Submitted:
06 December 2023
Posted:
07 December 2023
You are already at the latest version
Abstract
New [1,2]dithiolo[3,4-b]pyridine-5-carboxamides were synthesized through the reaction of dithiomalondianilide (N,N'-diphenyldithiomalondiamide) with 3-aryl-2-cyanoacrylamides or by a three-component reaction involving aromatic aldehydes, cyanoacetamide and dithiomalon- dianilide in the presence of morpholine. The structure of 6-amino-4-(2,4-dichlorophenyl)- 7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamide was con-firmed using X-ray crystallography. To understand the reaction mechanism in detail, density functional theory (DFT) calculations were performed with a Grimme B97-3c composite computa-tional scheme. The results revealed that the rate limiting step is a cyclization process leading to the closure of 1,4-dihydropyridine ring with an activation barrier of 28.8 kcal/mol. Some of dithiolo[3,4-b]pyridines exhibited moderate herbicide safening effects against 2,4-D . Additional-ly, ADMET parameters were calculated and molecular docking studies were performed to identify potential protein targets.
Keywords:
heterocyclization
; Michael addition
; dithiomalondiamides
; dithiolo[3
; 4-b]pyridines
; DFT calculations
; reaction mechanism studies
; herbicide safeners
; molecular docking
; ADMET properties.
1. Introduction
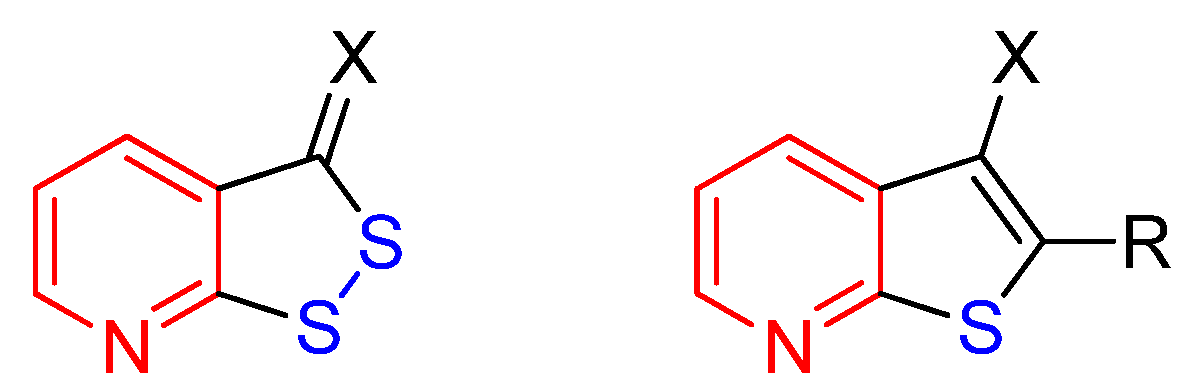
Due to unique properties, sulfur-containing heterocyclic compounds attract the interest of researchers. Thus, a number of derivatives of thiophene, 1,2-dithiol, thiazole, thiadiazole, thiopyran, thiadiazine, etc. have found application in medical chemistry, optoelectronics or as agrochemicals (for reviews see [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15]). The condensed heterocyclic system of [1,2]dithiolo[3,4-b]pyridine (Scheme 1) is of interest as a structural analog of other sulfur-containing heterocycles, thieno[2,3-b]pyridines. However, in contrast to the well studied thienopyridines (for reviews see [16,17,18,19,20,21]), only few papers have dealt with the synthesis and properties of [1,2]dithiolo[3,4-b]pyridines [22,23,24,25,26,27,28,29].
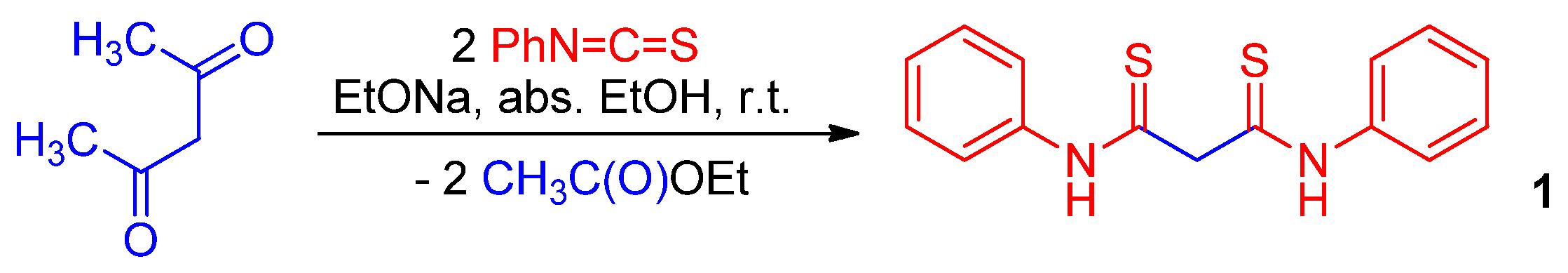
Recently, we have described the synthesis of functionalized [1,2]dithiolo[3,4-b]pyri- dines based on dithiomalondianilide 1 [30,31,32]. The starting thioamide 1 is readily available by the reaction of acetylacetone with phenyl isothiocyanate in the presence of sodium ethoxide [33,34] (Scheme 2).
Dithiomalondianilide 1 is actively used as an S,S- or S,N-bidentate complexing agent [35,36,37,38,39,40,41,42,43], lubricant [44,45] and corrosion inhibitor [46]. However, the synthetic potential of compound 1 as a reagent for heterocyclic chemistry has not yet been fully disclosed and heterocyclization reactions were reported in only few papers [47,48,49,50,51,52,53,54,55,56,57,58]. As cyclic disulfides, 1,2-dithiol derivatives are for special interest for biological studies since such disulfides are particularly prone to redox reactions (for chemistry and biological activity of disulfides see review papers [59,60,61,62,63]). Our interest in the chemistry of dithiomalondianilide and [1,2]dithiolo[3,4-b]pyridines has subsequently led us to examine the reaction of 1 with 2-cyanoacrylamides and to study the reaction mechanism in details.
2. Results and Discussion
2.1. Synthesis
We found that 3-aryl-2-cyanoacrylamides 2a-f react with dithiomalondianilide 1 in hot ethanol in the presence of morpholine to form the previously undescribed 6-amino-4-aryl-7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamides 3a-f in 37-54% yields (Scheme 3, Method A). Compounds 3 can also be prepared in a one-pot manner by reaction of aromatic aldehydes, cyanoacetamide and dithiomalondianilide 1 under similar conditions (Scheme 3, Method B); however, in this case the yields of products 3 are somewhat lower (22-43%, Table 1). The reaction proceeded smoothly in EtOH; other solvents such as i-PrOH or acetone gave rather unsatisfactory results. The nature of amine (morpholine, piperidine, triethylamine) does not significantly affect the yields of products. The prepared [1,2]dithiolo[3,4-b]pyridines 3a-f have chiral center C-4 and are racemic mixtures of (R)- and (S)-enantiomers.
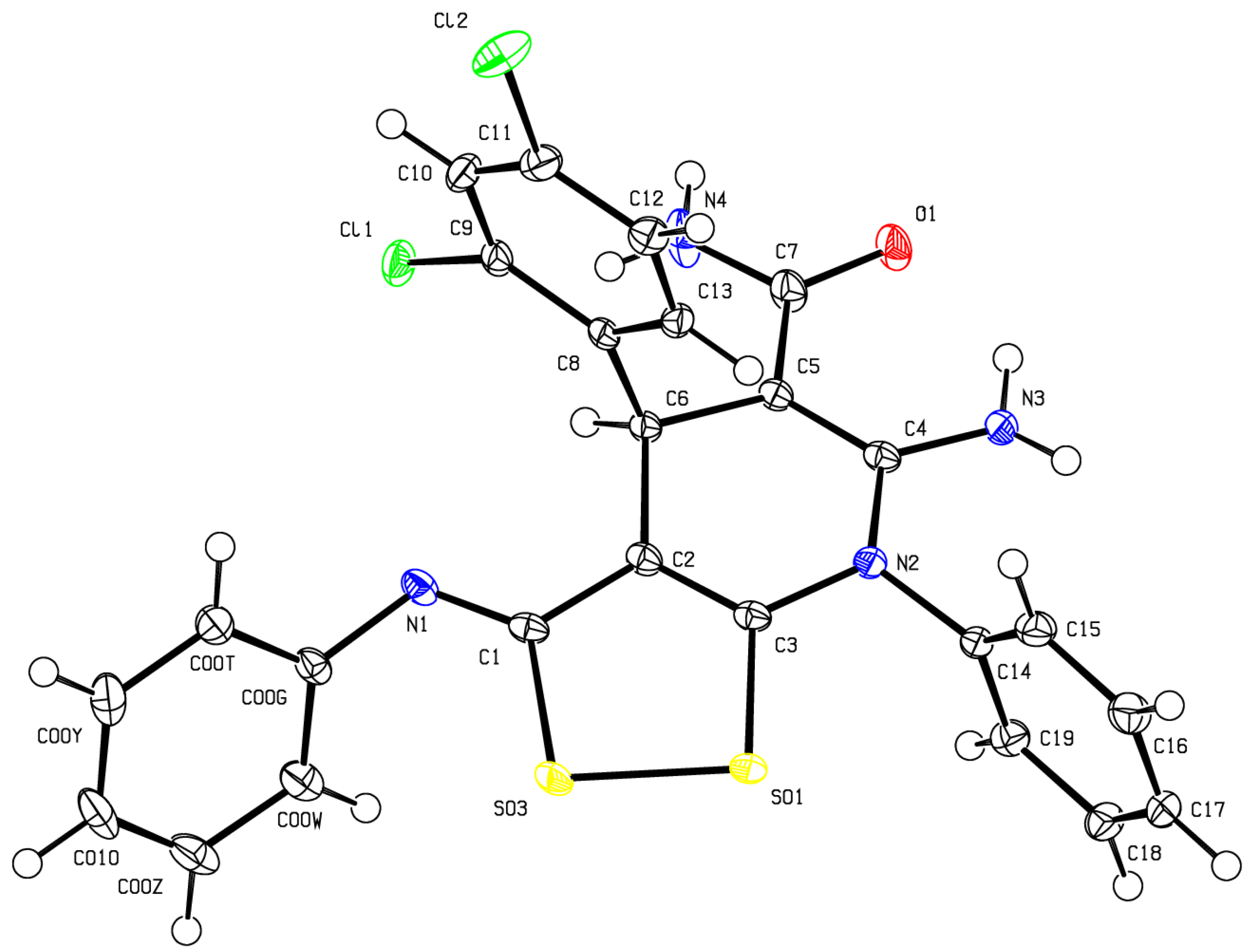
The structure of compounds 3 was was confirmed by FT-IR, HRMS and NMR spectroscopy (including 1H NMR, 13C DEPTQ, 1Н–1H COSY, 1Н–13С HSQC, 1Н–13С HMBC) (See Electronic Supplementary Material File). Also, the structure of 6-amino- 4-(2,4-dichlorophenyl)-7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamide 3d was confirmed by X-ray diffraction analysis (Figure 1).
Thus, the 1H NMR spectra of compounds 3 show characteristic signals of two non-equivalent phenyl groups, singlets of H-4 protons at δ 4.94-5.64 ppm, broadened peaks of NH2 (δ 6.26-6.63 ppm) and CONH2 (δ 7.30-7.58 ppm) protons. In the FTIR spectra of [1,2]dithiolo[3,4-b]pyridines 3, no C≡N absorption bands was observed, and the bands corresponding to amide C=O and C=N vibrations were detected. 13C NMR spectra revealed no C=S signals but C=N carbon signals appeared at δ = 162.1-163.3 ppm.
The compounds 3 are colored in shades of yellow (from canary yellow to mustard yellow), sparingly soluble in boiling acetone or ethyl acetate, soluble in DMF or DMSO, and insoluble in alcohols.
2.2. The studies of the reaction mechanism
In recent papers [30,31,32] we have described the preparation of [1,2]dithiolo[3,4-b]pyridines by reaction of dithiomalondianilide 1 with activated Michael substrates such as substituted acrylonitriles 4,5 (Scheme 4) but no detailed study of the reaction mechanism has been carried.
Here, we would like to consider a possible mechanism for the formation of the [1,2]dithiolo[3,4-b]pyridine ring system using the reaction of compound 1 with 2-cyanoacrylamides 2 as an example. It is obvious that an oxidizing agent is required for the successful formation of 1,2-dithiol ring. Two reagents, either air oxygen or an unsaturated nitrile, can play the role of oxidizing agent. In a number of studies concerning the synthesis of pyridine derivatives starting from unsaturated nitriles (e.g., arylmethylidene malononitriles 4) it was shown that partially saturated pyridine intermediate easily reacts with unsaturated nitriles to give α,β-saturated nitriles and corresponding oxidation products—nicotinonitriles [64,65,66,67,68] (Scheme 5):
It was reported that when unsaturated nitriles were taken in a two-fold excess, the yields of the target pyridines were increased sharply [67,68]. Although no formation of disulfides was observed in the above reactions, activated nitriles can potentially play the role of oxidizing agents for 1,2-dithiol ring closure reaction.
As a model reaction, we studied the reaction of 2-cyano-4-(methoxyphenyl)acryl- amide 2f with dithiomalondianilide 1 in the presence of morpholine in EtOH. Compound 2f was prepared beforehand by Knoevenagel reaction of 4-methoxybenzaldehyde with cyanoacetamide in the presence of morpholine in EtOH at 50 °C.
To elucidate the nature of the oxidizing agent, the reaction between 1 and 2f was carried out in four different variants:
- (1)
- under argon flow to exclude the effect of oxygen, at a ratio of 2f:1 = 2:1;
- (2)
- under air stream at the ratio 2f:1 = 1:1 (Table 1, entry 6);
- (3)
- under air atmosphere at a ratio of 2f:1 = 2:1;
- (4)
We found that in the reaction (1), only trace amounts of product 3f was detected by TLC and NMR after 3 hours. In the experiments (2) and (3) we observed the formation of product 3f precipitate, though in (3) TLC and HRMS showed the presence of considerable amounts of starting acrylamide 2f in the crude product. Finally, in the experiment (4) no formation of 3f was detected and the starting material remained unreacted. The yields of pure dithiolopyridine 3f in the experiments (2) and (3) were 54% and 55%%. Thus, no significant increase in the yields was observed when a twofold excess of acrylamide 2f was used. This fact indirectly negates the role of acrylamide 2f as an oxidant in this reaction. Overall, from the conducted experiments (1-4) we can conclude that a) air oxygen is the oxidizing agent in this reaction, and b) the formation of the pyridine ring precedes the oxidation to form the 1,2-dithiol fragment (since the product of dithiomalondianilide 1 oxidation, 1,2-dithiol 6 did not react with acrylamide 2f under reported conditions).
