D.; Casado, M.; Paoletti, P. Triheteromeric NMDA receptors: from structure to synaptic physiology. Curr Opin Physiol 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Snell LD, Johnson KM. Characterization of the inhibition of excitatory amino acid-induced neurotransmitter release in the rat striatum by phencyclidine-like drugs. J Pharmacol Exp Ther 1986, 238, 938–46. [Google Scholar]
- Burnashev, N.; Schoepfer, R.; Monyer, H.; Ruppersberg, J.P.; Günther, W.; Seeburg, P.H.; Sakmann, B. Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science 1992, 257, 1415–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Williams, Z.; Goodman, C.B.; Oriaku, E.T.; Harris, C.; Thomas, M.; Soliman, K.F.A. Effects of NMDA receptor inhibition by phencyclidine on the neuronal differentiation of PC12 cells. Neurotoxicology 2006, 27, 558–66. [Google Scholar] [CrossRef]
- Paoletti, P.; Ascher, P.; Neyton, J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci 1997, 17, 5711–25. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou. Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Sheng, M.; Cummings, J.; Roldan, L.A.; Jan, Y.N.; Jan, L.Y. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 1994, 368, 144–7. [Google Scholar] [CrossRef]
- Chazot, P.L.; Stephenson, F.A. Molecular dissection of native mammalian forebrain NMDA receptors containing the NR1 C2 exon: direct demonstration of NMDA receptors comprising NR1, NR2A, and NR2B subunits within the same complex. J Neurochem 1997, 69, 2138–44. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, Y.; Yasuda, R.P.; Dunah, A.W.; Wolfe, B.B. The majority of N-methyl-D-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B). Mol Pharmacol 1997, 51, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Al-Hallaq, R.A.; Conrads, T.P.; Veenstra, T.D.; Wenthold, R.J. NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J Neurosci 2007, 27, 8334–43. [Google Scholar] [CrossRef] [PubMed]
- Monyer, H.; Sprengel, R.; Schoepfer, R.; Herb, A.; Higuchi, M.; Lomeli, H.; Burnashev, N.; Sakmann, B.; Seeburg, P.H. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science 1992, 256, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, D.J.A.; Livesey, M.R.; Hardingham, G.E. Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 2013, 74, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Otaño, I.; Luján, R.; Tavalin, S.J.; Plomann, M.; Modregger, J.; Liu, X.-B.; Jones, E.G.; Heinemann, S.F.; Lo, D.C.; Ehlers, M.D. Endocytosis and synaptic removal of NR3A-containing NMDA receptors by PAC-SIN1/syndapin1. Nat Neurosci 2006, 9, 611–21. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wu, A.; Berglund, K.; Gu, X.; Jiang, M.Q.; Talati, J.; Zhao, J.; Wei, L.; Yu, S.P. Pathogenesis of sporadic Alzheimer’s disease by deficiency of NMDA receptor subunit GluN3A. Alzheimers Dement 2022, 18, 222–39. [Google Scholar] [CrossRef]
- Grand, T.; Abi Gerges, S.; David, M.; Diana, M.A.; Paoletti, P. Unmasking GluN1/GluN3A excitatory glycine NMDA receptors. Nat Commun 2018, 9, 4769. [Google Scholar] [CrossRef]
- Harney, S.C.; Jane, D.E.; Anwyl, R. Extrasynaptic NR2D-containing NMDARs are recruited to the synapse during LTP of NMDAR-EPSCs. J Neurosci 2008, 28, 11685–94. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G.; 2007. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med 2007, 13, 54–63. [Google Scholar] [CrossRef]
- Yoshioka, A.; Yamaya, Y.; Saiki, S.; Kanemoto, M.; Hirose, G.; Beesley, J.; Pleasure, D. Non-N-methyl-D-aspartate glutamate receptors mediate oxygen--glucose deprivation-induced oligodendroglial injury. Brain Res 2000, 854, 207–15. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; Steffens, H.; Schomburg, E.D.; Pérez-Samartín, A.; Pérez-Cerdá, F.; Bakhtiari, D.; Matute, C.; Löwel, S.; Griesinger, C.; Hirrlinger, J.; Kirchhoff, F.; Nave, K.-A. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 2016, 91, 119–32. [Google Scholar] [CrossRef] [PubMed]
- Palygin, O.; Lalo, U.; Pankratov, Y. Distinct pharmacological and functional properties of NMDA receptors in mouse cortical astrocytes. Br J Pharmacol 2011, 163, 1755–66. [Google Scholar] [CrossRef] [PubMed]
- Adamsky, A.; Kol, A.; Kreisel, T.; Doron, A.; Ozeri-Engelhard, N.; Melcer, T.; Refaeli, R.; Horn, H.; Regev, L.; Groysman, M.; London, M.; Goshen, I. Astrocytic activation generates de novo neuronal potentiation and memory enhancement. Cell 2018, 174, 59–71. [Google Scholar] [CrossRef]
- Perea, G.; Araque, A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science 2007, 317, 1083–6. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Papouin, T.; Oliet, S.H.R.; Rusakov, D.A. Long-term potentiation depends on release of D-serine from astrocytes. Nature 2010, 463, 232–6. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Koh, W.; Lee, C.J.; Pankratov, Y. The tripartite glutamatergic synapse. Neuropharmacology 2021, 199, 108758. [Google Scholar] [CrossRef]
- Raghunatha, P.; Vosoughi, A.; Kauppinen, T.M.; Jackson, M.F. Microglial NMDA receptors drive pro-inflammatory responses via PARP-1/TRMP2 signaling. Glia 2020, 68, 1421–34. [Google Scholar] [CrossRef]
- Clements, J.D.; Lester, R.A.; Tong, G.; Jahr, C.E.; Westbrook, G.L. The time course of glutamate in the synaptic cleft. Science 1992, 258, 1498–501. [Google Scholar] [CrossRef]
- Satarker, S.; Bojja, S.L.; Gurram, P.C.; Mudgal, J.; Arora, D.; Nampoothiri, M. Astrocytic glutamatergic transmission and its implications in neurodegenerative disorders. Cells 2022, 11, 1139. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, F.; Moon, C.; Schlüter, O.M.; Wang, H. Bi-directional regulation of CaMKIIα phosphorylation at Thr286 by NMDA receptors in cultured cortical neurons. J Neurochem 2012, 122, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, A.S.; Jenkins, M.A.; Banke, T.G.; Schousboe, A.; Makino, Y.; Johnson, R.C.; Huganir, R.; Traynelis, S.F. Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci 2011, 14, 727–35. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Gong, R.; Qin, L.; Bao, Y.; Fu, Y.; Gao, S.; Yang, H.; Ni, J.; Yuan, T.-F.; Lu, W. Trafficking of NMDA receptors is essential for hippocampal synaptic plasticity and memory consolidation. Cell Rep 2022, 40, 111217. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, M.; Kim, E.; Sheng, M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci 1996, 16, 2157–63. [Google Scholar] [CrossRef] [PubMed]
