Submitted:
03 January 2024
Posted:
04 January 2024
You are already at the latest version
Abstract
Menaquinone is one of the major lipoquinones involved in the respiratory chains of bacteria, which includes ubiquinone. Menaquinone is an essential component in the electron transfer process and ATP synthesis especially in some Gram-positive bacteria including Mycobacterium tuberculosis (M. Tb). However, mammals utilize ubiquinone in the analogous electron transfer processes, and do not synthesize menaquinone. In the biosynthesis of menaquinone, MenA is an important enzyme, which is involved in the conversion of 1,4-dihydroxy-2-napthoate to demethylmenaquinone via prenyl transferase activity. Therefore MenA is chosen as a drug target in determining the inhibitor. While designing and screening MenA inhibitors we observed that benzophenone derivatives showed high coordination with enzymes in transition state and interrupted the growth of M. Tb moderately. In addition, diphenyl ether derivatives also showed moderate MIC and IC50 results. Therefore, to improve the biological activity we synthesized a polymeric compound having both benzophenone and diphenyl ether groups. In addition, a hydrophilic functional group was introduced into the polymeric compound to enhance the biological activity. The synthesized polymeric inhibitor showed improved MIC and IC50 values to inhibit the M. tb infection.
Keywords:
Menaquinone
; inhibitor
; Mycobacterium tuberculosis
; MIC
; IC50
1. Introduction
Tuberculosis (TB) is the second largest infection in number of deaths around the world.[1] Approximately one third of the world population is thought to have latent tuberculosis infections. However, HIV-TB is arising fast and in addition, HIV is also stimulating latent TB to form active TB infection. In this order novel drugs are necessary to eradicate TB.[2] Most of the TB drugs were based on inhibiting the process of cell wall synthesis, and protease inhibitor [3] therefore a novel method of inhibiton is important for drug discovery and development.[4,5,6,7,8,9]
Traditional antibiotics are effective through few mechanisms of action that typically include inhibiting protein, cell wall, and nucleic acid synthesis. Specifically, antibiotics like penicillin and other β-lactams, as well as glycopeptides, act primarily upon cell wall synthesis. Tetracyclines, aminoglycosides, and macrolides inhibit protein synthesis; fluoroquinolones and rifamycins inhibit nucleic acid synthesis. However, and most importantly, microorganisms possess several different mechanisms through which they are able to circumvent these processes and, in turn, become resistant to antibiotics. Thus, the emergence and spread of the drug-resistant infectious diseases is a major obstacle for prevention and control efforts made by researchers. The increasing frequencies of drug-resistant bacteria have a tremendous effect on the field of drug development and have motivated scientists to explore new approaches and targets.[4,5,8,9,10,11]
Bacilli require electron transport chain components and ATP synthesis for survival. Recent studies have demonstrated that the electron transport system is a potential target for the development of antibacterial drugs. The lipoquinones are involved in the electron transport chain components of bacteria and it consists of menaquinone (MK; i.e., vitamin K) and ubiquinones(i.e., Coenzyme Q10). However, mammals require only ubiquinone for the same process and menaquinone is an important component in the electron transfer process in bacteria.[12] Lipid-soluble electron carriers, ubiquinone and menaquinones, play essential roles in electron transport-coupled ATP synthesis.[13,14] MK transfers electrons under both aerobic and anaerobic respiration in most Gram-positive bacteria, and ubiquinone is responsible for electron transfer under aerobic respiration in Gram-negative bacteria and MK under anaerobic condition in Gram-negative bacteria.[15,16] Because MK is not synthesized in humans, it is a potential target for the development of new antimicrobials against bacteria to overcome resistance.[2,4,5,8,17]
Also, menaquinones are the major lipoquinones of mycobacteria and many other gram positive bacteria [18]. Interestingly, biosynthesis of menaquinone is essential in mycobacteria.[17] Generally menaquinone was biosynthesized from chorismate, 1. In presence of MenF, MenD, MenC, MenE, MenB, chorismate is converted into 1,4-dihydroxy-2-napthoate, 2. 1,4-dihydroxy-2-napthoate, 2 undergoes decarboxylation and prenylation by MenA to produce demethylmenaquinone, 3. Eventually MenG methylates the demethylmenaquinone to form menaquinone, 4 (Figure 1). [4,5,6,7,8] Hence inhibition of MenA will interrupt the biosynthesis of menaquinone and prevent the growth of microorganisms. Therefore, MenA is chosen as a drug target to determine the inhibitor.[2,4,5,8,17]
Early reports reinforce the importance of specific enzymes in the MK biosynthetic pathway as potential targets for discovering novel molecules to fight multi-drug resistant Gram-positive pathogens using MK biosynthesis.[19,20,21,22] For instance, in 2012 Lu et al. reported on some inhibitors of 1,4-dihydroxy-2-naphthoyl-CoA synthase (MenB) in M. tb.[19] In a subsequent mechanism-based narration, o-succinylbenzoate-AMP-based analogues were synthesized and identified as inhibitors of acyl-CoA synthetase (MenE).[20,22] In 2007, benzophenone-based compounds were also synthesized and reported to show MenA inhibitory activities against Gram-positive bacteria and the Mycobacterium species.[23,24,25] These findings indicate that MenA is indeed a valid target against Gram-positive bacteria and Mycobacterium species.[23]
In addition to their potential as antibiotics, inhibitors of the menaquinone biosynthesis pathway could also be used as tools for studying the biology of bacteria.[2,4,5,8,17] These inhibitors could be used to investigate the roles of menaquinone and the pathway in the physiology and virulence of bacteria, providing valuable insights into the mechanisms of bacterial pathogenesis.
Hence, the biosynthesis of menaquinone is an essential pathway in the metabolism of many mycobacteria and Gram-positive bacteria. The pathway is responsible for the biosynthesis of a crucial component of the electron transport chain and is required for energy generation. Inhibitors of the menaquinone biosynthesis pathway have shown promise as potential drugs against drug-resistant strains of many bacteria including Gram-positive bacteria.[2,4,5,8,17]
In general, polymers that are active pharmaceutical ingredients can be also called as polymeric inhibitors.[26,27,28,29,30,31,32] Generally these are neither drug carriers nor prodrugs but drugs themselves. The high molecular weight polymeric inhibitors contain various functional characteristics to selectively recognize, coordinate with the enzymes in the disease causing species. In addition the high molecular weight of these polymeric inhibitors makes them systemically non-adsorbed, thus providing a long-term safety profile over traditional small molecule drug products.[26,27,28,29,30,31,32] Furthermore, the multiple functional groups incorporated in the polymer enhances the polyvalent binding interaction with the enzymes in bacteria,[33] which will be an added advantage for polymeric inhibitors over traditional small molecules.
In this regard, we designed a polymeric compound to inhibit the MenA enzyme of mycobacteria. The polymeric compound was also designed by taking into account the small molecule which showed reasonable inhibition individually by itself. The polymeric compound was synthesized and tested its characteristics as MenA inhibitor against M. tb.
