
Submitted:
31 August 2023
Posted:
01 September 2023
You are already at the latest version
Abstract
Addressing global environmental problem, water splitting to produce hydrogen fuel by solar energy is getting so much attention. In the water splitting, the essential problem to solve is the development of efficient catalysts for oxygen production. In this paper, having the prospect for a practical application of photocatalysts to artificial photosynthesis, molecular mechanisms in the current literature are briefly reviewed. At first, recent progress in the function of Mn cluster at the natural photosystem II is briefly described. The kinds of devices in which oxygen evolution reaction (OER) catalysts are used were designated; water electrolyzers, photoelectrodes, and photocatalysts. Some methods for analyzing molecular mechanism in OER catalysis, emphasized by FTIR method, are shown briefly. After describing common OER mechanisms, the molecular mechanisms are discussed for TiO2 and BiVO4 photoelectrodes with our novel data, followed by presenting of co-catalysts IrO2, RuO2, NiO2, and other metal oxides for OER catalysts. Recent reports describing OER catalysts of perovskites, layered double hydroxides (LDH), metal-organic frameworks (MOF), single atom catalysts, as well as metal complexes are reviewed. Finally, by comparing with natural photosystem, the requiring factors to improve the activity of the catalysts for artificial photosynthesis will be discussed.
Keywords:
oxygen evolution reaction
; catalysis
; mechanism
; artificial photosynthesis
; titanium oxide
; bismuth vanadate
; perovskite
; metal oxides
; Fourier transform infrared spectroscopy
; density functional theory
; metal oxide
; solar to hydrogen conversion efficiency
1. Introduction
The photosynthetic system using the solar energy has played a major role in the evolution of plants on the earth, which can oxidize water to generate molecular oxygen, then the global environment was greatly changed 2.4 billion years ago [1]. Before the generation of oxygen, living organism used hydrogen sulfide as an electron source, but over the course of about one billion years, living organism evolved and became to generate living organisms by using water as an electron source. Thus, the present earth environment contains oxygen. Among living organisms that use oxygen, Homo sapiens was born only about 0.2 million years ago. In recent years, the industrial revolution using fossil fuels changed rapidly the global environment. Then, to replace fossil fuels there is a need to produce hydrogen using solar energy as a chemical energy. In addition, it is becoming a reality that hydrogen is used to synthesize e-fuels by the reaction with CO2. In the generation of hydrogen by the water splitting, molecular oxygen should be evolved at the same time. However, it is not so easy to artificially achieve the photosynthetic oxygen generation by developing a novel system in a short period of time, which took one billion years to be systematized in the natural world. The oxidation of water to produce oxygen, i.e., oxygen evolution reaction (OER), involves transfers of four-electrons and four-protons, while the reduction of water to produce hydrogen, i.e., hydrogen evolution reaction (HER), is a reaction of two-electrons and two-protons. Actually the overpotential for OER is inevitably higher than that for HER, making it a very difficult reaction to achieve. In the OER process, chemical species in each oxidation step are unstable and called reactive oxygen species (ROS). They are OH radicals, hydrogen peroxide, and superoxide radical, which are probably produced in OER processes, because ROS were actually detected by the photocatalytic reactions at the semiconductor surface [2]. To sustain the global environment, the reduction of CO2 by solar light is also important reaction. However, the CO2 reduction catalysts are useful only when water is oxidized to generate oxygen at the same time [3]. Since the reaction is very important, there are a lot of research papers describing the strategy to improve the OER efficiency by several method, and the analysis of the reaction mechanism are also reported.
In this paper, molecular mechanisms of water oxidation catalysis are briefly reviewed, with anticipating a practical application to artificial photosynthetic systems with photocatalysts [4]. At first, the function of Mn cluster at the center of the photosystem II and the movement of proton to be released from water is briefly described. It is followed by the classification of catalysts for the OER employed, i.e., electrolyzer, photoelectrodes, and photocatalysts, in which the reaction mechanisms should have some difference. In the next section, the methods to investigate molecular mechanism of OER processes are brief described, where Fourier transfer infrared (FTIR) spectroscopy was mainly mentioned because it could detect molecular structure in the OER process. Next is the widespread common mechanisms of OER, followed by the molecular mechanism of OER for each catalyst which includes TiO2, BiVO4, SrTiO3 as semiconductor having the ability of direct excitation with photons. Especially for TiO2 and BiVO4, based on some unpublished data some discussions were added to review the reported data. Following part of this report devoted to the recent progress about molecular mechanism of OER for various catalysts; IrO2, RuO2, transition metal oxides, CoOx, NiOx, FeOx, mixed metal oxides, perovskites, layered dihydroxides (LDHs), metal organic frameworks (MOFs), metal complexes, single atom catalysts (SAC), and so on. Finally, the characteristic difference between natural photosystem and present OER catalysts are pointed out, and the requiring factors to improve the activity of the catalysts for artificial photosynthesis will be discussed.
2. Oxygen Evolution Mechanism of Photosystem II (PS II)
In photosynthesis of plant, acrossing the thylakoid membrane, a series of electron transfer and proton transfer takes place simultaneously to produce one molecular oxygen and four protons by the oxidation of two water molecules on the one side. On the other side of the membrane, high energy molecule is produced ATP from ADP in conjunction with the formation of an electron carrier, NADPH [5]. Oxygen is produced in light-driven water plastoquinone oxidoreductase enzyme which is known as photosystem II (PS II). At the reaction center of PS II, photo excitation energy is gathered with six pigments at the manganese-calcium-oxide based Mn4CaO5 cluster in the reaction center, and then four electrons and four protons are transferred [6,7].
The radiation-damage-free structure of PS II was successfully analyzed with femtosecond X-ray free-electron lasers (XFEL). On the absorption of second photon, sixth O atom is coordinated at the position of 1.5Å from the fifth O atom of the Mn4CaO5 cluster, forming O=O bond [8]. The sixth O comes from H2O which was introduced by flipping amine of the glutamic acid (Glu) residue [9].
Detailed time cause of electron transfer and proton transfer were analyzed by many spectroscopic methods, such as time-dependent fluorescence and transient absorption. The Mn4CaO5 cluster, at which molecular oxygen is generated, releases an electron to reduce the neighboring phenylalanine residue (Phe) [7]. Flash induced FTIR difference spectroscopy revealed that the electron further moves to an iron quinone complex due to a small energy difference [10]. EPR measurements forecasted that, at the last quasi-stable state in four step oxidation, peroxo group, O-O bond, is produced in the cluster, and immediately it turns into O2 molecule forming O vacancy [11]. Density functional theory (DFT) calculation suggested that, this O-O bond has the ionic state of [O2]3-, and then antiferromagnetic interaction with Mn ion causes to stabilize O-O group to make a unique low energy reaction path [12].
Application of time-resolved X-ray emission spectroscopy (XES) to each oxidation step revealed that oxidized tyrosin residue (Tyr) causes the fourth electron transfer generating MnIV in the cluster, all four Mn ions of the cluster became the valency of IV before the generation of O-O bond [13]. Structural analysis was further developed [14], time-resolved structure after the photon absorption suggested time cause of the position of ligands and the water path [15]. Time-resolved IR spectra of microsecond resolution and computational chemistry showed the reaction time of electron transfer and proton transfer of four steps after each photon absorption as shown in Figure 1 [16].
In the photosynthesis cycle in Figure 1, reaction process from S3’ to S4 was clarified recently as shown in Figure 2 [16]. (i) Immediately after the electron release from the CaMn4O5 cluster to Tyr, the proton coordinated to Glu moves to asparagine acid (Asp) residue by a tactical proton relay mechanism. (ii) O6 gets close to O5, and (iii) O-O bond is formed.
A new prototype of a semiartificial system is reported by anchoring PS II on polyethyleneimine-coated microporous carbon electrode. Practical application may have a difficulty in the low current density and durability [17]. Thus, the hybrid application may not be easy because PS II has been developed only to work in nature.
3. Devices for Oxygen Evolution
It has been reported since 18 century that application of electricity to the electrode meets with the dissociation of water to produce hydrogen and oxygen at the ratio of 2:1 [18]. This electrolysis reaction is practically used as water electrolyzer to produce oxygen in the present day. About 50 years ago, it was found that a TiO2 semiconductor electrode caused the formation of oxygen and hydrogen by irradiating light [19]. After the report of this photoelectrochemical method, particulate photocatalytic reaction was tried to photo-split water into hydrogen and oxygen with many semiconducting materials [4]. As the developments in the water splitting methods, this section is divided by three subheadings; water electrolyzers, photoelectrochemical cells, and particulate photocatalysts.
3.1. Water Electrolyzer
Currently, 95% of total hydrogen is formed by steam reforming of fossil fuels [20]. On the other hand, 5% of hydrogen in global market is produced by water electrolysis. The electricity capital expenses account for over 60% of H2 content for renewable electricity if the electrolyzer can keep sustainable operation for 4000 hours [21]. If the electricity could be served by solar panels, residual electricity caused by the monthly and dairy variations of sunlight could be used to store as the hydrogen energy. This system may actually contribute to the global environmental problems. From the view point of the global transportation of energy, green hydrogen, as solar driven chemical energy, should become indispensable in the near future and then water electrolysis will become widespread technology. Since OER process is significantly difficult by comparing with HER process as described above, industrial scale of OER devices becomes important to produce green hydrogen.
As the water electrolyzer, there are alkali system in which anode and cathode is separated by porous diaphragm [21], acidic system with proton exchange membrane [22,23,24], and solid oxide system with high temperature O2- conductor [25]. Water electrolysis is the reverse reaction of hydrogen-oxygen fuel cell where chemical energy is changed to electric energy. Therefore, the equipment configuration of the apparatuses with proton exchange membrane and solid electrolyte resemble the polymer electrolyte fuel cell (PEFC) and solid oxide fuel cell (SOFC), respectively. Alkaline water electrolyzer is conventionally used in industry, but the maximum current density is 400 mAcm-2 and the spending a long time to start the operation. Then, the alkaline system is not favorable to use the renewable energy with large fluctuation [25]. Since the solid electrolyte need high temperature, most of the researches aim to acid water electrolysis [26]. The U.S. Department of Energy announced a goal to reach the lifetime of 80,000 hours in 2050 [23].
Electrochemical water splitting system can be easily integrated with photovoltaic system, to serve a solar water splitting system. Actually, solar to hydrogen (STH) conversion efficiency of 30% has been reported by connecting an available 3 layer solar cell of InGaP/GaAs/GaInNAsS with an electrolyzer consisting of Pt black cathode and Ir black anode [27]. By integrating Pt and Ni on the each surface of GaInP/GainAs/Ge solar cell, the obtained photoelectrochemical cell showed 18% of STH efficiency at 15 suns [28]. By using a commercial available IrRu coated Ti anode connected to a Pt coated Si photocathode by perovskite solar cell, the STH efficiency was reported to be more than 17% [29]. Furthermore, by using a concentrated integrated photoelectrochemical device, kilowatt-scale solar hydrogen production system is reported to be realized [30]. Though higher STH efficiency has been reported, enlargement of the devices and the cost for the practical operation may become problem.
The OER catalysts for water electrolysis have been developed mainly in alkaline and acidic condition. Since photocatalytic water splitting is expected to take place mainly in neutral solution, the OER catalysts for electrolyzer may not be suitable to the photocatalytic system. However, water electrolysis in nearly neutral condition have been also developed, which contains the OER catalysts used as co-catalyst of photocatalysts [31].
3.2. Photoelectrochemical Cell
Photoelectrochemical (PEC) cells based on an interfacial electric field formed at a semiconductor-electrolyte junction aim to achieve solar-driven water electrolysis at low cost and with low complexity [32]. This is the method to produce hydrogen by solar energy without using solar cells, which is discovered 50 years ago [33]. PEC devices have been investigated for many semiconducting materials. The water electrolyzers stated above are operated at a high current of more than 1Acm-2. On the other hand, since the uncondensed solar flux is less than 10 mAcm-2, the operation condition of OER catalysts of PEC devices is different from that of electrolyzers. The materials which pose catalytic activity and durability under anodic polarization are limited to TiO2, WO3, BiVO4, α-Fe2O3, β-NiOOH, CuWO4 [34,35,36,37].
Photoelectrochemical reaction with semiconductor electrodes was investigated in connection with the research in the reaction mechanism at the surface of visible-light responsive photocatalysts; GaN [38], Ta3N5 [39,40], LaTiO2N [41]. When BiVO4 was used as photoanode, solar water splitting efficiency of 3.17% has been reported [42]. To increase the OER efficiency, loading of CoPi catalyst [43], and application of high valence iron ion, Fe4+, have been reported [44]. The STH efficiency of the device made of semiconductor photoelectrodes has not exceed that of the above mentioned combination device with a water electrolyzer and a photovoltaic cell. For solar-driven electron source, OER at SrTiO3 and BiVO4 photoanodes were used not only to the generation of hydrogen but also the reduction of CO2 to generate formic acid [45] and carbon monoxide [46], respectively.
3.3. Particulate Photocatalysts
Particulate semiconductor photocatalysts seem to act as a small device of electrolyzer combined solar cell [47]. However, reaction on the electrode surface are essentially different from that on the photocatalyst surface as discussed previously [48]. In the case of electrochemical reaction, electrons are supplied continuously from the electric source through lead wire. On the other hand in the case of photocatalysts and photoelectrode, interfacial electron transfer reactions occur only at the absorption of photons. Thus, to proceed four-electron reaction, the reaction step of OER should wait until the absorption of next photon. Besides, in electrochemical reaction, the electron transfer occurs at the electric field gradient formed at the surface of the electrode against the electrolyte in the solution, while in photocatalytic reaction the surface-trapped carrier generate the electric field gradient against the surface Fermi level [48]. Since the surface trapped carrier may recombine with the carrier generated by the next absorption of photon, the electron transfer reaction in photocatalysis should be rapid to overcome the rapid recombination reaction. Thus the stability of the reaction intermediate, ROS in the case of OER, is most important to accomplish the process. In the case of the PS II described above, the reaction intermediate was not formed but the photon energy is accumulated in the valency of Mn ions, and, on the third flash, O2 is released immediately after the O-O bond formation.
There is an extensive review about water splitting with semiconductor photocatalysts [49]. Application of photocatalysis to the reduction of CO2 has been compiled [50]. For practical testing of solar hydrogen generation, porous sheets of 100 m2 made of photocatalyst powder were employed where the STH efficiency was 0.76% [51]. To increase the STH efficiency, p-n junction photocatalysts were proposed by combining two semiconductors to avoid the recombination [52]. In the recent paper, by using Co3O4 as an OER co-catalyst on InGaN/GaN nanowire photocatalyst, STH of 9% in pure water, 7% in sea water have been reported [53]. In large scale photocatalytic water-splitting system, an STH efficiency of 6.2% was described [53]. In comparison with the photovoltaic-electrolyzer combination systems, photocatalytic systems have some strong points which contain the scalability to enlarge for industrial manufactures with abundant materials [49].
4. Methods for Investigating Mechanism
Molecular mechanism of OER in atomic level is conveniently predicted by means of calculation chemistry. However, it should be supported by appropriate experimental measurements. In the field of photocatalysis, FTIR and electron paramagnetic resonance (EPR) spectroscopies under the operation have been used to investigate the reaction mechanism. Operand/in situ measurements are recently utilized for other spectroscopy such as X-ray absorption spectroscopy (XAS) and X-ray emission spectroscopy (XES), especially for electrocatalysts [54,55]. In this section, such analytical methods for catalyst surface, including operand X-ray diffraction (XRD) measurements, electrochemical transmission microscopy (EC-TEM), will be described briefly.
4.1. Electrochemical analysis
Electrochemistry is conventional technique to investigate the electron transfer reaction at the solid surface. For kinetic analysis used are linear sweep voltammetry (LSV), cyclic voltammetry (CV), and electrochemical impedance spectroscopy (EIS). For the analysis of the catalytic activity, initial rise and slope of the Tafel plot of LSV were used. Initial rise is represented by overpotential at 10 mA cm-2, for example. The slope described in the unit of mV/dec is useful to microkinetic analysis, which can predict the rate-determining step in the four-step oxidation of OER [56,57,58]. In the cases of α-Fe2O3. BiVO4, TiO2 electrodes, when the density of holes is small, the OER rate depended first order on the holes, indicating that the first step of OER is rate-determining. On the other hand, when the hole density is large, the OER rate depend third order, indicating the third oxidation step of rate determining [59,60], which agrees with the prediction that the second oxidation step is the formation of stable H2O2. Furthermore, in CV measurements, isotope effect on the peak potential by using D2O [61] could be used to investigate the mechanism of OER. These electrochemical measurements could not be applied to investigate semiconductor particulate photocatalysts, because the current at the particle surface cannot easily measured.
4.2. Fourier Transform InfraRed (FTIR) spectroscopy
FTIR (or IR) spectroscopy is a technique to measure the vibrational mode of bonds between atoms and, different form the Raman spectroscopy, active only for the vibrational motions which can change dipole moment. Since the sensitivity is usually higher than the Raman spectroscopy, FTIR is the technique to obtain the key information for molecular mechanism of catalysis [62]. Especially for OER mechanism, FTIR could detect directly the adsorbed intermediate species, which is usually the ROS adsorbed on metal ions. To confine the detection area, the attenuated total reflectance (ATR) method could be used. The use of D2O and H218O, which causes the isotope shift of the signal as predicted by the computer simulation, is helpful to assign the signal. When linearly polarized IR light was used, the direction of the bond against the surface could be detected. Though FTIR spectroscopy is a traditional technique, a novel methods with a synchrotron radiation source has been reported [63].
In Table 1, listed are the reported position of FTIR signals for the OER intermediates adsorbed on several metal ions.
4.3. Raman spectroscopy
Raman spectroscopy can be used to detect vibration mode of the surface species similarly to the FTIR spectroscopy, and different from FTIR, in Raman spectroscopy, the signal of H2O could not be observed [92]. Therefore, it is useful for applying to operand detection of intermediate in OER. For example, the change of surface CoO to CoOOH in the OER reaction at Co3O4 surface have been reported [93]. Since the signal intensity is generally weak, surface enhanced Raman spectroscopy (SERS) with plasmonic metal particles and tip-enhanced Raman scattering (TERS) with plasmonic SPM (scanning probe microscope) tips have been utilized [62].
4.4. Electron Paramagnetic Resonance (EPR) spectroscopy
EPR spectroscopy is a unique method to directly detect paramagnetic chemical species with the unpaired electrons which is caused at the surface of catalysts in the both liquid and solid states [94,95]. Since the method is usually to detect electron spins, it is called electron spin resonance (ESR) in sometimes, or it should be called electron magnetic resonance (EMR) [96] by contrasting with NMR. Since metal atoms could have several spin states depending on the valency and coordination states, EPR is useful technique to detect the situation around the active site. In addition to metal ions, it could also be used for detecting radical species derived from organic compounds and inorganic molecules.
4.5. Electrochemical Transmission Electron Microscope (EC-TEM)
Though the main part of TEM should be in an ultra-high vacuum, when the sample holder is arranged to be very thin and well shielded from the vacuum, operando TEM image could be obtained [97,98]. Furthermore, electron beam refraction pattern could be acquired [98]. Actually, a phase transition under the polarization was reported [98]. When EC-TEM was combined with electron energy-loss spectroscopy (EELS) and energy dispersive spectroscopy (EDS), valence and distribution of positive ions under catalytic reaction could be investigated [99].
