
Submitted:
01 December 2023
Posted:
01 December 2023
You are already at the latest version
Abstract
Eosinophils in peripheral blood account for 0.3-5% of leukocytes, which is equivalent to 0.05-0.5 x 109/l. A count equal or above 0.5 x 109/l is considered eosinophilia, while a count equal or above 1.5 x 109/l is defined as hypereosinophilia. In bone marrow, eosinophilia is considered when eosinophils make up more than 6% of the total nuclear cells. In daily clinical practice, the most common causes of reactive eosinophilia are non- hematologic, whether non-neoplastic (allergic diseases, drugs, infections or immunological diseases) or neoplastic (solid tumors). Eosinophilia associated with a haematological malignancy may be reactive or secondary to the production of eosinophilopoietic cytokines, and this is mainly seen in lymphoid neoplasms (Hodgkin lymphoma, mature T-cell neoplasms, lymphoid variant of hypereosinophilic syndrome and B-acute lymphoblastic leukemia/lymphoma). Eosinophilia associated with a haematological malignancy may also be neoplastic or primary, derived from the malignant clone, usually in myeloid neoplasms or with origin in stem cell (myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions, acute myeloid leukemia with Core Binding Factor translocations, mastocytosis, myeloproliferative neoplasms, myelodysplastic/myeloproliferative neoplasms and myelodysplastic neoplasms). There are no concrete data in standardized cytological and cytometric procedures that could predict whether eosinophilia is reactive o clonal. The verification is usually indirect, based on the categorization of the accompanying hematologic malignancy. This review focuses on the broad differential diagnosis of haematological malignancies with eosinophilia.
Keywords:
Eosinophilia
; hematological neoplasm
; myeloid/lymphoid neoplasm with eosinophilia
; tyrosine kinase fusion genes
; acute leukemia
1. Introduction
1.1. The eosinophil
Eosinophils are granular leukocytes originating in the bone marrow. The term "eosinophil" was coined by Paul Ehrlich in 1879 when he observed that the granules of these cells stained with acidic dyes, specifically with eosin Y (tetrabromofluorescein), producing a striking bright orange color [1]. However, it is highly likely that other researchers had observed this cell before Ehrlich [2].
Eosinophils should be considered tissue cells, with their presence in the blood being only circumstantial. They originate in the bone marrow from a pluripotent stem cell, which gives rise to a pluripotent myeloid progenitor cell, and this, in turn, to a specific or committed precursor in the genesis of the eosinophil granulocyte. Cytokines encoded by genes located proximally on 5q31, such as interleukin 3 (IL-3), interleukin 5 (IL-5) (the most specific to the eosinophil lineage), and granulocyte-macrophage colony-stimulating factor (GM-CSF), act on them, regulating their maturation and differentiation [3]. Similarly, IL-33 is also crucial for the basal homeostasis of eosinophils, involved in their differentiation and activation through its interaction with Interleukin 1 receptor-like 1 (IL1RL1), also known as the ST2 receptor [4]. After entering the bloodstream (hours), they migrate to various tissues. They are attracted in response to chemokines, primarily those of the eotaxin family: eotaxin-1/CCL11, eotaxin-2/CCL24, eotaxin-3/CCL26, RANTES/CCL5, which activate the CCR3 receptor (C-C motif chemokine receptor 3) expressed on eosinophils. These chemokines are produced by fibroblasts, epithelial cells, endothelial cells, smooth muscle cells, T lymphocytes, and macrophages [5]. In the same way, complement C5a and lipid mediators have been linked as chemotactic factors [6]. Once in the tissues, eosinophils can survive for several weeks. They are preferentially present in the mucosa of the respiratory, digestive, and genitourinary tracts.
1.2. Eosinophil biological activity
Eosinophils process a single type of granulation, with heterogeneous content in their primordial stage (pre-eosinophil), which later condenses and crystallizes into what is referred to as specific granulation [7]." In terms of ultrastructure, these granules possess a central electrondense crystalline core surrounded by a matrix, and they are rich in cationic proteins that are highly toxic to parasites (helminths), as well as to bacteria, fungi, and viruses. The "major basic protein" is concentrated in the central crystalline core, while the matrix contains the "eosinophil-derived neurotoxin" or ribonuclease 2, the "cationic protein of the eosinophil" or ribonuclease 3, and the "eosinophil peroxidase" that generates potent oxidants. They also contain galectin-10, or "Charcot-Leyden protein," which can be observed when crystallized in bone marrow aspirates and in tissues affected by eosinophilic infiltration [8]. In addition, mediators such as cytokines, chemokines, growth factors, lipid mediators, as well as multiple receptors for these, and enzymes are found. Thus, they play an immunomodulatory role in innate and adaptive immune responses, have anti-inflammatory and antitumor activity, and mediate in the repair, remodeling, and maintenance of tissues and their homeostasis [9].
However, when eosinophils undergo massive and persistent activation, they can damage the tissues and organs in which they operate due to the toxicity of their content. This is what happens in hypereosinophilic syndromes or immediate hypersensitivity reactions (type I). A proinflammatory and prothrombotic state is generated, and transforming growth factor-beta and interleukins are released, promoting the proliferation and activation of fibroblasts, leading to subsequent fibrosis at the expense of the parenchyma [9].
1.3. Normal and Pathological Morphology of Eosinophils
In blood smears fixed with methanol and stained with Romanowsky stains (based on methylene blue, eosin, and their derivatives), the normal mature eosinophil reaches a size between 14 and 16 µm. The nucleus is bilobed, classically described as eyeglass-shaped, and sometimes trilobed. The cytoplasm, which appears translucent, is almost entirely occupied by a relatively thick, dense, orange granulation (0.5-1.5 µm) that, unlike basophils, does not overlap the nucleus. Vacuoles are not typically observed (Figure 1A,B).
Regarding cytochemical aspects, the presence of myeloperoxidase in normal eosinophils is demonstrated through enzymatic cytochemistry and an appropriate substrate. Indirectly, the "content" of myeloperoxidase can be deduced (though not proven) through staining with Sudan Black B, which is particularly soluble in neutral fats (triglycerides) and phospholipids, both of which are abundant in the granulation of neutrophils and eosinophils. These cell types can be distinguished by the thicker granulation of eosinophils (Figure 1C,D). The eosinophil exhibits an absence of esterases and both acid and alkaline phosphatases. The Periodic Acid-Schiff (PAS) reaction is negative, or with weak intergranular positivity, in contrast to the marked positivity of segmented neutrophils. Additionally, metachromasia with Toluidine Blue, which is characteristic in basophilic granulocytes, is not observed [3].
Morphological alterations of the eosinophil, regardless of whether eosinophilia is reactive or malignant, can include nuclear morphological abnormalities (absence of lobulation, hypersegmentation, or ring nuclei), defects in the cytoplasm (total or partial degranulation, vacuoles, persistent basophilia, visualization of pre-eosinophilic granulation, increased thickness of granules), size alteration (anisocytosis, gigantism, small forms), and maturation disharmonies, which encompass previous defects such as persistence of cytoplasmic basophilia in elements with advanced maturity [10] (Figure 2).
1.4. Flow cytometry of the eosinophil
By immunophenotype, the normal eosinophil is characterized by a pattern of elevated size (FSC) and complexity (SSC), reflecting its cytomorphological features. They originate in the bone marrow from pluripotent hematopoietic stem cells CD34+ CD117+. They express the panleukocytic marker CD45, and with differentiation, they lose the expression of CD34, CD117, and HLADR, acquiring CD11b, CD15, CD65, and cytoplasmic eosinophilic peroxidase (CyEPO). Eosinophils produce this peroxidase, which shares some homology with the myeloperoxidase found in neutrophil and monocyte leukocytes [11]. Other markers present in these cells include CD13, CD33, CD193 (CCR3), and the alpha subunit of the High-affinity IgE receptor (FcεR1α) [4,12]. In contrast to neutrophils, they exhibit slightly weaker expression of CD15 in the absence of CD16 (Figure 3) [12].
2. Eosinophilia
Eosinophils in peripheral blood constitute 0.3-5% of leukocytes, equivalent to 0.05-0.5 x 109/l. Eosinophilia is considered when their count is ≥ 0.5 x 109/l, and hypereosinophilia when it is ≥ 1.5 x 109/l. In a bone marrow aspirate, a normal rate is considered up to 6% and, in bone marrow biopsy, the definition of medullary eosinophilia has been proposed with ≥ 20% eosinophils [13,14].
Most cases of eosinophilia are not associated with hematologic diseases and are reactive. Among their causes are parasitic infections, allergies, medications, chronic inflammatory processes, etc. Solid tumors that can present with a leukemoid reaction featuring blood and/or bone marrow hypereosinophilia, mimicking a myeloproliferative neoplasm (MPN) (Figure 4), should also be included. Additionally, some of the new antitumor immunotherapies can cause eosinophilia with dysplasia [15,16].
Persistent eosinophilia associated with a hematologic disease can be reactive to the release of eosinopoietic cytokines (polyclonal or benign) or neoplastic (belonging to the malignant clone) Table 1 [17]. In practice, clonality is not directly determined on purified eosinophils. Instead, the evidence is indirect and is based on the underlying pathology and identified markers of clonality.
3. Hematological neoplasms associated with eosinophilia
3.1. Hematological neoplasms associated with reactive eosinophilia
3.1.1. Classical Hodgkin Lymphoma
The observation of pronounced and persistent reactive eosinophilia is an unusual finding in patients with B-cell lymphomas, except for Hodgkin lymphoma. Approximately 15% of these patients exhibit it, typically in a mild form. Histologically, it is better distinguished in the inflammatory infiltrate surrounding Reed-Sternberg cells (Figure 5). Inside these cells, IL-5 mRNA has been demonstrated by in situ hybridization. It is unknown whether these cells or the abundant Th2 lymphocytes present are the main source of eosinopoietic cytokines [18,19].
3.1.2. Mature T-cell neoplasms
Mature T-cell neoplasms associated with eosinophilia mostly derive from memory CD4+ T cells, which can produce eosinophilopoietic cytokines. Sometimes it is doubtful whether eosinophilia is due to the tumor cells themselves or to locally attracted reactive Th2 lymphocytes. In any case, proliferating eosinophils are not related to the malignant clone, and can be integrated into the inflammatory context that surrounds the tumor cells. In the setting of eosinophilia, the study of the immunophenotype of T lymphocytes is relevant, with the antibody against TRBC1 JOVI-1 having recently been incorporated and validated as a useful marker in the identification by flow cytometry of clonal T populations with a TCR alphaBeta [20].
The presence of eosinophilia is estimated in 17-25% of mycosis fungoides cases, and it is described with much higher frequency in Sezary syndrome, observed in 10 out of 13 patients (77%) [18,21]. In both mycosis fungoides and Sezary syndrome, clonal T lymphocytes commonly exhibit a CD4+ CD3+ phenotype in the absence of CD7 and CD26 expression. In the case of Sezary syndrome, a phenotype of central memory CD4 T lymphocytes (CD45RA- CD45RO+ CCR7+ CD27+ and CD62L+/-) is characteristic, unlike mycosis fungoides where its immunophenotype profile is that of an effector memory cell resident in the skin (CD45RA - CD45RO+, CCR7-, CD27- and CD62L-). Furthermore, it is accompanied by the expression of intense CD28+ and CD279 [22]. In nodal T-follicular helper cell lymphoma, angioimmunoblastic-type, eosinophilia is also common, in 32-50% of cases. This lymphoma originates from a follicular CD4 T cell with a phenotype that is characterized by weak or negative expression of CD3, with expression of CD10 and PD-1 (CD279) maintaining the expression of other pan-T markers such as CD5 (Figure 6) [23]. Adult T-cell leukemia/lymphoma follows in frequency with eosinophilia in 20% of cases. In this case, it is generally a CD4+CD8- T lymphocyte phenotype, although to a lesser extent they can be double positive (CD4+CD8+) or CD4-CD8+. Furthermore, the expression of CD25 and CCR4 is typical, the latter being essential as it is currently a therapeutic target [24]. More rarely, eosinophilia is found associated with other T lymphomas, although histologically it is frequently found with a peritumoral arrangement [18].
3.1.3. Lymphocyte variant hypereosinophilic syndrome
This is a rare variant of hypereosinophilic syndrome (HES), with eosinophilia due to overproduction eosinophilopoeitic cytokines secreted by clonal T lymphocytes with an abnormal immunophenotype. Persistent blood hypereosinophilia is related to tissue infiltration and organ damage. In a large review of 148 cases, the median age at diagnosis was 46 years-old and there was no difference by gender. The most common clinical manifestations were cutaneous (81%), while cardiovascular manifestations occurred in 11.5% of cases. The most frequent immunophenotype of T cells was CD3- CD4+. Although their behavior is indolent, they have an increased risk of transformation to T lymphoma (risk of transformation at 10-year: 19,9%) [25].
3.1.4. B Acute lymphoblastic leukemia/lymphoma
Significant eosinophilia during or prior to diagnosis is observed in less than 1% of B acute lymphoblastic leukemia/lymphoma (B-ALL). The expansion of eosinophils is considered reactive to the production of cytokines by lymphoblasts. Eosinophils can show dysplastic feature in morphology and by immunophenotype (Figure 7 and Figure 8).
Eosinophilia is often associated with B-ALL with t(5;14)(q31.1;q32.1);IGH::IL3, an entity initially defined in the 2008 World Health Organization (WHO) Classification. Actually, this entity is named as B-ALL with t(5;14)(q31.1;q32.3)/IL3::IGH in accordance with International Consensus Classification (ICC) 2022. It is a very rare subtype of B-ALL. In a review of 24 patients, this leukemia mainly affects adolescent or young adult males who suffer frequent manifestations derived from eosinophilia (neurological, thromboembolic, pulmonary and cutaneous) [26]. The relevance of eosinophilia in the blood and bone marrow can relativizes the percentage of blasts, which can be less than 20%. In addition, eosinophilia and PAX5 rearrangement (PAX5::GSDMA and PAX5::ZCCHC7) has also been found [27,28]; as well as the observation of a hyperdiploid karyotype with structural alteration [29].
3.1.5. T acute lymphoblastic leukemia/lymphoma
The appearance of eosinophilia in T acute lymphoblastic leukemia/lymphoma (T-ALL) is anecdotal, having been described in a case of near-early T precursor ALL with a t(5;7)(q31;q21)/CDK6::IL3, which may have a functional mechanism similar to the IGH::IL3 rearrangement of B-ALL [30].
3.2. Hematological malignancies associated with neoplastic or primary eosinophilia
3.2.1. Myeloid/lymphoid neoplasms with eosinophilia and defining gene rearrangement
The 2016 WHO classification recognized four entities with genetic rearrangements of PDGFRA (4q12), PDGFRB (5q32), FGFR1 (8p11.2) and as a provisional entity PCM1::JAK2 [t(8;9)(p22;p24.1)] [31]. These entities are recognized within the section of “Myeloid/lymphoid neoplasms with eosinophilia (M/LN-eo) and defining gene rearrangements” in the 5th edition 2022 WHO classification. While in the 2022 ICC the name of this section is “M/LN-eo and tyrosine kinase gene fusions” [32,33]. Both classifications include a modification in the PCM1::JAK2 category that is now called JAK2 rearrangement. Also, the latest classifications include three new entities: M/LN-eo with FLT3 rearrangement, M/LN-eo with ETV6::ABL1 fusion, and M/LN-eo with other tyrosine kinase gene fusions. They have in common a very low incidence, and the cell of origin is a pluripotent stem cell, hence the variability in the involvement of hematopoiesis. The most common presentation is as a chronic process resembling MPN or myeloproliferative/ myelodysplastic neoplasia (MDS/MPN). However, an aggressive presentation of disease can be observed at diagnosis or by progression, in the form of acute myeloid leukemia (AML), T-ALL and, more rarely, B-ALL or acute leukemia of ambiguous lineage. In some patients the same neoplasia occurs simultaneously chronically in the bone marrow and aggressively in the extramedullary involvement [34,35]. Currently, an association with a low-grade B lymphoid neoplasia has not been described. Singularly, when the clinical presentation is a B-ALL, the differential diagnosis must be made with B-ALL with BCR::ABL1-like features. In the latter case, the rearrangement is restricted to lymphoblasts, normally without eosinophilia, unlike an aggressive form of NLM-eo in which the rearrangement affects all hematopoietic lineages, with or without eosinophilia [35]. The pathophysiology of these diseases includes the expression of a fusion gene that involves PDGFRA, PDGFRB, FGFR1, JAK2, FLT3, ABL1 or other kinases giving rise to an aberrant constitutively activated tyrosine kinase.
The observation of eosinophilia in blood, bone marrow and, in certain cases, extramedullary involvement is usually a common sign that guides the diagnosis, although peripheral or tissue expression may be very mild or absent. When its presentation is as MPN or MDS/MPN, a certain degree of fibrosis in bone marrow frequently is observed. The vast majority of rearrangements can be diagnosed by fluorescence in situ hybridization (FISH) and suspected in cytogenetic study, except for FIP1L1::PDGFRA, which is cryptic. At diagnosis, reverse transcription polymerase chain reaction (RT-PCR) can also be used for the FIP1L1::PDGFRA rearrangement. Likewise, RT-PCR can be used to follow-up when the partner of any of them is identified. Exceptionally, in all groups there may be cryptic cases that require RNA sequencing for identification.
Significantly, the majority of patients are male, especially in PDGFRA, PDGFRB and, to a lesser extent, JAK2 rearrangements and FGFR1. It occurs at any age of life with a peak between 45-50 years, being a little earlier in the FGFR1 group [34,35,36,37]. The clinical manifestations and exploratory findings are diverse. In stable chronic forms, a constitutional syndrome can simply be seen, eventually with hepatosplenomegaly, and in case of morphology of chronic eosinophilic leukemia (CEL), organic involvement with fibrosis secondary to eosinophilic infiltration can be added. Obviously, the deterioration is more severe when it is AML or T-ALL, accompanied in this case by lymphadenopathy [34,35].
