
Submitted:
15 December 2023
Posted:
15 December 2023
You are already at the latest version
Abstract
Both brain-derived neurotrophic factor (BDNF) and glucocorticoids (GCs) have multiple roles in the various aspects of neurons, including cell survival and synaptic function. BDNF and its receptor TrkB express in the central nervous system (CNS) neurons extensively, and contribution of BDNF/TrkB system in neuronal function is evident, thus, its downregulation has been considered to be involved in the pathogenesis of Alzheimer’s disease (AD). GCs, a stress-related molecule, and glucocorticoid receptor (GR) are also considered to be associated with AD, in addition to mental disorders such as depression. Importantly, a growing body of evidence suggest a close relationship between BDNF/TrkB-mediated signaling and GCs/GR system in the CNS. Here, we introduce current studies on the interaction among neurotrophic system and stress in CNS neurons, and discuss their involvement in the pathophysiology of AD.
Keywords:
Brain-derived neurotrophic factor
; TrkB
; Intracellular signaling
; Synaptic plasticity
; Glucocorticoids
; GR
; depression
; Alzheimer’s disease
1. Introduction
In the mammalian brain, brain-derived neurotrophic factor, BDNF, the most extensively studied neurotrophin, has been recognized as critical player for promoting neuronal survival and differentiation, and regulating synaptic plasticity. A growing body of evidence indicate that protective effects of BDNF against neural damage in the central nervus system (CNS) is through activating TrkB, a high affinity receptor for BDNF, although its precursor molecule, proBDNF, binding to p75NTR that is a firstly identified common receptor for neurotrophins including nerve growth factor (NGF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4), also affects neuronal aspects such as neuronal survival and functions, negatively [1,2,3]. The critical involvement of BDNF/TrkB-mediated signaling, such as phospholipase CγPLCγ PI3k/Akt-, and ERK-signaling in the neuro- and glio-genesis, as well as in synaptic function is highlighted [4]. As expected, in addition to experimental research studies, clinical evidence also showed the alteration in BDNF levels in neurodegenerative diseases can be promising biomarker in most neurodegenerative conditions in neurodegenerative diseases including Alzheimer's disease (AD) [5]. Furthermore, it has been demonstrated that apoptotic elements, which are considered to be responsible for manifestations linked to the pathophysiology of AD, interact with various signaling molecules including BDNF/TrkB, and downstream signaling pathways [6]. And also, clinical and preclinical research demonstrate that the alteration of BDNF/TrkB-mediated signaling is involved in depression pathology [7].
In order to cope with a broad spectrum of stressful stimuli, the hypothalamic-pituitary-adrenal (HPA)-axis functions as critical neuroendocrine system. The regulation of blood concentrations of glucocorticoids (GCs), which are secreted from the adrenal glands on top of the kidneys, is achieved through the negative-feedback mechanism of the HPA-axis while abnormally increased CC levels may be induced under the chronic stressful condition and causes a dysfunction of brain. Such a dysregulation of the HPA-axis has been suggested to be involved in the pathogenesis of not only mental disorders but also AD [8]. As expected, a variety of studies show that GCs affects neurons and induce neurobiological changes (molecular and cellular levels) via activation of their receptors, glucocorticoid receptor (GR), or mineralocorticoid receptor (MR). Therefore, studies on an influence of GCs in neuronal aspects such as cell proliferation and cell survival, neurogenesis, synaptic function, genetic vulnerability, and epigenetic alterations are extensively performed to clarify the basis of brain diseases [9]. In this review, we introduce current studies concerning possible interaction of BDNF/TrkB system with GCs/receptors function in the CNS neurons. Furthermore, we discuss an involvement of the interplay of BDNF and GCs in the pathophysiology of AD.
2. BDNF/TrkB system in neuronal function
BDNF is a member of neurotrophin family which consists of NGF, NT-3, and NT-4, and is most widely distributed in the mammalian brain and has multiple roles in the synaptic plasticity. Each neurotrophin has high affinity receptor. NGF binds to TrkA with high affinity. Similarly, both BDNF and NT-4 bind to TrkB, and NT-3 also binds to TrkC, with high affinity. As a common receptor, p75NTR also associates with all neurotrophins, and contributes to neuronal responses such as cell death although the Trks have been considered to be involved in a variety of positive effects including cell survival and synaptic regulation [10]. Importantly, when BDNF binds to the high affinity receptor, TrkB, mainly, three signaling pathways including PLCγ PI3k/Akt-, and ERK-signaling are activated to affect the neuronal events such as synaptic transmission (See Figure 1). Because the BDNF/TrkB system has critical roles in the various brain regions including hippocampal and cortical areas which are required for learning and memory functions, it is demonstrated that downregulation of the BDNF/TrkB system has a link to the pathogenesis of various brain diseases including AD and mental disorders. In the embryonic and adult stages, an important contribution of the BDNF/TrkB to the neurogenesis is well recognized and the changed neurogenesis is also associated with pathophysiology of these brain diseases [11]. It is well known that these TrkB-dependent signaling pathways and resultant neural cell events are stimulated by mature (processed) BDNF. Firstly, BDNF molecule is translated as a precursor protein, proBDNF, and is further cleaved into the small mature molecule that binds to TrkB receptor with high affinity. In contrast, p75NTR functions as a high affinity receptor for proBDNF while being low affinity for mature neurotrophins including mature BDNF, and the p75NTR-mediated signaling exerts negative impact on neurons, including cell death induction and decreased synaptic function [1,2,3].
A role of TrkB.T1 isoform, an alternative splicing of TrkB, is also intensively studied because of its biological contribution in the CNS [12]. TrkB.T1 isoform has a truncated C-terminal domain and lacks the intracellular domains of the full-length receptor TrkB (TrkB.FL), resulting in loss of autophosphorylation activity which is required to trigger intracellular signaling. Recently, possible involvement of an imbalance between these TrkB isoforms in aberrant signaling and hyperpathia pain has been demonstrated [12]. Evidence suggests that the truncated receptor, TrkB.T1, is harmful when it is up-regulated in response to injury or inflammation although BDNF/TrkB (the FL type) system is essential for brain development and maintenance in adulthood, including contributing to cell survival promotion and positive regulation of synaptic function [12]. It is possible that various neuronal responses depend on the expression balance of pro/mature BDNF, various TrkB receptor isoforms, and p75NTR in the brain regions.
It was reported that a transplantation of neural stem cells (NSCs) after the hypoxic treatment increased neuronal survival of ChAT (choline acetyltransferase)-positive neurons in spinal cord injury (SCI) rat model. In the system, upregulation of growth factors including NT-3, glial cell-derived neurotrophic factor (GDNF), and BDNF was observed, suggesting a transplantation of NSCs received the hypoxic preconditioning to increase these trophic factors is an effective strategy for cell-based therapy in the treatment of SCI [13]. Interestingly, recent study has demonstrated the promotion of axonal remodeling and restoration of abnormal synaptic structures using adeno-associated virus (AAV) vectors carrying genes encoding BDNF or TrkB in the stroke model [14]. After the middle cerebral artery occlusion (MCAO), the stroke rats received a combination therapy with AAV-BDNF and AAV-TrkB showed significantly improved upper limb motor functional recovery and neurotransmission efficiency compared to AAV vector treatment alone. It was also revealed that increased levels of synapsin I, postsynaptic density protein 95 (PSD-95), and GAP-43 were achieved by the combination therapy, strongly suggesting an importance of the BDNF/TrkB system in the functional recovery in the injured CNS [14].
Glial population is also affected by BDNF. Astrocytes have a role in neuronal survival and differentiation, and support oligodendrocytes to maintain brain function. Recent report on astroglial functions has shown that the morphology, physiology and survival of both oligodendrocytes and neurons were affected by astrocytic Methyl-CpG-Binding Protein 2 (MeCP2). Interestingly, BDNF mRNA expression and secreted BDNF protein levels in astrocytes are changed after astroglial MeCP2 knockdown [15]. Datta et al. (2018) showed significant differences in dopaminergic neuronal cell survival co-cultured with midbrain astrocytes under 6-OHDA stress in comparison to hindbrain and forebrain astrocytes [16]. They also found that an increased secretion of BDNF in midbrain astrocytes was noted comparing with hindbrain and forebrain astrocytes in the presence of 6-OHDA, suggesting astrocytic BDNF contributes to the survival of dopaminergic neurons [16]. Surprisingly, Harley et al. (2021) reported that selective ablation of BDNF in microglia, that is resident immune cells in the CNS, enhanced the production of newborn neurons under inflammatory conditions in which LPS (lipopolysaccharide)-induced infection or traumatic injury of brain were conducted [17]. It was revealed that BDNF ablation in microglia interfered with self-renewal/proliferation caused the decrease of their overall density, implying an influence of the microglial BDNF on neurogenesis via regulating microglia population dynamics and states [17]. Various BDNF actions in both neuronal and/or glial cells should be further studied in a variety of brain regions.
3. Glucocorticoids and neuronal functions
It is well known that the function of the HPA-axis that is critically important to cope with stressful conditions, by which blood levels of GCs is strictly regulated, has been extensively studied as a dysregulation of the HPA-axis has been suggested to be one of the risk factors in the pathogenesis of mental disorders and AD [8]. Importantly, secreted GCs from the adrenal glands that is on top of the kidneys contributes to regulated blood levels of GCs via the negative-feedback mechanism of the HPA-axis. Since the HPA-axis is considered as the key endocrine system, which has a critical role for coordinating body-wide changes to survive against stress, a lot of studies is focusing on a relationship between depression and dysfunction of the HPA axis [18]. Recent evidence also shows changed neuronal function of corticotrophin releasing hormone (CRH) neurons in the paraventricular nucleus of the hypothalamus (PVN) is involved in stress related behaviors [18]. Generally, an activation of the HPA-axis which is required for a secretion of GCs acting on a variety of organs to cope with the stress during the process of adaptation for it invokes CRH release from PVN neurons.
Roles of GCs and their receptors including glucocorticoids receptor (GR, low affinity) and mineralocorticoid receptor (MR, high affinity) in the CNS neurons has been discussed since several neurobiological changes (genetic, epigenetic, molecular and cellular, levels) caused by released GCs/receptors under the chronic stress are closely associated with negative consequences including the psychiatric disorders, therefore, in vitro studies examining the impacts of GCs on neuronal aspects such as cell proliferation and survival, neurogenesis, neurotransmission, genetic vulnerability, epigenetic alterations, and inflammation, are very important [9]. Therefore, in this section, current studies concerning contributions of GCs/receptors to neuronal aspects has been introduced.
Recently, Wei et al. (2023) reported a high emotional reactivity of forebrain GR overexpression mice [19]. They showed that a prolonged duration of anxiety behavior, and upregulation of cFos co-labeling in the calbindin1+ glutamatergic neurons in the ventral hippocampus CA1 and in the DARPP-32 (dopamine- and cAMP-regulated phosphoprotein of M(r) 32 kDa) + glutamatergic neurons in the posterior basolateral amygdala after an optogenetic stimulation in the ventral dentate gyrus of GR overexpression mice [19]. A study by Dwyer et al. (2023) has showed that a novel neurosteroid, NTS-105, has beneficial effects against traumatic brain injury (TBI). Using organotypic hippocampal slice cultures, it was revealed that deficits in hippocampal LTP (long-term potentiation, an important form of synaptic plasticity) was prevented by post-traumatic administration of NTS-105, and the post-traumatic cell death was also decreased by NTS-105 treatment [20]. Interestingly, the novel neurosteroid inhibited activation of the MR and the androgen receptor, while it was not active at the GR [20]. We recently performed a social reward conditioned place preference (SCPP) test using AKR-, BALB/c-, and C57BL/6J-strain mail mice, and found significant anxiety-like and anhedonia-like behaviors in BALB/c mice with downregulation of GR in the nucleus accumbens (NAC), compared with those in AKR and C57BL/6Jstrains [21]. In cultured cortical and hippocampal neurons, GC exposure induced significant downregulation of GR expression and decreased synaptic functions including release of glutamate, while MR expression was unchanged [22,23]. Recently, McCann et al. (2021) reported the highest density of MRs and MR:GR ratio in the hippocampal CA2 area compared with all other subregions including CA1, CA3, and the dentate gyrus (DG) in adult mice, and found that the MR in CA2 area is required for the acquisition of area-specific gene and protein expression of pyramidal neurons [24].
