Submitted:
08 January 2024
Posted:
10 January 2024
You are already at the latest version
Abstract
Tumor necrosis factor (TNF) is a pleiotropic cytokine regulating many physiological and pathological immune-mediated processes. Although initially described as a tumor-cytotoxic agent, over time, it has also been recognized as an essential pro-inflammatory cytokine implicated in the pathogenesis of different autoimmune diseases, such as multiple sclerosis (MS). Several studies have shown an increase in TNF expression both in acute and chronic active MS brain lesions, as well as its involvement in maintaining a chronic inflammatory intrathecal process known as “compartmentalized inflammation”, which contributes to disease progression and disability accumulation in MS, even more than acute inflammatory activity. Indeed, high TNF levels were observed in the serum and cerebrospinal fluid of subjects with MS and correlated to disease severity, promoting axonal damage and, consequently, neurodegeneration. In this review we discuss the current knowledge of TNF and its receptors involvement in MS progression, focusing on disability progression independent of relapse activity (PIRA), meant as the disability accumulation evident in relapsing MS patients not related to acute inflammatory events.
Keywords:
TNF biology
; MS pathology
; PIRA
; MS lesions
; chronic compartmentalized inflammation
; neurodegeneration
; biomarkers
; disease progression. 1. Introduction
1. Introduction
Multiple sclerosis (MS) is a chronic immune-mediated and neurodegenerative disease of the central nervous system (CNS) affecting millions of people worldwide [1] and representing the most common cause of non-traumatic neurological disability in young adults [2].
MS is a complex multifactorial disease caused by complex gene–environment interactions and characterized by multiple pathological hallmarks, ranging from immune dysregulation and neuroinflammation to neurodegenerative mechanisms [3].
Several molecular changes, including the increase of cytokines, chemokines, nitric oxide, reactive oxygen species, glutamate, and free radicals, affect the pathogenesis and the disease course of MS [4].
MS clinical course is highly variable, heterogeneous and unpredictable at an individual level. Generally, it is characterized by transient and recurrent episodes of focal acute CNS inflammation early on, with complete or partial resolution (relapsing-remitting MS - RRMS) and, over time, by a prominent process of neurodegeneration, resulting in a late slow steady progressive accumulation of physical disability and cognitive impairment in absence of relapses (secondary progressive MS - SPMS) [5]. On the other hand, few cases reported a slow continuous neurological deterioration without relapse from the earliest stages of the disease (primary progressive MS - PPMS) [5,6].
Beyond this traditional phenotypic categorization, it is now clear that MS progresses along a continuum from RRMS to progressive MS (PrMS), with differing levels of neurologic reserve explaining phenotypic differences [7].
This emerging view of MS disease as a single-stage disorder, where all patients exhibit a progressive course since the disease onset, which can be overlapped by relapses [7], is supported by the new concept of progression independent of relapse activity (PIRA) [8]. The term PIRA, proposed by Kappos et al., refers to the progressive clinical deterioration occurring in many RRMS patients without signs of inflammatory activity [8]. This notion aligns with several previous observational studies that show disability accumulation is largely independent of superimposed focal inflammation and undetectable by conventional clinical-radiological parameters [9,10].
Although the frequency of PIRA has been reported within the first 5 years following the first MS-related clinical attack, its identification in clinical practice remains unclear due to the lack of standardized definitions (such as a time window after the last relapse) and/or measures to detect it (such as based on EDSS score or increase in composite measure) [11].
The mechanisms driving PIRA are not fully elucidated but are undoubtedly associated with smoldering inflammatory and neurodegenerative processes. In a recent, prospective, large sample size study, Cagol et al. showed that RRMS patients with PIRA (defined as a 6-month confirmed disability progression with no relapse during the 90-days before and the 180-days after the initial EDSS increase) exhibit more pronounced diffuse cerebral cortical volume loss [12]. This finding aligns with several studies demonstrating that grey matter (GM) atrophy is predictive of long-term accumulation of physical and cognitive disability [13] and conversion to PrMS [14].
Cerebral GM damage, which manifests as both focal cortical lesion(s) and diffuse cortical and deep GM atrophy, provides one of the best clinical correlations with irreversible disability accumulation [13,15] and topographically associated with aberrant tertiary B-cell-enriched lymphoid structures affecting the cerebral meninges [16]. The extent of meningeal immune infiltration correlates with the degree of subpial GM demyelination, microglial activation, and axonal loss [16,17,18,19].
MS patients with a progressive and severe course of the disease also display chronic active lesions (CALs), a subset of white matter (WM) lesions characterized by an inactive core surrounded by a “rim” of activated microglia [20,21,22]. CALs are associated with nearby persistent demyelination and axonal loss, even in the absence of blood-brain barrier (BBB) damage [20,21,22].
Molecular-neuropathological studies on progressive MS cases supported the hypothesis that soluble factors (chemokines and cytokines) produced by meningeal tertiary lymphoid structures and/or circulating immune cells, may diffuse throughout the cerebrospinal fluid (CSF) into the cortex, inducing brain damage either directly or indirectly through microglia activation [23]. In this regard, Kosa and colleagues recently found that CSF biomarkers associated with immune-related pathways correlate with clinical and imaging MS severity outcomes and predict future rates of disability accumulation [24].
All these findings suggest that chronic inflammation in the CNS continuously disturbs neuroaxonal homeostasis, leading to prominent neurodegeneration, even outside of MS relapses and especially at the progressive stage [25]. This confirmed that compartmentalized inflammation (involving CSF, meninges and parenchyma) is a major mechanism driving progressive multiple sclerosis.
Among the different cytokines found to increase in the CSF of MS patients [23], tumor necrosis factor (TNF) represents the major pro-inflammatory cytokine correlated with the degree of disability in patients with progressive MS [26].
Selmaj et al. have also provided significant evidence according to which an increase of TNF uniquely occurred locally within the CNS of MS cases and not in other neurodegenerative brain diseases, such as Alzheimer’s or Parkinson’s disease [27], further suggesting that the combination of inflammation, demyelination and neurodegeneration is an MS quite-specific process, as supported then also by a study by Fischer et al. [28]
TNF exerts its potent pro-inflammatory activity by the activation of TNF receptors type-1 (TNFR1) and type-2 (TNFR2) signalling [29,30]. Besides this inflammatory action, TNF has excitotoxic [31] and necro-apoptotic activities on oligodendrocytes [32,33] and neurons mainly through TNFR1 activation [34].
A post-mortem study has additionally revealed that an imbalance of TNF receptors type-1 (TNFR1) and type-2 (TNFR2) signalling plays a role in determining the severity of MS [35], demonstrating a strong correlation between compartmentalized inflammation and the high expression of genes involved in TNFR1 signal cascade [35].
This review summarizes the contribution of TNF and its receptors in MS progression and investigates their involvement in neurodegenerative mechanisms occurring during chronic inflammatory events.
2. TNF biology, cellular production and signalling pathways
The master pro-inflammatory cytokine, TNF, has been shown to have a broad spectrum of cellular effects, including inflammatory response, cellular activation, and programmed cell death [36]. TNF belongs to the TNF superfamily, which includes 19 ligands produced primarily by monocytes/macrophages but also by T and B lymphocytes, smooth muscle cells, adipocytes, osteoclasts and fibroblasts, although in smaller quantities [36,37].
TNF is expressed initially as a transmembrane protein (mTNF, 26 KDa 233-amino-acid), which requires proteolytic cleavage by the TNF converting enzyme (TACE) to release soluble TNF (sTNF, 17 kDa 157-amino-acid). mTNF and sTNF are produced by a wide range of peripheral and central immune cells, such as activated macrophages, effector CD4 and CD8 T cells and B lymphocytes and microglia as well as neurons, oligodendrocytes and astrocytes [38].
Both mTNF and sTNF are biologically active and exert their effects by modulating a complex signalling pathway with wide-ranging downstream responses through two distinct surface receptors belonging to the TNF receptor (TNFR) superfamilies (comprise 29 receptors): the TNF Receptor Superfamily Member 1A (TNFRSF1A-TNFR1; p55/60; CD120a) and TNF Receptor Superfamily Member 1B (TNFRSF1B-TNFR2; p75/80; CD120b) [36,37,38].
The two receptors differ significantly in structure, binding affinity, localization, function and signalling pathways activated [39,40].
TNFR1, expressed on the membrane of all cell types except erythrocytes, shows a high affinity towards sTNF, promoting both necrosis and apoptotic pathways as well as pro-inflammatory signalling [41], through its death domain (DD), which when activated by TNF binding, recruits the TNFR1-associated death domain (TRADD). TRADD can in turn recruit Fas-associated death domain (FADD) and receptor-interacting serine/threonine-protein kinase 1 (RIPK1), which can either lead to necroptosis trough RIPK3 and mixed lineage kinase domain-like pseudokinase (MLKL) activation, or apoptosis through caspase 8 and caspase 3 recruitment [42,43]. On the contrary, pro-inflammatory signalling is mediated by TNFR-associated factor 2 (TRAF2) activation of, mitogen-activated protein kinases (MAPKs), such as c-Jun-N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK), and transcription factor nuclear factor-κB (NFκB) [42].
