Submitted:
23 January 2024
Posted:
25 January 2024
You are already at the latest version
Abstract
In our previous studies, the marine-derived fungus Emericellopsis maritima BC17 was found to produce new eremophilane-type sesquiterpenoids on solid media. In order to explore its potential to produce more metabolites, E. maritima BC17 was subjected to a one strain-many compounds (OSMAC) analysis leading to the discovery of three new eremophilanes (1-3) and fourteen known derivatives (4-17) in the liquid media Czapek Dox and PDB. Their structures were established by extensive analyses of the 1D and 2D NMR, and HRESIMS data, as well as ECD data for the assignment of their absolute configurations. Antitumoral and antimicrobial activities of the isolated metabolites 1, 3, 11, and 15 were investigated. PR toxin 3-deacetyl (15) exhibited cytotoxic activity against HepG2, MCF-7, A549, A2058 and Mia PaCa-2 human cancer cell lines with IC50 values ranging from 2.5 to 14.7 µM. In addition, 15 exhibited selective activity against methicillin-sensitive S. aureus ATCC29213 at the highest concentration tested of 128 µg/mL.
Keywords:
Eremophilane
; Emericellopsis maritima
; marine-derived fungus
; OSMAC approach
; antimicrobial
; antitumoral
1. Introduction
Over the past decade, marine natural products have played an extremely important role in the process of new drug discovery and development [1]. Marine fungi represent relatively untapped sources for new active compounds, providing several new metabolites in recent years [2].
Emericellopsis is a genus of Ascomycota fungi within the order Hypocreales. These fungi have a wide environmental amplitude and universal distribution [3]. According to the different phylogeny and ecology, it has been categorized into two distinct groups, specifically marine and terrestrial clades [4].
Emericellopsis spp. are ubiquitous in the marine environment and can exist under extreme conditions such as temperature, pressure, salinity, and pH [3]. These fungi have also been isolated from different marine macroalgae and sponges [5,6] and have previously been studied due to their ability to produce peptides with antifungal [7], antitumoral [8], and antibiotic activity [9,10].
Specifically, E. maritima has previously been isolated from Atlantic sponges and the deep-sea [11,12], but no secondary metabolites had been published, so its study is an important starting point as a source of potential bioactive compounds.
The strain E. maritima BC17 was isolated from sediments collected along an intertidal gradient of the inner Bay of Cádiz, Spain [13]. Recently, the chemical investigation of extracts from the culture of this fungus on different solid media provided four new eremophilane-type sesquiterpenes, together with thirteen known derivatives [13]. Inspired by OSMAC strategy, in this study E. maritima BC17 was subjected to further investigation using liquid culture media in order to increase its chemical diversity. As a result, three undescribed eremophilanes (1-3) and fourteen known derivatives (4-17) were isolated using the liquid media Czapek Dox and potato dextrose broth (PDB). Antimicrobial and cytotoxic activities of the compounds 1, 3, 11, and 15 are also reported.
2. Results and Discussion
The fungus E. maritima BC17 was cultivated in the liquid culture media Czapek-Dox and PDB, and incubated for 14 or 28 days in surface and shaken conditions, as described in Materials and Methods. After extraction and purification by HPLC, in addition to fourteen known compounds (4-17), three new eremophilanes (1-3) were isolated and characterized. In particular, the undescribed eremophilanes 1-3 were only isolated from Czapek-Dox fermentation broth, which showed the highest chemical diversity.
Figure 1.
Eremophilanes (1-17) isolated from the strain Emericellopsis maritima BC17 cultivated in liquid culture media.
Figure 1.
Eremophilanes (1-17) isolated from the strain Emericellopsis maritima BC17 cultivated in liquid culture media.
The molecular formula for compound 1 was established as C12H16O4, deduced from the sodiated molecular ion [M+Na]+ (m/z 247.0933 [M+Na]+, calculated for C12H16O4Na 247.0946) observed in its HRESIMS (Figure S7). Its IR spectrum showed absorption bands characteristic for hydroxy (3447 cm−1) and enone carbonyl groups (1637 cm−1). The 1H NMR data (Table 1) indicated the presence of two methyl signals at δH 1.28 (d, J = 7.0 Hz) and 1.35 (s), and two olefinic signals at δH 6.17 (d, J = 1.5 Hz) and 6.30 (s). The 13C NMR data (Table 1) displayed twelve signals including one carbonyl (δC 183.9), four olefinic carbons (δC 124.4, 127.4, 147.9, 171.1), two methyls (δC 13.2, 22.0), one methylene (δC 37.5), other three methines, two of which were oxygenated (δC 43.4, 74.1, 74.6), and another quaternary carbon (δC 45.0).
Two double bonds and one carbonyl group accounted for three of five degrees of unsaturation, indicating the presence of two rings in the structure. The two-ring system was then established based on 1H−1H COSY (Figure S3), HSQC (Figure S4), and HMBC (Figure S5) correlations. Inspection of the 1H−1H COSY and HSQC data led to the assignment of a C(1)H2−C(2)H−C(3)H−C(4)H−C(15)H3 unit. The assigned spin system, together with HMBC correlations from H2-1 to C-3/C-5/C-9/C-10, from H-4 to C-14, and from H-15 to C-3/C-4/C-5, permitted the complete elucidation of ring A. HMBC correlations from H-6 to C-7/C-8/C-10/C-4/C-14 and from H-9 to C-1/C-5/C-7 allowed the elucidation of ring B. Thus, compound 1 was established as a nor-eremophilane sesquiterpene. Its 1H and 13C NMR spectra (Figures S1 and S2) were very similar to those of compound guignarderemophilane A (18), previously reported as a metabolite from the endophytic fungus Guignardia mangiferae [14], except for the absence of the acetyl group signals at C-3.
