
Submitted:
26 October 2023
Posted:
27 October 2023
You are already at the latest version
Abstract
This review summarizes the currently known biochemical neuroadaptive mechanisms of remote ischemic conditioning. In particular, it focuses on the significance of the pro-adaptive effects of remote ischemic conditioning, which allow prevention of neurological and cognitive impairments associated with hippocampal dysregulation after brain damage. The neuro-immuno-humoral pathway of transmitting a conditioning stimulus, as well as the molecular basis of the early and delayed phases of neuroprotection, including anti-apoptotic, antioxidant and anti-inflammatory components, are also outlined. Based on the close interplay between the effects of ischemia, especially mediated by interaction of HIF factors and steroid hormones, the involvement of the hypothalamic-pituitary-adrenocortical system in remote ischemic conditioning is also discussed.
Keywords:
ischemic conditioning
; brain
; hippocampus
; adaptation
; mechanisms of neuroprotection
; HIF-1
; HPA
1. Introduction
Ischemic preconditioning was first described in the heart as a reduction in the size of the myocardial infarction region provided by 5-minute coronary artery occlusion cycles before a 40-minute episode of ischemia [1]. It was then shown that brief episodes of occlusion-reperfusion of the circumflex coronary artery reduced damage caused by hourly occlusion of the adjacent descending artery [2]. Thus, a preconditioning can be remote, including occlusion and deocclusion of such arteries as the mesenteric, intestinal, renal and abdominal aorta, etc. [3,4]. Further, the concept of remote preconditioning was expanded by the application of short cycles of ischemia-reperfusion to the hind limb, demonstrating that intermittent ischemic impacts on an organ remote from the heart can provide significant cardioprotection [5]. Remote post-conditioning was first reported by Andreka and colleagues in a pig model of acute myocardial infarction resulting in a reduced infarct size when a 5-minute episode of ischemia-reperfusion was applied to the hind limb after the end of coronary artery occlusion [6]. Further studies revealed the protective effect of remote ischemic conditioning (RIC) on other vital organs, including the brain [7], kidneys [8], liver, lungs, gastrointestinal tract, skin, etc. [4].
Based on numerous clinical and experimental data, it can be concluded that RIC, like hypoxic or pharmacological conditioning, enhances the resistance of the brain and other organs to damaging effects not only of an ischemic nature, but causes a systemic reaction involving universal endogenous adaptive mechanisms and increases the adaptive potential of the organism as a whole. For example, in liver resections and transplants [9], heart and coronary artery surgery [10], kidney surgery and reconstructive microsurgery [11], remote intermittent ischemia reduces overall operational stress, diastolic blood pressure, incidence of strokes, inflammation, edema, neurological and cognitive impairment, reduces days of treatment in the intensive care unit and helps to increase cerebral blood flow and integrity of the blood-brain barrier.
2. Effects of RIC on the hippocampus
Since stroke is one of the main causes of neurological disability worldwide, and the main goal of its treatment is to save the penumbra by restoring blood flow and reducing nerve cell death, the neuroprotective properties of RIC are primarily evaluated by reducing the size of cerebral infarction. Studies in rodents show that RIC either before, during or after an ischemic episode, on average, can reduce infarct volume by 35-42% compared to controls, with its outcomes after short cycles of application being more pronounced in mice while in rats longer cycles were more effective [12,13]. While current approved therapies are based on restoration of blood flow and do not act directly on the brain parenchyma, RIC is effective not only against penumbra neuronal dysfunction, but has a positive effect on other regions and the brain as a whole, showing anti-apoptotic, antioxidant and anti-inflammatory effects (for review see [14]). For example, a 2-week post-RIC after experimental total brain hypoperfusion in a model of vascular cognitive impairment results in an improvement in cerebral blood flow and a decrease in neurodegenerative signs such as beta-amyloid accumulation, inflammation, and cell death in all areas of the animal brain [15]. It has been shown that limb pre-RIC before MCAO in rats reduces whole-brain mitochondrial and total oxidative stress, lipid peroxidation, oxidative DNA damage, and oxidative protein damage [16].
The hippocampus is considered to be more sensitive to ischemia than other brain regions. Thus, in animal models of transient global ischemia, which mimics the delayed neuronal death caused by cerebral ischemic stroke, a significant increase in neuronal apoptosis was observed in the CA1 and CA2 regions of the hippocampus [17]. Several studies have shown that remote ischemic limb conditioning effectively reduces neuronal apoptosis in this area of the hippocampus when applied shortly before an experimental stroke, as evidenced by DNA fragmentation and/or the formation of apoptotic bodies [18]. Very significant neuroprotective effect in the hippocampal region in rats was also observed with a single 20-minute limb RIC applied as a very delayed post-RIC 2 days after a global 10-minute ischemic stroke [19].
According to an interesting hypothesis, the hippocampus not only suffers from cerebral ischemia more than other regions but is also sensitive to any distant focal damage [20]. The development of post-stroke cognitive and psychopathological disorders is not directly related to the severity and localization of the primary brain lesion but is primarily due to the functional and structural changes in the hippocampus, a region of the brain that is selectively vulnerable to harmful external factors and responds to them with increased secretion of cytokines [21]. Moreover, ischemic and focal brain lesions induce excessive secretion of stress corticosteroid hormones that interact with hippocampal receptors, triggering signaling pathways leading to neuroinflammation and subsequent impairment of neurogenesis, neurodegeneration, depression, and dementia [22,23].
Based on the sensitivity of the hippocampus to distant injury, one can also assume its susceptibility to remote conditioning. Indeed, employment of remote ischemic postconditioning for the treatment of experimental traumatic brain injury in mice allowed to minimize neuronal changes in the CA1 area of the hippocampus and improve cognitive functions and motor coordination [24,25]. A significant improvement in sensorimotor functions with the use of delayed post-RIC has also been shown in a model of neonatal hypoxia-ischemia in rats [26]. In a classical experimental model of focal cerebral ischemia-reperfusion, pre-RIC also improved spatial learning and memory capacity, probably due to its protection of cholinergic neurons in the CA1 region of the hippocampus [27]. After application of post-RIC in this model, a reduction in infarct area was observed when RIC was applied up to 3 hours after stroke, but not when RIC was applied after 6 hours or 2 months, although the "hippocampal" behavioral outcome was improved in all cases [28]. In general, in the experiments with occlusion of the middle cerebral or carotid artery, hypothermic circulatory arrest, etc. RIC significantly improved neurological, functional, cognitive, and behavioral parameters, in which the central role belongs to the hippocampus. Normalization of the functions of the hippocampus can explain the effectiveness of RIC in rehabilitation therapy, in the prevention and treatment of neurological diseases of ischemic or inflammatory origin, in traumatic brain injuries, vascular cognitive impairment, dementia and Alzheimer's disease [13,29].
The effect of remote ischemia on the release of corticosteroids and the function of the hippocampus, as a regulator of various forms of behavioral plasticity, is in agreement with the anxiolytic and antidepressant effects of limb ischemia-reperfusion treatment recently identified in our laboratory [30,31,32]. RIC has been found to normalize behavioral and hormonal outcomes in models of depression and PTSD (post-traumatic stress disorder), effectively preventing development of the disorders when applied in pre- and post-injury regimens [30,31,32]. It should be noted that in the paradigms of severe hypobaric hypoxia, RIC neither reduces the risk of altitude sickness in subjects, nor prevents acute mountain sickness, cerebral edema or pulmonary edema, but facilitates rehabilitation [33]. In our experiments in rats, there was no increase in animal survival under the conditions of acute severe hypoxic exposure after RIC, but the rehabilitation period was significantly improved in the survived animals subjected to RIC. In particular, they did not display the symptoms of post-hypoxic pathology related to the malfunctioning of the hippocampus [unpublished data].
3. Neuro-immuno-humoral pathway
The transmission of the protective ischemia-reperfusion effect from the limb to the target organ involves simultaneously the nervous, circulatory, and immune systems, working in close interaction. The somatosensory system, spinal cord and autonomic nervous systems are involved in the neuronal pathway. The transmission there is carried out according to the type of the reflex - from sensory (afferent) neurons of the C-fibers to the integration center in the central nervous system and to the motor (efferent) neurons of the vagus nerve. This has been proven by experiments with nerve and spinal cord cutting [34,35,36], with application of nicotinic and opioid receptor antagonists [37,38], or blockers of the afferent fiber, autonomic ganglia and parasympathetic motor neurons [34,35,36,39], which canceled or reduced the protective effect of RIC, as well as by the experiments in which electrical or chemical neurostimulation partially reproduced it [35,36,40]. Moreover, the afferent nerve pathway can be activated by physiologically active substances in the blood, and vice versa, peripheral nerves can induce the synthesis of humoral factors. For example, activation of the vagus nerves, including indirect RIC, induces the synthesis and release of NO, nitrites, and glucagon-like peptide-1 (GLP-1) in visceral organs [41,42]. The non-selective TRPV1 cationic channels, expressed in the primary sensory nerves and regulated by physical or chemical stimuli, are activated during limb ischemia-reperfusion cycles and release neuropeptides, calcitonin-like peptide (CGRP), and substance P (SP) [43,44].
The release of biologically active substances into the bloodstream in response to RIC has also been demonstrated in various models. Today, among the main humoral factors for the realization of the protective effect of remote ischemia are listed adenosine, nitrite/NO, calcitonin gene-related peptide (CGRP), bradykinin, catecholamines (norepinephrine), microRNAs, endogenous opioids, free radicals, cytokines (IL-1α, IL-10) and chemokines (stromal factor 1α (SDF-1α)), GLP-1, apolipoprotein AI, hydrophobic peptides, prostanoids, endocannabinoids, leukotrienes, adrenomedullin, etc. [42,45,46]. It is assumed that some of them are transferred to the target organ by endogenous extracellular vesicular nanoparticles originating from the endothelium, hematopoietic cells or platelets [47]. Exosomes interact with target cells using a number of surface molecules. Because they carry chemokines and microRNAs, they can via HIF-1α reduce levels of IL-6 and TNFα, and via VEGF and NO synthases mediate angiogenesis, while via Hsp70 and Bim they inhibit apoptosis in target cells [48,49]. Many of the humoral factors (norepinephrine, biologically active peptides, NO, opioids, etc.) are able to bind to receptors in the nervous system, acting as neurotransmitters or modulators. One such example is interaction of GLP-1 with sensory fibers [50]. In plasma samples of patients exposed to RIC, an increase in the concentration of biogenic amines and amino acids, in particular glycine, which is an inhibitory neurotransmitter, was reported [51].
It has recently been shown that parasympathetic pathways stimulated by RIC provide organ protection not only by direct innervation, but also through immunomodulation, inhibiting cytokine production and integrin expression on neutrophils via the cholinergic anti-inflammatory pathway, but only in the presence of an intact and innervated spleen. RIC also reduces inflammation through the release of anti-inflammatory exosomes, affects circulating white blood cells, and immune precursors in the spleen [14,44,48,52].
4. Early phase of neuroprotection
For RIC, as for other types of conditioning effects, two phases of neuroprotection are characteristic. The early phase begins immediately after the conditioning stimulus and lasts for several hours, while the late phase occurs after 12-48 hours and persists for several days and even months (the "second window") [53,54]. The signals of alternating cycles of ischemia-reperfusion or occlusion-deocclusion enter the brain via the neuro-immuno-humoral pathway in the form of extracellular primary messengers (neurotransmitters, hormones, neuromodulators, cytokines and other RIC factors), initiating molecular events at the level of membrane receptors and ion channels [55]. Neuronal receptors, having received a signal, activate the calcium, phosphoinositide, and cyclic nucleotide systems of intracellular signal transduction in the form of corresponding secondary intracellular messengers (cAMP or cGMP, Ca2+, inositol triphosphate -PI3, NO, etc.), which activate or inhibit effector protein kinases and proteolytic enzymes, which, in turn, modulate functionally important proteins of ion channels, receptors, synaptic vesicles, cytoskeleton, mitochondria, etc. These processes form the basis of an early wave of adaptive responses [56,57,58].