- El-Husseini, A.E.; Schnell, E.; Chetkovich, D.M.; Nicoll, R.A.; Bredt, D.S. PSD-95 involvement in maturation of excitatory synapses. Science 2000, 290, 1364–8. [Google Scholar] [CrossRef]
- Lim, I.A.; Hall, D.D.; Hell, J.W. Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and syn-apse-associated protein 102. J Biol Chem 2002, 277, 21697–711. [Google Scholar] [CrossRef] [PubMed]
- Fiumelli, H.; Jabaudon, D.; Magistretti, P.J.; Martin, J.L. BDNF stimulates expression, activity and release of tissue-type plasminogen activator in mouse cortical neurons. Eur J Neurosci 1999, 1999 11, 1639–46. [Google Scholar] [CrossRef]
- Pang, P.T.; Teng, H.K.; Zaitsev, E.; Woo, N.T.; Sakata, K.; Zhen, S.; Teng, K.K.; Yung, W.H.; Hempstead, B.L.; Lu, B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 2004, 306, 487–91. [Google Scholar] [CrossRef] [PubMed]
- Mulkey, R.M.; Herron, C.E.; Malenka, R.C. An essential role for protein phosphatases in hippocampal long-term depression. Science 1993, 261, 1051–5. [Google Scholar] [CrossRef]
- Li, Z.; Jo, J.; Jia, J.-M.; Lo, S.-C.; Whitcomb, D.J.; Jiao, S.; Cho, K.; Sheng, M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 2010, 141, 859–71. [Google Scholar] [CrossRef]
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect Biol 2012, 4, a005710. [Google Scholar] [CrossRef] [PubMed]
- Bloodgood, B.L.; Giessel, A.J.; Sabatini, B.L. Biphasic synaptic Ca influx arising from compartmentalized elec-trical signals in dendritic spines. PLoS Biol 2009, 7, e1000190. [Google Scholar] [CrossRef] [PubMed]
- Lanté, F.; Cavalier, M.; Cohen-Solal, C.; Guiramand, J.; Vignes, M. Developmental switch from LTD to LTP in low frequency-induced plasticity. Hippocampus 2006, 16, 981–9. [Google Scholar] [CrossRef] [PubMed]
- Roche, K.W.; Standley, S.; McCallum, J.; Dune Ly, C.; Ehlers, M.D.; Wenthold, R.J. Molecular determinants of NMDA receptor internalization. Nat Neurosci 2001, 4, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Grosshans, D.R.; Clayton, D.A.; Coultrap, S.J.; Browning, M.D. LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat Neurosci 2002, 5, 27–33. [Google Scholar] [CrossRef]
- Sanz-Clemente, A.; Nicoll, R.A.; Roche, K.W. Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist 2013, 19, 62–75. [Google Scholar] [CrossRef]
- Yashiro, K.; Philpot, B.D. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 2008, 55, 1081–94. [Google Scholar] [CrossRef]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 2004, 304, 1021–4. [Google Scholar] [CrossRef]
- Bellone, C.; Nicoll, R.A. rapid bidirectional switching of synaptic NMDA receptors. Neuron 2007, 55, 779–85. [Google Scholar] [CrossRef]
- Dupuis, J.P.; Ladépêche, L.; Seth, H.; Bard, L.; Varela, J.; Mikasova, L.; Bouchet, D.; Rogemond, V.; Honnorat, J.; Hanse, E.; Groc, L. Surface dynamics of GluN2B-NMDA receptors controls plasticity of maturing glutamate synapses. EMBO J 2014, 33, 842–61. [Google Scholar] [CrossRef]
- Sanz-Clemente, A.; Matta, J.A.; Isaac, J.T.R.; Roche, K.W. Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron 2010, 67, 984–96. [Google Scholar] [CrossRef]
- Sanz-Clemente, A.; Gray, J.A.; Ogilvie, K.A.; Nicoll, R.A.; Roche, K.W. Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep 2013, 3, 607–14. [Google Scholar] [CrossRef]
- Borgdorff, A.J.; Choquet, D. Regulation of AMPA receptor lateral movements. Nature 2002, 417, 649–53. [Google Scholar] [CrossRef] [PubMed]
- Opazo, P.; Labrecque, S.; Tigaret, C.M.; Frouin, A.; Wiseman, P.W.; De Koninck, P.; Choquet, D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 2010, 67, 239–52. [Google Scholar] [CrossRef] [PubMed]
- Sumioka, A.; Yan, D.; Tomita, S. TARP phosphorylation regulates synaptic AMPA receptors through lipid bilayers. Neuron 2010, 66, 755–67. [Google Scholar] [CrossRef] [PubMed]
- Penn, A.C.; Zhang, C.L.; Georges, F.; Royer, L.; Breillat, C.; Hosy, E.; Petersen, J.D.; Humeau, Y.; Choquet, D.; 2017. Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Nature 2017, 549, 384–8. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Schrama, L.H.; Kamal, A.; Gispen, W.H.; Cattabeni, F.; Di Luca, M. Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. J Neurosci 2001, 21, 1501–9. [Google Scholar] [CrossRef]
- Gardoni, F.; Mauceri, D.; Fiorentini, C.; Bellone, C.; Missale, C.; Cattabeni, F.; Di Luca, M. CaMKII-dependent phosphorylation regulates SAP97/NR2A interaction. J Biol Chem 2003, 278, 44745–52. [Google Scholar] [CrossRef]
- Mauceri, D.; Gardoni, F.; Marcello, E.; Di Luca, M. Dual role of CaMKII-dependent SAP97 phosphorylation in mediating trafficking and insertion of NMDA receptor subunit NR2A. J Neurochem 2007, 100, 1032–46. [Google Scholar] [CrossRef]
- Tingley, W.G.; Ehlers, M.D.; Kameyama, K.; Doherty, C.; Ptak, J.B.; Riley, C.T.; Huganir, R.L. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem 1997, 272, 5157–66. [Google Scholar] [CrossRef]
- Scott, D.B.; Blanpied, T.A.; Swanson, G.T.; Zhang, C.; Ehlers, M.D.; 2001. An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci 2001, 21, 3063–72. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.B.; Blanpied, T.A.; Ehlers, M.D. Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 2003, 45, 755–67. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, S.; Nakazawa, T.; Tanimura, A.; Kiyama, Y.; Tezuka, T.; Watabe, A.M.; Katayama, N.; Yokoyama, K.; Inoue, Takeshi, Izumi-Nakaseko, H. ; Kakuta, S.; Sudo, K.; Iwakura, Y.; Umemori, H.; Inoue, Takafumi, Murphy, N.P.; Hashimoto, K.; Kano, M.; Manabe, T.; Yamamoto, T. Involvement of NMDAR2A tyrosine phosphory-lation in depression-related behaviour. EMBO J 2009, 28, 3717–29. [Google Scholar] [CrossRef]
