Submitted:
13 October 2023
Posted:
13 October 2023
You are already at the latest version
Abstract
The review concerns the chemical structures of all the triterpene glycosides from the most valuable commercially harvesting North Atlantic sea cucumber Cucumaria frondosa and their biological activities. The isolation and structural elucidation of 17 specific triterpene glycosides are described along with isolation and identification of two known glycosides. Antitumor, immunomodulatory, antithrombotic and anti-Alzheimer disease activities are also described. Frondoside A, the major component of the glycosidic fraction seems to be very perspective leader-compound for creation of useful medicinal preparations.
Keywords:
sea cucumbers
; Cucumaria frondosa
; triterpene glycosides
; frondosides
; frondoside A
; biological activity.
1. Introduction
The sea cucumbers are representatives of especial class of marine invertebrates, Holothuroidea, belonging to the phylum Echinodermata including also the starfish, brittle stars, sea urchins and sea lilies. Cucumaria frondosa (Gunner, 1767) belongs to the family Cucumariidae. The areal of this North Atlantic species lays from Massachusetts shallow waters to Canada including Newfoundland, Greenland, Iceland, Great Britain and Atlantic shore of Western Europe, as well as Norwegian, Barents Seas and Western part of Kara Sea [1,2]. Several specialists supposed that Cucumaria japonica from North Western Part of the Pacific Ocean is a subspecies of C. frondosa, namely C. frondosa japonica as it was noted in the recent article concerning supercritical CO2 extraction of saponins from C. japonica [3]. Such point of view is obsolete and incorrect. Levin et Gudimova (1997) have carefully compared morphological characteristics of C. japonica and C. frondosa including ossicles forms and concluded that they are really different independent species. Moreover, they have noted that the main components of glycosidic fractions contained different sugars in third position of carbohydrate chains, namely glucose for C. japonica and xylose for C. frondosa glycosides and specific set of aglycones for each species [4]. Because sea cucumber triterpene glycosides are good chemotaxonomical characteristic the differences in their structures independently confirms the distinctiveness of these species [5].
C. frondosa is a filter-feeding animal (sestonophage), i.e. it filters organic particles from sea water for feeding. That is why this species lives at rocky or pure sandy substrates washing with strong currents at the depths from 30 to 300 m. Due to this species lives at significant depths, C. frondosa may be collected only by dredging with different dredges or bottom trawls. The industrial harvesting of C. frondosa was started in 1988 in State of Maine (USA) by Peter Donald Collin created industrial technology of the sea cucumber processing resulted in the dried body walls obtaining. P.D. Collin collaborated with Japanese distributors operating at Chinese market, so the commercial result was good. As the methodology of processing has not been patented the technology was widely disseminated in State of Maine and Canada. Partial overfishing led to imposition of a seasonal limitations and personal quota for the harvesting [1].
As well as most of the other sea cucumber species C. frondosa is a source of triterpene glycosides having wide spectrum of biological activities caused both by their membranolytic properties and ability to interact with specific receptors. The glycosides include the triterpene lanostane aglycones and carbohydrate chains, preferably branched pentaosides composed of D-xylose, D-glucose, D-quinovose, and 3-O-methyl-D-glucose monosaccharide residues. The carbohydrate chain is attached only to C-3 of an aglycone. The most of aglycones possess an 18(20)-lactone being so-called holostane derivatives. There are a lot of reviews concerning sea cucumber glycosides and their properties mentioning the triterpene glycosides from C. frondosa [6,7,8,9,10,11,12,13].
However, there is no any article specially covered all the structures of C. frondosa triterpene glycosides and their biological properties. It this review, we intend to present comprehensive information concerning chemical structures of triterpene glycosides from C. frondosa and their biological activities.
2. Structures of Triterpene glycosides from C. frondosa
The first information concerning chemical structures of glycosides from C. frondosa was published by Canadian chemists Findlay and Daljeet (1984) [14] after analysis of the products of acid hydrolysis of glycosidic fraction isolated from the extract of this sea cucumber (Figure 1). Using NMR spectroscopy, they have found monoxyloside of native aglycone, a holostane derivative having 16-OAc-group and 7(8)-double bond (1), and bioside having the same aglycone, xylose residue as the first sugar and quinovose residue as terminal monosaccharide. The position of interglycosidic linkage wasn’t determined. Moreover, native aglycone 3 as well as its isomers 4 and 5 with 8(9)- and 9(11)-double bonds, respectively, have also been isolated. The derivatives 4 and 5 are the products of acid catalyzed migration of the double bonds from 7(8)- to 8(9)- and 9(11)-positions. The configuration of 16-OAc in all these compounds was erroneously determined as α based on chemical shift of H-17 that was deshielded. This was attributed as cis-relationship to the C-16 acetate.
Nevertheless, Elyakov et al. (1981) have reported the structure of cucumariogenin, a native aglycone of cucumarioside G1 from the sea cucumber Eupentacta (Cucumaria) fraudatrix having the same holostane nucleus based on the analysis of coupling constant between H-16 and H-17 and definitely described the configuration of 16-OAc as β [15]. Rodriguez and Riguera (1989) [16] have isolated four glycosides from Aslia (=Cucumaria) levewrrei having the aglycones with the same holostane nucleus and supported point of view of Findlay and Daljeet about configuration of 16-OAc as α and just repeated their error despite of new X-ray data published by Ilyin et al. (1985) [17] and undoubtly confirmed β-configuration of 16-OAc.
The structure of frondoside A (6), a major component of glycosidic fraction from C. frondosa (Figure 2), was elucidated by another group of Canadian chemists (Girard et al. 1990) [18]. The saponin was relatively easy isolated by hydrophobic chromatography on Amberlite XAD-20 followed by common low pressure column chromatography on silica gel using CHCl3/MeOH/H2O in different ratios as mobile phase. The authors postulated β-configuration of 16-OAc on the base of COSY artefact in 2D NOESY spectrum between H-16 and H-17 that is inadequate methodology. However, the question has already been resolved by the analysis of coupling constants between the same protons and X-ray data not only for cucumarioside G1 but also for lefevriosides A, A1, B, and C having the identical holostane nuclei. NMR spectral data of frondogenin (the aglycone of frondoside A) were coincident with those for aglycone holostane nuclei mentioned above glycosides [15,16,17]. The structure of carbohydrate chain of 6 was elucidated using extensive analysis of 2D NMR spectra such as 1H-1H COSY and NOESY, the last one spectrum also demonstrated the positions of interglycosidic linkages. These data good correlated with FABMS obtained at registration both in positive and in negative modes. The minimal necessary chemical transformations also were used including acid hydrolysis followed by derivatization of sugars for GLC-MS analysis and desulfation for comparison spectral characteristics of native glycoside and its desulfated derivatives for confirmation of the position of sulfate group. The first sugar in carbohydrate chain was sulfated xylose, the second – quinovose, the third – xylose, and the fourth (terminal) sugar – 3-O-methylglucose residue. Another terminal residue was xylose attached to C-2 of the quinovose [18].
Avilov et al. (1997) have isolated frondoside A (6) along with minor sulfated linear tetraoside, frondoside A1 (7), from the sample of C. frondosa collected near Murmansk shore of Barents Sea (Figure 2) [19]. The full coincidence of chemical structures of major component of the glycosidic fractions from C. frondosa collected near opposite shores of Atlantic Ocean confirms their taxonomical identity. The structure of 7 was elucidated by chemical correlation with glycoside 6. The Smith degradation of 6 eliminated terminal xylose residue from C-2 of quinovose residue yielded the spectrally pure frondoside A1 (7). It seems to be obvious that frondoside A1 (7) is biosynthetic precursor of frondoside A (6).
In continuation of their researches of C. frondosa Findlay et al. (1992) have isolated frondoside B, minor disulfated branched pentaoside having glycose as the third monosaccharide residue and holostane aglycone with 7(8)-double bond lacking an oxygen substituent at C-16 (Figure 3). The structure of the glycoside 8 was reliably elucidated using 1H-1H COSY, NOESY as well as 13C NMR and confirmed by FABMS (negative ion mode). But the authors could not find quasi-molecular ions obviously due to FAB is too hard mode of ionization.
The reported by the authors frondecaside 9 (Figure 3) seems to be a subject of science fiction. The authors reported that this oligosaccharide was isolated from the fraction adsorbed by XAD-2, desalted with water and eluted with MeOH. So polar substance having ten sugars and six sulfates could not be adsorbed by XAD-2 because it is too hydrophylic. The pentasaccharide carbohydrate chain seems to be correctly elucidated but the authors didn’t find a quasi-molecular ion in the FAB mass-spectrum. The cause of finding “frondecaside” seems to be obvious: the presence of great diversity of glycosides having different aglycones but the same trisulfated pentasaccharide branched chains. The signals of the carbohydrate chain were easy accumulated in the NMR spectra but the signals of aglycones became invisible because of low content of each component [20].
The mysterious of “frondecaside” was figured out later by Avilov et al. (1998) isolated the most polar subfraction of glycosidic fraction from C. frondosa collected in Barents Sea. Because it was impossible to separate the native glycosides by HPLC without special procedures invented later, the structure of 10 including the number and position of sulfate groups was established by comparison of the NMR spectra of impure glycoside 10 and pure desulfated derivative 11, which was obtained from the fraction enriched by frondoside C (10) by solvolytic dedulfation with mixture pyridine – dioxane followed by purification by HPLC (Figure 4). The structure of the branched pentasaccharide chains of 10 and 11 was easily elucidated by comparison its spectral data with those of cucumarioside A7-1 from Cucumaria japonica and its desulfated derivative. The carbohydrate moieties of frondoside C and cucumarioside A1-7 proved to be the same. The sequence of monosaccharide residues in carbohydrate chain of 11 was confirmed by fragmentation in the FABMS(+). The structure of lanostane aglycone (non-holostane derivative without an 18(20)-lactone) having 9(11)- and 24(25)-double bonds, (20R)-hydroxyl and (22R)-OAc group was determined using conventional 13C and 1H NMR spectroscopy as well as 1H-1H COSY and HMQC [21].
Yayli and Findlay (1999) have isolated minor frondoside D (12) differing from frondoside A (6) by the presence of (23R)-OH group (Figure 5). The structure of 12 was elucidated by 13C, 1H and 2D NMR spectroscopy and supported by FAB MS data [22].
In 2001 Yayli (already without Findley and any other co-authors) has reported about three new chemical structures of minor triterpene glycosides from C. frondosa (Figure 6). The structure of frondoside F (13) published in Indian Journal of Chemistry, too far from Canadian shore, where the sea cucumbers were collected, cannot be taken seriously. The author didn’t get quasi-molecular ion in the FABMS(+) and didn’t use FABMS(–) that is more appropriated mode of the ion registration for a trisulfated glycoside. The absence of adequate mass-spectrometry data does the structural evidence insufficient. The author claimed several chemical sensations including the absence of a double bond in the lanostane nucleus and α-configuration of H-9 that is impossible from biosynthetic point of view. He also proclaimed unprecedented an 18(22)-lactone but didn’t confirm its presence by NOESY correlation between H-17β and H-22β. The structure of trisulfated pentasaccharide chain identical to that of frondoside C was established correctly using 2D NMR procedures [23].
Yayli has also reported the elucidation of the structures of two minor monosulfated pentaosides having 16-keto group, frondosides E1 (14) and E2 (15), isolated as inseparable mixture (Figure 7). Glycoside 14 proposed to have 7(8)- and 25(26)-double bonds and glycoside 15 proposed to have 9(11)- and 24(25)-double bond. The carbohydrate chains were identical to those of cucumarioside A2-2 from Cucumaria japonia and frondoside E1 (14) was declared to be identical to cucumarioside A2-2. However, as the structures were elucidated using only NMR spectra of inseparable mixture, it seems to be impossible to determine certain combination of double bonds for each glycoside and the structure of 14 and 15 are only hypothetical with probability of 50% [23].
Hence, the general cause of creation of so doubted hypothetical structures as “frondecaside”, frondoside F, frondosides E1 and E2 was a difficulty of separation of complicated polar sulfated glycosides even at the use of reversed phase HPLC. The special dynamic modificators for HPLC mobile phases were necessary to resolve this problem [24].
