Submitted:
11 December 2023
Posted:
13 December 2023
You are already at the latest version
Abstract
The rate constants of elementary processes in the atom-diatom system O(3P)+H2(v), including the processes of vibrational relaxation and dissociation, were studied using the quasi-classical trajectory method. All calculations were carried out along the ground potential energy surface (PES) 3A′′ that was approximated by a neural network. Approximation data were obtained using ab initio quantum chemistry methods at the extended multi-configuration quasi-degenerate second-order perturbation theory XMCQDPT2 in a basis set limit. The calculated cross sections of the reaction channels are in good agreement with the literature data. A complete set of state-to-state rate constants was obtained for the metathesis reaction, the dissociation and VT relaxation of the H2 molecule upon collision with an O atom. According to this data Arrhenius approximations over a wide temperature range were obtained for the thermal rate constants of considered processes. Data obtained on the dissociation constants and VT relaxation of vibrationally excited H2 molecules can be used in constructing kinetic models describing the oxidation of hydrogen at high temperatures or highly nonequilibrium conditions.
Keywords:
Quasiclassical trajectory (QCT) method
; state-resolved rate constants
; hydrogen
; atom-molecule collisions
; dissociation
; vibrational energy relaxation
1. Introduction
Modeling combustion processes in the hydrogen-oxygen system has great practical innovation, since hydrogen is a promising fuel that can improve both the efficiency and environmental friendliness of gas turbine engines and installations. The problem of quantitative description of kinetics in the hydrogen-oxygen system remains unsolved, both with points of agreement between the results of various experiments, and with points of agreement between theoretical models and experiment [1]. The method in presented work to estimate the state-to-state reaction rate constants makes it possible to increase the efficiency of studying hydrogen combustion in advanced gas turbine engines. This approach based on molecular dynamics methods.
Numerical modeling of molecular dynamics is an actively developing field of science, which has become the de facto main method to obtain new information about the rate constants of physicochemical processes. The progress of molecular dynamics methods is due to the availability of powerful computers, the development of computational algorithms (in particular, the increasingly widespread use of neural networks), the ability to perform numerical simulation from the first principles and the absence in a numerical experiment of some natural limitations inherent in laboratory diagnostic methods. In particular, the methods of molecular dynamics allows to obtain the most complete description of the dependence of the probabilities of physicochemical processes on the quantum states (rotational, vibrational, electronic) of reacting particles in a wide temperature range. The complete sets of state-to-state rate constants acquired in this way can be used both to obtain averaged characteristics of the reacting system (for example, thermal rate constants), or used directly in kinetic models for thermally nonequilibrium gas observed, for example, in flows with shock waves [2,3,4,5,6,7,8,9] and detonation waves [10,11,12,13,14], flows in a supersonic nozzle [15,16,17], in gas discharges [18,19,20], during emission/absorption of intense radiation [21,22], in the upper layers of the Earth’s atmosphere [23]. Thus, the development of the methods for state-specific rate constants calculations and the generation of new data on rate constants is an important urgent task, to which this study is devoted.
The main difficulty in estimating the state-to-state rate constants of the reactions and the energy exchange processes is that it is impossible to apply simple estimation approaches (the model of solid spheres, the capture model, the theory of the transition state, etc.) without additional assumptions and empirical corrections. To obtain state-to-state rate constants, it is necessary to use dynamic calculation methods based either on solving the equations of classical mechanics (for example, the method of quasi-classical trajectories (QCT) [24]), or on solving the equations of quantum dynamics of the system under study (for example, the real wave packets method [25]). The latter group of methods is quite difficult to implement even for simple atom-diatom systems, therefore, the main method for estimating the state-to-state rate constants used in most studies is the QCT method [26,27,28,29,30,31,32]. Therefore, in this paper the implementation and application of the QCT method will be consider.
The implementation of the QCT method requires either a preliminary assignment of the entire potential energy surface (PES) of a system of colliding particles in some analytical form (PES can be obtained from ab initio calculations or based on the modelling multidimensional potentials, for example, LEPS potential [33]), or calculating the PES along each calculated trajectory "on the fly" by calculating the energy gradient at the trajectory point (usually by ab initio methods) [34]. In this paper, the first approach was used – the construction of an approximation of the PES based on data from ab initio calculations and subsequent calculations of the trajectory ensembles of interest, since the number of trajectories per ensemble should be large enough so that the relative statistical error in calculating the rate constants is less than 10%.
A separate task is to approximate the PES as a multidimensional function depending on the coordinates of the atomic nuclei of the reacting system, for which approximations based on the least squares method are often used, representing the PES function as a series of degrees of atomic coordinates [35]. An alternative approach is to use a neural network as a universal approximator. One of such methods is approximation by a neural network that accepts symmetrized coordinates as an input (PIP-NN — permutation invariant polynomials-neural network method) [36]. The use of such symmetrized coordinates makes it possible, with the existing indistinguishability in permutations of identical atoms in the reacting system, to reduce the number of PES points necessary for constructing an approximation.
The purpose of the present work is, first, to implement the QCT method for calculating the state-to-state rate constants in the atom-diatom A+BC test system using the PIP-NN method to approximate the PES of such a system, and, second, to obtain a complete set of state-resolved rate constants for the exchange reaction A+BC=AB+C, dissociation and VT (vibrational-translational) relaxation of BC molecules on A atoms. The system was chosen as such a test system, since it was previously studied in detail by molecular dynamics methods in the works of other authors [37,38,39,40,41], which will allow to validate our implementation of the QCT method. Nevertheless, in previous studies [37,38,39,40,41], the probabilities of dissociation and VT relaxation of H2 molecules on the O atom were not considered. So, the QCT method for these processes, as far as the authors know, is used for the first time. The obtained full set of state-to-state rate constants for system (including dissociation ant VT relaxation) can be important, for example, for describing the nonequilibrium processes in the Earth’s ozone layer, and approximations for thermal rate constants obtained on its basis can be used in kinetic models of combustion of hydrogen-containing fuels.
2. Methodology
2.1. PES approximation
All calculations of the PES of the system along the lower main surface were carried out at the level of the extended multi-configuration quasi-degenerate second-order perturbation theory XMCQDPT2 [42]. As an initial approximation of the wave function, the results of the calculation by the multi-configuration self-consistent field method with a dynamic choice of weights DW-CASSCF [43] was used. Such a choice of a multi-configuration calculation method is associated with the need for a smooth construction of the PES, which is most likely provided by the DW-CASSCF method (see Ref. [44] for details). The size of the active space specified in the DW-CASSCF method is 8 electrons distributed in 6 orbitals, i.e. the full valence space for the considered system. The main set of basis functions in all calculations is the family of Dunning basis sets with diffuse functions aug-cc-pVXZ (X=D, T, Q) [45]. Based on calculations in the basis sets aug-cc-pVXZ (X=D, T, Q), the energies were calculated at each point of the PES in the limit of the complete basic set aug-cc-pV∞Z using the Riemann zeta function [46]. All ab initio calculations were carried out in the Firefly QC v.8.2.0 software package [47], partially based on the GAMESS (US) [48] source code.
The obtained set of PES data was approximated using the PIP-NN method [36]. This approach to fit PES has important advantages: the universality of the method, the availability of neural network training tools (for example, TensorFlow [49], PyTorch [50], Keras [51] software packages, etc.), the availability to implement the analytical nuclear gradient at any point of the PES. At the same time, the main drawback of this approach is related to the peculiarity of the neural network device — for a neural network with a sigmoid activation function or a hyperbolic tangent, the main calculation time of the output neuron is spent on multiple calculations of this function, as well as matrix-vector multiplication operations.
