
Submitted:
27 September 2023
Posted:
28 September 2023
You are already at the latest version
Abstract
Fosmidomycin (FOS) is a naturally occurring compound active against the enzyme DXR in the MEP pathway, and using it as a template for lead structure design is an effective strategy in developing new active compounds. In this work, by replacing the hydroxamate unit of FOS with pyrazole, isoxazole and the related heterocycles that also have the metal ion binding affinity, while retaining the monophosphonic acid in FOS or replacing it with a bisphosphonic acid group, heterocycle-containing mono- and bisphosphonic acid compounds as FOS analogs were designed. The key steps involved in the facile synthesis of these FOS analogs include the Michael addition of diethyl vinylphosphonate or tetraethyl vinylidenebisphosphonate to β-dicarbonyl compounds and the subsequent cyclic condensation with hydrazine or hydroxylamine. Two additional isoxazolinone-bearing FOS analogs were synthesized by the Michaelis-Becker reaction with diethyl phosphite as a key step. The bioactivity evaluation on model plants demonstrated that several compounds have better herbicidal activities compared to FOS, with the most active compound showing a 3.7-fold inhibitory activity on Arabidopsis thaliana, while on the root and stalk of Brassica napus L. and Echinochloa crus-galli in pre-emergence inhibitory activity test, the activities of this compound were found to be 3.2- and 14.3-, 5.4- and 9.4-fold, respectively, and in post-emergency activity test on Amaranthus retroflexus and Echinochloa crus-galli, 2.2- and 2.0-fold inhibition activities were displayed. Despite the significant herbicidal activity, this compound exhibited a DXR inhibitory activity lower than that of FOS but comparable to that of other non-hydroxamate DXR inhibitors, and the dimethylallyl pyrophosphate rescue assay gave no statistical significance, suggesting a different target might be involved in the inhibiting process. This work demonstrates that using bioisosteric replacement can be considered a valuable strategy for discovering new FOS analogs that may have high herbicidal activity.
Keywords:
monophosphonic acid
; bisphosphonic acid
; heterocycle
; DXR
; herbicidal activity
1. Introduction
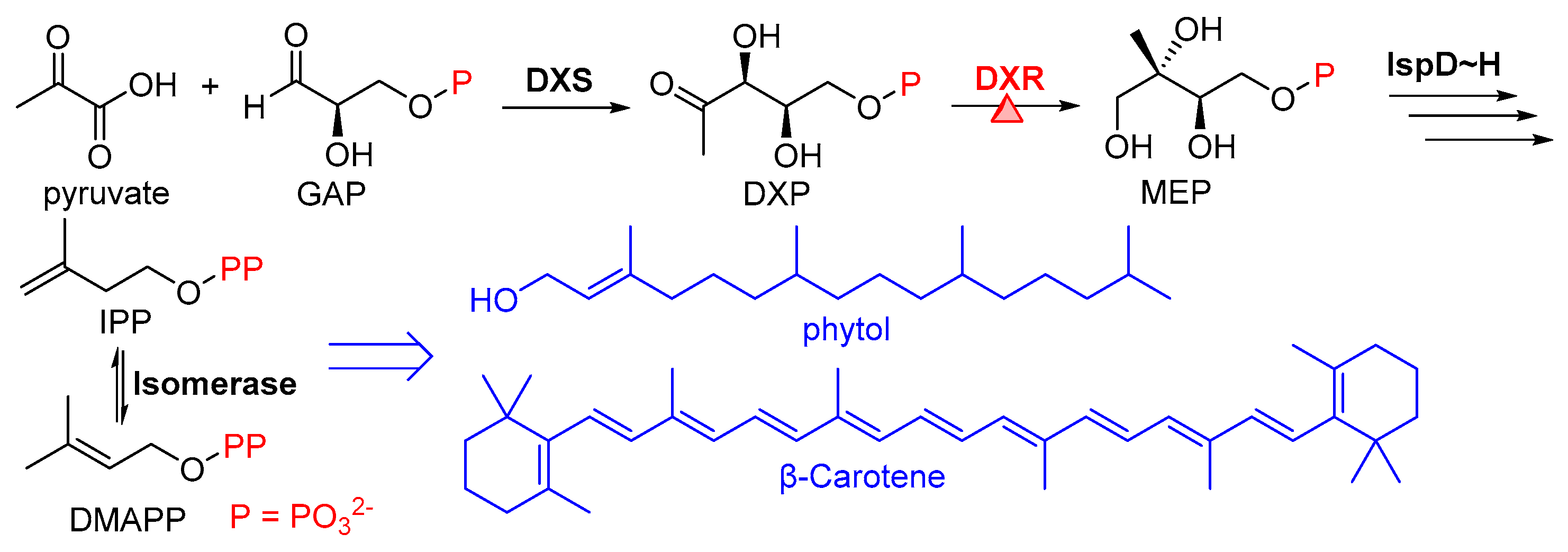
The 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway, which is widespread in bacteria and plants but absent in mammals [1], has gained significant attention in recent years since seven key enzymes are involved in this pathway (Figure 1), which can serve as new targets for developing novel herbicides and antibacterial agents [2,3]. The products of the MEP pathway, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), are crucial precursors for the synthesis of various isoprenoids [4]. For example, plants utilize IPP and DMAPP produced via the MEP pathway for synthesizing phytol, β-carotene, and other substances that are essential to the photosynthesis process and physiological regulation (Figure 1) [5]. The second enzyme in this pathway, 1-deoxy-D-xylulose 5-phosphate reductoisomerase, shortly as DXR and also known as IspC, which catalyzes the isomerization and reduction of 1-deoxy-D-xylulose 5-phosphate (DXP) to produce MEP [6], is the most encouraging target for the development of novel antibacterial and antimalarial drugs because two natural occurring products, fosmidomycin (FOS) and FR900098 (FR), have been found active in targeting this enzyme and the inhibition mechanism have been well documented [7,8]. Although DXR has been considered a promising target for herbicides [9,10], the research focusing on the herbicide discovery based on DXR is rather limited, making the development of DXR inhibitors as herbicidal lead compounds a work of great significance.
Figure 1.
The MEP pathway for synthesis of the isoprenoid precursors IPP and DMAPP that lead to two essential isoprenoid products phytol and β-carotene as examples in plants, with the function of DXR enzyme in converting DXP to MEP highlighted.
Figure 1.
The MEP pathway for synthesis of the isoprenoid precursors IPP and DMAPP that lead to two essential isoprenoid products phytol and β-carotene as examples in plants, with the function of DXR enzyme in converting DXP to MEP highlighted.
The two natural products FOS and FR, originally isolated from Streptomyces species [11,12], are structurally composed of three parts, phosphonic acid, hydroxamate, and the trimethylene linker, as shown in Figure 2A. FOS and FR are the substrate competitive inhibitors of DXR and have been well-characterized, showing a wide range of bioactivities including antibacterial and antimalarial effects [13]. The FOS and FR themselves, however, have some drawbacks that need to be overcome, such as low lipophilicity, poor pharmacokinetics, and low bioavailability, which impede their applications as clinic drugs [14]. Nevertheless, they have been extensively employed as the templates for developing new related drugs, and a variety of structural modifications on FOS have been carried out [15]. Among the most potent DXR inhibitors thus developed are those structurally similar to FOS, all containing the hydroxamate as the binding group to the metal ion in the DXR active site [13,14,15]. Unfortunately, the hydroxamate group not only exhibits metabolic instability, but also has strong side effects on various metal enzymes [16,17,18], even the approved hydroxamate containing drugs, such as vorinostat and panobinostat, tend to have low bioavailability [18]. Using non-hydroxamate groups to replace the hydroxamate group has become an effective method for improving the bioavailability of the related drugs [19,20].
Under this context, several works on using non-hydroxamate groups to replace the hydroxamate group of FOS and FR for developing DXR inhibitors have been reported [21,22,23,24] and their structures are given in Figure 2A. Structurally, these non-hydroxamate DXR inhibitors also consist of the three fragments: mono- or bisphosphonic acid, linker, and a non-hydroxamate group, but the interaction patterns of these compounds with the active site of DXR are different from FOS. For example, the catechol- and N-hydroxypyridone-containing FOS analogs were supposed to bind to the metal ion of the DXR active site but with moderate MtDXR inhibitory activities (IC50 = 41 and 53 μM, respectively, versus 0.08 μM for FOS) [21]. The crystal X-Ray analyses indicated that the pyridine and quinoline-containing FOS analogs are unable to bind with the metal ion, but induce conformational changes in the active site of DXR to create a lipophilic pocket accommodating the pyridine or quinoline group that leads to an inhibitory activity against EcDXR with IC50 as low as 0.84 μM [22]. Furthermore, some examples demonstrated that the bisphosphonate group is able to bind with the metal ion while the pyridine or isoquinoline group occupies the hydrophobic cavity in the active site of DXR, resulting in IC50 of 4 and 7 μM against EcDXR [23]. Although these non-hydroxamate inhibitors are inferior to FOS in DXR inhibitory activity, some of them still have similar or higher anti-infective activities compared to FOS possibly due to the improved bioavailability or the inhibition on other targets [21,22,23,24].
Figure 2.
Structures of two naturally occurring DXR inhibitors FOS and FR and reported non-hydroxamate DXR inhibitors (A), exemplified nitrogen-containing heterocycles in commercial herbicides (B), and the design strategy for heterocycle-containing mono- and bisphosphonic acids in this work (C).
Figure 2.
Structures of two naturally occurring DXR inhibitors FOS and FR and reported non-hydroxamate DXR inhibitors (A), exemplified nitrogen-containing heterocycles in commercial herbicides (B), and the design strategy for heterocycle-containing mono- and bisphosphonic acids in this work (C).