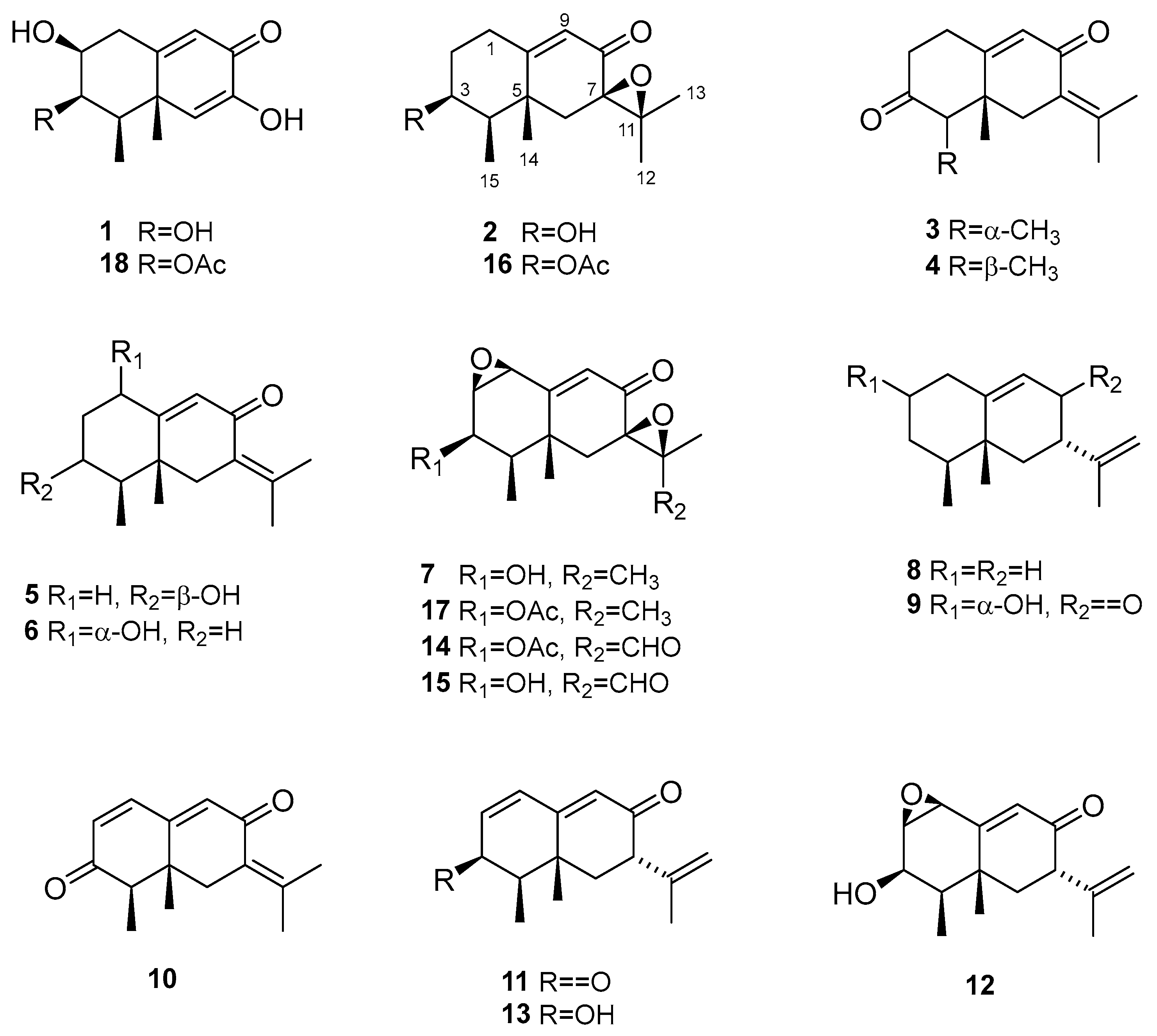
The relative configuration of 1 was determined by analysis of 1D NOESY (Figure S6a-g) correlations and the splitting patterns of related protons (Figure 2). The NOESY correlations between H-2, H-3 and H-4 revealed that they showed the same orientation. The NOE effect between the equatorial Me-15 and H-6 revealed that C5−C6 was equatorial and that Me-14 was axial. The correlations between Me-14 and Me-15 suggested they were in the syn conformation. These correlations permitted us to propose the relative configuration of 1 as shown in Figure 2.
Absolute stereochemistry of compound 1 was established by comparison of the experimental electronic circular dichroism (ECD) spectrum with the one obtained from quantum mechanical time-dependent density functional theory (TD-DFT) calculations for the (2S,3R,4R,5S) stereoisomer in the 200-400 nm region (Figure 3) [15]. Moreover, compound 1 showed an ECD spectrum similar to that of the previously reported acetylated compound guignarderemophilane A (18) [14]. Based on these data, the absolute configuration at C-2, -3, -4 and -5 was determined to be S, R, R, and S, respectively. Therefore, the structure of 1 was characterized and named as 3-deacetyl guignarderemophilane A.
Compound 2 was obtained as a colourless oil with the molecular formula C15H22O3, as determined by the HRESIMS peak at m/z 273.1446 [M+Na]+ (Figure S14), indicating five degrees of unsaturation. Its IR spectrum showed absorptions corresponding to a hydroxyl group (3496 cm−1) and an α,β-unsaturated keto group (1674 cm−1). The 13C NMR data (Table 1) displayed fifteen carbon resonances, some of which could be assigned to one keto group (δC 195.2), a C=C double bond (δC 123.6, 172.4), one 1,2-epoxy group (δC 64.7, 65.6), one oxygenated sp3 carbon (δC 70.8), and four methyl groups in the high-field region (δC 12.6, 19.1, 21.3, 24.5). These functionalities account for three degrees of unsaturation, thus hinting the bicyclic nature of 2. Analysis of the 1H NMR data (Table 1) revealed the presence of four methyl groups, including three singlets (δH 1.30, 1.36, 1.41) and one doublet (δH 1.15, J=7.1 Hz) in the high-field region. These data were characteristic of an eremophilane-type sesquiterpene, i.e., a cis-decaline ring with an isopropyl group (C-11/Me-12/Me-13) and two methyl groups, one connected to a methine group (CH-4) and the other one connected to a quaternary carbon (C-5), resulting in one doublet (Me-15) and one singlet (Me-14) signals. In addition, the signals for one olefinic proton (δH 5.92) and one oxymethine proton (δH 3.97) could be seen in the 1H NMR spectrum (Figure S8), which in combination with the signals observed in the 13C NMR spectrum (Figure S9) suggested the occurrence of one trisubstituted double bond, and one oxygenated methine group. The positions of these functional groups in the eremophilane skeleton were clarified by analysis of the HMBC spectrum (Figure S12). The presence of an epoxide moiety with 13C peaks at C-7 (δC 65.6) and C-11 (δC 64.7) was confirmed by HMBC correlations from Me-13 (δH 1.30) to C-7, C-11 and C-12 (δC 21.3), from H2-6 (δH 2.08 and 1.99) to C-7, C-10 (δC 172.4), C-11, and C-14 (δC 24.5), and from Me-12 (δH 1.41) to C-7, C-11, and C-13 (δC 19.1). Correlation between Me-15 (δH 1.15, d) and the oxygenated methine (δC 70.8) placed the hydroxyl substituent at C-3. The 1H and 13C NMR spectra of 2 (Figures S8 and S9) were very similar to those of compound 3-acetoxy-7(11)-epoxyeremophil-9-en-8-one (16), previously reported by our research’s group as an E. maritima BC17 metabolite from solid culture media [13], except for the absence of the acetyl group signals at C-3.
