Submitted:
28 November 2023
Posted:
28 November 2023
You are already at the latest version
Abstract
1,3 Dipolar cycloaddition of imidazolidine derivatives containing exocyclic double bonds is a convenient method of creating spiro-conjugated molecules with the promising anticancer active. In this work, the derivatives of parabanic acid (2-thioxoimidazolidine-4,5-diones and 5-aryliminoimidazolidine-2,4-diones) were firstly investigated as dipolarophiles in the reactions with nitrile imines. The generation of nitrile imines was carried out either by the addition of tertiary amine to hydrazonoyl chlorides «drop by drop» or using recently proposed diffusion mixing technique, which led to ~5-15% increasing of target compounds yields. It was found that the addition of nitrile imines to C=S or C=N exocyclic double bonds led to 1,2,4-thiazolines or triazolines and occurred regioselectively in accordance to the ratio of FMO coefficients of reactants. The yield of the resulting spiro-compound depended on the presence of alkyl substituents in nitrile imine structure and was significantly decreases at the reactions with imines with strong electron donor or electron-withdrawing groups. Some of the obtained compounds showed reasonable in vitro cytotoxicity. IC50 values were calculated for HCT116 (colon cancer) cells using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) test.
Keywords:
parabanic acids
; imines
; imidazolones
; spiro-compounds
; 1
; 3-dipolar cycloaddition
; nitrile imines.
1. Introduction
Nitrile imines are widely recognized for their high reactivity in 1,3-dipolar cycloaddition reactions, both in carbon-carbon multiple bonds and carbon-heteroatom bonds [1]. The reactions of nitrile imines with asymmetric dipolarophiles usually proceed regio- and chemoselectively. The addition of nitrile imine to the C=C double bond results in the formation of a pyrazolidine fragment, which is found in the compounds possessing a broad spectrum of biological activities, such as anti-inflammatory [2], antiviral, antimicrobial [3], analgesic, immunosuppressive, antibacterial [4], anticancer [5], antidepressant and neuroprotective [6] properties. The discovery of these properties has made the reactions of dipolar cycloaddition of nitrile imines a common tool in the synthesis of biologically active molecules and natural compounds [7].
Hydantoins exhibit a wide range of biological activity, among which antitumor, antibacterial, anticonvulsant, antiepileptic and antiarrhythmic properties, as well as action as muscle relaxants can be distinguished [8]. The introduction of methylidene derivatives of hydantoins and thiohydantoins in the 1,3-dipolar cycloaddition reaction enables to obtain spiro-conjugated compounds containing several heterocyclic pharmacophore fragments in one molecule [9,10,11]. Sterically constrained structure of spiro-conjugated molecules coupled with bioactive fragments suggests the appearance of noticeable cytotoxic properties in the resulting compounds and thereby causes interest not only for organic, but also for medical chemistry.
Despite the fact that 5-arylidene(thio)hydantoins have long been known as dipolarophiles [12,13], their analogues containing exocyclic C=N bonds have not been previously investigated in such reactions.
There are in the literature a few examples where compounds with an exocyclic C=N bonds were used as dipolarophiles. However, the available data demonstrated a good reactivity of the imino group in comparison with the trisubstituted bond in alkenes [14]. In all cases, the addition of nitrile imines to the C=N bond occurs regioselectively with the formation of 1,2,4-triazole [1].
In a recent work by S.H. Ungoren [15], it has been shown that the interaction of N,N-disubstituted parabanic acids with aryliminophosphoranes leads to the formation of 5-imino substituted hydantoins with a good yield. Among the data available in the literature on the chemical properties of 5-substituted hydantoins in the 1,3-dipolar cycloaddition reactions, we found no examples of the interaction of some 1,3-dipoles and an exocyclic bond C=N of such compounds.
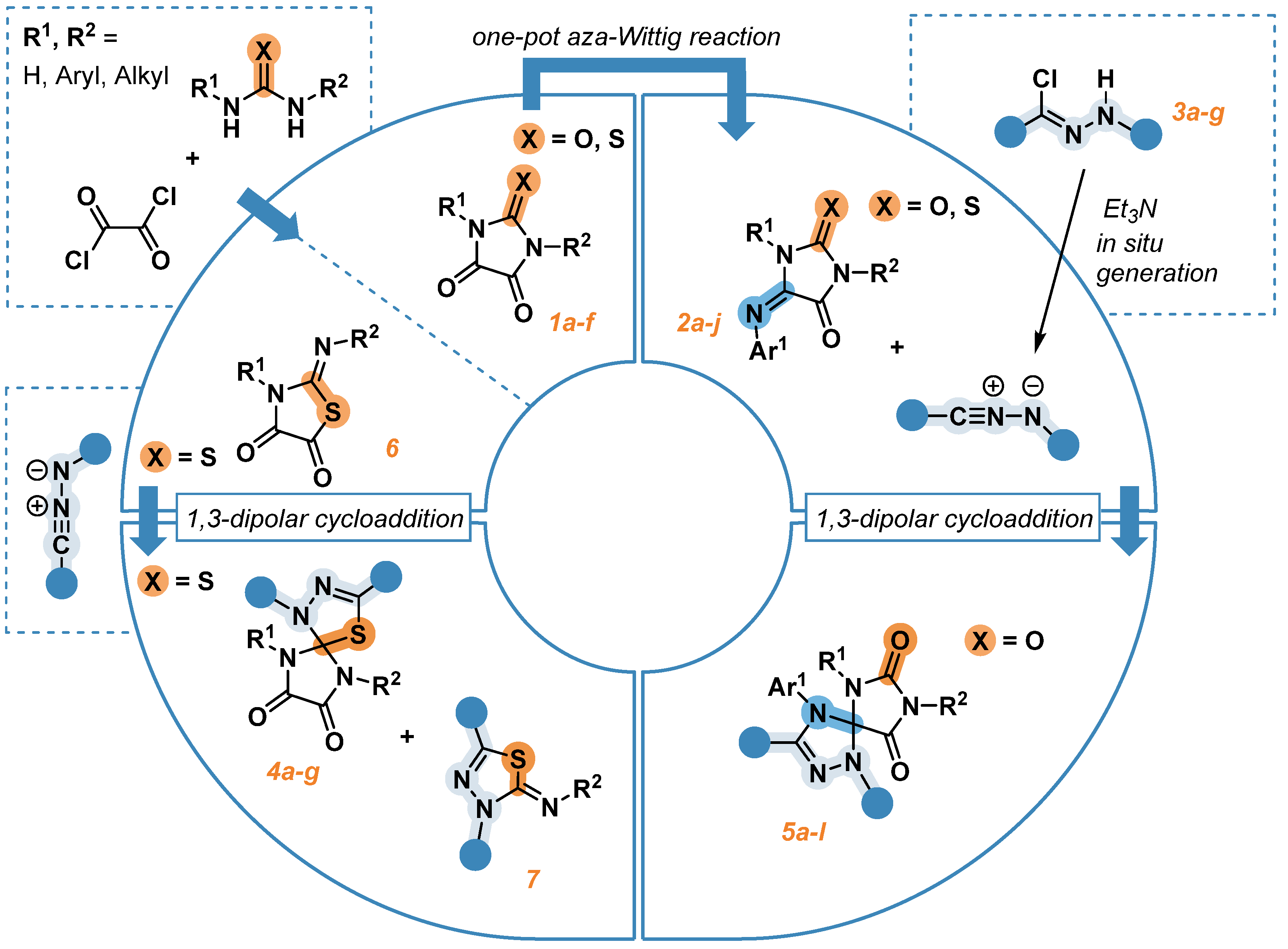
Therefore, in this paper we investigated the reactivity of N- and N,N’-substituted thioparabanic acids in reactions of 1,3-dipolar cycloaddition. Besides, a one-pot method was proposed for the synthesis of 5-arylimino-1,3-diphenylimidazolidine-4-ones, which were further used as dipolarophiles in reactions with nitrile imines. The transformations discussed in this article are summed up in Scheme 1.
2. Results and Discussion
2.1. Synthesis of the Imidazolidine-4,5-diones 1c-f
The preparation of substituted parabanic acids and their sulfur-containing analogues by the reaction of oxalyl chloride and corresponding thioureas was described more than a century ago [16], but for a long time the structure of the resulting product remained the subject of discussion [16,17,18]. The differences in the obtained results were caused by the possibility of formation not only of substituted thioparabanic acid II, but also of intermediate 2-iminothiazolidine-4,5-dione I, which in some cases could not be isolated (Scheme 2). At the same time, it was found that the isomerization of intermediate I into product II occurs quite easily, for example, when boiling in ethanol [18].
Investigating the reaction of diphenyl thiourea and oxalyl chloride, we found that the formation of a particular of the two isomeric products can be conveniently monitored using NMR spectroscopy. Since 1,3-diphenyl-2-thioxoimidazolidine-4,5-dione 1c has a plane of symmetry passing through the C=S bond and the middle of the C4-C5 bond, the protons of aromatic fragments are equivalent and present in the spectrum as two sets of signals. 3-Phenyl-2-(phenylimino)thiazolidine-4,5-dione 6 does not contain symmetry elements, so the signals of aromatic protons have a more complex multiplicity (Figure 1).
In many publications, data on the formation of isomeric products I and II (Scheme 2) as a result of the reaction of diphenyl thiourea and oxalyl chloride were presented implicitly [15,19,20], and we could not find a clear methodology that allows us to obtain exclusively isomer II. By conducting reactions of various thioureas with oxalyl chloride, we found that a thiazolidine-type product (I) was formed very easily. The product was obtained with a high yield after 1-2 hours of boiling the reaction mixture in an aprotic solvent with a low boiling point, such as dichloromethane. When boiling the reaction mixture in a more highly boiling solvent, such as acetonitrile, a noticeable amount of isomerized imidazolidine-type product (II) was formed in a few hours (about 10% after 2 hours). We also found that chromatographic separation of isomers 6 and 1c is not feasible, since 2-imino-thiazolidine-4,5-dione 6 was completely transformed into 2-thioxoimidazolidine-4,5-dione 1c passing through silica gel.
In contrast to the spectrally distinguishable compounds 1c and 6, the determination of the structure of products obtained from oxalyl chloride and non-symmetric thioureas (R1 ≠ R2) is a more difficult task, since the result of the reaction can be both an imidazolidine product II and two isomeric thiazolidines I. In this regard, imidazolidine-2,4,5-triones 1a-b and 2-thioxoimidazolidine-4,5-diones 1c-f were obtained according to two different synthetic strategies. The first ones were obtained boiling a mixture of oxalyl chloride and urea in DCM, followed by treatment with a solution of sodium bicarbonate to neutralize oxalyl chloride and hydrochloric acid residues. To obtain sulfur-containing products, we used acetonitrile as a solvent, and flash chromatography as a purification method, after which only imidazolidine-type products 1c-f were isolated. The yields of all compounds 1a-f were good to excellent with the exception of product 1f, containing an unsubstituted N3 atom (Table 1).
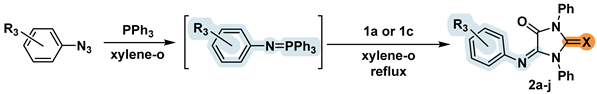
2.2. Synthesis of the 5-Aryliminoimidazolidine-2,4-diones 2a-l
In a recent paper by Ş.H. Ungoren and colleagues [15], it was reported that substituted parabanic acids react with phosphonium ylides to form 5-alkylidene and 5-imino substituted hydantoins. We modified this technique and carried out the one-pot aza-Wittig reaction without isolation of intermediate aryliminophosphoranes. Typically, a mixture of arylazide and triphenylphosphine in dry xylene was stirred at a temperature of about 50℃ until nitrogen release stopped (0.5 to 2h). Imidazoldione 1 was added to the phosphorus ylide formed in situ, after which the mixture was boiled for 2 hours. The product was isolated using column chromatography and, if necessary, additionally recrystallized from isopropanol. In all cases, imines 2a-j were obtained with good yields (Table 2). The resulting product C=N double bond configuration was attributed in accordance with the literature data on the structure of similar compounds [15].
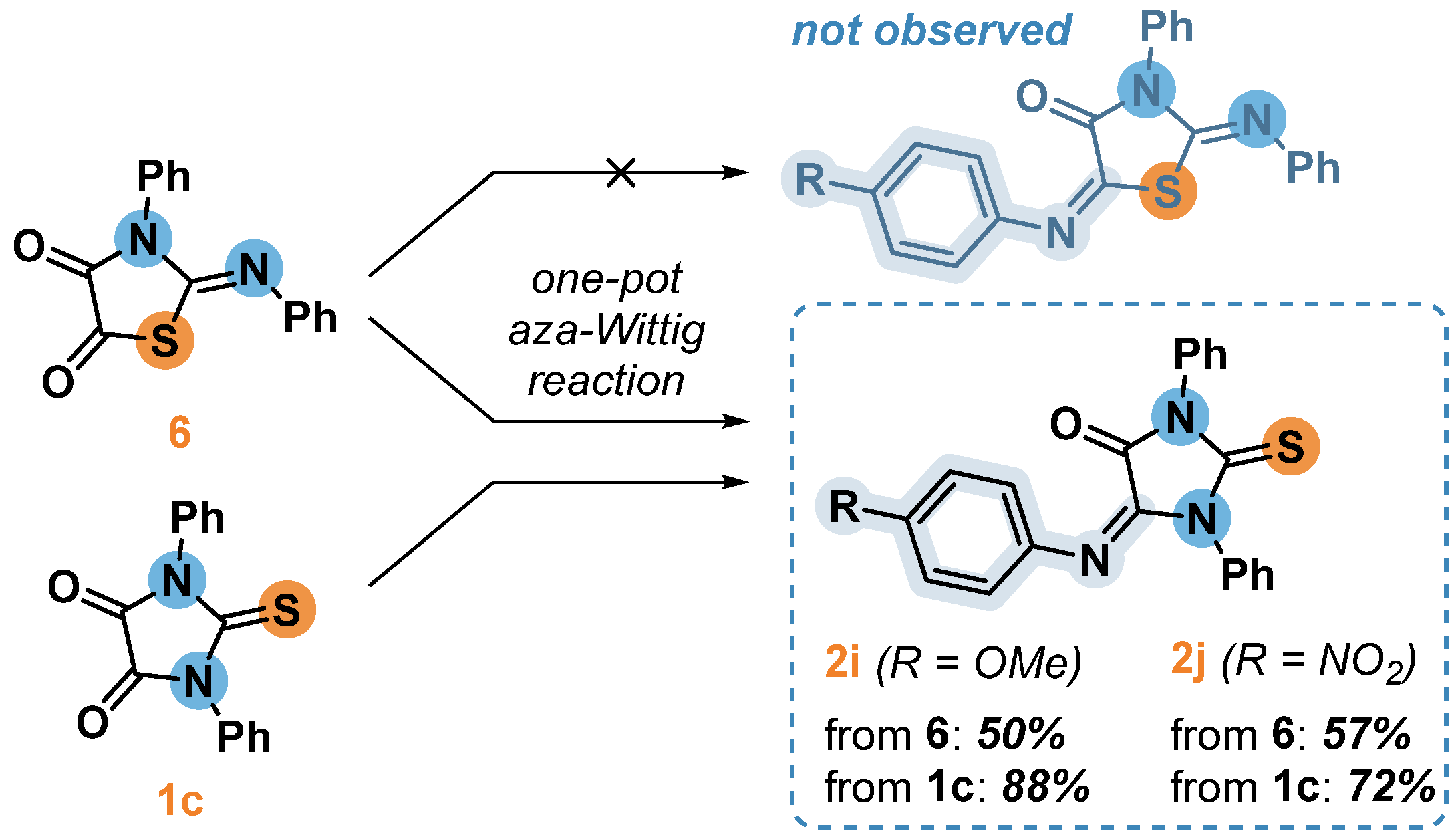
As previously described [20], Wittig reactions with thiazolidine 6 gives the corresponding trisubstituted alkene. Therefore, carrying out a similar aza-reaction to obtain thiazolidine with two C=N bonds simultaneously at positions C2 and C5 seemed quite real. Contrary to this, we unexpectedly found that compound 6 in the reactions with phosphonium ylides, formed in situ from PPh3 and corresponding arylazides, undergoes a one-pot aza-Wittig reaction to furnish the same product as it was formed at the identical conditions from thioparabanic acid 1c, but with a significantly lower yield. The presence of an exocyclic C=S bond in compounds 2i and 2j was indicated by the appearance of a carbon atom of the thiocarbonyl group signal at ~180 ppm in the 13C NMR spectrum. It remains uncertain whether the formation of the imidazolidine moiety was initiated during the reaction, or whether the isomerization of the thiazolidine cycle into imidazolidine occurred during chromatographic purification of imines 2i and 2j.