4.6. X-ray Photoelectron Spectroscopy (XPS)
XPS is used usually in vacuum because electrons cannot easily move in the air, therefore it is difficult to measure the surface of catalyst under water environment. When the sample surface is separated from vacuum condition with a thin membrane, the detection of XPS of samples under the normal pressure could be achieved [100].
4.7. X-ray Absorption Spectroscopy (XAS)
XAS can be used to detect the surrounding of the X-ray emitting metal. In the XAS method, X-ray absorption near-edge structure (XANES) gives the information of valence and electronic configuration, and extended X-ray absorption fine structure (EXAFS) is used to find out inter atomic distance, coordination number, disorder of the coordination shell [101,102,103]. For electrocatalysts, operand XAS observation have been reported [102,104,105].
4.8. X-ray Emission Spectroscopy (XES)
XES is a technique that probe the radiative transitions from an occupied upper shell to an unoccupied or partially occupied core orbital. Since the excitation to the upper shell is required to cause the transition from the upper shell, XES provides the complementary information of XAS. When the excitation is performed with XFEL or synchrotron radiation, time-resolved spectra could be obtained. Information about electronic structure, crystal field, and metal-ligand covalence could be obtained. When emission wavelength could be detected from soft X-ray to hard X-ray, measurable element spans from ligand elements, such as O, S, P, to transition metals and actinides [61]. The apparatus used for XES is applicable for the measurements of resonant inelastic X-ray scattering (RIXS) and high energy resolution fluorescence detected XAS (HERFD-XAS) [61].
4.9. Mössbauer spectroscopy
Mössbauer spectroscopy is a kind of resonance absorption techniques in which the gamma ray of radio isotopes is used as the light source. Since photon energy of gamma ray is very high, the sensitivity is high. The oxidation state, the spin state and symmetric property of ligands could be analyzed. However, nuclei which have a gamma-ray emitting isotope are limited to Fe, Sn, Ir, so on [106]. Recently, synchrotron resonance was used as the light source for the Mössbauer spectra of one of the actinide 161Dy [107]. Time-dependency of the ligand states [107] and the vibrational stets of ligand of 57Fe, was operando measured in CO2 reduction reaction [108].
5. Common mechanisms of OER catalysis
Figure 3(a,c,d) show the catalytic mechanisms with metal oxides for OER reaction where four protons and four electrons are removed from two H2O molecules to produce one O2 molecule. The mechanisms are classified by whether the lattice oxygen is included (d) or not included (a,c).
In the case that the lattice oxygen is not included, when the reaction proceeds only one metal ion as shown in Figure 3(a), it is called adsorbate evolution mechanism (AEM). While two adjacent metal ions contribute to proceed the OER as shown in Figure 3(c), it is called interaction of two metal-oxide entities (I2M) mechanism, oxide pair mechanism (OPM), or oxide coupling mechanism (OCM). On the other hand, when lattice oxygen is participated in the mechanism, it is inclusively called lattice oxygen mechanism (LOM), and Figure 3(d) is an example of the possible LOM processes [61,109].
On the assumption of AEM process, as shown in Figure 3(b), H2O is sequentially oxidized to *OH, *O, *OOH as the intermediates adsorbed on the surface, where * represents the adsorption site on the catalyst. The free energy can be calculated by computational method, usually density functional theory (DFT) calculation is employed. For example, the free energy of each intermediate for OER catalysis of TiO2, IrO2, and WO3 is shown in the literature [110]. Figure 3(b) schematically shows the free energy of each oxidation step in AEM process. Since the oxidation energy of water is 1.23 eV ×4, the position in the free energy of *+H2O and *+O2 is fixed. The overpotential η is calculated from the maximum difference of the free energy Δ Gn,max = max{Δ Gn (n=1,2,3,4)} in the four energy steps as the difference from 1.23 eV, η = Δ Gn,max -1.23 eV. As the results of the calculation, it was shown that the free energies of *OH and *OOH are linearly correlated, meaning the energy deference between Δ G*OH and Δ G*OOH , that is Δ G2+Δ G3 in Figure 3(b), is constant to be 3.2 eV. Therefore, when Δ G*O locates at the middle between Δ G*OOH and Δ G*OH the overpotential η becomes minimum. Therefore, when the expected overpotential η is plotted against Δ G2 (=Δ G*O - Δ G*OH ), η is expected to become minimum of 0.37 at Δ G2 =1.6 eV. This is named scaling relationship, but it is adopted only on the AEM assumption [111].
In the reaction model of LOM, one of the O atoms of oxygen molecule comes from lattice oxygen. Thus, the reaction mechanism shown in Figure 3(d) is one of the possible LOM processes. In Figure 4, three other possible LOM processes are shown [112]. In the oxygen-vacancy site mechanism (OVSM) shown in Figure 4(a), a hole attacks the surface lattice oxygen, followed by nucleophilic attack with a water molecule (WNA) causes the addition of OH group at the lattice O atom. In the next hole attacks causes the release of O2 and a proton. Next oxidation step cases a surface lattice OH from OH- in water. And with the fourth hole, surface OH release proton to return to the original surface O atom. In the single-metal-site mechanism (SMSM) shown in Figure 4(b), when a hole comes into surface lattice oxygen, surface OH becomes surface OO by releasing proton and making surface O vacancy, with the next hole, O2 released and the surface O vacancy was filled by OH. At the next step, surface OH at the metal is recovered, then at the next step, H of the surface lattice OH is removed to return to the original form. In the double-metal-site mechanism (DMSM) of Figure 4(c), the protons of the adjacent two bridged OH groups are removed by two holes and the M-OO-M bond is formed at the surface. At the next step, O2 is released and the vacancy is coved with surface OH at the attack of the fourth hole.
Beside four LOM processes described above, other LOM path can be drown depending on the kind of metals and the surface crystalline lattice structure. Based on the assumption of the process, the oxidation steps of Gibbs free energy in OER process can be theoretically calculated. It should be noted that removing of surface lattice oxygen in the LOM process suggests a high probability of dissolution of metal ions.
6. Molecular mechanism of OER
6.1. TiO2
History of TiO2 for photo induced water splitting and its molecular mechanism have been described in our previous report [113]. Abundant reports in which molecular mechanism of OER at TiO2 surface could be found. Research progress in the theoretical calculations for water splitting with TiO2 based photocatalysts were also reported [114,115]. Most of the theoretical calculation were performed for rutile (110) and (001) surfaces. Recently rutile (100) surface was investigated for OER activity assuming AEM process [116]. However, it has been shown experimentally that rutile powder consist of (110) and (011) or equivalent surfaces, and reduction occurs at (110) surface while oxidation occurs at (011) surface. Though anatase (101) surface is also used in the theoretical calculation, many experimental observations have indicated that rutile TiO2 is more active for OER than anatase TiO2 [2, 4]. This experimental conclusion could be explained by the authors as the surface Ti5C-Ti5C distance of rutile is shorter than that of anatase [117]. And the recent theoretical calculation supported this explanation by comparing anatase (101) surface with rutile (110) surface [118].
In our previous report, a molecule Ti5O19H16 modeling rutile (011) surface was used to investigate OER process [113]. As the result of the total energy calculation, surface Ti5C which can be coordinated by H2O could make a hydrogen bonding with facing bridged O tom. As shown in Figure 5, the first hole attacks the bridge O and then moves to the hydrogen bonding H2O molecule to form Ti-O• at the facing Ti5C that is the adsorbed OH radical. Second hole can attack Ti-O• or bridged O again to generate OH radical combined to Ti-O•, resulting in the formation of Ti-OOH species. This adsorbed hydroxyperoxo could stabilized by forming Ti-OO-Ti structure with a facing another Ti5C at the (011) surface. This calculation result suggested that the surface three Ti atoms concerning to the OER process are not laid on a line but the third Ti of Ti5C should be facing to the bridged O [113].
Two decades ago, Nakamura et al. reported a molecular mechanism of water oxidation at rutile powder as shown in Figure 6 [119,120], and, in our previous review for the ROS formation [2], their reports were referred in the discussion. In his mechanism, 3-coordinated bridging O at a step or kink of the surface is attacked by a hole, simultaneously WNA takes place to form Ti-O•, and then a pair of Ti-O• generate Ti-OO-Ti structure, and it becomes Ti-OOH [120]. Our prediction above suggests that the third surface Ti atom may contribute the following steps. The assignment of FTIR signals was added in this figure. Since the peak at 812 cm-1 was not clearly shown in their in situ spectra [119], Ti-O• may directly become Ti-OOH by the second hole.
The molecular mechanism in Figure 6 was obtained by the observations with the ATR-FTIR for rutile powder and the STM and photoluminescence for rutile single crystals of (100) and (110) facets. The assignment of FTIR spectra were performed for photoinduced O2 reduction under irradiation based on the isotope effect using D2O and H218O [75]. For the signal of Ti-OOH group, the isotope effect on the FTIR peaks are shown in Table 2. The calculated signal position and the isotope effect are also listed in Table 2, which was performed for the above mentioned (011) surface model. The signal assignment seem reasonable and the isotope effect are well simulated. In the calculation, the signal intensity for bending mode δ(OOH) is larger than that of the stretching mode ν(OO) and signal of bending mode was clearly shown in the report [75]. However, in the in situ FTIR spectra for water oxidation, the signal at larger than 1100 cm-1 in the spectrum was not shown [119]. The spectrum measured in H218O did not show the spreading near 1100 cm-1. Since the many chemical species have signals around 838 cm-1, the observation of Ti-OOH in the experiment may have some problem. They used Fe3+ ions for electron scavenger in the ATR-FTIR measurements, while in our study, when electron scavenger was oxygen, no peaks around 850 cm-1 was observed [117]. They concluded the involvement of lattice oxygen in the OER process, based on the small shift observed for the 838 cm-1 peak, and deduced the molecular mechanism in Figure 6. Since the assignment of the observed peak has ambiguity, the OER process in Figure 6 may have some problem.
It has had a consensus that first oxidation takes place at bridged oxygen Ti-O-Ti, and the reaction mechanism of OER was discussed [2]. Recently, Zhuang and Cheng [121] reported that at the rutile (110) surface, pKa of Ti5COH2 is larger than that of Ti5COH-, and then the coordination structure, Ti5CO2- exist stably in water and easily become Ti-O•. This results in calculation seems to contradict to the fact that isoelectric point of rutile TiO2 is around pH 6. The rutile (110) is not a minor surface in rutile powder as described obove. It should be noted that theoretical calculations can bring the result on the basis of the assumed model regardless of the experimental reality. Therefore, it should be careful to refer the molecular mechanism derived only from the theoretical calculations. The calculation should be used only for the case justified with the experimental results.
6.2. BiVO4
BiVO4 was discovered first as the semiconductor photocatalyst to have ability of water oxidation with visible light and the developed process was compiled in the report by Kudo et al. [122]. As the oxygen evolution photocatalysts, Mo-doped BiVO4 embedded into an Au layer was employed in a Z-scheme photocatalytic system to exhibit 1.1 % of STH efficiency [123]. Aiming to the utilization in photocatalytic oxidation, there are many reports which describe the effects of surface crystalline system, morphologies, hetero junctions, and so on [124].
Electrochemical impedance technique was applied to investigate the micro kinetics at BiVO4 photoelectrode and found the long-lived holes (0.1- 1 s). Two kinds of recombination paths were suggested; one is the fast recombination with photogenerated electrons and another is the recombination with BiVO4 bulk electrons [125]. By measuring the life-time of the photoluminescence, the recombination was found to occur in nano seconds, which is probably caused by the multiphonon transition with deep-energy defect [126]. Sub-bandgap is formed by the V-deficiency to accelerate the recombination rate [127].
To understand the reaction mechanism, there are many computational researches in literature. Walsh et al. concluded that valence band (VB) of O3p is coupled with Bi6s to rise the VB maximum, while conduction band (CB) consists of V3d and O2p and Bi6p. Therefore, adsorption of ROS is expected to take place at Bi site [128]. On the other hand, in a later report for (010) surface, O vacancy provides the V site as active site. That is, the presence of the O vacancy increased the adsorption energy of H2O, OH, O•, and the calculated free energy showed the decrease of barrier for spontaneous charge transfer to electrolyte [129]. Doping of Co2+ replacing with Bi3+, forming O vacancy, and H2O was replaced the vacancy, which has been calculated to cause decrease in the free energy by -0.28 eV [130]. At (001) surface without vacancy, OH radical is easily formed, on the other hand at (101) surface, where vacancies are generated by Mo/W doping, strong charge transfer to oxidation intermediates in OER takes place. This difference of the surface character causes charge transfer between (101) and (001) surfaces [131]. For (010) surface, surface O vacancy is important to the adsorption of water in the catalytic activity [132]. The effect of O vacancy at the subsurface on the charge accumulation in OER process was investigated. As a result, the O vacancy does not affect the photon acquisition nor energy transportation in the crystal [133].
Twin-structured BiVO4 was examined in the energy calculation [134]. The structure of rate-determining step was considered with energy calculations in the OER process for four models (two AEM and two LOM). It was concluded that a larger amount of twin structure causes a high OER activity [134].
Nikacevic et al. theoretically suggested some routes of OER at BiVO4 surface as shown in Figure 7. At (001) surface, 97 % of vacancy are coordinated with water, As shown in Pathway A in this figure, Bi-OO-V is formed as an intermediate, and O2 is evolved. On the other hand, only 0.05% of the O vacancy at the (011) surface was coordinated with water, but as shown in pathway B, through Bi-OOH structure, O2 is evolved as OER pathway B with byproduct of H2O2 as HPER pathway [135].
In the experimental research for the molecular mechanism of BiVO4 OER process, the surface interrogation scanning electrochemical microscopy technique was applied to W/Mo doped BiVO4 electrodes [136]. In this report, the generation of OH radical was at the ratio of 6% of the absorbed photons in the OER process [136]. However they detected the OH radical by the oxidation of IrCl62- for electrochemical monitoring. For non-doped BiVO4 electrodes, Nakabayashi et al. detected OH radical by trapping with fluorescent reagent and reported the yield of OH radical was 0.06 % of the photocurrent while almost 100 % was used to generate molecular oxygen [137]. FTIR spectra under the light irradiation was measured for the BiVO4 photoanode [138]. The in situ FTIR spectra measured in H218O and D2O are compiled in Figure 8. To analyze the isotope shift, DFT calculation with B3LYP/LanL2DZ method in Gausian03W was performed for model molecule (OH)4Bi-OOH and the isotope shift is shown in Table 3. When the calculated isotope shift was compared with the measured FTIR spectra in the literature [138], the reported assignment of the peak in D2O was found to be incorrect. The peak positions of δ(OOD) and ν(OO) should exchange in D2O. And then the large isotope shift in H218O shows that the both O in Bi-OOH originated from water and that the lattice oxygen of BiVO4 did not contain in the OER process. Thus, experimental observations of the presence of Bi-OOH and 100% yield of O2 show that the OER pathway B in Figure 7 is only the molecular mechanism of OER at the BiVO4 photoanode.
6.3. SrTiO3
Strontium titanate (SrTiO3 ) is a well-documented photocatalyst in both one-step and two-step (Z-scheme) water splitting systems as first reported in 1980 [142]. By using ultrafast sub-band-gap probe at 400 nm and white light in SrTiO3, one-electron intermediates were identified to be radicals located in Ti–O• (oxyl) and Ti–O•–Ti (bridge) groups with the initial radical formation time of 1.3 ps as shown in Figure 9 [143]. After photo triggering the water oxidation reaction from the Nb doped n-SrTiO3 surface, the microsecond decay of the intermediates affirms transition-state theory through two distinct time constants. The reaction conditions can be adjusted to allow selection between the two pathways, one characterized by a labile intermediate facing the electrolyte (the oxyl), and the other by a lattice oxygen (the bridge) [144]. Picosecond optical spectra of the Ti-OH population on lightly doped SrTiO3 are ordered by the surface hydroxylation. A Langmuir isotherm as a function of pH extracted an effective equilibrium constant relatable to the free energy difference of the first oxygen evolution reaction step [145].
Single crystalline SrTiO3 photoanodes were irradiated with a focused laser to spatially define the degradation. By characterizing the degradation using optical spectroscopy and electron microscopy, the material dissolution constitutes an upper bound of 6% of the charge passed in a pH 13 electrolyte, while for pH 7, it reaches 23%; the pH dependence is anti-correlated with the ultrafast population of trapped charge. Since a dominant lattice dissolution reaction as thermodynamically concomitant with the OER occurred, the reaction mechanism in in Ref [144] was not supported, though the photon flux seems very high in this experiment [146].
6.4. Ga2O3
On Ga2O3-based photocatalysts the overall water splitting mechanism at a molecular level was suggested, based on the ATR-FTIR investigations combining with the mass spectroscopy (MS) analysis. Different from those observed in other semiconductor photocatalysts, a direct hydroxyl radical formation mechanism was suggested. Where two adsorbed OH radicals became adsorbed H2O2 or adsorbed O22-, which caused the O2 evolution with other two holes [81].
On different α-Ga2O3 surfaces, namely (001), (100), (110), and (012), water adsorption and activation were explored by means of DFT calculations. The dissociative water adsorption on all the studied low-index surfaces are thermodynamically favorable, and the most preferentially exposed surface is (012). The computation for electrochemical model was used to investigate both the HER and the OER on α-Ga2O3 surfaces. The results indicate that the (100) and (110) surfaces are the most favorable ones for HER and OER, respectively, but they have low stability [147].
6.5. IrO2 and RuO2
Iridium oxide (IrOx) was used as the OER co-catalyst of Y2Ti2O5S2 photocatalyst for visible-light water splitting [148]. Ir-based catalysts are the catalysts of choice to date; nevertheless, their high price and scarcity have greatly hampered the widespread utilization of the proton exchange membrane water electrolysis technique [149]. On the other hand, ruthenium (Ru), at higher earth abundance and lower price, possesses superior catalytic activity to Ir; yet, it is prone to dissolution nature results in inferior stability that cannot be implemented in practical device [149]. The surface stability and dissolution of three prominent electro(photo)catalysts for water splitting: RuO2, IrO2, and TiO2 in the rutile phase was investigated by using a combination of ab initio steered molecular dynamics, enhanced sampling, and ab initio thermodynamics. A distinct site specificity in the dissolution of the RuO2(110) surface was identified, whereas no such surface site specificity exists for the IrO2(110) surface [150]. However, the mechanistic interplay between the OER and material degradation during water electrolysis is not yet well understood even for the most studied OER electrocatalysts such as IrO2 and RuO2 [151].
Anodically grown IrOX catalyst films were studied using Raman spectroscopy. In addition to deuteration and 18O substitution experiments, theoretical models were also constructed using DFT to interpret the experimental data. The material was found to be composed of [IrO6]n edge-sharing polyhedra (with n ≥ 3) and characterized over a large potential range (0.0–1.8 V). Ir centers are connected to each other via μ-O type oxygen linkages that allow for the Ir centers to electronically couple to each other. Oxidation of Ir3+ to Ir4+ at 0.7–1.2 V within a μ-O linked polymeric geometry resulted in a blue coloration of the material at high potentials. Theoretical calculations indicated that the optical transition responsible for the color is essentially an Ir to Ir charge transfer transition [152].