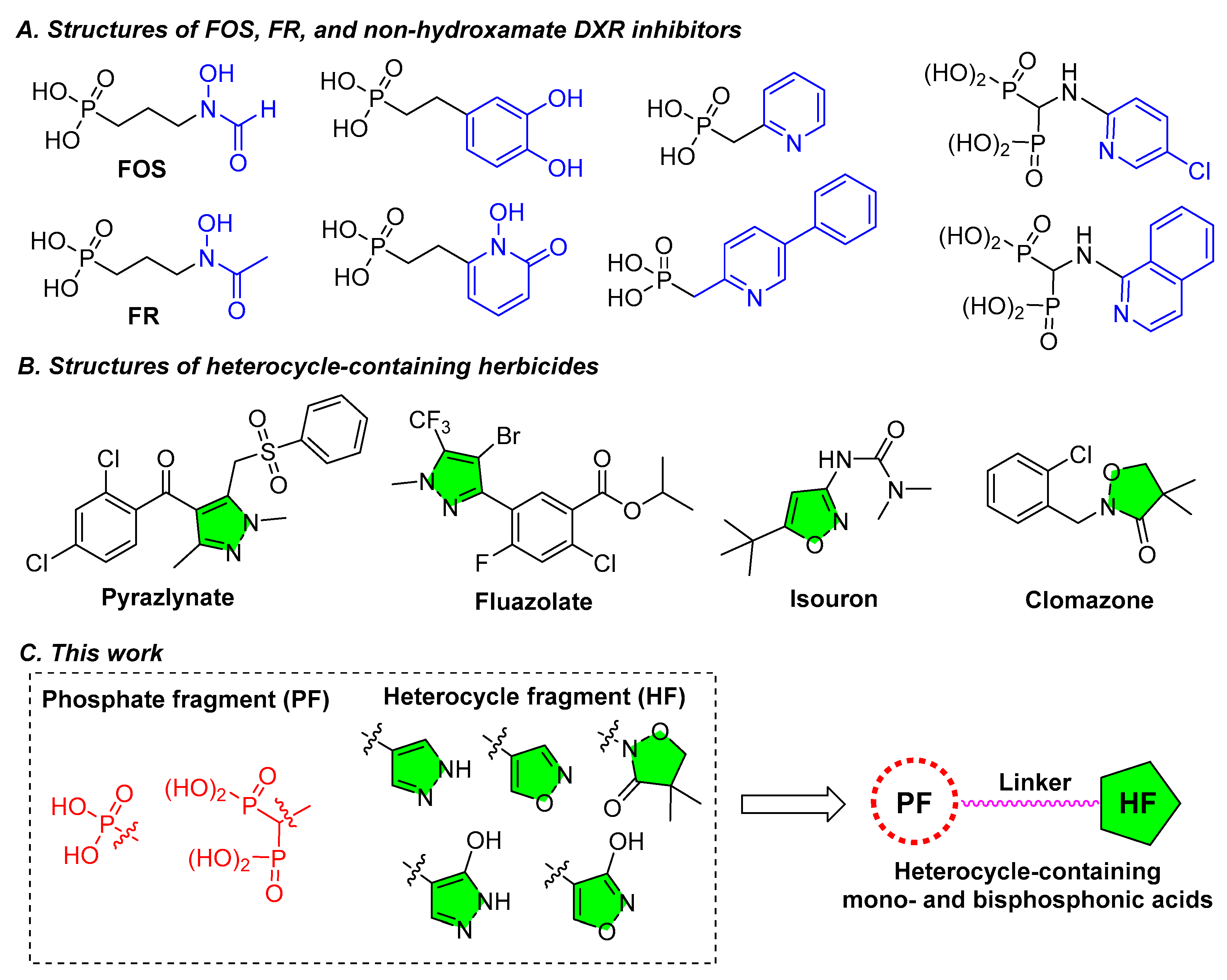
On the other hand, heterocycle-containing organophosphorus compounds are widespread in drugs, active molecules, functional materials, and pesticides, and heterocycles as important structural moieties have been extensively used in the design of agrochemicals due to their structural diversity and bioisosteric replacement ability [25]. For example, commercial herbicides such as pyrazlynate and fluazolate contain a pyrazole ring, isouron features the presence of isoxazole ring, and especially the herbicide clomazone, which contains an isoxazolinone and is activated by metabolism in weeds, targets the DXS enzyme, the first enzyme in the MEP pathway [26] (Figure 2B). In addition, nitrogen-containing heterocycles such as pyrazole, pyrazolone and isoxazole can form monodentate coordination with divalent metal ions and have been widely employed as active substructural moieties in the design of metalloenzyme inhibitors [27]. Considering the advantages of heterocycles in herbicide compounds design and their ability to bind metal ions, replacement of hydroxamate of FOS with a suitable heterocycle would provide an opportunity for developing DXR inhibitors with improved herbicidal activity while reducing the issue regarding low bioavailability of the hydroxamate group.
Given the drawbacks of hydroxamate group in DXR inhibitors, the advances of non-hydroxamate DXR inhibitors, and the bioisosteric role of nitrogen-containing heterocycles, in this work, heterocycle-containing mono- and bisphosphonic acid compounds were designed and their substructural complements and general structure are shown in Figure 2C. The phosphonic acid in FOS is either retained or substituted with a bisphosphonic acid group, and the hydroxamate is replaced with a heterocycle such as pyrazole, isoxazole or isoxazolinone. To synthesize these compounds, several key steps, such as the Michael addition of diethyl vinylphosphonate or tetraethyl vinylidenebisphosphonate to β-dicarbonyl compounds, the cyclic condensation with hydrazine or hydroxylamine, and the Michaelis-Becker reaction with diethyl phosphite, were employed. These compounds were then tested for their activities in inhibiting model plants and the DXR enzyme. Other techniques such as DMAPP rescue assay and molecular docking were also used to explore the possible action mechanism of active compounds.
2. Results and Discussion
2.1. Chemistry
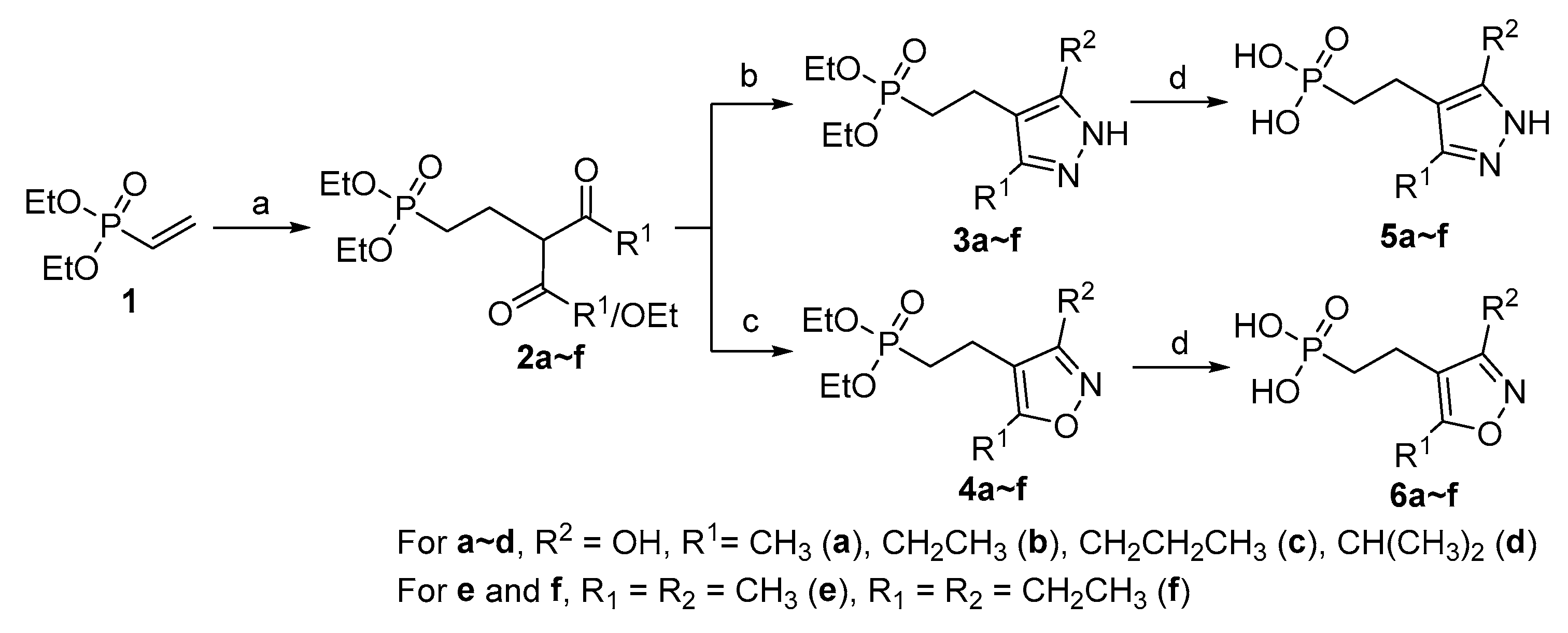
Synthesis routes. The cyclic condensation reaction of β-dicarbonyl compounds with hydrazine and hydroxylamine is one efficient method for synthesizing pyrazole and isoxazole derivatives [28]. Based on this reaction as a key step, in this work, a three-step synthesis route to the monophosphonic acid compounds 5 and 6 was designed as shown in Scheme 1. First, diethyl vinylphosphonate (1) as a Michael acceptor reacted with β-ketoesters or β-diketones in the presence of K2CO3 and benzyltriethylammonium chloride (TEBAC) to yield the intermediates 2. In the second step, 2 was used to react with hydrazine or hydroxylamine, respectively, giving the corresponding cyclization intermediates 3 and 4. Finally, the ethyl protecting groups were removed with TMSBr to yield the target products 5 and 6 in 70~82% overall yields.
Scheme 1.
Synthesis of the monophosphonic acid series 5 and 6. Reagents and conditions: (a) RCOCH2COOEt or RCOCH2COR, K2CO3, TEBAC, CH3CN, 80℃; (b) N2H4·H2O, EtOH, 75℃; (c) NH2OH·HCl, K2CO3, EtOH, 75℃; (d) i) TMSBr, CH2Cl2, 0℃ to r.t.; ii) THF/H2O, r.t.
Scheme 1.
Synthesis of the monophosphonic acid series 5 and 6. Reagents and conditions: (a) RCOCH2COOEt or RCOCH2COR, K2CO3, TEBAC, CH3CN, 80℃; (b) N2H4·H2O, EtOH, 75℃; (c) NH2OH·HCl, K2CO3, EtOH, 75℃; (d) i) TMSBr, CH2Cl2, 0℃ to r.t.; ii) THF/H2O, r.t.
