
Submitted:
03 October 2023
Posted:
05 October 2023
You are already at the latest version
Abstract
A warning sign for impending cardio-vascular events is not fully established. In the process to plaque rupture, formation of vulnerable plaque is important, and oxidized cholesterols play important role in its progression. Furthermore, the significance of vasa vasorum penetrating medial smooth muscle layer and rich in atheromatous lesion should be paid attention. Cardio-ankle vascular index (CAVI) is new arterial stiffness index of the arterial tree from the origin of the aorta to the ankle. CAVI reflects functional stiffness in addition to structural stiffness. Rapid rise of CAVI means medial smooth muscle cell contraction, and strangling vasa vasorum. We reported rapid rise of CAVI in peoples after big earthquake, following high frequency of cardiovascular events. And we met several cases who showed rapid rise of CAVI a few weeks or months before suffering cardiovascular events. To explain these sequences of events, we proposed a hypothesis; A rapid rise of CAVI means medial smooth muscle contraction, strangling vasa vasorum, leading ischemia, necrosis of vulnerable plaque, then, the plaque ruptures. In individuals having high CAVI, further rapid rise of CAVI might be a warning sign for impending cardiovascular events. In such case, treatments to decrease CAVI had better to be taken soon.
Keywords:
cholesterol oxidative products
; atheromatous lesion
; plaque rupture
; vasa vasorum
; cardio-ankle vascular index
Introduction
Arteriosclerotic disease is the major cause of mortality and morbidity among cardiovascular diseases not only in developed countries, but also in developing countries [1]. As to the formation of atherosclerotic lesions in the arterial wall, many hypotheses have been proposed. Cholesterol was first thought to be a cause of atheroma formation [2]. However, cholesterol itself is an important lipid component in the cell membrane. Then, denatured low density lipoproteins (LDL) containing cholesterol was proposed as a cause of form cell formation in atheroma [3,4,5]. Later, Ross et al proposed an injury response hypothesis [6] to explain intimal thickening composed of the synthetic type of smooth muscle cell (SMC) proliferation, leading to stenosis of the arterial cavity. However, stenosis was not necessarily observed in the occluded coronary artery by thrombus [7]. Then, plaque rupture theory was proposed [8]. In that theory, vulnerable plaque is formed subsequent to a chronic inflammatory reaction in the intima. In chronic inflammatory reaction, macrophages digest the surrounding tissues, thinning a covering cap. When thin cap ruptures, formed thrombus occlude the rumen of the artery, leading to myocardial infarction. However, the real causes to provoke inflammation in atheromatous lesion were not fully clarified, although many cytokines relating to inflammatory reactions were investigated. Oxysterol products are toxic to cells [9] Oxysterol products might be a ringleader to make vulnerable plaque.
On the other side, it is reported that atheromatous lesions are rich in vasa vasorum [10,11]. Ischemia of intimal atheromatous lesions produces angiogenic factors such as vascular endothelial growth factor; subsequently neovascularization develops from the adventitia into intimal atheroma through the medial smooth muscle layer to form a network of vasa vasorum. The significance of vasa vasorum penetrating through the medial smooth muscle layer into intimal plaque, has not been fully discussed.
Arterial stiffness is composed of structural stiffness and also functional stiffness. Pulse wave velocity (PWV) has been used as an index reflecting arterial stiffness [12]. However, PWV is essentially affected by blood pressure at the measuring time [13]; thus, PWV is inappropriate as an index to assess functional stiffness. Recently, the cardio-ankle vascular index (CAVI) was proposed as an index of arterial stiffness of the arterial tree from the origin of the aorta to the ankle. CAVI is not affected by blood pressure at the measuring time [14,15]. CAVI reflects functional stiffness as well as structural stiffness [16,17]. Functional stiffness reflects medial smooth muscle contraction.
By the way, a predictive sign for impending cardio-vascular events is not established yet, although many risk factors were mentioned for the formation of atherosclerosis and of vulnerable plaque. It is known that cardiovascular events occurred frequently after natural disasters such as big earth quakes [18] and sporting events [19]. We measured CAVI of the people living 300km far away from epicenter of the great East Japan Earthquake in 2011. CAVI values of the healthy people and also of the people with atherosclerotic diseases were transiently enhanced [18]. In addition, we met several cases, who showed rapid rise of CAVI several weeks or months before suffering cardiovascular events. To explain these sequences, we proposed new hypothesis explaining the processes to plaque rupture; A rapid rise of CAVI means medial smooth muscle contraction, strangling vasa vasorum, leading ischemia and necrosis. Finally, plaque rupture occurs [20].
In this review, the role of oxidized cholesterol in the formation of atheromatous lesions in the intima, leading to vulnerable plaque is discussed. Then, the meanings of vasa vasorum rich in atheromatous lesion and penetrating medial smooth muscle layer from adventitia and of rapid CAVI rise which reflects arterial smooth muscle contraction, were discussed. Then, the process to propose the hypothesis that medial smooth muscle contraction induces plaque rupture, was introduced. Finally, various treatments to improve CAVI were reviewed.
1. Reconsideration concerning the formation of vulnerable plaque
The formation of atherosclerotic lesions has been studied by many researchers, and several hypotheses have been proposed [2,3,4,5,6,7]. One important component is believed to be cholesterol, as cholesterol has been found to be the main deposited compound in human atheromatous lesions in the artery [2]. Cholesterol is carried by LDL, intermediate-density lipoproteins (IDL) and/or small dense LDL and is believed to enter the intimal lesion via intimal endothelial cells [3]. There, denatured LDL is taken up by macrophages, and the macrophages are converted to foam cells via the scavenger pathway, making a lipid pool in the intima [4,5]. Then, an inflammatory reaction occurs and vulnerable plaque is formed, leading to plaque rupture [7,8]. In this process of progression of atherosclerosis, how the lipid pool was formed in the adjacent to intimal lamina, and how the inflammatory reaction occurred in the lipid pool must be reconsidered to understand the process to plaque rupture.
1). The formation of the lipid pool in the intima of the arterial wall:
One first question is the location of the lipid pool in the arterial wall. By human pathological studies showed that cholesterol deposition in the artery is not observed just under the intimal endothelial cell layer [21,22]. The lipid pool is situated deep in the intima, adjacent to the internal lamina. Cholesterol-rich lipoproteins such as LDL, IDL and/or small dense LDL in the blood had been generally thought to enter through the intimal endothelial layer from the lumen of the artery [23.24]. If the main entrance of cholesterol-rich lipoproteins was the surface of the endothelial layer of the lumen of the artery, the lipid pool would be formed just under the endothelial layer. However, why the cholesterol pool develops in the deep area adjacent to the internal lamina in the intima, not just beneath the endothelial layer? Recently, several studies have reported that vasa vasorum exist in the arterial wall and become abundant in advanced atherosclerotic lesions [25,26], but the significance of vasa vasorum has not been fully discussed. Nourishment of arteries had been believed mainly by diffusion from the lumen of the vessel. However, recently it has also been reported that cholesterol-rich lipoproteins and nutrients are transported by vasa vasorum from the adventitia and enter the arterial wall [27] (Figure 1). The vasa vasorum originate at the adventitia, penetrate the medial smooth muscle layer and internal lamina, and then reach the intimal area. If cholesterol-carrying lipoproteins, such as LDL, IDL and small dense LDL, are carried by vasa vasorum from the adventitia to the intima through the medial smooth muscle layers, it is easily accepted that a lipid pool composed of cholesterol is formed near the internal lamina at the deep intimal layer.
