Submitted:
21 November 2023
Posted:
22 November 2023
You are already at the latest version
Abstract
An efficient stereoselective three-component reaction for the synthesis of spiro[4H-chromene-3,3’-oxindole] derivatives was realized through an organocatalyzed cascade Knoevenagel/Michael/cyclization reaction using a quinidine-derived squaramide as the catalyst. Under the optimized conditions, the reactions of isatins, malononitrile, and sesamol yield the desired spirooxindoles in good yields (75–87%) and moderate to high ee values (up to 90% ee). Two pairs of selected enantiomers were subjected to evaluate their antiproliferative activities on three types of human cancer cell lines using the MTT assay. The results indicated that stereoselectivity and electrical effect showed significant impact on activity. Enanotiomer R-3f exhibited optimal cytotoxic activity against U2OS cell line, which was close to the inhibitory activity of positive control, Adriamycin.
Keywords:
enantioselective
; cascade Knoevenagel/Michael/cyclization
; quinidine-squaramide
; spirooxindoles
1. Introduction
Chromenes are important core scaffolds in various natural products and pharmaceutical molecules that exhibit a wide spectrum of biological activities, such as anti-inflammatory, antitumour, antimicrobial, antitoxin and antimalarial properties [1,2,3,4,5,6,7,8,9]. Specifically, chromenes structure with chiral centres in numerous natural products have been proven to have diverse pharmacological effects (Figure 1) [10,11,12].
In addition, optically active spirooxindole derivatives containing heterocyclic frameworks have attracted tremendous attention because of their prominent activities and wide utility as synthetic intermediates for alkaloids and clinical drugs [13,14,15,16]. Over past years, the significant progress has been made in the stereoselective construction of spirooxindole [17,18,19,20,21,22,23,24,25,26]. Considering the potential biological activity of both chromenes and spirooxindole structures, we think splicing two fragments into hybrid derivatives may result in a series of structurally and biologically significant new molecules. In recent years, various attempts have also been made for the synthesis of spirooxindoles with fused 4H-chromenes using different catalysts [27,28,29,30,31,32]. In 2012, Wang group developed a rosin thiourea catalysed reaction of coumarins with isatylidene malononitriles [28]. Afterwards, Zhao reported quinine-thiourea catalysed three-component cascade reactions of isatins, malononitrile, and 2-hydroxynaphthalene-1,4- diones [29]. In 2016, Khan et. al. disclosed the first enantioselective addition of naphthols and sesamol to indolylidene cyanoacetate derived from isatins organocatalysed by quinine -thiourea[30]. Additionally, Zhou [31] and Abdolmohammadi [32] respectively reported Knoevenagel/Michael/Cyclization of isatins, malononitrile, and sesamol, respectively. However, the methods reported in two studies did not involve asymmetric synthesis and resulted in racemic spirooxindoles. Despite the great developments, to the best of our knowledge, to date, the enantioselective one-pot, cascade reaction of isatins, malononitrile, and sesamol has not been explored.
We herein first reported the enantioselective domino Knoevenagel/Michael/ cyclization of isatins, malononitrile, and sesamol by employing the cinchona alkaloids, Takemoto’s catalyst, proline, diphenyl aminoethanol and phosphoric acid derivatives 1a-1m (Figure 2).
2. Results and Discussion
We first applied the catalysts 1a–1m in the cascade reaction of N-benzylisatin(2a), malononitrile and sesamol to screen the optimal catalyst. The reaction was carried out with CH2Cl2 as a solvent in the presence of 10 mol% of catalysts at room temperature for 24 h (Table 1). Catalysts 1a-1l preceded the reaction smoothly to give the desired product 3a in 68-85% yields with 5-40% ees while phosphoric acid 1m failed to catalyse the reaction (entry 13). Among of them, quinidine-squaramide 1f was optimal in terms of the yield and enantioselectivity (entry 6) while Takemoto’s catalysts and proline derivative showed poor asymmetric induction.
To improve the enantioselectivity of the transformation, we investigated a variety of different reaction conditions (Table 2). The survey of solvents showed that CHCl3 was optimal in terms of the yield and enantioselectivity (entry 2). The screening of catalyst loading showed that a 10 mol% equivalent of 1f was optimal. Reduction of the catalyst loading to 5 mol% led to an obviously decrease in enantioselectivity and yield (entry 8 vs. entry 2), and 20 mol% loading offered no improvement in the asymmetric induction, albeit with a slightly improved yield (entry 9 vs. entry 2). When the reaction temperature was lowered from rt to 0 °C, the enantioselectivity of the product was improved to 58%, and the yield could be remained at the same level by prolonging the reaction time (entry 10). Next, a further temperature drop to −20 °C caused a significant decrease in both enantioselectivity (40% ee) and yield (68%). Furthermore, diluting the reaction concentration by half was detrimental to the yield and enantiocontrol (entry 12 vs. entry 10). Adding 4 Å molecular sieves (MS) led to a slightly higher ee value of 61% and increased yield (entry 13 vs. entry 10). Subsequently, the reaction was carried out in dry CHCl3 under anaerobic conditions, and the ee value of the product was not improved (entry 14 vs. entry 13). Based on these experiments, the optimized conditions were determined to be CHCl3 as the solvent with a 10 mol% loading of catalyst 1f in the presence of 4 Å MS (200 mg) at 0 °C.
With the optimized conditions in hand, we explored the scope and general applicability of the protocol. A wide range of substituted isatins were evaluated, as shown in Table 3. A wide range of isatins bearing various substituents on the phenyl ring, such as halogens, methyl groups and methoxyl groups, were tolerated, giving the desired products in good yields (75-87%) with 45-90% ees, except 5-nitro substituted isatin, which produced 3i with only 10% ee (entry 9). Therefore, the enantioselectivity was obviously affected by the substituted position on the phenyl ring of isatins. The reaction of 5-Br-substituted N-Bn-isatin afforded the optimal enantiomeric excess (90%ee, entry 6). Then, a 0.5 mmol scale asymmetric domino reaction of 2f was conducted under the optimum conditions, and product 3f was obtained in 82% yield with 85% ee (data in parentheses, entry 6). In the case of isatin and N-methylisatin as substrates, decreased ee values were obtained (entries 13, 14 vs. entry 1).
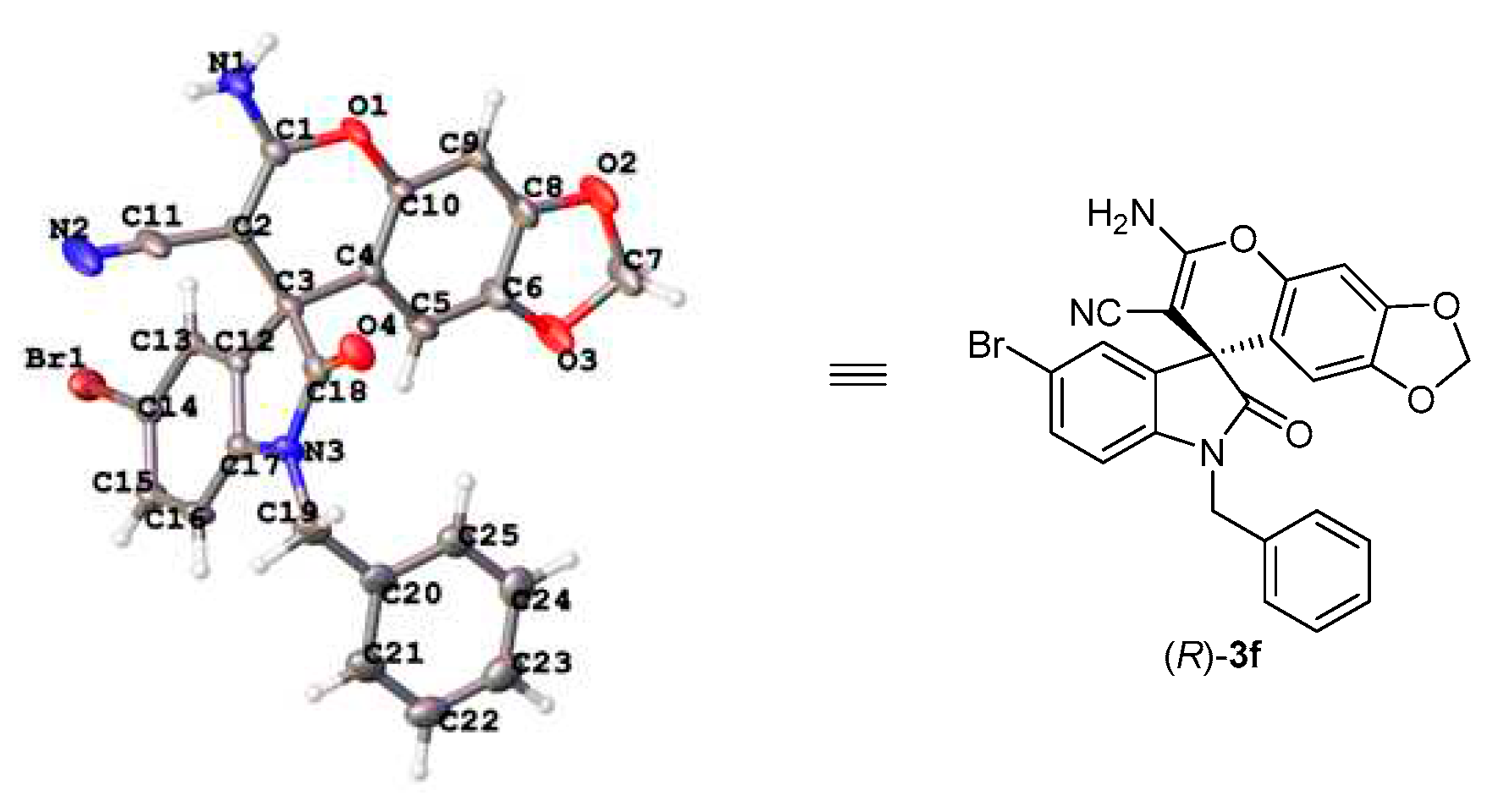
The absolute configuration of spiro[4H-chromene-3,3’-oxindole] products 3 were unambiguously assigned as R according to the X-ray crystal structure analysis of 3f (Figure 3) [33] .
