Submitted:
07 December 2023
Posted:
08 December 2023
You are already at the latest version
Abstract
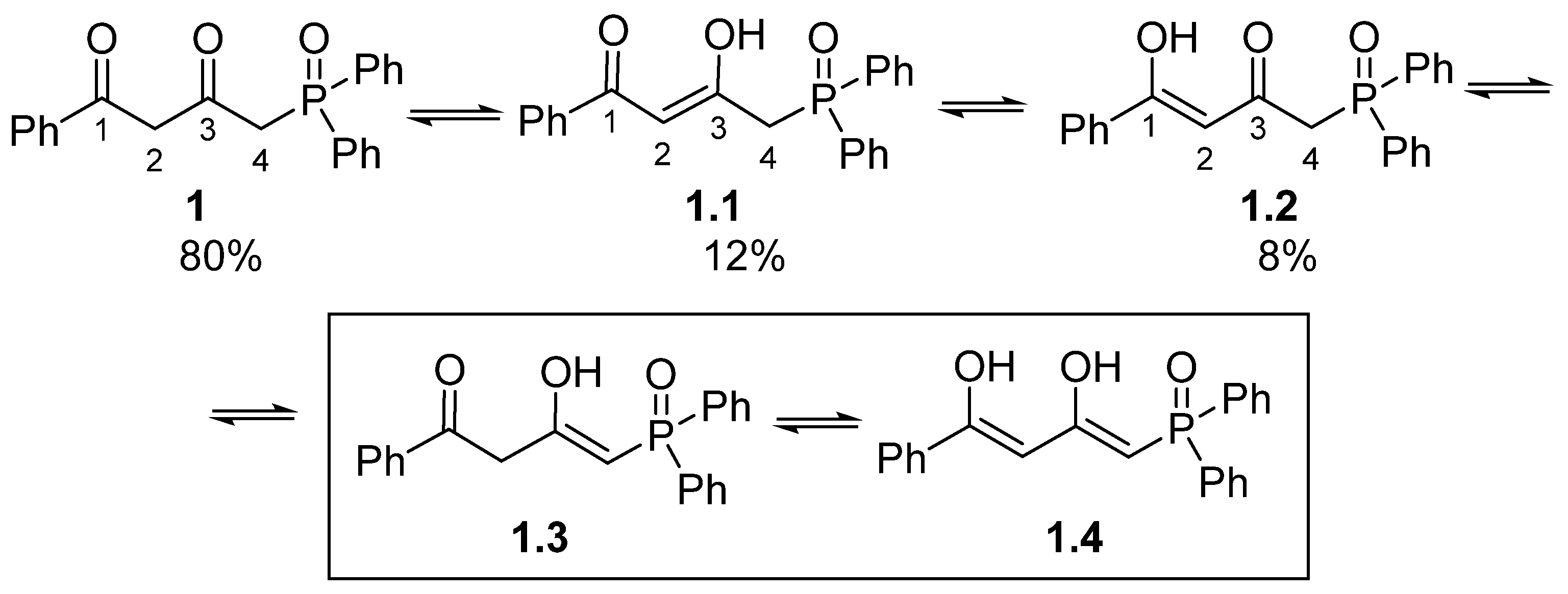
Novel 4-(diphenylphosphoryl)-1-phenylbutane-1,3-dione was synthesized by the reaction of diketone enamine with diphenylchloro phosphine followed by oxidation and hydrolysis of the enamine phosphine formed. Tautomeric transformation of the diketone moiety of phosphine oxide was investigated by 1H NMR. Three out of five possible tautomers were observed in the 4-(diphenylphosphoryl)-1-phenylbutane-1,3-dione solution. The major tautomer was the form with a diketone fragment.
Keywords:
phosphine oxides
; ligands
; lanthanides
; triketones
; keto-enolic tautomerism
1. Introduction
Phosphine oxides with an additional functional group are effective ligands for preparing complexes with lanthanides [1] with promising photophysical properties for fabricating OLEDs [2,3,4] or detecting various compounds [5]. Another class of well-studied ligands for complex synthesis with lanthanides are β-diketones [6]. Their functionalization with more coordination groups of various nature leads to more complex ligands with interesting and perspective coordination properties [7,8]. Moreover, both phosphine oxides and β-diketones are used as co-ligands for obtaining various luminescent complexes with lanthanides [9,10] for biomedical application [11]
We report here the synthesis and keto-enolic tautomerism of 4-(diphenylphosphoryl)-1-phenylbutane-1,3-dione.