PDGFRA and PDGFRB rearrangements are very sensitive to first- and second-generation tyrosine kinase inhibitors (imatinib and dasatinib). Responses with these inhibitors are also described in the ETV6::ABL1 fusion. Cases of FLT3 rearrangement may be sensitive to FLT3 inhibitors; while in those of FGFR1 and JAK2 the response to therapeutic targets is much lower, being the allogeneic transplantation of hematopoietic progenitors the curative option [38].
3.2.1.1. Myeloid/lymphoid neoplasm with PDGFRA rearrangement
This is the most frequent among M/LN-eo and defining gene rearrangement. The annual incidence has been estimated at around 0.18 cases per million inhabitants [39]. Within the rarity of these neoplasms, the most observed rearrangement is the FIP1L1-PDGFRA fusion, a cryptic translocation due to an interstitial deletion in 4q12. This category also includes variant translocations of PDGFRA with other genes such as KIF5B [40], CDK5RAP2 [41], STRN, ETV6 [42] BCR [43], TNKS2 [44] and FOXP1 [45].
At diagnosis, it is usual the presentation as MPN, almost always chronic eosinophilic leukemia, with the eosinophils frequently being dysmorphic, although they can be practically normal or with minimal dysplasia. Eosinophilia is a generalized trait (close to 100%), normally very high, although there may be a selection bias in some series that only included patients with eosinophilia. Isolated cases lacking relevant eosinophilia have been described [46]. Generally, the bone marrow is hypercellular, with an increase in eosinophils that can be normal or dysmorphic; there may also be an increase of blast (<20%), and an increase in dispersed mast cells is common or, more rarely, forming loose clusters, which may present atypical spindle-shaped morphology and usually express CD25. Histologically, the finding of moderate reticulin fibrosis is characteristic [39]. This presentation can occur with or without simultaneous extramedullary involvement by myeloid sarcoma, T-ALL, or rarely B lymphoblastic lymphoma [47]. Chronic forms (CEL or other MPN) can also progress to the blast phase, mainly to AML or to T-ALL. Progression to B-ALL is exceptional, with two cases having been described in adult subjects, one with FIP1L1-PDGFRA rearrangement and another with BCR-PDGFRA; the first of them presented during diagnosis with MPN with eosinophilia and T lymphoblastic lymphoma and a year later developed B-ALL [48]. The second case presented as MPN in the chronic phase, developing B-ALL a month later [49].
Other less common presentations may include systemic mastocytosis (SM) with eosinophilia, AML, T-ALL, or other MPN, often associated with peripheral eosinophilia. Its presentation as B-ALL is not described except for a single case, without specifying its characteristics, within a series of cases of Ph-like B-ALL [50]. Although infrequently, a subclass of cases could present with extramedullary involvement without concomitant bone marrow involvement [34]. Anecdotal presentations include a patient with three synchronous hematologic malignancies driven by the FIP1L1::PDGFRA rearrangement: cutaneous T lymphoma, lymphoblastic lymphoma in lymph node, and MPN in bone marrow [51]. Likewise, some association with papulosis lymphomatoid has been described [52].
In the context of FIP1L1::PDGFRA rearrangements, 2 series stand out with 151 and 78 patients, respectively [36,39]. In the largest series, a retrospective study of 151 patients in the chronic phase, the mean age at diagnosis was 49 years ( ±12) and 95% was male. Although a fraction of patients (17%) were asymptomatic, the remaining majority presented general symptoms such as asthenia, weight loss and persistent fever. Up to 72% of patients may manifest at least one symptom related to hypereosinophilia: skin manifestations (pruritus, dermal lesions), pulmonary (cough, restrictive disease related to fibrosis), cardiac (sometimes as severe as endomyocardial fibrosis, acute myocarditis, heart and mitral failure, or intracardiac thrombus formation), neurological (mainly ischemic stroke), gastrointestinal (eosinophilic gastroenteritis) and/or vascular (venous thromboembolism and arterial thrombosis). In approximately half of the cases, splenomegaly was demonstrated at diagnosis, which may be accompanied by hepatomegaly. All patients present eosinophilia and in up to 31% it is the only finding. Anemia (24%), mild thrombopenia (28%), and variable leukocytosis, often with neutrophilia (20%), but also with monocytosis (16%), basophilia (13%) or lymphopenia (12%), are also described. The observation of lymphocytosis is unusual. Serum levels of vitamin B12 and tryptase are regularly elevated, being much less common an increase in IgE [39].
German Registry for Disorders of Eosinophils and Mast Cells (GREM) described 78 patients with FIP1L1::PDGFRA gene fusion, 65 patients (83%) presenting in the chronic phase and 13 cases (17%) in the blast phase. In addition, 4 patients developed a blast phase during follow-up. Fourteen out of the 17 cases that developed acute leukemia were of the myeloid line (4 extramedullary cases) and the remaining 3 cases were of the lymphoid line (all of them extramedullary disease) [36].
In pediatric age, this rearrangement is extremely rare, with the male predominance being less pronounced. Five of the 11 cases described were girls. Nine cases were MPN with eosinophilia type CEL, one an T-ALL and the last one extramedullary B-ALL with MPN with eosinophilia in bone marrow [47].
Virtually almost all patients with PDGFRA rearrangements respond to imatinib, with a sensitivity 100 times greater than BCR::ABL1, hence the importance of its early detection for the prevention of organ and tissue damage [53].
3.2.1.2. Myeloid/lymphoid neoplasm with PDGFRB rearrangement
PDGFRB rearrangement follow in order of frequency to the PDGFRA rearrangement. Both types of neoplasms share a marked parallelism in their pronounced dominance in men, median age of presentation in adults aged 49–53 years (range 20–80), clinical and cytological presentation, and in their excellent response to imatinib. The most expressive difference between the two neoplasms lies in the lower incidence and amount of eosinophilia in M/LN-eo with PDGFRB rearrangement, which was limited to 50–58% of cases, and even absent in up to 21–25% of cases [36,54]. Organic involvement is not significant, except for splenomegaly, which is described in up to 83% of affected patients and which may be accompanied by hepatomegaly [54].
More than 40 different partners of PDGFRB rearrangement have been described, with ETV6 being the most frequent. In this case, the presentation as chronic myelomonocytic leukemia (CMML) with eosinophilia and t(5;12)(q32;p13.2)(22) is typical. Most of PDGFRB rearrangements are detected by alterations in the karyotype that affect 5q32 and by a break-apart FISH probe, although exceptionally this rearrangement can be cryptic and require RNA-sequencing for diagnosis. The high availability of partners seems to influence the heterogeneity of presentation in the chronic phase. This partner availability is greater than in individuals with FIP1L1-PDGFRA and may present as MPN or MDS/MPN (more like CMML and less like atypical chronic myeloid leukemia) (Figure 9). PDGFRB rearrangement can also present as acute leukemia, at diagnosis or in progression, typically as AML and rarely as T lymphoblastic lymphoma. In exceptional cases, progression as angioimmunoblastic T cell lymphoma has been described [54].
In a series of 135 adult patients with M /LN-eo), 26 cases corresponded to the subcategory of PDGFRB rearrangements. Of these cases, 22 (85%) presented as chronic phase, while four cases (15%) were in blast phase, with one patient developing a blast phase during follow-up. Of the five cases that developed acute leukemia, two were of myeloid lineage (one extramedullary case) and three cases of lymphoid lineage (two extramedullary, one a T-cell lymphoma) [36]. At least nine pediatric cases were reported [55,56,57,58,59,60], with no differences by gender.
Responses with durable remissions have been described to low doses of imatinib [54].
3.2.1.3. Myeloid/lymphoid neoplasm with FGFR1 rearrangement
The cases with this rearrangement are particularly rare, with around 100 cases having been reported. As with the previous subtypes, presentation is highly heterogeneous. However, the common link is the demonstration of the rearrangement of FGFR1 (chromosome 8p11). These rearrangements are usually observed by cytogenetics, although at least three cases have been described with cytogenetically cryptic FGFR1 detected by FISH or RNA sequencing [61]. At least 14 partner genes have been reported. The most frequently observed are ZMYM2 in t(8;13)(p11;q12), followed by BCR, CEP110, and FGFR1OP (FOP). While a certain tendency for the “partner” to influence the phenotype of the disease has been observed, this is not always the case. Somatic mutations are common, especially in RUNX1. The age range of the patients is wide (1–87 years), with a median age in adults in the largest series between 46 and 51 years [62,63]. Unlike PDGFRA and PDGFRB rearrangements, men displayed only a slight predominance of FGFR1 rearrangement – slightly above 50% [62,63].
M/LN-eo with FGFR1 rearranged present most frequently as MPN with or without concomitant involvement by acute leukemia or lymphoblastic lymphoma. Blood eosinophilia is common, ranging from 50% to 80% (assessed in short series of patients) [35,36,63]. A high tendency towards blastic transformation, AML, B or T-ALL, or mixed phenotype leukemia was observed [64,65,66]. Presentations have been described as B-ALL with BCR::FGFR1 rearrangement and additional cytogenetic alterations. When acute leukemia is treated and remitted, MPN appears with leukocytosis, splenomegaly, and isolated persistence of the FGFR1 rearrangement [65].
About 27 cases are described with this rearrangement that present as acute leukemia of a mixed phenotype, ambiguous lineage or switching lineage in the evolution (eight cases), without eosinophilia in up to 21% of the cases [61], similar to the cases with BCR::ABL and KMT2A rearrangements. Occasionally, patients with M/LN-eo and FGFR1 rearrangement associated with systemic mastocytosis were seen, although an activating mutation of KIT D816V was not always confirmed. These cases should be classified as systemic mastocytosis with an associated hematological malignancy [67].
A highly significant difference of this group of patients compared to PDGFRA and PDGFRB rearrangements is their lack of response to first- and second-generation tyrosine kinase inhibitors. The clinical course of the FGFR1 rearrangement cases is usually aggressive, with rapid progression to blast crisis and a short period of patient survival. Aggressive chemotherapy and allogeneic transplantation of hematopoietic progenitors are considered the best curative option. In isolated cases, clinical trials with other therapeutic targets, such as pemigatinib, ponatinib, sorafenib or olverembatinib, have demonstrated some clinical efficacy [74].
3.2.1.4. Myeloid/lymphoid neoplasm with PCM1::JAK2 rearrangement
In addition to PCM1::JAK2 o t(8;9)(p22;p24), ETV6::JAK2 and BCR::JAK2 stand out for their similar behavior. Thus, in the fifth edition of the WHO Classification and the ICC, this subtype has been redefined as NLM-eo with JAK2 rearrangement [32,75]. To date, approximately 100 cases of M/LN-eo with PCM1::JAK2 rearrangement or variants have been described. In a review of 66 cases with PCM1::JAK2 rearrangement [76], the mean age of presentation was 47 years (range 6–86 years), with 77% being men. The most common presentation was chronic, as MPN or MDS/MPN, with eosinophilia in 75% of patients, and often with erythroid proliferation dysplastic. Other less frequent presentations were as acute leukemia (AML or B/T ALL), or blast crisis from a previous MPN, or a minority as cutaneous T-cell lymphoma. When this subtype presents as B-ALL, a differential diagnosis must be made with Ph-like B-ALL, although rearrangements with certain “partners” (SSBP2, PAX5, RFX3, USP25 and ZNF274) are normally considered Ph-like B-ALL. Survival is highly variable, depending on presentation. Target therapy with JAK2 inhibitors, such as ruxolitinib, may be of benefit [77] but allogeneic transplantation of hematopoietic progenitors is considered the only curative option [37].
3.2.1.5. Myeloid/lymphoid neoplasms with eosinophilia and defining gene rearrangement: new entities
In 2022, both the fifth edition of the WHO Classification and the ICC recognized the following three new groups within the NLM-eo and tyrosine kinase (TK) fusion genes category:
Myeloid /lymphoid neoplasms with FLT3/t(v;13q1212) rearrangements
These neoplasms are rare, just over 30 cases have been described in the literature, including a multicenter study with 12 patients and case report descriptions, [78,79]. The most frequent fusion partner is ETV6 (12p13), while the following have also been described: ZMYM2, TRIP11, SPTBN1, GOLGB1, CCDC88C, MYO18A, and BCR. This subtype predominates in men, although, with an M:F ratio (1.9:1), not as marked as in other types of M/LN-eo. The age at diagnosis is highly variable, with an age range from eight months to 80 years. Eosinophilia is frequent, although absent in some cases [78,80]. Regarding presentation, these patients often manifest as MPN-eo or MDS/NMP. Extramedullary involvement is common, including T-lymphoblastic lymphoma, mixed-phenotype acute leukemia, myeloid sarcoma, and, rarely, B-ALL. Coexistence of extramedullary disease and chronic form in the bone marrow have also been described [35]. Pediatric cases are anecdotal, but at least three cases have been described [78,81,82].
FLT3/t(v;13q1212) rearrangements are largely detectable by cytogenetics (chromosome 13q12) and FISH but can be cryptic (like TRIP11), which would require RNA-sequencing. The importance of their recognition lies in the fact that they respond to FLT3 inhibitors.
Myeloid/lymphoid neoplasms with ETV6::ABL1/t(9;12)(q34.1;p13.2) fusion
This category currently only includes one ABL1 partner, which is ETV6. At least 13 other partners of ABL1 have been described, but most cases present as Ph-like B-ALL or de novo T-ALL. These cases are therefore not included in this category [83,84].
Because the ETV6::ABL1 rearrangement shares the constitutive activation of the same tyrosine kinase as the BCR::ABL1 rearrangement, it has the peculiarity of being the one that most closely resembles chronic myeloid leukemia (CML), although it is much less frequent. The myeloid neoplasm with ETV6::ABL1 rearrangement typically presents in chronic phase with a CML- like morphology. The presence of eosinophilia is almost constant and basophilia is common. As in CML, there may be progression to blast crisis (myeloid or lymphoid, mostly B lineage and a minority T) or rarely present as AML. When the ETV6::ABL1 rearrangement presents as B-ALL, it most often corresponds to a Ph-like B-ALL, and eosinophilia is almost never present.
In the series by Zaliova and collaborators (own and a review of the literature) that includes 44 cases, up to 22 cases were ALL (21 being Ph-like ALL, 21 B-ALL and one T-ALL) and 22 myeloid neoplasms (18 MPN cases and four AML cases). The median age in the 18 cases described in the chronic phase is 51 (range 24–72 years), with a predominance in men: The male to female ratio is 2.4:1. In the same series, children and young adults with this rearrangement presented more like acute leukemias and older adults more like MPN [85].
Most of these rearrangements are produced by an insertion of either ABL1 in ETV6 or ETV6 in ABL1. A translocation t(9;12) is less frequent since both genes would be transcribed in the opposite direction in this case, and an inversion of one of the two genes would also be required. Thus, its detection by cytogenetics is usually cryptic, and it is diagnosed by FISH with break-apart probes for ABL1 (more useful when the insertion is of ABL1 in ETV6) and for ETV6 (more useful when the insertion is of ETV6 in ABL1). This rearrangement can also be suspected when probes such as BCR::ABL or ETV6::RUNX1 produce extra signals for ABL1 or ETV6. RNA-sequencing can also confirm this rearrangement and provide a diagnosis if it had been previously overlooked.
In the chronic phase, these patients respond to tyrosine kinase inhibitors, such as imatinib, or second and third generation ones. Just like in CML, the response is poor in the blast phase.
Myeloid/lymphoid neoplasms with other tyrosine kinase gene fusions
These gene fusions affect other tyrosine kinase genes not included in the previous categories, have hematopoietic lineage plasticity and eosinophilia, and present similarly. Some of these rearrangements are ETV6::FGFR2 [86], ETV6::LYN [87], ETV6::NTRK3 [88], RANBP2::ALK [89], BCR::RET, and FGFR1OP::RET [90].
3.2.2. Core Binding Factor acute myeloid leukemias
3.2.2.1. LMA with inv (16)( p13.1q22) o t(16;16)(p13.1;q22); CBFB::MYH11
The diversity of these leukemias presents a myelomonocytic cytology, sometimes purely monocytic. The percentage of eosinophils in the blood is usually normal – although eosinophilia may exist as these leukemias are usually hyperleukocytic – and eosinophils do not present relevant dysplasia. In the bone marrow examination, eosinophilia is usually frank, with unmature elements. Thick and dark violet-purple granules are characteristic (Figure 10) [91,92].
At times, the presence of eosinophils, less than 6%, is observed in a blastic hypercellularity environment. In our series of 13 cases, we observed blood eosinophilia in 6/13 (46%) and marrow eosinophilia in 10/13 (77%). In exceptional cases, bone marrow eosinophilia becomes massive, without expression in the blood (Figure 11).
Cytochemically, positivity is typical both with the PAS reaction and in the enzymatic demonstration of chloracetate esterase (Figure 12). In the immunophenotypic pattern, the presence of blasts with myeloid differentiation is common, as is another subtype of blasts with commitment to monocytes according to their markers [93].
3.2.2.2. LMA with t(8; 21)( q22;q22.1); RUNX1::RUNX1T1
These leukemias are known for their peculiar morphology, with large Auer rods in the blasts and marked neutrophil dysgranulopoiesis. Eosinophilia, both in the blood and in the marrow, is much less evident than in the previous group. In a series of 165 patients collected by the Spanish Hematological Cytology Group [94], blood eosinophilia was observed in 7/148 cases (4.7%) and marrow eosinophilia in 22/137 (16%); (unpublished data). The eosinophils are less dysplastic and without thick and dark red-violet granulation (Figure 13). The chlorocetate esterase is negative, and the PAS reaction may be negative or positive [91,95,96]. In the immunophenotype, most common is the presence of immature blasts with expression of CD34 and CD117, with expression of myeloid markers, such as CD13, CD33 and lymphoid markers, especially CD19 and CD56 [93].
3.2.3. Mastocytosis
In mastocytosis, a clonal proliferation of eosinophils can be observed that would have its origin in the same neoplastic precursor (multilineage involvement due to the KIT mutation).
The 2022 WHO classification includes peripheral and/or central eosinophilia among the signs of myeloproliferation and/or myelodysplasia that are part of the B findings of systemic mastocytosis (SM). However, this is only the case when no reactive cause is identified and criteria for associated hematological neoplasia are not met [32,97].
