Submitted:
29 December 2023
Posted:
03 January 2024
You are already at the latest version
Abstract
Bipyridine and its related compounds are starting materials or precursors for a variety of valuable substances, such as biologically active molecules, ligands for catalysts, photosensitizers, viologens, and supramolecular architectures. Therefore, it is important to classify the synthesis methods and know their characteristics. Representative examples include the methods using homo- and heterocoupling of pyridine derivatives in the presence of catalyst. Because bipyridine compounds have a high ability to coordinate with metal centers, a decrease in catalytic activity and yield is often observed in the reaction system. To address this issue, the review provides insight into recent advances in bipyridine synthesis using metal complexes under both homogeneous and heterogeneous conditions. Moreover, the review also sheds light on strategies for bipyridine synthesis involving sulfur and phosphorous compounds. These alternative pathways offer promising avenues for overcoming challenges associated with traditional-catalyzed methods, providing a more comprehensive understanding of the synthesis landscape.
Keywords:
Suzuki coupling
; Stille coupling
; Negishi coupling
; Ullmann coupling
; Wurtz coupling
; electrochemical method
1. Introduction
Bipyridines and their derivatives have been extensively used as fundamental components in various applications, including biologically active molecules, ligands in transition-metal catalysis, photosensitizers, viologens, supramolecular structures (Figure 1) [1-11]. Many synthetic methods for the preparation of bipyridines have been developed. Due to the low conversion rate and severe reaction conditions of conventional methods, various methods are still being developed. The problem is that the product has a high ability to coordinate with the metal center, resulting in a decrease in catalytic activity. Although there are several reviews on the synthesis of bipyridine derivatives [12,13], this review aims to categorize recent research findings, focusing on metal-catalyzed cross-coupling reactions (including Suzuki, Negishi, and Stille couplings), metal-catalyzed homo-coupling reactions (Ullmann coupling and Wurtz coupling), electrochemical methods, and other innovative techniques and provides a comprehensive overview of recent advancements in the synthesis of bipyridines, emphasizing the diverse strategies employed to overcome challenges associated with conventional methods.
2. Metal-catalyzed cross-coupling (Suzuki, Negishi, and Stille coupling reactions)
2.1. Suzuki coupling reaction in homogeneous catalytic system
Among transition-metal cross-coupling reactions, Suzuki coupling is a particularly attractive route to construct C(sp2)‒C(sp2) bonds and has been widely used for the synthesis of bipyridine structure [14,15,16,17,18,19,20]. Although numerous palladium catalysts were employed in Suzuki coupling, a significant drawback is the tendency of the coordination of bipyridine products with the palladium metal, resulting in a decrease in catalytic activity. The most crucial factor in achieving the goal is the design of a catalytic system. Several recent representative examples are illustrated in Figure 2.
Matondo et al and Cashman et al independently reported Suzuki coupling between pyridyl boronic acids and bromopyridines in the presence of Pd(PPh3)4 and Na2CO3, which is typical conditions of Suzuki coupling (Figure 2(a)) [21,22]. However, the yields of products were moderate (≈ 50‒65%) and high loading of Pd source (> 10mol%) was required. This may be due to the aforementioned decrease in catalytic activity during the reaction. Aiming to improve catalytic systems, Wu et al reported Suzuki coupling of 3-pyridine boronic pinacol ester with pyridyl halides with cyclopalladated ferrocenylimine catalyst (Figure 2(b)) [23]. This palladium catalyst exists stably in the atmosphere, and bipyridine derivatives can be synthesized in high yield without inert gas. Lee et al. reported the synthesis of bipyridines using Suzuki coupling with an imidazolium salt employed as the ligand for the palladium catalyst (Figure 2(c)) [24]. A turnover number (TON) of up to 850,000 was observed in the coupling reaction. Thus, boronic acids with 3- and 4-pyridyl groups are stable and can be used to synthesize a variety of compounds via Suzuki coupling. Pharmaceutical bipyridine-based compounds, such as milrinone, was synthesized using these methods. However, in these reports only 3- or 4-pyridylboronic acid derivatives are applicable.
In contrast, 2-pyridyl boronic acid derivatives show poor stability [25], and it is difficult to introduce 2-pyridyl groups into aromatic rings due to low efficiency in the catalytic reaction [26]. Therefore, the preparation of stable unsubstituted 2-pyridylboron derivatives remains a synthetic challenge. Miyaura et al addressed this challenge by the synthesis of bipyridines by the coupling between [2-PyB(OCH2)3CHCH3]M (M = K+, Na+, or Li+) and 2-bromopyridines (Figure 2(d)) [27]. Similarly, Stevens et al reported Suzuki coupling of 2-pyridineboronic acid N-phenyldiethanolamine ester with bromopyridines (Figure 2(e)) [28]. The boronic ester has demonstrated stability during prolonged storage and is now commercially available. Couplings were performed with 5 mol% PdCl2(PPh3)2 and 1.1 equivalent of boronic ester relative to the bromopyridine to afford 2,2'-bipyridine-type products in good yields. Yamamoto et al reported that Suzuki coupling of tetrabutylammonium 2-pyridylborate salts with chloropyridines can produce the corresponding bipyridine products with PdCl2(dcpp) (dcpp: 1,3-bis(dicyclohexylphosphino)propane) in good to excellent yield (Figure 2(f)) [29]. In this reaction, addition of N-methyl ethanolamine led to an increase in the yield. The reactivity varies with the type of 2-pyridylborate cation. The tetrabutyl ammonium cation exhibits a greater accelerating effect in the reaction, and the order of reactivity is as follows: Li+ < Na+ < K+ < Cs+ < Bu4N+. This method enables the efficient synthesis of many 2,2’-bipyridine-type molecules. The addition of CuI as a co-catalyst was effective in increasing the yield of 2,2’-bipyridine. The exact role of CuI in this reaction is not known; however, such an effect of copper salts has been successfully employed in analogous coupling reactions of heteroarylboron compounds. In this case, addition of CuI improves the yield of products.
2.2. Negishi and Stille coupling reactions in homogeneous catalytic system
Stille coupling is a synthetic method using organotin compounds [30,31]. Stille coupling is highly reactive, and the characteristic is that it may progress even in systems where progression does not occur with the Suzuki coupling [32]. However, a notable drawback of Stille coupling is the toxicity and unpleasant nature of orgasnostannyl compounds. Schubert et al reported that Stille-type cross-coupling procedures are utilized to prepare various 2,2’-bipyridines (Figure 3(a)) [33]. Terpyridine derivatives can be synthesized using the techniques, which are accessible as valuable chelating ligands. Wu et al reported Stille coupling between 3- or 2-stannylpyridines and bromopyridines catalyzed by 1 mol% cyclopalladated ferrocenylimine with tricyclohexylphosphine in the presence of CuI (additive) and CsF (base) (Figure 3(b)) [34]. Padwa and Wang et al also contributed to this field, presenting the synthesis of bipyridines through the Stille reaction between stannylated pyridines and bromopyridines in the presence of PdCl2(PPh3)2 (Figure 3(c)) [35]. Biquinolinyl compounds can be synthesized by the use of 2-quinolinyl stannanes and 2-bromo substituted quinolines in the same reaction conditions. The approach can be extended to the synthesis of lavendamycin and analogues starting from functionalized quinolines and β-carboline building blocks. Although Stille coupling is useful in the synthesis of bipyridine derivatives, the organotin molecules as reactants are high toxic.
In related research, bipyridines can also be synthesized by Negishi coupling as an alternative synthetic method reported by Bilhel. Particularly, good results are obtained when PdBr(Ph)(PPh3)2 is employed as a catalyst (Figure 3(d)) [36]. The catalyst, termed a post-oxidative addition precatalyst, can be easily prepared with high stability against air and moisture. The catalyst showed the high activity in Negishi coupling [37]. Yin et al and Carrick et al reported the preparation of 2,2’-bipyridine derivatives by Negishi coupling between 2-pyridyl zinc halides and bromopyridines in the presence of Pd(dba)2 and XPhos [38,39]. The method complements current reactions for the coupling of 2-pyridyl organometallic reagents and is anticipated to find utility in drug discovery and development efforts. Quéguiner et al reported Negishi coupling between (3-(diisopropylcarbamoyl)-4-methoxypyridin-2-yl)zinc(II) chloride and 2-bromopyridine in the presence of catalytic amount of Pd(PPh3)4 (Figure 3(f)). The reaction proceeds in THF under reflux for 20 h. The coupling product can be converted into caerulomycin B or caerulomycin C, which are useful for STAT1 signaling inhibitors and immunosuppressants, showing their applications in medicinal chemistry [40].
2.3. Other cross-coupling reaction in homogeneous catalytic system
In this section, I introduce the synthesis of bipyridines using metal catalysts, which cannot be classified as Suzuki coupling, Stille coupling, or Negishi coupling. Gooβen and other researchers have demonstrated the synthesis of biaryls and heterobiaryls via decarboxylative cross-coupling of aromatic carboxylates [41,42]. Use this method, Chennamanei et al reported microwave-assisted Pd-catalyzed decarboxylative cross-coupling of pyridyl carboxylates with bromopyridines (Figure 4(a)) [43]. 1,10-Phenathroline provided an improvement of the yield, which showed bite angle of bidentate N,N-ligand played a critical role in the decarboxylation process. Especially, they succeeded the decarboxylative cross-coupling of both 3-pyridyl and 4-pyridyl carboxylates with aryl bromides for the synthesis of 3- or 4-arylpyridines, which are of interest in pharmacological applications.
Expanding on this approach, Kumar and Joshi et al. have recently developed dinuclear palladium pincer complexes incorporating Pd‒Pd bond based on N,N,Se-ligands. Bipyridine compounds can be synthesized in good yield by combining decarboxylation of picolinic acid and C‒H activation of pyridine using this complex as a catalyst (Figure 4(b)) [44]. In this reaction Ag2O was used as an oxidant, resulted in the formation of Ag(I)‒pyridine. 2,2':6',2''-Terpyridine can be synthesized by employing 2,6-pyridinecarboxylic acid in place of picolinic acid. This study not only introduces an efficient route to bipyridine synthesis but also demonstrates the adaptability of this methodology with the use of different starting materials and application in synthetic contexts.
Willis et al reported the synthesis of bipyridine derivatives by the coupling of pyridine sulfinate with bromopyridines (Figure 4(c)) [45]. In this reaction, β-nitrile pyridylsulfones worked as efficient base-labile latent sulfinate reagents in Pd-catalyzed cross-coupling reaction. The acrylonitrile by-product was not observed and is assumed to be removed by evaporation. The scope of electrophilic partners is broad, displaying a good tolerance of multiple functional groups, and substitution patterns, delivering the desired cross-coupled products in good to high yields. The method allowed access to a diverse range of 2-arylpyridines and demonstrated the application of this reaction to medicinal molecules. As related study, they also reported that 2-pyridyl sulfinate salts (sodium or lithium) were effective coupling partners in Pd-catalyzed cross-coupling reaction with bromopyridines (Figure 4(d)) [46]. The scope of halide partner is considerable and allows the preparation of a broad range of bipyridine derivatives. They showed that application of the method to medicinally relevant molecules is possible, including in the context of library synthesis. These two studies not only expand the scope of bipyridine synthesis but also highlight the versatility and practicality of these methods in the field of medicinal chemistry.
Transition-metal catalyzed C‒H arylation of heteroarenes has recently emerged as a promising strategy. Based on the idea, Yu et al reported the synthesis of dipyridines through Pd-catalyzed non-directed C-3 arylation of pyridine (Figure 4(e)) [47]. They could synthesize 3,3’-bipyridine and 5-(pyridine-3-yl)pyrimidine in good yield using the method. The utility of the method has been demonstrated in a concise synthesis of the pyridine-based drug molecules. Mongin et al reported Pd-catalyzed cross-coupling between bromopyridines and lithium tri(3-quinolinyl)magnesite, which was prepared by the bromine-magnesium exchange of 3-bromoquinoline and Bu3MgLi [48]. Using the method, functionalized quinoline derivatives can be prepared in moderate yield (Figure 4(e)). Zhou reported the cross-coupling of pyridyl aluminum reagents with pyridyl bromides using Pd(OAc)2 and (o-tolyl)3P. The reactions proceeded efficiently at rt without base. Although the scope was limited to unsubstituted pyridyl aluminum reagents, the yields of products were good [49]. Serrano et al reported homo and hetero coupling of bromopyridines by neophylpalladacycle (Figure 4(g)) [50]. They reported the reaction between 2-BrPy and 2-Br-6-(C3H5O2)-Py gave a mixture of 2,2’-bipyridine, 6-(1,3-dioxolan-2-yl)-2,2’-bipyridine, and 6,6’-di(1,3-dioxolan-2-yl)-2,2’-bipyridine in a ratio of 3:1:2. The ratio was determined by the measurement of 1H NMR.