In the framework of the PIP-NN method, before constructing and training a neural network that plays the role of a universal approximator, it is necessary to transform the Cartesian coordinates of atomic nuclei into symmetrized coordinates. Since the system corresponds to the AB2 type, the general form of symmetrized coordinates is already known via Morse coordinates [52]:
Here is a dimensional coefficient equal to 1-2 Å−1. In general, it is necessary to solve a combinatorial problem with respect to permutations of atoms and the corresponding permutations of interatomic distances. Software packages such as MAGMA [53] and SINGULAR [54], which are able to determine permutation groups based on a given permutation matrix, help in solving these problems. The coordinates obtained in this way within the PIP-NN approach are fed to the input of the neural network, and at the output the neural network gives the potential energy value.
In this paper, to approximate the PES of the system, a neural network of the multilayer perceptron type with two hidden layers was trained, 45 neurons being on the first hidden layer, 55 on the second. The RMS error after training was about 10 meV.
2.2. Rate constants estimation
The direct calculation of the rate constants by the QCT method is based on a statistical analysis of the outcomes of trajectory ensembles with specified conditions. We will briefly describe the stages of all calculations that eventually lead to estimates of the rate constants of interest, according to the methodology from [24,55,56].
First of all, it is necessary to convert the Cartesian coordinates of the atomic nuclei into Jacobi coordinates for the atom–diatomic molecule system. In the case of the system, these are the three coordinates of the H1 atom relative to the H2 atom, the three coordinates of the atom relative to the center of mass of the H1 H2 molecule, and the three coordinates of the center of mass of the entire H1H2 system. After the corresponding linear transformation, the Hamiltonian of the system will take the form:
Here are appropriate reduced masses, is the total mass of the entire system, or are the Jacobi coordinates, and or are the generalized momentum corresponding to , is the potential energy of the system at the point with the coordinates . The resulting Hamiltonian (2) allows to compose the equations of motion of Hamiltonian mechanics, which were then solved numerically using an explicit one-step Runge-Kutta-Merson method of the 4th order of accuracy with a variable integration step.
Each ensemble of trajectories was set by initial conditions, expressions for which are described in detail in [24,55,56]. Here we only note that the calculation of the reaction cross section was carried out without scanning over the impact parameter, but each ensemble of trajectories was set only by the vibrational and rotational numbers of the H2 molecule and the kinetic energy of the incoming atom.
Also, to construct the initial conditions and for subsequent analysis of each trajectory, it is necessary to know the vibrational-rotational levels of H2 and OH molecules, but the approximation of the anharmonic Morse potential may be inaccurate when describing upper vibrational-rotational levels. Therefore, the energy levels of diatomic molecules were determined by numerically solving the radial component of the stationary Schrödinger equation for a diatomic molecule with the LEVEL16 program [57]. However, it is worth noting that this program, alongside with bound states, also calculates the energy levels of quasi-bound states. Despite the fact that some of the quasi-bound states may be metastable and have a lifetime of more than a day, such states will not be considered within the framework of the QCT method, and the trajectories that led to such an outcome will be attributed to the process of dissociation.
Thus, it becomes possible to calculate the cross sections of the following reactions in this system:
The reaction cross section was calculated using the ratio of the number of trajectories that led to the required outcome to the total number of trajectories of the ensemble N according to the formula from [24,55,56]:
with the statistical error
Here, is the maximum value of the impact parameter at which the probability of a particular process (reaction, dissociation or VT relaxation process) is above zero, E is the kinetic energy of the incoming atom. In all calculations, the total number of trajectories that compose one ensemble was equal to 20000, and the highest kinetic energy of the atom was equal to 9 eV.
On the basis of the obtained cross sections the rate constants and their error were calculated by the formulas:
Here and are the electronic statistical weights of the studied PES and reagents, respectively, is the Boltzmann constant. The integrals of formulas (6) were calculated numerically with piecewise linear interpolation of the reaction cross section :
where and . Such an approximation of the reaction cross section makes it possible to calculate the rate constant by simple summation according to the following formula (the error of such a numerical approach is many times less than the statistical error of trajectory calculations, usually amounting to 5-15% [55]):
From the obtained vibrational-rotational state-to-state rate constants (6) it is possible to determine the vibrational state-resolved rate constants (assuming that the translational and rotational degrees of freedom of the reagents and the products are in equilibrium) and the thermally equilibrium rate constant as follows:
where is the normalized population of the H2 energy level with the vibrational and rotational numbers v and J, N is the number of vibrational levels of the molecule H2, is number of rotational levels in the v-th vibrational level.
3. Results and discussion
The methodology described above for obtaining state-to-state rate constants and its specific implementation naturally needs validation. As validation data, the results of QCT calculations of rate constants or calculations using direct modeling of quantum dynamics performed by other authors, as well as data on rate constants borrowed from experimental works can be used. For the considered system, the most widely presented in the literature are the state-to-state rate constants for the process. So, in Figure 1 the cross sections for the reaction from our QCT estimates are presented in comparison with the similar QCT results from [38]. One can notice a slight overestimation of cross sections in our modelling compared to [38]. This may be due to the fact that the energy barrier of the reaction H+OH within the framework of the transition state theory for the PES obtained in this work turned out to be slightly lower (=11.5 kcal/mole) than the corresponding value from [38] (=13 kcal/mole).
Figure 2 shows a comparison of the state-to-state rate constants found by the QCT method in this work and obtained on the basis of quantum dynamics calculations in [41]. The above comparison shows a good qualitative agreement of the results of the two computational studies. Nevertheless, for reaction channels with a quantum number , the difference in the values of the rate constants determined by different methods can be significant, and in the temperature range K can reach an order of magnitude. This discrepancy may be due to the fact that the QCT method cannot take into account quantum effects, such as the tunnel effect and the over-barrier reflection.
A comparison of the thermally equilibrium reaction rate constant, calculated in this work via the QCT method and obtained in [41] on the basis of quantum dynamics calculations, is shown in Figure 3. In addition, Figure 3 shows an estimate of this rate constant obtained using the non-variational transition state theory adjusted for the tunnel effect and various Arrhenius approximations of the rate constant used in the kinetic models [58,59,60]. One can notice an expected overestimation of the rate constant by the transition state theory, while the QCT method gives a closer values, although somewhat higher at K, relative to the known data. Such an overestimation has the same cause as in the case of the reaction cross section — lowering the energy barrier at the XMCQDPT2/aug-cc-pV∞Z level of theory. The discrepancy in the values of the rate constant at K is explained by the different temperature ranges for which the Arrhenius approximation was obtained. In the temperature range K, the thermally equilibrium rate constant of the exchange reaction obtained in this work can be approximated with good accuracy by the Arrhenius dependence (cm3/(mole s)):
For the dissociation reaction and the VT relaxation process of H2 molecules in collision with O atoms, there is no data on rate constants in the literature (as far as the authors know). Nevertheless, in order to validate the calculation method, it is interesting to compare the estimates of the rate constants of these processes obtained by the QCT method with the available information on the rates of dissociation and VT relaxation of hydrogen molecules in collisions with other partners. For the dissociation process, such a comparison is presented in Figure 4. It turns out that the equilibrium rate constant of the dissociation of H2 obtained in this work significantly exceeds those with the different partners available in the literature. However, at temperatures K, the dissociation rate constant of hydrogen upon collision with O atoms coincides by the order of magnitude with the dissociation rate constants in collision with H and H2O. The thermally equilibrium rate constant of H2 dissociation obtained in this work in the temperature range K can be described with good accuracy using the double Arrhenius dependence (cm3/(mole s)):
Figure 5 shows a comparison of the VT relaxation time VT of H2 molecules in collisions with various partners. Note that for the processes of hydrogen relaxation on H, H2 and O, the relaxation time was determined using the VT rate constant of the transition from the first excited vibrational level to the ground state:
Here is the VT relaxation rate constant, is the characteristic vibrational temperature. It can be seen from Figure 5 that according to the results of calculations performed in this work, the vibrational relaxation of H2 proceeds faster on the O atom than on the Ar atom, but slower than on the H atoms, what is consistent with the qualitative expectations of the dependence of the relaxation time of the molecule on the mass of the collisional partner, expressed, for example, in the well-known Millikan–White formula [65]. At the same time, the results of the calculation using the QCT method predict a more effective VT relaxation of H2 in collisions with O atoms than in collisions with molecular hydrogen. It can be assumed that this effect is due to the fact that a molecule as a collisional partner, in addition to translational degree of freedom, has rotational and vibrational ones, unlike a single atom.