2. Results and Discussion
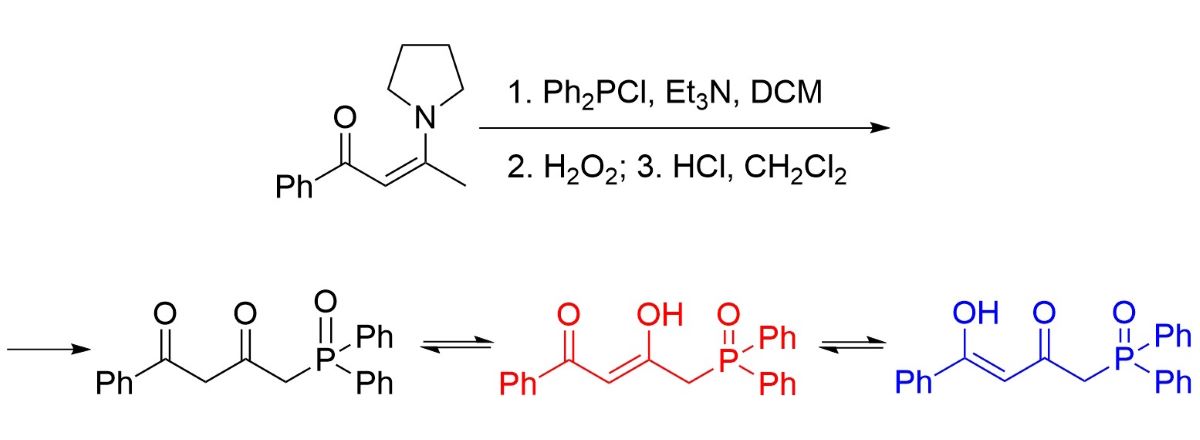
The synthesis was performed based on acetophenone 1 by the technique described for enamines of acetoacetic acid (Scheme 1). [12]. By condensing 1 with ethyl acetate, diketone 2 was obtained [13]. Then diketone 2 was transformed in enamine 3 at the reaction with pyrrolidine [14]. The subsequent reaction with diphenylchlorophosphine intermediately forming phosphine 5 followed by oxidation and hydrolysis have led to the target phosphine oxide with a 50% yield. Synthesis of compound 1 at the reaction of 1-(diphenylphosphoryl)propan-2-one with methyl benzoate over lithium diisopropylamide is mentioned in the recent study of [15] as an intermediate. However, phosphine oxide 1 was not isolated, and its spectral characteristics were not defined either. Moreover, the synthetic route presented herein is distinguished by using simpler techniques and readily available reagents.
Structure of compound 1 was studied by 1H NMR and 13C spectroscopy (Figure 1).