2.4. Cross-coupling reaction in heterogeneous catalytic system
Heterogeneous catalytic systems have significant advantages, including easy production purification and the ability for repeated use. Representative examples are shown in Figure 5. Vici et al reported Ni/Al2O3‒SiO2 (50 mol%) or Pd/Al2O3 (5 mol%) afforded 2,2’-bipyridine products through Negishi-coupling between 2-pyridyl zinc bromide and 2-bromopyridine derivatives (Figure 5(a)) [51]. The yields were dramatically enhanced under the irradiation of microwave (300 W). The reaction completed within 1 h. In the catalytic system, no coupling product was obtained when pyridyl magnesium bromides were used instead of pyridyl zinc reagents.
Several nanoparticles have been reported to have high catalytic activity on their surfaces and a high TON value. The most studied catalysts are palladium nanoparticles. When used as a catalyst, it is necessary to protect the surface of nanoparticles with a protective agent. A variety of materials have been explored for the purpose, including polymers, dendrimers, surfactants, and organic ligands as surface stabilizing molecules. Here, we will introduce an example in which it is held on a carrier that can be easily recovered and reused. Gros and Fort reported Suzuki coupling of polystyrene-supported 2-pyridylboron reagent with bromopyridines leading to bipyridines in high yield (Figure 5(b)) [52]. The polymer-supported material is the stable source of 2-pyridylboranate. The study opens new perspectives for the Suzuki coupling in combinatorial chemistry for the preparation of bipyridine derivatives. The ease of recovery and reusability of supported catalysts shows practical value and is consistent with the growing interest in sustainable synthesis methodologies.
3. Metal-catalyzed homo-coupling reaction (Wurtz coupling and Ullmann coupling)
Wurtz reaction is a good technique to obtain symmetrical bipyridines [53]. Wurtz coupling typically involves reacting organic halides with sodium dispersion (Figure 6(a)). In this reaction, bipyridines can be synthesized by reacting pyridines with Na dispersion and then reacting with an excess amount of oxidizing agent (Figure 6(b)) [54,55]. The reaction mechanism is shown in Figure 6(c). This method contributes a valuable tool to access diverse bipyridine derivatives.
As a result of the unamiable sodium metal condition, the SET approach toward bipyridines has been overlooked. McMullin, Dawson, and Lu et al reported the synthesis of room temperature stable electride reagent K+(LiHMDS)e‒ (HMDS: 1,1,1,3,3,3-hexamethyldisilazide). The material was easily prepared from potassium metal and LiHMDS via mechanochemical ball milling at 20 mmol scale. The reagent is versatile in mediating the facile transition-metal free pyridine C‒H activation and C‒C coupling (Figure 6(d)) [56]. As a related research, Mandal et al reported the synthesis of bipyridines through transition-metal free C‒H functionalization employing a bis-phenalenyl compound and K(Ot-Bu) (Figure 10(f)) [57]. The reaction mechanism involves a single electron transfer (SET) from a phenalenyl-based radical to generate a reactive pyridyl radical from the halogenated pyridine, which forms a C(sp2)‒C(sp2) bond with pyridine through SET. The presence of organic radicals was confirmed by ESR measurement. Since the yield of the biheteroaryl compound is moderate in this method, it is necessary to consider methods to improve the yield.
As an application of Wurtz coupling, Fort et al. synthesized polyhalogenated 4,4'-bipyridine by coupling 4-lithiodihalopyridine with an oxidizing agent (I2 or MnO2) (Figure 6(f)) [58]. The reaction mechanism was studied by isolation and characterization of several byproducts. The drawback of these methods is that the bipyridine derivative cannot be synthesized unless there are multiple halogen substituents in the pyridine ring with moderate yields.
Transition-metal catalyzed homocoupling of Grignard reagents is one of the most efficient synthetic methods for the construction of symmetrical bipyridyl backbones as an improved method of Wurtz coupling approach [59]. To date, many reports have been published for coupling reactions using metal reagents. The problem encountered with these methods is two-step synthetic route, by which organometallic compounds were initially prepared and isolated, followed by a subsequent conversion into bipyridine products in the presence of an oxidant as a separate reaction. Demand for stoichiometric amounts of organic oxidant limits the use for large-scale preparation. Bhat et al reported metal-catalyzed procedure for the homocoupling of Grignard reagent prepared in situ to give symmetrical bipyridines in a single step [60]. Low-valent metal species is generated in the presence of Grignard reagent in situ [61]. The reaction was performed in the presence of oxygen as an oxidant. The reaction mechanism was shown in Figure 6(g). When the absorption spectrum of the reaction solution was measured, a peak derived from peroxo-M(III) species was observed at 420 nm. This chemical species is considered to be the key chemical species in the transformation. It should be noted that the reaction system is tolerant to chloro-, nitro-, cyano- and hetero-aryl functionalities and afford good to high yields of symmetrical biaryls with a minimum amount of catalyst.
Ullmann coupling is a valuable technique to obtain symmetrical bipyridines [62,63]. The original and convenient route to synthesize the symmetric bipyridines is the stoichiometric copper-mediated homocoupling of aryl halides [64,65,66,67,68,69,70,71]. Figure 7 shows the reaction mechanism when copper metal is used as a typical example. In this case, two possible reaction mechanisms are considered: a radical process and an anion process. It is unknown in detail which mechanism is responsible for the progression. The use of high temperatures (neat and > 200 °C), poor substrate scope, and need to use stoichiometric amounts of copper reagent has limited the utility of these reactions. Despite these limitations, the Ullmann coupling remains a significant method for obtaining symmetrical bipyridines. Advances in reaction conditions and exploration of alternative methodologies may further enhance the practicality and efficiency of this synthetic route.
It has been reported that bipyridine compounds can be synthesized in good yield by performing two oxidative additions of halogenated pyridine in the presence of a palladium catalyst and a reducing agent. Recent representative examples are shown in Figure 8. For example, the combination of Pd(OAc)2 and piperazine in DMF at 140 °C facilitated the homocoupling of bromopyridines (Figure 8(a)) [72]. Although the reaction required high temperature (140 °C), it is operationally straightforward and good substrate compatibility.
Lee et al reported that treatment of bromopyridines in the presence of Pd(OAc)2 with indium and LiCl efficiently produced bipyridines through homo coupling in good to excellent yield (Figure 8(b)) [73]. Although the mechanism of the coupling reactions based on bimetallic system has not been established, the key point of the transformation proceeded via a direct transfer from indium to palladium(II) species. Zhang and Yang reported bimetallic Ullmann coupling of bromopyridines in the presence of stoichiometric copper powder and a catalytic amount of Pd(OAc)2 (Figure 8(c)). The catalytic system showed good tolerance to different functional groups in good yield under relatively mild conditions [74]. The coupling process was promoted via radicals generated by redox interaction between Cu(0) and Pd(IV) species in the heating system. The results indicated the robust tolerance of the method for bromopyridines with different functional groups and various symmetric bipyridines were efficiently prepared with good chemical yields. Carrick et al reported the synthesis of 2,2’-bipyridines and bis-1,2,4-triazinyl-2,2’-bipyridines via Pd-catalyzed Ullmann type reaction in the presence of Zn, Cu(I), and TMEDA (Figure 8(d)) [75]. The transformation suggests a synergistic transformation dependent on cooperativity of Pd(II), Zn(0), and Cu(I). The prepared bipyridine derivatives were considered in separation experiments of spent nuclear fuel, emphasizing the practical applications of the synthetic methodology. These processes highlight the unique reactivity achieved through the use of bimetallic systems and provide new avenues for bipyridine synthesis.
Tanaka et al reported PdCl2(PhCN)2-promoted reductive coupling of bromopyridines proceeded smoothly to afford the corresponding bipyridines in the presence of TDAE (tetrakis(dimethylamino)ethylene) as organic reductant in good yield (Figure 8(e)) [76]. TDAE has a mild reducing ability and hardly reduces functional groups. The reductive coupling reaction would be initiated with reduction of Pd(II) with TDAE generating Pd(0) species. While the homocoupling of 2-bromopyridine and 4-bromopyridine progressed in this reaction, no reaction occurred with 3-bromopyridine.
Several research groups investigated reaction systems utilizing alcohol as both a solvent and reducing agent. For example, Fen et al reported the synthesis of bipyridine via Pd-catalyzed reductive homocoupling in 1,4-butanediol under air (Figure 8(f)). The reaction proceeded in the presence of 0.01mol% of Pd(OAc)2 as a catalyst, utilizing 1,4-butanediol as O,O-ligand, solvent, and reductant. Therefore, no extra reducing agents and ligand are required in the catalytic system [77]. The low loading of Pd catalyst and mild reaction conditions show the superiority of the method. Zhang et al reported that Pd(dppf)-catalyzed reductive homocoupling of bromopyridine or iodopyridine in 3-pentanol. XPS studies indicated that the oxidation of 3-pentanol is involved with the in situ regeneration of the reductive Pd0(dppf) active species. During the reaction, 3-pentanol works as reducing agent, and is converted to 3-pentanone. The catalytic system is very simple, and elimination of external additive simplify the product separation and purification processes (Figure 5(g) [78].
Recently, examples of Pd-catalyzed homocoupling of halopyridines were reported without the use of a reducing agent [79,80,81]. For example, Deschong demonstrated various coupling reactions using Pd catalysts with high catalytic activity (Figure 8(h)) [82]. In their study, 2-iodopyridine was successfully converted to 2,2'-dipyridyl in good yield in the presence of Pd(dba)2, P(t-Bu)2(o-biphenyl) and (i-Pr)2NEt. However, when employing the same conditions with 2-bromopyridine, only trace amounts of the coupled product were obtained.
It has been reported that this reaction proceeds even with a cost-effective nickel catalyst. Traditionally, reductive coupling with stoichiometric amounts of hydrated NiCl2, PPh3, and Zn according to Tiecco, Testaferri et al afforded bipyridines in good yield (Figure 8(i)) [83]. They observed that application of catalytic method to the synthesis of bipyridines from halopyridines led to low yields of bipyridines due to competing reductive dehalogenation of the substrates. Duan et al reported a facile synthetic approach for symmetrical and unsymmetrical 2,2′-bipyridines through the Ni-catalyzed reductive couplings of 2-halopyridines (Figure 8(j)) [84]. The couplings were efficiently catalyzed by NiCl2·6H2O without the use of external ligands to give 2,2’-bipyridines in high yield. 3,3’-Bipyridines could not be synthesized by the catalytic systems. The result suggested that the product, 2,2’-bipyridine derivatives, acted as ligands for nickel(II), facilitating the smooth progress of the coupling reaction. A variety of 2,2′-bipyridines was efficiently synthesized.
Fan and Song reported that 2,2’,6,6’-tetramethyl-4,4’-bipyridine was obtained by the homocoupling of 4-bromo-2,6-dimethylpyridine under mild conditions with NiBr2(PPh3)2, Et4NI, and zinc powder in high yield [85]. They further investigated electrochemical properties of the viologen derivatives on 2,2’,6,6’-positions of the 4,4’-bipyridine core rings. Many coordinating pyridine derivatives can be synthesized. The examples provided highlight strategic opportunities to acquire diverse complexants for the definition structure–activity relationships in separations systems.
Caubene et al reported effective Ullmann coupling of pyridyl halides using Ni catalyst prepared from NaH/ t-BuONa/ Ni(OAc)2/ PPh3 (Figure 8(k)) [86]. The optimal component ratio was determined as 4:2:1:4. DME and t-BuONa were found to be best solvent and activating alkoxides, respectively. In this reaction, the reduced product as a side reaction was suppressed to about 20%.
Dehydrogenative coupling of functionalized pyridines with direct C‒H bond activation appears as a promising alternative from an economic and environmental perspectives. Suzuki et al reported that diruthenium tetrahydrido complex, Cp*Ru(μ-H)4RuCp*, catalyzed dimerization of 4-substituted pyridines. The reaction proceeds through the cleavage of C‒H bonds with the Ru complex (Figure 8(l)) [87]. In this reaction, the 2-position of pyridines was the reaction site, producing corresponding bipyridine derivatives. The reactivity of dehydrogenative coupling changes depending on the substituent on pyridine ring, and higher pKa resulted in the product in good yield. No byproduct, such as terpyridine, was formed in the transformation. They isolated bis(μ-pyridyl) and μ-η2:η2-bipyridine coordinated Ru complexes as the intermediates in the catalytic cycles.