Silchenko et al. (2005) also have isolated the series of glycosides, frondosides A2-1 (16), A2-2 (17), A2-3 (18), and A2-6 (19), having monosulfated pentasaccharide chain with glycose as a third monosaccharide residue differing from frondoside A (6) having xylose residue in this position (Figure 8). The glycosides were isolated from C. frondosa collected near the shore of State of Maine, USA. The subfration of monosulfated glycosides was isolated by common way using hydrophobic chromatography on Polychrom-1 (powderized Teflon) followed by flash chromatography on silica gel. The subsequent HPLC on a Diasphere-110 C18 (4.6×250 mm) column using different ratios of MeOH/H2O as mobile phase with Na2HPO4∙12H2O (1M water solution) as a dynamic modificator followed by HPLC on a chiral column Chiradex 5 µm using the same modificator allowed to separate inseparable earlier glycosidic mixture. The aglycones of glycosides 16 and 19 have opposite combination of doble bonds in the nucleus and side chain than hypothetical frondosides E1 (14) and E2 (15). Hence their structures probably were elucidated erroneously. Glycoside 16 has a 24(25)-double bond and 16-keto group. Glycoside 17 possesses 24-keto-group conjugated with 25(26)-double bond while glycoside 18 has 23(24)-E-double bond. The glycosides 17 and 18 doesn’t have any substituents at C-16. Glycoside 19 has 9(11)- and 25(26)-double bonds and 16-ketogroup [25].
In continuation of these investigations Silchenko et al. using HPLC on reversed phase column with dynamic modificator in mobile phase have isolated frondosides A2-4 (20), A2-7 (21), and A2-8 (22), monosulfated branched pentaosides also having glucose residue as a third sugar in charbohydrate chain (Figure 9). Their structures were elucidated using 2D NMR procedures and HR MALDI TOF MS.
Frondosides A2-7 (21) and A2-8 (22) are isomers and differ from each other only by the position of a double bond in nucleus of their non-holostane-type aglycones. These glycosides have 18-hydroxy group, (22R)-OAc and 24(25)-double bond. Glycoside 20 has common holostane aglycone with 7(8)- and 24(25)-double bonds [26].
Later Silchenko et al. (2007) using the same HPLC methodology with dynamic modificators have isolated eight individual trisulfated branched pentaosides (23–27 were new and 10, 29 and 30 were earlier known) from C. frondosa collected near the shore of State of Maine (USA) (Figure 10). Frondosides A7-1 (23) and A7-2 (24) are holostane derivatives with 7(8)- and 24(25)-double bonds differing by the presence / absence of 16-keto-group. Frondoside A7-3 (25) is non-holostane derivative lacking an 18(20)-lactone having CH3-18 and 20R-hydroxyl instead bearing also 22R-OH in the side chain. Frondoside A7-4 (26) is an isomer of the glycoside 25 by the position of double bond in the aglycone nucleus [7(8)- instead of 9(11)-double bond]. Isofrondoside C (27) is 7(8)-isomer of known frondoside C (10) by the position of double bond in the aglycone nucleus. In order to confirm the position of all the sulfate groups the glycoside 27 was desulfated. Comparison of 13C NMR spectra of 27 and its desulfated derivative 28 confirmed all the positions of sulfate groups because corresponding shifting effects of sulfate groups were registered. Frondoside C (10) was firstly isolated as individual substance as well as isokoreoside A (30) earlier found in Cucumaria conicospermium. Known earlier from Cucumaria koreaensis koreoside A (29) is 22,23,24,25,26,27-hexa-nor-lanostane derivative with 20-keto-group and 7(8)-double bond was also found in C. frondosa being the isomer of isokoreoside A (30) by intranuclear double bond position [27].
Hence 17 new triterpene glycosides were isolated from C. frondosa along with two earlier known compounds, their structures were reliably established. Six of them are non-holostane derivatives having no lactone ring. The characteristic feature of glycosides biosynthesis in C. frondosa is the co-presence of the aglycones with two different positions (7(8)- and 9(11)-) of intranuclear double bond. All the glycosides were isolated as individual substances due to first using for their separation HPLC on reversed phase columns with special salt additives to mobile phases, so called dynamic modificators. The major component of glycosidic fraction is frondoside A (6) that may be isolated as individual substance even without using of HPLC. That is why the major part of investigations concerning biological activities of triterpene glycosides of C. frondosa were carried out on frondoside A.
3. General Ways of Biosynthesis of Triterpene Glycosides in Cucumaria frondosa
The biosynthesis of sea cucumber triterpene glycosides is of mosaic (combinatorial) type, due to carbohydrate chains and aglycones are formed independently and create a biosynthetic network (Figure 11). There are two main branches in the biosynthesis of carbohydrate chains of C. frondosa glycosides. The first one is the path leading to monosulfated branched pentaosides having a xylose as the third monosaccharide unit. The second one is the path leading to monosulfated branched pentaosides with glucose as the third monosaccharide unit and additional sulfate groups (by three).
Probably, two different oxidosqualencyclases performing the formation of lanostane precursors having either 7(8)- or 9(11)-double bonds participate in the biosynthesis of aglycones of C. frondosa (Figure 12). That is why the pairs of isomers having either 7(8)- or 9(11)-unsaturation such as frondosides A7-3 (25) and A7-4 (26), frondoside C (10) and isofrondoside C (27), koreoside A (29) and isokoreoside A (30) were revealed.
There are two main directions of transformations of the both types of aglycones precursors: to holostane derivatives (having 18(20)-lactone) and to lanostane derivatives without a lactone (Figure 12). Acetylation of C-16 is characteristic for the holostane aglycones with 7(8)-double bond only. In the precursors having 16-keto-group the side chains may be modified only by double bonds but not oxygen-containing substituents. The side chain cleavage in the process of biosynthesis occurs only in trisulfated non-holostane glycosides indicating, on the one hand, that this step is a final and on the other hand that such norlanostane trisulfated glycosides can play a hormone-like function while frondoside A (6) most likely performs protective role against predators.
4. Biological Activities of Triterpene Glycosides from C. frondosa
The major part of works concerning activities of individual triterpene glycosides from C. frondosa are dedicated to different aspects of antitumor action.
The detail investigation of the activity of frondoside A against pancreatic cancer was carried out by Li et al. (2008) [28]. The authors have found by measuring of 3H-thymidine incorporation in the cells that frondoside A inhibited proliferation of AsPC-1 human pancreatic cancer cells. The glycoside also induced apoptotic morphological changes (increase of sub-G0/G1 cell population) in the studied cell culture that was confirmed by annexin V binding and TUNEL assay. Using Western blotting analysis, the authors revealed frondoside A induced apoptosis through the mitochondrial pathway and caspase cascade activation. The antitumor action of frondoside A was confirmed by in vivo experiments, which demonstarted the inhibition of growth of AsPC-1 xenografts in athymic mice.
Al Shemali et al. (2014) [29] have studied synergic effects of frondoside A and gemcitabine, a convential cytotoxic anticancer drug against AsPC-1 and S2013 human pancreatic cancer cell lines. They have found that growth inhibition was significantly greater for the drug combinations than their effects at the studied doses. The combination was tested in vivo using the athymic mouse model. The xenografts were allowed to form the tumours for one month before the administration of the drugs alone or in combination. At the lowest dose tested, the combination was significantly more effective than any drugs alone. They concluded that the combination may have good perspective for a cure of pancreatic cancer patients.
Al Shemali et al. (2016) [30] have carried out comparative study of frondoside A (6), frondoside B (8), and frondoside C (really the mixture of trisulfated pentaosides) as well as the aglycone of frondoside A. Frondoside A was the most effective inhibitor of the growth of pancreatic cancer cells while frondoside B was less effective. Similar results were obtained during in vivo experiments on cancer xenografts in nude mice. The authors also showed the all the studied substances were not active at the oral administration.
Nguen at al (2017) [31] have found that frondoside A (6) suppressed the PAK1-dependent growth of A549 lung cancer as well as pancreatic cancer cells.
Jin et al. (2009) [32] have studied activity of frondoside A (6) against leukemic cells comparing this activity with that of cucumarioside A2-2 from Cucumaria japonica. The last glycoside is similar to frondoside A but has glucose insdead of xylose as the third sugar in branched pentasaccharide chain and 16-keto-group instead of 16β-O-acetic group. The both glycosides induced apoptosis of leukemic cells but by different mechanisms. Cucumarioside A2-2 induced caspase-dependent apoptosis while frondoside A caspase-independent one. The authors have tested induction of apoptosis at cytotoxic concentrations (IC100) haven’t taken into account that the glycosides are strong membranolytics. So, the quantitative interpretation of double staining area in annexin-FITC flow cytometry as “late apoptosis” only seems to be wrong.
Sajwani et al. (2017) [33] have investigated the effects of frondoside A against acute leukemia CCRF-CEM, HL-60 and THP-1 cell lines alone and in combination with conventional drugs. CCRF-CEM cells were very sensitive to frondoside A while HL-60 and THP1 were less sensitive. Frondoside A induced synergic anticancer effects with all the studied conventional drugs.
Al Marzouqi et al. (2011) [34] have studied the effects of frondoside A (6) on human estrogen receptor-negative MDA-MB-231 breast cancer cell line including survival, migration and invasion in vitro. The MCF10-A cells of normal human mammary epithelium were used as control. Frondoside A decreased the viability of the cancer cells with EC50 of 2.5 μM while the epithelium cells were more resistant to frondoside A (EC50 of 5 μM). It was shown that frondoside A induced apoptosis via increase of the sub-G1 (apoptotic) cell fraction through the activation of p53, and followed by activation of the caspases 9 and 3/7 cell death pathways in the cancer cells. Moreover, frondoside A inhibited tumor cells migration and invasion. Authors also showed the glycoside significantly decreased the growth of MDA-MB-231 tumor xenografts in athymic mice. The side toxic effects were absent in these in vivo experiments. The authors found that frondoside A has synergic effect against breast cancer cells in a composition with paclitaxel, a conventional chemotherapeutic agent and concluded that frondoside A may be a therapeutic agent for breast cancer.
Park et al. (2012) [35] demonstrated frondoside A significantly inhibited TPA-induced colony formation, invasion and migration of MBA-MB-231 human breast cancer cells. These in vitro experiments were carried out at non-cytotoxic concentrations. Frondoside A suppressed TPA-induced MMP-9 enzymatic activity, both secretion and expression probably via the suppression of AP-1 and NF-κB signaling pathways. This effect also correlated with increased expression of TIMP-1 and TIMP-2. Frondoside A decreased the activation of the PI3K/Akt, ERK1/2, and p38 MAPK signals. All these data allowed thee authors to conclude the anti-metastatic effect of frondoside A against human breast cancer cells. These results indicate the possible use of frondoside A as an antimetastatic agent for breast cancer.
Ma et al. (2012) [36] showed that frondoside A has strong antimetastatic activity. Frondoside A was i.p. administrated to mice bearing mammary gland-implanted tumors. The inhibition of spontaneous tumor metastasis to the lungs was observed as result of this experiment. Frondoside A modulated the functions of the EP receptors participated in promotion of metastasis. Cyclooxygenase-2 activity in many cancer cells promotes tumor growth and metastasis by producing PGE(2) which acts on the prostaglandin E receptors, preferably EP4 and EP2. The authors have found that frondoside A antagonizes EP2 and EP4 prostaglandin E receptors. The glycoside inhibited tritium labeled PGE(2) binding to recombinant EP2 or EP4-expressing cells at low μM concentrations. Moreover, EP4 or EP2-linked activation of intracellular cAMP and EP4-mediated ERK1/2 activation were also blocked by frondoside A. These results revealed that antimetastatic activity of frondoside A may be a novel mechanism of action.
Attoub et al. (2019) [37] have found that flavonoid compound butein at the IC25 and IC50 concentration enhances the anti-cancer effect of frondoside A against lung cancer cells (A549 and LNM35) and the breast cancer cells (MDA-MB-231 and T47D) in vitro.
Sulaiman et al. (2021) [38] explored the anti-cancer potential of butein, a biologically active flavonoid, on two major solid tumors, namely, A549 lung and MDA-MB-231 breast cancer cells alone and in combination with frondoside A. They have demonstrated that combination of butein with frondoside A induced additive effects on inhibiting of A549 and MDA-MB-231 cellular viability, induction of activity of caspase 3/7, colony growth inhibition, and cellular migration and invasion inhibition. Such combination induced a synergistic effect on the inhibition of HUVECs migration in vitro. The authors concluded that such combination may have the relevance of these compounds' combination in lung and breast cancer therapy.