TNFR2, expressed only in fewer cell types (neurons, oligodendrocytes, microglia and T lymphocytes), mediates local homeostatic effects, such as cell survival, tissue regeneration and inflammation through preferentially binding mTNF [44,45]. Unlike TNFR1, TNFR2 does not have a death domain. Nevertheless, a recent study has shown that under some circumstances TNFR2 signalling also has pro-apoptotic effects, by amplifying TNFR1-mediated stimulation of apoptosis or cooperating in the binding of TNF to TNFR1 [38,46]. However, the mechanism of TNFR2-mediated cell death is still unclear and homeostasis and cell survival remain the primary functions exerted by TNFR2-mediated signalling through TRAF (1/2) activation of MAPKs (JNK and ERK), protein kinase B (Akt), and NFκB [42].
Therefore, albeit in different ways, both TNFR1 and TNFR2signalling, may lead to NF-κB and MAPK activation, increasing the expression of inflammatory genes encoding chemokines and cytokines (including TNF itself) [39,40] as well as inducing anti-apoptotic transcriptional programs promoting cell survival, cell proliferation and cell differentiation [47,48].
This duality of TNFR signalling, which can induce both survival and cell death, probably depends on the cellular environment, the relative surface levels of TNFR1 and TNFR2, and their cellular activation status (Figure 1). However, the effects of altering the TNFR1/TNFR2 balance in normal and altered physiology remain to be understood [49].
3. Potential pathological implications of TNF/TNFRs impairment in MS and EAE
3.1. Neuroinflammation
TNF exerts pleiotropic functions playing a crucial role in several immune-mediated conditions, including rheumatoid arthritis [50], systemic lupus erythematosus [51] and Crohn’s disease [52]. As a potent mediator of inflammation, principally via TNFR1 signalling, TNF is considered the major cytokine involved in the pathogenesis of MS [29,30].
A relevant action of TNF is to activate T lymphocytes, enhancing their proliferation and recruitment and increasing pro-inflammatory cytokine production in the CNS by inducing the activation of NF-κB signaling pathways [53]. TNF-dependent T cell activation contributes to blood-brain barrier (BBB) damage via meningeal mast cells secondary activation and, therefore, promotes further inflammatory cell influx with consequent myelin and neuronal damage [54,55].
Not surprisingly, elevated production of TNF is found in MS patients [27,56] and in experimental autoimmune encephalomyelitis (EAE), the most used murine model of MS [56].
High TNF levels were found in active demyelinating lesions [26] as well as in the serum and CSF of MS patients [57,58,59], correlating with disease severity [59,60,61,62]. In EAE mice, TNF mRNA expression is upregulated in the CNS in parallel with disease progression, its external administration increases EAE severity [56,63] since it appears to be involved in immune cell (macrophages and T cells) activation and infiltration into the CNS [62].
The use of EAE transgenic mice for TNF and TNFRs has significantly contributed to understanding TNF's pathological role in MS [45].
TNF-gene knock-out (KO) EAE mice showed a milder disease course due to reduced leukocyte intrathecal trafficking and BBB permeability compared to wild-type (WT) EAE mice [64]. This evidence suggests that alterations in the TNF signaling are involved in the (early) pathological MS mechanisms typical of MS that occur in the CNS [64].
Beyond the cytokine, several studies have also investigated the role of the TNFRs in MS pathology. Specifically, TNFR1 KO EAE mice showed a reduction of immunopathological signs and symptoms of the disease compared to WT mice, whereas TNFR2 KO EAE mice showed more severe disease symptoms, an enhanced T cell infiltration in the CNS and diffuse demyelination [65].
Intriguingly, a crucial role of TNFR2 has recently been demonstrated in regulatory T cell (Treg) biology, promoting their proliferation and expansion [66]. Tregs are essential in maintaining immune homeostasis and preventing autoimmunity [63,64,65,66,67,68,69]. Not surprisingly, an impaired functional suppression of Tregs in response to autoreactive T cells is typically reported in MS [70]. In line with this, a recent study on Treg-restricted TNFR2 deficiency mice with induced EAE developed an aggressive disease, indicating the critical protective role of TNFR2 signalling [71]. However, the significance of intrinsic TNFR2 signalling in Treg cells in vivo remains incompletely defined [71].
3.2. Neurodegeneration, Demyelination and Remyelination
In addition to immune cell activation and infiltration, TNF signalling is involved in neurodegenerative processes. In particular, TNF promotes neuronal excitotoxicity and oligodendrocyte death, acting directly on neurons and glia, through TNFRs, with a further TNF release [42,72].
An elegant study by Centonze et al. has shown that increased concentration of TNF released by activated microglia induces changes in the expression and physiological properties of glutamate AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors (AMPRs) and NMDA (N-methyl-D-aspartate) receptors (NMDARs) in EAE mice [73]. Specifically, TNF acting on neuronal TNFR1 receptors causes excitotoxicity by increasing the surface expression of AMPARs and the activation of NMDARs, prolonging the duration of glutamate post-synaptic response [73].
In addition to excitotoxicity, TNF/TNFR1 signalling is involved in triggering oligodendrocyte apoptosis [32]. Consistent with this, TNF overexpressing transgenic mice developed spontaneous demyelinating lesions similar to those seen in MS [32,74,75]. On the contrary, TNF through TNFR2 facilitates remyelination by promoting oligodendrocyte differentiation in EAE [76]. Furthermore, TNFR2 KO mice develop more severe EAE motor disease than WT mice [76,77,78]. In particular, TNFR2 conditional KO EAE mice, a novel transgenic mouse with selective TNFR2 ablation in oligodendrocytes, have shown that altered TNFR2 signalling results in impaired remyelination [78].
These findings suggest that TNF could have a bimodal action depending on the receptor subunit involved in its signalling cascade. TNFR1 signalling plays a harmful role in MS, while when TNF acts via TNFR2 it exerts a protective action resulting in an attenuation of aggressive course of the disease in EAE. In MS, TNF (through enhanced TNFR1- and weak TNFR2-signalling) contributes to disease pathogenesis and progression, leading to inflammatory demyelination, remyelination failure, and neuronal functional damage via synaptic damage.
4. TNF and meningeal inflammation.
Increased levels of pro-inflammatory cytokines and cytotoxic mediators are found in the CSF of MS patients [23]; specifically, CSF levels of TNF were correlated with the degree of disability in patients with PrMS [23,26,79,80] but not detected in patients with other neurodegenerative diseases [27]. This increase is determined by immune cell infiltration into the CNS; in particular, lymphocytes and macrophages enter the brain through the perivascular space and meninges, where they release cytokines and chemokines that trigger glial cells and neurons to release additional inflammatory mediators, such as IL-1β, TNF, and IFN-ɣ [81]. The resulting mounting intrathecal neuroinflammation induces a local and chronic immune response that alters synaptic transmission and neuroaxonal homeostasis [17], leading to an increasingly inflammatory environment in the CSF, which bathes the cortical layers [23,79]. In this regard, a strong correlation was found between CSF/meningeal inflammation and the degree of cortical damage, microglial activation, and axonal loss [17,23,79]. Chronic inflammation causes GM damage in MS from the earliest stages of the disease. It leads to disability accumulation independently from acute inflammation due to the decreased capacity of the compensatory mechanisms [17,25,82,83]. Early cortical GM damage is indeed related to a more severe and rapid disease course in terms of disability progression and cognitive impairment [25].
In this regard, Kosa and colleagues have recently demonstrated that neuroinflammation increases with MS progression, identifying specific inflammatory pathways correlated with MS progression, which include both innate and adaptive immunity of Th17 (IL17, GM-CSF and IL6), Th1 (IFNγ and TNF) and Th2 (IL13 and IL4) phenotype [24]. Moreover, Magliozzi et al. showed that meningeal inflammation specifically alters the balance between TNFR1 pro-cell death and TNFR1/TNFR2 pro-cell survival signalling, causing a more severe disease manifestation from the early stages [35]. In addition, this study not only confirmed the elevated TNF levels in the CSF of MS patients at the time of diagnosis but also showed a greater overexpression of the TNFR1 gene in MS patients, especially in cortical GM tissues of progressive subjects [35]. These results are in line with a recent study by Picon and colleagues that provides substantial evidence for TNF-mediated activation of necroptotic signaling via TNFR1 in cortical neurons of progressive MS cases [34]. In fact, the study demonstrates an increased expression of multiple steps in the TNF/TNFR1 signaling pathway leading to necroptosis, including the key proteins TNFR1, FADD, RIPK1, RIPK3 and MLKL [33].