2. Materials and Methods
Starting materials were purchased from Aldrich chemicals. Flash chromatography was performed using EM Science silica gel 60 (230-400 mesh). All glassware was oven dried, assembled hot and cooled under a stream of nitrogen before use. Reactions with air sensitive materials were carried out by standard syringe techniques. 1H-NMR were recorded on a Varian Unity/Inova-400 NB (400 MHz). Chemical shifts are reported in parts per million (ppm) down field from TMS, using residual CDCl3 (7.27 ppm) as an internal standard. Data are reported as follows: Chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of a doublet, m = multiplet, br = broad), coupling constants and integration. 13C-NMR was recorded in a Varian Unity/Inova-400 NB (100 MHz). Using broadband proton decoupling. Chemical shifts are reported in parts per million (ppm) downfield from TMS, using the middle resonance of CDCl3 (77.0 ppm) as an internal standard. Mass Spectra (MS-ESI) were obtained from Mass Spectrometry Lab, Chemistry Department, Colorado state university.
2.1. Synthesis of Benzophenone Derivative, 5
5 was synthesized as described previously.[2] Briefly, the phenolic group of benzophenone was reacted with dibromo-octane to give the aryl bromo compound in good yield. To a stirred solution of (phenyl)2,4-dihydroxyphenyl)methanone (1 eq.) in DMF were added K2CO3 (5 eq.) and 1,8-dibromooctane (1.5 eq.). After 12h, the reaction mixture was quenched with water and extracted with ethyl acetate. The combined extracts were washed with brine, dried over sodium sulphate, filtered and concentrated in vaccuo. The compound was purified by silica gel chromatography using hexane: EtOAc (30:1) solvent mixture to provide 85% yield of alkylated benzophenone.
Alkylated benzophenone: 1H-NMR (CDCl3, 300 MHz): δ 1.42 (m, 8H), 1.86 (m, 4H), 3.41 (t, 2H, J = 6.6 Hz), 4.01 (t, 2H, J = 6.3 Hz), 6.39 (d, 1H, J = 9.0 Hz), 6.50 (s, 1H), 7.53 (m, 4H), 7.62 (d, 2H, J = 8.2 Hz), 12.7 (s, 1H). 13C-NMR (CDCl3, 100 MHz): δ 25.8, 27.9, 28.6, 28.8, 29.0, 32.7, 33.9, 68.3, 101.4, 107.7, 112.9, 128.2, 128.8, 131.4, 135.1, 138.2, 165.7, 166.3.
2.2. Benzophenone N-Allylamine, 5
The alkylated benzophenone (1 eq.) was dissolved in 1mL of THF and followed by addition of allylamine (any amine) (1.5 eq.). The reaction mxture was stirred at rt for 3 days and after completion of reaction (confirmed by TLC), it was concentrated and loaded in silica gel column. 5 was purified by silica gel column chromatography using 3-5% ( methanol:chloroform) mixture to give 75 % yield of final product.
1H-NMR (CDCl3, 300 MHz): δ 1.43 (m, 8H), 1.90 (m, 4H), 2.26 (s, 3H), 2.39 (t, 2H, J = 6.3 Hz), 3.06 (d, 2H, J = 6.3 Hz), 4.06 (m, 2H), 5.19 (m, 2H), 5.90 (m, 1H), 6.40 (d, 1H, J = 6.6 Hz), 6.50 (s, 1H), 7.55 (m, 4H), 7.64 (m, 2H), 12.70 (s, 1H). 13C-NMR (CDCl3, 100 MHz): δ 23.4, 25.9, 27.4, 28.9, 29.2, 29.4, 33.2, 41.7, 57.0, 67.4, 98.4, 101.5, 107.8, 128.3, 128.8, 131.4, 135.2, 166.3.
2.3. Synthesis of Alkylated Phenol Bromide Compounds, 7
2.45 mmol of 4-(4-flouro phenyl)phenol, 6 was dissolved in DMF (4 mL) followed by addition of 2.7 mmol of dibromo-octane. The reaction mixture was cooled to 0 ºC and 2.7 mmol of potassium carbonate solid was added and the mixture was allowed to warm to RT and stirred for 12 h. After completion of the reaction (confirmed by TLC), the reaction mixture was quenched by adding saturated ammonium chloride solution, slightly acidified by 10 % HCl solution and extracted with ethyl acetate, dried and evoprated. The crude mixture is purified by silica gel column chromatography using 6 –25 % (EtOAc : Hexane) solvent mixture and gave 85 % yield of alkylated phenol bromide,7.
1H-NMR (CDCl3, 300 MHz): δ 1.45 (m, 8H), 1.82 (m, 4H), 3.42 (t, 2H, J = 6.9 Hz), 3.94 (t, 2H, J = 6.3 Hz), 6.92 (m, 8H).
2.4. Synthesis of Diphenyl Ether N-Allyl Compounds, 8
The alkylated phenol bromide, 0.057 mmol was dissolved in 1mL of THF and followed by addition of 0.57 mmol allylamine (any amine) addition. The reaction was carried out at rt for 3 days and after completion (confirmed by TLC), the reaction mixture was concentrated and purified by silica gel column chromatography using 3-5% ( methanol: chloroform) mixture to yield 75% of final compounds.
1H-NMR (CDCl3, 300 MHz): δ 1.49 (m, 8H), 1.78 (m, 4H), 2.26 (s, 3H), 2.38 (t, 2H, J = 7.8 Hz), 3.04 (d, 2H, J = 6.0 Hz), 3.94 (t, 2H, J = 6.6 Hz), 5.19 (m, 2H), 5.89 (m, 1H), 6.93 (m, 8H).
2.5. Synthesis of Polymeric Alkyl Bromide, 10
Poly(3,3′,4,4′-benzophenone tetra carboxylic anhydride-co-4,4′-oxydianiline/1,3-phenylenediamine, amic acid solution, 9 was reacted with dibromooctane in DMF in presence of potassium carbonate for 12 hours. After completion of the reaction (confirmed by TLC), the reaction mixture was filtered washed with hexane and dried.
2.6. Synthesis of Polymeric Alkyl Allyamine, 11
The dried product, 10 was dissolved in DMF and 1.2 eq. of N-methyl allylamine was added and allowed to react for 12 hours. After completion of the reaction (confirmed by TLC), the reaction mixture was filtered washed with hexane and dried. After purification of the crude mixture the compound was heated at 40 ºC for 12 hours to form water insoluble polymeric inhibitor and electron micrographs were taken before and after reacting with enzymes. For MIC and IC50 determination the polymeric inhibitor was ground into powder and dissolved in DMSO, where it looked like clear solution after sonication.