It is recognized that females have higher incidences of affective disorders including anxiety, PTSD, and major depression although mechanism under the sex biases remain unclear. Montgomery et al. (2023) showed an acute activation of the HPA-axis by clozapine-N-oxide (CNO) in the transgenic mice expressing the Gq-coupled Designer Receptor that are exclusively stimulated by Designer Drugs (DREADD) receptor hM3Dq in corticotropin-releasing factor (CRF, or CRH) neurons, and found that a novel sex-specific dissociation of GCs levels by the CRH activation method [25]. Although hM3Dq-expressing males displayed physiological stress sensitivity including reductions in body and thymus weights, the corticosterone levels in response to CNO in the hM3Dq-females is greater than that in males. The hM3Dq-female animals also showed significant increases in baseline and fear-conditioned freezing behaviors, suggesting possible contribution of activation of CRH neurons to sex-specific behaviors [25]. You et al. (2023) used predator scent stimulus (PSS) paradigm where mice were exposed to a volatile predator cue (e.g., cat odor), as psychological stressor, and found that appetite changes were caused by PSS [26]. Interestingly, in their system, proenkephalin (Penk)-expressing lateral hypothalamic (LHPenk) neurons contribute to overconsumption of high-fat diet (HFD) eating because silencing of LHPenk neurons normalizes the HFD overconsumption caused by PSS. They also found that an elevated corticosterone has a role in reactivity of GR-containing LHPenk neurons to HFD as an inhibition of the GR resulted in suppression of PSS-induced responses, demonstrating a critical role of physiological/hormonal alterations in appetite regulation via affecting the feeding-related hypothalamic circuit functions [26].
As mentioned above, GCs, the principal effectors of stress, is associated with multiple signaling in neurons, because a variety of expression of membrane and nuclear receptor subtypes [27]. It has been suggested that both GR and MR, for GCs, are localized to cell membrane, cytosol, and nucleus, resulting in multiple intracellular signaling and in different time scales regulation of synaptic function. Interestingly, growing evidence also demonstrated that rapid actions of acute stress-induced GCs, including changes in synaptic transmission and neuronal excitability, depend on activation of membrane GRs and MRs [27].
Markedly, a growing body of evidence demonstrate that impaired mitochondrial function is associated with major depressive disorder, and a relationship between the mitochondrial dysfunction and GCs stress is also important to understand a pathogenesis of depressive disorder [28]. Indeed, studies have demonstrated a critical influence of mitochondria system in the cognitive processes in the mature brain via regulating NSC fate during neurodevelopment [29], and a role of mitochondrial function in the NAc, and of CRH neurons in anxiety and stress [30]. Collectively, an influence of the HPA-axis system including CRH and GCs in the CNS neurons is evident, therefore, how the altered function of both mitochondrial and GCs stress system affect the BDNF/TrkB system is very interesting.
4. Glucocorticoid stress, BDNF, and neuronal functions
Evidence has suggested an involvement of altered blood concentration of GCs in the pathogenesis of various brain diseases in human. In addition, in adult mammals and humans, it has been demonstrated that the neuroplasticity, which is controlled through various mechanisms, is influenced by GC stress. As expected, regulated levels of blood GCs are closely associated with brain function through affecting hippocampal plasticity, including glutamatergic neurotransmission, neurogenesis, systems of neurotrophic factors, microglia, and astrocytes, neuroinflammation, and so on [31]. In addition to the amygdala and hippocampus, the prefrontal cortices are involved in corticolimbic system, and are perturbed by abnormal function of BDNF/TrkB-related signaling and activity of GR [32]. The long-term effects of developmental stressors on the BDNF/TrkB system and GR activity in the prefrontal cortices has been also argued to understand possible pathogenesis of neuropsychiatric illnesses including depression [32]. For example, a recent study has demonstrated an involvement of BDNF-mediated GR function in the long-term memory retention. Arango-Lievano et al. (2019) found that either the lack of GR-phosphorylation (PO4) which was caused via mutating the BDNF-dependent PO4 sites or decreased secretion of BDNF using the BDNF-Val66Met mutant resulted in reduced maintenance of newly formed postsynaptic dendritic spines in the cortex and caused the malfunction in memory retention in mice [33]. An interplay of BDNF/TrkB system with the HPA-axis has central roles in healthy function of brain, and it is probably that changed interaction of BDNF/TrkB-signaling with GR activity is involved in brain dysfunction.
As useful experimental strategy, excess GC exposure using animals have been extensively performed. Using postnatal Dairy Bull Calves, an examination on leptin concentrations and brain development markers including BDNF after the exogenous GC administration has been carried out. McCarty et al. (2023) has showed decreased expression of BDNF, FGF1 (fibroblast growth factor), and FGF2 in the hypothalamic of Holstein bulls at 5 days after intravenously infusion of cortisol within 4 hours of parturition followed by a second infusion 24 hours postpartum, with reduced leptin levels in blood and cerebrospinal fluid [34]. Moreover, mice received neonatal dexamethasone (DEX, a synthetic steroid) exposure exhibited depressive-like behaviors as well as downregulation of BDNF protein. Chen et al (2020) et al. performed a daily intraperitoneal injection of DEX for postnatal 1 (P1) to P7 to examine an impact of early-life DEX exposure on expression levels of BDNF in the hippocampus [35]. Although the neonatal DEX (ND) treatment leaded to depressive-like behaviors in the adulthood (P90-P110), the expression of hippocampal BDNF did not show any change. Interestingly, after ND exposure, hippocampal BDNF levels in the perinatal period and puberty (P40) were significantly downregulated, suggesting a disturbance of BDNF/TrkB system during the neuronal development [35]. A lot of evidence has showed downregulation of BDNF system after GC stress [8] (see Figure1).
Using AKR-, BALB/c-, and C57BL/6J-strain male mice, we examined depression-like behaviors, social reward responses, and BDNF expression [21]. Interestingly, among them, the BALB/c line exhibited the highest anxiety-like and anhedonia-like behaviors, and also displayed increased responses to social rewards in SCPP, with downregulated GR, and no changed protein levels of MR, TrkB, and p75 in the NAC, suggesting possible involvement of NAC GR activity in an increased social reward response and anxiety [21]. In contrast, after preadolescent rats were exposed to an enriched environment (EE) and combined exercise training (CET), it was revealed that hippocampal GR was significantly increased in both EE and CET groups [36]. Furthermore, hippocampal BDNF and VEGF (vascular endothelial growth facto) were also increased by EE and CET. The serum corticosterone (CORT) levels were decreased in EE rats; however, serum insulin-like growth factor-1 (IGF-1) were increased only in CET rats [36].
BDNF has pivotal roles in neurite outgrowth and expression of synaptic molecules which are essential for pre- and/or post-synaptic function in the CNS neurons. Therefore, it is probable that GCs affect the BDNF-dependent neuronal function. Previously, we reported that DEX exposure inhibited the BDNF-dependent neurite outgrowth and expression of synaptic proteins in cultured hippocampal neurons [37]. In the system, although ERK-signaling is important for the action of the BDNF-dependent neurite outgrowth and synaptic protein upregulation, DEX decreased the action by the BDNF via repressing ERK signaling [37]. Recently, Restelli et al. (2023) has showed possible role of Sprouty4 in the negative impact of GCs on the BDNF function [38]. Using PC12 cells, they showed the promotion of transcription of Sprouty4, which is a regulatory molecule repressing ERK signaling stimulated by NGF, after DEX treatment. Importantly, the knockdown of Sprouty4 or an induction of dominant negative Sprouty4 (Y53A), reversed the DEX-dependent inhibition in the NGF/TrkA-promoted ERK activation [38]. They also found that expression of Sprouty4 was upregulated by DEX in hippocampal neurons, and the knockdown of hippocampal Sprouty4 attenuated the negative influence of DEX on neurite formation, and ERK activation stimulated by BDNF, suggesting that Sprouty4 is involved in the inhibitory effects of GCs in the neurotrophin function [38]. It was also reported that retinoic acid (RA) signaling through retinoic acid receptor α (RARα) is involved the regulation of the HPA-axis [39,40]. Ke et al. (2019) examined possible effects of Ro41-5253, a selective RARα antagonist, using a depression animal model [39]. They found that an increased sucrose preference in sucrose preference test (SPT), numbers of crossing and rearing in open field test (OFT), and reduced immobility time in forced swimming test (FST) by Ro41-5253 treatment in depression rats. Furthermore, the antagonist reduced serum levels of CORT, and increased BDNF, PSD95, synaptophysin (SYP) and MAP2 in the hippocampus of depression rats, suggesting that antidepressant-like effects of Ro41-5253 on depression rats is via improving hyperactivity of the HPA-axis and hippocampal neuronal dysfunction [39].
Because growing evidence has also demonstrated that the Val66Met variation in human BDNF, where a valine (Val) amino acid is replaced with a methionine (Met) amino acid at position 66 of the BDNF protein, is associated with susceptibility to mental disorders including depression [41], the differences in neuronal function due to the Val66Met variation is very interesting. Previous reports suggest that the Val66Met variation of BDNF results in an alteration of activity-dependent secretion and intracellular trafficking of the neurotrophin [42,43]. Recently, using animal models, changed responses of the variation to acute stress has been also shown. Musazzi et al. (2022) exposed adult male BDNFVal/Met and BDNFVal/Val knock-in mice to an acute restraint stress for 30 min and investigated levels of both BDNF and CORT [44]. They found that the presynaptic release of glutamate, phosphorylation of cyclic-AMP response element-binding protein (CREB), and levels of c-fos (an immediate early gene) in the hippocampus of BDNFVal/Met were higher than those in BDNF Val/Val animals although plasma CORT concentration, and nuclear GR expression and its phosphorylation in both BDNFVal/Met and BDNFVal/Val mice were similar [44]. Another recent report has suggested that an adolescent GC stress leaded to sex-specific disruptions to fear extinction and GABAergic system due to the Val66Met genotype of BDNF [45]. During late adolescence, both male and female Val/Val and Met/Met mice received CORT through their drinking water. It was revealed that the CORT exposure selectively abolished fear extinction of female Met/Met animals while any effect of sex, genotype, or treatment was not observed for recovery of fear [45]. When examining three types of interneurons including parvalbumin, somatostatin, and calretinin in the amygdala nuclei, changed cell density of somatostatin-positive population in female (but not male) animals by Val66Met genotype, and altered calretinin-positive cell density in female (but not male) by CORT treatment were observed although parvalbumin-positive cell density was not changed by the CORT treatment or genotype [45].