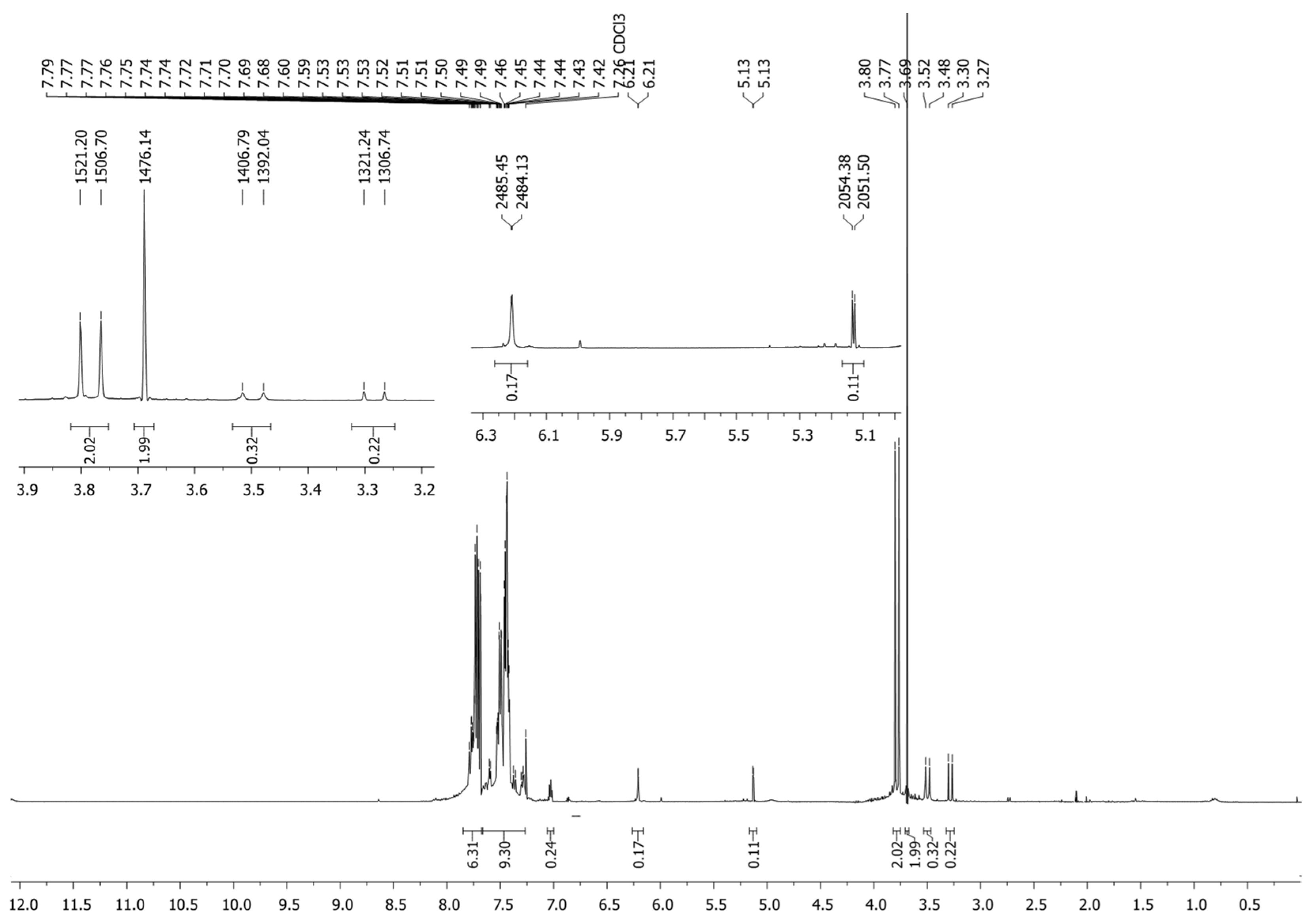
It is common knowledge that keto-enol tautomerism is characteristic of β-diketones and enol tautomer is often preferable in non-hydrogen bonding solvents [6,7,8,16]. In contrast, phosphine oxide 1 was isolated as a mixture of tautomers with prevailing diketo-form, the content of which as about 80% (Scheme 2). Mono-enol tautomers 1.1 and 1.2 made 8 and 12 %, respectively. Doublet H4 at 3.64 ppm (2JPH = 14.5 Hz) and the H2 proton singlet at 3.55 ppm confirm the presence of tautomer 1 as the main one in the mixture. At the same time, the H4 protons of minor tautomers 1.1 and 1.2, as well, show as doublets in the 1H NMR spectrum (3.36 ppm, 2JPH = 14.7 Hz and 3.14 ppm, 2JPH = 14.5 Hz). Presence of the H4 proton doublets in alpha-position relative to phosphorus points clearly to the absence of any prototropism involving them. Signals of the H2 protons located in tautomers 1.1 and 1.2 at the sp2-hybrid carbon atom manifest themselves in weaker fields (6.07 ppm, 4JPH = 1.7 Hz, and 4.99 ppm, 4JPH = 2.9 Hz). Protons of the hydroxy groups of enols 1.1 and 1.2 were not registered in the spectra, probably due to low intensity or deuterium exchange with CDCl3 (For full 1H NMR spectrum (0-18.2 ppm), please see ESI). Tautomers 1.3 and 1.4 were not registered spectrally. Thus, three of five possible tautomers are realized for compound 1.