2.7. MIC Determination:
96-well U-bottom assay plates were seeded with 5 x 104 CFU in a volume of 0.1 mL/well for M. Tb H37Rv assays. The compound was serially diluted in DMSO as 1:2 dilutions in a separate 96-well plate and then transferred to the assay plate. The final concentration of DMSO in the assay plates did not exceed 2.5%. Plates were incubated at 37°C for 10 days. O.D.600 readings were taken every 3-4 days, with a final reading taken at 10 days for M.tb. Sample wells exhibiting 20% or less of the average O.D.600 readings of the untreated control wells were considered to contain a drug concentration resulting in inhibition. Alamar Blue dye solution was added to the wells at the end of the incubation period to a final concentration of 10% (v/v) and assessed after 48 hours to determine the point of activity. For protein binding determination, 10% or 45% of normal mouse serum was included in a duplicate set of MIC assay plates. Each MIC assay included untreated controls, medium blanks, and control compounds.[17,34,35]
2.8. IC50 Determination:
The enzymatic activity was characterized using membrane fractions prepared from M. tuberculosis as described previously.[17,35,36] Briefly, M. Tb (H37Rv) was grown to mid-log phase in glycerol alanine salts medium, washed with saline and harvested by centrifugation. The resulting pellet was irradiated for 18 h at 2,315 Rads/min using a JL Shepard instrument with a 137Cs source. This exposure was calculated and carried to kill 100% of the bacteria but retain 90% of enzyme activity. The washed cell pellet was resuspended in homogenization buffer containing 50 mM MOPS (pH 7.9), 0.25 M sucrose, 10 mM MgCl and 5 mM 2-mercaptoethanol and disrupted by probe sonication on ice with a Sanyo Soniprep 150 (10 cycles of 60 sec on and 90 sec off). The resulting suspension was centrifuged at 27,000 x g for 15 min. The pellet was discarded and the supernatant was centrifuged at 100,000 x g for 1 h in a Beckman Ti70.1 rotor. The pellet (membranes) was resuspended in homogenization buffer, divided into aliquots and frozen at -70ºC. The protein concentrations were estimated by conventional BCA protein assay kit (Pierce, Rockford, IL). MenA assays were conducted as described before.[37] Briefly, assay mixtures contained 500 µM DHNA, 10 µM[3H]-farnesyl diphosphate (American Radiolabeled Chemicals, St. Louis, MO), 5 mM MgCl2 and 0.1% CHAPS in 100 mM MOPS (pH 8) and an appropriate amount of membrane protein including the known amount of inhibitor. After completion of reaction, it was stopped by the addition of 1 ml of 0.1 M AcOH in MeOH. The resulting mixture was extracted twice with 3 ml of hexanes and the combined extracts were evaporated to dryness under N2 and redissolved in CHCl3: MeOH (2:1). An aliquot was taken for liquid scintillation counting and the remaining material was subjected to TLC on silica gel plates, which were developed in hexane: Et2O (95:5). Radioactive spots on the thin layer plated were located and quantitated using a Bioscan System 200 Imaging Scanner.[17,35] IC50 values were calculated for the inhibitors and compared to the MIC values.
3. Results
Presently, multiple drug resistance and extensively drug resistance by pathogenic bacteria has been increasing. In this regard a novel approach and drug target is necessary.[1,38,39] MenA is found to be a leading drug target and its involvement in electron transfer process is also unique.[4,5,6,7,8] Therefore determining MenA inhibitors was shown more interest and new compounds and polymer where synthesized.
Benzophenone derivatives as MenA inhibitors has been well studied.[2] Here, we synthesized one of the known benzophenone derivative, 5 by the reported procedure (Figure 2). Briefly, the phenolic group of benzophenone was reacted with dibromo octane and subsequently reacted with N-methyl allyl amine. The synthesized benzophenone derivative, 5 was tested against M.tb and showed MIC of 12. 5 µg/ml and IC50 of 5.5 µg/ml. Even though the synthesized compound showed biological activity, it wasn’t significant enough to move forward.
To improve the biological activity of the synthesized compound, we introduced diphenyl ether group in the backbone (Figure 3). The diphenyl ether substrate also contained flouro group and free hydroxyl group to increase the biological activity of the compounds by increasing the ability to coordinate with enzyme. The toxicity of the benzophenone compound also could be reduced by changing the functional group from carbonyl to ether. The final compound, 8 was synthesized by reacting the phenolic hydroxyl group, 6 with dibromo-octane in presence of potassium carbonate with high yield. The other terminal of the bromo-octane, 7 was reacted with N-methyl allyl amine to synthesis the diphenyl ether amino compound, 8. The synthesized compound, 8 was again tested against M. tb and showed MIC of 6.2 µg/ml and IC50 of 12.9 µg/ml (Table 1). Interestingly, the diphenyl ether derivative, 8 showed MIC of 6.2 µg/ml and benzophenone derivative, 5 showed IC50 of 5.5 µg/ml therefore we designed a polymer compound containing both benzophenone and diphenyl ether structure embedded in the backbone.
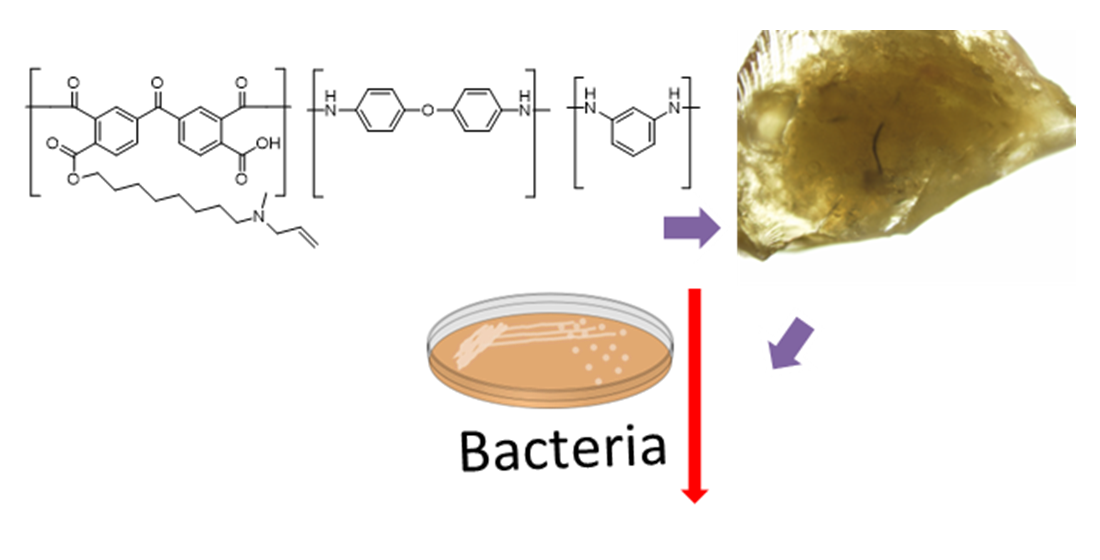
Recently, discovery of polymeric drugs are growing therefore we designed a novel polymeric compound 11 (Figure 4).[26,27,28,29,30,31,32] As well as from our results, benzophenone, 5 and diphenyl ether, 8 backbone based compounds showed reasonably good growth inhibition, therefore we incorporated the benzophenone and diphenyl ether groups in the polymeric compound , 11. Briefly, poly (3,3′,4,4′-benzophenone tetra carboxylic anhydride-co-4,4′-oxydianiline/1,3-phenylenediamine, amic acid, 9 solution was alkylated with dibromooctane in DMF using potassium carbonate. Followed by amination using N-methyl allylamine. The synthesized compound, 11 was tested against M. tb and observed 1 µg/ml minimum inhibitory concentration and IC50 of 1 µg/ml (Table 2). The surface morphology of the material was studied by electron microscopy and before heating the polymer it looked like amorphous powder. To study the activity of enzymes on polymers, we made solid polymers by heating. After heating it looked like shining crystals. Whereas the reacted polymer showed a black stain on its surface, where the enzyme could have reacted and changed the morphology of the polymer as shown in the Figure 5. The synthesized high molecular weight polymeric inhibitors contains various functional characteristics to selectively recognize, coordinate with the M.tb MenA. In addition the high molecular weight of these polymeric inhibitors, 11 could make them systemically non-adsorbed, thus providing a long-term safety profile over the small molecule (5 and 8) drug products. Furthermore, the multiple functional groups incorporated in the polymeric inhibitor, 11 could enhance the polyvalent binding interaction with MenA enzyme thus enhancing the activity.