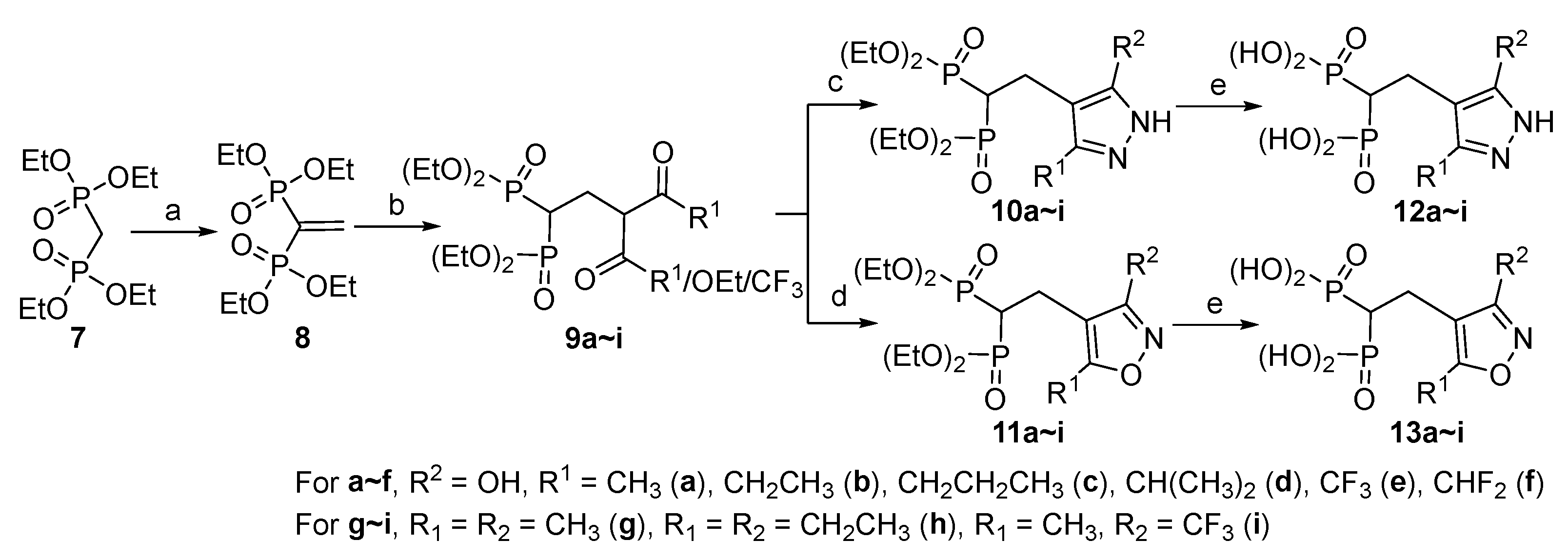
A similar synthesis route was used to access the bisphosphonic acid series 12 and 13, as shown in Scheme 2. First, a Michael acceptor tetraethyl vinylidenebisphosphonate (VBP) was synthesized through the reaction of tetraethyl methylenebisphosphonate (7) with paraformaldehyde in methanol and followed by dehydration with the catalysis of TsOH [29]. Then, tetraethyl VBP (8) reacted with β-ketoesters or β-diketones to give the intermediates 9 using lithium bis(trimethylsilyl)amide (LiHMDS) as base. Cyclic condensation with hydrazine or hydroxylamine followed by deprotection gave the target compounds 12 and 13 in 75%~88% overall yields.
Scheme 2.
Synthesis of the bisphosphonic acid series 12 and 13. Reagents and conditions: (a) i) (CHO)n, Et2NH, MeOH, 65℃; (ii) TsOH, toluene, 110℃; (b) RCOCH2COOEt or RCOCH2COR, LiHMDS, THF, 0℃ to r.t.; (c) N2H4·H2O, EtOH, 75℃; (d) NH2OH·HCl, K2CO3, EtOH, 75℃; (e) i) TMSBr, CH2Cl2, 0℃ to r.t.; ii) THF/H2O, r.t.
Scheme 2.
Synthesis of the bisphosphonic acid series 12 and 13. Reagents and conditions: (a) i) (CHO)n, Et2NH, MeOH, 65℃; (ii) TsOH, toluene, 110℃; (b) RCOCH2COOEt or RCOCH2COR, LiHMDS, THF, 0℃ to r.t.; (c) N2H4·H2O, EtOH, 75℃; (d) NH2OH·HCl, K2CO3, EtOH, 75℃; (e) i) TMSBr, CH2Cl2, 0℃ to r.t.; ii) THF/H2O, r.t.
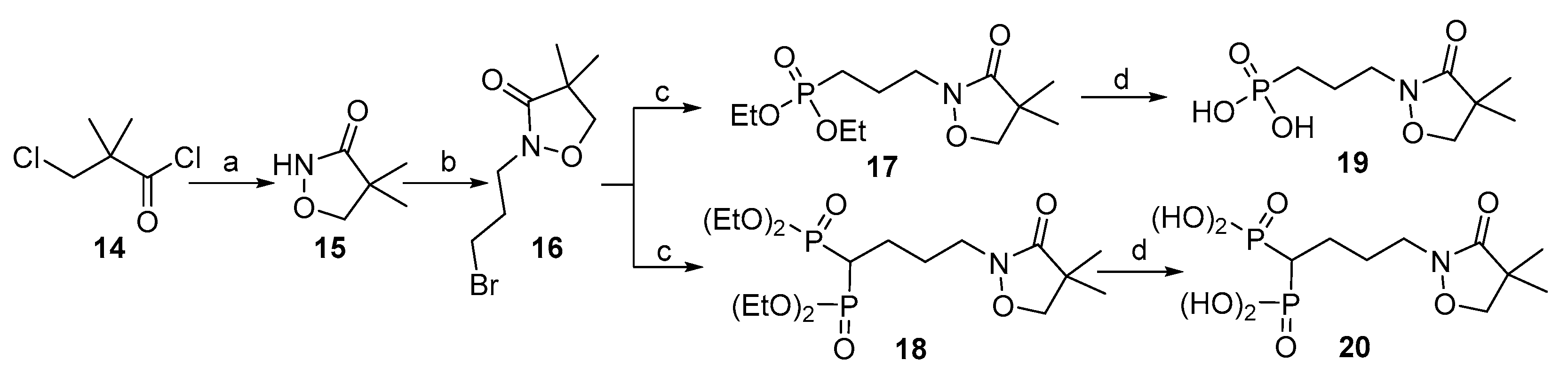
The synthesis route for isoxazolinone-containing compounds 19 and 20 is different from the previous ones and the Michaelis-Becker reaction as a key step was used, as shown in Scheme 3. In this route, with 3-chloropivaloyl chloride (14) as the starting material, and through the amidation with hydroxylamine followed by cyclization [30], the intermediate 4,4-dimethylisoxazolidin-3-one (15) was obtained. Nucleophilic substitution on 15 with 1,3-dibromopropane in the presence of NaH yielded the brominated intermediate 16. Next, the Michaelis-Becker reaction of 16 with diethyl phosphite (1) and methylenebisphosphonate (7) was performed to afford the intermediates 17 and 18, respectively, in the presence of Cs2CO3 and tetrabutylammonium iodide (TBAI). The subsequent deprotection of the ethyl protecting groups using TMSBr gave the target compounds 19 and 20 in yields of 62% and 55%, respectively.
Scheme 3.
Synthesis of the target compounds 19 and 20. (a) i) NH2OH·HCl, NaOH, H2O, 0℃ to r.t.; (ii) NaOH, Na2CO3, H2O, 45℃; (b) Br(CH2)3Br, NaH, DMF, 0℃ to r.t.; (c) HPO(OEt)2 or 7, Cs2CO3, TBAI, DMF, r.t.; (d) i) TMSBr, CH2Cl2, 0℃ to r.t.; ii) THF/H2O, r.t.
Scheme 3.
Synthesis of the target compounds 19 and 20. (a) i) NH2OH·HCl, NaOH, H2O, 0℃ to r.t.; (ii) NaOH, Na2CO3, H2O, 45℃; (b) Br(CH2)3Br, NaH, DMF, 0℃ to r.t.; (c) HPO(OEt)2 or 7, Cs2CO3, TBAI, DMF, r.t.; (d) i) TMSBr, CH2Cl2, 0℃ to r.t.; ii) THF/H2O, r.t.
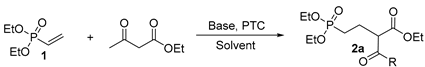
Optimization of reaction conditions. In the synthesis of the target compounds, the facile connection of the phosphonate group with a heterocycle through a C-P bond is a key step, especially when a suitable linker length has to be considered in terms of the structural feature of the naturally occurring DXR-active products FOS and FR. The most common methods for simple C-P bond formation include the Michaelis-Arbuzov reaction and the Michaelis-Becker reaction [31,32], but both of the reactions require harsh reaction conditions and the yields are usually low. Another important strategy for synthesizing phosphonate-containing compounds involves the use of vinylphosphonate as Michael acceptor to react with a Michael donor such as ketone, β-ketoester, and Grignard reagent [33,34,35]. This type of synthesis tends to be facile and mild in reaction conditions, but the subsequent utilization of the functional groups of the Michael donors for heterocycle construction is seldom reported. In addition, the reports on the reaction of vinylphosphonate and vinylidenebisphosphonate as Michael acceptors are rather few perhaps due to the relatively low reactivity in this type of Michael addition as compared to the analogous vinylcarboxylate Michael acceptors. In our case, this type of Michael addition was exploited with the reaction of vinylphosphonate and vinylidenebisphosphonate with β-dicarbonyl compounds as Michael donors, which could cyclize with hydrazine and hydroxylamine to form heterocycles, as shown in Scheme 1 and Scheme 2. Hence, the optimization of the reaction conditions for Michael addition between vinylphosphonates (1) and β-dicarbonyls appeared to be crucial for the target compound synthesis.
