Submitted:
25 November 2023
Posted:
27 November 2023
You are already at the latest version
Abstract
Milroy's disease (MD), also known as hereditary lymphedema type I, is a rare autosomal dominantly inherited primary lymphedema. It accounts for approximately twenty percent of all primary lymphedemas. MD typically presents clinically with bilateral lower extremity lymphedema. Our case report is about a rare case of MD presenting with unilateral lymphedema in both upper and lower extremities in a patient admitted to our clinic. A 24-year-old woman who had been under observation for lymphedema since she was 8 years old was diagnosed with MD by physical examination, venous Doppler ultrasound, whole body lymphoscintigraphy, and molecular genetic testing in our clinic, and partial improvement was achieved with Complete Decongestive Therapy, the gold standard treatment for lymphedema. Genetic counseling services may be especially useful for the patient and family. Although MD is rarely seen in the literature, many more studies are needed to identify carrier variants in the community.
Keywords:
Hereditary lymphedema type I
; inherited primary lymphedema
; Milroy's Disease
; Complete Decongestive Therapy
1. Introduction
Milroy's disease(MD), also known as hereditary lymphedema type I, first described by Milroy in 1892 is a primary lymphedema inherited through autosomal dominant transmission [1]. It can manifest either at birth or later in life. In the United States, primary lymphedemas occur in 1:10,000 individuals, accounting for approximately twenty percent of all primary lymphedemas, with around 200 reported cases in the literatüre [2].
The most common cause of MD is a mutation in the Fms-related tyrosine kinase 4 (FLT4) gene [3]. The FLT4 gene plays a crucial role in the production of a protein called vascular endothelial growth factor receptor 3 (VEGFR3), which is essential for the development of the lymphatic system. Lymphatic vessels with mutations in VEGFR3 are also referred to as congenital aplasia of lymphatic vessels [4]. Issues in the transport of lymphatic fluid result in the accumulation of lymphedema within bodily tissues.
MD typically presents clinically with lymphedema in the bilateral lower extremities. Besides lymphedema, it can lead to severe complications such as intestinal lymphangiectasia, cellulitis, bacterial infections on the dorsum of the foot, and pleural effusion [5]. The degree of symptoms can vary within families, and some carriers of the condition may remain asymptomatic [6].
Prior studies have shown that due to FTL4 mutations, there is a reduction in lymphatic vessel density by approximately 51-61% in the lower extremities and around 26-33.6% in the upper extremities [7]. Therefore, albeit rare, lymphedema can be observed in both the upper and lower extremities [8]. A multidisciplinary approach involving geneticists, physiatrists, neonatologists, dermatologists, and surgical teams is crucial for the treatment and follow-up of these cases.
Our case will be related to the rare occurrence of MD presenting with unilateral lymphedema in both the upper and lower extremities in a patient who presented to our clinic.
2. Case Report
A 24-year-old female patient, married and a homemaker, with a history of two miscarriages, had never experienced a full-term pregnancy. She had been under observation for lymphedema since the age of 8. Her aunt also had a history of lymphedema and recurrent miscarriages. Over the last four years, the patient has been using oral contraceptives (OCs). She reported that her swelling and edema had increased further after starting OCs and sought our advice and treatment options.