All these results support the hypothesis that neurodegeneration in MS is mainly driven by chronic inflammation in the CNS, with a preponderant involvement of activated TNF/TNFR1 signalling.
5. TNF and MS lesions
Degenerative processes include demyelination, axonal injury and neuron loss, resulting in multifocal WM lesions and diffuse GM damage in subpial and subventricular regions close to CSF and meninges [84]. Significant upregulation of TNF and TNFR1 was found in white matter (WM) and subpial GM lesions [85].
WM lesions can be classified as active, chronic active (CALs; smouldering, slowly expanding, mixed active/inactive), remyelinating, and chronic inactive lesions [86]. Active lesions develop from normal appearing white matter (NAWM) and are characterized by areas of demyelination and activated macrophages and microglia. These lesions can remyelinate in the presence of activated microglia or evolve into CALs or inactive lesions [87]. CALs are characterized by a demyelinated hypocellular nucleus and rims of iron-laden activated microglia [88,89], while inactive lesions are well-defined areas of demyelination and axonal degeneration in the absence of inflammation [20,75,87].
Chronic compartmentalized inflammation leads to the formation of CALs, which increase in number as the disease progresses [28,90,91]. In fact, they represent more than half of all focal WM lesions especially in progressive MS patients [92], depicting a relevant pathological finding associated with a severe disease course mediated by neuroaxonal damage, in the absence of superimposed acute inflammatory activity [20,88,91].
TNF was identified in CALs but absent in inactive lesions with a consistent immunoreactivity reported principally in activated microglia and T cells at the lesion's edge [27,89]. In addition, a seminal study by Jackle et al. explored the immunological-molecular profile of CALs; through microarray analysis, they found an upregulation of different genes associated with immune functions including those for TNF and its receptors indicating its central role in the formation of CALs [93]. Specifically, the TNFR1 gene showed an almost 5-fold increased transcript expression in these lesion types [93].
TNF and TNFR1 also increased in cortical lesions [35]. GM damage, including cortical lesions and atrophy, is already present in the early disease phase [25,75,94,95], becoming more prominent during its progression [96]. Early cortical involvement is related to a more severe and rapid disease course in terms of disability progression and cognitive impairment. The transcriptional profile of chronic subpial GM lesions isolated from MS brain samples with prominent meningeal inflammation is consistent with skewing toward a detrimental, pro-inflammatory environment and microglia phenotype [85]. Such evidence is also supported by the above-mentioned study by Magliozzi et al., which demonstrates in subpial GM lesions of progressive MS cases higher levels of TNFR1 exclusively, but not TNFR2 [35]. Overall, these studies highlight TNF/TNFR1 signalling as potential future therapeutic targets for mitigating the impact of both CALs and GM lesions in MS.
6. TNF and PIRA
According to the newly proposed categorization, the different clinical MS phenotypes (RRMS, SPMS, PPMS, and PRMS) identified in 1996 [97] are summarized in relapsing–remitting disease versus progressive disease [98]. Both clinical forms of MS appear to reflect the same underlying disease process characterized by neuroinflammation and subsequent neurodegeneration [99,100] present in all MS lesions across the entire disease course [99,100,101,102,103,104]. In this context, compartmentalized neuroinflammation appears crucial in the onset and progression of neurodegenerative mechanisms that result in axonal loss and brain atrophy [105], strongly correlated with long-term functional and cognitive disability [106].
Several studies have also proven the association between focal inflammatory activity and diffuse and regional atrophic changes [12,107,109]. Specifically, MS lesions appear to cause brain volume loss through direct inflammatory damages leading to myelin and axonal loss and, indirectly, tissue loss following Wallerian degeneration [12].
In addition, evidence from neuropathological, imaging, and biomarker studies suggests a more continuous axonal loss across all clinically defined stages of MS, both in early and relapsing MS rather than in more advanced and progressive stages [8].
The classic RRMS/PrMS subdivision has been overcome since the emergence of a new concept of MS, the evidence of progression independent of relapse activity (PIRA) [12].
PIRA represents the first and main event responsible for irreversible disability accumulation in adult patients with RRMS, which occurs from 80% to 90% [8]. PIRA is already present in the early phases of disease and may even occur during disease-modifying treatments (DMTs) [12,106,109,110]. Two similar important studies investigating PIRA in early MS showed that about one-fourth of patients with RRMS may develop PIRA during the first ten years of the disease [111,112]. Patients who developed their first PIRA event very early in the disease course showed an unfavorable prognosis [112].
PIRA occurs in roughly 5% of all patients with RRMS per annum, causing at least 50% of all disability accrual events in typical RRMS [113]. In this regard, a recent study confirms that up to 50% of the disability accumulation in adult patients with RRMS is not associated with evident relapses [114]. Relapses may mask disease progression and the loss of function over time may be as gradual in some patients that the patient or physician does not notice them; this would explain why PIRA is underestimated in RRMS [114].
Furthermore, patients with PIRA show significantly increased GM atrophy and CALs number, providing additional important evidence of the association between PIRA and diffuse neurodegeneration [12].
Currently, there are no specific biomarkers that can identify PIRA conditions. Overall, the only biomarker of ongoing neuronal damage taken into consideration, is serum neurofilament light chain (sNfLs). However, its association with long-term clinical outcomes or its ability to reflect slow and diffuse neurodegenerative damage in MS is not completely clear [115]. This lack of clarity is probably due to unstable measurements subject to physiological changes such as age or body mass index fluctuations [115,116].
In addition, although early treatment with DMTs is considered effective in preventing the transition to the progressive phase [115], several recent observational studies failed to confirm a beneficial association of DMT with PIRA [113,118,119].
Hence, the need to find a biological target that specifically reflects current and future prognostic disability and irreversible CNS tissue damage due to PIRA represents an urgent need.
7. Anti-TNF therapy and their potential use for PIRA
Based on TNF’s strong pro-inflammatory activity, several anti-inflammatory drugs targeting TNF signalling have been developed and approved for treating inflammatory diseases, such as Crohn’s disease, ankylosing spondylitis, and rheumatoid arthritis. Specifically, five TNF blockers are available for clinical use: infliximab, adalimumab, golimumab, certolizumab and etanercept. There is anti-TNF serum based on either anti-TNF antibodies (infliximab, adalimumab, golimumab and certolizumab) or TNFR fusion proteins (etanercept) that act as antagonists by blocking TNF (both mTNF and sTNF) interactions with TNFRs [120]. Despite being considered relatively safe and effective for the above-mentioned diseases, serious effects associated with immune suppression have been reported in MS [121,122]. In particular, a clinical trial of infliximab showed unfavorable results, whit increased disease activity and MRI lesion load, proving the association of TNF inhibitors with CNS demyelination [121,123]. Although the relationship between TNF blockers and demyelination remains uncertain, it is easy to speculate that it is probably since these blockers are not selective, i.e., they block the interaction TNFR1, which has a primarily pro-inflammatory action, and TNFR2, which has a primarily protective effect. This confirms the crucial role of TNF in the CNS, which exerts both potent pro-inflammatory effects (via TNFR1) and essential protective functions (via TNFR2) under pathological conditions [29,62]. Specifically, TNF through TNFR2 signalling modulates the reactivity of self-reactive T cells to self-antigens, promoting the expansion of Tregs cells and, subsequently, the preservation of myelin-oligodendrocytes [84]. Selective inhibition of TNFR1 and selective activation of TNFR2 through the use and even discovery of new antagonist and agonist antibodies could represent a new molecular target for developing therapeutic agents in MS [45]. In this regard, a recent preclinical study showed that treating atrosab, a human monovalent antibody against TNFR1 developed for treating inflammatory diseases, reduces disease severity. This preclinical evidence seems promising for finding novel effective drugs for MS and perhaps PIRA in the future [29].