- Grau, C.; Arató, K.; Fernández-Fernández, J.M.; Valderrama, A.; Sindreu, C.; Fillat, C.; Ferrer, I.; de la Luna, S.; Altafaj, X. ; 2014. DYRK1A-mediated phosphorylation of GluN2A at Ser1048 regulates the surface expression and channel activity of GluN1/GluN2A receptors. Front Cell Neurosci 8, 331. [CrossRef]
- Lee, C.-C.; Chang, C.-P.; Lin, C.-J.; Lai, H.-L.; Kao, Y.-H.; Cheng, S.-J.; Chen, H.-M.; Liao, Y.-P.; Faivre, E.; Buée, L.; Blum, D.; Fang, J.-M.; Chern, Y. Adenosine augmentation evoked by an ENT1 inhibitor improves memory impairment and neuronal plasticity in the APP/PS1 mouse model of Alzheimer’s disease. Mol Neurobiol 2018, 55, 8936–52. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Komai, S.; Tezuka, T.; Hisatsune, C.; Umemori, H.; Semba, K.; Mishina, M.; Manabe, T.; Yamamoto, T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J Biol Chem 2001, 276, 693–99. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; Greengard, P. Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci 2005, 8, 1051–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, B.; Li, G.; Chen, L.; Chen, L. Simvastatin enhances NMDA receptor GluN2B expression and phosphorylation of GluN2B and GluN2A through increased histone acetylation and Src signaling in hippo-campal CA1 neurons. Neuropharmacology 2016, 107, 411–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Meng, Z.-X.; Chen, Y.-Z.; Li, Y.-P.; Zhou, H.-Y.; Yang, M.; Zhao, T.-T.; Gong, Y.-L.; Wu, Y.; Liu, T. Enriched environment enhances histone acetylation of NMDA receptor in the hippocampus and improves cognitive dysfunction in aged mice. Neural Regen Res 2020, 15, 2327–34. [Google Scholar] [CrossRef]
- Morris, R.G.; Anderson, E.; Lynch, G.S.; Baudry, M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature 1986, 319, 774–6. [Google Scholar] [CrossRef]
- Davis, S.; Butcher, S.P.; Morris, R.G. The NMDA receptor antagonist D-2-amino-5-phosphonopentanoate (D-AP5) impairs spatial learning and LTP in vivo at intracerebral concentrations comparable to those that block LTP in vitro. J Neurosci 1992, 12, 21–34. [Google Scholar] [CrossRef]
- Lodge, D.; Watkins, J.C.; Bortolotto, Z.A.; Jane, D.E.; Volianskis, A. The 1980s: d-AP5, LTP and a decade of NMDA receptor discoveries. Neurochem Res 2019, 44, 516–30. [Google Scholar] [CrossRef]
- Gozzi, A.; Herdon, H.; Schwarz, A.; Bertani, S.; Crestan, V.; Turrini, G.; Bifone, A. Pharmacological stimulation of NMDA receptors via co-agonist site suppresses fMRI response to phencyclidine in the rat. Psychopharmacology (Berl) 2008, 201, 273–84. [Google Scholar] [CrossRef]
- Li, J.-H.; Vicknasingam, B.; Cheung, Y.-W.; Zhou, W.; Nurhidayat, A.W.; Jarlais, D.C.D.; Schottenfeld, R. To use or not to use: an update on licit and illicit ketamine use. Subst Abuse Rehabil 2011, 2, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Pothula, S.; Kato, T.; Liu, R.-J.; Wu, M.; Gerhard, D.; Shinohara, R.; Sliby, A.-N.; Chowdhury, G.M.I.; Behar, K.L.; Sanacora, G.; Banerjee, P.; Duman, R.S. Cell-type specific modulation of NMDA receptors triggers antidepressant actions. Mol Psychiatr 2021, 26, 5097–111. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Brown, K.A.; Georgiou, P.; Yuan, P.; Zarate, C.A.; Thompson, S.M.; Gould, T.D. NMDA Receptor Activation-Dependent Antidepressant-Relevant Behavioral and Synaptic Actions of Ketamine. J Neurosci 2023, 43, 1038–50. [Google Scholar] [CrossRef]
- Maurice, T.; Su, T.P.; Parish, D.W.; Nabeshima, T.; Privat, A. PRE-084, a sigma selective PCP derivative, attenuates MK-801-induced impairment of learning in mice. Pharmacol Biochem Behav 1994, 49, 859–69. [Google Scholar] [CrossRef] [PubMed]
- Hubert, J.P.; Delumeau, J.C.; Glowinski, J.; Prémont, J.; Doble, A. Antagonism by riluzole of entry of calcium evoked by NMDA and veratridine in rat cultured granule cells: evidence for a dual mechanism of action. Br J Pharmacol 1994, 113, 261–7. [Google Scholar] [CrossRef]
- Kretschmer, B.D.; Kratzer, U.; Schmidt, W.J. Riluzole, a glutamate release inhibitor, and motor behavior. Naunyn Schmiedebergs Arch Pharmacol 1998, 358, 181–90. [Google Scholar] [CrossRef]
- Lamanauskas, N.; Nistri, A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur J Neurosci 2008, 27, 2501–14. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 2000, 47, 351–4. [Google Scholar] [CrossRef]
- Sircar, R.; Rappaport, M.; Nichtenhauser, R.; Zukin, S.R.; 1987. The novel anticonvulsant MK-801: a potent and specific ligand of the brain phencyclidine/sigma-receptor. Brain Res 1987, 435, 235–40. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, I.J.; Hughes, N.; Carroll, C.B.; Brotchie, J.M. Reversal of parkinsonian symptoms by intrastriatal and systemic manipulations of excitatory amino acid and dopamine transmission in the bilateral 6-OHDA lesioned marmoset. Behav Pharmacol 1995, 6, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.J.; Huang, H.; Pritchett, D.B.; Lynch, D.R. Interactions between ifenprodil and the NR2B subunit of the N-Methyl-D-aspartate receptor. J Biol Chem 1996, 271, 9603–11. [Google Scholar] [CrossRef] [PubMed]
- Sarre, S.; Lanza, M.; Makovec, F.; Artusi, R.; Caselli, G.; Michotte, Y. In vivo neurochemical effects of the NR2B selective NMDA receptor antagonist CR 3394 in 6-hydroxydopamine lesioned rats. Eur J Pharmacol 2008, 584, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. The NMDA receptor antagonist memantine as a symptomatological and neuro-protective treatment for Alzheimer’s disease: preclinical evidence. Int J Geriat Psychiatr 2003, 18, S23–S32. [Google Scholar] [CrossRef] [PubMed]
- Murakawa-Hirachi, T.; Mizoguchi, Y.; Ohgidani, M.; Haraguchi, Y.; Monji, A. Effect of memantine, an an-ti-Alzheimer’s drug, on rodent microglial cells in vitro. Sci Rep 2021, 11, 6151. [Google Scholar] [CrossRef] [PubMed]
- Blanpied, T.A.; Clarke, R.J.; Johnson, J.W. ;. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J Neurosci 2005, 25, 3312–22. [Google Scholar] [CrossRef]
- Block, F.; Schwarz, M. Dextromethorphan reduces functional deficits and neuronal damage after global ischemia in rats. Brain Res 1996, 74, 153–9. [Google Scholar] [CrossRef]