The signal from the ischemic limb can stimulate G-protein-bound receptors on the surface of nerve cells, which is accompanied by activation of the protein kinases type C, type B (Akt), cytoplasmic tyrosine kinase (Trk) and MAP kinases. Kinases C and B are inhibited by phosphorylation of glycogen synthetase kinase-3β (GSK3β), which is one of the key mechanisms for keeping nonspecific mitochondrial pores in a closed state, preventing loss of proton gradient and separation of oxidation and phosphorylation, organelle damage, intracellular edema, calcium overload and apoptosis. The cardio- and neuroprotective effects of ischemic conditioning are also mediated via phosphorylation by serine and threonine kinases of ATP-dependent potassium channels of sarcolemma and mitochondrial membranes leading to their opening. This prevents the formation of mitochondrial pores and is associated with an increase in the generation of superoxide radicals in the respiratory chain [59,60]. Bradykinin, binding to B2 receptors (BDKRB2), participates in remote conditioning as an endogenous cytoprotective mediator, providing the triggering of the mitochondrial anti-apoptotic pathway through activation of PI3K/Akt/eNOS signaling and regulation of redox status through the release of NO [56,57]. Rapid RIC-mediated energy-saving effects are also due to the activation and then increase in the level of adenosine monophosphate-activated protein kinase (AMPK), a regulator of cellular energy homeostasis, which can be activated, for example, by a factor inhibiting macrophage migration (MIF), the level of which rises in plasma after ischemia [61,62]. In addition, the formation of neuronal tolerance after conditioning involves the sequential activation of the VEGF receptor, which has tyrosine kinase activity [63].
In addition to phosphorylation, nitrosylation of key mitochondrial proteins mediates the rapid cytoprotective effect of nitrite (humoral factor RIC) by reducing the generation of mitochondrial reactive oxygen species (ROS), which contributes to the preservation of the closed pore of mitochondria and inhibits the release of cytochrome C [64]. Acetylcholine provides a rapid anti-inflammatory effect through nicotinic receptors, by which it inhibits the release of TNFα from macrophages and inhibits the polymerization of F-actin, which is critical for the expression on the surface of circulating neutrophils of β2-integrin CD11b, which regulates their adhesion and migration [44].
Inhibition and suppression of expression, as a result of RIC, of the water channel protein aquaporin (AQP), located in the terminal legs of astrocytes, prevents an increase in transmembrane water flow, cytotoxic edema of astrocytes and an increase in the permeability of the BBB. There is also a general improvement in functional neurological recovery after stroke due to the suppression of AQP4 in astrocytes [65]. Another molecular target of the decongestant action of remote post-conditioning is intracellular matrix metallopeptidase 9 (MMP-9), which destroys the components of dense contacts between endothelial cells [66].
The "second window" of protection, as opposed to the early phase, depends on gene expression and protein synthesis, which are triggered by kinases through the induction of transcription factors to form the long-term stability of brain neurons [54,67]. Some proteins may be involved in both phases of neuroprotection, e.g. intracellular messenger and transcription factor STAT3 (signal transmitter and transcription activator 3). Activation of membrane receptors gp130, TNFR2, S1PR in the nervous system, by ligands such as IL-6 and 10, sphingosine-1-phosphate S1P, TNFα, lipoproteins, melatonin, erythropoietin, insulin and leptin lead to phosphorylation, mainly by JAK kinase, of the STAT3 factor. Activated by the conditioning signal, P-STAT3 is dimerized and moved to the nucleus, where it regulates the transcription of the genes of antioxidant, anti-apoptotic and proangiogenic protective proteins Bcl-xl, cytochrome C 2 oxidase subunit (COX2), cytokine signalling suppressor 3 (SOCS3), MCL-1, SOD2, VEGF, metallothionein, etc. As a result, it reduces neuronal inflammation and apoptosis. STAT3 is not only a transcription factor, but also non-genomically regulates the functions of mitochondria, endoplasmic reticulum and lysosomes. Phosphorylation and other modifications allow STAT3 to interact with GRIM19 (a component of mitochondrial complex I) and TOM20 (translocase of the outer membrane) to enter the mitochondria. There, STAT3 interacts with several proteins, promoting Ca2+ homeostasis, maintaining electron transport chain activity, increasing ATP levels, and reducing ROS production by inhibiting mitochondrial pore opening [68].
5. Delayed anti-apoptotic mechanisms
The mechanisms by which remote conditioning reduces cerebral infarction include, first of all, inhibition of cellular apoptosis, both internal, mediated by mitochondria, and external, triggered by death receptors [69,70].
The main intracellular molecular signaling pathways activated by RIC and mediating its neuroprotective effects are the Akt, mTOR, MAPK, PKC, and TLR4 pathways [71]. The PI3K/Akt pathway is an important signaling mediator that regulates cell survival by inhibiting the processes of apoptosis and growth [72]. The mTOR signaling pathway plays a central role in cellular metabolism, differentiation, development, autophagy, and survival [73]. The MAPK family is a major family of regulatory kinases that transform extracellular signals into cellular responses, participating in many physiological and pathological processes, and playing a controversial role in the mechanisms of RIC [74]. PKC is a family of serine/threonine protein kinases, of which δPKC and εPKC are closely related to cerebral damage. Remote ischemic pre- and post-conditioning due to ROS-mediated inhibition of the endogenous signaling cascade of δPKC activation causes the cleavage of δPKC, the activity of which usually contributes to cell death, and phosphorylation of εPKC, which promotes the survival of neurons. TLR4 is an important mediator of the innate immune response, mediates neuroinflammation and is involved in ischemic tolerance. RIC inhibits TLR4 mRNA and suppresses TLR4 overexpression, which can attenuate inflammatory cell infiltration, reduce heart attack volume, and improve neurobehavioral function [75].
The RIC stimulus inhibits the translocation of the poly-ADP-ribose polymerase (PARP) from the nucleus to the mitochondria, which prevents the release of mitochondrial apoptosis-inducing factor (AIF), an electron transport flavoprotein that itself plays an important role in the survival and death of neuronal cells, and also, in turn, activates the enzyme poly-ADP-ribose polymerase-1 (PARP-1), which is also responsible for various neurological disorders. In addition, distancing ischemia-reperfusion blocks the physical interaction of nuclear AIF with cyclophilin A and histone H2AX, which, in combination with inhibition of AIF / PARP pathways, prevents chromatin condensation and DNA fragmentation of neuronal cells [76].
RIC has been shown to attenuate cerebral damage in animals by suppressing the expression of TRAIL mRNA (an apoptosis-inducing ligand associated with tumor necrosis factor released by glia, damaged neurons, and white blood cells), thereby preventing TRAIL from binding to death receptors (DR) to form a death-inducing signaling complex (DISC) that triggers neuronal apoptosis through activation of caspase-8, which activates caspase-3, etc. [77]. Also, limb ischemia inhibits neuronal apoptosis by directly attenuating the activation of caspase-3 and TRAIL death receptors [78]. In addition, RIC enhances mRNA expression of the anti-apoptotic inhibitory protein cFLIP, which, as a close isoform of caspase-8, can instead physically bind to the catalytic site of the FAS-associated death domain (FADD), thereby preventing the formation of another DISC and suppressing the transmission of apoptotic signals [77].
Cycles of limb RIC promote neuronal survival by leveling the ischemia-reperfusion-induced increase in levels of lipocalin Lcn-2, a protein that regulates inflammation, iron metabolism and cell death, reducing its content in the brain and the number of Lcn-2-positive astrocytes [79]. This prevents the interaction of Lcn-2 with its receptor (carrier of organic cations) on the membrane of the neuron, which reduces the neuronal expression of the pro-apoptotic Bcl-2 family, the proapoptotic gene Bim, an inducer of post-ischemic neuronal death [80]. Also, when using ischemic conditioning, a decrease in the expression of pro-apoptotic proteins Bax, Bid and caspase-3, and an increase in anti-apoptotic Bcl-2 and Bcl-xl were demonstrated, which inhibits the opening of the mitochondrial pore and effectively reduces apoptosis.
Brain-derived neurotrophic factor BDNF is another likely mediator of RIC-induced neuroprotection. BDNF binds to TrkB receptors, mobilizing TrkB kinase to activate the MAPK/ERK and PI3K/Akt signaling cascades that promote neuronal differentiation and survival. BDNF induced by ROS activates HIF-1α, Nrf2 and their gene programs, and Nrf2, in turn, can initiate BDNF expression [81]. The PI3K/Akt signaling pathway was also shown to regulate expression of murine E3-ubiquitin ligase MDM2 which when phosphorylated, stabilizes in the nucleus and interacts with p53, which leads to destabilization of p53, thus preventing neuronal apoptosis [82].
The prevention of neuronal damage is also played by the mechanism of microautophagy, which allows removal of dysfunctional cellular components, especially of damaged mitochondria, preventing the release of cytochrome c and the transmission of death signals. Distancing ischemia affects the expression of checkpoint proteins of cellular autophagy/apoptosis, for example, reduces plasma levels of HMGB1, a protein secreted by immune cells as a cytokine mediator of inflammation, but can, on the contrary, stimulate cytosolic HMGB1, which regulates apoptosis, protecting the autophagy proteins beclin-1 and ATG5 from calpain-mediated cleavage [83,84]. Initiation of the opioid receptor/PI3K/AKT/GSK3β signaling pathway under RIC can also lead to phosphorylation of Bcl-2 and breakdown of the Bcl-2/Beclin-1 complex, which plays an important role in stimulating autophagy and reducing mitochondrial damage in conditioned rats after cerebral ischemia [85]. Also, when using RIC, there is a predominance of the conjugated form of phosphatidylethanolamine of the microtubule-bound protein LC3-II, which is recruited to the membranes of the autophagosome, via its cytosolic form LC3-I, leading to a decrease in sequestosome 1 levels and suppression of mTOR kinase, an increase in beclin-1 and hemoxygenase-1 (HO1), which mediates pro-autophagic signaling and prevention of cell death [85,86,87].
6. Antioxidant and anti-inflammatory mechanisms
One of the main, closely interrelated mechanisms for preventing neurological dysfunction after focal cerebral injuries by means of remote ischemic exposure is the reduction of oxidative stress in neurons which can be detected at the level of malondialdehyde and 8-hydroxy-2-deoxyguanosine, as well as of neuroinflammation detected at the levels of myeloperoxidase, TNFα, IL-1 and IL-6 [88,89,90].