Several Ullmann couplings using heterogeneous catalysts have been also reported. Sakurai et al reported bimetallic gold-palladium alloy nanoclusters as an effective catalyst for Ullmann coupling of chloropyridines under ambient conditions (Figure 8(m)) [88]. The Ullmann coupling product was not observed when monometallic Au and/or Pd cluster was used as a catalyst. In contrast to conventional transition metal catalysts, 2-chloropyridine was found to be highly reactive as compared to 2-bromopyridine. From the UV-vis and ICP-AES measurements, a significant amount of leached Pd(II) was observed in the coupling with 2-bromopyridine as compared with 2-chloropyridine. This observation suggests that the leaching process may be a crucial factor in diminishing reactivity. Ren et al reported that light-induced oxidative half-reaction of water splitting is effectively coupled with reduction of bromopyridines (Figure 8(n)) [89]. The present strategy allows various aryl bromides to undergo smoothly the reductive coupling with Pd/g-C3N4 (graphite phase carbon nitride) as the photocatalyst, giving a pollutive reductant-free method for synthesizing bipyridine skeletons. Moreover, the use of green visible-light energy endows this process with additional advantages including mild conditions and good functional group tolerance. Although this method has some disadvantages such as a use of environmentally unfriendly dioxane, and the addition of Na2CO3, it can guide chemists to use water as a reducing agent to develop clean procedures for various organic reactions. The utilization of readily available and non-toxic water by photocatalytic water splitting is highly attractive in green chemistry.
4. Electrochemical methods
An electrochemical method is one of the most promising synthetic approaches from an environmental perspective. The method is important to avoid the toxicity and cost especially in the synthesis of pharmaceutical molecules. However, few effective methods of bipyridine synthesis are reported [90]. Navarro et al reported nickel-catalyzed electro reductive homocoupling of bromopyridines in an undivided cell using Zn or Fe anode. By optimizing the reaction conditions, they efficiently synthesized heterocoupling products (Figure 9(a) left) [91,92]. The method is simple and efficient with isolated yield up to 98% using N,N-dimethylformamide as the solvent. They also explored heterocoupling reactions, and statistical yield were observed in the hetrocoupling. For example, a reasonable isolated product yield of 6,6’’-dimethyl-2,2’:6’,2’’-terpyridine was observed in the reaction between 2,6-dichloropyridine and 2-bromo-6-methylpyridine. The catalytic cycle was shown in Figure 9(a) right. The advantage of the method is that PPh3 ligand is not required, and that active Ni(0) is generated using constant current density. However, it is not controlled by a complete electrode reaction.
Lennox et al reported the electrochemical approach of bipyridine derivatives synthesis from N,N’-dipyridylureas (Figure 9(b)) [93]. In their method, the urea and graphite electrode were added to the cathodic H-cell, while graphite electrode was added to anodic H-cell. DMF was used as solvent. The reaction was subjected to electrolysis at 3 Fmol-1 and ‒6 mA under N2 atmosphere. The electrochemical transformation is important in that it is an intermolecular reaction. In their case, sterically hindered samples are also suitable for the synthesis of bipyridine derivatives. The conformational alignment of the arenes in the N,N’-diaryl urea intermediates promote C‒C bond formation following single-electron reduction. The method represented complementary reactivity to most metal-catalyzed coupling reaction. The procedure is operationally simple and represents an improvement over known synthesis of the disubstituted bipyridine.
5. Other methods
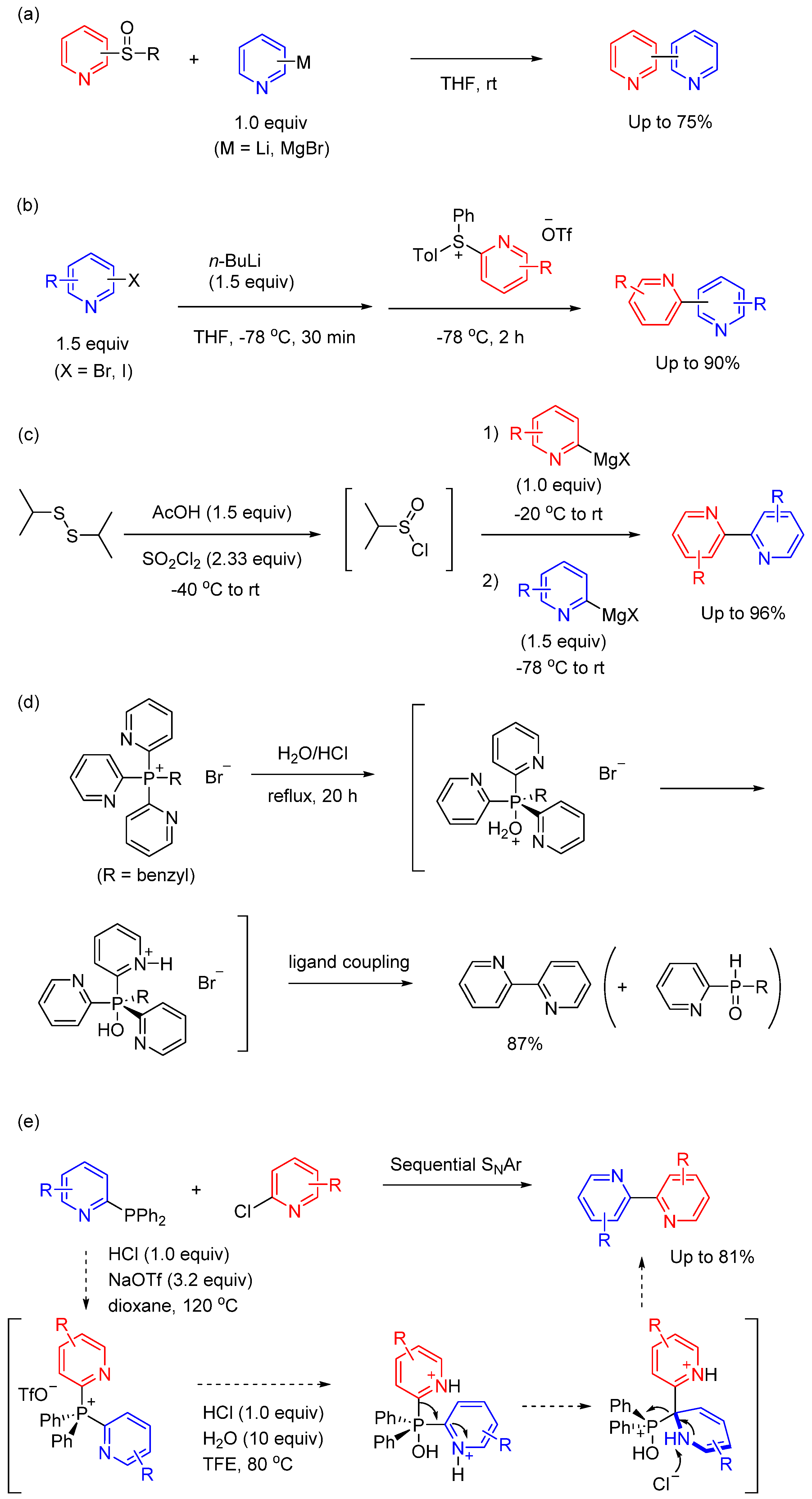
In this section, we discuss synthetic methods that cannot be classified as sections 1‒4. Development of alternative methods such as transition-metal free systems for cross-coupling remains a desirable attempt. From this point of view, sulfur-mediated organic synthesis has been a rich area for C‒C bond formation [94,95]. For example, a series of studies by Oae and Furukawa demonstrated that the addition of pyridyl lithium (or pyridyl magnesium bromide) to pyridyl aryl sulfoxides led to the formation of bipyridine derivatives via sulfurane intermediates. (Figure 10(a)) [96,97]. The mechanism for the formation of bipyridyls can be rationally explained in terms of an initial attack of the Grignard reagent on the sulfinyl sulfur atom to afford the sulfurane as an intermediate from which the two pyridyl groups couple selectively while the phenyl (or tolyl) group on the sulfoxide does not participate in the reaction. The selective C‒C bond formation gives precision to the process of mechanism. Several sulfur-mediated bipyridine synthesis have been reported that further improve this approach [98,99,100].
Recently, McGarrigle et al disclosed the synthesis of pyridylsulfonium salts and their application in the preparation of bipyridine derivatives through ligand coupling reaction [101]. The key intermediate sulfonium salts were obtained by Cu(OTf)2-catalyzed S-selective arylation of p-tolylpyridyl sulfide with Ph2IOTf. To demonstrate the synthetic utility, the resulting pyridylsulfonium salts were employed in a scalable transition-metal-free coupling protocol, yielding functionalized bipyridines with remarkable functional group tolerances (Figure 10(b)). This modular methodology permits selective introduction of functional groups from commercially available pyridyl halides, facilitating the synthesis of both symmetrical and unsymmetrical 2,2’- and 2,3’-bipyridines. Importantly, the bipyridine in this case was formed via a sulfuran intermediate, highlighting the utility of this method in obtaining structurally diverse bipyridine compounds. Qin et al presented a sulfinyl(IV) chloride-mediate cross-coupling involving two pyridyl Grignard reagents (Figure 10(c)) [102]. The intermediate of this transformation, isopropyl sulfinyl(IV) chloride, can be readily obtained from diisopropyl disulfide. Addition of successive pyridyl nucleophiles to sulfinyl (IV) chloride facilitates the formation of a trigonal bipyramidal sulfurane intermediate. Subsequent reductive elimination results in the production of bispyridyl products in a practical and efficient manner. A large number of functional groups are tolerated under the reaction conditions, allowing rapid access to molecular complexity. In contrast to transition metal-catalyzed couplings, this reaction is uniquely suited for the preparation of Lewis basic substrates which are difficult to couple under classical conditions.
Phosphorus-mediated C‒C bond formation has attracted much attention. Especially, the synthesis of bipyridines via a phosphorus-ligand coupling reaction has been investigated and the feasibility of this process using a variety of different precursors has been demonstrated [103,104]. Oae’s research revealed that treating tri(2-pyridyl)benzyl phosphonium bromide with acidic water resulted in the formation of 2,2’-bipyridine in good yield (Figure 5(d) [105]. No formation of 2-benzylpyridine was observed in the reaction. The result suggested that benzyl group showed no chance to come at the axial position in the intermediate in aqueous conditions. Consequently, this method is excellent for synthesizing symmetrical bipyridine compounds. Inspired by the method, McNally et al reported a strategy to form bipyridines by coupling pyridylphophines with chrolopyridines. The reaction proceeds via a tandem SNAr-ligand-coupling sequence (Figure 5(e)) [106]. The synthetic process is as follows: heating phosphine and chropyridine in dioxane with HCl and NaOTf forms the bis-heterocyclic phosphonium salt. Subsequently, the further addition of HCl and H2O in TFE (trifluoroethanol) proceed the ligand coupling reaction. A diverse set of bis-azine biaryl products can be formed in good to excellent yield, including substitution patterns such as 2,2’-bipyridines, that are challenging for traditional metal-catalyzed approaches. Abundant chloroazines, simple protocols, and valuable bispyridine products make the approach useful for medicinal chemists.
6. Summary
This review provides a comprehensive overview of recent advancements in the synthesis of bipyridine derivatives. The synthetic methods are categorized as metal-catalyzed heterocoupling, Ullmann coupling and Wurtz coupling, electrochemical approaches, and other methods. The development of efficient synthetic methods for bipyridine derivatives holds promise for the synthesis of diverse functional materials. Each synthetic approach highlighted in this review exhibits unique characteristics, and continued efforts are directed towards synthetic methods that are applicable to the range of bipyridine derivatives. Future challenges lie in implementing these methods in an industrial scale to ensure efficiency of the synthesis process. Continuous exploration and optimization of these methodologies are critical to bring out the full potential of bipyridine derivatives in the synthesis of advanced functional materials.