In a series of 2,350 patients with mastocytosis, eosinophilia was reported in 6.8% and hypereosinophilia in 3.1%, mainly associated with advanced forms. Eosinophilia is frequently seen in bone marrow aspirates and biopsies, even in cases without significant peripheral eosinophilia [98,99]. In exceptional cases, eosinophils can become phagocytosed by the mast cells of an MS, perhaps as a sign of malignancy [100].
In advanced MS with eosinophilia, a differential diagnosis must be made with M/LN- eo and defining gene rearrangements because the coexistence of both is exceptional [101]. It should be noted that in the M/LN- eo an increase in mast cells with aberrant expression of CD25 with or without expression of CD2 may occur. However, dense multifocal infiltrates do not form, and they do not present the KIT mutation [32]. Another frequently described marker (80%) in MS mast cells is CD30 [102,103], for which no conclusive descriptions currently exist in the M/ LN- eo. To diagnose the presence of both entities, it is necessary to demonstrate the rearrangement of an M/LN- eo and meet MS criteria.
3.2.4. Myeloproliferative neoplasms
MPN can present with eosinophilia that requires the ruling out of associated MS or M/LN-eo.
3.2.5. Chronic myeloid leukemia
In the chronic phase, eosinophilia may be present in peripheral blood and bone marrow. Unusual presentation forms have been identified, and they are recognized as “eosinophilic variants of CML” with intense eosinophilia similar to chronic eosinophilic leukemia (CEL). Approximately six cases have been described, with a median age lower than that of CML, most without splenomegaly, and with frequent cutaneous manifestations and vascular symptoms [104].
3.2.6. Chronic eosinophilic leukemia
CEL (2022 WHO), or CEL, not otherwise specified (ICC 2022) is an extremely rare disorder. CEL is accompanied by peripheral hypereosinophilia and significant infiltration of the marrow and different organs. Cases with other MPN or MPN/MDS, NLM- eo, MDS, mastocytosis and AML with CBF translocations are excluded. The diagnostic criteria for CEL have been updated in 2022 WHO classification. In addition to eosinophilia (on at least two occasions over an interval of at least four weeks), the criteria include evidence of clonality and abnormal bone marrow morphology. The WHO has eliminated the increase in blasts (≥ 5% in the bones marrow and/ or ≥ 2% in the peripheral blood) as an alternative CEL criterion to clonality. This finding is maintained along with the rest of the criteria in the ICC 2022 [32,75].
Organic involvement due to eosinophil infiltration in the absence of abnormal bone marrow morphology, blastosis and/or genetic clonality points to idiopathic HES. In the same circumstances, if organic damage is absent, idiopathic hypereosinophilia would be considered.
One of the best studied series is from the Mayo Clinic, with 17 patients diagnosed according to 2016 WHO criteria (median 63 years, male 88%). Most patients presented with systemic symptoms due to digestive, cardiac, and pulmonary involvement, as well as splenomegaly and hepatomegaly. The blood count showed leukocytosis with eosinophilia (median 6.4 x 109/l) and moderate anemia. Eosinophils presented dysplasia in half of the cases. In bone marrow, an increase in the M:E ratio 5:1 (47%), eosinophilia (median 43%), dysmegakaryopoiesis (41.2%), fibrosis (17%), and one case with blasts (>5%) was described. Cytogenetic alterations were described in 15/17 patients and mutations in ASXL1 in two patients. The prognosis was poor, with a median survival of 16 months and progression to acute leukemia in only three cases [105].
3.2.7. Myeloproliferative / myelodysplastic neoplasms
MDS/MPN can present with eosinophilia. In these cases, an associated systemic mastocytosis must be ruled out and/or, less frequently, a M/LN-eo.
3.2.8. Myelodysplastic neoplasms
The finding of eosinophilia in MDS is unusual and requires reactive causes to be ruled out. A retrospective series of 288 patients described marrow eosinophilia in 36 (12.5%) patients, of which only 18% had peripheral eosinophilia. The usual assumption is that eosinophilia is part of the neoplastic clone. The most common cytogenetic abnormalities are alterations of chromosome 7, complex karyotypes, and isochromosome 17q. A worse survival of these patients is reported, but it could be mediated by the underlying cytogenetics [106].
4. Conclusions
* Eosinophilia associated with a hematological malignancy can be reactive or secondary to the production of cytokines eosinophilopoietic. This type of eosinophilia is mainly observed in lymphoid neoplasms. Eosnophilia can also be neoplastic and primary derived from the malignant clone, usually in myeloid or stem cells neoplasms.
* No data collected in cytological and cytometric studies can predict whether eosinophilia is reactive or clonal. The verification is indirect and supported by the categorization of the accompanying hematological neoplasm.
* In the presence of eosinophilia, flow cytometry can identify a clonal T lymphoid population, mast cells with an aberrant phenotype, and blasts from an acute leukemia.
Funding
This research received no external funding
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ehrlich, P. (1879). Uber die spezifischen Granulationen des Blutes. Arch Anat Physiol., 571-579.
- Kay AB. The early history of the eosinophil. Clin Exp Allergy. 2015 Mar;45(3):575-82. [CrossRef] [PubMed]
- Woessner Casas S, Florensa Brich L. La citología óptica en el diagnóstico hematológico. 5ª. Ed. Madrid: Acción Médica S. A.; 2006.
- Johnston LK, Bryce PJ. Understanding Interleukin 33 and Its Roles in Eosinophil. Development. Front Med (Lausanne). 2017 May 2;4:51. [CrossRef]
- Varricchi G, Galdiero MR, Loffredo S, Lucarini V, Marone G, Mattei F, Marone G, Schiavoni G. Eosinophils: The unsung heroes in cancer? Oncoimmunology. 2017 Nov 13;7(2):e1393134. [CrossRef]
- DiScipio RG, Schraufstatter IU. The role of the complement anaphylatoxins in the recruitment of eosinophils. Int Immunopharmacol. 2007 Dec 20;7(14):1909-23. [CrossRef] [PubMed]
- Melo RCN, Weller PF. Contemporary understanding of the secretory granules in human eosinophils. J Leukoc Biol. 2018 Jul;104(1):85-93. [CrossRef]
- Acharya KR, Ackerman SJ. Eosinophil granule proteins: form and function. J Biol Chem. 2014 Jun 20;289(25):17406-15. [CrossRef]
- Kanda A, Yasutaka Y, Van Bui D, Suzuki K, Sawada S, Kobayashi Y, Asako M, Iwai H. Multiple Biological Aspects of Eosinophils in Host Defense, Eosinophil-Associated Diseases, Immunoregulation, and Homeostasis: Is Their Role Beneficial, Detrimental, Regulator, or Bystander? Biol Pharm Bull. 2020;43(1):20-30. [CrossRef] [PubMed]
- Goasguen JE, Bennett JM, Bain BJ, Brunning R, Zini G, Vallespi MT, Tomonaga M, Locher C; International Working Group on Morphology of MDS. The role of eosinophil morphology in distinguishing between reactive eosinophilia and eosinophilia as a feature of a myeloid neoplasm. Br J Haematol. 2020 Nov;191(3):497-504. [CrossRef]
- Zederbauer M, Furtmüller PG, Brogioni S, Jakopitsch C, Smulevich G, Obinger C. Heme to protein linkages in mammalian peroxidases: impact on spectroscopic, redox and catalytic properties. Nat Prod Rep. 2007 Jun;24(3):571-84. [CrossRef] [PubMed]
- Orfao A, Matarraz S, Pérez-Andrés M, et al. Immunophenotypic dissection of normal hematopoiesis. J Immunol Methods. 2019;475:112684. [CrossRef]
- Bain, BJ. Eosinophilic leukaemias and the idiopathic hypereosinophilic syndrome. Br J Haematol. 1996 Oct;95(1):2-9. [PubMed]
- Valent P, Klion AD, Horny HP, Roufosse F, Gotlib J, Weller PF, Hellmann A, Metzgeroth G, Leiferman KM, Arock M, Butterfield JH, Sperr WR, Sotlar K, Vandenberghe P, Haferlach T, Simon HU, Reiter A, Gleich GJ. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012 Sep;130(3):607-612.e9. [CrossRef]
- Delgado-Serrano J, Morales-Camacho RM, Caballero-Velázquez T, García-Canale S, Vargas MT, Prats-Martín C. Eosinophils engulfing platelets and with ring-shaped nuclei in nivolumab-associated eosinophilia. Br J Haematol. 2020 Mar;188(6):812. [CrossRef] [PubMed]
- Morales-Camacho RM, Prats-Martín C. Eosinophils with ring-shaped nuclei in a patient treated with adalimumab. Blood. 2019 Jan 3;133(1):101. [CrossRef] [PubMed]
- Valent P, Degenfeld-Schonburg L, Sadovnik I, Horny HP, Arock M, Simon HU, Reiter A, Bochner BS. Eosinophils and eosinophil-associated disorders: immunological, clinical, and molecular complexity. Semin Immunopathol. 2021 Jun;43(3):423-438. [CrossRef]
- Roufosse F, Garaud S, de Leval L. Lymphoproliferative disorders associated with hypereosinophilia. Semin Hematol. 2012 Apr;49(2):138-48. [CrossRef] [PubMed]
- Samoszuk M, Nansen L. Detection of interleukin-5 messenger RNA in Reed-Sternberg cells of Hodgkin's disease with eosinophilia. Blood. 1990 Jan 1;75(1):13-6. [PubMed]
- Muñoz-García N, Lima M, Villamor N, Morán-Plata FJ, Barrena S, Mateos S, Caldas C, Balanzategui A, Alcoceba M, Domínguez A, Gómez F, Langerak AW, van Dongen JJM, Orfao A, Almeida J. Anti-TRBC1 Antibody-Based Flow Cytometric Detection of T-Cell Clonality: Standardization of Sample Preparation and Diagnostic Implementation. Cancers (Basel). 2021 Aug 30;13(17):4379. [CrossRef]
- Tancrède-Bohin E, Ionescu MA, de La Salmonière P, Dupuy A, Rivet J, Rybojad M, Dubertret L, Bachelez H, Lebbé C, Morel P. Prognostic value of blood eosinophilia in primary cutaneous T-cell lymphomas. Arch Dermatol. 2004 Sep;140(9):1057-61. [CrossRef] [PubMed]
- Pulitzer MP, Horna P, Almeida J. Sézary syndrome and mycosis fungoides: An overview, including the role of immunophenotyping. Cytometry B Clin Cytom. 2021 Mar;100(2):132-138. [CrossRef]
- Pu Q, Qiao J, Liu Y, Cao X, Tan R, Yan D, Wang X, Li J, Yue B. Differential diagnosis and identification of prognostic markers for peripheral T-cell lymphoma subtypes based on flow cytometry immunophenotype profiles. Front Immunol. 2022 Nov 18;13:1008695. [CrossRef]
- Tamaki T, Karube K, Sakihama S, Tsuruta Y, Awazawa R, Hayashi M, Nakada N, Matsumoto H, Yagi N, Ohshiro K, Nakazato I, Kitamura S, Nishi Y, Miyagi T, Yamaguchi S, Nakachi S, Morishima S, Masuzaki H, Takahashi K, Fukushima T, Wada N. A Comprehensive Study of the Immunophenotype and its Clinicopathologic Significance in Adult T-Cell Leukemia/Lymphoma. Mod Pathol. 2023 Aug;36(8):100169. [CrossRef] [PubMed]
- Shi Y, Wang C. What we have learned about lymphocytic variant hypereosinophilic syndrome: A systematic literature review. Clin Immunol. 2022 Apr;237:108982. [CrossRef] [PubMed]
- Fournier B, Balducci E, Duployez N, Clappier E, Cuccuini W, Arfeuille C, Caye-Eude A, Delabesse E, Bottollier-Lemallaz Colomb E, Nebral K, Chrétien ML, Derrieux C, Cabannes-Hamy A, Dumezy F, Etancelin P, Fenneteau O, Frayfer J, Gourmel A, Loosveld M, Michel G, Nadal N, Penther D, Tigaud I, Fournier E, Reismüller B, Attarbaschi A, Lafage-Pochitaloff M, Baruchel A. B-ALL With t(5;14)(q31;q32); IGH-IL3 Rearrangement and Eosinophilia: A Comprehensive Analysis of a Peculiar IGH-Rearranged B-ALL. Front Oncol. 2019 Dec 10;9:1374. [CrossRef]
- McClure BJ, Heatley SL, Rehn J, Breen J, Sutton R, Hughes TP, Suppiah R, Revesz T, Osborn M, White D, Yeung DT, White DL. High-risk B-cell acute lymphoblastic leukaemia presenting with hypereosinophilia and acquiring a novel PAX5 fusion on relapse. Br J Haematol. 2020 Oct;191(2):301-304. [CrossRef] [PubMed]
- Kwon A, Fuda F, Gagan J, John S, Aquino V, Chen W. Rare circulating lymphoblasts with striking eosinophilia: A rare case of B-lymphoblastic leukemia with PAX5::ZCCHC7. Am J Hematol. 2023 Jun;98(6):989-990. [CrossRef] [PubMed]
- Ferruzzi V, Santi E, Gurdo G, Arcioni F, Caniglia M, Esposito S. Acute Lymphoblastic Leukemia with Hypereosinophilia in a Child: Case Report and Literature Review. Int J Environ Res Public Health. 2018 Jun 4;15(6):1169. [CrossRef]
- Pierini V, Bardelli V, Giglio F, Arniani S, Matteucci C, Pellanera F, Quintini M, Crescenzi B, Bruno A, Sabattini E, Falcinelli E, Ruggeri L, Ronchi P, Ponzoni M, Ciceri F, Mecucci C, La Starza R. A Novel t(5;7)(q31;q21)/CDK6::IL3 in Immature T-cell Acute Lymphoblastic Leukemia With IL3 Expression and Eosinophilia. Hemasphere. 2022 Oct 19;6(11):e795. [CrossRef]
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J (Eds): WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition). IARC: Lyon 2017.
- Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, Bejar R, Berti E, Busque L, Chan JKC, Chen W, Chen X, Chng WJ, Choi JK, Colmenero I, Coupland SE, Cross NCP, De Jong D, Elghetany MT, Takahashi E, Emile JF, Ferry J, Fogelstrand L, Fontenay M, Germing U, Gujral S, Haferlach T, Harrison C, Hodge JC, Hu S, Jansen JH, Kanagal-Shamanna R, Kantarjian HM, Kratz CP, Li XQ, Lim MS, Loeb K, Loghavi S, Marcogliese A, Meshinchi S, Michaels P, Naresh KN, Natkunam Y, Nejati R, Ott G, Padron E, Patel KP, Patkar N, Picarsic J, Platzbecker U, Roberts I, Schuh A, Sewell W, Siebert R, Tembhare P, Tyner J, Verstovsek S, Wang W, Wood B, Xiao W, Yeung C, Hochhaus A. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022 Jul;36(7):1703-1719. [CrossRef]
- Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, Wang SA, Bagg A, Barbui T, Branford S, Bueso-Ramos CE, Cortes JE, Dal Cin P, DiNardo CD, Dombret H, Duncavage EJ, Ebert BL, Estey EH, Facchetti F, Foucar K, Gangat N, Gianelli U, Godley LA, Gökbuget N, Gotlib J, Hellström-Lindberg E, Hobbs GS, Hoffman R, Jabbour EJ, Kiladjian JJ, Larson RA, Le Beau MM, Loh ML, Löwenberg B, Macintyre E, Malcovati L, Mullighan CG, Niemeyer C, Odenike OM, Ogawa S, Orfao A, Papaemmanuil E, Passamonti F, Porkka K, Pui CH, Radich JP, Reiter A, Rozman M, Rudelius M, Savona MR, Schiffer CA, Schmitt-Graeff A, Shimamura A, Sierra J, Stock WA, Stone RM, Tallman MS, Thiele J, Tien HF, Tzankov A, Vannucchi AM, Vyas P, Wei AH, Weinberg OK, Wierzbowska A, Cazzola M, Döhner H, Tefferi A. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022 Sep 15;140(11):1200-1228. [CrossRef]
- Kim AS, Pozdnyakova O. SOHO State of the Art Updates and Next Questions | Myeloid/Lymphoid Neoplasms with Eosinophilia and Gene Rearrangements: Diagnostic Pearls and Pitfalls. Clin Lymphoma Myeloma Leuk. 2022 Sep;22(9):643-651. [CrossRef] [PubMed]
- Pozdnyakova O, Orazi A, Kelemen K, King R, Reichard KK, Craig FE, Quintanilla-Martinez L, Rimsza L, George TI, Horny HP, Wang SA. Myeloid/Lymphoid Neoplasms Associated With Eosinophilia and Rearrangements of PDGFRA, PDGFRB, or FGFR1 or With PCM1-JAK2. Am J Clin Pathol. 2021 Feb 4;155(2):160-178. [CrossRef] [PubMed]
- Metzgeroth G, Steiner L, Naumann N, Lübke J, Kreil S, Fabarius A, Haferlach C, Haferlach T, Hofmann WK, Cross NCP, Schwaab J, Reiter A. Myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions: reevaluation of the defining characteristics in a registry-based cohort. Leukemia. 2023 Sep;37(9):1860-1867. [CrossRef]
- Saft L, Kvasnicka HM, Boudova L, Gianelli U, Lazzi S, Rozman M. Myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase fusion genes: A workshop report with focus on novel entities and a literature review including paediatric cases. Histopathology. 2023 Dec;83(6):829-849. [CrossRef] [PubMed]
- Shomali W, Gotlib J. World Health Organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022 Jan 1;97(1):129-148. [CrossRef] [PubMed]
- Rohmer J, Couteau-Chardon A, Trichereau J, Panel K, Gesquiere C, Ben Abdelali R, Bidet A, Bladé JS, Cayuela JM, Cony-Makhoul P, Cottin V, Delabesse E, Ebbo M, Fain O, Flandrin P, Galicier L, Godon C, Grardel N, Guffroy A, Hamidou M, Hunault M, Lengline E, Lhomme F, Lhermitte L, Machelart I, Mauvieux L, Mohr C, Mozicconacci MJ, Naguib D, Nicolini FE, Rey J, Rousselot P, Tavitian S, Terriou L, Lefèvre G, Preudhomme C, Kahn JE, Groh M; CEREO and GBMHM collaborators. Epidemiology, clinical picture and long-term outcomes of FIP1L1-PDGFRA-positive myeloid neoplasm with eosinophilia: Data from 151 patients. Am J Hematol. 2020 Nov;95(11):1314-1323. [CrossRef] [PubMed]
- Score J, Curtis C, Waghorn K, Stalder M, Jotterand M, Grand FH, Cross NC. Identification of a novel imatinib responsive KIF5B-PDGFRA fusion gene following screening for PDGFRA overexpression in patients with hypereosinophilia. Leukemia. 2006 May;20(5):827-32. [CrossRef] [PubMed]
- Walz C, Curtis C, Schnittger S, Schultheis B, Metzgeroth G, Schoch C, Lengfelder E, Erben P, Müller MC, Haferlach T, Hochhaus A, Hehlmann R, Cross NC, Reiter A. Transient response to imatinib in a chronic eosinophilic leukemia associated with ins(9;4)(q33;q12q25) and a CDK5RAP2-PDGFRA fusion gene. Genes Chromosomes Cancer. 2006 Oct;45(10):950-6. [CrossRef] [PubMed]
- Curtis CE, Grand FH, Musto P, Clark A, Murphy J, Perla G, Minervini MM, Stewart J, Reiter A, Cross NC. Two novel imatinib-responsive PDGFRA fusion genes in chronic eosinophilic leukaemia. Br J Haematol. 2007 Jul;138(1):77-81. [CrossRef] [PubMed]
- Baxter EJ, Hochhaus A, Bolufer P, Reiter A, Fernandez JM, Senent L, Cervera J, Moscardo F, Sanz MA, Cross NC. The t(4;22)(q12;q11) in atypical chronic myeloid leukaemia fuses BCR to PDGFRA. Hum Mol Genet. 2002 Jun 1;11(12):1391-7. [CrossRef] [PubMed]
- Chalmers ZR, Ali SM, Ohgami RS, Campregher PV, Frampton GM, Yelensky R, Elvin JA, Palma NA, Erlich R, Vergilio JA, Chmielecki J, Ross JS, Stephens PJ, Hermann R, Miller VA, Miles CR. Comprehensive genomic profiling identifies a novel TNKS2-PDGFRA fusion that defines a myeloid neoplasm with eosinophilia that responded dramatically to imatinib therapy. Blood Cancer J. 2015 Feb 6;5(2):e278. [CrossRef]
- Sugimoto Y, Sada A, Shimokariya Y, Monma F, Ohishi K, Masuya M, Nobori T, Matsui T, Katayama N. A novel FOXP1-PDGFRA fusion gene in myeloproliferative neoplasm with eosinophilia. Cancer Genet. 2015 Oct;208(10):508-12. [CrossRef] [PubMed]
- Jain N, Khoury JD, Pemmaraju N, Kollipara P, Kantarjian H, Verstovsek S. Imatinib therapy in a patient with suspected chronic neutrophilic leukemia and FIP1L1-PDGFRA rearrangement. Blood. 2013 Nov 7;122(19):3387-8. [CrossRef] [PubMed]
- Akiely R, Almasri F, Almasri N, Abu-Ghosh A. Case Report: Pediatric myeloid/lymphoid neoplasm with eosinophilia and PDGFRA rearrangement: The first case presenting as B-lymphoblastic lymphoma. Front Pediatr. 2022 Dec 14;10:1059527. [CrossRef]
- Huang Q, Snyder DS, Chu P, Gaal KK, Chang KL, Weiss LM. PDGFRA rearrangement leading to hyper-eosinophilia, T-lymphoblastic lymphoma, myeloproliferative neoplasm and precursor B-cell acute lymphoblastic leukemia. Leukemia. 2011 Feb;25(2):371-5. [CrossRef] [PubMed]
- Trempat P, Villalva C, Laurent G, Armstrong F, Delsol G, Dastugue N, Brousset P. Chronic myeloproliferative disorders with rearrangement of the platelet-derived growth factor alpha receptor: a new clinical target for STI571/Glivec. Oncogene. 2003 Aug 28;22(36):5702-6. [CrossRef] [PubMed]
- Roberts KG, Gu Z, Payne-Turner D, McCastlain K, Harvey RC, Chen IM, Pei D, Iacobucci I, Valentine M, Pounds SB, Shi L, Li Y, Zhang J, Cheng C, Rambaldi A, Tosi M, Spinelli O, Radich JP, Minden MD, Rowe JM, Luger S, Litzow MR, Tallman MS, Wiernik PH, Bhatia R, Aldoss I, Kohlschmidt J, Mrózek K, Marcucci G, Bloomfield CD, Stock W, Kornblau S, Kantarjian HM, Konopleva M, Paietta E, Willman CL, Mullighan CG. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J Clin Oncol. 2017 Feb;35(4):394-401. [CrossRef]
- Krigstein M, Menzies A, Fay K, Lukeis R, Cheung K, Parker A. FIP1L1::PDGFRA fusion driving three synchronous haematological malignancies. Pathology. 2023 Dec;55(7):1040-1044. [CrossRef] [PubMed]
- Bellani V, Croci GA, Bucelli C, Maronese CA, Alberti S, Iurlo A, Cattaneo D. Lymphomatoid papulosis associated with myeloid neoplasm with eosinophilia and FIP1L1::PDGFRA rearrangement: Successful imatinib treatment in two cases. J Dermatol. 2023 Oct;50(10):1330-1334. [CrossRef] [PubMed]
- Baccarani M, Cilloni D, Rondoni M, Ottaviani E, Messa F, Merante S, Tiribelli M, Buccisano F, Testoni N, Gottardi E, de Vivo A, Giugliano E, Iacobucci I, Paolini S, Soverini S, Rosti G, Rancati F, Astolfi C, Pane F, Saglio G, Martinelli G. The efficacy of imatinib mesylate in patients with FIP1L1-PDGFRalpha-positive hypereosinophilic syndrome. Results of a multicenter prospective study. Haematologica. 2007 Sep;92(9):1173-9. [CrossRef] [PubMed]
- Jawhar M, Naumann N, Schwaab J, Baurmann H, Casper J, Dang TA, Dietze L, Döhner K, Hänel A, Lathan B, Link H, Lotfi S, Maywald O, Mielke S, Müller L, Platzbecker U, Prümmer O, Thomssen H, Töpelt K, Panse J, Vieler T, Hofmann WK, Haferlach T, Haferlach C, Fabarius A, Hochhaus A, Cross NCP, Reiter A, Metzgeroth G. Imatinib in myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRB in chronic or blast phase. Ann Hematol. 2017 Sep;96(9):1463-1470. [CrossRef] [PubMed]
- Darbyshire PJ, Shortland D, Swansbury GJ, Sadler J, Lawler SD, Chessells JM. A myeloproliferative disease in two infants associated with eosinophilia and chromosome t(1;5) translocation. Br J Haematol. 1987 Aug;66(4):483-6. [CrossRef] [PubMed]
- Wilkinson K, Velloso ER, Lopes LF, Lee C, Aster JC, Shipp MA, Aguiar RC. Cloning of the t(1;5)(q23;q33) in a myeloproliferative disorder associated with eosinophilia: involvement of PDGFRB and response to imatinib. Blood. 2003 Dec 1;102(12):4187-90. [CrossRef] [PubMed]
- Li Z, Yang R, Zhao J, Yuan R, Lu Q, Li Q, Tse W. Molecular diagnosis and targeted therapy of a pediatric chronic eosinophilic leukemia patient carrying TPM3-PDGFRB fusion. Pediatr Blood Cancer. 2011 Mar;56(3):463-6. [CrossRef] [PubMed]
- Abraham S, Salama M, Hancock J, Jacobsen J, Fluchel M. Congenital and childhood myeloproliferative disorders with eosinophilia responsive to imatinib. Pediatr Blood Cancer. 2012 Nov;59(5):928-9. [CrossRef] [PubMed]
- Berking AC, Flaadt T, Behrens YL, Yoshimi A, Leipold A, Holzer U, Lang P, Quintanilla-Martinez L, Schlegelberger B, Reiter A, Niemeyer C, Strahm B, Göhring G. Rare and potentially fatal - Cytogenetically cryptic TNIP1::PDGFRB and PCM1::FGFR1 fusion leading to myeloid/lymphoid neoplasms with eosinophilia in children. Cancer Genet. 2023 Apr;272-273:29-34. [CrossRef] [PubMed]
- Wang SC, Yang WY. Myeloid neoplasm with eosinophilia and rearrangement of platelet-derived growth factor receptor beta gene in children: Two case reports. World J Clin Cases. 2021 Jan 6;9(1):204-210. [CrossRef]
- McKeague SJ, O'Rourke K, Fanning S, Joy C, Throp D, Adams R, Harvey Y, Keng TB. Acute leukemia with cytogenetically cryptic FGFR1 rearrangement and lineage switch during therapy: A case report and literature review. Am J Clin Pathol. 2023 Oct 19:aqad135. [CrossRef] [PubMed]
- Strati P, Tang G, Duose DY, Mallampati S, Luthra R, Patel KP, Hussaini M, Mirza AS, Komrokji RS, Oh S, Mascarenhas J, Najfeld V, Subbiah V, Kantarjian H, Garcia-Manero G, Verstovsek S, Daver N. Myeloid/lymphoid neoplasms with FGFR1 rearrangement. Leuk Lymphoma. 2018 Jul;59(7):1672-1676. [CrossRef] [PubMed]
- Hernández-Boluda JC, Pereira A, Zinger N, Gras L, Martino R, Nikolousis E, Finke J, Chinea A, Rambaldi A, Robin M, Saccardi R, Natale A, Snowden JA, Tsirigotis P, Vallejo C, Wulf G, Xicoy B, Russo D, Maertens J, Daguindau E, Lenhoff S, Hayden P, Czerw T, McLornan DP, Yakoub-Agha I. Allogeneic hematopoietic cell transplantation in patients with myeloid/lymphoid neoplasm with FGFR1-rearrangement: a study of the Chronic Malignancies Working Party of EBMT. Bone Marrow Transplant. 2022 Mar;57(3):416-422. [CrossRef] [PubMed]
- Wakim JJ, Tirado CA, Chen W, Collins R. t(8;22)/BCR-FGFR1 myeloproliferative disorder presenting as B-acute lymphoblastic leukemia: report of a case treated with sorafenib and review of the literature. Leuk Res. 2011 Sep;35(9):e151-3. [CrossRef] [PubMed]
- Haslam K, Langabeer SE, Kelly J, Coen N, O'Connell NM, Conneally E. Allogeneic Hematopoietic Stem Cell Transplantation for a BCR-FGFR1 Myeloproliferative Neoplasm Presenting as Acute Lymphoblastic Leukemia. Case Rep Hematol. 2012;2012:620967. [CrossRef]
- Baldazzi C, Iacobucci I, Luatti S, Ottaviani E, Marzocchi G, Paolini S, Stacchini M, Papayannidis C, Gamberini C, Martinelli G, Baccarani M, Testoni N. B-cell acute lymphoblastic leukemia as evolution of a 8p11 myeloproliferative syndrome with t(8;22)(p11;q11) and BCR-FGFR1 fusion gene. Leuk Res. 2010 Oct;34(10):e282-5. [CrossRef] [PubMed]
- Duckworth CB, Zhang L, Li S. Systemic mastocytosis with associated myeloproliferative neoplasm with t(8;19)(p12;q13.1) and abnormality of FGFR1: report of a unique case. Int J Clin Exp Pathol. 2014 Jan 15;7(2):801-7.
- Macdonald D, Aguiar RC, Mason PJ, Goldman JM, Cross NC. A new myeloproliferative disorder associated with chromosomal translocations involving 8p11: a review. Leukemia. 1995 Oct;9(10):1628-30. [PubMed]
- Macdonald D, Reiter A, Cross NC. The 8p11 myeloproliferative syndrome: a distinct clinical entity caused by constitutive activation of FGFR1. Acta Haematol. 2002;107(2):101-7. [CrossRef] [PubMed]
- Nakayama H, Inamitsu T, Ohga S, Kai T, Suda M, Matsuzaki A, Ueda K. Chronic myelomonocytic leukaemia with t(8;9)(p11;q34) in childhood: an example of the 8p11 myeloproliferative disorder? Br J Haematol. 1996 Mar;92(3):692-5. [CrossRef] [PubMed]
- van den Berg H, Kroes W, van der Schoot CE, Dee R, Pals ST, Bouts TH, Slater RM. A young child with acquired t(8;9)(p11;q34): additional proof that 8p11 is involved in mixed myeloid/T lymphoid malignancies. Leukemia. 1996 Jul;10(7):1252-3. [PubMed]
- Wong WS, Cheng KC, Lau KM, Chan NP, Shing MM, Cheng SH, Chik KW, Li CK, Ng MH. Clonal evolution of 8p11 stem cell syndrome in a 14-year-old Chinese boy: a review of literature of t(8;13) associated myeloproliferative diseases. Leuk Res. 2007 Feb;31(2):235-8. [CrossRef] [PubMed]
- Zhang WW, Habeebu S, Sheehan AM, Naeem R, Hernandez VS, Dreyer ZE, López-Terrada D. Molecular monitoring of 8p11 myeloproliferative syndrome in an infant. J Pediatr Hematol Oncol. 2009 Nov;31(11):879-83. [CrossRef] [PubMed]
- Yan Y, Qu S, Liu J, Li C, Yan X, Xu Z, Qin T, Jia Y, Pan L, Gao Q, Jiao M, Li B, Gale RP, Xiao Z. Olverembatinib for myeloid/lymphoid neoplasm associated with eosinophilia and FGFR1 rearrangement. Leuk Lymphoma. 2023 Sep;64(9):1605-1610. [CrossRef] [PubMed]
- Tzankov A, Reichard KK, Hasserjian RP, Arber DA, Orazi A, Wang SA. Updates on eosinophilic disorders. Virchows Arch. 2023 Jan;482(1):85-97. [CrossRef] [PubMed]
- Kaplan HG, Jin R, Bifulco CB, Scanlan JM, Corwin DR. PCM1-JAK2 Fusion Tyrosine Kinase Gene-Related Neoplasia: A Systematic Review of the Clinical Literature. Oncologist. 2022 Aug 5;27(8):e661-e670. [CrossRef]
- Rumi E, Milosevic JD, Casetti I, Dambruoso I, Pietra D, Boveri E, Boni M, Bernasconi P, Passamonti F, Kralovics R, Cazzola M. Efficacy of ruxolitinib in chronic eosinophilic leukemia associated with a PCM1-JAK2 fusion gene. J Clin Oncol. 2013 Jun 10;31(17):e269-71. [CrossRef] [PubMed]
- Tang G, Tam W, Short NJ, Bose P, Wu D, Hurwitz SN, Bagg A, Rogers HJ, Hsi ED, Quesada AE, Wang W, Miranda RN, Bueso-Ramos CE, Medeiros LJ, Nardi V, Hasserjian RP, Arber DA, Orazi A, Foucar K, Wang SA. Myeloid/lymphoid neoplasms with FLT3 rearrangement. Mod Pathol. 2021 Sep;34(9):1673-1685. [CrossRef] [PubMed]
- Venable ER, Gagnon MF, Pitel BA, Palmer JM, Peterson JF, Baughn LB, Hoppman NL, Greipp PT, Ketterling RP, Patnaik MS, Kelemen K, Xu X. A TRIP11:: FLT3 gene fusion in a patient with myeloid/lymphoid neoplasm with eosinophilia and tyrosine kinase gene fusions: a case report and review of the literature. Cold Spring Harb Mol Case Stud. 2023 Mar 24;9(1):a006243. [CrossRef]
- Schoelinck J, Gervasoni J, Guillermin Y, Beillard E, Pissaloux D, Chassagne-Clement C. T cell phenotype and lack of eosinophilia are not uncommon in extramedullary myeloid/lymphoid neoplasms with ETV6::FLT3 fusion: a case report and review of the literature. Virchows Arch. 2023 Nov 20. [CrossRef] [PubMed]
- Spitzer B, Dela Cruz FS, Ibanez Sanchez GD, Zhang Y, Xiao W, Benayed R, Markova A, Rodriguez-Sanchez MI, Bouvier N, Roshal M, Kung AL, Shukla N. ETV6-FLT3-positive myeloid/lymphoid neoplasm with eosinophilia presenting in an infant: an entity distinct from JMML. Blood Adv. 2021 Apr 13;5(7):1899-1902. [CrossRef]
- Munthe-Kaas MC, Forthun RB, Brendehaug A, Eek AK, Høysæter T, Osnes LTN, Prescott T, Spetalen S, Hovland R. Partial Response to Sorafenib in a Child With a Myeloid/Lymphoid Neoplasm, Eosinophilia, and a ZMYM2-FLT3 Fusion. J Pediatr Hematol Oncol. 2021 May 1;43(4):e508-e511. [CrossRef] [PubMed]
- Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood. 2017 Nov 9;130(19):2064-2072. [CrossRef]
- De Braekeleer E, Douet-Guilbert N, Rowe D, Bown N, Morel F, Berthou C, Férec C, De Braekeleer M. ABL1 fusion genes in hematological malignancies: a review. Eur J Haematol. 2011 May;86(5):361-71. [CrossRef] [PubMed]
- Zaliova M, Moorman AV, Cazzaniga G, Stanulla M, Harvey RC, Roberts KG, Heatley SL, Loh ML, Konopleva M, Chen IM, Zimmermannova O, Schwab C, Smith O, Mozziconacci MJ, Chabannon C, Kim M, Frederik Falkenburg JH, Norton A, Marshall K, Haas OA, Starkova J, Stuchly J, Hunger SP, White D, Mullighan CG, Willman CL, Stary J, Trka J, Zuna J. Characterization of leukemias with ETV6-ABL1 fusion. Haematologica. 2016 Sep;101(9):1082-93. [CrossRef]
- Carll T, Patel A, Derman B, Hyjek E, Lager A, Wanjari P, Segal J, Odenike O, Fidai S, Arber D. Diagnosis and treatment of mixed phenotype (T-myeloid/lymphoid) acute leukemia with novel ETV6-FGFR2 rearrangement. Blood Adv. 2020 Oct 13;4(19):4924-4928. [CrossRef]
- Telford N, Alexander S, McGinn OJ, Williams M, Wood KM, Bloor A, Saha V. Myeloproliferative neoplasm with eosinophilia and T-lymphoblastic lymphoma with ETV6-LYN gene fusion. Blood Cancer J. 2016 Apr 8;6(4):e412. [CrossRef]
- Kralik JM, Kranewitter W, Boesmueller H, Marschon R, Tschurtschenthaler G, Rumpold H, Wiesinger K, Erdel M, Petzer AL, Webersinke G. Characterization of a newly identified ETV6-NTRK3 fusion transcript in acute myeloid leukemia. Diagn Pathol. 2011 Mar 15;6:19. [CrossRef]
- Maesako Y, Izumi K, Okamori S, Takeoka K, Kishimori C, Okumura A, Honjo G, Akasaka T, Ohno H. inv(2)(p23q13)/RAN-binding protein 2 (RANBP2)-ALK fusion gene in myeloid leukemia that developed in an elderly woman. Int J Hematol. 2014 Feb;99(2):202-7. [CrossRef] [PubMed]
- Ballerini P, Struski S, Cresson C, Prade N, Toujani S, Deswarte C, Dobbelstein S, Petit A, Lapillonne H, Gautier EF, Demur C, Lippert E, Pages P, Mansat-De Mas V, Donadieu J, Huguet F, Dastugue N, Broccardo C, Perot C, Delabesse E. RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia. 2012 Nov;26(11):2384-9. [CrossRef] [PubMed]
- Naymagon L, Marcellino B, Mascarenhas J. Eosinophilia in acute myeloid leukemia: Overlooked and underexamined. Blood Rev. 2019 Jul;36:23-31. [CrossRef] [PubMed]
- Larson RA, Williams SF, Le Beau MM, Bitter MA, Vardiman JW, Rowley JD. Acute myelomonocytic leukemia with abnormal eosinophils and inv(16) or t(16;16) has a favorable prognosis. Blood. 1986 Dec;68(6):1242-9. [PubMed]
- Ouyang J, Goswami M, Peng J, Zuo Z, Daver N, Borthakur G, Tang G, Medeiros LJ, Jorgensen JL, Ravandi F, Wang SA. Comparison of Multiparameter Flow Cytometry Immunophenotypic Analysis and Quantitative RT-PCR for the Detection of Minimal Residual Disease of Core Binding Factor Acute Myeloid Leukemia. Am J Clin Pathol. 2016 Jun;145(6):769-77. [CrossRef]
- Raya José María, Gómez-Hernando Marta, Guijarro Francesca, Alonso Esther, Arnan Montserrat, Senent Leonor, Vicente Ana Isabel, Borrego Asunción, Gómez-Casares María Teresa, González-González Carmen, Ondarra Laida, Saumell Silvia, Montoro María Julia, Abío María de la O, Daza Sonia, Orna Elisa, Caballero María del Mar, Ródenas Isabel, Lo Riso Laura, De Miguel Carlos, Blanco Angela, Ricard María Pilar, Roldán Verónica, Martín-Santos Taida, Grupo Español de Citología Hematológica. (2021). PO-115: Leucemia aguda mieloide con t(8,21); RUNX1-RUNX1T1: análisis multicéntrico retrospectivo de 165 pacientes. LXIII Congreso Nacional de la SEHH XXXVII Congreso Nacional de la SETH Pamplona, 14-16 de octubre, 2021. Haematologica. Volume 106 OCTOBER 2021 - S2. ISSN 0390-6078.