4. Materials and Methods
All reagents were used as purchased from Sigma-Aldrich or Acros Chemicals without further purification. Solvents were purified by standard procedures before use. All reactions were run under an argon atmosphere unless in aqueous media.
NMR experiments were carried out with 400 MHz [400 MHz (1H), 162 MHz (31P)] or 600 MHz [600 MHz (1H), 243 MHz (31P),] spectrometers equipped with a pulsed gradient unit capable of producing magnetic field pulse gradients in the z-direction of 53.5 G cm–1. All spectra were acquired in a 5 mm gradient inverse broadband probe head. Chemical shifts (d) are expressed in parts per million, relative to the residual 1H signal of CDCl3, and the signals are designated as follows: s, singlet; d, doublet; t, triplet; m, multiplet. Coupling constants (J) are in hertz (Hz). The ESI MS measurements were performed using an AmazonX ion trap mass spectrometer (Bruker Daltonic GmbH, Germany) in the positive or negative mode in the mass range of 70–3000. IR spectra were recorded with a Bruker Tensor-27 instrument for the samples in KBr pellets. The elemental analysis was carried out on a CHNS analyzer EuroEA3028-HT-OM (Eurovector SpA, Italy). The samples were weighed on Sartorius CP2P (Germany) microbalances in tin capsules. Callidus 4.1 software was used to perform quantitative measurements and evaluate the data received.
1-phenylbutane-1,3-dione (2)
Prepared according to the previously described procedure [13]. Yellow oil. Yield 85%. Exists as a mixture of keto/enol tautomers with 1:10 ratio. 1H NMR (400 MHz, CDCl3) (enol tautomer) δ 7.90 – 7.86 (m, 2H), 7.55 – 7.49 (m, 1H), 7.45 (dd, J = 8.3, 6.7 Hz, 2H), 6.18 (s, 1H), 2.20 (s, 3H).
1-phenyl-3-(pyrrolidin-1-yl)but-2-en-1-one (3)
Prepared by previously described procedure [14] from 1g (6.2 mmol) of 2 and 0.5 ml (0.44 g, 6.2 mmol) of pyrrolidine in 10 ml of benzene. Yiled 1.257g (94 %), brown oil, which solidifies while standing. Spectral data are in consistent with the previously described [17] 1H NMR (600 MHz, CDCl3) δ 7.86 (dd, J = 8.0, 1.6 Hz, 2H), 7.42 – 7.37 (m, 3H), 5.61 (s, 1H), 3.53 (brs, 2H), 3.37 (brs, 2H), 2.68 (s, 3H), 2.00 (brs, 4H). Compound 3 was used in the next step without purification.
4-(diphenylphosphoryl)-1-phenylbutane-1,3-dione (diketo-tautomer)
To a stirred solution of compound 3 (0.65 g, 3 mmol) and triethylamine (4.16 ml, 0.303 g, 3 mmol) in CH2Cl2 (5 ml) under an argon atmosphere Ph2PCl (0.55 ml, 0.661 g, 3 mmol) was added dropwise and stirred at room temperature for 24 h. Then (NH2)2CO·H2O2 (0.282 g, 3 mmol) was added in one portion and the mixture stirred for additional 1 h. The reaction mixture was washed with water and 10 ml of 5% HCl solution was added. After 2d the water was separated, and organic phase was dried under Na2SO4 and concentrated on rotary evaporator. Crude product was purified by dry-column flash chromatography on Schott’s funnel (silica gel 230-400 mesh), using gradient elution (eluent from EtOAc : Hexane 4 :1 to EtOAc). Yield 0.542 g (50%)/ Pale yellow gum. Exists as a mixture of keto/enol tautomers 1, 1.1 and 1.2 with 20:3:2 ratio respectively. IR (thin film) 3434 (s, br), 2924 (w, sh), 1710 (w), 1603 (s, br), 1439 (s), 1224 (w), 1194 (s), 1116 (m), 1101 (m), 720 (m), 692 (s), 521 (s) cm 1H NMR (400 MHz, CDCl3) δ 7.74 – 7.71 (m, 3H, ArH), 7.71 – 7.67 (m, 3H, ArH), 7.55 – 7.47 (m, 3H, ArH), 7.47 – 7.39 (m, 6H, ArH), 6.21 (d, J = 1.6 Hz, 1H, H2 of tautomer 1.1), 5.13 (d, J = 3.0 Hz, 1H, H2 of tautomer 1.2), 3.78 (d, J = 14.6 Hz, 2H, H4), 3.69 (s, 2H, H2), 3.50 (d, J = 14.7 Hz, 2H, H4 of tautomer 1.1), 3.28 (d, J = 14.5 Hz, 2H, H4 of tautomer 1.1). 