For an Ir oxide nanocluster catalyst system, a surface hydroperoxide, Ir-OOH, as an intermediate of OER has been detected by recording FTIR spectra of the OO vibrational mode at 830 cm–1. The detection was achieved upon oxidation of water under pulsed excitation of a visible light sensitizer [Ru(bpy)3]2+ [82]. The OER mechanism of IrOx was investigated based on charge accumulation. The valence change of Ir is more favorable than O–O bond formation. In situ evanescent wave spectroscopy revealed that an intermediate assignable to Ir5+ with oxygen ligands in opposite spin served as the precursor of OER regardless of pHs (2 to 12), as the generation of this species was not related to valence changes of Ir. The results confirmed that charge accumulation was not rate-limiting for OER on IrOX, which is a key mechanistic difference between IrOx and less-efficient 3d metal electrocatalysts [153]. Time-resolved operando spectroelectrochemistry was employed to investigate the redox-state kinetics of IrOX electrocatalyst films for both water and hydrogen peroxide oxidation. Three different redox species involving Ir3+, Ir3x+, Ir4+, and Ir4y+ were identified spectroscopically. A first-order reaction mechanism was suggested for H2O2 oxidation driven by Ir4+ states, and a higher-order reaction mechanism involving the cooperative interaction of multiple Ir4y+ states for water oxidation [154].
On calcined and uncalcined IrO2, operando XAS spectroscopy was utilized to study the OER under different protocols. At the elevated OER potentials above 1.5 V, stronger Ir−Ir interactions were observed, which were more dominant in the calcined [155].
With first-principles calculations integrated with implicit solvation at constant potentials, the detailed atomistic reaction mechanism of OER was examined for the IrO2(110) surface. The surface phase diagram was determined, and the possible reaction pathways including kinetic barriers, and computed reaction rates were explored based on the micro kinetic models [156]. The classical mechanism at the IrO2(110) surface was reconsidered. The OER follows a bi-nuclear mechanism with adjacent top surface oxygen atoms as fixed adsorption sites, whereas the Ir atoms underneath play an indirect role and maintain their saturated 6-fold oxygen coordination at all stages of the reaction. The oxygen molecule is formed, via an Ir–OOOO–Ir transition state, by association of the outer oxygen atoms of two adjacent Ir–OO surface entities, leaving two intact Ir–O entities at the surface behind [157].
An IrO2 nanoribbon of monoclinic phase, which is distinct from tetragonal rutile IrO2, was provided by a molten-alkali mechanochemical method. The intrinsic catalytic activity of IrO2 nanoribbon was higher than that of rutile IrO2 due to the low d band center of Ir in this special monoclinic phase structure, as confirmed by DFT calculations [158]. Ultrasmall Pd@Ir core–shell nanoparticles (5 nm) with 3 atomic layer of iron carbon nanotubes were constructed as an exceptional bifunctional electrocatalyst in acidic water splitting. Due to the core–shell structure, strain generated at hetero interfaces leads to an up shifted d band center of Ir atoms contributing to a 62-fold better mass activity than commercial IrO2; besides, the electronic hybridization suppresses the electrochemical dissolution of Ir; as a result, robust stability was also achieved [159].
IrXRu1−xO2, x = 1, 0.6, 0.3 and 0, was prepared by the hydrolysis synthesis, and a mechanistic study of the OER was reported. The polarization curves recorded at pHs of 0 to 3 could be well fitted to a model consisting of a series of concerted electron-proton transfer reactions (mononuclear mechanism). It was suggested that the third or fourth step is rate-determining for RuO2 and IrO2, respectively [160].
For single-crystal RuO2(110) in acidic electrolyte the surface structural changes as a function of potential were investigated by in situ surface X-ray scattering measurements with DFT calculations. The redox peaks at 0.7, 1.1 and 1.4 V vs. RHE could be attributed to the surface transitions associated with the successive deprotonation of –H2O on the coordinately unsaturated Ru sites and hydrogen adsorbed to the bridging oxygen sites. At potentials relevant to the OER, an –OO species on the unsaturated Ru sites was detected, which was stabilized by a neighboring –OH group on the unsaturated Ru site or bridge site. A new OER pathway, where the deprotonation of the –OH group is used to stabilize –OO, was found to be rate-limiting [161]. For the RuO2(110) surface, DFT method with considering a possible magnetic effects on the electronic configuration was applied for calculating the thermodynamic stability of possible O versus OH terminations and their effect on the free energies of the OER steps. The magnetic moment of RuO2 supplies an important contribution to obtaining a low overpotential and to its insensitivity to the exact O versus OH coverage of RuO2 (110) surface [162].
The OER kinetics on RuO2 rutile (110), (100), (101), and (111) orientations were experimentally investigated, finding (100) the most active. The potential involvement of lattice oxygen in the OER mechanism was assessed with online electrochemical mass spectrometry, which showed no evidence of oxygen exchange on these oriented facets in acidic or basic electrolytes, suggesting lattice oxygen is not exchanged in catalyzing OER on crystalline RuO2 surfaces [163].
Rh doping for RuO2 and surface oxygen vacancies to precisely regulate unconventional OER reaction path via the Ru–O–Rh active sites have been reported. Quasi in situ/operando characterizations demonstrated the recurrence of reversible oxygen species under working potentials for enhanced activity and durability. It was theoretically revealed that Rh-RuO2 passes through a more optimal reaction path of lattice oxygen mediated mechanism-oxygen vacancy site mechanism. The synergistic interaction of defects and Ru–O–Rh active sites causes the *O formation with the rate-determining step, breaking the barrier limitation (*OOH) suggested by the traditional AEM process [164].
DFT calculations for RuO2 demonstrated that the LOM can give rise to higher OER activity than the AEM at the active sites involving structural defects, both intrinsic and extrinsic. Although the AEM is preferred for the perfect (110) and (211) surfaces, the formation of metal vacancies due to catalyst dissolution may lead to much lower OER overpotentials for the LOM. By screening several metal impurities in RuO2, the dopants such as Ni and Co can promote the LOM over the AEM even for the perfectly structured surfaces [165]. Transition metal (TM)-doped rutile RuO2 with different ratios of TM and Ru were discussed through DFT calculation with Hubbard U correction (+ U). In low TM doping concentration, the evolved O2 is generated through the AEM, and the OER activity is limited by the scaling relationship of OER intermediates. In higher TM doping concentration, the evolved O2 is generated through the LOM for Cu- or Ni-doped RuO2. The distribution of Ru 4d and O 2p orbitals and the adsorption energy of H and O were found to be the major factors that affect the conversion of AEM into LOM [151].
Dispersing RuO2 over defective TiO2 enriched with oxygen vacancies (RuO2/D-TiO2) was reported with an electronic structure modulating strategy. Synergetic (spectro-) electrochemistry and theoretical simulations revealed a continuous band structure at the interface between RuO2 and defective TiO2, as well as a lowered energetic barrier for *OOH formation, which are accountable for the largely enhanced acidic OER kinetics [166]. The effect of titanium substitution at different concentrations within nanoscale RuO2, Ru1-XTiXO2 (x = 0–50 at. %), on the structure, was reported for the OER activity and stability using combined experiments and theory [167]. For MRuOX solid solution (M = Ce4+, Sn4+, Ru4+, Cr4+), the stability was customized by controlling the Ru charge. A scalable single cell electrolyzer using SnRuOX anode and a polymer electrolyte membrane conveyed an ever smallest degradation rate during a 1300 h operation at 1Acm−2 [168]. For the sulfate-functionalized RuFeOx (S-RuFeOx) catalyst in proton exchange membrane of water electrolyzer was investigated for OER activity and stability, because RuO2 shows relatively poor stability. Coupled with the Fe cation doping, S-RuFeOx displayed a remarkable OER performance [169].
6.6. Perovskite as electrocatalysts
Perovskite-type oxide nanocrystals (AxByOz), which possess distinct thermal stability, ionic conductivity and electron mobility properties, have attracted increasing interest as efficient OER catalysts [170]. The electronic structure of perovskite-type nanocrystal plays a decisive role in electrocatalytic performance, the orbital filling, metal-oxygen hybridization, and electron correlations of perovskite-type oxide nanocrystals for high-performance OER catalysis were systematically investigated [170]. By using soft X-ray emission and absorption spectroscopies, perovskite OER catalysts were analyzed for the partial density of states on an absolute energy scale. The decreasing the solid-state charge-transfer energy of perovskite can change the mechanisms of the OER from electron-transfer-limited to proton–electron-coupled, to proton-transfer-limited reactions [171]. For electrocatalysts, the perovskite catalysts with noble-metals exhibit a smallest overpotential in various types of catalysts as shown in Figure 10 [172].
On various LaXSr1−xCoO3−δ as OER catalyst, a general strategy was demonstrated for steering the two mechanisms, AEM and LOM. By delicately controlling the oxygen defect contents, the dominant OER mechanism can be arbitrarily transformed between AEM-LOM-AEM accompanied by a volcano-type activity variation trend. Experimental and computational evidence explicitly revealed that the phenomenon is due to the fact that the increased oxygen defects alter the lattice oxygen activity with a volcano-type trend and preserve the Co0 state for preferably OER [173].
For NdNiO3, link between structural anisotropy and the OER catalytic activity was established by DFT calculation. The NdNiO3 with (100), (110), and (111) orientations display similar oxidative states and metal–oxygen covalence characteristics, but distinct OER activities in experimental results were the order of (100) > (110) > (111). DFT results confirm that film orientation is a critical determinant of the reaction mechanism. The OER on (100)-surface favors proceeding via a LOM. In contrast, the reaction on (110)- and (111)-surfaces followed the AEM. The anisotropic oxygen vacancy formation energy and stability are strongly correlated to the reaction mechanism and performance [174]. On LaNiO3 epitaxial thin films, electrochemical-scanning tunneling microscopy (EC-STM) was used to directly observe structural dynamics during the OER. Based on comparison of dynamic topographical changes in different compositions, reconstruction of surface morphology originated from transition of Ni species on the surface termination during the OER was proposed [175]. The change in surface topography was induced by Ni(OH)2/NiOOH redox transformation by quantifying STM images [175].
On La1-xNiO3 perovskite electrocatalysts, direct O–O coupling promoted the OER activity at the interfacial active sites for decorated Ag (x) nanoparticles. The theoretical calculation revealed that oxygen evolution via the dual-site mechanism with direct O–O coupling becomes more favorable than that via the conventional AEM. At x=0.05, the electrocatalyst showed 20 times higher mass activity before and 74 times after an accelerated durability test than that of the IrO2 electrocatalyst [176].
Ca2–xIrO4 nanocrystals exhibited very high stability of about 62 times that of benchmark IrO2. Lattice-resolution images and surface-sensitive spectroscopies demonstrated the Ir-rich surface layer with high relative content of Ir5+ sites, which is responsible for the high activity and long-term stability. Combining operando IR spectroscopy with XAS method, key intermediates of Ir6+=O and Ir6+OO– on Ir-based oxides electrocatalysts were observed, and they were stable even just from 1.3 V vs. RHE. DFT calculations indicated that the catalytic activity of Ca2IrO4 is enhanced remarkably after surface Ca leaching, and Ir=O and IrOO– intermediates can be stabilized on positively charged active sites of Ir-rich surface layer [63]. Layered perovskite Sr2IrO4 was chemically exfoliated into protonated colloidal nanosheets with an undamaged perovskite framework. This OER catalyst exhibited about 10 times higher activity than the IrO2 catalyst film. As shown in Figure 11. DFT calculation indicated that electrons from inner Ir atom to the surface was observed in IrO2 (e) but not in the case of perovskite nanosheet (d), then the free energy of O* is moderate as shown in (a) compared with the case of IrO2 (b). Thus, the structural hydroxyl groups on the surface of protonated nanosheets participate in the catalytic cycle [177].
6.7. Transition metal (TM) compounds
Multicomponent transition metal oxides and (oxy)hydroxides are the most promising OER catalysts due to their low cost, adjustable structure, high electrocatalytic activity, and outstanding durability. Co-, Ni-, and Fe-based OER catalysts have been considered to be potential candidates to replace noble metals, especially for electrocatalysts, due to their tunable 3d electron configuration and spin state, versatility in terms of crystal and electronic structures, as well as abundance in nature [23]. The latest advances in the rational design of the related OER electrocatalysts and the modulation of the electronic structure of active sites were comprehensively summarized, besides brief overview about the mechanisms of OER and the theory and calculation criteria [178].
Wang, et al. reviewed the fundamental understanding of the electronic structure of low-cost TM oxide-based catalysts for electrochemical OER, and its relationship with the catalytic activity and the reaction mechanism was discussed [179]. Feng, et al. reviewed the relationship between TMs and OER catalyst activity, and then, the mechanism of synthesis strategy in different types of TMs-based catalysts was summarized [180]. Guo, et al. reviewed the state-of-the-art amorphous transition metal-based OER electrocatalysts, involving oxides, hydroxides, sulfides, phosphides, borides, and their composites, and then the practical application and theoretical modeling of the OER mechanisms in the OER were presented [181]. Though transition metal phosphides often exhibit an excellent HER activity, the OER catalytic performance is not outstanding. Huang et al. reviewed the strategies for preparing highly active OER catalysts of transition metal phosphides [182].
The early transition metals (Ti, V, and Cr) can form very stable M=O units, while the late transition metals (Ni and Cu) can only theoretically form unstable M=O structures. On the other hand, for Mn, Fe, and Co, the metal-oxo motif switch between two valence tautomers in the form of Mn+1=O2– and Mn–O•–. The former with an electrophilic oxygen atom can proceed via the water nucleophilic-attack (WNA) acid-base pathway to form the O–O bond, whereas the latter favors the oxygen radical coupling pathway for O–O bond formation [183].
6.7.1. CoOx
CoOOH was selected as the OER co-catalyst of aluminum-doped strontium titanate (SrTiO:Al) photocatalyst to attain almost unity in the internal quantum efficiency of UV induced water splitting with Rh/Cr co-catalyst for HER [184]. The recent progress of Co3O4-based electrocatalytic materials for the acidic OER was presented with particular reference to the catalytic mechanism and guidelines for the design principles from both experimental and theoretical perspectives [185]. Afterward, emerging strategies were outlined to improve the catalytic performance of Co3O4-based acidic OER catalysts, including phase engineering, component regulation with doping, composite with carbon-based materials, and multi-phase hybridization [185].
For the application of Co oxides to photocatalysts, operando XPS measurements were performed. The catalyst undergoes chemical-structural transformations as a function of the applied anodic potential, with complete conversion of the Co(OH)2 and partial conversion of the spinel Co3O4 phases to CoO(OH) under precatalytic electrochemical conditions. This interpretation revealed that the presence of Co(OH)2 enhances catalytic activity by promoting transformations to CoO(OH) [186]. To study the mechanism of OER on cobalt oxyhydroxide (CoOOH), operando X-ray absorption and Raman spectroscopy revealed that a Co(IV) species, CoO2 , is the dominating resting state of the catalyst. Oxygen isotope exchange experiments showed that a cobalt superoxide species is an active intermediate in the OER. This intermediate is formed concurrently to the oxidation of CoOOH to CoO2. Combing spectroscopic and electrokinetic data, the rate-determining step of the OER was identified as the release of dioxygen from the superoxide intermediate [86].
By using water-in-salt electrolyte, the water activity was systematically tuned and the mechanism as a function of applied potentials in water electrolysis was probed. The mechanism is sensitive to the applied potential. The Co-OO-Co bond forms via an intramolecular oxygen coupling mechanism at low potentials, whereas it proceeds through a water nucleophilic attack (WNA) mechanism by forming Co-OOH at high potentials [187].
The morphology-dependent analysis for well-defined crystalline cobalt oxyhydroxides CoOOH revealed that the active sites are exclusively located at lateral facets rather than basal facets. Theoretical calculations show that the coordinately unsaturated cobalt sites of lateral facets upshift the O 2p-band center closer to the Fermi level, thereby enhancing the covalency of Co-O bonds to yield the reactivity [188]. The sequential oxidation kinetics with Co3O4 nanoparticles involving multi-active sites for water oxidation in OER catalytic cycle were resolved by applying quasi-operando transient absorption spectroscopy to a typical photosensitization with Ru-dye and sacrificial electron donor. The Co(IV) intermediate distribution plays a determining role in OER activity and results in the slow overall OER kinetics [189]. The redox process between Co(III) and Co(IV) species does not follow a proton-coupled electron transfer mechanism that is thought to be common prior to the OER, but it involves a proton decoupled electron transfer, clarified by isotope labeling experiments and in situ electrostatic modulation [190].
Oxygen vacancy (Vo) rich environment facilitates the reconstruction of Co3O4 to the Co(OH)2 intermediate with proton vacancies (Co(II)Ox(OH)y), which is favorable for the formation of the active species of Co-OOH. Correlative operando Raman spectra characterizations and electrokinetic analyses indicated that a moderate Vo density can switch the O–O bond formation pathway, from a water nucleophilic attack (WNA) to an intramolecular nucleophilic attack pathway, which is more kinetically favorable for water oxidation [191]. As shown in Figure 12(b) with O vacancy, at step 3, three protons and one electron are removed to form Co-OOH. At the step 4, CoIII sites of Co-OOH are oxidized to CoIV which can be deprotonated (step 8) . by hole attack oxo ligand CoIV=O forms a Co-O-O triangle (step 9), and then becomes CoII-OO• (step 10). At the next oxidation (step 10), O2 is released and CoII back to CoIII with the coordination of water. I2M process was excluded the experimental results of using H218O isotope [191].
Amorphous CoOOH layer architecture was loaded onto the surface of TiO2. Tafel analysis, EIS, and CV methods showed that the carrier transfer barrier within the electrode and the transition of CoIIIOOH to CoIVOOH have the dominating effects on the photoelectrochemical performance. Theoretical calculation revealed that the interface between the CoOOH and TiO2 improves the electronic-transfer ability among Co sites [192]. Amorphous CoOOH layers are electrochemically synthesized on the surface of various cobalt sulfides CoSα, and found the decrease in the intermolecular energy gap. The decrease in the energy gap accelerates the formation of OER-active high-valent CoIV species [193].
Co3O4 nanocrystals anchored on carbon nitride nanofiber (CNF) were prepared and found that the OER activity under visible light increased by 124 times, where heterogeneous kinetics is improved based on a synergistic effect between its binary components for charge separation and the facet (222) exposure of Co3O4 nanocrystals. DFT calculations revealed that oxygen vacancies at (222) facet lead to a reduction of the bandgap of the nanocrystals [194].
The water oxidation with Co cubane cluster Co4O4(OAc)4(py)4 as the catalysis was examined by time-resolved rapid scan ATR FTIR spectroscopy, the μ-peroxido structure Co-OO-Co was established as the intermediate. Where the one-electron oxidized cubane was the sole source of charge which was driven either in alkaline solution by a visible light sensitizer or in hydroxide (OH–) containing acetonitrile solution [85].
The simulations on the OER mechanism were performed and, in addition, the influence of Fe substitution was examined. CoIV in the pristine cobalt(oxy)hydroxide promotes the efficient formation of an active O radical intermediate followed by intramolecular O−O coupling. In the case of Fe substitution, the early oxidation of FeIII to FeIV promotes the electrophilic character in the reaction center, reducing the proton affinity of the surface-bound hydroxyl moieties [195].