With these preliminary data in hands, we investigated the reaction mechanism in more detail. Quantum chemical study of the mechanism was performed using ORCA 5.0.4 software package [70,71]. The search of transition states, determination of reaction trajectories, calculations of vibrational frequencies and Gibbs free energy were examined through Density Functional Theory (DFT) with Grimme’s B97-3c composite calculation scheme [72,73] based on the combination of B97 GGA functional and def2-mTZVP basis set with the D3BJ dispersion correction [74]. Transition states were searched using relaxed scan and nudged elastic band (NEB) methods [75]. The found geometry of transition states was confirmed by the presence of an imaginary vibrational frequency corresponding to the reaction coordinate. All the calculations were performed with non-specific solvation using the СРСМ model (ethanol as the solvent) [76]. ChemCraft 1.8 software was used to visualize molecular geometry and vibrational frequencies. According to the results of quantum chemical studies, we proposed the mechanism of [1,2]dithiolo[3,4-b]pyridine system formation (Scheme 7).
As we can see, the entire bicyclic ring system formation process can be divided into two main stages: heterocyclization to form dihydropyridine ring and oxidative cyclization to form cyclic disulfide moiety. Since the reaction was carried out in the presence of excessive base (morpholine), we considered the model system of dithiomalondianilide 1 + the Michael acceptor (2-cyano-3-(4-methoxyphenyl)acrylamide 2f) + two molecules of morpholine at the first stage. An oxygen molecule was added to the model system in the second stage. In order to determine the optimal geometry of the initial state, the most stable conformation of dithiomalondianilide 1 was determined.
The first step of the cascade reaction mechanism involves deprotonation of the C42 atom (Scheme 7, Figure 2) of dithiomalondianilide 1 molecule followed by the Michael addition of resulting anion to the C55 atom of 2-cyano-3-(4-methoxyphenyl)acrylamide 2f. Next, deprotonation of N51 atom in the resulted Michael adduct I-2 leads to relatively stable dianion I-3. Then intramolecular nucleophilic attack at nitrile carbon atom C61 occurs to close a pyridine ring. This process proceeds synchronously with the protonation of С≡N nitrogen atom N64. Thus formed anion I-4 then undergoes secondary protonation at N64 atom to form intermediate I-5. The molecular structures of intermediates (I) and transition states (TS) are given in Figure 2 and Figure 3. The energy profile of this process is shown in Figure 4. See also gif animations of imaginary vibrations of transition states TS1-TS9 in the Supporting information.
The oxidation of intermediate I-5 (Scheme 7, Figure 3) involves initial deprotonation at C42 atom followed by addition of molecular oxygen at S54 atom to form a peroxysulfenic acid (intermediate I-6) (Figure 5). After deprotonation of CSNHPh nitrogen, an intramolecular nucleophilic substitution of hydroperoxide anion at S31 sulfur atom occurs to form disulfide bond of the final product 3f. In the process of nucleophilic substitution, the hydroperoxide anion is protonated to release hydrogen peroxide. The energy profile of this process is shown in Figure 6.
Overall, at the first stage of dihydropyridine ring formation the highest activation energy (28.8 kcal/mol) corresponds to the intramolecular cyclization occurred at cyano group. Apparently, this is the rate limiting step for the entire process since the highest activation barrier at the second stage (formation of disulfide bond/1,2-dithiol ring closure) is somewhat lower (23.6 kcal/mol).
2.3. ADMET calculations and molecular docking studies
Despite the relatively poor study of dithiolopyridines and their properties, it is known that some 3H-[1,2]dithiolo[3,4-b]pyridines showed antimycobacterial activity [27] and moderate antibacterial activity [22]. As close structural analogs/bioisosters of thieno[2,3-b]pyridines, [1,2]dithiolo[3,4-b]pyridines are of interest as promising candidates for bioscreening. The compounds 3a-f were evaluated for compliance with Lipinski’s “rule of five” [77], and ADMET parameters were estimated using the free online resources: SwissADME website (http://www.swissadme.ch/), GUSAR service (http://www.way2drug.com/gusar/acutoxpredict.html), OSIRIS Property Explorer service (https://www.organic-chemistry.org/prog/peo/), AdmetSAR website (http://lmmd.ecust.edu.cn/admetsar2).
The analysis for compliance with Lipinski’s “rule of five” is intended to predict drug-likeness and bioavailability for an oral therapeutic agent using physicochemical properties. The following parameters were calculated: cLogP (logarithm of the distribution coefficient between n-octanol and water, log(coctanol/cwater), solubility (log S), topological polar surface area (TPSA), a number of toxicological characteristics—risks of side effects (mutagenic, oncogenic, reproductive effects), similarity parameter with known drugs (drug-likeness), as well as a general assessment of the pharmacological potential of the compound (drug score). Since (R)- and (S)-stereoisomers of 3 revealed the same values in the in silico calculations, the results presented in the Table S12 (Electronic Supplementary Material file) apply equally to both enantiomers.
As we can see from the Table S12, for all compounds except 3b and 3d the cLogP values do not exceed 5.0 to indicate the expected good absorption and permeability [77]. However, the compounds 3 mostly do not fit the Lipinski’s rule of five in terms of molecular weight (MW ≥ 500 Da). Also the compounds 3 showed calculated poor solubility (logS < -4) and do not pass the Veber filter [78] (TPSA ≥ 140 Å2). The results of ADMET calculations are given in the Tables S13–S24 (Electronic Supplementary Material file). In general, good gastrointestinal absorption and blood-brain barrier permability were predicted for all the compounds 3a-f. However, the calculations revealed probable hepatotoxicity and mitochondrial toxicity. At the same time, low acute oral toxicity was predicted. The calculation using the GUSAR service assigns all compounds to categories 4 (LD50 >300–2000 mg/kg) or 5 (LD50 >2000–5000 mg/kg) according to OECD criteria [79]. Overall, compounds 3 do not meet certain oral availability criteria as lead molecules—e.g., poor solubility, some violations for Ghose filter (MW > 480, molar refractivityMR > 130), Veber filter (violation: TPSA > 140 Å2), Egan filter (violation: TPSA > 131.6 Å2), Muegge filter (violation: TPSA > 150 Å2), insaturation peremeter (fraction of Csp3 < 0.25), etc.
For both (R)- and (S)-enantiomers for each of 3H-[1,2]dithiolo[3,4-b]pyridines 3a-f, most likely protein targets were predicted using with the GalaxyWEB server (https://galaxy.seoklab.org/index.html) [80,81]. We used GalaxySagittarius-AF, a newest protein-ligand docking protocol drug-like compounds using AlphaFold models. The 3D structures of (R)- and (S)-enantiomers of compounds 3a-f were pre-optimized by molecular mechanics in the MM2 force field for geometry optimization and energy minimization. The molecular docking studies were performed in binding compatability prediction mode, using PDB + AlphaFold models and Re-ranking using docking mode. Table S25 (Supplementary materials file) shows the docking results for 25 target-ligand complexes for both isomers of 3a-f with the minimum binding free energy ΔGbind and the best protein-ligand interaction score. The predicted protein targets are marked using identifiers in the Protein Data Bank (PDB) and in the UniProt database.
As we can see from Table S25, for both (R)- and (S)-enantiomers of 3H-[1,2]dithiolo[3,4-b]pyridines 3a-f, a similar pool of protein targets is predicted but the scoring function values are different. The most common targets for 3H-[1,2]dithiolo[3,4-b]pyridines 3a-f are human trypsin 1 (PDB ID 1trn_A) with ΔGbind values ranging from -19.65 kcal/mol to -21.415 kcal/mol, urokinase-type plasminogen activator (PDB ID 3khv) with ΔGbind = -21.003…-24.09 kcal/mol, coagulation factor VII (PDB ID 4yt6_H) with ΔGbind = -18.771…-21.267 kcal/mol, focal adhesion kinase 1 (FAK1, PDB ID 4gu6_A) with ΔGbind = -23.786…-25.831 kcal/mol, proto-oncogene tyrosine-protein kinase LCK (PDB ID 3ac8_A) with ΔGbind = -21.315…-23.956 kcal/mol, serine/threonine-protein kinase B-raf (PDB ID 4mnf) with ΔGbind = -20.983…-24.744 kcal/mol, sterile alpha motif and leucine zipper containing kinase ZAK (PDB ID 7yaw) with ΔGbind = -23.539…-25.157 kcal/mol. One of the protein-ligand complexes was visualized using the GalaxySagittarius-AF software package and shown in Figure 7.
2.4. Agrochemical studies
Recently, some 3-aminothieno[2,3-b]pyridines [82,83,84] and related 3-imino-3H- [1,2]dithiolo[3,4-b]pyridines [32] were recognized as effective 2,4-D herbicide safeners and plant growth regulators.
2,4-D (2,4-dichlorophenoxyacetic acid) is a herbicide which is elatively low-toxic for humans and actively used for chemical weeding of cultivated plants [85]. Chemical weeding of crops is an important element in the protection of agricultural crops from weeds, because weeds reduce the yield of the most important crops by 15-25%. However, it should be noted that herbicides as plant-killing agents are toxic not only for weeds. This also applies to sunflower, for which widely used 2,4-dichlorophenoxyacetic acid is highly toxic. Thus, if the dose of 0.5-0.8 kg/ha on the active ingredient 2,4-D is recommended for weed control in crops of resistant cereals, for sunflower the dose as small as 15-18 g/ha of the active ingredient leads to a 40–60% reduction in the crop yield.
One possible solution to this problem is to use so-called herbicide antidotes, or herbicide safeners. Herbicide safeners (for reviewes see [86,87,88]) are agrochemicals that able to neutralize phytotoxins in plants by different mechanisms thus protecting crop plants from herbicide injury. Herbicide safeners are harmless to crop plants, but do not affect the activity of herbicides against weeds.
The compounds 3b, 3d and 3f as close structural analogs of known active antidotes [32,82,83,84] were examined as herbicide safeners on sunflower seedlings using the reported procedure [82]. The antidote effect A was determined as a ratio of hypocotyls (or roots) length of sunflower seedlings in the “herbicide + antidote” experiments to the length in the reference group (where the seedlings were treated with 2,4-D only) (Equation (1)):
where Lexp is an organ length (mm) in the group of seedlings treated with herbicide and antidote, and Lref is an organ length (mm) in the reference group of sunflower seedlings.
А = (Lexp/Lref)×100%,
We found that dithiolopyridines 3b, 3d and 3f exhibited weak to moderate 2,4-D antidote effect in the laboratory experiments (Table 2).
As we can see from the Table 2, 3H-[1,2]dithiolo[3,4-b]pyridines 3b, 3d and 3f as herbicide safeners reduced the negative effects of 2,4-D on sunflower seedling hypocotyls by 16-32% and by 16-33% on sunflower seedling roots.
3. Materials and Methods
1H and 13C DEPTQ NMR spectra and 2D NMR experiments were recorded in solutions of DMSO-d6 on a Bruker AVANCE-III HD (Bruker BioSpin AG, Fällanden, Switzerland) and Agilent 400/MR (Agilent Technologies, US) instruments (at 400 MHz for 1H and 101 MHz for 13C nuclei). Residual solvent signals were used as internal standards, in DMSO-d6 (2.49 ppm for 1H, and 39.50 ppm for 13C nuclei). Single crystal X-ray diffraction analysis of compound 3d was performed on an automatic four-circle diffractometer Agilent Super Nova, Dual, Cu at zero, Atlas S2. High-resolution mass spectra (HRMS) were registered with a Bruker MaXis Impact (Bruker Daltonics, Bremen, Germany) spectrometer (electrospray ionization, using HCO2Na–HCO2H for calibration). The samples were dissolved in MeCN under moderate heating (37-38 °C) and ultrasonication. See Electronic Supplementary Material file for NMR, FTIR, HRMS spectral charts and X-ray analysis data.
FT-IR spectra were measured on a Bruker Vertex 70 instrument (Bruker Optics GmbH & Co. KG, Ettlingen, Germany) equipped with an ATR sampling module. Elemental analyses were carried out using a Carlo Erba 1106 Elemental Analyzer (Carlo Erba Strumentazione, Cornaredo, Italy). Reaction progress and purity of isolated compounds were controlled by TLC on Sorbfil-A plates (Imid Ltd., Krasnodar, Russia), eluent—acetone:hexane 2:1 or ethyl acetate. Developed TLC plates were stained with UV-light and iodine vapors.
N,N′-Diphenyldithiomalondiamide (dithiomalondianilide) 1 was prepared from acetylacetone and phenyl isothiocyanate as reported [33,34]. (E)-3-Aryl-2-cyanoacrylamides 2a-f were prepared by reaction of 2-cyanoacetamide with aromatic aldehydes in the presence of catalytic amounts of morpholine in EtOH at 40-50 °С [89] or in water in the presence of surfactants [90]. All other reagents and solvents were purchased from commercial vendors (BioInLabs, Rostov-on-Don, Russia) and used as received.
General procedure for the preparation of 6-amino-4-aryl-7-phenyl-3-(phenyl- imino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamides 3a-f (Method A). A vial was charged, under air, with dithiomalondianilide 1 (500 mg, 1.75 mmol), 3-aryl-2-cyanoacrylamide 2a-f (1.75 mmol) and EtOH (15 mL). A mixture was treated with morpholine (0.23 mL, 1.75 mmol) at 40-50 °C (internal temperature) under vigorous stirring. Complete dissolution of starting materials and formation of a deep-yellow solution occurred for a very short time (a matter of minutes). Usually, a yellow precipitate begins to separate within half an hour. The reaction mixture was stirred for another 2-3 h (TLC control) and left to stand at room temperature until crystallization was complete. The precipitated product was filtered off, washed with EtOH and purified either by flash chromatography (silica gel, eluent–acetone) or by recrystallization from acetone or acetone–EtOH mixtures to give pure dithiolopyridines 3a-f as yellow crystalline solids.