In the glutamatergic neuronal function, the act of receptors in the postsynaptic sites are important, and the contribution of BDNF and GCs to the receptor regulation is interesting issue. Recently, plasticity-related receptor expression and phosphorylation at the synaptic surface after the exposure to CORT and BDNF has been reported [46]. Using slices from the sensorimotor cortex exposed to BDNF, CORT, or the simultaneous application of both BDNF and CORT (BDNF+CORT) for 30 min, and immunoblotting for NMDA-type or AMPA-type receptors were performed. Although CORT application increased NMDA-type (GluN2A, B) and AMPA-type (GluA1) receptor phosphorylation, BDNF preferentially upregulated surface levels of these glutamate receptor subunits. Interestingly, BDNF+CORT induced the phosphorylation of receptors and failed to change synaptic surface expression levels of these receptor subunits [46].
Gong et al. (2019) has reported a proteomic profiling of the neural cells using depressive model caused by CORT exposure. Their proteomic profiles of mouse neuronal C17.2 stem cells in vitro and brains obtained from the depressive-like mice caused by CORT displayed downregulated expression of mitochondrial oxidative phosphorylation-related proteins by CORT, in addition to reduced BDNF and GR, which were reversed by treatment with berberine, a chemical found in some plants including coptidis rhizome [47]. Lu et al. (2021) reported a downregulation of Nr3c1 (encoding GR) by cytoplasmic polyadenylation element-binding protein 3 (CPEB3) using a genome-wide screen of CPEB3-bound transcripts [48]. Interestingly, they also fund that traumatic intensity-dependent PTSD-like fear memory in the CPEB3-KO mice, and that hippocampal BDNF downregulation was associated with increased levels of GR during fear extinction in the CPEB3-KO animals, suggesting possible role of GR-BDNF signaling in the fear extinction [48].
As shown above, a growing body of evidence indeed demonstrate a close relationship between BDNF dysfunction and the chronic stress. An approach concerning the influence of an acute stressor on the BDNF levels in humans is very interesting because amount of cortisol concentration under an acute physical stress [49] and possibility of neuronal activity regulation by GCs [50] were reported. Hermann et al. (2021) examined the serum levels of BDNF of healthy young males who received the Trier Social Stress Test (TSST), as there is a possibility that an effect of acute psychosocial stress is dependent on the species [51]. The acute stress significantly increased the serum BDNF concentration, in addition to increased cortisol levels, compared with control [51]. It is possible that acute or moderate stressful condition is suitable for an increased function of brain as BDNF is upregulated although possible mechanism under a transition from the upregulation to the downregulation in BDNF expression levels when receiving stressor stimulation at several levels should be revealed.
Using meta-analysis and systematic reviews, available literatures in which BDNF and cortisol analysis was performed, possible involvement of Val66Met polymorphism of BDNF in the interplay between BDNF- and cortisol-contribution in mediating neuronal survival and synaptic plasticity has been examined [52]. Remarkedly, the integrated results from literatures demonstrate that BDNF and cortisol play distinct roles, respectively, in the physiology of the brain, and that their physiological actions are integrated through GR dynamics. More importantly, the Val66Met polymorphism of BDNF seems to affect individual cortisol responsivity to stress [52], strongly suggesting a critical contribution of BDNF system and GR-signaling in neuronal functions. In the future, precise understanding of intracellular interaction of BDNF- and GR-signaling will further clarify biological systems on the neurotrophin-dependent synaptic plasticity and, more importantly, will hit on a clue of new therapeutic targets for neurodegenerative and psychiatric diseases.
5. The Interplay of BDNF and Glucocorticoids in AD
5.1. The Role of BDNF in AD
AD is a multifactorial neurodegenerative disorder characterized by progressive cognitive decline, synaptic dysfunction, and memory impairment. While the disease's hallmark pathological features include the accumulation of amyloid-beta (Aβ) plaques and neurofibrillary tangles composed of hyperphosphorylated tau, increasing attention has been directed towards understanding the role of BDNF dysregulation in AD pathophysiology [8,53].
A growing body of evidence suggests that individuals with AD exhibit significant alterations in BDNF levels. Notably, numerous studies have reported reduced BDNF expression in the brains of AD patients, particularly in regions susceptible to AD pathology, such as the hippocampus and cortex [54,55]. These changes are indicative of disrupted BDNF homeostasis in AD. In addition to the reduced BDNF in the patients’ brains, the clinical relevance of altered serum BDNF levels in AD becomes apparent when examining their associations with cognitive decline [56]. Lower serum BDNF levels have been consistently correlated with cognitive impairment and the severity of AD symptoms [57,58]. These suggest that BDNF alterations may be an important factor contributing to the clinical manifestations of the disease.
Interestingly, the Val66Met polymorphism in the BDNF gene has been reported to be associated with AD. Recent studies have suggested that individuals carrying the Met allele may have an increased risk of developing AD and may exhibit more severe cognitive decline compared to those with the Val/Val genotype [59,60]. This genetic variation can affect the function and secretion of BDNF [42,61], and it has been associated with alterations in brain structure and function, as well as various neurological and psychiatric conditions, including AD [41,62].
The mechanisms behind BDNF dysregulation in AD are complex and not fully understood. One prominent factor contributing to decreased BDNF levels is the neurotoxicity associated with Aβ plaques [63]. Aβ oligomers are known to interfere with BDNF signaling pathways and, thus, compromise BDNF availability in the brain [64]. Moreover, mature BDNF levels were observed to be decreased following intracerebroventricular injection of Aβ1–42 in the rodent hippocampus [65,66]. Additionally, an increase in the proBDNF/mature BDNF ratio was shown after treatment with Aβ25–35 [67,68]. These findings suggest that the proteolytic cleavage of BDNF was also affected by Aβ25–35. Interestingly, fibrillary Aβ25–35 has been shown to selectively elevate the mRNA level of TrkB-T1, a dominant negative regulator of the full-length TrkB receptor [69].
Neuroinflammation is another key player in the dysregulation of BDNF. In AD, chronic neuroinflammatory processes, characterized by the activation of microglia and astrocytes, lead to increased production of pro-inflammatory cytokines such as interleukin-1 (IL-1) [70]. These inflammatory mediators can negatively affect BDNF expression and signaling [71,72]. For example, a study demonstrated that repeated intracerebroventricular (i.c.v.) injection of IL-1 caused a decrease in BDNF mRNA expression [73]. IL-1β was also found to compromise the neurotrophic effects provided by BDNF through suppressing the PI3-K/Akt signaling [74].
The interaction between BDNF and other molecules central to AD pathogenesis, including tau protein and apolipoprotein E (ApoE), further complicates the picture of BDNF dysregulation in the disease. Recently, Barbereau et al. (2020) demonstrated that the expression of the human Tau-P301L mutation in zebrafish neurons results in a decrease in BDNF expression, while it does not have an impact on TrkB expression [75]. Moreover, a significant decrease in the mRNA level of BDNF in the frontal cortex was observed five days after the administration of aggregated tau protein into the fourth lateral ventricle of C57Bl/6J mice [76].
The APOE gene is the most significant genetic risk factor for late-onset AD, which is the most common form of the disease [77]. Having the ApoE4 allele is associated with an increased risk of developing AD, while having the ApoE2 allele is associated with a decreased risk. ApoE3 is considered neutral in terms of AD risk [78]. Interestingly, ApoE4 has been shown to enhance the nuclear translocation of histone deacetylases (HDACs) in human neurons, leading to a reduction in BDNF gene transcription, while ApoE3 increases histone 3 acetylation and upregulates the expression of BDNF [79]. Moreover, the presence of both ApoE4 and BDNF Val66Met polymorphisms is correlated with a more severe impairment in egocentric navigation and greater atrophy in the medial temporal lobe regions among individuals with amnestic Mild Cognitive Impairment (aMCI) [80]. Pietzuch et al. (2021) also suggested that the interactions between APOE and BDNF polymorphisms may have an impact on the maintenance of functional brain connections in older adults and could potentially represent a vulnerable phenotype associated with the progression of AD [81].
Understanding these molecular interactions is crucial for unraveling the complex relationship between BDNF and AD pathology. It is worth noting that these insights do not imply a direct causative link but rather a complex interplay among multiple factors.
5.2. The Role of Glucocorticoids in AD
The role of glucocorticoids in the pathophysiology of AD is also a subject of growing interest and significance. The central concept of understanding the molecular relationship between glucocorticoids and AD is the presence of GRs in the brain. These receptors, primarily located in the hippocampus and prefrontal cortex, are known to mediate the effects of GCs on various physiological processes, including metabolism, immune response, and neural plasticity [82,83].
Recent research has demonstrated the expression of GRs in key brain regions links to the vulnerability to AD pathology [84]. The interaction between GCs and these receptors is complex. Under normal physiological conditions, GCs play a crucial role in synaptic plasticity, cognitive function, and memory consolidation [85]. However, chronic exposure to elevated GC levels, a common consequence of chronic stress, can lead to GR dysregulation [86]. This dysregulation may result in a hyperactive stress response and potentially contribute to AD pathogenesis [86].
Aβ plaques and hyperphosphorylated tau tangles are hallmarks of AD pathology. Studies have unveiled intriguing links between GCs and the accumulation of these toxic proteins [87,88]. Notably, GCs appear to influence both levels of Aβ and tau through distinct mechanisms. Research indicates that GCs can promote the production of Aβ peptides, particularly the more aggregation-prone Aβ42 isoform [89]. This effect may be mediated through the modulation of enzymes involved in Aβ production, such as β-secretase [90,91]. Furthermore, GCs have been implicated in impairing the clearance of Aβ from the brain, potentially by interfering with several Aβ-degrading proteases, such as insulin-degrading enzyme and matrix metalloproteinase-9 [92].
In the context of tau pathology, GCs have been associated with the hyperphosphorylation of tau protein [88]. Chronic GC exposure is known to activate kinases responsible for tau phosphorylation, including GSK3, CDK5, and ERK1/2, leading to the formation of neurofibrillary tangles [93,94,95,96]. This tau pathology is closely tied to synaptic dysfunction and neurodegeneration in AD [88].
Neuroinflammation is another crucial element in understanding the role of GCs in AD. Microglia, the resident immune cells of the CNS, play a pivotal role in maintaining brain homeostasis and responding to pathological insults. Recent research has revealed a multifaceted connection between GCs and microglia activation [84,97]. Chronic exposure to elevated GC levels, as seen in conditions of chronic stress, may result in an overactivation of microglia [98]. This hyperactivity can lead to a pro-inflammatory state, characterized by the release of pro-inflammatory cytokines, reactive oxygen species (ROS), and other neurotoxic molecules [98]. Such neuroinflammatory responses are detrimental to neuronal health and have been closely associated with the progression of AD [98]. Additionally, GCs can modulate microglial phenotype. Under normal conditions, microglia can exhibit both pro-inflammatory (M1) and anti-inflammatory (M2) phenotypes, depending on the context [99]. Dysregulated GC signaling has been shown to skew microglia toward the pro-inflammatory state, exacerbating neuroinflammation in AD [99]. Moreover, microglia-mediated degradation of Aβ, that is crucial for Aβ clearance, may be impaired under conditions of GC dysregulation [100].
Oxidative stress is another prominent feature of neuroinflammation and AD pathophysiology. GCs have been implicated in modulating oxidative stress pathways, further linking them to neuroinflammatory processes [101]. Elevated GC levels can promote the generation of ROS and reduce the brain's antioxidant defenses [102]. This imbalance can lead to oxidative damage of cells, proteins, and lipids [103]. Oxidative stress not only directly contributes to neurodegeneration but also exacerbates neuroinflammation by activating microglia and promoting the release of pro-inflammatory mediators [104]. Furthermore, the oxidative damage inflicted by GC-induced oxidative stress may also play a role in the formation of Aβ plaques and tau tangles [105]. Oxidatively modified proteins are more prone to aggregation and may contribute to the seeding and propagation of Aβ and tau pathology [105].