The relative configuration of 2 was determined by analysis of 1D NOESY correlations (Figure S13a-f). The H-1 (δH 2.90) and Me-14 protons showed strong NOE correlations with each other, indicating that H-1β and Me-14 were in axial orientations on the same face of ring A. The NOE correlations between H-3 and H-1 (δH 2.16) and H-4 confirmed that these protons were on the opposite side from H-1β/Me-14. NOE correlations between Me-14 and Me-15 indicated both methyls were on the same face. The β-orientation of the epoxide at C-7 and C-11 in 2 was evidenced by the NOE effect from Me-12 to H-6α (δH 2.08) (Figure 2).
The ECD spectrum of 2 was very similar to that obtained for the previously reported compound 16 [13]. In addition, the ECD curve for the (3S,4R,5R,7R) stereoisomer, calculated with the TD-DFT theoretical method, matched well with the experimental ECD spectrum of 2 (Figure 3). As a result, its absolute stereochemistry was assigned as (3S,4R,5R,7R)-3-hydroxy-7(11)-epoxyeremophil-9-en-8-one (2).
Compound 3 had a molecular formula of C15H20O2, as deduced from HRESIMS (Figure S21), which was consistent with six degrees of unsaturation. The 13C NMR data (Table 1) displayed fifteen signals, including two carbonyl groups (δC 190.5, 212.1), four olefinic carbons (δC 127.0, 128.2, 146.1, 162.6), four methyls (δC 11.0, 22.8, 23.1, 25.1), three methylenes (δC 29.0, 37.4, 37.5), another methine (δC 53.6), and an additional quaternary carbon (δC 42.2). The 1H NMR data (Table 1) showed four methyl groups, including one singlet (δH 1.20) and three doublets (δH 1.10, J=7.0 Hz; 1.85, J=1.4 Hz; 2.15, J=2.0 Hz) in the high-field region.
NMR data (Table 1) of 3 revealed a great similarity with those of isopetasone (4) [16].However, the 1H and 13C NMR spectra (Figures S15 and S16) of both compounds differed significantly in the shift of the signals corresponding to H-1β (δH 2.87 in 3 and 2.72 in 4), H-6α (δH 2.49 in 3 and 2.82 in 4), Me-14 (δC/H 25.1/1.20 in 3 and 18.5/0.96 in 4), and Me-15 (δC 11.0 in 3 and 7.4 in 4). This suggested a possible difference in the stereochemistry of these methyls. The NOESY correlation between H-1 (δH 2.87) and Me-14 (δH 1.20) (Figures S20a and 20d) revealed that this methyl group was axial, while the correlation from Me-14 to H-4 (δH 2.60) (Figure S20d) positioned the latter to the equatorial site, indicating that Me-14 and H-4 shared the same β-configuration (Figure 2), in contrast to all eremophilanes described in this work. Hence, compound 3 was elucidated to be 4-epiisopetasone.
This stereochemistry was confirmed by comparison of the predicted ECD spectrum from TD-DFT calculations with the experimental one of compound 3. The calculated spectrum reproduced well the experimental data (Figure 3), confirming the 4S and 5R absolute configuration of 3.
In view of these results, it is noteworthy that the new metabolites could only be isolated under surface culture conditions and with Czapek-Dox medium. In particular, compound 3 belongs to a different stereochemical series than all eremophilanes previously isolated from the E. maritima BC17 strain. This indicates that the OSMAC approach is useful for accessing cryptic metabolites that would not be obtained under standard laboratory conditions.
The other isolated compounds 4-17 were identified by comparing the spectroscopic data with the literature: isopetasone (4) [17], (+)-3-epiisopetasol (5) [18], 1α-hydroxydehydrofukinone (6) [19], eremofortin A alcohol (7) [20], (+)-aristolochene (8) [21], 7-hydroxy-4a,5-dimethyl-3-prop-1-en-2-yl-3,4,5,6,7,8-hexahydronaphthalen-2-one (9) [22], warburgiadione (10) [17], (1R,7S,8aR)-1,8a-dimethyl-7-(prop-1-en-2-yl)-1,7,8,8a-tetrahydronaphthalene-2,6-dione (11) [23], eremofortine B (12) [24], 3β-hydroxy-7β-eremophil-1(2),9(10),11(12)-trien-8-one (13) [25], PR toxin (14) [24], PR toxin 3-deacetyl (15) [26,27], (3S)-acetoxy-7(11)-epoxyeremophil-9-en-8-one (16) [13], and eremofortine A (17) [24]. Compounds 11, 12, and 15 are reported for the first time as fungal metabolites from E. maritima BC17 in this work, indicating the usefulness of the OSMAC strategy to increase the chemical diversity of the strain under study.
PR toxin 3-deacetyl (15) was previously chemically synthesised from PR toxin (14) by Wei et al [27]. In order to confirm the configuration at C-3 of 15, this compound was acetylated with acetic anhydride and p-toluenesulfonic acid to give a product whose spectroscopic data coincided with those found in literature for PR toxin (14) [24]. This has allowed us to confirm the absolute configuration of compound 15 and report it for the first time as a fungal metabolite.
The undescribed isolated metabolites 1 and 3 and the known compounds 11 and 15 were evaluated for antimicrobial and cytotoxic activities. They were tested in triplicates against a panel of 8 human pathogens, including both Gram-negative and Gram-positive bacteria (Escherichia coli ATCC25922, Pseudomonas aeruginosa PAO-1, Acinetobacter baumannii ATCC19606, Klebsiella pneumoniae ATCC700603, methicillin-resistant Staphylococcus aureus MB5393, and sensitive S. aureus ATCC29213), yeast (Candida albicans ATCC64124) and fungal (Aspergillus fumigatus ATCC46645) strains. No antimicrobial activity was detected for these metabolites against the human pathogenic strains assayed, except for compound 15 that exhibited selective activity against methicillin-sensitive S. aureus ATCC29213 at the highest concentration tested of 128 µg/mL (Table 2).
Antitumoral activity of these compounds (1, 3, 11, and 15) was also tested against liver (HepG2, ATCC HB-8065), breast (MCF-7, ATCC HTB-22), lung (A549, ATCC CCL-185), skin (A2058, ATCC CRL-3601), and pancreas (Mia PaCa-2, ATCC CRL-1420) human cancer cells using MTT test. Methyl methanesulfonate (MMS) 4mM was used as positive control of cell death and doxorubicin was included as known chemotherapeutic agent.
In previous studies, in vitro tests of compound 11, isolated from dry root of Valeriana jatamansi, indicated that it played a restraining effect to human brain malignant glioblastoma U251 cell by inhibiting cell proliferation and inducing cell apoptosis [23]. However, in this work we did not detect antiproliferative activity of this metabolite, nor of compounds 1 and 3, against the human cancer cells assayed. Among the eremophilanes tested, only compound 15 was active against all of the tested cell lines with IC50 values ranging from 2.5 to 14.7 µM (Table 3). In a previous study, we reported that the PR toxin (14) inhibited the same human cancer cells tested with IC50 values in the range of 3.75-33.44 µg/mL [13]. These results confirm that the aldehyde group at C-12 present in both compounds, 14 and 15, is directly related to their biological activity.