General procedure for the preparation of 6-amino-4-aryl-7-phenyl-3-(phenyl- imino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamides 3a-f (Method B). Morpholine (0.23 mL, 1.75 mmol) was added to a mixture of 2-cyanoacetamide (150 mg, 1.75 mmol) and substituted benzaldehyde (1.75 mmol) in EtOH (6-8 mL). The reaction mixture was stirred at ~60 °C for 5 min, then dithiomalondianilide 1 (500 mg, 1.75 mmol) and EtOH (10 mL) were added. The resulted mixture was stirred at 40-50 °C for 2-3 h and monitored by TLC until the starting reagents were consumed. After the reaction was complete, the reaction mixture was left to stand at room temperature for complete crystallization. The precipitated product was filtered off, washed with EtOH and purified as described above to give pure dithiolopyridines 3a-f as yellow crystalline solids.
6-Amino-4-(2-nitrophenyl)-7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]-pyridine-5-carboxamide (3a). The yield was 54% (method A) or 43% (method B). 1H NMR (400 MHz, DMSO-d6): 5.64 (s, 1H, H-4), 6.63 (br s, 2H, NH2), 6.73 (d, 3J = 7.5 Hz, 2H, H-2 H-6 Ph), 7.03-7.07 (m, 1H, H-4 Ph), 7.28-7.32 (m, 2H, H-3 H-5 Ph), 7.44-7.49 (m, 1H, H-4 Ar), 7.58 (br s, CONH2), 7.64-7.69 (m, 5H, Ph), 7.78-7.83 (m, 3H, Ar). 13C NMR (101 MHz, DMSO-d6): 34.6 (C-4), 77.6 (C-5), 111.1 (C-3a), 120.0 (2C, C-2 C-6 Ph), 123.4 (C-3 Ar), 124.2 (C-4 Ph), 127.9 (C-4 Ar), 129.4 (2C, C-3 C-5 Ph), 130.2 (CH Ph), 130.4 (CH Ph), 131.0 (C-6 Ar), 134.2 (C-5 Ar), 135.5 (C-1 Ph), 140.0 (C-1 Ar), 148.5 (C-NO2), 150.5 (C-1 Ph), 152.0 (C-6), 154.4 (C-7a), 163.3 (C-3), 171.1 (CONH2). FTIR, νmax, cm-1: 3439, 3350, 3171, 3119, 3063 (N-H, C-H); 1668 (C=O); 1645 (C=N); 1533 (NO2 asym); 1357 (NO2 sym). HRMS (ESI) m/z: calculated for C25H20N5O3S2 [M+H]+: 502.1008, found 502.1023 (Δ 2.99 ppm).
6-Amino-4-(4-chlorophenyl)-7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamide (3b). The yield was 40% (method A) or 33% (method B). 1H NMR (400 MHz, DMSO-d6): 5.24 (s, 1H, H-4), 6.55 (br s, 2H, NH2), 6.84 (d, 3J = 7.5 Hz, 2H, H-2 H-6 Ph), 7.07-7.10 (m, 1H, H-4 Ph), 7.30-7.39 (m, 6H, H-3 H-5 Ph, 2H Ar, CONH2 overlapped), 7.43 (d, 3J = 8.3 Hz, 2H, H Ar), 7.60–7.67 (m, 5H, Ph). 13C NMR (101 MHz, DMSO-d6): 38.6 (C-4), 78.0 (C-5), 112.4 (C-3a), 120.0 (2C, C-2 C-6 Ph), 124.2 (C-4 Ph), 127.8 (2C, CH Ar), 129.5 (2C, CH Ar), 129.7 (2C, C-3 C-5 Ph), 130.1 (CH Ph), 130.5 (CH Ph), 131.0 (CH Ph), 135.4 (C–Cl), 135.6 (C-1 Ph), 146.6 (C-1 Ar), 150.8 (C-1 Ph), 152.0 (C-6), 154.5 (C-7a), 163.1 (C-3), 171.3 (CONH2). FTIR, νmax, cm-1: 3406, 3300, 3151, 3055 (N-H, C-H); 1662 (C=O, C=N). HRMS (ESI) m/z: calculated for C25H20ClN4OS2 [M+H]+: 491.0767, found 491.0780 (Δ 2.65 ppm).
6-Amino-4-(3-nitrophenyl)-7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamide (3c). The yield was 37% (method A) or 27% (method B). 1H NMR (400 MHz, DMSO-d6): 5.32 (s, 1H, H-4), 6.58 (br s, 2H, NH2), 6.89 (d, 3J = 7.5 Hz, 2H, H-2 H-6 Ph), 7.06-7.10 (m, 1H, H-4 Ph), 7.32-7.36 (m, 2H, H-3 H-5 Ph), 7.40 (br s, CONH2), 7.58-7.67 (m, 6H, H-5 Ar + Ph), 8.01 (br d, 3J = 7.5 Hz, H-6 Ar), 8.08 (dd, 3J = 8.2 Hz, 3J = 1.4 Hz, H-4 Ar), 8.58-8.59 (m, 1H, H-2 Ar). 13C NMR (101 MHz, DMSO-d6): 38.7 (C-4), 78.7 (C-5), 111.2 (C-3a), 120.0 (2C, C-2 C-6 Ph), 121.3 (С-4 Ar), 122.6 (C-2 Ar), 124.3 (C-4 Ph), 129.4 (С-5 Ar), 129.7 (2C, C-3 C-5 Ph), 130.0 (CH Ph), 130.4 (CH Ph), 130.9 (CH Ph), 134.1 (C-6 Ar), 135.7 (C-1 Ph), 147.3 (С-1 Ar), 148.0 (C-NO2), 150.9 (C-1 Ph), 151.4 (C-6), 154.7 (C-7a), 163.2 (C-3), 171.2 (CONH2). FTIR, νmax, cm-1: 3493, 3458, 3444, 3402, 3342, 3211, 3057 (N-H, C-H); 1647 (C=O, C=N), 1575 (NO2 asym); 1348 (NO2 sym). HRMS (ESI) m/z: calculated for C25H20N5O3S2 [M+H]+: 502.1008, found 502.1023 (Δ 2.99 ppm).
6-Amino-4-(2,4-dichlorophenyl)-7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamide (3d). The yield was 39% (method A) or 31% (method B). According to NMR, after recrystallization from acetone–EtOH mixture the product appeared as solvate 3d:EtOH = 2:1. 1H NMR (400 MHz, DMSO-d6): 1.05 (t, 3J = 6.8 Hz, 3H, EtOH), 3.43-3.46 (m, 2H, EtOH), 4.33 (t, 3J = 5.0 Hz, 1H, EtOH), 5.31 (s, 1H, H-4), 6.26 (br s, 2H, NH2), 6.79 (d, 3J = 7.5 Hz, 2H, H-2 H-6 Ph), 7.02-7.06 (m, 1H, H-4 Ph), 7.28-7.31 (m, 4H, H-3 H-5 Ph, CONH2), 7.39 (dd, 3J = 8.2 Hz, 4J = 1.7 Hz, H-5 Ar), 7.48 (d, 4J = 1.7 Hz, H-3 Ar), 7.60-7.64 (m, 5H, Ph), 7.73 (d, 3J = 8.2 Hz, H-6 Ar). 13C NMR (101 MHz, DMSO-d6): 18.5 (CH3 EtOH), 38.7 (C-4), 56.0 (CH2 EtOH), 77.9 (C-5), 109.8 (C-3a), 120.0 (2C, C-2 C-6 Ph), 124.1 (C-4 Ph), 126.9 (C-5 Ar), 128.6 (C-3 Ar), 129.5 (2C, C-3 C-5 Ph), 130.29 (CH Ph), 130.32 (CH Ph), 130.9 (CH Ph), 131.4 (C-4 Ar), 133.0 (C–Cl Ar), 133.9 (C-6 Ar), 135.6 (C-1 Ph), 141.4 (C-1 Ar), 150.3 (C-1 Ph), 151.0 (C-6), 154.9 (C-7a), 162.1 (C-3), 171.3 (CONH2). FTIR, νmax, cm-1: 3473, 3363, 3333, 3144, 3051 (N-H, C-H); 1707 (C=O), 1633 (C=N). HRMS (ESI) m/z: calculated for C25H18Cl2N4NaOS2 [M+Na]+: 547.0197, found 547.0174 (Δ 4.2 ppm).
6-Amino-4-(3,4-dimethoxyphenyl)-7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamide (3e). The yield was 40% (method A) or 22% (method B). 1H NMR (400 MHz, DMSO-d6): 3.79 (s, 3Н, MeO–Ar), 3.84 (s, 3Н, MeO–Ar), 5.00 (s, 1H, H-4), 6.30 (br s, 2H, NH2), 6.82–6.90 (m, 5H, H Ph, H Ar), 7.06-7.10 (m, 1H, H-4 Ph), 7.28-7.33 (m, 4H, H-3 H-5 Ph, CONH2), 7.55–7.66 (m, 5H, H Ar).13C NMR (101 MHz, DMSO-d6): 38.0 (C-4), 54.9 (ArOCH3), 55.0 (ArOCH3), 79.1 (C-5), 110.8 (C-3a), 111.2 (CH Ar), 112.0 (CH Ar), 118.9 (CH Ar), 120.1 (2C, C-2 C-6 Ph), 124.3 (C-4 Ph), 129.6 (2C, C-3 C-5 Ph), 129.9 (2C, CH Ph), 130.4 (2C, CH Ph), 130.9 (C-4 Ph), 135.5 (C-1 Ph), 137.2 (C-1 Ar), 147.7 (C–OMe), 148.3 (C–OMe), 150.4 (C-1 Ph), 151.5 (C-6), 154.0 (C-8a), 163.0 (C=N), 171.1 (C=O). FTIR, νmax, cm-1: 3475, 3388, 3267, 3178, 3047 (N-H, C-H); 1668 (C=O), 1633 (C=N). Elemental Analysis (С27Н24N4O3S2, M 516,63): calculated (%): C, 62.77; H, 4.68; N, 10.84; found (%): C, 62.57; H, 4.90; N, 10.79.
6-Amino-4-(4-methoxyphenyl)-7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamide (3f). The yield was 54% (method A) or 35% (method B). 1H NMR (400 MHz, DMSO-d6): 3.74 (s, 3Н, MeO–Ar), 4.94 (s, 1H, H-4), 6.26 (br s, 2H, NH2), 6.81 (d, 3J = 7.5 Hz, 2H, H-2 H-6 Ph), 6.93 (d, 3J = 8.4 Hz, 2H, H-Ar), 7.04-7.08 (m, 1H, H-4 Ph), 7.29-7.37 (m, 6H, H-3 H-5 Ph, H Ar, CONH2), 7.55–7.63 (m, 5H, H Ar). 13C DEPTQ NMR (101 MHz, DMSO-d6): 37.9 (C-4), 54.9 (OCH3), 78.0 (C-5), 113.0 (C-3a), 113.5 (2C, CH Ar), 120.1 (2C, C-2 C-6 Ph), 124.3 (C-4 Ph), 128.0 (2C, CH Ar), 129.5 (2C, C-3 C-5 Ph), 130.0 (2C, CH Ph), 130.3 (2C, CH Ph), 131.0 (C-4 Ph), 135.5 (C-1 Ph), 138.9 (C-1 Ar), 150.7 (C-1 Ph), 152.0 (C-6), 153.9 (C-8a), 157.5 (C–OMe), 163.1 (C=N), 171.1 (C=O). HRMS (ESI) m/z: calculated for C26H22N4O2S2 [M+H]+: 487.1262, found 487.1276 (Δ 2.87 ppm).
X-ray studies for single crystals of 3d.
Single crystals of 6-amino-4-(2,4-dichlorophenyl)-7-phenyl-3-(phenylimino)- 4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamide 3d (C25H18Cl2N4OS2, M 525.45 g/mol) were prepared by slow evaporation of saturated solution in DMSO. The crystal was kept at 100.01(10) K during data collection. Using Olex2 [91], the structure was solved with the olex2.solve structure solution program using Charge Flipping and refined with the SHELXL [92] package using Gauss-Newton minimisation.
The crystals are monoclinic, space group I2/a (no. 15), at 100.01(11) K: a = 11.97350(10) Å, b = 12.96920(10) Å, c = 34.4739(2) Å, α = 90°, β = 90.7890(10)°, γ = 90°, V = 5352.83(7) Å3, Z = 8, μ(Cu Kα) = 3.837 mm-1, Dcalc = 1.304 g/cm3, F(000) = 2160.0, 29542 reflections measured (7.282° ≤ 2Θ ≤ 153.23°), 5572 unique (Rint = 0.0263, Rsigma = 0.0158) which were used in all calculations. The final R1 was 0.0352 (I > 2σ(I)) and wR2 was 0.0939 (all data).
A full set of crystallographic data has been deposited in the Cambridge Crystallographic Data Center (CCDC 2310349).
Herbicide safening effect studies.
Germinated sunflower seeds (cv. Master) with 2-4 mm long embryo roots were placed in a solution of 2,4-D (10–3% by weight) for 1 h to achieve 40‒60% inhibition of hypocotyls growth. After treatment the seedlings were washed with pure water and placed into a solution of the corresponding compound 3b, 3d and 3f (concentrations 10–2, 10–3, 10–4 or 10–5% by weight, “herbicide + antidote” experiments). After 1 h the seedlings were washed with pure water and placed on paper stripes (10×75 cm, 20 seeds per stripe). The stripes were rolled and placed into beakers with water (50 cm3). The reference group of seedlings (“herbicide” experiments) was kept in 2,4-D solution (10–3%) for 1 h and then in water for 1 h. The “control” seedlings were kept in water for 2 h. The temperature of all solutions was maintained at 28 °C. The seedlings were then thermostated for 3 days at 28 °С. Each experiment was performed in triplicate, 20 seeds were used in each experiment. The results are given in Table 2.
4. Conclusions
In summary, we have synthesized new [1,2]dithiolo[3,4-b]pyridine-5-carboxamides by base-promoted reaction of dithiomalondianilide with 3-aryl-2-cyanoacrylamides. The structure of the compounds was confirmed by spectral data and X-ray diffraction analysis. The mechanism of formation of dithiolopyridine bicyclic ring system was examined in detail through DFT calculations with Grimme’s B97-3c composite scheme based on the combination of B97 GGA functional and def2-mTZVP basis set with the D3BJ dispersion correction. It was shown that the rate-limiting step is the closure of pyridine ring, whereas further oxidation and closure of 1,2-dithiol ring with air oxygen has a lower energy barrier.