- Dere, E.; Topic, B.; De Souza Silva, M.A.; Fink, H.; Buddenberg, T.; Huston, J.P. ; 2003. NMDA-receptor antagonism via dextromethorphan and ifenprodil modulates graded anxiety test performance of C57BL/6 mice. Behav Pharmacol 14, 245–9. [CrossRef]
- Kemppainen, P.; Waltimo, A.; Waltimo, T.; Könönen, M.; Pertovaara, A.; 1997. Differential effects of noxious conditioning stimulation of the cheek by capsaicin on human sensory and inhibitory masseter reflex responses evoked by tooth pulp stimulation. J Dent Res 1997, 76, 1561–8. [Google Scholar] [CrossRef]
- Weinbroum, A.A.; Gorodetzky, A.; Nirkin, A.; Kollender, Y.; Bickels, J.; Marouani, N.; Rudick, V.; Meller, I. Dextromethorphan for the reduction of immediate and late postoperative pain and morphine consumption in orthopedic oncology patients: a randomized, placebo-controlled, double-blind study. Cancer 2002, 95, 1164–70. [Google Scholar] [CrossRef]
- Weinbroum, A.A.; Bender, B.; Nirkin, A.; Chazan, S.; Meller, I.; Kollender, Y. Dextromethorphan-associated epidural patient-controlled analgesia provides better pain- and analgesics-sparing effects than dextrome-thorphan-associated intravenous patient-controlled analgesia after bone-malignancy resection: a randomized, placebo-controlled, double-blinded study. Anesth Analg 2004, 98, 714–22. [Google Scholar] [CrossRef]
- Williams, K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol 1993, 44, 851–9. [Google Scholar]
- Matsunaga, S.; Kishi, T.; Nomura, I.; Sakuma, K.; Okuya, M.; Ikuta, T.; Iwata, N. The efficacy and safety of memantine for the treatment of Alzheimer’s disease. Exp Opin Drug Safety 2018, 17, 1053–61. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.R.; Pizon, A.F.; Brooks, D.E. Dextromethorphan-induced serotonin syndrome. Clin Toxicol 2008, 46, 771–3. [Google Scholar] [CrossRef] [PubMed]
- Guyot, M.C.; Hantraye, P.; Dolan, R.; Palfi, S.; Maziére, M.; Brouillet, E.; 1997. Quantifiable bradykinesia, gait abnormalities and Huntington’s disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience 1997, 79, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Greenamyre, J.T. Alternative excitotoxic hypotheses. Neurology 1992, 42, 733–8. [Google Scholar] [CrossRef]
- Ingelsson, M.; Fukumoto, H.; Newell, K.L.; Growdon, J.H.; Hedley-Whyte, E.T.; Frosch, M.P.; Albert, M.S.; Hyman, B.T.; Irizarry, M.C. Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 2004, 62, 925–31. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 2006, 27, 1372–84. [Google Scholar] [CrossRef]
- Fonte, C.; Smania, N.; Pedrinolla, A.; Munari, D.; Gandolfi, M.; Picelli, A.; Varalta, V.; Benetti, M.V.; Brugnera, A.; Federico, A.; Muti, E.; Tamburin, S.; Schena, F.; Venturelli, M. Comparison between physical and cognitive treatment in patients with MIC and Alzheimer’s disease. Aging (Albany NY) 2019, 11, 3138–55. [Google Scholar] [CrossRef]
- Simpson, J.E.; Ince, P.G.; Lace, G.; Forster, G.; Shaw, P.J.; Matthews, F.; Savva, G.; Brayne, C.; Wharton, S.B.; 2010. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging 2010, 31, 578–90. [Google Scholar] [CrossRef]
- Wei, W.; Nguyen, L.N.; Kessels, H.W.; Hagiwara, H.; Sisodia, S.; Malinow, R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci 2010, 13, 190–6. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 2011, 31, 6627–38. [Google Scholar] [CrossRef] [PubMed]
- Companys-Alemany, J.; Turcu, A.L.; Vázquez, S.; Pallàs, M.; Griñán-Ferré, C. Glial cell reactivity and oxidative stress prevention in Alzheimer’s disease mice model by an optimized NMDA receptor antagonist. Sci Rep 2022, 12, 17908. [Google Scholar] [CrossRef] [PubMed]
- Esposito, Z.; Belli, L.; Toniolo, S.; Sancesario, G.; Bianconi, C.; Martorana, A. ; 2013. Amyloid β, Glutamate, Exci-totoxicity in Alzheimer’s Disease: Are We on the Right Track? CNS Neurosci Ther 19, 549–55. [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; Eckert, A.; Staufenbiel, M.; Hardeman, E.; Götz, J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–97. [Google Scholar] [CrossRef] [PubMed]
- Vitolo, O.V.; Sant’Angelo, A.; Costanzo, V.; Battaglia, F.; Arancio, O.; Shelanski, M. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci USA 2002, 99, 13217–21. [Google Scholar] [CrossRef]
- Bordji, K.; Becerril-Ortega, J.; Nicole, O.; Buisson, A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-ß production. J Neurosci 2010, 30, 15927–42. [Google Scholar] [CrossRef]
- Sze, S.C.; Wong, C.K.; Yung, K.K. Modulation of the gene expression of N-methyl-D-aspartate receptor NR2B subunit in the rat neostriatum by a single dose of specific antisense oligodeoxynucleotide. Neurochem Int 2001, 39, 319–27. [Google Scholar] [CrossRef]
- Bi, H.; Sze, C.-I. N-Methyl-d-aspartate receptor subunit NR2A and NR2B messenger RNA levels are altered in the hippocampus and entorhinal cortex in Alzheimer’s disease. J Neurol Sci 2002, 200, 11–8. [Google Scholar] [CrossRef]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Differential expression of N-methyl-D-aspartate receptor NR2 isoforms in Alzheimer’s disease. J Neurochem 2004, 90, 913–9. [Google Scholar] [CrossRef]
- Mishizen-Eberz, A.J.; Rissman, R.A.; Carter, T.L.; Ikonomovic, M.D.; Wolfe, B.B.; Armstrong, D.M. Biochemical and molecular studies of NMDA receptor subunits NR1/2A/2B in hippocampal subregions throughout pro-gression of Alzheimer’s disease pathology. Neurobiol Dis 2004, 15, 80–92. [Google Scholar] [CrossRef]
- Tsang, S.W.Y.; Vinters, H.V.; Cummings, J.L.; Wong, P.T.-H.; Chen, C.P.L.-H.; Lai, M.K.P. Alterations in NMDA receptor subunit densities and ligand binding to glycine recognition sites are associated with chronic anxiety in Alzheimer’s disease. Neurobiol Aging 2008, 29, 1524–32. [Google Scholar] [CrossRef]
- Schmitt, F.A.; Ashford, W.; Ernesto, C.; Saxton, J.; Schneider, L.S.; Clark, C.M.; Ferris, S.H.; Mackell, J.A.; Schafer, K.; Thal, L.J. The severe impairment battery: concurrent validity and the assessment of longitudinal change in Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord, 2.