8. Discussion and conclusions
TNF is considered the major cytokine involved in the pathogenesis of MS [29,30]. TNF exerts its pleiotropic effects by interacting with its receptors (TNFRs), TNFR1 and TNFR2, inducing respectively survival or cell death signals. TNFR1 signalling appears to be involved in the induction of neuroinflammatory processes, while TNFR2 is involved in neuroprotection and sustains homeostasis processes. The relative levels of TNFR1 and TNFR2 on the cellular surface and their activation status determine the complexity of the TNF/TNFRs signal. In MS, the alterations in TNFR1/TNFR2 balance have been confirmed [49] and associated with a more severe MS course, since the earliest stages of the disease [35]. In this regard, the use of transgenic mouse models has significantly contributed to understanding the pathological role of TNF in MS [45]. TNF/TNFR1 KO mice showed a milder disease course [64,65], whereas TNFR2 KO mice showed more severe EAE symptoms and diffuse demyelination [65]. Moreover, the selective ablation of TNFR2 in oligodendrocytes resulted in impaired remyelination [78]. Several studies confirmed that TNF/TNFR1 signalling mediates necroptosis and apoptosis in oligodendrocytes [32,33] and causes neuronal excitotoxicity [73] and necroptosis in cortical neurons [76], leading to inflammatory demyelinating processes and neurodegeneration. Demyelination and neurodegeneration are associated with the formation and expansion of typical pathological lesions, contributing to MS pathogenesis and progression. It is, therefore, not surprising that TNF and TNFR1 have been found overexpressed at the level of CALs and GM lesions in MS patients [27,35,89,93], especially in those with progressive MS [35]. In addition, increased levels of TNF were detected in the CSF of MS patients [23] to correlate with the degree of disability in patients with progressive disease [23,26,79,80]. TNF increase in the CSF is indeed associated with a chronic compartmentalized inflammation, which causes GM damage from the early stages, leading to more severe and rapid disease in terms of disability accumulation, disability progression and cognitive impairment [17,25]. All these results (summarized in Table 1) suggest a possible role of TNF/TNFR1 signal activation in disease progression independent of acute inflammation and in a decrease of compensatory mechanisms following a neuronal insult [17,25,82,83].
Progression independent of relapse activity, PIRA, is the main contributor to irreversible disability accumulation since the early phases of the disease and along the entire disease course [8,12,111–113]. However, disease progression can be extremely gradual and slow compared to relapses, making PIRA underestimated, not easily recognized [114], and, consequently, untreatable, although no recent observational studies confirm a beneficial association of DMT with PIRA [113,118,119].
Likewise, the TNF/ TNFRs blockers, currently available for the treatment of several inflammatory diseases, are ineffective but also harmful for MS patients [121,122]. TNF inhibitors' failure in MS therapy could be due to their unselective action, which, in attempting to inhibit TNFR1 signalling, fails to preserve the neuroprotective and cell survival processes associated with TNFR2 signalling.
For this reason, the use of selective modulators of TNFRs through TNFR2 activation and/or TNFR1 silencing (i.e., atrosab) represents a valid potential therapeutic option. Alterations in TNF/TNFRs signalling seem to be implicated not only in inflammatory but also in neurodegenerative processes, suggesting TNF and its receptors could be possible prognostic and therapeutic targets of PIRA.
Author Contributions
Conceptualization, V.M. and M.C.; writing—original draft preparation, V.M.; writing—review and editing, F.C.; visualization, E.T; M.G.; M.B.; S.Z.; F.V.; V.C.; D.M.; A.T.; supervision, M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Browne P, Chandraratna D, Angood C, Tremlett H, Baker C, Taylor BV, Thompson AJ. Atlas of Multiple Sclerosis 2013: A growing global problem with widespread inequity. Neurology. 2014 Sep 9;83(11):1022-4. [CrossRef]
- Koch-Henriksen N, Sørensen PS. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol. 2010 May;9(5):520-32. [CrossRef]
- Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, Rocca MA. Multiple sclerosis. Nat Rev Dis Primers. 2018 Nov 8;4(1):43. Erratum in: Nat Rev Dis Primers. 2018 Nov 22;4(1):49. [CrossRef]
- Wan ECK. Cellular and Molecular Mechanisms in the Pathogenesis of Multiple Sclerosis. Cells. 2020 Oct 1;9(10):2223. [CrossRef]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008 Oct 25;372(9648):1502-17. [CrossRef]
- Vollmer TL, Nair KV, Williams IM, Alvarez E. Multiple Sclerosis Phenotypes as a Continuum: The Role of Neurologic Reserve. Neurol Clin Pract. 2021 Aug;11(4):342-351. [CrossRef]
- Scalfari, A. MS can be considered a primary progressive disease in all cases, but some patients have superimposed relapses - Yes. Mult Scler. 2021 Jun;27(7):1002-1004. [CrossRef]
- Kappos L, Wolinsky JS, Giovannoni G, Arnold DL, Wang Q, Bernasconi C, Model F, Koendgen H, Manfrini M, Belachew S, Hauser SL. Contribution of Relapse-Independent Progression vs Relapse-Associated Worsening to Overall Confirmed Disability Accumulation in Typical Relapsing Multiple Sclerosis in a Pooled Analysis of 2 Randomized Clinical Trials. JAMA Neurol. 2020 Sep 1;77(9):1132-1140. [CrossRef]
- Cree BAC, Hollenbach JA, Bove R, Kirkish G, Sacco S, Caverzasi E, Bischof A, Gundel T, Zhu AH, Papinutto N, Stern WA, Bevan C, Romeo A, Goodin DS, Gelfand JM, Graves J, Green AJ, Wilson MR, Zamvil SS, Zhao C, Gomez R, Ragan NR, Rush GQ, Barba P, Santaniello A, Baranzini SE, Oksenberg JR, Henry RG, Hauser SL. Silent progression in disease activity-free relapsing multiple sclerosis. Ann Neurol. 2019 May;85(5):653-666. [CrossRef]
- Cree BA, Gourraud PA, Oksenberg JR, Bevan C, Crabtree-Hartman E, Gelfand JM, Goodin DS, Graves J, Green AJ, Mowry E, Okuda DT, Pelletier D, von Büdingen HC, Zamvil SS, Agrawal A, Caillier S, Ciocca C, Gomez R, Kanner R, Lincoln R, Lizee A, Qualley P, Santaniello A, Suleiman L, Bucci M, Panara V, Papinutto N, Stern WA, Zhu AH, Cutter GR, Baranzini S, Henry RG, Hauser SL. Long-term evolution of multiple sclerosis disability in the treatment era. Ann Neurol. 2016 Oct;80(4):499-510. [CrossRef]
- Tur C, Carbonell-Mirabent P, Cobo-Calvo Á, Otero-Romero S, Arrambide G, Midaglia L, Castilló J, Vidal-Jordana Á, Rodríguez-Acevedo B, Zabalza A, Galán I, Nos C, Salerno A, Auger C, Pareto D, Comabella M, Río J, Sastre-Garriga J, Rovira À, Tintoré M, Montalban X. Association of Early Progression Independent of Relapse Activity With Long-term Disability After a First Demyelinating Event in Multiple Sclerosis. JAMA Neurol. 2023 Feb 1;80(2):151-160. [CrossRef]
- Cagol A, Schaedelin S, Barakovic M, Benkert P, Todea RA, Rahmanzadeh R, Galbusera R, Lu PJ, Weigel M, Melie-Garcia L, Ruberte E, Siebenborn N, Battaglini M, Radue EW, Yaldizli Ö, Oechtering J, Sinnecker T, Lorscheider J, Fischer-Barnicol B, Müller S, Achtnichts L, Vehoff J, Disanto G, Findling O, Chan A, Salmen A, Pot C, Bridel C, Zecca C, Derfuss T, Lieb JM, Remonda L, Wagner F, Vargas MI, Du Pasquier R, Lalive PH, Pravatà E, Weber J, Cattin PC, Gobbi C, Leppert D, Kappos L, Kuhle J, Granziera C. Association of Brain Atrophy With Disease Progression Independent of Relapse Activity in Patients With Relapsing Multiple Sclerosis. JAMA Neurol. 2022 Jul 1;79(7):682-692. [CrossRef]
- Filippi M, Preziosa P, Copetti M, Riccitelli G, Horsfield MA, Martinelli V, Comi G, Rocca MA. Gray matter damage predicts the accumulation of disability 13 years later in MS. Neurology. 2013 Nov 12;81(20):1759-67. [CrossRef]
- Scalfari A, Romualdi C, Nicholas RS, Mattoscio M, Magliozzi R, Morra A, Monaco S, Muraro PA, Calabrese M. The cortical damage, early relapses, and onset of the progressive phase in multiple sclerosis. Neurology. 2018 Jun 12;90(24):e2107-e2118. [CrossRef]