The calculation of ADMET parameters for the new compounds was carried out. It was shown that dithiolopyridines 3 do not fully meet the criteria of oral bioavailability. The results of molecular docking studies and search for possible protein targets for new new [1,2]dithiolo[3,4-b]pyridine-5-carboxamides show that the compounds are of interest as promising antitumor agents or as regulators of blood coagulation factor VII. Finally, in contrast to close structural analogs, [1,2]dithiolo[3,4-b]pyridine-5-carboxamides 3 were found to exhibit only moderate herbicide safening effects against 2,4-D in the laboratory experiments with sunflower seedlings.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
V.V.D.—conceptualization, supervision, investigation, data analysis, funding acquisition, writing (original draft, review and editing); A.V.B.—conceptualization, quantum-chemical studies, writing—review and editing; A.E.S.—investigation; A.Z.T.—data analysis; V.K.V.—data analysis; E.A.V.—investigation; V.D.S.—agrochemical studies; N.A.A.—data analysis; I.V.A.—supervision, data analysis. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, grant number 22-23-00458, https://rscf.ru/en/project/22-23-00458/.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
File Electronic Supplementary Material.pdf containing X-Ray crystallography data, 1H and 13C NMR, 2D NMR 1H-13C HSQC and 1H-13C HMBC, FTIR, HRMS spectral charts (Figures S1–S22, Tables S1–S25).
Acknowledgments
This work was carried out using the equipment of the Common Use Center ‘Diagnostics of Nanomaterials Structure and Properties’ and ‘Environmental Analysis Center’, Kuban State University, Krasnodar.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Devendar, P.; Yang, G.F. Sulfur-containing agrochemicals. Top. Curr. Chem. 2017, 375, 82. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Jiang, X. Synthesis and perspective of organosulfur chemicals in agrochemicals. Advanced Agrochem. 2023, 2, 3–14. [Google Scholar] [CrossRef]
- Pathania, S.; Narang, R.K.; Rawal, R.K. Role of sulphur-heterocycles in medicinal chemistry: An update. Eur. J. Med. Chem. 2019, 180, 486–508. [Google Scholar] [CrossRef] [PubMed]
- Sosnovskikh, V.Y. Synthesis and properties of 2,3-heteroannulated thiochromones-hetero analogs of thioxanthone. Chem. Heterocycl. Compd. 2019, 55, 103–125. [Google Scholar] [CrossRef]
- Taubert, K.; Kraus, S.; Schulze, B. Isothiazol-3(2H)-Ones, Part I: Synthesis, reactions and biological activity. J. Sulfur Chem. 2002, 23, 79–121. [Google Scholar] [CrossRef]
- Kletskov, A.V.; Bumagin, N.A.; Zubkov, F.I.; Grudinin, D.G.; Potkin, V.I. Isothiazoles in the design and synthesis of biologically active substances and ligands for metal complexes. Synthesis 2020, 52, 159–188. [Google Scholar] [CrossRef]
- Shafran, Y.; Glukhareva, T.; Dehaen, W.; Bakulev, V. Recent developments in the chemistry of 1,2,3-thiadiazoles. Adv. Heterocycl. Chem. 2018, 126, 109–172. [Google Scholar]
- Podlesný, J.; Bureš, F. Thienothiophene scaffolds as building blocks for (opto)electronics. Organics 2022, 3, 446–469. [Google Scholar] [CrossRef]
- Larionova, N.A.; Shestopalov, A.M.; Rodinovskaya, L.A.; Zubarev, A.A. Synthesis of biologically active heterocycles via a domino sequence involving an SN2/Thorpe–Ziegler Reaction Step. Synthesis 2022, 54, 217–245. [Google Scholar] [CrossRef]
- Konovalov, A.I.; Antipin, I.S.; Burilov, V.A.; Madzhidov, T.I.; Kurbangalieva, A.R.; Nemtarev, A.V.; Solovieva, S.E.; Stoikov, I.I.; Mamedov, V.A.; Zakharova, L.Ya.; Gavrilova, E.L.; Sinyashin, O.G.; Balova, I.A.; Vasilyev, A.V.; Zenkevich, I.G.; Krasavin, M.Yu.; Kuznetsov, M.A.; Molchanov, A.P.; Novikov, M.S.; Nikolaev, V.A.; Rodina, L.L.; Khlebnikov, A.F.; Beletskaya, I.P.; Vatsadze, S.Z.; Gromov, S.P.; Zyk, N.V.; Lebedev, A.T.; Lemenovskii, D.A.; Petrosyan, V.S.; Nenaidenko, V.G.; Negrebetskii, V.V.; Baukov, Yu.I.; Shmigol’, T.A.; Korlyukov, A.A.; Tikhomirov, A.S.; Shchekotikhin, A.E.; Traven’, V.F.; Voskresenskii, L.G.; Zubkov, F.I.; Golubchikov, O.A.; Semeikin, A.S.; Berezin, D.B.; Stuzhin, P.A.; Filimonov, V.D.; Krasnokutskaya, E.A.; Fedorov, A.Yu.; Nyuchev, A.V.; Orlov, V.Yu.; Begunov, R.S.; Rusakov, A.I.; Kolobov, A.V.; Kofanov, E.R.; Fedotova, O.V.; Egorova, A.Yu.; Charushin, V.N.; Chupakhin, O.N.; Klimochkin, Yu.N.; Osyanin, V.A.; Reznikov, A.N.; Fisyuk, A.S.; Sagitullina, G.P.; Aksenov, A.V.; Aksenov, N.A.; Grachev, M.K.; Maslennikova, V.I.; Koroteev, M.P.; Brel’, A.K.; Lisina, S.V.; Medvedeva, S.M.; Shikhaliev, Kh.S.; Suboch, G.A.; Tovbis, M.S.; Mironovich, L.M.; Ivanov, S.M.; Kurbatov, S.V.; Kletskii, M.E.; Burov, O.N.; Kobrakov, K.I.; Kuznetsov, D.N. Modern Trends of Organic Chemistry in Russian Universities. Russ. J. Org. Chem. 2018, 54, 157–371. [Google Scholar]
- Rakitin, O.A. Synthesis and reactivity of 3H-1, 2-dithiole-3-thiones. Molecules 2021, 26, 3595. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Aggarwal, N.; Marwaha, M.G.; Deep, A.; Chopra, H.; Matin, M.M.; Roy, A.; Emran, T.B.; Mohanta, Y.K.; Ahmed, R.; Mohanta, T.K.; Saravanan, M.; Marwaha, R.K.; Al-Harrasi, A. Thiazolidin-2,4-Dione Scaffold: An Insight into Recent Advances as Antimicrobial, Antioxidant, and Hypoglycemic Agents. Molecules 2022, 27, 6763. [Google Scholar] [CrossRef] [PubMed]
- Shainyan, B.A.; Zhilitskaya, L.V.; Yarosh, N.O. Synthetic Approaches to Biologically Active C-2-Substituted Benzothiazoles. Molecules 2022, 27, 2598. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, H.; Suárez, M.; Albericio, F. Thiadiazines, N, N-heterocycles of biological relevance. Molecules 2012, 17, 7612–7628. [Google Scholar] [CrossRef] [PubMed]
- Dotsenko, V.V.; Frolov, K.A.; Krivokolysko, S.G. Synthesis of partially hydrogenated 1,3,5-thiadiazines by Mannich reaction. Chem. Heterocycl. Comp. 2015, 51, 109–127. [Google Scholar] [CrossRef]
- Litvinov, V.P. The chemistry of 3-cyanopyridine-2(1H)-chalcogenones. Russ. Chem. Rev. 2006, 75, 577–599. [Google Scholar] [CrossRef]
- Bakhite, E.A.-G. Recent trends in the chemistry of thienopyridines. Phosphorus, Sulfur, Silicon Relat. Elem. 2003, 178, 929–992. [Google Scholar] [CrossRef]
- Litvinov, V.P.; Dotsenko, V.V.; Krivokolysko, S.G. Thienopyridines: synthesis, properties, and biological activity. Russ. Hcem. Bull. Int. Ed. 2005, 54, 864–904. [Google Scholar] [CrossRef]
- Litvinov, V.P.; Dotsenko, V.V.; Krivokolysko, S.G. The chemistry of thienopyridines. Adv. Heterocycl. Chem. 2007, 93, 117–178. [Google Scholar]
- Sajadikhah, S.S.; Marandi, G. Recent approaches to the synthesis of thieno[2,3-b]pyridines (microreview). Chem. Heterocycl. Compd. 2019, 55, 1171–1173. [Google Scholar] [CrossRef]
- Dotsenko, V.V.; Buryi, D.S.; Lukina, D.Yu.; Krivokolysko, S.G. Recent advances in the chemistry of thieno[2,3-b]pyridines 1. Methods of synthesis of thieno[2,3-b]pyridines. Russ. Chem. Bull. Int. Ed. 2020, 69, 1829–1858. [Google Scholar] [CrossRef]
- Pregnolato, M.; Terreni, M.; Ubiali, D.; Pagani, G.; Borgna, P.; Pastoni, F.; Zampollo, F. 3H-[1,2]Dithiolo[3,4-b]pyridine-3-thione and its derivatives. Synthesis and antimicrobial activity. Il Farmaco 2000, 55, 669–679. [Google Scholar] [CrossRef]
- Salvetti, R.; Martinetti, G.; Ubiali, D.; Pregnolato, M.; Pagani, G. 1,2-Dithiolan-3-ones and derivatives structurally related to leinamycin. Synthesis and biological evaluation. Il Farmaco 2003, 58, 995–998. [Google Scholar] [CrossRef]
- Martinez-Merino, V.; Gil, M.J.; Gonzalez, A.; Zabalza, J.M.; Navarro, J.; Manu, M.A. New 5-substituted derivatives of ethyl 2,3-dihydro-3-oxoisothiazolo[5,4-b]pyridine-2-acetate. Heterocycles 1994, 38, 333–344. [Google Scholar] [CrossRef]
- Borgna, P.; Pregnolato, M.; Invernizzi, A.G.; Mellerio, G. On the reaction between 3H-1,2-dithiolo[3,4-b]pyridine-3-thione and primary alkyl and arylalkylamines. J. Heterocycl. Chem. 1993, 30, 1079–1084. [Google Scholar] [CrossRef]
- Davis, R.C.; Grinter, T.J.; Leaver, D.; O’Neil, R.M.; Thomson, G.A. The dithiole series. Part 8. Synthesis of ring-fused 1,2-dithiolylium and isothiazolium salts from complexes containing cyclopalladated ligands. J. Chem. Soc. Perkin Trans. 1 1990, 2881–2887. [Google Scholar] [CrossRef]
- Pagani, G.; Pregnolato, M.; Ubiali, D.; Terreni, M.; Piersimoni, C.; Scaglione, F.; Fraschini, F.; Rodríguez Gascón, A.; Pedraz Muñoz, J.L. Synthesis and in vitro anti-mycobacterium activity of N-alkyl-1,2-dihydro-2-thioxo-3-pyridinecarbothioamides. Preliminary toxicity and pharmacokinetic evaluation. J. Med. Chem. 2000, 43, 199–204. [Google Scholar] [CrossRef]
- Ubiali, D.; Pagani, G.; Pregnolato, M.; Piersimoni, C.; Pedraz Muñoz, J.L.; Rodríguez Gascón, A.; Terreni, M. New N-Alkyl-1,2-dihydro-2-thioxo-3-pyridinecarbothioamides as antituberculous agents with improved pharmacokinetics. Bioorg. Med. Chem. Lett. 2002, 12, 2541–2544. [Google Scholar] [CrossRef]
- Ogurtsov, V.A.; Karpychev, Y.V.; Rakitin, O.A. Synthesis of 1-[(1,2-dithiol-3-ylidene)methyl]pyrrolo[1,2-a]pyrazines and 2-[(1,2-dithiol-3-ylidene)methyl]pyridines from 1,2-dithiole-3-thiones. Russ. Chem. Bull., Int. Ed. 2013, 62, 1076–1079. [Google Scholar] [CrossRef]
- Dotsenko, V.V.; Krivokolysko, S.G.; Frolov, K.A.; Chigorina, E.A.; Polovinko, V.V.; Dmitrienko, A.O.; Bushmarinov, I.S. Synthesis of [1,2]dithiolo[3,4-b]pyridines via the reaction of dithiomalondianilide with arylmethylidenemalononitriles. Chem. Heterocycl. Comp. 2015, 51, 389–392. [Google Scholar] [CrossRef]
- Dotsenko, V.V.; Aksenov, A.V.; Sinotsko, A.E.; Varzieva, E.A.; Russkikh, A.A.; Levchenko, A.G.; Aksenov, N.A.; Aksenova, I.V. The reactions of N,N′-diphenyldithiomalondiamide with arylmethylidene Meldrum’s acids. Int. J. Mol. Sci. 2022, 23, 15997. [Google Scholar] [CrossRef]
- Dotsenko, V.V.; Sinotsko, A.E.; Strelkov, V.D.; Varzieva, E.A.; Russkikh, A.A.; Levchenko, A.G.; Temerdashev, A.Z.; Aksenov, N.A.; Aksenova, I.V. Alkyl 4-aryl-6-amino-7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxy- lates: Synthesis and Agrochemical Studies. Molecules 2023, 28, 609. [Google Scholar] [CrossRef]
- Sinotsko, A.E.; Bespalov, A.V.; Pashchevskaya, N.V.; Dotsenko, V.V.; Aksenov, N.A.; Aksenova, I.V. N,N′-Diphenyldithiomalonodiamide: Structural Features, Acidic Properties, and In Silico Estimation of Biological Activity. Russ. J. Gen. Chem. 2021, 91, 2136–2150. [Google Scholar] [CrossRef]
- Barnikow, G.; Kath, V.; Richter, D. Isothiocyanate. II. N,N′-Aryl-substituierte Dithiomalonsäurediamide. J. Prakt. Chem. 1965, 30, 63–66. [Google Scholar] [CrossRef]