Attoub et al. (2013) [39] have found that frondoside A caused reduction in viability of LNM35, A549, and NCI-H460-Luc2 lung cancer cell lines, MDA-MB-435 and MCF-7 (breast cancer cell lines), and HepG2 hepatoblastoma cell line through a caspase 3/7-dependent cell death pathway. Moreover, frondoside A induced inhibition of cell migration, invasion and angiogenesis in vitro. Frondoside A decreased the growth, the angiogenesis and lymph node metastasis of LNM35 tumor xenografts in athymic mice. Frondoside A also prevented basal and bFGF induced angiogenesis in the CAM angiogenesis assay. Frondoside A also enhanced the inhibition of lung tumor growth by cisplatin, a conventional antitumor drug. These findings made frondoside A a promising therapeutic agent against lung cancer.
Wang et al. (2014) [40] have studied cytotoxicity of four frondosides in comparison with a series of glycosides from Indo-West Pacific sea cucumber Holothuria scabra by MTT method. It was shown that minor tetraoside frondoside A1 (7), frondoside A (6), an acid form of frondoside A (named by the authors as frondoside A6), and 24-dehydroderivative of acid form of frondoside A were active against three tumor cell lines including HepG2 hepatocellular carcinoma, HeLa cervical cancer, K562 leukemic lymphoma. HL-7702 human liver normal cells were used as control. All the studied glycosides from C. frondosa with the exception of different forms of frondoside A, revealed the better selective cytotoxicity against tumor cells than the glycosides from H. scabra. The authors have found that there was no significant difference in cytotoxic activity against HL-7702 cells between 7 and 6, the cytotoxicity of 7 on three tumor cells lines was more significant than that of frondoside A (6). Therefore, the cytotoxicity of tetraoside (7) was more selective than that of branched pentaoside (6) in neoplastic vs. normal cells. The finding that the cytotoxic potential of studied glycosides against tumor cells could be decreased by the presence of fifth terminal sugar branching the second monosaccharide residue of tetrasaccharide chain was in good agreement with earlier observations both for hemolytic and cytotoxic activities of the glycosides against tumor cells.
Dyshlovoy et al. (2017) [41] investigated the efficacy of frondoside A (6) on eight cell lines of Burkitt lymphoma. Glycoside 6 revealed cytotoxic action against all tested cell lines including multiresistant CA46 cells. Frondoside A induced caspases- and p53-independent apoptosis, which was characterized by decreased expression of antiapoptotic survivin and Bcl-2, mitochondria targeting (release of cytochrome C, HtrA2/Omi and the apoptosis-inducing factor (AIF), and alteration of production of ROS) as well as translocation of AIF to the nuclei. Moreover, the inhibition of pro-survival autophagy was found. The conventional chemotherapy is not effective for Burkitt lymphoma because nonfunctional caspases-dependent apoptosis pathways inactivates p53 mutations and induces pro-survival autophagy. Frondoside A may be a promising candidate for the treatment of refractory or relapsed Burkitt lymphoma having resistances to standard methods of therapy.
Dyshlovoy et al. (2016) [42] have studied efficacy, toxicity and mechanism of action of frondoside A (6) on castration-resistant prostate cancer (CRPC) cell lines both in vitro and in vivo. Glycoside 6 revealed high effectiveness against human prostate cancer cells, while non-malignant cells possessed by less sensitivity. It was interesting that proliferation and colony formation of cells having resistibility against enzalutamide and abiraterone were also inhibited by 6. This glycoside induced the arrest of cell type specific cell cycle and induction both caspase-dependent and independent apoptosis. The glycoside has induced Up-regulation or induction of several pro-apoptotic proteins (Bax, Bad, PTEN), cleavage of PARP and caspase-3 and down-regulation of survivin and Bcl-2 anti-apoptotic proteins. Global proteome analysis has shown the regulation by 6 of proteins involved in metastases formation, invasion of tumor cells, and apoptosis, including keratin 81, CrkII, IL-1β and cathepsin B. Frondoside A caused also the inhibition of pro-survival autophagy of the cancer cells. In the experiments in vivo, glycoside 6 inhibited tumor growth of DU145 and PC-3 cells with decreasing of lung metastasis, circulation of the tumor cells in the peripheral blood, increased the quantity of lymphocytes in the treated animals that also indicated an immunomodulatory effect of frondoside A. The authors concluded that all found effects of frondoside A may allow to create a promising new drug.
Dyshlovoy et al. (2017) [43] have investigated anticancer activity of frondoside A (6) on p53-wild type and p53-deficient human urothelial carcinoma cell lines T-24, RT4, RT112, TCCSUP, H T-1197, and 486p. They found that frondoside A has higher cytotoxicity in urothelial carcinoma cells (IC50 from 0.55 to 2.33 μM) than cisplatin. Induction of apoptosis by frondoside A was linked with the regulation of several pro-apoptotic factors including PARP, caspase-3, -8, and -9, p21, Bax, fragmentation of DNA, and externalization of phosphatidylserine. Remarkably, the inhibition of p53, as well as caspase inhibition, did not decrease apoptotic activity of 6 in contrast with cisplatin. Frondoside A inhibited pro-survival autophagy that is well known mechanism of drug resistance in urothelial carcinoma. It also showed synergistic action with cisplatin and gemcitabine.
Menchinskaya et al. (2013) [44] earlier have found another mechanism of multidrug resistance that could be blocked by frondoside A (6) and its complexes with cholesterol through inhibition of membrane transport P-glycoprotein in ascite form of mouse Ehrlich carcinoma cells. Glycoside prevented by this way an efflux of fluorescent probe Calcein from the cells. The authors concluded that the blocking of P-glycoprotein activity resulted in decrease of multidrug resistance, and the glycoside and its complex with cholesterol may be potential inhibitors of multidrug resistance in tumor cells.
Ru et al. (2023) [45] have demonstrated the efficacy of frondoside A (6) against bladder cancer. They have found anti-proliferative, anti-invasive, anti-angiogenic, and potent immunomodulatory effects. Moreover, the authors revealed synergic effects of 6 with CpG-ODN oligodeoxynucleotide which also has potent anti-cancer effects against different tumors including bladder cancer. Frondoside A and CpG-ODN separately and in combination revealed combined effects on human bladder cancer UM-UC-3 cells viability, migration, apoptosis, and cell cycle in vitro and in experiments in vivo decreased the growth of UM-UC-3 tumor xenografts. There were no any toxic side-effects while traditional chemotherapeutic agents EPI (etoposide, cisplatin and ifosfamide) caused weight loss in nude mice.The authors found that 6 decreased the viability of UM-UC-3 bladder cancer cells, inhibited cell migration, and induced the apoptosis leading to the increasing of the sub-G1 (apoptotic) cell fraction in a concentration-dependent manner. The authors concluded that the use of frondoside A alone or in combination with CpG-ODN may be a promising therapeutic strategy against bladder cancer.
Attoub et al. (2018) [46] have studied the effect of frondoside A (6) alone and in combination with the standard cytotoxic drugs oxaliplatin and 5-fluorouracil (5-FU) against human colon cancer cell lines including HCT-116, HT-29, and HCT8/S11. They found that frondoside A, oxaliplatin, and 5-FU cause a concentration- and time-dependent inhibition of proliferation of HT-29 colon cancer cells, HCT8/S11 and HCT-116 cells. Frondoside A was significantly more active than oxaliplatin and 5-FU. The combinative use of low concentrations of these preparations in vitro revealed the increasing the inhibition of proliferation and colony growth induced by oxaliplatin or 5-FU with frondoside A. The authors found that the inhibition of phosphorylation of ERK1/2 by frondoside A in combination with 5-FU or oxaliplatin was significantly enhanced. They showed that frondoside A and 5-FU, when used alone, may also induce apoptosis, while their pro-apoptotic effect is dramatically enhanced at the use in combination. The authors found that apoptosis induction in colon cancer cells by frondoside A may be a result of the inhibition of phosphorylation of the survival kinase AKT followed by caspase-3 activation, poly(ADP-ribose)polymerase (PARP) inactivation, and consequence of DNA damage, as it was deduced by the increase of ϒ-H2AX level. The authors concluded that frondoside A may have a significance in colon cancer therapy at the use in combination with oxaliplatin and 5-FU.
Xue et al. (2021) [47] showed that frondoside A (6) effectively inhibits MYC-driven medulloblastoma model without side effects through suppression of MYC expression and its downstream gene targets in medulloblastoma cells that may be a potential mechanism of frondoside A inhibitory effects on MYC-driven cancers. Intratumoral administration of 6 in orthotopic xenografts of MYC-driven medulloblastoma induced cytotoxicity in tumor xenografts, significantly increased the survival of tumor-bearing animals, and stimulated the recruitment of microglia/macrophages and T-lymphocytes to the tumors. The decreasing of MYC level allowed to predict frondoside A potency against glioblastoma and non–small cell lung cancer. The authors informed that their preclinical results justify the clinical translation of frondoside A in treating of MYC-driven MB and other human cancers.
Kalinin et al. (1992) [48] found that frondoside A (6) has strong effects on mouse erythrocytes including K+ efflux followed by hemolysis. So strong membranolytic action may be also one of the causes of cytotoxic activity of frondoside A against other types of cells including tumor one.
Tangrodchanapong et al. (2020) [49] investigated the effects of frondoside A (6) on amyloid-β aggregation and proteotoxicity. They showed that frondoside A delayed the worm paralysis of transgenic Caenorhabditis elegans induced by amyloid-β aggregation. Moreover, the number of amyloid-β plaque deposits worms’ tissues was strongly decreased. Frondoside A was more effective than ginsenoside-Rg3, a control ginseng glycoside. Immunobloting revealed that the level of small oligomers as well as high molecular weights of amyloid-β species in the transgenic C. elegans were strongly decreased after the treatment with frondoside A, while the level of amyloid-β monomers was not changed. The authors found that frondoside A also protected the worms from oxidative stress. The glycoside 6 rescued chemotaxis dysfunction in a transgenic strain of C. elegans whose neurons express amyloid-β. The authors concluded that frondoside A could protect against the toxicity induced by amyloid-β by suppressing amyloid-β oligomers formation and may be useful for treatment against Alzheimer’s disease.
Because frondoside A (6) inhibits the phosphatidylinositol-3-kinase (PI3K) pathway in different cancer cells [28,33,35] that is also crucially involved in platelet activation, Ampofo et al. (2020) [50] investigated the influence of the glycoside 6 on thrombus formation. Frondoside A did not change the platelets viability, but reduced surface expression of P-selectin (CD62P) and glycoprotein (GP)IIb/IIIa activation in the platelets after agonist stimulation. They also found that this process was controlled by downregulation of PI3K-dependent Akt and the phosphorylation of extracellular-stimuli-responsive kinase (ERK). Frondoside A strongly increased the complete vessel occlusion time in the mouse dorsal skinfold chamber model of photochemically induced thrombus formation and also the tail vein bleeding time. The authors concluded that frondoside A may suppress thrombus formation and may serve as a novel lead compound for the development of antithrombotic drugs.
Aminin et al. (2008) [51] have found strong immunomodulatory activity of frondoside A (6) in subtoxic doses. This glycoside demonstrated following effects in vitro: stimulation of lysosomal activity in peritoneal mouse macrophages at the doses of 0.1–0.38 μg/mL, increased macrophage phagocytosis of Staphylococcus aureus at the dose of 0.001 μg/mL, stimulated formation of reactive oxygen species in macrophages at the dose of 0.001 μg/mL. In vivo frondoside A (6) stimulated an increase in the number of antibody plaque-forming cells (normally B-cells in spleen) with a maximal effect at the dose of of 0.2 μg per mouse and increased lysosomal activity of mouse peritoneal macrophages. Frondoside A had low effect on the production of immunoglobulin M (IgM) after immunization with sheep erythrocytes in mice and didn’t significantly increase the IgM and IgG antibody levels in ovalbumin-immunized mice. The authors concluded that frondoside A is a specific immunostimulant of cell-based immunity including phagocytosis without a strong effect on humoral immune activity or adjuvant properties. Therefore, frondoside A may provide curative and/or preventive treatment options against diseases resistant to humoral immune protection where excessive production of non-functional antibodies may be a part of pathology.