Postischemic neuroinflammation is known to cause an imbalance between oxidative stress and antioxidant systems, and excessive accumulation of free radicals leads to oxidative damage to proteins, lipids and nucleic acids, i.e. oxidative stress contributes to the progression of ischemic neuronal insufficiency after cerebral damage. RIC prevents these disorders primarily by activating antioxidant systems by triggering the expression of proteins such as Nrf2 and HO1, quinone oxidoreductase 1 (NQO1), and superoxide dismutase (SOD). The use of RIC contributes to the movement of the transcription factor Nrf2 from the cytosol to the nucleus, where it binds to the DNA promoter antioxidant response element ARE, initiating the transcription of antioxidant cytoprotective proteins and enzymes – the key components of the antioxidant systems of glutathione and thioredoxine, and enzymes for the regeneration of NADP, ROS [91]. Those, in turn, mediate a significant decrease in the level of nitrotyrosine, P22 (catalytic subunit of NADPH oxidase) mRNA, xanthine oxidase, which play a fundamental role in maintaining redox homeostasis [92]. Secondly, RIC is able to reduce the production of superoxide by reversing the activity of endothelial NO synthase (eNOS), which, in the presence of cofactors of calmodulin, tetrahydrobiopterin (BH 4) produces mainly NO, and with their lack or decrease in the affinity, the "uncoupled” eNOS produces mainly superoxide [93]. Regulation of eNOS is also important for hemodynamics, as NO is a potent vasodilator and improves microcirculation and counteracts reperfusion damage by reducing ROS production. Moderate NO formation during RICs induces cerebroprotective adaptations. Moreover, through the PI3K/Akt signaling pathway in astrocytes it activates the synthesis of the glutamate-1 transporter (GLT-1), which removes glutamate from the synaptic cleft and prevents excitotoxicity [76,94].
Another important mechanism by which RIC can limit free radical oxidation is, clearly demonstrated in the liver where RIC in pre- and post-conditioning modes significantly reduces damage-induced levels of ALT, AST, TNFα, IL-6 and hepatic MDA in serum, as well as the formation of nitrotyrosine, and stimulates the activity of hepatic SOD, glutathione and glutathione peroxidase and the expression of HO1 [89]. Antioxidant mechanisms include activation by remote ischemia of G protein-coupled A1 adenosine receptors, which provides neuroprotection by regulating inflammation by lowering serum TNFα and NO levels and modulating oxidative stress by maintaining antioxidant levels, including SOD, NO and glutathione formation, and increasing ATP levels [95].
In experiments with ischemia or administration of lipopolysaccharides, it has been shown that RIC affects the key steps leading to systemic inflammation, in particular, suppresses the activation of nuclear factor NF-κB, significantly reduces the levels of IL-1, IL-6, TNF-α and IFN-γ both in plasma and in the brain, suppresses expression of ICAM-1 and VCAM-1 adhesion genes, while modulating the expression of hypoxia-induced factor HIF-1α and markedly increasing levels of HO1, which leads to the strengthening of the blood-brain barrier and a decrease in the infiltration of pro-inflammatory immune cells [96]. Inhibition of NF-κB-dependent pro-inflammatory pathways is considered the key event in preventing neuroinflammatory damage. In particular, blocking the release of cytokines can be carried out by reducing the NF-κB-mediated production of NLRP3 inflammasome. This can be achieved by a characteristic for RIC decrease in plasma of alarmin HMGB1, which triggers the secretion of pro-inflammatory cytokines through the IκB or ERK activation pathways induced by binding of NF-κB to TLR4 or RAGE receptors [48]. On the other hand, the association of TNFα or HMGB1 with TLR4 and the subsequent activation of the JAK/STAT transcription factors leads to activation of NF-κB, which affects the mitochondrial pore and synthesis of cytosolic HMGB1, i.e. promotes cell survival. RIC also mediates the decrease of cytokines by suppressing the myeloperoxidase (MPO) pathway, which is associated with an increased influx of neutrophils in the area of inflammation and, therefore, promotes the release of pro-inflammatory cytokines from them [16,44,83].
Ischemia-induced factor Nrf2 and AMPK kinase may promote the acquisition by brain microglial immune cells of the anti-inflammatory M2 phenotype, as opposed to pro-inflammatory M1 phenotype [97]. Damaged neurons release ATP and UTP, which act on purine receptors, causing microglia to differentiate towards the M1 phenotype. M1 microglia promote inflammation and disrupt axonal regrowth by releasing pro-inflammatory cytokines and NO, but they also clear cellular debris. In contrast, M2 microglia produce anti-inflammatory cytokines, VEGF, BDNF, platelet-derived growth factor (PDGF) and progranulin, which together suppress inflammation and promote axonal proliferation, angiogenesis, oligodendrogenesis, and remyelination. Preventing this phenotypic shift from M2 to M1 may contribute to recovery from stroke [98].
Suppression of inflammatory reactions by intermittent ischemia is also mediated by chemokines, in particular by a decrease in the content of monocyte chemoattractant protein-1 (MCP-1) and the intensity of selective recruitment of monocytes mediated by it [52,57]. In a model of spinal cord injury in rodents, the administration of a RIC humoral factor, SDF-1α, reduced the production of inflammasomal IL-1, IL-18, TNFα and NLRP3, suggesting that it also has anti-inflammatory effects [99]. Endocannabinoids also mediate the protective effect of ischemic conditioning through their CB1/ CB2 receptors, the activation of which causes a decrease in the formation of ROS, chemotaxis and activation of inflammatory cells, as well as a decrease in internal body temperature, increased neuroprotective signaling and an increase in coronary and cerebral dilatation, which prevents post-stroke motility disorders [100]. For the role of other potential RIC mediators, it is worth considering prostaglandins which, among various functions, have toxic properties in acute and chronic neurological conditions. It was reported that inhibition of the G-protein-bound prostaglandin F receptor of PGF2α in the CNS via attenuating intracellular calcium levels, improves neurobehavioral function and reduces infarct volume in mice after ischemia [101].
In models of cerebral ischemia, RIC causes an increase in the volume of the spleen, the percentage of cytotoxic T cells in it, an increase in the number of circulating B lymphocytes, the number of colonies of non-inflammatory monocytes (CD43 + / CD172a +), and at the same time a decrease in the content of cytotoxic T cells and natural killer cells in the brain. It is assumed that in the splenic axis of protection, an important role can be played by the anti-inflammatory cytokine IL-10, which regulates the amplitude of the cytokine response, and the levels of which are increased markedly by RIC [52].
6. Role of HIF-1α and steroid hormones
Preclinical studies have proven that activation of the HIF-1α pathway, a transcription factor that plays an important role in response to hypoxia and regulation of inflammation, energy metabolism, neurogenesis and apoptosis, plays a crucial role for the neuroprotective effects of RIC [102]. The use of a remote ischemic stimulus was shown to significantly increase HIF-1α mRNA and protein levels, leading to reduced cerebral damage, whereas administration of HIF-1α antagonists eliminated the neuroprotective effect of RIC [16]. Inactivation of the HIF-1α subunit expression leads to increased brain damage and decreased survival after ischemia and to a more pronounced learning disorder and decreased neurogenesis in the post-ischemic period [103].
In addition to hypoxia, HIF-1α can be induced by NF-κB, growth factors (IGF-1, PDGF), cytokines (TNFα and IL-1), and ROS. HIF-1α controls expression of more than 700 different target genes that mediate both adaptive and pathological processes. In neurons and astrocytes, it controls production of the protective cytokine erythropoietin which regulates apoptosis and autophagy, synaptic processes and neurogenesis, as well as the inflammatory response, by reducing the expression of cyclo-oxygenase 2 and iNOS and suppressing microglial activation [104,105].
HIF-1α stimulates the transcription of the vascular growth factor VEGF, which reduces the content of active caspase-3 in the hippocampus, modulates the expression of genes that are involved in glucose metabolism, for example, glucose transporter-1 (GLUT1) and lactate dehydrogenase A (LDH-A) [106]. HIF-1α at the transcriptional level induces the expression of hypoxia-sensitive microRNAs that regulate the stability and translation of mRNA by binding to a 3' non-coding region, resulting in degradation of the target mRNA and lower levels of the corresponding proteins [107]. HIF-1α subunits are involved in the regulation of anti-apoptotic factors Bcl-2 and Mcl-1, the induction of Bcl-xL and the suppression of pro-apoptotic factors Bid, Bax and Bak [108]. HIF-1α is also associated with the regulation of mitochondrial functions by influencing hexokinase, including the control of hexokinase II expression, which catalyzes the first stage of glycolysis and can suppress apoptosis by binding to a potential-dependent anionic channel on the mitochondrial membrane [109]. Today, the use of hypoxic conditioning that leads to activation of HIF-1α-induced cytoprotective mechanisms for reduction of the severity of the disease in with COVID-19 patients is actively discussed [110]. However, the role of HIF in stroke remains controversial because it is highly associated with the duration and severity of ischemia and as such might activate both protective and pathogenic mechanisms which requires different therapeutic strategies [104,105]. For example, HIF-1 during the early acute phase of the hypoxic response triggers a cascade of cerebral events associated with the suppression of the pentose phosphate pathway [111]. HIF-1α can also trigger p53-induced apoptosis through direct protein interaction of the oxygen-dependent degradation domain of HIF-1α with p53, stabilizing the latter, and through interaction with the modulator of function p53 Mdm2 [112,113]. The effect of HIF-1 on the induction of apoptosis also depends on the severity of hypoxia. Under moderate hypoxia or brief ischemia, HIF-1 has a predominantly protective effect by inducing the expression of anti-apoptotic proteins. During the periods of low oxygen pressure, HIF-1α mediates a shift in mitochondrial metabolism toward anaerobic glycolysis, which induces the production of pyruvate dehydrogenase 1 (PDK1) kinase and restricts the entry of acetyl-CoA into the tricarboxylic acid cycle. However, in differentiated macrophages, this metabolic change leads to increased synthesis of cytokines, such as IL-1β and IL-18, via the NF-κB pathway [102,105,113]. HIF-1α also can modulate the activity of NF-κB participating in the regulation of the PI3K/Akt pathway. It is possible that repeated stimulation of HIF-1α causes a separation of cytokine synthesis and immune tolerance; as with other TLR4-dependent pathways [96,104,114].
HIF-1α is an essential component of the pathways controlling cellular metabolism and plays an important role in regulating the effector functions of immune cells. In addition, HIF-1α is crucial for the maturation of dendritic cells and for the activation of T cells. HIF-1α is induced in LPS-activated macrophages, where it is crucially involved in glycolysis and induction of pro-inflammatory genes, especially IL-1β [115]. The mechanism of LPS-stimulated induction of HIF-1α involves succinate, which inhibits prolyl hydroxylase and prevents degradation of HIF-1α. Moreover, activated pyruvate kinase M2 interacts with HIF-1α and promotes its function. In another critical type of inflammatory cell, Th17 cells, HIF-1α acts through the retinoic acid-bound orphan receptor-γt to control their differentiation. Thus, HIF-1α acts as a key reprogrammer of inflammatory cell metabolism that activates expression of inflammatory genes [116].
In addition to the direct modulation of inflammation, HIF-1α is an important regulator of steroid synthesis. Its expression in adrenal cells dramatically affects the synthesis of hormones with systemic consequences [117]. HIF-1α deficiency causes an increase in the levels of enzymes responsible for steroidogenesis and a corresponding increase in circulating steroids, which leads to changes in cytokine levels and in the profile of circulating mature hematopoietic cells. Overexpression of HIF-1α mediates the insufficiency of steroid production due to impaired transcription of essential enzymes. Such abrupt or sustained changes affect many organ systems, and in particular sensitive areas of the brain.