On the basis of the absolute stereochemistry of 3f, a plausible transition state model is proposed. As shown in Scheme 1, isatin (2a) first reacts with malononitrile to yield the isatylidenemalononitrile intermediate, which is fixed and activated by the neighbouring two squaramide hydrogen atoms through double H-bonding, while sesamol is activated by an interaction between the tertiaryamine moiety of 1f and the hydroxy group of sesamol. Then, re-face addition of the electron-rich sesamol to the electron-deficient isatylidenemalononitrile generates the Michael adduct intermediate, which is further transformed to the final R-3f through subsequent intramolecular cyclization and tautomerization.
To broaden the scope of nucleophiles, electron-rich phenols (3,4-dimethoxy, 3,5-dimethoxy substituents), 1-naphthol and 4-hydroxyindole were reacted with N-benzylisatin and malononitrile under the screened catalyst conditions. Corresponding Spiro[4H-Chromene -3,3′-oxindoles] derivatives were smoothly obtained in good yields (Fig. 4). Among of them, 3,4-dimethoxy and 3,5-dimethoxy phenols have similar structures to sesamol. However, a moderate ee value (54%ee) was obtained in the reaction of 3,4-dimethoxy phenol as the nucleophile, while a low ee value (27%ee) was produced with 3,5-dimethoxy phenol as the reactant. Therefore, the substituted position of the OMe group had an important effect on the enantioselectivity. Furthermore, the reaction of 4-hydroxyindole obtained 56% enantioselectivity, while 1-naphthol as a nucleophile was unfavourable for the stereoselectivity of the reaction (17%ee).
Considering the anticancer activity of various isatin derivatives [34,35,36,37,38,39], we plan to study the antiproliferative activities of the obtained products. Moreover, to investigate the impact of the chiral centre and electrical effect on activity, we chose 5-Br-1-Bn (electron-withdrawing) and 5-Me-1-Bn (electron-donating) isatin-derived products 3f and 3g with relatively high ee values for the activity test. Two pairs of enantiomers were catalysed by optical isomers 1e and 1f (as shown in Figure 2), and the racemic products were catalysed by DABCO. The structures of enantiomers and their ee vaules are shown in Figure 5.
The antiproliferative activities of selected compounds were evaluated on three types of human cancer cell lines, human prostate cancer cells (C42), human cervical cancer cells (HeLa) and human osteosarcoma cells (U2OS) using the MTT [3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyl tetrazolium bromide] assay. It is evident from the results that most of the test compounds have shown cytotoxic activity on all tested cell lines in a concentration- dependent manner (Table 4).
It is apparent from the results that these derivatives are found to be more potent on C42 and U2OS cell lines, followed by HeLa cells. Furthermore, the chiral factor showed an obvious impact on activity. R-configuration compounds were found to be more effective than S-configuration or racemic products (entry 1 vs entries 2 and 3 and entry 4 vs entries 5 and 6). In addition, the electrical effect exhibited a certain impact on the activity, and the 5-Br-substituted derivative was found to be more potent than the 5-CH3-substituted product (entries 1, 2, and 3 vs entries 4, 5, and 6). From the screened compounds, compound R-3f exhibited excellent cytotoxic activity against the U2OS cell line (IC50: 6.73 μg/mL), which was close to the inhibitory activity of the positive control (Adriamycin, IC50: 4.72 μg/mL).
3. Conclusion
In summary, we have described the first enantioselective domino Michael/cyclization reaction of isatins, malononitrile, and sesamol organocatalysed by quinidine-derived squaramide to synthesize spiro [4H-chromene -3,3’-oxindole] derivatives in good yield with up to 90% enantioselectivity. The absolute configuration of compound 3f was ascertained as an R-isomer on the basis of X-ray crystallographic analysis. At present, the obtained ee values are not good, and further optimization of the catalyst and reaction conditions is underway. Two pairs of synthesized enantiomers were subjected to evaluation of their cytotoxic properties against different human cancer cell lines. The results indicated that stereoselectivity and electrical effects had obvious impacts on biological activity. Among of them, R-3f exhibited optimal cytotoxic activity against the U2OS cell line, which was close to the inhibitory activity of the positive control, Adriamycin. Therefore, spiro [4H-chromene-3,3’-oxindole] derivatives could be developed as antitumour candidate compounds after further research.
4. Materials and Methods
4.1. Chemistry
The 1H NMR spectra were recorded on a 500 MHz for 1 H and at 125 MHz for 13 C NMR, using DMSO–d6 as a solvent. The chemical shifts were reported in ppm, and the residual nondeuterated solvent (DMSO) as internal standard (2.5 and 39.52 ppm, respectively). The splitting patterns of the signals were reported as s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; and m, multiplet. High-resolution mass spectra (HRMS) were measured on a triple TOF 5600+ mass spectrometer equipped with an electrospray ionization (ESI) source in the negative-ion mode. The enantiomeric excess (ee) values of the products were determined by chiral HPLC, using Daicel Chiralpak AD columns (4.6 mm*250 mm). The reactions were monitored by thin layer chromatography (TLC). Purifications by column chromatography were conducted over silica gel (200–300 mesh). The organocatalysts 1a–1m were purchased from Daicel chiral technologies (China) company, which almostly have purity of 98% and stereoselectivity of 99%.
4.2. General Procedure for the Enantioselective Knoevenagel/Michael/cyclization reaction of isatins, malononitrile and sesamol
To a tube, mixture of isatins (0.1 mmol), malononitrile (0.1 mmol), sesamol (0.1 mmol) and organocatalyst 1f (0.01 mmol) in CHCl3 (1.0 mL) was added 4 Å molecular sieves (30 mg). The resulting mixture was stirred at 0 oC for 24 hours. After the reaction was finished (monitored by TLC), the reaction directly poured into a column chromatography on silica gel with hexane/EtOAc (4:1) as eluent to afford the products 3a–p, and 4-7. Among of 20 products, there are 12 new compounds. The enantiomeric ratio was determined by HPLC analysis on a chiral ChiralPak AD column. Experimental data can be found in Supplementary Materials.
3a:whith solid, mp: 80.5-81.5 oC; 1H NMR (500 MHz, DMSO-d6) δ 7.38 – 7.26 (m, 6H), 7.25 (d, J = 4.5 Hz, 2H), 7.14 (dd, J = 7.5, 1.0 Hz, 1H), 7.07 (td, J = 7.5, 1.0 Hz, 1H), 6.99 (d, J = 8.0 Hz, 1H), 6.79 (s, 1H), 5.99 (dd, J = 13.5, 1.0 Hz, 2H), 5.82 (s, 1H), 5.01 (d, J = 16.0 Hz, 1H), 4.88 (d, J = 16.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 177.4, 161.5, 147.9, 144.5, 143.6, 142.3, 136.1, 133.6, 129.2, 128.6, 127.5, 127.2, 124.7, 123.5, 118.6, 112.1, 109.7, 104.4, 102.1, 98.2, 53.48, 50.3, 43.2. [α]D25= +1 (c 0.50, MeOH)(61% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 10.7 min (major), 21.5 min (minor) .
3b: whith solid, mp: 235.5-236.6 oC; 1H NMR (500 MHz, DMSO-d6) δ 7.43 – 7.35 (m, 2H), 7.35 – 7.25 (m, 6H), 7.05 (dd, J = 8.0, 0.5 Hz, 1H), 7.02 – 6.95 (m, 1H), 6.79 (s, 1H), 6.01 (dd, J = 3.5, 1.0 Hz, 2H), 6.00 (s, 1H), 5.08 (d, J = 16.0 Hz, 1H), 4.87 (d, J = 16.0 Hz, 1H);13C NMR (126 MHz, DMSO-d6) δ 176.5, 161.6, 147.9, 144.5, 144.0, 143.9, 135.6, 130.8, 130.2, 129.1, 128.5, 127.5, 127.1, 123.6, 118.2, 109.6, 108.7, 103.9, 102.0, 98.0, 51.5, 50.9, 43.5; HRMS (ESI) m/z: [M+Na]+ calcd for C25H16ClN3O4Na 480.0727; found 480.0722; [α]D25= -4 (c 0.61, MeOH)(65% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 15.3 min (major), 26.1 min (minor) .
3c: whith solid, mp: 263.7-264.6 oC; 1H NMR (500 MHz, DMSO-d6) δ 7.38 (d, J = 7.0 Hz, 2H), 7.33 (t, J = 7.0 Hz, 2H), 7.30 – 7.25 (m, 3H), 7.25 – 7.18 (m, 2H), 7.02 (dd, J = 7.5, 1.0 Hz, 1H), 6.78 (s, 1H), 6.01 (s, 2H), 5.97 (s, 1H), 5.07 (d, J = 16.0 Hz, 1H), 4.87 (d, J = 16.0 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ 176.7, 161.7, 148.0, 144.5, 144.2, 144.2, 135.7, 131.1, 130.6, 128.6, 127.6, 127.2, 126.8, 119.3, 118.4, 109.5, 109.3, 104.0, 102.1, 98.0, 52.0, 51.4, 43.4; HRMS (ESI) m/z: [M+Na]+ calcd for C25H16BrN3O4Na: 524.0222; found: 524.0229; [α]D25= -3 (c 0.55, MeOH)(50% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 15.8 min (minor), 25.9 min (major) .