2). How does the lipid pool developed in the intima make progress to vulnerable plaque -The role of oxidized cholesterol for provoking inflammatory reaction -
Deposited cholesterol itself might not be toxic, because cholesterol is a natural organic compound and a part of the cell membrane, and is also ingredients of hormones and bile as mentioned above. Then, how does the cholesterol pool form and invade the surrounding area? One possible explanation is based on oxidized cholesterols. In atherosclerotic lesions, there are many oxidized cholesterols, such as 7α-ketocholesterol and 7α-hydroxycholesterol [28,29]. Those oxysterols are generated by various oxidative stresses, such as diabetes mellitus, smoking and aging during deposition in the lipid pool [30,31]. Furthermore, oxysterols are enzymatically produced [32]. The toxicity of oxysterols has been investigated in several studies. Deposited lipid containing oxysterol induces migration of SMCs [33], whereas 7-ketocholesterol enhances MMP-9 activity in THP-1 cells [34]; it also induces inflammation and angiogenesis [35]. Thus, oxysterols themselves might be the main cause to provoke an inflammatory reaction in atheromatous lesions. Furthermore, Ohtsuka et al reported that deposited lipid containing oxysterol induces apoptosis of SMCs [36] (Figure 2A). Additionally, 7-ketocholeterol induces apoptosis of cultured arterial SMCs [37]. We also observed apoptosis of SMCs in human coronary artery (Figure 2B). These results suggest that the cholesterol pool might expand by extinguishing the surrounding SMCs by inducing apoptosis in the intimal region, rendering the complicated lesion fragile. From these findings, it seems that the risk of cholesterol for arteriosclerosis is not only caused by serum LDL or IDL cholesterol levels, but also due to the amount of oxidized cholesterols in the intima. It is likely that those oxidized cholesterols in the lipid pool emerge into circulation. Hitsumoto et al reported that the serum level of 7-ketocholestrol is related to the progression of coronary atherosclerosis [38]. Endo et al reported that serum 7-ketocholesterol is significantly higher in multiple risk factors subjects (39.5 ng/ml) compared to non-multiple risk factors subjects (30.1 ng/ml). Furthermore, 7-ketocholesterol is increased according to the number of concurrent coronary risk factors [39]. From these observations and phenomena, anti-oxidative therapy in addition to lowering serum cholesterol levels must be strictly promoted to prevent the progression of atherosclerosis
Chronic kidney disease is one of the arteriosclerotic diseases, and it is known that oxidative stress influences its progression and complications. However, the role of antioxidants has not been clarified. It is known that probucol, a cholesterol lowering agent that also lower HDL-cholesterol, has antioxidant activity [40]. We administered probucol to patients with diabetic nephropathy and followed up for 5 years. Probucol administration decreased LDL-cholesterol. Furthermore, probucol prevented the progression of CKD and decreased the proportion of patients requiring hemodialysis, compared to non-administered patients [41]. Yamashita et al reported that probucol administration to familiar hypercholesterolemic patients decreases the incidence of cardiovascular events in Japan, despite the accompanying decrease in HDL-cholesterol [42]. Once again, the importance of antioxidative therapy for the prevention of arteriosclerotic diseases must be re-emphasized.
3). Neovascularization of vasa vasorum in the arterial wall
Recent observations have confirmed the presence of vasa vasorum and neocapillaries in atherosclerotic plaques as mentioned before [25]. This angiogenic process can be initiated by hypoxia in the intima [26]. Hypoxia is induced by impairment of oxygen diffusion from lumina by intimal thickening; this hypoxic condition induces expression and release of angiogenic factors (e.g. VEGF) [43]. Thus, the vasa vasorum density is higher in atherosclerotic prone areas, and neovascularization facilitates the supply of nutrients and oxygen to intimal lesion [27], as illustrated in Figure 1. This neovascularization also augments further lipid deposition, macrophage infiltration, and intimal SMC proliferation.
The features of vasa vasorum are that it is derived from the adventitia, passes through medial smooth muscle layers, and reaches the intimal lesion. As a resultant, blood flows into the intimal layer via vasa vasorum, and the sprouted neo-capillaries become under the control of the contractional state of medial smooth muscle.
As it is not clear how much blood is carried by vasa vasorum to the intimal atheromatous lesion, we observed the surgical endarterectomy of the carotid artery in a patient suffering from cervical artery stenosis. Just after peeling off the proliferative intimal layer of the stenotic carotid artery, the surface of denuded medial SMC layer was immediately covered with blood, which exuded from the medial smooth muscle layer. This blood was clearly transported by vasa vasorum penetrating the medial SMC layers from the adventitia (Figure 3A). During this operation, when the surface of the denuded medial smooth muscle layer was covered with a sheet of gauze dipped in saline, the bleeding on the surface of the smooth muscle layer did not stop, however, when this surface was covered with noradrenaline-dipped gauze, the bleeding stopped (Figure 3B) [20]. This study and observation series revealed that blood in the atheromatous lesion of the intima was transported by vasa vasorum which penetrated the medial smooth muscle layer. Based on these observations, it is noteworthy that the blood supply into the intimal atheromatous lesion was controlled by the status of medial smooth muscle contraction.
The next questions are how to guess 1) the degree of the atheromatous lesion, and 2) the blood supply condition into the intimal atheromatous lesion from the adventitia via vasa vasorum, by non-invasive measurement.
2. The meaning of a rapid increase in CAVI
1). What is CAVI?
To know the stages of arteriosclerosis noninvasively is difficult. Arterial stiffness indicates the degree of arteriosclerosis and several indices reflecting arterial stiffness have been proposed. PWV reflects arterial stiffness and can be easily calculated by dividing the length of the artery by the time during the pulse spreading from one end to the other end. Various kinds of PWV such as cfPWV [44] and baPWV [45] have been presented in the many papers published in the last 30 years. However, PWV depends on blood pressure at the measuring time [13]. Therefore, PWV is not suitable to measure accurate arterial stiffness, and as such, it has been difficult to clarify vascular function using PWV.
More recently, CAVI was proposed as a new arterial stiffness index of the arterial tree from the origin of the aorta to the ankle [14]. The equation was derived from the stiffness parameter β [46] and modified using the Bramwell-Hill equation [47]. The notable feature of CAVI is its independency from blood pressure at measuring time, because theoretically stiffness parameter β is independent from blood pressure. This has also been proven in clinical studies using α blocker, doxazosin and β blocker [16]. When the β-blocker metoprolol decreases blood pressure, CAVI remains changed, whereas, when doxazosin decreases blood pressure, CAVI also decreases; these experiments indicate that CAVI is independent on blood pressure at measuring time, and reflects functional stiffness based on the status of SMC contraction.
2). CAVI reflects the degree of atherosclerosis
The cut-off value of CAVI for arteriosclerosis was tentatively defined as 9. CAVI increases with age, and it is reported to be high in arteriosclerotic patients with coronary artery disease, cerebral infarction and CKD [15]. Figure 4 shows pictures of various atherosclerotic stages of the aorta. CAVI=7.3 indicates a nearly normal aorta in a 50-year-old woman. CAVI =11.0 represents far advanced stages of atherosclerosis. Furthermore, patients with the most coronary risk factors, such as hypertension, diabetes mellitus, hypercholesterolemia, sleep apnea syndrome, and metabolic syndrome, showed a significantly high CAVI [15]. The above findings indicate that CAVI reflects the structural stiffness of the arterial tree.
3). CAVI also reflects functional stiffness of the artery
An interesting feature of CAVI is that it reflects functional stiffness, which is derived from the contractional state of arterial smooth muscles. This conclusion can be deduced by the following studies. The administration of the adrenaline receptor α-blocker, doxazosin decreases CAVI [16]. Nitroglycerin administration also decreases CAVI among control subjects and patients with arteriosclerotic diseases [17]. The latter evidence indicates that the medial smooth muscle layer, even in advanced stages of atherosclerosis, retains the ability to contract or dilate in response to surrounding stimuli. Septic conditions decrease CAVI, accompanied by a decrease in blood pressure [48]. Further, various studies on the effects of various agents using rabbits showed rapid changes in CAVI. For example, nitroglycerin administration decreases CAVI of rabbits, as in humans [17,49]. Sakuma et al, reported that angiotensin II acutely increases arterial stiffness as monitored by CAVI in anesthetized rabbits [50]. Miyazaki et al reported that enhanced intracranial pressure provokes enhancement of blood pressure and CAVI in rabbits [51]. Taken together, those results indicate that CAVI reflects contraction or dilation of arterial smooth muscle; a rapid change in CAVI indicates the changes in arterial smooth muscle contraction.
4). CAVI just after huge natural disaster and stress
There were several papers reporting that much more cardiovascular events occurred immediately after huge disasters, sometimes accompanying high blood pressure [52] . Trichopoulos et al reported that psychological stress induced fatal heart attacks at the time of the earthquake [53]. Dobson et al reported that the number of heart attacks increased after the Newcastle earthquake [54]. Leor et al also reported that sudden cardiac death was triggered by an earthquake [55]. Regarding the cause of those earthquake-related sequence of cardiovascular events, psychological stress was discussed, however, the precise mechanism for such events was not fully evaluated. The huge earthquake “Great East Japan earthquake” occurred in the northeastern part of Japan in 2011. At our research center, Toho University Sakura Medical Center, in Chiba located 300 km from the epicenter, we measured CAVI of healthy colleagues working in our hospital just after the earthquake, and 2 and 4 weeks later. Compared with the value just after the earthquake, CAVI was decreased after 2 and 4 weeks. Moreover, patients with arteriosclerotic diseases who came to our hospital periodically showed a transient increase in CAVI at 1 week after the earthquake, and a decrease after a few months [18]. In the several days following the earthquake, the number of patients suffering from brain hemorrhage who visited our hospital increased by two-fold compared with before the earthquake. The number of deaths in Sakura City during the few months following the earthquake was 30% higher compared with the same time frame in the years leading up to the disaster [18]. The severe disaster provoked cerebrovascular death, accompanied by a rapid rise in CAVI.