- Tariot, P.N.; Farlow, M.R.; Grossberg, G.T.; Graham, S.M.; McDonald, S.; Gergel, I.; for the Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil. A randomized controlled trial. J Am Med Assoc 2004, 291, 317–24. [Google Scholar] [CrossRef] [PubMed]
- Mimica, N.; Presecki, P. Side effects of approved antidementives. Psychiatr Danub 2009, 21, 108–13. [Google Scholar]
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and Incidence of Huntington’s Disease: An Updated Systematic Review and Meta-Analysis. Mov Disord 2022, 37, 2327–35. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Albin, R.L.; Anderson, K.D.; D’Amato, C.J.; Penney, J.B.; Young, A.B. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA 1988, 85, 5733–7. [Google Scholar] [CrossRef]
- Foroud, T.; Gray, J.; Ivashina, J.; Conneally, P.M. Differences in duration of Huntington’s disease based on age at onset. J Neurol Neurosurg Psychiatr 1999, 66, 52–6. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Beck, C.A.; Darwin, K.; Nichols, P.; Brocht, A.F.D.; Biglan, K.M.; Shoulson, I.; Huntington Study Group COHORT Investigators. Natural history of Huntington disease. JAMA Neurol 2013, 70, 1520–30. [Google Scholar] [CrossRef]
- Duyao, M.; Ambrose, C.; Myers, R.; Novelletto, A.; Persichetti, F.; Frontali, M.; Folstein, S.; Ross, C.; Franz, M.; Abbott, M. ; 1993. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet 4, 387–392. [CrossRef]
- McKinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ozlu, M.I.; Serafin-Molina, K.; Risher, W.C.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, S.; Yin, H.H.; Eroglu, C. Huntingtin is required for normal excit-atory synapse development in cortical and striatal circuits. J Neurosci 2014, 34, 9455–72. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb Perspect Med 2017, 7, a024240. [Google Scholar] [CrossRef]
- Sun, Y.; Savanenin, A.; Reddy, P.H.; Liu, Y.F. Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J Biol Chem 2001, 276, 24713–8. [Google Scholar] [CrossRef]
- Young, A.B.; Greenamyre, J.T.; Hollingsworth, Z.; Albin, R.; D’Amato, C.; Shoulson, I.; Penney, J.B. NMDA receptor losses in putamen from patients with Huntington’s disease. Science 1988, 241, 981–3. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Young, A.B.; Penney, J.B.; Handelin, B.; Balfour, R.; Anderson, K.D.; Markel, D.S.; Tourtellotte, W.W.; Reiner, A. Abnormalities of striatal projection neurons and N-methyl-D-aspartate receptors in pre-symptomatic Huntington’s disease. N Engl J Med 1990, 322, 1293–8. [Google Scholar] [CrossRef]
- Cepeda, C.; Ariano, M.A.; Calvert, C.R.; Flores-Hernández, J.; Chandler, S.H.; Leavitt, B.R.; Hayden, M.R.; Levine, M.S.; 2001. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res 2001, 66, 525–39. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.J.; Levine, M.S. Changes in expression of N-methyl-D-aspartate receptor subunits occur early in the R6/2 mouse model of Huntington’s disease. Dev Neurosci 2006, 28, 230–8. [Google Scholar] [CrossRef] [PubMed]
- Jarabek, B.R.; Yasuda, R.P.; Wolfe, B.B. Regulation of proteins affecting NMDA receptor-induced excitotoxicity in a Huntington’s mouse model. Brain 2004, 127, 505–16. [Google Scholar] [CrossRef] [PubMed]
- Faideau, M.; Kim, J.; Cormier, K.; Gilmore, R.; Welch, M.; Auregan, G.; Dufour, N.; Guillermier, M.; Brouillet, E.; Hantraye, P.; Déglon, N.; Ferrante, R.J.; Bonvento, G. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum Mol Genet 2010, 19, 3053–67. [Google Scholar] [CrossRef] [PubMed]
- Heng, M.Y.; Detloff, P.J.; Wang, P.L.; Tsien, J.Z.; Albin, R.L. In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J Neurosci 2009, 29, 3200–5. [Google Scholar] [CrossRef]
- Lujan, B.; Liu, X.; Wan, Q. Differential roles of GluN2A- and GluN2B-containing NMDA receptors in neuronal survival and death. Int J Physiol Pathophysiol Pharmacol 2012, 4, 211–8. [Google Scholar]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 2002, 5, 405–14. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 2010, 11, 682–96. [Google Scholar] [CrossRef]
- Jiang, H.; Poirier, M.A.; Liang, Y.; Pei, Z.; Weiskittel, C.E.; Smith, W.W.; DeFranco, D.B.; Ross, C.A. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol Dis 2006, 23, 543–51. [Google Scholar] [CrossRef] [PubMed]
- Saft, C.; Burgunder, J.-M.; Dose, M.; Jung, H.H.; Katzenschlager, R.; Priller, J.; Nguyen, H.P.; Reetz, K.; Reilmann, R.; Seppi, K.; Landwehrmeyer, G.B. Symptomatic treatment options for Huntington’s disease (guidelines of the German Neurological Society). Neurol Res Pract 2023, 61. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Neuropathology of Parkinson disease. Parkinsonism Relat Disord 2018, 46 Suppl 1, S30–3. [Google Scholar] [CrossRef]