13C NMR (101 MHz, CDCl3) δ 195.85 (d, J = 5.6 Hz), 167.02, 140.37, 132.48 (overlapped on the next signal), 132.47 (d, J = 2.9 Hz), 131.89, 131.38 (d, J = 104.0 Hz), 131.12, 130.87 (d, J = 10.0 Hz), 128.86 (d, J = 12.5 Hz), 128.86 (overlapped on the previous signal) 50.83, 46.88 (d, J = 56.5 Hz). 31P NMR (243 MHz, CDCl3) δ 30.41 (1.1), 29.30 (1), 27.07 (1.2). Anal. Calcd for C22H19O3P (%): C, 72.92; H, 5.29; P, 8.55 . Found (%): C 72.63; H 5.41; P 8.47. ESI-MS m/z = 363.17 [M + H]+
Supplementary Materials
Supplementary Materials: The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: 1H NMR spectra of tautomeric mixture of compound 1 (CDCl3, 400 MHz); Figure S2: 13C NMR spectra of tautomeric mixture of compound 1 (101 MHz, CDCl3); Figure S3: 13C NMR (dept) spectra of tautomeric mixture of compound 1 (101 MHz, CDCl3); Figure S4: 31P {1H} NMR spectra of tautomeric mixture of compound 1 (243 MHz, CDCl3).
Author Contributions
“Conceptualization, D.T. and R.Z. A.M. and V.M.; methodology, X.X.; validation, and Z.Z.; formal analysis, R.Z.; investigation, E.B.; resources, V.F.; data curation, A.D.; writing—original draft preparation, D.T.; writing—review and editing, A.M.; visualization, D.T.; supervision, V.F. and A.M.; project administration, A.D.; funding acquisition, R.Z. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
The authors thank the Russian Science Foundation (grant 22-23-00853) for financial support. The authors are grateful to the staff of the Distributed Spectral-Analytical Center of Shared Facilities for Study of Structure, Composition and Properties of Substances and Materials of Federal Research Center of Kazan Scientific Center of Russian Academy of Sciences» for their research and assistance in discussing the results.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Platt, A.W.G. Lanthanide Phosphine Oxide Complexes. Coord. Chem. Rev. 2017, 340, 62–78. [Google Scholar] [CrossRef]
- Aslandukov, A.N.; Utochnikova, V. V.; Goriachiy, D.O.; Vashchenko, A.A.; Tsymbarenko, D.M.; Hoffmann, M.; Pietraszkiewicz, M.; Kuzmina, N.P. The Development of a New Approach toward Lanthanide-Based OLED Fabrication: New Host Materials for Tb-Based Emitters. Dalt. Trans. 2018, 47, 16350–16357. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.-H.; Cheng, C.-H. A Highly Efficient Universal Bipolar Host for Blue, Green, and Red Phosphorescent OLEDs. Adv. Mater. 2010, 22, 2468–2471. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liang, Q.; Han, C.; Zhang, J.; Xu, H. A Phosphanthrene Oxide Host with Close Sphere Packing for Ultralow-Voltage-Driven Efficient Blue Thermally Activated Delayed Fluorescence Diodes. Adv. Mater. 2017, 29, 1700553. [Google Scholar] [CrossRef] [PubMed]