Understanding these cellular mechanisms is vital in appreciating the intricate relationship between GC dysregulation and neuroinflammation in AD. Dysregulated GCs signaling can create a microenvironment that favors neuroinflammation and oxidative stress, both of which are detrimental to neuronal health. However, it is essential to recognize that while GCs are a piece of this puzzle, they do not act in isolation. Genetic factors, other environmental stressors, and the interplay between different pathological processes in AD must also be considered in the complex pathophysiology of this disease.
5.3. Crosstalk Between BDNF and GCs
As shown in the first half of this review, BDNF and GCs exert a multifaceted interplay in the brain. BDNF signaling is integral to neuronal survival, synaptic plasticity, and cognitive function. GCs, on the other hand, are central to the body's response to stress and inflammation. Evidence suggests that these two systems interact at various levels [106], providing a platform for further exploration. It is well-known that GCs modulate BDNF gene expression [107]. A study indicate that GR interact with the regulatory sequences in the BDNF gene promoter, thereby suppressing BDNF transcription [107]. There is also evidence that GCs can diminish the secretion of BDNF protein, impacting the presence of BDNF in the extracellular environment and its interaction with TrkB receptors [108]. The intracellular signaling pathways associated with the BDNF/TrkB system including PLCγ, PI3K/Akt, and MAPK cascades, have been found to be influenced by GCs [22,109,110]. This modulation results in altered activation of downstream effectors that play a role in structural and functional plasticity within brain regions relevant to AD, which in turn contributes to cognitive decline. These nuanced interactions above are believed to be crucial in understanding the impact of chronic stress and BDNF dysregulation on AD pathology.
BDNF, through its signaling pathways, may counteract some of the adverse effects of GCs in the brain [111]. BDNF is known to promote neuroprotection, synaptic plasticity, and neuronal survival, potentially mitigating the neuronal damage caused by excessive GC exposure [112,113]. This neuroprotective role of BDNF is of particular interest in the context of AD, where GC-related neurotoxicity is a concern. The balance between BDNF and GCs in modulating synaptic plasticity is a topic of ongoing research.
5.4. BDNF and GCs as Therapeutic Targets in AD
Given the pivotal role of BDNF in neuronal survival, synaptic plasticity, and cognitive function, BDNF dysregulation presents an attractive target for therapeutic interventions in AD. Strategies aimed at restoring BDNF levels and function, either by promoting its production or enhancing its signaling, hold promise in mitigating the cognitive deficits associated with AD (Table 1).
Several experimental approaches, such as BDNF mimetics [114,115,116,117,118,119], gene therapy [120,121], and lifestyle interventions like physical activity [122,123,124], are being explored as potential ways to address BDNF dysregulation and its consequences. BDNF mimetics are compounds that mimic the actions of BDNF or enhance its receptor binding, promoting neuroprotection and synaptic plasticity. One of these BDNF-enhancing compounds is 7,8-dihydroxyflavone (7,8-DHF), a selective TrkB agonist, as demonstrated by Wurzelmann et al. in 2017 [114]. Additionally, Yuk-Gunja-Tang (YG), a Korean traditional medicine, has the capacity to enhance the endogenous expression of BDNF [117]. Numerous preclinical studies have provided evidence of the effectiveness of these agents in animal models of AD [114,115,116,117,118,119]. In addition to the pharmacological approaches, lifestyle interventions such as physical exercise and cognitive stimulation have been shown to boost BDNF levels in the brain [124]. A meta-analysis of randomized controlled trials, as reported by Jia et al. in 2019, indicated that exercise interventions were linked to significant enhancements in global cognitive function among patients with mild-to-moderate AD [125]. Likewise, research has shown that exercise training can enhance BDNF expression, cognitive function and stimulate neuroplasticity in animal models of AD [122].
Recognizing the influence of GC dysregulation in AD pathophysiology opens up possibilities for novel therapeutic interventions. Strategies that aim to modulate GC activity represent a promising approach (Table 1). These include pharmacological interventions to normalize cortisol levels or reduce the sensitivity of glucocorticoid receptors. 11β-HSD1 is a pivotal enzyme responsible for the intracellular conversion of inactive cortisone into its active form, cortisol in humans (or 11-dehydrocortisone into corticosterone in rodents). Consequently, inhibiting 11β-HSD1 results in a decrease in cortisol levels in humans (or CORT levels in rodents) [126,127]. Sooy et al. (2010) showed that UE1961, an inhibitor of 11β-HSD1, demonstrated a significant enhancement in spatial memory performance in aged mice [128]. Moreover, the researchers showed that the administration of another inhibitor, UE2316, led to a decrease in Aβ plaques within the cortex of aged Tg2576 mice [129]. This reduction was concurrent with an elevation in insulin-degrading enzyme (one of the Aβ-degrading proteases) levels, which, in turn, resulted in memory improvements [129]. In addition to 11β-HSD1 inhibitors, selective GR modulators (GRMs) are designed to specifically inhibit GR activity in AD. A study demonstrated that treatment with CORT108297, one of the GRMs, led to a reduction in the levels of APP C-terminal fragments in the 3xTg-AD mouse mode [130]. Another study also showed that mice that were administered CORT108297 via intraperitoneal injection exhibited a complete reversal of memory deficits, as evaluated through the T maze test [131].
Besides pharmacological approaches, non-pharmacological interventions aimed at fostering resilience to stress and improving cognitive function could potentially offer benefits in the management of AD [132]. Stress management techniques, such as mindfulness-based interventions and cognitive-behavioral therapy, may be explored as preventive measures in individuals at risk of AD [127]. These approaches could reduce stress-related GR fluctuations and potentially delay disease onset [133,134]. Lifestyle modifications, including regular physical exercise, a balanced diet, and adequate sleep, may also have a significant impact on GC regulation [135]. These factors can help maintain a healthy stress response system and mitigate the effects of chronic stress on neuroinflammation, oxidative stress, and Aβ/Tau pathology [134,136].
BDNF and GCs stand as pivotal factors in the pathophysiology of AD. Their roles in cognitive function, synaptic plasticity, and Aβ/Tau pathology underscore their significance in understanding the disease and their potential as therapeutic targets. Investigating the intricate relationship between BDNF and GCs in AD remains an active area of research, offering hope for novel interventions aimed at mitigating cognitive decline and improving the lives of individuals affected by this challenging neurodegenerative disorder.

Table 1.
Therapeutic approaches targeting BDNF and glucocorticoid on AD models.
| Targets | Types | AD models | Treatments | Therapeutic Effects | References |
|---|---|---|---|---|---|
| BDNF | BDNF Mimetics | 5XFAD mouse | Oral administration of R13 (a TrkB receptor agonist) at doses of 7.25, 21.8, and 43.6 mg/kg for a duration of 3 months | Prevention of Aβ deposition, alleviation of the loss of hippocampal synapses, and improvement in memory deficits | [115] |
| 3XTg mouse | Daily oral administration of 3 mg/kg of CF3CN, a TrkB receptor agonist | Reduction of brain Aβ levels and recovery of cognitive functions | [116] | ||
| Rat subjected to elevated intraocular pressure through microbead injections | Intraperitoneal administration of 5 mg/kg of 7,8-DHF for a duration of 8 weeks | Downregulation of Aβ levels | [118] | ||
| C57BL/6J mouse injected with scopolamine (1 mg/kg) | Oral administration of Yuk-Gunja-Tang at a dosage of 150 mg/kg per day for a duration of 14 weeks | Improvement of memory function as assessed by the Y-maze, novel object recognition, and passive avoidance tests | [117] | ||
| Primary rat hippocampus neurons and HT-22 cell treated with Aβ 1-42 | 7.8-DHF were introduced with lipofectamine 3000 | Enhanced GAP-43 protein expression, reduced amyloidogenesis, ROS levels and caspase-3 activity | [119] | ||
| Gene Therapy | Transgenic mouse carrying the APP Swedish (K670M) and Indiana (V717F) | Introducing the BDNF gene lentivirally into the entorhinal cortex at the age of 2 months | Recovery of neuronal loss and hippocampal-dependent contextual fear conditioning | [120] | |
| P301L mutant tau mouse | Injections of AAV-BDNF to ventricles at the age of 3 months | Mitigation of behavioral deficits, prevention of neuronal loss, alleviation of synaptic degeneration, and reduction of neuronal abnormality | [121] | ||
| Physical Exercise | APP/PS1 mouse | Voluntary running on a running wheel for a period of 3 weeks | Upregulation of BDNF and the α-secretase processing of APP, leading to a reduction in the production of Aβ peptides | [122] | |
| 3XTg mouse | Training on a rodent motor-driven treadmill with a frequency of 5 days per week for a duration of 12 weeks | Reduced levels of Aβ plaque burden and neuroinflammation, as well as the alleviation of mitochondrial dysfunction | [123] | ||
| Wistar rat | Voluntary running on a treadmill for 40 minutes daily over a period of 6 days | Improved learning and memory abilities, accompanied by the upregulation of both BDNF and TrkB | [124] | ||
| Glucocorticoid | 11β-HSD1 Inhibitor | C57BL/6J mouse (24-month-old) | Intraperitoneal administration of UE1961 at a dose of 10 mg/kg, twice daily, for a duration of 10 days | Improvement of spatial memory performance in the Y-maze | [128] |
| Tg2576 mouse | Continuous infusions of UE2316 at a rate of 10 mg/kg/day for a duration of 29 days | Downregulation of Aβ plaques in the cerebral cortex, upregulation of insulin-degrading enzyme levels, and improvement in memory | [129] | ||
| GRM | 3XTg mouse | Subcutaneous administration of CORT108297 at a dose of 1.2 mg per day for a duration of 21 days | Downregulation of the levels of APP C-terminal fragments | [130] | |
| Rat injected intracerebroventricularly with Aβ25–35 | Intraperitoneal injection of CORT108297 at a dose of 20 mg/kg for a duration of 1 week | Recovery of hippocampal amyloid-β peptide generation, attenuation of neuroinflammation and apoptotic processes, restoration of hippocampal levels of synaptic markers, and improvement in cognitive function | [131] | ||
| Physical Exercise | NMRI mouse injected with streptozotocin (0.2 mg/mouse) | 4-week swimming exercise program (days 8 to 35) | Improved cognitive function, decreased anxiety- and depression-like behavior, increased BDNF levels, decreased hippocampal glutamate and TNF-α | [133] | |
| APP/PS1 mouse | Resistance exercise (climbing a ladder with a progressive overload), every other day, for 4 weeks | Reduction of Aβ plaques in the hippocampus, decreased plasma corticosterone levels, recovery of the behavioral dificits | [134] | ||
6. Conclusion
AD, characterized by the progressive loss of cognitive function and memory, remains a significant global health challenge, affecting millions of individuals worldwide. Despite decades of research, our understanding of the intricate mechanisms underlying this debilitating condition is far from complete. While the exact cause of AD is not fully understood, there are several known risk factors. Age is the most significant risk factor, with the risk increasing with age. A family history of the disease, genetic factors (such as specific mutations in the APP, PSEN1, and PSEN2 genes), and certain lifestyle factors (such as stress) can also influence one's risk.
Although its etiology remains multifactorial and complex, an emerging body of evidence suggests that neurotrophic factors and the intricate interplay with the stress hormone cortisol (GC) may play a pivotal role in AD pathogenesis. BDNF, a crucial neurotrophin, plays a central role in maintaining and promoting neuronal health, plasticity, and survival. Recent studies have demonstrated a significant reduction in BDNF levels in the brains of AD patients. Moreover, chronic stress and dysregulated GC signaling have been implicated in AD progression. Elevated cortisol levels, commonly associated with chronic stress, can lead to neuronal toxicity and impair synaptic plasticity.