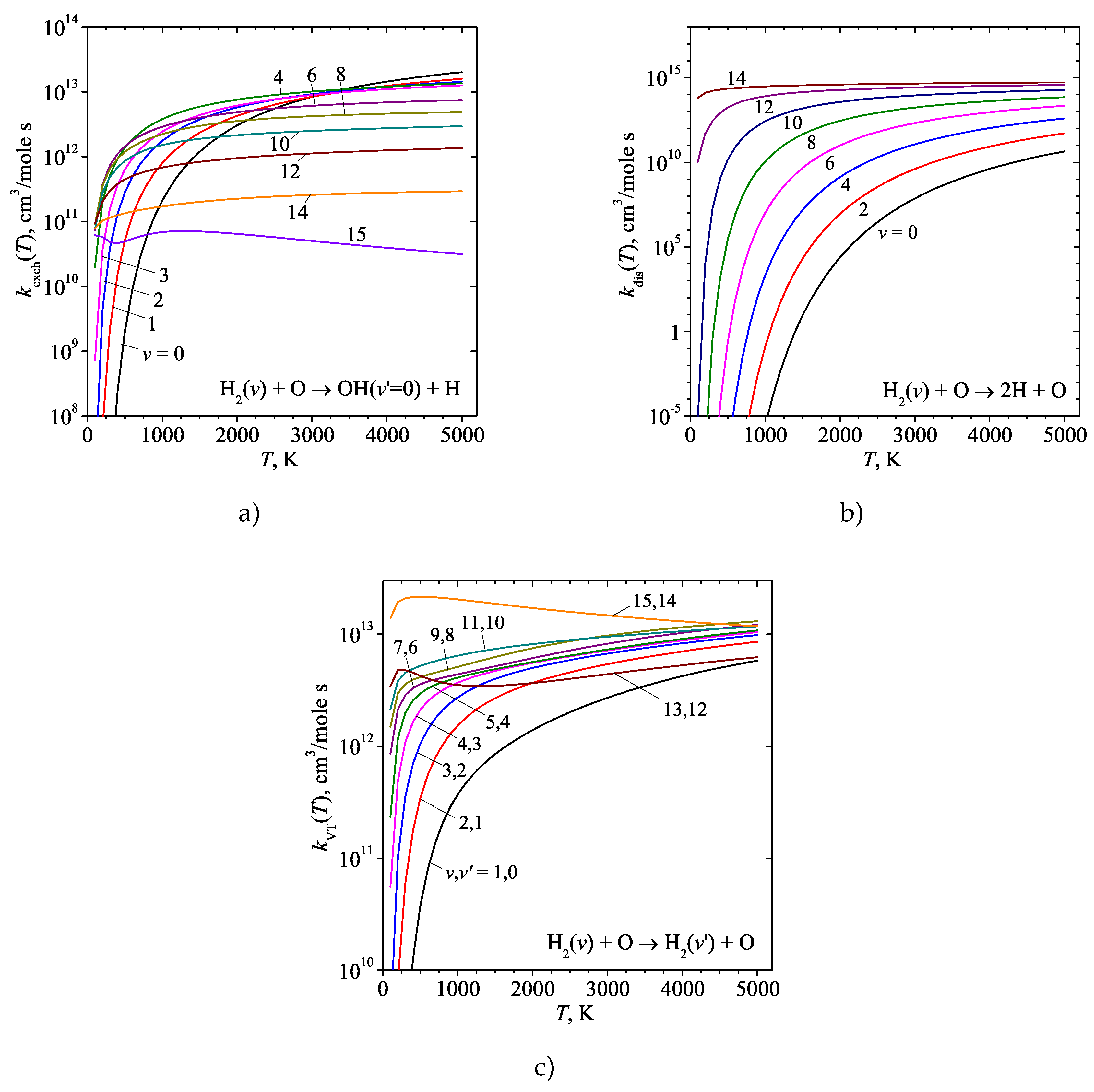
The lack of data in the literature on the probabilities of dissociation and VT relaxation of H2 molecules in collision with O atoms is due to the fact that the exchange reaction O+H2 H+OH is considered as a more probable outcome of the collision of H2 and O and hence is more important for kinetic models. The rate constants calculated in this work for these processes are shown in Figure 6. The comparison shows that for H2 molecules in the ground vibrational state, the exchange reaction is indeed a much more probable process than dissociation. However, for vibrationally excited H2 molecules, the rate constants of dissociation and VT relaxation upon collision with O can be comparable or even many times higher than the corresponding exchange reaction constants. Thus, the data obtained in this paper on the state-to-state rate constants for dissociation and VT relaxation of molecular hydrogen can be an important addition for the kinetic models describing hydrogen oxidation at high translational temperatures or under essentially nonequilibrium conditions.
4. Conclusion
In this study, an implementation of the QCT method was created for atom–diatom systems, the feature of which is the approximation of the PES of the system by a neural network. Using this implementation, cross sections and state-to-state rate constants for exchange reactions, dissociation and VT relaxation processes were calculated for the system. Based on the data on the state-to-state rate constants for exchange and dissociation reactions, Arrhenius approximations of thermally equilibrium rate constants over a wide temperature range were obtained.
It was shown that the cross sections and rate constants obtained in this work for the exchange reactions are in a good qualitative agreement with the results of the QCT and the quantum dynamics method calculations presented in the works of other authors [37,38,41]. In addition, the thermally equilibrium rate constant obtained for the exchange reaction is in good agreement with the corresponding rate constants used in various kinetic models [58,59,60].
As far as the authors are aware, there are no reliable data on the rate constants of dissociation and VT relaxation of H2 molecules in collision with O atoms in the literature, and the study of these processes by the QCT method was performed in this work for the first time. A comparative analysis of the obtained state-to-state rate constants showed that when H2 on the lower vibrational levels () collides with an O atom, the probability of its dissociation is negligible compared to the probability of an exchange reaction (however, this difference decreases with increasing temperature). But for the upper vibrational levels of hydrogen molecule, the rate constants of dissociation and VT relaxation of H2 on O are comparable or may even exceed the corresponding state-resolved rate constants of the exchange reaction. Thus, the rate constants of dissociation and VT relaxation of H2 on O obtained in this work can be useful in constructing kinetic models describing hydrogen oxidation under thermally nonequilibrium conditions or at high temperatures.
The results of this work make it possible to increase the efficiency of modeling processes in gas-turbine power plant using promising types of fuel, such as methane-hydrogen mixtures or pure hydrogen, including at the high combustion temperature inherent in hydrogen fuels.
In the future, it is planned to refine the calculation of the rate constants of energy exchange processes in this system, since in this work the interaction of the atom and the H2 molecule along the other two terms of this system was not taken into account. In addition, due to the versatility of our PES approximation method, the created computational complex can be easily adapted for other triatomic systems and other configurations of collisions, for example, for a recombination reaction A+B+C.
Author Contributions
Alexey V. Pelevkin: conceptualization, methodology, software, writing—review and editing. Ilya V. Arsentiev: data curation, writing—review and editing, visualization. Ilya N. Kadochnikov: conceptualization, methodology, validation, visualization, original draft preparation. Ivan A. Zubrilin: funding acquisition, project administration, conceptualization, supervision. Evgeny P. Filinov: formal analysis, validation. Denis V. Yakushkin: writing—review and editing, visualization.