3. Materials and Methods
3.1. General Experimental Procedures
Melting points were measured with a Reichert-Jung Kofler block. ECD spectra were recorded on a JASCO J-1500 CD spectrometer. Optical rotations were determined with a JASCO P-2000 polarimeter. IR spectra were recorded on a PerkinElmer Spectrum BX FT-IR spectrophotometer and reported as wavenumber (cm—1). TLC was performed on Merck Kiesegel 60 Å F254, 0.25 mm thick. Silica gel 60 PF254 (60-100 mesh, VWR) was used for column chromatography. HPLC was performed with a Hitachi/Merck L-6270 apparatus equipped with an UV-VIS detector (L 4250) and a differential refractometer detector (RI-7490). LiChroCART LiChrospher Si 60 (5µm, 250 mm × 4 mm), LiChroCART LiChrospher Si 60 (10µm, 250 mm × 10 mm), and ACE 5 SIL (5 µm, 250 mm × 4.6 mm id) columns were used for isolation experiments. 1H and 13C NMR measurements were recorded on Bruker 400, 500, and 700 MHz spectrometers with SiMe4 as the internal reference. Chemical shifts are expressed in ppm (δ), referenced to CDCl3 (Eurisotop, Saint-Aubiu, France, δH 7.25, δC 77.0) and CD3OD (Eurisotop, Saint-Aubiu, France, δH 3.30, δC 49.0). COSY, HSQC, HMBC, and NOESY experiments were performed using standard Bruker pulse sequence. NMR assignments were made using a combination of 1D and 2D techniques. High-Resolution Mass Spectroscopy (HRMS) was performed either with a double-focusing magnetic sector mass spectrometer in a QTOF mass spectrometer in the positive-ion ESI mode.
3.2. Fungal Material
The strain used in this work, E. maritima BC17, was isolated from sediment samples collected along an intertidal gradient of Bay of Cádiz, Spain [13]. This culture is deposited at the University of Cádiz, Mycological Herbarium Collection (UCA). Conidial stock suspensions were maintained viable in 80% glycerol at -40°C.
3.3. General Culture Conditions
E. maritima BC17 strain was grown in Petri dishes with PDA medium for one week at 25°C under white light (day light lamp). Then, 5 pieces of agar (~1cm) containing mycelium were added to Roux bottles for surface cultures or Erlenmeyer flasks for shaken cultures with PDB (Condalab, Madrid, Spain) or Czapek-Dox medium (50 g glucose, 1 g yeast extract, 5 g K2HPO4, 2 g NaNO3, 0.5 g MgSO4·7H2O, 0.01 g FeSO4·7H2O, 1L of water and pH adjusted to 6.5-7.0). The shaken cultures were incubated in an orbital shaker at 120 rpm.
Fermentation continued at 25°C under continuous white light for 14 or 28 days and then the mycelium was filtered. The broth was extracted three times with ethyl acetate and the organic extract dried over dry Na2SO4. The solvent was then evaporated and the residue chromatographed, first on a silica gel column and then by HPLC with an increasing gradient of ethyl acetate to n-hexane.
3.4. Surface Culture Fermentation
Czapek-Dox Medium Fermentation
E. maritima BC17 strain was incubated in 26 Roux bottles (150 mL per bottle) for 14 and 28 days. Evaporation of the solvent at reduced pressure gave the extracts as yellow oils, 746 mg (from 14 days of fermentation) and 316 mg (from 28 days of fermentation).
Chromatography of the extract fermented for 14 days gave, in addition to the known compounds 2-phenylethanol (22.0 mg), tyrosol (10.0 mg), 4 (3.5 mg), 5 (3.8 mg), 6 (2.0 mg), 7 (1.0 mg), and 8 (49.1 mg), the new metabolites 1 (1.0 mg), 2 (0.8 mg), and 3 (1.0 mg). Purification of the extract fermented for 28 days afforded the known compounds 2-phenylethanol (31.0 mg), tyrosol (32.0 mg), 4 (10.9 mg), 5 (10.6 mg), 6 (2.9 mg), 8 (1.8 mg), 9 (1.0 mg), and 10 (1.5 mg), and the new metabolites 2 (1.2 mg), and 3 (2.0 mg).
3-Deacetyl guignarderemophilane A (1): purified through semipreparative HPLC (n-hexane:EtOAc:Acetone 55:45:5, flow 2.0 mL/min, tR = 31 min). White solid; mp 64.7°C; -33.7° (c 1.7, MeOH); ECD (MeOH) λ (Δε) 214 (5.74), 256 (-2.71), 288 (-2.03) nm; IR νmax (cm-1) 3447, 2932, 1637, 1448, 655; HRMS(ESI+) calcd. for C12H16O4Na [M+Na]+ 247.0946, found 247.0933; 1H and 13C NMR data in Table 1; gHMBC (selected correlations) H-1α → C-3, C-5, C-9, C-10; H-1β → C-2, C-9, C-10; H-4α → C-5, C-14, C-15; H-6 → C-4, C-7, C-8, C-10, C-14; H-9 → C-1, C-5, C-7; Me-14β → C-4, C-5, C-6, C-10; Me-15β → C-3, C-4, C-5.
(3S,4R,5R,7R)-3-Hydroxy-7(11)-epoxyeremophil-9-en-8-one (2): purified through semipreparative HPLC (n-hexane:EtOAc:Acetone 65:30:5, flow 2.5 mL/min, tR = 51 min). Colourless oil. -25.4° (c 1.3, CHCl3); ECD (MeOH) λ (Δε) 205 (0.76), 220 (-3.12), 268 (-2.56), 331 (2.31) nm; IR νmax (cm-1) 3496, 2850, 1674; HRMS(ESI+) calcd. for C15H22O3Na [M+Na]+ 273.1467, found 273.1446; 1H and 13C NMR data in Table 1; gHMBC (selected correlations) H-1β → C-9, C-10; H2-6 → C-4, C-5, C-7, C-10, C-11, C-14; H-9 → C-1, C-5, C-7; Me-12 → C-7, C-11, C-13; Me-13 → C-7, C-11, C-12; Me-14β → C-4, C-5, C-6, C-10; Me-15β → C-3, C-4, C-5.