- Barnikow, G.; Kunzek, H. Nickel-Chelate von N,N′-Diaryl-dithiomalonsäure-diamiden. Z. Chem. 1966, 6, 343–343. [Google Scholar] [CrossRef]
- Peyronel, G.; Pellacani, G.C.; Benetti, G.; Pollacci, G. Nickel(II) complexes with dithiomalonamide and NN′-diphenyldithiomalonamide. J. Chem. Soc. Dalton Trans. 1973, 879–882. [Google Scholar] [CrossRef]
- Pellacani, G.C. Palladium(II) complexes with dithiomalonamide and N,N′-diphenyldithiomalonamide. Can. J. Chem. 1974, 52, 3454–3458. [Google Scholar] [CrossRef]
- Pellacani, G.C.; Peyronel, G.; Pollacci, G.; Coronati, R. Zinc(II) complexes of dithiomalonamide, N,N′-dimethyl- and N,N′-diphenyl-dithiomalonamide: ZnLX2 (X = Cl, Br, I) and ZnL2(ClO4)2. J. Inorg. Nucl. Chem. 1976, 38, 1619–1621. [Google Scholar] [CrossRef]
- Pellacani, G.C.; Peyronel, G.; Malavasi, W.; Menabue, L. Antimony and bismuth trihalide complexes of dithiomalonamide, N,N′-dimethyl- and N,N′-diphenyl-dithiomalonamide. J. Inorg. Nucl. Chem. 1977, 39, 1855–1857. [Google Scholar] [CrossRef]
- Pal, T.; Ganguly, A.; Maity, D.S.; Livingstone, S.E. N,N′-diphenyldithiomalonamide as a gravimetric reagent for nickel and cobalt. Talanta 1986, 33, 973–977. [Google Scholar] [CrossRef]
- Pal, T.; Ganguly, A.; Pal, A. Spectrophotometric determination of cobalt and nickel with N,N′-diphenyldithiomalonamide and elucidation of structures of the compounds. J. Ind. Chem. Soc. 1988, 65, 655–657. [Google Scholar]
- Battaglia, L.P.; Bonamartini Corradi, A.; Marzotto, A.; Menabue, L.; Pellacani, G.C. Nickel(II) and palladium(II) complexes of dithiomalonamides: Ligands which favor the formation of π-conjugation systems in the coordination. J. Crystallogr. Spectrosc. Res. 1988, 18, 101–112. [Google Scholar] [CrossRef]
- Battaglia, L.P.; Bonamartini Corradi, A.; Marzotto, A.; Menabue, L.; Pellacani, G.C. Co-ordinative abilities of ligands which favour S,S chelation: copper(I) halide complexes of N,N′-diphenyldithiomalonamide. The crystal and molecular structure of bis(N,N′-diphenyldithiomalonamide)copper(I) iodide–methanol (2/1). J. Chem. Soc. Dalton Trans 1988, 1713–1718. [Google Scholar] [CrossRef]
- Singh, T.; Singh, R.; Verma, V.K. Evaluation of 2,4-dithiomalonamides as extreme pressure additives in the four-ball test. Proc. Inst. Mech. Eng., Part D: J. Automob. Eng. 1990, 204, 103–107. [Google Scholar] [CrossRef]
- Singh, T. Tribochemistry and EP activity assessment of Mo-S complexes in lithium-base greases. Adv. Tribol. 2008, 2008, 947543. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, M.M. Substituted dithiomalonamides as inhibitor for the corrosion of AISI 304SS in phosphoric acid – hydrochloric acid mixture. Anti-Corros. Methods Mater. 1993, 40, 4–7. [Google Scholar] [CrossRef]
- Schmidt, U. Synthesen mit den Thioamiden der Malonsäure, I. 3,5-Diamino-dithiyliumsalze. Ein neuer pseudoaromatischer Fünfring mit 1,2-Stellung der S-Atome. Chem. Ber. 1959, 92, 1171–1176. [Google Scholar] [CrossRef]
- Barnikow, G. Isothiocyanate, XIV. 3.5-Bis-arylamino-1.2-dithioliumsalze aus N.N′-Diaryl-dithiomalonsäure-diamiden. Chem. Ber. 1967, 100, 1389–1393. [Google Scholar] [CrossRef]
- Nizovtseva, T.V.; Komarova, T.N.; Nakhmanovich, A.S.; Larina, L.I.; Lopyrev, V.A.; Kalistratova, E.F. Reaction of Dithiomalonamide and Dianilide with α-Acetylene Ketones. Russ. J. Org. Chem. 2002, 38, 1205–1207. [Google Scholar] [CrossRef]
- Nizovtseva, T.V.; Komarova, T.N.; Nakhmanovich, A.S.; Larina, L.I.; Lopyrev, V.A. Synthesis of 1,3-dithiinium salts. Arkivoc 2003, xiii, 191–195. [Google Scholar] [CrossRef]
- Elokhina, V.N.; Yaroshenko, T.I.; Nakhmanovich, A.S.; Larina, L.I.; Amosova, S.V. Reaction of dithiomalonic acid dianilide with substituted acetylenic ketones. Russ. J. Gen. Chem. 2006, 76, 1916–1918. [Google Scholar] [CrossRef]
- Volkova, K.A.; Nakhmanovich, A.S.; Elokhina, V.N.; Yaroshenko, T.I.; Larina, L.I.; Shulunova, A.M.; Amosova, S.V. Reaction of dithiomalonic acid dianilide with methyl propiolate. Russ. J. Org. Chem. 2007, 43, 768–770. [Google Scholar] [CrossRef]
- Obydennov, K.L.; Golovko, N.A.; Kosterina, M.F.; Pospelova, T.A.; Slepukhin, P.A.; Morzherin, Yu.Yu. Russ. Chem. Bull. Int Ed. 2014, 63, 1330–1336. [CrossRef]
- Beckert, R.; Gruner, M. Regioselektive Cyclisierung von Thiocarbonsäureamiden mit Bisimidoylchloriden – Synthese von Thiazolidinen mit einer Ketenacetal-Substruktur. Z. Naturforsch. B: Chem. Sci. 1997, 52, 1245–1250. [Google Scholar] [CrossRef]
- Barnikow, G. Isothiocyanate, X. Pyrazole und Thiophene aus N.N′-Diaryl-dithiomalonsäure-diamiden. Liebigs Ann. Chem. 1966, 700, 46–49. [Google Scholar] [CrossRef]
- Degorce, S.; Jung, F.H.; Harris, C.S.; Koza, P.; Lecoq, J.; Stevenin, A. Diversity-orientated synthesis of 3,5-bis(arylamino)pyrazoles. Tetrahedron Lett. 2011, 52, 6719–6722. [Google Scholar] [CrossRef]
- Dotsenko, V.V.; Sinotsko, A.E.; Varzieva, E.A.; Buryi, D.S.; Vasilin, V.K.; Aksenov, N.A.; Aksenova, I.V. Reaction of N,N′-diphenyldithiomalondiamide with aromatic aldehydes and cyanoacetamide. Russ. J. Gen. Chem. 2023, 93, 2518–2522. [Google Scholar] [CrossRef]
- Dotsenko, V.V.; Sinotsko, A.E.; Varzieva, E.A.; Chigorina, E.A.; Aksenov, N.A.; Aksenova, I.V. N,N′-Diphenyldithiomalon- amide as methylene active thioamide: A first synthesis of stable Michael adducts. Russ. J. Gen. Chem. 2022, 92, 2530–2535. [Google Scholar] [CrossRef]
- Koval, I.V. The chemistry of disulfides. Russ. Chem. Rev. 1994, 63, 735–750. [Google Scholar] [CrossRef]
- Witt, D. Recent developments in disulfide bond formation. Synthesis 2008, 2008, 2491–2509. [Google Scholar] [CrossRef]
- Mandal, B.; Basu, B. Recent advances in S–S bond formation. RSC Adv. 2014, 4, 13854–13881. [Google Scholar] [CrossRef]
- Chiu, J.; Hogg, P.J. Allosteric disulfides: Sophisticated molecular structures enabling flexible protein regulation. J. Biol. Chem. 2019, 294, 2949–5908. [Google Scholar] [CrossRef]
- Tyler, T.J.; Durek, T.; Craik, D.J. Native and Engineered Cyclic Disulfide-Rich Peptides as Drug Leads. Molecules 2023, 28, 3189. [Google Scholar] [CrossRef] [PubMed]
- Brunskill, J.S.A. Some cyano-amides and dicyano-glutaconimides derived from pyridine aldehydes. J. Chem. Soc. Perkin Trans. 1 1972, 2946–2950. [Google Scholar] [CrossRef]
- Shestopalov, A.M.; Rodinovskaya, L.A.; Zubarev, A.A.; Nesterov, V.N.; Ugrak, B.I.; Dutova, T.Y. Synthesis and domino reactions of polymethylene-3-cyanopyridine-2 (1Н)-thiones. J. Heterocycl. Chem. 2020, 57, 913–922. [Google Scholar] [CrossRef]
- Chigorina, E.A. 1-(Cyanoacetyl)-3,5-dimethylpyrazole as an effective alternative to cyanoacetic ester in the synthesis of 2, 6-dioxopiperidine-3, 5-dicarbonitrile derivatives. Chem. Heterocycl. Comp. 2013, 49, 574–585. [Google Scholar] [CrossRef]
- Soto, J.L.; Seoane, C.; Zamorano, P.; Cuadrado, F.J. A convenient synthesis of N-amino-2-pyridones. Synthesis 1981, 1981, 529–530. [Google Scholar] [CrossRef]
- Seoane, C.; Soto, J.L.; Zamorano, P. Preparation of substituted 1,6-diamino-2-oxopyridines. Org. Prep. Proced. Int. 1984, 16, 393–400. [Google Scholar] [CrossRef]
- Grabenko, A.D.; Kulaeva, L.N.; Pel’kis, P.S. Research on a number of substituted arylamides of dithiocarboxylic acids: XV. Oxidation of dithiomalonic acid arylamides to substituted 1,2-dithiols. Chem. Heterocycl. Compd. 1974, 10, 806–809. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Bannwarth, C.; Hansen, A.; Grimme, S. B97-3c: A revised low-cost variant of the B97-D density functional method. J. Chem. Phys. 2018, 148, 064104. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Brandenburg, J.G. Simplified DFT methods for consistent structures and energies of large systems. J. Phys.: Condens. Matter 2018, 30, 213001. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Delivery Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- OECD series on testing and assessment. Number 24. Guidance document on acute oral toxicity testing. Avail. URL is: https://one.oecd.org/document/env/jm/mono(2001)4/en/pdf.
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucl. Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Seok, C.; Baek, M.; Steinegger, M.; Park, H.; Lee, G.R.; Won, J. Accurate protein structure prediction: what comes next? Biodesign 2021, 9, 47–50. [Google Scholar] [CrossRef]
- Dotsenko, V.V.; Buryi, D.S.; Lukina, D.Y.; Stolyarova, A.N.; Aksenov, N.A.; Aksenova, I.V.; Strelkov, V.D.; Dyadyuchenko, L.V. Substituted N-(thieno[2,3-b]pyridine-3-yl)acetamides: synthesis, reactions, and biological activity. Monatsh. Chem. 2019, 150, 1973–1985. [Google Scholar] [CrossRef]
- Dotsenko, V.V.; Muraviev, V.S.; Lukina, D.Y.; Strelkov, V.D.; Aksenov, N.A.; Aksenova, I.V.; Krapivin, G.D.; Dyadyuchenko, L.V. Reaction of 3-Amino-4,6-diarylthieno[2,3-b]pyridine-2-carboxamides with ninhydrin. Russ. J. Gen. Chem. 2020, 90, 948–960. [Google Scholar] [CrossRef]
- Buryi, D.S.; Dotsenko, V.V.; Aksenov, N.A.; Aksenova, I.V.; Krivokolysko, S.G.; Dyadyuchenko, L.V. Synthesis and properties of 4,6-dimethyl-5-pentyl-2-thioxo-1,2-dihydropyridine-3-carbonitrile and 3-amino-4,6-dimethyl-5-pentylthieno[2,3-b]pyridi- nes. Russ. J. Gen. Chem. 2019, 89, 1575–1585. [Google Scholar] [CrossRef]
- Peterson, M.A.; McMaster, S.A.; Riechers, D.E.; Skelton, J.; Stahlman, P.W. 2,4-D past, present, and future: a review. Weed Technol. 2016, 30, 303–345. [Google Scholar] [CrossRef]
- Chkanikov, N.D.; Spiridonov, Y.Y.; Khalikov, S.S.; Muzafarova, A.M. Antidotes for reduction of phytotoxicity of the residues of sulfonylurea herbicides. INEOS OPEN 2019, 2, 145–152. [Google Scholar] [CrossRef]
- Deng, X. Current advances in the action mechanisms of safeners. Agronomy 2022, 12, 2824. [Google Scholar] [CrossRef]
- Jia, L.; Jin, X.Y.; Zhao, L.X.; Fu, Y.; Ye, F. Research progress in the design and synthesis of herbicide safeners: A review. J. Agric. Food Chem. 2022, 70, 5499–5515. [Google Scholar] [CrossRef]
- Dotsenko, V.V.; Krivokolysko, S.G.; Rusanov, E.B.; Gutov, A.V.; Litvinov, V.P. Synthesis of 3-(4-chlorophenyl)oxirane- 2,2-dicarboxamide. Russ. Chem. Bull. 2007, 56, 1470–1473. [Google Scholar] [CrossRef]
- Bezgin, D.A.; Ershov, O.V.; Ievlev, M.Y.; Belikov, M.Y.; Bardasov, I.N. Aqueous-Phase Synthesis and Solid-Phase Fluorescence of 3-(Methoxyphenyl)-2-cyanoacrylamides. Russ. J. Org. Chem. 2018, 54, 1100–1102. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinementand analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
Scheme 1.
The structures of [1,2]dithiolo[3,4-b]pyridines and thieno[2,3-b]pyridines.
Scheme 2.
Preparation of dithiomalondianilide 1.
Scheme 3.
Synthesis of the compounds 3.