Furthermore, we encountered a patient, whose CAVI rapidly increased, then, suffered from brain hemorrhage 2 weeks later. Another patient showed increased CAVI, then, suffered from myocardial infarction 4 months later. One patient suffered from aortic dissecting aneurysm one month after a rapid rise in CAVI. While these cases might be just incidental, they may point to the importance of CAVI as an indicator. Large prospective studies are needed to confirm the relationship between a rapid rise in CAVI and subsequent cardio-cerebrovascular events. However, if the relationship between cardiovascular events and the rapid rise in CAVI is confirmed, the following hypothesis might be proposed.
3. Smooth muscle cell contraction hypothesis for plaque rupture: The role of rapid rise in CAVI
The proposed mechanism for the occurrence of cardiovascular events after the rapid rise in CAVI in cases showing high CAVI, is as follows: at first, an atheromatous lesion is formed by infiltration of cholesterol-rich lipoproteins such as LDL, IDL and small dense LDL in lipid pool. Then, deposited cholesterols become oxidative products due to oxidative stress. The resultant oxysterols act as a toxic substance in the intima, which provokes inflammatory reactions such as stimulation of macrophages, promoting migration of SMCs and inducting apoptosis of SMCs, and eventually leading to the elongation of the lipid pool. Furthermore, the inflammatory reaction and anoxia induced by intima thickening stimulate neovascularization. Then, the vasa vasorum become enriched. At this stage, CAVI gradually increases. A high CAVI means the presence of vulnerable plaque in the arterial tree and coronary artery. These processes of arteriosclerotic lesion formation and subsequent SMC contraction and plaque rupture are illustrated in Figure 1 and Figure 5, respectively.
When a huge natural disaster, such as a severe earthquake and major psychological stress occur for people with a high CAVI, a further rapid rise in CAVI is observed. This means that medial SMCs contract. This contraction of SMC strangles the vasa vasorum which penetrate the medial SMC layer, consequently, the blood supply to the intimal atheromatous lesions ceases. This process leads to vulnerable plaque ischemia and necrosis. Subsequently, the rupture of vulnerable plaque occurs. Considering brain arteries in those with a high CAVI, a rapid rise in CAVI might mean disruption of the blood supply to brain arteries, leading to necrosis of brain arteries, following brain hemorrhage. In case of coronary arteries in those with a high CAVI, a rapid rise in CAVI means a lack of blood supply to vulnerable plaque. Plaque rupture causes thrombus to form in the lumen of the artery, leading to myocardial infarction. In case of the aorta, a rapid rise in CAVI causes strangulation of vasa vasorum and cessation of the blood supply to the intima, leading to necrosis of the intimal layer. Intimal necrosis renders dissection of the aorta. Osada et al reported that aortic dissection in the outer third of the media is related to the significance of vasa vasorum in the triggering process [56]. If the authors dare to state tentative values, a basal value of CAVI >10 may expect to have advanced stages of arteriosclerosis with vulnerable plaque, which is close to the mean CAVI value (7.84) in Japanese + 2 x standard deviation (2x1.07). Enhanced ΔCAVI of >0.7 would correspond to two fold the variation coefficient (3.7%) of CAVI measurement [14]. Thus, we proposed that a rapid rise in CAVI (ΔCAVI >0.7) in those with basal CAVI (>10) might be a prodrome of serious cardiovascular events in the near future.
Based on the “SMC contraction hypothesis of plaque rupture” which we previously proposed [20], monitoring of CAVI in daily life might be useful to elicit a caution of the life-threatening risk for occurrence of cardiovascular events in a very near future, although further prospective studies are needed to confirm this hypothesis.
4. Treatments to improve CAVI
The aforementioned sequences of events occurring in the arteriosclerotic lesion might support the “SMC contraction hypothesis for plaque rupture”. The next question is how to prevent a rapid rise in CAVI, which induces plaque rupture in advanced stages. To date, several treatment or methods to improve CAVI have been reported [15]. Among them, main treatments were summarized in Table 1.
1). Life style changes
Weight reduction decreases CAVI in obese diabetic patients. Continuous positive airway pressure therapy for sleep apnea syndrome decreases CAVI [refer to 15]. Waon therapy, in which patients are placed in a dry sauna at 60 degree for 15 minutes, improves CAVI [57].
2). Controlling high blood pressure
Antihypertensives such as angiotensin-receptor blockers (olmesartan), and calcium channel blockers (cilnidipine and efonidipine) are reported to decrease CAVI [refer to 15].
3). Control of diabetic mellitus
The response of CAVI to glucose-lowering treatment depends on the type of agent. Pioglitazone, the rapid-acting insulin, dipeptidyl peptidase 4 inhibitor, anagliptin, and the new sulfonylurea, glimepiride, are reported to decrease CAVI [refer to 15].
4). Among lipid lowering agents
Among the many available statins, pitavastatin decreases CAVI. Bezafibrate and eicosapentanoic acid are also reported to decrease CAVI [refer to 15].
5). Health Supplements
As for supplements, sirtuin enhancers, resveratrol and s-equal are reported to decrease CAVI in a double-blind study. CAVI might be a useful index to evaluate the effects of supplements [refer to 15].
Further studies are necessary to confirm that decreased CAVI improves the risk of mortality and morbidity.
Conclusions and Future Directions
To prevent the progression of atherosclerosis, antioxidant therapy should be re-emphasized. The roles of vasa vasorum in supplying the blood to arteriosclerotic lesion, and of medial smooth muscle contraction in the progression of vulnerable plaque might be much paid attention. As an index reflecting medial smooth muscle contraction, a rapid rise of CAVI might be a warning sigh for impending cardiovascular events.
In order to forecast impending cardiovascular events, the periodic monitoring of CAVI is recommended.
To prevent the cardiovascular events, a rapid rise of CAVI should be treated with various treatments, especially relieving mental stress. To strengthen this hypothesis, large prospective studies are necessary.
Author Contributions
Shirai K and Shimizu K wrote the manuscript. All authors have read and have agreed to the published version of the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created nor generated in this manuscript.
Acknowledgements
We would like to express our thanks to the following men and women for contributing to establish our hypothesis. Members of Internal Medicine, Toho University Sakura Medical Center: Mrs. Fusako Watanabe, Mrs. Noriko Ishihara, Mr. Hitoshi Watanabe, Mr. Takeyoshi Murano, Dr. Shoichiro Hashiguchi, Dr. Yo Miyashita, Dr. Itoh Yoshiaki, Dr Mitsuya Tozuka, Dr. Tomokazu Oyama, Dr. Hiroshi Ozaki, Dr. Kei Endo, Dr. Haruki Imamura, Dr. Takahiro Nakagami Department of Clinical Pathology, Toho University Sakura Medical Center: Dr. Noriaki Kameda, Dr. Hiroyuki Hiruta Department of Pharmacology and Therapeutics, Faculty of Pharmaceutical Sciences, Toho University, Japan: Yoshinobu Nagasawa Faculty of Pharmaceutical Sciences, Toho University, Japan: Prof. Takehiko Yajima, Associate Prof. Muneo Morishita.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Anitschkow, N.; Chalatow, S. Ueber experimentelle cholesterinsteatose und ihre bedeutung für die entstehung einiger pathologischer prozesse. Zentralbl. Allg. Pathol. Anat. 1913, 24, 1–9. (in German). [Google Scholar]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Witztum, J.L. History of discovery: Oxidized low-density lipoprotein and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2311–2316. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Lipoprotein metabolism in the macrophage: Implications for cholesterol deposition in atherosclerosis. Ann. Rev. Biochem. 1983, 52, 223–261. [Google Scholar] [CrossRef]