- De Pablo-Fernández, E.; Lees, A.J.; Holton, J.L.; Warner, T.T. Prognosis and neuropathologic correlation of clinical subtypes of Parkinson disease. JAMA Neurol 2019, 76, 470–9. [Google Scholar] [CrossRef] [PubMed]
- Nagatsua, T.; Sawadab, M. L-Dopa therapy for Parkinson’s disease: past, present, and future. Parkinsonism Relat Disord 2009, 15 Suppl 1, S3–8. [Google Scholar] [CrossRef]
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.-O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.; Brambilla, R.; Fisone, G.; Jon Stoessl, A.; Bourdenx, M.; Engeln, M.; Navailles, S.; De Deurwaerdère, P.; Ko, W.K.D.; Simola, N.; Morelli, M.; Groc, L.; Rodriguez, M.-C.; Gurevich, E.V.; Quik, M.; Morari, M.; Mellone, M.; Gardoni, F.; Tronci, E.; Guehl, D.; Tison, F.; Crossman, A.R.; Kang, U.J.; Steece-Collier, K.; Fox, S.; Carta, M.; Angela Cenci, M.; Bézard, E. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog Neurobiol 2015, 132, 96–168. [Google Scholar] [CrossRef] [PubMed]
- Marti, M.; Paganini, F.; Stocchi, S.; Mela, F.; Beani, L.; Bianchi, C.; Morari, M. Plasticity of glutamatergic control of striatal acetylcholine release in experimental parkinsonism: opposite changes at group-II metabotropic and NMDA receptors. J Neurochem 2003, 84, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.C.; Gunasekar, P.G.; Borowitz, J.L.; Isom, G.E. Dopamine-induced apoptosis is mediated by oxidative stress and Is enhanced by cyanide in differentiated PC12 cells. J Neurochem 2000, 74, 2296–304. [Google Scholar] [CrossRef]
- Jourdain, V.A.; Morin, N.; Grégoire, L.; Morissette, M.; Di Paolo, T. Changes in glutamate receptors in dyskinetic parkinsonian monkeys after unilateral subthalamotomy. J Neurosurg 2015, 123, 1383–93. [Google Scholar] [CrossRef]
- Verhagen Metman, L.; Blanchet, P.J.; van den Munckhof, P.; Del Dotto, P.; Natté, R.; Chase, T.N. A trial of dextromethorphan in parkinsonian patients with motor response complications. Mov Disord 1998, 13, 414–7. [Google Scholar] [CrossRef]
- Rascol, O.; Fabbri, M.; Poewe, W. Amantadine in the treatment of Parkinson's disease and other movement disorders. Lancet Neurol 2021, 20, 1048–56. [Google Scholar] [CrossRef] [PubMed]
- Uitti, R.J.; Rajput, A.H.; Ahlskog, J.E.; Offord, K.P.; Schroeder, D.R.; Ho, M.M.; Prasad, M.; Rajput, A.; Basran, P. Amantadine treatment is an independent predictor of improved survival in Parkinson’s disease. Neurology 1996, 46, 1551–6. [Google Scholar] [CrossRef] [PubMed]
- Merello, M.; Nouzeilles, M.I.; Cammarota, A.; Leiguarda, R. Effect of memantine (NMDA antagonist) on Parkinson’s disease: a double-blind crossover randomized study. Clin Neuropharmacol 1999, 22, 273–76. [Google Scholar] [PubMed]
- Dembitsky, V.M.; Gloriozova, T.A.; Poroikov, V.V. Pharmacological profile of natural and synthetic compounds with rigid adamantane-based scaffolds as potential agents for the treatment of neurodegenerative diseases. Biochem Biophys Res Commun 2020, 529, 1225–41. [Google Scholar] [CrossRef] [PubMed]
- Samnick, S.; Ametamey, S.; Leenders, K.L.; Vontobel, P.; Quack, G.; Parsons, C.G.; Neu, H.; Schubiger, P.A. Electrophysiological study, biodistribution in mice, and preliminary PET evaluation in a rhesus monkey of 1-amino-3-[18F]fluoromethyl-5-methyl-adamantane (18F-MEM): a potential radioligand for mapping the NMDA-receptor complex. Nucl Med Biol 1998, 25, 323–30. [Google Scholar] [CrossRef] [PubMed]
- Ametamey, S.M.; Samnick, S.; Leenders, K.L.; Vontobel, P.; Quack, G.; Parsons, C.G.; Schubiger, P.A. Fluorine-18 radiolabelling, biodistribution studies and preliminary PET evaluation of a new memantine derivative for imaging the NMDA receptor. J Recept Signal Transduct Res 1999, 19, 129–41. [Google Scholar] [CrossRef] [PubMed]
- Ametamey, S.M.; Bruehlmeier, M.; Kneifel, S.; Kokic, M.; Honer, M.; Arigoni, M.; Buck, A.; Burger, C.; Samnick, S.; Quack, G.; Schubiger, P.A. PET studies of 18F-memantine in healthy volunteers. Nucl Med Biol 2002, 29, 227–31. [Google Scholar] [CrossRef]
- Weissman, A.D.; Casanova, M.F.; Kleinman, J.E.; De Souza, E.B. PCP and sigma receptors in brain are not altered after repeated exposure to PCP in humans. Neuropsychopharmacology 1991, 4, 95–102. [Google Scholar]
- Salabert, A.-S.; Fonta, C.; Fontan, C.; Adel, D.; Alonso, M.; Pestourie, C.; Belhadj-Tahar, H.; Tafani, M.; Payoux, P. Radiolabeling of [18F]-fluoroethylnormemantine and initial in vivo evaluation of this innovative PET tracer for imaging the PCP sites of NMDA receptors. Nucl Med Biol 2015, 42, 643–53. [Google Scholar] [CrossRef]
- Salabert, A.-S.; Mora-Ramirez, E.; Beaurain, M.; Alonso, M.; Fontan, C.; Tahar, H.B.; Boizeau, M.L.; Tafani, M.; Bardiès, M.; Payoux, P. Evaluation of [18F]FNM biodistribution and dosimetry based on whole-body PET im-aging of rats. Nucl Med Biol 2018, 59, 1–8. [Google Scholar] [CrossRef]