3d whith solid, mp: 192.7-193.3 oC ; 1H NMR (500 MHz, DMSO-d6) δ 7.46 – 7.22 (m, 7H), 7.13 (ddd, J = 10.5, 9.0, 2.5 Hz, 2H), 6.99 (dd, J = 8.5, 4.0 Hz, 1H), 6.80 (s, 1H), 6.00 (dd, J = 10.5, 1.0 Hz, 2H), 5.88 (s, 1H), 5.02 (d, J = 16.0 Hz, 1H), 4.87 (d, J = 16.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 177.4, 161.5, 160.1, 158.2, 148.0, 144.6, 143.6, 138.6, 135.9, 135.4, 135.3, 128.7, 127.6, 127.2, 118.6, 115.8, 115.6, 112.7, 112.5, 111.4, 110.9, 110.8, 104.4, 102.1, 98.3, 53.1, 50.7, 43.3; [α]D25= +1 (c 0.58, MeOH)(60% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 13.7 min (major), 23.8 min (minor) .
3e: whith solid, mp: 188.7-190.0 oC ; 1H NMR (500 MHz, DMSO-d6) δ 7.37-7.26 (m, 8H), 7.25 (d, J = 2.0 Hz, 1H), 7.01 (d, J = 8.5 Hz, 1H), 6.80 (s, 1H), 6.01 (dd, J = 13.5, 1.0 Hz, 2H), 5.91 (s, 1H), 5.04 (d, J = 16.0 Hz, 1H), 4.86 (d, J = 16.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 177.1, 161.4, 148.0, 144.6, 143.6, 141.2, 135.7, 135.6, 129.2, 128.6, 127.6, 127.1, 124.8, 118.5, 111.4, 111.1, 104.4, 102.1, 98.3, 52.9, 50.5, 43.3; HRMS (ESI) m/z: [M+Na]+ calcd for C25H16ClN3O4Na 480.0727; found 480.0724; [α]D25= -1 (c 0.52, MeOH)(50% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 12.9 min (major), 21.1 min (minor) .
3f: whith solid, mp: 285.4-286.7 oC ;1H NMR (500 MHz, DMSO-d6) δ 7.48 (dd, J = 8.5, 2.0 Hz, 1H), 7.40 – 7.22 (m, 8H), 6.95 (d, J = 8.5 Hz, 1H), 6.79 (s, 1H), 6.00 (d, J = 14.0 Hz, 2H), 5.91 (s, 1H), 5.03 (d, J = 16.0 Hz, 1H), 4.85 (d, J = 16.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 176.9, 161.3, 147.9, 144.5, 143.5, 141.6, 135.9, 135.6, 131.9, 128.5, 127.4, 127.3, 127.0, 118.3, 115.1, 111.7, 111.1, 104.3, 102.0, 98.2, 53.0, 50.4, 43.2; HRMS (ESI) m/z: [M+Na]+ calcd for C25H16BrN3O4Na: 524.0222; found : 524.0228; [α]D25= -7 (c 0.65, MeOH)(90% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 13.8 min (major), 22.8 min (minor).
3g: whith solid, mp: 130.4-131.2 oC ;1H NMR (500 MHz, DMSO-d6) δ 7.36 – 7.21 (m, 7H), 7.07 (dd, J = 8.0, 1.0 Hz, 1H), 6.95 (s, 1H), 6.86 (d, J = 8.0 Hz, 1H), 6.78 (s, 1H), 5.98 (dd, J = 14.5, 1.0 Hz, 2H), 5.81 (s, 1H), 4.98 (d, J = 16.0 Hz, 1H), 4.84 (d, J = 16.0 Hz, 1H), 2.22 (s, 3H);13C NMR (125 MHz, DMSO-d6) δ 177.3, 161.4, 147.8, 144.5, 143.6, 139.9, 136.2, 133.8, 132.7, 129.5, 128.6, 127.5, 127.2, 125.1, 118.7, 112.2, 109.5, 104.5, 102.1, 98.2, 53.6, 50.3, 43.2, 20.5; [α]D25= -5 (c 0.63, MeOH)(70% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 13.7 min (major), 24.3 min (minor) .
3h: whith solid, mp: 191.8-192.5 oC ;1H NMR (500 MHz, DMSO-d6) δ 7.39 – 7.20 (m, 7H), 6.90 (d, J = 8.5 Hz, 1H), 6.85 (dd, J = 8.5, 2.5 Hz, 1H), 6.78 (s, 1H), 6.74 (d, J = 2.5 Hz, 1H), 6.00 (dd, J = 9.0, 1.0 Hz, 2H), 5.82 (s, 1H), 4.97 (d, J = 16.0 Hz, 1H), 4.85 (d, J = 16.0 Hz, 1H), 3.67 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 177.1, 161.4, 156.2, 147.8, 144.5, 143.5, 136.1, 135.5, 134.8, 128.6, 127.4, 127.1, 118.6, 113.9, 112.0, 111.3, 110.3, 104.4, 102.0, 98.2, 55.5, 53.5, 50.7, 43.2; HRMS (ESI) m/z: [M+Na]+ calcd for C26H19N3O5Na 476.1222; found 476.1227; [α]D25= +1 (c 0.52, MeOH)(57% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 19.5 min (major), 38.4 min (minor) .
3i: whith solid, mp: 148.7-150.1 oC ;1H NMR (500 MHz, DMSO-d6) δ 8.27 (dd, J = 8.5, 2.0 Hz, 1H), 8.01 (d, J = 2.0 Hz, 1H), 7.44 (s, 2H), 7.39 – 7.28 (m, 5H), 7.25 (d, J = 8.5 Hz, 1H), 6.83 (s, 1H), 6.06 (s, 1H), 6.00 (d, J = 13.5 Hz, 2H), 5.15 (d, J = 16.0 Hz, 1H), 4.95 (d, J = 16.0 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ 177.8, 161.5, 148.3, 148.2, 144.6, 143.7, 143.6, 135.2, 134.5, 128.6, 127.6, 127.0, 126.3, 120.1, 118.2, 110.3, 110.2, 104.6, 102.1, 98.3, 52.5, 50.3, 43.6; HRMS (ESI) m/z: [M+Na]+ calcd for C25H16N4O6Na 491.0968; found 491.0975; [α]D25= -2 (c 0.59, MeOH)(10% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 16.7 min (major), 34.6 min (minor) .
3j: whith solid, mp: 242.8-243.2 oC ;1H NMR (500 MHz, DMSO-d6) δ 7.41 – 7.25 (m, 7H), 7.17 (d, J = 8.0 Hz, 1H), 7.11 (dt, J = 8.0, 1.5 Hz, 2H), 6.79 (s, 1H), 6.00 (dd, J = 10.0, 1.0 Hz, 2H), 5.92 (s, 1H), 5.03 (d, J = 16.0 Hz, 1H), 4.88 (d, J = 16.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 177.4, 148.0, 144.6, 143.9, 135.7, 132.3, 128.6, 127.6, 127.1, 126.2, 123.2, 111.4, 110.0, 104.5, 102.1, 98.2, 50.0, 43.2; HRMS (ESI) m/z: [M+Na]+ calcd for C25H16ClN3O4Na 480.0727; found 480.0721; [α]D25= +1 (c 0.60, MeOH)(55% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 12.7 min (major), 17.9 min (minor) .
3k: whith solid, mp: 221.5-223.0 oC ;1H NMR (500 MHz, DMSO-d6) δ 7.37 – 7.24 (m, 8H), 7.16 (d, J = 7.0 Hz, 1H), 7.10 (t, J = 7.5 Hz, 1H), 6.80 (s, 1H), 6.13 (s, 1H), 6.01 (d, J = 6.0 Hz, 2H), 5.27 (s, 2H);13C NMR (125 MHz, DMSO-d6) δ 178.1, 161.4, 148.0, 144.7, 143.6, 138.5, 137.4, 136.8, 131.4, 128.5, 127.0, 126.0, 124.9, 124.1, 118.6, 114.7, 111.5, 104.9, 102.2, 98.2, 53.4, 50.5, 44.8; [α]D25= +3 (c 0.48, MeOH)(55% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 0.8 mL/min, 254 nm), tR= 17.5 min (major), 52.7 min (minor) .
3l: whith solid, mp: 256.5-257.3 oC ; 1H NMR (500 MHz, DMSO-d6) δ 7.48 (d, J = 8.0 Hz, 1H), 7.43 – 7.22 (m, 7H), 7.19 (d, J = 7.5 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 6.80 (s, 1H), 6.12 (s, 1H), 6.01 (d, J = 5.0 Hz, 2H), 5.37 (d, J = 17.0 Hz, 1H), 5.27 (d, J = 17.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 178.1, 161.3, 147.9, 144.6, 143.5, 139.8, 137.3, 137.0, 134.7, 128.3, 126.8, 125.9, 125.1, 124.5, 118.4, 111.5, 104.7, 102.0, 98.1, 53.5, 50.0, 44.4; HRMS (ESI) m/z: [M+Na]+ calcd for C25H16BrN3O4Na: 524.0222; found : 524.0226; [α]D25= +1 (c 0.45, MeOH)(67% ee); HPLC (Chiralpak AD, hexane:iPrOH = 80:20, 1.0 mL/min, 254 nm), tR= 24.2 min (major), 87.6 min (minor) .