- Haider L, Prados F, Chung K, Goodkin O, Kanber B, Sudre C, Yiannakas M, Samson RS, Mangesius S, Thompson AJ, Gandini Wheeler-Kingshott CAM, Ciccarelli O, Chard DT, Barkhof F. Cortical involvement determines impairment 30 years after a clinically isolated syndrome. Brain. 2021 Jun 22;144(5):1384-1395. [CrossRef]
- Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, Serafini B, Aloisi F, Roncaroli F, Magliozzi R, Reynolds R. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011 Sep;134(Pt 9):2755-71. [CrossRef]
- James RE, Schalks R, Browne E, Eleftheriadou I, Munoz CP, Mazarakis ND, Reynolds R. Persistent elevation of intrathecal pro-inflammatory cytokines leads to multiple sclerosis-like cortical demyelination and neurodegeneration. Acta Neuropathol Commun. 2020 May 12;8(1):66. [CrossRef]
- Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130:1089–1104. [CrossRef]
- Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010;68:477–493. [CrossRef]
- Absinta M, Sati P, Masuzzo F, Nair G, Sethi V, Kolb H, Ohayon J, Wu T, Cortese ICM, Reich DS. Association of Chronic Active Multiple Sclerosis Lesions With Disability In Vivo. JAMA Neurol. 2019 Dec 1;76(12):1474-1483. Erratum in: JAMA Neurol. 2019 Dec 1;76(12):1520. [CrossRef]
- Absinta M, Lassmann H, Trapp BD. Mechanisms underlying progression in multiple sclerosis. Curr Opin Neurol. 2020 Jun;33(3):277-285. [CrossRef]
- Ahmed SM, Fransen NL, Touil H, Michailidou I, Huitinga I, Gommerman JL, Bar-Or A, Ramaglia V. Accumulation of meningeal lymphocytes correlates with white matter lesion activity in progressive multiple sclerosis. JCI Insight. 2022 Mar 8;7(5):e151683. [CrossRef]
- Magliozzi R, Howell OW, Nicholas R, Cruciani C, Castellaro M, Romualdi C, Rossi S, Pitteri M, Benedetti MD, Gajofatto A, Pizzini FB, Montemezzi S, Rasia S, Capra R, Bertoldo A, Facchiano F, Monaco S, Reynolds R, Calabrese M. Inflammatory intrathecal profiles and cortical damage in multiple sclerosis. Ann Neurol. 2018 Apr;83(4):739-755. [CrossRef]
- Kosa P, Barbour C, Varosanec M, Wichman A, Sandford M, Greenwood M, Bielekova B. Molecular models of multiple sclerosis severity identify heterogeneity of pathogenic mechanisms. Nat Commun. 2022 Dec 12;13(1):7670. [CrossRef]
- Calabrese M, Magliozzi R, Ciccarelli O, Geurts JJ, Reynolds R, Martin R. Exploring the origins of grey matter damage in multiple sclerosis. Nat Rev Neurosci. 2015 Mar;16(3):147-58. [CrossRef]
- Sharief MK, Hentges R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med. 1991 Aug 15;325(7):467-72. [CrossRef]
- Selmaj K, Raine CS, Cannella B, Brosnan CF. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J Clin Invest. 1991 Mar;87(3):949-54. [CrossRef]
- Fischer MT, Wimmer I, Höftberger R, Gerlach S, Haider L, Zrzavy T, Hametner S, Mahad D, Binder CJ, Krumbholz M, Bauer J, Bradl M, Lassmann H. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain. 2013 Jun;136(Pt 6):1799-815. [CrossRef]
- Zahid M, Busmail A, Penumetcha SS, Ahluwalia S, Irfan R, Khan SA, Rohit Reddy S, Vasquez Lopez ME, Mohammed L. Tumor Necrosis Factor Alpha Blockade and Multiple Sclerosis: Exploring New Avenues. Cureus. 2021 Oct 17;13(10):e18847. [CrossRef]
- Navikas, V. & Link, H. Cytokines and the pathogenesis of multiple sclerosis [Review]. J. Neurosci. Res. 45, 322–333 (1996).
- Olmos G, Lladó J. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014;2014:861231. [CrossRef]
- Akassoglou K, Bauer J, Kassiotis G, Pasparakis M, Lassmann H, Kollias G, Probert L. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: models for multiple sclerosis with primary oligodendrogliopathy. Am J Pathol. 1998 Sep;153(3):801-13. [CrossRef]
- Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg H, Geng J, Py B, Zhou W, Amin P, Berlink Lima J, Qi C, Yu Q, Trapp B, Yuan J. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015 Mar 24;10(11):1836-49. [CrossRef]
- Picon C, Jayaraman A, James R, Beck C, Gallego P, Witte ME, van Horssen J, Mazarakis ND, Reynolds R. Neuron-specific activation of necroptosis signaling in multiple sclerosis cortical grey matter. Acta Neuropathol. 2021 Apr;141(4):585-604. [CrossRef]
- Magliozzi R, Howell OW, Durrenberger P, Aricò E, James R, Cruciani C, Reeves C, Roncaroli F, Nicholas R, Reynolds R. Meningeal inflammation changes the balance of TNF signalling in cortical grey matter in multiple sclerosis. J Neuroinflammation. 2019 Dec 7;16(1):259. [CrossRef]
- De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, Piccio L, Raychaudhuri S, Tran D, Aubin C, Briskin R, Romano S; International MS Genetics Consortium; Baranzini SE, McCauley JL, Pericak-Vance MA, Haines JL, Gibson RA, Naeglin Y, Uitdehaag B, Matthews PM, Kappos L, Polman C, McArdle WL, Strachan DP, Evans D, Cross AH, Daly MJ, Compston A, Sawcer SJ, Weiner HL, Hauser SL, Hafler DA, Oksenberg JR. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009 Jul;41(7):776-82. [CrossRef]
- Holbrook J, Lara-Reyna S, Jarosz-Griffiths H, McDermott M. Tumour necrosis factor signalling in health and disease. F1000Res. 2019 Jan 28;8:F1000 Faculty Rev-111. [CrossRef]
- Caminero A, Comabella M, Montalban X. Role of tumour necrosis factor (TNF)-α and TNFRSF1A R92Q mutation in the pathogenesis of TNF receptor-associated periodic syndrome and multiple sclerosis. Clin. Exp. Immunol. 2011, 166, 338–345.
- McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008 Oct 17;5:45. [CrossRef]
- Beutler B, Cerami A. The biology of cachectin/TNF--a primary mediator of the host response. Annu Rev Immunol. 1989;7:625-55. [CrossRef]
- Popa C, Netea MG, van Riel PL, van der Meer JW, Stalenhoef AF. The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res. 2007 Apr;48(4):751-62. [CrossRef]
- Maguire AD, Bethea JR, Kerr BJ. TNFα in MS and Its Animal Models: Implications for Chronic Pain in the Disease. Front Neurol. 2021 Dec 6;12:780876. [CrossRef]
- Liu Y, Liu T, Lei T, Zhang D, Du S, Girani L, et al.. RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review). Int J Mol Med. 201. 44:771–86. 10.3892/ijmm.2019.4244.
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996 Nov 1;274(5288):787-9. [CrossRef]
- Fresegna D, Bullitta S, Musella A, Rizzo FR, De Vito F, Guadalupi L, Caioli S, Balletta S, Sanna K, Dolcetti E, Vanni V, Bruno A, Buttari F, Stampanoni Bassi M, Mandolesi G, Centonze D, Gentile A. Re-Examining the Role of TNF in MS Pathogenesis and Therapy. Cells. 2020 Oct 14;9(10):2290. [CrossRef]
- Tartaglia LA, Rothe M, Hu YF, Goeddel DV. Tumor necrosis factor's cytotoxic activity is signaled by the p55 TNF receptor. Cell. 1993 Apr 23;73(2):213-6. [CrossRef]
- Hayden MS, Ghosh S. Regulation of NF-κB by TNF family cytokines. Semin Immunol. 2014 Jun;26(3):253-66. [CrossRef]
- Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol. 2014 Jun;26(3):237-45. [CrossRef]
- Brenner, D., Blaser, H. & Mak, T. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 15, 362–374 (2015). [CrossRef]
- Barton A, John S, Ollier WE, Silman A, Worthington J. Association between rheumatoid arthritis and polymorphism of tumor necrosis factor receptor II, but not tumor necrosis factor receptor I, in Caucasians. Arthritis Rheum. 2001 Jan;44(1):61-5. [CrossRef]
- Komata T, Tsuchiya N, Matsushita M, Hagiwara K, Tokunaga K. Association of tumor necrosis factor receptor 2 (TNFR2) polymorphism with susceptibility to systemic lupus erythematosus. Tissue Antigens. 1999 Jun;53(6):527-33. [CrossRef]
- Sashio H, Tamura K, Ito R, Yamamoto Y, Bamba H, Kosaka T, Fukui S, Sawada K, Fukuda Y, Tamura K, Satomi M, Shimoyama T, Furuyama J. Polymorphisms of the TNF gene and the TNF receptor superfamily member 1B gene are associated with susceptibility to ulcerative colitis and Crohn's disease, respectively. Immunogenetics. 2002 Mar;53(12):1020-7. [CrossRef]
- Scheurich P, Thoma B, Ucer U, Pfizenmaier K. Immunoregulatory activity of recombinant human tumor necrosis factor (TNF)-alpha: induction of TNF receptors on human T cells and TNF-alpha-mediated enhancement of T cell responses. J Immunol. 1987 Mar 15;138(6):1786-90.