Scheme 3.
Aza-Wittig reaction of isomers 1c and 6.
2.3. 1,3-Dipolar Cycloaddition of Nitrile Imines to 2-Thioxoimidazolidine-4,5-diones 1c-f and 3-Phenyl-2-(phenylimino)thiazolidine-4,5-dione 6
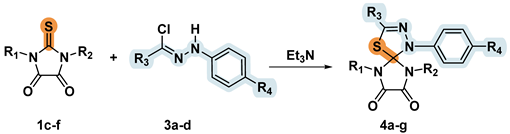
Despite the widespread recognition of 1,3-dipolar cycloaddition of thiohydantoins and their 5-methylidene/arylidene substituted analogues for creating spiro-linked compounds with 1,3,4-thiadiazoline fragment [10,11,12], there are no examples in the literature of 5-oxothiohydantoins (thioparabanic acids) being used as dipolarophiles. Nitrile imines are highly active 1,3-dipoles, so they are usually generated in situ from the corresponding precursors. To obtain nitrile imines, we used hydrazonoyl chlorides 3a-g, which are readily available by the method [11]. Our laboratory has recently developed the so-called “diffusion reagent mixing technique”, which allows gradual generate 1,3-dipoles in solution and effectively suppress the processes of their unwanted dimerization [21]. In many cases, diffusion mixing technique makes it possible to significantly increase the yield of the target [3+2]-cycloaddition products and especially effective in cases of interaction with highly reactive dipoles. For the reactions of 2-thioxoimidazolidine-4,5-diones 1c-f with nitrile imines, we compared two synthetic approaches, which differed in the way of introducing in the reaction mixture of a base generating a dipole from hydrazonoyl chloride 3a-d. In the first? "classical" method, the base solution was added drop by drop to a reaction mixture containing a dipolarophile 1c-f and a dipole precursor 3a-d. In the second approach, we used the equipment for the diffusion reagent mixture (see [15] and Supplementary Information), whereby the volatile base spontaneously diffused into a solution of reagents.
Both methods of reagent mixing enabled us to get spiro compounds 4a-g with a good yields (Table 3).
It was found that the utilization of the diffusion mixing technique resulted in the target product yields comparable or exceeded the result obtained by the amine adding drop by drop. Compound 4c was obtained with a yield near quantitative using both methods. The only exception was compound 4b, whose yield when using the diffusion mixing technique was significantly lower due to the formation of a by-product 7 (Scheme 4).
It is known that in several cases [11,22] the interaction of nitrile imines and cyclic dipolarophiles led to the destruction of the initial imidazolidine cycle instead of the creation of a spiro-jointed product. Probably, in our case, as in the above publications, the presence of a strong electrone acceptor (nitro group) in the dipole structure stabilizes the zwitterionic intermediate III, leading to the formation of the product 7 (Scheme 4). At the same time, the fragmentation of the molecule apparently occurs only when two aromatic fragments are present in the structure of the thioparabanic acid (no formation of a by-product was observed when products 4f and 4g were obtained). An analogous influence of the nature of substituents on the tendency to fragmentation was previously observed in the formation of spiro compounds from 5-arylidene thiohydantoins and thiosemicarbazides [23]. The structure of product 7 was proposed based on a match between the spectral data obtained by us with those described in the literature [24].
Optimizing the conditions for the reaction of nitrile imines and 2-thioxoimidazolidine-4,5-diones, we found out that the yield of the target spiro-conjugated product 4a-g for most substrates was primarily influenced by the electronic properties of the substituents and didn't depend much on the solvent selection. The situation dramatically changed when product 7 with a disrupted imidazolidine fragment could be formed as a result of the reaction (Table 4). TLC analysis of the reaction mixture revealed that compound 6 in high-polar solvents was either formed in trace amounts (Entry 2), or was absent from the mixture (Entry 1). Furthermore, the use of slightly less polar acetone or chloroform in the reaction led to the formation of a large amount of by-product 7, in some cases even exceeding the amount of the target spiro compound 4b (Entry 4 and 5). The yield of product 4b slightly increased when the reaction mixture cooled (Entry 3), but the significance of this factor was negligible.
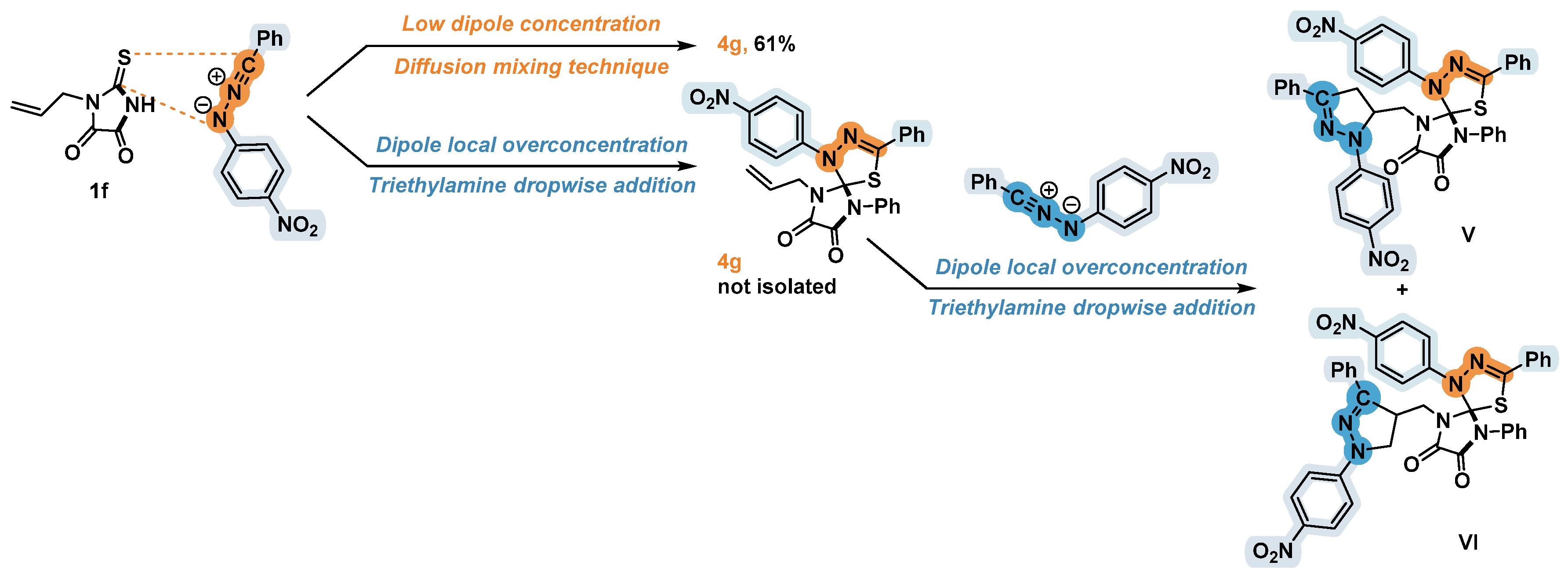
The monosubstituted double bond –CH=CH2 is known to be capable of reacting easily in the 1,3-dipolar cycloaddition, but in most cases the thiocarbonyl group is significantly more active dipolarophile than the carbon-carbon double bond [7]. The difference in the reactivity of C=S and C=C groups made it possible to synthesize the compound 4g, the reaction product of the thioparabanic acid 1f containing an allyl substituent at the nitrogen atom. By employing the diffusion mixing technique, we managed to obtain the product 4g in a good yield (Table 3), while carrying out this reaction by adding triethylamine drop by drop led to the formation of a mixture of compounds that could not be qualitatively separated by chromatography. Based on the analysis of the spectral data obtained for this mixture, we assumed that the dropwise addition of a very dilute triethylamine solution to 1f and hydrazonoyl chloride 3b led to a local overconcentration of the generated nitrile imine, which was sufficient to attach the second dipole molecule to the already formed spiro compound 4g (Scheme 5). Presumably, the addition of nitrile imine to the carbon-carbon double bonds of the allyl substituent of the product 4g occurred analogously to reactions with monosubstituted olefins [1] and led to the formation of two regioisomers V and VI very similar in their chromatographic properties. In the case of diffusion, the unstable 1,3-dipole was present in the solution at a low concentration sufficient for reaction with only one most active C=X bond.
Scheme 5.
The proposed products of 1-allyl-2-thioxoimidazolidine-4,5-dione 1f and hydrazonoyl chloride 3b reaction.
Scheme 5.
The proposed products of 1-allyl-2-thioxoimidazolidine-4,5-dione 1f and hydrazonoyl chloride 3b reaction.
Earlier it was reported that the cycloaddition of nitrile imines to the C=N bond of 2-iminothiazolidine-4-ones makes it possible to obtain a spiro-joined product containing the unaffected thiazolidine cycle [25] (Scheme 6 (a)). Meanwhile, the reactions of diphenyl nitrile imine with 5-arylidene substituted 2-iminothiazolidine-4-one resulted in the formation of a product of the sequential addition of two dipole molecules along both exocyclic double bonds C=N and C=S, which were formed as a result of the rearrangement of the intermediate spiro compound [26] (Scheme 6 (b)).
In our study, when the compound 6 was introduced into the reaction with nitrile imines, we established that the sulfur atom tendency to isomerize to an exocyclic position prevailed over the reactivity of the 2-arylimino fragment. The presence of a second carbonyl group in the structure of compound 6 facilitated a process similar to that occurring during isomerization in 1c, which was described in [18]. It appears that, unlike the process described in the article [26], interaction with the dipole leads to a cleavage of the S-C5 bond, instead of the S-C2 bond (Scheme 7).
Based on the obtained results, it can be concluded that 2-thioxoimidazolidine-4,5-diones 1c-f possess the capability to undergo the 1,3-dipolar cycloaddition with nitrile imines obtained in situ from the corresponding hydrazonoyl chlorides 3a-d. As a result, heterocycles 4a-g consisting of spiro-conjugated imidazolidine and 1,2,4-thiadiazoline fragments were formed. The nitrile imine generation can be accomplished through both the "classical" method of adding the base drop by drop, and the previously proposed diffusion mixing technique. The use of the latter enabled the site-selective attachment of the dipole to the C=S bond of 2-thioxoimidazolidine-4,5-dione 1f in the presence of a second dipolarophilic double bond in the molecule and to obtain the compound 4g. The formation of imidazolidine cycle fragmentation product 7 was observed when spiro compounds were obtained from 1,3-diphenyl-2-thioxoimidazolidine-4,5-dione 1c and nitrile imine generated by diffusion mixing technique. Cycloaddition of nitrile imines to 3-phenyl-2-(phenylimino)thiazolidine-4,5-dione 6 resulted in the same products 4a-d as were obtained from 1,3-diphenyl-2-thioxoimidazolidine-4,5-dione 1c.
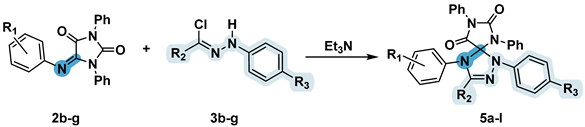
2.4. 1,3-Dipolar Cycloaddition of Nitrile Imines to 5-Arylimino-1,3-diphenylimidazolidine-2,4,5-triones
5-Arylimino-1,3-diphenylimidazolidine-2,4,5-triones 2b-g were introduced into reactions with nitrile imines obtained in situ from hydrazonoyl halides 3b-g (Table 5). Two alternative techniques were also used to generate dipoles, diffusion mixing and triethylamine dropwise addition (detailed description see in Section 2.3). Spiro compounds 5a-l were obtained in a moderate-to-high yield, which, however, was lower on average compared to those obtained for compounds 4.
The diffusion mixing technique revealed a higher yield of the spiro compounds than when triethylamine was added drop by drop, except for products 5f and 5j obtained from imine 2e (R1 = 4-Me). The yield of the cycloaddition product was highly influenced by the electronic properties of the substituents in the initial imine – the presence of a strong donor or acceptor substituent at the R1 position led to a sharp decrease in the yield of the compounds 5b, 5g-i. The obtained results indicate that the C=N group of 5-arylimino-1,3-diphenylimidazolidine-2,4-diones, is in most cases less active than the 2-thiocarbonyl group of compounds 1. When imines 2h-j containing a couple of reactive C=S and C=N bonds were introduced into the reactions with hydrazonoyl chlorides, we observed the formation of a complex mixture of products, presumably consisting of spiro compounds obtained as a results of the addition of a dipole along one of the C=S or C=N bonds, as well as the addition products of two dipole molecules along both of these bonds with two possible diastereomers formation.
Considering the cycloaddition reactions of nitrile imines from the point of frontier molecular orbital (FMO) theory, they are referred to as dipoles, reactions with which can be controlled both by the interaction between the highest occupied molecular orbital of the dipole (HOMOdipole) and the lowest unoccupied molecular orbital of the dipolarophile (LUMOdipolarophile), and by the interaction between the LUMO of the dipole and the HOMO of the dipolarophile [27]. Despite there exists a limited quantity of calculated data regarding the reactions of these dipoles with imines, it is widely acknowledged that the regioselectivity of the 1,3-dipolar cycloaddition is governed by on the relative disposition of dipole and dipolarophile frontier orbitals [27]. Both with the control of the HOMOimine-LUMOdipole gap and with the overlap of the LUMOdipole-HOMOimine, the reaction leads to the formation of a 1,3,4-triazole fragment [28]. In all the cases examined, we observed a regioselective addition of nitrile imines to the C=N double bond of 5-arylimino-1,3-diphenylimidazolidine-2,4-diones, which corresponds to theoretical principle regarding the course of such reactions. The structures of compounds 5с, 5g and 5i were confirmed by X-ray diffraction analysis for the single crystals (Figure 2).
The presence of substituents with pronounced electron-donating or electron-acceptor effects in the dipolarophile structure changes the energy of the frontier molecular orbitals, raising it (if the substituent has donor properties) or decreasing it (acceptor). As a result, one of the ways of overlapping the dipole and the dipolarophile frontier orbitals becomes more preferable [27]. Upon analyzing the correlation between the yield of the resulting spiro compounds and the electronic properties of substituents in aromatic fragments, we found a number of patterns:
- The presence of substituents with a strong mehomeric effects in the aromatic ring of imine 2 decreased the yield of the spiro compound, regardless of whether the substituent was a donor or acceptor. For example, the yield decreased in the sequence 5c (R1 = 4-Br) > 5g (R1 = 4-OMe) > 5b (R1 = 4-NO2) (Table 5).
- The introduction of a donor substituent into the aromatic fragment at the terminal carbon atom of nitrile imine increased the yield of the cycloaddition product. For example, the yields of the products 5c (R1 = 4-Br, R2 = 4-Me-C6H4) and 5g (R1 = 4-OMe, R2 = 4-Me-C6H4) were higher than those of 5h (R1 = 4-Br, R2 = Ph) and 5i (R1 = 4-OMe, R2 = Ph), respectively (Table 5).
The side processes of the dipole dimerization, which are typical for the 1,3-dipolar cycloaddition, were suppressed during the diffusion addition of amine by generating a dipole at low concentration, and with the addition of a base dropwise due to adding its dilute solution. Therefore, the decrease in the yields of target spiro compounds 5b, 5g-i (Table 5) is presumably attributed to the low imine reactivity, instead of other factors impeding the dipole addition. In these cases, the conversion of the initial imines remained low even after several days of conducting the reaction, while the yield of the product changed insignificantly with an increase of the amount of hydrazonoyl chlorides or triethylamine added to the reaction.