3m: whith solid, mp: 130.8-131.8 oC ; 1H NMR (500 MHz, DMSO-d6) δ 10.55 (s, 1H), 7.27 (td, J = 7.5, 1.5 Hz, 1H), 7.16 (s, 2H), 7.05 (d, J = 6.0 Hz, 1H), 7.01 (td, J = 7.5, 1.0 Hz, 1H), 6.93 (d, J = 7.5 Hz, 1H), 6.76 (s, 1H), 5.99 (dd, J = 7.5, 1.0 Hz, 2H), 5.88 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 178.4, 148.4, 146.6, 143.0, 139.4, 133.2, 128.6, 123.7, 121.1, 120.1, 109.0, 106.8, 100.7, 97.4, 75.1; [α]D25= -2 (c 0.51, MeOH)(45% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 11.8 min (major), 15.7 min (minor) .
3n: whith solid, mp: 260.9-262.3 oC ;1H NMR (500 MHz, DMSO) δ 7.38 (ddd, J = 8.0, 6.5, 2.5 Hz, 1H), 7.20 (s, 2H), 7.15 – 7.07 (m, 3H), 6.77 (s, 1H), 5.98 (dd, J = 4.5, 1.0 Hz, 2H), 5.91 (s, 1H), 3.19 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 177.1, 161.4, 147.7, 144.5, 143.6, 143.3, 133.6, 129.2, 124.4, 123.3, 118.5, 112.2, 109.1, 104.8, 102.0, 98.1, 53.6, 50.2, 26.4; [α]D25= -2 (c 0.50, MeOH)(50% ee); HPLC (Chiralpak AS, hexane:iPrOH = 90:10, 1.5 mL/min, 254 nm), tR= 54.9 min (minor), 61.6 min (major) .
3o: whith solid, mp: 187.9-188.8 oC ;1H NMR (500 MHz, DMSO-d6) δ 10.70 (s, 1H), 7.46 (dd, J = 8.3, 2.1 Hz, 1H), 7.27 – 7.20 (m, 3H), 6.90 (d, J = 8.5 Hz, 1H), 6.76 (s, 1H), 6.00 (dd, J = 9.5, 1.0 Hz, 2H), 5.98 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 178.2, 161.2, 147.8, 144.5, 143.5, 141.1, 136.6, 131.9, 131.4, 128.5, 127.4, 118.3, 112.0, 111.4, 104.4, 101.9, 98.1, 53.3, 50.8; HRMS (ESI) m/z: [M+Na]+ calcd for C18H10BrN3O4Na: 433.9752; found: 433.9756; [α]D25= -7 (c 0.62, MeOH)(72% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 9.6 min (major), 11.4 min (minor) .
3p: whith solid, mp: 260.7-261.2 oC ;1H NMR (500 MHz, DMSO-d6) δ 10.45 (s, 1H), 7.22 – 7.00 (m, 3H), 6.89-6.75 (m, 3H), 5.99 (d, J = 8.5 Hz, 2H), 5.88 (s, 1H), 2.23 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 178.8, 161.2, 147.7, 144.4, 143.5, 139.3, 134.6, 131.6, 129.4, 125.2, 118.6, 112.4, 109.8, 104.6, 102.0, 98.1, 53.9, 50.7, 20.6; [α]D25= -5 (c 0.56, MeOH) (59% ee); HPLC (Chiralpak AD, hexane:iPrOH = 70:30, 0.8 mL/min, 254 nm), tR= 9.6 min (major), 11.4 min (minor) .
4: whith solid, mp: 191.5-192.8 oC ; 1H NMR (500 MHz, DMSO-d6) δ 7.41 – 7.25 (m, 7H), 7.23 (s, 2H), 7.13 (d, J = 7.0 Hz, 1H), 7.07 (t, J = 7.5 Hz, 2H), 6.71 (s, 1H), 5.73 (s, 1H), 4.96 (q, J = 15.5 Hz, 2H), 3.78 (s, 3H), 3.32 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 177.3, 161.5, 149.6, 145.9 142.9, 142.2, 136.2, 133.5, 129.1, 128.6, 128.5, 127.4, 127.2, 124.6, 123.4, 118.5, 110.8, 109.5, 108.3, 100.8, 55.8, 55.7, 53.7, 49.9, 43.0; HRMS (ESI) m/z: [M+Na]+ calcd for C26H20N3O4Na: 462.1430; found: 462.1437; [α]D25= -5 (c 0.46, MeOH) (55% ee); HPLC (Chiralpak AD, hexane: iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 17.3 min (major), 33.7 min (minor).
5: whith solid, mp: 288.3-288.5 oC ; 1H NMR (500 MHz, DMSO-d6) δ 7.41 – 7.25 (m, 7H), 7.23 (s, 2H), 7.13 (d, J = 7.0 Hz, 1H), 7.07 (t, J = 7.5 Hz, 2H), 6.71 (s, 1H), 5.73 (s, 1H), 4.96 (q, J = 15.5 Hz, 2H), 3.78 (s, 3H), 3.32 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 177.2, 160.5, 160.0, 157.4, 150.4, 142.4, 136.4, 134.7, 128.4, 128.0, 127.7, 127.3, 123.1, 122.6, 118.2, 108.6, 101.4, 95.6, 93.4, 56.0, 55.8, 55.6, 47.4, 43.4; HRMS (ESI) m/z: [M+Na]+ calcd for C26H20N3O4Na: 462.1430; found: 462.1438; [α]D25= -4 (c 0.53, MeOH) (32% ee); HPLC (Chiralpak AS, hexane: iPrOH = 70:30, 1.0 mL/min, 254 nm), tR= 11.0 min (major), 20.5 min (minor).
6: whith solid, mp: 247.5-248.8 oC ; 1H NMR (500 MHz, DMSO-d6) δ 8.30 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.73 – 7.68 (m, 1H), 7.66 – 7.62 (m, 1H), 7.60 – 7.52 (m, 3H), 7.39 (d, J = 7.5 Hz, 2H), 7.32 (ddd, J = 16.0, 12.0, 7.5 Hz, 4H), 7.18 (d, J = 7.0 Hz, 1H), 7.06 (dd, J = 14.0, 7.5 Hz, 2H), 6.48 (d, J = 8.5 Hz, 1H), 5.05 (d, J = 16.0 Hz, H), 4.95 (d, J = 16.0 Hz, H);13C NMR (126 MHz, DMSO-d6) δ 177.4, 161.2 143.7, 142.4, 136.0, 133.8, 133.1, 129.3, 128.6, 127.7, 127.5, 127.2, 125.0, 124.5, 123.6, 122.9, 120.8, 114.6, 109.7, 50.5, 43.2; HRMS (ESI) m/z: [M+Na]+ calcd for C28H19N3O2Na: 452.1375; found: 452.1371; [α]D25= -1 (c 0.49, MeOH) (20% ee); HPLC (Chiralpak AS, hexane: iPrOH = 70:30, 0.8 mL/min, 254 nm), tR= 13.1 min (minor), 18.0 min (major).
7: whith solid, mp: 292.2-292.5 oC ; 1H NMR (500 MHz, DMSO-d6) δ 11.39 (s, 1H), 7.45 – 7.40 (m, 1H), 7.37 (d, J = 7.0 Hz, 2H), 7.33 (t, J = 7.5 Hz, 2H), 7.27 (ddd, J = 9.0, 6.5, 1.5 Hz, 4H), 7.11 (d, J = 6.5 Hz, 1H), 7.04 (t, J = 7.5 Hz, 2H), 6.98 (d, J = 8.0 Hz, 1H), 6.51 (t, J = 2.0 Hz, 1H), 6.08 (d, J = 8.5 Hz, 1H), 4.96 (dd, J = 35.4, 16.0 Hz, 2H);13C NMR (125 MHz, DMSO-d6) δ 178.1, 161.6, 142.3, 141.5, 136.7, 136.2, 134.8, 128.9, 128.6, 127.5, 127.2, 126.2, 124.8, 123.4, 119.0, 118.7, 116.6, 109.5, 109.0, 108.7, 97.6, 54.3, 50.2, 43.0; [α]D25= -1 (c 0.55, MeOH) (56% ee); HPLC (Chiralpak IA, hexane: iPrOH = 70:30, 1.5 mL/min, 254 nm), tR= 8.0 min (minor), 15.2 min (major).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Copies of 1H and 13C-NMR spectra and HPLC trace of products are available online at www.mdpi.com/xxx/s1. Figure S1-S40: NMR spectra of compounds 3a-3p and 4-7; Figure S41-80: HPLC trace of compounds 3a-3p and 4-7.