With regard to therapeutic implications, focusing on BDNF and GC signaling pathways holds significant promise as a potential strategy for the management of AD. Pharmacological interventions designed to either enhance BDNF levels or reduce GC signaling have demonstrated their potential in both preclinical and clinical studies of AD. Non-pharmacological strategies, including physical exercise, have also shown beneficial impacts on BDNF and GC levels and could potentially serve as complementary treatments for the disease. However, it's worth noting that there are still several challenges and research gaps that need to be addressed. For instance, it is essential to prudently address safety concerns and potential side effects linked to BDNF interventions. Given that BDNF has diverse effects on various cell types within the CNS and peripheral tissues through the activation of multiple signaling pathways, achieving the desired therapeutic outcomes without inducing adverse reactions can be difficult. Furthermore, the clinical translation of BDNF as a therapeutic agent has been constrained by challenges related to its ability to penetrate the blood-brain barrier (BBB). Neurotrophic factors including BDNF are macromolecules, which renders them unable to cross the BBB when administered peripherally. Therefore, developing a drug delivery system capable of targeting specific areas within the brain is essential for the effective administration of neurotrophic factors and addressing their high-molecular-weight nature. Variability in the response to BDNF- or GC-targeting treatment among individuals with AD represents another limitation. Various factors, including genetic variations, disease stage, and comorbidities, can influence the effectiveness of the therapies. It is crucial to explore the complex interactions between BDNF, stress, and other neurobiological pathways associated with the disorder.
In conclusion, this review illuminates the intricate relationship between BDNF and GC dysregulation in the pathophysiology of AD. As the global burden of AD continues to rise, it is imperative that we decipher the molecular intricacies driving this condition. By targeting BDNF and GC dysregulation, we may unlock novel therapies to improve the quality of life for AD patients and potentially alter the course of this devastating disease. Future research in this area promises to provide a deeper understanding of these mechanisms and pave the way for innovative treatment strategies, offering hope to millions affected by AD.
Author Contributions
TN and RK wrote and edited the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Grant-in-Aid for Scientific Research (C) (JSPS KAKENHI 20K06857) (TN) of the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and from the Takeda Science Foundation (TN). This study was also supported by Japanese Society of Inherited Metabolic Disease/Sanofi LSD Research Grant (RK), and the Grant-in-Aid for Young Scientists (JSPS KAKENHI 23K07772) (RK).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or reported in this review article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Meeker, R.B.; Williams, K.S. The p75 neurotrophin receptor: at the crossroad of neural repair and death. Neural Regen Res 2015, 10, 721-725. [CrossRef]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci 2005, 25, 5455-5463. [CrossRef]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep 2014, 7, 796-806. [CrossRef]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell Mol Neurobiol 2018, 38, 579-593. [CrossRef]
- Pisani, A.; Paciello, F.; Del Vecchio, V.; Malesci, R.; De Corso, E.; Cantone, E.; Fetoni, A.R. The Role of BDNF as a Biomarker in Cognitive and Sensory Neurodegeneration. J Pers Med 2023, 13. [CrossRef]
- Kumari, S.; Dhapola, R.; Reddy, D.H. Apoptosis in Alzheimer's disease: insight into the signaling pathways and therapeutic avenues. Apoptosis 2023, 28, 943-957. [CrossRef]
- Autry, A.E. Function of brain-derived neurotrophic factor in the hypothalamus: Implications for depression pathology. Front Mol Neurosci 2022, 15, 1028223. [CrossRef]
- Numakawa, T.; Kajihara, R. Involvement of brain-derived neurotrophic factor signaling in the pathogenesis of stress-related brain diseases. Front Mol Neurosci 2023, 16, 1247422. [CrossRef]
- Bassil, K.; Krontira, A.C.; Leroy, T.; Escoto, A.I.H.; Snijders, C.; Pernia, C.D.; Pasterkamp, R.J.; de Nijs, L.; van den Hove, D.; Kenis, G.; et al. In vitro modeling of the neurobiological effects of glucocorticoids: A review. Neurobiol Stress 2023, 23, 100530. [CrossRef]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol Histopathol 2010, 25, 237-258. [CrossRef]
- Numakawa, T.; Odaka, H. Brain-derived neurotrophic factor and neurogenesis. In Factors Affecting Neurodevelopment, Elsevier: 2021; pp 121-131.
- Cao, T.; Matyas, J.J.; Renn, C.L.; Faden, A.I.; Dorsey, S.G.; Wu, J. Function and Mechanisms of Truncated BDNF Receptor TrkB.T1 in Neuropathic Pain. Cells 2020, 9. [CrossRef]
- Fan, W.L.; Liu, P.; Wang, G.; Pu, J.G.; Xue, X.; Zhao, J.H. Transplantation of hypoxic preconditioned neural stem cells benefits functional recovery via enhancing neurotrophic secretion after spinal cord injury in rats. J Cell Biochem 2018, 119, 4339-4351. [CrossRef]
- Wang, J.; Cai, Y.; Sun, J.; Feng, H.; Zhu, X.; Chen, Q.; Gao, F.; Ni, Q.; Mao, L.; Yang, M.; et al. Administration of intramuscular AAV-BDNF and intranasal AAV-TrkB promotes neurological recovery via enhancing corticospinal synaptic connections in stroke rats. Exp Neurol 2023, 359, 114236. [CrossRef]
- Buch, L.; Lipi, B.; Langhnoja, J.; Jaldeep, L.; Pillai, P.P.; Prakash, P. Role of astrocytic MeCP2 in regulation of CNS myelination by affecting oligodendrocyte and neuronal physiology and axo-glial interactions. Exp Brain Res 2018, 236, 3015-3027. [CrossRef]
- Datta, I.; Ganapathy, K.; Razdan, R.; Bhonde, R. Location and Number of Astrocytes Determine Dopaminergic Neuron Survival and Function Under 6-OHDA Stress Mediated Through Differential BDNF Release. Mol Neurobiol 2018, 55, 5505-5525. [CrossRef]
- Harley, S.B.R.; Willis, E.F.; Shaikh, S.N.; Blackmore, D.G.; Sah, P.; Ruitenberg, M.J.; Bartlett, P.F.; Vukovic, J. Selective Ablation of BDNF from Microglia Reveals Novel Roles in Self-Renewal and Hippocampal Neurogenesis. J Neurosci 2021, 41, 4172-4186. [CrossRef]
- Stanton, L.M.; Price, A.J.; Manning, E.E. Hypothalamic corticotrophin releasing hormone neurons in stress-induced psychopathology: Revaluation of synaptic contributions. J Neuroendocrinol 2023, 35, e13268. [CrossRef]
- Wei, Q.; Kumar, V.; Moore, S.; Li, F.; Murphy, G.G.; Watson, S.J.; Akil, H. High emotional reactivity is associated with activation of a molecularly distinct hippocampal-amygdala circuit modulated by the glucocorticoid receptor. Neurobiol Stress 2023, 27, 100581. [CrossRef]
- Dwyer, M.K.R.; Amelinez-Robles, N.; Polsfuss, I.; Herbert, K.; Kim, C.; Varghese, N.; Parry, T.J.; Buller, B.; Verdoorn, T.A.; Billing, C.B., Jr.; et al. NTS-105 decreased cell death and preserved long-term potentiation in an in vitro model of moderate traumatic brain injury. Exp Neurol 2023, 371, 114608. [CrossRef]
- Chiba, S.; Numakawa, T.; Murata, T.; Kawaminami, M.; Himi, T. Enhanced social reward response and anxiety-like behavior with downregulation of nucleus accumbens glucocorticoid receptor in BALB/c mice. J Vet Med Sci 2023, 85, 30-39. [CrossRef]
- Numakawa, T.; Kumamaru, E.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Kunugi, H. Glucocorticoid receptor interaction with TrkB promotes BDNF-triggered PLC-gamma signaling for glutamate release via a glutamate transporter. Proc Natl Acad Sci U S A 2009, 106, 647-652. [CrossRef]
- Numakawa, T.; Odaka, H.; Adachi, N.; Chiba, S.; Ooshima, Y.; Matsuno, H.; Nakajima, S.; Yoshimura, A.; Fumimoto, K.; Hirai, Y.; et al. Basic fibroblast growth factor increased glucocorticoid receptors in cortical neurons through MAP kinase pathway. Neurochem Int 2018, 118, 217-224. [CrossRef]
- McCann, K.E.; Lustberg, D.J.; Shaughnessy, E.K.; Carstens, K.E.; Farris, S.; Alexander, G.M.; Radzicki, D.; Zhao, M.; Dudek, S.M. Novel role for mineralocorticoid receptors in control of a neuronal phenotype. Mol Psychiatry 2021, 26, 350-364. [CrossRef]
- Montgomery, K.R.; Bridi, M.S.; Folts, L.M.; Marx-Rattner, R.; Zierden, H.C.; Wulff, A.B.; Kodjo, E.A.; Thompson, S.M.; Bale, T.L. Chemogenetic activation of CRF neurons as a model of chronic stress produces sex-specific physiological and behavioral effects. Neuropsychopharmacology 2023. [CrossRef]
- You, I.J.; Bae, Y.; Beck, A.R.; Shin, S. Lateral hypothalamic proenkephalin neurons drive threat-induced overeating associated with a negative emotional state. Nat Commun 2023, 14, 6875. [CrossRef]
- Dos-Santos, R.C.; Sweeten, B.L.W.; Stelly, C.E.; Tasker, J.G. The Neuroendocrine Impact of Acute Stress on Synaptic Plasticity. Endocrinology 2023, 164. [CrossRef]
- Ciubuc-Batcu, M.T.; Stapelberg, N.J.C.; Headrick, J.P.; Renshaw, G.M.C. A mitochondrial nexus in major depressive disorder: Integration with the psycho-immune-neuroendocrine network. Biochim Biophys Acta Mol Basis Dis 2023, 1870, 166920. [CrossRef]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat Rev Neurosci 2019, 20, 34-48. [CrossRef]
- Daviu, N.; Bruchas, M.R.; Moghaddam, B.; Sandi, C.; Beyeler, A. Neurobiological links between stress and anxiety. Neurobiol Stress 2019, 11, 100191. [CrossRef]
- Gulyaeva, N.V. Glucocorticoids Orchestrate Adult Hippocampal Plasticity: Growth Points and Translational Aspects. Biochemistry (Mosc) 2023, 88, 565-589. [CrossRef]
- Barfield, E.T.; Gourley, S.L. Prefrontal cortical trkB, glucocorticoids, and their interactions in stress and developmental contexts. Neurosci Biobehav Rev 2018, 95, 535-558. [CrossRef]
- Arango-Lievano, M.; Borie, A.M.; Dromard, Y.; Murat, M.; Desarmenien, M.G.; Garabedian, M.J.; Jeanneteau, F. Persistence of learning-induced synapses depends on neurotrophic priming of glucocorticoid receptors. Proc Natl Acad Sci U S A 2019, 116, 13097-13106. [CrossRef]
- McCarty, K.J.; Pratt, S.L.; Long, N.M. Effects of Exogenous Glucocorticoid Infusion on Appetitic Center Development in Postnatal Dairy Bull Calves. Animals (Basel) 2023, 13. [CrossRef]
- Chen, Q.; Wang, F.; Zhang, Y.; Liu, Y.; An, L.; Ma, Z.; Zhang, J.; Yu, S. Neonatal DEX exposure leads to hyperanxious and depressive-like behaviors as well as a persistent reduction of BDNF expression in developmental stages. Biochem Biophys Res Commun 2020, 527, 311-316. [CrossRef]
- Rostami, S.; Haghparast, A.; Fayazmilani, R. The downstream effects of forced exercise training and voluntary physical activity in an enriched environment on hippocampal plasticity in preadolescent rats. Brain Res 2021, 1759, 147373. [CrossRef]
- Kumamaru, E.; Numakawa, T.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Niyaz, M.; Kudo, M.; Kunugi, H. Glucocorticoid prevents brain-derived neurotrophic factor-mediated maturation of synaptic function in developing hippocampal neurons through reduction in the activity of mitogen-activated protein kinase. Mol Endocrinol 2008, 22, 546-558. [CrossRef]