4. Discussion
Mycobacteria and Gram-positive bacteria are a significant cause of morbidity and mortality worldwide. Many of these bacteria, such as M. tuberculosis, Staphylococcus aureus and Streptococcus pneumoniae, are responsible for a wide range of infections, including tuberculosis, pneumonia, sepsis, and endocarditis. The emergence of drug-resistant strains of these bacteria has become a major public health concern and a significant barrier to the successful treatment of these infections.
One essential pathway in the metabolism of many Gram-positive bacteria is the biosynthesis of menaquinone, a type of vitamin K2 that is required for electron transport and energy generation through ATP synthesis. The menaquinone biosynthesis pathway is an attractive target for drug discovery as it is essential for the survival of many bacteria, including drug-resistant strains.[2,4,5,8,17]
The menaquinone biosynthesis pathway consists of several steps that convert precursors into the final product, menaquinone. In most bacteria, the pathway begins with the condensation of chorismate and isochorismate to form 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate (SEPHCHC) using MenD, which is then converted into 2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate (SHCHC) by the enzyme MenH. The next step is catalyzed by the enzyme MenC, MenE, MenB and MenX, which converts SHCHC into 1,4-dihydroxy-2-naphthoate (DHNA). DHNA is then converted into 2-demethyl-menaquinone by MenA. Demethyl menaquinone is subsequently converted into menaquinone by the enzyme MenG (Figure 1).[2,4,5,8,17]
The menaquinone biosynthesis pathway is an attractive target for drug discovery, as many of the enzymes involved in the pathway are essential for the survival of many bacteria, including drug-resistant strains. Inhibitors of these enzymes have shown promise as potential drugs against Gram-positive bacteria, including drug-resistant strains.
For example, the MenD enzyme has been targeted by the antibiotic fosfomycin, which inhibits the enzyme by covalently binding to its active site. Fosfomycin has been shown to have potent activity against Gram-positive bacteria, including drug-resistant strains of S. aureus and S. pneumoniae.
Another enzyme in the menaquinone biosynthesis pathway that has been targeted by antibiotics is MenE. MenE/MenB catalyzes the conversion of o-succinylbenzoate (OSB) into 1,4-dihydroxy-2-naphthoate (DHNA), a crucial step in the pathway. The antibiotic platensimycin inhibits MenE by binding to its active site, thereby preventing the conversion of OSB to DHNA.[20,22] Platensimycin has been shown to have potent activity against Gram-positive bacteria, including drug-resistant strains of S. aureus.
In addition to the menaquinone biosynthesis inhibitors potential as antibiotics, they could also be used as tools for studying the biology of bacteria.[2,4,5,8,17] These inhibitors could be used to investigate the roles of menaquinone and its intermediates and the pathway in the physiology and virulence of bacteria, to provide valuable insights into the mechanisms of bacterial pathogenesis.
Hence, the biosynthesis of menaquinone is an essential pathway in the metabolism of many mycobacteria and Gram-positive bacteria. The pathway is responsible for the biosynthesis of a crucial component of the electron transport chain menaquinone and is required for energy generation and ATP synthesis. Inhibitors of the menaquinone biosynthesis pathway have shown promise as potential drugs against drug-resistant strains of mycobacteria and Gram-positive bacteria.[2,4,5,8,17]
As discussed in our results, benzophenone compound, 5 showed good activity and diphenyl ether compound, 8 also showed good bacterial growth inhibition, hence, we incorporated the benzophenone and diphenyl ether group compounds in the polymeric compound, 11 (Figure 6) and observed good activity against both bacteria and MenA enzyme. The synthesized compound, 11 was tested against M. tb and observed 1 µg/ml minimum inhibitory concentration and IC50 of 1 µg/ml. The surface morphology of the material was studied by electron microscopy and initially, the polymer looked like an amorphous powder. However, after heating the polymer, it was a shiny crystal and on reaction with enzyme, it turned to a black stain on its surface, where the enzyme could have reacted and changed the morphology of the polymer. The synthesized high molecular weight polymeric inhibitors contains various functional characteristics to selectively recognize and coordinate with the M.tb MenA and enhace polyvalent binding interaction with MenA. These polymeric inhibitors, 11 could make them systemically non-adsorbed, thus providing a long-term safety profile over the small molecule (5 and 8) drug products as mentioned before. These studies emphasises that designing a combination of two active compounds could enhance the actvitiy of the compound and making a polymer compound/inhibitor also enhances the activity of the compound. These results were confirmed by targeting M. tuberculosis and M. Tb MenA.
5. Conclusion
The synthesized and characterized benzophenone derivative, 5 was tested against M.tb and showed MIC of 12. 5 µg/ml and IC50 of 5.5 µg/ml. Similarly the synthesized and characterized diphenyl ether derivative, 8 showed MIC 6.2 µg/ml and IC50 12.9 µg/ml. To enhance the biological activity of the inhibitor both benzophenone and diphenyl ether structures were incorporated in the polymeric backbone and a novel polymer containing benzophenone and diphenyl ether moiety was designed to inhibit the MenA in M. tb. The designed N- methyl allylamine linked poly (3,3′,4,4′-benzophenone tetra carboxylic anhydride-co-4,4′-oxydianiline/1,3-phenylenediamine, amic acid, 11 was synthesized in good yield for the first time and tested against M. tb. The new polymer, 11 showed MIC of 1 µg/ml and IC50 of 1 µg/ml , which is moderately better compared to its small molecule inhibitors 5 and 8. In vitro optimization of the polymeric inhibitor is currently in progress.
Funding
We would like to thank Nebraska Research Initiative Faculty Award to P.N. for funding.
Acknowledgments
We would like to thank Markus Kincaid from CSU for assisting the IC50 assay.