- Swirsky DM, Li YS, Matthews JG, Flemans RJ, Rees JK, Hayhoe FG. 8;21 translocation in acute granulocytic leukaemia: cytological, cytochemical and clinical features. Br J Haematol. 1984 Feb;56(2):199-213. [CrossRef] [PubMed]
- Haferlach T, Bennett JM, Löffler H, Gassmann W, Andersen JW, Tuzuner N, Casslleth PA, Fonatsch C, Schoch C, Schlegelberger B, Becher R, Thiel E, Ludwig WD, Sauerland MC, Heinecke A, Büchner T. Acute myeloid leukemia with translocation (8;21). Cytomorphology, dysplasia and prognostic factors in 41 cases. AML Cooperative Group and ECOG. Leuk Lymphoma. 1996 Oct;23(3-4):227-34. [CrossRef] [PubMed]
- Valent P, Akin C, Hartmann K, Alvarez-Twose I, Brockow K, Hermine O, Niedoszytko M, Schwaab J, Lyons JJ, Carter MC, Elberink HO, Butterfield JH, George TI, Greiner G, Ustun C, Bonadonna P, Sotlar K, Nilsson G, Jawhar M, Siebenhaar F, Broesby-Olsen S, Yavuz S, Zanotti R, Lange M, Nedoszytko B, Hoermann G, Castells M, Radia DH, Muñoz-Gonzalez JI, Sperr WR, Triggiani M, Kluin-Nelemans HC, Galli SJ, Schwartz LB, Reiter A, Orfao A, Gotlib J, Arock M, Horny HP, Metcalfe DD. Updated Diagnostic Criteria and Classification of Mast Cell Disorders: A Consensus Proposal. Hemasphere. 2021 Oct 13;5(11):e646. [CrossRef]
- Kluin-Nelemans HC, Reiter A, Illerhaus A, van Anrooij B, Hartmann K, Span LFR, Gorska A, Niedoszytko M, Lange M, Scaffidi L, Zanotti R, Bonadonna P, Perkins C, Elena C, Malcovati L, Shoumariyeh K, von Bubnoff N, Parente R, Triggiani M, Schwaab J, Jawhar M, Caroppo F, Fortina AB, Brockow K, Zink A, Fuchs D, Kilbertus A, Yavuz AS, Doubek M, Mattsson M, Hagglund H, Panse J, Sabato V, Aberer E, Niederwieser D, Breynaert C, Várkonyi J, Kennedy V, Lortholary O, Jakob T, Hermine O, Rossignol J, Arock M, Gotlib J, Valent P, Sperr WR. Prognostic impact of eosinophils in mastocytosis: analysis of 2350 patients collected in the ECNM Registry. Leukemia. 2020 Apr;34(4):1090-1101. [CrossRef]
- Kovalszki A, Weller PF. Eosinophilia in mast cell disease. Immunol Allergy Clin North Am. 2014 May;34(2):357-64. [CrossRef]
- Morales-Camacho RM, Villanueva-Herraiz S, Prats-Martín C, Borrero JJ, Bernal R, Vargas MT. Eosinophil phagocytosis in advanced systemic mastocytosis with eosinophilia. Br J Haematol. 2018 Jun;181(5):578. [CrossRef] [PubMed]
- Moshref Razavi H, Yu R. Myeloid neoplasm with PDGFRB rearrangement, presenting as systemic mastocytosis-chronic eosinophilic leukemia. Am J Hematol. 2022 May;97(5):668-669. [CrossRef] [PubMed]
- Leguit RJ, Wang SA, George TI, Tzankov A, Orazi A. The international consensus classification of mastocytosis and related entities. Virchows Arch. 2023 Jan;482(1):99-112. [CrossRef] [PubMed]
- Morgado JM, Perbellini O, Johnson RC, Teodósio C, Matito A, Álvarez-Twose I, Bonadonna P, Zamò A, Jara-Acevedo M, Mayado A, Garcia-Montero A, Mollejo M, George TI, Zanotti R, Orfao A, Escribano L, Sánchez-Muñoz L. CD30 expression by bone marrow mast cells from different diagnostic variants of systemic mastocytosis. Histopathology. 2013 Dec;63(6):780-7. [CrossRef] [PubMed]
- Langabeer SE. The eosinophilic variant of chronic myeloid leukemia. EXCLI J. 2021 Nov 26;20:1608-1609. [CrossRef]
- Morsia E, Reichard K, Pardanani A, Tefferi A, Gangat N. WHO defined chronic eosinophilic leukemia, not otherwise specified (CEL, NOS): A contemporary series from the Mayo Clinic. Am J Hematol. 2020 Jul;95(7):E172-E174. [CrossRef] [PubMed]
- Matsushima T, Handa H, Yokohama A, Nagasaki J, Koiso H, Kin Y, Tanaka Y, Sakura T, Tsukamoto N, Karasawa M, Itoh K, Hirabayashi H, Sawamura M, Shinonome S, Shimano S, Miyawaki S, Nojima Y, Murakami H. Prevalence and clinical characteristics of myelodysplastic syndrome with bone marrow eosinophilia or basophilia. Blood. 2003 May 1;101(9):3386-90. [CrossRef] [PubMed]
Figure 1.
A. Bilobed eosinophil. B. Trilobed eosinophil. C. Eosinophil with high myeloperoxidase content. D. Eosinophil myelocyte which is Sudan Black B positive.
Figure 1.
A. Bilobed eosinophil. B. Trilobed eosinophil. C. Eosinophil with high myeloperoxidase content. D. Eosinophil myelocyte which is Sudan Black B positive.
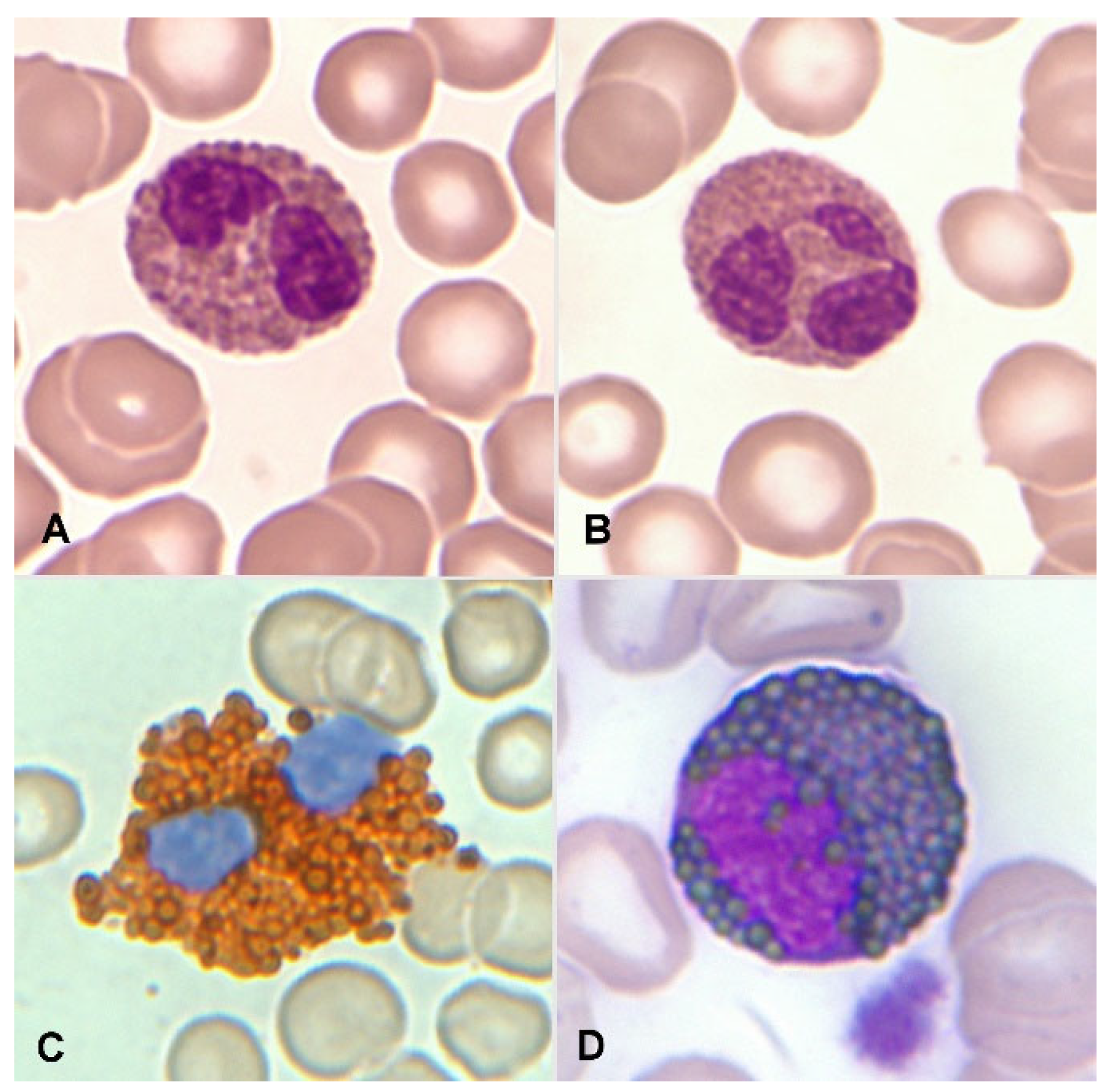
Figure 2.
Bone marrow aspirate. May-Grünwald-Giemsa (MGG), x1.000. Dysplastic eosinophils. A. Giant form with a small non-lobulated nucleus. B. Nuclear hypersegmentation. C. Mature eosinophil with a round non-lobulated nucleus and hypogranularity. D. Vacuolar images. E. Persistence of cytoplasmic basophilia in a large band eosinophil, F. Metamyelocyte with thick pre-eosinophilic granulation arranged peripherally.
Figure 2.
Bone marrow aspirate. May-Grünwald-Giemsa (MGG), x1.000. Dysplastic eosinophils. A. Giant form with a small non-lobulated nucleus. B. Nuclear hypersegmentation. C. Mature eosinophil with a round non-lobulated nucleus and hypogranularity. D. Vacuolar images. E. Persistence of cytoplasmic basophilia in a large band eosinophil, F. Metamyelocyte with thick pre-eosinophilic granulation arranged peripherally.
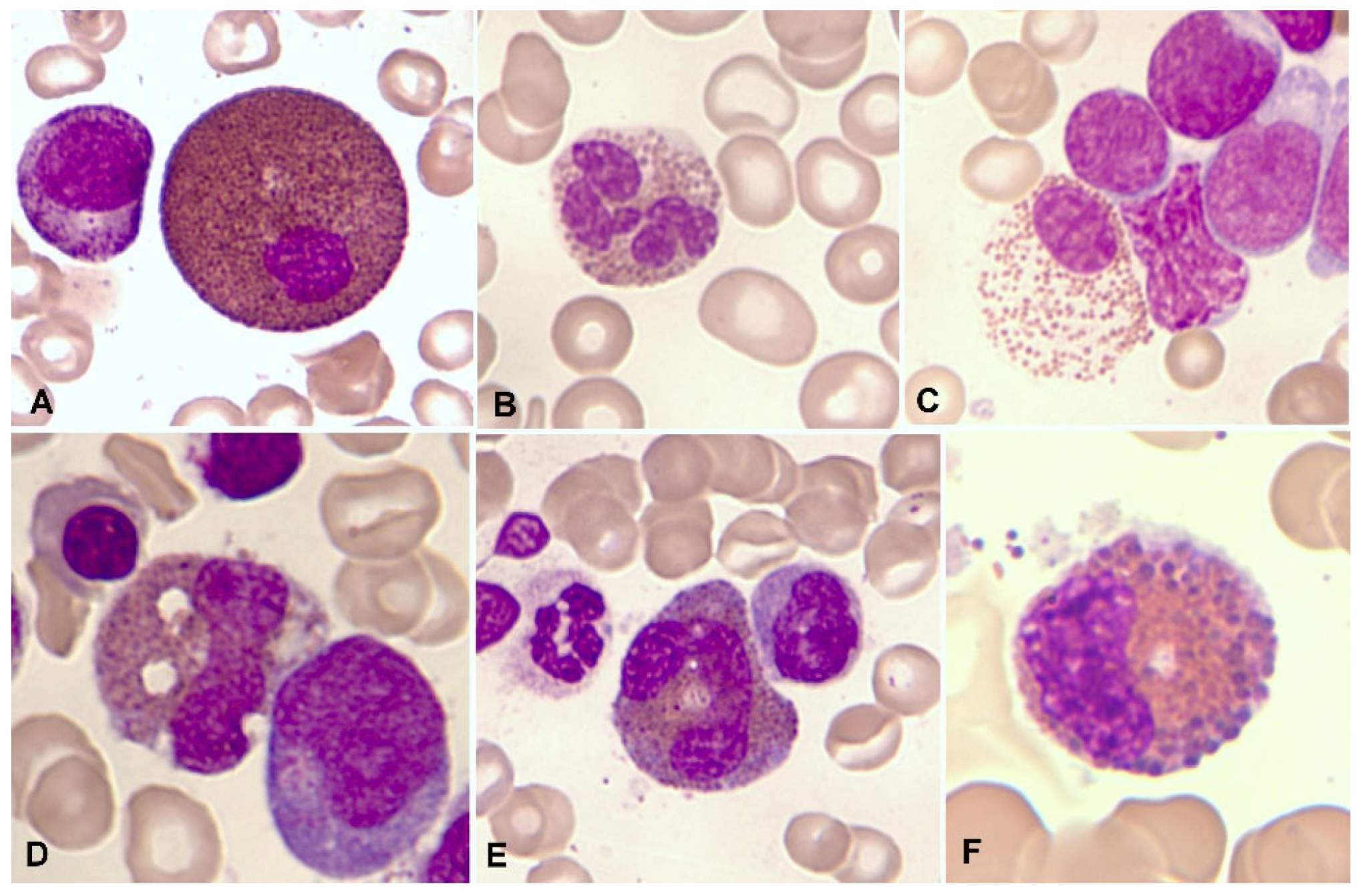
Figure 3.
Representation of normal eosinophils (in orange) by flow cytometry in peripheral blood.