The study of the effects of RIC on the regulation of the hypothalamic-pituitary-adrenocortical axis (HPA) which is the main hormonal stress-response system of the organism, is of undoubted theoretical and practical interest, but is currently insufficiently studied. Despite the insufficiency of the experimental data, it is logical to assume the participation of glucocorticoid hormones in the mechanisms of remote conditioning for several reasons. First, there is a significant cross-interaction between the effects of hypoxia/ischemia and glucocorticoids (GCs) on homeostasis and the regulation of cellular responses to oxygen deficiency, stress, and inflammation [118,119]. Glucocorticoid receptors (GRs) and HIFs, whose participation in the effects of RIC has been unequivocally proven, can be colocalized in the same compartments of the nucleus, and probably interact directly. For example, in juvenile zebrafish, HIF-1α suppresses the GR response to exogenous glucocorticoids, reduces cortisol levels by inhibiting proopiomelanocortin (POMC) expression and blocking intracellular GR transcriptional activity. GCs, by contrast, stabilize HIF through pVHL degradation [120]. The functional role of the HIF-1α factor in the regulation of GR mRNA and protein expression, and the associated GC activity, has been demonstrated, and, conversely, HIF-dependent gene expression is enhanced by ligand-dependent activation of GRs (for review see [121]. HIF and GCs exert a direct and cell-type-specific effect on each other, enhancing or suppressing the transcription of the N3RC1 and HIF genes, respectively, since the promoter of the N3RC1 gene contains the HRE region and the promoter of the HIF-1α GRE element [122]. HIF and GRs can compete for promoters of effector proteins, for example, in the pulmonary epithelium where the decrease in the anti-inflammatory effect of GCs in hypoxia can be explained by the binding of HIF-1α to HRE present in the promoter of histone deacetylase 2 (HDAC2), which is usually recruited by activated GRs to suppress NF-κB-mediated transcription of pro-inflammatory proteins [123]. These factors can act synergistically. For example, hypoxia increases the expression of the GC-inducible protein GILZ (glucocorticoid-induced leucine zipper) common in the cells of the immune system, which can suppress the activation of macrophages, NF-κB-dependent production of pro-inflammatory cytokines and inflammatory mediators. In addition, HIF-1α is able to physically interact with GILZ, which explains the suppression of hypoxia-induced expression of COX-2 by a synthetic GC, dexamethasone [124]. Administration of dexamethasone weakens the activity of HIF-1α in hypoxia by reducing its binding to DNA and HRE activity due to the difficulty of nuclear translocation of HIF-1α. Hypoxia, in turn, can cause a decrease in the levels of GR mRNA and protein and inhibit nuclear translocation of GR, that weakens the anti-inflammatory effect of dexamethasone [118,119,125].
An argument in favor of involving HPA in the effects of RIC can be the fact that GC has a certain neuroprotective effect, weakening the inflammatory response in ischemic damage due to a significant decrease in the production of TNFα, inhibition of cleaved caspase-3, activation of phosphorylated Akt and effect on the VEGF pathway. Dexamethasone and prednisolone are routinely used to treat asthma and ischemic lesion as well neonatal hypoxia, to prevent the symptoms of altitude sickness. In these cases, they mainly act as anti-inflammatory drugs, but also reduce the permeability and vasoconstriction, improving blood oxygenation and redox balance [118]. In our recent experiments on pharmacological preconditioning with dexamethasone, a significant hypoxia-protective effect of GCs was also demonstrated [126]. We have shown a significant effect of hypoxia, especially after conditioning by moderate hypoxia, on the function of HPA and on the number of GR in the brain of experimental animals [32,127].
On the other hand, neuroprotective or neurotoxic outcomes are determined by the dosage and duration of exposure to steroid hormones. For example, high levels of GCs cause the accumulation of glutamate in the brain, thereby mediating cytotoxicity and neuronal damage, enhancing apoptotic cell death by overexpressing cyclin-dependent kinase 5 (CDK5) [128]. Chronic hypercortisolemia or its recurrent excessive release cause nerve cell damage and dendrite atrophy, reduce neurogenesis in the hippocampus, and disrupt synaptic plasticity by affecting gene transcription, cell signaling, modulation of synapse structure, transmission, and glia function [20]. Mechanisms of the neuronal damaging action of critical doses of GCs include regulation of NMDA and AMPA receptors; decreased activity of glucose transporters; regulation of BDNF synthesis and its interaction with the glutamate system; increased currents through the Ca2+ channels; activation of the MAPK/Elk-1 pathway with phosphorylation and acetylation of histone H3 and induction of c-Fos and Egr-1; activation of microglia with the release of cytokines and alarmin HMGB1 with subsequent signal transmission via TLR2/TLR4 and NLRP3 priming; induction of pro-inflammatory hippocampal genes; stimulation of the amyloidogenic pathway; hypoactivity of GH; violation of the redox balance [23].
According to current views [20,23], excess of corticosteroids after focal brain damage, especially in patients with dysregulation of HPA and an abnormal stress response, via interaction with GRs of the hippocampus causes molecular, functional and structural changes, leading to cognitive and mental disorders. Limb ischemia/reperfusion, being a mild stressor, and, having some mechanisms common to conditioning effects, can attenuate the level of steroid hormones and the function of HPA. In our experiments with modeling depression and PTSD in rats, RIC normalized the basal level of GCs in the blood of animals and modulated the HPA, preventing its dysregulation by a feedback mechanism [30,32]. With a high probability, RIC is also able to positively affect the functioning of HPA under conditions of hypoxia, ischemia and brain damage, preventing excessive secretion of corticosteroid hormones, overactivation of GCs in the hippocampus and its "remote" damage, which undoubtedly deserves further investigation. Particularly interesting results are expected when RIC is applied for the prevention and correction of cognitive and depressive disorders after stroke or ischemic attacks against the background of previous dysregulation of HPA.
Further studies of the mechanisms of implementation of the adaptive effects of remote ischemic conditioning on the brain and other tissues will expand the range of clinical applications of this technique.
7. Conclusion
Rapidly accumulating evidence clearly demonstrates that remote ischemic conditioning is a powerful tool for preventing brain damage after stroke or traumas of various nature as well as against progression of chronic pathologies. Although there is still scope for further research in this area of neurology it is certain that RIC has positive effects on neural repair via activation of neurogenesis, regeneration of axons and dendritic networks, synaptogenesis, myelination, angiogenesis and BBB functions. Development of further remote ischemic pre- and postconditioning protocols will give clinicians new instruments for treating various neurological disorders.
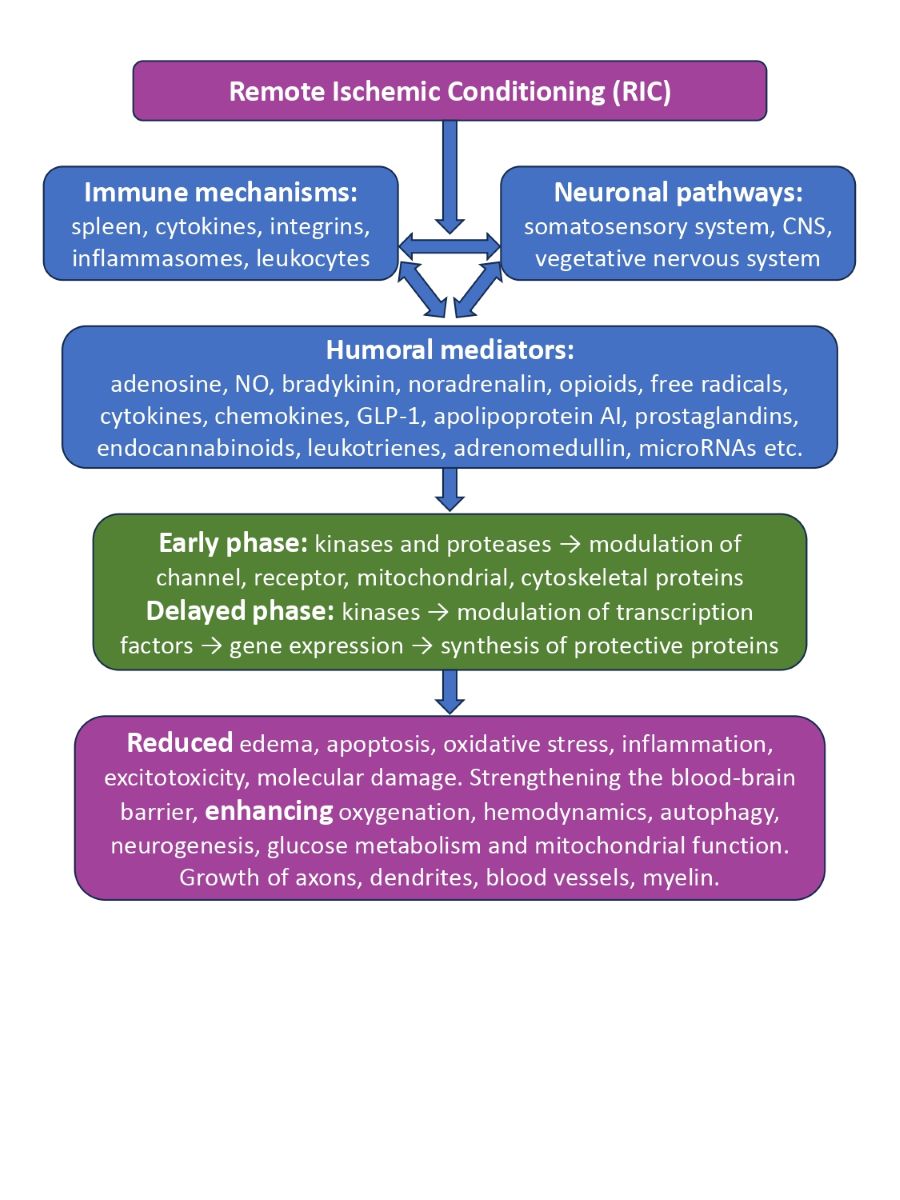
Figure 1.
General scheme for triggering and implementing the neuroprotective effects of remote ischemic conditioning.
Figure 1.
General scheme for triggering and implementing the neuroprotective effects of remote ischemic conditioning.