Aminin et al. (2009) [52] have studied the effect frondoside A (6) on mouse splenocytes in vitro using the monitoring of differential protein expression by 2D gel analysis followed by MALDI ToF MS and nano LC-ESI Q-ToF MS/MS. Differential image analysis of the gels showed that approximately thirty protein spots were differentially expressed. They included NSFL1 cofactor p47 and hnRNP K (down-regulated), as well as Septin-2, NADH dehydrogenase [ubiquinone] iron–sulfur protein 3, and GRB2-related adaptor protein 2 (up-regulated). Immuno-assay analysis confirmed the above data. The glycoside also stimulated the proliferation of splenocytes. The authors concluded that holothurian triterpene glycosides including frondoside A expressed immuno-stimulatory effects by enhancing the natural cellular defense barrier that is necessary to fight different pathogens.
5. Conclusions
The sea cucumber Cucumaria frondosa is an amazing gift of nature for humankind due to this species is mass and widely distributed and biosynthesizing in significant amount so unique metabolite as frondoside A. The structures of the other 19 triterpene glycosides found in C. frondosa are also interesting but mainly for chemists. These compounds formed as result of mosaic type biosynthesis have preserved in their structures the steps of biosynthetic pathways allowing to trace them. Additionally, the differences in chemical structures illustrates the performing of diverse functions in the producing animals. Moreover the glycosides are usefull for the resolving of taxonomical issues of the sea cucumbers.
The major part of articles dedicated to various biological activities and physiological action of individual triterpene glycoside from this sea cucumber – frondoside A. This is a predominant component of glycosidic fraction which may be easy isolated even without the use of HPLC. Since C. frondosa is mass commercially harvesting species of sea cucumbers there are good perspectives for the isolation of enough amount of frondoside A from the processing cooking water of C. frondosa for development of pharmaceutical preparations. Frondoside A seems to be effective for treatment of so dangerous tumors as pancreatic cancer, acute leukemia, leukemic lymphoma, breast cancer, mammary gland-implanted tumor, lung cancer, hepatocellular carcinoma, cervical cancer, Burkitt lymphoma, castration-resistant prostate cancer, urothelial carcinoma, human colon cancer, and MYC-driven medulloblastoma. Frondoside A have strong membranolytic action causing mouse erythrocyte hemolysis and necrotic death of other cells with sterol-containing membranes. However, the most of anticancer effects were found in sub-cytotoxic doses. Frondoside A induced anticancer effects in vivo and in vitro via influence on very different cell signaling paths including caspase-depending and caspase independent induction of apoptosis. This glycoside may effectively decrease the multidrug resistance by blocking the activity of membrane transport P-glycoprotein or, in any cases, through inhibition of pro-survival autophagy. Frondoside A also significantly enforced the activities of conventional cancer chemotherapy drugs such as cisplatin, gemcitabine, EPI (etoposide, cisplatine and ifosamide). The most of researchers studied the antitumor effects of frondoside A hihgly estimated the perspectives of using the glycoside alone and in combination with conventional drugs in the treatment of different types of cancer.
Additionally to anticancer properties, frondoside A may be perspective leader-compound for a treatment of Alzheimer’s disease, as antithrombotic preparation, and especially as stimulator of cell immunity without significant influence on humoral immunity that also may be applied for treatment of diseases resistant to humoral immune protection.
Author Contributions
Conceptualization, V.I.K.; writing—original draft preparation, V.I.K. and A.S.S.; writing—review and editing, V.I.K., A.S.S. and S.A.A.; funding acquisition, V.I.K. and A.S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, grant number 23-13-00078. The APC was funded by V.I.K. and A.S.S. from APC discount vouchers.
Institutional Review Board Statement
Not applicable.
Acknowledgments
The authors are very appreciative to Dr. Natalia V. Ivanchina (G.B. Elyakov Pacific Institute of Bioorganic Chemistry, Vladivostok, Russia) for reading the text and useful discussion.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hamel, J.-F.; Mercier, A. Population status, fisheries and trade of sea cucumbers in temperate areas of the Northern Hemisphere. In Sea cucumbers. A global review of fisheries and trade. FAO Fisheries and Aquaculture Technical Paper. No. 516; Toral-Granda, V., Lovatelli, A., Vasconcellos, M., Eds.; FAO: Rome, Italy, 2008; pp. 257–291. [Google Scholar]
- Dvoretsky, A.G.; Dvoretsky, V.G. Cucumaria in Russian waters of the Barents Sea: biological aspects and aquaculture potential. Front. Mar. Sci. 2021, 8, 613453. [Google Scholar] [CrossRef]
- Zakharenko, A.; Romanchenko, D.; Thinh, P.D.; Pikula, K.; Hang, C.T.T.; Yuan, W.; Xia, X.; Chaika, V.; Chernyshev, V.; Zakharenko, S.; Razgonova, M.; Chung, G.; Golokhvast, K. Features and advantages of supercritical CO2. Extraction of sea cucumber Cucumaria frondosa japonica Semper, 1868. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Levin, V.S.; Gudimova, E.N. Taxonomic interrelations of holothurians Cucumaria frondosa and C. japonica (Dendrochirotida, Cucumariidae). Zool. Zhurn. 1997, 75, 575–584. [Google Scholar]
- Avilov, S.A.; Kalinin, V.I.; Smirnov, A.V. Use of triterpene glycosides for resolving taxonomic problems in the sea cucumber genus Cucumaria (Holothorioidea, Echinodermata). Biochem. Syst. Ecol. 2004, 32, 715–733. [Google Scholar] [CrossRef]
- Mondol, M.A.M.; Shin, H.J.; Rahman, M.A. Sea cucmber glycosides: chemical structures, producing species and important biological properties. Marine Drugs 2017, 15. [Google Scholar] [CrossRef]
- Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Stonik, V.A. Non-holostane aglycones of sea cucumber triterpene glycosides. Structure, biosynthesis, evolution. Steroids 2019, 147, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, Y.; Franko, C.M.M. Acetylated triterpene glycosides and their biological activity from Holothurioidea reported in the past six decades. Marine Drugs 2016, 14, 147. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, N.B.; Mohammed, A.; Rao, C.V. Sea cucumbers metabolites as potent anti-cancer agents. Marine Drugs 2015, 13, 2909–2923. [Google Scholar] [CrossRef] [PubMed]
- Aminin, D.L.; Menchinskaya, E.S.; Pisliagin, E.A.; Silchenko, A.S; Avilov, S.A.; Kalinin, V.I. Anticancer activity of sea cucumber triterpene glycosides. Marine Drugs 2015, 6, 1202–1223. [Google Scholar] [CrossRef]
- Sajwani, F.H. Frondoside A is a potential anticancer agent from sea cucumbers. J. Cancer Res. Ther. 2019, 15, 953–960. [Google Scholar] [CrossRef]
- Hossain, A.; Dave, D; Shahidi, F. Northern sea cucumber (Cucumaria frondosa): A potential candidate for functional food, nutraceutical, and pharmaceutical sector. Marine Drugs 2020, 18, 274. [Google Scholar] [CrossRef] [PubMed]
- Fagbohun, O.F.; Joseph, J.S.; Oriyomi, O.V.; Rupasinghe, H.P.V. Saponins of North Atlantic sea cucumber: chemistry, health benefits, and future prospectives. Marine Drugs 2023, 21, 262. [Google Scholar] [CrossRef] [PubMed]
- Findlay, J.; Daljeet, A. Frondogenin, a new aglycone from the sea cucumber Cucumaria frondosa. J. Nat. Prod. 1984, 47, 320–324. [Google Scholar] [CrossRef]
- Elyakov, G.B.; Stonik, V.A.; Afiyatullov, S.S.; Kalinovsky, A.I.; Sharypov, V.F.; Korotkikh, L.Y. Native genins from glycosides of holothurians. Dokl. AN SSSR 1981, 259, 1367–1369. [Google Scholar]
- Rodriguez, J.; Riguera, R. Lefevreosides: four novel triterpenoid glycosides from the sea cucumber Cucumaria levevrei. J. Chem. Research (M) 1989, 2620–2636. [Google Scholar]
- Ilyin, S.G.; Reshetniak, M.V.; Afiyatullov, S.S.; Stonik, V.A.; Elyakov, G.B. Crystalic and molecular structure of diacetate of holost-8(9)-en-3α,16β-diol. Dokl. AN SSSR 1985, 284, 356–359. [Google Scholar]
- Girard, M.; Belanger, J.; ApSimon, J.W.; Garneau, F.-X.; Harvey, C.; Brisson, J.-R. Frondoside A. A novel triterpene glycoside from the holothurian Cucumaria frondosa. Can. J. Chem. 1990, 1990. 68, 11–18. [Google Scholar] [CrossRef]
- Avilov, S.A.; Kalinin, V.I.; Drozdova, O.A.; Kalinovsky, A.I.; Stonik, V.A. , Gudimova E.N. Triterpene glycosides from the holothurian Cucumaria frondosa. Chem. Nat. Comp. 1993, 29, 216–218. [Google Scholar] [CrossRef]
- Findlay, J.A.; Yayli, N.; Radics, L. Novel sulfated oligosaccharides from the sea cucumber Cucumaria frondosa. J. Nat. Prod. 1992, 55, 93–101. [Google Scholar] [CrossRef]
- Avilov, S.A.; Drozdova, O.A.; Kalinin, V.I.; Kalinovsky, A.I.; Stonik, V.A.; Gudimova, E.N.; Riguera, R.; Jimenez, C. Frondoside C, a new nonholostane triterpene glycoside from the sea cucumber Cucumaria frondosa: structure and cytotoxicity of its desulfated derivative. Can. J. Chem. 1998, 76, 137–141. [Google Scholar] [CrossRef]
- Yayli, N.; Findlay, J. A triterpenoid saponin from Cucumaria frondosa. Phytochemistry 1999, 50, 135–138. [Google Scholar] [CrossRef]
- Yayli, N. Minor saponins from the sea cucumber Cucumaria frondosa. Indian J. Chem. 2001, 40B, 399–404. [Google Scholar]
- Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I. Separation procedures for complicated mixtures of sea cucumber triterpene glycosides with isolation of individual glycosides, their comparison with HPLC/MS metabolomic approach, and biosynthetic interpretation of the obtained structural data. In Studies in Natural Products Chemistry; Rachman, A.U., Ed.; Elsevier Science B.V.: Amsterdam, The Netherlands, 2022; Volume 72, pp. 103–146. [Google Scholar]