It also should be noted that when the reacting nitrile imines were stabilized by the single aromatic substituent (compounds 4d in Table 4, 5k and 5l in Table 5) the difference between the yields of the spiro compound 5 was more significant using different dipole generating techniques than for compounds 4, where the cycloaddition of nitrile imine occurred to the C=S bond. For instance, the difference in the yields of the compound 5k obtained by diffusion mixing and by triethylamine dropwise addition was nearly twice as large as for the product 4d and reached almost 30%. This disparity may be caused by the facile dimerization of the dipole obtained from the hydrazonoyl halide 3d, with a relatively low reactivity of the dipolarophile. For the compound 5l, the difference in yields of target spiro-compounds at different methods of dipole generation was not so significant (16%), apparently due to the greater activity of the C=N bond.
The surprising result was that substituents in the aromatic fragment of imine 2 did not have a significant steric effect on the yields of 5c (R1 = 4-Br), 5d (R1 = 2-Br) and 5e (R1 = 2-Cl), which were comparable for both methods of dipole generation (dropwise or diffusion mixing). Despite this, the NMR spectra of the isolated products 5d and 5e demonstrated two sets of signals, indicating the formation of two isomers. These isomers in the case of compound 5d were separated by column chromatography (see Subsection 3.5), in contrast to the isomers of 5e, distinguishable by TLC analysis, but hardly separable preparatively. It was also demonstrated using NMR 1H, that in the solution individual isomers of compound 5d were gradually transformed into an isomeric mixture. In about a month, they reached an equilibrium ratio coincided with that had been previously determined for the reaction mixture. If necessary, this isomers mixture may be repeatedly separated. Based on [29] the observed phenomena can be explained by the antropoisomerism conferred by the repulsive interactions of ortho-substituent R1 (halogen atom) and the aromatic substituent R2 at the neighboring nitrogen atom of the triazoline cycle (Figure 3).
In conclusion, it is worth noting that the 1,3-dipolar cycloaddition of nitrile imines to compounds 2 containing an exocyclic bond C=N resulted in the formation of a heterocyclic product 5 with the junked imidazolidine and 1,2,4-triazoline fragments. The yields of the products 5were generally good to excellent, except for those obtained from imines 2f and 2g, which substituents had pronounced electron-donating or electron-acceptor properties. The formation of a complex mixtures comprising the products of the addition of 1 or 2 nitrile imine molecules was observed for the sulfur-containing dipolarophiles 2h-j. The structures of compounds 5с, 5g and 5i were confirmed by X-ray diffraction analysis. In the reactions of nitrile imine formed from hydrazonoyl halide 3e and dipolarophiles 2b and 2d containing ortho-substituents in arylimino fragments, products 5d and 5e were obtained as a mixture of antropoisomers.
2.5. Synthesis and 1,3-Dipolar Cycloaddition Reactions of the Imines 2k и 2l Obtained from Unsimmetrical Ureas
Trying to obtain imine from non-symmetrically substituted imidazoletrione 1b, we found that both neighboring C=O group may undergoes the reaction, thus giving a mixture of products (Scheme 2). In contrast to the chemoselective aza-Wittig reaction of the carbonyl group at N-Alkyl claimed in [15], in our case the major product contained an imino-group in a position adjacent to N-Ph. The isomeric products 2k and 2l had almost identical chromatographic properties and were indistinguishable for TLC-analysis at CHCl3 elution (this eluent was used in [15]). But the difference between 2k and 2l retention factors became noticeable in less polar systems (for example, hexane-EtOAc or hexane-DCM) and was maximum in the hexane-Et2O system that we used for separation (see Subsection 3.3). Since X-ray diffraction analysis of 2k and 2l turned out to be unrealizable, a structural organization of the imines was proposed based on the structures of the spiro compounds 5m and 5n obtained by their reaction with hydrazonoyl chloride 3b. The aza-Wittig reaction chemoselectivity observed for substrate 1b can be attributed to partial delocalization of the lone pair of electrons on nitrogen by an aromatic fragment, which enhanced the carbonyl activity of the C5 atom nearby. The ratio of the isolated products (2k:2l ≈ 1:3) corresponded with the ratio of isomers estimated by 1H NMR spectrum of aliquot taken from the reaction mixtures before separation.
Scheme 5.
Synthesis of 5m and 5n. Isolated yields are presented.
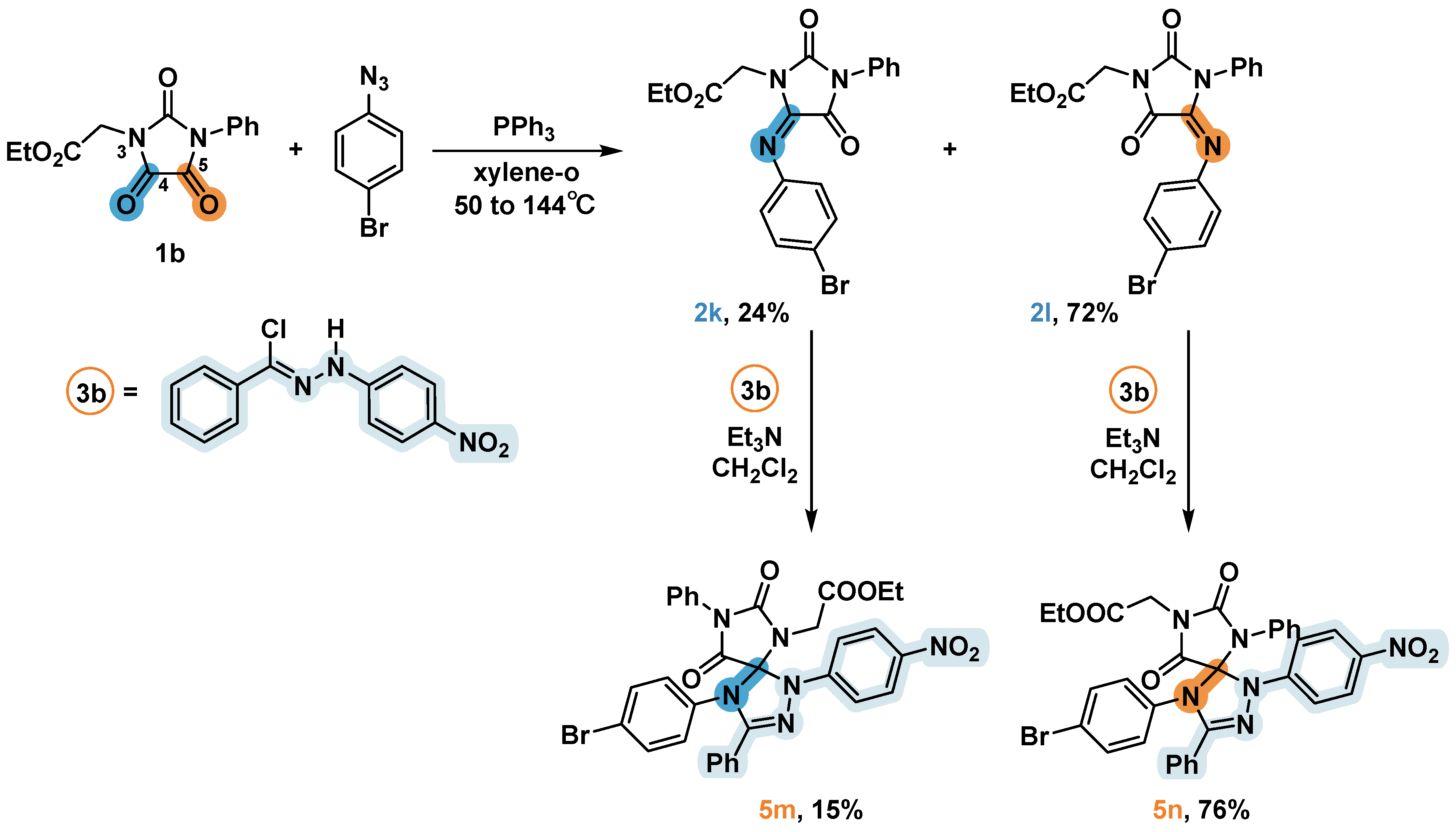
Compounds 2k and 2l were further reacted with hydrazonoyl chloride 3b to form corresponding products 5m and 5n (triethylamine was added dropwise), the structure of which was established on the basis of NMR spectra data. In the 1H NMR spectrum of the compound 5m, the signals of the CH2-group protons at the nitrogen atom of the imidazolidine cycle are presented as two doublets at 4.32 and 4.10 ppm with a coupling constant value of 17.6 Hz (see Supplemental information), which may be due to the difference in the local surrounding of these protons, which are located near the spiro connection. The aliphatic region of the compound 5n 1H NMR spectrum was almost identical to the original imine 2l, which indicates the remoteness of this substituent from other fragments of the molecule.
The activity of compounds 2k and 2l in 1,3-dipolar cycloaddition reactions ьфн be generalized as follows. On the one hand, at the identical preparative procedures the yield of 5n was significantly higher to that obtained in the reaction of the same hydrazonoyl chloride 3b and imine containing only aromatic substituents (Yielddrop (5h) = 50% vs 76% for 5n). On the other hand, the compound 5m was obtained with a low yield of 15%. At the same time, we observed a very low conversion of imine 2k, which was isolated unchanged at the end of the reaction.
The replacement of phenyl by the -CH2COOEt group in imine 2c led to an increase of the nitrogen atom N3 lone electron pair delocalization between neighboring carbon atoms C2 and C4 in both compounds (2k and 2l). Consequently, if the fragment C=N is located at C4 (imine 2k), the electron density on this atom increases significantly more than in compound 2l, where the arylimino group is located at atom C5 (due to two factors: first, this atom is more distant from N3, and second, N3 it turns out to be surrounded by two C=O groups delocalizing its electron density). This small increase in the electron density of the C=N bond could contribute to the optimal overlap between the MO imine and the dipole, thereby dramatically raising the yield of the cycloaddition product.
2.6. Biological Evaluation of Selected Spiro-Compounds
Cytotoxicity of some spiro derivatives of both structural types 4 and 5 was evaluated using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) test in human colorectal carcinoma cell line HCT116 (Table 6). Overall, the selected compounds demonstrated moderate cytotoxic activity with IC50 values in the range of ~25-45 µM.
The obtained cytotoxicity results allow to make some preliminary conclusions about the effect of substituents in aromatic fragments on the activity of compounds. Firstly, type 4 compounds with a NO2-group in one of the aromatic fragments showed slightly better results (compounds 4a, 4b and 4f) compared to 4e. On the other hand, compound 5j, which does not contain NO2-groups, showed lower IC50 values compared to 5c and 5f. When comparing the IC50 values for compounds 4b and 4f, it can be noticed that the combination of a substituted aromatic fragment and a CH2COOEt group at nitrogen atoms is somewhat more preferable than the presence of two phenyls. It can also be pointed out that the presence of a halogen atom in the R1 position of the compound 5c led to a slight increase in the cytotoxicity compared to 5f (R1 = CH3). As can be seen from the IC50 measurements, the cytotoxicity of the presented compounds belongs to the region of micromolar concentrations, which is a good, but insufficient value compared to the known lead compounds. This result may be explained by the high lipophilicity of compounds containing from 3 to 5 aromatic fragments within a relatively small volume of the molecule.
3. Materials and Methods
3.1. General Information
The reagents were purchased from commercial sources, being used without further purification. All solvents used were purified and dehydrated as described in [30]. Reactions were checked by thin layer chromatography (TLC) analysis using silica plates with a fluorescent indicator (254 nm) and visualized with a UV lamp.
The nuclear magnetic experiments were recorded using two different spectrometers, BrukerAvance (Bruker Optik GmbH, Ettlingen, Germany) and Agilent 400-MR (Agilent Technologies, Santa Clara, CA, US) operating at 400 MHz for 1H and 100 MHz for 13C nuclei. Chemical shifts were reported in delta (δ) units (ppm), relative to the residual peak of solvents (ref: CDCl3/DMSO-d6, 1H: 7.26/2.50 ppm; 13C: 77.16/39.52 ppm) and coupling constants (J) in Hz.
The IR spectra were recorded using a Thermo Nicolet iS5 FT-IR Spectrometer (Thermo Electron Scientific Instruments LLC, Madison, WI USA) with an ATR module.
Electrospray ionization high-resolution mass spectra were recorded in positive ion mode on a TripleTOF 5600+ quadrupole time-of-flight mass spectrometer (ABSciex, Concord, Vaughan, ON, Canada) equipped with a DuoSpray ion source. The following MS parameters were applied: capillary voltage 5.5 kV; nebulizing and curtain gas pressures—15 and 25 psi, respectively; ion source temperature—ambient; declustering potential 20 V; m/z range 100–1200. Elemental compositions of the detected ions were determined based on accurate masses and isotopic distributions using Formula Finder software (ABSciex, Concord, ON, Canada). The maximum allowed deviation of the experimental molecular mass from the calculated one was 5 ppm.
The X-Ray data were collected via STOE diffractometer Pilatus100K detector (DECTRIS AG, Baden, Switzerland), Cu Kα (1.54086Å) radiation, rotation method mode. STOE X-AREA software was used for cell refinement and data reduction. Data collection and image processing were performed with X-Area 1.67 (STOE & Cie GmbH, Darmstadt, Germany, 2013). Intensity data were scaled with LANA (part of X-Area) in order to minimize differences in intensities of symmetry-equivalent reflections (multi-scan method). CCDC 2310170 JK269 (compound 5i), CCDC 2310171 JK314-2 (compound 5g) and CCDC 2310172MB-13 (compound 5c) contain the supplementary crystallographic data for this paper. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif website.
3.2. General Procedure for the Synthesis of the Imidazolidine-3,4,5-triones 1a-b and 3-Phenyl-2-(phenylimino)thiazolidine-4,5-dione 6
Oxalyl chloride (1.3 eq) was added dropwise to a stirring solution of urea or thiourea (1 eq) in DCM (0.1 M). The resulting mixture was heated at reflux for 2 h, then cooled to room temperature and washed with brine. The aqueous phase was separated and washed twice with small portions of DCM. Combined organic extracts were dried over anhydrous Na2SO4 and evaporated at reduced pressure to give a solid precipitate of the product.
1,3-Diphenylimidazolidine-2,4,5-trione (1a) was obtained from 1,3-diphenylurea (2.12 g, 10 mmol) and oxalyl chloride (1.65 g, 13 mmol) as white solid (2.64 g, 99%). 1H NMR (400 MHz, CDCl3) δ 7.57 – 7.52 (m, 4H, Ar), 7.51 – 7.45 (m, 6H, Ar).
Ethyl 2-(2,4,5-trioxo-3-phenylimidazolidin-1-yl)acetate (1b) was obtained from ethyl (phenylcarbamoyl)glycinate (1.11 g, 5 mmol) and oxalyl chloride (0.83 g, 6.5 mmol) as white solid (1.37 g, 99%). 1H NMR (400 MHz, CDCl3) δ 7.55 – 7.49 (m, 2H, Ar), 7.48 – 7.41 (m, 3H, Ar), 4.49 (s, 2H, NCH2), 4.28 (q, J = 7.1 Hz, 2H, COOCH2), 1.32 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 166.0, 155.7, 155.3, 152.2, 129.8, 129.6, 129.3, 125.8, 62.7, 40.0, 14.2. HRMS (ESI): calcd for C13H12N2O5 (M+Na)+ 299.0638, found 299.0637.
3-Phenyl-2-(phenylimino)thiazolidine-4,5-dione (6) was obtained from 1,3-diphenylthiourea (2.28 g, 10 mmol) and oxalyl chloride (1.65 g, 13 mmol) as yellow solid (2.80 g, 99%). 1H NMR (400 MHz, CDCl3) δ 7.60 – 7.53 (m, 2H, Ar), 7.53 – 7.48 (m, 1H, Ar), 7.48 – 7.42 (m, 2H, Ar), 7.39 – 7.32 (m, 2H, Ar), 7.23 – 7.18 (m, 1H, Ar), 6.94 – 6.89 (m, 2H, Ar)
3.3. General Procedure for the Synthesis of the 2-Thioxoimidazolidine-4,5-diones 1c-f
Oxalyl chloride (1.3 eq) was added dropwise to a stirring solution of thiourea (1 eq) in acetonitrile (0.1 M). The resulting mixture was heated at reflux for 6 h. The solvent was removed at reduced pressure and the residue was purified by flash column chromatography on silica gel using CHCl3 as eluent.