- Ferrero Restelli, F.; Federicci, F.; Ledda, F.; Paratcha, G. Sprouty4 at the crossroads of Trk neurotrophin receptor signaling suppression by glucocorticoids. Front Mol Neurosci 2023, 16, 1090824. [CrossRef]
- Ke, Q.; Li, R.; Cai, L.; Wu, S.D.; Li, C.M. Ro41-5253, a selective antagonist of retinoic acid receptor α, ameliorates chronic unpredictable mild stress-induced depressive-like behaviors in rats: Involvement of regulating HPA axis and improving hippocampal neuronal deficits. Brain Res Bull 2019, 146, 302-309. [CrossRef]
- Chen, X.N.; Meng, Q.Y.; Bao, A.M.; Swaab, D.F.; Wang, G.H.; Zhou, J.N. The involvement of retinoic acid receptor-alpha in corticotropin-releasing hormone gene expression and affective disorders. Biol Psychiatry 2009, 66, 832-839. [CrossRef]
- Wang, Y.; Li, O.; Li, N.; Sha, Z.; Zhao, Z.; Xu, J. Association between the BDNF Val66Met polymorphism and major depressive disorder: a systematic review and meta-analysis. Front Psychiatry 2023, 14, 1143833. [CrossRef]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257-269. [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front Cell Neurosci 2019, 13, 363. [CrossRef]
- Musazzi, L.; Tornese, P.; Sala, N.; Lee, F.S.; Popoli, M.; Ieraci, A. Acute stress induces an aberrant increase of presynaptic release of glutamate and cellular activation in the hippocampus of BDNF(Val/Met) mice. J Cell Physiol 2022, 237, 3834-3844. [CrossRef]
- Raju, S.; Notaras, M.; Grech, A.M.; Schroeder, A.; van den Buuse, M.; Hill, R.A. BDNF Val66Met genotype and adolescent glucocorticoid treatment induce sex-specific disruptions to fear extinction and amygdala GABAergic interneuron expression in mice. Horm Behav 2022, 144, 105231. [CrossRef]
- Thacker, J.S.; Mielke, J.G. The combined effects of corticosterone and brain-derived neurotrophic factor on plasticity-related receptor phosphorylation and expression at the synaptic surface in male Sprague-Dawley rats. Horm Behav 2022, 145, 105233. [CrossRef]
- Gong, Q.; Yan, X.J.; Lei, F.; Wang, M.L.; He, L.L.; Luo, Y.Y.; Gao, H.W.; Feng, Y.L.; Yang, S.L.; Li, J.; et al. Proteomic profiling of the neurons in mice with depressive-like behavior induced by corticosterone and the regulation of berberine: pivotal sites of oxidative phosphorylation. Mol Brain 2019, 12, 118. [CrossRef]
- Lu, W.H.; Chao, H.W.; Lin, P.Y.; Lin, S.H.; Liu, T.H.; Chen, H.W.; Huang, Y.S. CPEB3-dowregulated Nr3c1 mRNA translation confers resilience to developing posttraumatic stress disorder-like behavior in fear-conditioned mice. Neuropsychopharmacology 2021, 46, 1669-1679. [CrossRef]
- Wahl, P.; Mathes, S.; Köhler, K.; Achtzehn, S.; Bloch, W.; Mester, J. Acute metabolic, hormonal, and psychological responses to different endurance training protocols. Horm Metab Res 2013, 45, 827-833. [CrossRef]
- Jeanneteau, F.; Chao, M.V. Are BDNF and glucocorticoid activities calibrated? Neuroscience 2013, 239, 173-195. [CrossRef]
- Hermann, R.; Schaller, A.; Lay, D.; Bloch, W.; Albus, C.; Petrowski, K. Effect of acute psychosocial stress on the brain-derived neurotrophic factor in humans - a randomized cross within trial. Stress 2021, 24, 442-449. [CrossRef]
- de Assis, G.G.; Gasanov, E.V. BDNF and Cortisol integrative system - Plasticity vs. degeneration: Implications of the Val66Met polymorphism. Front Neuroendocrinol 2019, 55, 100784. [CrossRef]
- Numakawa, T.; Kajihara, R. Neurotrophins and Other Growth Factors in the Pathogenesis of Alzheimer's Disease. Life (Basel) 2023, 13. [CrossRef]
- Du, Y.; Wu, H.T.; Qin, X.Y.; Cao, C.; Liu, Y.; Cao, Z.Z.; Cheng, Y. Postmortem Brain, Cerebrospinal Fluid, and Blood Neurotrophic Factor Levels in Alzheimer's Disease: A Systematic Review and Meta-Analysis. J Mol Neurosci 2018, 65, 289-300. [CrossRef]
- Garzon, D.; Yu, G.; Fahnestock, M. A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer's disease parietal cortex. J Neurochem 2002, 82, 1058-1064. [CrossRef]
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C. Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer's Disease (AD): A Systematic Review and Meta-Analysis. Int J Mol Sci 2019, 20. [CrossRef]
- Angelucci, F.; Veverova, K.; Katonová, A.; Vyhnalek, M.; Hort, J. Serum PAI-1/BDNF Ratio Is Increased in Alzheimer's Disease and Correlates with Disease Severity. ACS Omega 2023, 8, 36025-36031. [CrossRef]
- Mori, Y.; Tsuji, M.; Oguchi, T.; Kasuga, K.; Kimura, A.; Futamura, A.; Sugimoto, A.; Kasai, H.; Kuroda, T.; Yano, S.; et al. Serum BDNF as a Potential Biomarker of Alzheimer's Disease: Verification Through Assessment of Serum, Cerebrospinal Fluid, and Medial Temporal Lobe Atrophy. Front Neurol 2021, 12, 653267. [CrossRef]
- Lim, Y.Y.; Laws, S.M.; Perin, S.; Pietrzak, R.H.; Fowler, C.; Masters, C.L.; Maruff, P. BDNF VAL66MET polymorphism and memory decline across the spectrum of Alzheimer's disease. Genes Brain Behav 2021, 20, e12724. [CrossRef]
- Bessi, V.; Mazzeo, S.; Bagnoli, S.; Padiglioni, S.; Carraro, M.; Piaceri, I.; Bracco, L.; Sorbi, S.; Nacmias, B. The implication of BDNF Val66Met polymorphism in progression from subjective cognitive decline to mild cognitive impairment and Alzheimer's disease: a 9-year follow-up study. Eur Arch Psychiatry Clin Neurosci 2020, 270, 471-482. [CrossRef]
- del Toro, D.; Canals, J.M.; Ginés, S.; Kojima, M.; Egea, G.; Alberch, J. Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J Neurosci 2006, 26, 12748-12757. [CrossRef]
- Brown, D.T.; Vickers, J.C.; Stuart, K.E.; Cechova, K.; Ward, D.D. The BDNF Val66Met Polymorphism Modulates Resilience of Neurological Functioning to Brain Ageing and Dementia: A Narrative Review. Brain Sci 2020, 10. [CrossRef]
- Eggert, S.; Kins, S.; Endres, K.; Brigadski, T. Brothers in arms: proBDNF/BDNF and sAPPα/Aβ-signaling and their common interplay with ADAM10, TrkB, p75NTR, sortilin, and sorLA in the progression of Alzheimer's disease. Biol Chem 2022, 403, 43-71. [CrossRef]
- Zheng, Z.; Sabirzhanov, B.; Keifer, J. Oligomeric amyloid-{beta} inhibits the proteolytic conversion of brain-derived neurotrophic factor (BDNF), AMPA receptor trafficking, and classical conditioning. J Biol Chem 2010, 285, 34708-34717. [CrossRef]
- Yan, P.; Xue, Z.; Li, D.; Ni, S.; Wang, C.; Jin, X.; Zhou, D.; Li, X.; Zhao, X.; Chen, X.; et al. Dysregulated CRTC1-BDNF signaling pathway in the hippocampus contributes to Aβ oligomer-induced long-term synaptic plasticity and memory impairment. Exp Neurol 2021, 345, 113812. [CrossRef]
- Zhang, L.; Fang, Y.; Lian, Y.; Chen, Y.; Wu, T.; Zheng, Y.; Zong, H.; Sun, L.; Zhang, R.; Wang, Z.; et al. Brain-derived neurotrophic factor ameliorates learning deficits in a rat model of Alzheimer's disease induced by aβ1-42. PLoS One 2015, 10, e0122415. [CrossRef]
- Angelucci, F.; Čechová, K.; Průša, R.; Hort, J. Amyloid beta soluble forms and plasminogen activation system in Alzheimer's disease: Consequences on extracellular maturation of brain-derived neurotrophic factor and therapeutic implications. CNS Neurosci Ther 2019, 25, 303-313. [CrossRef]
- Gerenu, G.; Martisova, E.; Ferrero, H.; Carracedo, M.; Rantamäki, T.; Ramirez, M.J.; Gil-Bea, F.J. Modulation of BDNF cleavage by plasminogen-activator inhibitor-1 contributes to Alzheimer's neuropathology and cognitive deficits. Biochim Biophys Acta Mol Basis Dis 2017, 1863, 991-1001. [CrossRef]
- Jerónimo-Santos, A.; Vaz, S.H.; Parreira, S.; Rapaz-Lérias, S.; Caetano, A.P.; Buée-Scherrer, V.; Castrén, E.; Valente, C.A.; Blum, D.; Sebastião, A.M.; et al. Dysregulation of TrkB Receptors and BDNF Function by Amyloid-β Peptide is Mediated by Calpain. Cereb Cortex 2015, 25, 3107-3121. [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 2021, 17, 157-172. [CrossRef]
- Patterson, S.L. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1β, BDNF and synaptic plasticity. Neuropharmacology 2015, 96, 11-18. [CrossRef]
- Zhang, J.C.; Yao, W.; Hashimoto, K. Brain-derived Neurotrophic Factor (BDNF)-TrkB Signaling in Inflammation-related Depression and Potential Therapeutic Targets. Curr Neuropharmacol 2016, 14, 721-731. [CrossRef]
- Song, C.; Zhang, Y.; Dong, Y. Acute and subacute IL-1β administrations differentially modulate neuroimmune and neurotrophic systems: possible implications for neuroprotection and neurodegeneration. J Neuroinflammation 2013, 10, 59. [CrossRef]
- Tong, L.; Balazs, R.; Soiampornkul, R.; Thangnipon, W.; Cotman, C.W. Interleukin-1 beta impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol Aging 2008, 29, 1380-1393. [CrossRef]
- Barbereau, C.; Yehya, A.; Silhol, M.; Cubedo, N.; Verdier, J.M.; Maurice, T.; Rossel, M. Neuroprotective brain-derived neurotrophic factor signaling in the TAU-P301L tauopathy zebrafish model. Pharmacol Res 2020, 158, 104865. [CrossRef]
- Oreshko, A.S.; Rodnyy, A.Y.; Bazovkina, D.V.; Naumenko, V.S. Effects of central administration of the human Tau protein on the Bdnf, Trkb, p75, Mapt, Bax and Bcl-2 genes expression in the mouse brain. Vavilovskii Zhurnal Genet Selektsii 2023, 27, 342-348. [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol 2019, 15, 501-518. [CrossRef]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205-221. [CrossRef]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Aβ Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J Neurosci 2015, 35, 7538-7551. [CrossRef]
- Laczó, J.; Cechova, K.; Parizkova, M.; Lerch, O.; Andel, R.; Matoska, V.; Kaplan, V.; Matuskova, V.; Nedelska, Z.; Vyhnalek, M.; et al. The Combined Effect of APOE and BDNF Val66Met Polymorphisms on Spatial Navigation in Older Adults. J Alzheimers Dis 2020, 78, 1473-1492. [CrossRef]
- Pietzuch, M.; Bindoff, A.; Jamadar, S.; Vickers, J.C. Interactive effects of the APOE and BDNF polymorphisms on functional brain connectivity: the Tasmanian Healthy Brain Project. Sci Rep 2021, 11, 14514. [CrossRef]
- Viho, E.M.G.; Buurstede, J.C.; Mahfouz, A.; Koorneef, L.L.; van Weert, L.; Houtman, R.; Hunt, H.J.; Kroon, J.; Meijer, O.C. Corticosteroid Action in the Brain: The Potential of Selective Receptor Modulation. Neuroendocrinology 2019, 109, 266-276. [CrossRef]
- Koning, A.; Buurstede, J.C.; van Weert, L.; Meijer, O.C. Glucocorticoid and Mineralocorticoid Receptors in the Brain: A Transcriptional Perspective. J Endocr Soc 2019, 3, 1917-1930. [CrossRef]
- Pedrazzoli, M.; Losurdo, M.; Paolone, G.; Medelin, M.; Jaupaj, L.; Cisterna, B.; Slanzi, A.; Malatesta, M.; Coco, S.; Buffelli, M. Glucocorticoid receptors modulate dendritic spine plasticity and microglia activity in an animal model of Alzheimer's disease. Neurobiol Dis 2019, 132, 104568. [CrossRef]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci 2011, 13, 22-37. [CrossRef]
- Dioli, C.; Papadimitriou, G.; Megalokonomou, A.; Marques, C.; Sousa, N.; Sotiropoulos, I. Chronic Stress, Depression, and Alzheimer's Disease: The Triangle of Oblivion. Adv Exp Med Biol 2023, 1423, 303-315. [CrossRef]
- Klyubin, I.; Ondrejcak, T.; Hu, N.-W.; Rowan, M.J. Glucocorticoids, synaptic plasticity and Alzheimer's disease. Current Opinion in Endocrine and Metabolic Research 2022, 25, 100365.