4-Epiisopetasone (3): purified through semipreparative HPLC (n-hexane:EtOAc 80:20, flow 2.5 mL/min, tR = 83 min). Colourless oil. +83.3° (c 0.3, CHCl3); ECD (MeOH) λ (Δε) 213 (2.74), 246 (6.13), 282 (-2.80) nm; IR νmax (cm-1) 2924, 1717, 1660, 1458; HRMS(ESI+) calcd. for C15H21O2 [M+H]+ 233.1542, found 233.1543; 1H and 13C NMR data in Table 1; gHMBC (selected correlations) H-1β → C-2, C-9, C-10; H-4β → C-5, C-14, C-15; H-9 → C-1, C-5, C-7; Me-12 → C-7, C-11, C-13; Me-13 → C-7, C-11, C-12; Me-14β → C-4, C-5, C-6, C-10; Me-15α → C-3, C-4, C-5.
PDB Medium Fermentation
E. maritima BC17 strain was incubated in 26 Roux bottles (150 mL per bottle) for 14 and 28 days. Evaporation of the solvent at reduced pressure afforded the extracts as yellow oils, 331 mg (from 14 days of fermentation) and 261 mg (from 28 days of fermentation).
Purification of the extracts only afforded tyrosol (8 mg for 14 days and 18.0 mg for 28 days) and 2-phenylethanol (88 mg for 14 days and 80.0 mg for 28 days).
3.5. Shaken Culture Fermentation
Czapek-Dox Medium Fermentation
The strain E. maritima BC17 was fermented in 1.4 L of Czapek-Dox liquid medium (200 mL per flask) for 14 days at 25°C and 120 rpm under continuous white light. Each Erlenmeyer flask was inoculated with 5 PDA discs (9 mm) from a 7 days-old culture. The resulting extract of 388 mg was chromatographed and subsequently purified employing HPLC to yield the known compounds 4 (2.8 mg), 5 (4.0 mg), 6 (4.0 mg), 7 (0.6 mg), 9 (10.0 mg), 10 (2.0 mg), 11 (1.2 mg), 12 (1.6 mg), 13 (6.0 mg), 14 (0.5 mg), and 15 (9.2 mg).
PDB Medium Fermentation
The strain E. maritima BC17 was fermented in 3.2 L of PDB (200 mL per flask) for 14 days at 25°C and 120 rpm under continuous white light. Each Erlenmeyer flask was inoculated with 5 PDA discs (9 mm) from a 7 days-old culture. The resulting extract of 168 mg was chromatographed and subsequently purified by HPLC to yield the known compounds 4 (8.0 mg), 9 (0.2 mg), 16 (0.5 mg), and 17 (1.0 mg).
3.6. Acetylation of compound 15
Compound 15 (0.97 mg, 0.0035 mmol) was treated with a small crystal of p-toluenesulfonic acid in 2 mL of acetic anhydride. The reaction was stirred for 24 h at room temperature, neutralized with H2O and Na2CO3 and extracted with ethyl acetate. The organic layer was dried over dry Na2SO4 and the solvent eliminated by distillation under reduced pressure. Finally, the product was purified using an analytical normal-phase HPLC column (LiChrospher Si 60, 250 x 4 mm, 5 µm) applying an isocratic method (n-hexane:EtOAc 70:30, 1 mL/min) to yield 14 (0.25 mg, 0.0008 mmol, 22% yield, tR 31 min).
3.7. Computational Details of EDC Calculations
The molecular structure analysis of compounds 1-3 employed the semiempirical PM6 method [28]. Quantum mechanical computations were carried out using the Gaussian 16 package [29]. A comprehensive geometric optimization was conducted with Density Functional Theory (DFT) employing B3LYP functionals [30,31] and the 6−311+G(2d,p) basis set. Following this, calculations were executed to determine energies, oscillator strengths, and rotational strengths for the initial 20 electronic excitations, employing the TD-DFT methodology [32,33]. The solvent effect (methanol) was considered in the calculations using the Polarizable Continuum Model (PCM) with the Implicit Solvation Energy (IEF) approach [34,35,36]. To replicate the ECD spectrum of the conformer, a Gaussian function was utilized with a half-bandwidth of 0.33 eV.
3.8. In Vitro Antimicrobial Assays
The antimicrobial activities of compounds 1, 3, 11, and 15 were evaluated against six bacterial and two fungal human pathogens. Antibacterial susceptibility of the compounds was tested against A. baumannii ATCC19606, E. coli ATCC25922, P. aeruginosa PAO-1, K. pneumoniae ATCC700603, and methicillin-resistant S. aureus MB5393, and sensitive S. aureus ATCC29213, while antifungal activity was tested against A. fumigatus ATCC46645 and C. albicans ATCC64124 following previously described methodologies [37]. Briefly, each compound was serially diluted in 100% DMSO with a dilution factor of 2 to provide 10 final assay concentrations in all antimicrobial assays. The MIC was defined as the lowest concentration of the compound that inhibited ≥90% of the growth of a microorganism after overnight incubation. Genedata Screener software, version 18.0.4-Standard (Genedata, Inc., Basel, Switzerland) was used to process and analyze the data and to calculate the RZ’ factor, which predicts the robustness of an assay [37]. In all experiments performed in this work, the RZ’ factor obtained was between 0.89 and 0.98.
3.9. In Vitro Antitumoral Assays
The tumor human cell lines used were liver HepG2 (ATCC HB-8065), breast MCF-7 (ATCC HTB-22), lung A549 (ATCC CCL-185), skin A2058 (ATCC CRL-3601) and pancreas Mia PaCa-2 (ATCC CRL-1420). Purified compounds were dissolved in DMSO 100% at 9 mM (compound 1), 34.4 mM (compound 3), 6.4 mM (compound 11), and at 28.8 mM (compound 15). The compounds were assayed for cytotoxicity using MTT test. The MTT test is a colorimetric assay for measuring the activity of enzymes that reduce 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a yellow tetrazole, to formazan dyes, giving a purple color. Briefly, after 24 h, seeded cells were treated with compounds at a maximum dilution of 1 / 200 from the stocks in 10 points of 2-fold dilution per triplicate, for 72 h. MMS (Sigma Aldrich, St. Louis, Missouri, United States), at 4mM, was used as a positive control of cell death, DMSO 0.5% as a negative control and doxorubicin (Sigma Aldrich) was included as a known chemotherapeutic agent. After the incubation period, the plates containing treated cells were washed with 200 µL of phosphate-buffered saline (PBS) 1X (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4). Then, MTT dye (Thiazolyl blue tetrazolium bromide, ACROS Organics BV, Geel, Belgium) was added at 0.5 mg/mL and plates were incubated for 2 h. The supernatant was then removed and 100 µL of DMSO 100% were added to each well in order to dissolve the resulting formazan precipitates. Finally, absorbance was measured at 570 nm with an EnVision Multilabel Plate Reader (PerkinElmer, Waltham, USA). Data obtained was analyzed using Genedata Screener Software inhibitory curves fit to a Hill Equation model to calculate IC50 and the confidence intervals at 95%.