Funding
The work was supported by project FSSS-2022-0019, implemented within the framework of the federal project “Development of human capital in the interests of regions, industries and the research and development sector”, and consequently “New laboratories were created, including those under the guidance of young promising researchers”.
Institutional Review Board Statement
In this section, you should add the Institutional Review Board Statement and approval number, if relevant to your study. You might choose to exclude this statement if the study did not require ethical approval. Please note that the Editorial Office might ask you for further information. Please add “The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of NAME OF INSTITUTE (protocol code XXX and date of approval).” for studies involving humans. OR “The animal study protocol was approved by the Institutional Review Board (or Ethics Committee) of NAME OF INSTITUTE (protocol code XXX and date of approval).” for studies involving animals. OR “Ethical review and approval were waived for this study due to REASON (please provide a detailed justification).” OR “Not applicable” for studies not involving humans or animals.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data available upon request.
Acknowledgments
The authors are grateful to their colleagues Alexander S. Sharipov and Boris I. Loukhovitski for stimulating discussions.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| QCT | quasiclassical trajectory |
| PES | Potential energy surface |
| VT relaxation | vibrational-translational relaxation |
| PIP-NN | permutation invariant polynomials-neural network |
| XMCQDPT2 | extended multi-configuration quasi-degenerate second-order perturbation theory |
| DW-CASSCF | dynamically weighted complete active space self-consistent field method |
References
- Skrebkov, O.V.; Kostenko, S.S.; Smirnov, A.L. Vibrational nonequilibrium in reaction hydrogen with oxygen (review, in press). Technical Physics.
- Park, C. Review of chemical-kinetic problems of future NASA missions. I-Earth entries. Journal of Thermophysics and Heat transfer 1993, 7, 385–398. [Google Scholar] [CrossRef]
- Capitelli, M.; Armenise, I.; Gorse, C. State-to-state approach in the kinetics of air components under re-entry conditions. J. Thermophys. Heat Transfer 1997, 11, 570–578. [Google Scholar] [CrossRef]
- Treanor, C.E.; Adamovich, I.V.; Williams, M.J.; Rich, J.W. Kinetics of nitric oxide formation behind shock waves. Journal of Thermophysics and Heat Transfer 1996, 10, 193–199. [Google Scholar] [CrossRef]
- Kustova, E.V.; Nagnibeda, E.A.; Shevelev, Y.D.; Syzranova, N.G. Comparison of different models for non-equilibrium CO2 flows in a shock layer near a blunt body. Shock Waves 2011, 21, 273–287. [Google Scholar] [CrossRef]
- Surzhikov, S.; Reynier, P.; Seller, G.; Taccogna, F. Radiative aerothermodynamics of entering space vehicles: toward the use of state-to-state approach. Open Plasma Phys. J 2014, 7, 127–154. [Google Scholar] [CrossRef]
- Kadochnikov, I.N.; Loukhovitski, B.I.; Starik, A.M. Kinetics of plasmachemical processes in the expanding flow of nitrogen plasma. Phys. Scr. 2013, 88, 058306 (12pp). [Google Scholar] [CrossRef]
- Gimelshein, S.F.; Wysong, I.J.; Fangman, A.J.; Andrienko, D.A.; Kunova, O.V.; Kustova, E.V.; Morgado, F.; Garbacz, C.; Fossati, M.; Hanquist, K.M. Kinetic and continuum modeling of high-temperature air relaxation. Journal of Thermophysics and Heat Transfer 2022, 36, 870–893. [Google Scholar] [CrossRef]
- Aiken, T.T.; Boyd, I.D. Analysis of Critical Rate Processes for Ionization in Shock-Heated Air. AIAA AVIATION 2023 Forum, 2023, p. 3330.
- Voelkel, S.; Masselot, D.; Varghese, P.L.; Raman, V. Analysis of Hydrogen-Air Detonation Waves with Vibrational Nonequilibrium. AIP Conf. Proc. 2016, 1786, 070015. [Google Scholar]
- Shi, L.; Shen, H.; Zhang, P.; Zhang, D.; Wen, C. Assessment of vibrational non-equilibrium effect on detonation cell size. Combustion Science and Technology 2017, 189, 841–853. [Google Scholar] [CrossRef]
- Kadochnikov, I.N.; Arsentiev, I.V. Modelling of vibrational nonequilibrium effects on the H2–air mixture ignition under shock wave conditions in the state-to-state and mode approximations. Shock Waves 2020, 30, 491–504. [Google Scholar] [CrossRef]
- Skrebkov, O.V.; Kostenko, S.S.; Smirnov, A.L. Vibrational nonequilibrium and reaction heat effect in diluted hydrogen-oxygen mixtures behind reflected shock waves at 1000<T<1300 K. International Journal of Hydrogen Energy 2020, 45, 3251–3262. [Google Scholar]
- Smirnov, V.N.; Vlasov, P.A. Effects of hydrocarbon impurities, vibrational relaxation, and boundary-layer-induced pressure rise on the ignition of H2–O2–Ar mixtures behind reflected shock waves. International Journal of Hydrogen Energy 2021, 46, 9580–9594. [Google Scholar] [CrossRef]
- Colonna, G.; Capitelli, M. Self-consistent model of chemical, vibrational, electron kinetics in nozzle expansion. Journal of thermophysics and heat transfer 2001, 15, 308–316. [Google Scholar] [CrossRef]
- Guy, A.; Bourdon, A.; Perrin, M.Y. Consistent multi-internal-temperatures models for nonequilibrium nozzle flows. Chemical Physics 2013, 420, 15–24. [Google Scholar] [CrossRef]
- Zidane, A.; Haoui, R.; Sellam, M.; Bouyahiaoui, Z. Numerical study of a nonequilibrium H2-O2 rocket nozzle flow. International Journal of Hydrogen Energy 2019, 44, 4361–4373. [Google Scholar] [CrossRef]
- Starikovskiy, A.; Aleksandrov, N. Plasma-assisted ignition and combustion. Prog. Energy Combust. Sci. 2013, 39, 61–110. [Google Scholar] [CrossRef]
- Popov, N.A. Kinetics of plasma-assisted combustion: effect of non-equilibrium excitation on the ignition and oxidation of combustible mixtures. Plasma Sources Sci. Technol. 2016, 25, 043002(31pp). [Google Scholar] [CrossRef]
- Pietanza, L.D.; Colonna, G.; Capitelli, M. Activation of vibrational-induced CO2 dissociation in cold non-equilibrium plasma. Plasma Physics and Controlled Fusion 2023, 65, 044004. [Google Scholar] [CrossRef]
- Kadochnikov, I.N.; Arsentiev, I.V.; Loukhovitski, B.I.; Sharipov, A.S. State-to-state vibrational kinetics of diatomic molecules in laser-induced ignition of a syngas-air mixture: Modeling study. Chemical Physics 2022, 562, 111669. [Google Scholar] [CrossRef]
- Mankelevich, Y.A.; Rakhimova, T.V.; Voloshin, D.G.; Chukalovskii, A.A. Vibrationally Excited Ozone in Kinetics of O/N/Ar Mixtures after Ozone Photolysis. Russian Journal of Physical Chemistry A 2023, 97, 1033–1045. [Google Scholar] [CrossRef]
- Capitelli, M.; Ferreira, C.M.; Gordiets, B.F.; Osipov, A.I. Plasma kinetics in atmospheric gases; Vol. 31, Springer Series on Atomic, Optical, and Plasma Physics, Springer-Verlag, Berlin, 2000.
- Truhlar, D.G.; Muckerman, J.T. Reactive scattering cross sections III: Quasiclassical and semiclassical methods. In Atom-Molecule Collision Theory: A Guide for the Experimentalist; Springer, 1979; pp. 505–566.