- Du, F.; Yu, Q.; Swerdlow, R.H.; Waites, C.L. Glucocorticoid-driven mitochondrial damage stimulates Tau pathology. Brain 2023, 146, 4378-4394. [CrossRef]
- Kulstad, J.J.; McMillan, P.J.; Leverenz, J.B.; Cook, D.G.; Green, P.S.; Peskind, E.R.; Wilkinson, C.W.; Farris, W.; Mehta, P.D.; Craft, S. Effects of chronic glucocorticoid administration on insulin-degrading enzyme and amyloid-beta peptide in the aged macaque. J Neuropathol Exp Neurol 2005, 64, 139-146. [CrossRef]
- Ding, S.; Yang, L.; Huang, L.; Kong, L.; Chen, M.; Su, Y.; Li, X.; Dong, X.; Han, Y.; Li, W.; et al. Chronic glucocorticoid exposure accelerates Aβ generation and neurotoxicity by activating calcium-mediated CN-NFAT1 signaling in hippocampal neurons in APP/PS1 mice. Food Chem Toxicol 2022, 168, 113407. [CrossRef]
- Siegel, G.; Gerber, H.; Koch, P.; Bruestle, O.; Fraering, P.C.; Rajendran, L. The Alzheimer's Disease γ-Secretase Generates Higher 42:40 Ratios for β-Amyloid Than for p3 Peptides. Cell Rep 2017, 19, 1967-1976. [CrossRef]
- Wang, Y.; Li, M.; Tang, J.; Song, M.; Xu, X.; Xiong, J.; Li, J.; Bai, Y. Glucocorticoids facilitate astrocytic amyloid-β peptide deposition by increasing the expression of APP and BACE1 and decreasing the expression of amyloid-β-degrading proteases. Endocrinology 2011, 152, 2704-2715. [CrossRef]
- Sotiropoulos, I.; Catania, C.; Riedemann, T.; Fry, J.P.; Breen, K.C.; Michaelidis, T.M.; Almeida, O.F. Glucocorticoids trigger Alzheimer disease-like pathobiochemistry in rat neuronal cells expressing human tau. J Neurochem 2008, 107, 385-397. [CrossRef]
- Sotiropoulos, I.; Catania, C.; Pinto, L.G.; Silva, R.; Pollerberg, G.E.; Takashima, A.; Sousa, N.; Almeida, O.F. Stress acts cumulatively to precipitate Alzheimer's disease-like tau pathology and cognitive deficits. J Neurosci 2011, 31, 7840-7847. [CrossRef]
- Dey, A.; Hao, S.; Wosiski-Kuhn, M.; Stranahan, A.M. Glucocorticoid-mediated activation of GSK3β promotes tau phosphorylation and impairs memory in type 2 diabetes. Neurobiol Aging 2017, 57, 75-83. [CrossRef]
- Yi, J.H.; Brown, C.; Whitehead, G.; Piers, T.; Lee, Y.S.; Perez, C.M.; Regan, P.; Whitcomb, D.J.; Cho, K. Glucocorticoids activate a synapse weakening pathway culminating in tau phosphorylation in the hippocampus. Pharmacol Res 2017, 121, 42-51. [CrossRef]
- Milligan Armstrong, A.; Porter, T.; Quek, H.; White, A.; Haynes, J.; Jackaman, C.; Villemagne, V.; Munyard, K.; Laws, S.M.; Verdile, G.; et al. Chronic stress and Alzheimer's disease: the interplay between the hypothalamic-pituitary-adrenal axis, genetics and microglia. Biol Rev Camb Philos Soc 2021, 96, 2209-2228. [CrossRef]
- Merighi, S.; Nigro, M.; Travagli, A.; Gessi, S. Microglia and Alzheimer's Disease. Int J Mol Sci 2022, 23. [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front Aging Neurosci 2022, 14, 815347. [CrossRef]
- Harris-White, M.E.; Chu, T.; Miller, S.A.; Simmons, M.; Teter, B.; Nash, D.; Cole, G.M.; Frautschy, S.A. Estrogen (E2) and glucocorticoid (Gc) effects on microglia and A beta clearance in vitro and in vivo. Neurochem Int 2001, 39, 435-448. [CrossRef]
- Sharma, V.K.; Singh, T.G. Navigating Alzheimer's Disease via Chronic Stress: The Role of Glucocorticoids. Curr Drug Targets 2020, 21, 433-444. [CrossRef]
- Sato, H.; Takahashi, T.; Sumitani, K.; Takatsu, H.; Urano, S. Glucocorticoid Generates ROS to Induce Oxidative Injury in the Hippocampus, Leading to Impairment of Cognitive Function of Rats. J Clin Biochem Nutr 2010, 47, 224-232. [CrossRef]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life (Basel) 2020, 10. [CrossRef]
- Wei, P.; Yang, F.; Zheng, Q.; Tang, W.; Li, J. The Potential Role of the NLRP3 Inflammasome Activation as a Link Between Mitochondria ROS Generation and Neuroinflammation in Postoperative Cognitive Dysfunction. Front Cell Neurosci 2019, 13, 73. [CrossRef]
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative Stress Occurs Prior to Amyloid Aβ Plaque Formation and Tau Phosphorylation in Alzheimer's Disease: Role of Glutathione and Metal Ions. ACS Chem Neurosci 2023, 14, 2944-2954. [CrossRef]
- Jeanneteau, F.; Borie, A.; Chao, M.V.; Garabedian, M.J. Bridging the Gap between Brain-Derived Neurotrophic Factor and Glucocorticoid Effects on Brain Networks. Neuroendocrinology 2019, 109, 277-284. [CrossRef]
- Chen, H.; Lombès, M.; Le Menuet, D. Glucocorticoid receptor represses brain-derived neurotrophic factor expression in neuron-like cells. Mol Brain 2017, 10, 12. [CrossRef]
- Suri, D.; Vaidya, V.A. Glucocorticoid regulation of brain-derived neurotrophic factor: relevance to hippocampal structural and functional plasticity. Neuroscience 2013, 239, 196-213. [CrossRef]
- Lambert, W.M.; Xu, C.F.; Neubert, T.A.; Chao, M.V.; Garabedian, M.J.; Jeanneteau, F.D. Brain-derived neurotrophic factor signaling rewrites the glucocorticoid transcriptome via glucocorticoid receptor phosphorylation. Mol Cell Biol 2013, 33, 3700-3714. [CrossRef]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophic Factor and Glucocorticoid Stress in Neurogenesis. Int J Mol Sci 2017, 18. [CrossRef]
- Numakawa, T.; Adachi, N.; Richards, M.; Chiba, S.; Kunugi, H. Brain-derived neurotrophic factor and glucocorticoids: reciprocal influence on the central nervous system. Neuroscience 2013, 239, 157-172. [CrossRef]
- Radecki, D.T.; Brown, L.M.; Martinez, J.; Teyler, T.J. BDNF protects against stress-induced impairments in spatial learning and memory and LTP. Hippocampus 2005, 15, 246-253. [CrossRef]
- Ju, L.; Yang, J.; Zhu, T.; Liu, P.; Yang, J. BDNF-TrkB signaling-mediated upregulation of Narp is involved in the antidepressant-like effects of (2R,6R)-hydroxynorketamine in a chronic restraint stress mouse model. BMC Psychiatry 2022, 22, 182. [CrossRef]
- Wurzelmann, M.; Romeika, J.; Sun, D. Therapeutic potential of brain-derived neurotrophic factor (BDNF) and a small molecular mimics of BDNF for traumatic brain injury. Neural Regen Res 2017, 12, 7-12. [CrossRef]
- Chen, C.; Wang, Z.; Zhang, Z.; Liu, X.; Kang, S.S.; Zhang, Y.; Ye, K. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer's disease. Proc Natl Acad Sci U S A 2018, 115, 578-583. [CrossRef]
- Liao, J.; Chen, C.; Ahn, E.H.; Liu, X.; Li, H.; Edgington-Mitchell, L.E.; Lu, Z.; Ming, S.; Ye, K. Targeting both BDNF/TrkB pathway and delta-secretase for treating Alzheimer's disease. Neuropharmacology 2021, 197, 108737. [CrossRef]
- Pak, M.E.; Yang, H.J.; Li, W.; Kim, J.K.; Go, Y. Yuk-Gunja-Tang attenuates neuronal death and memory impairment via ERK/CREB/BDNF signaling in the hippocampi of experimental Alzheimer's disease model. Front Pharmacol 2022, 13, 1014840. [CrossRef]
- Gupta, V.; Chitranshi, N.; Gupta, V.; You, Y.; Rajput, R.; Paulo, J.A.; Mirzaei, M.; van den Buuse, M.; Graham, S.L. TrkB Receptor Agonist 7,8 Dihydroxyflavone is Protective Against the Inner Retinal Deficits Induced by Experimental Glaucoma. Neuroscience 2022, 490, 36-48. [CrossRef]