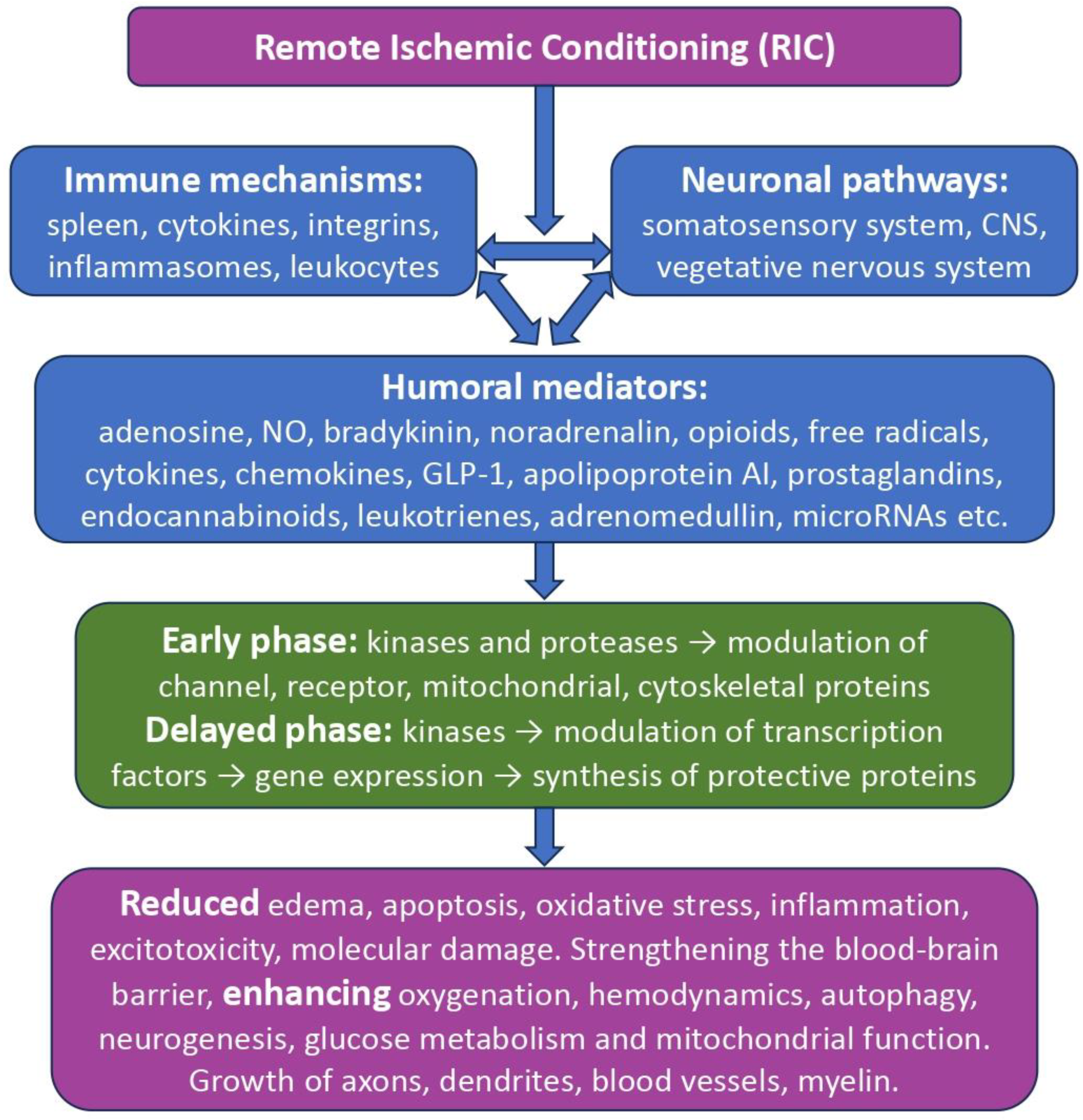
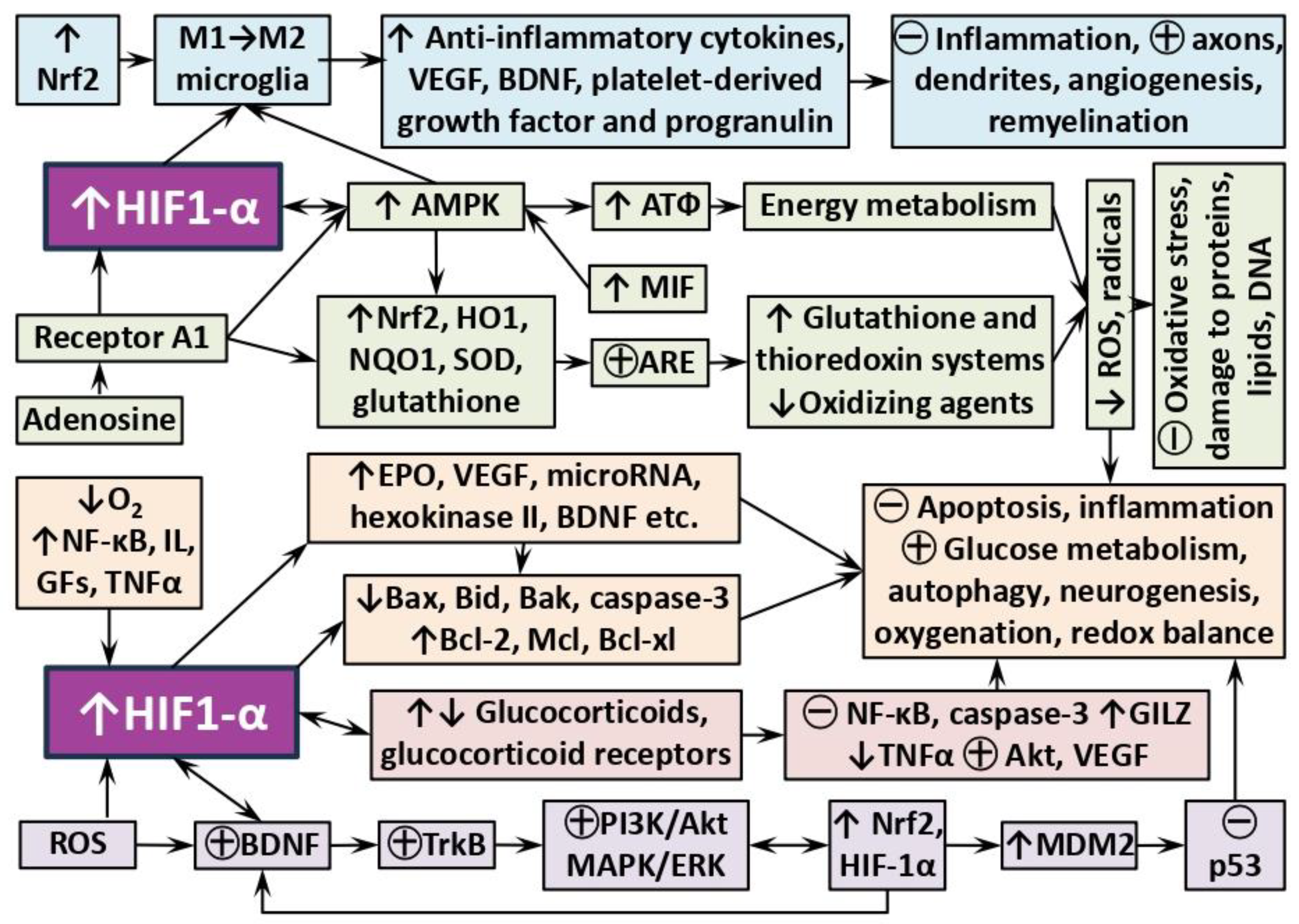
Figure 2.
Some examples of HIF-dependent neuroadaptive mechanisms of remote ischemic conditioning. ↑ – increase, synthesis; ↓ – decrease; ㊉ − activation, strengthening; ㊀ – inhibition, weakening. Explanations and abbreviations in the text.
Figure 2.
Some examples of HIF-dependent neuroadaptive mechanisms of remote ischemic conditioning. ↑ – increase, synthesis; ↓ – decrease; ㊉ − activation, strengthening; ㊀ – inhibition, weakening. Explanations and abbreviations in the text.
Author Contributions
All authors participated in writing and editing the manuscript and agree with its content.
Funding
This study was supported by the State Program GP-47 "Scientific and Technological Development of the Russian Federation" (2019-2030).
Conflicts of Interest
Authors report no conflict of interest.
References
- Murry, C.E.; Jennings, R.B.; Reimer, K. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Przyklenk, K.; Bauer, B.; Ovize, M.; Kloner, R.A.; Whittaker, P. Regional ischemic 'preconditioning' protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1983, 87, 893–899. [Google Scholar] [CrossRef]
- Malhotra, S.; Naggar, I.; Stewart, M.; Rosenbaum, D.M. Neurogenic pathway mediated remote preconditioning protects the brain from transient focal ischemic injury. Brain Res. 2011, 1386, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Candilio, L.; Malik, A.; Hausenloy, D.J. Protection of organs other than the heart by remote ischemic conditioning, J. Cardiovasc. Med. (Hagerstown) 2013, 14, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, R.K.; Mortensen, U.M.; White, P.A.; Kristiansen, S.B.; Schmidt, M.R.; Hoschtitzky, J.A.; Vogel, M.; Sorensen, K.; Redington, A.N.; MacAllister, R. Transient limb ischemia induces remote ischemic preconditioning in vivo. Circulation 2002, 106, 2881–2883. [Google Scholar] [CrossRef]
- Andreka, G.; Vertesaljai, M.; Szantho, G.; Font, G.; Piroth, Z.; Fontos, G.; Juhasz, E.D.; Szekely, L.; Szelid, Z.; Turner, M.S.; Ashrafian, H.; Frenneaux, M.P.; Andreka, P. Remote ischaemic postconditioning protects the heart during acute myocardial infarction in pigs. Heart 2007, 93, 749–752. [Google Scholar] [CrossRef]
- Dave, K.R.; Saul, I.; Prado, R.; Busto, R.; Perez-Pinzon, M.A. Remote organ ischemic preconditioning protect brain from ischemic damage following asphyxial cardiac arrest. Neurosci. Let. 2006, 404, 170–175. [Google Scholar] [CrossRef]
- Kadkhodaee, M.; Seifi, B.; Najafi, A.; Sedaghat, Z. First report of the protective effects of remote per- and postconditioning on ischemia/reperfusion-induced renal injury. Transplantation 2011, 92, e55. [Google Scholar] [CrossRef]
- Stankiewicz, R.; Grąt, M. Direct, remote and combined ischemic conditioning in liver surgery. World J. Hepatol. 2021, 13, 533–542. [Google Scholar] [CrossRef]
- Cho, Y.J.; Kim, W.H. Perioperative Cardioprotection by Remote Ischemic Conditioning. Int. J. Mol. Sci. 2019, 20, 4839. [Google Scholar] [CrossRef]
- Sogorski, A.; Harati, K.; Kapalschinski, N.; Daigeler, A.; Hirsch, T.; Lehnhardt, M.; Goertz, O.; Kolbenschlag, J. Remote Ischemic Conditioning - Endogenous Tissue Protection and its Possible Applications in Surgery. 2018. Zentralbl. Chir. 2018, 143, 42–49, (article in German). [Google Scholar] [CrossRef]
- Torres-Querol, C.; Quintana-Luque, M.; Arque, G.; Purroy, F. Preclinical evidence of remote ischemic conditioning in ischemic stroke, a metanalysis update. Sci. Rep. 2021, 11, 23706. [Google Scholar] [CrossRef]
- Mollet, I.; Marto, J.P.; Mendonça, M.; Baptista, M.V.; Vieira, H.L.A. Remote but not Distant: a Review on Experimental Models and Clinical Trials in Remote Ischemic Conditioning as Potential Therapy in Ischemic Stroke. Mol. Neurobiol. 2022, 59, 294–325. [Google Scholar] [CrossRef]
- Mollet, I.; Martins, C.; Ângelo-Dias, M.; Carvalho, A.S.; Aloria, K.; Matthiesen, R.; Viana-Baptista, M.; Borrego, L.M.; Vieira, H.L.A. Pilot study in human healthy volunteers on the mechanisms underlying remote ischemic conditioning (RIC) - Targeting circulating immune cells and immune-related proteins. J. Neuroimmunol. 2022, 367, 577847. [Google Scholar] [CrossRef]
- Khan, M.B.; Hoda, M.N.; Vaibhav, K.; Giri, S.; Wang, P.; Waller, J.L.; Ergul, A.; Dhandapani, K.M.; Fagan, S.C.; Hess, D.C. Remote ischemic postconditioning: harnessing endogenous protection in a murine model of vascular cognitive impairment. Transl. Stroke Res. 2015, 6, 69–77. [Google Scholar] [CrossRef]
- Xia, M.; Ding, Q.; Zhang, Z.; Feng, Q. Remote Limb Ischemic Preconditioning Protects Rats Against Cerebral Ischemia via HIF-1α/AMPK/HSP70 Pathway. Cell. Mol. Neurobiol. 2017, 37, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.; Wieloch, T.; Smith, M.L. Brain damage in a mouse model of global cerebral ischemia. Effect of NMDA receptor blockade. Brain Res. 2003, 982, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.G.; Li, W.B.; Li, Q.J.; Chen, X.L.; Liu, H.Q.; Feng, R.F.; Ai, J. Limb ischemic preconditioning attenuates apoptosis of pyramidal neurons in the CA1 hippocampus induced by cerebral ischemia-reperfusion in rats. Sheng Li Xue Bao [Acta physiologica Sinica] 2004, 55, 407–412. [Google Scholar]
- Burda, R.; Danielisova, V.; Gottlieb, M.; Nemethova, M.; Bonova, P.; Matiasova, M.; Morochovic, R.; Burda, J. (2014). Delayed remote ischemic postconditioning protects against transient cerebral ischemia/reperfusion as well as kainate-induced injury in rats. Acta Histochem. 2014, 116, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, N.V. Biochemical Mechanisms and Translational Relevance of Hippocampal Vulnerability to Distant Focal Brain Injury: The Price of Stress Response. Biochemistry (Moscow) 2019, 84, 1306–1328. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, M.V.; Moiseeva, Y.V.; Zhanina, M.Y.; Lazareva, N.A.; Gulyaeva, N.V. A Comparative Study of Koizumi and Longa Methods of Intraluminal Filament Middle Cerebral Artery Occlusion in Rats: Early Corticosterone and Inflammatory Response in the Hippocampus and Frontal Cortex. Int. J. Mol. Sci. 2021, 22, 13544. [Google Scholar] [CrossRef] [PubMed]
- Shishkina, G.T.; Kalinina, T.S.; Gulyaeva, N.V.; Lanshakov, D.A.; Dygalo, N.N. Changes in Gene Expression and Neuroinflammation in the Hippocampus after Focal Brain Ischemia: Involvement in the Long-Term Cognitive and Mental Disorders. Biochemistry (Moscow) 2021, 86, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, N.V.; Onufriev, M.V.; Moiseeva, Y.V. Ischemic Stroke, Glucocorticoids, and Remote Hippocampal Damage: A Translational Outlook and Implications for Modeling. Front. Neurosci. 2021, 15, 781964. [Google Scholar] [CrossRef] [PubMed]
- Pandit, V.; Khan, M.; Zakaria, E.R.; Largent-Milnes, T.M.; Hamidi, M.; O'Keeffe, T.; Vanderah, T.W.; Joseph, B. Continuous remote ischemic conditioning attenuates cognitive and motor deficits from moderate traumatic brain injury. J. Trauma Acute Care Surg. 2018, 85, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Sandweiss, A.J.; Azim, A.; Ibraheem, K.; Largent-Milnes, T.M.; Rhee, P.; Vanderah, T.W.; Joseph, B. Remote ischemic conditioning preserves cognition and motor coordination in a mouse model of traumatic brain injury. J. Trauma Acute Care Surg. 2017, 83, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Drunalini Perera, P.N.; Hu, Q.; Tang, J.; Li, L.; Barnhart, M.; Doycheva, D.M.; Zhang, J.H.; Tang, J. Delayed remote ischemic postconditioning improves long term sensory motor deficits in a neonatal hypoxic ischemic rat model. PloS One 2014, 9, e90258. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Lu, Y.; Zhang, Y.; Li, Y.; Jiang, L. Remote ischemic preconditioning improves spatial learning and memory ability after focal cerebral ischemia-reperfusion in rats. Perfusion 2013, 28, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Yan, Z.; Wei, D.; Gao, X.; Chen, X.; Zhao, H. Limb remote ischemic postconditioning protects against focal ischemia in rats. Brain Res. 2009, 1288, 88–94. [Google Scholar] [CrossRef]
- Vasdekis, S.N.; Athanasiadis, D.; Lazaris, A.; Martikos, G.; Katsanos, A.H.; Tsivgoulis, G.; Machairas, A.; Liakakos, T. The role of remote ischemic preconditioning in the treatment of atherosclerotic diseases. Brain Behav. 2009, 3, 606–616. [Google Scholar] [CrossRef]