Funding
This work was financially supported in part by the Tonen General Sekiyu Research/Development Encouragement & Scholarship Foundation and a Grant-in-Aid for Scientific Research (C) (no. JP22K05250) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Acknowledgments
The author thanks Tonen General Sekiyu Research/Development Encouragement & Scholarship Foundation and a Grant-in-Aid for Scientific Research (C) (no. JP22K05250) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kaes, C.; Katz, A.; Hosseini, M. W. Bipyridine: The Most Widely Used Ligand. A Review of Molecules Comprising at Least Two 2,2‘-Bipyridine Units. Chem. Rev. 2000, 100, 3553–3590. [Google Scholar] [CrossRef] [PubMed]
- Hassan, J.; Sevignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl−Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction. Chem. Rev. 2002, 102, 1359–1470. [Google Scholar] [CrossRef] [PubMed]
- Newkome, G. R.; Patri, A. K.; Holder, E.; Schubert, U. S. Synthesis of 2,2’-Bipyridines: Versatile Building Blocks for Sexy Architectures and Functional Nanomaterials. Eur. J. Org. Chem. 2004, 235–254. [Google Scholar] [CrossRef]
- Schubert, U. S.; Kersten, J. L.; Pemp, A. E.; Eisenbach, C. D.; Newkome, G. R. A New Generation of 6,6′-Disubstituted 2,2′-Bipyridines: Towards Novel Oligo(bipyridine) Building Blocks for Potential Applications in Materials Science and Supramolecular Chemistry. Eur. J. Org. Chem. 1998, 2573–2581. [Google Scholar]
- Mathieu, J.; Fraisse, B.; Lacour, D.; Ghermani, N.; Montaigne, F.; Marsura, A. An Original Supramolecular Helicate from a Bipyridine–Bipyrazine Ligand Strand and NiII by Self-Assembly. Eur. J. Inorg. Chem. 2006, 133–136. [Google Scholar]
- Liu, B.; Yu, W. L.; Pei, J.; Liu, S. Y.; Lai, Y. H.; Huang, W. Design and Synthesis of Bipyridyl-Containing Conjugated Polymers: Effects of Polymer Rigidity on Metal Ion Sensing. Macromolecules 2001, 34, 7932–7940. [Google Scholar] [CrossRef]
- Balzani, V.; Juris, A. Photochemistry and photophysics of Ru(II) polypyridine complexes in the Bologna group. From early studies to recent developments. Coord. Chem. Rev. 2001, 211, 97–115. [Google Scholar] [CrossRef]
- Blakemore, D. “Synthetic Methods in Drug Discovery”, The Royal Society of Chemistry, 2016, vol. 1, pp. 1‒69.
- Constable, E. C.; Housecroft, C. E. The Early Years of 2,2′-Bipyridine—A Ligand in Its Own Lifetime. Molecules 2019, 24, 3951. [Google Scholar] [CrossRef] [PubMed]
- Chelucci, G.; Thummel, R. P. Chiral 2,2′-Bipyridines, 1,10-Phenanthrolines, and 2,2′:6′,2′′-Terpyridines: Syntheses and Applications in Asymmetric Homogeneous Catalysis. Chem. Rev. 2002, 102, 3129–3170. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, N. C. Chiral 2,2’-bipyridines: ligands for asymmetric induction. J. Chem. Soc. Perkin Trans. 1 2002, 1831–1842. [Google Scholar] [CrossRef]
- Campeau, L.-C. Fagnou, K. Applications of and alternatives to π-electron-deficient azine organometallics in metal catalyzed cross-coupling reactions. Chem. Soc. Rev. 2007, 36, 1058–1068. [Google Scholar] [CrossRef]
- Hapke, M.; Brandt, L.; Lützen, A. Versatile tools in the construction of substituted 2,2’-bipyridines—cross-coupling reactions with tin, zinc and boron compounds. Chem. Soc. Rev. 2008, 37, 2782–2797. [Google Scholar] [CrossRef]
- Miyamura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Metal-catalyzed cross-coupling reactions, Diederich, F. Stang, P. J. Eds. Wiley-VCH, Weinheim, Germany, 1998.
- Stanforth, S. P. Catalytic cross-coupling reactions in biaryl synthesis. Tetrahedron 1998, 54, 263–303. [Google Scholar] [CrossRef]
- Suzuki, A. Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles, 1995–1998. J. Organomet. Chem. 1999, 576, 147–168. [Google Scholar] [CrossRef]
- Kotha, S.; Lahiri, K.; Kashinath, D. Recent applications of the Suzuki–Miyaura cross-coupling reaction in organic synthesis. Tetrahedron 2002, 58, 9633–9695. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamamoto, Y. Cross-coupling Reactions of Organoboranes: An Easy Method for C‒C Bonding. Chem. Lett. 2011, 40, 894–901. [Google Scholar] [CrossRef]
- Miyaura, N. Organoboron Compounds. Top Curr. Chem. 2002, 219, 11–59. [Google Scholar]
- Matondo, H.; Ouhaja, N.; Souirti, S.; Baboulene, M. Oxidation and Suzuki Cross-Coupling Reaction from Azaheteroarylboronic Acids. Main Group Met. Chem. 2002, 25, 163–167. [Google Scholar] [CrossRef]
- Denton, T. T.; Zhang, X.; Cashman, J. R. 5-Substituted, 6-Substituted, and Unsubstituted 3-Heteroaromatic Pyridine Analogues of Nicotine as Selective Inhibitors of Cytochrome P-450 2A6. J. Med. Chem. 2005, 48, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, L.; Song, M.; Mak, T. C. W.; Wu, Y. Highly efficient cyclopalladated ferrocenylimine catalyst for Suzuki cross-coupling reaction of 3-pyridylboronic pinacol ester with aryl halides. J. Organomet. Chem. 2006, 691, 1301–1306. [Google Scholar] [CrossRef]
- Kumar, M. R.; Park, K.; Lee, S. Synthesis of Amido-N-imidazolium Salts and their Applications as Ligands in Suzuki–Miyaura Reactions: Coupling of Hetero- aromatic Halides and the Synthesis of Milrinone and Irbesartan. Adv. Synth. Catal. 2010, 352, 3255–3266. [Google Scholar] [CrossRef]
- Matondo et al. reported 2-pyridylboronic acid by the hydrolysis of 2-pyridyl(tristrimethylsilyl)borate. See: Motondo, H.; Souirti, S.; Baboulène, M. Improved Synthesis of Azaheteroarylboronic Acids Using tris-Trimethylsilylborate Under Mild Conditions. Synth. Commun. 2003, 33, 795–800. [CrossRef]
- Cool, X. A. F.; de Gombert, A.; McKnight, J.; Pantaine, L. R. E.; Wills, M. C. The 2-Pyridyl Problem: Challenging Nucleophiles in Cross-Coupling Arylations. Angew. Chem. Int. Ed. 2021, 60, 11068–11091. [Google Scholar]
- Yamamoto, Y.; Takizawa, M.; Yu, X.-Q.; Miyaura, N. Palladium-Catalyzed Cross-Coupling Reaction of Heteroaryltriolborates with Aryl Halides for Synthesis of Biaryls. Heterocycles 2010, 80, 359–368. [Google Scholar] [CrossRef]
- Jones, N. A.; Antoon, J. W.; Bowie, Jr. A. L.; Borak, J. B.; Stevens, E. P. Synthesis of 2,2′-bipyridyl-type compounds via the Suzuki-Miyaura cross-coupling reaction. J. Heterocyclic Chem. 2007, 44, 363–367. [Google Scholar] [CrossRef]
- Sakashita, S.; Takizawa, M.; Sugai, J.; Ito, H.; Yamamoto, Y. Tetrabutylammonium 2-Pyridyltriolborate Salts for Suzuki-Miyaura Cross-Coupling Reactions with Aryl Chlorides. Org. Lett. 2013, 15, 4308–4311. [Google Scholar] [CrossRef]
- Stille, J. A. Palladium-katalysierte Kupplungsreaktionen organischer Elektrophile mit Organozinn-Verbindungen. Angew. Chem. 1986, 98, 504–519. [Google Scholar] [CrossRef]
- Negishi, E.; King, A. O.; Okukado, N. Selective carbon-carbon bond formation via transition metal catalysis. 3. A highly selective synthesis of unsymmetrical biaryls and diarylmethanes by the nickel- or palladium-catalyzed reaction of aryl- and benzylzinc derivatives with aryl halides. J. Org. Chem. 1977, 42, 1821–1823. [Google Scholar] [CrossRef]
- For example, see: Kumazawa, K. ; Yamanoi, Y.; Yoshizawa, M.; Kusukawa, T.; Fujita, M. A palladium(II)-clipped aromatic sandwich. Angew. Chem. Int. Ed. 2004, 43, 5936–5940. [Google Scholar] [CrossRef] [PubMed]
- Heller, M.; Schubert, U. S. Functionalized 2,2‘-Bipyridines and 2,2‘:6‘,2‘ ‘-Terpyridines via Stille-Type Cross-Coupling Procedures. J. Org. Chem. 2002, 67, 8269–8272. [Google Scholar] [CrossRef]
- Ma, G.; Leng, Y.; Wu, Y.; Wu, Y. Facile synthesis of 3-arylpyridine derivatives by palladacycle-catalyzed Stille cross-coupling reaction. Tetrahedron 2013, 69, 902–909. [Google Scholar] [CrossRef]
- G. Verniest, X. Wang, N. De Kimpe, A. Padwa, Heteroaryl Cross-Coupling as an Entry toward the Synthesis of Lavendamycin Analogues: A Model Study. J. Org. Chem. 2010, 75, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-Q.; Schmitt, M.; Bihel, F. POxAP Precatalysts and the Negishi Cross-Coupling Reaction. Synthesis 2020, 52, 51–59. [Google Scholar] [CrossRef]