Conflicts of Interest
The authors declare no competing financial interests.
References
- Narayanasamy, P., H. Eoh, P. J. Brennan, and D. C. Crick. 2010. Synthesis of 4-diphosphocytidyl-2-C-methyl-D-erythritol 2-phosphate and kinetic studies of Mycobacterium tuberculosis IspF. Chemistry & biology 17: 117-122. [CrossRef]
- Kurosu, M., P. Narayanasamy, K. Biswas, R. Dhiman, and D. C. Crick. 2007. Discovery of 1,4-dihydroxy-2-naphthoate [corrected] prenyltransferase inhibitors: new drug leads for multidrug-resistant gram-positive pathogens. Journal of medicinal chemistry 50: 3973-3975.
- Brennan, P. J., and D. C. Crick. 2007. The cell-wall core of Mycobacterium tuberculosis in the context of drug discovery. Curr Top Med Chem 7: 475-488. [CrossRef]
- Choi, S. R., J. Frandsen, and P. Narayanasamy. 2017. Novel long-chain compounds with both immunomodulatory and MenA inhibitory activities against Staphylococcus aureus and its biofilm. Scientific reports 7: 40077. [CrossRef]
- Choi, S. R., M. A. Larson, S. H. Hinrichs, A. M. Bartling, J. Frandsen, and P. Narayanasamy. 2016. Discovery of bicyclic inhibitors against menaquinone biosynthesis. Future Med Chem 8: 11-16. [CrossRef]
- Choi, S. R., M. A. Larson, S. H. Hinrichs, and P. Narayanasamy. 2016. Development of potential broad spectrum antimicrobials using C-symmetric 9-fluorenone alkyl amine. Bioorganic & medicinal chemistry letters 26: 1997-1999. [CrossRef]
- Choi, S. R., and P. Narayanasamy. 2021. Synthesis, optimization, in vitro and in vivo study of bicyclic substituted amine as MenA inhibitor. Bioorganic & medicinal chemistry letters 47: 128203. [CrossRef]
- Choi, S. R., and P. Narayanasamy. 2023. In Vitro and In Vivo Antimicrobial Activity of an Oxidative Stress-Mediated Bicyclic Menaquinone Biosynthesis Inhibitor against MRSA. ACS Infect Dis 9: 2016-2024. [CrossRef]
- Choi, S. R., Narayanasamy, P. 2024. Investigating Novel IspE Inhibitors of the MEP Pathway in Mycobacterium. Microorganisms 12: 18. [CrossRef]
- Edagwa, B., Y. Wang, and P. Narayanasamy. 2013. Synthesis of azide derivative and discovery of glyoxalase pathway inhibitor against pathogenic bacteria. Bioorganic & medicinal chemistry letters 23: 6138-6140. [CrossRef]
- Edagwa, B. J., and P. Narayanasamy. 2013. Synthesis of chirally pure 1-deoxy-d-xylulose-5-phosphate : A substrate for IspC assay to determine inhibitor. Chemical sciences journal 4.
- Shineberg, B., and I. G. Young. 1976. Biosynthesis of bacterial menaquinones: the membrane-associated 1,4-dihydroxy-2-naphthoate octaprenyltransferase of Escherichia coli. Biochemistry 15: 2754-2758. [CrossRef]
- Kurosu, M., and E. Begari. 2010. Vitamin K2 in electron transport system: are enzymes involved in vitamin K2 biosynthesis promising drug targets? Molecules 15: 1531-1553.
- Bentley, R., and R. Meganathan. 1982. Biosynthesis of vitamin K (menaquinone) in bacteria. Microbiological Reviews 46: 241-280.
- Schutz, M., M. Brugna, E. Lebrun, F. Baymann, R. Huber, K.-O. Stetter, G. Hauska, R. Toci, D. Lemesle-Meunier, P. Tron, C. Schmidt, and W. Nitschke. 2000. Early evolution of cytochrome bc complexes. Journal of Molecular Biology 300: 663-675. [CrossRef]
- Unden, G., and J. Bongaerts. 1997. Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochimica et Biophysica Acta, Bioenergetics 1320: 217-234. [CrossRef]
- Dhiman, R. K., S. Mahapatra, R. A. Slayden, M. E. Boyne, A. Lenaerts, J. C. Hinshaw, S. K. Angala, D. Chatterjee, K. Biswas, P. Narayanasamy, M. Kurosu, and D. C. Crick. 2009. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Molecular microbiology 72: 85-97. [CrossRef]
- Honaker, R. W., R. K. Dhiman, P. Narayanasamy, D. C. Crick, and M. I. Voskuil. 2010. DosS responds to a reduced electron transport system to induce the Mycobacterium tuberculosis DosR regulon. Journal of bacteriology 192: 6447-6455. [CrossRef]
- Li, X., N. Liu, H. Zhang, S. E. Knudson, H.-J. Li, C.-T. Lai, C. Simmerling, R. A. Slayden, and P. J. Tonge. 2011. CoA Adducts of 4-Oxo-4-phenylbut-2-enoates: Inhibitors of MenB from the M. tuberculosis Menaquinone Biosynthesis Pathway. ACS Medicinal Chemistry Letters 2: 818-823.
- Lu, X., H. Zhang, J. Tonge Peter, and S. Tan Derek. 2008. Mechanism-based inhibitors of MenE, an acyl-CoA synthetase involved in bacterial menaquinone biosynthesis. Bioorganic & medicinal chemistry letters 18: 5963-5966.
- Dhiman, R. K., S. Mahapatra, R. A. Slayden, M. E. Boyne, A. Lenaerts, J. C. Hinshaw, S. K. Angala, D. Chatterjee, K. Biswas, P. Narayanasamy, M. Kurosu, and D. C. Crick. 2009. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from nonreplicating persistence. Molecular Microbiology 72: 85-97. [CrossRef]
- Lu, X., R. Zhou, I. Sharma, X. Li, G. Kumar, S. Swaminathan, J. Tonge Peter, and S. Tan Derek. 2012. Stable analogues of OSB-AMP: potent inhibitors of MenE, the o-succinylbenzoate-CoA synthetase from bacterial menaquinone biosynthesis. Chembiochem : a European journal of chemical biology 13: 129-136.
- Kurosu, M., P. Narayanasamy, K. Biswas, R. Dhiman, and D. C. Crick. 2007. Discovery of 1,4-Dihydroxy-2-naphthoate Prenyltransferase Inhibitors: New Drug Leads for Multidrug-Resistant Gram-Positive Pathogens. J. Med. Chem. 50: 3973-3975. [CrossRef]
- Debnath, J., S. Siricilla, B. Wan, C. Crick Dean, J. Lenaerts Anne, G. Franzblau Scott, and M. Kurosu. 2012. Discovery of selective menaquinone biosynthesis inhibitors against Mycobacterium tuberculosis. Journal of medicinal chemistry 55: 3739-3755.