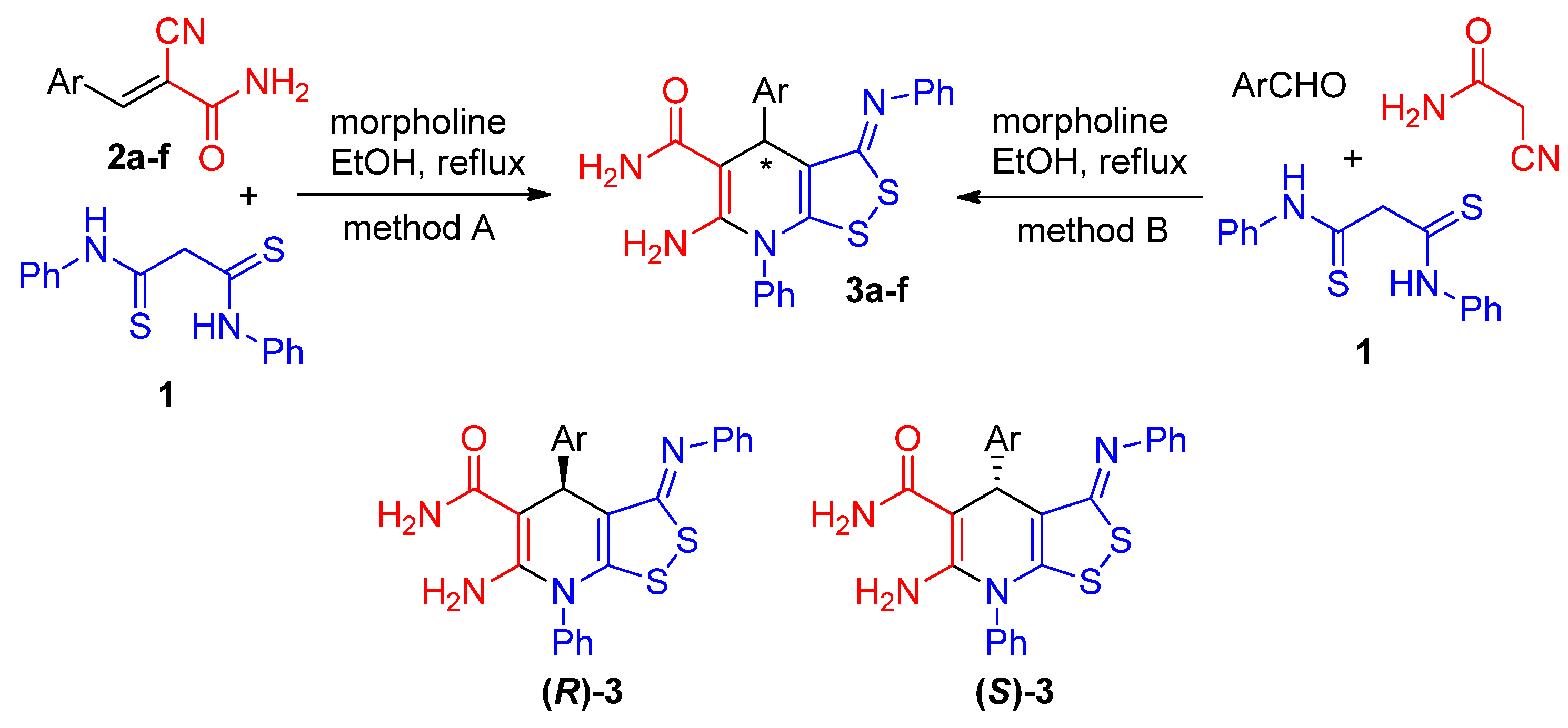
Figure 1.
ORTEP drawing of X-ray structure for 6-amino-4-(2,4-dichlorophenyl)-7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamide 3d (CCDC deposition number 2310349).
Figure 1.
ORTEP drawing of X-ray structure for 6-amino-4-(2,4-dichlorophenyl)-7-phenyl-3-(phenylimino)-4,7-dihydro-3H-[1,2]dithiolo[3,4-b]pyridine-5-carboxamide 3d (CCDC deposition number 2310349).
Scheme 4.
Preparation of [1,2]dithiolo[3,4-b]pyridines by Michael-type reaction of dithiomalondianilide 1 with activated alkenes as Michael acceptors.
Scheme 4.
Preparation of [1,2]dithiolo[3,4-b]pyridines by Michael-type reaction of dithiomalondianilide 1 with activated alkenes as Michael acceptors.
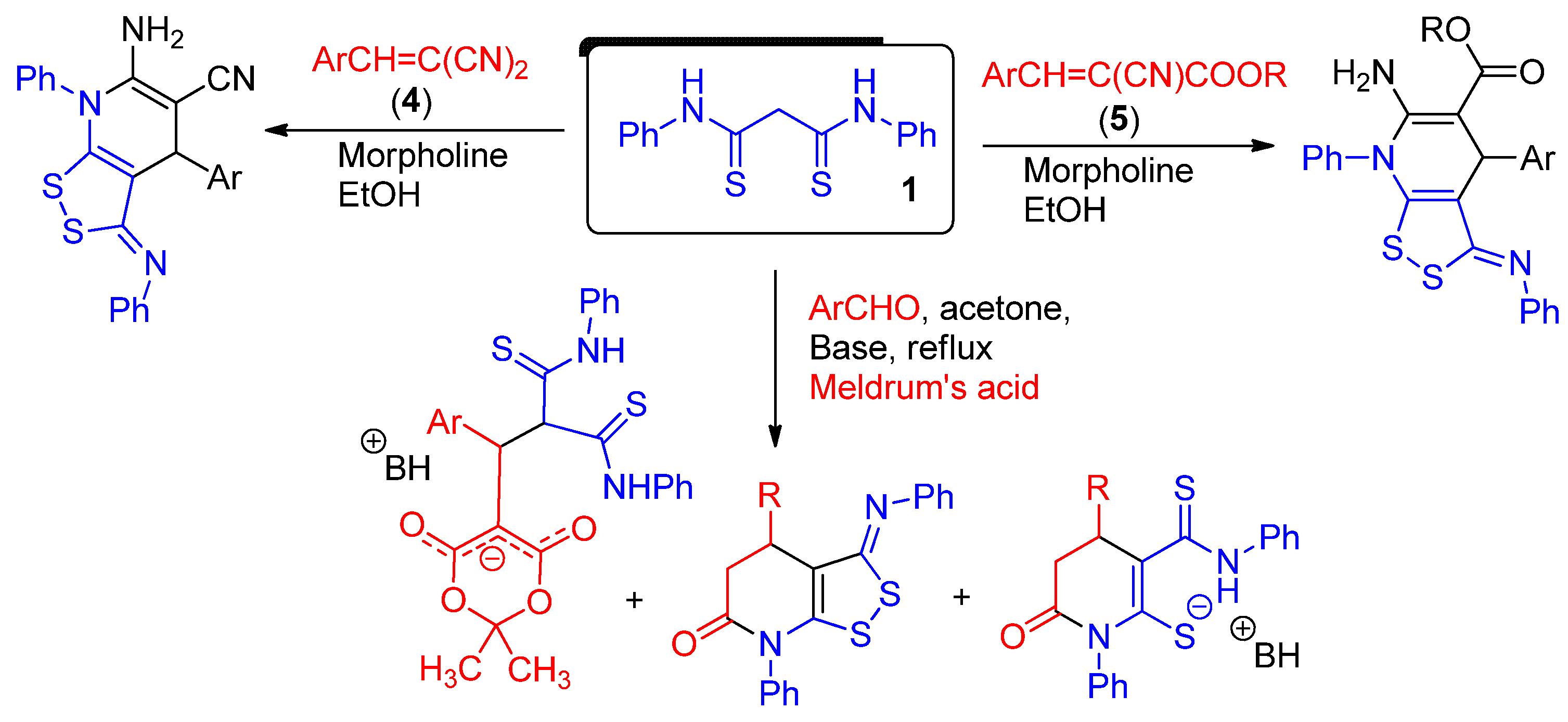
Scheme 5.
The reactions involving unsaturated nitriles both as building blocks for pyridine synthesis and as oxidizing agents.
Scheme 5.
The reactions involving unsaturated nitriles both as building blocks for pyridine synthesis and as oxidizing agents.
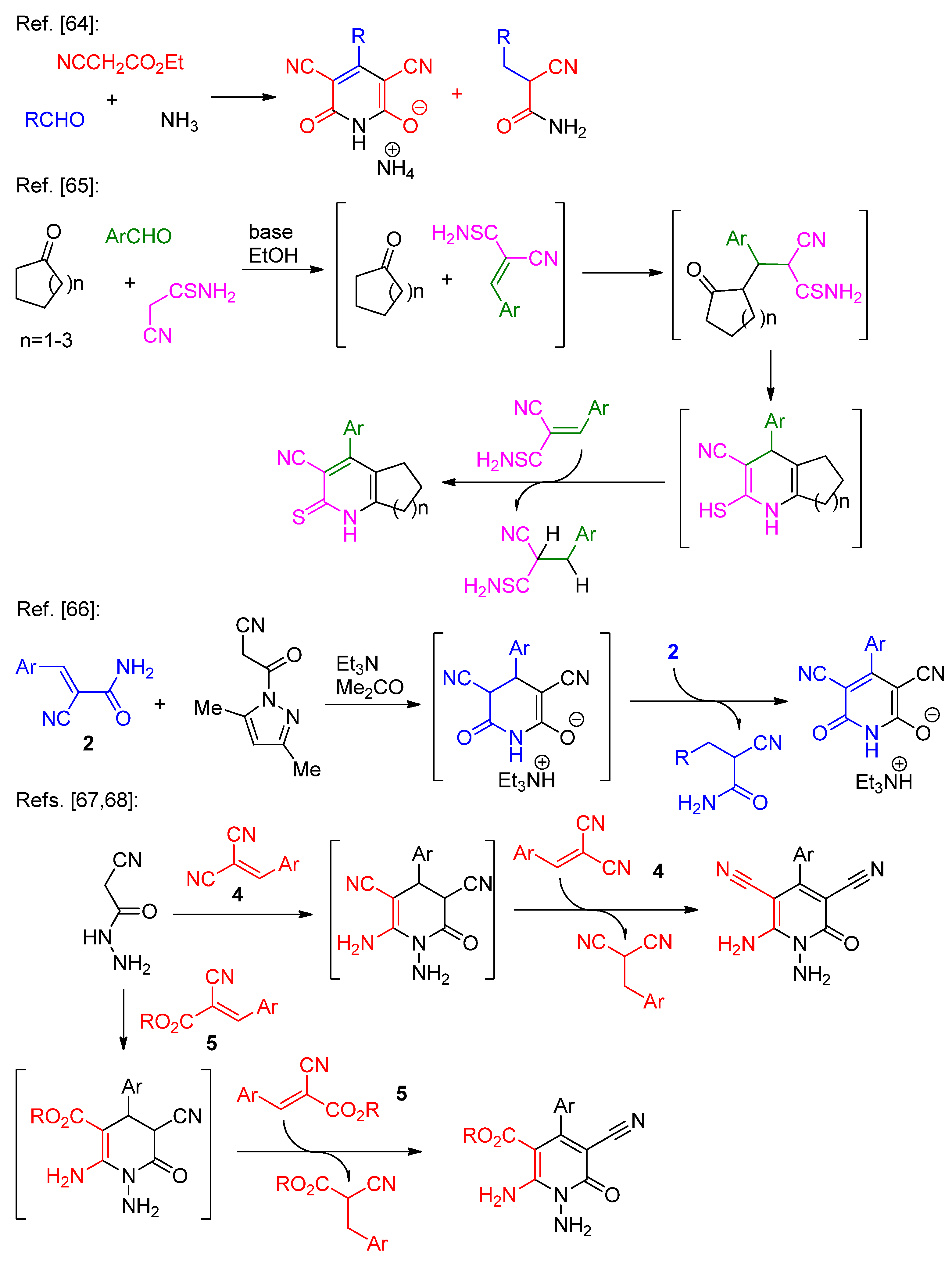
Scheme 6.
Preparation of 1,2-dithiol 6 by oxidation of dithiomalondianilide 1.
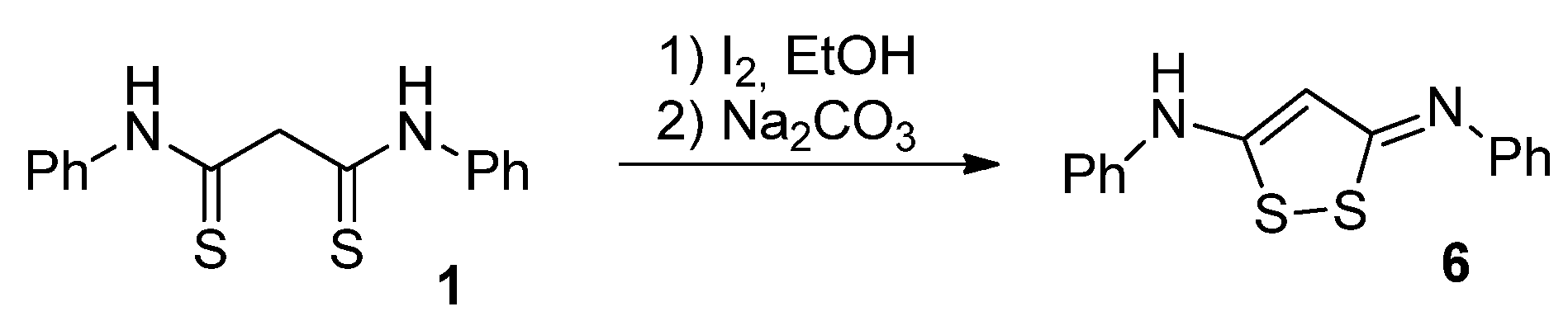
Scheme 7.
The proposed mechanism of [1,2]dithiolo[3,4-b]pyridine ring system formation.
Figure 2.
Molecular structures of intermediates I-1, I-2 and transition states TS1, TS2 (geometry and energy optimized at the B97-3c level).
Figure 2.
Molecular structures of intermediates I-1, I-2 and transition states TS1, TS2 (geometry and energy optimized at the B97-3c level).