- Tang, S. Q.; Bricard, J.; Schmitt, N.; Bihel, F. Fukuyama Cross-Coupling Approach to Isoprekinamycin: Discovery of the Highly Active and Bench-Stable Palladium Precatalyst POxAP. Org. Lett. 2019, 21, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Luzung, M. R.; Patel, J. S.; Yin, J. A Mild Negishi Cross-Coupling of 2-Heterocyclic Organozinc Reagents and Aryl Chlorides. J. Org. Chem. 2010, 75, 8330–8332. [Google Scholar] [CrossRef]
- Downs, R. P.; Chin, A.-L.; Dean, K. M.; Carrick, J. D. Synthesis of Functionalized Hemi-1,2,4-triazinyl-[2,2′]-bipyridines via Telescoped Condensation of [2,2′]-bipyridinyl-6-carbonitrile. J. Heterocycl. Chem. 2017, 54, 3008–3014. [Google Scholar] [CrossRef]
- Mongin, F.; Trécourt, F.; Gervais, B.; Mongin, O.; Quéguiner, G. First Synthesis of Caerulomycin B. A New Synthesis of Caerulomycin C. J. Org. Chem. 2002, 67, 3272–3276. [Google Scholar] [CrossRef]
- Rodriguez, N.; Gooβen, L. J. Decarboxylative coupling reactions: a modern strategy for C–C-bond formation. Chem. Soc. Rev. 2011, 40, 5030–5048. [Google Scholar] [CrossRef] [PubMed]
- Cornella, J.; Larrosa, I. Decarboxylative Carbon–Carbon Bond-Forming Transformations of (Hetero)aromatic Carboxylic Acids. Synthesis 2012, 44, 653–676. [Google Scholar] [CrossRef]
- Chennamanneni, L. R.; William, A.D.; Johannes, C. W. Palladium-catalyzed decarboxylative cross-coupling of 3-pyridyl and 4-pyridyl carboxylates with aryl bromides. Tetrahedron Lett. 2015, 56, 1293–1296. [Google Scholar] [CrossRef]
- Singh, S.; Shinde, V. N.; Kumar, S.; Meena, N.; Bhuvanesh, N.; Rangan, K.; Kumar, A.; Joshi, H. Mono and Dinuclear Palladium Pincer Complexes of NNSe Ligand as a Catalyst for Decarboxylative Direct C‒H Hetroarylation of (Hetero)arenes. Chem Asian J. 2023, 18, e202300628. [Google Scholar] [CrossRef]
- Cook, X. A. F.; Pantaine, L. R. E.; Blakemore, D. C.; Moses, I. B.; Sach, N. W.; Shavnya, A.; Willis, M. C. Base-Activated Latent Heteroaromatic Sulfinates as Nucleophilic Coupling Partners in Palladium-Catalyzed Cross-Coupling Reactions. Angew. Chem. Int. Ed. 2021, 60, 22461–22468. [Google Scholar] [CrossRef] [PubMed]
- Markovic, T.; Rocke, B. N.; Blakemore, D. C.; Mascitti, V.; Willis, M. C. Pyridine sulfinates as general nucleophilic coupling partners in palladium-catalyzed cross-coupling reactions with aryl halides. Chem. Sci. 2017, 8, 4437–4442. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Gao, G.-L.; Edmunds, A. J. F.; Worthington, P. A.; Morris, J. A.; Yu, J.-Q. Ligand-Promoted C3-Selective Arylation of Pyridines with Pd Catalysts: Gram-Scale Synthesis of (±)-Preclamol. J. Am. Chem. Soc. 2011, 133, 19090–19093. [Google Scholar] [CrossRef]
- Dumouchel, S.; Mongin, F.; Trécourt, F.; Quéguiner, G. Synthesis and reactivity of lithium tri(quinolinyl)magnesates. Tetrahedron 2003, 59, 8629–8640. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, L.; Li, Y.; Xie, T.; Zhou, S. Synthesis of Heteroaryl Compounds through Cross-Coupling Reaction of Aryl Bromides or Benzyl Halides with Thienyl and Pyridyl Aluminum Reagents. J. Org. Chem. 2014, 79, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Nicasio-Collazo, J.; Wrobel, K.; Wrobel, K.; Serrano, O. Csp2–Br bond activation of Br-pyridine by neophylpalladacycle: formation of binuclear seven-membered palladacycle and bipyridine species. New J. Chem. 2017, 41, 8729–8733. [Google Scholar] [CrossRef]
- Moore, L. R.; Vicic, D. A. A. Heterogeneous-Catalyst-Based, Microwave-Assisted Protocol for the Synthesis of 2,2’-Bipyridines. Chem. Asian J. 2008, 3, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Gros, P.; Doudouh, A.; Fort, Y. New polystyrene-supported stable source of 2-pyridylboron reagent for Suzuki couplings in combinatorial chemistry. Tetrahedron Lett. 2004, 45, 6239–6241. [Google Scholar] [CrossRef]
- Wurtz, A. Sur Une Nouvelle Classes de Radicaux rganiques. Ann. Chem. Phys. 1855, 44, 275–313. [Google Scholar]
- Hunig, S.; Wehner, I. A Convenient Synthesis of 2,2′,6,6′-Tetramethyl-4,4′-bipyridine and Its Oxidation to 2,2′,6,6′-Tetracarboxy-4,4′-bipyridine. Synthesis 1989, 552–554. [Google Scholar] [CrossRef]
- Yamanoi, Y.; Terasaki, N.; Miyachi, M.; Inoue, Y.; Nishihara, H. Enhanced photocurrent production by photosystem I with modified viologen derivatives. Thin Solid Films, 2012, 520, 5123–5127. [Google Scholar] [CrossRef]
- Davison, N.; Quirk, J. A.; Tuna, F.; Collison, D.; McMullin, C. L.; Michaels, H.; Morritt, G. H.; Waddell, P. G.; Gould, J. A.; Freitag, M.; Dawson, J. A.; Lu, E. A room-temperature-stable electride and its reactivity: Reductive benzene/pyridine couplings and solvent-free Birch reductions. Chem 2023, 9, 576–591. [Google Scholar] [CrossRef]
- Banik, A.; Paira, R.; Shaw, B. K.; Vijaykumar, G.; Mandal, S. K. Open-Shell Phenalenyl in Transition Metal-Free Catalytic C–H Functionalization. J. Org. Chem. 2018, 83, 3236–3244. [Google Scholar] [CrossRef] [PubMed]
- Abboud, M.; Mamane, V.; Aubert, E.; Lecomte, C.; Fort, Y. Synthesis of Polyhalogenated 4,4’-Bipyridines via a Simple Dimerization Procedure. J. Org. Chem. 2010, 75, 3224–3231. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, H.; Shi, W.; Lei, A. Bond Formations between Two Nucleophiles: Transition Metal Catalyzed Oxidative Cross-Coupling Reactions. Chem. Rev. 2011, 111, 1780–1824. [Google Scholar] [CrossRef]
- Bhat, A. P. I.; Bhat, B. R. Single-step oxidative homocoupling of aryl Grignard reagents via Co(II), Ni(II) and Cu(II) Complexes under air. Appl. Organometal. Chem. 2014, 28, 383–388. [Google Scholar] [CrossRef]
- Ramnial, T.; Taylor, S. A.; Clyburne, J. A. C.; Walsby, C. J. Grignard reagents in ionic solvents: electron transfer reactions and evidence for facile Br–Mg exchange. Chem. Commun. 2007, 2066–2068. [Google Scholar] [CrossRef]
- Ullmann, F.; Bielecki, J. Ueber Synthesen in der Biphenylreihe. Chem. Ber. 1901, 34, 2174–2185. [Google Scholar] [CrossRef]
- Ullmann, F.; Meyer, G. M.; Loewenthal, O.; Gilli, E. Ueber symmetrische Biphenylderivate. Liebigs Ann. Chem. 1904, 332, 38–81. [Google Scholar] [CrossRef]
- Newkome, G. R.; Patri, A. K.; Holder, E.; Schubert, U. S. Synthesis of 2,2’-Bipyridines: Versatile Building Blocks for Sexy Architectures and Functional Nanomaterials. Eur. J. Org. Chem. 2004, 235–254. [Google Scholar] [CrossRef]
- Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl−Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction. Chem. Rev. 2002, 102, 1359–1470. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T. D.; Crouch, R. D. Cu, Ni, and Pd Mediated Homocoupling Reactions in Biaryl Syntheses: The Ullmann Reaction. Org. React. 2004, 63, 265–555. [Google Scholar] [CrossRef]
- Fanta, P. E. The Ullmann Synthesis of Biaryls, 1945-1963. Chem. Rev. 1964, 64, 613–632. (d) Fanta, P. E. The Ullmann Synthesis of Biaryls. Synthesis 1974, 9–21. [CrossRef]
- Larock, R. C. Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2nd ed.; Wiley-VCH, New York, 1999.
- Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan P., C. Copper catalysed Ullmann type chemistry: from mechanistic aspects to modern development. Chem. Soc. Rev. 2014, 43, 3525–3550. [Google Scholar] [CrossRef] [PubMed]
- Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl−Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction. Chem. Rev. 2002, 102, 1359–1470. [Google Scholar] [CrossRef] [PubMed]
- Iyoda, M.; Otsuka, H.; Sato, K.; Nisato, N.; Oda, M. Homocoupling of Aryl Halides Using Nickel(II) Complex and Zinc in the Presence of Et4NI. An Efficient Method for the Synthesis of Biaryls and Bipyridines. Bull. Chem. Soc. Jpn. 1990, 63, 80–87. [Google Scholar] [CrossRef]