6.7.2. NiOx
For the nascent ultra-small NiOOH particles (<3 nm), the thermodynamics of Ni dissolution was calculated by using first-principles theory at a near-neutral pH range, and the mechanism of OER on the γ-NiOOH surface was clarified. It was concluded that (i)∼4% Ni cations on the surface of γ-NiOOH dissolve at pH = 7 and 1.73 V vs. RHE; (ii) on the pristine γ-NiOOH surface, OER proceeds via the “lattice peroxide” mechanism (*H2O → *OH → *O–OlattH* → O–Olatt → O2) with an overpotential of 0.70 V; (iii) in the presence of Ni cationic vacancies, OER proceeds via the “hydroperoxide” mechanism (*OH + *H2O → *2OH → *OOH → O2) with an overpotential of 0.40 V [196].
For NiOOH-based materials, light-triggered reversible geometric conversion between octahedron (NiO6) and square planar (NiO4) was proposed. The unit cell was undergo to achieve electronic states with alternative metal and oxygen characters throughout the oxygen evolution process. Utilizing this electron transfer pathway can bypass the potential limiting steps, that is, O–O bonding in AEM and deprotonation in LOM. As a result, the electrocatalysts that operate through this route showed superior activity compared with previously reported electrocatalysts [197,198].
By incorporating Fe and V into Ni(OH)2 lattices, OER activity was improved. X-ray photoelectron/absorption spectroscopies revealed the synergistic interaction between Fe/V dopants and Ni in the host matrix, which subtly modulates local coordination environments and electronic structures of the Fe/V/Ni cations. Further, in-situ XAS analyses manifested contraction of metal–oxygen bond lengths in the activated catalyst, with a short V–O bond distance. DFT calculations indicated that the V site of the Fe/V co-doped nickel (oxy)hydroxide gave near-optimal binding energies of OER intermediates and had lower overpotential compared with Ni and Fe sites [199]. A series of Mn-, Co-, Fe-, and Zn-doped nickel oxides was investigated by using operando UV−vis spectroscopy coupled with time-resolved stepped potential spectroelectrochemistry. The Ni2+/Ni3+ redox peak potential was found to shift anodically from Mn- < Co- < Fe-< Zn-doped samples, suggesting a decrease in oxygen binding energetics from Mn- to Zn-doped samples. The OER kinetics had a second-order dependence on the density of these oxidized species, suggesting a chemical rate-determining step involving coupling of two oxo species. The intrinsic turnover frequency per oxidized species exhibits a volcano trend with the binding energy of oxygen on the Ni site, having a maximum activity for the Fe-doped sample as shown in Figure 13. For Ni centers that bind oxygen too strongly (Mn- and Co-doped oxides), OER kinetics is limited by O−O coupling and oxygen desorption, while for Ni centers that bind oxygen too weakly (Zn-doped oxides), OER kinetics is limited by the formation of oxo groups [200].
Oxygen vacancy-enriched porous NiO/ln2O3 nanofibers (Vo–NiO/ln2O3@NFs) was fabricated for efficient OER electrocatalysis. Abundant Vo modulated the electronic configuration of the catalyst for altering the adsorption of intermediates to reduce the OER overpotential and promote O* formation, upshifting the d band center of metal centers near the Fermi level, and also increasing the electrical conductivity and enhancing the OER reaction kinetics simultaneously. In situ Raman spectra suggested that the Vo can render the NiO/ln2O3 more easily reconstructible on the surface during the OER course [201].
DFT +U calculations revealed that Ir-doping of a β-NiOOH(001) surface enhanced the electric conductivity while also activating an oxygen site involving three Ni atoms to realize a remarkably low OER overpotential of only η = 0.46 V, much lower than the oxygen site involving three Ni atoms in pristine β-NiOOH (η = 0.66 V) [202]. Since theoretical calculations predicted that Co, Rh, and Ir dopants would lead to low overpotentials to improve OER activity of Ni-based hydroxides, an experimental confirmation on the altered OER activities for a series of metals (Mo, W, Fe, Ru, Co, Rh, Ir) doped into γ-NiOOH has been reported [203]. The in situ electrical conductivity for metal doped γ-NiOOH correlated well with the trend in enhanced OER activities. The DFT calculations, which suggested that the intrinsic connections to the double exchange interaction between adjacent metal ions with various d orbital occupancies, rationalized the experimental results, serving as an indicator for the key metal-oxo radical character [203].
6.7.3. FeOx
Recent advancement and progress initializing Fe-based OER electrocatalysts with different supporting materials, including carbon-based materials, layered double hydroxides, Prussian blue analogous, metal–organic frameworks, were reviewed by Xiong, et al. [204]. In the review, the OER mechanism and some typical OER electrochemical parameters of Fe-based electrocatalysts supported on various supporting materials from the experimental and theoretical viewpoint were highlighted. Some challenges and expectations for promoting the catalytic performance were described [204].
In photoelectrochemical (PEC) water oxidation on hematite (α-Fe2O3), the mechanism of the subsequent rate-limiting O–O bond formation step was investigated by rate law analysis based on EIS measurements and probing the reaction intermediates with operando FTIR spectroscopy. Distinct reaction orders of ∼1 and ∼2 were observed in near-neutral and highly alkaline environments, respectively. The unity rate law in near-neutral pH regions suggests a mechanism of water nucleophilic attack (WNA) to –Fe=O to form the O–O bond. Operando observation of a surface superoxide species by FTIR further confirmed this pathway. In highly alkaline regions, coupling of adjacent surface trapped holes (I2M) becomes the dominant mechanism. While both are operable at intermediate pHs, mechanism switch from I2M to WNA induced by local pH decrease was observed at high photocurrent level as shown in Figure 14 [89]. In the recent report, transient photocurrent measurements for hematite photoanodes, revealed that the OER rate has a third-order dependence on the surface hole density. A mechanism wherein the reaction proceeds by accumulating oxidizing equivalents through a sequence of one-electron oxidations of surface hydroxy groups was proposed. The key O–O bond formation step occurs by the dissociative chemisorption of a hydroxide ion involving three oxyl sites [205].
Polycrystalline γ-FeO(OH), synthesized at room temperature, was used as a stable, although reactive, anode for OER, and electrokinetic studies were performed to unravel the OER pathway [206]. The cell temperature, hydroxyl ion concentration, and the cation of the supporting electrolyte were varied, and the influence of external bias on the OER activity was recorded. Tafel slope, and charge-transfer resistance values at high temperatures up to 65 °C, which unambiguously highlights the influence of the thermodynamic barrier and electron transfer kinetics. The faster OER kinetics on polycrystalline γ-FeO(OH) can also be attributed to an appreciably low activation energy, where variation of the electrolyte concentration indicated a first-order dependence on [OH-]. Deuterium isotope effect implicated the dissociation of hydroxyl ions on the polycrystalline γ-FeO(OH) as the rate-determining step. The direct effect of cations such as Li, Na, and K of the electrolyte on OER indicated a weak interaction of the cations with the surface-active [FeIII-OH] species [206].
Fe3O4 with oxygen vacancies (Fe3O4-VO) was synthesized via Ar ion irradiation method and its OER activity was greatly improved by properly modulating the electron density around Fe atoms, which were evaluated with XANES and EXAFS methods. DFT results indicated the enhancement in desorption of the *OOH groups which significantly reduced the OER reaction barrier. Fe3O4-Vo catalyst showed an overpotential of better than commercial RuO2 at high potential [207].
Ni, Co, Yb doped–FeOOH nanorod arrays grown directly on a carbon cloth (CC) are synthesized by a simple one-step hydrothermal method. The doped Ni2+ and Co2+ can occupy Fe2+ and Fe3+ sites in FeOOH, increasing the concentration of oxygen vacancies and the doped Yb3+ with a larger ionic radius can occupy the interstitial sites, which leads to more edge dislocations. The oxygen vacancies and edge dislocations greatly enrich the active sites in FeOOH/CC. In addition, DFT calculations confirmed that doping of Ni2+, Co2+, and Yb3+ modulates the electronic structure of the main active Fe sites, bringing its d-band center closer to the Fermi level and reducing the Gibbs free energy change of the rate-determining step of the OER [208].
6.7.4. MnOx
Nature uses a Mn cluster for water oxidation in PS II, and thus, water oxidation using Mn clusters is interesting in artificial water-splitting systems. An ultra-thin manganese oxide (MnOX) was selected as co-catalyst to modify the surface of BiVO4 photoanode by a spray pyrolysis method [209]. The PEC measurements demonstrated that the surface charge transport efficiency strikingly increased by the MnOX modification. After applying Ar plasma on the BiVO4/MnOx sample, the transport efficiency further increased and it was around 7 times higher comparing with that of pristine BiVO4 samples. The remarkable PEC performance could be attributed to the increased charge carrier density, extended carrier lifetime and additional exposed Mn active sites on the BiVO4 surface [209].
An α-Mn2O3/FTO electrocatalyst was used in nonaqueous (CH3CN and DMF) and aqueous 0.1 M KPi (pH 7.0) solutions for kinetic studies of heterogeneous water oxidation. The rate of water oxidation was first order in catalyst concentration and in H2O concentration. The square wave and cyclic voltammetry measurements revealed the stepwise proton-coupled electron transfer oxidations of the active MnII–OH2 site to MnIII–OH and then to MnIV=O and finally an electron transfer oxidation of MnIV=O to MnV=O species. The MnV=O species undergoes a rate-limiting O atom transfer to H2O to give a MnIII–OOH2 species that, in turn, undergoes further oxidations to release O2 [61].
A Mn–K cluster was investigated for electrochemical water oxidation. By using XAS, SEM, TEM, XRD, FTIR spectroscopy, and electrochemical methods, it was revealed that conversion into nanosized Mn oxide occurred for the cluster, and the nanosized Mn oxides are the true catalyst for water oxidation [210].
The Mn3O4 nanocatalyst, which exhibits superb catalytic activity for water oxidation under neutral conditions, was analyzed for the complex capacitance. By the change in Mn valence between MnII and MnIV, charge was accumulated on the catalyst surface prior to the rate-determining O−O bond forming step. The dissipation ratio was proposed for understanding the energy balance between charge accumulation and charge consumption for chemical O−O bond formation [211]. In Mn3O4 nanoparticles, a profile imaging technique was exploited to understand the correlation between surface atomic structures and the OER. The surface structures of Mn3O4 nanoparticles were changed by the reaction and the surface Mn ions were reconstructed. The commonly considered active sites were disappeared from the reconstructed planes, whereas Mn ions were still exposed at the edges of nanoparticles. Thus, the surface reconstructions can deactivate low-index surfaces of Mn oxides in the OER process, which was further validated by DFT calculations [212].
An MnVII = O intermediate during electrocatalytic water oxidation by a c-disordered δ-MnOx was identified as an onset-potential-dependent reduction peak at 0.93 V. This intermediate is proved to be highly reactive and much more oxidative than permanganate ion. Thus, a new catalytic mechanism for water oxidation catalyzed by Mn oxides was proposed with involvement of the MnVII =O intermediate in a resting state and the MnIV-O-MnVII =O as a real active species for O-O bond formation. Figure 15 shows the proposed catalytic cycle, involving MnVII=O, in MnOx-catalyzed water-oxidation reaction. The overall mechanistic process involves charge accumulation (S0/S3), charge rearrangement (S3/S4), active-state formation (S4/S4’ ), and oxygen evolution (S4’ /S0) [68].
6.7.5. Mixed metal oxides
Two or three transition metals are mixed to form oxides of high electrocatalytic performance for water electrolyzers at a low cost. An NiFe oxide catalyst was employed as the anode catalyst with an NiMo oxide cathode catalyst with a high-performance perovskite-Si tandem solar cell, achieving a record 20% STH efficiency [213]. Nickel ferrite, NiFe2O4, and cobalt ferrite, CoFe2O4, are efficient and promising anode catalyst materials in the field of electrochemical water splitting.
In Ni-Fe water oxidation electrocatalysts, Ni is likely not an active site for water oxidation, because Ni cannot achieve high-oxide states in aqueous environments at relevant potentials [214]. For the OER of NiFeOxHy, addition of Co2+ cation increased the current density by 32.7% by the cation transport effect [215]. Using operando XAS , it was revealed that Ni oxidizes from the initial +2 oxidation state to +3/+4 state [216]. For Ni-Fe (oxy)hydroxides, in situ monitoring of the Fe active site number and turn-frequency number provided important insights into the activity degradation/regeneration caused by Fe dissolution/adsorption as well as a site-dependent activity and stability [217]. In the case of NiFe2O4, an Fe-site-assisted LOM pathway as the preferred OER mechanism was predicted. On the other hand, in the case of CoFe2O4, an Fe-site-assisted LOM pathway and a Co-site-assisted AEM pathway could both play a role [218].
Amorphous/crystalline NiFe2O4 induced by vanadium doping showed a superior electrocatalyst and long-term stability [219]. For amorphous Ni-Fe mixed metal oxides, analysis of the XAS revealed local structural transitions. A dual-site OER reaction mechanism was proposed, in which potential and rate-determining steps occur at Ni and Fe sites, respectively [220].
An Fe/Ca-based bimetallic oxide, CaFe2O4 , exhibited outstanding OER activity in alkaline media. DFT calculations suggested an unconventional mechanism via direct formation of O–O bonds between two oxygen intermediates, which are adsorbed on a multi-iron site on the catalyst surface [221].
On spinel NiCo2O4 abundant Co defects were preferentially produced by tuning the M–O bond length. Theoretical calculations and experiments proved that Al doping elongated the Co–O bond and promoted ionization of Co under plasma treatment [222]. Spinel Co2MnO4 showed higher OER activity, most probably due to the ideal binding energies of the oxygen evolution reaction intermediates [223].
For modulated NiFeX and FeCoX (X = W, Mo, Nb, Ta, Re and MoW) oxyhydroxide catalysts, in situ and ex situ soft and hard XAS were used to characterize the oxidation transition, and facilitate the lower OER overpotential [224]. (Co–Fe–Pb)Ox in acidic solutions through a cobalt-selective self-healing mechanism was investigated. The kinetics of the process was investigated by soft XAS and it was revealed that low concentrations of Co2+ in the solution stabilize the catalytically active Co(Fe) sites [225].
6.8. Layered Double Hydroxide (LDH)
LDH are emerging catalyst materials with inner layer water molecules and higher anion exchange capacity. They have been extensively used as electrocatalytic materials owing to their high specific surface area, environmental friendliness, lower cost, and non-toxicity [226]. A kind of LDHs itself may become photocatalysts for water splitting. The electronic properties, such as band structure, bandgap energy (Eg), density of states (DOS), and band edge placement, for MIIMIII-LDHs (MIII =Mg, Co, Ni and Zn; MIII=Al and Ga) were calculated by using the DFT + U method. The band structures of Mg and Zn-based LDHs and Co and Ni-based LDHs are responsive to ultraviolet (Eg > 3.1 eV) and visible light (Eg< 3.1 eV), respectively. The DOS calculations revealed that the photogenerated hole localizes on the surface hydroxyl group of LDHs, facilitating the oxidization of a water molecule without a long transportation route. The band edge placements of NiGa-, CoAl-, ZnAl-, and NiAl-LDHs have a driving force (0.965 eV, 0.836 eV, 0.667 eV, and 0.426 eV, respectively) toward oxygen evolution. In the experimental observations, only CoAl-LDH was an efficient oxygen evolution photocatalyst, agreeing well with the theoretical prediction [227].
For NiFe-LDH and Ni-LDH, the critical role of superficial oxygen vacancies (VO) in enhancing the electronic transport was discussed based on the electrochemical analysis by correlating with electrocatalytic activities [228]. In-situ conversion process to yield monolayer of Ni(OH)2 on electrodes was presented and the dynamic active site of the monolayer promoted OER process. Doping with Co caused the oscillation of Ni and Co valence states in NiCo hydroxide. This study defined an in-situ conversion process to yield monolayer LDH and fundamental understanding of the origin of the active sites in monolayer LDHs for the OER [229]. Direct spectroscopic evidence for the different active sites in Fe-free and Fe-containing Ni oxides was reported for ultrathin LDHs samples. 18O-labeling experiments in combination with in situ Raman spectroscopy were employed to probe the role of lattice oxygen as well as an active oxygen species, NiOO-, in the catalysts. It was found that lattice oxygen is involved in the OER for Ni and NiCo-LDHs, but not for NiFe and NiCoFe-LDHs. Moreover, NiOO- is a precursor to oxygen for Ni and NiCo-LDHs, but not for NiFe and NiCoFe- LDHs [78]. For M-doped Ni-based LDH (M =Ni, Co, and Fe), OER mechanism was investigated theoretically for the reaction processes of AEM, LOM and IMOC(= intramolecular oxygen coupling) mechanis. Theory predicted overpotential, Tafel slopes and finding were in agreement with the observation. As the result, depending on the applied potential, reaction mechanism changed [230]. Besides electrocatalysts, NiFe-LDH may be used as the flexible electrode of Zinc-Air batteries [231].
For Cu–NiFe–LDH electrocatalyst, a novel magnetic FeIII site spin-splitting strategy was suggested [232]. The electronic structure and spin states of the FeIIII sites are effectively induced by the Jahn–Teller effect of Cu2+. The theoretical calculations and operando ATR FTIR revealed that the facilitation for the O–O bond formation accelerated the production of O from OH and improved the OER activity [232]. For as-prepared sulfated Co-NiFe-LDH nanosheets, the kinetic energy barrier of the O–O coupling is significantly reduced. The formation of M-OOH on the active site at low overpotential was directly confirmed in 1 M KOH solution by in situ Raman and charge transfer fitting results. In weakly alkaline environment of 0.1 M KOH, a sequential proton–electron transfer mechanism replaces the concerted proton–electron transfer mechanism, and the proton transfer step becomes the rate-determining step (RDS) as illustrated in Figure 16 [233].
A Pt-induced NiFe-LDH (Pt-NiFe LDH) nanosheet was synthesized and large current density electrodes could be achieved in OER as well as HER [234]. At NiO/NiFe-LDH, the adsorption energy of *OH and *OOH can be adjusted independently, so as to bypass the scaling relationship and achieve high catalytic performance [235]. ZnO nanoparticles are uniformly distributed on the NiFe-LDH nanoflowers, which are prepared uniformly on the three-dimensional porous Ni foam. The active sites change from Fe cations to Ni cations during OER and the OER dynamics was significantly improved [236]. Hierarchical bimetal nitride/hydroxide (NiMoN/NiFe LDH) array exhibits the industrially required current density. In-situ electrochemical spectroscopy reveals that hetero interface facilitates dynamic structure evolution to optimize electronic structure. Operando EIS measurement implied the accelerated OER kinetics and intermediate evolution due to fast charge transport. For OER mechanism the combination of theoretical and experimental studies revealed that as-activated NiMoN/NiFe-LDH follows LOM process with accelerated kinetics [237].
NiFe LDH@Ni3S2 heterostructure as an efficient bifunctional electrocatalyst for overall water splitting was prepared. 3D porous heterostructure arrays caused the good electrocatalytic activity with a low Tafel slope [238]. By incorporating a semiconductor CdS/CdSe-MoS2 and NiFe-LDH for the OER, the as-prepared photoelectrode required a potential lower than the theoretical water splitting potential. Operando XAS measurements revealed that the formation of highly oxidized Ni species under illumination provides large photocurrent gains [239]. Hetero structures of LDH with graphitic carbon nitride (g-C3N4) stand as promising photo- and electro-catalysts for water oxidation and reduction. Mechanisms involved in electrocatalytic, photocatalytic, and photoelectrocatalytic water splitting processes reviewed with the necessary insights on the material [240]. By taking CuTi-LDH@g-C3N4 and Bi2O2CO3/NiFe-LDH@g-C3N4 as examples, the importance of heterojunctions and interfacial chemistry in the water splitting mechanism was explained in detail [240].