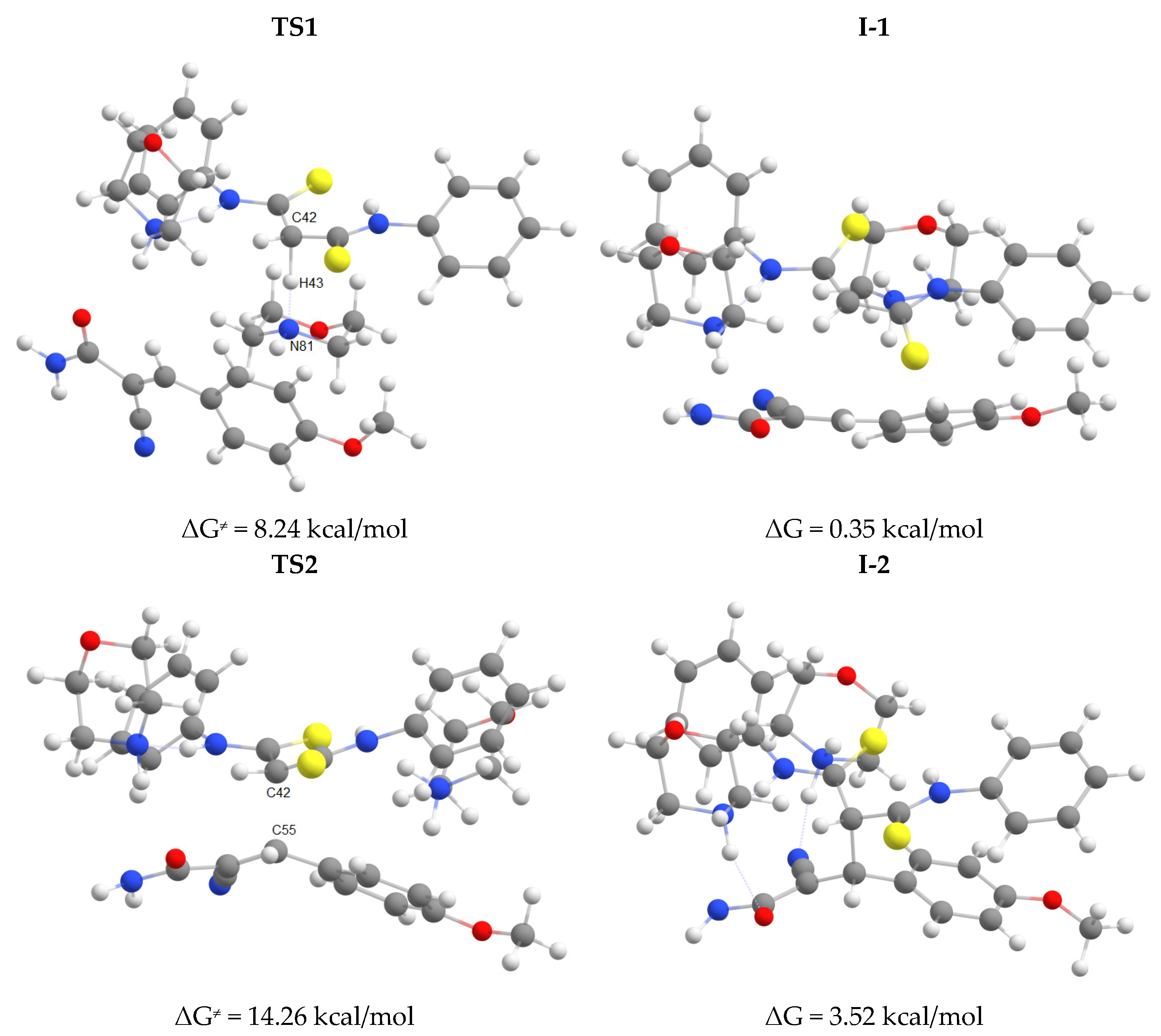
Figure 3.
Molecular structures of intermediates I-3–I-5 and transition states TS3–TS5 (geometry and energy optimized at the B97-3c level).
Figure 3.
Molecular structures of intermediates I-3–I-5 and transition states TS3–TS5 (geometry and energy optimized at the B97-3c level).
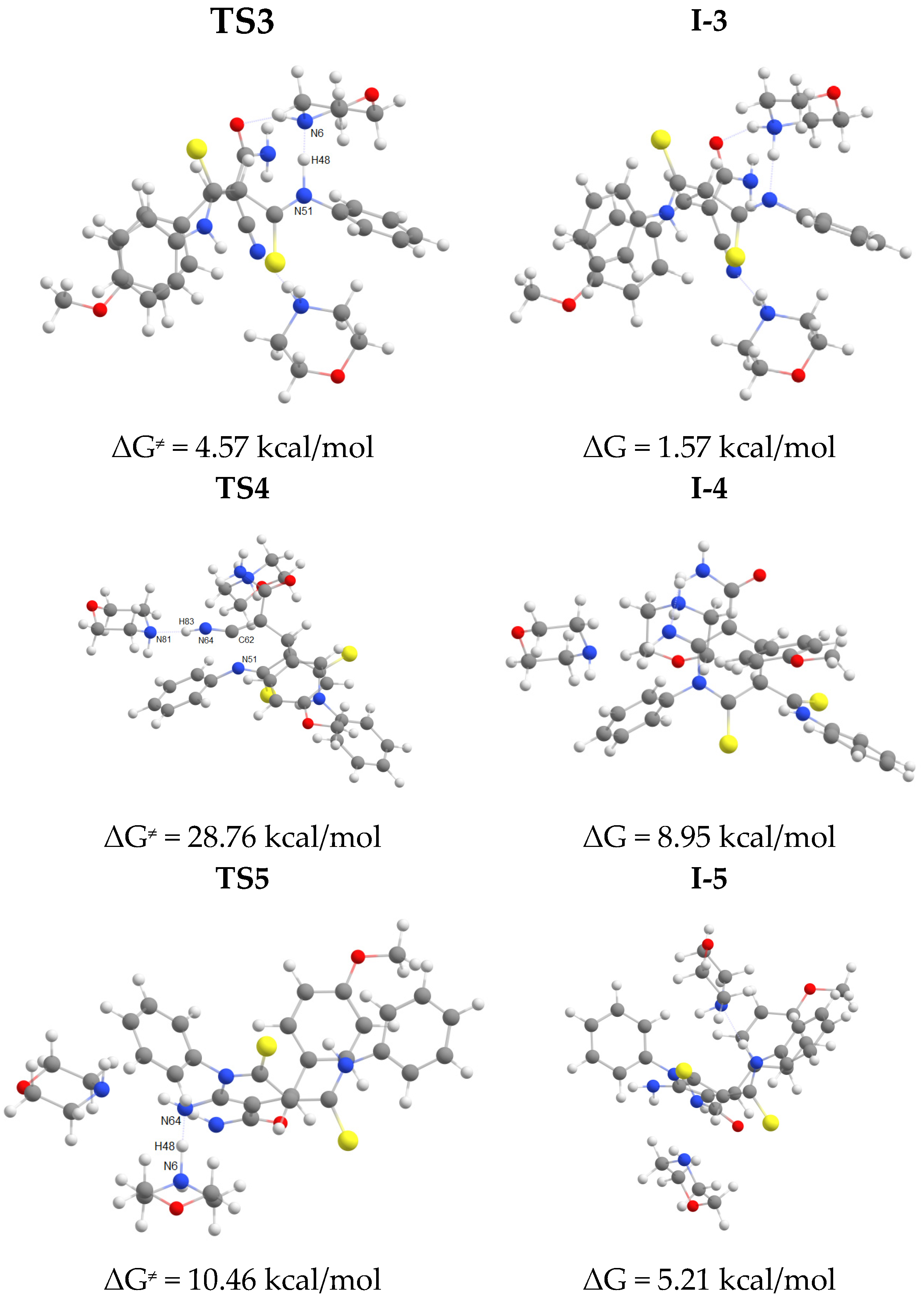
Figure 4.
Energy profile for pyridine ring formation in EtOH (B97-3c calculations).
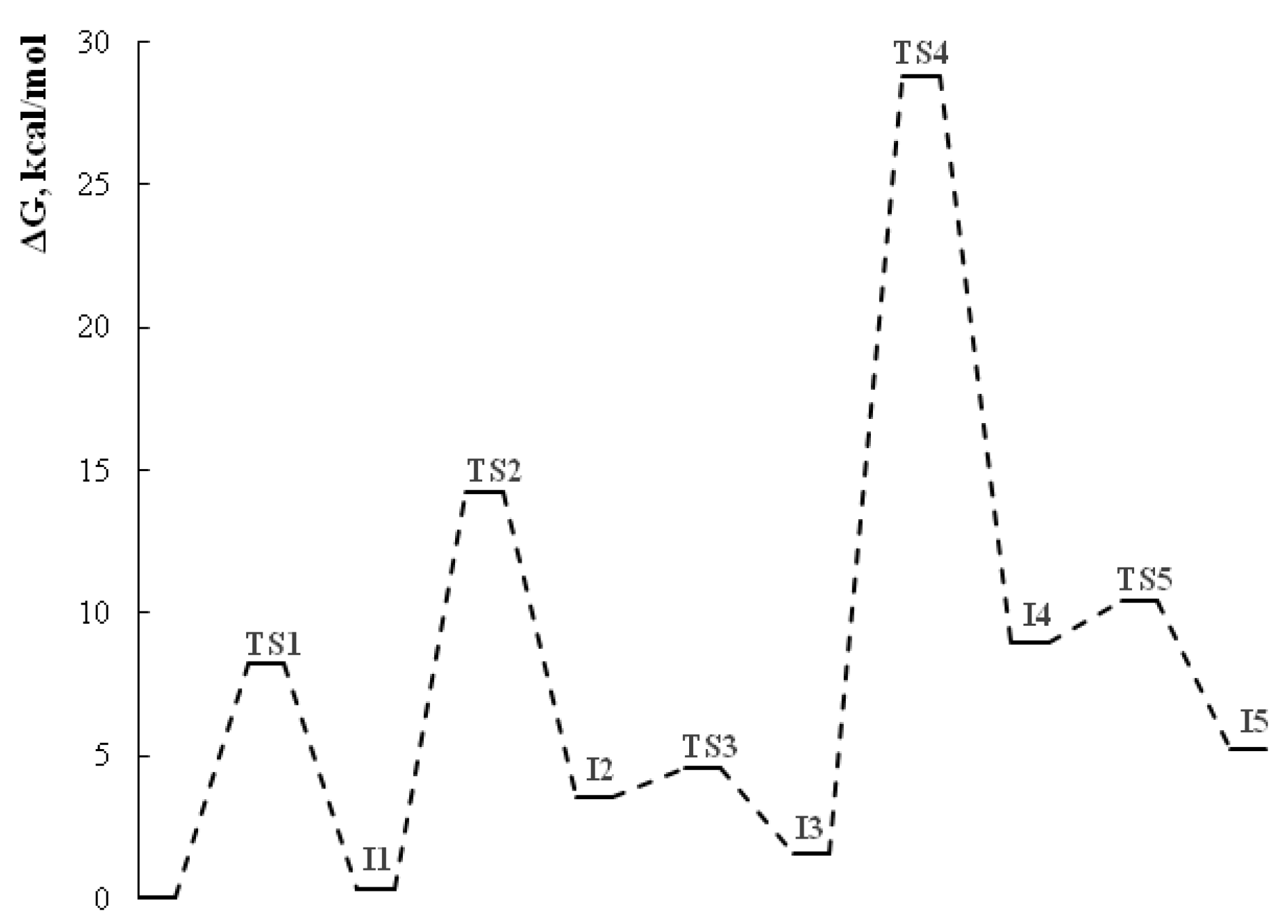
Figure 5.
Molecular structures of intermediates I-6–I-8, dithiolopyridine 3f and transition states TS6–TS8 (geometry and energy optimized at the B97-3c level).
Figure 5.
Molecular structures of intermediates I-6–I-8, dithiolopyridine 3f and transition states TS6–TS8 (geometry and energy optimized at the B97-3c level).