1,3-Diphenyl-2-thioxoimidazolidine-4,5-dione (1c) was obtained from 1,3-diphenylthiourea (2.28 g, 10 mmol) and oxalyl chloride (1.65 g, 13 mmol) as orange solid (2.68 g, 95%). 1H NMR (400 MHz, CDCl3) δ 7.59 – 7.50 (m, 6H, Ar), 7.42 – 7.37 (m, 4H, Ar).
Ethyl 2-(3-(4-ethoxyphenyl)-4,5-dioxo-2-thioxoimidazolidin-1-yl)acetate (1d) was obtained from ethyl ((4-ethoxyphenyl)carbamothioyl)glycinate (0.07 g, 0.25 mmol) and oxalyl chloride (0.04 g, 0.33 mmol) as beige solid (0.08 g, 93%). 1H NMR (400 MHz, CDCl3) δ 7.25 – 7.20 (m, 2H, Ar), 7.02 – 6.97 (m, 2H, Ar), 4.76 (s, 2H, NCH2), 4.27 (q, J = 7.1 Hz, 2H, COOCH2CH3), 4.07 (q, J = 7.0 Hz, 2H, OCH2CH3), 1.44 (t, J = 7.0 Hz, 3H, OCH2CH3), 1.31 (t, J = 7.2 Hz, 3H, COOCH2CH3). 13C NMR (101 MHz, CDCl3) δ 180.0, 165.9, 160.0, 154.8, 154.4, 129.2, 123.7, 115.3, 63.9, 62.5, 42.7, 14.9, 14.2. HRMS (ESI): calcd for C15H16N2O5S (M+Na)+ 359.0672, found 359.0675.
Ethyl 2-(3-(4-methoxyphenyl)-4,5-dioxo-2-thioxoimidazolidin-1-yl)acetate (1e) was obtained from ethyl ((4-methoxyphenyl)carbamothioyl)glycinate (0.11 g, 0.40 mmol) and oxalyl chloride (0.07 g, 0.52 mmol) as beige solid (0.10 g, 77%). 1H NMR (400 MHz, CDCl3) δ 7.26 – 7.22 (m, 2H), 7.04 – 6.99 (m, 2H), 4.77 (s, 2H, NCH2), 4.27 (q, J = 7.1 Hz, 2H, COOCH2CH3), 3.85 (s, 3H, OCH3), 1.31 (t, J = 7.1 Hz, 3H, COOCH2CH3). 13C NMR (101 MHz, CDCl3) δ 179.9, 165.9, 160.6, 154.8, 154.4, 129.2, 123.9, 114.9, 62.6, 55.7, 42.7, 14.2. HRMS (ESI): calcd for C14H14N2O5S (2M+Na)+ 667.1139, found 667.1138.
1-Allyl-2-thioxoimidazolidine-4,5-dione (1f) was obtained from 1-allylthiourea (2.42 g, 21 mmol) and oxalyl chloride (3.42 g, 27 mmol) as yellowish solid (1.67 g, 47%). 1H NMR (400 MHz, CDCl3) δ 9.11 (br.s, 1H, NH), 5.83 (ddt, J = 16.4, 10.1, 6.0 Hz, 1H, CH), 5.34 – 5.27 (m, 2H, =CH2), 4.54 (d, J = 6.0 Hz, 2H, NCH2). 13C NMR (101 MHz, DMSO) δ 178.0, 155.7, 155.0, 129.3, 120.2, 43.7. HRMS (ESI): calcd for C6H6N2O2S (M+Na)+ 193.0042, found 193.0042.
3.4. General Procedure for the Synthesis of the 5-Aryliminoimidazolidine-2,4-diones 2a-l
Triphenylphosphine (1.1 eq) and arylazide (1.1 eq) solution in xylene (0.1 M) was stirred at 50℃ for 0.5-2 hours (until nitrogen release becomes visually undetectable) and charged with imidazolidine-4,5-dione 1a-c (1 eq). The reaction mixture was refluxed for 6 hours and then was left for 16 hours with stirring without additional heating. The solvent was removed in vacuo and the resulting oily mass was purified by flash chromatography with CHCl3 as eluent. Compounds 2a-j were additionally recrystallized from isopropyl alcohol. Compounds 2k and 2l were isolated by column chromatography with hexane-Et2O system as eluent (gradient elution 80/20 to 55/45).
(E)-5-((4-Chlorophenyl)imino)-1,3-diphenylimidazolidine-2,4-dione (2a) was obtained from 1a (0.10 g, 0.4 mmol), 1-azido-4-chlorobenzene (0.07 g, 0.44 mmol) and PPh3 (0.12 g, 0.44 mmol) as orange solid (0.12 g, 79%). 1H NMR (400 MHz, CDCl3) δ 7.57 – 7.51 (m, 4H, Ar), 7.50 – 7.40 (m, 6H, Ar), 7.28 (d, J = 8.5 Hz, 2H, Ar), 6.94 (d, J = 8.5 Hz, 2H, Ar). 13C NMR (101 MHz, CDCl3) δ 153.3, 152.1, 144.6, 131.4, 131.0, 130.4, 130.2, 129.6, 129.4, 129.3, 128.9, 128.9, 128.8, 127.2, 126.0, 125.9, 121.5. HRMS (ESI): calcd for C21H14ClN3O2 (M+H)+ 376.0847, found 376.0852.
(E)-5-((2-Chlorophenyl)imino)-1,3-diphenylimidazolidine-2,4-dione (2b) was obtained from 1a (0.40 g, 1.5 mmol), 1-azido-2-chlorobenzene (0.25 g, 1.65 mmol) and PPh3 (0.43 g, 1.65 mmol) as yellow solid (0.34 g, 60%). 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 7.9 Hz, 2H, Ar), 7.56 (t, J = 7.7 Hz, 2H, Ar), 7.47 (d, J = 4.2 Hz, 5H, Ar), 7.40 (d, J = 8.2 Hz, 2H, Ar), 7.21 (t, J = 7.6 Hz, 1H, Ar), 7.06 (t, J = 7.6 Hz, 1H, Ar), 6.95 (d, J = 8.0 Hz, 1H, Ar). 13C NMR (101 MHz, CDCl3) δ 153.2, 152.1, 144.0, 141.5, 131.3, 130.2, 129.6, 129.4, 129.3, 128.9, 127.2, 127.2, 125.9, 125.4, 120.6. HRMS (ESI): calcd for C21H14ClN3O2 (2M+Na)+ 773.1441, found 773.1443.
(E)-5-((4-Bromophenyl)imino)-1,3-diphenylimidazolidine-2,4-dione (2c) was obtained from 1a (0.39 g, 1.45 mmol), 1-azido-4-bromobenzene (0.32 g, 1.60 mmol) and PPh3 (0.42 g, 1.60 mmol) as yellow solid (0.37 g, 60%). 1H NMR (400 MHz, CDCl3) δ 7.57 – 7.53 (m, 4H, Ar), 7.51 – 7.45 (m, 5H, Ar), 7.42 (d, J = 8.3 Hz, 3H, Ar), 6.87 (d, J = 8.2 Hz, 2H, Ar). 13C NMR (101 MHz, CDCl3) δ 153.2, 152.0, 145.1, 139.9, 131.7, 131.4, 130.2, 129.4, 129.3, 128.9, 128.9, 127.2, 125.9, 121.8, 118.1. HRMS (ESI): calcd for C21H14BrN3O2 (M+H)+ 420.0342, found 420.0344.
(E)-5-((2-Bromophenyl)imino)-1,3-diphenylimidazolidine-2,4-dione (2d) was obtained from 1a (0.19 g, 0.71 mmol), 1-azido-2-bromobenzene (0.15 g, 0.78 mmol) and PPh3 (0.20 g, 0.78 mmol) as yellow solid (0.23 g, 76%). 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.9 Hz, 2H, Ar), 7.63 (d, J = 8.0 Hz, 1H, Ar), 7.58 (t, J = 7.8 Hz, 2H, Ar), 7.53 – 7.45 (m, 5H, Ar), 7.43 – 7.39 (m, 1H, Ar), 7.29 (t, J = 7.6 Hz, 1H, Ar), 7.06 – 6.97 (m, 2H, Ar). 13C NMR (101 MHz, CDCl3) δ 153.0, 145.3, 132.7, 131.3, 130.2, 129.4, 129.3, 128.9, 127.8, 127.2, 125.9, 125.7, 120.4, 114.1. HRMS (ESI): calcd for C21H14BrN3O2 (M+Na)+ 442.0162, found 442.0167.
(E)-1,3-Diphenyl-5-(p-tolylimino)imidazolidine-2,4-dione (2e) was obtained from 1a (0.27 g, 1.00 mmol), 1-azido-4-methylbenzene (0.15 g, 1.10 mmol) and PPh3 (0.29 g, 1.10 mmol) as light orange solid (0.35 g, 84%). 1H NMR (400 MHz, CDCl3) δ 7.61 – 7.52 (m, 4H, Ar), 7.52 – 7.44 (m, 5H, Ar), 7.43 – 7.38 (m, 1H, Ar), 7.13 (d, J = 7.9 Hz, 2H, Ar), 6.92 (d, J = 7.9 Hz, 2H, Ar), 2.33 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 153.2, 143.4, 139.4, 134.8, 130.4, 129.3, 129.2, 128.8, 128.8, 128.7, 127.3, 126.1, 120.2, 120.1, 120.1, 21.1. HRMS (ESI): calcd for C22H17N3O2 (M+H)+ 356,1394, found 356.1396.
(E)-5-((4-Methoxyphenyl)imino)-1,3-diphenylimidazolidine-2,4-dione (2f) was obtained from 1a (0.53 g, 2.00 mmol), 1-azido-4-methoxybenzene (0.33 g, 2.20 mmol) and PPh3 (0.58 g, 2.20 mmol) as orange solid (0.48 g, 65%). 1H NMR (400 MHz, DMSO-d6) δ 7.58 – 7.54 (m, 4H, Ar), 7.52 (d, J = 7.6 Hz, 2H, Ar), 7.48 – 7.44 (m, 4H, Ar), 7.02 (d, J = 8.7 Hz, 2H, Ar), 6.84 (d, J = 8.8 Hz, 2H, Ar), 3.72 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 156.4, 153.9, 152.4, 140.7, 139.0, 132.5, 131.0, 129.0, 128.9, 128.5, 128.2, 127.9, 127.0, 122.3, 113.5, 55.2. HRMS (ESI): calcd for C22H17N3O3 (M+H)+ 372.1343, found 372.1347.
(E)-5-((4-Nitrophenyl)imino)-1,3-diphenylimidazolidine-2,4-dione (2g) was obtained from 1a (0.53 g, 2.00 mmol), 1-azido-4-nitrobenzene (0.36 g, 2.20 mmol) and PPh3 (0.58 g, 2.20 mmol) as light yellow solid (0.58 g, 75%). 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 2H, Ar), 7.57 (m, 3H, Ar), 7.52 – 7.38 (m, 7H, Ar), 7.03 (s, 2H, Ar). 13C NMR (101 MHz, CDCl3) δ 153.4, 152.8, 152.0, 144.5, 140.3, 131.1, 130.0, 129.5, 129.4, 129.2, 129.1, 127.1, 125.8, 124.8, 120.2. HRMS (ESI): calcd for C21H14N4O4 (M+H)+ 409.0907, found 409.0911.
(E)-5-((4-bromophenyl)imino)-1,3-diphenyl-2-thioxoimidazolidin-4-one (2h) was obtained from 1c (0.50 g, 1.77 mmol), 1-azido-4-bromobenzene (0.39 g, 1.94 mmol) and PPh3 (0.51 g, 1.94 mmol) as yellow solid (0.64 g, 82%). 1H NMR (400 MHz, CDCl3) δ 7.60 – 7.55 (m, 2H, Ar), 7.50 (q, J = 9.2, 8.3 Hz, 6H, Ar), 7.43 – 7.36 (m, 4H, Ar), 6.91 (d, J = 8.3 Hz, 2H, Ar). 13C NMR (101 MHz, CDCl3) δ 179.2, 153.1, 144.5, 133.8, 132.0, 131.7, 129.8, 129.7, 129.5, 129.4, 128.9, 128.3, 125.4, 122.4, 118.7. HRMS (ESI): calcd for C22H17N3O2S (2M+Na)+ 797.1975, found 797.1986.
(E)-5-((4-Methoxyphenyl)imino)-1,3-diphenyl-2-thioxoimidazolidin-4-one (2i) was obtained from 1c (0.71 g, 2.50 mmol), 1-azido-4-methoxybenzene (0.41 g, 2.75 mmol) and PPh3 (0.72 g, 2.75 mmol) as red solid (0.86 g, 88%). 1H NMR (400 MHz, CDCl3) δ 7.58 – 7.53 (m, 2H, Ar), 7.53 – 7.46 (m, 6H, Ar), 7.40 (d, J = 7.4 Hz, 2H, Ar), 7.17 (d, J = 6.5 Hz, 2H, Ar), 6.83 (d, J = 6.6 Hz, 2H, Ar), 3.78 (s, 3H, OCH3). 13C NMR (101 MHz, CDCl3) δ 178.9, 158.3, 153.1, 139.3, 137.6, 134.2, 132.3, 129.6, 129.3, 129.0, 128.4, 123.9, 113.7, 55.5. HRMS (ESI): calcd for (M+H)+ 388.1114, found 388.1122.
(E)-5-((4-Nitrophenyl)imino)-1,3-diphenyl-2-thioxoimidazolidin-4-one (2j) was obtained from 1c (0.47 g, 1.66 mmol), 1-azido-4-nitrobenzene (0.30 g, 1.83 mmol) and PPh3 (0.48 g, 1.83 mmol) as light yellow solid (0.48 g, 72%). 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 2H, Ar), 7.64 – 7.46 (m, 8H, Ar), 7.41 (d, J = 7.4 Hz, 2H, Ar), 7.08 (s, 2H, Ar). 13C NMR (101 MHz, CDCl3) δ 179.2, 152.3, 144.6, 141.1, 133.3, 131.8, 129.8, 129.5, 129.4, 128.7, 128.1, 124.6, 120.4. HRMS (ESI): calcd for C21H14N4O3S (M+H)+ 403.0859, found 403.0864.
Ethyl (E)-2-(5-((4-bromophenyl)imino)-2,4-dioxo-3-phenylimidazolidin-1-yl)acetate (2k) was obtained from 1b (0.46 g, 1.66 mmol), 1-azido-4-bromobenzene (0.36 g, 1.83 mmol) and PPh3 (0.48 g, 1.83 mmol) as orange solid (0.17 g, 24%). 1H NMR (400 MHz, CDCl3) δ 7.48 – 7.43 (m, 3H, Ar), 7.43 – 7.37 (m, 4H, Ar), 6.92 – 6.87 (m, 2H, Ar), 4.59 (s, 2H, NCH2), 4.29 (q, J = 7.2 Hz, 2H, COOCH2), 1.33 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 166.8, 153.5, 152.6, 144.6, 139.3, 131.8, 130.2, 129.3, 128.9, 125.9, 122.3, 118.5, 62.3, 40.7, 14.3. HRMS (ESI): calcd for C19H16BrN3O4 (M+Na)+ 452.0216, found 452.0222.
Ethyl (E)-2-(4-((4-bromophenyl)imino)-2,5-dioxo-3-phenylimidazolidin-1-yl)acetate (2l) was obtained from 1b (0.46 g, 1.66 mmol), 1-azido-4-bromobenzene (0.36 g, 1.83 mmol) and PPh3 (0.48 g, 1.83 mmol) as yellow solid (0.52 g, 72%). 1H NMR (400 MHz, CDCl3) δ 7.54 – 7.48 (m, 4H, Ar), 7.46 – 7.39 (m, 3H, Ar), 6.84 (d, J = 8.5 Hz, 2H, Ar), 4.38 (s, 2H, NCH2), 4.25 (q, J = 7.1 Hz, 2H, COOCH2), 1.30 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 166.4, 153.9, 152.4, 144.9, 140.3, 131.8, 131.3, 129.4, 128.9, 127.2, 122.0, 118.3, 62.5, 39.7, 14.2. HRMS (ESI): calcd for C19H16BrN3O4 (M+Na)+ 452.0216, found 452.0220.