Author Contributions
Liming Wang and Hongwen Mu are co-first authors; they contributed equally to this work. They performed the experiments, acquired and analyzed the original data. Yuhong Sun conducted instrumental analysis. Y. Jin supervised the experiment, analyzed and checked all the data, wrote the draft and revised manuscript. Wei Zhang designed the research plan, provided funding supporting. All authors have given approval to the final version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of Jilin province (No. 20230101226JC), the Department of Education of Jilin province (No. JJKH20240595KJ), and the Department of Education of Jilin province (No. JT53101022010).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Xiao, Z.-P.; Peng, Z.-Y.; Dong, J.-J.; He, J.; Ouyang, H.; Feng, Y.-T.; Lu, C.-L.; Lin, W.-Q.; Wang, J.-X.; Xiang, Y.-P.; Zhu, H.-L. Synthesis, Structure−Activity Relationship Analysis and Kinetics Study of Reductive Derivatives of Flavonoids as Helicobacter Pylori Urease Inhibitors. Eur. J. Med. Chem. 2013, 63, 685–695. [Google Scholar] [CrossRef]
- Yee, E. M. H.; Pasquier, E.; Iskander, G.; Wood, K.; Black, D. S.; Kumar, N. Synthesis of Novel Isoflavene- Propranolol Hybrids as Anti-Tumor Agents. Bioorg. Med. Chem. 2013, 21, 1652–1660. [Google Scholar] [CrossRef]
- Eiffe, E.; Pasquier, E.; Kavallaris, M.; Herbert, C.; Black, D. S.; Kumar, N. Synthesis, Anticancer and Anti- Inflammatory Activity of Novel 2-Substituted Isoflavenes. Bioorg. Med. Chem, 2014; 22, 5182–5193. [Google Scholar] [CrossRef]
- Chen, Y.; Cass, S. L.; Kutty, S. K.; Yee, E. M. H.; Chan, D. S. H.; Gardner, C. R.; Vittorio, O.; Pasquier, E.; Black, D. S.; Kumar, N. Synthesis, Biological Evaluation and Structure−Activity Relationship Studies of Isoflavene Based Mannich Bases with Potent Anti-Cancer Activity. Bioorg. Med. Chem. Lett. 2015, 25, 5377–5383. [Google Scholar] [CrossRef]
- Renko, D.; Provot, O.; Rasolofonjatovo, E.; Bignon, J.; Rodrigo, J.; Dubois, J.; Brion, J. D.; Hamze, A.; Alami, M. Rapid Synthesis of 4- Arylchromenes from Ortho-Substituted Alkynols: A Versatile Access to Restricted Isocombretastatin A-4 Analogues as Antitumor Agents. Eur. J. Med. Chem. 2015, 90, 834–844. [Google Scholar] [CrossRef]
- Yee, E. M. H.; Brandl, M. B.; Black, D. S.; Vittorio, O.; Kumar, N. Synthesis of Isoflavene- Thiosemicarbazone Hybrids and Evaluation of Their Anti-Tumor Activity. Bioorg. Med. Chem. Lett. 2017, 27, 2454–2458. [Google Scholar] [CrossRef]
- Malik, N.; Zhang, Z.; Erhardt, P. Total Synthesis of (±)-Glyceollin II and a Dihydro Derivative. J. Nat. Prod. 2015, 78, 2940–2947. [Google Scholar] [CrossRef]
- Muharini, R.; Díaz, A.; Ebrahim, W.; Mandi, A.; Kurtan, T.; Rehberg, N.; Kalscheuer, R.; Hartmann, R.; Orfali, R. S.; Lin, W.; Liu, Z.; Proksch, P. Antibacterial and Cytotoxic Phenolic Metabolites from the Fruits of Amorpha Ruticosa. J. Nat. Prod. 2017, 80, 169–180. [Google Scholar] [CrossRef]
- Rueda-Zubiaurre, A.; Yahiya, S.; Fischer, O. J.; Hu, X.; Saunders, C. N.; Sharma, S.; Straschil, U.; Shen, J.; Tate, E. W.; Delves, M. J.; Baum, J.; Barnard, A.; Fuchter, M. J. Structure−Activity Relationship Studies of a Novel Class of Transmission Blocking Antimalarials Targeting Male Gametes. J. Med. Chem. 2020, 63, 2240–2262. [Google Scholar] [CrossRef]
- Chen, M,-S.; Yu, S.-J. Characterization of Lipophilized Monomeric and Oligomeric Grape Seed Flavan-3-ol Derivatives. J. Agric. Food. Chem. 2017, 65, 8875-8883. [CrossRef]
- Hu, B.; Liu, X.; Zhang, C.; Zeng, X. Food Macromolecule Based Nanodelivery Systems for Enhancing the Bioavailability of Polyphenols. J. Food. Drug. Anal. 2017, 25, 3–15. [Google Scholar] [CrossRef]
- Cheenpracha, S.; Karalai, C.; Ponglimanont, C.; Kanjana-Opas, A. Candenatenins, A. Phenolic Compounds from the Heartwood of Dalbergia Candenatensis. J. Nat. Prod. 2009, 72, 1395–1398. [Google Scholar] [CrossRef]
- Singh, G. S.; Desta, Z. Y. Isatins as Privileged Molecules in Design and Synthesis of Spiro Fused Cyclic Frameworks. Chem. Rev. 2012, 112, 6104–6155. [Google Scholar] [CrossRef]
- Williams, R. M.; Cox, R. J. Paraherquamides, Brevianamides, and Asperparalines:Laboratory Synthesis and Biosynthesis. An Interim Report. Acc. Chem. Res. 2003, 36, 127–139. [Google Scholar] [CrossRef]
- Yang, Y.-T.; Zhu, J.-F.; Liao, G.; Xu, H.-J.; Yu, B. The Development of Biologically Important Spirooxindoles as New Antimicrobial Agents. Curr. Med. Chem. 2018, 25, 2233–2244. [Google Scholar] [CrossRef]
- Panda, S. S.; Jones, R. A.; Bachawala, P.; Mohapatra, P. P. Spirooxindoles as Potential Pharmacophores. Mini. Rev. Med. Chem. 2017, 17, 1515–1536. [Google Scholar] [CrossRef]
- Chen, W.-B.; Wu, Z.-J.; Pei, Q.-L.; Cun, L.-F.; Zhang, X.-M.; Yuan, W.-C. Highly Enantioselective Construction Of Spiro, [4H-Pyran-3.30 -Oxindoles] Through a Domino Knoevenagel/Michael/Cyclization Sequence Catalyzed by Cupreine. Org. Lett. 2010, 12, 3132–3135. [Google Scholar] [CrossRef]
- Pellissier, H.; Recent Developments in Asymmetric Organocatalytic Domino Reactions. Recent Developments in Asymmetric Organocatalytic Domino Reactions. Adv. Synth. Catal. 2012, 354, 237–294. [CrossRef]
- Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C. F. Organocatalytic Asymmetric Assembly Reactions: Synthesis of Spirooxindoles via Organocascade Strategies. ACS Catal. 2014, 4, 743–762. [Google Scholar] [CrossRef]
- Santos, M. M. M. Recent Advances in the Synthesis of Biologically Active Spirooxindoles. Tetrahedron 2014, 70, 9735–9757. [Google Scholar] [CrossRef]
- Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Bifunctional AmineSquaramides: Powerful Hydrogen-Bonding Organocatalysts for Asymmetric Domino/Cascade Reactions. Adv. Synth. Catal. 2015, 357, 253–281. [Google Scholar] [CrossRef]
- Ardkhean, R.; Caputo, D. F. J.; Morrow, S. M.; Shi, H.; Xiong, Y.; Anderson, E. A. Cascade Polycyclizations in Natural Product Synthesis. Chem. Soc. Rev. 2016, 45, 1557–1569. [Google Scholar] [CrossRef]
- Mei, G.-J.; Shi, F. Catalytic Asymmetric Synthesis of Spirooxindoles: Recent Developments. Chem. Commun. 2018, 54, 6607–6621. [Google Scholar] [CrossRef]
- Chanda, T.; Zhao, J. C.-G. Recent Progress in Organocatalytic Asymmetric Domino Transformations. Adv. Synth. Catal. 2018, 360, 2–79. [Google Scholar] [CrossRef]
- Mohammadi Ziarani, G.; Moradi, R.; Lashgari, N. Asymmetric Synthesis of Chiral Oxindoles Using Isatin as Starting Material. Tetrahedron 2018, 74, 1323–1353. [Google Scholar] [CrossRef]
- Konda, S.; Jakkampudi, S.; Arman, H. D.; Zhao, J.C.-G. Enantioselective Synthesis of Spiro[4H- pyran-3,3-oxindole] Derivatives Catalyzed by Cinchona Alkaloid Thioureas: Significant Water Effects on the Enantioselectivity. Synthetic Commun. 2019, 49, 2971–2982. [Google Scholar] [CrossRef]
- Tan, F.; Lu, L.-Q.; Yang, Q.-Q.; Guo, W.; Bian, Q.; Chen, J.-R.; Xiao, W.-J. Enantioselective Cascade Michael Addition/ Cyclization Reactions of 3-Nitro-2H-Chromenes with 3-Isothiocyanato Oxindoles: Efficient Synthesis of Functionalized Polycyclic Spirooxindoles. Chem. Eur. J. 2014, 20, 3415–3420. [Google Scholar] [CrossRef]
- Jiang, X.; Sun, Y.; Yao, J.; Cao, Y.; Kai, M.; He, N.; Zhang, X.; Wang, Y.; Wang, R. Core Scaffold-Inspired Concise Synthesis of Chiral Spirooxindole-Pyranopyrimidines with Broad- Spectrum Anticancer Potency. Adv. Synth. Catal. 2012, 354, 917–925. [Google Scholar] [CrossRef]
- Zhao, H.-W.; Li, B.; Tian, T.; Meng, W.; Yang, Z.; Song, X.-Q.; Chen, X.-Q.; Pang, H.-L. Highly Enantioselective Synthesis of Chiral Pyranonaphthoquinone-Fused Spirooxindoles through Organocatalytic Three-Component Cascade Reactions. Eur. J. Org. Chem. 2015, 2015, 3320–3326. [Google Scholar] [CrossRef]
- Kumari, P.; Nandi, S.; Kumar, G.; Khan,N. H.; Kureshy,R. I.; Abdi, S. H. R.; Eringathodi, S.; Bajaj, H. C. Construction of Highly Enantioselective Spiro-oxindole Derivatives with Fused Chromene via Organocascade Catalysis. RSC Adv. 2016, 6, 52384–52390. [CrossRef]
- Yang, C.; Yang, J.; Guo F. -M.; Zhao, Z.; Feng, T.-T.; Liu X. -L.; Zhou Y. Synthesis of Novel Spiro-Fused Pyranyl Benzo[d] [1,3]dioxol Oxindoles. Chin. J. Synth. Chem. 2015, 23, 599–602. [CrossRef]
- Abdolmohammadi, S.; Rasouli Nasrabadi, S. R.; Dabiri, M. R.; Banihashemi Jozdani, S. M. TiO2 Nanoparticles Immobilized on Carbon Nanotubes: An Efficient Heterogeneous Catalyst in Cyclocondensation Reaction of Isatins with Malononitrile and 4-Hydroxycoumarin or 3, 4- Methylenedioxyphenol under Mild Reaction Conditions. Appl Organometal Chem. 2020, e5462. [Google Scholar] [CrossRef]
- CCDC-2291478 (3f) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via. www.ccdc.cam.ac.uk/data_request/cif.