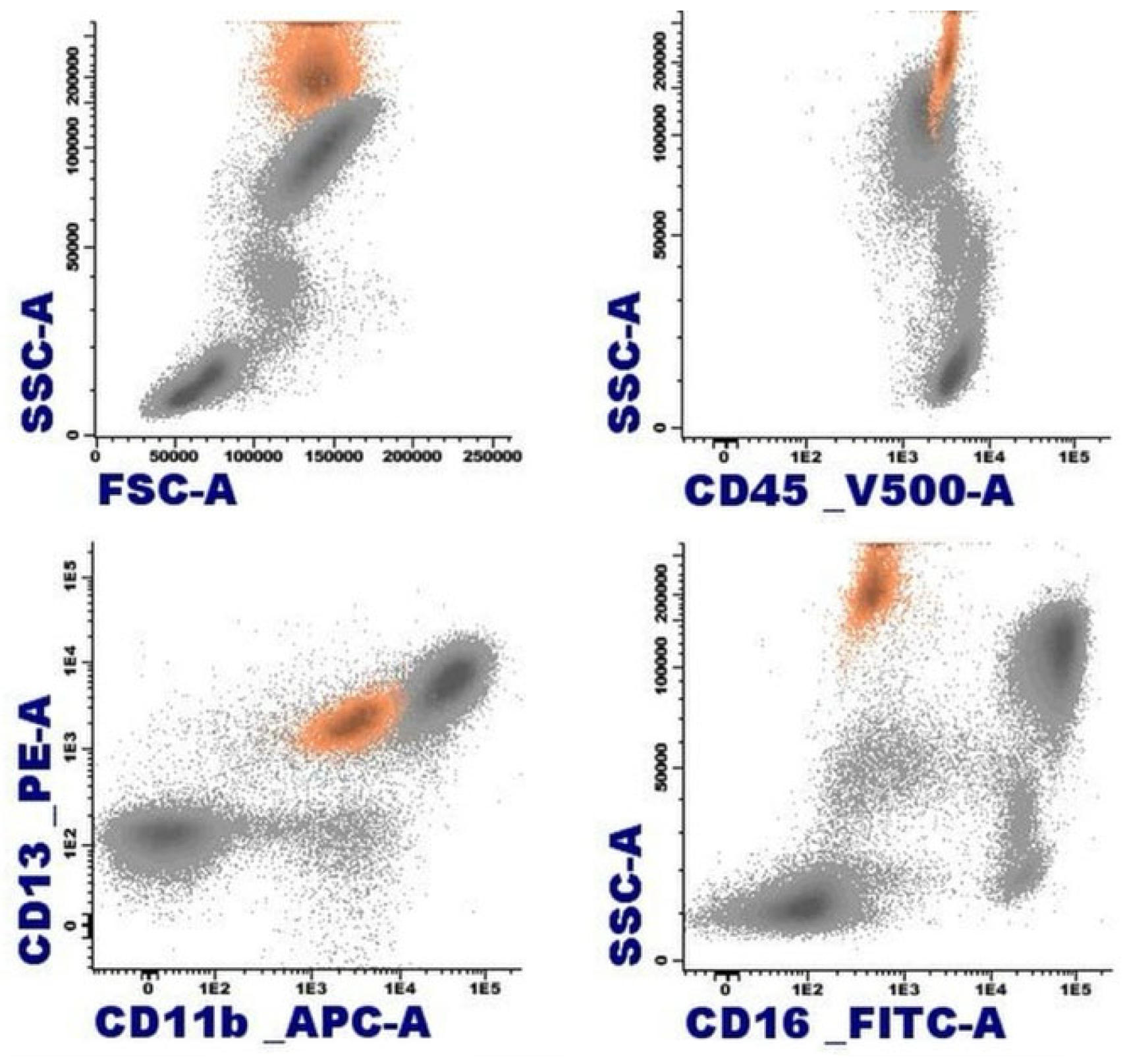
Figure 4.
Leukemoid reaction with accompanying hypereosinophilia concurrently with the diagnosis and treatment of cervical cancer. A. Peripheral blood smear showing abundant eosinophils (MayGrünwald-Giemsa [MGG], x1.000). B. Hypercellular bone marrow aspirate with scattered eosinophils (MGG, x1.000). C. Chart displaying elevated eosinophil counts in the blood, which sharply decrease following tumor remission.
Figure 4.
Leukemoid reaction with accompanying hypereosinophilia concurrently with the diagnosis and treatment of cervical cancer. A. Peripheral blood smear showing abundant eosinophils (MayGrünwald-Giemsa [MGG], x1.000). B. Hypercellular bone marrow aspirate with scattered eosinophils (MGG, x1.000). C. Chart displaying elevated eosinophil counts in the blood, which sharply decrease following tumor remission.
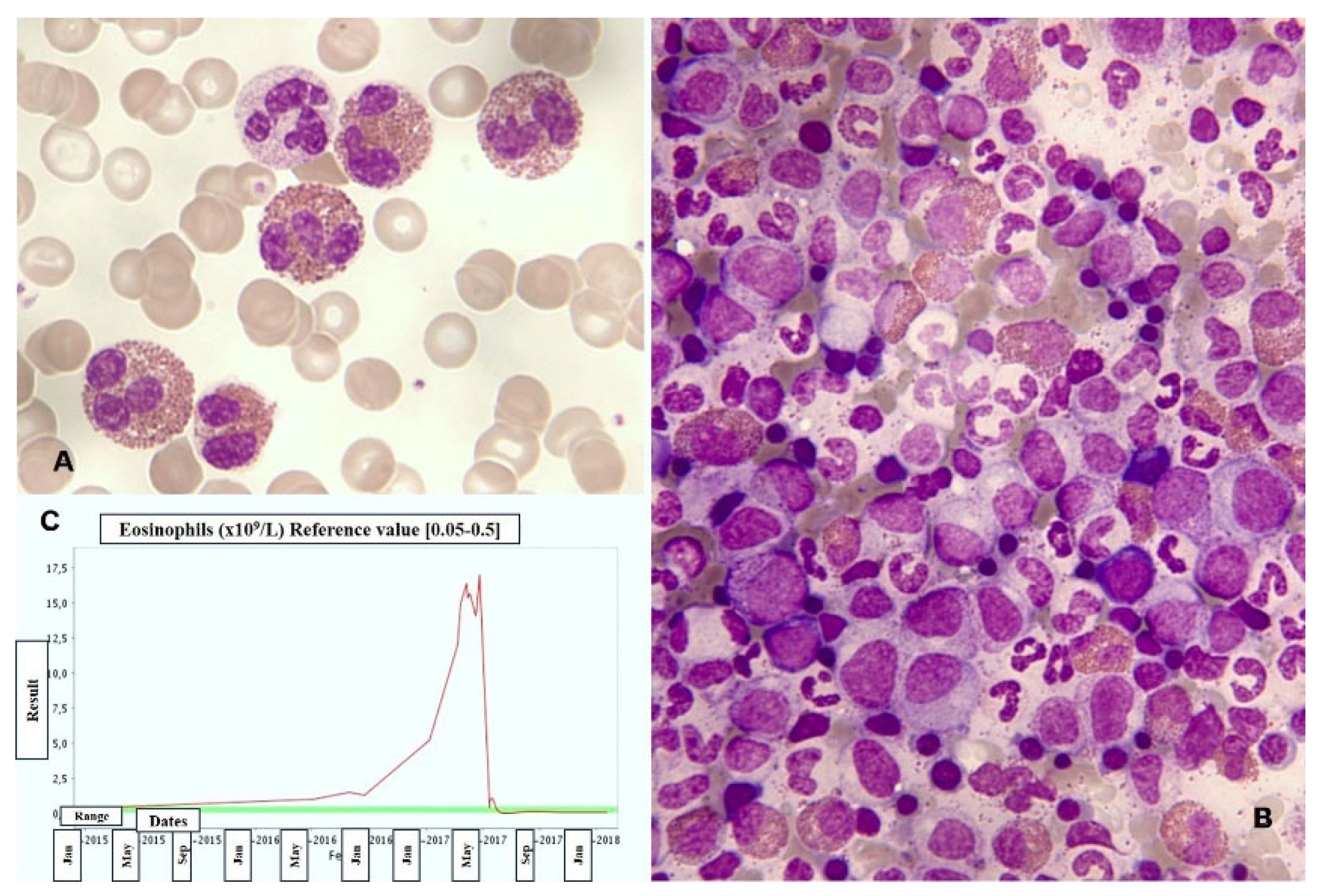
Figure 5.
Bone marrow aspirate. May-Grünwald-Giemsa, x1.000. Patient with Hodgkin's lymphoma presenting infiltrative nodules in the bone marrow with an eosinophilic component. A. Hodgkin cell with a large irregular nucleolus. B. Reed-Sternberg-like cell with mirror nucleus. C. Bone marrow biopsy. Hematoxylin and eosin, x630. Mononuclear cells with prominent nucleoli and infiltration of eosinophils.
Figure 5.
Bone marrow aspirate. May-Grünwald-Giemsa, x1.000. Patient with Hodgkin's lymphoma presenting infiltrative nodules in the bone marrow with an eosinophilic component. A. Hodgkin cell with a large irregular nucleolus. B. Reed-Sternberg-like cell with mirror nucleus. C. Bone marrow biopsy. Hematoxylin and eosin, x630. Mononuclear cells with prominent nucleoli and infiltration of eosinophils.
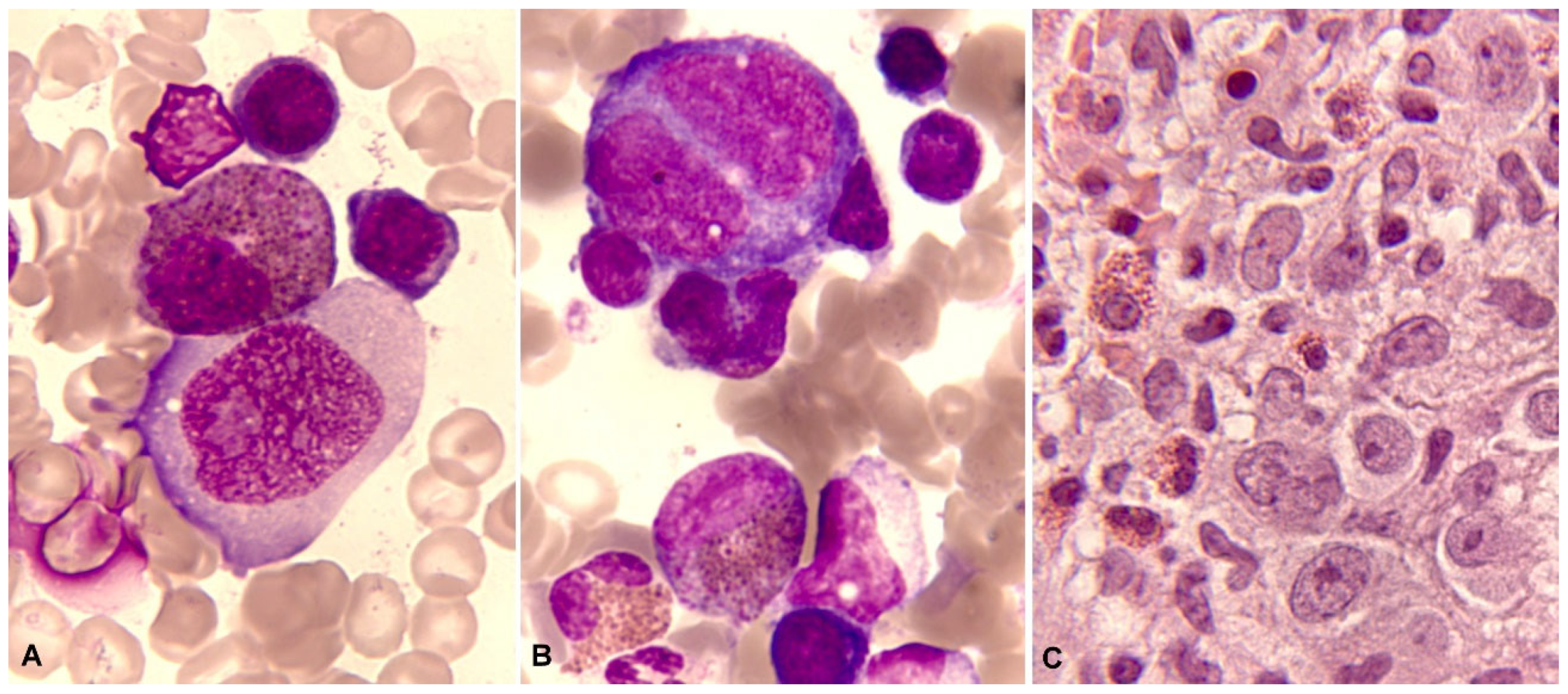
Figure 6.
Characteristic immunophenotype of angioimmunoblastic T lymphoma (AITL) with eosinophilia is illustrated in a representative case: light blue, T cells; blue, abnormal T cells; orange, eosinophils; purple, plasma cells. A. Abnormal T cells express CD5 bright and lack CD3 or show dim expression. B. CD4+ CD3-/+ weak subset. C. Differentiation markers of T cells show effector memory phenotype (CD45RA- CD197-/+weak). D. Positive expression of CD279. E. CD10 specific pattern of AITL; abnormal T cells show a clonal pattern according to expression of TRBC1 (TRBC1 negative). .
Figure 6.
Characteristic immunophenotype of angioimmunoblastic T lymphoma (AITL) with eosinophilia is illustrated in a representative case: light blue, T cells; blue, abnormal T cells; orange, eosinophils; purple, plasma cells. A. Abnormal T cells express CD5 bright and lack CD3 or show dim expression. B. CD4+ CD3-/+ weak subset. C. Differentiation markers of T cells show effector memory phenotype (CD45RA- CD197-/+weak). D. Positive expression of CD279. E. CD10 specific pattern of AITL; abnormal T cells show a clonal pattern according to expression of TRBC1 (TRBC1 negative). .
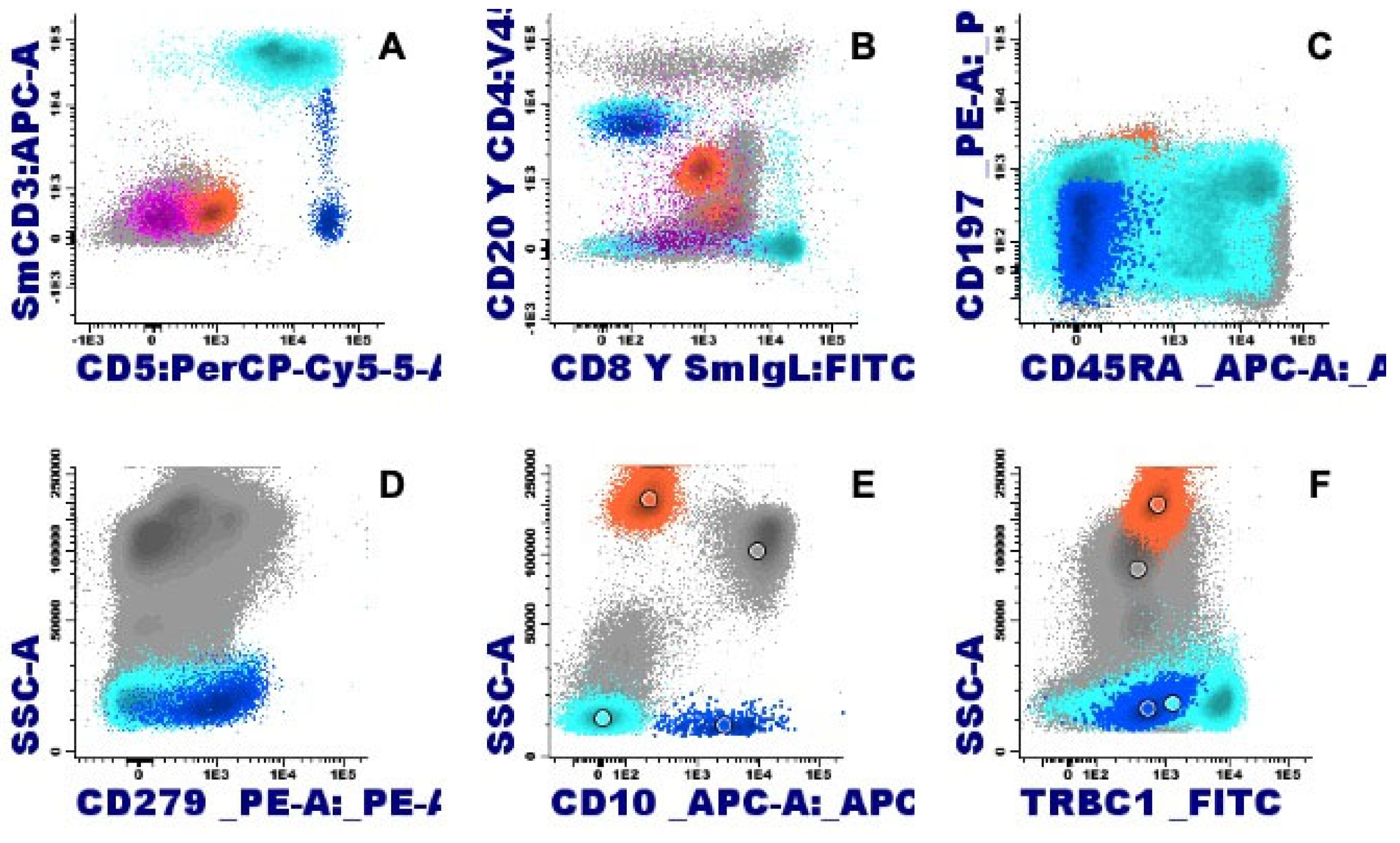
Figure 7.
Bone marrow aspirate. May-Grünwald-Giemsa, x1.000. B-acute lymphoblastic leukemia. B. Blasts accompanied by dysplastic eosinophils characterized by their large size, hypogranularity, and nuclear immaturity.
Figure 7.
Bone marrow aspirate. May-Grünwald-Giemsa, x1.000. B-acute lymphoblastic leukemia. B. Blasts accompanied by dysplastic eosinophils characterized by their large size, hypogranularity, and nuclear immaturity.