- Kassiotis G, Pasparakis M, Kollias G, Probert L. TNF accelerates the onset but does not alter the incidence and severity of myelin basic protein-induced experimental autoimmune encephalomyelitis. Eur J Immunol. 1999 Mar;29(3):774-80. [CrossRef]
- Russi AE, Walker-Caulfield ME, Guo Y, Lucchinetti CF, Brown MA. Meningeal mast cell-T cell crosstalk regulates T cell encephalitogenicity. J Autoimmun. 2016 Sep;73:100-10. [CrossRef]
- Begolka WS, Vanderlugt CL, Rahbe SM, Miller SD. Differential expression of inflammatory cytokines parallels progression of central nervous system pathology in two clinically distinct models of multiple sclerosis. J Immunol. 1998 Oct 15;161(8):4437-46.
- Vladić A, Horvat G, Vukadin S, Sucić Z, Simaga S. Cerebrospinal fluid and serum protein levels of tumour necrosis factor-alpha (TNF-alpha) interleukin-6 (IL-6) and soluble interleukin-6 receptor (sIL-6R gp80) in multiple sclerosis patients. Cytokine. 2002 Oct 21;20(2):86-9. [CrossRef]
- Maimone D, Gregory S, Arnason BG, Reder AT. Cytokine levels in the cerebrospinal fluid and serum of patients with multiple sclerosis. J Neuroimmunol. 1991 Apr;32(1):67-74. [CrossRef]
- Williams SK, Maier O, Fischer R, Fairless R, Hochmeister S, Stojic A, Pick L, Haar D, Musiol S, Storch MK, Pfizenmaier K, Diem R. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS One. 2014 Feb 28;9(2):e90117. [CrossRef]
- Rieckmann P, Albrecht M, Ehrenreich H, Weber T, Michel U. Semi-quantitative analysis of cytokine gene expression in blood and cerebrospinal fluid cells by reverse transcriptase polymerase chain reaction. Res Exp Med (Berl). 1995;195(1):17-29. [CrossRef]
- van Oosten BW, Barkhof F, Scholten PE, von Blomberg BM, Adèr HJ, Polman CH. Increased production of tumor necrosis factor alpha, and not of interferon gamma, preceding disease activity in patients with multiple sclerosis. Arch Neurol. 1998 Jun;55(6):793-8. [CrossRef]
- Kemanetzoglou E, Andreadou E. CNS Demyelination with TNF-α Blockers. Curr Neurol Neurosci Rep. 2017 Apr;17(4):36. PMID: 28337644; PMCID: PMC5364240. [CrossRef]
- Valentin-Torres A, Savarin C, Hinton DR, Phares TW, Bergmann CC, Stohlman SA. Sustained TNF production by central nervous system infiltrating macrophages promotes progressive autoimmune encephalomyelitis. J Neuroinflammation. 2016 Feb 22; 13:46. [CrossRef]
- Körner H, Lemckert FA, Chaudhri G, Etteldorf S, Sedgwick JD. Tumor necrosis factor blockade in actively induced experimental autoimmune encephalomyelitis prevents clinical disease despite activated T cell infiltration to the central nervous system. Eur J Immunol. 1997 Aug;27(8):1973-81. [CrossRef]
- Suvannavejh GC, Lee HO, Padilla J, Dal Canto MC, Barrett TA, Miller SD. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35-55)-induced experimental autoimmune encephalomyelitis. Cell Immunol. 2000 Oct 10;205(1):24-33. [CrossRef]
- Yang S, Wang J, Brand DD, Zheng SG. Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front Immunol. 2018 Apr 19;9:784. [CrossRef]
- Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006 Jul 1;108(1):253-61. [CrossRef]
- Chen X, Subleski JJ, Kopf H, Howard OM, Männel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol. 2008 May 15;180(10):6467-71. [CrossRef]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003 Feb 14;299(5609):1057-61. [CrossRef]
- Sospedra M, Martin R. Immunology of Multiple Sclerosis. Semin Neurol. 2016 Apr;36(2):115-27. [CrossRef]
- Atretkhany KN, Mufazalov IA, Dunst J, Kuchmiy A, Gogoleva VS, Andruszewski D, Drutskaya MS, Faustman DL, Schwabenland M, Prinz M, Kruglov AA, Waisman A, Nedospasov SA. Intrinsic TNFR2 signaling in T regulatory cells provides protection in CNS autoimmunity. Proc Natl Acad Sci U S A. 2018 Dec 18;115(51):13051-13056. [CrossRef]
- Dopp JM, Mackenzie-Graham A, Otero GC, Merrill JE. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol. 1997 May;75(1-2):104-12. [CrossRef]
- Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A, Musella A, D'Amelio M, Cavallucci V, Martorana A, Bergamaschi A, Cencioni MT, Diamantini A, Butti E, Comi G, Bernardi G, Cecconi F, Battistini L, Furlan R, Martino G. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci. 2009 Mar 18;29(11):3442-52. [CrossRef]
- Probert L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience. 2015 Aug 27;302:2-22. [CrossRef]
- Lucchinetti CF, Brück W, Rodriguez M, Lassmann H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity on pathogenesis. Brain Pathol. 1996 Jul;6(3):259-74. [CrossRef]
- hher R, Padutsch T, Bracchi-Ricard V, Murphy KL, Martinez GF, Delguercio N, Elmer N, Sendetski M, Diem R, Eisel ULM, Smeyne RJ, Kontermann RE, Pfizenmaier K, Bethea JR. Exogenous activation of tumor necrosis factor receptor 2 promotes recovery from sensory and motor disease in a model of multiple sclerosis. Brain Behav Immun. 2019 Oct;81:247-259. [CrossRef]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001 Nov;4(11):1116-22. [CrossRef]
- Madsen PM, Motti D, Karmally S, Szymkowski DE, Lambertsen KL, Bethea JR, Brambilla R. Oligodendroglial TNFR2 Mediates Membrane TNF-Dependent Repair in Experimental Autoimmune Encephalomyelitis by Promoting Oligodendrocyte Differentiation and Remyelination. J Neurosci. 2016 May 4;36(18):5128-43. [CrossRef]
- Magliozzi R, Scalfari A, Pisani AI, Ziccardi S, Marastoni D, Pizzini FB, Bajrami A, Tamanti A, Guandalini M, Bonomi S, Rossi S, Mazziotti V, Castellaro M, Montemezzi S, Rasia S, Capra R, Pitteri M, Romualdi C, Reynolds R, Calabrese M. The CSF Profile Linked to Cortical Damage Predicts Multiple Sclerosis Activity. Ann Neurol. 2020 Sep;88(3):562-573. [CrossRef]
- Javor J, Shawkatová I, Ďurmanová V, Párnická Z, Čierny D, Michalik J, Čopíková-Cudráková D, Smahová B, Gmitterová K, Peterajová Ľ, Bucová M. TNFRSF1A polymorphisms and their role in multiple sclerosis susceptibility and severity in the Slovak population. Int J Immunogenet. 2018 Jul 16. [CrossRef]
- Kalafatakis I, Karagogeos D. Oligodendrocytes and Microglia: Key Players in Myelin Development, Damage and Repair. Biomolecules. 2021 Jul 20;11(7):1058. [CrossRef]
- Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015 Sep 15;15(9):545-58. [CrossRef]
- Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012 Nov 5;8(11):647-56. [CrossRef]
- Healy LM, Stratton JA, Kuhlmann T, Antel J. The role of glial cells in multiple sclerosis disease progression. Nat Rev Neurol. 2022 Apr;18(4):237-248. [CrossRef]
- Veroni C, Serafini B, Rosicarelli B, Fagnani C, Aloisi F, Agresti C. Connecting Immune Cell Infiltration to the Multitasking Microglia Response and TNF Receptor 2 Induction in the Multiple Sclerosis Brain. Front Cell Neurosci. 2020 Jul 7; 14:190. [CrossRef]
- Kuhlmann T, Ludwin S, Prat A, Antel J, Brück W, Lassmann H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017 Jan;133(1):13-24. [CrossRef]
- Prineas JW, Kwon EE, Cho ES, Sharer LR, Barnett MH, Oleszak EL, Hoffman B, Morgan BP. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol. 2001 Nov;50(5):646-57. [CrossRef]
- Calvi A, Clarke MA, Prados F, Chard D, Ciccarelli O, Alberich M, Pareto D, Rodríguez Barranco M, Sastre-Garriga J, Tur C, Rovira A, Barkhof F. Relationship between paramagnetic rim lesions and slowly expanding lesions in multiple sclerosis. Mult Scler. 2023 Mar;29(3):352-362. [CrossRef]
- Hofman FM, Hinton DR, Johnson K, Merrill JE. Tumor necrosis factor identified in multiple sclerosis brain. J Exp Med. 1989 Aug 1;170(2):607-12. [CrossRef]
- Elkjaer ML, Frisch T, Reynolds R, Kacprowski T, Burton M, Kruse TA, Thomassen M, Baumbach J, Illes Z. Molecular signature of different lesion types in the brain white matter of patients with progressive multiple sclerosis. Acta Neuropathol Commun. 2019 Dec 11;7(1):205. [CrossRef]