- Baranova, K.A.; Zenko, M.Y. Applying remote ischemic pre- and postconditioning for the correction of experimental depression. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova, 2019, 119, 72–80. [Google Scholar] [CrossRef]
- Baranova, K.A.; Zenko, M.Y. Anxiolytic effect of remote ischemic pre- and post-conditioning in the model of post-traumatic stress disorder. Zh. Vyssh. Nervn. Deyat. Im I.P. Pavlova. 2018, 68, 663–672, (article in Russian). [Google Scholar] [CrossRef]
- Baranova, K.A.; Pivina, S.G.; Rybnikova, E.A. The Anxiolytic Effects of Moderate Hypoxia and Remote Ischemia in the Posttraumatic Stress Disorder Model Are Accompanied by Modification of Functioning of the Hypothalamic–Pituitary–Adrenal Axis. Neurochem. J. 2018, 12, 130–134. [Google Scholar] [CrossRef]
- Molano Franco, D.; Nieto Estrada, V.H.; Gonzalez Garay, A.G.; Martí-Carvajal, A.J.; Arevalo-Rodriguez, I. Interventions for preventing high altitude illness: Part 3. Miscellaneous and non-pharmacological interventions. Cochrane Database Syst. Rev. 2019, 4, CD013315. [Google Scholar] [CrossRef] [PubMed]
- Steensrud, T.; Li, J.; Dai, X.; Manlhiot, C.; Kharbanda, R.K.; Tropak, M.; Redington, A. Pretreatment with the nitric oxide donor SNAP or nerve transection blocks humoral preconditioning by remote limb ischemia or intra-arterial adenosine. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1598–H1603. [Google Scholar] [CrossRef]
- Basalay, M.; Barsukevich, V.; Mastitskaya, S.; Mrochek, A.; Pernow, J.; Sjöquist, P.-O.; Ackland, G.L.; Gourine, A.V.; Gourine, A. Remote ischaemic pre- and delayed postconditioning—similar degree of cardioprotection but distinct mechanisms. Exp. Physiol. 2012, 97, 908–917. [Google Scholar] [CrossRef]
- Donato, M.; Buchholz, B.; Rodríguez, M.; Pérez, V.; Inserte, J.; García-Dorado, D.; Gelpi, R.J. Role of the parasympathetic nervous system in cardioprotection by remote hind limb ischaemic preconditioning. Exp. Physiol. 2013, 98, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.T.; Lu, Y.; Mei, B.; Xia, Z.; Irwin, M.G. Cardioprotection from remote preconditioning involves spinal opioid receptor activation. Life Sci. 2012, 91, 860–865. [Google Scholar] [CrossRef]
- Han, R.; Zhang, G.; Qiao, X.; Guo, Y.; Sun, L.; Li, J.; Gao, C.; Sun, X. α7 Nicotinic Acetylcholine Receptor Mediates the Neuroprotection of Remote Ischemic Postconditioning in a Rat Model of Asphyxial Cardiac Arrest. J. Surg. Res. 2020, 246, 6–18. [Google Scholar] [CrossRef]
- Mastitskaya, S.; Marina, N.; Gourine, A.; Gilbey, M.P.; Spyer, K.M.; Teschemacher, A.G.; Kasparov, S.; Trapp, S.; Ackland, G.L.; Gourine, A.V. Cardioprotection evoked by remote ischaemic preconditioning is critically dependent on the activity of vagal pre-ganglionic neurones. Cardiovasc. Res. 2012, 95, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, B.; Kelly, J.; Muñoz, M.C.; Bernatené, E.A.; Méndez Diodati, N.; González Maglio, D.H.; Dominici, F.P.; Gelpi, R.J. Vagal stimulation mimics preconditioning and postconditioning of ischemic myocardium in mice by activating different protection mechanisms. Am. J. Physiol. Circ. Physiol. 2018, 97, 908–917. [Google Scholar] [CrossRef]
- Rassaf, T.; Totzeck, M.; Hendgen-Cotta, U.B.; Shiva, S.; Heusch, G.; Kelm, M. Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ. Res. 2014, 114, 1601–1610. [Google Scholar] [CrossRef]
- Basalay, M.V.; Mastitskaya, S.; Mrochek, A.; Ackland, G.L.; Gutierrez del Arroyo, A.; Sanchez, J.; Sjoquist, P.O.; Pernow, J.; Gourine, A.V.; Gourine, A. Glucagon-like peptide-1 (GLP-1) mediates cardioprotection by remote ischaemic conditioning. Cardiovasc. Res. 2016, 112, 669–676. [Google Scholar] [CrossRef]
- Gao, Y.; Song, J.; Chen, H.; Cao, C.; Lee, C. TRPV1 activation is involved in the cardioprotection of remote limb ischemic postconditioning in ischemia-reperfusion injury rats. Biochem. Biophys. Res. Commun. 2015, 463, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- May, S.M.; Chiang, E.; Reyes, A.; Martir, G.; Patel, A.; Karmali, S.; Patel, S.; West, S.; Del Arroyo, A.G.; Gourine, A.V.; Ackland, G.L. Neuromodulation of innate immunity by remote ischaemic conditioning in humans: Experimental cross-over study, Brain Behav. Immun. Health. 2021, 16, 100299. [Google Scholar] [CrossRef] [PubMed]
- Prokudina, E.S.; Maslov, L.N.; Tsibulnikov, S.Y.; Singh, N.; Klim, V.S.; Skryabina, A.А. The role of humoral factors in the remote preconditioning of the heart. Russ. Fiziol. Zh. Im. I.M. Sechenova. 2019, 105, 416–436, (article in Russian). [Google Scholar] [CrossRef]
- Rehni, A.K.; Singh, N.; Jaggi, A.S. Possible involvement of insulin, endogenous opioids and calcitonin gene-related peptide in remote ischaemic preconditioning of the brain. Yakugaku zasshi: J. Pharm. Society. 2007, 127, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Riquelme, J.A.; Zheng, Y.; Vicencio, J.M.; Lavandero, S.; Yellon, D.M. Endothelial cells release cardioprotective exosomes that may contribute to ischaemic preconditioning. Sci. Rep. 2018, 8, 15885. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.; Davidson, S.M.; Yellon, D.M. Does remote ischaemic conditioning reduce inflammation? A focus on innate immunity and cytokine response. Basic Res. Cardiol. 2021, 116, 12. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Huang, M.; Wu, J.; Jiang, Q.; Zheng, X. Exosomes isolated from the plasma of remote ischemic conditioning rats improved cardiac function and angiogenesis after myocardial infarction through targeting Hsp70. Aging (Albany NY). 2020, 12, 3682–3693. [Google Scholar] [CrossRef]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef]
- Chao de la Barca, J.M.; Bakhta, O.; Kalakech, H.; Simard, G.; Tamareille, S.; Catros, V.; Callebert, J.; Gadras, C.; Tessier, L.; Reynier, P.; Prunier, F.; Mirebeau-Prunier, D. Metabolic Signature of Remote Ischemic Preconditioning Involving a Cocktail of Amino Acids and Biogenic Amines. J. Am. Heart. Assoc. 2016, 5, e003891. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Jiang, W.; Liu, Z.; Li, F.; Yang, J.; Zhao, Y.; Ran, Y.; Meng, Y.; Ji, X.; Geng, X.; Du, H.; Hu, X. Splenic responses play an important role in remote ischemic preconditioning-mediated neuroprotection against stroke. J. Neuroinflammation. 2018, 15, 167. [Google Scholar] [CrossRef]
- Malhotra, S.; Naggar, I.; Stewart, M.; Rosenbaum, D.M. Neurogenic pathway mediated remote preconditioning protects the brain from transient focal ischemic injury. Brain Res. 2011, 1386, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Ren, C.; Chen, X.; Zhao, H. The chronic protective effects of limb remote preconditioning and the underlying mechanisms involved in inflammatory factors in rat stroke. PloS One 2012, 7, e30892. [Google Scholar] [CrossRef]
- Zhou, G.; Li, M.H.; Tudor, G.; Lu, H.T.; Kadirvel, R.; Kallmes, D. Remote Ischemic Conditioning in Cerebral Diseases and Neurointerventional Procedures: Recent Research Progress. Front. Neurol. 2018, 9, 339. [Google Scholar] [CrossRef]
- Sharma, D.; Maslov, L.N.; Singh, N.; Jaggi, A.S. Remote ischemic preconditioning-induced neuroprotection in cerebral ischemia-reperfusion injury: Preclinical evidence and mechanisms. Eur. J. Pharmacol. 2020, 883, 173380. [Google Scholar] [CrossRef]
- Chen, G.; Thakkar, M.; Robinson, C.; Doré, S. Limb Remote Ischemic Conditioning: Mechanisms, Anesthetics, and the Potential for Expanding Therapeutic Options. Front. Neurol. 2018, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Sprick, J.D.; Mallet, R.T.; Przyklenk, K.; Rickards, C.A. Ischaemic and hypoxic conditioning: potential for protection of vital organs. Exp. Physiol. 2019, 104, 278–294. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, H.; Takahashi, T.; Hsieh, J.; Liao, J.; Steinberg, G.K.; Zhao, H. The Akt signaling pathway contributes to postconditioning's protection against stroke; the protection is associated with the MAPK and PKC pathways. J. Neurochem. 2008, 105, 943–955. [Google Scholar] [CrossRef]
- Zhou, Y.; Fathali, N.; Lekic, T.; Ostrowski, R.P.; Chen, C.; Martin, R.D.; Tang, J.; Zhang, J.H. Remote limb ischemic postconditioning protects against neonatal hypoxic-ischemic brain injury in rat pups by the opioid receptor/Akt pathway. Stroke. 2011, 42, 439–444. [Google Scholar] [CrossRef]