- Silchenko, A.S.; Avilov, S.A.; Antonov, A.S.; Kalinovsky, A.I.; Dmitrenok, P.S.; Kalinin, V.I.; Stonik, V.A.; Woodward, C.; Collin, P.D. Glycosides from the sea cucumber Cucumaria frondosa III. Structure of frondosides A2-1, A2-2, A2-3 and A2-6, four new minor monosulfated triterpene glycosides. Can. J. Chem. 2005, 83, 21–27. [Google Scholar] [CrossRef]
- Silchenko, A.S.; Avilov, S.A.; Antonov, A.S.; Kalinovsky, A.I.; Dmitrenok, P.S.; Kalinin, V.I.; Woodward, C.; Collin, P.D. Glycosides from the sea cucumber Cucumaria frondosa. IV. Structure of frondosides A2-4, A2-7, and A2-8, three new minor monosulfaed triterpene glycosides. Can. J. Chem. 2005, 83, 2120–2126. [Google Scholar] [CrossRef]
- Silchenko, A.S.; Avilov, S.A.; Kalinovsky, A.I.; Dmitrenok, P.S.; Kalinin, V.I. , Morre, J.; Deinzer, M.L.; Woodward, C.; Collin, P.D. Glycosides from the North Atlantic sea cucumber Cucumaria frondosa V – Structures of five new minor trisulfated triterpene oligoglycosides, frondosides A7-1, A7-3, A7-4, and isofrondoside C. Can. J. Chem. 2007, 85, 626–636. [Google Scholar] [CrossRef]
- Li, X.; Roginsky, A.B.; Ding, X.Z.; Woodward, C. ; Collin. P.; Newman, R.A.; Bell, R.H.Jr.; Adrian, T.E. Review of the apoptosis pathways in pancreatic cancer and the anti-apoptotic effects of the novel sea cucumber compound, Frondoside A. Ann. N. Y. Acad. Sci. 2008, 1138, 181–198. [Google Scholar] [CrossRef]
- Al Shemaili, J.; Mensah-Brown, E.; Parekh, K.S.; Thomas, A.; Attoub, S.; Hellman, B.; Nyberg, F.; Adem, A.; Collin, P.; Adrian, T.E. Frondoside A enhances the antiproliferative effects of gemcitabine in pancreatic cancer. Europ. J. Cancer 2014, 50, 1391–1398. [Google Scholar] [CrossRef]
- Al Shemaili, J.; Parekh, K.A.; Newman, R.A.; Hellman, B.; Woodward, C.; Adem, A.; Collin, P.; Adrian, T.E. Pharmacokinetics in mouse and comparative effects of frondosides in pancreatic cancer. Marine Drugs 2016, 14, 115. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.C.Q.; Yoshimura, K. ; Kumazawa,S.; Tawata, S.; Maruta, H. Frondoside A from sea cucumber and nymphaeols from Okinawa propolis: Natural anti-cancer agents that selectively inhibit PAK1 in vitro. Drug Discov. Therapeut. 2017, 11, 110–114. [Google Scholar] [CrossRef]
- Jin, J.O.; Shastina, V.V.; Shin, S.W.; Xu, Q.; Park, J.I.; Rasskazov, V.A.; Avilov, S.A.; Fedorov, S.N.; Stonik, V.A.; Kwak, J.Y. Differential effects of triterpene glycosides, frondoside A and cucumarioside A2-2 isolated from sea cucumbers on caspase activation and apoptosis of human leukemia cells. FEBS Lett. 2009, 18, 697–702. [Google Scholar] [CrossRef]
- Sajwani, F.H.; Collin, P.; Adrian, T.E. Frondoside A potentiates the effects of conventional therapeutic agents in acute leukemia. Leuk. Res. 2017, 63, 98–108. [Google Scholar] [CrossRef]
- Al Marzouqi, N.; Iratni, R.; Nemmar, A.; Arafat, K.; Ahmed Al Sultan, M.; Yasin, J.; Collin, P.; Mester, J.; Adrian, T.E.; Attoub, S. Frondoside A inhibits human breast cancer cell survival, migration, invasion and the growth of breast tumor xenografts. Eur. J. Pharmacol. 2011, 1, 668–25. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, Y.H.; Kim, Y.; Lee, S.J. Frondoside A has an anti-invasive effect by inhibiting TPA-induced MMP-9 activation via NF-κB and AP-1 signaling in human breast cancer cells. Int. J. Oncol. 2012, 41, 933–940. [Google Scholar] [CrossRef]
- Ma, X.; Kundu, N.; Collin, P.D.; Goloubeva, O.; Fulton, A.M. Frondoside A inhibits breast cancer metastasis and antagonizes prostaglandin E receptors EP4 and EP2. Breast Cancer Res. Treat. 2012, 132, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Attoub, S.; Sulaiman, S.; Arafat, K. 14P. Butein inhibits solid tumors cell viability, colony, and tumor growth via STAT3 signaling pathway and enhance the anti-cancer effects of Frondoside-A and camptothecin. Ann. Oncol. 2019, 30, i6. [Google Scholar] [CrossRef]
- Sulaiman, S.; Arafat, K.; Al-Azawi, A.M.; AlMarzooqi, N.A.; Snah, L.; Attoub, S. Butein and frondoside-A combination exhibits additive anti-cancer effects on tumor cell viability, colony growth, and invasion and synergism on endothelial cell migration. Int. J. Mol. Sci. 2021, 31, 431. [Google Scholar] [CrossRef] [PubMed]
- Attoub, S.; Arafat, K.; Gelaude, A.; Al Sultan, M.A.; Bracke, M. ; Collin, P, Takahashi, T.; Adrian, T.E.; De Wever, O. Frondoside A suppressive effects on lung cancer survival, tumor growth, angiogenesis, invasion, and metastasis. PLoS ONE 2013, 8, 53087. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Han, H.; Chen, X.; Yi, Y.; Sun, H. Cytotoxic and apoptotic-inducing activity of triterpene glycosides from Holothuria scabra and Cucumaria frondosa against HepG2 cells. Marine Drugs 2014, 12, 4274–4290. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Rast, S.; Hauschild, J.; Otte, K.; Alsdorf, W.H.; Madanchi, R.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Dierlamn, J.; Honecker, F.; Stonik, V.A.; Bokemeyer, C.; von Amsberg, G. ; Frondoside A induces AIF-associated caspase-independent apoptosis in Burkitt’s lymphoma cells. Leuk. Limph. 2017, 58, 2905–2915. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Menchinskaya, E.S.; Venz, S.; Rast, S.; Amann, K.; Hauschild, J.; Otte, K.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Alsdorf, W.; Madanchi, R.; Bokemeyer, C.; Schumacher, U.; Walther, R.; Aminin, D.L.; Fedorov, S.N.; Shubina, L.K.; Stonik, V.A.; Balabanov, S.; Honecker, F.; von Amsberg, G. The marine triterpene glycoside frondoside A exhibits activity in vitro and in vivo in prostate cancer. Mol. Cancer Biol. 2016, 138, 2450–2465. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Madanchi, R.; Hauschild, J.; Otte, K.; Alsdorf, W.H.; Schumacher, U.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Honecker, F.; Stonik, V.A.; Bokemeyer, C.; von Amsberg, G. The marine triterpene glycoside frondoside A induces p53-independent apoptosis and inhibits autophagy in urothelial carcinoma cells. BMC Cancer 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Menchinskaya, E.S.; Aminin, D.L.; Avilov, S.A.; Silchenko, A.S.; Andryjashchenko, P.V.; Kalinin, V.I.; Stonik, V.A. Inhibition of tumor cells multidrug resistance by cucumarioside A2–2, frondoside А and their complexes with а cholesterol. Nat. Prod. Commun. 2013, 8, 1377–1380. [Google Scholar] [PubMed]
- Ru, R.; Chen, G.; Liang, X.; Cao, X.; Yuan, L.; Meng, M. Sea cucumber derived triterpenoid glycoside frondoside A: a potential anti-bladder cancer drug. Nutrients 2023, 15, 378. [Google Scholar] [CrossRef]
- Attoub, S.; Arafat, K.; Khalaf, T.; Sulaiman, S.; Iratni, R. Frondoside A enhances the anti-cancer effects of oxaliplatin and 5-fluorouracil on colon cancer cells. Nutrients 2018, 10, 560. [Google Scholar] [CrossRef]
- Xue, Y.; Fu, Y.; Zhao, F.; Gui, G.; Li, Y.; Rivero-Hinojosa, S.; Liu, G.; Li, Y.; Xia, S.; Eberhart, C.G.; Ying, M. Frondoside A inhibits an MYC-driven medulloblastoma model derived from human-induced pluripotent stem cells. Mol. Cancer. Ther. 2021, 20, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, V.I.; Volkova, O.V.; Likhatskaya, G.N.; Prokofieva, N.G.; Agafonova, I.G.; Anisimov, M.M.; Kalinovsky, A.I.; Avilov, S.A.; Stonik, V.A. Hemolytic activity of trterpene glycosides from cucumariidae family holothurians and evolution of this group of toxins. J. Nat. Toxins. 1992, 1, 17–30. [Google Scholar]
- Tangrodchanapong, T.; Sobhon, P.; Meemon, K. Frondoside A Attenuates amyloid-β proteotoxicity in transgenic Caenorhabditis elegans by suppressing its formation. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Ampofo, E.; Spater, T.; Nalbach, L.; Menger, M.D.; Laschke, M.W. The marine-derived triterpenoid frondoside A inhibits thrombus formation. Marine Drugs 2020, 18, 111. [Google Scholar] [CrossRef]
- Aminin, D.L.; Agafonova, I.G.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Stonik, V.A.; Collin, P.D.; Woodward, C. Immunomodulatory properties of frondoside A, a major triterpene glycoside from the North Atlantic commercially harvested sea cucumber Cucumaria frondosa. J. Med. Food 2008, 11, 443–453. [Google Scholar] [CrossRef]
- Aminin, D.L.; Koy, C.; Dmitrenok, P.S.; Müller-Hilke, B.; Koczan, D.; Arbogast, B.; Silchenko, A.S.; Kalinin, V.I. Avilov, S.A.; Stonik, V.A.; Collin, P.D.; Thiesen, H.-J. Deinzer, M.L.; Glocker, M.O. Immunomodulatory effects of holothurian triterpene glycosides on mammalian splenocytes determined by mass spectrometric proteome analysis. J. Proteomics 2009, 72, 886–906. [Google Scholar] [CrossRef]
Figure 1.
Products of partial acid hydrolysis of glycosidic fraction of Cucumaria frondosa. 1 – β-D-xylopyranoside of the native aglycone; 2 – β-D-quinovopyranosyl-(1→?)-β-D-xylopyranoside of the native aglycone; 3 – frondogenin, a native aglycone; 4 and 5 – products of acid catalyzed double bond migration in native aglycone. The configuration of 16-OAc is erroneously determined as α.
Figure 1.
Products of partial acid hydrolysis of glycosidic fraction of Cucumaria frondosa. 1 – β-D-xylopyranoside of the native aglycone; 2 – β-D-quinovopyranosyl-(1→?)-β-D-xylopyranoside of the native aglycone; 3 – frondogenin, a native aglycone; 4 and 5 – products of acid catalyzed double bond migration in native aglycone. The configuration of 16-OAc is erroneously determined as α.
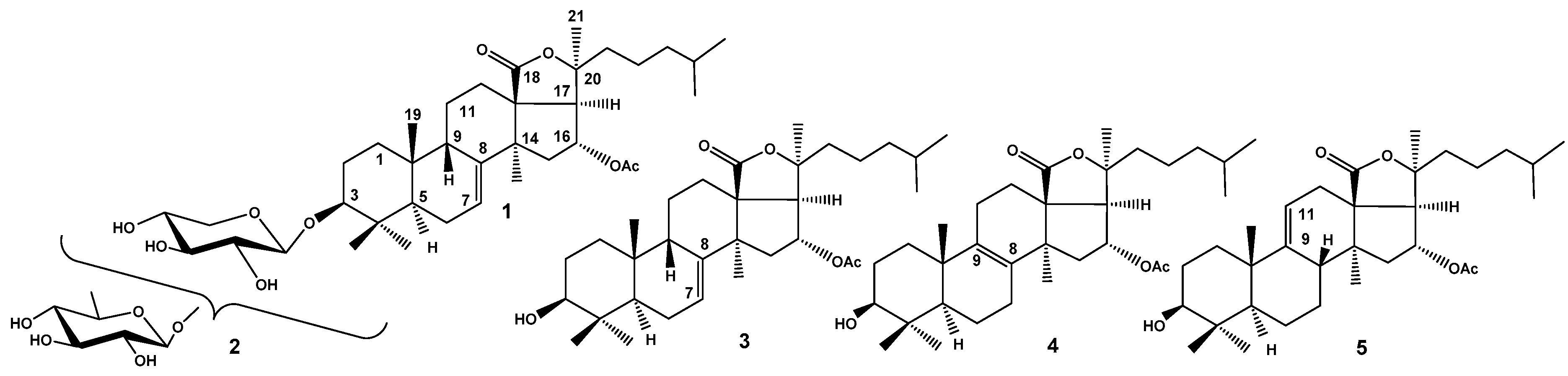
Figure 2.
The structures of frondoside A (6), major component of the glycosidic fraction of C. frondosa and its minor precursor frondoside A1 (7).
Figure 2.
The structures of frondoside A (6), major component of the glycosidic fraction of C. frondosa and its minor precursor frondoside A1 (7).