- Gray, S.K.; Balint-Kurti, G.G. Quantum dynamics with real wave packets, including application to three-dimensional (J=0) D+H2=HD+H reactive scattering. The Journal of chemical physics 1998, 108, 950–962. [Google Scholar] [CrossRef]
- Gorse, C.; Capitelli, M.; Bacal, M.; Bretagne, J.; Laganà, A. Progress in the non-equilibrium vibrational kinetics of hydrogen in magnetic multicusp H-ion sources. Chemical physics 1987, 117, 177–195. [Google Scholar] [CrossRef]
- Pogosbekian, M.J.; Sergievskaia, A.L.; Losev, S.A. Verification of theoretical models of chemical exchange reactions on the basis of quasiclassical trajectory calculations. Chemical Physics 2006, 328, 371–378. [Google Scholar] [CrossRef]
- Esposito, F.; Armenise, I.; Capitelli, M. N–N2 state to state vibrational-relaxation and dissociation rates based on quasiclassical calculations. Chemical Physics 2006, 331, 1–8. [Google Scholar] [CrossRef]
- Bender, J.D.; Valentini, P.; Nompelis, I.; Paukku, Y.; Varga, Z.; Truhlar, D.G.; Schwartzentruber, T.; Candler, G.V. An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of N2+N2 dissociation reactions. The Journal of chemical physics 2015, 143. [Google Scholar] [CrossRef] [PubMed]
- Andrienko, D.A.; Boyd, I.D. State-specific dissociation in O2–O2 collisions by quasiclassical trajectory method. Chemical Physics 2017, 491, 74–81. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Jaffe, R.L.; Schwenke, D.W.; Panesi, M. Construction of a coarse-grain quasi-classical trajectory method. I. Theory and application to N2–N2 system. The Journal of chemical physics 2018, 148. [Google Scholar] [CrossRef]
- Vargas, J.; Monge-Palacios, M.; Lacoste, D.A. State-Specific Dissociation and Inelastic Rate Constants for Collisions of H2 with H and He. Journal of Thermophysics and Heat Transfer 2023. pp. 1–12. [Google Scholar]
- Kuntz, P.J.; Nemeth, E.M.; Polanyi, J.C.; Rosner, S.D.; Young, C.E. Energy distribution among products of exothermic reactions. II. Repulsive, mixed, and attractive energy release. The Journal of Chemical Physics 1966, 44, 1168–1184. [Google Scholar] [CrossRef]
- Sharipov, A.S.; Loukhovitski, B.I. Energy disposal into the vibrational degrees of freedom of bimolecular reaction products: Key factors and simple model. Chemical Physics 2021, 544, 111098. [Google Scholar] [CrossRef]
- Dawes, R.; Thompson, D.L.; Guo, Y.; Wagner, A.F.; Minkoff, M. Interpolating moving least-squares methods for fitting potential energy surfaces: Computing high-density potential energy surface data from low-density ab initio data points. The Journal of chemical physics 2007, 126. [Google Scholar] [CrossRef]
- Jiang, B.; Li, J.; Guo, H. Potential energy surfaces from high fidelity fitting of ab initio points: The permutation invariant polynomial-neural network approach. International Reviews in Physical Chemistry 2016, 35, 479–506. [Google Scholar] [CrossRef]
- Braunstein, M.; Adler-Golden, S.; Maiti, B.; Schatz, G. Quantum and classical studies of the O(3P)+H2(v=0-3, j=0)→OH+H reaction using benchmark potential surface. Technical Report, 2003; AIR FORCE RESEARCH 116 LAB EDWARDS AFB CA SPACE AND MISSILE PROPULSION DIV. [Google Scholar]
- Braunstein, M.; Adler-Golden, S.; Maiti, B.; Schatz, G. Quantum and classical studies of the O(3P)+H2(v=0–3, j=0)→OH+H reaction using benchmark potential surfaces. The Journal of chemical physics 2004, 120, 4316–4323. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Rosa, C.; Brandão, J. Theoretical studies on the O(3P)+H2→OH+H reaction. Chemical physics letters 2006, 418, 250–254. [Google Scholar] [CrossRef]
- Xu, Z.H.; Zong, F.J.; Han, B.R.; Dong, S.H.; Liu, J.Q.; Ji, F. Effects of a reagent’s rotational and vibrational excitations on reaction O(3P)+H2(n= 0, 3, j= 0, 3, 5, 7, 9, 12, 15)→OH+H. Chinese Physics B 2012, 21, 093103. [Google Scholar] [CrossRef]
- Veselinova, A.; Agúndez, M.; Goicoechea, J.R.; Menéndez, M.; Zanchet, A.; Verdasco, E.; Jambrina, P.; Aoiz, F.J. Quantum study of reaction O(3P)+H2(v,j)→OH+H: OH formation in strongly UV-irradiated gas. Astronomy & Astrophysics 2021, 648, A76. [Google Scholar]
- Granovsky, A.A. Extended Multi-Configuration Quasi-Degenerate Perturbation Theory: The New Approach to Multi-state Multi-Reference Perturbation Theory. J. Chem. Phys. 2011, 134, 214113. [Google Scholar] [CrossRef] [PubMed]
- Deskevich, M.P.; Nesbitt, D.J. Dynamically weighted multiconfiguration self-consistent field: Multistate calculations for F+H2O→HF+OH reaction paths. J. Chem. Phys. 2004, 120, 7281. [Google Scholar] [CrossRef] [PubMed]
- Pelevkin, A.V.; Loukhovitski, B.I.; Sharipov, A.S. Reaction of the N Atom with Electronically Excited O2 Revisited: A Theoretical Study. The Journal of Physical Chemistry A 2021, 125, 8294–8312. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning Jr., T. H.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Lesiuk, M.; Jeziorski, B. Complete basis set extrapolation of electronic correlation energies using the Riemann zeta function. Journal of Chemical Theory and Computation 2019, 15, 5398–5403. [Google Scholar] [CrossRef] [PubMed]
- Granovsky, A.A. Firefly V. 8.2.0. http://classic.chem.msu.su/gran/firefly/index.html, 2016.
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; Windus, T.L.; Dupuis, M.; Montgomery, J.A. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Goodfellow, I.; Harp, A.; Irving, G.; Isard, M.; Jia, Y.; Jozefowicz, R.; Kaiser, L.; Kudlur, M.; Levenberg, J.; Mané, D.; Monga, R.; Moore, S.; Murray, D.; Olah, C.; Schuster, M.; Shlens, J.; Steiner, B.; Sutskever, I.; Talwar, K.; Tucker, P.; Vanhoucke, V.; Vasudevan, V.; Viégas, F.; Vinyals, O.; Warden, P.; Wattenberg, M.; Wicke, M.; Yu, Y.; Zheng, X. TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems, 2015. Software available from tensorflow.org.
- Paszke, A.; Gross, S.; Chintala, S.; Chanan, G.; Yang, E.; DeVito, Z.; Lin, Z.; Desmaison, A.; Antiga, L.; Lerer, A. Automatic differentiation in PyTorch. NIPS-W, 2017.
- Chollet, Francois and others. Keras. https://github.com/fchollet/keras, 2015. GitHub.
- Braams, B.J.; Bowman, J.M. Permutationally invariant potential energy surfaces in high dimensionality. International Reviews in Physical Chemistry 2009, 28, 577–606. [Google Scholar] [CrossRef]
- Bosma, W.; Cannon, J.; Playoust, C. The Magma algebra system I: The user language. Journal of Symbolic Computation 1997, 24, 235–265. [Google Scholar] [CrossRef]
- Decker, W.; Greuel, G.M.; Pfister, G.; Schönemann, H. Singular 4-1-2—A computer algebra system for polynomial computations. http://www. singular. uni-kl. de, 2019. [Google Scholar]
- Polak, L.S.; Goldenberg, M.J.; Levickij, A.A. Vychislitel’nye metody v himicheskoj kinetike [Computational methods in chemical kinetics]; Nauka, 1984.