- Kurosu, M., and D. C. Crick. 2009. MenA is a promising drug target for developing novel lead molecules to combat Mycobacterium tuberculosis. Medicinal Chemistry 5: 197-207. [CrossRef]
- Choi, S. R., B. E. Britigan, D. M. Moran, and P. Narayanasamy. 2017. Gallium nanoparticles facilitate phagosome maturation and inhibit growth of virulent Mycobacterium tuberculosis in macrophages. PLoS ONE 12: e0177987. [CrossRef]
- Choi, S. R., B. E. Britigan, and P. Narayanasamy. 2017. Ga(III) Nanoparticles Inhibit Growth of both Mycobacterium tuberculosis and HIV and Release of Interleukin-6 (IL-6) and IL-8 in Coinfected Macrophages. Antimicrobial agents and chemotherapy 61: e02505-02516. [CrossRef]
- Choi, S. R., B. E. Britigan, and P. Narayanasamy. 2019. Iron/Heme Metabolism-Targeted Gallium(III) Nanoparticles Are Active against Extracellular and Intracellular Pseudomonas aeruginosa and Acinetobacter baumannii. Antimicrobial agents and chemotherapy 63: e02643-02618. [CrossRef]
- Choi, S. R., B. E. Britigan, and P. Narayanasamy. 2019. Treatment of Virulent Mycobacterium tuberculosis and HIV Coinfected Macrophages with Gallium Nanoparticles Inhibits Pathogen Growth and Modulates Macrophage Cytokine Production. mSphere 4. [CrossRef]
- Choi, S. R., B. E. Britigan, and P. Narayanasamy. 2022. Synthesis and in vitro analysis of novel gallium tetrakis(4-methoxyphenyl)porphyrin and its long-acting nanoparticle as a potent antimycobacterial agent. Bioorganic & medicinal chemistry letters 62: 128645. [CrossRef]
- Choi, S. R., B. E. Britigan, B. Switzer, T. Hoke, D. Moran, and P. Narayanasamy. 2018. In Vitro Efficacy of Free and Nanoparticle Formulations of Gallium(III) meso-Tetraphenylporphyrine against Mycobacterium avium and Mycobacterium abscessus and Gallium Biodistribution in Mice. Molecular pharmaceutics 15: 1215-1225. [CrossRef]
- Choi, S. R., G. A. Talmon, B. E. Britigan, and P. Narayanasamy. 2021. Nanoparticulate beta-Cyclodextrin with Gallium Tetraphenylporphyrin Demonstrates in Vitro and in Vivo Antimicrobial Efficacy against Mycobacteroides abscessus and Mycobacterium avium. ACS Infect Dis.
- Sundararajan, G., and N. Prabagaran. 2001. A new polymer-anchored chiral catalyst for asymmetric Michael addition reactions. Org Lett 3: 389-392. [CrossRef]
- Gruppo, V., C. M. Johnson, K. S. Marietta, H. Scherman, E. E. Zink, D. C. Crick, L. B. Adams, I. M. Orme, and A. J. Lenaerts. 2006. Rapid microbiologic and pharmacologic evaluation of experimental compounds against Mycobacterium tuberculosis. Antimicrobial agents and chemotherapy 50: 1245-1250. [CrossRef]
- Dhiman, R. K., V. Pujari, J. M. Kincaid, M. A. Ikeh, T. Parish, and D. C. Crick. 2019. Characterization of MenA (isoprenyl diphosphate:1,4-dihydroxy-2-naphthoate isoprenyltransferase) from Mycobacterium tuberculosis. PLoS One 14: e0214958. [CrossRef]
- Crick, D. C., M. C. Schulbach, E. E. Zink, M. Macchia, S. Barontini, G. S. Besra, and P. J. Brennan. 2000. Polyprenyl phosphate biosynthesis in Mycobacterium tuberculosis and Mycobacterium smegmatis. Journal of bacteriology 182: 5771-5778. [CrossRef]
- Leppik, R. A., P. Stroobant, B. Shineberg, I. G. Young, and F. Gibson. 1976. Membrane-associated reactions in ubiquinone biosynthesis. 2-Octaprenyl-3-methyl-5-hydroxy-6-methoxy-1,4-benzoquinone methyltransferase. Biochim Biophys Acta 428: 146-156. [CrossRef]
- Narayanasamy, P., and D. C. Crick. 2008. Enantiomeric Synthesis of 2-C-Methyl-D-Erythritol 2, 4-Cyclodiphosphate. Heterocycles 76: 243-247. [CrossRef]
- Narayanasamy, P., H. Eoh, and D. C. Crick. 2008. Chemoenzymatic synthesis of 4-diphosphocytidyl-2-C-methyl-D-erythritol: A substrate for IspE. Tetrahedron letters 49: 4461-4463. [CrossRef]
Figure 1.
Biosynthetic pathway for synthesis of menaquinone, 4. Biosynthesis of menaquinone from chorismite using MenF, D,C,E,B, A and G.
Figure 1.
Biosynthetic pathway for synthesis of menaquinone, 4. Biosynthesis of menaquinone from chorismite using MenF, D,C,E,B, A and G.
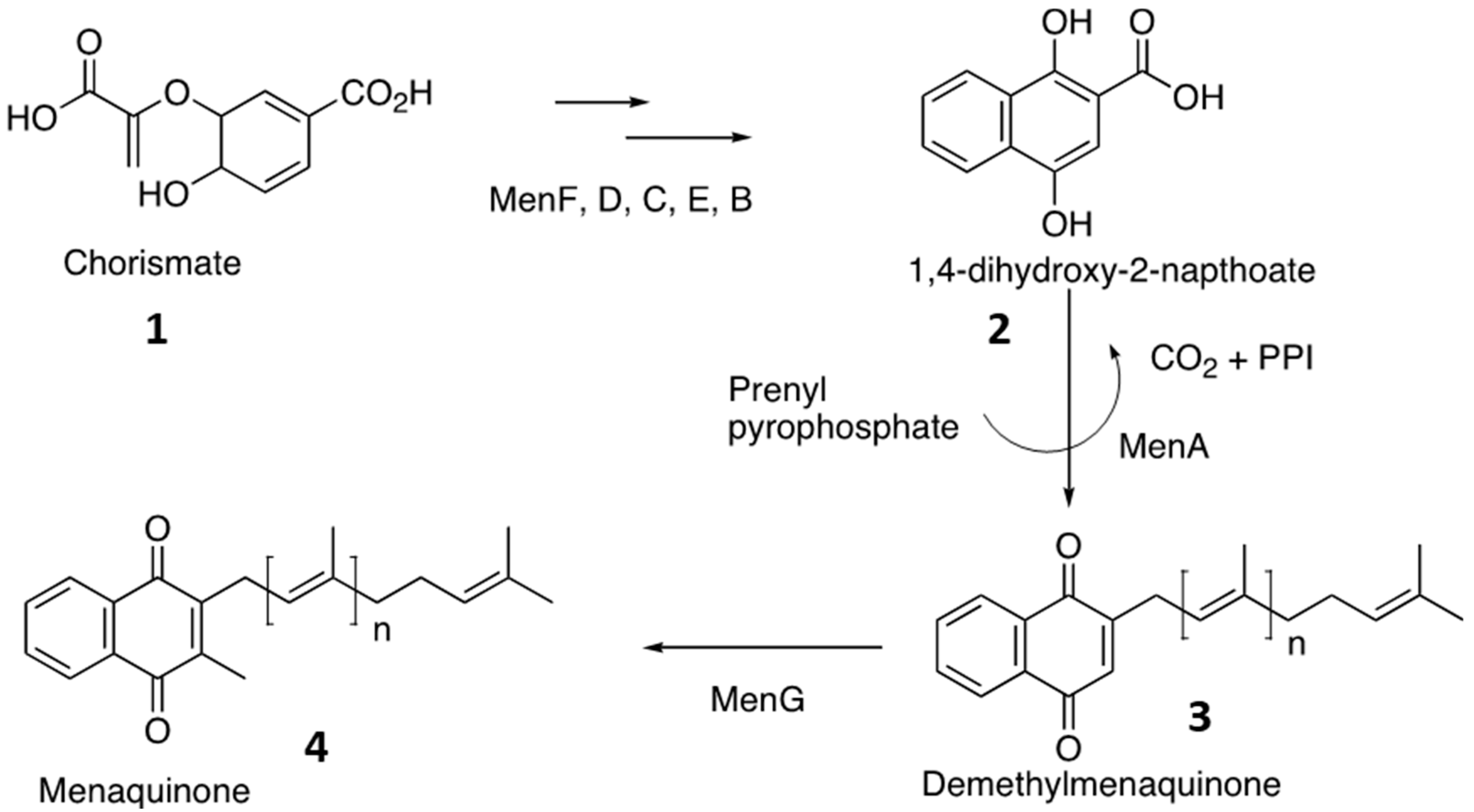
Figure 2.