6.9. Metal–Organic Framework (MOF)
MOF derived materials have been demonstrated with high surface area, porous structure, increased electron transport, accessible active sites, and tailorable properties. And then they may provide a new route for designing of the catalysts with excellent electrochemical water oxidation activity [241]. Singh, et al. discussed the MOF-derived electrocatalysts by electronic structure modulation, since the recent studies of MOF derived electrocatalysts mainly focused on the morphological development, crystal structure modulation, facet engineering, and enhancement of the electrochemical surface area [241]. As the controlling factors for efficient OER, discussed were eg orbital filling, metal–oxygen covalency, mixed valency of metal ions, octahedral vs tetrahedral occupancy of the metal ions, and vacancy engineering. To improve the OER activity and stability of the MOF catalysts, tuning the electronic properties by interfacial modulation, surface overlayer, Fermi level manipulation, self-supported strategy, and heterostructure formation have been addressed [241].
FeNi–tannic acid coordination crystal was in situ grown on Ni foam ((FeNi)–Tan/NF) to directly catalyze the OER, and it exhibited predominant electrocatalytic OER activity [242]. A series of MOFs; FeM-MOF (M = Fe, Co, Ni, Zn, Mn; H4L = 3,3,5,5’-azoxybenzenetetracarboxylic acid) were synthesized under a simple and mild condition. Among of them, the FeCo-MOF catalyst exhibits an extremely low overpotential and small Tafel slope in an alkaline electrolyte for OER, which had far exceeded the commercial catalyst IrO2 [243].
The surface reconstruction phenomenon of MOF-based nanomaterial electrocatalysts for OER was summarized, and the effects of structural and compositional transformation on the catalytic activity were discussed because many MOF-based catalysts inevitably undergo irreversible surface reconstruction during the redox process [244]. The causes and conditions of surface reconstruction and its influence on OER performance were also discussed, demonstrating the structure–activity relationship between surface reconstruction and catalytic performance [244]. A facile impregnation method through an ion-exchange process to fabricate Fe-doped Co-BDC nanosheets (Fe@Co-BDC NSs, BDC=benzenedicarboxylic) was proposed, though most of the related reports focus on the hydrothermal methods to prepare the mixed-metal MOFs. For an efficient OER electrocatalyst, the morphology change and electronic structure of Fe@Co-BDC NSs were important to represent significant enhancement in the activity [245].
For MOF-based catalysts, in situ or operando Raman spectroscopic studies is useful to identify the adsorption sites, defect sites, structural or spin transitions, reaction centers, intermediates, and so on. Sunil et al. reviewed the current researches for OER mechanism with Raman spectroscopy in probing the structure, guest adsorption, catalytic activity, and reaction mechanisms of MOFs [246]. MOFs may become alternative OER catalysts, because in situ self-reconstruction from MOFs to (oxy)hydroxides could be performed in alkaline electrolytes. Thus, Fe-doped Co-MOF nanosheets were prepared and utilized straightforwardly as OER electrocatalysts. CoFe-layered bimetallic hydroxides (CoFe-LDHs) with abundant active sites were obtained from in situ conversion of Co-MOF/Fe after etching by the KOH electrolyte, which are generally actual active species. Meanwhile, the introduction of Fe ions will also produce a synergistic effect that greatly improves the electrocatalytic OER performance [247].
A new MOF with well-defined Co–Mo dual sites, HZIF-2-CoMo, (HZIF=Hybrid zeolitic imidazolate framework), was reported, which can promote the OER process through an unconventional Mo/Co dual-site relay mechanism. Theoretical calculations suggested that the Mo and Co sites stabilize the *OH and *OOH intermediates, respectively, and that the unique Co–O–Mo configuration induces the formation of a Co–O*–Mo transition intermediate, remarkably reducing the reaction free energy. As a result, HZIF-2-CoMo showed one of the best OER electrocatalysts reported [248]. Two-dimensional cobalt ion (Co2+) and benzimidazole (bIm) based zeolite-imidazole framework nanosheets were reported as exceptionally efficient electrocatalysts for the OER. Liquid-phase ultrasonication was applied to exfoliate a [Co4(bIm)16] zeolite-imidazole framework (ZIF), named as ZIF-9(III) phase, into nanoscale sheets. ZIF-9(III) is selectively prepared through simple mechanical grinding of cobalt nitrate and benzimidazole in the presence of a small amount of ethanol. The electrochemical and physicochemical characterization data supported the assignment of the OER activity of the exfoliated nanosheet derived material to nitrogen coordinated cobalt oxyhydroxide N4CoOOH sites, following a mechanism known for Co-porphyrin and related systems as shown in Figure 17 [249].
6.10. Metal Complexes
Since the CaMn4O5 cluster in PS II can catalyze the OER with a very low overpotential, it is expected that multinuclear water oxidation catalysts possess superior OER performances. Inspired by the CaMn4O5 cluster in PS II, some multinuclear complexes were synthesized that could catalyze water oxidation [250]. Mimicking the Mn4 cluster, artificial Mn4 oxide cluster was proposed with the DFT calculation. The cluster may act OER catalyst as the theoretically exhibited ability of electron and proton transfers [251]. Since degradation of organic ligands are inevitable character for molecular catalysts, the ligands which poses the ability for electron transfer come under consideration [250]. By referring the molecular mechanism of PS II, redox-active ligands were employed for metal complex catalysts and then an achievement of current efficiency of 100%, overpotential of 200-300 mV, and the turnover frequency of 100s-1 has been reported [250].
Metal complex catalysts of artificial OER were investigated, at the beginning, for mainly with Ru, Mn, and Ir. However, recent investigations shift to non-precious natural abundant metals, such as Fe, Co, Ni, and Cu, and grow with understanding of proton-coupled electron transfer and O-O bond formation [252]. Recently, some mononuclear heterogeneous catalysts showed a high OER activity, which testified that mononuclear active sites with suitable coordination surroundings could also catalyze water oxidation efficiently. Though some mononuclear molecular complexes for OER show high water oxidation activity, it cannot be excluded that the high activity arises from the formation of dimeric species. The development of mono-/multinuclear homo- and heterogeneous catalysts for water oxidation was focused in a review paper [253]. This review also provided active sites and possible catalytic mechanisms of OER on the mono-/multinuclear catalysts [253]. Karmakar summarized the progress in research on molecular catalysts based on transition metals in the homogeneous phase with emphasizing the current mechanistic understanding of the water oxidation reaction. The factors that influence the character of water oxidation catalysts, such as reaction conditions, attached ligands, and transition metal centers, have been discussed as well [254].
6.10.1. Mn complexes
An electrocatalytic water oxidation reaction with MnIII tris(pentafluorophenyl) corrole (MTPC) in propylene carbonate (PC) was reported. O2 was generated at the MnV/IV potential with hydroxide, but a more anodic potential was required to evolve O2 with only water. With a synthetic MnV(O) complex of MTPC, a second order rate constant was determined with hydroxide, whereas its reaction with water occurred much more slowly. Significantly, during the electrolysis of MTPC with water, a MnIV-peroxo species was identified with various spectroscopic methods, including UV–vis, EPR, and FTIR spectroscopies. Isotope-labeling experiments confirmed that both O atoms of this peroxo species are derived from water, suggesting the involvement of the WNA (water nucleophilic attack) mechanism in water oxidation. DFT calculations suggested that the nucleophilic attack of hydroxide on MnV(O) and also WNA to 1e--oxidized MnV(O) are feasibly involved in the catalytic cycles but that direct WNA to MnV(O) is not likely to be the main O–O bond formation pathway in the electrocatalytic OER by MTPC [72].
6.10.2. Fe complexes
A pentanuclear Fe complex was reported as efficiently and the robustly OER catalyst with a turnover frequency of 1,900 s-1, which is about three orders of magnitude larger than that of other Fe-based catalysts. Electrochemical analysis confirmed the redox flexibility of the system, characterized by six different oxidation states between FeII5 and FeIII5 , where the FeIII5 state is active for oxidizing water. Quantum chemistry calculations indicated that the presence of adjacent active sites facilitates O–O bond formation with a low reaction barrier [255].
The kinetics of water oxidation by K2FeO4 has been reinvestigated by UV/Vis spectrophotometry from pH 7–9 in 0.2 M phosphate buffer. The rate of reaction was found to be second-order in both [FeO42−] and [H+]. These results are consistent with a proposed mechanism in which the first step involves the initial equilibrium protonation of FeO42− to give FeO3(OH)−, which then undergoes rate-limiting O−O bond formation. Analysis of the O2 isotopic composition for the reaction in H218O suggests that the predominant pathway for water oxidation is intramolecular O−O coupling. DFT calculations supported the proposed mechanism [256].
6.10.3. Cu complexes
With a family of CuII o-phenylene bis-oxamidate complexes, the reactivity sequence for the OER was found to be a function of the substitution pattern on the periphery of the aromatic ring. In-situ EPR, FTIR, and spectroelectrochemistry suggested that ligand-centered oxidations were preferred over Cu-centered oxidations. The resonance Raman spectroelectrochemical study revealed the accumulation of a bis-imine bound CuII superoxide species under catalytic turnover as the reactive intermediate, which provides the evidence for the O–O bond formation during the OER process [79]. Bearing the redox-active HL ligand, CuII(HL)(OTf)2 (HL = Hbbpya = N,N-bis(2,2’-bipyrid-6-yl)amine) was investigated for an OER catalyst. Thus Cu catalyst was found to be active as an OER catalyst at pH 11.5, at which the deprotonated complex [CuII(L–)(H2O)]+ is the predominant species in solution. The overall OER mechanism was found to be initiated by two proton-coupled electron-transfer steps. Kinetically, a first-order dependence in the catalyst, a zeroth-order dependence in the phosphate buffer were found. A computational study supported the formation of a Cu–oxyl intermediate, [CuII(L•)(O•)(H2O)]+. From this intermediate onward, formation of the O–O bond proceeds via a single-electron transfer from an approaching hydroxide ion to the ligand. Throughout the mechanism, the CuII center is proposed to be redox-inactive [257].
The mechanism of OER catalyzed by a mononuclear Cu complex in alkaline conditions was studied by DFT calculations and shown in Figure 18. Firstly, a water molecule coordinating with the copper center of the complex of CuII generates the complex of CuII–H2O which undergoes two proton-coupled electron transfer processes to produce intermediate (•L–CuII–O•) which can be described as a CuII center interacting with a ligand radical antiferromagnetically and an oxyl radical ferromagnetically. The oxidation process occurs mainly on the ligand moiety, which can trigger O–O bond formation via the WNA mechanism. The attacking water transfers one of the protons to the HPO42− coupled with an electron transfer to the ligand radical, which generates a transient •OH interacting with the oxyl radical and H2PO4−. Then, the O–O bond is formed through the direct coupling of the oxyl radical and the OH radical. The triplet di-oxygen could be released after two oxidation processes. The O–O bond formation was suggested to be the rate-limiting step. Thus the Cu complex catalyzes water oxidation with the help of a redox non-innocent ligand and HPO42− [258].
6.11. Single atom catalysts (SACs)
During the past few years, several research efforts have also unveiled the immense potential of SACs for the OER. Excellent OER performance has been demonstrated for both noble metal and non-noble metal SACs with lab-scale electrodes. The long-term stability of promising SACs is less well explored though, and considerable efforts are still required to assess this under technologically relevant conditions [259].
Atomically dispersed Ir atoms incorporated into spinel Co3O4 lattice as an acidic OER catalyst were reported to exhibit excellent activity and stability for water oxidation. FTIR observation of *OOH indicates that the AEM rather than the LOM dominates the OER process [83]. An electrocatalyst with Ru-atom-array patches supported on α-MnO2 (Ru/MnO2) for the OER was investigated. Ru/MnO2 showed a high activity and outstanding stability with small degradation after 200 h operation. Operando vibrational and mass spectroscopy measurements were performed to probe the reaction intermediates and gaseous products for validating the OER pathway, suggesting a mechanism that involves only *O and *OH species as intermediates. This mechanism allows direct O–O radical coupling for O2 evolution, i.e., OPM process in Figure 3(c) was suggested. First-principles calculations confirmed the cooperative catalysis mechanism with a reduced energy barrier. Time-dependent elemental analysis demonstrated the occurrence of the in-situ dynamic cation exchange reaction during the OER which is the key for triggering the reconstruction of Ru atoms into the ordered array with high durability [80].
A macromolecule-assisted SAC providing high-density Co single atoms (10.6 wt % Co SAC) in a pyridinic N-rich graphenic network was reported [260]. The highly porous carbon network with increased conjugation and vicinal Co site decoration significantly enhanced the electrocatalytic OER in 1 M KOH with more than 300 h stability. Operando X-ray absorption near-edge structure demonstrated the formation of electron-deficient Co-O coordination intermediates, accelerating OER kinetics [260].
A representative set of 11 transition metal atoms (Sc, V, Ti, Cr, Mn, Fe, Co, Ni, Cu, Pd, Pt) anchored on nitrogen-doped graphene were considered by means of DFT calculations[261]. The most of them form stable superoxo and peroxo intermediates when they react with molecular oxygen, which has a direct impact on the OER. Thus, in the corresponding microkinetic models, this step of the reaction cannot be neglected. Representative Gibbs energy profile is shown in Figure 19. Depending on the transition metal atom, the inclusion of the superoxo/peroxo complexes in the analysis of the reaction profile can change the kinetics by several orders of magnitude [261]. To clarify the assignment of the intermediates, a DFT technique was applied to a set of 30 SACs made by ten metal atoms (Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Pd and Pt) anchored on three widely used 2D carbon-based materials, i.e., graphene, nitrogen-doped graphene and carbon nitride. On SACs it has been generally assumed that OER occurs via formation of three intermediates, *OH, *O, and *OOH species, they could be referred as M(OH), M(O), and M(OOH). In all cases, however, other intermediates M(OH)2, M(O)(OH), M(O)2, and M(O2) were formed with higher stabilities [262].
Compared with SACs, the dual-atom catalysts (DACs) are attracting more attraction including higher metal loading, more versatile active sites, and excellent catalytic activity. Several general synthetic strategies and structural characterization methods of DACs were introduced and the involved oxygen catalytic mechanisms were discussed in the review paper [263]. Dual-atom catalysts, particularly those with heteronuclear active sites, have the potential to outperform the well-established single-atom catalysts for oxygen evolution reaction. a large-scale DFT is employed to explore the feasibility of *O-*O coupling mechanism, which can circumvent the scaling relationship with improving the catalytic performance of N-doped graphene supported Fe-, Co-, Ni-, and Cu-containing heteronuclear dual-atom catalysts [264].
6.12. Effect of surface functionalization
It has been found that an addition of carbonate salts to Pt-loaded TiO2 suspensions led to highly efficient stoichiometric photocatalytic decomposition of liquid water into H2 and O2. Since a high concentration of carbonate ions is essential for the catalytic photodecomposition of water, it was considered that the carbonate species aid desorption of O2 from the TiO2 surface [265]. To elucidate the effect of carbonates, DFT calculations are performed to study the photoinduced H2O and H2CO3 oxidation mechanisms on TiO2 and BiVO4 [266]. The computational results verify that the adsorbed H2CO3 molecule is easily photo-oxidized compared with the adsorbed H2O molecule, facilitating the formation of the peroxide intermediate and improving O2 evolution and H2O2 production [266].
The use of surface functionalization with phosphate ion groups (Pi) enhances the interfacial proton transfer. As the results, the Pi functionalization on La0.5Sr0.5CoO3−δ and LaCoO3 gave rise to a significant enhancement of the OER activity when compared to La0.5Sr0.5CoO3−δ and LaCoO3. It was demonstrated that the surface functionalization by Pi enhanced the activity when the OER kinetics is limited by the proton transfer as shown in Figure 20. By Sr2+ cation substitution, O 2p band closed to the Fermi level, assisting the deprotonation step from –OOH to -OO•. Thus, depending on the position of Fermi level, the function of Pi changed from keeping water near the surface to assisting deprotonation reaction [267].
The hydrated cobalt phosphate (CoPi) co-catalyst was investigated for O–O bond and OOH formation, based on the conventional experimental findings. Theoretical calculations of hydrated CoPi cluster models elucidated the roles of phosphate as a source of oxygen and deliverer of protons, both of which result in the spontaneous formation of an O–O bond after the release of two electrons and two protons. The calculations also show that OOH formation proceeds subsequently depending on the spin electronic states of the hydrated CoPi surface, and O2 formation then spontaneously progresses after the release of two electrons and two protons [268].
A proton acceptor, TA2− (terephthalate ion) can mediate proton transfer pathways by preferentially accepting protons, which optimizes the O−H adsorption/activation process and reduces the kinetic barrier for O−O bond formation. A proton-transfer-promotion mechanism for OER electrocatalysts with FeO6/NiO6 units is proposed by in situ Raman spectroscopy, catalytic tests, and theoretical calculations [269].
The strong adsorption of hexadecyltrimethylammonium cations on the surface of electrocatalysts provides the increased absolute number of OH− ions near the electrocatalyst surface, which effectively promotes the OER performance of electrocatalysts, such as Fe1−yNiyS2@Fe1−xNixOOH micro platelets and SrBaNi2Fe12O22 powders [270].
7. Perspectives
In the abundant researches articles of the OER developments, the catalysts for electrolyzers (electrocatalysts) are dominant because they are actually working in industries. These catalysts for electrolyzer may not directly applied for artificial photosynthesis using photoelectrodes or photocatalysts. However, the molecular mechanism is applicable under the consideration of the difference in the reaction conditions. Since the electrochemical OER catalysts are used with electrolyte, they work in alkaline or acid solution. However, photoelectrodes and photocatalysts are usually used in moderate pH values. Then, the most attention should be paid to the pH effects on the reaction mechanism when electrocatalysts are consulted.
Nature assembled PS II with CaMn4O5 cluster as the ultimate OER catalysts. Compared with the present artificial catalysts, the molecular mechanism of the PS II catalytic reaction is different in the following issues. (i) Immediately after the O-O bond formation O2 is released from the cluster, while the lifetime of the *OO- or *OO* in artificial catalysts is so long to be easily detected. Therefore, the catalysts, of which O-O intermediate species can be detected, may not be really generalized as a good catalyst. (ii) In the CaMn4O5 cluster, the four transition metals share the each positive hole to give four MnIV ions. In the case of artificial OER catalysts, one or two metal ions participate the four-electron reaction. Therefore, intermediate species of OER should be receive the next hole at the same metal ions. Namely, the catalysts of AEM process may have a difficulty to improve the activity. Then, a catalysts with more than one metal atoms appear promising in the future. (iii) The pathways of proton and water molecules are controlled in the PS II process. To keep the pathways, the surroundings of the Mn4 cluster is rather hydrophobic. The artificial OER catalysts are usually surrounded by water molecules and whole surface of the catalysts can adsorb water molecules. To improve OER activity of the artificial catalysts, controlling the proton-transfers may become important, and it could be achieved by surface functionalization of the present catalysts. The description in Section 6.12 may provide a clue to the improvement of the OER activity.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
I thank the Library of Kyoto University for accessing to the literature.
Conflicts of Interest
The author declares no conflict of interest.