- Abdel-Halim, H.M.; Jaafreh, S.M. Reaction rate constants from classical trajectories of atom-diatomic molecule collisions. Zeitschrift für Naturforschung A 2008, 63, 159–169. [Google Scholar] [CrossRef]
- Le Roy, R.J. LEVEL: A computer program for solving the radial Schrödinger equation for bound and quasibound levels. Journal of Quantitative Spectroscopy and Radiative Transfer 2017, 186, 167–178. [Google Scholar] [CrossRef]
- Varga, T.; Olm, C.; Nagy, T.; Zsély, I.G.; Valkó, É.; Pálvölgyi, R.; Curran, H.J.; Turányi, T. Development of a joint hydrogen and syngas combustion mechanism based on an optimization approach. International journal of chemical kinetics 2016, 48, 407–422. [Google Scholar] [CrossRef]
- Starik, A.M.; Sharipov, A.S.; Titova, N.S. Intensification of syngas ignition through the excitation of CO molecule vibrations: a numerical study. Journal of Physics D: Applied Physics 2010, 43, 245501. [Google Scholar] [CrossRef]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Hampson, J.R.F.; Kerr, J.A.; Troe, J. Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry. Supplement IV. J. Phys. Chem. Ref. Data 1992, 21, 1125–568. [Google Scholar] [CrossRef]
- Tsang, W.; Hampson, R.F. Chemical Kinetic Data Base for Combustion Chemistry. J. Phys. Chem. Ref. Data. 1986, 15, 1087–1280. [Google Scholar] [CrossRef]
- Cohen, N.; Westberg, K.R. Chemical kinetic data sheets for high-temperature chemical reactions. Journal of physical and chemical reference data 1983, 12, 531–590. [Google Scholar] [CrossRef]
- Dove, J.E.; Teitelbaum, H. The vibrational relaxation of H2. I. Experimental measurements of the rate of relaxation by H2, He, Ne, Ar, and Kr. Chemical Physics 1974, 6, 431–444. [Google Scholar] [CrossRef]
- Cacciatore, M.; Capitelli, M.; Billing, G. Vibration-to-translation energy exchanges in H2 colliding with highly vibrationally excited H2 molecules. Chemical physics letters 1989, 157, 305–308. [Google Scholar] [CrossRef]
- Millikan, R.C.; White, D.R. Systematics of vibrational relaxation. J. Chem. Phys. 1963, 39, 3209–3213. [Google Scholar] [CrossRef]
Figure 1.
The reaction cross section along the lower PES, calculated according to our methodology (solid lines with error bars) and using the QCT method from [38] (markers signed Braunstein&Adler-Golden2004).
Figure 1.
The reaction cross section along the lower PES, calculated according to our methodology (solid lines with error bars) and using the QCT method from [38] (markers signed Braunstein&Adler-Golden2004).
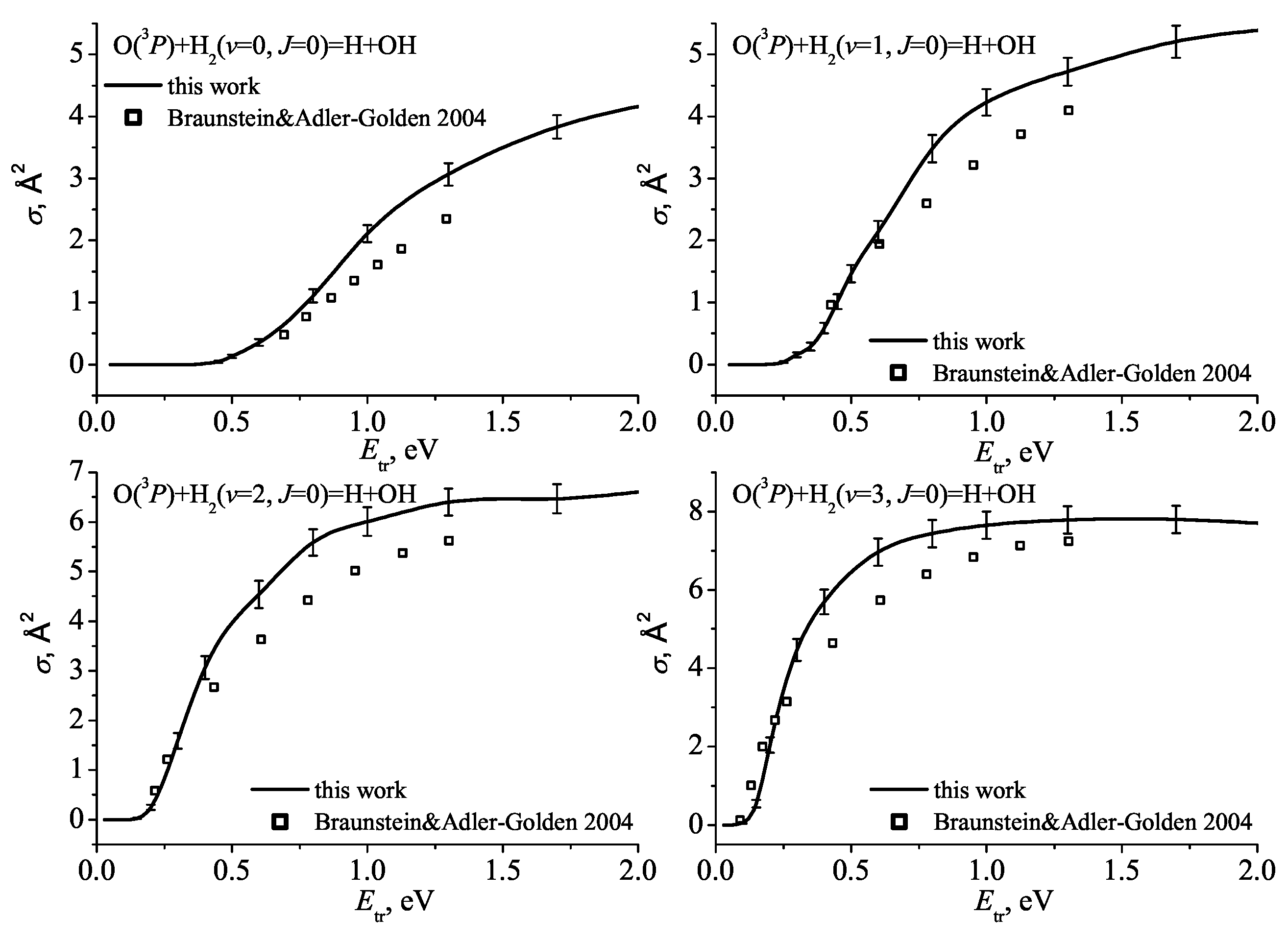
Figure 2.
Comparison of the state-to-state reaction rate calculated in this work by the QCT method (dashed lines) and obtained in [41] using quantum dynamics calculations.
Figure 2.
Comparison of the state-to-state reaction rate calculated in this work by the QCT method (dashed lines) and obtained in [41] using quantum dynamics calculations.