- R. Zairov, A. Dovzhenko, N. Terekhova, T. Kornev, Y. Zhou, Z. Huang, D. Tatarinov, G. Nizameeva, R.R. Fayzullin, A.T. Gubaidullin, T. Salikhova, F. Enrichi, V.F. Mironov, A. Mustafina, Phosphineoxide-Chelated Europium(III) Nanoparticles for Ceftriaxone Detection, Nanomaterials. 13 (2023). [CrossRef]
- Aromí, G.; Gamez, P.; Reedijk, J. Poly Beta-Diketones: Prime Ligands to Generate Supramolecular Metalloclusters. Coord. Chem. Rev. 2008, 252, 964–989. [Google Scholar] [CrossRef]
- Vigato, P.A.; Peruzzo, V.; Tamburini, S. The Evolution of β-Diketone or β-Diketophenol Ligands and Related Complexes. Coord. Chem. Rev. 2009, 253, 1099–1201. [Google Scholar] [CrossRef]
- Clegg, J.K.; Li, F.; Lindoy, L.F. Oligo-β-Diketones as Versatile Ligands for Use in Metallo-Supramolecular Chemistry: Recent Progress and Perspectives. Coord. Chem. Rev. 2022, 455, 214355. [Google Scholar] [CrossRef]
- Xu, H.; Zhu, R.; Zhao, P.; Xie, L.-H.; Huang, W. Photophysical and Electroluminescent Properties of a Series of Monochromatic Red-Emitting Europium-Complexed Nonconjugated Copolymers Based on Diphenylphosphine Oxide Modified Polyvinylcarbazole. Polymer (Guildf). 2011, 52, 804–813. [Google Scholar] [CrossRef]
- Raj, D.B.A.; Francis, B.; Reddy, M.L.P.; Butorac, R.R.; Lynch, V.M.; Cowley, A.H. Highly Luminescent Poly(methyl Methacrylate)-Incorporated Europium Complex Supported by a Carbazole-Based Fluorinated β-Diketonate Ligand and a 4,5-Bis(diphenylphosphino)-9,9-Dimethylxanthene Oxide Co-Ligand. Inorg. Chem. 2010, 49, 9055–9063. [Google Scholar] [CrossRef] [PubMed]
- Zairov, R.R.; Dovzhenko, A.P.; Sapunova, A.S.; Voloshina, A.D.; Tatarinov, D.A.; Nizameev, I.R.; Gubaidullin, A.T.; Petrov, K.A.; Enrichi, F.; Vomiero, A.; et al. Dual Red-NIR Luminescent Eu Yb Heterolanthanide Nanoparticles as Promising Basis for Cellular Imaging and Sensing. Mater. Sci. Eng. C 2019, 105, 110057. [Google Scholar] [CrossRef] [PubMed]
- Kostyuk, A.N.; Svyaschenko, Y. V; Volochnyuk, D.M.; Lysenko, N. V; Tolmachev, A.A.; Pinchuk, A.M. Structural Sensitivity in Phosphorylation of Enamines—Derivatives of β-Aminocrotonic Acid with Diphenylchlorophosphine. Tetrahedron Lett. 2003, 44, 6487–6491. [Google Scholar] [CrossRef]
- Inagaki, S.; Saito, K.; Suto, S.; Aihara, H.; Sugawara, A.; Tamura, S.; Kawano, T. Synthesis of 5-Aryl-3(2 H )-Furanones Using Intramolecular Cyclization of Sulfonium Salts. J. Org. Chem. 2018, 83, 13834–13846. [Google Scholar] [CrossRef] [PubMed]
- A General Synthesis Of 4-Isoxazolecarboxylic Esters: Ethyl 3-Ethyl-5-Methyl-4-Isoxazolecarboxylate. Org. Synth. 1973, 53, 59. [CrossRef]
- Maass, J.S.; Wilharm, R.K.; Luck, R.L.; Zeller, M. Photoluminescent Properties of Three Lanthanide Compounds of Formulae LnCl3(diphenyl((5-Phenyl-1H-Pyrazol-3-Yl)methyl)phosphine oxide)2, Ln = Sm, Eu and Tb: X-Ray Structural, Emission and Vibrational Spectroscopies, DFT and Thermogravimetric Studies. Inorganica Chim. Acta 2018, 471, 481–492. [Google Scholar] [CrossRef]
- Allen, G.; Dwek, R.A. An N.m.r. Study of Keto–enol Tautomerism in β-Diketones. J. Chem. Soc. B 1966, 161–163. [CrossRef]
- Zheng, Y.; Liu, Z.-W.; Li, T.; Li, X.; Li, S.-H. KIO 3 -Mediated γ-C(sp 3 )–H Sulfenylation of Enaminones. Org. Lett. 2022, 24, 7533–7537. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Synthesis of 4-(diphenylphosphoryl)-1-phenylbutane-1,3-dione.