- Chen, M.; Hu, J.; Tang, X.; Zhu, Q. Piperazine as an Inexpensive and Efficient Ligand for Pd-Catalyzed Homocoupling Reactions to Synthesize Bipyridines and Their Analogues. Curr. Org. Synth. 2019, 16, 173–180. [Google Scholar] [CrossRef]
- Lee, K.; Lee, P. H. Efficient homo-coupling reactions of heterocyclic aromatic bromides catalyzed by Pd(OAc)2 using indium. Tetrahedron Lett. 2008, 49, 4302–4305. [Google Scholar] [CrossRef]
- Li, C.; Shi, Y.; Chen, Q.; Zhang, K.; Yang, G. Copper Powder and Pd(II) Salts Triggered One-Pot Aromatic Halide Homocoupling via a Radical Pathway. J. Org. Chem. 2023, 88, 2306–2313. [Google Scholar] [CrossRef]
- Waters, G. D.; Carrick, J. D. Convergent access to bis-1,2,4-triazinyl-2,2’-bipyridines (BTBPs) and 2,2’-bipyridines via a Pd catalyzed Ullmann-type reaction. RSC Adv. 2020, 10, 10807–10815. [Google Scholar] [CrossRef]
- Kuroboshi, M.; Waki, Y.; Tanaka, H. Palladium-Catalyzed Tetrakis(dimethylamino)ethylene-Promoted Reductive Coupling of Aryl Halides. J. Org. Chem. 2003, 68, 3938–3942. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, L.; Feng, W. Facile palladium–catalyzed homocoupling of aryl halides using 1,4-butanediol as solvent, reductant and O,O-ligand. ChemistrySelect 2016, 1, 630–634. [Google Scholar] [CrossRef]
- Zeng, M.; Du, Y.; Shao, L.; Qi, C.; Zhang, X.-M. Palladium-Catalyzed Reductive Homocoupling of Aromatic Halides and Oxidation of Alcohols. J. Org. Chem. 2010, 75, 2556–2563. [Google Scholar] [CrossRef] [PubMed]
- Penalva, V.; Hassan, J.; Lavenot, L.; Gozzi, C.; Lemaire, M. Direct homocoupling of aryl halides catalyzed by palladium. Tetrahedron Lett. 1998, 39, 2559–2560. [Google Scholar] [CrossRef]
- Hassan, J.; Penalva, V.; Lavenot, L.; Gozzi, C.; Lemaire, M. Catalytic alternative of the Ullmann reaction. Tetrahedron 1998, 54, 13793–13804. [Google Scholar] [CrossRef]
- Torii, S.; Tanaka, H.; Morisaki, K. Pd(0)-catalyzed electro-reductive coupling of aryl halides. Tetrahedron Lett. 1985, 26, 1655–1658. [Google Scholar] [CrossRef]
- Manoso, A. S.; DeShong, P. Improved Synthesis of Aryltriethoxysilanes via Palladium(0)-Catalyzed Silylation of Aryl Iodides and Bromides with Triethoxysilane. J. Org. Chem. 2001, 66, 7449–7455. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Tingoli, M.; Chianelli, D.; Montanucci, M. A Convenient Synthesis of Bipyridines by Nickel-Phosphine Complex-Mediated Homo Coupling of Halopyridines. Synthesis 1984, 736–738. [Google Scholar] [CrossRef]
- Liao, L.-Y.; Kong, X.-R.; Duan, X.-F. Reductive Couplings of 2-Halopyridines without External Ligand: Phosphine-Free Nickel-Catalyzed Synthesis of Symmetrical and Unsymmetrical 2,2’-Bipyridines. J. Org. Chem. 2014, 79, 777–782. [Google Scholar] [CrossRef]
- Li, H.; Fan, H.; Hu, B.; Hu, L.; Chang, G.; Song, J. Spatial Structure Regulation: A Rod-Shaped Viologen Enables Long Lifetimein Aqueous Redox Flow Batteries. Angew. Chem. Int. Ed. 2021, 60, 26971–26977. [Google Scholar] [CrossRef]
- Vanderesse, R.; Lourak, M.; Fort, Y.; Caubere, P. Activation of reducing agents. Sodium hydride containing complex reducing agents 23. Symmetrical coupling of nitrogen-containing heterocyclic halides. Tetrahedron Lett. 1986, 27, 5483–5486. [Google Scholar] [CrossRef]
- Dhital, R. N.; Kamonsatikul, C.; Somsook, E.; Sakurai, H. Bimetallic gold–palladium alloy nanoclusters: an effective catalyst for Ullmann coupling of chloropyridines under ambient conditions. Catal. Sci. Technol. 2013, 3, 3030–3035. [Google Scholar] [CrossRef]
- Kawashima, T.; Takao, T.; Suzuki, H. Dehydrogenative Coupling of 4-Substituted Pyridines Catalyzed by Diruthenium Complexes. J. Am. Chem. Soc. 2007, 129, 11006–11007. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Guo, Y.; An, W.; Ren, Y.-L.; Qin, Y.; Niu, C.; Zheng, X. Coupling photocatalytic water oxidation with reductive transformations of organic molecules. Nat. Commun. 2022, 13, 6186. [Google Scholar] [CrossRef] [PubMed]
- Cassol, T. M.; Demnitz,F. W. J.; Navarro, M.; Neves, Eduardo A. d. Two convenient and high-yielding preparations of 6,6’-dimethyl-2,2’-bipyridine by homocoupling of 6-bromopicoline. Tetrahedron Lett. 2000, 41, 8203–8206. [Google Scholar] [CrossRef]
- Oliveira, J. L.; Silva, M. J.; Florêncio, T.; Urgin, K.; Sengmany, S.; Léonel, E.; Nédélec, J.-Y.; Navarro, M. Electrochemical coupling of mono and dihalopyridines catalyzed by nickel complex in undivided cell. Tetrahedron 2012, 68, 2383–2390. [Google Scholar] [CrossRef]
- de Franḉa, K. W. R.; Marcelo Navarro, M.; Léonel, É.; Durandetti, M.; Nédélec. J.-Y. Electrochemical Homocoupling of 2-Bromomethylpyridines Catalyzed by Nickel Complexes. J. Org. Chem. 2002, 67, 1838–1842. [Google Scholar] [CrossRef] [PubMed]
- Stammers, E.; Parsons, C. D.; Clayden, J.; Lennox, A. J. J. Electrochemical synthesis of biaryls by reductive extrusion from N,N’-diarylureas. Nat. Commun. 2023, 14, 4561. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D.; Klose, I.; Oost, R.; Neuhaus, J.; Maulide, N. Bond-Forming and -Breaking Reactions at Sulfur(IV): Sulfoxides, Sulfonium Salts, Sulfur Ylides, and Sulfinate Salts. Chem. Rev. 2019, 119, 8701–8780. [Google Scholar] [CrossRef]
- Foubelo, F.; Yus, M. Functionalised Organolithium Compounds by Sulfur-Lithium Exchange. Chem. Soc. Rev. 2008, 37, 2620–2633. [Google Scholar] [CrossRef]
- Furukawa, N.; Shibutani, T.; Fujihara, H. Preparation of pyridyl Grignard reagents and cross coupling reactions with sulfoxides bearing azaheterocycles. Tetrahedron Lett. 1987, 28, 5845–5848. [Google Scholar] [CrossRef]
- Kawai, T.; Furukawa, N.; Oae, S. A Convenient Preparation of Bipyridines through Ligand Coupling Reaction with σ-Sulfurane Formed by Treatment of Methyl 2-Pyridyl Sulfoxide with Grignard Reagents. Tetrahedron Lett. 1984, 25, 2549–2552. [Google Scholar] [CrossRef]
- Oae, S.; Uchida, Y. Ligand-Coupling Reactions of Hypervalent Species. Acc. Chem. Res. 1991, 24, 202–208. [Google Scholar] [CrossRef]
- Oae, S. Ligand coupling reactions of hypervalent species. Pure Appl. Chem. 1996, 68, 805–812. [Google Scholar] [CrossRef]
- Oae, S.; Inubushi, Y.; Yoshihara, M. Thionyl Chloride—A Good Ligand Coupling Reagent. Phosphorous Sulfur Silicon Relat. Elem. 1995, 103, 101–110. [Google Scholar] [CrossRef]
- Duong, V. K.; Horan, A. M.; McGarrigle, E. M. Synthesis of Pyridylsulfonium Salts and Their Application in the Formation of Functionalized Bipyridines. Org. Lett. 2020, 22, 8451–8457. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Tsien, J.; Qin, T. Sulfur(IV)-Mediated Unsymmetrical Heterocycle Cross-Couplings. Angew. Chem. Int. Ed. 2020, 59, 7372–7376. [Google Scholar] [CrossRef] [PubMed]
- Mann, F. G.; Watson, J. Conditions of Salt Formation in Polyamines and Kindred Compounds. Salt Formation in the Tertiary 2-Pyridylamines, Phosphines, and Arsines. J. Org. Chem. 1948, 13, 502–531. [Google Scholar] [CrossRef]
- Newkome, G. R.; Hager, D. C. A New Contractive Coupling Procedure. Convenient Phosphorus Expulsion Reaction. J. Am. Chem. Soc. 1978, 100, 5567–5568. [Google Scholar] [CrossRef]
- Uchida, Y.; Kozawa, H.; Oae, S. Formation of 2,2’-bipyridyl by ligand coupling on the phosphorous atom. Tetrahedron Lett. 1989, 30, 6365–6368. [Google Scholar] [CrossRef]
- Boyle, B. T.; Hilton, M. C.; McNally, A. Nonsymmetrical Bis-Azine Biaryls from Chloroazines: A Strategy Using Phosphorus Ligand-Coupling. J. Am. Chem. Soc. 2019, 141, 15441–15449. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Examples of functional materials containing bipyridine core.
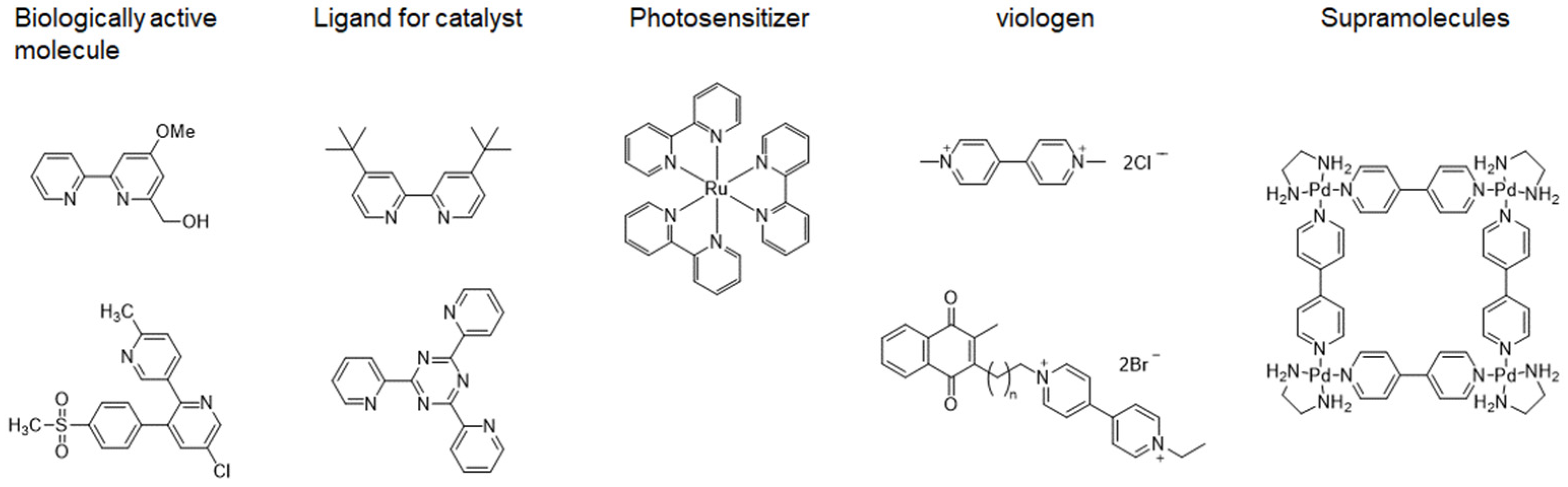
Figure 2.
Representative synthesis of bipyridine-type structures using Suzuki coupling.
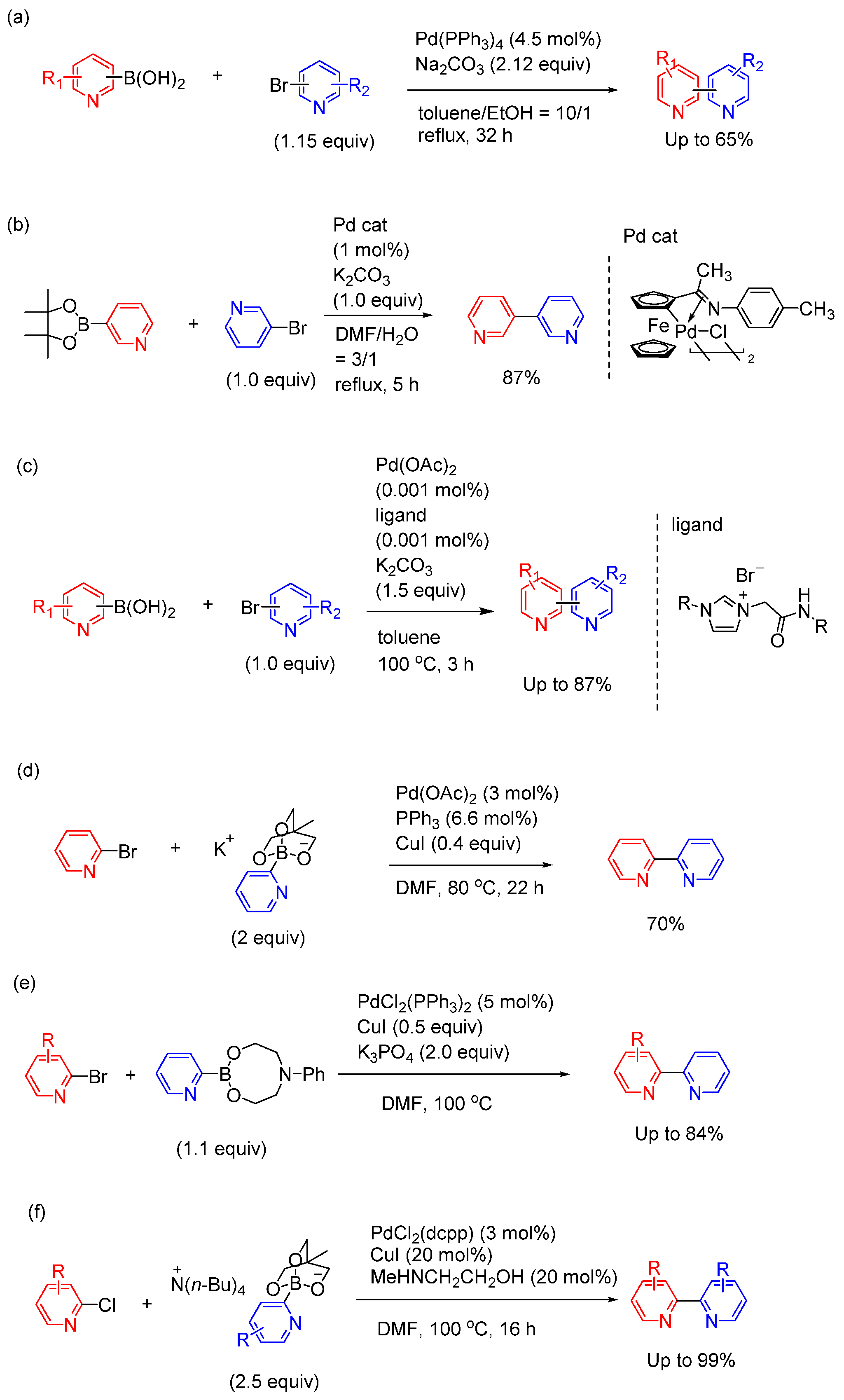
Figure 3.
Synthesis of bipyridine. (a)‒(c) Stille coupling. (d)‒(f) Negishi coupling.
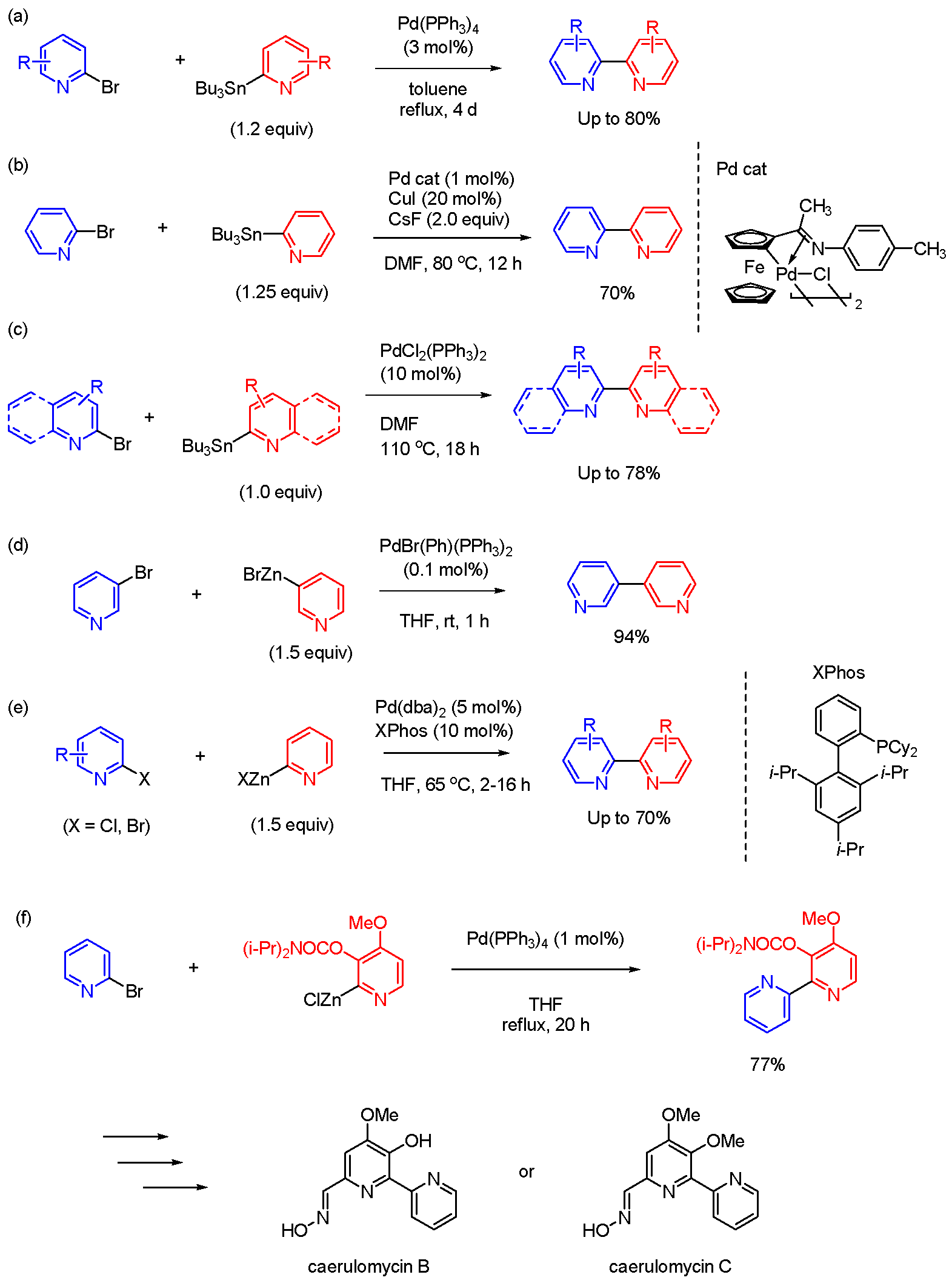
Figure 4.