- Prathima, P. S.; Rajesh, P.; Rao, J. V.; Kailash, U. S.; Sridhar, B.; Rao, M. M. “On Water” Expedient Synthesis of 3-Indolyl- 3- Hydroxy Oxindole Derivatives and Their Anticancer Activity in Vitro. Eur. J. Med. Chem. 2014, 84, 155–159. [Google Scholar] [CrossRef]
- Huong, T. T. L.; Dung,D.T. M.; Huan,N. V.; Cuong, L. V.; Hai, P. T.; Huong, L.T. T.; Kim, J.; Kim, Y. G.; Han,S. B.; Nam, N. H. Novel N-Hydroxybenzamides Incorporating 2-Oxoindoline with Unexpected Potent Histone Deacetylase Inhibitory Effects and Antitumor Cytotoxicity. Bioorg. Chem. 2017, 71, 160- 169. [CrossRef] [PubMed]
- Kumar, S.; Saha, S. T.; Gu, L.; Palma, G.; Perumal, S.; Singh-Pillay, A.; Singh, P.; Anand, A.; Kaur, M. ; Kumar, V 1H-1,2,3-Triazoletethered Nitroimidazole-Isatin Conjugates: Synthesis, Docking, and Anti-Proliferative Evaluation against Breast Cancer. ACS Omega, 2018, 3, 12106–12113. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M. R.; Alagumuthu, M.; Dhayabaran, V. V. N-Substituted Hydroxynaphthalene Imino-oxindole Derivatives as New Class of PI3-Kinase Inhibitor and Breast Cancer Drug: Molecular Validation and Structure-Activity Relationship Studies. Chem. Biol. Drug Des. [CrossRef]
- Diao, Q. -P.; Guo, H.; Wang, G. -Q. Design, Synthesis, and in Vitro Anticancer Activities of Diethylene Glycoltethered Isatin -1,2,3-Triazole-Coumarin Hybrids. J Heterocycl Chem. 2019, 56, 1667 -1671. [CrossRef]
- Shirai, F.; Mizutani, A.; Yashiroda, Y.; Tsumura, T.; Kano, Y.; Muramatsu, Y.; Chikada, T.; Yuki, H.; Niwa, H.; Sato, S.; Washizuka, K.; Koda, Y.; Mazaki, Y.; Jang, M. K.; Yoshida, H.; Nagamori, A.; Okue, M.; Watanabe, T.; Kitamura, K.; Shitara, E.; Honma, T.; Umehara,T.; Shirouzu, M.; Fukami, T.; Seimiya, H.; Yoshida, M.; Koyama, H. Design and Discovery of an Orally Efficacious Spiroindolinone-Based Tankyrase Inhibitor for the Treatment of Colon Cancer. J. Med. Chem. 2020, 63, 4183 -4204. [CrossRef]
Figure 1.
Examples of biologically active compounds containing chiral chromenes.
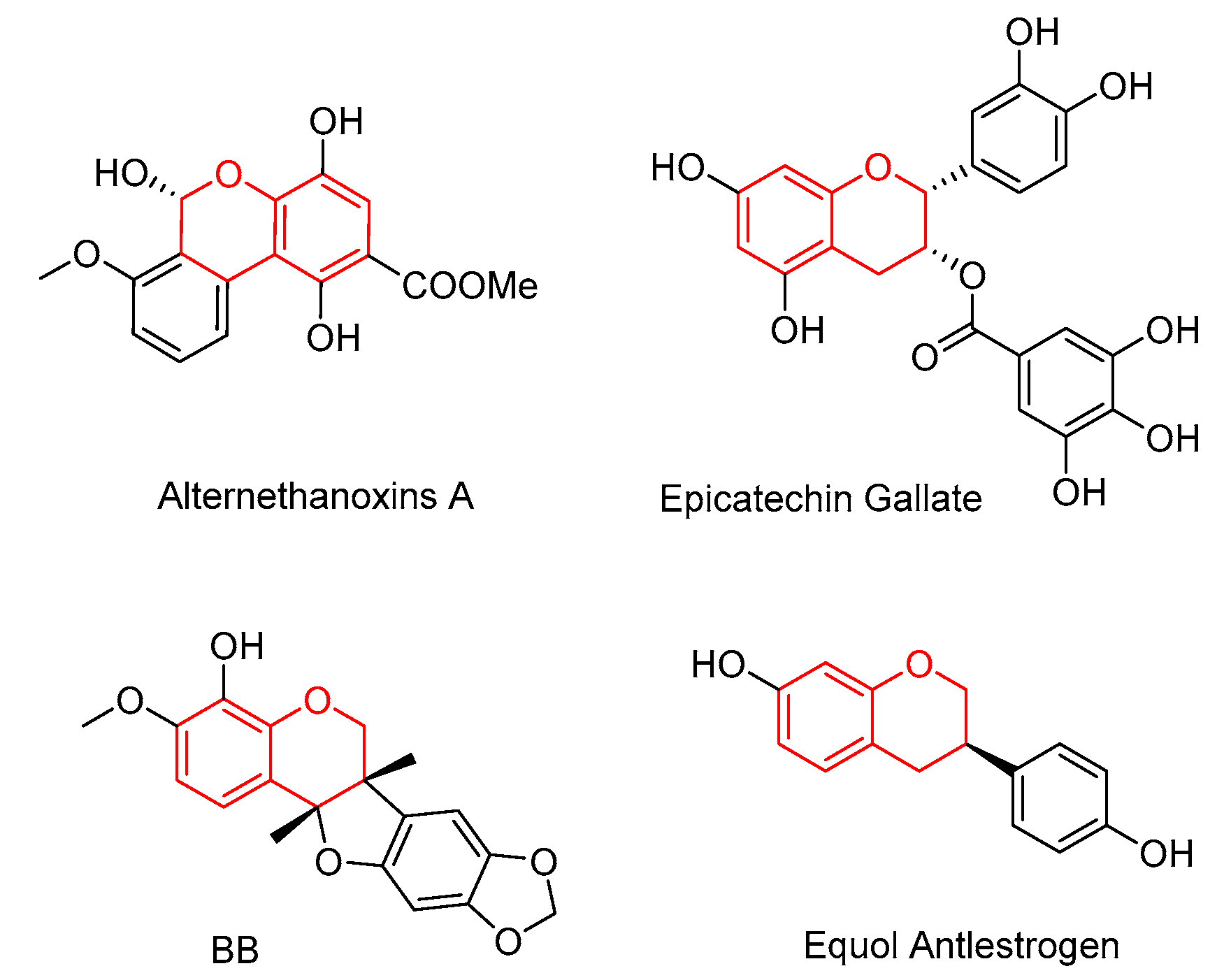
Figure 2.
The structure of screened organocatalysts (1a–1m).