- Beaurain, M.; Talmont, F.; Pierre, D.; Péran, P.; Boucher, S.; Hitzel, A.; Rols, M.-P.; Cuvillier, O.; Payoux, P.; Salabert, A.-S. Pharmacological characterization of [18F]-FNM and evaluation of NMDA receptors activation in a rat brain injury model. Mol Imaging Biol 2023, 25, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 2006, 63, 856–64. [Google Scholar] [CrossRef] [PubMed]
- aan het Rot, M.; Collins, K.A.; Murrough, J.W.; Perez, A.M.; Reich, D.L.; Charney, D.S.; Mathew, S.J. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry 2010, 67, 139–45. [Google Scholar] [CrossRef]
- Amat, J.; Dolzani, S.D.; Tilden, S.; Christianson, J.P.; Kubala, K.H.; Bartholomay, K.; Sperr, K.; Ciancio, N.; Watkins, L.R.; Maier, S.F. ; 2016. Previous ketamine produces an enduring blockade of neurochemical and behavioral effects of uncontrollable stress. J Neurosci 36, 153–61. [CrossRef]
- Brachman, R.A.; McGowan, J.C.; Perusini, J.N.; Lim, S.C.; Pham, T.H.; Faye, C.; Gardier, A.M.; Mendez-David, I.; David, D.J.; Hen, R.; Denny, C.A. Ketamine as a prophylactic against stress-induced depressive-like behavior. Biol Psychiatr 2016, 79, 776–86. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.C.; LaGamma, C.T.; Lim, S.C.; Tsitsiklis, M.; Neria, Y.; Brachman, R.A.; Denny, C.A. Prophylactic ketamine attenuates learned fear. Neuropsychopharmacology 2017, 42, 1577–89. [Google Scholar] [CrossRef]
- Brady, K.T.; Killeen, T.K.; Brewerton, T.; Lucerini, S. Comorbidity of psychiatric disorders and posttraumatic stress disorder. J Clin Psychiatr 2000, 61 Suppl 7, 22–32. [Google Scholar]
- Davidson, J.R.; Rothbaum, B.O.; van der Kolk, B.A.; Sikes, C.R.; Farfel, G.M. Multicenter, double-blind comparison of sertraline and placebo in the treatment of posttraumatic stress disorder. Arch Gen Psychiatr 2001, 58, 485–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.K.; Le Pen, G.; Eckmier, A.; Rubinstenn, G.; Jay, T.M.; Denny, C.A. Fluoroethylnormemantine, a novel derivative of memantine, facilitates extinction learning without sensorimotor deficits. Int J Neuropychopharmacol 2021, 24, 519–31. [Google Scholar] [CrossRef]
- Chen, B.K.; Luna, V.M.; Shannon, M.E.; Hunsberger, H.C.; Mastrodonato, A.; Stackmann, M.; McGowan, J.C.; Ru-binstenn, G.; Denny, C.A. Fluoroethylnormemantine, a novel NMDA receptor antagonist, for the prevention and treatment of stress-induced maladaptive behavior. Biol Psychiatr 2021, 90, 458–72. [Google Scholar] [CrossRef]
- Maurice, T.; Phan, V.-L.; Privat, A. The anti-amnesic effects of sigma1 (σ1) receptor agonists confirmed by in vivo antisense strategy in the mouse. Brain Res 2001, 898, 113–21. [Google Scholar] [CrossRef]
- Couly, S.; Denus, M.; Bouchet, M.; Rubinstenn, G.; Maurice, T. Anti-amnesic and neuroprotective effects of fluoroethylnormemantine in a pharmacological mouse model of Alzheimer’s disease. Int J Neuropsychopharmacol 2020, 24, 142–57. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Slunt, H.H.; Ratovitski, T.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng 2001, 17, 157–65. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; Younkin, S.G.; Borchelt, D.R. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum Mol Genet 2004, 13, 159–70. [Google Scholar] [CrossRef]
Figure 1.
N-methyl-D-aspartate (NMDA) receptor and its ligand binding sites. (A) NMDAR to the resting potential blocks by Mg2+ with its ligand binding sites. (B) Activation of NMDAR by AMPAR depolarization and binding of both glutamate and glycine/D-serine. This activation results in the opening of the channel and allows voltage-dependent flow of Mg2+ out of the NMDAR and entrance of massive Ca2+ into the cell inducing activation of PSD95 and synaptic plasticity. (C) NMDAR antagonist binds PCP site and partially blocks the influx of Ca2+ thus preventing neuronal membrane depolarization. This preventing further propagation of the neuronal signal, neurotransmitter release and downstream signaling mechanisms.
Figure 1.
N-methyl-D-aspartate (NMDA) receptor and its ligand binding sites. (A) NMDAR to the resting potential blocks by Mg2+ with its ligand binding sites. (B) Activation of NMDAR by AMPAR depolarization and binding of both glutamate and glycine/D-serine. This activation results in the opening of the channel and allows voltage-dependent flow of Mg2+ out of the NMDAR and entrance of massive Ca2+ into the cell inducing activation of PSD95 and synaptic plasticity. (C) NMDAR antagonist binds PCP site and partially blocks the influx of Ca2+ thus preventing neuronal membrane depolarization. This preventing further propagation of the neuronal signal, neurotransmitter release and downstream signaling mechanisms.
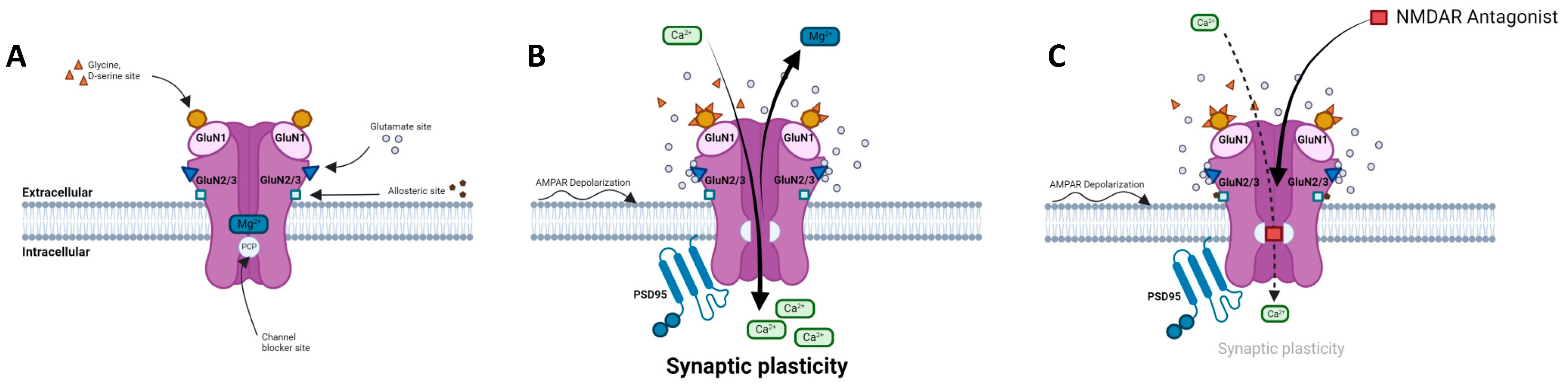
Figure 2.