- Luchetti S, Fransen NL, van Eden CG, Ramaglia V, Mason M, Huitinga I. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol. 2018 Apr;135(4):511-528. [CrossRef]
- Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I, Mandrekar J, Bramow S, Metz I, Brück W, Lassmann H, Lucchinetti CF. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. 2015 Nov;78(5):710-21. [CrossRef]
- Jäckle K, Zeis T, Schaeren-Wiemers N, Junker A, van der Meer F, Kramann N, Stadelmann C, Brück W. Molecular signature of slowly expanding lesions in progressive multiple sclerosis. Brain. 2020 Jul 1;143(7):2073-2088. [CrossRef]
- Wegner C, Esiri MM, Chance SA, Palace J, Matthews PM. Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology. 2006 Sep 26;67(6):960-7. [CrossRef]
- Kutzelnigg A, Lassmann H. Cortical lesions and brain atrophy in MS. J Neurol Sci. 2005 Jun 15;233(1-2):55-9. [CrossRef]
- Amato MP, Bartolozzi ML, Zipoli V, Portaccio E, Mortilla M, Guidi L, Siracusa G, Sorbi S, Federico A, De Stefano N. Neocortical volume decrease in relapsing-remitting MS patients with mild cognitive impairment. Neurology. 2004 Jul 13;63(1):89-93. [CrossRef]
- Klineova S, Lublin FD. Clinical Course of Multiple Sclerosis. Cold Spring Harb Perspect Med. 2018 Sep 4;8(9):a028928. [CrossRef]
- Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, Wolinsky JS, Balcer LJ, Banwell B, Barkhof F, Bebo B Jr, Calabresi PA, Clanet M, Comi G, Fox RJ, Freedman MS, Goodman AD, Inglese M, Kappos L, Kieseier BC, Lincoln JA, Lubetzki C, Miller AE, Montalban X, O'Connor PW, Petkau J, Pozzilli C, Rudick RA, Sormani MP, Stüve O, Waubant E, Polman CH. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014 Jul 15;83(3):278-86. [CrossRef]
- Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009 May;132(Pt 5):1175-89. [CrossRef]
- Hauser SL, Cree BAC. Treatment of Multiple Sclerosis: A Review. Am J Med. 2020 Dec;133(12):1380-1390.e2. [CrossRef]
- Doinikow B. Über De- und Regenerationserscheinungen an Achsenzylindern bei der multiplen Sklerose. Z ges Neurol Psych 1915; 27: 151–178.
- Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis. Brain 1997; 120: 393–399.
- Kuhlmann T, Moccia M, Coetzee T, Cohen JA, Correale J, Graves J, Marrie RA, Montalban X, Yong VW, Thompson AJ, Reich DS; International Advisory Committee on Clinical Trials in Multiple Sclerosis. Multiple sclerosis progression: time for a new mechanism-driven framework. Lancet Neurol. 2023 Jan;22(1):78-88. [CrossRef]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998 Jan 29;338(5):278-85. [CrossRef]
- Kapica-Topczewska K, Collin F, Tarasiuk J, Czarnowska A, Chorąży M, Mirończuk A, Kochanowicz J, Kułakowska A. Assessment of Disability Progression Independent of Relapse and Brain MRI Activity in Patients with Multiple Sclerosis in Poland. J Clin Med. 2021 Feb 19;10(4):868. [CrossRef]
- Del Negro I, Pez S, Gigli GL, Valente M. Disease Activity and Progression in Multiple Sclerosis: New Evidences and Future Perspectives. J Clin Med. 2022 Nov 9;11(22):6643. [CrossRef]
- Radue EW, Barkhof F, Kappos L, Sprenger T, Häring DA, de Vera A, von Rosenstiel P, Bright JR, Francis G, Cohen JA. Correlation between brain volume loss and clinical and MRI outcomes in multiple sclerosis. Neurology. 2015 Feb 24;84(8):784-93. [CrossRef]
- Wang C, Barnett MH, Yiannikas C, Barton J, Parratt J, You Y, Graham SL, Klistorner A. Lesion activity and chronic demyelination are the major determinants of brain atrophy in MS. Neurol Neuroimmunol Neuroinflamm. 2019 Jul 16;6(5):e593. [CrossRef]
- Ostini C, Bovis F, Disanto G, Ripellino P, Pravatà E, Sacco R, Padlina G, Sormani MP, Gobbi C, Zecca C. Recurrence and Prognostic Value of Asymptomatic Spinal Cord Lesions in Multiple Sclerosis. J Clin Med. 2021 Jan 26;10(3):463. [CrossRef]
- Kapica-Topczewska K, Collin F, Tarasiuk J, Czarnowska A, Chorąży M, Mirończuk A, Kochanowicz J, Kułakowska A. Assessment of Disability Progression Independent of Relapse and Brain MRI Activity in Patients with Multiple Sclerosis in Poland. J Clin Med. 2021 Feb 19;10(4):868. [CrossRef]
- Portaccio E, Bellinvia A, Fonderico M, Pastò L, Razzolini L, Totaro R, Spitaleri D, Lugaresi A, Cocco E, Onofrj M, Di Palma F, Patti F, Maimone D, Valentino P, Confalonieri P, Protti A, Sola P, Lus G, Maniscalco GT, Brescia Morra V, Salemi G, Granella F, Pesci I, Bergamaschi R, Aguglia U, Vianello M, Simone M, Lepore V, Iaffaldano P, Filippi M, Trojano M, Amato MP. Progression is independent of relapse activity in early multiple sclerosis: a real-life cohort study. Brain. 2022 Aug 27;145(8):2796-2805. [CrossRef]
- Tur C, Carbonell-Mirabent P, Cobo-Calvo Á, Otero-Romero S, Arrambide G, Midaglia L, Castilló J, Vidal-Jordana Á, Rodríguez-Acevedo B, Zabalza A, Galán I, Nos C, Salerno A, Auger C, Pareto D, Comabella M, Río J, Sastre-Garriga J, Rovira À, Tintoré M, Montalban X. Association of Early Progression Independent of Relapse Activity With Long-term Disability After a First Demyelinating Event in Multiple Sclerosis. JAMA Neurol. 2023 Feb 1;80(2):151-160. [CrossRef]
- Müller J, Cagol A, Lorscheider J, Tsagkas C, Benkert P, Yaldizli Ö, Kuhle J, Derfuss T, Sormani MP, Thompson A, Granziera C, Kappos L. Harmonizing Definitions for Progression Independent of Relapse Activity in Multiple Sclerosis: A Systematic Review. JAMA Neurol. 2023 Nov 1;80(11):1232-1245. [CrossRef]
- Lublin FD, Häring DA, Ganjgahi H, Ocampo A, Hatami F, Čuklina J, Aarden P, Dahlke F, Arnold DL, Wiendl H, Chitnis T, Nichols TE, Kieseier BC, Bermel RA. How patients with multiple sclerosis acquire disability. Brain. 2022 Sep 14;145(9):3147-3161. [CrossRef]
- Bittner S, Oh J, Havrdová EK, Tintoré M, Zipp F. The potential of serum neurofilament as biomarker for multiple sclerosis. Brain. 2021 Nov 29;144(10):2954-2963. [CrossRef]
- Khalil M, Pirpamer L, Hofer E, Voortman MM, Barro C, Leppert D, Benkert P, Ropele S, Enzinger C, Fazekas F, Schmidt R, Kuhle J. Serum neurofilament light levels in normal aging and their association with morphologic brain changes. Nat Commun. 2020 Feb 10;11(1):812. [CrossRef]
- Zou H, Li R, Hu H, Hu Y, Chen X. Modulation of Regulatory T Cell Activity by TNF Receptor Type II-Targeting Pharmacological Agents. Front Immunol. 2018 Mar 26;9:594. [CrossRef]
- Brown JWL, Coles A, Horakova D, Havrdova E, Izquierdo G, Prat A, Girard M, Duquette P, Trojano M, Lugaresi A, Bergamaschi R, Grammond P, Alroughani R, Hupperts R, McCombe P, Van Pesch V, Sola P, Ferraro D, Grand'Maison F, Terzi M, Lechner-Scott J, Flechter S, Slee M, Shaygannejad V, Pucci E, Granella F, Jokubaitis V, Willis M, Rice C, Scolding N, Wilkins A, Pearson OR, Ziemssen T, Hutchinson M, Harding K, Jones J, McGuigan C, Butzkueven H, Kalincik T, Robertson N; MSBase Study Group. Association of Initial Disease-Modifying Therapy With Later Conversion to Secondary Progressive Multiple Sclerosis. JAMA. 2019 Jan 15;321(2):175-187. [CrossRef]
- Bsteh G, Hegen H, Altmann P, et al. Retinal layer thinning is reflecting disability progression independent of relapse activity in multiple sclerosis. Mult Scler J Exp Transl Clin. 2020;6(4):2055217320966344. [CrossRef]
- Graf J, Leussink VI, Soncin G, et al. Relapse-independent multiple sclerosis progression under natalizumab. Brain Commun. 2021;3(4):fcab229. [CrossRef]
- van Oosten BW, Barkhof F, Truyen L, Boringa JB, Bertelsmann FW, von Blomberg BM, Woody JN, Hartung HP, Polman CH. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. 1996 Dec;47(6):1531-4. [CrossRef]
- Kemanetzoglou E, Andreadou E. CNS Demyelination with TNF-α Blockers. Curr Neurol Neurosci Rep. 2017 Apr;17(4):36. [CrossRef]
- Bosch X, Saiz A, Ramos-Casals M; BIOGEAS Study Group. Monoclonal antibody therapy-associated neurological disorders. Nat Rev Neurol. 2011 Mar;7(3):165-72. [CrossRef]
Figure 1.