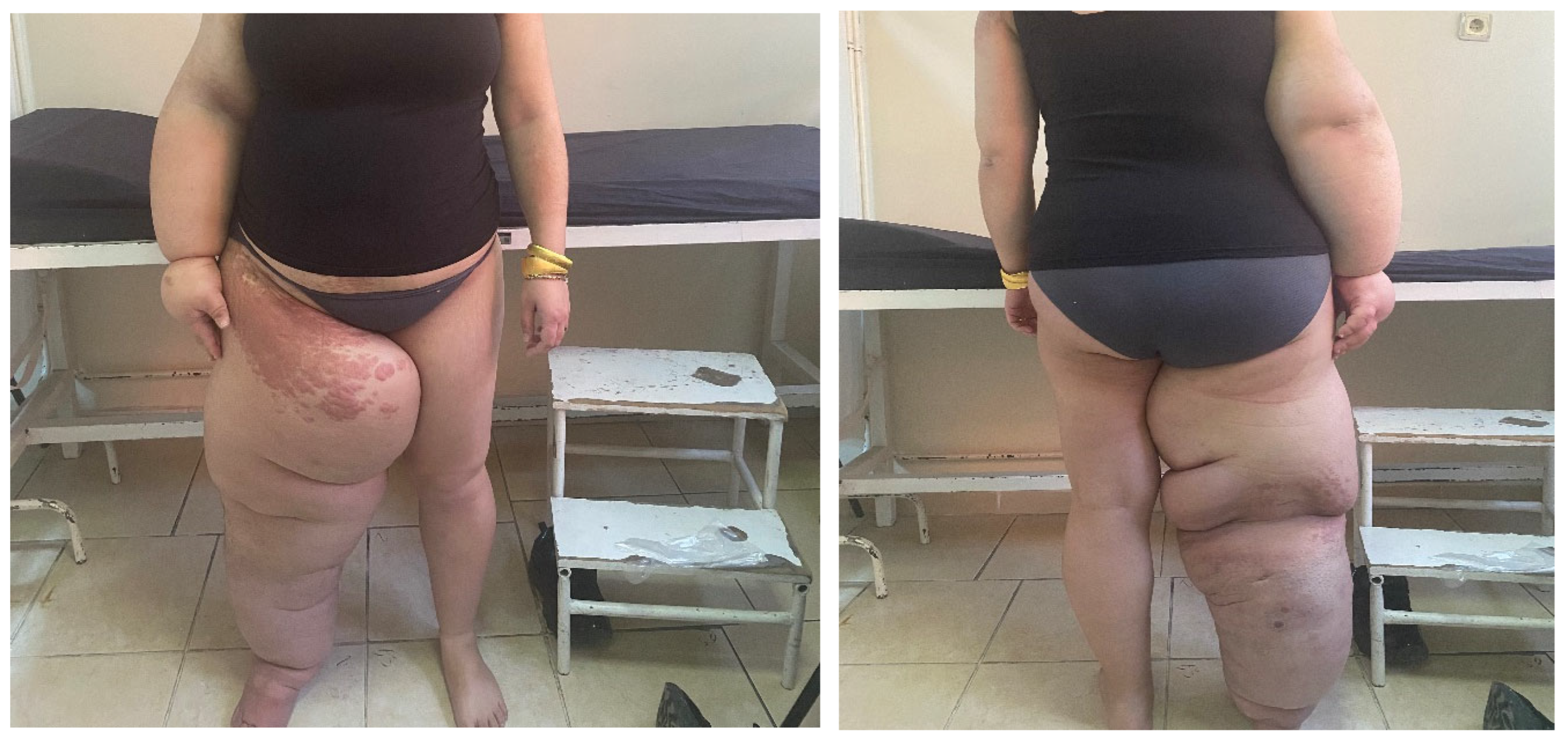
During the patient's examination, there were signs of swelling, increased circumference, and tense fibrous bands in the right lower extremity, right upper extremity, and genital area. Additionally, hyperkeratotic, papillomatous lesions were observed in the right inguinal region and the anterior surface of the right elbow (see Figure 1). The patient did not report any pain upon palpation. Lymphedema was assessed using extremity volume measurement, with a volume difference of 1954 ml and a percentage difference of 81% for the lower extremities. For the upper extremities, the volume difference was 1116 ml with a percentage difference of 57.6%.
Venous Doppler ultrasound of the lower extremities revealed reflux flow at the saphenofemoral junction, while the upper extremity venous Doppler results were normal.
A whole-body lymphoscintigraphy was performed, which failed to visualize the right main lymphatic channel, right pelvic, and right axillary lymph nodes. These findings were consistent with grade IV lymphatic drainage disorder in the lymphoscintigraphy.