The reaction of vinylphosphonate 1 with acetoacetate as a model reaction was first optimized and the results are shown in Table 1. When strong bases were used, such as t-BuOK, NaH, and NaOEt, and the reaction performed in THF or EtOH at room temperature for 6 h, the yields of the Michael addition were determined to be 60, 45, and 40%, respectively, for the isolated product 2a (entries 1~3), and heating (entry 4) and expending the reaction time in the case of t-BuOK as base gave no increase in yield. Since these conditions gave no ideal yields, we had to sought after alternative methods. It was reported that for a typical Michael addition reaction, a moderately strong base such as Cs2CO3 or K2CO3 could be beneficial with the assistance of a phase-transfer catalyst (PTC) [36]. Indeed, when TBAI or TEBAC used as PTC and Cs2CO3 or K2CO3 as base, the yield was significantly increased (entries 5~8), with the best yield of 90% for the reaction performed with TEBAC as PTC and K2CO3 as base (entry 8). For other β-ketoesters and β-diketones, the reaction proceeded similarly and gave the corresponding intermediate 2 in yields of 87~92%. On the other hand, vinylidenebisphosphonate as the Michael acceptor tended to react faster with β-ketoesters and β-diketones using LiHMDS as base under the reaction conditions indicated in Scheme 2. This higher reactivity of vinylidenebisphosphonate than vinylphosphonate is due to its higher electrophilicity cause by the double electron-withdrawing effect of the two phosphonate groups on the vinyl unit. The data of yield of these target compounds are given in the Supporting Information.

Table 1.
Table 1. Optimization of the reaction conditions for Michael additiona.
| Entry | Base | PTC | Solvent | Temp (℃) | Yield (%)b |
| 1 | t-BuOK | -- | THF | r.t. | 60 |
| 2 | NaH | -- | THF | r.t. | 45 |
| 3 | NaOEt | -- | EtOH | r.t. | 40 |
| 4 | t-BuOK | -- | THF | 66 | 62 |
| 5 | Cs2CO3 | TBAI | CH3CN | 80 | 80 |
| 6 | Cs2CO3 | TEBAC | CH3CN | 80 | 85 |
| 7 | K2CO3 | TBAI | CH3CN | 80 | 83 |
| 8 | K2CO3 | TEBAC | CH3CN | 80 | 90 |
a Reaction conditions: 1 (1 mmol), ethyl acetoacetate (1 mmol), base (1 mmol), PTC (0.1 mmol), solvent (5 mL), 6 h. bYield of isolated product.
2.2. Arabidopsis Growth Inhibitory Activity
An early report described that FOS as a DXR enzyme inhibitor can disrupt the biosynthesis of essential isoprenoids for chlorophyll and carotenoids, leading to bleaching and developmental arrest in Arabidopsis thaliana (Arabidopsis) [37]. Thus, using FOS as the reference compound, the activity of the synthesized compounds against the growth of Arabidopsis was screened at an initial concentration of 100 mg/L, and the data are provided in Supporting Information. For compounds with inhibition rates higher than 50% at the initial concentration of 100 mg/L, their median effective concentration (EC50) values were determined. In the total 32 compounds, 10 compounds were found to have inhibition rates higher than 50% against Arabidopsis at the initial concentration, with their EC50 ranging from 7.8 to 88.3 mg/L, as shown in Table 2. Among them, the series 13 compounds, including 13a, 13d, 13e, and 13f, performed better in inhibitory activity than FOS, and compound 13e has the highest activity with EC50 down to 7.8 mg/L, a 3.7-fold activity of FOS, which has an EC50 value of 27.5 mg/L.
Table 2.
Median effective concentrations of target compounds against Arabidopsis growth.
| Comp | EC50 (mg/L)a | Comp | EC50 (mg/L) | Comp | EC50 (mg/L) |
| 5a | >100 | 6f | >100 | 13b | 37.7±2.8 |
| 5b | >100 | 12a | >100 | 13c | 48.4±5.3 |
| 5c | >100 | 12b | >100 | 13d | 23.1±1.9 |
| 5d | >100 | 12c | >100 | 13e | 7.8±1.2 |
| 5e | >100 | 12d | >100 | 13f | 8.7±1.3 |
| 5f | >100 | 12e | >100 | 13g | >100 |
| 6a | 28.6±3.9 | 12f | >100 | 13h | >100 |
| 6b | >100 | 12g | >100 | 13i | 40.7±2.9 |
| 6c | >100 | 12h | >100 | 19 | >100 |
| 6d | 45.7±4.8 | 12i | 88.3±4.3 | 20 | >100 |
| 6e | >100 | 13a | 21.6±3.8 | FOS | 27.5±3.1 |
a EC50 values of inhibition on Arabidopsis are presented as mean ± SD.
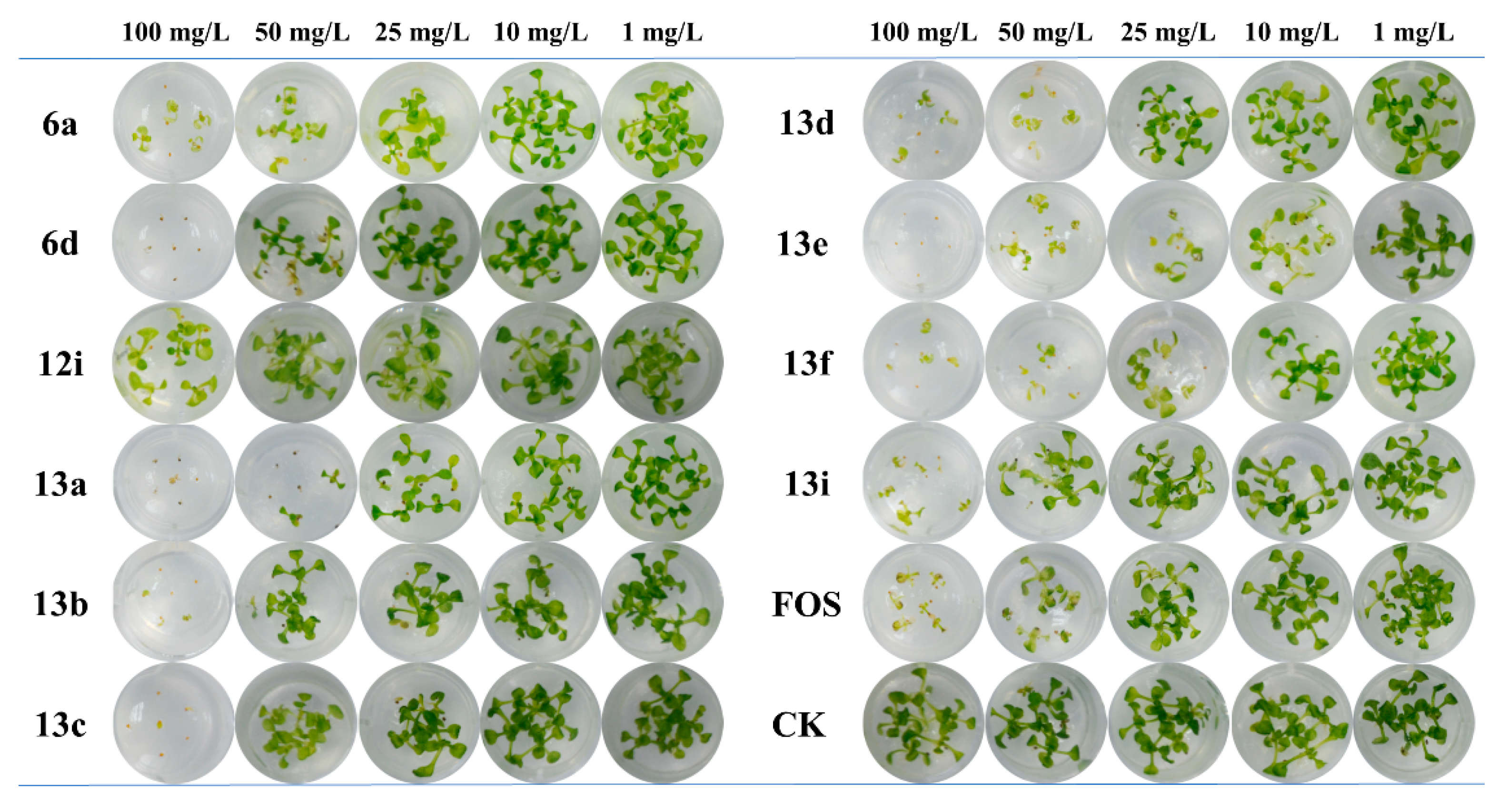
The phenotype of Arabidopsis treated with FOS and the 10 compounds at concentrations of 100, 50, 25, 10, and 1 mg/L was further tested by monitoring with digital camera and the results are shown in Figure 3. It can be seen that FOS and all the test compounds exhibit concentration-dependent growth inhibition and bleaching on Arabidopsis, suggesting the effect may arise from the inhibition on a certain pathway in the plant chloroplast, just as FOS does. At a high applied concentration of 100 mg/L, most compounds could even completely inhibit the germination of Arabidopsis. Notably, two compounds, 13e and 13f, performed superior in Arabidopsis growth inhibition and bleaching effect to the control FOS at all tested concentrations.
Figure 3.
Inhibition and bleaching effects of the 10 active compounds on Arabidopsis.
2.3. Pre-emergence Herbicidal Activity
Two model plants, dicot Brassica napus L. (BN) and monocot Echinochloa crus-galli (EC), were tested for the pre-emergence herbicidal activity of all the synthesized compounds using a standard Petri dish method [38]. The EC50 values collected on the inhibition of root and stalk for the 10 active compounds plus the 2 isoxazolinone-containing compounds 19 and 20 are shown in Table 3 for discussion, while the data for the other compounds are given in Supporting Information. All 12 compounds in Table 3 displayed pre-emergence herbicidal activities with the EC50 values comparable or superior to that of FOS, and especially, compound 13e has EC50 of 10.7 and 2.3 mg/L and compound 13f has EC50 values of 17.9 and 5.8 mg/L, on BN root and stalk, respectively, while FOS only has the corresponding EC50 of 34.7 and 32.9 mg/L. The 2 isoxazolinone-containing compounds 19 and 20, which although have no obvious effects on Arabidopsis, behaved better in inhibition of these two model plants, with the compound 20 more powerful than FOS in the inhibitory EC50 of BN stalk as 8.2 versus 32.9 mg/L.
Table 3.
Pre-emergence herbicidal activities of the 10 active compounds plus the compounds 19 and 20 on the root and stalk of BN and ECa.
Table 3.