References
- Hocke, K. Oxygen in the Earth System, Oxygen 2023, 3, 287–299. 3. [CrossRef]
- Nosaka, Y.; Nosaka, A.Y. Generation and detection of reactive oxygen species in photocatalysis. Chem. Rev. 2017, 117, 11302–11336. [Google Scholar] [CrossRef]
- Yoshino, S.; Takayama, T.; Yamaguchi, Y.; Iwase, A.; Kudo, A. CO2 reduction using water as an electron donor over heterogeneous photocatalysts aiming at artificial photosynthesis. Acc. Chem. Res. 2022, 55, 966–977. [Google Scholar] [CrossRef]
- Nosaka, Y.; Nosaka, A. Introduction to Photocatalysis: From Basic Science to Applications, Royal Society of Chemistry: Cambridge, UK. 2016; 272p. [CrossRef]
- Oliver, N.; Avramov, A.P.; Nürnberg, D.J.; Dau, H.; Burnap, R.L. From manganese oxidation to water oxidation: assembly and evolution of the water-splitting complex in photosystem II. Photosynthesis Res. 2022, 152, 107–133. [Google Scholar] [CrossRef]
- Najafpour, M.M.; Zaharieva, I.; Zand, Z.; Hosseini, S.M.; Kouzmanova, M.; Hołyńska, M.; Tranca, I.; Larkum, A.W.; Shen, J.-R.; Allakhverdiev, S.I. Water-oxidizing complex in photosystem II: Its structure and relation to manganese-oxide based catalysts. Coord. Chem. Rev. 2020, 409, 213183. [Google Scholar] [CrossRef]
- Yoneda, Y.; Arsenault, E.A.; Yang, S., Jr.; Orcutt, K.; Iwai, M.; Fleming, G.R. The initial charge separation step in oxygenic photosynthesis. Nat. Commun. 2022, 13, 2275. [Google Scholar] [CrossRef]
- Suga, M.; Akita, F.; Sugahara, M.; Kubo, M.; Nakajima, Y.; Nakane, T.; Yamashita, K.; Umena, Y.; Nakabayashi, M.; Yamane, T.; et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature, 2017, 543, 131–135. [Google Scholar] [CrossRef]
- Suga, M.; Akita, F.; Yamashita, K.; Nakajima, Y.; Ueno, G.; Li, H.; Yamane, T.; Hirata, K.; Umena, Y.; Yonekura, S.; et al. An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an X-ray free-electron laser. Science, 2019, 366, 334–338. [Google Scholar] [CrossRef]
- Kato, Y.; Noguchi, T. Redox properties and regulatory mechanism of the iron-quinone electron acceptor in photosystem II as revealed by FTIR spectroelectrochemistry. Photosynthesis Res. 2022, 152, 135–151. [Google Scholar] [CrossRef]
- Corry, T.A.; O’Malley, P.J. Evidence of O–O Bond formation in the final metastable S3 state of nature’s water oxidizing complex implying a novel mechanism of water oxidation. J. Phys. Chem. Lett. 2018, 9, 6269–6274. [Google Scholar] [CrossRef]
- Rummel, F.; O’Malley, P.J. How nature makes O2: An electronic level mechanism for water oxidation in photosynthesis. J. Phys. Chem. B 2022, 126, 8214–8221. [Google Scholar] [CrossRef]
- Davis, K.M.; Sullivan, B.T.; Palenik, M.C.; Yan, L.; Purohit, V.; Robison, G.; Kosheleva, I.; Henning, R.W.; Seidler, G.T.; Pushkar, Y. Rapid evolution of the photosystem II electronic structure during water splitting. Phys. Rev. X 2018, 8, 041014. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.S.; Makita, H.; Bogacz, I.; Fuller, F.; Bhowmick, A.; Hussein, R.; Ibrahim, M.; Zhang, M.; Chatterjee, R.; Cheah, M.H.; et al. Capturing the sequence of events during the water oxidation reaction in photosynthesis using XFELs. FEBS Lett. 2023, 597, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, A.; Hussein, R.; Bogacz, I.; Simon, R.S.; Ibrahim, M.; Chatterjee, R.; Doyle, M.D.; Cheah, M.H.; Fransson, T.; Chernev, P.; et al. Structural evidence for intermediates during O2 formation in photosystem II. Nature, 2023, 617, 629. [Google Scholar] [CrossRef]
- Greife, P.; Schönborn, M.; Capone, M.; Assunção, R.; Narzi, D.; Guidoni, L.; Dau, H. The electron–proton bottleneck of photosynthetic oxygen evolution. Nature, 2023, 617, 623. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, H.; Sibbons, J.; Sun, H.; Wang, H.; Wang, S. Photoelectrochemical water oxidation and longevous photoelectric conversion by a photosystem II electrode. Adv. Energy. Mater. 2021, 11, 2100911. [Google Scholar] [CrossRef]
- de Levie, R. The electrolysis of water. J. Electroanal. Chem. 1999, 476, 92–92. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K.; Kikuchi, S. Photosensitized electrolytic oxidation at TiO2 semiconductor electrodes (in Japanese). J. Chem. Soc. Japan. 1969, 72, 108–113. [Google Scholar] [CrossRef]
- Longden, T.; Beck, F.J.; Jotzo, F.; Andrews, R.; Prasad, M. ‘Clean’ hydrogen? – Comparing the emissions and costs of fossil fuel versus renewable electricity based hydrogen. Appl. Energy 2022, 306(Part B), 118145. [Google Scholar] [CrossRef]
- ul Haq, T.; Haik, Y. A roadmap towards sustainable anode design for alkaline water electrolysis. Appl. Catal. B: Environ. 2023, 334, 122853. [Google Scholar] [CrossRef]
- An, L.; Wei, C.; Lu, M.; Liu, H.; Chen, Y.; Scherer, G.G.; Fisher, A.C.; Xi, P.; Xu, Z.J.; Yan, C.-H. Recent development of oxygen evolution electrocatalysts in acidic environment. Adv. Mater. 2021, 33, 2006328. [Google Scholar] [CrossRef]
- Yu, M.; Budiyanto, E.; Tgysgz, H. Principles of water electrolysis and recent progress in cobalt-, nickel-, and iron-based oxides for the oxygen evolution reaction. Angew. Chem. Int. Ed. 2022, 61, e202103824. [Google Scholar] [CrossRef]
- Ma, Q.; Mu, S. Acidic oxygen evolution reaction: Mechanism, catalyst classification, and enhancement strategies. Interdiscip. Mater. 2023, 2, 53–90. [Google Scholar] [CrossRef]
- Vinodh, R.; Kalanur, S.S.; Natarajan, S.K.; Pollet, B.G. Recent advancements of polymeric membranes in anion exchange membrane water electrolyzer (AEMWE): A critical review. Polymers 2023, 15, 2144. [Google Scholar] [CrossRef]
- Lin, Y.; Dong, Y.; Wang, X.; Chen, L. Electrocatalysts for the oxygen evolution reaction in acidic media. Adv. Mater. 2022, 35, 2210565. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Seitz, L.C.; Benck, J.D.; Huo, Y.; Chen, Y.; Wei, J.; Ng, D.; Bilir, T.; Harris, J.S.; Jaramillo, T.F. Solar water splitting by photovoltaic-electrolysis with a solar-to-hydrogen efficiency over 30%. Nat. Commun. 2016, 7, 13237. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Al-Shankiti, I.; Ziani, A.; Wehbe, N.; Idriss, H. A stable integrated photoelectrochemical reactor for H2 production from water attains a solar-to-hydrogen efficiency of 18% at 15 suns and 13% at 207 suns. Angew. Chem. Int. Ed. 2020, 59, 14802–14808. [Google Scholar] [CrossRef] [PubMed]
- Karuturi, S.K.; Shen, H.; Sharma, A.; Beck, F.J.; Varadhan, P.; Duong, T.; Narangari, P.R.; Zhang, D.; Wan, Y.; He, J.-H.; et al. Over 17% efficiency stand-alone solar water splitting enabled by perovskite-silicon tandem absorbers. Adv. Energy Mater. 2020, 10, 2000772. [Google Scholar] [CrossRef]
- Holmes-Gentle, I.; Tembhurne, S.; Suter, C.; Haussener, S. Kilowatt-scale solar hydrogen production system using a concentrated integrated photoelectrochemical device. Nat. Energy, 2023, 8, 586–596. [Google Scholar] [CrossRef]
- Anantharaj, S.; Aravindan, V. Developments and perspectives in 3d transition-metal-based electrocatalysts for neutral and near-neutral water electrolysis. Adv. Energy Mater. 2020, 10, 1902666. [Google Scholar] [CrossRef]
- Mayer, M.T. Photovoltage at semiconductor–electrolyte junctions. Curr. Opinion Electrochem. 2017, 2, 104–110. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature, 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Mesa, C.A.; Francàs, L.; Yang, K.R.; Garrido-Barros, P.; Pastor, E.; Ma, Y.; Kafizas, A.; Rosser, T.E.; Mayer, M.T.; Reisner, E.; et al. Multihole water oxidation catalysis on hematite photoanodes revealed by operando spectroelectrochemistry and DFT. Nat. Chem. 2020, 12, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Rajan, A.G.; Martirez, J.M.P.; Carter, E.A. Why do we use the materials and operating conditions we use for heterogeneous (photo)electrochemical water splitting? ACS Catal. 2020, 10, 11177–11234. [Google Scholar] [CrossRef]
- Tang, R.; Zhou, S.; Zhang, Z.; Zheng, R.; Huang, J. Engineering nanostructure–interface of photoanode materials toward photoelectrochemical water oxidation. Adv. Mater. 2021, 33, 2005389. [Google Scholar] [CrossRef]
- Cendula, P.; Bedoya-Lora, F.E.; Prabhakar, R.R. Semiconductor catalysts for oxygen and hydrogen evolution reactions. ACS Appl. Energy Mater. 2022, 5, 14593–14604. [Google Scholar] [CrossRef]
- Ertem, M.Z.; Kharche, N.; Batista, V.S.; Hybertsen, M.S.; Tully, J.C.; Muckerman, J.T. Photoinduced water oxidation at the aqueous GaN (101̅0) interface: Deprotonation kinetics of the first proton-coupled electron-transfer step. ACS Catal. 2015, 5, 2317–2323. [Google Scholar] [CrossRef]
- Higashi, T.; Nishiyama, H.; Suzuki, Y.; Sasaki, Y.; Hisatomi, T.; Katayama, M.; Minegishi, T.; Seki, K.; Yamada, T.; Domen, K. Transparent Ta3N5 photoanodes for efficient oxygen evolution toward the development of tandem cells. Angew. Chem. Int. Ed. 2019, 58, 2300–2304. [Google Scholar] [CrossRef]
- Pihosh, Y.; Minegishi, T.; Nandal, V.; Higashi, T.; Katayama, M.; Yamada, T.; Sasaki, Y.; Seki, K.; Suzuki, Y.; Nakabayashi, M.; et al. Ta3N5-nanorods enabling highly efficient water oxidation via advantageous light harvesting and charge collection. Energy Environ. Sci., 2020, 13, 1519–1530. [Google Scholar] [CrossRef]
- Ma, Z.; Pie˛tak, K.; Pia˛tek, J.; DeMoulpied, J.R.; Rokicin´ska, A.; Kus´trowski, P.; Dronskowski, R.; Zlotnik, S.; Coridan, R.H.; Slabon, A. Semi-transparent quaternary oxynitride photoanodes on GaN underlayers. Chem. Commun., 2020, 56, 13193. [Google Scholar] [CrossRef]
- Huang, D.; Wang, K.; Li, L.; Feng, K.; An, N.; Ikeda, S.; Kuang, Y.; Ng, Y.; Jiang, F. 3.17% efficient Cu2ZnSnS4–BiVO4 integrated tandem cell for standalone overall solar water splitting. Energy Environ. Sci., 2021, 14, 1480–1489. [Google Scholar] [CrossRef]
- Pan, Z.; Chen, S.; Katayama, K. Roles of surface states in cocatalyst-loaded hematite photoanodes for water oxidation, J. Phys. Chem. C 2023, 127, 3904–3909. [Google Scholar] [CrossRef]
- Jeon, T.H.; Han, S.; Kim, B.; Park, C.; Kim, W.; Park, H.; Choi, W. High-valent iron redox-mediated photoelectrochemical water oxidation. ACS Energy Lett. 2022, 7, 59–66. [Google Scholar] [CrossRef]
- Morikawa, T.; Sato, S.; Sekizawa, K.; Suzuki, T.M.; Arai, T. Solar-driven CO2 reduction using a semiconductor/molecule hybrid photosystem: From photocatalysts to a monolithic artificial leaf. Acc. Chem. Res. 2022, 55, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Andrei, V.; Jagt, R.A.; Rahaman; Lari, M. L.; Lazarov, V.K.; MacManus-Driscoll, J.L.; Hoye, R.L.Z.; Reisner, E. Long-term solar water and CO2 splitting with photoelectrochemical BiOI–BiVO4 tandems. Nat. Mater. 2022, 21, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Takanabe, K. Photocatalytic water splitting: Quantitative approaches toward photocatalyst by design. ACS Catal. 2017, 7, 8006–8022. [Google Scholar] [CrossRef]
- Nosaka, Y.; Nosaka, A.Y. Intrinsic nature of photocatalysis by comparing with electrochemistry. Phys. Chem. Chem. Phys. 2020, 22, 7146–7154. [Google Scholar] [CrossRef]
- Wang, Q.; Domen, K. Particulate photocatalysts for light-driven water splitting: Mechanisms, challenges, and design strategies. Chem. Rev. 2020, 120, 919–985. [Google Scholar] [CrossRef]
- Yoshino, S.; Takayama, T.; Yamaguchi, Y.; Iwase, A.; Kudo, A. CO2 reduction using water as an electron donor over heterogeneous photocatalysts aiming at artificial photosynthesis. Acc. Chem. Res. 2022, 55, 966–977. [Google Scholar] [CrossRef]
- Nishiyama, H.; Yamada, T.; Nakabayashi, M.; Maehara, Y.; Yamaguchi, M.; Kuromiya, Y.; Nagatsuma, Y.; Tokudome, H.; Akiyama, S.; Watanabe, T.; et al. Photocatalytic solar hydrogen production from water on a 100-m2 scale. Nature 2021, 598, 304–307. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, K.; Xiang, Y.; Wang, J.; Lin, W.; Guo, M.; Ma, G. Facet-oriented assembly of Mo:BiVO4 and Rh:SrTiO3 particles: Integration of p–n conjugated photo-electrochemical system in a particle applied to photocatalytic overall water splitting. ACS Catal. 2022, 12, 2415–2425. [Google Scholar] [CrossRef]
- Zhou, P.; Navid, I.A.; Ma, Y.; Xiao, Y.; Wang, P.; Ye, Z.; Zhou, B.; Sun, K.; Mi, Z. Solar-to-hydrogen efficiency of more than 9% in photocatalytic water splitting. Nature, 2023, 613, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Li, C.M.; Wang, D. Mechanisms of water oxidation on heterogeneous catalyst surfaces. Nano Res. 2021, 14, 3446–3457. [Google Scholar] [CrossRef]
- Zhao, Y.; Saseendran, D.P.A.; Huang, C.; Triana, C.A.; Marks, W.R.; Chen, H.; Zhao, H.; Patzke, G.R. Oxygen evolution/reduction reaction catalysts: From in situ monitoring and reaction mechanisms to rational design. Chem. Rev. 2023, 123, 6257–6358. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 13801. [Google Scholar] [CrossRef]
- Zhang, J.; Tao, H.B.; Kuang, M.; Yang, H.B.; Cai, W.; Yan, Q.; Mao, Q.; Liu, B. Advances in thermodynamic-kinetic model for analyzing the oxygen evolution reaction. ACS Catal. 2020, 10, 8597–8610. [Google Scholar] [CrossRef]
- Corby, S.; Rao, R.R.; Steier, L.; Durran, J.R. The kinetics of metal oxide photoanodes from charge generation to catalysis. Nat. Rev. Mater. 2021, 6, 1136–1155. [Google Scholar] [CrossRef]
- Francàs, F.; Corby, S.; Selim, S.; Lee, D.; Mesa, C.A.; Godin, R.; Pastor, E.; Stephens, I.E.L.; Choi, K.-S.; Durrant, J.R. Spectroelectrochemical study of water oxidation on nickel and iron oxyhydroxide electrocatalysts, Nat. Commun. 2019, 10, 5208. [Google Scholar] [CrossRef]
- Franc`as, L.; Selim, S.; Corby, S.; Lee, D.; Mesa, C.A.; Pastor, E.; Choi, K.-S.; Durrant, J.R. Water oxidation kinetics of nanoporous BiVO4 photoanodes functionalised with nickel/iron oxyhydroxide electrocatalysts. Chem. Sci. 2021, 12, 7442. [Google Scholar] [CrossRef]
- Zahran, Z.N.; Mohamed, E.A.; Naruta, Y. Kinetic and mechanism of heterogeneous water oxidation by α-Mn2O3 sintered on FTO electrode. ACS Catal. 2016, 6, 4470–4476. [Google Scholar] [CrossRef]
- Xu, Z.; Liang, Z.; Guo, W.; Zou, R. In situ/operando vibrational spectroscopy for the investigation of advanced nanostructured electrocatalysts. Coord. Chem. Rev. 2021, 436, 213824. [Google Scholar] [CrossRef]
- Li, N.; Cai, L.; Gao, G.; Lin, Y.; Wang, C.; Liu, H.; Liu, Y.; Duan, H.; Ji, Q.; Hu, W.; et al. Operando direct observation of stable water-oxidation intermediates on Ca2–XIrO4 nanocrystals for efficient acidic oxygen evolution. Nano Lett. 2022, 22, 6988–6996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; de Respinis, M.; Frei, H. Time-resolved observations of water oxidation intermediates on a cobalt oxide nanoparticle catalyst. Nat. Chem. 2014, 6, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Zhou, W.; Zhang, H.; Zhou, W.; Zhao, X.; Li, Y.; Liu, M.; Cheng, W.; Liu, Q. Dynamic evolution of solid–liquid electrochemical interfaces over single-atom active sites. J. Am. Chem. Soc. 2020, 142, 12306–12313. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, X.; Li, Y.; Xu, Y.; Su, H.; Che, W.; He, J.; Zhang, H.; Liu, M.; Zhou, W.; et al. In situ construction of flexible V-Ni redox centers over Ni-based MOF nanosheet arrays for electrochemical water oxidation. Small Methods 2021, 5, 2100573. [Google Scholar] [CrossRef]
- Chu, H.-A.; Hillier, W.; Law, N.A.; Babcock, G.T. Vibrational spectroscopy of the oxygen-evolving complex and of manganese model compounds. Biochim. Biophys. Acta 2001, 1503, 69–82. [Google Scholar] [CrossRef]
- Zhang, B.; Daniel, Q.; Fan, L.; Liu, T.; Meng, Q.; Sun, L. Identifying MnVII-oxo species during electrochemical water oxidation by manganese oxide. iScience, 2018, 4, 144–152. [Google Scholar] [CrossRef]