TNF signalling. TNF signalling is mediated by two isoforms (mTNF and sTNF) exerting their effects by modulating a complex signalling pathway through two distinct surface TNF receptors: TNFR1 and TNFR2. TNFR1 shows a high affinity towards sTNF, which once bound recruits TRADD. TRADD binds FADD and RIPK1, leading to necroptosis through RIPK3 and MLKL activation, or apoptosis through caspase8 and caspase 3 activation. On the other hand, mTNF interacts with TNFR2, inducing inflammation and homeostasis through TRAF1/2 activation of JNK, ERK MAPKs, Akt and/ or NFκB.
Figure 1.
TNF signalling. TNF signalling is mediated by two isoforms (mTNF and sTNF) exerting their effects by modulating a complex signalling pathway through two distinct surface TNF receptors: TNFR1 and TNFR2. TNFR1 shows a high affinity towards sTNF, which once bound recruits TRADD. TRADD binds FADD and RIPK1, leading to necroptosis through RIPK3 and MLKL activation, or apoptosis through caspase8 and caspase 3 activation. On the other hand, mTNF interacts with TNFR2, inducing inflammation and homeostasis through TRAF1/2 activation of JNK, ERK MAPKs, Akt and/ or NFκB.
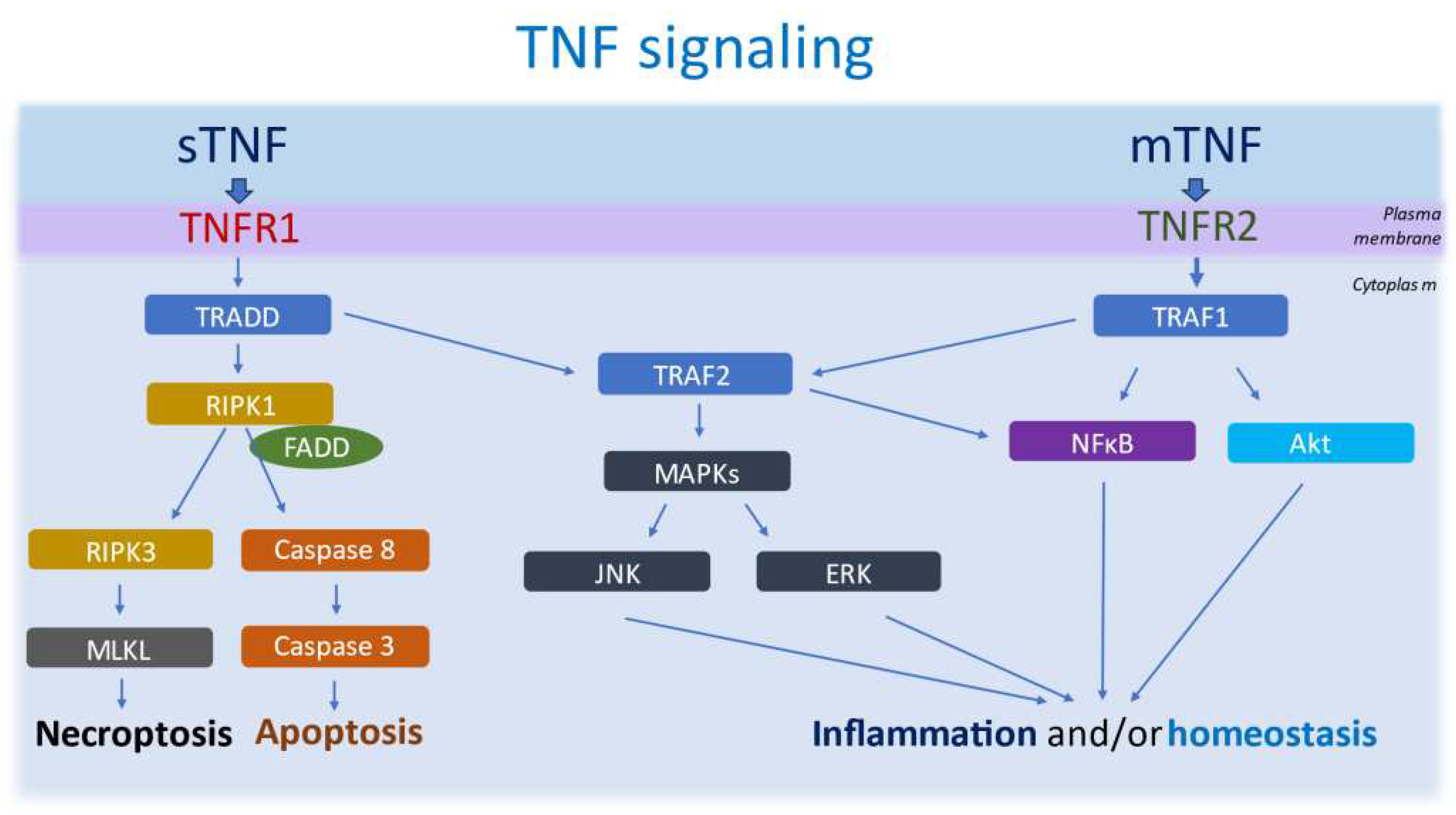

Table 1.
Studies on the role of TNF in MS and EAE.
| Type Cells Involved | Effects | Effects | Relevance | Relevance in MS | References | ||
|---|---|---|---|---|---|---|---|
| MS patients | TNF in the MS CSF | Macrophages, T cells | Infiltration of activated macrophages/T cells in the brain parenchyma | MS lesions formation | Inflammation | Disability accumulation | [53,54,55] |
| Neurons, Glial cells | Activation of neurons and glial cells | MS lesions formation | Neuroinflammation | [23,26,79,80] | |||
| TNF in MS Lesions | Neurons | Cortical lesionsand Atrophy | GM damage | Neurodegeneration | Disability progression | [35] | |
| Microglia, T cells | Chronic active lesions | WM lesions | Demyelination | [93] | |||
| EAE model | EAE mice | Macrophages, T cells | Infiltration of activated macrophages/T cells inthe brain parenchyma | WM lesions | Neuroinflammation | Diffuse Demyelination | [56] |
| Neurons | AMPAR/NMDAR Overexpression | Neuronal Excitotoxicity | Neurodegeneration | Severe Disease Symptoms | [73] | ||
| EAE TNF KO mice | Macrophages, T cells, Neurons, Glial cells | Infiltration reduction by macrophages and T cells; No AMPAR/NMDAR Overexpression; Enhanced Tregs cells | Neuronal Homeostasis | Neuroprotection | Severe Disease Symptoms | [64] | |
| EAE TNFR1 KO mice | Macrophages, T cells, Neurons, Glial cells | No AMPAR/NMDAR OverexpressionEnhanced Tregs cells | Remyelination Increase and Neurodegeneration Reduction | Neuroprotection | Reduction of disease signs and clinical symptoms | [65] | |
| EAE TNFR2 KO mice | T regs | Suppression of T regs in response to autoreactive T cells | Impaired Remyelination | Demyelination | Aggressive Disease | [70,71] | |
| Oligodendrocytes | Oligodendrocytes Death | Impaired Remyelination | Demyelination | Severe Disease Symptoms; Enhanced T cells infiltration in the CNS; Diffuse Demyelination | [65,78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