Pre-emergence herbicidal activities of the 10 active compounds plus the compounds 19 and 20 on the root and stalk of BN and ECa.
| Comp | EC50 (mg/L) | |||
| BN | EC | |||
| root | stalk | root | stalk | |
| 6a | 17.3 | 10.5 | 24.0 | 18.9 |
| 6d | 67.5 | 38.2 | 40.1 | 25.7 |
| 12i | 36.6 | 21.7 | 53.1 | 47.7 |
| 13a | 26.9 | 8.5 | 27.1 | 10.2 |
| 13b | 38.8 | 20.2 | 29.9 | 35.3 |
| 13c | 27.7 | 46.9 | 43.9 | 45.8 |
| 13d | 25.2 | 11.0 | 27.2 | 13.5 |
| 13e | 10.7 | 2.3 | 7.4 | 4.1 |
| 13f | 17.9 | 5.8 | 9.7 | 3.6 |
| 13i | 40.5 | 33.5 | 32.1 | 34.6 |
| 19 | 79.3 | 39.2 | >100 | 88.0 |
| 20 | 34.6 | 8.2 | 40.0 | 25.2 |
| FOS | 34.7 | 32.9 | 40.2 | 38.4 |
a BN, Brassica napus L.; EC, Echinochloa crus-galli.
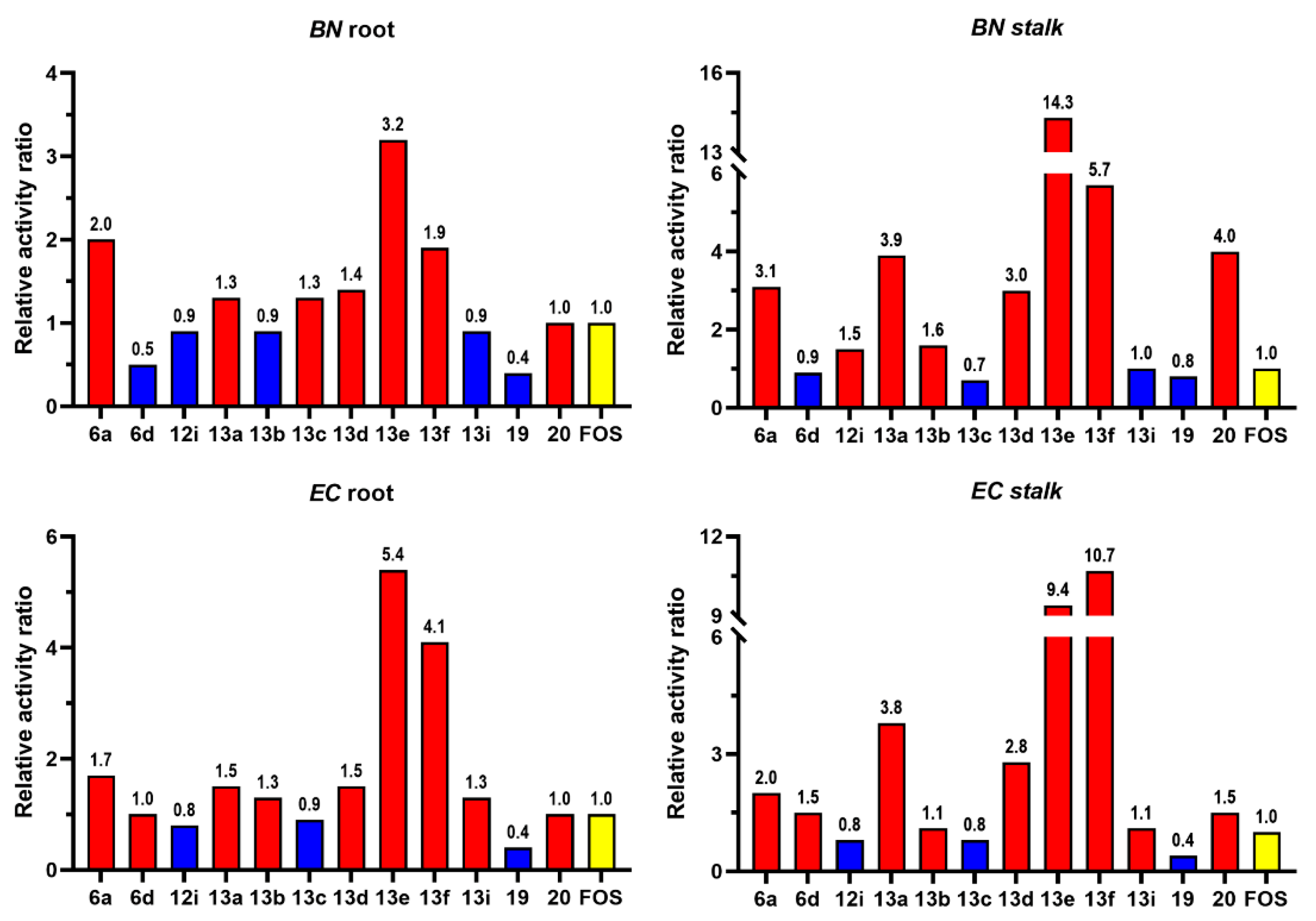
To give a better illustration of the activity difference of the 12 compounds along with FOS, four column graphs, representing separately the four sets of relative activity data on root and stalk of BN and EC with respect to that of FOS, are given in Figure 4. It can be seen from Figure 4A~B that compounds 6a, 13a, 13d~f, and 20 are more active on the root and stalk of BN than FOS, which are more evident on the inhibition of BN stalk, with 13e and 13f having 14.3- and 5.7-fold relative activities, respectively, with respect to that of FOS. For the inhibition on EC, as shown in Figure 4C~D, most of the compounds exhibited a stronger inhibitory activity than FOS. Among them, compounds 13e and 13f also have the strongest inhibitory effects on the root and stalk of EC, with the relative activities of 5.4- and 10.7-fold, respectively, with respect to that of FOS. It is also noted that both 19 and 20 have the similar activities on the inhibition of the root of BN and EC to FOS, but the compound 20 has a much stronger inhibitory effect on the stalk of BN and EC than FOS, with 4.0 and 1.5-fold relative activities, respectively, as compared to FOS.
Figure 4.
Relative activities of the 10 active compounds plus the compounds 19 and 20 on the root and stalk of the BN and EC.
Figure 4.
Relative activities of the 10 active compounds plus the compounds 19 and 20 on the root and stalk of the BN and EC.
2.4. Post-emergence Herbicidal Activity
In terms of the results from the inhibition assays on Arabidopsis and on the pre-emergence model plants BN and EC, six compounds, 6a, 13a, 13d~f, and 20, were selected for the post-emergence herbicidal activity evaluation on representative weeds Amaranthus retroflexus (AR) and Echinochloa crus-galli (EC). The spray dose was 300 g ai/ha, and the herbicidal effects were evaluated by visual observation [38] and fresh weighing [39], and the results are provided in Table 4. All the test compounds, including FOS, have post-emergence inhibitory activities on the weeds. In general, all compounds behaved better on AR than on EC. Among them, 13e showed the best herbicidal activity with the visual evaluation levels of +++ for AR and ++ for EC, which are superior to FOS that has the levels of + for both weeds. The inhibition rates of 13e on AR and EC determined by fresh weighing are 70.3% and 53.5%, respectively, 1.2- and 1.0-fold more active than FOS. In addition, compound 13f also demonstrated similar post-emergence herbicidal activity to 13e, and much better than FOS in the inhibition activity against AR and EC.
Table 4.
Post-emergence herbicidal activities of the 6 selected compounds against AR and ECa.
| Comp | Visual evaluationb | Loss of weight (%) | ||
| AR | EC | AR | EC | |
| 6a | + | + | 35.6 | 30.8 |
| 13a | ++ | + | 52.3 | 35.8 |
| 13d | ++ | + | 45.7 | 34.9 |
| 13e | +++ | ++ | 70.3 | 53.5 |
| 13f | +++ | ++ | 61.8 | 48.1 |
| 20 | + | + | 23.6 | 22.9 |
| FOS | + | + | 31.5 | 27.4 |
a AR, Amaranthus retroflexus; EC, Echinochloa crus-galli. b Activity grade (percentage of inhibition): “++++”: ≥ 80%; “+++”: ≥ 60%; “++”: ≥ 40%; “+”: ≥ 20%; “−”: < 20%.
2.5. DXR Inhibitory Activity
After evaluating the herbicidal activity, the synthesized compounds were also tested for their inhibitory activity against recombinant E. coli DXR (EcDXR) according to the literature method [40], and the preliminary screening was performed at a compound concentration of 100 μM. For compounds with an inhibition rate (InR) higher than 50%, their IC50 values were determined by using a series of successively decreasing concentration. As shown in Table 5, most of the target compounds showed weak or no activity against DXR at a concentration of 100 μM, except the two 3-hydroxyisoxazole-containing bisphosphonate compounds, 13a and 13e, which have inhibitory rates of 60.2% and 54.9%, respectively, with IC50 of 77.9 and 106.7 μM. These IC50 values are much larger than the IC50 0.38 μM of FOS, a value determined in this work that is comparable to the literature value [40], demonstrating that 13a and 13e may be the weak inhibitors of DXR. But due to their high plant inhibitory effects and their low DXR activities, these compounds may have other targets involved in the inhibition process. Another possibility that cannot be excluded is that the deviations might arise from some unknown factors, such as the difference in homology between the plant-origin DXR and EcDXR, and the different molecular structure parameters involved in the complex processes of absorption, distribution, metabolism, and excretion during the action.
Table 5.
Table 5. Inhibition activities of all the synthesized compounds on DXR enzyme.
| Comp | InR (%)a | Comp | InR (%) | Comp | InR (%) |
| 5a | 11.9 | 6f | 11.7 | 13b | 27.4 |
| 5b | 12.9 | 12a | 13.7 | 13c | 28.4 |
| 5c | 6.1 | 12b | 21.9 | 13d | 14.0 |
| 5d | 17.7 | 12c | 32.3 | 13e | 54.9 |
| 5e | 23.0 | 12d | 12.4 | 13f | 24.4 |
| 5f | 32.4 | 12e | 22.8 | 13g | 7.3 |
| 6a | 15.8 | 12f | 6.1 | 13h | 10.3 |
| 6b | 19.5 | 12g | 11.6 | 13i | 30.3 |
| 6c | 7.5 | 12h | 2.3 | 19 | 22.5 |
| 6d | 17.3 | 12i | 27.1 | 20 | 29.5 |
| 6e | 12.1 | 13a | 60.2 | FOS | 98.7 |
a InR value was determined by measuring the DXR activity at a compound concentration of 100 μM.