- Zandi, O.; Hamann, T. Determination of photoelectrochemical water oxidation intermediates on haematite electrode surfaces using operando infrared spectroscopy. Nat. Chem. 2016, 8, 778–783. [Google Scholar] [CrossRef]
- Herlihy, D.; Waegele, M.; Chen, X.; Pemmaraju, C.D.; Prendergast, D.; Cuk, T. Detecting the oxyl radical of photocatalytic water oxidation at an n-SrTiO3/aqueous interface through its subsurface vibration. Nat. Chem. 2016, 8, 549–555. [Google Scholar] [CrossRef]
- Su, H.; Zhou, W.; Zhou, W.; Li, Y.; Zheng, L.; Zhang, H.; Liu, M.; Zhang, X.; Sun, X.; Xu, Y.; et al. In-situ spectroscopic observation of dynamic coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 2021, 12, 6118. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.-P.; Guo, M.; Lv, B.; Guo, K.; Jin, X.; Zhang, W.; Lee, Y.-M.; Fukuzum, S.; Nam, W.; Cao, R. Identifying intermediates in electrocatalytic water oxidation with a manganese corrole complex. J. Am. Chem. Soc. 2021, 143, 14613–14621. [Google Scholar] [CrossRef]
- Vivek, J.P.; Berry, N.G.; Zou, J.; Nichols, R.J.; Hardwick, L.J. In situ surface-enhanced infrared spectroscopy to identify oxygen reduction products in nonaqueous metal–oxygen batteries. J. Phys. Chem. C 2017, 121, 19657–19667. [Google Scholar] [CrossRef]
- Lin, W.; Frei, H. Photochemical and FT-IR probing of the active site of hydrogen peroxide in Ti silicalite sieve. J. Am. Chem. Soc. 2002, 124, 9292–9298. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Imanishi, A.; Murakoshi, K.; Nakato, Y. In situ FTIR studies of primary intermediates of photocatalytic reactions on nanocrystalline TiO2 films in contact with aqueous solutions. J. Am. Chem. Soc. 2003, 125, 7443–7450. [Google Scholar] [CrossRef]
- Zhou, Z.; Kong, Y.; Tan, H.; Huang, Q.; Wang, C.; Pei, Z.; Wang, H.; Liu, Y.; Wang, Y.; Li, S.; et al. Cation-vacancy-enriched nickel phosphide for efficient electrosynthesis of hydrogen peroxides. Adv. Mater. 2022, 34, 2106541. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Zhao, X.; Su, H.; Tang, F.; Che, W.; Zhang, H.; Liu, Q. Lattice-strained metal–organic-framework arrays for bifunctional oxygen electrocatalysis. Nat. Energy 2019, 4, 115–122. [Google Scholar] [CrossRef]
- Lee, S.; Banjac, K.; Lingenfelder, M.; Hu, X. Oxygen isotope labeling experiments reveal different reaction sites for the oxygen evolution reaction on nickel and nickel iron oxides. Angew. Chem. Int. Ed. 2019, 58, 10295–10299. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Ghatak, A.; Ro, Y.; Guillot, R.; Halime, Z.; Aukauloo, A.; Dey, A. Ligand radical mediated water oxidation by a family of copper o-phenylene bis-oxamidate complexes. Inorg. Chem. 2021, 60, 9442–9455. [Google Scholar] [CrossRef]
- Lin, C.; Li, J.; Li, X.; Yang, S.; Luo, W.; Zhang, Y.; Kim, S.-H.; Kim, D.-H.; Shinde, S.S.; Li, Y.-F.; Liu, Z.-P.; Jiang, Z.; Leel, J.-H. In-situ reconstructed Ru atom array on α-MnO with enhanced performance for acidic water oxidation. Nat Catal. 2021, 4, 1012–1023. [Google Scholar] [CrossRef]
- Chen, T.; Ding, Q.; Wang, X.; Feng, Z.; Li, C. Mechanistic studies on photocatalytic overall water splitting over Ga2O3-based photocatalysts by operando MS-FTIR spectroscopy. J. Phys. Chem. Lett. 2021, 12, 6029–6033. [Google Scholar] [CrossRef]
- Sivasankar, N.; Weare, W.W.; Frei, H. Direct observation of a hydroperoxide surface intermediate upon visible light-driven water oxidation at an Ir oxide nanocluster catalyst by rapid-scan FT-IR spectroscopy. J. Am. Chem. Soc. 2011, 133, 12976–12979. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Y.; Mu, X.; Wu, Z.; Jin, X.; Li, J.; Xu, Y.; Yang, L.; Xi, X.; Jang, H.; et al. Spinel-anchored iridium single atoms enable efficient acidic water oxidation via intermediate stabilization effect. ACS Catal. 2023, 13, 3757–3767. [Google Scholar] [CrossRef]
- Shao, M.H.; Adzic, R.R. Spectroscopic identification of the reaction intermediates in oxygen reduction on gold in alkaline solutions. J. Phys. Chem. B 2005, 109, 16563–16566. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Frei, H. Observation of O-O bond forming step of molecular Co4O4 cubane catalyst for water oxidation by rapid-scan FT-IR spectroscopy. ACS Catal. 2020, 10, 2138–2147. [Google Scholar] [CrossRef]
- Moysiadou, A.; Lee, S.; Hsu, C.-S.; Chen, H.M.; Hu, X. Mechanism of oxygen evolution catalyzed by cobalt oxyhydroxide: Cobalt superoxide species as a key intermediate and dioxygen release as a rate-determining step. J. Am. Chem. Soc. 2020, 142, 11901–11914. [Google Scholar] [CrossRef]
- Hunter, B.M.; Thompson, N.B.; Müller, A.M.; Rossman, G.R.; Hill, M.G.; Winkler, J.R.; Gray, H.B. Trapping an iron(VI) water-splitting intermediate in nonaqueous media. Joule 2018, 2, 747–763. [Google Scholar] [CrossRef]
- Hu, C.; Hu, Y.; Fan, C.; Yang, L.; Zhang, Y.; Li, H.; Xie, W. Surface-enhanced Raman spectroscopic evidence of key intermediate species and role of NiFe dual-catalytic center in water oxidation. Angew. Chem. Int. Ed. 2021, 60, 19774–19778. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Liu, A.; Chen, C.; Song, W.; Zhao, J. Rate-limiting O–O bond formation pathways for water oxidation on hematite photoanode. J. Am. Chem. Soc. 2018, 140, 3264–3269. [Google Scholar] [CrossRef]
- Shao, M.-H.; Liu, P.; Adzic, R.R. Superoxide anion is the intermediate in the oxygen reduction reaction on platinum electrodes. J. Am. Chem. Soc. 2006, 128, 7408–7409. [Google Scholar] [CrossRef]
- Li, W.; Fu, H.; Cao, Y.; Wang, H.; Hao Yu, H.; Qiao, Z.; Liang, H.; Peng, F. Mn3O4@C nanoparticles supported on porous carbon as bifunctional oxygen electrodes and their electrocatalytic mechanism. ChemElectroChem, 2019, 6, 359–368. [Google Scholar] [CrossRef]
- Hess, C. New advances in using Raman spectroscopy for the characterization of catalysts and catalytic reactions. Chem. Soc. Rev. 2021, 50, 3519–3564. [Google Scholar] [CrossRef]
- Huang, J.; Sheng, H.; Ross, R.D.; Han, J.; Wang, X.; Song, B.; Jin, S. Modifying redox properties and local bonding of Co3O4 by CeO2 enhances oxygen evolution catalysis in acid. Nat. Commun. 2021, 12, 3036. [Google Scholar] [CrossRef] [PubMed]
- Brückner, A. In situ electron paramagnetic resonance: A unique tool for analyzing structure–reactivity relationships in heterogeneous catalysis. Chem. Soc. Rev. 2010, 39, 4673–4684. [Google Scholar] [CrossRef] [PubMed]
- Brückner, A. In situ EPR spectroscopy in heterogeneous catalysis: Stepchild or ray of hope? Chem. Ingenieur Technik, 2014, 86, 1871–1882. [Google Scholar] [CrossRef]
- Chiesa, M.; Giamello, E. On the role and applications of electron magnetic resonance techniques in surface chemistry and heterogeneous catalysis. Catal. Lett. 2021, 151, 3417–3436. [Google Scholar] [CrossRef]
- Balaghi, S.E.; Mehrabani, S.; Mousazade, Y.; Bagheri, R.; Sologubenko, A.S.; Song, Z.; Patzke, G.R.; Najafpour, M.M. Mechanistic understanding of water oxidation in the presence of a copper complex by in situ electrochemical liquid transmission electron microscopy. ACS Appl. Mater. Interfaces, 2021, 13, 19927–19937. [Google Scholar] [CrossRef]
- Peña, N.O.; Ihiawakrim, D.; Han, M.; Lassalle-Kaiser, B.; Carenco, S.; Sanchez, C.; Laberty-Robert, C.; Portehault, D.; Ersen, O. Morphological and structural evolution of Co3O4 nanoparticles revealed by in situ electrochemical transmission electron microscopy during electrocatalytic water oxidation. ACS Nano 2019, 13, 11372–11381. [Google Scholar] [CrossRef]
- Zhao, G.; Yao, Y.; Lu, W.; Liu, G.; Guo, X.; Tricoli, A.; Zhu, Y. Direct observation of oxygen evolution and surface restructuring on Mn2O3 nano catalysts using in situ and ex situ transmission electron microscopy. Nano Lett. 2021, 21, 7012–7020. [Google Scholar] [CrossRef]
- Mom, R.; Frevel, L.; Velasco-Vélez, J.-J.; Plodinec, M.; Knop-Gericke, A.; Schlögl, R. The oxidation of platinum under wet conditions observed by electrochemical X-ray photoelectron spectroscopy. J. Am. Chem. Soc. 2019, 141, 6537–6544. [Google Scholar] [CrossRef]
- van Oversteeg, C.H.M.; Doan, H.Q.; de Groot, F.M.F.; Cuk, T. In situ X-ray absorption spectroscopy of transition metal based water oxidation catalysts. Chem. Soc. Rev. 2017, 46, 102–125. [Google Scholar] [CrossRef]
- Beaumont, S.K. Soft XAS as an in situ technique for the study of heterogeneous catalysts. Phys. Chem. Chem. Phys. 2020, 22, 18747–18756. [Google Scholar] [CrossRef]
- Cutsail III, G.E.; DeBeer, S. Challenges and opportunities for applications of advanced X-ray spectroscopy in catalysis research. ACS Catal. 2022, 12, 5864–5886. [Google Scholar] [CrossRef]
- Park, J.; Cho, J. Advances in understanding mechanisms of perovskites and pyrochlores as electrocatalysts using in-situ X-ray absorption spectroscopy. Angew. Chem. Int. Ed. 2020, 59, 15314–15324. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-J.; Zhu, Y.; Wang, J.; Chen, H.-C.; Tung, C.-W.; Chu, Y.-C.; Chen, H.M. In situ X-ray diffraction and X-ray absorption spectroscopy of electrocatalysts for energy conversion reactions. J. Mater. Chem. A 2020, 8, 19079–19112. [Google Scholar] [CrossRef]
- Zeng, Y.; Li, X.; Wang, J.; Sougrati, M.T.; Huang, Y.; Zhang, T.; Liu, B. In situ/operando Mössbauer spectroscopy for probing heterogeneous catalysis. Chem Catal. 2021, 1, 1215–1233. [Google Scholar] [CrossRef]
- Scherthan, L.; Schmidt, S.F.M.; Auerbach, H.; Hochdörffer, T.; Wolny, J.A.; Bi, W.; Zhao, J.; Hu, M.Y.; Toellner, T.; Alp, E.E.; et al. 161Dy time-domain synchrotron Mössbauer spectroscopy for investigating single-molecule magnets incorporating Dy ions. Angew. Chem. Int. Ed. 2019, 58, 3444–3449. [Google Scholar] [CrossRef]
- Kunze, S.; Grosse, P.; Lopez, M.B.; Sinev, I.; Zegkinoglou, I.; Mistry, H.; Timoshenko, J.; Hu, M.Y.; Zhao, J.; Alp, E.E.; Chee, S.W.; Cuenya, B.R. Operando NRIXS and XAFS investigation of segregation phenomena in Fe-Cu and Fe-Ag nanoparticle catalysts during CO2 electroreduction. Angew. Chem. Int. Ed. 2020, 59, 22667–22674. [Google Scholar] [CrossRef]
- Craig, M.J.; Coulter, G.; Dolan, E.; Soriano-López, J.; Mates-Torres, E.; Schmitt, W.; García-Melchor, M. Universal scaling relations for the rational design of molecular water oxidation catalysts with near-zero overpotential. Nat. Commun. 2019, 10, 4993. [Google Scholar] [CrossRef]
- Siahrostami, S.; Li, G.-L.; Viswanathan, V.; Nørskov, J.K. One- or two-electron water oxidation, hydroxyl radical, or H2O2 evolution. J. Phys. Chem. Lett. 2017, 8, 1157–1160. [Google Scholar] [CrossRef]
- Man, I.C.; Su, H.-Y.; Calle-Vallejo, F.; Hansen, H.A.; Martínez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem, 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Zhang, N.; Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 2021, 14, 4647–4671. [Google Scholar] [CrossRef]
- Nosaka, Y. Water photo-oxidation over TiO2—History and reaction mechanism. Catalysts 2022, 12, 1557. [Google Scholar] [CrossRef]
- Wang, D.; Sheng, T.; Chen, J.; Wang, H.-F.; Hu, P. Identifying the key obstacle in photocatalytic oxygen evolution on rutile TiO2. Nat. Catal. 2018, 1, 291–299. [Google Scholar] [CrossRef]
- Li, B.; Wu, S.; Gao, X. Theoretical calculation of a TiO2-based photocatalyst in the field of water splitting: A review. Nanotechnol. Rev. 2020, 9, 85. [Google Scholar] [CrossRef]
- Malik, A.S.; Fredin, L.A. Unraveling the water oxidation mechanism on a stoichiometric and reduced rutile TiO2(100) surface using first-principles calculations. J. Phys. Chem. C 2023, 127, 3444–3451. [Google Scholar] [CrossRef]
- Kakuma, Y.; Nosaka, A.Y.; Nosaka, Y. Difference of TiO2 photocatalytic mechanism between rutile and anatase studied by the detection of active oxygen and surface species in water. Phys. Chem. Chem. Phys. 2015, 17, 18691–18698. [Google Scholar] [CrossRef]
- Li, F.; Chen, J.-F.; Gong, X.-Q.; Hu, P.; Wang, D. Subtle structure matters: The vicinity of surface Ti5C cations alters the photooxidation behaviors of anatase and rutile TiO2 under aqueous environments. ACS Catal. 2022, 12, 8242–8251. [Google Scholar] [CrossRef]
- Nakamura, R.; Nakato, Y. Primary intermediates of oxygen photoevolution reaction on TiO2 (rutile) particles, revealed by in situ FTIR absorption and photoluminescence measurements. J. Am. Chem. Soc. 2004, 126, 1290–1298. [Google Scholar] [CrossRef]
- Nakamura, R.; Okamura, T.; Ohashi, N.; Imanishi, A.; Nakato, Y. Molecular mechanisms of photoinduced oxygen evolution, PL emission, and surface roughening at atomically smooth (110) and (100) n-TiO2 (rutile) surfaces in aqueous acidic solutions. J. Am. Chem. Soc. 2005, 127, 12975–12983. [Google Scholar] [CrossRef]
- Zhuang, Y.-B.; Cheng, J. Deciphering the anomalous acidic tendency of terminal water at rutile(110)–water interfaces. J. Phys. Chem. C 2023, 127, 10532–10540. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef]
- Wang, Q.; Hisatomi, T.; Jia, Q.; Tokudome, H.; Zhong, M.; Wang, C.; Pan, Z.; Takata, T.; Nakabayashi, M.; Shibata, N.; Li, Y.; et al. Scalable water splitting on particulate photocatalyst sheets with a solar-to-hydrogen energy conversion efficiency exceeding 1. Nat. Mater 2016, 15, 611–5. [Google Scholar] [CrossRef]
- Rather, R.A.; Mehta, A.; Lu, Y.; Valant, M.; Fang, M.; Liu, W. Influence of exposed facets, morphology and hetero-interfaces of BiVO4 on photocatalytic water oxidation: A review. Int. J. Hydrogen Energy, 2021, 46, 21866–21888. [Google Scholar] [CrossRef]
- Ma, Y.; Pendlebury, S.R.; Reynal, A.; Formal, F.L.; Durrant, J.R. Dynamics of photogenerated holes in undoped BiVO4 photoanodes for solar water oxidation. Chem. Sci. 2014, 5, 2964–2973. [Google Scholar] [CrossRef]
- Abdellaoui, I.; Islam, M.M.; Remeika, M.; Higuchi, Y.; Kawaguchi, T.; Harada, T.; Budich, C.; Maeda, T.; Wada, T.; Ikeda, S.; Sakurai, T. Photocarrier recombination dynamics in BiVO4 for visible light-driven water oxidation. J. Phys. Chem. C 2020, 124, 3962–3972. [Google Scholar] [CrossRef]
- Tran-Phu, T.; Fusco, Z.; Bernardo, I.D.; Lipton-Duffin, J.; Toe, C.Y.; Daiyan, R.; Gengenbach, T.; Lin, C.-H.; Bo, R.; Nguyen, H.T.; et al. Understanding the role of vanadium vacancies in BiVO4 for efficient photoelectrochemical water oxidation. Chem. Mater. 2021, 33, 3553–3565. [Google Scholar] [CrossRef]
- Walsh, A.; Yan, Y.; Huda, M.N.; Al-Jassim, M.M.; Wei, S.-H. Band edge electronic structure of BiVO4: Elucidating the role of the Bi s and V d orbitals. Chem. Mater. 2009, 21, 547–551. [Google Scholar] [CrossRef]
- Hu, J.; Zhao, X.; Chen, W.; Su, H.; Chen, Z. Theoretical insight into the mechanism of photoelectrochemical oxygen evolution reaction on BiVO4 anode with oxygen vacancy. J. Phys. Chem. C 2017, 121, 18702–18709. [Google Scholar] [CrossRef]
- Liu, G.; Li, F.; Zhu, Y.; Lia, J.L.; Sun, L. Cobalt doped BiVO4 with rich oxygen vacancies for efficient photoelectrochemical water oxidation. RSC Adv. 2020, 10, 28523. [Google Scholar] [CrossRef]
- Liu, T.; Liu, R.; Li, Q.; Yang, J. Theoretical insight into the role of defects and facets in the selectivity of products in water oxidation over bismuth vanadate (BiVO4). ACS Sustainable Chem. Eng. 2020, 8, 1980–1988. [Google Scholar] [CrossRef]
- Wang, W.; Strohbeen, P.J.; Lee, D.; Zhou, C.; Kawasaki, J.K.; Choi, K.-S.; Liu, M.; Galli, G. The role of surface oxygen vacancies in BiVO4. Chem. Mater. 2020, 32, 2899–2909. [Google Scholar] [CrossRef]
- Steinitz-Eliyahu, R.; Hernangómez-Pérez, D.; Hegner, F.S.; Nikačević, P.; López, N.; Refaely-Abramson, S. Mixed excitonic nature in water-oxidized BiVO4 surfaces with defects. Phys. Rev. Mater. 2022, 6, 065402. [Google Scholar] [CrossRef]