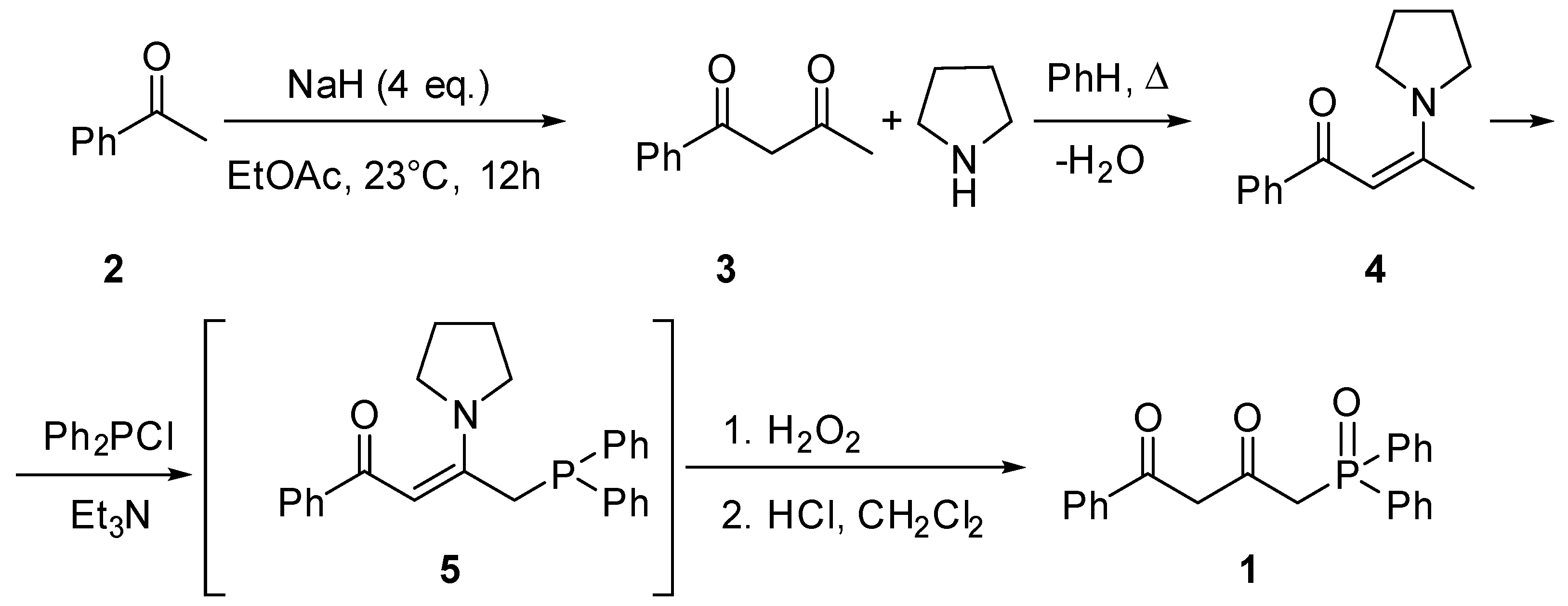
Figure 1.
1H NMR spectra of tautomeric mixture of compound 1 (CDCl3, 400 MHz).
Scheme 2.
Tautomeric transformations of 4-(diphenylphosphoryl)-1-phenylbutane-1,3-dione in solution.
Scheme 2.
Tautomeric transformations of 4-(diphenylphosphoryl)-1-phenylbutane-1,3-dione in solution.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