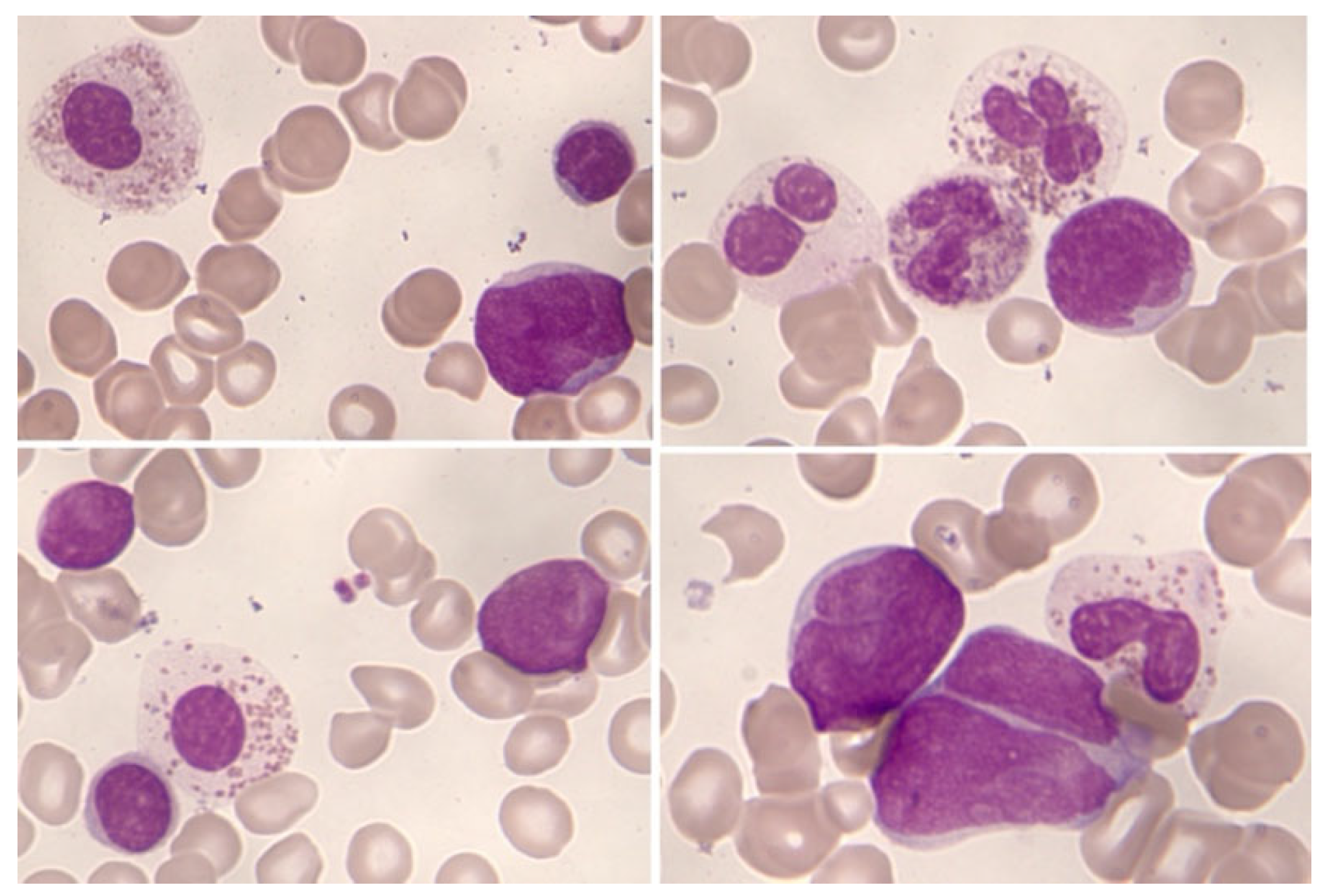
Figure 8.
B acute lymphoblastic leukemia (B-ALL) with eosinophilia by Flow cytometry (bone marrow): In orange, eosinophils are represented with varying degrees of granulation, mostly hypogranular, making identification challenging (heterogeneous SSC, mostly low). Eosinophils are characterized by intense expression of CD45 and weakly positive for CD15; the immature B-cell population, highlighted in green, corresponds to a common B-cell acute lymphoblastic leukemia (ALL-B), characterized by weak expression of CD19, CD34, CD45, and positive for CD10, and negative for CD15/CD65.
Figure 8.
B acute lymphoblastic leukemia (B-ALL) with eosinophilia by Flow cytometry (bone marrow): In orange, eosinophils are represented with varying degrees of granulation, mostly hypogranular, making identification challenging (heterogeneous SSC, mostly low). Eosinophils are characterized by intense expression of CD45 and weakly positive for CD15; the immature B-cell population, highlighted in green, corresponds to a common B-cell acute lymphoblastic leukemia (ALL-B), characterized by weak expression of CD19, CD34, CD45, and positive for CD10, and negative for CD15/CD65.
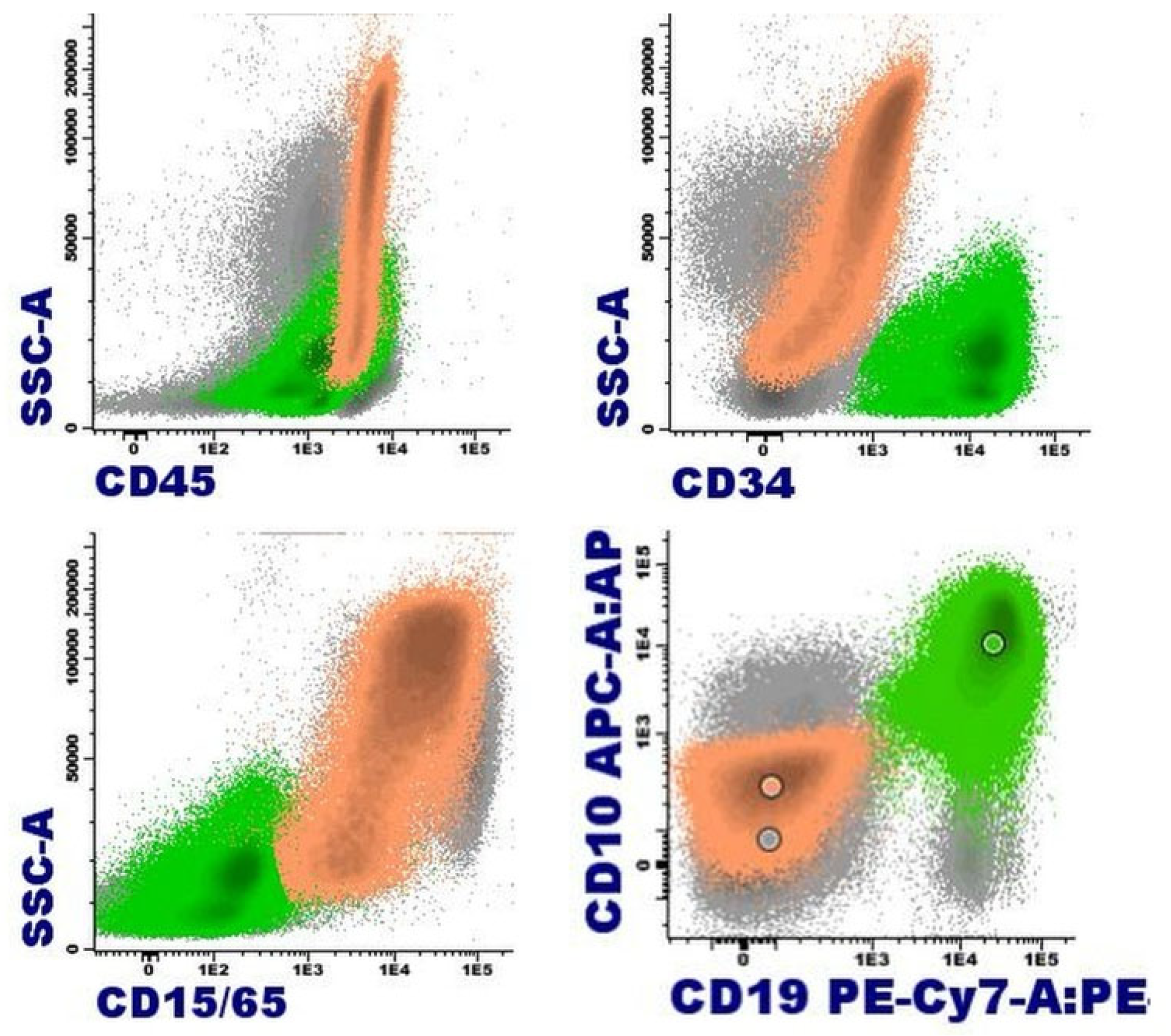
Figure 9.
Patient with M/lN-eo with PDGFRB rearrangement (Courtesy or Dr. Marina Gómez Rosa, Virgen de Valme hospital, Seville). May-Grünwald-Giemsa. A. Peripheral blood: myelemia and left shift, B. Hypercellular bone marrow (aspirate) with eosinophilia. Bone marrow biopsy: D. Inmunohistochemistry with CD117 reveals an increase in scattered mast cells. E. Bone marrow biopsy showing Grade 1 reticulin fibrosis by the World Health Organization criteria. E. Bone marrow karyotype: 46,XY, t(5;10)(q32;q21)[20]. F. Break-apart probe for PDGFRB rearrangement: a rearrangement of PDGFRB is observed.
Figure 9.
Patient with M/lN-eo with PDGFRB rearrangement (Courtesy or Dr. Marina Gómez Rosa, Virgen de Valme hospital, Seville). May-Grünwald-Giemsa. A. Peripheral blood: myelemia and left shift, B. Hypercellular bone marrow (aspirate) with eosinophilia. Bone marrow biopsy: D. Inmunohistochemistry with CD117 reveals an increase in scattered mast cells. E. Bone marrow biopsy showing Grade 1 reticulin fibrosis by the World Health Organization criteria. E. Bone marrow karyotype: 46,XY, t(5;10)(q32;q21)[20]. F. Break-apart probe for PDGFRB rearrangement: a rearrangement of PDGFRB is observed.
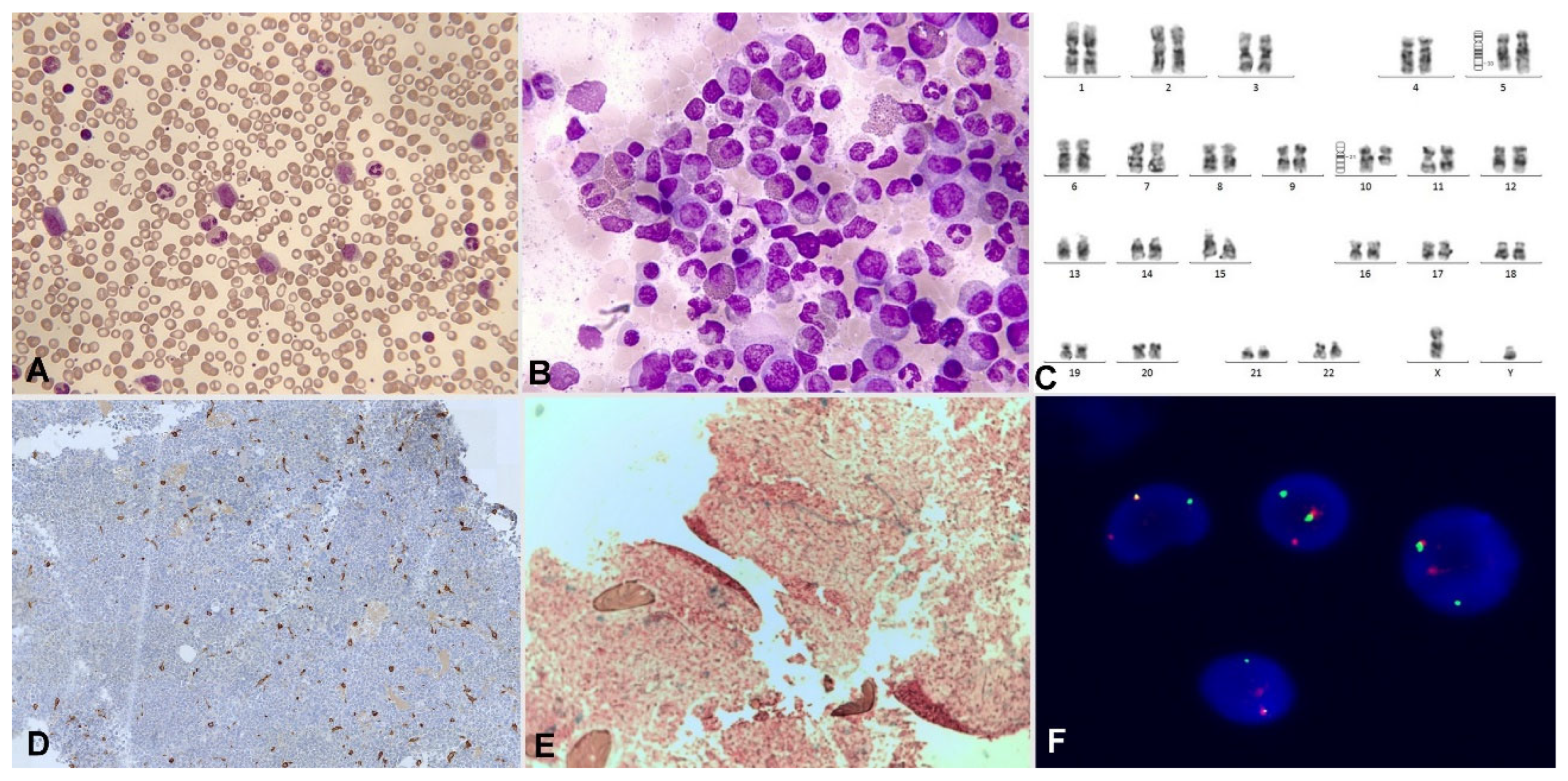
Figure 10.
Bone marrow aspirate. May-Grünwald- Giemsa, x1.000. Acute myeloid leukemia with inv(16)(p13.1q22). In both images, dysmorphic eosinophils with thick reddish-violet granules are evident; the one on the left highlights the intensity of eosinophilia.
Figure 10.
Bone marrow aspirate. May-Grünwald- Giemsa, x1.000. Acute myeloid leukemia with inv(16)(p13.1q22). In both images, dysmorphic eosinophils with thick reddish-violet granules are evident; the one on the left highlights the intensity of eosinophilia.
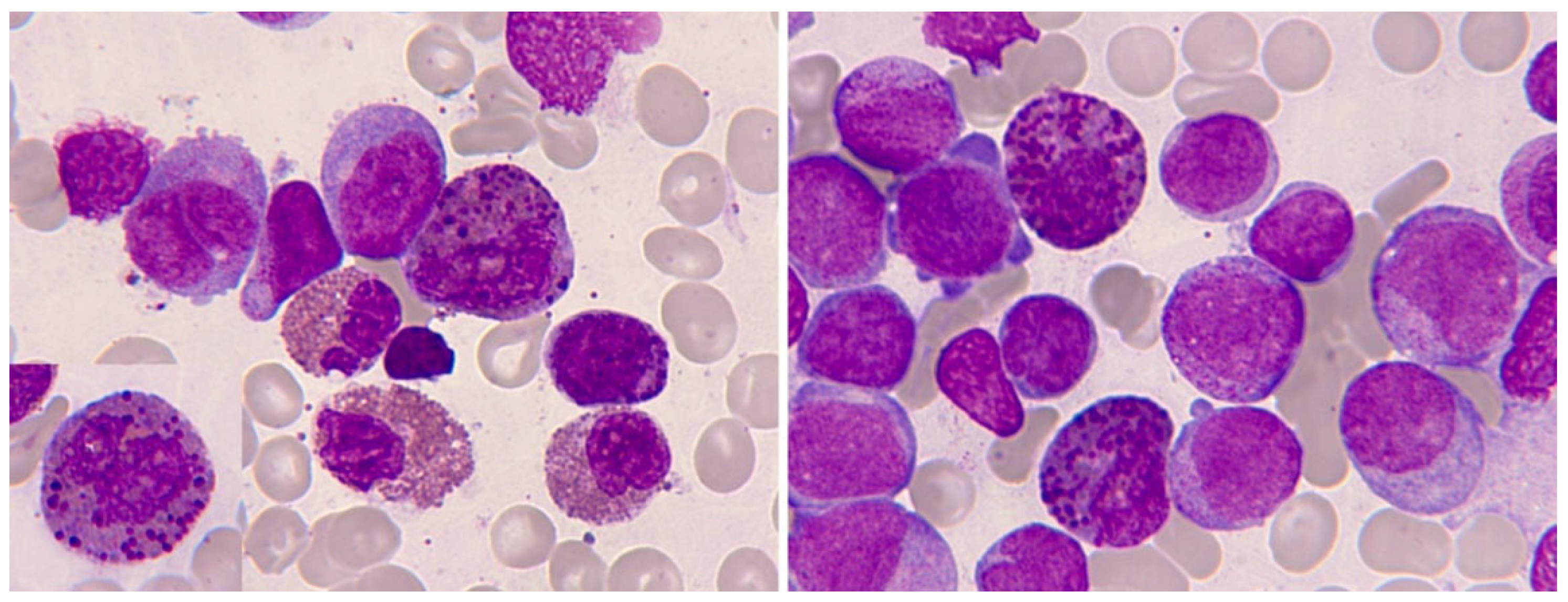
Figure 11.
Bone marrow aspirate. May-Grünwald-Giemsa, x1.000. Oligoblastic acute myeloid leukemia with inv(16)(p13.1q22). A. Cytological image with a blast containing a long Auer rod, surrounded by eosinophils. B. Pseudo-Chédiak-Higashi inclusions. C. The histological image clearly shows massive medullary eosinophilia. D. Bone marrow karyotype: 46,XY,inv(16)(p13.1q22)[3]/46,idem,t(1;18)(p22;p11.2).