- Ross, R.; Glomset, J. The pathogenesis of atherosclerosis. N. Engl. J. Med. 1976, 295, 369–377, 420–425. [Google Scholar] [CrossRef]
- Muller, J.E.; Tofler, G.H.; Stone, P.H. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation. 1989, 79, 733–743. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the Mechanisms of Acute Coronary Syndromes. The “Vulnerable Plaque” and Superficial Erosion. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef]
- Vejux A, Lizard G. Cytotoxic effects of oxysterols associated with human diseases: Induction of cell death (apoptosis and/or oncosis), oxidative and inflammatory activities, and phospholipidosis. Mol Aspects Med. 2009, 30, 153–70. [Google Scholar] [CrossRef]
- Sedding, D.G.; Boyle, E.C.; Demandt, J.A.F.; Sluimer, J.C.; Dutzmann, J.; Haverich, A.; Bauersachs, J. Vasa Vasorum Angiogenesis: Key Player in the Initiation and Progression of Atherosclerosis and Potential Target for the Treatment of Cardiovascular Disease. Front. Immunol. 2018, 9, 706. [Google Scholar] [CrossRef]
- Doyle, B.; Caplice, N. Plaque neovascularization and antiangiogenic therapy for atherosclerosis. J. Am. Coll. Cardiol. 2007, 49, 2073–2080. [Google Scholar] [CrossRef]
- Asmar, R. Pulse wave velocity principles and measurement. In Arterial Stiffness and Pulse Wave Velocity; Asmar, R., O’Rourke, M.F., Safar, M., Eds.; Elsevier: Amsterdam, Netherlands, 1999; pp. 25–55. [Google Scholar]
- Nye, E.R. The effect of blood pressure alteration on the pulse wave velocity. Br. Heart J. 1964, 266, 261–265. [Google Scholar] [CrossRef]
- Shirai, K.; Utino, J.; Otsuka, K.; Takata, M. A novel blood pressure-independent arterial wall stiffness parameter; cardio-ankle vascular index (CAVI). J. Atheroscler. Thromb. 2006, 13, 101–107. [Google Scholar] [CrossRef]
- Saiki, A.; Ohira, M.; Yamaguchi, T.; Nagayama, D.; Shimizu, N.; Shirai, K.; Tatsuno, I. New Horizons of Arterial Stiffness Developed Using Cardio-Ankle Vascular Index (CAVI). J. Atheroscler. Thromb. 2020, 8, 732–748. [Google Scholar] [CrossRef] [PubMed]
- Shirai, K.; Song, M.; Suzuki, J.; Kurosu, T.; Oyama, T.; Nagayama, D.; Miyashita, Y.; Yamamura, S.; Takahashi, M. Contradictory effects of β1- and α1-aderenergic receptor blockers on cardio-ankle vascular stiffness index (CAVI) - The independency of CAVI from blood pressure. J. Atheroscler. Thromb. 2011, 18, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Shimizu, K.; Takahashi, M.; Tatsuno, I.; Shirai, K. The Effect of Nitroglycerin on Arterial Stiffness of the Aorta and the Femoral-Tibial Arteries. J. Atheroscler. Thromb. 2017, 10, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Takahashi, M.; Shirai, K. A huge earthquake hardened arterial stiffness monitored with cardio-ankle vascular index. J. Atheroscler. Thromb. 2013, 20, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Leeka J, Schwartz BG, Kloner RA. Sporting events affect spectators' cardiovascular mortality: it is not just a game..Am J Med. 2010, 123, 972–7. [CrossRef]
- Shimizu K, Takahashi M, Sato S, Saiki A, Nagayama D, Harada M, Miyazaki C, Takahara A, Shirai K. Rapid Rise of Cardio-Ankle Vascular Index May Be a Trigger of Cerebro-Cardiovascular Events: Proposal of Smooth Muscle Cell Contraction Theory for Plaque Rupture.Vasc Health Risk Manag. 2021, 17, 37–47. [CrossRef]
- Shiomi, M. The History of the WHHL Rabbit, an Animal Model of Familial Hypercholesterolemia (I) - Contribution to the Elucidation of the Pathophysiology of Human Hypercholesterolemia and Coronary Heart Disease. J. Atheroscler. Thromb. 2020, 27, 105–118. [Google Scholar] [CrossRef]
- Otsuka, F.; Kramer, M.C.; Woudstra, P.; Yahagi, K.; Ladich, E.; Finn, A.V.; de Winter, R.J.; Kolodgie, F.D.; Wight, T.N.; Davis, H.R.; Joner, M.; Virmani, R. Natural Progression of Atherosclerosis from Pathologic Intimal Thickening to Late Fibroatheroma in Human Coronary Arteries: A Pathology Study. Atherosclerosis. 2015, 241, 772–782. [Google Scholar] [CrossRef]
- Abumrad, N.A.; Cabodevilla, A.G.; Samovski, D.; Pietka, T.; Basu, D.; Goldberg, I.J. Endothelial Cell Receptors in Tissue Lipid Uptake and Metabolism. Circ. Res. 2021, 128, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Dabagh, M.; Jalali, P.; Tarbell, J.M. The transport of LDL across the deformable arterial wall: the effect of endothelial cell turnover and intimal deformation under hypertension. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H983–96. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.K.; Heistad, D.D. [The vasa vasorum of the arteries.] J. Mal. Vasc. 1996, 21 (Suppl. C), 266–269 (in French), 266–269. (in French). [Google Scholar]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–24. [Google Scholar] [CrossRef]
- Subbotin, V.M. Neovascularization of coronary tunica intima (DIT) is the cause of coronary atherosclerosis. Lipoproteins invade coronary intima via neovascularization from adventitial vasa vasorum, but not from the arterial lumen: a hypothesis. Theor. Biol. Med. Model. 2012, 9, 11. [Google Scholar] [CrossRef]
- Brown, A.J.; Jessup, W. Oxysterols and atherosclerosis. Atherosclerosis. 1999, 142, 1–28. [Google Scholar] [CrossRef]
- Poli, G.; Sottero, B.; Gargiulo, S.; Leonarduzzi, G. Cholesterol oxidation products in the vascular remodeling due to atherosclerosis. Mol. Aspects. Med. 2009, 30, 180–189. [Google Scholar] [CrossRef]
- Garcia-Cruset, S.; Carpenter, K.L.; Guardiola, F.; Stein, B.K.; Mitchinson, M.J. Oxysterol profiles of normal human arteries, fatty streaks and advanced lesions. Free Radic. Res. 2001, 35, 31–41. [Google Scholar] [CrossRef]
- Griffiths, W.J.; Yutuc, E.; Abdel-Khalik, J.; Crick, P.J.; Hearn, T.; Dickson, A.; Bigger, B.W.; Hoi-Yee Wu, T.; Goenka, A.; Ghosh, A.; Jones, S.A.; Covey, D.F.; Ory, D.S.; Wang, Y. Metabolism of Non-Enzymatically Derived Oxysterols: Clues from sterol metabolic disorders. Free Radic. Biol. Med. 2019, 144, 124–133. [Google Scholar] [CrossRef]
- Brown, A.J.; Watts, G.F.; Burnett, J.R.; Dean, R.T.; Jessup, W. Sterol 27-hydroxylase acts on 7-ketocholesterol in human atherosclerotic lesions and macrophages in culture. J. Biol. Chem. 2000, 275, 27627–33. [Google Scholar] [CrossRef] [PubMed]
- Oyama, T.; Miyashita, Y.; Kinoshita, K.; Watanabe, H.; Shirai, K.; Yagima, T. Effect of deposited lipids in atheromatous lesions on the migration of vascular smooth muscle cells. J. Atheroscler. Thromb. 2002, 9, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, H.; Miyashita, Y.; Watanabe, H.; Shirai, K. Enhancement of MMP-9 activity in THP-1 cells by 7-ketocholesterol and its suppression by the HMG-CoA reductase inhibitor Fluvastatin. J. Atheroscler. Thromb. 2005, 12, 308–314. [Google Scholar] [CrossRef]
- 35.Amaral, J.; Lee, J.W.; Chou, J.; Campos, M.M.; Rodríguez, I.R. 7-Ketocholesterol induces inflammation and angiogenesis in vivo: a novel rat model. PLoS One. 2013, 8, e56099. [Google Scholar] [CrossRef]