2.6. DMAPP Rescue and Molecule Docking
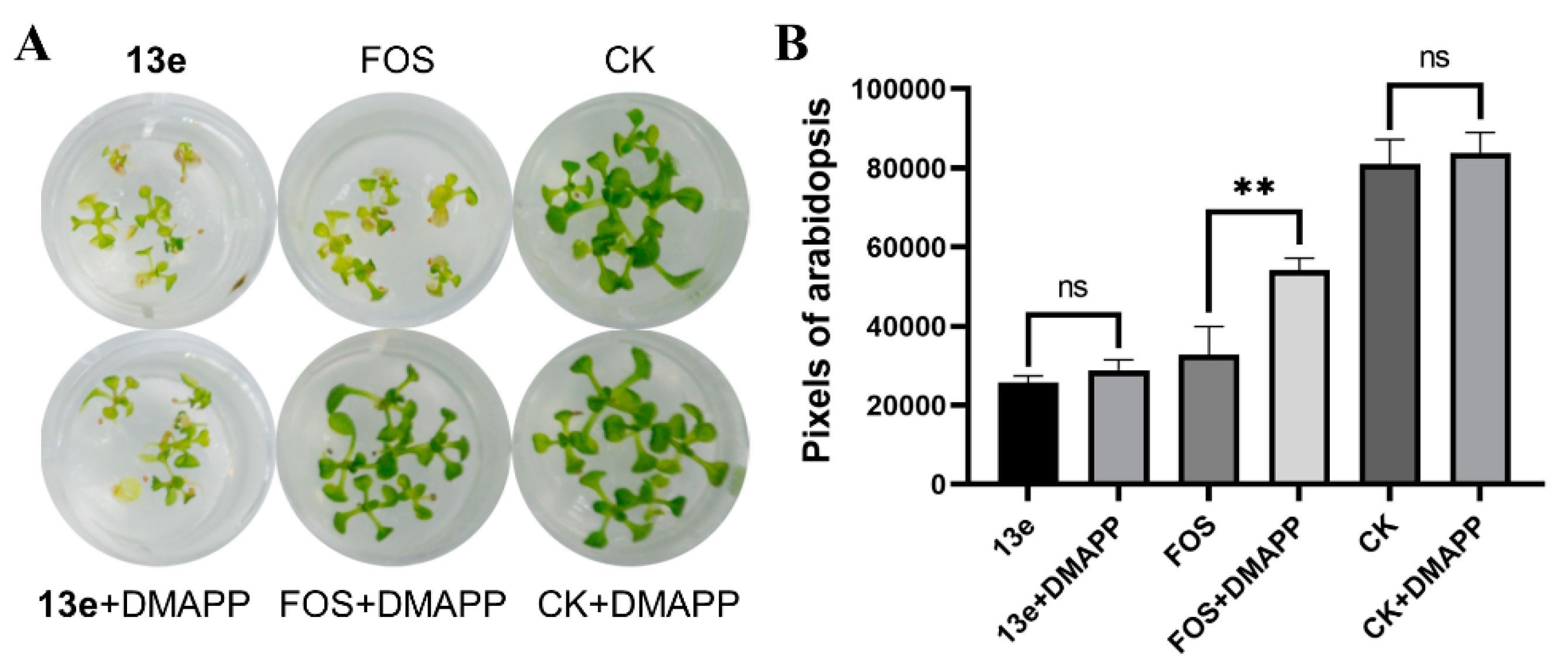
DMAPP rescue. To explore whether the active compounds are acting on the DXR enzyme in the MEP pathway, a DMAPP rescue assay was conducted using the most active compound 13e and the control FOS. The principle of DMAPP rescue is that if the growth inhibition of Arabidopsis is caused by the inhibition of DXR enzyme, adding exogenous DMAPP as an intermediate downstream of DXR should restore the growth of Arabidopsis. As shown in Figure 5, the developmental arrest and bleaching of Arabidopsis by FOS treatment were rescued by the addition of DMAPP, with the green channel pixel values of the Arabidopsis images increased from 32,789 to 54,164, representing a 1.65-fold rescue with evident statistical significance. On the contrary, the addition of DMAPP to the compound 13e treatment group did not show a significant increase in pixel values, indicating that 13e may act on an herbicidal target outside of the MEP pathway.
Figure 5.
Rescue of bleaching and developmental arrest in Arabidopsis by adding exogenous DMAPP. (A) Images of 13e- and FOS-treated Arabidopsis before and after rescue with DMAPP, along with the blank control CK. The concentrations of 13e and FOS were 20 mg/L and 40 mg/L, respectively, and the DMAPP concentration was 150 mg/L. (B) Green channel pixel values of Arabidopsis images after different treatments. **p < 0.01, ns: no significance.
Figure 5.
Rescue of bleaching and developmental arrest in Arabidopsis by adding exogenous DMAPP. (A) Images of 13e- and FOS-treated Arabidopsis before and after rescue with DMAPP, along with the blank control CK. The concentrations of 13e and FOS were 20 mg/L and 40 mg/L, respectively, and the DMAPP concentration was 150 mg/L. (B) Green channel pixel values of Arabidopsis images after different treatments. **p < 0.01, ns: no significance.
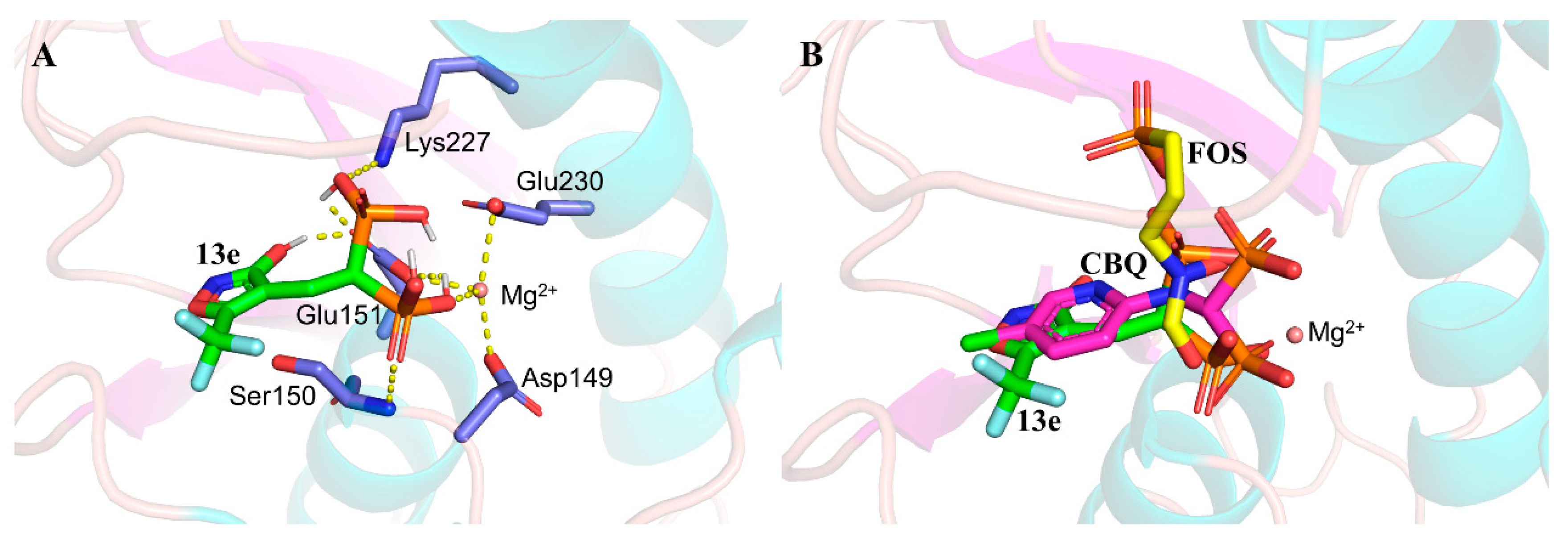
Molecule docking. To further assess the possibility of the mode of action of the active compounds on the target enzyme DXR, molecular docking of compound 13e was performed using Autodock Vina software. Given the structural similarity between the reported pyridine-containing bisphosphonate (CBQ, shown in Figure 2A) and 13e, the complex crystal structure of CBQ with EcDXR (PDB ID: 1T1R) was chosen as a template for docking [23]. The interactions between compound 13e and the surrounding residues of the DXR active site are depicted in Figure 6A. The bisphosphonate group of 13e forms hydrogen bonding with Ser150, Glu151 and Lys227 within the bond length range of < 3.0 Å, while the 3-hydroxyisoxazole also interacts with Glu151 to form an additional hydrogen bond. In addition, 13e demonstrates a binding mode to Mg2+ in the DXR active site similar to that of CBQ, through the coordination of one hydroxyl oxygen of the bisphosphonate group. In Figure 6B, the binding conformations of 13e and CBQ could be superimposed well, with the bisphosphonate group locating in the Mg2+ binding site, and the heterocycle part occupying the hydrophobic cavity, both of which are different from FOS that uses its hydroxamate group to bind with the metal enzyme. It is worthy of mentioning that 13e and CBQ have comparable relative activities with respect to FOS in DXR inhibition, but with much inferior IC50 as compared to that of FOS [23,40]. This tendency to give low inhibition effects for 13e and CBQ may arise from the fact that their binding conformations are reversed as compared to that of FOS, which uses the hydroxamate moiety to coordinate with the metal ion and the monophosphonate to bind with the surrounding residues at the active site of DXR enzyme.
Figure 6.
The docked conformation of 13e with DXR enzyme and the interactions with the surrounding residues (A) and the conformational superimposition of 13e with CBQ and FOS in the DXR active site (B). Key residues are shown as slate sticks, the hydrogen bonds and coordinate bonds are highlighted in yellow dashed lines, and the Mg2+ ion is presented as a wheat sphere.
Figure 6.
The docked conformation of 13e with DXR enzyme and the interactions with the surrounding residues (A) and the conformational superimposition of 13e with CBQ and FOS in the DXR active site (B). Key residues are shown as slate sticks, the hydrogen bonds and coordinate bonds are highlighted in yellow dashed lines, and the Mg2+ ion is presented as a wheat sphere.