Structure of benzophenone N-allylamine compound, 5.
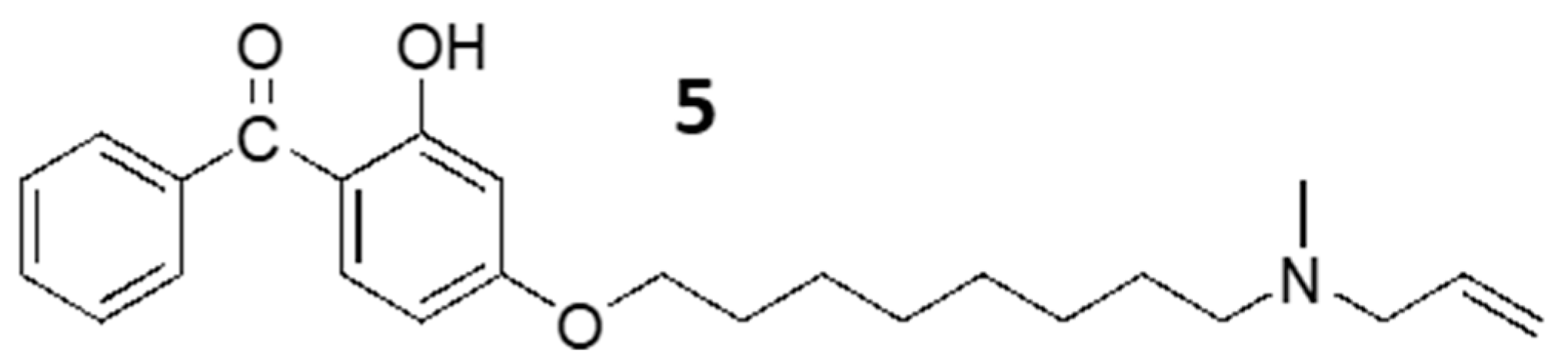
Figure 3.
Synthesis of diphenyl ether allyl amine compound, 8. Diphenyl ether was alkylated using dibromo octane and K2CO3 followed by amination using N-methyl allylamine.
Figure 3.
Synthesis of diphenyl ether allyl amine compound, 8. Diphenyl ether was alkylated using dibromo octane and K2CO3 followed by amination using N-methyl allylamine.
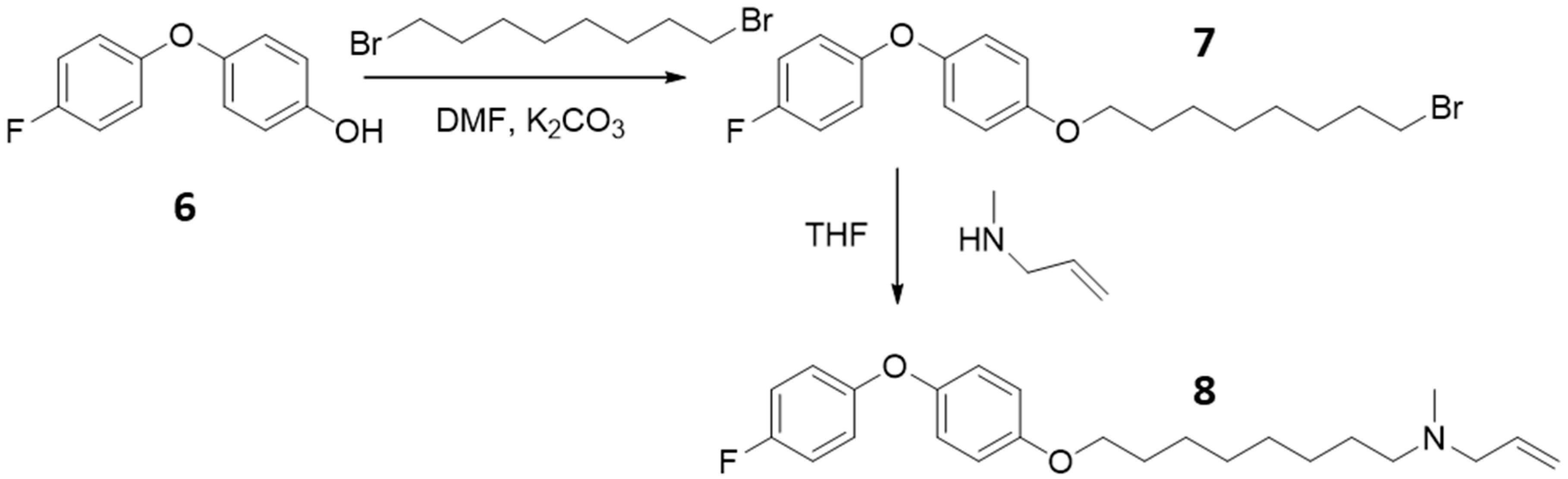
Figure 4.
Synthesis of polymeric inhibitor, 11. Poly (3,3′,4,4′-benzophenone tetra carboxylic anhydride-co-4,4′-oxydianiline/1,3-phenylenediamine, amic acid, 9 was alkylated using dibromo octane and K2CO3 followed by amination using N-methyl allylamine.
Figure 4.