- 36.Ohtsuka, M.; Miyashita, Y.; Shirai, K. Lipids deposited in human atheromatous lesions induce apoptosis of human vascular smooth muscle cells. J. Atheroscler. Thromb. 2006, 13, 256–262. [Google Scholar] [CrossRef]
- Miyashita, Y.; Ozaki, H.; Koide, N.; Otsuka, M.; Oyama, T.; Itoh, Y.; Mastuzaka, T.; Shirai, K. Oxysterol-induced apoptosis of vascular smooth muscle cells is reduced by HMG-CoA reductase inhibitor, pravastatin. J. Atheroscler. Thromb. 2002, 9, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Hitsumoto, T. : Takahashi, M.: Iizuka, T.: Shirai, K. Clinical significance of serum 7-ketocholesterol concentrations in the progression of coronary atherosclerosis. J. Atheroscler. Thromb. 2009, 16, 363–370. [Google Scholar] [CrossRef]
- Endo, K. : Oyama, T.: Saiki, A.: Ban, N.: Ohira, M.: Koide, N.: Murano, T.: Watanabe, H.: Nishii, M.: Miura, M.: Sekine, K.: Miyashita, Y.: Shirai, K. Determination of serum 7-ketocholesterol concentrations and their relationships with coronary multiple risks in diabetes mellitus. Diabetes Res. Clin. Pract. 2008, 80, 63–68. [Google Scholar] [CrossRef]
- Zimetbaum, P.; Eder, H.; Frishman, W. Probucol: pharmacology and clinical application. J. Clin. Pharmacol. 1990, 30, 3–9. [Google Scholar] [CrossRef]
- Endo, K.; Saiki, A.; Yamaguchi, T.; Sakuma, K.; Sasaki, H.; Ban, N.; Kawana, H.; Nagayama, D.; Nagumo, A.; Ohira, M.; Oyama, T.; Murano, T.; Miyashita, Y.; Yamamura, S.; Suzuki, Y.; Shirai, K.; Tatsuno, I. Probucol suppresses initiation of chronic hemodialysis therapy and renal dysfunction-related death in diabetic nephropathy patients: Sakura study. J. Atheroscler. Thromb. 2013, 20, 494–502. [Google Scholar] [CrossRef]
- Yamashita, S.; Arai, H.; Bujo, H.; Masuda, D.; Ohama, T.; Ishibashi, T.; Yanagi, K.; Doi, Y.; Nakagawa, S.; Yamashiro, K.; Tanabe, K.; Kita, T.; Matsuzaki, M.; Saito, Y.; Fukushima, M.; Matsuzawa, Y; PROSPECTIVE Study Group. Probucol Trial for Secondary Prevention of Atherosclerotic Events in Patients with Coronary Heart Disease (PROSPECTIVE). J. Atheroscler. Thromb. 2021, 28, 103–123. [Google Scholar] [CrossRef]
- Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN Role of lipids and intraplaque hypoxia in the formation of neovascularization in atherosclerosis. Ann Med. 2017, 49, 661–677. [CrossRef]
- Laurent, S.; Cockcroft, J.; Van Bortel, L.; et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur. Heart J. 2006, 27, 2588–2605. [Google Scholar] [CrossRef] [PubMed]
- Yamashina, A.; Tomiyama, H.; Takeda, K.; Tsuda, H.; Arai, T.; Hirose, K.; Koji, Y.; Hori, S.; Yamamoto, Y. Validity, reproducibility, and clinical significance of noninvasive brachial-ankle pulse wave velocity measurement. Hypertens. Res. 2002, 25, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Handa, H.; Nagasawa, S.; Okumura, A.; Moritake, K. Stiffness and elastic behavior of human intracranial and extracranial arteries. J. Biomech. 1980, 13, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Bramwell, J.C.; Hill, A.V. Velocity of the pulse wave in man. Proc. Roy. Soc. B: Biol. 1922, 93, 298–306. [Google Scholar]
- Nagayama, D.; Imamura, H.; Endo, K.; Saiki, A.; Sato, Y.; Yamaguchi, T.; Watanabe, Y.; Ohira, M.; Shirai, K.; Tatsuno, I. Marker Of Sepsis Severity Is Associated With The Variation In Cardio-Ankle Vascular Index (CAVI) During Sepsis Treatment. Vasc. Health Risk Manag. 2019, 15, 509–516. [Google Scholar] [CrossRef]
- Chiba, T.; Sakuma, K.; Komatsu, T.; Cao, X.; Aimoto, M.; Nagasawa, Y.; Shimizu, K.; Takahashi, M.; Hori, Y.; Shirai, K.; Takahara, A. Physiological role of nitric oxide for regulation of arterial stiffness in anesthetized rabbits. J. Pharmacol. Sci. 2019, 139, 42–45. [Google Scholar] [CrossRef]
- Sakuma, K.; Shimoda, A.; Shiratori, H.; Komatsu, T.; Watanabe, K.; Chiba, T.; Aimoto, M.; Nagasawa, Y.; Hori, Y.; Shirai, K.; Takahara, A. Angiotensin II acutely increases arterial stiffness as monitored by cardio-ankle vascular index (CAVI) in anesthetized rabbits. J. Pharmacol. Sci. 2019, 140, 205–209. [Google Scholar] [CrossRef]
- Miyazaki, C.; Shimizu, K.; Nagasawa, Y.; Chiba, T.; Sakuma, K.; Aimoto, M,; Yamamoto, T. ; Takahashi, M.; Sugo, N.; Takahara, A.; Shirai, K.J. Effects of Enhanced Intracranial Pressure on Blood Pressure and the Cardio-Ankle Vascular Index in Rabbits. Atheroscler. Thromb. 2021, 28, 1241–1249. [Google Scholar] [CrossRef]
- 52. Kario K, Matsuo T, Kobayashi H, Yamamoto K, Shimada K. Earthquake-induced potentiation of acute risk factors in hypertensive elderly patients: possible triggering of cardiovascular events after a major earthquake. J Am Coll Cardiol. [CrossRef]
- Trichopoulos, D.; Zavitsanos, X.; Katsouyanni, K.; Tzonou, A.; Dalla-Vorgia, P. Psychological stress and fatal heart attack: the Athens (1981) earthquake natural experiment. Lancet. 1983, 321, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Dobson, A.J.; Alexander, H.M.; Malcolm, J.A.; Steele, P.L.; Miles, T.A. Heart attacks and the Newcastle earthquake. Med. J. Aust. 1991, 155, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Leor, J.; Poole, W.K.; Kloner, R.A. Sudden cardiac death triggered by an earthquake. N. Engl. J. Med. 1996, 334, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Osada, H.; Kyogoku, M.; Ishidou, M.; Morishima, M.; Nakajima, H. Aortic dissection in the outer third of the media: what is the role of the vasa vasorum in the triggering process? Eur. J. Cardiothorac. Surg. 2013, 43, e82–88. [Google Scholar] [CrossRef]
- Nakagami, T.; Shimizu, K.; Hirano, K.; Kiyokawa, H.; Iwakawa, M.; Ikeda, Y.; Akiba, T.; Terayama, K.; Ogawa, A.; Shirai, K. Cardio-Ankle Vascular Index Reflects the Efficacy of Waon Therapy in Heart Failure Patients. Int. Heart J. 2022, 63, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Formation of arteriosclerotic lesion: Stage Ⅰ. Phase 1: Cholesterol rich lipoproteins such as LDL, IDL and small dense LDL are carried by vasa vasorum and enter into deep intimal area. There, cholesterols are oxidized, and several oxidative products are produced. Oxysterols are toxic and do damage to the surrounding tissues. For example, 7-ketocholeterol induces apoptosis of smooth muscle cells and expands the lipid pool. Phase 2: Oxysterols induced inflammatory reaction and macrophages infiltrate. Smooth muscle cell migrated from the media to the intima, and proliferate to make intimal thickening. Phase 3: Vasa vasorum develops from adventitia into intimal lesion through medial smooth muscle layer. CAVI increased as arteriosclerosis develops. CAVI, cardio-ankle vascular index; IDL, intermediate-density lipoproteins; LDL, low-density lipoprotein.
Figure 1.
Formation of arteriosclerotic lesion: Stage Ⅰ. Phase 1: Cholesterol rich lipoproteins such as LDL, IDL and small dense LDL are carried by vasa vasorum and enter into deep intimal area. There, cholesterols are oxidized, and several oxidative products are produced. Oxysterols are toxic and do damage to the surrounding tissues. For example, 7-ketocholeterol induces apoptosis of smooth muscle cells and expands the lipid pool. Phase 2: Oxysterols induced inflammatory reaction and macrophages infiltrate. Smooth muscle cell migrated from the media to the intima, and proliferate to make intimal thickening. Phase 3: Vasa vasorum develops from adventitia into intimal lesion through medial smooth muscle layer. CAVI increased as arteriosclerosis develops. CAVI, cardio-ankle vascular index; IDL, intermediate-density lipoproteins; LDL, low-density lipoprotein.