2.7. Discussion on Structure-Activity Relationships
In this work, the bioactivity evaluation of the synthesized compounds has been performed on Arabidopsis, pre-emergency model plants Brassica napus L. and Echinochloa crus-galli, post-emergency plants Amaranthus retroflexus and Echinochloa crus-galli, and the recombinant DXR enzyme. Several compounds demonstrated better herbicidal activities compared to FOS, although they might have a different target involved in the inhibiting process. The inhibition phenotype analyses revealed that the molecular structure of the synthesized compounds has a significant influence on the inhibition of different plants, and on Arabidopsis the structure-activity relationship is of representativeness.
In terms of the molecular structure and the Arabidopsis inhibitory activity, several trends could be derived. One is from the influence of the number of phosphate fragment, and bisphosphonates tended to have higher herbicidal activities on Arabidopsis than the monophosphonates, as illustrated in the EC50 ratios for the two bis-/monophosphonate pairs 13a/6a and 13d/6d, 21.6/28.6 mg/L and 23.1/45.7 mg/L, respectively. The second trend comes from the influence of heterocycle unit. For example, the 3-hydroxyisoxazole-containing monophosphonates 6a and 6d displayed moderate inhibitory activities with EC50 of 28.6 and 45.7 mg/L, while other heterocycle-containing monophosphonates gave low or even no inhibition effects. The 3-hydroxyisoxazole-containing bisphosphonates 13a~f performed even better, showing moderate to good activity with EC50 from 7.8 to 48.4 mg/L. The third trend is from the influence of fluorine-containing substituent, and -CF3 (for 13e) and -CHF2 (for 13f) on 3-hydroxyisoxazole have the highest activities, with EC50 as low as 7.8 and 8.7 mg/L, respectively, outweighing all other compounds.
Finally, compounds 19 and 20, which contain the isoxazolinone fragment like the active component of the commercial herbicide clomazone, only exhibited a weak inhibitory effect on Arabidopsis. Clomazone targets the first enzyme DXS in the MEP pathway as a type of propesticide, which is activated by metabolic oxidation in the plant to form ketoclomazone [26]. It is speculated that compounds 19 and 20 are unable to undergo this kind of metabolism in the plant, thereby giving no activation to display an inhibitory role.
The structure-activity relationship summarized on the Arabidopsis inhibition is also applicable to that of the inhibition on other model plants. For example, 3-hydroxyisoxazole-containing 6a and 13a demonstrated good inhibitory activity not only on Arabidopsis but also on other tested model plants; introducing -CF3 and -CHF2 onto the 3-hydroxyisoxazole further improved the herbicidal activity on all plants, especially in the case of compound 13e, which has one -CF3 group on 3-hydroxyisoxazole and displayed the best activity among all the synthesized compounds.
3. Materials and Methods
3.1. Instruments and Reagents
1H NMR, 13C NMR, and 31P NMR spectra were recorded on a Varian Mercury-Plus 400 MHz spectrometer. HRMS were acquired using an Agilent 6224 TOF LC/MS instrument. All reagents and solvents used were of analytical or chemical purity. FOS was synthesized with reference to the method reported by our lab [41].
3.2. Synthesis
The detailed synthesis procedures and characterization data of target compounds are given in the Supporting information.
3.3. Biological Assays
3.3.1. Arabidopsis Growth Inhibition Assay
The Arabidopsis inhibitory activity was evaluated using 24-well plates [42], and the specific cultivation conditions are given in Supporting Information. After 10 days of cultivation, the plates were photographed with a digital camera, and the Adobe Photoshop software was used to determine the green channel pixel value of Arabidopsis image in each well. The inhibition rate was calculated by comparing the pixel values of the compound treatment group to that of the blank group. The initial test concentration was 100 mg/L, and for compounds with inhibition rates higher than 50%, the concentrations of 50, 25, 10 and 1 mg/L were further tested to calculate their EC50 values.
3.3.2. Pre-Emergence Herbicidal Inhibition Assay
The pre-emergence activity of the compounds against monocot Echinochloa crus-galli (EC) and dicot Brassica napus L. (BN) was evaluated using the standard Petri dish test [38]. The compounds were dissolved in DMF, emulsified with Tween-80, and diluted with water to form stock solutions, each of which was further diluted to a gradient of 100, 50, 25, 10, and 1 mg/L for test. The test solutions were added to Petri dishes lined with a filter paper and on which 10 seeds of EC and BN were placed. After cultured in an intelligent climate chamber with a humidity of 65% at 26 °C for 7 days, the corresponding EC50 values were calculated.
3.3.3. Post-Emergence Herbicidal Inhibition Assay
The post-emergence activity of compounds against EC and Amaranthus retroflexus (AR) was determined at a dose of 300 g ai/ha. The model plants initially grow as seedlings, and a few days prior to treatment, five seedlings with similar growth conditions for the plant were transplanted into one pot containing nutrient soil. The test solutions were prepared by dissolving the compounds in water containing DMF and Tween-80 and sprayed when the plant grew to the 1~2 leaf stage. The herbicidal activity was evaluated by visual observation [38] and fresh weighing [39] after two weeks of treatment.
3.3.4. DXR Enzyme Inhibition Assay
The cloning and expression of EcDXR was performed using a method previously reported [40], and the enzyme inhibition activity was evaluated by monitoring the oxidation of NADPH in the enzyme catalysis process using a microplate reader. The detailed protocol is given in Supporting Information.
3.3.5. DMAPP Rescue
The DMAPP rescue on Arabidopsis was conducted using the afore-mentioned culture conditions. The Arabidopsis seeds were divided into three groups labeled as FOS, 13e, and blank control, and the test concentrations of FOS and 13e were 40 and 20 mg/L, respectively. To one of the two species in each group an additional 150 μg of DMAPP (Aladdin) was added. After 10 days of cultivation, the green channel pixel values of Arabidopsis in each well were measured. A two-tailed paired Student's t-test was used to compare the two species within the same group, and P < 0.05 indicates a statistically significant difference.
3.4. Molecular Docking
The co-crystal structure of EcDXR with bisphosphonate compound (PDB ID: 1T1S) was used as the docking template, and the molecular docking of 13e to the EcDXR active site was performed by Autodock vina software. The docking employed the semi-flexible docking mode of the Vina force field, with the coordinate values of Mg2+ as the docking center. According to the docking affinity data and by visually inspecting the docking conformations using the PyMOL software, the best binding conformation of 13e was determined and superimposed with that of CBQ and FOS in the EcDXR active site for comparative analysis.
4. Conclusions
In this work, heterocycle-containing mono- and bisphosphonic acid compounds as FOS analogs were designed by replacing the hydroxamate unit of FOS with various heterocycles while retaining the monophosphonic acid in FOS or replacing it with a bisphosphonic acid group. These compounds were facilely synthesized with the key steps including the Michael addition of diethyl vinylphosphonate or tetraethyl vinylidenebisphosphonate with β-dicarbonyls, and the subsequent cyclic condensation with hydrazine or hydroxylamine. Two additional isoxazolinone-bearing FOS analogs were also synthesized by the Michaelis-Becker reaction as a key step. With the optimization of the key steps, the target compounds were obtained in high yields. The bioactivity evaluation on Arabidopsis and the pre- and post-emergence herbicidal tests on model plants revealed that some of the compounds have activities higher or comparable to that of FOS, and one compound, namely 13e, was identified to have the best activity, for example, on Arabidopsis with a 3.7-fold inhibitory activity enhancement compared to the control FOS. The molecule docking suggested that 13e could interact with the active site of DXR in a different binding manner from FOS, while the DMAPP rescue assay failed to indicate that the DXR enzyme is the target of 13e, implying that a different target might be involved in the inhibiting process. With the finding of valuable compounds with activity higher than FOS and the established facile synthesis method, though, this work demonstrated a valuable strategy of bioisosteric replacement for discovering new FOS analogs with high herbicidal activity.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
X.W. synthesized the target compounds, performed the activity test, and made a draft of this manuscript; M.B. expressed the DXR enzyme and screened the enzyme inhibitory activity; Z.Y. made some synthesis of target compounds and some biological evaluation; D.J. directed all the experimental work, and A.Z. provided the subject support and the working thought and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Acknowledgements
The authors gratefully acknowledge the financial supports of the National Natural Science Foundation of China (22277038), the National Key Research and Development Program for Special Projects (2022YFA1207400), and in part by the Fundamental Research Funds for the Central Universities from China (2022CXZZ101).
Conflicts of Interest
The authors declare no competing financial interest.