3.5. General Procedure for the Synthesis of the Spirocyclic Products 4a-g and 5a-n
Diffusion mixing technique (a): Small vial (15 ml volume, diameter 1.3 cm) equipped with a magnetic bar was charged with a mixture of dipolarophile 1 or 2 (1 eq, 0.150 mmol) and hydrazonoyl chloride 3 (1.1 eq, 0.165 mol) in 4 ml of chloroform and then placed in bigger vial (50 ml volume, diameter 3.5 cm) containing triethylamine (35.85 mmol, 5 ml). The outer vial was tightly closed with a lid and the reaction mixture was stirred at room temperature for 24–48 h.
Triethylamine dropwise addition (b): A solution of TEA (1.2 eq) in DCM (3 mL) was slowly added for 30 min to a stirring solution dipolarophile 1 or 2 (1 eq, 0.150 mmol) and hydrazonoyl chloride 3 (1.1 eq, 0.165 mol) in 4 ml of DCM. After the addition, the reaction mixture was stirred at room temperature for 24–48 h.
In both cases after the reactions were completed (TCL-control) the solvent was removed in vacuo and the residue was purified by column chromatography on silica gel with EtOAc/hexane or DCM as eluent.
3-(4-Chloro-3-nitrophenyl)-1,6,9-triphenyl-4-thia-1,2,6,9-tetraazaspiro[4.4]non-2-ene-7,8-dione (4a) was obtained from 1c and 3a as pale yellow solid (96%a/88%b). 1H NMR (400 MHz, DMSO-d6) δ 8.12 (dd, J = 1.9, 0.7 Hz, 1H, Ar), 7.72 – 7.66 (m, 2H, Ar), 7.52 (dd, J = 8.8, 7.3 Hz, 2H, Ar), 7.42 (m, 8H, Ar), 7.27 – 7.20 (m, 5H, Ar). 13C NMR (101 MHz, DMSO-d6) δ 156.3, 148.1, 140.5, 139.4, 132.8, 132.3, 130.1, 130.1, 129.8, 129.5, 129.4, 127.7, 125.8, 124.3, 121.9, 117.4, 112.1. HRMS (ESI): calcd for C28H18ClN5O4S (M+H)+ 578.0660, found 578.0665.
1-(4-Nitrophenyl)-3,6,9-triphenyl-4-thia-1,2,6,9-tetraazaspiro[4.4]non-2-ene-7,8-dione (4b) was obtained from 1c and 3b as beige solid (44%a/91%b). 1H NMR (400 MHz, CDCl3) δ 8.33 – 8.27 (m, 2H, Ar), 7.55 – 7.51 (m, 2H, Ar), 7.46 – 7.42 (m, 2H, Ar), 7.42 – 7.33 (m, 9H, Ar), 7.25 – 7.20 (m, 4H, Ar). 13C NMR (101 MHz, CDCl3) δ 155.0, 151.7, 148.8, 145.2, 144.4, 131.4, 130.0, 130.0, 129.2, 126.6, 124.9, 124.7, 120.8, 120.8. HRMS (ESI): calcd for C28H19N5O4S (M+H)+ 522.1231, found 522.1226.
3-(4-Bromophenyl)-1,6,9-triphenyl-4-thia-1,2,6,9-tetraazaspiro[4.4]non-2-ene-7,8-dione (4c) was obtained from 1c and 3c as white solid (99%a,b). 1H NMR (400 MHz, CDCl3) δ 7.46 – 7.42 (m, 2H, Ar), 7.42 – 7.37 (m, 2H, Ar), 7.36 – 7.31 (m, 8H, Ar), 7.30 – 7.27 (m, 2H, Ar), 7.26 – 7.19 (m, 5H, Ar). 13C NMR (101 MHz, CDCl3) δ 155.78, 140.64, 140.00, 132.55, 132.00, 129.99, 129.65, 129.51, 128.98, 127.55, 127.52, 124.79, 124.43, 118.21, 112.00. HRMS (ESI): calcd for C28H19BrN4O2S (M+H)+ 555.0485, found 555.0487.
3-Methyl-1,6,9-triphenyl-4-thia-1,2,6,9-tetraazaspiro[4.4]non-2-ene-7,8-dione (4d) was obtained from 1c and 3d as white solid (88%a/74%b). 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.32 (m, 8H, Ar), 7.26 – 7.20 (m, 6H, Ar), 7.14 (t, J = 7.3 Hz, 1H, Ar), 2.00 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 155.8, 140.9, 139.2, 132.8, 129.9, 129.6, 129.4, 127.6, 124.1, 117.7, 112.0, 16.5. HRMS (ESI): calcd for C23H18N4O2S (2M+Na)+ 851.2193, found 851.2186.
Ethyl 2-(3-(4-bromophenyl)-9-(4-ethoxyphenyl)-7,8-dioxo-1-phenyl-4-thia-1,2,6,9-tetraazaspiro[4.4]non-2-en-6-yl)acetate (4e) was obtained from 1d and 3c as beige solid (77%a/49%b). 1H NMR (400 MHz, CDCl3) δ 7.48 (d, J = 8.5 Hz, 2H, Ar), 7.39 – 7.32 (m, 4H, Ar), 7.24 (d, J = 8.4 Hz, 2H, Ar), 7.18 (t, J = 7.3 Hz, 1H, Ar), 7.08 (d, J = 9.0 Hz, 2H, Ar), 6.79 (d, J = 9.0 Hz, 2H, Ar), 4.31 (d, J = 17.6 Hz, 1H, NCH2), 4.28 – 4.13 (m, 2H, COOCH2CH3), 4.01 – 3.90 (m, 3H, NCH2+OCH2CH3), 1.34 (t, J = 7.0 Hz, 3H, OCH2CH3), 1.24 (t, J = 7.1 Hz, 3H, COOCH2CH3). 13C NMR (101 MHz, CDCl3) δ 166.1, 159.6, 156.4, 155.4, 140.4, 140.0, 132.1, 129.9, 129.0, 128.9, 127.6, 125.2, 124.5, 124.2, 118.8, 115.2, 111.0, 63.7, 62.4, 41.9, 14.7, 14.1. HRMS (ESI): calcd for C28H25BrN4O5S (M+H)+ 609.0802, found 609.0803.
Ethyl 2-(9-(4-methoxyphenyl)-1-(4-nitrophenyl)-7,8-dioxo-3-phenyl-4-thia-1,2,6,9-tetraazaspiro[4.4]non-2-en-6-yl)acetate (4f) was obtained from 1e and 3b as beige solid (93%a/94%b). 1H NMR (400 MHz, CDCl3) δ 8.25 (d, J = 9.3 Hz, 2H, Ar), 7.58 – 7.52 (m, 2H, Ar), 7.47 – 7.39 (m, 5H, Ar), 7.03 (d, J = 9.0 Hz, 2H, Ar), 6.82 (d, J = 9.0 Hz, 2H, Ar), 4.39 (d, J = 17.5 Hz, 1H, NCH2), 4.20 (qq, J = 10.8, 7.1 Hz, 2H, COOCH2CH3), 4.06 (d, J = 17.5 Hz, 1H, NCH2), 3.74 (s, 3H, OCH3), 1.24 (t, J = 7.1 Hz, 3H, COOCH2CH3). 13C NMR (101 MHz, CDCl3) δ 165.8, 160.5, 145.7, 131.4, 129.4, 129.2, 129.0, 126.7, 125.7, 123.8, 117.0, 115.1, 110.6, 62.6, 55.6, 42.1, 14.2. HRMS (ESI): calcd for C27H23N5O7S (M+Na)+ 584.1210, found 584.1217.
6-Allyl-1-(4-nitrophenyl)-3-phenyl-4-thia-1,2,6,9-tetraazaspiro[4.4]non-2-ene-7,8-dione (4g) was obtained from 1f and 3b as light yellow solid (61%a). 1H NMR (400 MHz, CDCl3) δ 8.53 – 8.39 (m, 4H, Ar), 7.96 – 7.91 (m, 2H, Ar), 7.62 – 7.53 (m, 3H, Ar), 7.51 – 7.44 (m, 1H, NH), 5.90 (ddt, J = 17.2, 10.2, 5.6 Hz, 1H, CH), 5.29 – 5.18 (m, 2H, =CH2), 4.06 – 4.00 (m, 2H, NCH2). 13C NMR (101 MHz, CDCl3) δ 168.9, 167.4, 160.2, 158.2, 146.7, 143.6, 133.4, 133.4, 132.4, 129.6, 128.8, 127.1, 124.9, 124.7, 117.1, 42.2. HRMS (ESI): calcd for C19H15N5O4S (2M+Na)+ 841.1582, found 841.1582.
4-(4-Bromophenyl)-1,3,6,8-tetraphenyl-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5a) was obtained from 2d and 3e as beige solid (71%a/62%b). 1H NMR (400 MHz, CDCl3) δ 7.53 – 7.50 (m, 2H, Ar), 7.47 – 7.42 (m, 3H, Ar), 7.42 – 7.38 (m, 3H, Ar), 7.38 – 7.35 (m, 4H, Ar), 7.35 – 7.26 (m, 7H, Ar), 7.15 – 7.11 (m, 2H, Ar), 7.07 – 7.02 (m, 1H, Ar), 6.98 – 6.93 (m, 2H, Ar). 13C NMR (101 MHz, CDCl3) δ 165.0, 151.8, 146.6, 141.4, 135.7, 133.9, 132.9, 130.6, 130.0, 129.7, 129.5, 129.5, 129.0, 128.7, 128.6, 127.9, 127.6, 126.3, 126.1, 124.9, 122.7, 121.9, 115.6, 97.5. HRMS (ESI): calcd for C34H24BrN5O2 (M+H)+ 614.1186, found 614.1193.
1,4-Bis(4-nitrophenyl)-6,8-diphenyl-3-(p-tolyl)-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5b) was obtained from 2g and 3f as orange solid (40%a/13%b). 1H NMR (400 MHz, CDCl3) δ 8.28 – 8.23 (m, 2H, Ar), 8.20 – 8.16 (m, 2H, Ar), 7.52 – 7.44 (m, 3H, Ar), 7.39 – 7.34 (m, 4H, Ar), 7.33 – 7.27 (m, 3H, Ar), 7.25 – 7.18 (m, 6H, Ar), 7.13 (d, J = 8.1 Hz, 2H, Ar), 2.35 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 164.0, 151.3, 147.8, 146.5, 145.5, 142.1, 141.9, 141.8, 132.8, 130.1, 130.0, 129.9, 129.8, 129.6, 128.6, 128.0, 126.6, 126.1, 125.7, 125.2, 125.1, 122.2, 113.6, 95.5, 21.6. HRMS (ESI): calcd for C35H25N7O6 (M+Na)+ 662.1759, found 662.1765.
4-(4-Bromophenyl)-1-(4-nitrophenyl)-6,8-diphenyl-3-(p-tolyl)-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5c) was obtained from 2d and 3f as beige solid (90%a/85%b). 1H NMR (400 MHz, DMSO-d6) δ 8.34 (d, J = 9.4 Hz, 2H, Ar), 7.69 (d, J = 8.7 Hz, 2H, Ar), 7.55 – 7.50 (m, 3H, Ar), 7.49 – 7.43 (m, 4H, Ar), 7.39 – 7.35 (m, 3H, Ar), 7.33 (d, J = 8.2 Hz, 2H, Ar), 7.23 – 7.17 (m, 4H, Ar), 7.15 (d, J = 8.6 Hz, 2H, Ar), 2.29 (s, 3H,CH3). 13C NMR (101 MHz, DMSO-d6) δ 163.5, 148.2, 145.2, 140.7, 135.0, 133.1, 132.5, 129.9, 129.9, 129.8, 129.6, 129.5, 128.9, 128.8, 128.6, 127.9, 126.8, 126.5, 126.1, 122.1, 121.5, 113.1, 95.3, 21.0. HRMS (ESI): calcd for C35H25BrN6O4 (M+Na)+ 695.1013, found 695.1015.
4-(2-Bromophenyl)-1-(4-nitrophenyl)-6,8-diphenyl-3-(p-tolyl)-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5d) was obtained from 2c and 3f as yellow solid (84%a/83%b). Mixture of isomers I and II (1:2.3 in CDCl3). Isomer I: 1H NMR (400 MHz, CDCl3) δ 8.20 – 8.15 (m, 2H, Ar), 7.68 – 7.65 (m, 1H, Ar), 7.53 – 7.49 (m, 2H, Ar), 7.47 – 7.41 (m, 5H, Ar), 7.29 – 7.24 (m, 2H, Ar), 7.23 – 7.17 (m, 4H, Ar), 7.17 – 7.11 (m, 6H, Ar), 2.34 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 163.6, 151.6, 150.0, 145.8, 141.4, 141.3, 137.1, 134.2, 133.5, 133.2, 130.6, 130.5, 129.7, 129.6, 129.4, 129.2, 128.7, 127.8, 127.7, 126.0, 125.9, 125.1, 123.3, 122.9, 113.2, 96.2, 21.6. HRMS (ESI): calcd for C35H25BrN6O4 (M+Na)+ 695.1013, found 695.1016. Isomer II: 1H NMR (400 MHz, CDCl3) δ 8.24 – 8.19 (m, 2H, Ar), 7.60 – 7.55 (m, 2H, Ar), 7.45 – 7.39 (m, 6H, Ar), 7.34 – 7.27 (m, 5H, Ar), 7.26 – 7.21 (m, 3H, Ar), 7.10 – 7.04 (m, 4H, Ar), 2.33 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 163.5, 151.5, 149.0, 145.6, 141.3, 141.1, 135.6, 134.9, 133.4, 130.8, 130.4, 129.8, 129.6, 129.5, 129.3, 128.9, 128.8, 128.0, 127.3, 126.1, 126.0, 125.1, 124.6, 124.1, 113.3, 95.5, 21.7. HRMS (ESI): calcd for C35H25BrN6O4 (M+Na)+ 695.1013, found 695.1022.
4-(2-Chlorophenyl)-1-(4-nitrophenyl)-6,8-diphenyl-3-(p-tolyl)-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5e) was obtained from 2a and 3f as orange solid (87%a/81%b). Mixture of isomers I and II (1:1.75 in DMSO-d6). 1H NMR (400 MHz, DMSO-d6) δ 8.33 (d, J = 9.3 Hz, 2HII, Ar), 8.28 (d, J = 9.2 Hz, 2HI, Ar), 7.88 (d, J = 7.8 Hz, 1HII, Ar), 7.71 – 7.64 (m, 1HI+1HII, Ar), 7.56 – 7.45 (m, 5HI+7HII, Ar), 7.44-7.35 (m, 3HI+3HII, Ar), 7.35-7.30 (m, 5HI+4HII, Ar), 7.29-7.26 (m, 1HI, Ar), 7.23 (d, J = 8.0 Hz, 2HI, Ar), 7.19-7.12 (m, 3HI+2HII, Ar), 7.06 – 7.02 (m, 2HII, Ar), 2.29 (s, 3HI+3HII, CH3). 13C NMR (101 MHz, DMSO-d6) δ 163.1, 163.0, 150.9, 150.7, 149.4, 148.7, 145.2, 145.0, 141.2, 141.0, 140.6, 140.5, 134.4, 133.1, 132.9, 132.6, 131.9, 131.7, 131.7, 131.3, 131.1, 130.9, 130.0, 129.8, 129.5, 129.5, 129.4, 129.3, 129.3, 128.8, 128.7, 128.6, 127.9, 127.3, 126.9, 126.8, 126.7, 126.5, 126.3, 126.0, 125.1, 123.4, 122.7, 113.1, 113.1, 113.0, 95.8, 95.1, 40.2, 40.0, 39.7, 39.5, 39.3, 39.1, 38.9, 21.0, 20.8. HRMS (ESI): calcd for C35H25ClN6O4 (M+Na)+ 651.1518, found 651.1527.
1-(4-Nitrophenyl)-6,8-diphenyl-3,4-di-p-tolyl-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5f) was obtained from 2e and 3f as beige solid (71%a/88%b). 1H NMR (400 MHz, DMSO-d6) δ 8.32 (d, J = 9.0 Hz, 2H, Ar), 7.52 (d, J = 8.0 Hz, 3H, Ar), 7.46 (dd, J = 14.1, 6.4 Hz, 4H, Ar), 7.39 – 7.31 (m, 5H, Ar), 7.28 (d, J = 7.9 Hz, 2H, Ar), 7.18 (d, J = 7.9 Hz, 2H, Ar), 7.07 (d, J = 7.6 Hz, 4H, Ar), 2.32 (s, 3H, CH3), 2.28 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 163.7, 150.8, 148.7, 145.4, 140.8, 140.5, 138.6, 132.8, 132.8, 130.5, 130.0, 129.7, 129.4, 129.4, 128.3, 127.8, 127.1, 126.8, 126.4, 125.6, 122.4, 112.9, 95.6, 20.9, 20.6. HRMS (ESI): calcd for C36H28N6O4 (M+Na)+ 631.2064, found 631.2072.