- Xia, M.; Ding, Q.; Zhang, Z.; Feng, Q. Remote Limb Ischemic Preconditioning Protects Rats Against Cerebral Ischemia via HIF-1α/AMPK/HSP70 Pathway, Cell. Mol. Neurobiol. 2017, 37, 1105–1114. [Google Scholar] [CrossRef]
- Wang, C.; Zuo, B.; Wu, X. The Role of Macrophage Migration Inhibitory Factor in Remote Ischemic Postconditioning. Can. J. Cardiol. 2019, 35, 501–510. [Google Scholar] [CrossRef]
- Wick, A.; Wick, W.; Waltenberger, J.; Weller, M.; Dichgans, J.; Schulz, J.B. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J. Neurosci. 2002, 22, 6401–6407. [Google Scholar] [CrossRef]
- Rassaf, T.; Totzeck, M.; Hendgen-Cotta, U.B.; Shiva, S.; Heusch, G.; Kelm, M. Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ. Res. 2014, 114, 1601–1610. [Google Scholar] [CrossRef]
- Li, S.; Hu, X.; Zhang, M.; Zhou, F.; Lin, N.; Xia, Q.; Zhou, Y.; Qi, W.; Zong, Y.; Yang, H.; Wang, T. Remote ischemic post-conditioning improves neurological function by AQP4 down-regulation in astrocytes. Behav. Brain Res. 2015, 289, 1–8. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, X.; Sun, Y.; Wang, B.; Zhou, C.; Luo, Y.; Ge, P. Ischemic postconditioning decreases cerebral edema and brain blood barrier disruption caused by relief of carotid stenosis in a rat model of cerebral hypoperfusion. PloS One. 2013, 8, e57869. [Google Scholar] [CrossRef]
- Wang, Q.; Wills, M.; Li, F.; Geng, X.; Ding, Y. Remote ischemic conditioning with exercise (RICE) promotes functional rehabilitation following ischemic stroke. Neurol. Res. 2021, 43, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Comità, S.; Femmino, S.; Thairi, C.; Alloatti, G.; Boengler, K.; Pagliaro, P.; Penna, C. (2021) Regulation of STAT3 and its role in cardioprotection by conditioning: focus on non-genomic roles targeting mitochondrial function. Basic Res. Cardiol. 2021, 116, 56. [Google Scholar] [CrossRef]
- Liang, W.; Lin, C.; Yuan, L.; Chen, L.; Guo, P.; Li, P.; Wang, W.; Zhang, X. Preactivation of Notch1 in remote ischemic preconditioning reduces cerebral ischemia-reperfusion injury through crosstalk with the NF-κB pathway. J. Neuroinflammation 2019, 16, 181. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Li, L.; Mo, X.; Zhang, L.; Xie, Y.; Guo, Q.; Wang, Y. Non-invasive remote limb ischemic postconditioning protects rats against focal cerebral ischemia by upregulating STAT3 and reducing apoptosis. Int. J. Mol. Med. 2014, 34, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Ma, W.; Liu, K.P.; Yang, J.W.; Wang, X.B.; Wu, Z.; Zhang, T.; Wang, J.W.; Liu, W.; Liu, J.; Liang, Y.; Zhang, X.K.; Li, J.J.; Guo, J.H.; Li, L.Y. Advances in intervention methods and brain protection mechanisms of in situ and remote ischemic postconditioning. Metab. Brain. Dis. 2021, 36, 53–65. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.C.; Xian, X.H.; Li, W.B.; Li, L.; Yan, C.Z.; Li, Q.J.; Zhang, M. Activation of p38 MAPK participates in brain ischemic tolerance induced by limb ischemic preconditioning by up-regulating HSP 70. Exp. Neurol. 2010, 224, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Yan, W.; Zhou, J.; Pei, H.; Zhao, R. Per- and post-remote ischemic conditioning attenuates ischemic brain injury via inhibition of the TLR4/MyD88 signaling pathway in aged rats. Exp. Brain Res. 2021, 239, 2561–2567. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Xu, W.; Chen, J.; Zhang, X.; Shi, L.; Ren, C. Remote limb preconditioning protects against ischemia-induced neuronal death through ameliorating neuronal oxidative DNA damage and parthanatos. J. Neurol. Sci. 2016, 366, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, G.C.; Schaan, A.P.; Cabral, G.F.; Santana-da-Silva, M.N.; Pinto, P.; Vidal, A.F.; Ribeiro-Dos-Santos, Â. A Cell's Fate: An Overview of the Molecular Biology and Genetics of Apoptosis. Int. J. Mol. Sci. 2019, 20, 4133. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Jin, W.; Zhang, X.; Chen, J.; Ren, C. Remote Limb Preconditioning Generates a Neuroprotective Effect by Modulating the Extrinsic Apoptotic Pathway and TRAIL-Receptors Expression. Cell. Mol. Neurobiol. 2017, 37, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jha, M.K.; Suk, K. Lipocalin-2 in the Inflammatory Activation of Brain Astrocytes. Crit. Rev. Immunol. 2015, 35, 77–84. [Google Scholar] [CrossRef]
- Liu, M.; Chen, J.; Zhang, S.; Ren, C. Downregulation of lipocalin-2 and Bim expression after remote limb preconditioning in the ischemic rat brain. Brain Res. 2018, 1679, 1–9. [Google Scholar] [CrossRef]
- Yao, W.; Lin, S.; Su, J.; Cao, Q.; Chen, Y.; Chen, J.; Zhang, Z.; Hashimoto, K.; Qi, Q.; Zhang, J.C. Activation of BDNF by transcription factor Nrf2 contributes to antidepressant-like actions in rodents. Transl. Psychiatry 2021, 11, 140. [Google Scholar] [CrossRef]
- Barrio, E.; Vecino, R.; Sánchez-Morán, I.; Rodríguez, C.; Suárez-Pindado, A.; Bolaños, J.P.; Almeida, A.; Delgado-Esteban, M. Preconditioning-Activated AKT Controls Neuronal Tolerance to Ischemia through the MDM2-p53 Pathway. Int. J. Mol. Sci. 2021, 22, 7275. [Google Scholar] [CrossRef]
- Wang, J.; Han, D.; Sun, M.; Feng, J. A Combination of Remote Ischemic Perconditioning and Cerebral Ischemic Postconditioning Inhibits Autophagy to Attenuate Plasma HMGB1 and Induce Neuroprotection Against Stroke in Rat. J. Mol. Neurosci. 2016, 58, 424–431. [Google Scholar] [CrossRef]
- Han, Z.; Cao, J.; Song, D.; Tian, L.; Chen, K.; Wang, Y.; Gao, L.; Yin, Z.; Fan, Y.; Wang, C. Autophagy is involved in the cardioprotection effect of remote limb ischemic postconditioning on myocardial ischemia/reperfusion injury in normal mice, but not diabetic mice. PloS One. 2014, 9, e86838. [Google Scholar] [CrossRef]
- Qi, Z.; Dong, W.; Shi, W.; Wang, R.; Zhang, C.; Zhao, Y.; Ji, X.; Liu, K.J.; Luo, Y. Bcl-2 phosphorylation triggers autophagy switch and reduces mitochondrial damage in limb remote ischemic conditioned rats after ischemic stroke. Transl. Stroke Res. 2015, 6, 198–206. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, J.; Xiong, X.; Xu, Y.; Zhang, H.; Huang, C.; Tian, Y.; Jiao, C.; Wang, X.; Li, X. Remote ischemic preconditioning protects against liver ischemia-reperfusion injury via heme oxygenase-1-induced autophagy. PloS One 2014, 9, e98834. [Google Scholar] [CrossRef]
- Rohailla, S.; Clarizia, N.; Sourour, M.; Sourour, W.; Gelber, N.; Wei, C.; Li, J.; Redington, A.N. Acute, delayed and chronic remote ischemic conditioning is associated with downregulation of mTOR and enhanced autophagy signaling. PloS One 2014, 9, e111291. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, H.; Wang, T.; He, J.; Wei, J.; Wang, T.; Dong, J. Remote limb ischemic post-conditioning attenuates ischemia-reperfusion injury in rat skin flapby limiting oxidative stress. Acta Cir. Bras. 2016, 31, 15–21. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, Z.; Liu, S.; Bi, J.; Zhang, J.; Du, L.; Ding, X.; Liu, C. Remote ischemic conditioning protects against acetaminophen-induced acute liver injury in mice. Hepatol. Res. 2017, 47, 234–245. [Google Scholar] [CrossRef]
- Kim, Y.H.; Yoon, D.W.; Kim, J.H.; Lee, J.H.; Lim, C.H. Effect of remote ischemic post-conditioning on systemic inflammatory response and survival rate in lipopolysaccharide-induced systemic inflammation model. J. Inflamm. (Lond). 2014, 11, 16. [Google Scholar] [CrossRef]
- Li, P.; Su, L.; Li, X.; Di, W.; Zhang, X.; Zhang, C.; He, T.; Zhu, X.; Zhang, Y.; Li, Y. Remote limb ischemic postconditioning protects mouse brain against cerebral ischemia/reperfusion injury via upregulating expression of Nrf2, HO-1 and NQO-1 in mice. Int. J. Neurosci. 2016, 126, 552–559. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Chen, G.; Yang, J.; Lu, G.; Guo, J.; Dou, Y. Limb remote ischemic post-conditioning reduces brain reperfusion injury by reversing eNOS uncoupling. Ind. J. Exp. Biol. 2014, 52, 597–605. [Google Scholar]
- He, N.; Jia, J.J.; Li, J.H.; Zhou, Y.F.; Lin, B.Y.; Peng, Y.F.; Chen, J.J.; Chen, T.C.; Tong, R.L.; Jiang, L.; Xie, H.Y.; Zhou, L.; Zheng, S.S. Remote ischemic perconditioning prevents liver transplantation-induced ischemia/reperfusion injury in rats: Role of ROS/RNS and eNOS. World J. Gastroenterol. 2017, 23, 830–841. [Google Scholar] [CrossRef]