References
- Frank, A.; Groll, M. The methylerythritol phosphate pathway to isoprenoids. Chem. Rev. 2017, 117, 5675–5703. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chang, W.C.; Xiao, Y.; Liu, H.W.; Liu, P. Methylerythritol phosphate pathway of isoprenoid biosynthesis. Annu. Rev. Biochem. 2013, 82, 497–530. [Google Scholar] [CrossRef]
- Masini, T.; Kroezen, B.S.; Hirsch, A.K. Druggability of the enzymes of the non-mevalonate-pathway. Drug Discov. Today 2013, 18, 1256–1262. [Google Scholar] [CrossRef]
- Pu, X.; Dong, X.; Li, Q.; Chen, Z.; Liu, L. An Update on the function and regulation of methylerythritol phosphate and mevalonate pathways and their evolutionary dynamics. J. Integr. Plant Biol. 2021, 63, 1211–1226. [Google Scholar] [CrossRef]
- Vranová, E.; Coman, D.; Gruissem, W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef] [PubMed]
- Hoeffler, J.F.; Tritsch, D.; Grosdemange-Billiard, C.; Rohmer, M. Isoprenoid biosynthesis via the methylerythritol phosphate pathway. Mechanistic investigations of the 1-deoxy-D-xylulose 5-phosphate reductoisomerase. Eur. J. Biochem. 2002, 269, 4446–4457. [Google Scholar] [CrossRef] [PubMed]
- Koppisch, A.T.; Fox, D.T.; Blagg, B.S.J.; Poulter, C.D. E. E. Coli MEP synthase: steady-state kinetic analysis and substrate binding. Biochemistry 2001, 41, 236–243. [Google Scholar] [CrossRef]
- Yajima, S.; Hara, K.; Iino, D.; Sasaki, Y.; Kuzuyama, T.; Ohsawa, K.; Seto, H. Structure of 1-deoxy-D-xylulose 5-phosphate reductoisomerase in a quaternary complex with a magnesium ion, NADPH and the antimalarial drug fosmidomycin. Acta Crystallogr. F. 2007, 63, 466–470. [Google Scholar] [CrossRef]
- Lichtenthaler, H.K.; Zeidler, J.; Schwender, J.; Müller, C. The non-mevalonate isoprenoid biosynthesis of plants as a test system for new herbicides and drugs against pathogenic bacteria and the malaria parasite. Z. Naturforsch. C. 2000, 55, 305–313. [Google Scholar] [CrossRef]
- Possell, M.; Ryan, A.; Vickers, C.E.; Mullineaux, P.M.; Hewitt, C.N. Effects of fosmidomycin on plant photosynthesis as measured by gas exchange and chlorophyll fluorescence. Photosynth. Res. 2010, 104, 49–59. [Google Scholar] [CrossRef]
- Okuhara, M.; Kuroda, Y.; Goto, T.; Okamoto, M.; Terano, H.; Kohsaka, M.; Aoki, H.; Imanaka, H. Studies on new phosphonic acid antibiotics. III. Isolation and characterization of FR-31564, FR-32863 and FR-33289. J. Antibiot. 1980, 33, 24–28. [Google Scholar] [CrossRef]
- Okuhara, M.; Kuroda, Y.; Goto, T.; Okamoto, M.; Terano, H.; Kohsaka, M.; Aoki, H.; Imanaka, H. Studies on new phosphonic acid antibiotics. I. FR-900098, isolation and characterization. J. Antibiot. 1980, 33, 13–17. [Google Scholar] [CrossRef]
- Masini, T.; Hirsch, A.K.H. Development of inhibitors of the 2C-Methyl-D-erythritol 4-Phosphate (MEP) pathway enzymes as potential anti-infective agents. J. Med. Chem. 2014, 57, 9740–9763. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, S.; Sundriyal, S. Non-hydroxamate inhibitors of 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR): A critical review and future perspective. Eur. J. Med. Chem. 2021, 213, 113055. [Google Scholar] [CrossRef]
- Knak, T.; Abdullaziz, M.A.; Höfmann, S.; Alves Avelar, L.A.; Klein, S.; Martin, M.; Fischer, M.; Tanaka, N.; Kurz, T. Over 40 years of fosmidomycin drug research: A comprehensive review and future opportunities. Pharmaceuticals 2022, 15, 1553. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Kozikowski, A.P. Why hydroxamates may not Be the best histone deacetylase inhibitors-what some may have forgotten or would rather forget? ChemMedChem 2015, 11, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Hermant, P.; Bosc, D.; Piveteau, C.; Gealageas, R.; Lam, B.; Ronco, C.; Roignant, M.; Tolojanahary, H.; Jean, L.; Renard, P.Y.; Lemdani, M.; Bourotte, M.; Herledan, A.; Bedart, C.; Biela, A.; Leroux, F.; Deprez, B.; Deprez-Poulain, R. Controlling plasma stability of hydroxamic acids: a MedChem toolbox. J. Med. Chem. 2017, 60, 9067–9089. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar]
- Cohen, S.M. A bioinorganic approach to fragment-based drug discovery targeting metalloenzymes. Acc. Chem. Res. 2017, 50, 2007–2016. [Google Scholar] [CrossRef]
- Chen, A.Y.; Adamek, R.N.; Dick, B.L.; Credille, C.V.; Morrison, C.N.; Cohen, S.M. Targeting metalloenzymes for therapeutic intervention. Chem. Rev. 2018, 119, 1323–1455. [Google Scholar] [CrossRef]
- Andaloussi, M.; Lindh, M.; Björkelid, C.; Suresh, S.; Wieckowska, A.; Iyer, H.; Karlén, A.; Larhed, M. Substitution of the phosphonic acid and hydroxamic acid functionalities of the DXR inhibitor FR900098: an attempt to improve the activity against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2011, 21, 5403–5407. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Endo, K.; Kato, M.; Cheng, G.; Yajima, S.; Song, Y. Structures of 1-deoxy-D-xylulose-5-phosphate reductoisomerase/lipophilic phosphonate complexes. ACS Med. Chem. Lett. 2010, 2, 165–170. [Google Scholar] [CrossRef]
- Yajima, S.; Hara, K.; Sanders, J.M.; Yin, F.; Ohsawa, K.; Wiesner, J.; Jomaa, H.; Oldfield, E. Crystallographic structures of two bisphosphonate: 1-deoxyxylulose-5-phosphate reductoisomerase complexes. J. Am. Chem. Soc. 2004, 126, 10824–10825. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Sundriyal, S.; Rubio, V.; Shi, Z.Z.; Song, Y. Coordination chemistry based approach to lipophilic inhibitors of 1-deoxy-D-xylulose-5-phosphate reductoisomerase. J. Med. Chem. 2009, 52, 6539–6542. [Google Scholar] [CrossRef] [PubMed]
- Lamberth, C. Heterocyclic chemistry in crop protection. Pest. Manag. Sci. 2013, 69, 1106–1114. [Google Scholar] [CrossRef]
- Mueller, C.; Schwender, J.; Zeidler, J.; Lichtenthaler, H.K. Properties and inhibition of the first two enzymes of the non-mevalonate pathway of isoprenoid biosynthesis. Biochem. Soc. T. 2000, 28, 792–793. [Google Scholar] [CrossRef]
- Li, G.; Su, Y.; Yan, Y.H.; Peng, J.Y.; Dai, Q.Q.; Ning, X.L.; Zhu, C.L.; Fu, C.; McDonough, M.A.; Schofield, C.J.; Huang, C.; Li, G.B. MeLAD: an integrated resource for metalloenzyme-ligand associations. Bioinformatics 2019, 36, 904–909. [Google Scholar] [CrossRef]
- Padmaja, A.; Reddy, G.S.; Venkata Nagendra Mohan, A.; Padmavathi, V. Michael adducts-source for biologically potent heterocycles. Chem. Pharm. Bul. 2008, 56, 647–653. [Google Scholar] [CrossRef]
- Bulman Page, P.C.; Moore, J.P.; Mansfield, I.; McKenzie, M.J.; Bowler, W.B.; Gallagher, J.A. Synthesis of bone-targeted oestrogenic compounds for the inhibition of bone resorption. Tetrahedron 2001, 57, 1837–1847. [Google Scholar] [CrossRef]
- Chang J.H. Herbicidal 3-isoxazolidinones and hydroxamic acids. US 4405357, 1983-09-20.
- Adeyemi, C.M.; Faridoon; Isaacs, M. ; Mnkandhla, D.; Hoppe, H.C.; Krause, R.W.M.; Kaye, P.T. Synthesis and antimalarial activity of N-benzylated (N-arylcarbamoyl)alkyl-phosphonic acid derivatives. Bioorgan. Med. Chem. 2016, 24, 6131–6138. [Google Scholar] [CrossRef]
- Chofor, R.; Risseeuw, M.; Pouyez, J.; Johny, C.; Wouters, J.; Dowd, C.; Couch, R.; Van Calenbergh, S. Synthetic fosmidomycin analogues with altered chelating moieties do not inhibit 1-deoxy-D-xylulose 5-phosphate reductoisomerase or Plasmodium falciparum growth in vitro. Molecules 2014, 19, 2571–2587. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Wahl, H.; Papadopoulos, K. Asymmetric Michael additions via SAMP/RAMP hydrazones enantioselective synthesis of 2-substituted 4-oxophosphonates. Liebigs Ann. 1995, 7, 1177–1184. [Google Scholar] [CrossRef]
- Dutta, S.; Malla, R.K.; Bandyopadhyay, S.; Spilling, C.D.; Dupureur, C.M. Synthesis and kinetic analysis of some phosphonate analogs of cyclophostin as inhibitors of human acetylcholinesterase. Bioorgan. Med. Chem. 2010, 18, 2265–2274. [Google Scholar] [CrossRef] [PubMed]
- Lolli, M.L.; Lazzarato, L.; Di Stilo, A.; Fruttero, R.; Gasco, A. Michael addition of Grignard reagents to tetraethyl ethenylidenebisphosphonate. J. Organomet. Chem. 2002, 650, 77–83. [Google Scholar] [CrossRef]
- Zare, A.; Hasaninejad, A.; Parhami, A.; Zare, A.R.M.; Khalafi Nezhad, A. Microwave-assisted michael addition of amides to alpha, beta-unsaturated esters under solvent-free conditions. Pol. J. Chem. 2008, 82, 1059–1066. [Google Scholar]
- Sauret-Güeto, S.; Botella-Pavía, P.; Flores-Pérez, U.; Martínez-García, J.F.; San Román, C.; León, P.; Boronat, A.; Rodríguez-Concepción, M. Plastid cues posttranscriptionally regulate the accumulation of key enzymes of the methylerythritol phosphate pathway in Arabidopsis. Plant Physiol. 2006, 141, 75–84. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Y.; Wang, X.; Duan, L.; Duan, J.; Li, W.; Zhang, A. Synthesis and evaluation of halogenated 5-(2-Hydroxyphenyl)pyrazoles as pseudilin analogues targeting the enzyme IspD in the methylerythritol phosphate pathway. J. Agric. Food Chem. 2020, 68, 3071–3078. [Google Scholar] [CrossRef]
- Wang, Y.E.; Yang, D.; Huo, J.; Chen, L.; Kang, Z.; Mao, J.; Zhang, J. Design, synthesis, and herbicidal activity of thioether containing 1,2,4-triazole schiff bases as transketolase inhibitors. J. Agric. Food Chem. 2021, 69, 11773–11780. [Google Scholar] [CrossRef]
- Kuntz, L.; Trisch, D.; Grosdemange-Billiard, C.; Hemmerlin, A.; Willem, A.; Bach, T.; Rohmer, M. Isoprenoid biosynthesis as a target for antibacterial and antiparasitic drugs: phosphonohydroxamic acids as inhibitors of deoxyxylulose phosphate reducto-isomerase. Biochem J. 2005, 386, 127–135. [Google Scholar] [CrossRef]
- Wu, X.; Ping, H.; Song, C.; Duan, J.; Zhang, A. Optimization synthesis of phosphorous-containing natural products fosmidomycin and FR900098. Phosphorus Sulfur. 2023, 198, 446–452. [Google Scholar] [CrossRef]
- Corral, M.G.; Leroux, J.; Stubbs, K.A.; Mylne, J.S. Herbicidal properties of antimalarial drugs. Sci Rep-UK. 2017, 7, 45871. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