4. Conclusions
In this research, three previously undescribed eremophilanes (1-3) and fourteen known eremophilane derivatives (4-17) were isolated and identified from the fungal strain E. maritima BC17 cultivated in the liquid culture media Czapek Dox and PDB, in order to increase its chemical diversity. In a previous report, we investigated the solid media extracts of this fungus providing four new eremophilane-type sesquiterpenes, together with thirteen known derivatives [13]. Particularly, new eremophilanes 1-3 were only isolated from the culture medium Czapek-Dox, which showed the highest chemical diversity. Compound 3 belongs to a different stereochemical series to that of all eremophilanes previously isolated from the strain under study. These results confirm that the OSMAC approach is a simple, quick and effective strategy in enhancing the chemo-diversity of marine natural products by activating silent gene clusters.
Furthermore, compounds 1, 3, 11 and 15 were tested for antimicrobial and cytotoxic activities. Compound 15 exhibited cytotoxic activity against HepG2, MCF-7, A549, A2058 and Mia PaCa-2 human cancer cell lines with IC50 values ranging from 2.5 to 14.7 µM, with the aldehyde group at C-12 being responsible for its biological activity as in 14 [13].
Supplementary Materials
1D and 2D NMR and HRESIMS spectra of compounds 1-3.
Author Contributions
Conceptualization, R.D.-P. and J.A.; methodology, R.D.-P. and C.P.; investigation, J.R.V.-S. and C.P.; cytotoxicity assays, M.C.R.; antimicrobial assays, P.S. and M.d.l.C.; ECD calculations, D.Z. and J.S.M.; writing—original draft preparation, J.A.; writing—review and editing, J.A., R.D.-P. and J.R.V.-S.; supervision, J.A. and R.D.-P.; project administration, J.A. and R.D.-P.; funding acquisition, J.A. and R.D.-P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was co-financed by the 2014–2020 ERDF Operational Programme and by the Department of Economy, Knowledge, Business and University of the Regional Government of Andalusia. Project reference: FEDER-UCA18-105749.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in Supplementary Material.
Acknowledgments
J.R.V.-S. thanks the Spanish Ministry of Universities for agrant of the National Programme FPU 2022.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Agrawal, S.; Dufossé, L.; Deshmukh, S.K. Antibacterial metabolites from an unexplored strain of marine fungi Emericellopsis minima and determination of the probable mode of action against Staphylococcus aureus and methicillin-resistant S.aureus. Biotechnol. Appl. Biochem. 2023, 70, 120–129. [Google Scholar] [CrossRef]
- Xu, L.; Meng, W.; Cao, C.; Wang, J.; Shan, W.; Wang, Q. Antibacterial and antifungal compounds from marine fungi. Mar. Drugs 2015, 13, 3479–3513. [Google Scholar] [CrossRef]
- Grum-Grzhimaylo, A.A.; Georgieva, M.L.; Debets, A.J.M.; Bilanenko, E.N. Are alkalitolerant fungi of the Emericellopsis lineage (Bionectriaceae) of marine origin? IMA Fungus 2013, 4, 213–228. [Google Scholar] [CrossRef]
- Zuccaro, A.; Summerbell, R.C.; Gams, W.; Schroers, H.-J.; Mitchell, J.I. A new Acremonium species associated with Fucus spp., and its affinity with a phylogenetically distinct marine Emericellopsis clade. Stud. Mycol. 2004, 50, 283–297. [Google Scholar]
- Gonçalves, M.F.M.; Vicente, T.F.L.; Esteves, A.C.; Alves, A. Novel halotolerant species of Emericellopsis and Parasarocladium associated with macroalgae in an estuarine environment. Mycologia 2020, 112, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Wiese, J.; Ohlendorf, B.; Blümel, M.; Schmaljohann, R.; Imhoff, J.F. Phylogenetic identification of fungi isolated from the marine sponge Tethya aurantium and identification of their secondary metabolites. Mar. Drugs 2011, 9, 561–585. [Google Scholar] [CrossRef] [PubMed]
- Kuvarina, A.E.; Gavryushina, I.A.; Kulko, A.B.; Ivanov, I.A.; Rogozhin, E.A.; Georgieva, M.L.; Sadykova, V.S. The emericellipsins A-E from an alkalophilic fungus Emericellopsis alkalina show potent activity against multidrug-resistant pathogenic fungi. J. Fungi 2021, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Rogozhin, E.; Sadykova, V. A lipoaminopeptaibol secreted by alkalophilic fungus Emericellopsis alkalina demonstrates a strong cytotoxic effect against tumor cell lines. In Proceedings of The 2nd Molecules Medicinal Chemistry Symposium (MMCS): Facing Novel Challenges in Drug Discovery, Barcelona, Spain, 15–17 May 2019; MDPI: Basel, Switzerland, 2019; 22. [Google Scholar] [CrossRef]