- Hu, S.; Dong, H.; Zhang, H.; Wang, S.; Hou, L.; Chen, S.; Zhang, J.; Xiong, L. Noninvasive limb remote ischemic preconditioning contributes neuroprotective effects via activation of adenosine A1 receptor and redox status after transient focal cerebral ischemia in rats. Brain Res. 2012, 1459, 81–90. [Google Scholar] [CrossRef]
- Corcoran, S.E.; and O'Neill, L.A. HIF1α and metabolic reprogramming in inflammation. J. Clin. Invest. 2016, 126, 3699–3707. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Y.; Xu, Y.; Ruan, W.; Wang, H.; Zhang, Y.; Saavedra, J.M.; Zhang, L.; Huang, Z.; and Pang, T. A Dual AMPK/Nrf2 Activator Reduces Brain Inflammation After Stroke by Enhancing Microglia M2 Polarization. Antioxid. Redox Signal. 2018, 28, 141–163. [Google Scholar] [CrossRef]
- Zendedel, A.; Johann, S.; Mehrabi, S.; Joghataei, M.T.; Hassanzadeh, G.; Kipp, M.; and Beyer, C. Activation and Regulation of NLRP3 Inflammasome by Intrathecal Application of SDF-1a in a Spinal Cord Injury Model. Mol. Neurobiol. 2016, 53, 3063–3075. [Google Scholar] [CrossRef]
- Pacher, P.; Haskó, G. Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br. J. Pharmacol. 2008, 153, 252–262. [Google Scholar] [CrossRef]
- Kim, Y.T.; Moon, S.K.; Maruyama, T.; Narumiya, S.; Doré, S. Prostaglandin FP receptor inhibitor reduces ischemic brain damage and neurotoxicity. Neurobiol. Dis. 2012, 48, 58–65. [Google Scholar] [CrossRef]
- Leu, T.; Schützhold, V.; Fandrey, J.; Ferenz, K.B. When the Brain Yearns for Oxygen. Neurosignals 2019, 27, 50–61. [Google Scholar] [CrossRef]
- Carrica, L.; Li, L.; Newville, J.; Kenton, J.; Gustus, K.; Brigman, J.; Cunningham, L.A. Genetic inactivation of hypoxia inducible factor 1-alpha (HIF-1α) in adult hippocampal progenitors impairs neurogenesis and pattern discrimination learning. Neurobiol. Learn. Mem. 2019, 157, 79–85. [Google Scholar] [CrossRef]
- Mitroshina, E.V.; Savyuk, M.O.; Ponimaskin, E.; Vedunova, M.V. Hypoxia-Inducible Factor (HIF) in Ischemic Stroke and Neurodegenerative Disease. Front. Cell. Dev. Biol. 2021, 9, 703084. [Google Scholar] [CrossRef]
- Barteczek, P.; Li, L.; Ernst, A.S.; Böhler, L.I.; Marti, H.H.; Kunze, R. Neuronal HIF-1α and HIF-2α deficiency improves neuronal survival and sensorimotor function in the early acute phase after ischemic stroke. J. Cereb. Blood Flow Metab. 2017, 37, 291–306. [Google Scholar] [CrossRef]
- Jin, X.; Kuang, Y.; Li, L.; Li, H.; Zhao, T.; He, Y.; Di, C.; Kang, J.; Yuan, L.; Yu, B.; Li, Q. A positive feedback circuit comprising p21 and HIF-1α aggravates hypoxia-induced radioresistance of glioblastoma by promoting Glut1/LDHA-mediated glycolysis. FASEB J. 2022, 36, e22229. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, L.; Liu, J.; Jin, Y.; Lin, Z.; Du, S.; Fu, Z.; Chen, T.; Qin, Y.; Sui, F.; Jiang, Y. HIF-1α/microRNA-128-3p axis protects hippocampal neurons from apoptosis via the Axin1-mediated Wnt/β-catenin signaling pathway in Parkinson's disease models. Aging 2020, 12, 4067–4081. [Google Scholar] [CrossRef]
- Sasabe, E.; Tatemoto, Y.; Li, D.; Yamamoto, T.; Osaki, T. Mechanism of HIF-1alpha-dependent suppression of hypoxia-induced apoptosis in squamous cell carcinoma cells. Cancer Sci. 2005, 96, 394–402. [Google Scholar] [CrossRef]
- Lambert, C.M.; Roy, M.; Robitaille, G.A.; Richard, D.E.; Bonnet, S. HIF-1 inhibition decreases systemic vascular remodelling diseases by promoting apoptosis through a hexokinase 2-dependent mechanism. Cardiovasc. Res. 2010, 88, 196–204. [Google Scholar] [CrossRef]
- Serebrovska, Z.O.; Chong, E.Y.; Serebrovska, T.V.; Tumanovska, L.V.; Xi, L. ; Hypoxia, HIF-1α, and COVID-19: from pathogenic factors to potential therapeutic targets. Acta Pharmacol. Sin. 2020, 41, 1539–1546. [Google Scholar] [CrossRef]
- Vetrovoy, O.; Sarieva, K.; Lomert, E.; Nimiritsky, P.; Eschenko, N.; Galkina, O.; Lyanguzov, A.; Tyulkova, E.; Rybnikova, E. Pharmacological HIF1 Inhibition Eliminates Downregulation of the Pentose Phosphate Pathway and Prevents Neuronal Apoptosis in Rat Hippocampus Caused by Severe Hypoxia. J. Mol. Neurosci. 2020, 70, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.S.; Kim, H.; Lee, S.M.; Kim, S.T.; Cho, E.J.; Youn, H.D. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol. Cell 2006, 22, 395–405. [Google Scholar] [CrossRef]
- Suzuki, H.; Tomida, A.; Tsuruo, T. Dephosphorylated hypoxia-inducible factor 1alpha as a mediator of p53-dependent apoptosis during hypoxia. Oncogene 2001, 20, 5779–5788. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liao, X.Y.; Pan, M.X.; Tang, J.C.; Chen, S.F.; Zhang, Y.; Lu, P.X.; Lu, L.J.; Zou, Y.Y.; Qin, X.P.; Bu, L.H.; Wan, Q. Glycine Exhibits Neuroprotective Effects in Ischemic Stroke in Rats through the Inhibition of M1 Microglial Polarization via the NF-κB p65/Hif-1α Signaling Pathway. J. Immunol. 2019, 202, 1704–1714. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; Zheng, L.; Gardet, A.; Tong, Z.; Jany, S.S.; Corr, S.C.; Haneklaus, M.; Caffrey, B.E.; Pierce, K.; Walmsley, S.; Beasley, F.C.; Cummins, E.; Nizet, V.; Whyte, M.; Taylor, C.T.; Lin, H.; Masters, S.L.; Gottlieb, E.; Kelly, V.P.; Clish, C.; Auron, P.E.; Xavier, R.J.; O'Neill, L.A. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, A.F.; O'Neill, L.A.J. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Watts, D.; Stein, J.; Meneses, A.; Bechmann, N.; Neuwirth, A.; Kaden, D.; Krüger, A.; Sinha, A.; Alexaki, V.I.; Luis Gustavo Perez-Rivas, Kircher, S.; Martinez, A.; Theodoropoulou, M.; Eisenhofer, G.; Peitzsch, M.; El-Armouche, A.; Chavakis, T.; Wielockx, B. HIF1α is a direct regulator of steroidogenesis in the adrenal gland. Cell. Mol. Life Sci. 2021, 78, 3577–3590. [CrossRef] [PubMed]
- Vanderhaeghen, T.; Beyaert, R.; Libert, C. Bidirectional Crosstalk Between Hypoxia Inducible Factors and Glucocorticoid Signalling in Health and Disease. Front. Immunol. 2021, 12, 684085. [Google Scholar] [CrossRef]
- Baranova, K.A.; Mironova, V.I.; Rybnikova, E.A.; Samoilov, M.O. Characteristics of the transcription factor HIF-1α expression in the rat brain during the development of a depressive state and the antidepressive effects of hypoxic preconditioning. Neurochem. J. 2010, 4, 35–40. [Google Scholar] [CrossRef]
- Marchi, D.; Santhakumar, K.; Markham, E.; Li, N.; Storbeck, K.H.; Krone, N.; Cunliffe, V.T.; van Eeden, F. Bidirectional crosstalk between Hypoxia-Inducible Factor and glucocorticoid signalling in zebrafish larvae. PLoS Genet. 2020, 16, e1008757. [Google Scholar] [CrossRef]
- Rybnikova, E.; Nalivaeva, N. Glucocorticoid-Dependent Mechanisms of Brain Tolerance to Hypoxia. Int. J. Mol. Sci. 2021, 22, 7982. [Google Scholar] [CrossRef]
- Leonard, M.O.; Godson, C.; Brady, H.R.; Taylor, C.T. Potentiation of glucocorticoid activity in hypoxia through induction of the glucocorticoid receptor. J. Immunol. 2005, 174, 2250–2257. [Google Scholar] [CrossRef]
- Charron, C.E.; Chou, P.C.; Coutts, D.; Kumar, V.; To, M.; Akashi, K.; Pinhu, L.; Griffiths, M.; Adcock, I.M.; Barnes, P.J.; Ito, K. Hypoxia-inducible factor 1alpha induces corticosteroid-insensitive inflammation via reduction of histone deacetylase-2 transcription. J. Biol. Chem. 2009, 284, 36047–36054. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, Y.Y.; Song, X.L.; Cai, H.Y.; Chen, J.C.; Song, L.N.; Yang, R.; Lu, J. Upregulations of glucocorticoid-induced leucine zipper by hypoxia and glucocorticoid inhibit proinflammatory cytokines under hypoxic conditions in macrophages. J. Immunol. 2012, 188, 222–229. [Google Scholar] [CrossRef]
- Wagner, A.E.; Huck, G.; Stiehl, D.P.; Jelkmann, W.; Hellwig-Bürgel, T. Dexamethasone impairs hypoxia-inducible factor-1 function. Biochem. Biophys. Res. Commun. 2008, 372, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Rybnikova, E.A.; Baranova, K.A.; Zenko, M.Y.; Churilova, A.V.; Stupin, K.V. Comparative analysis of various modes of preconditioning to increase high altitude tolerance. Integr. Physiol. 2022, 3, 348–358. [Google Scholar] [CrossRef]
- Baranova, K.A. Preconditioning by Moderate Hypoxia Increases the Amount of Corticosteroid Receptors in the Rat Brain in a Model of Depression. Neurochem. J. 2020, 14, 279–285. [Google Scholar] [CrossRef]
- Dallas, M.; Boycott, H.E.; Atkinson, L.; Miller, A.; Boyle, J.P.; Pearson, H.A.; Peers, C. Hypoxia suppresses glutamate transport in astrocytes. The Journal of neuroscience: the official journal of the Society for Neuroscience 2007, 27, 3946–3955. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