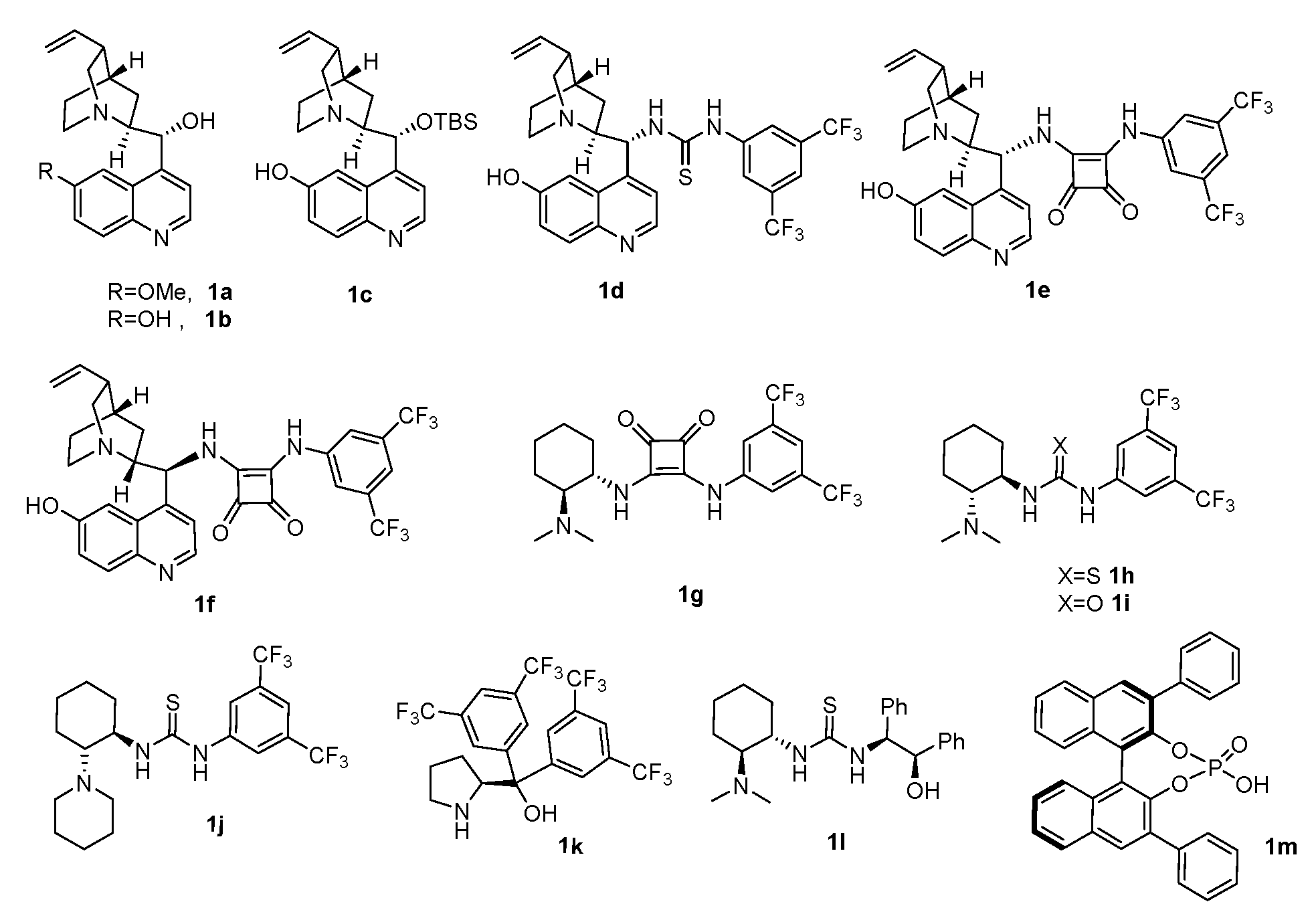
Figure 3.
X-ray crystal structure of 3f.
Scheme 1.
Proposed transition state for the formation of 3f.
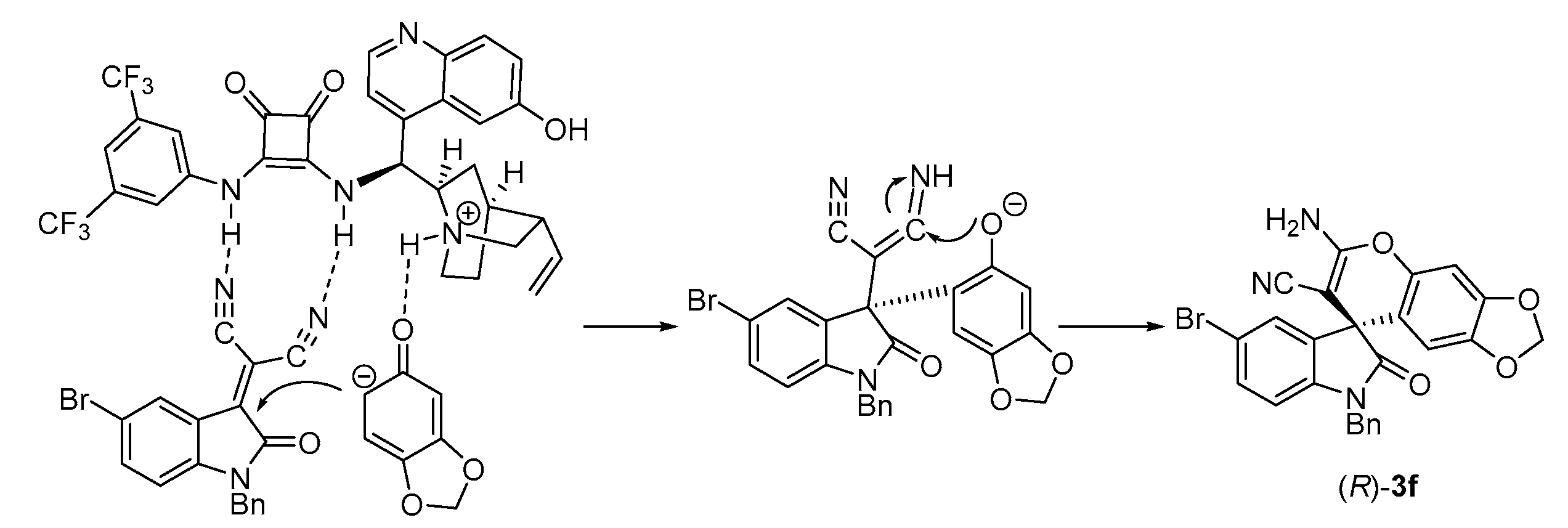
Figure 4.
Expansion of the scope of nucleophiles
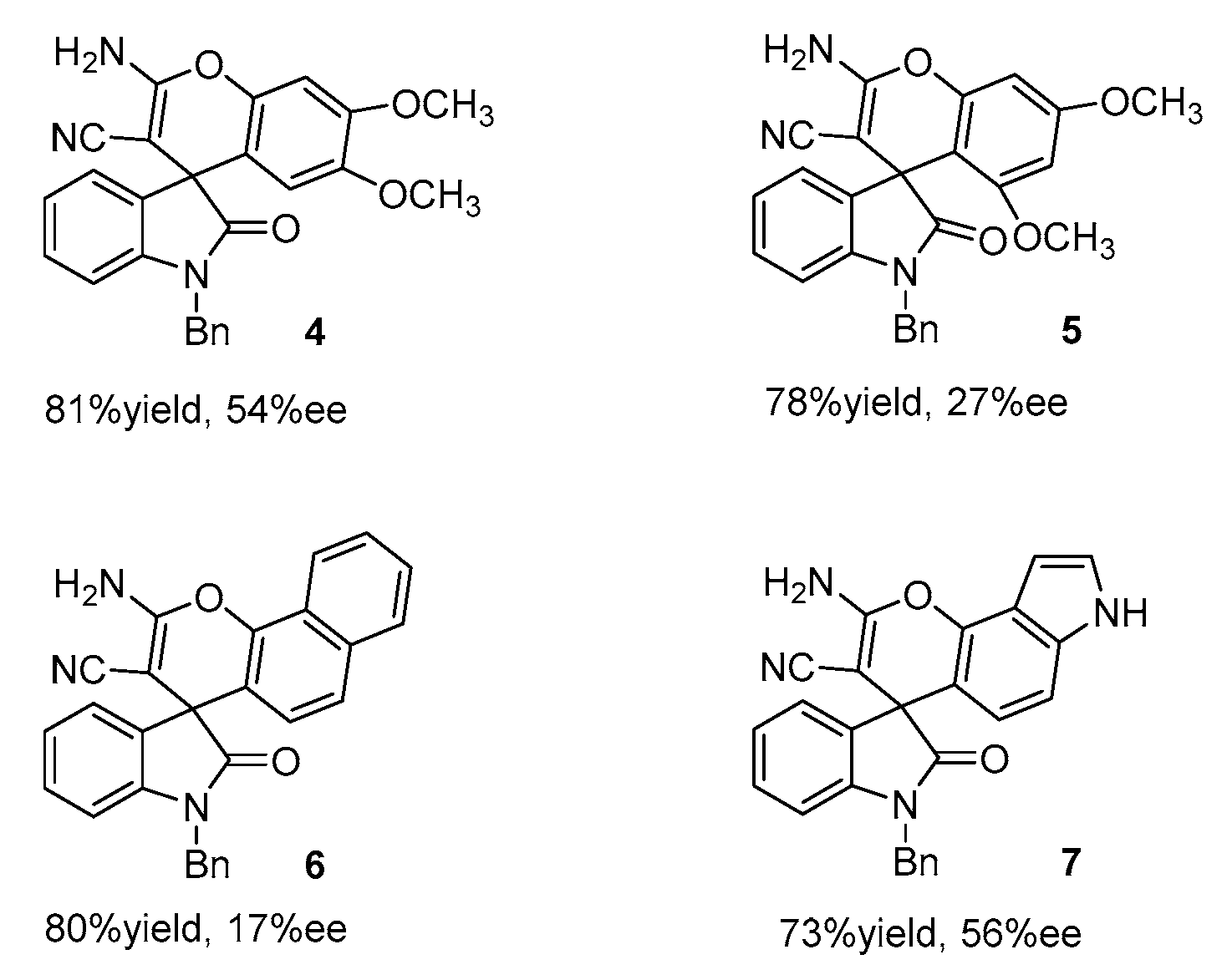
Figure 5.