Figure 11.
Bone marrow aspirate. May-Grünwald-Giemsa, x1.000. Oligoblastic acute myeloid leukemia with inv(16)(p13.1q22). A. Cytological image with a blast containing a long Auer rod, surrounded by eosinophils. B. Pseudo-Chédiak-Higashi inclusions. C. The histological image clearly shows massive medullary eosinophilia. D. Bone marrow karyotype: 46,XY,inv(16)(p13.1q22)[3]/46,idem,t(1;18)(p22;p11.2).
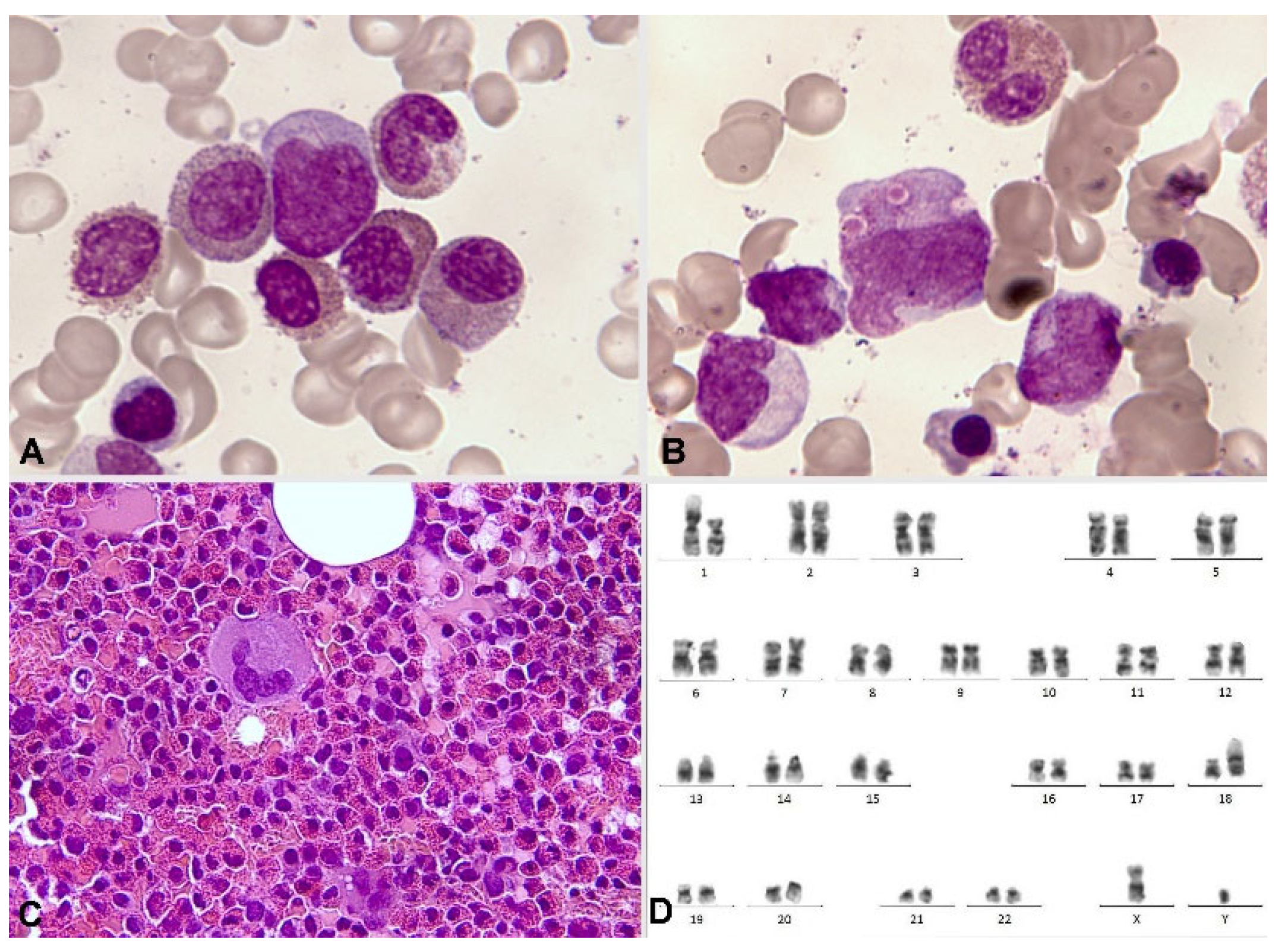
Figure 12.
Bone marrow aspirate. May-Grünwald-Giemsa, x1.000. Acute myeloid leukemia with inv(16)(p13.1q22). A. Eosinophil with thick dark pre-eosinophilic granules. B. Positive periodic acid-Schiff (PAS) reaction in immature eosinophils, in contrast to the negativity in an immature segmented neutrophil. C. Two immature eosinophils showing marked chloroacetate esterase activity.
Figure 12.
Bone marrow aspirate. May-Grünwald-Giemsa, x1.000. Acute myeloid leukemia with inv(16)(p13.1q22). A. Eosinophil with thick dark pre-eosinophilic granules. B. Positive periodic acid-Schiff (PAS) reaction in immature eosinophils, in contrast to the negativity in an immature segmented neutrophil. C. Two immature eosinophils showing marked chloroacetate esterase activity.
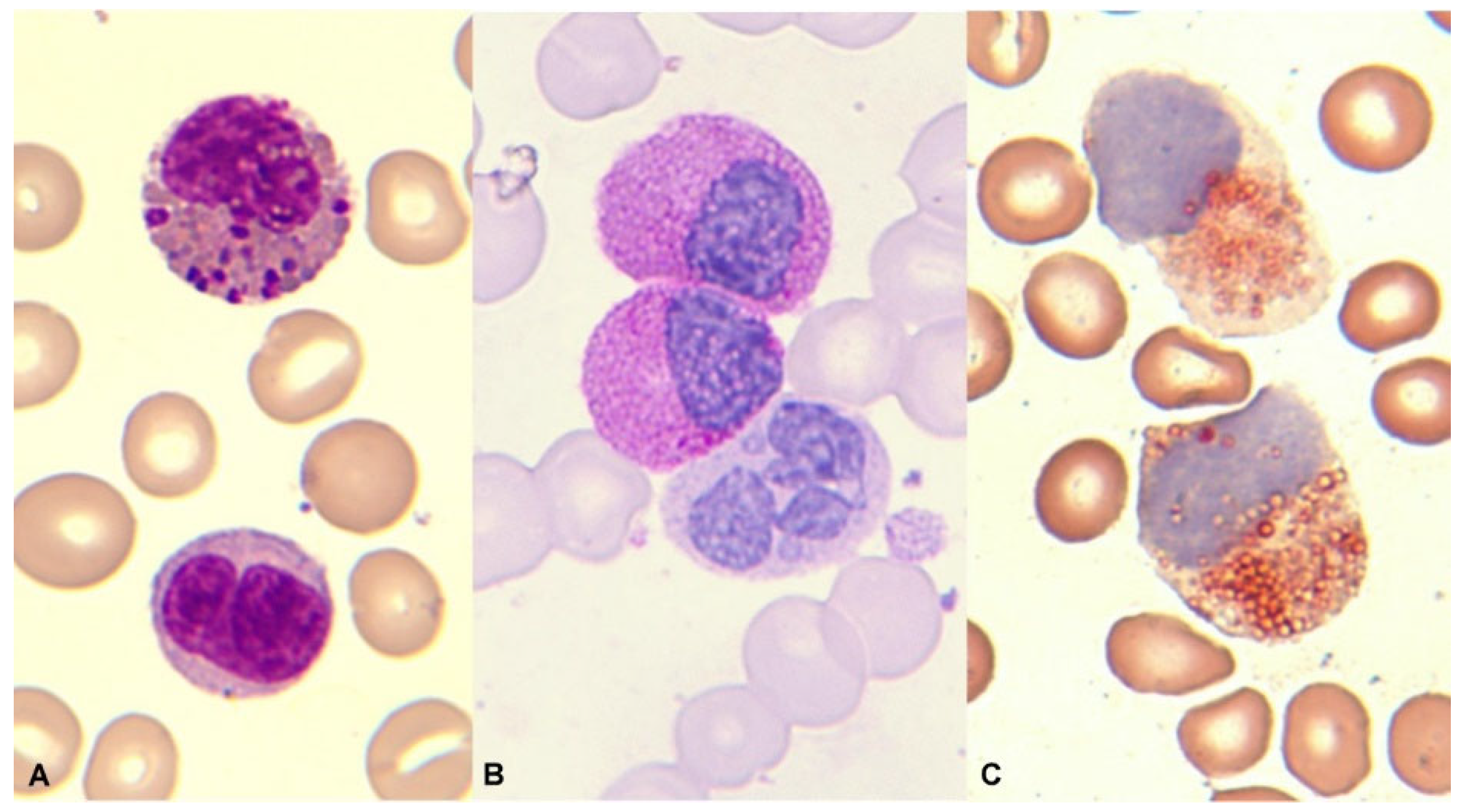
Figure 13.
Bone marrow aspirate. May-Grünwald-Giemsa, x1.000. Acute myeloid myeloid with t(8;21)(q22;q22.1). A. Granulocytic dysplasia with peripheral basophilia reinforcement. B. Dysplastic eosinophils with increased granulation thickness. C. Eosinophils with mirror nuclei.
Figure 13.
Bone marrow aspirate. May-Grünwald-Giemsa, x1.000. Acute myeloid myeloid with t(8;21)(q22;q22.1). A. Granulocytic dysplasia with peripheral basophilia reinforcement. B. Dysplastic eosinophils with increased granulation thickness. C. Eosinophils with mirror nuclei.
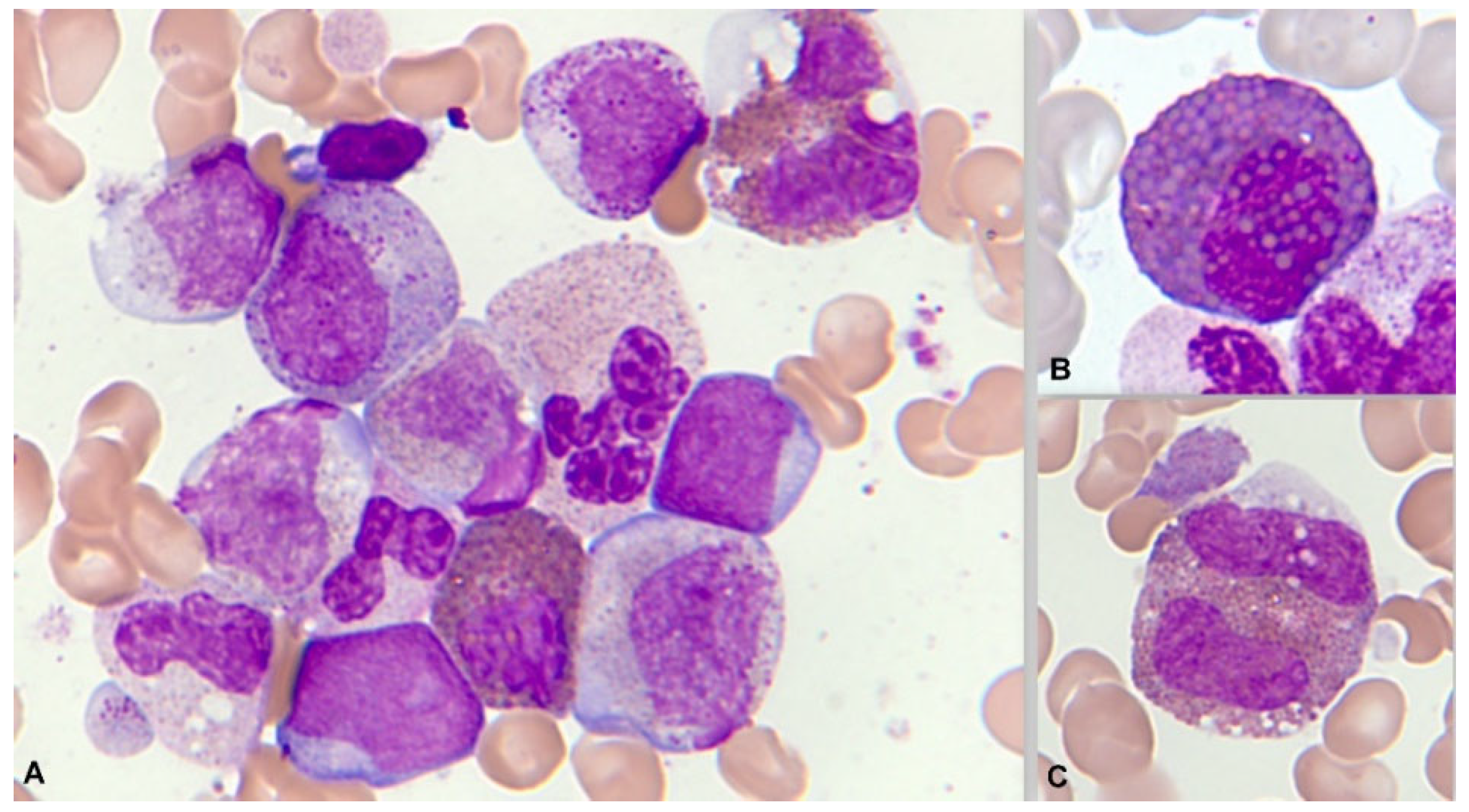
Figure 14.
Bone marrow aspirate. May-Grünwald- Giemsa, x1.000. Systemic mastocytosis associated with chronic myelomonocytic leukemia with eosinophilia. A. Hypogranular spindle-shaped mast cell with bilobed nucleus, three eosinophils, and two promonocytic forms. B. Phagocytosis of eosinophil by mast cell, completed. C. Phagocytosis of eosinophil by mast cell, in progress.
Figure 14.
Bone marrow aspirate. May-Grünwald- Giemsa, x1.000. Systemic mastocytosis associated with chronic myelomonocytic leukemia with eosinophilia. A. Hypogranular spindle-shaped mast cell with bilobed nucleus, three eosinophils, and two promonocytic forms. B. Phagocytosis of eosinophil by mast cell, completed. C. Phagocytosis of eosinophil by mast cell, in progress.
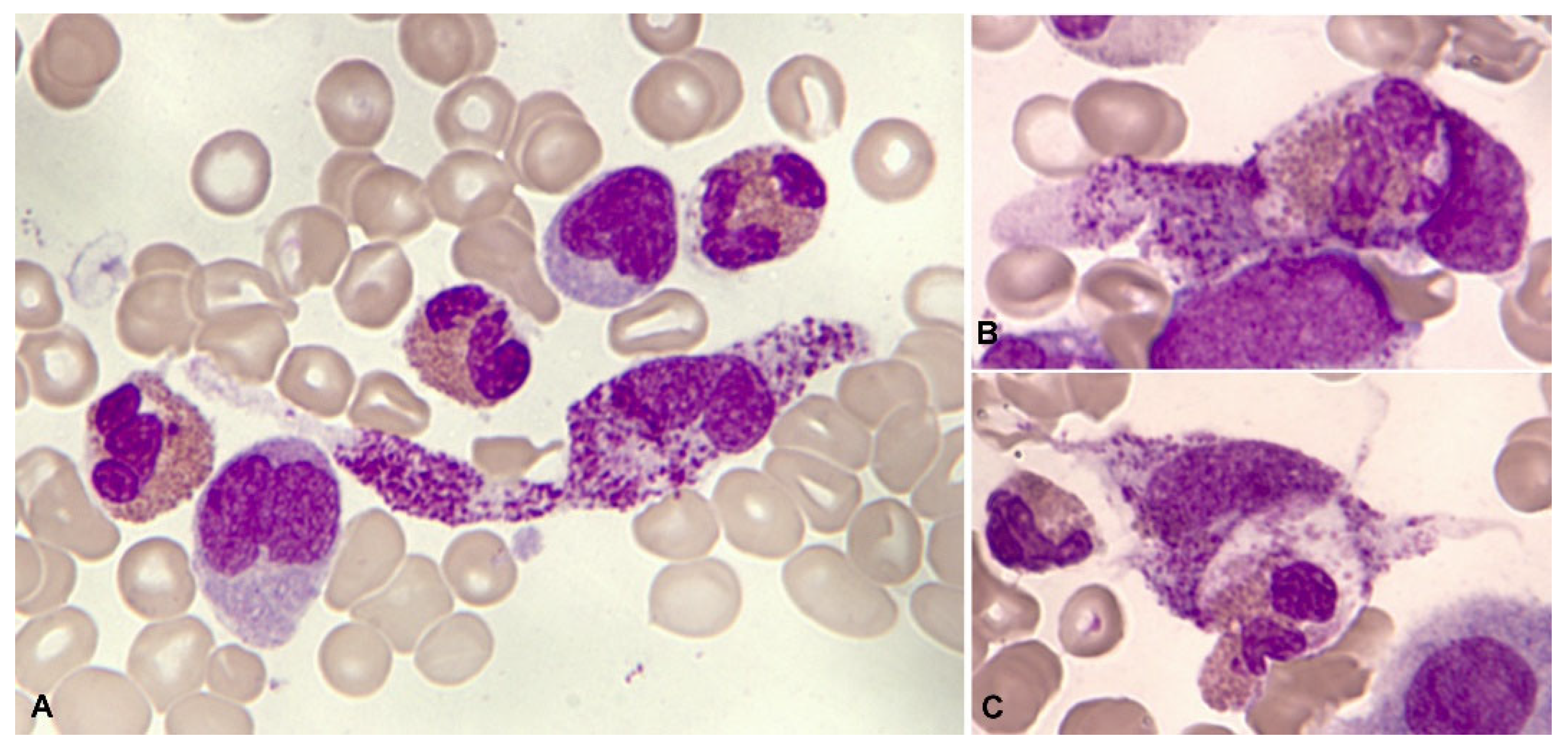

Table 1.
 |
* In these disorders, eosinophilia is usually reactive to the release of eosinopoietic cytokines (polyclonal or benign). ** In these disorders, eosinophils are usually neoplastic (belonging to the malignant clone).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