4-(4-Methoxyphenyl)-1-(4-nitrophenyl)-6,8-diphenyl-3-(p-tolyl)-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5g) was obtained from 2f and 3f as orange solid (52%a/38%b). 1H NMR (400 MHz, DMSO-d6) δ 8.31 (d, J = 9.5 Hz, 2H, Ar), 7.55 – 7.50 (m, 3H, Ar), 7.50 – 7.43 (m, 4H, Ar), 7.39 – 7.31 (m, 5H, Ar), 7.19 (d, J = 7.5 Hz, 2H, Ar), 7.13 (d, J = 8.4 Hz, 2H, Ar), 7.10 – 7.06 (m, 2H, Ar), 7.04 (d, J = 9.3 Hz, 2H, Ar), 3.76 (s, 3H, OCH3), 2.28 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 163.8, 159.3, 150.9, 148.9, 145.4, 140.9, 140.4, 133.0, 130.0, 129.7, 129.5, 129.4, 129.4, 129.1, 128.2, 127.8, 127.6, 126.8, 126.4, 125.3, 122.3, 115.2, 112.9, 95.8, 55.6, 21.0. HRMS (ESI): calcd for C36H28N6O5 (M+H)+ 625.2194, found 625.2199.
4-(4-Bromophenyl)-1-(4-nitrophenyl)-3,6,8-triphenyl-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5h) was obtained from 2d and 3b as beige solid (63%a/50%b). 1H NMR (400 MHz, CDCl3) δ 8.28 – 8.22 (m, 2H, Ar), 7.50 – 7.46 (m, 3H, Ar), 7.46 – 7.41 (m, 4H, Ar), 7.40 – 7.33 (m, 5H, Ar), 7.30 (m, 5H, Ar), 7.16 – 7.11 (m, 2H, Ar), 6.99 – 6.93 (m, 2H, Ar). 13C NMR (101 MHz, CDCl3) δ 164.3, 151.5, 148.7, 145.7, 141.6, 134.8, 133.3, 133.2, 130.9, 130.2, 129.9, 129.7, 129.5, 129.0, 128.9, 128.2, 128.1, 126.1, 126.0, 125.4, 124.7, 122.8, 113.3, 96.0. HRMS (ESI): calcd for C34H23BrN6O4 (M+Na)+ 681.0856, found 681.0853.
4-(4-Methoxyphenyl)-1-(4-nitrophenyl)-3,6,8-triphenyl-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5i) was obtained from 2b and 3b as beige solid (60%a/50%b). 1H NMR (400 MHz, DMSO-d6) δ 8.34 – 8.30 (m, 3H, Ar), 7.54 – 7.51 (m, 3H, Ar), 7.50 – 7.48 (m, 2H, Ar), 7.47 – 7.43 (m, 5H, Ar), 7.40 – 7.36 (m, 4H, Ar), 7.14 (d, J = 8.6 Hz, 2H, Ar), 7.10 – 7.07 (m, 2H, Ar), 7.04 (d, J = 9.1 Hz, 2H, Ar), 3.76 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 163.9, 159.4, 150.9, 148.9, 145.4, 140.6, 133.0, 130.9, 130.0, 129.8, 129.5, 129.5, 129.1, 128.9, 128.2, 127.9, 127.5, 126.9, 126.4, 125.4, 125.3, 115.3, 113.0, 95.9, 55.6. HRMS (ESI): calcd for C35H26N6O5 (M+H)+ 611.2037, found 611.2041.
3-(4-Chlorophenyl)-1,6,8-triphenyl-4-(p-tolyl)-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5j) was obtained from 2e and 3g as beige solid (57%a/93%b). 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 7.7 Hz, 2H, Ar), 7.44 – 7.40 (m, 2H, Ar), 7.40 – 7.34 (m, 4H, Ar), 7.34 – 7.27 (m, 6H, Ar), 7.24 – 7.21 (m, 2H, Ar), 7.13 – 7.09 (m, 4H, Ar), 7.05 – 7.01 (m, 1H, Ar), 6.97 (d, J = 8.3 Hz, 2H, Ar), 2.34 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 165.1, 151.9, 146.2, 141.4, 138.6, 135.8, 134.1, 133.5, 130.7, 130.4, 129.7, 129.4, 129.4, 129.1, 128.9, 128.8, 127.4, 127.3, 126.3, 125.2, 124.9, 122.4, 115.3, 97.8, 21.2. HRMS (ESI): calcd for C35H26ClN5O2 (M+H)+ 584.1848, found 584.1849.
3-Methyl-4-(4-nitrophenyl)-1,6,8-triphenyl-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5k) was obtained from 2g and 3d as orange solid (88%a/59%b). 1H NMR (400 MHz, CDCl3) δ 8.27 – 8.22 (m, 2H, Ar), 7.46 – 7.37 (m, 7H, Ar), 7.36 – 7.31 (m, 3H, Ar), 7.23 – 7.19 (m, 4H, Ar), 7.15 – 7.11 (m, 2H, Ar), 7.05 (m, 1H, Ar), 2.03 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 165.5, 151.5, 147.1, 144.1, 141.7, 140.6, 133.9, 130.4, 129.8, 129.6, 129.5, 129.2, 128.1, 127.7, 126.0, 125.3, 124.8, 122.9, 115.9, 96.9, 12.1. HRMS (ESI): calcd for C29H22N6O4 (M+H)+ 519.1775, found 519.1772.
4-(4-Bromophenyl)-3-methyl-1,6,8-triphenyl-1,2,4,6,8-pentaazaspiro[4.4]non-2-ene-7,9-dione (5l) was obtained from 2d and 3d as orange solid (73%a/57%b). 1H NMR (400 MHz, CDCl3) δ 7.54 – 7.50 (m, 2H, Ar), 7.49 – 7.45 (m, 2H, Ar), 7.45 – 7.36 (m, 5H, Ar), 7.35 – 7.28 (m, 3H, Ar), 7.23 – 7.19 (m, 2H, Ar), 7.15 – 7.10 (m, 2H, Ar), 7.03 – 6.98 (m, 1H, Ar), 6.96 – 6.90 (m, 2H, Ar), 1.93 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 165.9, 151.6, 145.4, 142.0, 134.3, 133.3, 133.2, 130.6, 130.6, 129.7, 129.4, 129.0, 127.2, 126.2, 124.6, 123.4, 122.3, 115.3, 97.1, 11.6. HRMS (ESI): calcd for C29H22BrN5O2 (M+H)+ 552.1030, found 552.1023.
Ethyl 2-(4-(4-bromophenyl)-1-(4-nitrophenyl)-7,9-dioxo-3,8-diphenyl-1,2,4,6,8-pentaazaspiro[4.4]non-2-en-6-yl)acetate (5m) was obtained from 2k and 3b as orange solid (15%b). 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 9.2 Hz, 2H, Ar), 7.51 – 7.47 (m, 3H, Ar), 7.46 – 7.43 (m, 2H, Ar), 7.42 – 7.40 (m, 2H, Ar), 7.37 – 7.33 (m, 3H, Ar), 7.29 (d, J = 9.2 Hz, 2H, Ar), 7.22 (d, J = 8.1 Hz, 2H, Ar), 7.09 – 7.02 (m, 2H, Ar), 4.32 (d, J = 17.6 Hz, 1H, NCH2), 4.21 – 4.07 (m, 3H, NCH2+COOCH2CH3), 1.19 (t, J = 7.1 Hz, 3H, COOCH2CH3). 13C NMR (101 MHz, CDCl3) δ 167.4, 164.0, 152.4, 148.8, 146.0, 141.8, 135.6, 133.1, 131.0, 130.1, 129.6, 129.3, 128.9, 128.7, 128.2, 125.9, 125.7, 125.5, 122.5, 113.7, 95.4, 62.3, 41.0, 14.1. HRMS (ESI): calcd for C32H25BrN6O6 (M+Na)+ 691.0911, found 691,0913.
Ethyl 2-(4-(4-bromophenyl)-1-(4-nitrophenyl)-7,9-dioxo-3,6-diphenyl-1,2,4,6,8-pentaazaspiro[4.4]non-2-en-8-yl)acetate (5n) was obtained from 2l and 3b as orange solid (76%b). 1H NMR (400 MHz, DMSO-d6) δ 8.24 – 8.19 (m, 2H, Ar), 7.60 – 7.56 (m, 2H, Ar), 7.46 – 7.41 (m, 2H, Ar), 7.41 – 7.37 (m, 5H, Ar), 7.37 – 7.31 (m, 3H, Ar), 7.30 – 7.26 (m, 2H, Ar), 7.03 – 6.97 (m, 2H, Ar), 4.56 – 4.45 (m, 2H, NCH2), 4.20 (qd, J = 7.0, 1.4 Hz, 2H, COOCH2CH3), 1.21 (t, J = 7.1 Hz, 3H, COOCH2CH3). 13C NMR (101 MHz, DMSO-d6) δ 167.0, 164.1, 151.1, 148.3, 144.9, 140.7, 134.3, 132.9, 132.3, 130.9, 129.8, 129.0, 128.8, 128.5, 127.9, 126.0, 125.7, 125.0, 121.4, 113.5, 95.3, 61.9, 14.0. HRMS (ESI): calcd for C32H25BrN6O6 (M+Na)+ 691.0911, found 691.0913.
3.6. Reagents for MTT Test
Sigma-Aldrich (Schnelldorf, Germany) provided 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Trypan blue, phosphate-buffered saline (PBS), and dimethyl sulfoxide (DMSO) were purchased from PanEco (Moscow, Russia). Fetal bovine serum was obtained from HyClone (Logan, UT, USA), along with flasks and plates purchased from Nunc (Moscow, Russia).
3.7. Cell Lines and Cytotoxicity Evaluation
Human colon adenocarcinoma cell line (HCT116) was purchased from ATCC (Manassas, VA, USA). Cancer cells were routinely grown in RPMI 1640 culture medium, supplemented with 10% fetal bovine serum, glutamine, and 100 U/mL penicillin. HCT116 cell line was grown in flasks in RPMI 1640 fresh culture medium with supplements at 37 oC and 5% CO2. Cells were grown as monolayer cultures, and the cells in the exponential growth phase were trypsinized and suspended in RPMI 1640 medium supplemented.
3.8. Cytotoxicity
To evaluate the cytotoxicity of compounds in vitro, we placed cells in (4 ̶ 7) × 103 cells/mL concentrations in 96-well culture plates for 24 h. Cells were counted after treatment with Trypan blue solution (0.4%). They were then exposed to different concentrations of compounds in two-fold serial (50-100 µM) dilutions in pre-incubated cells at 37 oC for 72 h. In control wells with untreated cells, only (DMCO + PBS) were added. Cell viability was measured by the standard MTT test [31]. The absorbance was measured at 540 nm using a Multiskan™ FC microplate photometer and Skanlt software 6.1 RE for microplate reader, both from Thermo Scientific (Waltham, MA, USA).
3.9. Statistical Analysis
In vitro experiments were carried out in triplicate. Graphpad prism version 9.0 was used to determine the IC50. The data of IC50 are presented as mean ± standard deviation (SD).
4. Conclusions
A series of new spiro derivatives containing pyrazoline and imidazoledione fragments were synthesized by 1,3-dipolar cycloaddition of nitrile imines, which were generated in situ from hydrazonoyl chlorides, to the parabanic acid derivatives. It was shown that the cycloaddition reactions proceed regioselectively in all cases, regardless of the presence of electron-donating or electron-withdrawing substituents in the 1,3-dipole molecules. The study of the 1,3-dipolar cycloaddition reaction of nitrile imines by DFT calculation methods showed that 1,3-dipoles act as nucleophiles in the reaction with 3-phenyl-5-methylidenehydantoin, and the 1,3-dipolar cycloaddition reaction with normal electronic demands (NED) should have been realized.
The reactions of 2-thioxoimidazolidine-4,5-diones and 5-arylimino-1,3-diphenylimidazolidine-2,4,5-triones with nitrile imines were carried out using two alternative synthetic approaches, which differed in the method of adding a base generating a dipole from hydrazonoyl chloride ("classic" dropwise addition method and a diffusion mixing technique). It was found that the diffusion mixing technique typically gives the same or better yield of the target product compared to the addition of amine drop by drop.
Cytotoxycity of some the obtained spiro derivatives was evaluated using MTT test in human colorectal carcinoma cell line HCT116. The tested compounds demonstrated moderate cytotoxic activity with IC50 values in the range of ~25-45 µM.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, E.K.B. and M.E.K.; methodology, M.E.F.; validation, D.E.S., Y.K.G. and V.A.R.; formal analysis, Y.K.G.; investigation, J.V.K., V.T.T., L.M.P., M.E.F.. and V.A.T.; resources, Y.K.G.; data curation, M.E.K.; writing—original draft J.V.K.; supervision, E.K.B.; project administration, M.E.F.; funding acquisition, E.K.B. All authors have read and agreed to the published version of the manuscript; investigation, A.S.P, V.A.Y. and V.S.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, grant number 21-13-00023, and as in part of the NMR and X-ray study by the M.V. Lomonosov Moscow State University Program of Development.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are available from the authors upon request.