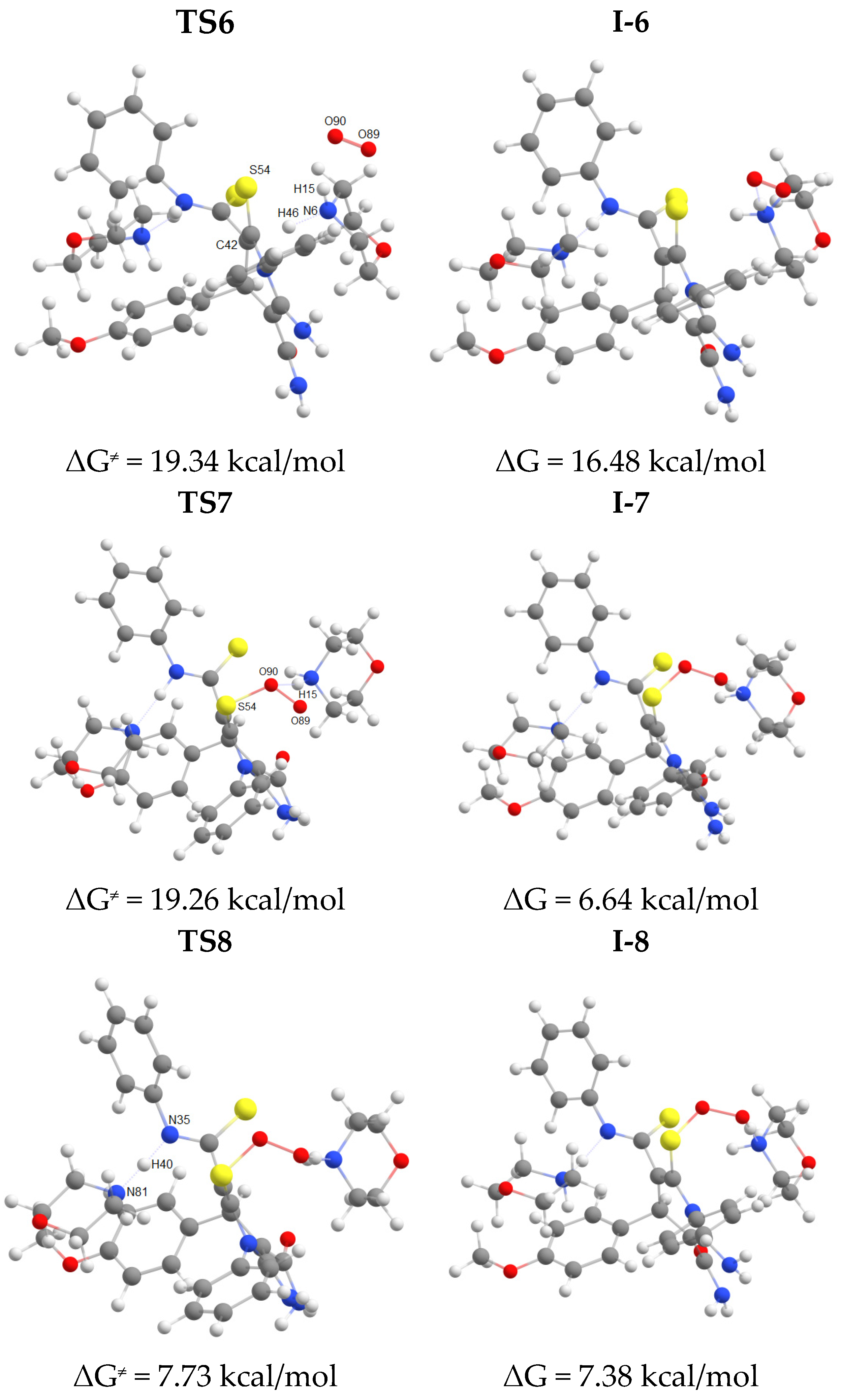
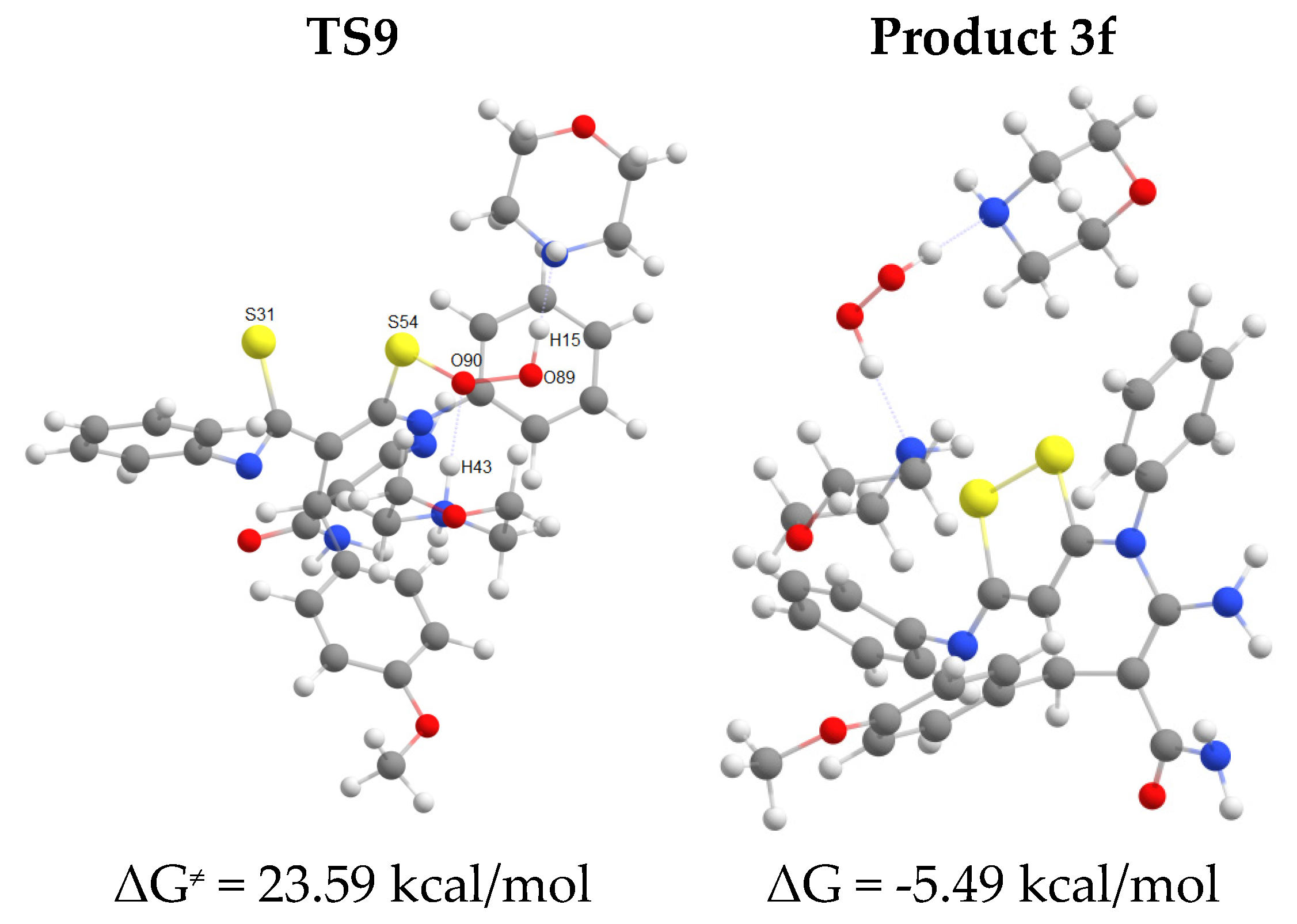
Figure 6.
Energy profile for disulfide bond formation in EtOH (B97-3c calculations).
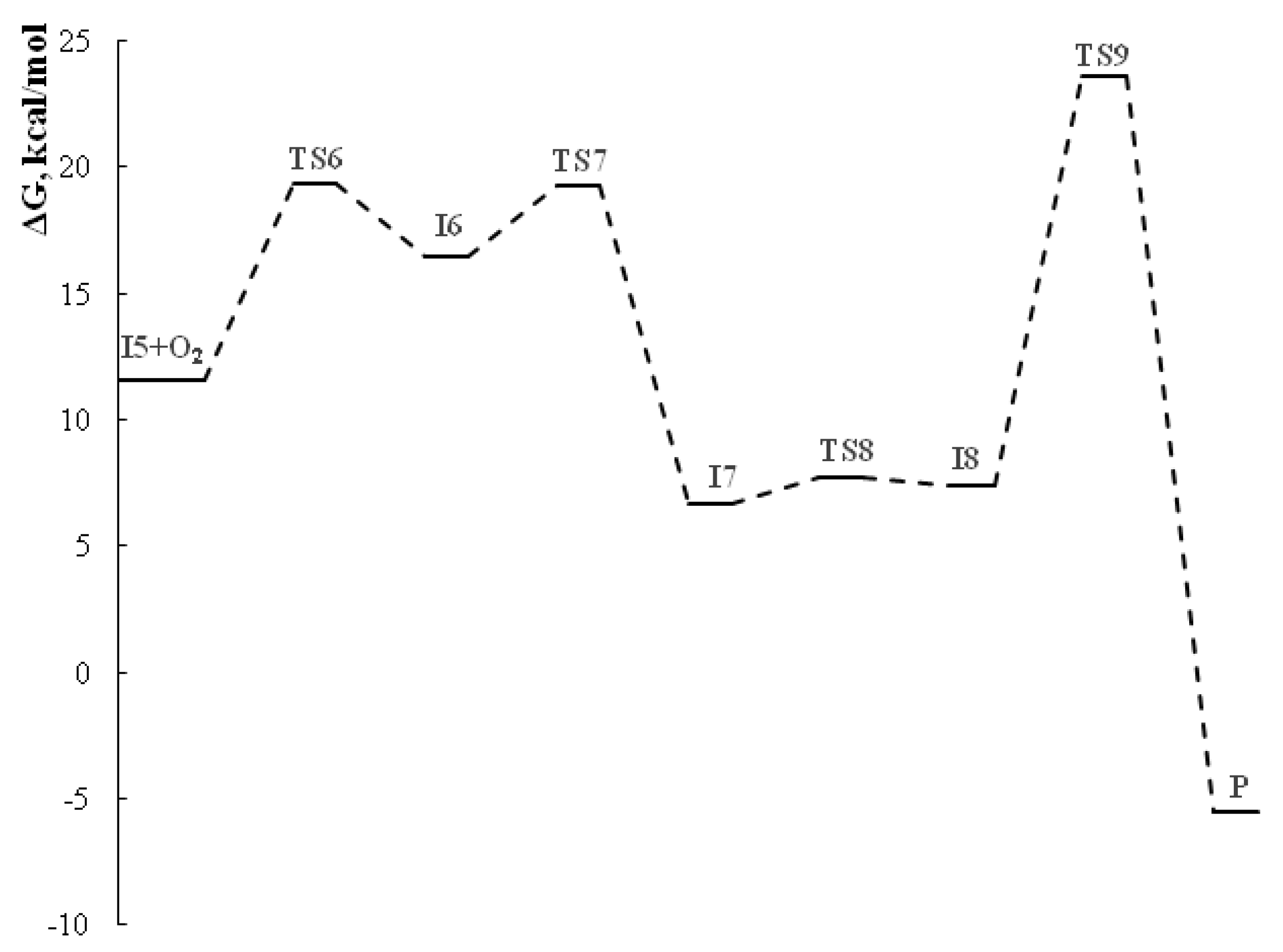
Figure 7.
Position of compound (R)-3a in the site of coagulation factor VII (PDB ID 4yt6_H) by molecular docking.
Figure 7.
Position of compound (R)-3a in the site of coagulation factor VII (PDB ID 4yt6_H) by molecular docking.
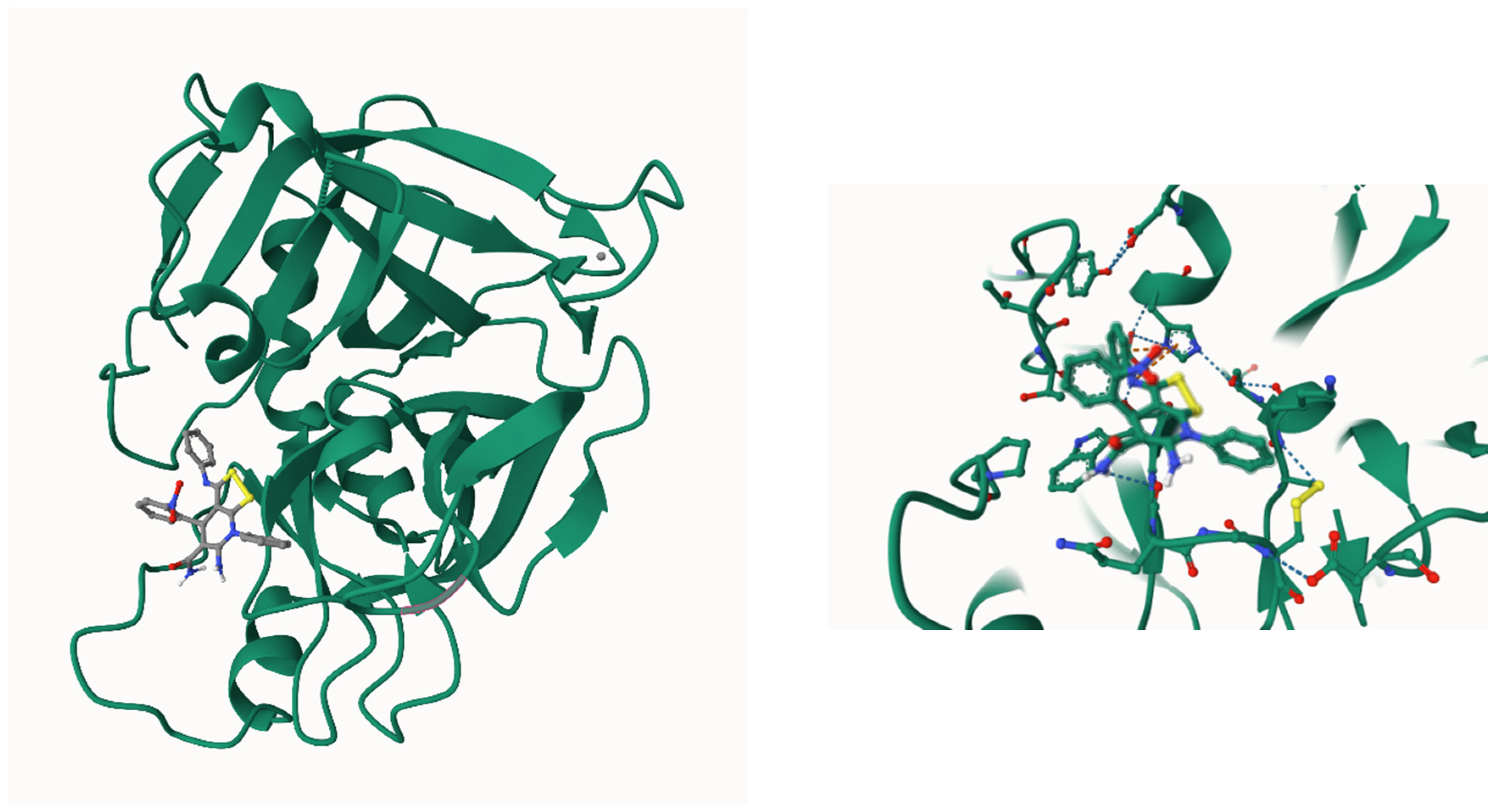

Table 1.
The structure and yields of compounds 3.
| Entries | Product | Methods | Yields, % |
|---|---|---|---|
| entry 1 | 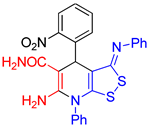 3a 3a |
A B |
54 43 |
| entry 2 | 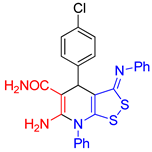 3b 3b |
A B |
40 33 |
| entry 3 | 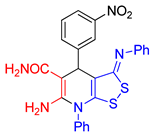 3c 3c |
A B |
37 27 |
| entry 4 | 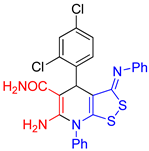 3d 3d |
A B |
39 31 |
| entry 5 | 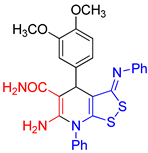 3e 3e |
A B |
40 22 |
| entry 6 | 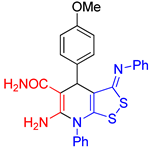 3f 3f |
A B |
54 35 |
Table 2.
The antidote effects of the compounds 3b, 3d and 3f with respect to herbicide 2,4-D.
| N | compound | Organ | Antidote effect A at different concentrations, % 1 | |||
|---|---|---|---|---|---|---|
| 10-2 | 10-3 | 10-4 | 10-5 | |||
| 1 | 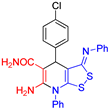 3b 3b |
roots | 123 | 121 | 121 | 116 |
| hypocotyls | 127 | 130 | 124 | 120 | ||
| 2 | 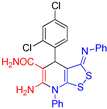 3d 3d |
roots | 130 | 131 | 123 | 128 |
| hypocotyls | 132 | 127 | 120 | 128 | ||
| 3 | 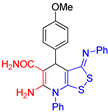 3f 3f |
roots | 133 | 127 | 132 | 130 |
| hypocotyls | 132 | 132 | 116 | 119 | ||
1 The differences are reliable at P = 0.95.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