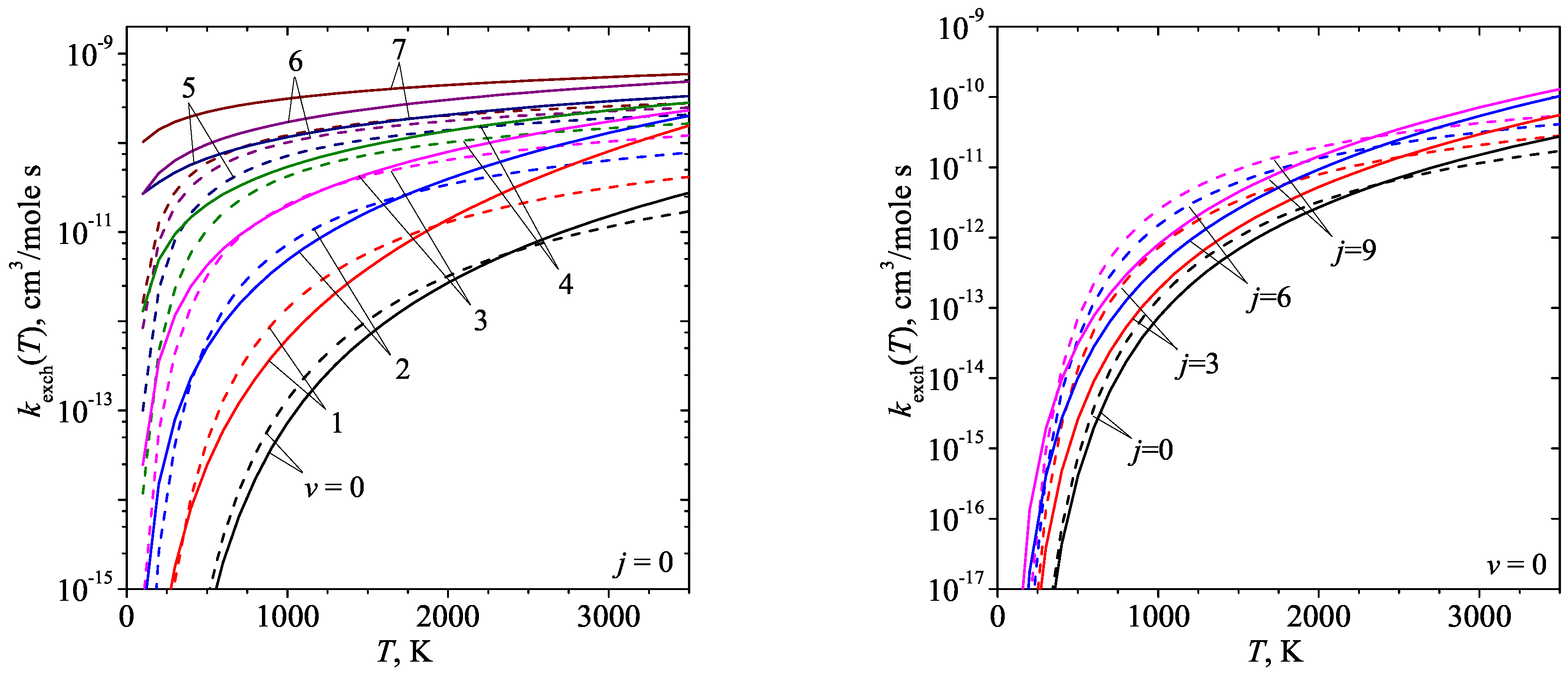
Figure 3.
Thermal rate constant of the reaction O+H2 H+OH along the lower PES, obtained on the basis of the QCT method and the non-variational transition state theory, as well as the data on the reaction rate constant available in the literature (Veselinova2021 [41], Varga2016 [58], Starik2010 [59], Baulch1992 [60]).
Figure 3.
Thermal rate constant of the reaction O+H2 H+OH along the lower PES, obtained on the basis of the QCT method and the non-variational transition state theory, as well as the data on the reaction rate constant available in the literature (Veselinova2021 [41], Varga2016 [58], Starik2010 [59], Baulch1992 [60]).
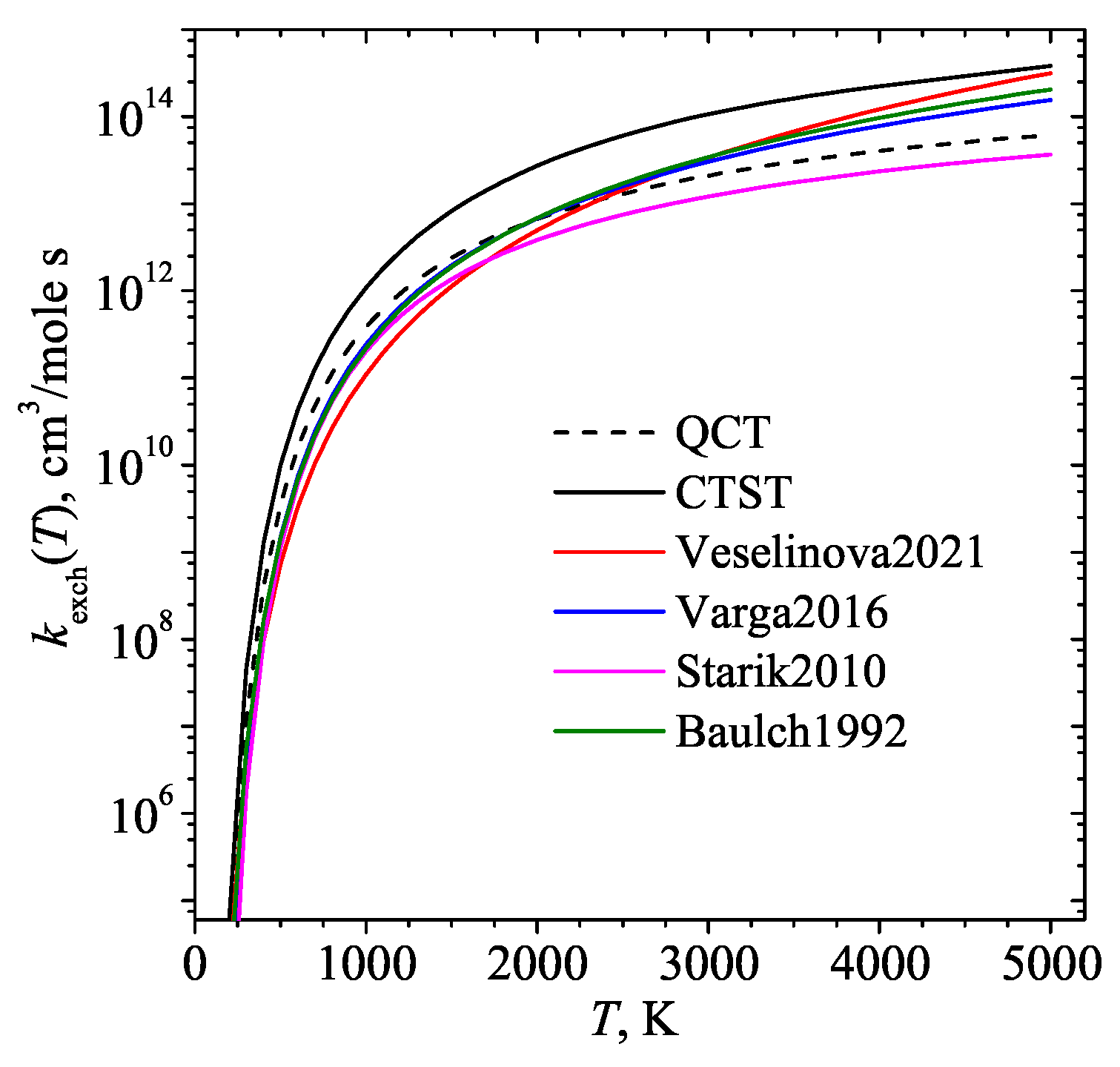
Figure 4.
Comparison of the thermal H2 + O = 2H + O dissociation rate constant calculated in this paper with hydrogen dissociation rate constants presented in other works (Baulch1992 [60], Tsang1986 [61], Cohen1983 [62]).