Synthesis of bipyridine derivatives using other cross-coupling reaction in homogeneous catalytic system.
Figure 4.
Synthesis of bipyridine derivatives using other cross-coupling reaction in homogeneous catalytic system.
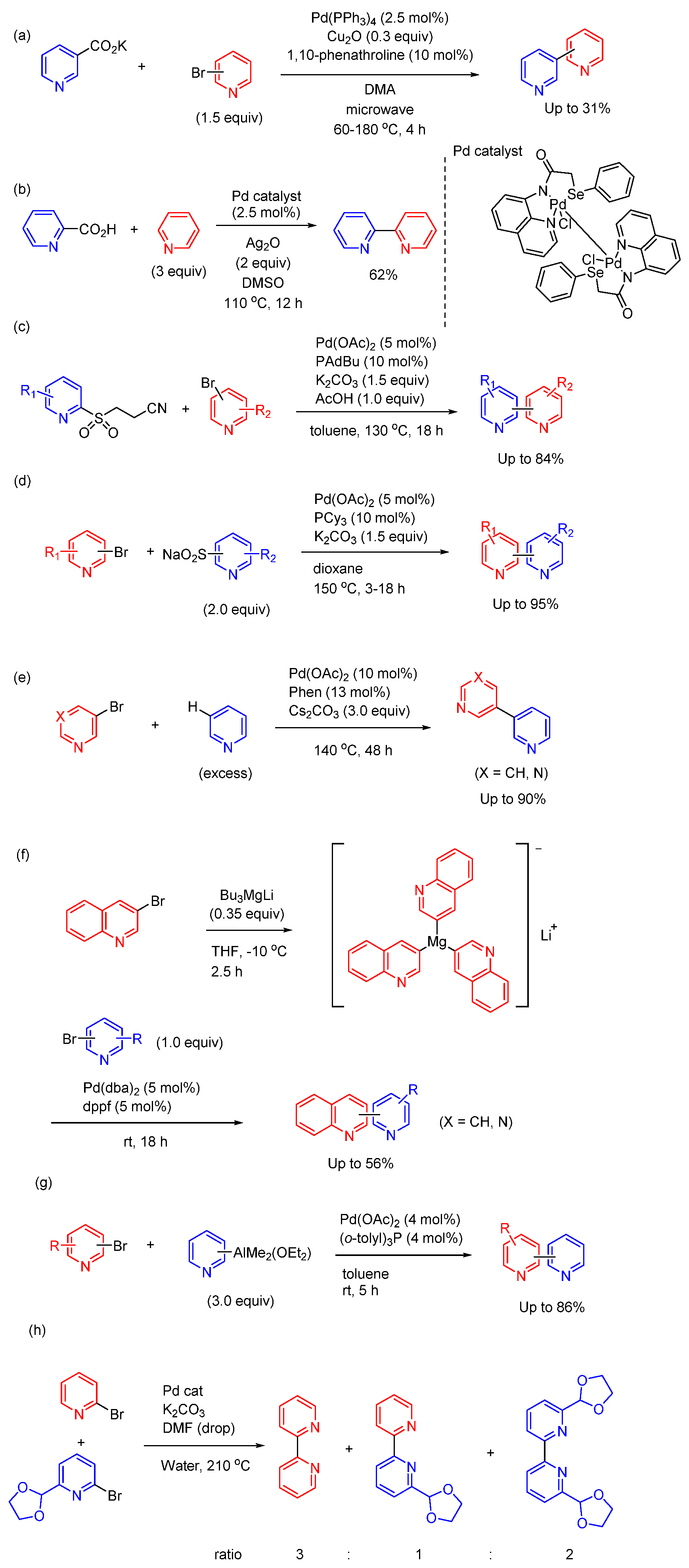
Figure 5.
(a) Negishi coupling with Pd/Al2O3 under the microwave irradiation. (b) Suzuki coupling with polystyrene-supported 2-pyridylboron reagent.
Figure 5.
(a) Negishi coupling with Pd/Al2O3 under the microwave irradiation. (b) Suzuki coupling with polystyrene-supported 2-pyridylboron reagent.
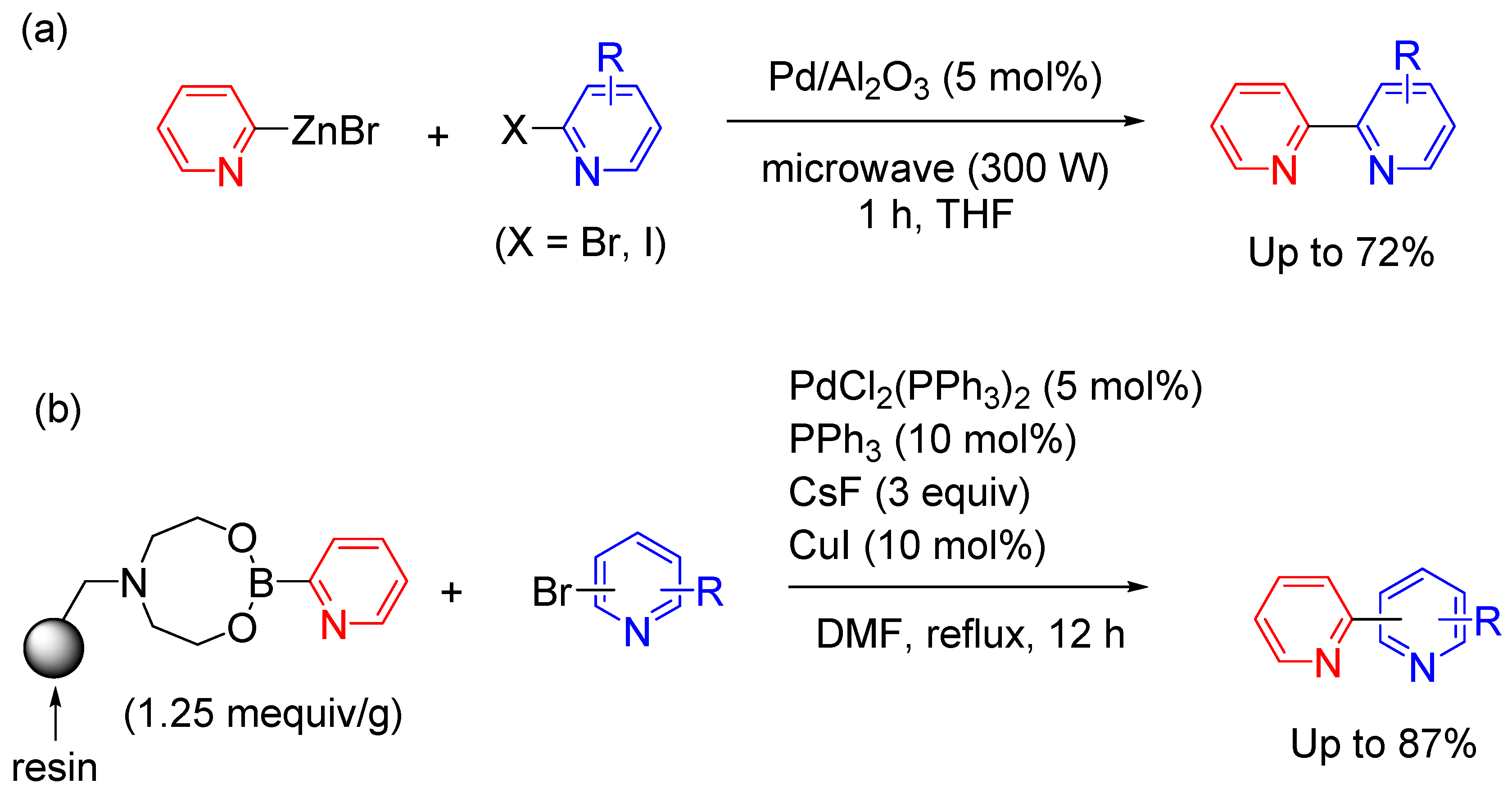
Figure 6.
(a) Reaction mechanism of Wurtz coupling of aryl halides using Na dispersion. (b) Wurtz coupling of pyridine derivatives using Na dispersion and oxidant. (c) Reaction mechanism of Wurtz coupling of pyridine. (d) Synthesis of 4,4’-bipyridine with stable electride reagent K+(LiHMDS)e‒. (e) C‒H functionalization employing a bis-phenalenyl compound and K(Ot-Bu). (f) Synthesis of 4,4’-bipyridine derivatives via dimerization process. (g) Proposed mechanism for the metal catalyzed homocoupling reaction of Grignard reagents in the presence of oxygen.
Figure 6.
(a) Reaction mechanism of Wurtz coupling of aryl halides using Na dispersion. (b) Wurtz coupling of pyridine derivatives using Na dispersion and oxidant. (c) Reaction mechanism of Wurtz coupling of pyridine. (d) Synthesis of 4,4’-bipyridine with stable electride reagent K+(LiHMDS)e‒. (e) C‒H functionalization employing a bis-phenalenyl compound and K(Ot-Bu). (f) Synthesis of 4,4’-bipyridine derivatives via dimerization process. (g) Proposed mechanism for the metal catalyzed homocoupling reaction of Grignard reagents in the presence of oxygen.
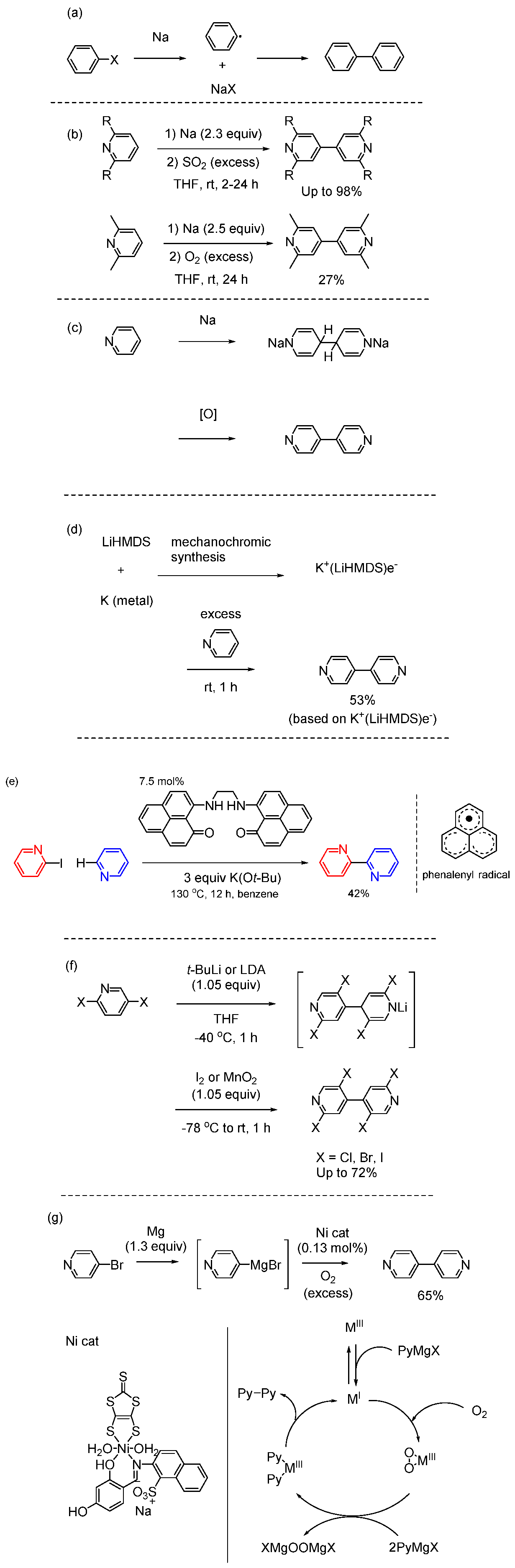
Figure 7.