The tripartite synapse comprising pre- and post-synaptic glutamatergic neurons, astrocyte and interneuron in (A) a healthy brain and (B) AD patient brain. Glutamatergic excitotoxicity is induced by accumulation of amyloid ß aggregates in the synaptic cleft, interacting with post-synaptic NMDARs and astrocytic EAATs, leading to synaptic loss and impaired physiological and behavioral responses.
Figure 2.
The tripartite synapse comprising pre- and post-synaptic glutamatergic neurons, astrocyte and interneuron in (A) a healthy brain and (B) AD patient brain. Glutamatergic excitotoxicity is induced by accumulation of amyloid ß aggregates in the synaptic cleft, interacting with post-synaptic NMDARs and astrocytic EAATs, leading to synaptic loss and impaired physiological and behavioral responses.
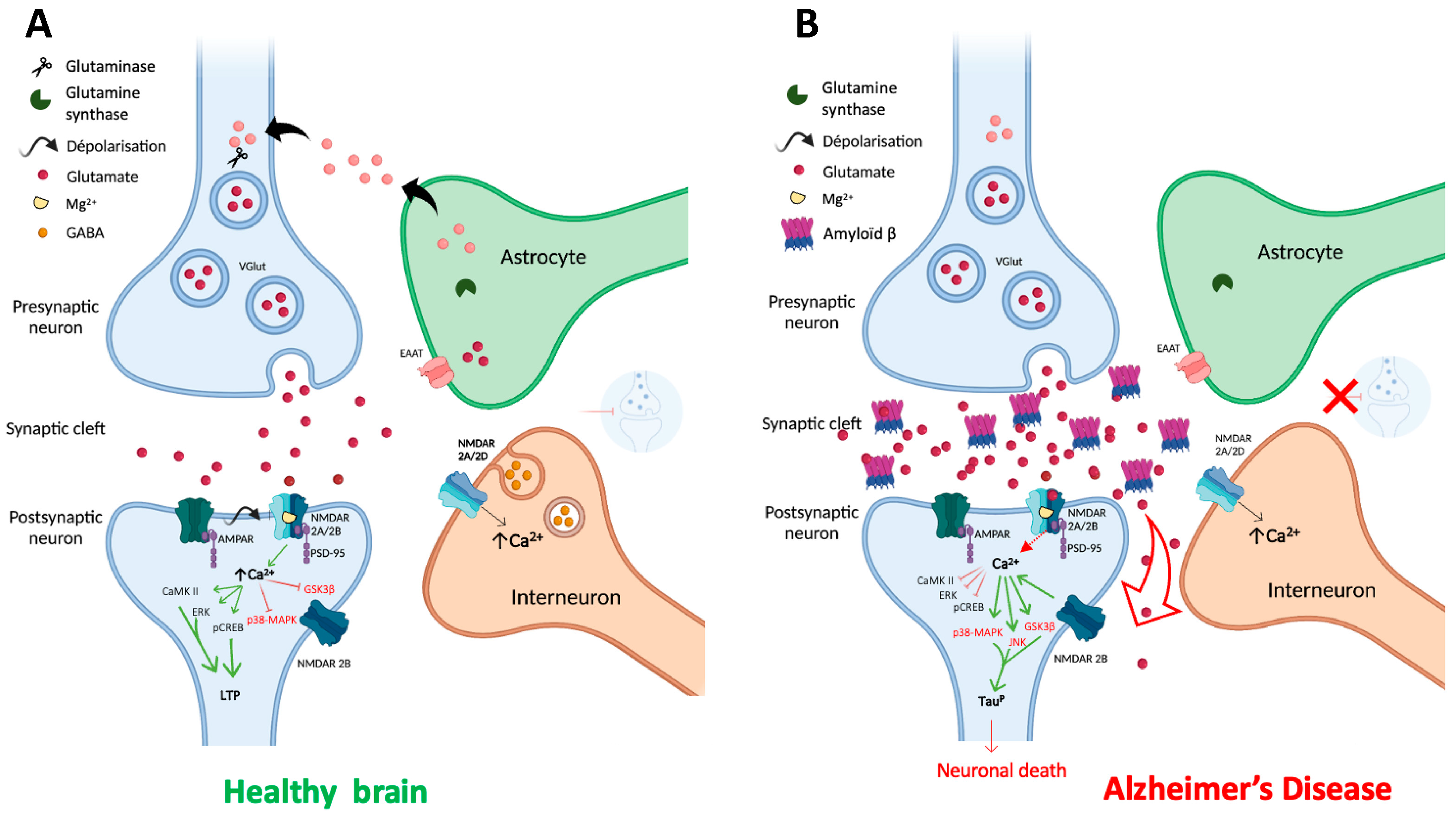

Table 1.
Listing of the reference NMDAR modulators.
| Compounds | Modulator | Selectivity | Action | References |
|---|---|---|---|---|
| D-AP5 | Hight selective competitive antagonist | GluN2A | Inhibits excitatory response. Impacts behavioural learning, blocks plasticity (LTP). | [90,91,92] |
| Riluzole | Competitive antagonist | GluN1/GluN2B | Indirect block of NMDAR. Protect from motor deficit | [98,99,100] |
| Phencyclidine | Selective uncompetitive antagonist | GluN2B/D(PCP site) | Induces psychotic and dissociative schizophrenia-like symptoms. Impairs NMDAR neurotransmission in vivo. | [93] |
| Ketamine | Uncompetitive antagonist | GluN2B/D(PCP site) | Applied in post-synapse : inhibits excitatory pyramidal neuron in extra-synaptic GluN2B. Applied in pre-synapse: inhibits GluN2D in interneuron (induces disinhibition of glutamate release in post-synapse). Up-regulates hippocampal AMPARs (GluA1/GluA2). antidepressant. | [95,101] |
| Dizocilpine | Uncompetitive antagonist | GluN2B/D(PCP site) | Anticonvulsant, antidepressant. Induces memory impairments. | [97,102] |
| Ifenprodil | Uncompetitive antagonist | GluN1/GluN2B | Blocks GluN2B (140-fold preference for NR2B over NR2A subunits). Induces an inhibition of GluN2R receptor currents. anti-Parkinsonian effect. | [103,104,105] |
| Memantine | Uncompetitive antagonist | GluN1/GluN2B | Blocks GluN2B extra-synaptic and induces glutamatergic excitotoxicity. Used for moderate-to-severe AD. | [106,107] |
| Amantadine | Uncompetitive antagonist | GluN1/GluN2B | Blocks GluN1/GluN2B by accelerating channel closure during channel block. Used as antiparkinsonian drug | [108] |
| Dextrometorphan | Uncompetitive antagonist | GluN2A | Blocks GluN2A subunit. Prevent neuronal damage and modulates pain sensation | [109,110,111,112,113] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