Conflicts of Interest
The authors declare no conflict of interest
References
- Jamieson, C.; Livingstone, K. The Nitrile Imine 1,3-Dipole; Springer International Publishing: Cham, 2020; ISBN 978-3-030-43480-9. [Google Scholar] [CrossRef]
- Mantzanidou, M.; Pontiki, E.; Hadjipavlou-Litina, D. Pyrazoles and Pyrazolines as Anti-Inflammatory Agents. Molecules 2021, 26, 3439. [Google Scholar] [CrossRef]
- Chalkha, M.; Akhazzane, M.; Moussaid, F.Z.; Daoui, O.; Nakkabi, A.; Bakhouch, M.; Chtita, S.; Elkhattabi, S.; Housseini, A.I.; El Yazidi, M. Design, Synthesis, Characterization, in Vitro Screening, Molecular Docking, 3D-QSAR, and ADME-Tox Investigations of Novel Pyrazole Derivatives as Antimicrobial Agents. New J. Chem. 2022, 46, 2747–2760. [Google Scholar] [CrossRef]
- Viveka, S.; Dinesha; Shama, P.; Nagaraja, G.K.; Ballav, S.; Kerkar, S. Design and Synthesis of Some New Pyrazolyl-Pyrazolines as Potential Anti-Inflammatory, Analgesic and Antibacterial Agents. Eur. J. Med. Chem. 2015, 101, 442–451. [Google Scholar] [CrossRef]
- Wang, X.; Pan, Y.; Huang, X.; Mao, Z.; Wang, H. A Novel Methodology for Synthesis of Dihydropyrazole Derivatives as Potential Anticancer Agents. Org. Biomol. Chem. 2014, 12, 2028–2032. [Google Scholar] [CrossRef]
- Ahsan, M.J.; Ali, A.; Ali, A.; Thiriveedhi, A.; Bakht, M.A.; Yusuf, M.; Salahuddin; Afzal, O.; Altamimi, A.S.A. Pyrazoline Containing Compounds as Therapeutic Targets for Neurodegenerative Disorders. ACS Omega 2022, 7, 38207–38245. [Google Scholar] [CrossRef]
- S. Shawali, A. Chemoselectivity in 1,3-Dipolar Cycloaddition Reactions of Nitrilimines with Multifunctionalized Dipolarophiles. Curr. Org. Chem. 2014, 18, 598–614. [Google Scholar] [CrossRef]
- Cho, S.; Kim, S.-H.; Shin, D. Recent Applications of Hydantoin and Thiohydantoin in Medicinal Chemistry. Eur. J. Med. Chem. 2019, 164, 517–545. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.J.A.; Nunes, R.C.; Amaral, J.D.; Gonçalves, L.M.; Rodrigues, C.M.P.; Moreira, R.; Santos, M.M.M. Spirotriazoline Oxindoles: A Novel Chemical Scaffold with in Vitro Anticancer Properties. Eur. J. Med. Chem. 2017, 140, 494–509. [Google Scholar] [CrossRef] [PubMed]
- Yavari, I.; Taheri, Z.; Sheikhi, S.; Bahemmat, S.; Halvagar, M.R. Synthesis of Thia- and Thioxo-Tetraazaspiro[4.4]Nonenones from Nitrile Imines and Arylidenethiohydantoins. Mol. Divers. 2021, 25, 777–785. [Google Scholar] [CrossRef]
- Filkina, M.E.; Baray, D.N.; Beloglazkina, E.K.; Grishin, Y.K.; Roznyatovsky, V.A.; Kukushkin, M.E. Regioselective Cycloaddition of Nitrile Imines to 5-Methylidene-3-Phenyl-Hydantoin: Synthesis and DFT Calculations. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Hassaneen, H.M.; Daboun, H.A.; Abdelhadi, H.A.; Abdel-Reheim, N.A. Site Selectivity and Regiochemistry of Nitrilimines. Cycloadditions to 1, 3- Diphenyl-2-Thiono-4-Imidazolidinone and Its 5-Phenylmethylene Derivatives. Phosphorus. Sulfur. Silicon Relat. Elem. 1995, 107, 269–273. [Google Scholar] [CrossRef]
- Ivanenkov, Y.A.; Kukushkin, M.E.; Beloglazkina, A.A.; Shafikov, R.R.; Barashkin, A.A.; Ayginin, A.A.; Serebryakova, M.S.; Majouga, A.G.; Skvortsov, D.A.; Tafeenko, V.A.; et al. Synthesis and Biological Evaluation of Novel Dispiro-Indolinones with Anticancer Activity. Molecules 2023, 28, 1325. [Google Scholar] [CrossRef] [PubMed]
- Farag, A.M.; Dawood, K.M.; Khedr, N.A. Regioselective Synthesis of Novel 4,4′-and 5,5′-Bi-(1,2,4- Triazole) Derivatives. J. Chem. Res. 2007, 2007, 472–474. [Google Scholar] [CrossRef]
- Üngören, Ş.H.; Kani, İ.; Günay, A. A Facile Protocol for the Preparation of 5-Alkylidene and 5-Imino Substituted Hydantoins from N,N′-Disubstituted Parabanic Acids. Tetrahedron Lett. 2012, 53, 4758–4762. [Google Scholar] [CrossRef]
- Biltz, H.; Topp, E. Synthesis of Parabanic and Substituted Parabanic Acids. Berichte der Dtsch. Chem. Gesellschaft 1913, 46, 1387–1404. [Google Scholar] [CrossRef]
- Stoffel, P.J. The Preparation of Parabanic Acids from 1,1,3-Trisubstituted Ureas via a Hofmann Elimination Reaction. J. Org. Chem. 1964, 29, 2794–2796. [Google Scholar] [CrossRef]
- Ulrich, H.; Sayigh, A.A.R. The Reaction of Oxalyl Chloride with Substituted Ureas and Thioureas. J. Org. Chem. 1965, 30, 2781–2783. [Google Scholar] [CrossRef]
- Watanabe, N.; Hamano, M.; Todaka, S.; Asaeda, T.; Ijuin, H.K.; Matsumoto, M. Diphenylparabanic Acid as a Synthon for the Synthesis of α-Diketones and α-Ketocarboxylic Acids. J. Org. Chem. 2012, 77, 632–639. [Google Scholar] [CrossRef]
- Üngören, Ş.H.; Albayrak, S.; Günay, A.; Yurtseven, L.; Yurttaş, N. A New Method for the Preparation of 5-Acylidene and 5-Imino Substituted Rhodanine Derivatives and Their Antioxidant and Antimicrobial Activities. Tetrahedron 2015, 71, 4312–4323. [Google Scholar] [CrossRef]
- Shybanov, D.E.; Filkina, M.E.; Kukushkin, M.E.; Grishin, Y.K.; Roznyatovsky, V.A.; Zyk, N. V.; Beloglazkina, E.K. Diffusion Mixing with a Volatile Tertiary Amine as a Very Efficient Technique for 1,3-Dipolar Cycloaddition Reactions Proceeding via Dehydrohalogenation of Stable Precursors of Reactive Dipoles. New J. Chem. 2022, 46, 18575–18586. [Google Scholar] [CrossRef]
- Yavari, I.; Taheri, Z.; Naeimabadi, M.; Bahemmat, S.; Halvagar, M.R. A Convenient Synthesis of Tetrasubstituted Pyrazoles from Nitrile Imines and 2-(Thioxothiazolidin-5-Ylidene)Acetates. Synlett 2018, 29, 918–921. [Google Scholar] [CrossRef]
- Ghasempour, L.; Asghari, S.; Tajbakhsh, M.; Mohseni, M. Preparation of New Spiropyrazole, Pyrazole and Hydantoin Derivatives and Investigation of Their Antioxidant and Antibacterial Activities. Chem. Biodivers. 2021, 18. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, Q.; Zhao, Q.; Yu, W.; Chang, J. Synthesis of 2-Imino-1,3,4-Thiadiazoles from Hydrazides and Isothiocyanates via Sequential Oxidation and P(NMe2)3-Mediated Annulation Reactions. Org. Lett. 2020, 22, 4378–4382. [Google Scholar] [CrossRef]
- Dalloul, H.M. Synthesis of Spiro-Heterocycles with the Thiazolidinone Moiety from Nitrilimines. J. Chinese Chem. Soc. 2009, 56, 196–201. [Google Scholar] [CrossRef]
- Liu, B.; Li, X.F.; Liu, H.C.; Yu, X.Y. Unexpected Nitrilimine Cycloaddition of Thiazolo[3,2-a]Pyrimidine Derivatives. Tetrahedron Lett. 2013, 54, 6952–6954. [Google Scholar] [CrossRef]
- Houk, K.N.; Sims, J.; Watts, C.R.; Luskus, L.J. Origin of Reactivity, Regioselectivity, and Periselectivity in 1,3-Dipolar Cycloadditions. J. Am. Chem. Soc. 1973, 95, 7301–7315. [Google Scholar] [CrossRef]
- Bojilova, A.; Rodios, N.A.; Tsoleridis, C.A.; Alexandrou, N.E. The Reaction of 1-( N -phenacylidene)Amino-1,2,3-triazoles with Diphenylnitrilimine. J. Heterocycl. Chem. 1990, 27, 735–738. [Google Scholar] [CrossRef]
- Shybanov, D.E.; Kukushkin, M.E.; Hrytseniuk, Y.S.; Grishin, Y.K.; Roznyatovsky, V.A.; Tafeenko, V.A.; Skvortsov, D.A.; Zyk, N. V.; Beloglazkina, E.K. [4+2]-Cycloaddition to 5-Methylidene-Hydantoins and 5-Methylidene-2-Thiohydantoins in the Synthesis of Spiro-2-Chalcogenimidazolones. Int. J. Mol. Sci. 2023, 24, 5037. [Google Scholar] [CrossRef]
- Tietze, L.F.; Eicher, T. Reaktionen Und Synthesen Im Organisch-chemischen Praktikum Und Forschungslaboratorium; 1991; ISBN 3527308741.
- Bank, U.; Reinhold, D.; Ansorge, S. [Measurement of cellular activity with the MTT test. Optimization of the method]. Allerg. Immunol. (Leipz.) 1991, 37, 119–123. [Google Scholar] [PubMed]
Scheme 1.
Synthetic approaches to the regioselective 1,3-dipolar cycloaddition reaction of nitrile imines to thioparabanic acids and 5-imino substituted hydantoins.
Scheme 1.
Synthetic approaches to the regioselective 1,3-dipolar cycloaddition reaction of nitrile imines to thioparabanic acids and 5-imino substituted hydantoins.
Scheme 2.
Reaction of disubstituted thiourea and oxalyl chloride.
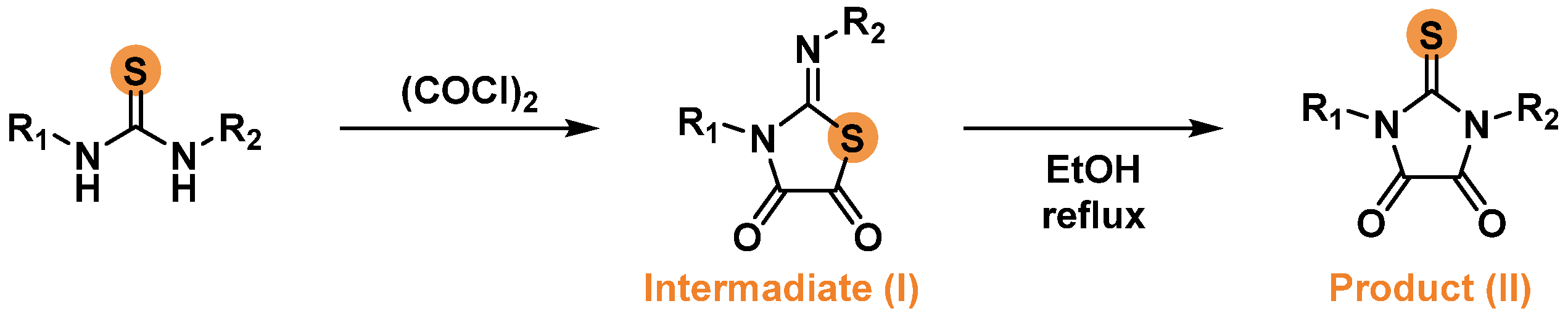
Figure 1.
Fragments of 1H NMR spectra of 1,3-diphenyl-2-thioxoimidazolidine-4,5-dione 1c and 3-phenyl-2-(phenylimino)thiazolidine-4,5-dione 6.
Figure 1.
Fragments of 1H NMR spectra of 1,3-diphenyl-2-thioxoimidazolidine-4,5-dione 1c and 3-phenyl-2-(phenylimino)thiazolidine-4,5-dione 6.
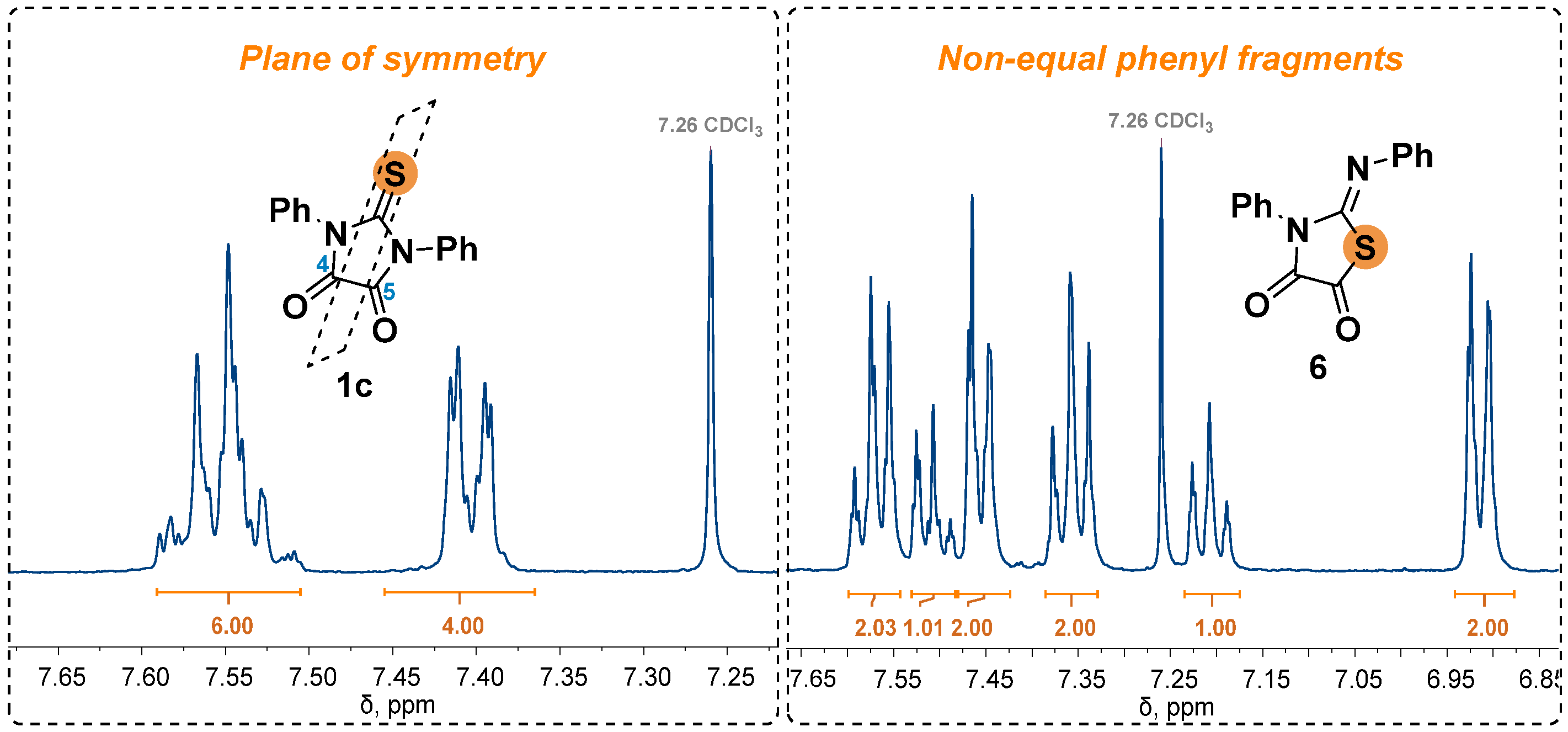
Scheme 4.
The proposed mechanism of 1,3-diphenyl-2-thioxoimidazolidine-4,5-dione 1с and nitrile imine interaction.
Scheme 4.
The proposed mechanism of 1,3-diphenyl-2-thioxoimidazolidine-4,5-dione 1с and nitrile imine interaction.
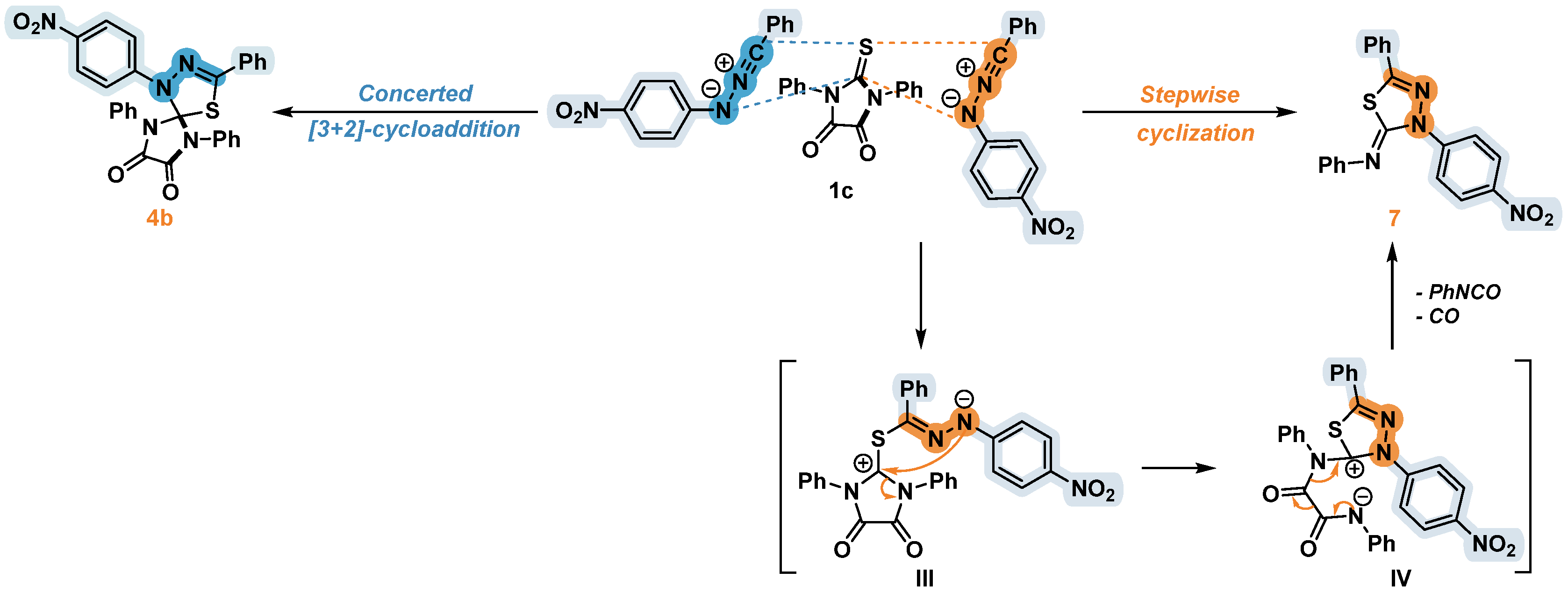
Scheme 6.