- Inostroza, A.; Lara, L.; Paz, C.; Perez, A.; Galleguillos, F.; Hernandez, V.; Becerra, J.; González-Rocha, G.; Silva, M. Antibiotic activity of Emerimicin IV isolated from Emericellopsis minima from Talcahuano Bay, Chile. Nat. Prod. Res. 2018, 32, 1361–1364. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Saha, S. The genus Simplicillium and Emericellopsis: A review of phytochemistry and pharmacology. Biotechnol. Appl. Biochem. 2022, 69, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- Batista-García, R.A.; Kumar, V.V.; Ariste, A.; Tovar-Herrera, O.E.; Savary, O.; Peidro-Guzmán, H.; González-Abradelo, D.; Jackson, S.A.; Dobson, A.D.W.; Sánchez-Carbente, M. del R.; et al. Simple screening protocol for identification of potential mycoremediation tools for the elimination of polycyclic aromatic hydrocarbons and phenols from hyperalkalophile industrial effluents. J. Environ. Manage. 2017, 198, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bovio, E.; Garzoli, L.; Poli, A.; Prigione, V.; Firsova, D.; McCormack, G.P.; Varese, G.C. The culturable mycobiota associated with three atlantic sponges, including two new species: Thelebolus balaustiformis and T. spongiae. Fungal Syst. Evol. 2018, 1, 141–167. [Google Scholar] [CrossRef]
- Virués-Segovia, J.R.; Millán, C.; Pinedo, C.; González-Rodríguez, V.E.; Papaspyrou, S.; Zorrilla, D.; Mackenzie, T.A.; Ramos, M.C.; de la Cruz, M.; Aleu, J.; et al. New eremophilane-type sesquiterpenes from the marine sediment-derived fungus Emericellopsis maritima BC17 and their cytotoxic and antimicrobial activities. Mar. Drugs 2023, 21, 634. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Qu, J.; Ma, S.; Zang, C.; Zhang, Y.; Yu, S. Eremophilane sesquiterpenes and polyketones produced by an endophytic Guignardia fungus from the toxic plant Gelsemium elegans. J. Nat. Prod. 2015, 78, 2149–2154. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-C.; Ferreira, D.; Ding, Y. Determination of absolute configuration of natural products: Theoretical calculation of Electronic Circular Dichroism as a tool. Curr. Org. Chem. 2010, 14, 1678–1697. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, K.; Izuta, I.; Oka, H.; Sakaguchi, R. Total synthesis of (±)-Isopetasol, (±)-3-Epiisopetasol, and (±)-Warburgiadion. Tetrahedron Lett. 1974, 15, 2187–2190. [Google Scholar] [CrossRef]
- Brooks, C.J.W.; Draffan, G.H. Sesquiterpenoids of Warburgia species. I. Warburgin and warburgiadione. Tetrahedron 1969, 25, 2865–2885. [Google Scholar] [CrossRef]
- Sumarah, M.W.; Puniani, E.; Sørensen, D.; Blackwell, B.A.; Miller, J.D. Secondary metabolites from anti-insect extracts of endophytic fungi isolated from Picea rubens. Phytochemistry 2010, 71, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Bohlmann, F.; Knoll, K.H. Naturally occurring terpene derivatives. 200. Two new eremophilane derivatives from Senecio suaveolens. Liebigs Ann. der Chemie 1979, 470–472. [Google Scholar] [CrossRef]
- Moreau, S.; Cacan, M.; Lablache-Combier, A. Eremofortin C, a new metabolite obtained from Penicillium roqueforti cultures and from biotransformation of PR toxin. J. Org. Chem. 1977, 42, 2632–2634. [Google Scholar] [CrossRef]
- Demyttenaere, J.C.R.; Adams, A.; Van Belleghem, K.; De Kimpe, N.; König, W.A.; Tkachev, A.V. De novo production of (+)-Aristolochene by sporulated surface cultures of Penicillium roqueforti. Phytochemistry 2002, 59, 597–602. [Google Scholar] [CrossRef]
- Daengrot, C.; Rukachaisirikul, V.; Tansakul, C.; Thongpanchang, T.; Phongpaichit, S.; Bowornwiriyapan, K.; Sakayaroj, J. Eremophilane sesquiterpenes and diphenyl thioethers from the soil fungus Penicillium copticola PSU-RSPG138. J. Nat. Prod. 2015, 78, 615–622. [Google Scholar] [CrossRef]
- Fan, S. Eremophilane type sesquiterpenoid compound and medical application for treating glioma. China, CN105503556 A, 20 March 2016. [Google Scholar]
- Moreau, S.; Biguet, J.; Lablache-Combier, A.; Baert, F.; Foulon, M.; Delfosse, C. Structures and stereochemistry of the sesquiterpenes of Penicillium roqueforti, PR toxin, and eremofortins A, B, C, D and E. Tetrahedron 1980, 36, 2989–2997. [Google Scholar] [CrossRef]
- Lin, A.; Wu, G.; Gu, Q.; Zhu, T.; Li, D. New eremophilane-type sesquiterpenes from an Antarctic deep-sea derived fungus, Penicillium sp. PR19 N-1. Arch. Pharm. Res. 2014, 37, 839–844. [Google Scholar] [CrossRef]
- Capasso, R.; Borrelli, V.; Iacobellis, N.S.; Basile, G. Preparation of derivatives of phomenone and PR toxin to study structure-biological activity correlations of eremophilanic sesquiterpenes. Ann. della Fac. di Sci. Agrar. della Univ. degli Stud. di Napoli, Portici, IV 1986, 20, 20–28. [Google Scholar]
- Wei, R.; Schnoes, H.K.; Hart, P.A.; Strong, F.M. The structure of PR toxin, a mycotoxin from Penicillium roqueforti. Tetrahedron 1975, 31, 109–114. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: Theoretical bases, computational implementation, and numerical applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ravipati, A.S.; Koyyalamudi, S.R.; Jeong, S.C.; Reddy, N.; Bartlett, J.; Smith, P.T.; de la Cruz, M.; Monteiro, M.C.; Melguizo, Á.; et al. Anti-Fungal and anti-bacterial activities of ethanol extracts of selected traditional Chinese medicinal herbs. Asian Pac. J. Trop. Med. 2013, 6, 673–681. [Google Scholar] [CrossRef]
Figure 2.
Selected NOESY correlations exhibited by compounds 1-3.
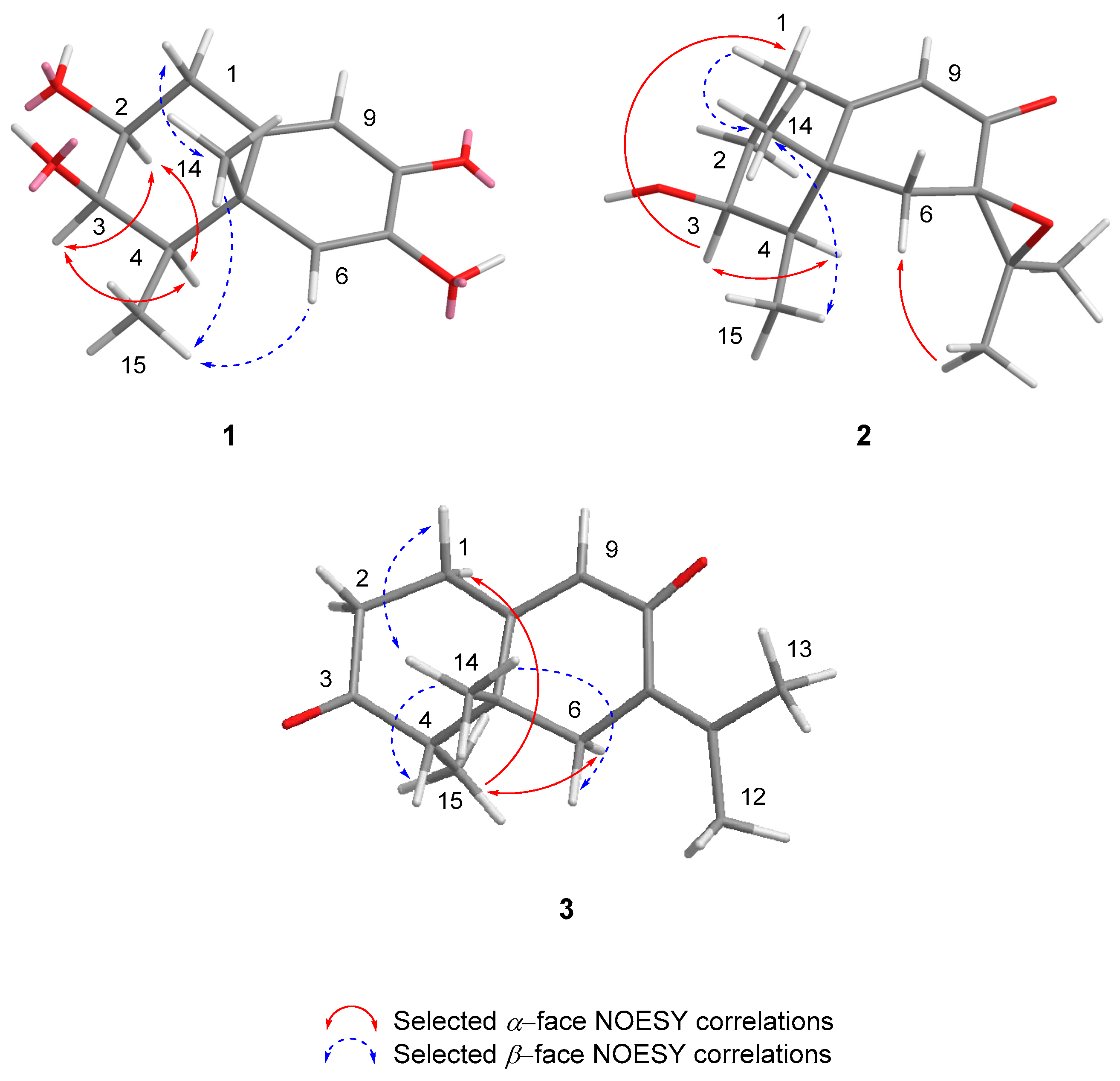
Figure 3.