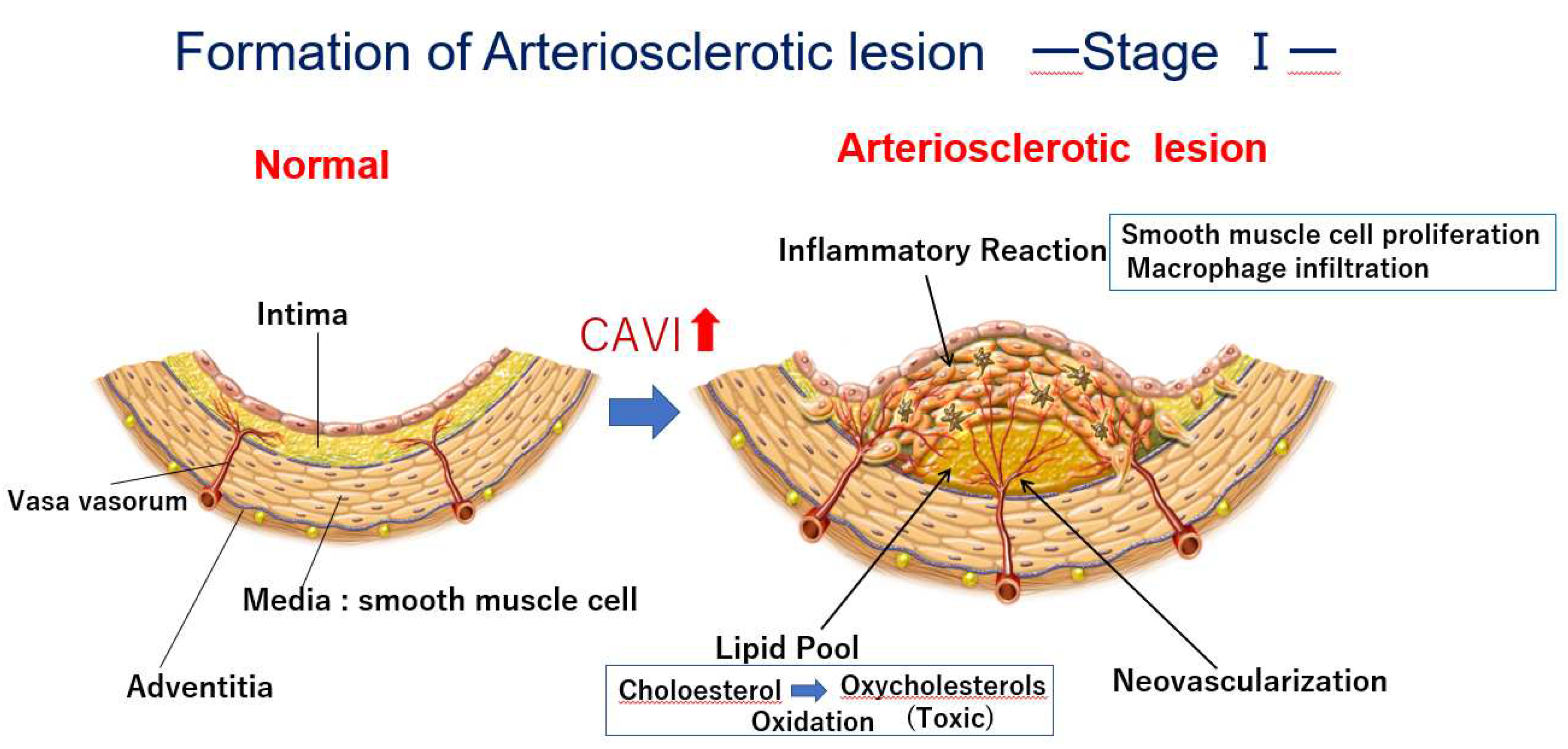
Figure 2.
Oxysterol-induced apoptosis of smooth muscle cells. A) Oxysterol rich fraction-induced apoptosis of cultured smooth muscle cells. B) Smooth muscle cells are observed to fall into apoptosis in human arteriosclerotic lesion.
Figure 2.
Oxysterol-induced apoptosis of smooth muscle cells. A) Oxysterol rich fraction-induced apoptosis of cultured smooth muscle cells. B) Smooth muscle cells are observed to fall into apoptosis in human arteriosclerotic lesion.
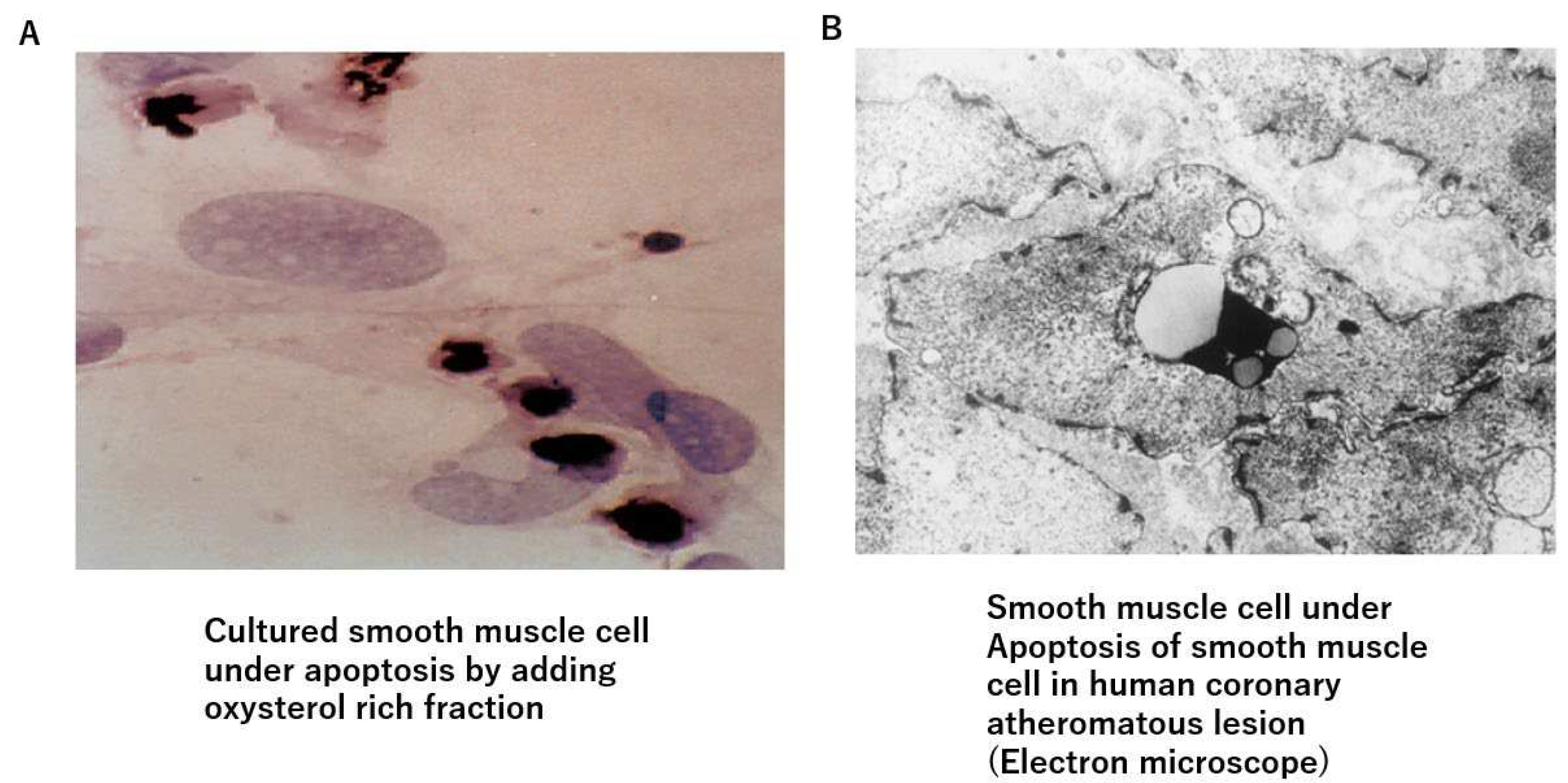
Figure 3.
A) At endarterectomy of the carotid artery, the intimal atheromatous layer could be peeled away from the medial smooth muscle cell layer. The denuded surface of medial smooth muscle layer was promptly covered with the blood, indicating that the intimal arteriosclerotic lesion was supplied by blood with vasa vasorum, which penetrated through the medial smooth muscle layer from the adventitia. Figure 3. Bleeding at the surface of needed medial layer of arteriosclerotic artery at endarterectomy. B) When the surface of the smooth muscle cell layers was covered with a sheet of gauze dipped with norepinephrine, the blood exuding from the medial smooth muscle layer was stopped, indicating that contraction of the medial smooth muscle stopped blood supply from the adventitia and caused ischemia of the intimal lesion. The medial smooth muscle contraction would bring a rapid increase in CAVI.
Figure 3.
A) At endarterectomy of the carotid artery, the intimal atheromatous layer could be peeled away from the medial smooth muscle cell layer. The denuded surface of medial smooth muscle layer was promptly covered with the blood, indicating that the intimal arteriosclerotic lesion was supplied by blood with vasa vasorum, which penetrated through the medial smooth muscle layer from the adventitia. Figure 3. Bleeding at the surface of needed medial layer of arteriosclerotic artery at endarterectomy. B) When the surface of the smooth muscle cell layers was covered with a sheet of gauze dipped with norepinephrine, the blood exuding from the medial smooth muscle layer was stopped, indicating that contraction of the medial smooth muscle stopped blood supply from the adventitia and caused ischemia of the intimal lesion. The medial smooth muscle contraction would bring a rapid increase in CAVI.