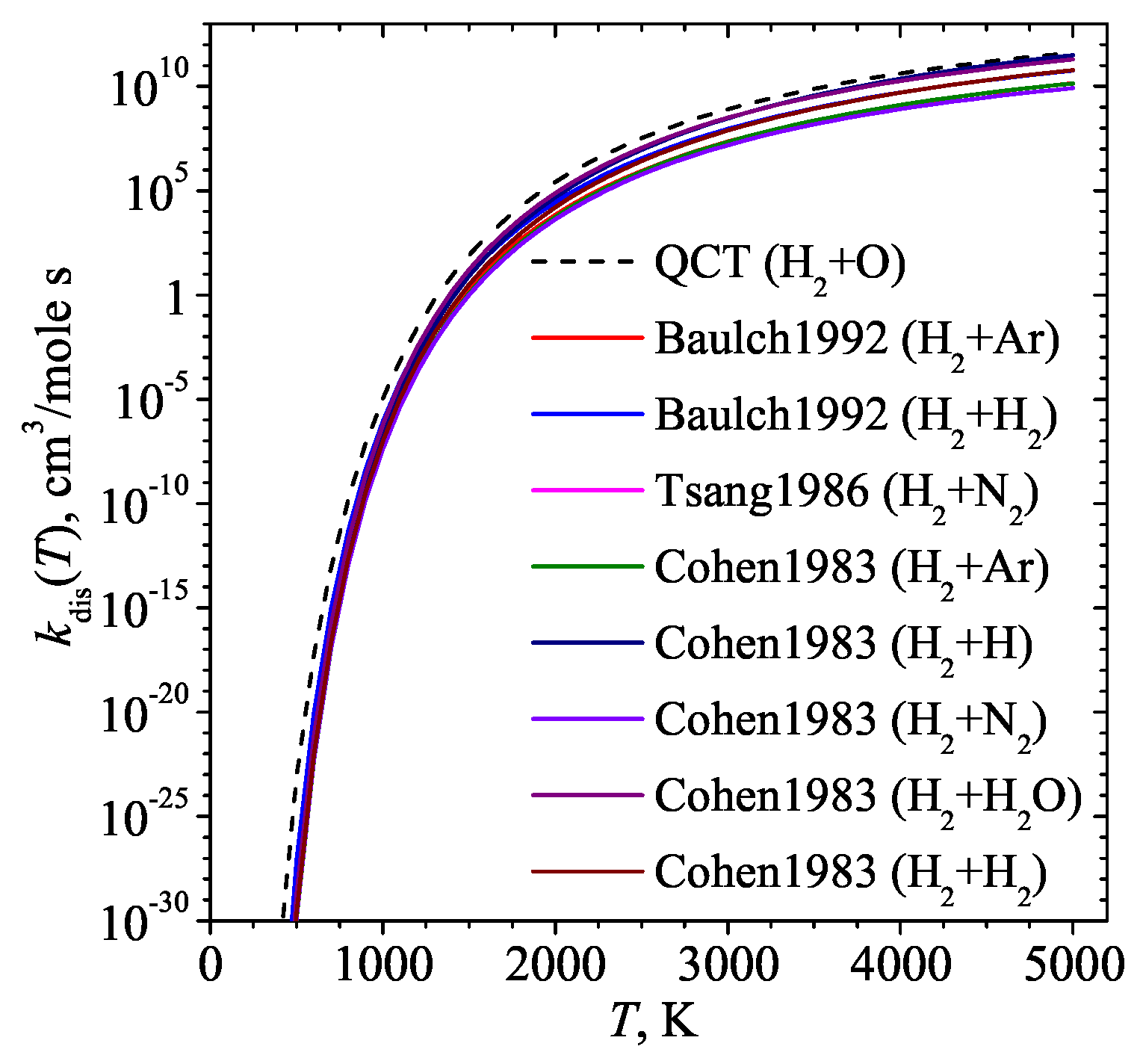
Figure 5.
Vibrational relaxation time of H2 molecules on various collisional partners. Dashed lines are experimental data (Dove1974 [63]), solid lines are calculation results (Cacciatore1989 [64], Gorse1987 [26]).
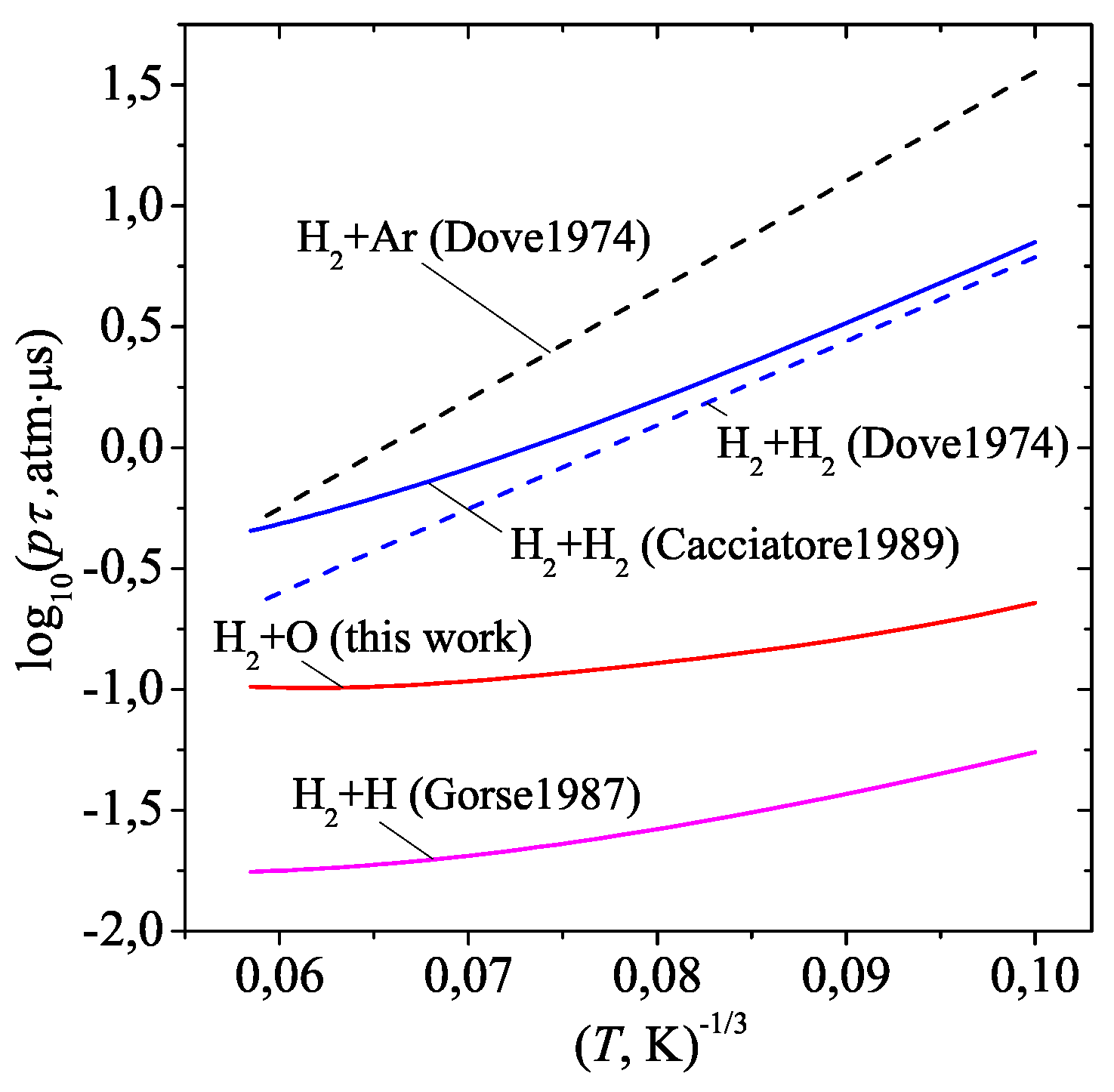
Figure 6.
The state-to-stare rate constants of the exchange reaction (a), dissociation (b) and VT relaxation (c) of H2 molecules in collision with O atoms calculated with the QCT method.
Figure 6.
The state-to-stare rate constants of the exchange reaction (a), dissociation (b) and VT relaxation (c) of H2 molecules in collision with O atoms calculated with the QCT method.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