Experimental and calculated ECD spectra for compounds 1-3. Calculations were performed with the conformers shown in Figure 2.
Figure 3.
Experimental and calculated ECD spectra for compounds 1-3. Calculations were performed with the conformers shown in Figure 2.

Table 1.
1H and 13C NMR spectroscopic data for compounds 1-3.
| 1 | 2 | 3 | ||||
| Position | δH, Mult (J in Hz)a | δC, Typeb | δH, Mult (J in Hz)c | δC, Typed | δH, Mult (J in Hz)e | δC, Typed |
| 1α | 2.45, dd (12.0, 4.5) | 37.5, CH2 | 2.16, m | 28.0, CH2 | 2.55, m | 29.0, CH2 |
| 1β | 2.90, td (12.0, 1.5) | 2.90, m | 2.87, dddd (14.7, 11.3, 7.6, 1.8) | |||
| 2α | 3.53, ddd (12.0, 4.5, 3.2) | 74.6, CH | 1.67, m | 37.2, CH2 | 2.19, ddt (14.2, 4.7, 2.3) | 37.4, CH2 |
| 2β | - | 2.16, m | 2.50, m | |||
| 3α | 3.70, td (3.2, 3.0) | 74.1, CH | 3.97, brs | 70.8, CH | - | 212.1, C |
| 3β | - | - | - | |||
| 4 | 1.41, qd (7.0, 3.0) | 43.4, CH | 1.75, qd (7.1, 3.3) | 43.7, CH | 2.60, qd (7.0, 3.3) | 53.6, CH |
| 5 | - | 45.0, C | - | 42.2, C | - | 42.2, C |
| 6α | 6.30, s | 127.4, CH | 2.08, d (15.0) | 38.1, CH2 | 2.34, d (15.0) | 37.5, CH2 |
| 6β | - | 1.99, d (15.0) | 2.49, d (15.0) | |||
| 7 | - | 147.9, C | - | 65.6, C | - | 127.0, C |
| 8 | - | 183.9, C | - | 195.2, C | - | 190.5, C |
| 9 | 6.17, d (1.5)- | 124.4, CH | 5.92, d (1.3)- | 123.6, CH | 5.90, d (1.8)- | 128.2, CH |
| 10 | 171.1, C | 172.4, C | 162.6, C | |||
| 11 | - | - | - | 64.7, C | - | 146.1, C |
| 12 | - | - | 1.41, s | 21.3, CH3 | 1.85, d (1.4) | 23.1, CH3 |
| 13 | - | - | 1.30, s | 19.1, CH3 | 2.15, d (2.0) | 22.8, CH3 |
| 14 | 1.35, s | 22.0, CH3 | 1.36, s | 24.5, CH3 | 1.20, s | 25.1, CH3 |
| 15 | 1.28, d (7.0) | 13.2, CH3 | 1.15, d (7.1) | 12.6, CH3 | 1.10, d (7.0) | 11.0, CH3 |
a 500 MHz, CD3OD; b 125 MHz, CD3OD; c 400 MHz, CDCl3; d 125 MHz, CDCl3; e 500 MHz, CDCl3.
Table 2.
Minimum inhibitory concentration (MIC) of compounds 1, 3, 11, and 15 against eight human pathogens.
Table 2.
Minimum inhibitory concentration (MIC) of compounds 1, 3, 11, and 15 against eight human pathogens.
| MIC (µg/mL) | ||||||||
| Compound | A. fumigatus ATCC46645 | C. albicans ATCC64124 | K. pneumonia ATCC700603 | E. coli ATCC25922 | MSSA ATCC29213 | MRSA MB5393 | A. baumannii ATCC19606 | P. aeruginosa PAO-1 |
| 1 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 |
| 3 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 |
| 11 | >24 | >24 | >24 | >24 | >24 | >24 | >24 | >24 |
| 15 | >128 | >128 | >128 | >128 | 128 | >128 | >128 | >128 |
Table 3.
Half-maximal inhibitory concentration (IC50) of compounds 1, 3, 11, and 15 of cell viability in five tumor cell lines. Confidence intervals at 95% are shown in brackets as calculated with the Genedata© Screener software, version 18.0.4-Standard.
Table 3.
Half-maximal inhibitory concentration (IC50) of compounds 1, 3, 11, and 15 of cell viability in five tumor cell lines. Confidence intervals at 95% are shown in brackets as calculated with the Genedata© Screener software, version 18.0.4-Standard.
| IC50 (µM) | |||||
| Compound | HepG2 | MCF7 | A549 | A2058 | Mia PaCa-2 |
| 1 | >45 | >45 | >45 | >45 | >45 |
| 3 | >172 | >172 | >172 | >172 | >172 |
| 11 | >33 | >33 | >33 | >33 | >33 |
| 15 | 7.6 (6.8-7.9) | 4.7 (4.0-5.4) | 14.7 (12.6-17.3) | 12.2 (10.8-14.0) | 2.5 (2.2-2.9) |
| Doxorubicin | 0.21 (0.19-0.23) | 0.21 (0.16-0.28) | 0.90 (0.70-1.00) | 0.10 (0.08-0.13) | 0.43 (0.36-0.50) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