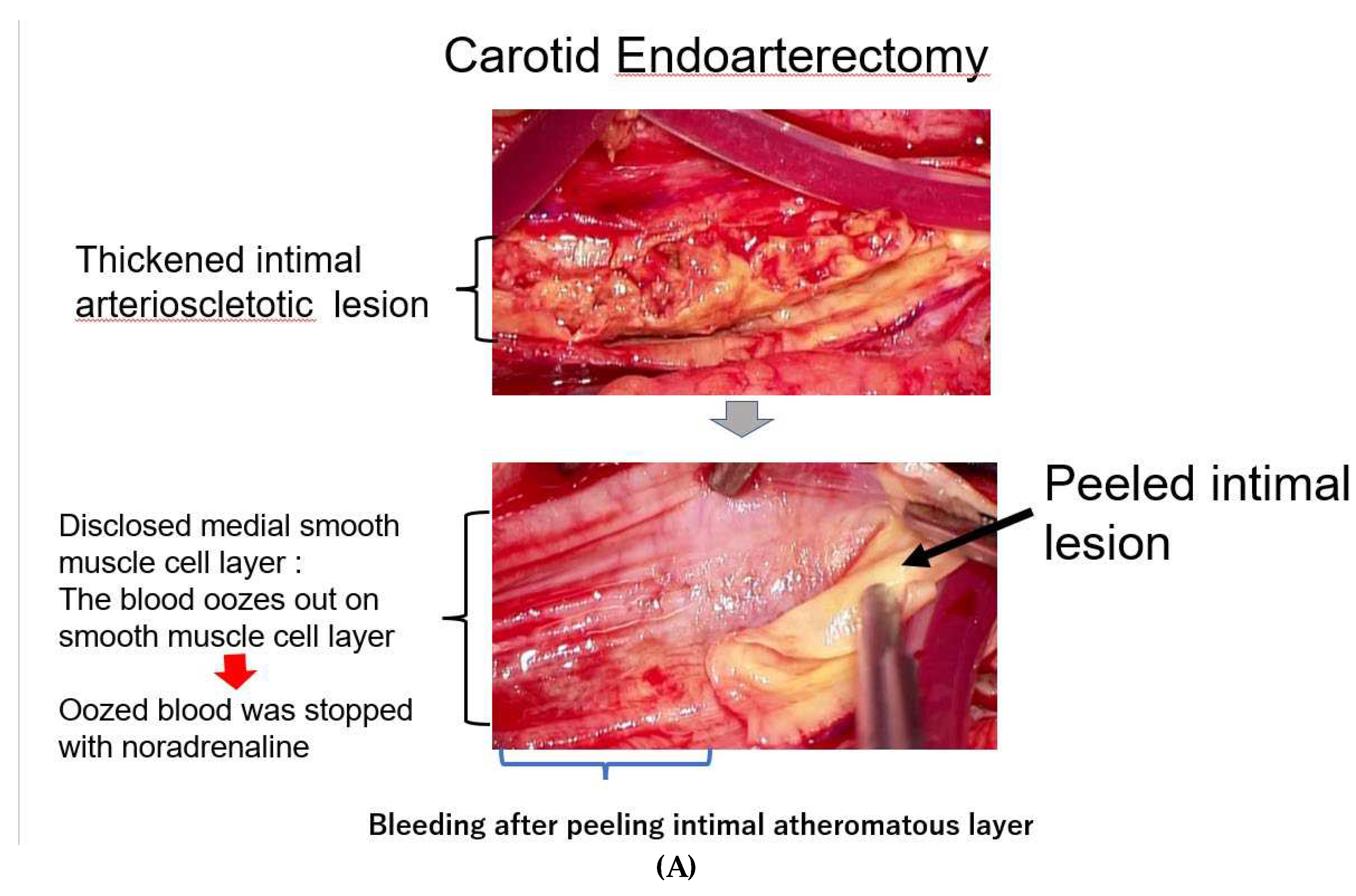
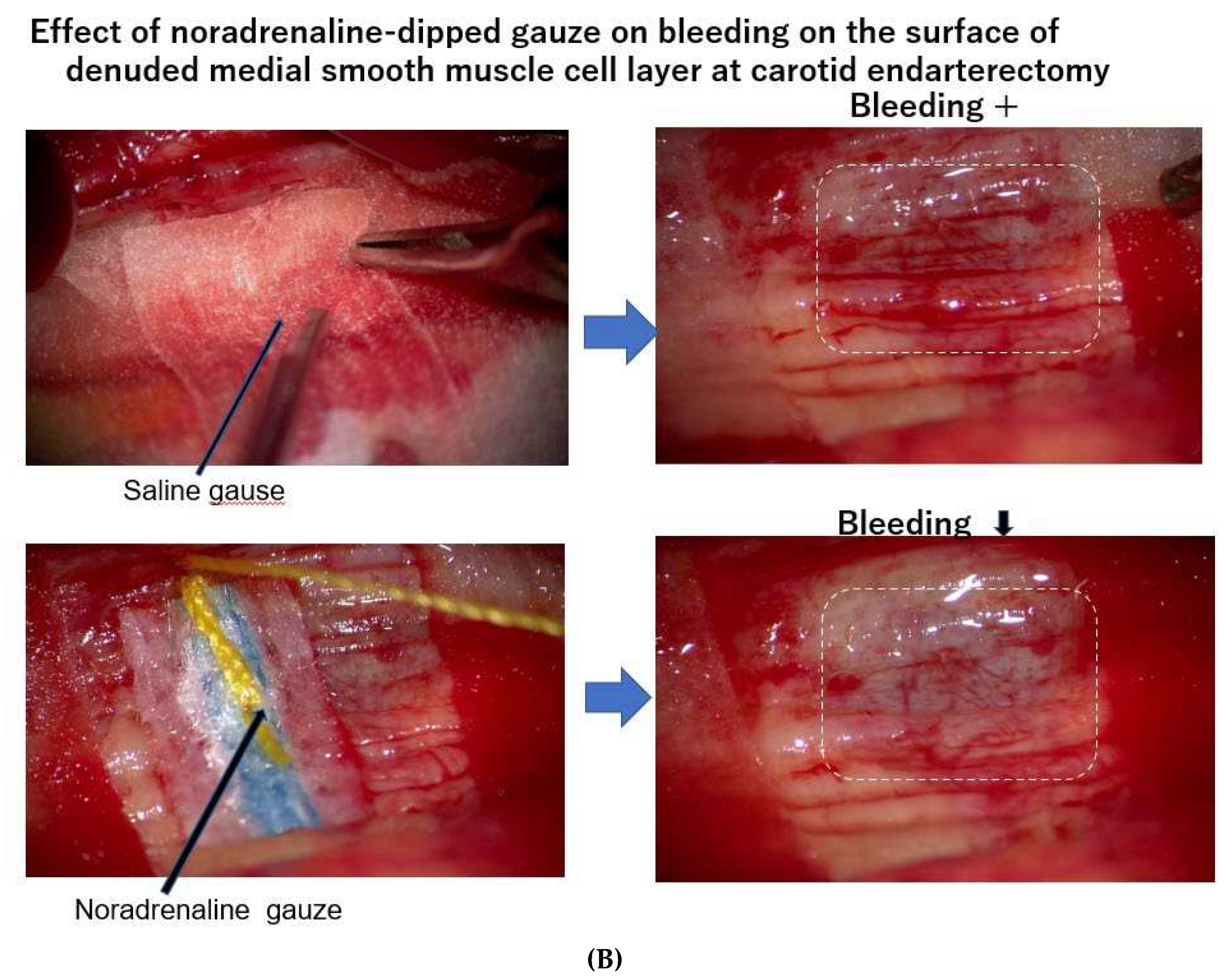
Figure 4.
Various stages of atherosclerosis of the aortae and CAVI. A) Aorta of a 50-year-old woman. CAVI was 7.3, which is a nearly normal level (-0.5SD). B) Aorta of a 74-year-old man. CAVI was11.0, which is high for his age (+2SD). C) Aorta of a 76-year-old man. CAVI was11.0, which is high for his age (+2SD). High CAVI value over +2SD from average value for ages might indicates the presence of arteriosclerosis with vulnerable plaque. CAVI: cardio-ankle vascular index.
Figure 4.
Various stages of atherosclerosis of the aortae and CAVI. A) Aorta of a 50-year-old woman. CAVI was 7.3, which is a nearly normal level (-0.5SD). B) Aorta of a 74-year-old man. CAVI was11.0, which is high for his age (+2SD). C) Aorta of a 76-year-old man. CAVI was11.0, which is high for his age (+2SD). High CAVI value over +2SD from average value for ages might indicates the presence of arteriosclerosis with vulnerable plaque. CAVI: cardio-ankle vascular index.