- Lee, Y.J.; Jeong, Y.J.; Kang, E.J.; Kang, B.S.; Lee, S.H.; Kim, Y.J.; Kang, S.S.; Suh, S.W.; Ahn, E.H. GAP-43 closely interacts with BDNF in hippocampal neurons and is associated with Alzheimer's disease progression. Front Mol Neurosci 2023, 16, 1150399. [CrossRef]
- Nagahara, A.H.; Mateling, M.; Kovacs, I.; Wang, L.; Eggert, S.; Rockenstein, E.; Koo, E.H.; Masliah, E.; Tuszynski, M.H. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J Neurosci 2013, 33, 15596-15602. [CrossRef]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer's disease. Transl Psychiatry 2016, 6, e907. [CrossRef]
- Nigam, S.M.; Xu, S.; Kritikou, J.S.; Marosi, K.; Brodin, L.; Mattson, M.P. Exercise and BDNF reduce Aβ production by enhancing α-secretase processing of APP. J Neurochem 2017, 142, 286-296. [CrossRef]
- Kim, D.; Cho, J.; Kang, H. Protective effect of exercise training against the progression of Alzheimer's disease in 3xTg-AD mice. Behav Brain Res 2019, 374, 112105. [CrossRef]
- Xu, L.; Zhu, L.; Zhu, L.; Chen, D.; Cai, K.; Liu, Z.; Chen, A. Moderate Exercise Combined with Enriched Environment Enhances Learning and Memory through BDNF/TrkB Signaling Pathway in Rats. Int J Environ Res Public Health 2021, 18. [CrossRef]
- Jia, R.X.; Liang, J.H.; Xu, Y.; Wang, Y.Q. Effects of physical activity and exercise on the cognitive function of patients with Alzheimer disease: a meta-analysis. BMC Geriatr 2019, 19, 181. [CrossRef]
- Canet, G.; Hernandez, C.; Zussy, C.; Chevallier, N.; Desrumaux, C.; Givalois, L. Is AD a Stress-Related Disorder? Focus on the HPA Axis and Its Promising Therapeutic Targets. Front Aging Neurosci 2019, 11, 269. [CrossRef]
- Watermeyer, T.; Robb, C.; Gregory, S.; Udeh-Momoh, C. Therapeutic implications of hypothalamic-pituitaryadrenal-axis modulation in Alzheimer's disease: A narrative review of pharmacological and lifestyle interventions. Front Neuroendocrinol 2021, 60, 100877. [CrossRef]
- Sooy, K.; Webster, S.P.; Noble, J.; Binnie, M.; Walker, B.R.; Seckl, J.R.; Yau, J.L. Partial deficiency or short-term inhibition of 11beta-hydroxysteroid dehydrogenase type 1 improves cognitive function in aging mice. J Neurosci 2010, 30, 13867-13872. [CrossRef]
- Sooy, K.; Noble, J.; McBride, A.; Binnie, M.; Yau, J.L.; Seckl, J.R.; Walker, B.R.; Webster, S.P. Cognitive and Disease-Modifying Effects of 11β-Hydroxysteroid Dehydrogenase Type 1 Inhibition in Male Tg2576 Mice, a Model of Alzheimer's Disease. Endocrinology 2015, 156, 4592-4603. [CrossRef]
- Baglietto-Vargas, D.; Medeiros, R.; Martinez-Coria, H.; LaFerla, F.M.; Green, K.N. Mifepristone alters amyloid precursor protein processing to preclude amyloid beta and also reduces tau pathology. Biol Psychiatry 2013, 74, 357-366. [CrossRef]
- Pineau, F.; Canet, G.; Desrumaux, C.; Hunt, H.; Chevallier, N.; Ollivier, M.; Belanoff, J.K.; Givalois, L. New selective glucocorticoid receptor modulators reverse amyloid-β peptide-induced hippocampus toxicity. Neurobiol Aging 2016, 45, 109-122. [CrossRef]
- da Costa Daniele, T.M.; de Bruin, P.F.C.; de Matos, R.S.; de Bruin, G.S.; Maia Chaves, C.J.; de Bruin, V.M.S. Exercise effects on brain and behavior in healthy mice, Alzheimer's disease and Parkinson's disease model-A systematic review and meta-analysis. Behav Brain Res 2020, 383, 112488. [CrossRef]
- Bashiri, H.; Enayati, M.; Bashiri, A.; Salari, A.A. Swimming exercise improves cognitive and behavioral disorders in male NMRI mice with sporadic Alzheimer-like disease. Physiol Behav 2020, 223, 113003. [CrossRef]
- Campos, H.C.; Ribeiro, D.E.; Hashiguchi, D.; Glaser, T.; Milanis, M.D.S.; Gimenes, C.; Suchecki, D.; Arida, R.M.; Ulrich, H.; Monteiro Longo, B. Neuroprotective effects of resistance physical exercise on the APP/PS1 mouse model of Alzheimer's disease. Front Neurosci 2023, 17, 1132825. [CrossRef]
- Irazoki, E.; Contreras-Somoza, L.M.; Toribio-Guzmán, J.M.; Jenaro-Río, C.; van der Roest, H.; Franco-Martín, M.A. Technologies for Cognitive Training and Cognitive Rehabilitation for People With Mild Cognitive Impairment and Dementia. A Systematic Review. Front Psychol 2020, 11, 648. [CrossRef]
- Cutuli, D.; Decandia, D.; Giacovazzo, G.; Coccurello, R. Physical Exercise as Disease-Modifying Alternative against Alzheimer's Disease: A Gut-Muscle-Brain Partnership. Int J Mol Sci 2023, 24. [CrossRef]
Figure 1.
Relationship between BDNF/TrkB system, glucocorticoids, and HPA-axis. Neurotrophins including nerve growth factor (NGF), BDNF, neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4) are generated as mature forms from the precursor proneurotrophins after receiving proteolytic cleavage. Precursors bind to p75NTR with a high affinity while mature forms bind to Trks preferentially, and contribute to cell survival, regulating synaptic plasticity, and neuroprotection via activating intracellular signaling pathways (MAPK/ERK-, phospholipase Cγ (PLCγ)-, PI3K/Akt-pathways). Especially, BDNF/TrkB system is involved in a variety of neuronal events in the CNS. On the other hand, released glucocorticoids (GCs) through activating of the Hypothamic-Pituitry-Adrenal-axis (HPA-axis) also has a role in the CNS function, and its levels are regulated via a negative-feedback action in the HPA-axis. GCs is an important molecule to cope with stressful conditions, however, abnormally increased levels of GCs under the chronic stress exerts negative impact on neurons, including blockade of the BDNF/TrkB system and/or directly suppression of neuronal functions. CRH, corticotropin-releasing hormone; ACTH, adrenocorticotropic hormone.
Figure 1.
Relationship between BDNF/TrkB system, glucocorticoids, and HPA-axis. Neurotrophins including nerve growth factor (NGF), BDNF, neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4) are generated as mature forms from the precursor proneurotrophins after receiving proteolytic cleavage. Precursors bind to p75NTR with a high affinity while mature forms bind to Trks preferentially, and contribute to cell survival, regulating synaptic plasticity, and neuroprotection via activating intracellular signaling pathways (MAPK/ERK-, phospholipase Cγ (PLCγ)-, PI3K/Akt-pathways). Especially, BDNF/TrkB system is involved in a variety of neuronal events in the CNS. On the other hand, released glucocorticoids (GCs) through activating of the Hypothamic-Pituitry-Adrenal-axis (HPA-axis) also has a role in the CNS function, and its levels are regulated via a negative-feedback action in the HPA-axis. GCs is an important molecule to cope with stressful conditions, however, abnormally increased levels of GCs under the chronic stress exerts negative impact on neurons, including blockade of the BDNF/TrkB system and/or directly suppression of neuronal functions. CRH, corticotropin-releasing hormone; ACTH, adrenocorticotropic hormone.
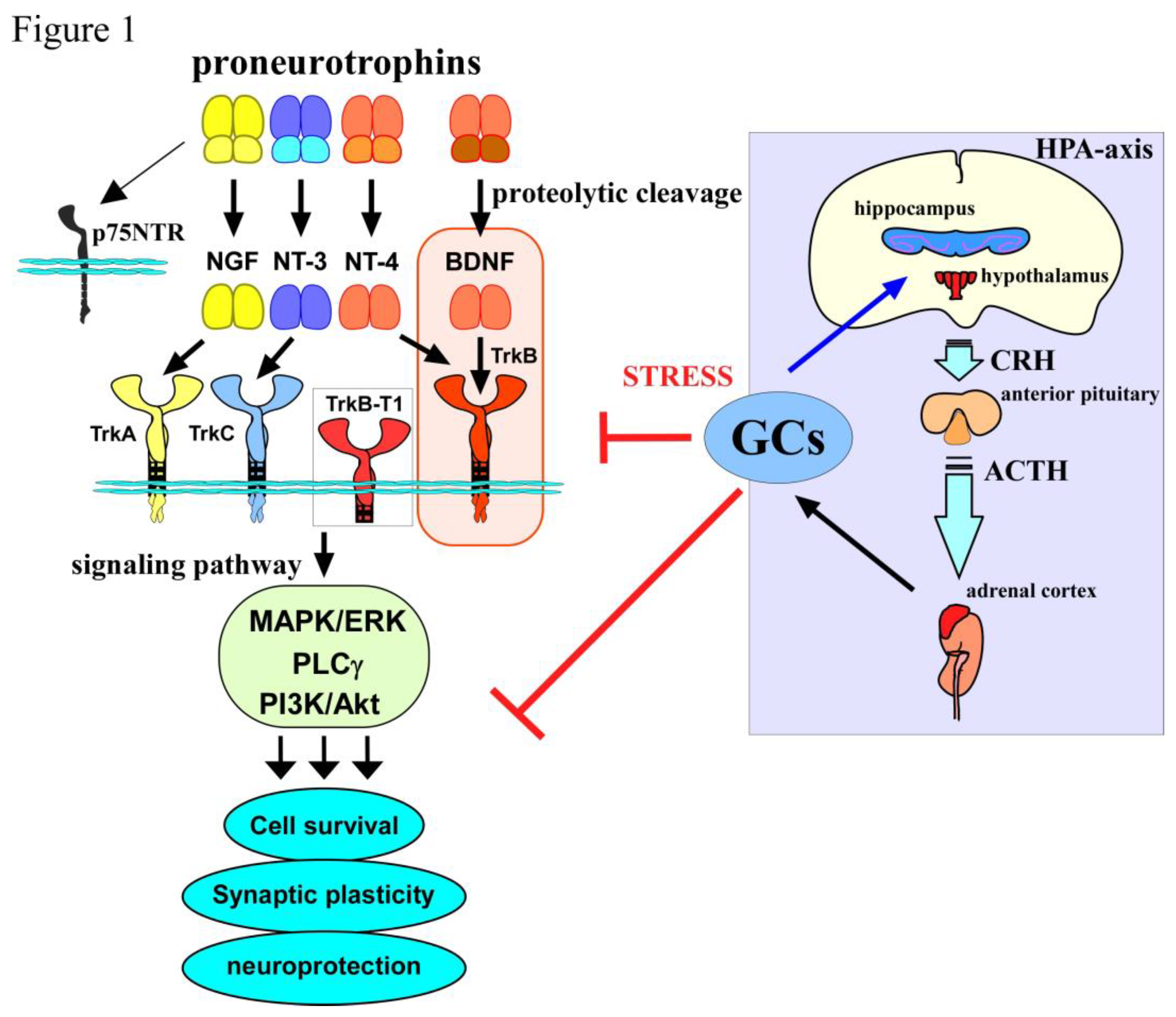
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