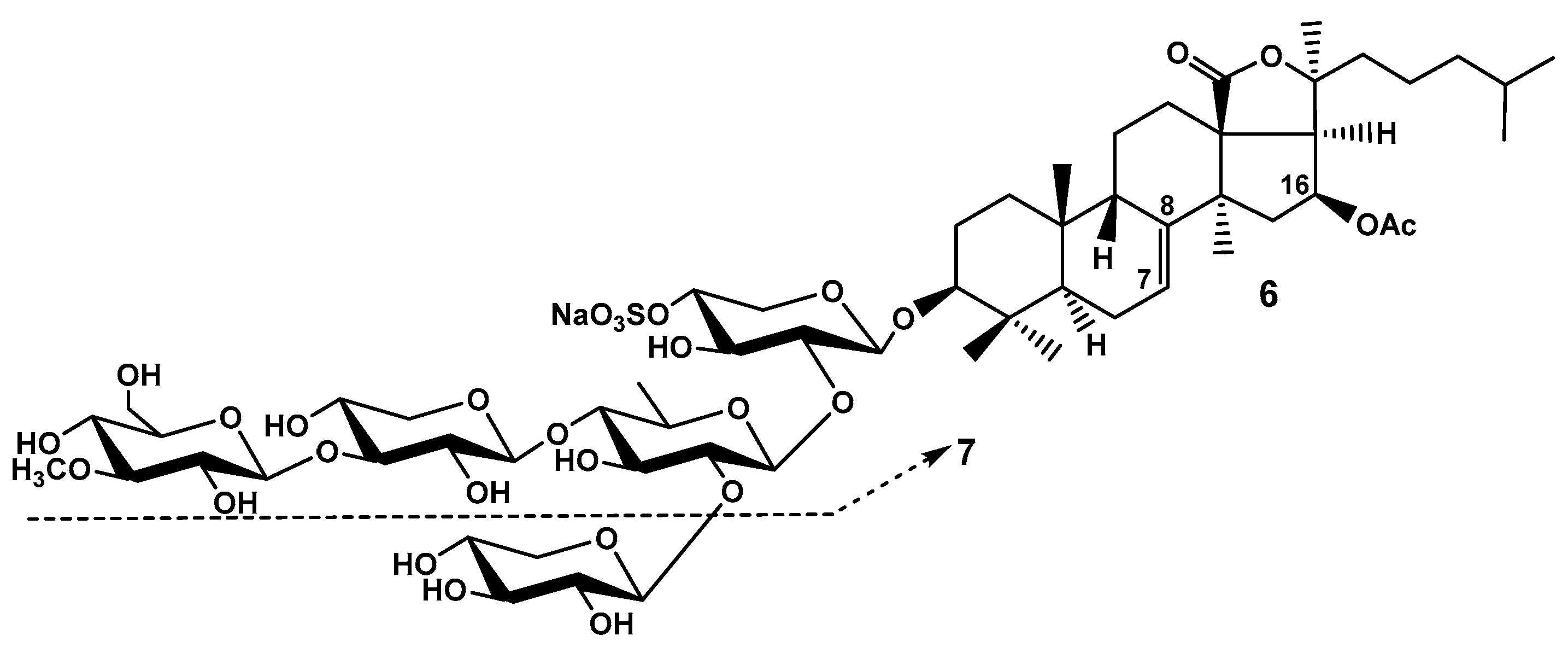
Figure 3.
Frondoside B (8) – a disulfated branched pentaoside having no 16-OAc in the aglycone and “frondecaside” (9) –a hypothetic dimeric hexasulfated decasaccharide.
Figure 3.
Frondoside B (8) – a disulfated branched pentaoside having no 16-OAc in the aglycone and “frondecaside” (9) –a hypothetic dimeric hexasulfated decasaccharide.
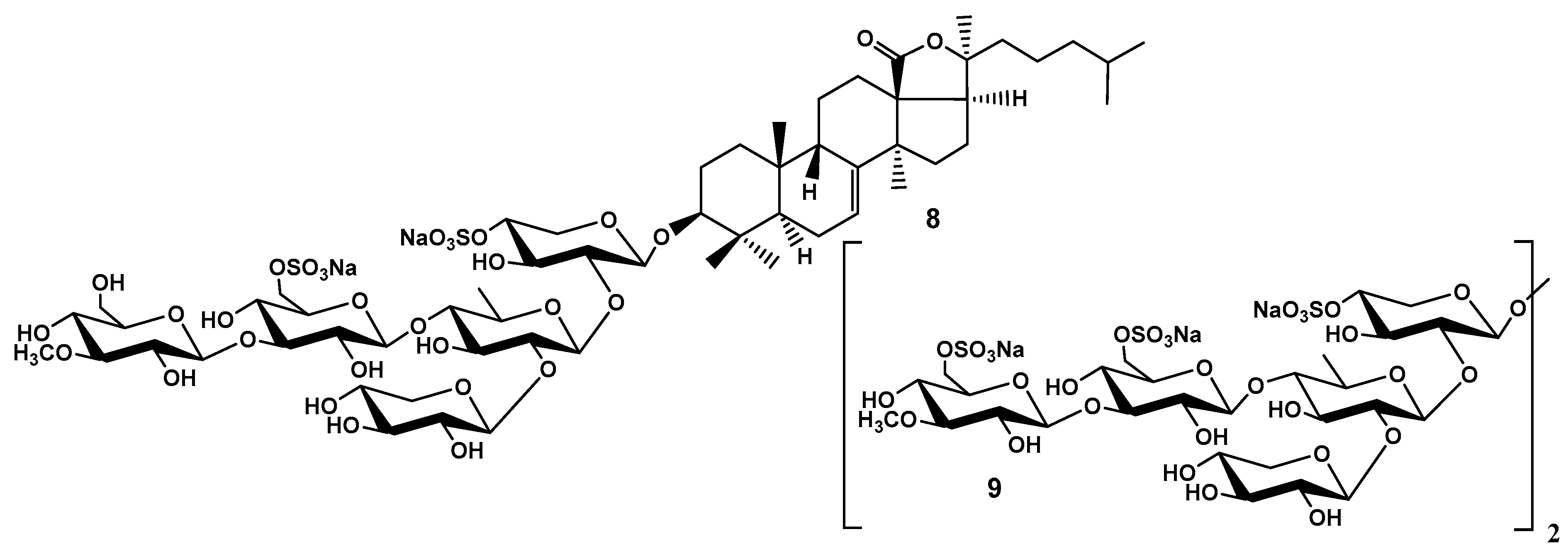
Figure 4.
Frondoside C (10) – the main component of the most polar glycosidic subfraction from C. frondosa identified in unseparable glycoside mixture homogenous by carbohydrate chains and isolated as desulfated derivative 11.
Figure 4.
Frondoside C (10) – the main component of the most polar glycosidic subfraction from C. frondosa identified in unseparable glycoside mixture homogenous by carbohydrate chains and isolated as desulfated derivative 11.
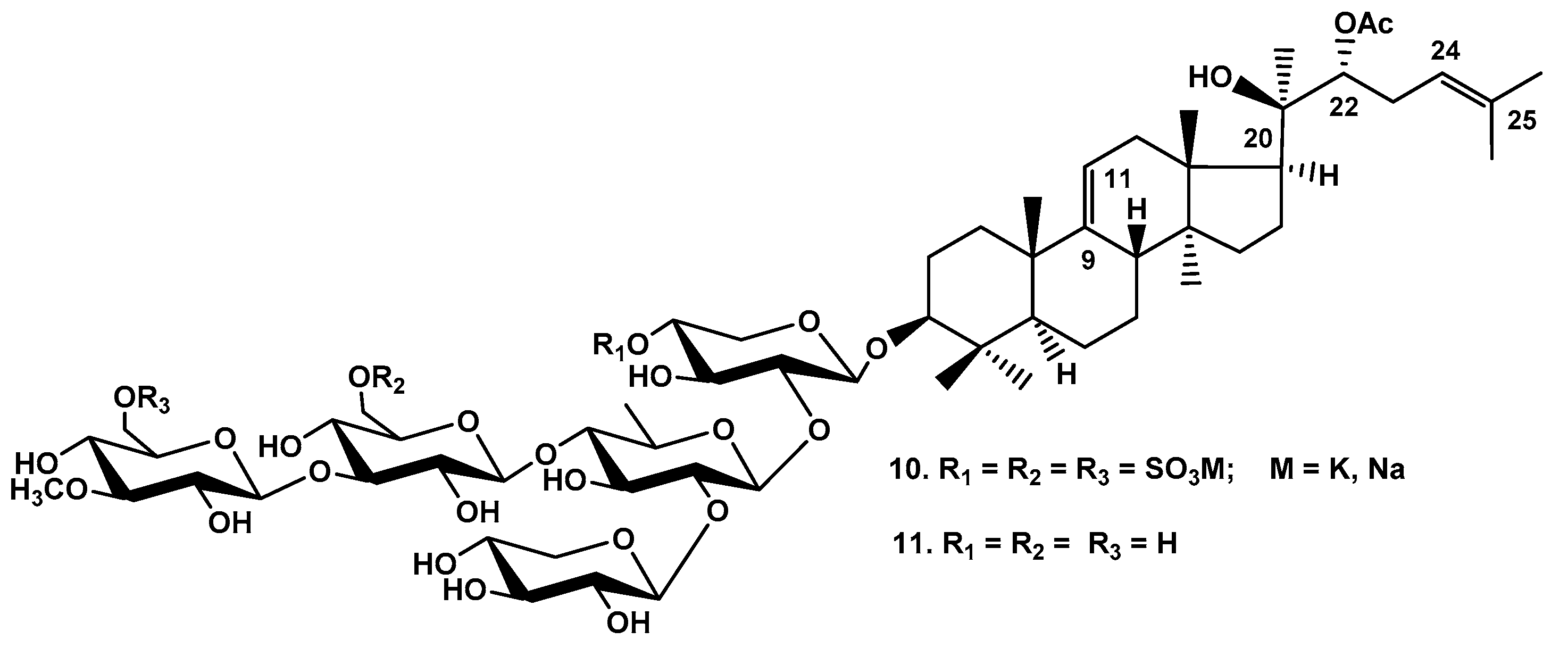
Figure 5.
Frondoside D (12), minor monosulfated triterpene glycoside from C. frondosa.
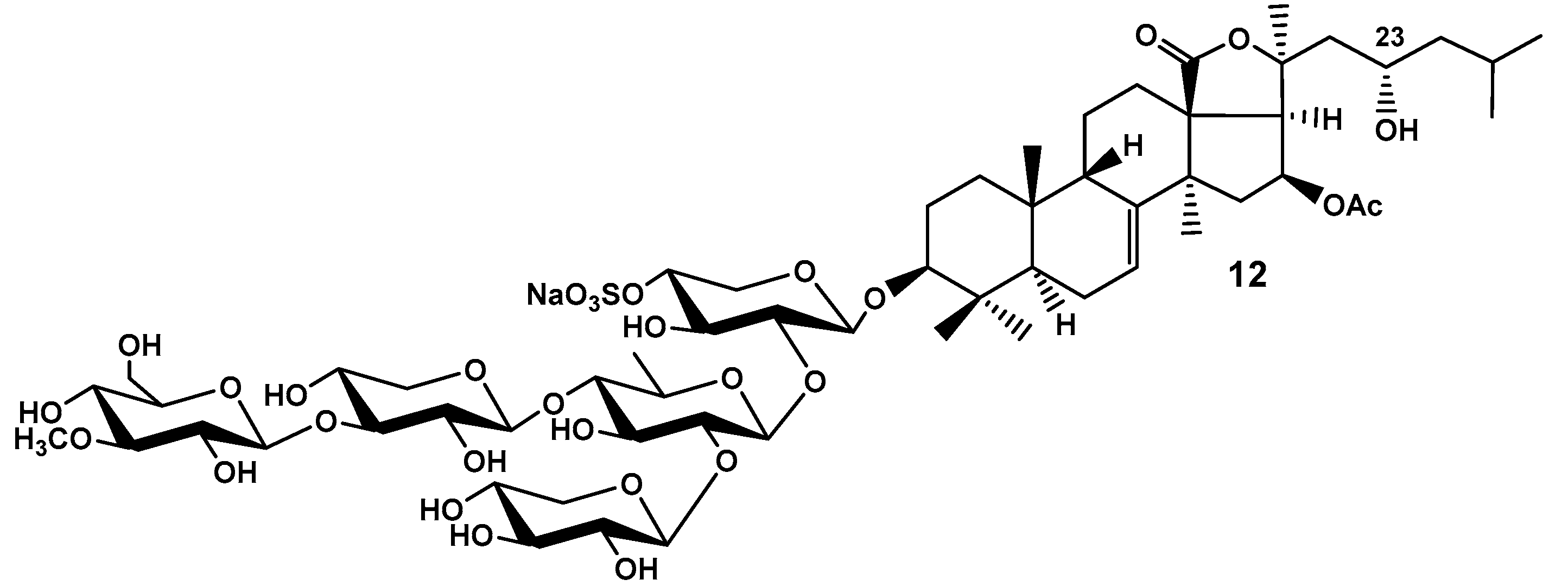
Figure 6.
Frondoside F (13), hypothetical minor triterpene glycoside from C. frondosa.