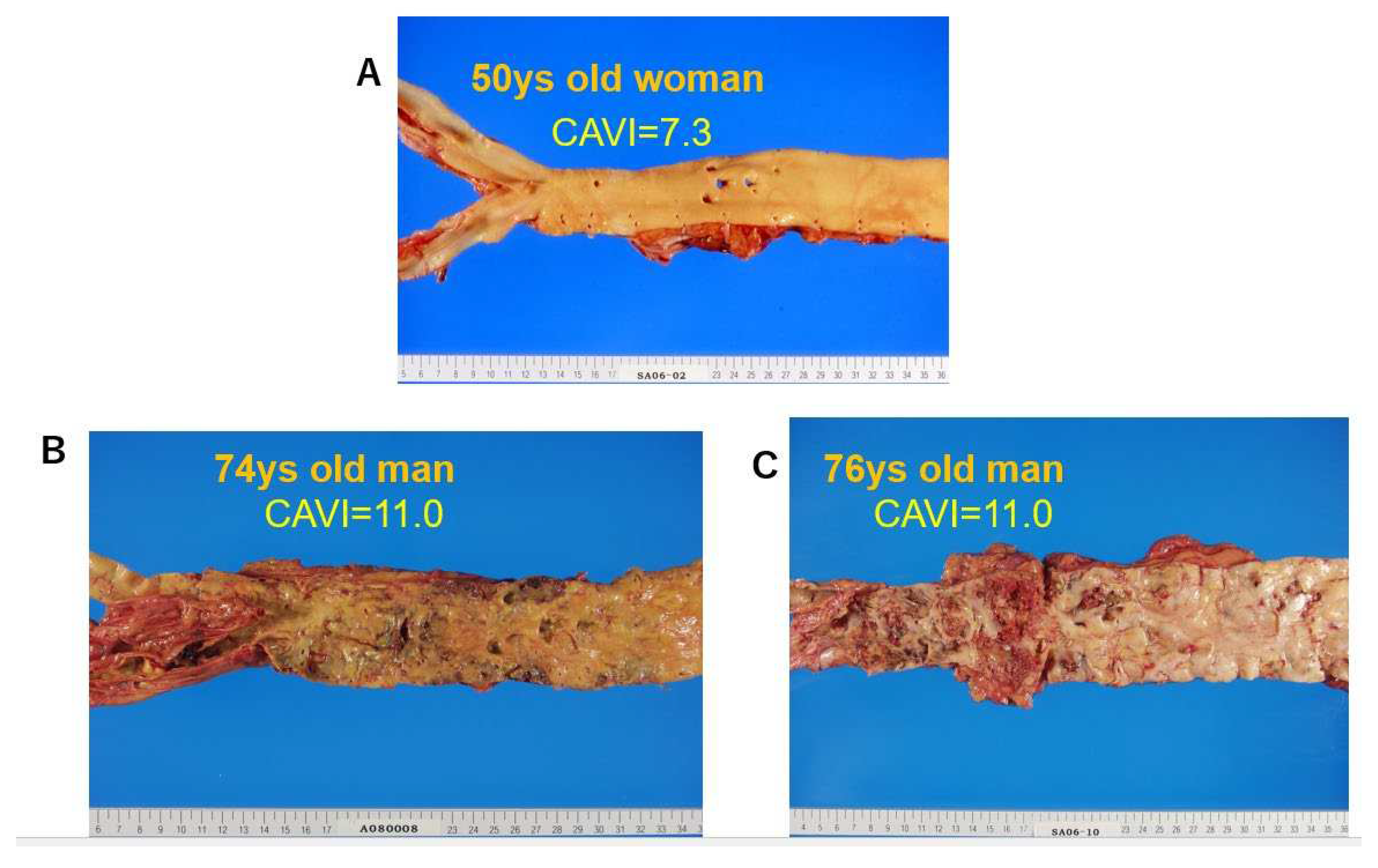
Figure 5.
The process to plaque rupture of vulnerable plaque triggered by medial smooth muscle contraction monitored with a rapid rise in CAVI: Stage 2. When a rapid rise of CAVI was observed, medial smooth muscle contraction occurs. Then, the vasa vasorum are strangled and blood supply to intimal lesion is stopped, which causes ischemia and necrosis of vulnerable plaque, following plaque rupture. Cardiovascular events such as cerebral bleeding, myocardial infarction and dissecting aneurysm in the aorta might happen in a near future.
Figure 5.
The process to plaque rupture of vulnerable plaque triggered by medial smooth muscle contraction monitored with a rapid rise in CAVI: Stage 2. When a rapid rise of CAVI was observed, medial smooth muscle contraction occurs. Then, the vasa vasorum are strangled and blood supply to intimal lesion is stopped, which causes ischemia and necrosis of vulnerable plaque, following plaque rupture. Cardiovascular events such as cerebral bleeding, myocardial infarction and dissecting aneurysm in the aorta might happen in a near future.
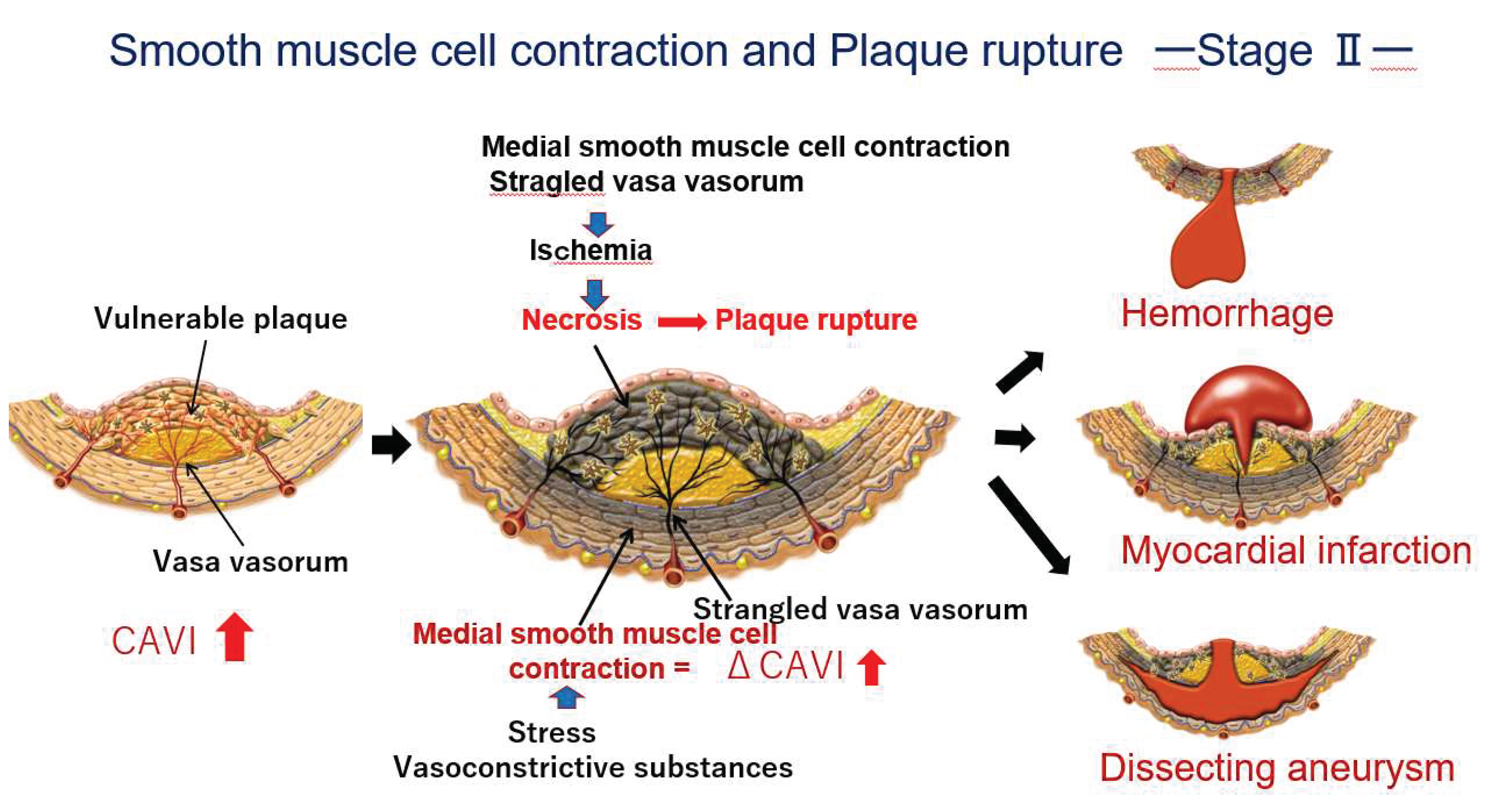

Table 1.
Improving treatments and medicines for high CAVI.
 |
ARB, angiotensin receptor blocker; CCB, calcium channel blocker; CPAP, continuous positive airway pressure; SAS, sleep apnea syndrome..
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