Reaction mechanism of Ullmann coupling using copper metal. (a) Radical process. (b) Anion process.
Figure 7.
Reaction mechanism of Ullmann coupling using copper metal. (a) Radical process. (b) Anion process.
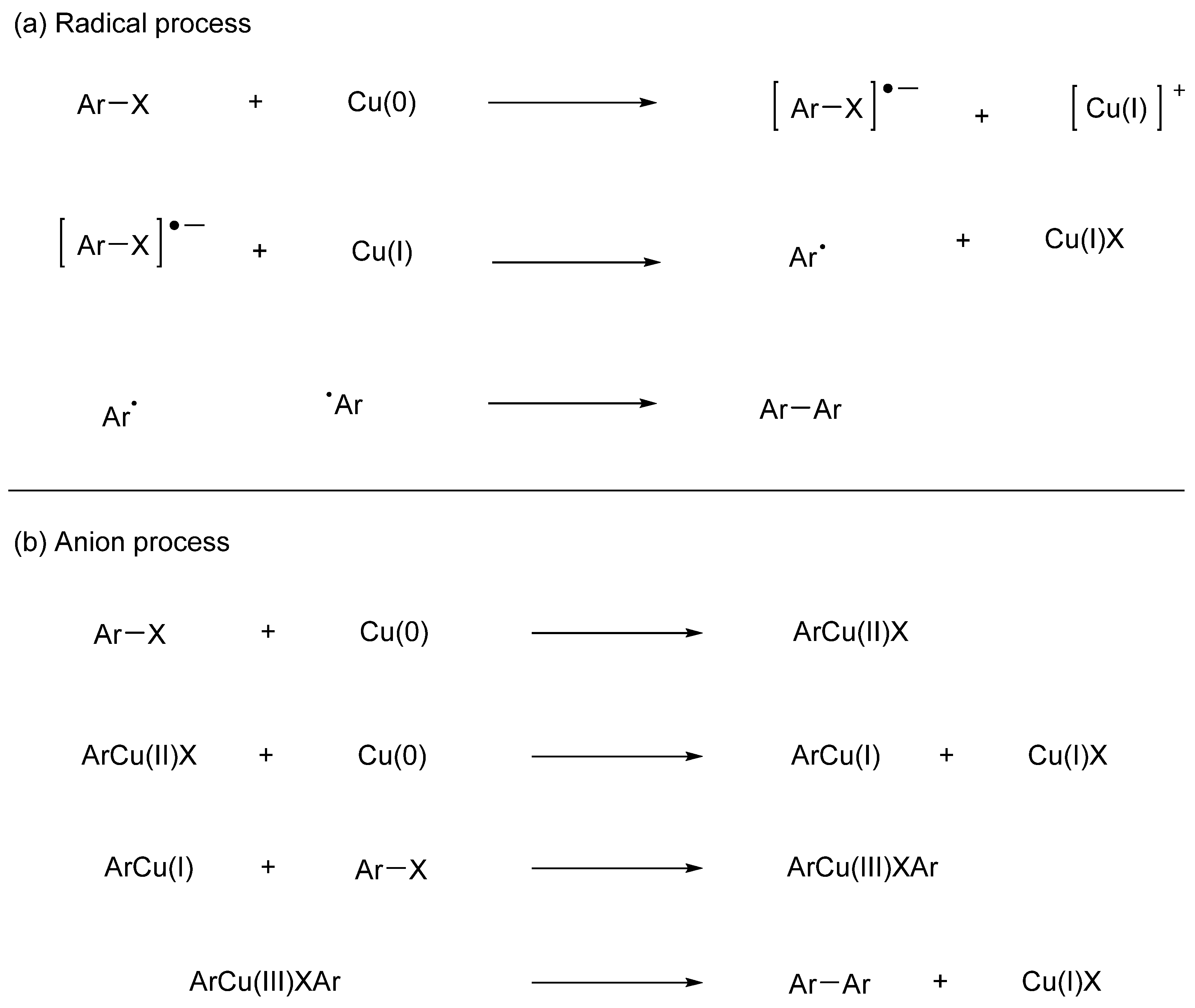
Figure 8.
Representative examples of homocoupling of pyridine derivatives in the presence of catalysts.
Figure 8.
Representative examples of homocoupling of pyridine derivatives in the presence of catalysts.
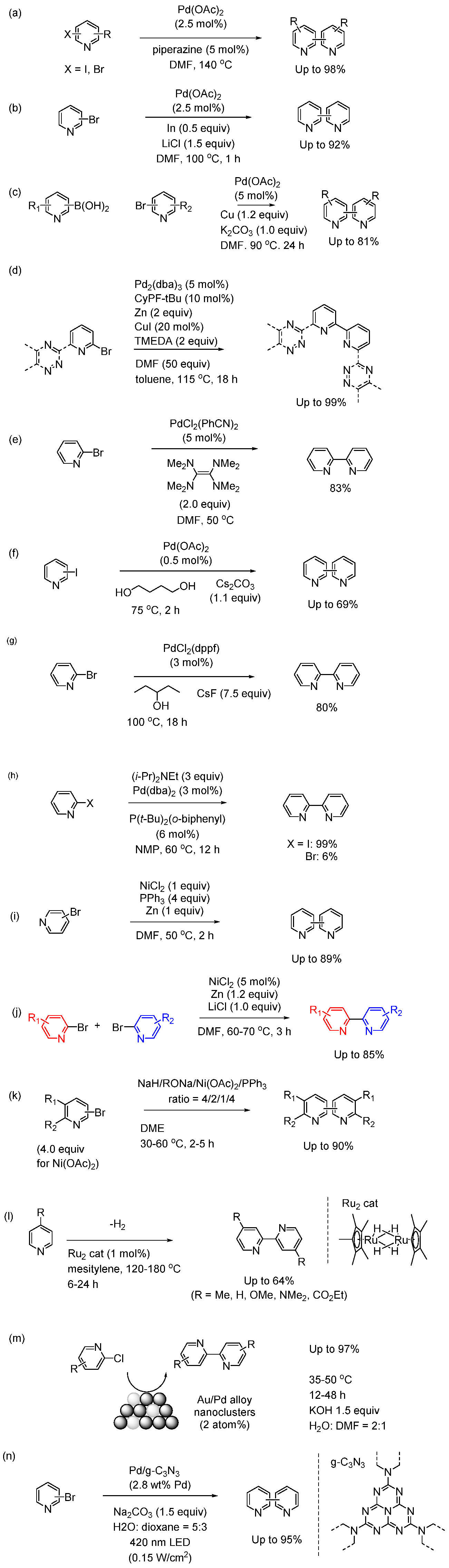
Figure 9.
(a) Coupling of 2-bromopyridines. DMF, TBABF, Fe or Zn anode, rt (left) Reaction mechanism (right). (b) Electrochemical intramolecular coupling reaction of pyridyl urea derivatives. Gr: graphite electrode. (c) Reductive coupling of bromopyridines under photoirradiation.
Figure 9.
(a) Coupling of 2-bromopyridines. DMF, TBABF, Fe or Zn anode, rt (left) Reaction mechanism (right). (b) Electrochemical intramolecular coupling reaction of pyridyl urea derivatives. Gr: graphite electrode. (c) Reductive coupling of bromopyridines under photoirradiation.
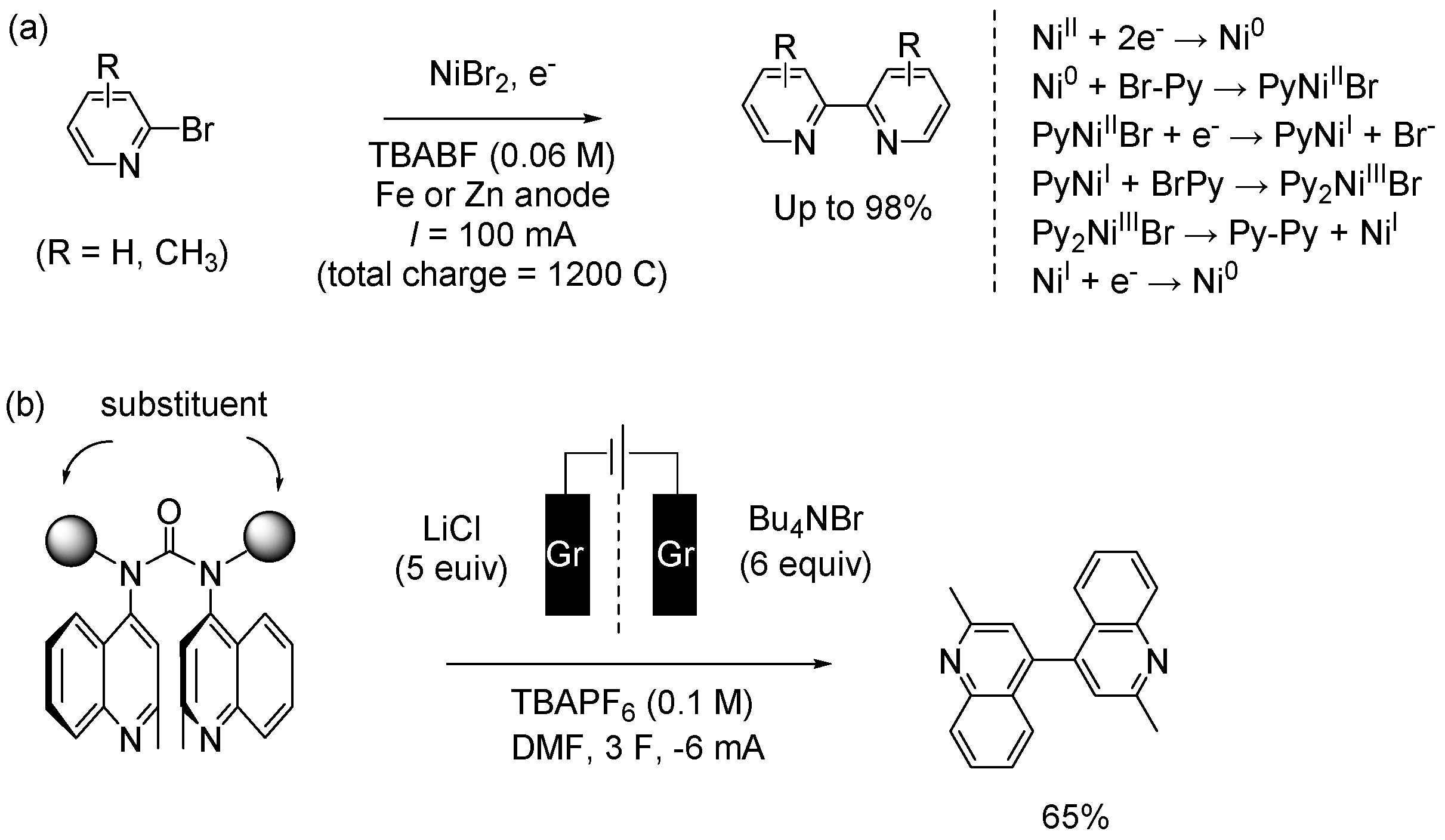
Figure 10.
(a)‒(c) Sulfur-mediated synthesis of bipyridine derivatives. (d),(e) Phosphorous-mediated synthesis of bipyridine derivatives.
Figure 10.
(a)‒(c) Sulfur-mediated synthesis of bipyridine derivatives. (d),(e) Phosphorous-mediated synthesis of bipyridine derivatives.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