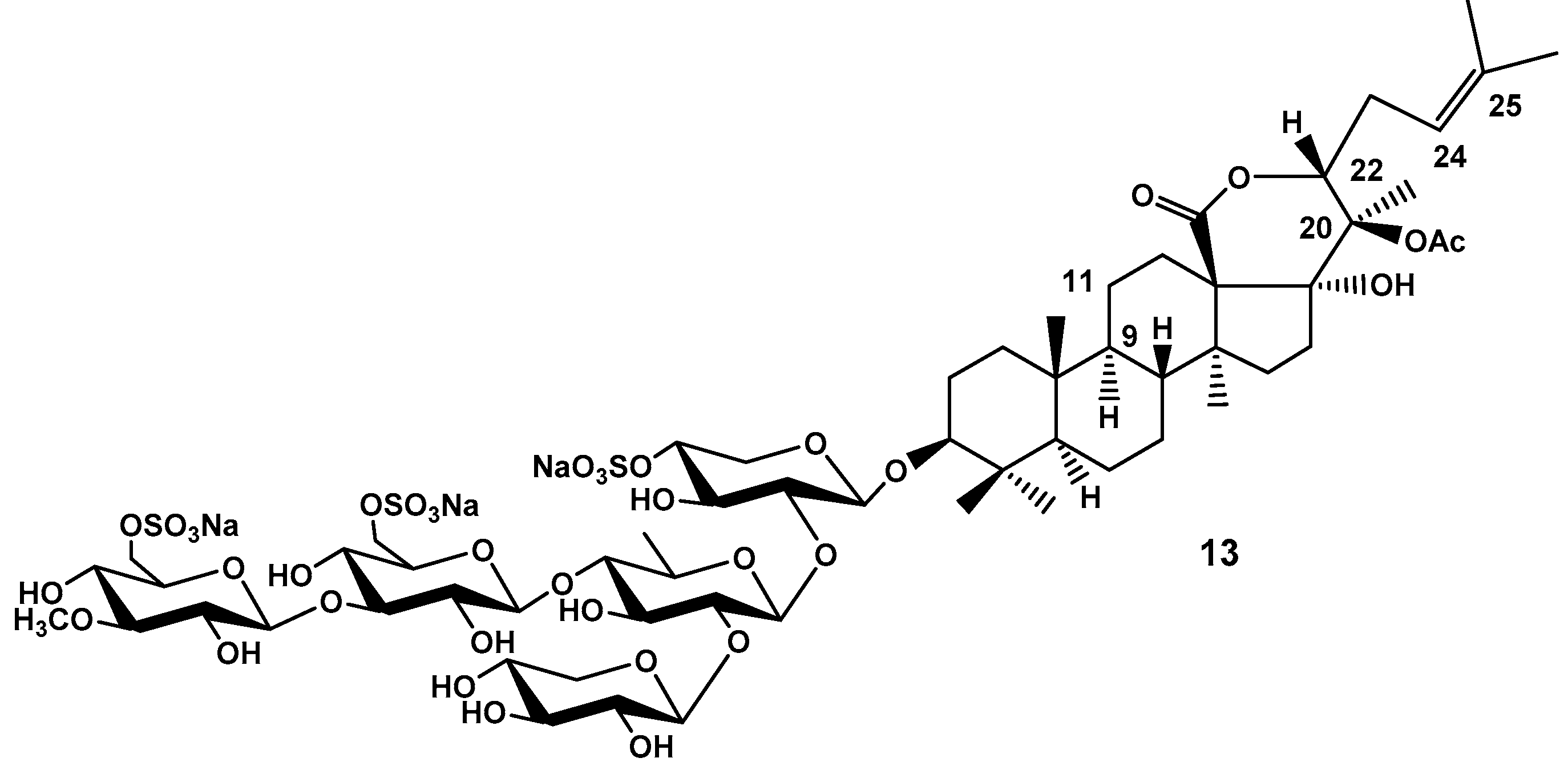
Figure 7.
Frondosides E1 (14) and E2 (15), minor hypothetical glycosides from C. frondosa, isolated as inseparable mixture.
Figure 7.
Frondosides E1 (14) and E2 (15), minor hypothetical glycosides from C. frondosa, isolated as inseparable mixture.
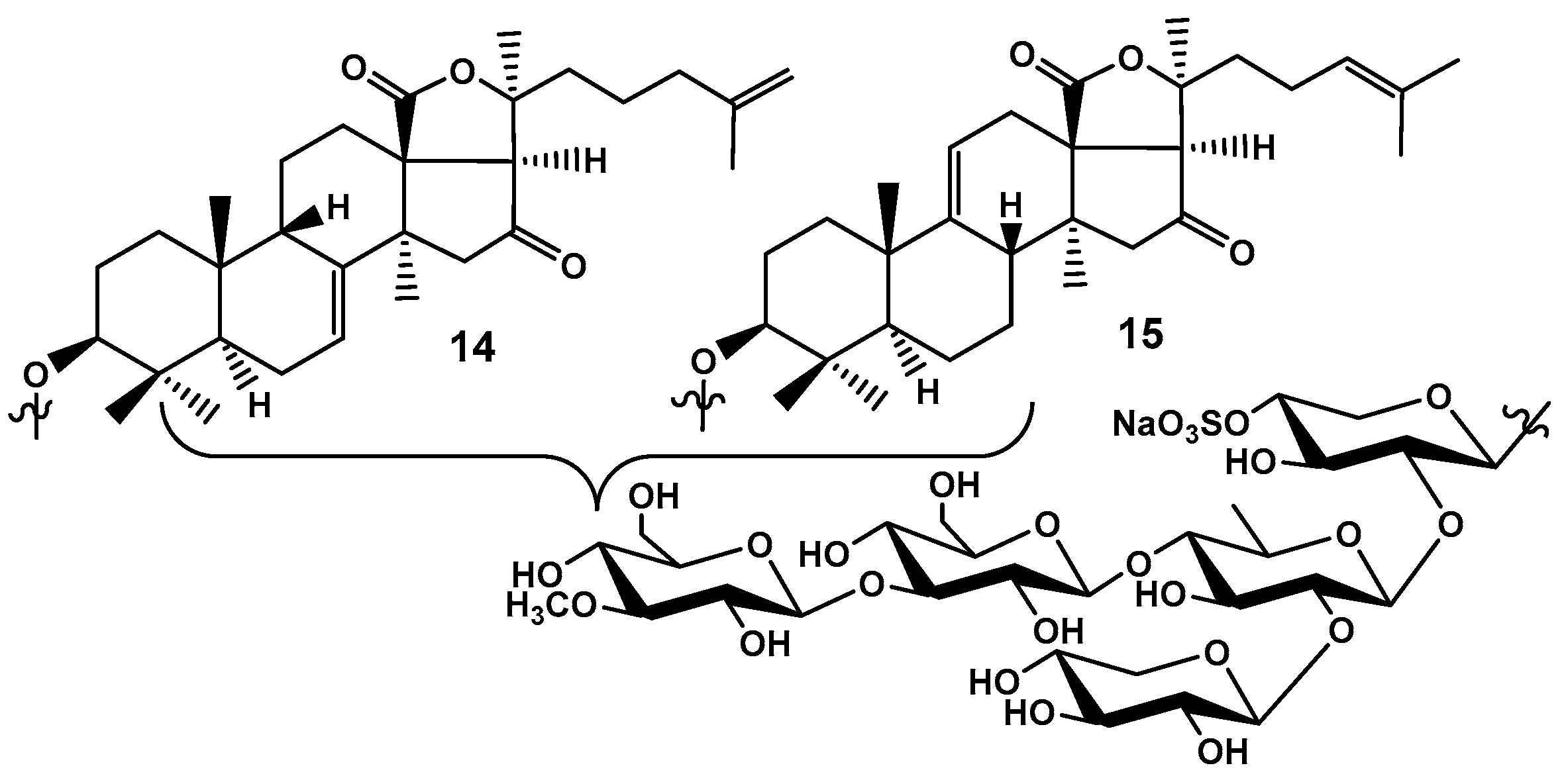
Figure 8.
Frondosides A2-1 (16), A2-2 (17), A2-3 (18), and A2-6 (19), minor monosulfated branched pentaosides isolated from C. frondosa using dynamic modificators for HPLC mobile phase and a chiral column.
Figure 8.
Frondosides A2-1 (16), A2-2 (17), A2-3 (18), and A2-6 (19), minor monosulfated branched pentaosides isolated from C. frondosa using dynamic modificators for HPLC mobile phase and a chiral column.
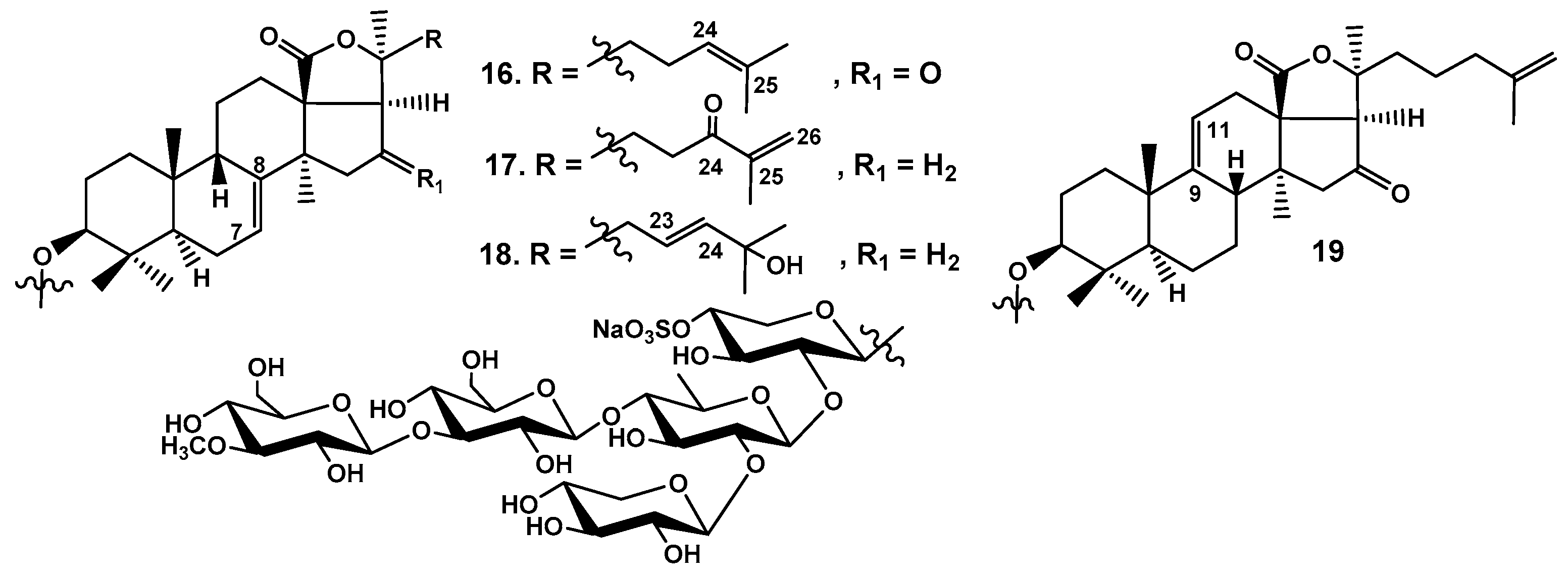
Figure 9.
Frondosides A2-4 (20), A2-7 (21), A2-8 (22), minor monosulfated branched pentaosides isolated from C. frondosa using a dynamic modificator for HPLC mobile phase and a chiral column.
Figure 9.
Frondosides A2-4 (20), A2-7 (21), A2-8 (22), minor monosulfated branched pentaosides isolated from C. frondosa using a dynamic modificator for HPLC mobile phase and a chiral column.