Molecular genetic testing identified a mutation in the FLT4 gene located on chromosome 5q35. Based on these findings, a diagnosis of MD was established.
The patient underwent a 3-week treatment program which included skincare, manual lymphatic drainage, dual-layer bandaging, diaphragmatic exercises, lymphedema exercises, walking exercises (Complete Decongestive Therapy), and a ketogenic diet for weight loss. Additionally, the patient received recommendations from dermatology for papillomatous lesions and from cardiovascular surgery regarding venous insufficiency. After completing the treatment, compression garments for the right upper and lower extremities were prescribed following measurements and fittings. The volume difference in the lower extremities decreased to 1092 ml, with a percentage difference of 68%, after complete decongestive therapy. In the upper extremities, the volume difference decreased to 940 ml, with a percentage difference of 48.5%, after ketogenic diet therapy.
3. Conclusion
In the treatment of lymphedema that develops in MD, the main goal is to reduce swelling and circumference discrepancies and to prevent infectious factors like lymphangitis. Genetic analysis, venous Doppler ultrasound, and lymphoscintigraphy are important diagnostic tools in this context [9]. The gold standard treatment for lymphedema is Complete Decongestive Therapy[10].This therapy encompasses compression treatments, lymphedema exercises, and proper skincare. Genetic counseling services can be particularly beneficial for the patient and their family. Although MD is rarely seen in the literature, there is a need for a much larger number of studies to detect carrier variants within the community.
References
References
- Milroy, W.F. An undescribed variety of hereditary edema. New York Med. J. 1892, 12. [Google Scholar]
- Bissonnette, B.; Luginbuehl, I.; Engelhardt, T. Syndromes: Rapid Recognition and Perioperative Implications, 2nd ed.; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar]
- Evans, A.L.; Brice, G.; Sotirova, V.; Mortimer, P.; Beninson, J.; Burnand, K.; et al. Mapping of primary congenital lymphedema to the 5q35.3 region. Am. J. Hum. Genet. 1999, 64, 547–55. [Google Scholar] [CrossRef] [PubMed]
- Ghalamkarpour, A.; Debauche, C.; Haan, E.; Van Regemorter, N.; Sznajer, Y.; Thomas, D.; et al. Sporadic in utero generalized edema caused by mutations in the lymphangiogenic genes VEGFR3 and FOXC2. J. Pediatr. 2009, 155, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Alitalo, K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell 2010, 140, 460–476. [Google Scholar] [CrossRef] [PubMed]
- Ferrell, R.E.; Levinson, K.L.; Esman, J.H.; Kimak, M.A.; Lawrence, E.C.; Barmada, M.M.; Finegold, D.N. Hereditary lymphedema: evidence for linkage and genetic heterogeneity. Hum. Mol. Genet. 1998, 7, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- Mellor, R.H.; Hubert, C.E.; Stanton, A.W.; Tate, N.; Akhras, V.; Smith, A.; et al. Lymphatic dysfunction, not aplasia, underlies Milroy disease. Microcirculation 2010, 17, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Connell, F.; Brice, G.; Mortimer, P. Phenotypic characterization of primary lymphedema. Ann. N. Y. Acad Sci. 2008, 1131, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Sarica, M.; Gordon, K.; van Zanten, M.; Heenan, S.D.; Mortimer, P.S.; Irwin, A.G.; et al. Lymphoscintigraphic Abnormalities Associated with Milroy Disease and Lymphedema-Distichiasis Syndrome. Lymphat. Res. Biol. 2019, 17, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Forner-Cordero, I.; Muñoz-Langa, J.; Forner-Cordero, A.; DeMiguel-Jimeno, J.M. Predictive factors of response to decongestive therapy in patients with breast-cancer-related lymphedema. Ann. Surg Oncol. 2010, 17, 744–751. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Hereditary Lymphedema Type 1: Milroy Disease.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