Synthesis of polymeric inhibitor, 11. Poly (3,3′,4,4′-benzophenone tetra carboxylic anhydride-co-4,4′-oxydianiline/1,3-phenylenediamine, amic acid, 9 was alkylated using dibromo octane and K2CO3 followed by amination using N-methyl allylamine.
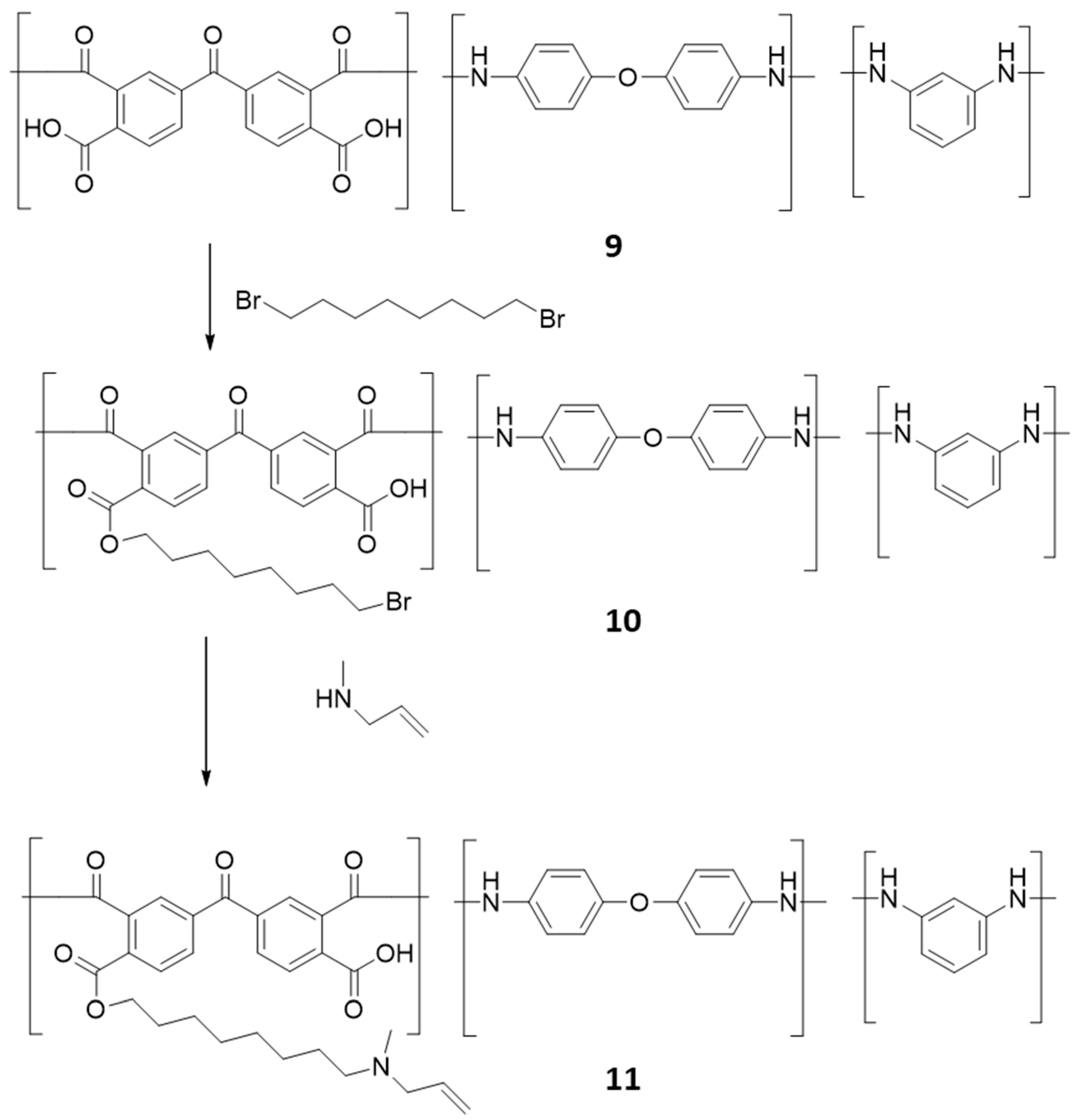
Figure 5.
Electron microscopic picture of polymeric inhibitor, 11.

Figure 6.
Schematic diagram of designing polymer.
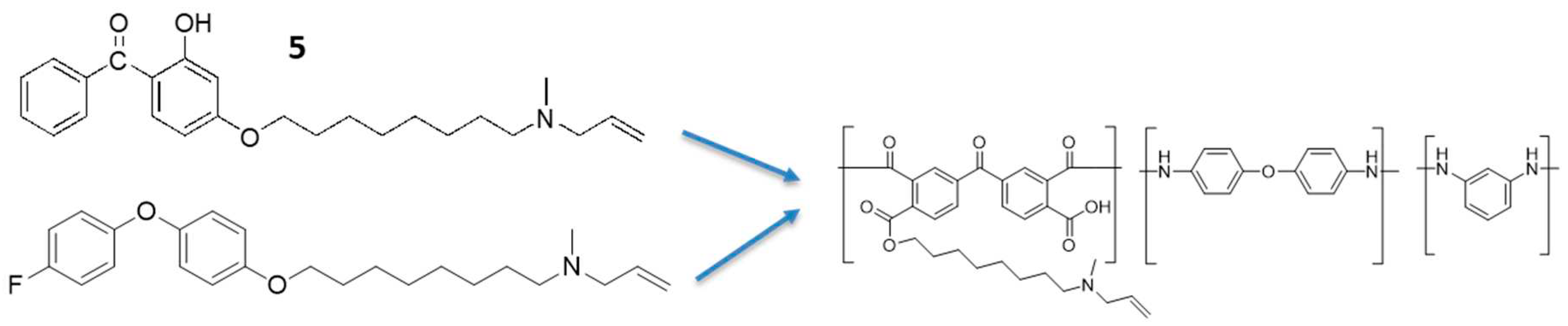

Table 1.
The minimum inhibitory concentration (MIC) against bacteria and Enzymatic activity (IC50) against MenA for lead compound.
Table 1.
The minimum inhibitory concentration (MIC) against bacteria and Enzymatic activity (IC50) against MenA for lead compound.
| MIC (µg/mL) | IC50 | ||
|---|---|---|---|
| Compound | M.tb | M.tb MenA | |
| 8 | 6.2 | 12.9 | |
M.tb – Mycobacterium tuberculosis (H37Rv).
Table 2.
The minimum inhibitory concentration (MIC) against bacteria and Enzymatic activity (IC50) against MenA for polymer compound.
Table 2.
The minimum inhibitory concentration (MIC) against bacteria and Enzymatic activity (IC50) against MenA for polymer compound.
| MIC (µg/mL) | IC50 | ||
|---|---|---|---|
| Compound | M.tb | M.tb MenA | |
| 11 | 1.0 | 1.0 | |
M.tb – Mycobacterium tuberculosis (H37Rv).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