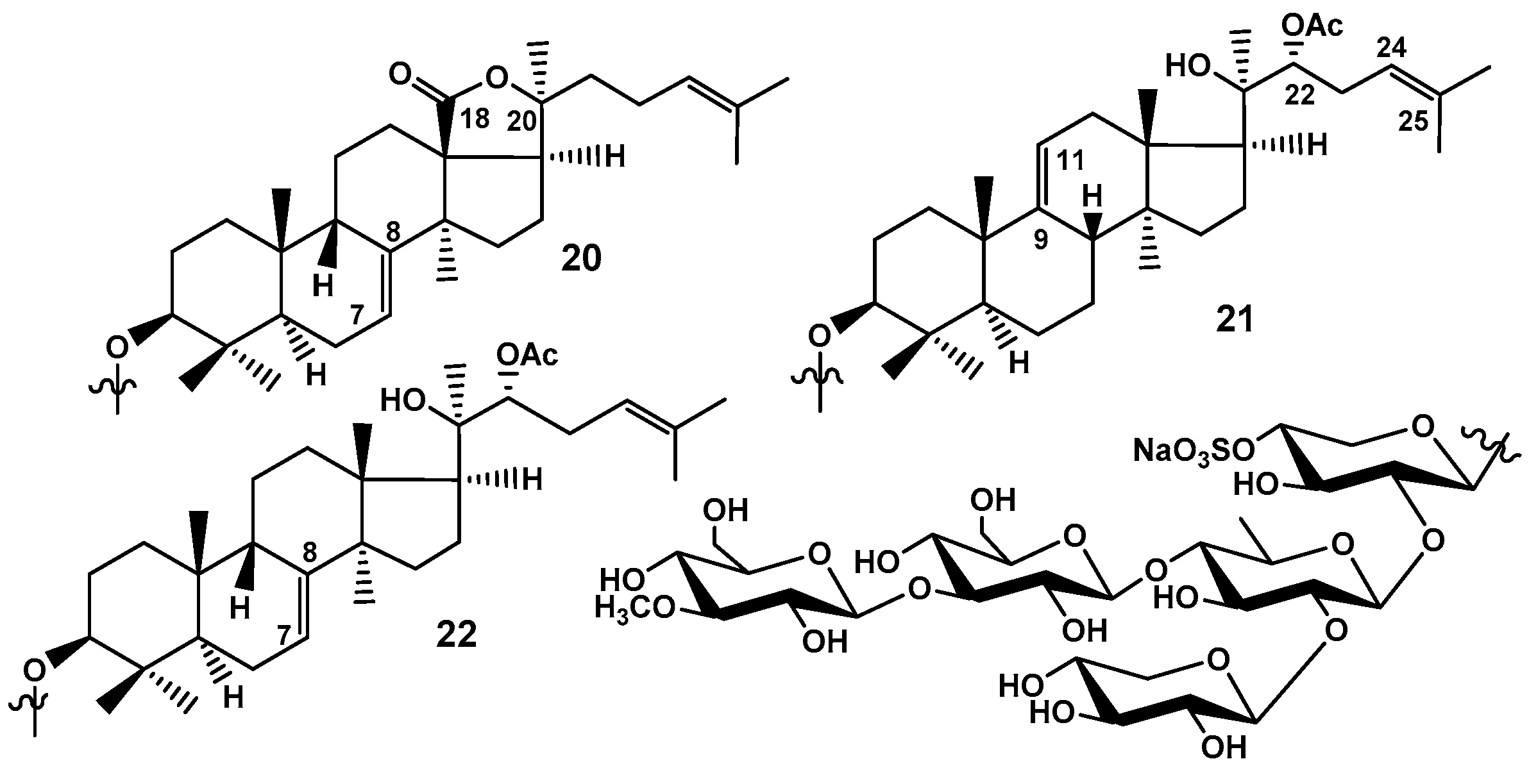
Figure 10.
Minor trisulfated branched pentaosides isolated from C. frondosa: 23 – frondoside A7-1; 24 – frondoside A7-2; 25 – frondoside A7-3; 26 – frondoside A7-4; 27 – isofrondoside C; 28 – desulfated derivative obtained from isofrondoside C; 10 – frondoside C firstly isolated as individual native substance; 29 – koreoside A isolated earlier from Cucumaria koreaensis; 30 – isokoreoside A earlier found in Cucumaria conicospermium and firstly isolated as native compound.
Figure 10.
Minor trisulfated branched pentaosides isolated from C. frondosa: 23 – frondoside A7-1; 24 – frondoside A7-2; 25 – frondoside A7-3; 26 – frondoside A7-4; 27 – isofrondoside C; 28 – desulfated derivative obtained from isofrondoside C; 10 – frondoside C firstly isolated as individual native substance; 29 – koreoside A isolated earlier from Cucumaria koreaensis; 30 – isokoreoside A earlier found in Cucumaria conicospermium and firstly isolated as native compound.
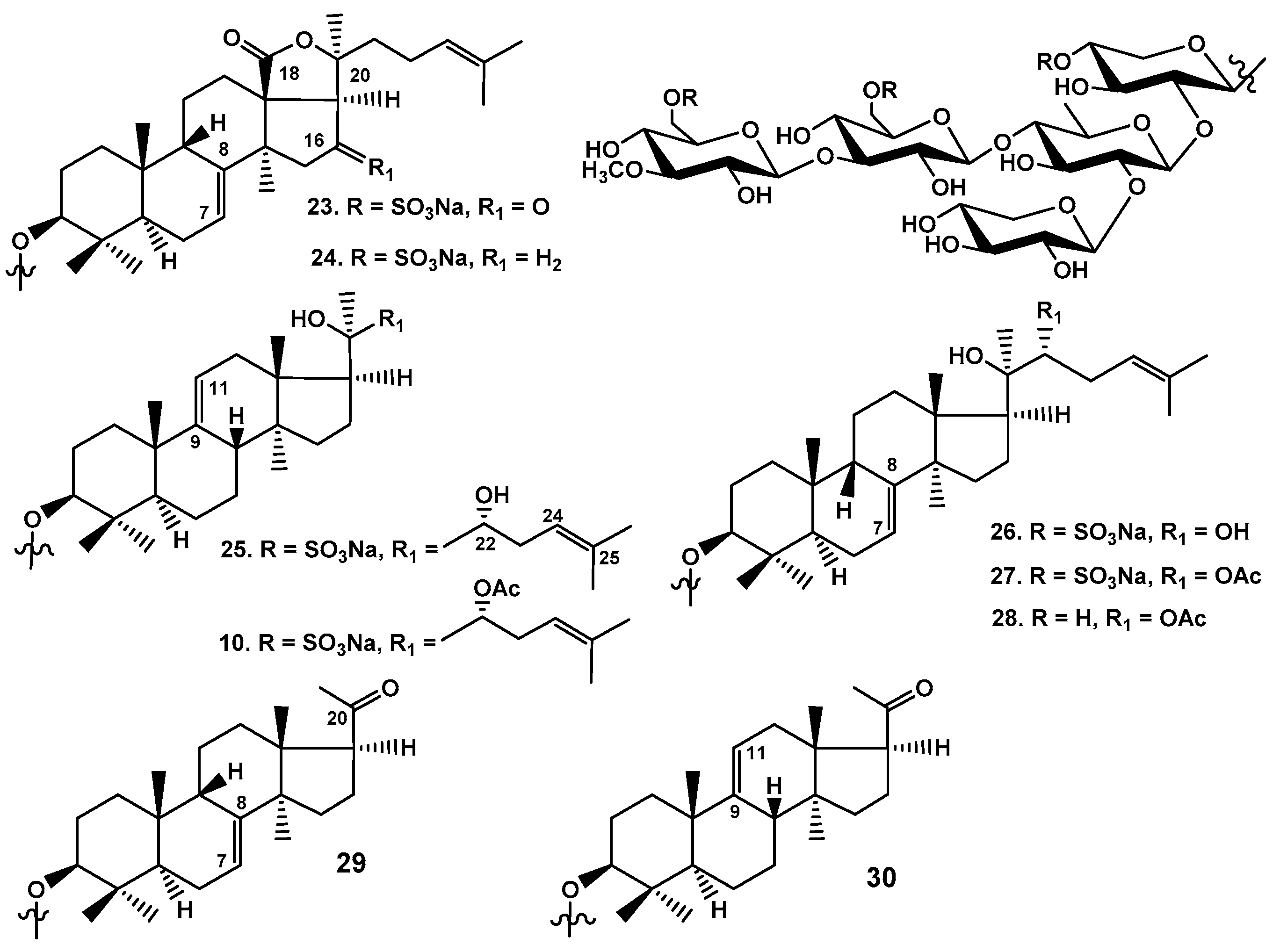
Figure 11.
Scheme of biosynthesis of main groups of carbohydrate chains in triterpene glycosides of C. frondosa. The left branch is the path to the glycosides with a xylose residue as the third sugar. The right branch is the path to the glycosides having glucose as the third monosaccharide residue and additional sulfate groups.
Figure 11.
Scheme of biosynthesis of main groups of carbohydrate chains in triterpene glycosides of C. frondosa. The left branch is the path to the glycosides with a xylose residue as the third sugar. The right branch is the path to the glycosides having glucose as the third monosaccharide residue and additional sulfate groups.
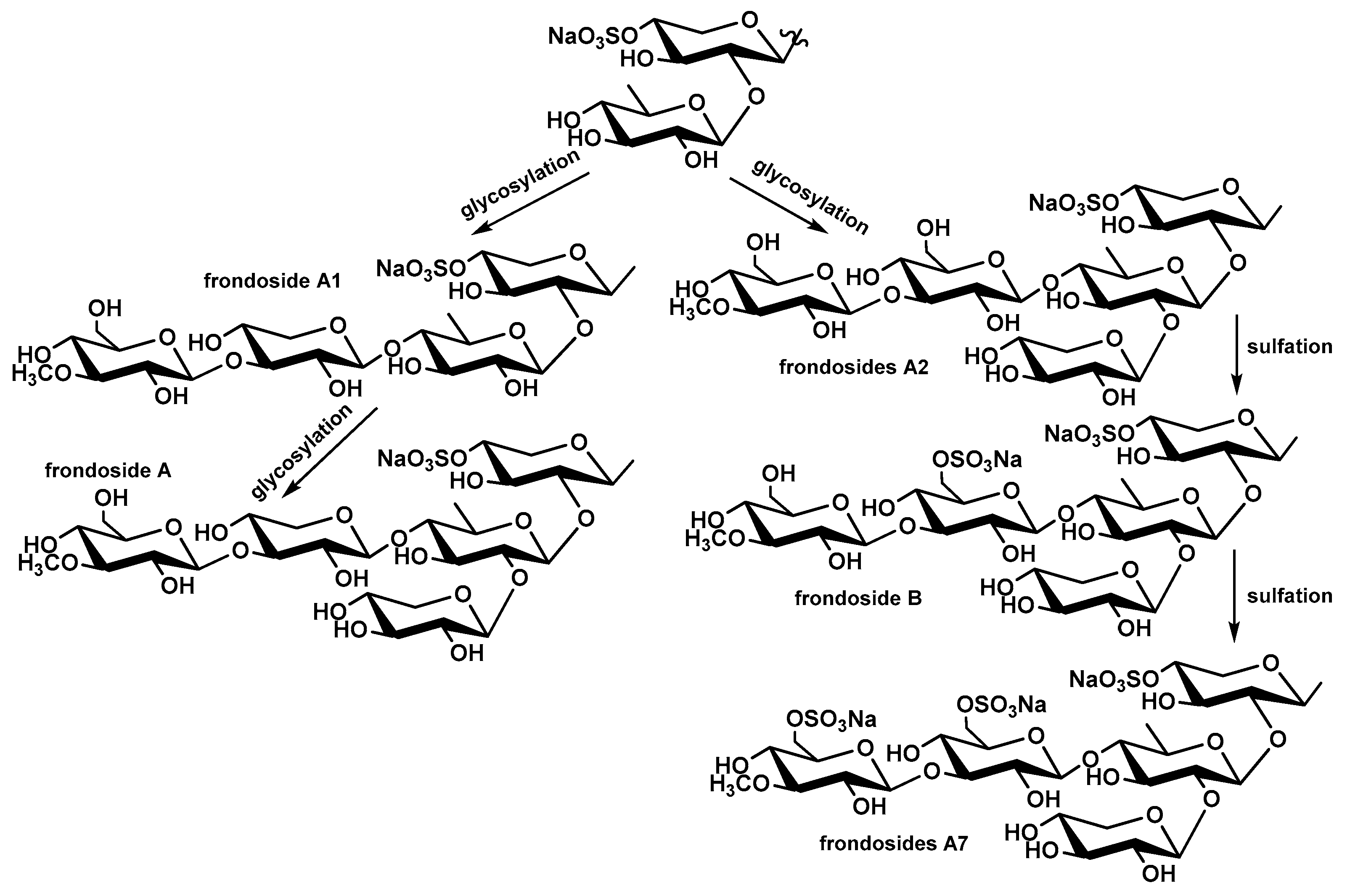
Figure 12.
The general paths of aglycone biosynthesis in triterpene glycosides of C. frondosa from lanostane precursors with 7(8)- or 9(11)-double bonds with formation of corresponding holostane [with an 18(20)-lactone] and non-holostane [having no a lactone] derivatives.
Figure 12.
The general paths of aglycone biosynthesis in triterpene glycosides of C. frondosa from lanostane precursors with 7(8)- or 9(11)-double bonds with formation of corresponding holostane [with an 18(20)-lactone] and non-holostane [having no a lactone] derivatives.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