Reactions of nitrilimines with 2-iminothiazolidine-4-one (a) and its 5-arylidene derivative (b) [25,26].
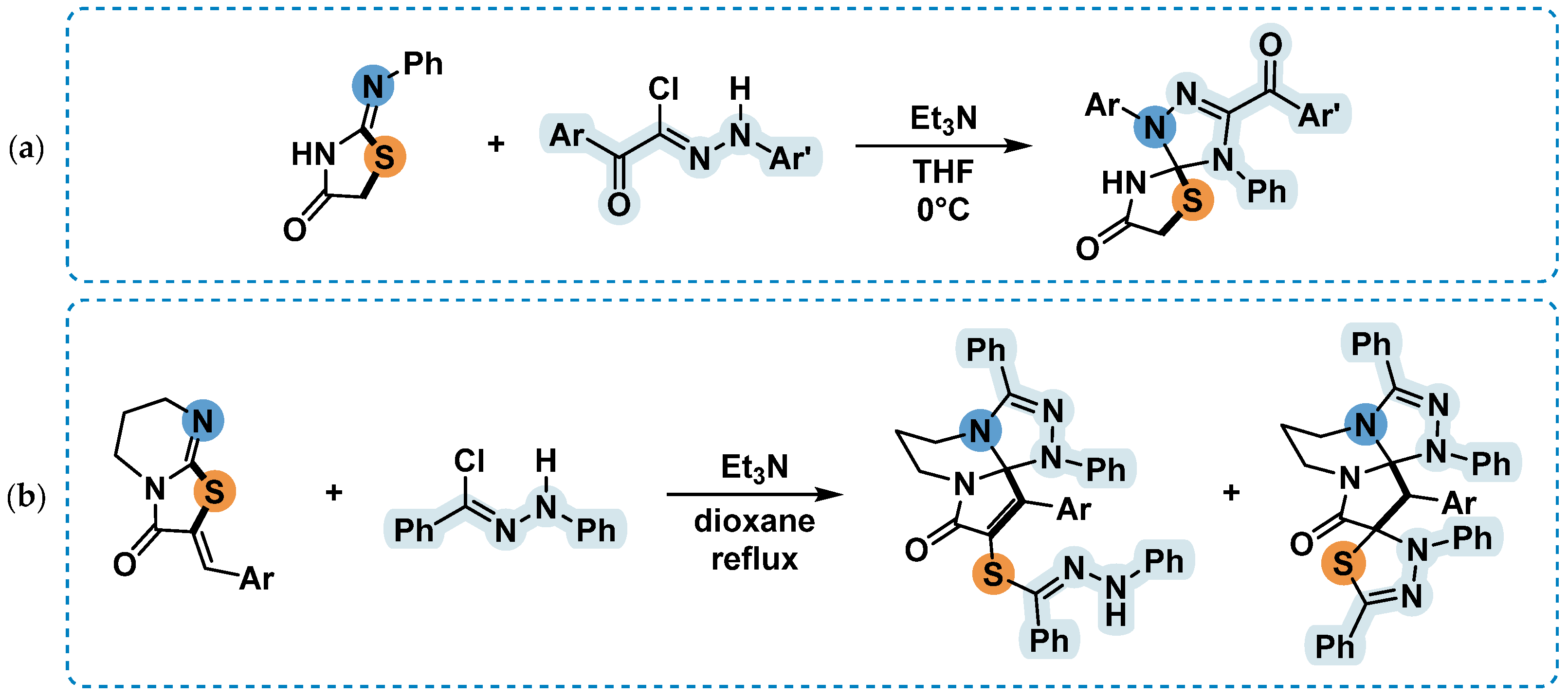
Scheme 7.
The proposed mechanism of 3-phenyl-2-(phenylimino)thiazolidine-4,5-dione 6 and nitrile imine interaction.
Scheme 7.
The proposed mechanism of 3-phenyl-2-(phenylimino)thiazolidine-4,5-dione 6 and nitrile imine interaction.
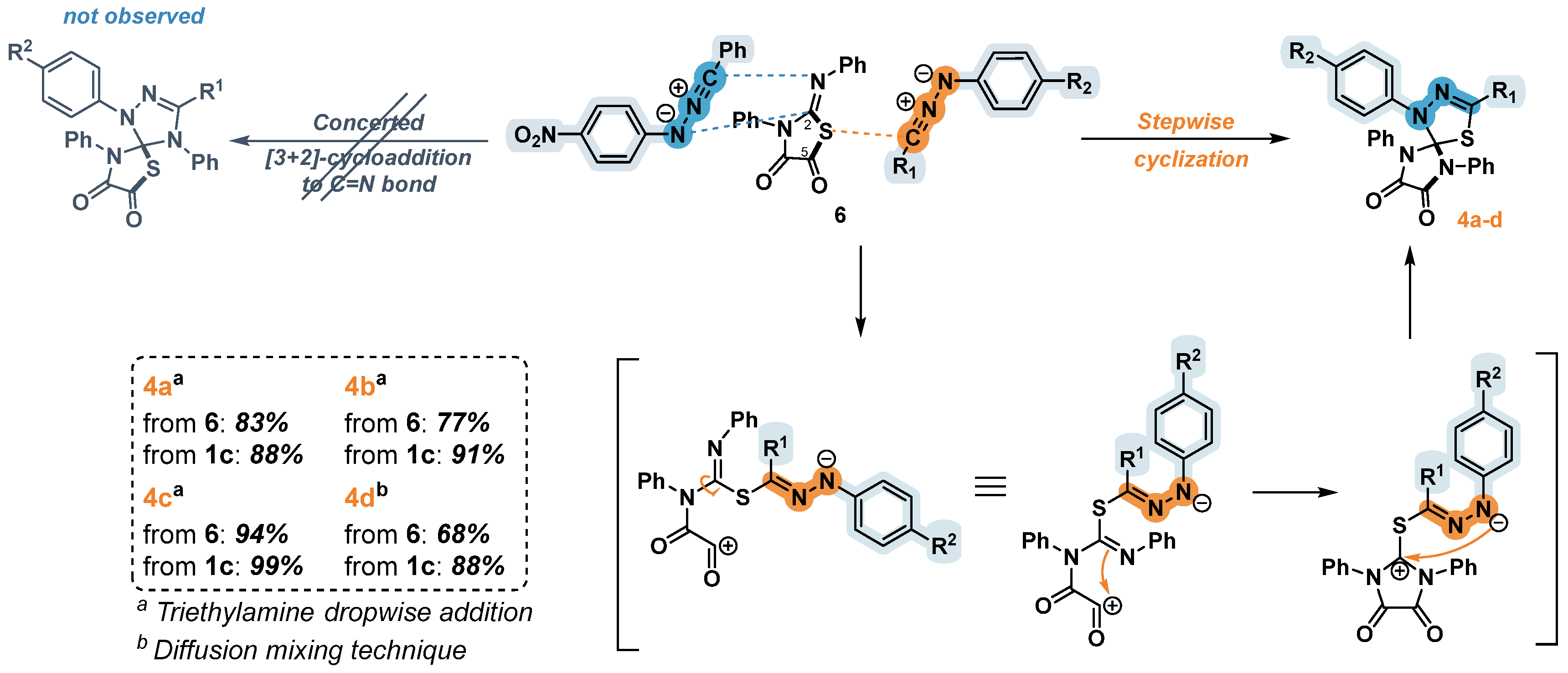
Figure 2.
Molecular structures of compounds (a) 5с; (b) 5g; (c) 5i.
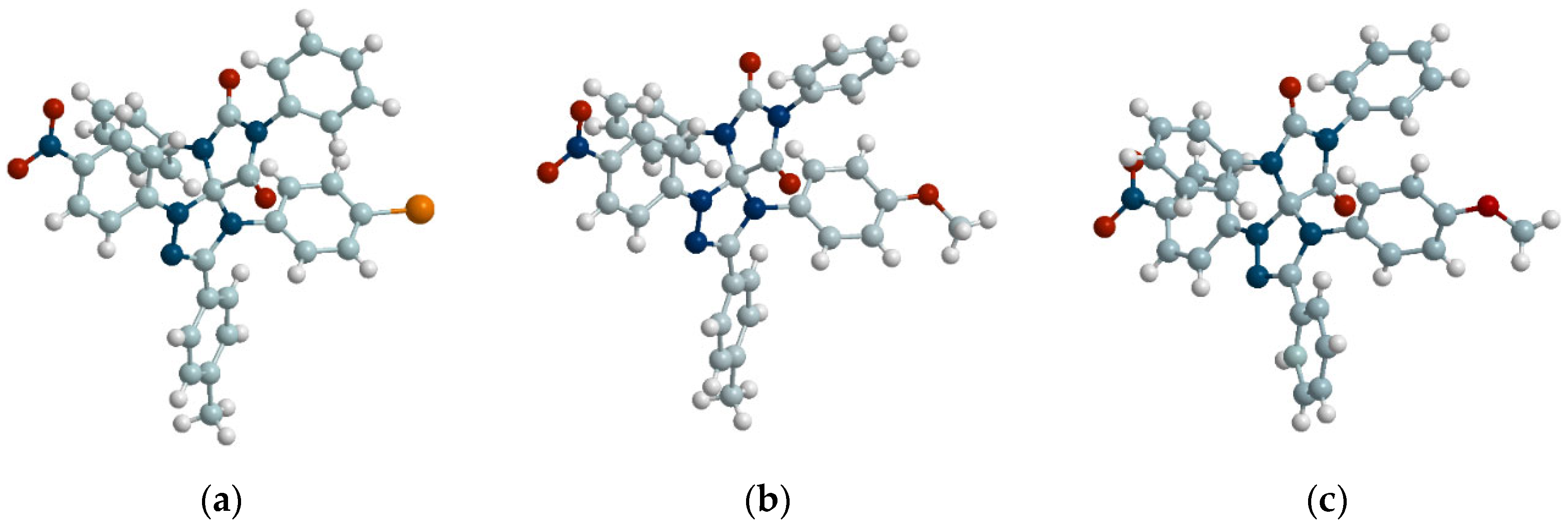
Figure 3.
Hindered rotation in compounds 5d and 5e caused by presence of ortho-substituents in aryl fragments.
Figure 3.
Hindered rotation in compounds 5d and 5e caused by presence of ortho-substituents in aryl fragments.

Table 1.
Synthesis of imidazolidine-4,5-diones 1a-f.
 | ||||
| Compound№ | X | R1 | R2 | Yield a, % |
| 1a b | O | Ph | Ph | 99 |
| 1b b | O | Ph | CH2COOEt | 99 |
| 1c c | S | Ph | Ph | 98 |
| 1d c | S | 4-EtO-C6H4 | CH2COOEt | 93 |
| 1e c | S | 4-MeO-C6H4 | CH2COOEt | 77 |
| 1f c | S | All | H | 47 |
a Isolated yield; b DCM, reflux 2h; c CH3CN, reflux 6h, purified by flash chromatography.
Table 2.
Synthesis of imines 2a-j.
 | |||
| Compound№ | X | R3 | Yield a, % |
| 2a | O | 4-Cl | 79 |
| 2b | O | 2-Cl | 60 |
| 2c | O | 4-Br | 60 |
| 2d | O | 2-Br | 76 |
| 2e | O | 4-Me | 84 |
| 2f | O | 4-OMe | 65 |
| 2g | O | 4-NO2 | 75 |
| 2h | S | 4-Br | 82 |
| 2i | S | 4-OMe | 88 |
| 2j | S | 4-NO2 | 72 |
a Isolated yield.
Table 3.
Reaction of 2-thioxoimidazolidine-4,5-diones 1c-f and hydrazonoyl chlorides 3a-d.
 | ||||||
| Compound№ | R1 | R2 | R3 | R4 | Diffusion mixing | Dropwise addition |
| Yield a, % | Yield a, % | |||||
| 4a | Ph | Ph | 3-NO2-4-Cl-C6H3 | H | 96 | 88 |
| 4b | Ph | Ph | Ph | 4-NO2 | 44 | 91 |
| 4c | Ph | Ph | 4-Br-C6H4 | H | 99 | 99 |
| 4d | Ph | Ph | Me | H | 88 | 74 |
| 4e | 4-EtO-C6H4 | CH2COOEt | 4-Br-C6H4 | H | 77 | 49 |
| 4f | 4-MeO-C6H4 | CH2COOEt | Ph | 4-NO2 | 93 | 94 |
| 4g | All | H | Ph | 4-NO2 | 61 | - b |
a Isolated yield; b Yield was not defined.
Table 4.
The influence of reaction conditions on the formation of a by-product.
| Entry | Solvent | T | Yielda of 4b, % | Yielda of 7,% |
| 1 | CH3OH | rt | 60 | - |
| 2 | CH3CN | rt | 75 | <1b |
| 3 | CHCl3 | -17 ℃ | 49 | 32 |
| 4 | CHCl3 | rt | 44 | 35 |
| 5 | (CH3)2CO | rt | 34 | 45 |
a Isolated yield, diffusion mixing technique; b Traces.
Table 5.
Reaction of 5-arylimino-1,3-diphenylimidazolidine-2,4-diones 2b-g and hydrazonoyl chlorides 3b-g.
Table 5.
Reaction of 5-arylimino-1,3-diphenylimidazolidine-2,4-diones 2b-g and hydrazonoyl chlorides 3b-g.
 | |||||
| Compound № | R1 | R2 | R3 | Diffusion mixing | Dropwise addition |
| Yield a, % | Yield a, % | ||||
| 5a | 4-Br | Ph | H | 71 | 62 |
| 5b | 4-NO2 | 4-Me-C6H4 | NO2 | 40 | 13 |
| 5с | 4-Br | 4-Me-C6H4 | NO2 | 90 | 85 |
| 5d | 2-Br | 4-Me-C6H4 | NO2 | 84 | 83 |
| 5e | 2-Cl | 4-Me-C6H4 | NO2 | 87 | 81 |
| 5f | 4-Me | 4-Me-C6H4 | NO2 | 71 | 88 |
| 5g | 4-OMe | 4-Me-C6H4 | NO2 | 52 | 38 |
| 5h | 4-Br | Ph | NO2 | 62 | 50 |
| 5i | 4-OMe | Ph | NO2 | 60 | 50 |
| 5j | 4-Me | 4-Cl-C6H4 | H | 57 | 93 |
| 5k | 4-NO2 | Me | H | 88 | 59 |
| 5l | 4-Br | Me | H | 73 | 57 |
a Isolated yield.
Table 6.
Cytotoxicity monitoring results (MTT test, cell line HCT116).
| Compound№ | IC50, µM | R1 | R2 | R3 | R4 | Structure type |
| 4a | 24,75±0,01 | Ph | Ph | 3-NO2-4-Cl-C6H3 | H |  |
| 4b | 35,04±0,06 | Ph | Ph | Ph | 4-NO2 | |
| 4e | 48,51±0,03 | 4-EtO-C6H4 | CH2COOEt | 4-Br-C6H4 | H | |
| 4f | 29,61±0,08 | 4-MeO-C6H4 | CH2COOEt | Ph | 4-NO2 | |
| 5c | 40,99±0,01 | 4-Br | 4-Me-C6H4 | NO2 | - | 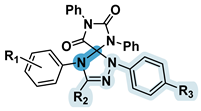 |
| 5f | 43,41±0,01 | 4-Me | 4-Me-C6H4 | NO2 | - | |
| 5j | 35,07±0,01 | 4-Me | 4-Cl-C6H4 | H | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