The structures of two pairs of enantiomers
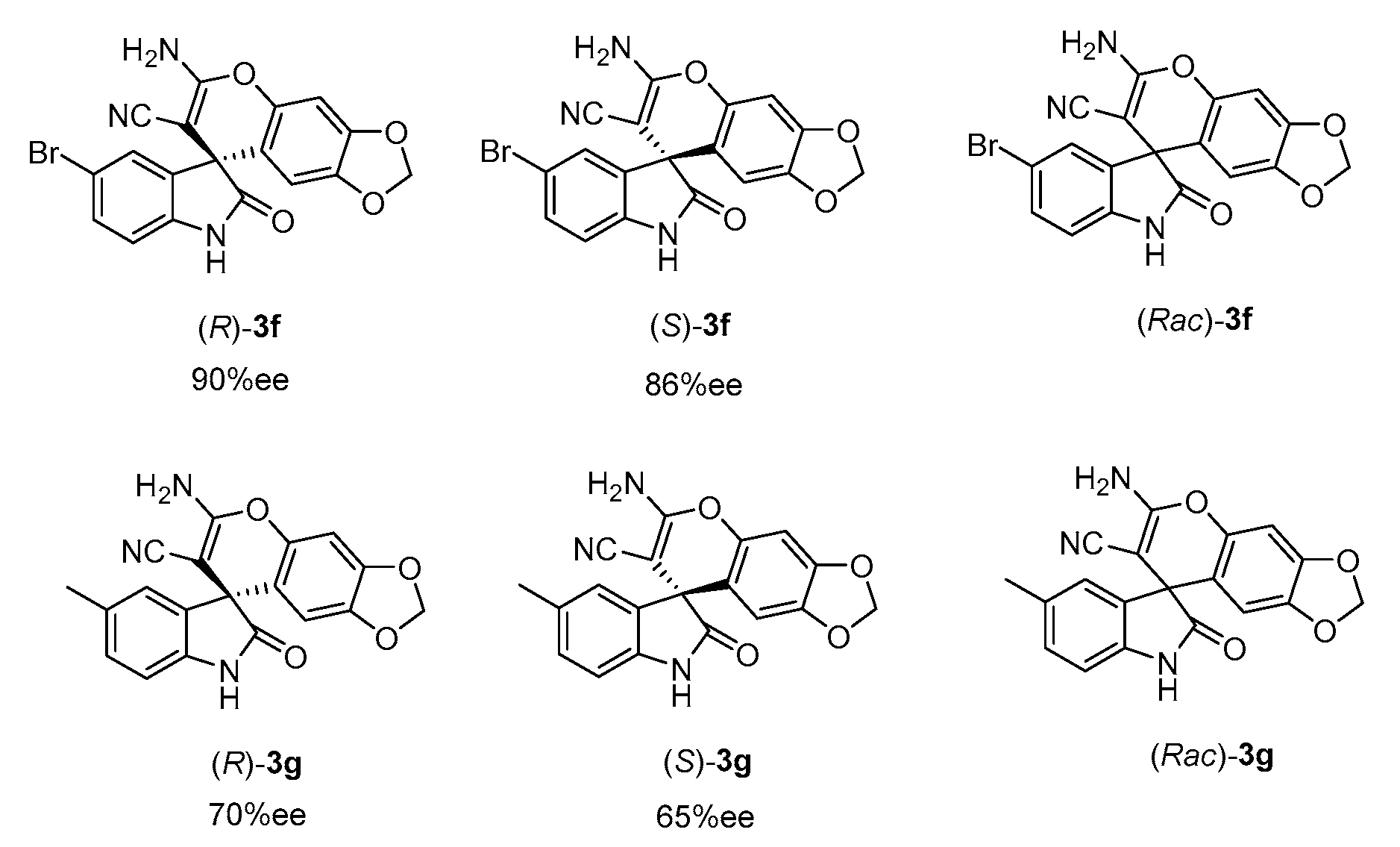

Table 1.
Asymmetric Knoevenagel/Michael/cyclization reaction of isatin 2a, malononitrile and sesamol catalysed by 1a-k a.
Table 1.
Asymmetric Knoevenagel/Michael/cyclization reaction of isatin 2a, malononitrile and sesamol catalysed by 1a-k a.
 | |||
|---|---|---|---|
| Entry | Catalyst | Yield (%)b | %eec |
| 1 | 1a | 78 | 10 |
| 2 | 1b | 76 | 12 |
| 3 | 1c | 80 | 10 |
| 4 | 1d | 81 | 29 |
| 5 | 1e | 83 | 38 |
| 6 | 1f | 85 | 40 |
| 7 | 1g | 82 | 26 |
| 8 | 1h | 80 | 20 |
| 9 | 1i | 68 | 5 |
| 10 | 1j | 73 | 12 |
| 11 | 1k | 74 | 12 |
| 12 | 1l | 72 | 9 |
| 13 | 1m | - | - |
aReaction condition:N-benzylisatin (0.1 mmol) , malononitrile (0.1 mmol), sesamol (0.1 mmol) and catalyst 1f ( 0.01 mmol ) in CH2Cl2 (1.0 mL) at rt . bisolated yield. c Determined by HPLC analysis (Chiralpak AD).
Table 2.
Screening of reaction conditions for the asymmetric cascade reaction catalysed by 1f a.
| Entry. | Solvent | Temperature | Catalyst. Amount (% mmol) |
Yield (%)b | %eec |
|---|---|---|---|---|---|
| 1 | CH2Cl2 | rt | 10 | 85 | 40 |
| 2 | CHCl3 | rt | 10 | 84 | 42 |
| 3 | (CH2)2Cl2 | rt | 10 | 76 | 28 |
| 4 | Et2O | rt | 10 | 78 | 13 |
| 5 | THF | rt | 10 | 78 | 16 |
| 6 | PhMe | rt | 10 | 81 | 16 |
| 7 | EtOAc | rt | 10 | 75 | 10 |
| 8 | CHCl3 | rt | 5 | 71 | 35 |
| 9 | CHCl3 | rt | 20 | 88 | 36 |
| 10 | CHCl3 | 0 | 10 | 83 | 58 |
| 11 | CHCl3 | -20 | 10 | 68 | 40 |
| 12d | CHCl3 | 0 | 10 | 83 | 52 |
| 13e | CHCl3 | 0 | 10 | 85 | 61 |
| 14f | CHCl3 | 0 | 10 | 87 | 60 |
aReaction condition:N-benzylisatin (0.10 mmol), sesamol (0.10 mmol), malononitrile (0.10 mmol) and 1f in solvent (1 mL). bisolated yield. c Determined by HPLC analysis (ChiralpakAD). d 2 mL of solvent. e 4Å MS (about 30 mg). f 4-methylbenzoic acid(0.01 mmol). g 4-nitrobenzoic acid(0.01 mmol). f with 4Å MS (about 30 mg) in anhydrous and anaerobic condition.
Table 3.
Generality of the enantioselective Knoevenagel/Michael/cyclization reaction of isatins, malononitrile and sesamol a.
Table 3.
Generality of the enantioselective Knoevenagel/Michael/cyclization reaction of isatins, malononitrile and sesamol a.
 | ||||
|---|---|---|---|---|
| Entry | R1, R2 | Product | Yield (%)b | %eec |
| 1 | H, Bn(2a) | 3a | 85 | 61 |
| 2 | 4-Cl, Bn(2b) | 3b | 83 | 65 |
| 3 | 4-Br, Bn(2c) | 3c | 77 | 50 |
| 4 | 5-F, Bn(2d) | 3d | 81 | 60 |
| 5 | 5-Cl, Bn(2e) | 3e | 85 | 50 |
| 6 | 5-Br, Bn(2f) | 3f | 87(82) d | 90(85)d |
| 7 | 5-Me, Bn(2g) | 3g | 81 | 70 |
| 8 | 5-OMe, Bn(2h) | 3h | 79 | 57 |
| 9 | 5-NO2, Bn(2i) | 3i | 84 | 10 |
| 10 | 6-Cl, Bn(2j) | 3j | 82 | 55 |
| 11 | 7-Cl, Bn(2k) | 3k | 75 | 55 |
| 12 | 7-Br, Bn(2l) | 3l | 80 | 67 |
| 13 | H, H(2m) | 3m | 79 | 45 |
| 14 | H, Me(2n) | 3n | 78 | 50 |
| 15 | 5-Br, H(2o) | 3o | 83 | 72 |
| 16 | 5-Me, H(2p) | 3p | 81 | 59 |
aReaction condition: isatins (0. 10 mmol), sesamol (0.10 mmol), malononitrile (0.10 mmol) and catalyst 1f(0.01 mmol) in CHCl3(1 mL) at 0 oC. bisolated yield. c Determined by HPLC analysis (Chiralpak AD). dThe data in parentheses are the yield and ee value from reaction of 0.5 mmol scale.
Table 4.
In vitro cytotoxicity of Spiro[4H-Chromene -3,3′- oxindoles] derivatives against C42, HeLa & U2OS human cancer cells by MTT assay.
Table 4.
In vitro cytotoxicity of Spiro[4H-Chromene -3,3′- oxindoles] derivatives against C42, HeLa & U2OS human cancer cells by MTT assay.
| Entry | compounds | IC50 valuesa (μg/mL) | ||
|---|---|---|---|---|
| C42 | HeLa | U2OS | ||
| 1 | R-3f | 11.57 | 16.32 | 6.73 |
| 2 | S-3f | 23.83 | 74.69 | 62.31 |
| 3 | Racemic-3f | 15.32 | 55.52 | 26.39 |
| 4 | R-3g | 12.22 | 47.97 | 17.98 |
| 5 | S-3g | 31.88 | 225.62 | 73.10 |
| 6 | Racemic-3g | 18.48 | 171.91 | 44.26 |
| 7 | Adriamycin | 6.02 | 1.48 | 4.72 |
a IC50 is defined as the concentration, which results in a 50% decrease in cell number as compared with that of the control cultures in the absence of an inhibitor and were calculated using the respective regression analysis. b Adriamycin was employed as positive control.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