- Huang, M.; He, W.; Xu, Z.; Zhu, H. Enhanced catalytic mechanism of twin-structured BiVO4. J. Phys. Chem. Lett. 2021, 12, 10610–10615. [Google Scholar] [CrossRef] [PubMed]
- Nikačević, P.; Hegner, F.S.; Galán-Mascarós, J.R.; López, N. Influence of oxygen vacancies and surface facets on water oxidation selectivity toward oxygen or hydrogen peroxide with BiVO4. ACS Catal. 2021, 11, 13416–13422. [Google Scholar] [CrossRef]
- Park, H.S.; Leonard, K.C.; Bard, A.J. Surface interrogation scanning electrochemical microscopy (SISECM) of photoelectrochemistry at a W/Mo-BiVO4 semiconductor electrode. J. Phys. Chem. C 2013, 117, 12093–12102. [Google Scholar] [CrossRef]
- Nakabayashi, Y.; Nishikawa, M.; Saito, N.; Terashima, C.; Fujishima, A. Significance of hydroxyl radical in photoinduced oxygen evolution in water on monoclinic bismuth vanadate. J. Phys. Chem. C 2017, 121, 25624–25631. [Google Scholar] [CrossRef]
- Nakabayashi, Y.; Suzuki, N.; Terashima, C.; Fujishima, A. In situ infrared analysis for the process of water photo-oxidation on monoclinic bismuth vanadate. J. Phys. Chem. C 2021, 125, 18579–18587. [Google Scholar] [CrossRef]
- Sun, Q.; Ren, K.; Qi, L. Boosting the performance of BiVO4 photoanodes by the simultaneous introduction of oxygen vacancies and cocatalyst via photoelectrodeposition. ACS Appl. Mater. Interfaces 2022, 14, 37833–37842. [Google Scholar] [CrossRef]
- Pan, L.; Wu, J.; Xu, X.; Lv, F.; Chen, Y.; Guo, L. Photoelectrochemical performance of bismuth vanadate photoanode for water splitting under concentrated light irradiation. Int. J. Hydrogen Energy, 2023, 48, 13479–13488. [Google Scholar] [CrossRef]
- Majumder, S.; Su, X.; Kim, K.H. Effective strategy of incorporating Co3O4 as a co-catalyst onto an innovative BiVO4/Fe2TiO5 core-shell heterojunction for effective photoelectrochemical water-splitting application. Surf. Interfaces. 2023, 39, 102936. [Google Scholar] [CrossRef]
- Avcıoǧlu, C.; Avcıoǧlu, S.; Bekheet, M.F.; Gurlo, A. Photocatalytic overall water splitting by SrTiO3: Progress report and design strategies. ACS Appl. Energy Mater. 2023, 6, 1134–1154. [Google Scholar] [CrossRef]
- Chen, X.; Choing, S.N.; Aschaffenburg, D.J.; Pemmaraju, C.D.; Prendergast, D.; Cuk, T. The formation time of Ti–O• and Ti–O•–Ti radicals at the n-SrTiO3/aqueous interface during photocatalytic water oxidation. J. Am. Chem. Soc. 2017, 139, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Aschaffenburg, D.J.; Cuk, T. Selecting between two transition states by which water oxidation intermediates decay on an oxide surface. Nat. Catal. 2019, 2, 820–827. [Google Scholar] [CrossRef]
- Vinogradov, I.; Singh, S.; Lyle, H.; Paolino, M.; Mandal, A.; Rossmeisl, J.; Cuk, T. Free energy difference to create the M-OH• intermediate of the oxygen evolution reaction by time-resolved optical spectroscopy. Nat. Mater. 2022, 21, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Lyle, H.; Singh, S.; Magnano, E.; Nappini, S.; Bondino, F.; Yazdi, S.; Cuk, T. Assessing and quantifying thermodynamically concomitant degradation during oxygen evolution from water on SrTiO3. ACS Catal. 2023, 13, 8206–8218. [Google Scholar] [CrossRef]
- Zhou, X.; Hensen, E.J.M.; van Santen, R.A.; Li, C. DFT simulations of water adsorption and activation on low-index α-Ga2O3 surfaces. Chem. Eur. J. 2014, 20, 6915–6926. [Google Scholar] [CrossRef]
- Wang, Q.; Nakabayashi, M.; Hisatomi, T.; Sun, S.; Akiyama, S.; Wang, Z.; Pan, Z.; Xiao, X.; Watanabe, T.; Yamada, T.; et al. Oxysulfide photocatalyst for visible-light-driven overall water splitting. Nat. Mater. 2019, 18, 827–832. [Google Scholar] [CrossRef]
- Yu, H.; Ge, J. Recent advances in Ru-based electrocatalysts toward acid electrochemical water oxidation. Curr. Opinion Electrochem. 2023, 39, 101296. [Google Scholar] [CrossRef]
- Raman, A.S.; Vojvodic, A. Providing atomistic insights into the dissolution of rutile oxides in electrocatalytic water splitting. J. Phys. Chem. C 2022, 126, 922–932. [Google Scholar] [CrossRef]
- Liu, S.; Chang, Y.; He, N.; Zhu, S.; Wang, L.; Liu, X. Competition between lattice oxygen and adsorbate evolving mechanisms in rutile Ru-based oxide for the oxygen evolution reaction. ACS Appl. Mater. Interf. 2023, 15, 20563–20570. [Google Scholar] [CrossRef]
- Pavlovic, Z.; Ranjan, C.; Gao, Q.; van Gastel, M.; Schlögl, R. Probing the structure of a water-oxidizing anodic iridium oxide catalyst using Raman spectroscopy. ACS Catal. 2016, 6, 8098–8105. [Google Scholar] [CrossRef]
- Ooka, H.; Yamaguchi, A.; Takashima, T.; Hashimoto, K.; Nakamura, R. Efficiency of oxygen evolution on iridium oxide determined from the pH dependence of charge accumulation. J. Phys. Chem. C 2017, 121, 17873–17881. [Google Scholar] [CrossRef]
- Bozal-Ginesta, C.; Rao, R.R.; Mesa, C.A.; Liu, X.; Hillman, S.A.J.; Stephens, I.E.L.; Durrant, J.R. Redox-state kinetics in water-oxidation IrOX electrocatalysts measured by operando spectroelectrochemistry. ACS Catal. 2021, 11, 15013–15025. [Google Scholar] [CrossRef]
- Czioska, S.; Boubnov, A.; Escalera-López, D.; Geppert, J.; Zagalskaya, A.; Röse, P.; Saraçi, E.; Alexandrov, V.; Krewer, U.; Cherevko, S.; Grunwaldt, J.-D. Increased Ir−Ir interaction in iridium oxide during the oxygen evolution reaction at high potentials probed by operando spectroscopy. ACS Catal. 2021, 11, 10043–10057. [Google Scholar] [CrossRef]
- Ping, Y.; Nielsen, R.J.; Goddard III, W.A. The reaction mechanism with free energy barriers at constant potentials for the oxygen evolution reaction at the IrO2(110) surface. J. Am. Chem. Soc. 2017, 139, 149–155. [Google Scholar] [CrossRef]
- Binninger, T.; Doublet, M.-L. The Ir–OOOO–Ir transition state and the mechanism of the oxygen evolution reaction on IrO2(110). Energy Environ. Sci. 2022, 15, 2519. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Yin, K.; Ji, Y.; Zhu, W.; Fan, Z.; Li, Y.; Zhong, J.; Shao, M.; Kang, Z.; Shao, Q. Iridium oxide nanoribbons with metastable monoclinic phase for highly efficient electrocatalytic oxygen evolution. Nat. Commun. 2023, 14, 1248. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Chang, C.; Luo, F.; Yang, Z. Modulation in the d band of Ir by core–shell construction for robust water splitting electrocatalysts in acid. ACS Appl. Mater. Interfaces. 2023, 15, 20081–20088. [Google Scholar] [CrossRef] [PubMed]
- Reksten, A.H.; Thuv, H.; Seland, F.; Sunde, S. The oxygen evolution reaction mechanism at IrXRu1-XO2 powders produced by hydrolysis synthesis. J. Electroanal. Chem. 2018, 819, 547–561. [Google Scholar] [CrossRef]
- Rao, R.R.; Kolb, M.J.; Halck, N.B.; Pedersen, A.F.; Mehta, A.; You, H.; Stoerzinger, K.A.; Feng, Z.; Hansen, H.A.; Zhou, H.; et al. Towards identifying the active sites on RuO2(110) in catalyzing oxygen evolution. Energy Environ. Sci., 2017, 10, 2626–2637. [Google Scholar] [CrossRef]
- Liang, Q.; Bieberle-Hütter, A.; Brocks, G. Anti-ferromagnetic RuO2: A stable and robust OER catalyst over a large range of surface terminations. J. Phys. Chem. C 2022, 126, 1337–1345. [Google Scholar] [CrossRef]
- Stoerzinger, K.A.; Diaz-Morales, O.; Kolb, M.; Rao, R.R.; Frydendal, R.; Qiao, L.; Wang, X.R.; Halck, N.B.; Rossmeisl, J.; Hansen, H.A.; et al. Orientation-dependent oxygen evolution on RuO2 without lattice exchange. ACS Energy Lett. 2017, 2, 876–881. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, R.; Ding, Y.; Zhang, B.; Li, H.; Bai, B.; Li, M.; Cui, Y.; Xiao, J.; Wu, Z.-S. Unraveling oxygen vacancy site mechanism of Rh-doped RuO2 catalyst for long-lasting acidic water oxidation. Nat. Commun. 2023, 14, 1412. [Google Scholar] [CrossRef] [PubMed]
- Zagalskaya, A.; Alexandrov, V. Role of defects in the interplay between adsorbate evolving and lattice oxygen mechanisms of the oxygen evolution reaction in RuO2 and IrO2. ACS Catal. 2020, 10, 3650–3657. [Google Scholar] [CrossRef]
- Wang, X.; Wan, X.; Qin, X.; Chen, C.; Qian, X.; Guo, Y.; Xu, Q.; Cai, W.-B.; Yang, H.; Jiang, K. Electronic structure modulation of RuO2 by TiO2 enriched with oxygen vacancies to boost acidic O2 evolution. ACS Catal. 2022, 12, 9437–9445. [Google Scholar] [CrossRef]
- Godínez-Salomón, J.F.; Ospina-Acevedo, F.; Albiter, L.A.; Bailey, K.O.; Naymik, Z.G.; Mendoza-Cruz, R.; Balbuena, P.B.; Rhodes, C.P. Titanium substitution effects on the structure, activity, and stability of nanoscale ruthenium oxide oxygen evolution electrocatalysts: Experimental and computational study. ACS Appl. Nano Mater. 2022, 5, 11752–11775. [Google Scholar] [CrossRef]
- Shi, Z.; Li, J.; Wang, Y.; Liu, S.; Zhu, J.; Yang, J.; Wang, X.; Ni, J.; Jiang, Z.; Zhang, L.; Wang, Y.; Liu, C.; Xing, W.; Ge, J. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 2023, 14, 843. [Google Scholar] [CrossRef]
- Xue, Y.; Fang, J.; Wang, X.; Xu, Z.; Zhang, Y.; Lv, Q.; Liu, M.; Zhu, W.; Zhuang, Z. Sulfate-functionalized RuFeOx as highly efficient oxygen evolution reaction electrocatalyst in acid. Adv. Funct. Mater. 2021, 31, 2101405. [Google Scholar] [CrossRef]
- Shang, C.; Xiao, X.; Xu, Q. Coordination chemistry in modulating electronic structures of perovskite-type oxide nanocrystals for oxygen evolution catalysis. Coord. Chem. Rev. 2023, 485, 215109. [Google Scholar] [CrossRef]
- Hong, W.T.; Stoerzinger, K.A.; Lee, Y.-L.; Giordano, L.; Grimaud, A.; Johnson, A.M.; Hwang, J.; Crumlin, E.J.; Yange, W.; Shao-Horn, Y. Charge-transfer-energy-dependent oxygen evolution reaction mechanisms for perovskite oxides. Energy Environ. Sci. 2017, 10, 2190–2200. [Google Scholar] [CrossRef]
- Shi, Z.; Wang, X.; Ge, J.; Liu, C.; Xing, W. Fundamental understanding of the acidic oxygen evolution reaction: Mechanism study and state-of-the-art catalysts. Nanoscale, 2020, 12, 13249–13275. [Google Scholar] [CrossRef]
- Lu, M.; Zheng, Y.; Hu, Y.; Huang, B.; Ji, D.; Sun, M.; Li, J.; Peng, Y.; Si, R.; Xi, P.; Yan, C.-H. Artificially steering electrocatalytic oxygen evolution reaction mechanism by regulating oxygen defect contents in perovskites. Sci. Adv. 2022, 8, eabq3563. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Huang, J.; Zhu, Y.; Zhou, H.; Hu, Z.; Liao, Y.-K.; Lai, Y.-H.; Chen, C.-T.; Chu, Y.-H.; Zhang, K.H.L.; Fu, X.; Zuo, F.; Li, J.; Sun, Y. Structural anisotropy determining the oxygen evolution mechanism of strongly correlated perovskite nickelate electrocatalyst. ACS Sustainable Chem. Eng. 2021, 9, 4262–4270. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, C.-R.; Ding, T.-Y.; Gu, J.; Yan, J.-W.; Cheng, J.; Zhang, K.H.L. Direct observation of the dynamic reconstructed active phase of perovskite LaNiO3 for the oxygen evolution reaction. Chem. Sci., 2023, 14, 5906. [Google Scholar] [CrossRef]
- Lee, S.; Kishore, M.R.A.; Kim, D.; Kang, H.; Chun, J.; Oh, L.S.; Park, J.H.; Kim, H.J.; Yoo, J.S.; Lim, E. Direct O–O coupling promoted the oxygen evolution reaction by dual active sites from Ag/LaNiO3 interfaces. ACS Appl. Energy Mater. 2022, 5, 14658–14668. [Google Scholar] [CrossRef]
- Chen, H.; Shi, L.; Sun, K.; Zhang, K.; Liu, Q.; Ge, J.; Liang, X.; Tian, B.; Huang, Y.; Shi, Z.; Wang, Z.; Zhang, W.; Liu, M.; Zou, X. Protonated iridate nanosheets with a highly active and stable layered perovskite framework for acidic oxygen evolution. ACS Catal. 2022, 12, 8658–8666. [Google Scholar] [CrossRef]
- Han, J.; Guan, J. Multicomponent transition metal oxides and (oxy)hydroxides for oxygen evolution. Nano Res. 2023, 16, 1913–1966. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, K.H.L.; Hofmann, J.P.; de la Pe˜naO'Shea, V.A.; Oropeza, F.E. The electronic structure of transition metal oxides for oxygen evolution reaction. J. Mater. Chem. A 2021, 9, 19465. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, H.; Wang, X.; Hu, C.; Jing, H.; Cheng, J. Role of transition metals in catalyst designs for oxygen evolution reaction: A comprehensive review Int. J. Hydrogen Energ. 2022, 47, 17946–17970. [Google Scholar] [CrossRef]
- Guo, T.; Li, L.; Wang, Z. Recent development and future perspectives of amorphous transition metal-based electrocatalysts for oxygen evolution reaction. Adv. Energy Mater. 2022, 12, 2200827. [Google Scholar] [CrossRef]
- Huang, C.-J.; Xu, H.-M.; Shuai, T.-Y.; Zhan, Q.-N.; Zhang, Z.-J.; Li, G.-R. A review of modulation strategies for improving catalytic performance of transition metal phosphides for oxygen evolution reaction. App. Catal. B: Environ. 2023, 325, 122313. [Google Scholar] [CrossRef]
- Zhang, W.; Cao, R. Switching the O–O bond formation mechanism by controlling water activity. Chem 2021, 7, 1981–1992. [Google Scholar] [CrossRef]
- Takata, T.; Jiang, J.; Sakata, Y.; Nakabayashi, M.; Shibata, N.; Nandal, V.; Seki, K.; Hisatomi, T.; Domen, K. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 2020, 581, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, R.; Guo, M.; Zhang, Q. Recent progress of advanced Co3O4-based materials for electrocatalytic oxygen evolution reaction in acid: From rational screening to efficient design. Int. J. Hydrogen Energ. 2023, in press. [Google Scholar] [CrossRef]
- Favaro, M.; Yang, J.; Nappini, S.; Magnano, E.; Toma, F.M.; Crumlin, E.J.; Yano, J.; Sharp, I.D. Understanding the oxygen evolution reaction mechanism on CoOX using operando ambient-pressure X-ray photoelectron spectroscopy. J. Am. Chem. Soc. 2017, 139, 8960–8970. [Google Scholar] [CrossRef]
- Lang, C.; Li, J.; Yang, K.R.; Wang, Y.; He, D.; Thorne, J.E.; Croslow, S.; Dong, Q.; Zhao, Y.; Prostko, G.; et al. Observation of a potential-dependent switch of water-oxidation mechanism on Co-oxide-based catalysts. Chem 2021, 7, 2101–2117. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, Q.; Ju, S.; Hsu, C.-S.; Chen, H.M.; Zhang, D.; Song, F. Identifying the geometric catalytic active sites of crystalline cobalt oxyhydroxides for oxygen evolution reaction. Nat. Commun. 2022, 13, 6650. [Google Scholar] [CrossRef]
- Kang, W.; Wei, R.; Yin, H.; Li, D.; Chen, Z.; Huang, Q.; Zhang, P.; Jing, H.; Wang, X.; Li, C. Unraveling sequential oxidation kinetics and determining roles of multi-cobalt active sites on Co3O4 catalyst for water oxidation. J. Am. Chem. Soc. 2023, 145, 3470–3477. [Google Scholar] [CrossRef]
- Lin, Y.; Yu, L.; Tang, L.; Song, F.; Schlögl, R.; Heumann, S. In situ identification and time-resolved observation of the interfacial state and reactive intermediates on a cobalt oxide nanocatalyst for the oxygen evolution reaction. ACS Catal. 2022, 12, 5345–5355. [Google Scholar] [CrossRef]
- Zhou, D.; Li, F.; Zhao, Y.; Wang, L.; Zou, H.; Shan, Y.; Fu, J.; Ding, Y.; Duan, L.; Liu, M.; Sun, L.; Fan, K. Mechanistic regulation by oxygen vacancies in structural evolution promoting electrocatalytic water oxidation. ACS Catal. 2023, 13, 4398–4408. [Google Scholar] [CrossRef]
- Ren, X.; Ji, Y.; Zhai, Y.; Yuan, N.; Ding, J.; Li, Y.; Yan, J.; Liu, S.F. Self-assembled CoOOH on TiO2 for enhanced photoelectrochemical water oxidation. J. Energy Chem. 2021, 60, 512–521. [Google Scholar] [CrossRef]
- Yao, N.; Wang, G.; Jia, H.; Yin, J.; Cong, H.; Chen, S.; Luo, W. Intermolecular energy gap-induced formation of high-valent cobalt species in CoOOH surface layer on cobalt sulfides for efficient water oxidation. Angew. Chem. Int. Ed. 2022, 61, e202117178. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Li, H.; Xia, Y.; Wang, L.; Yin, K.; Wei, Y.; Liu, X.; Luo, S. Co3O4 nanocrystals with oxygen vacancy-rich and highly reactive (222) facet on carbon nitride scaffolds for efficient photocatalytic oxygen evolution. ACS Appl. Mater. Interfaces 2020, 12, 44608–44616. [Google Scholar] [CrossRef]
- Bhattacharyya, K.; Auer, A.A. Oxygen evolution reaction electrocatalysis on cobalt(oxy)hydroxide: role of Fe impurities. J. Phys. Chem. C 2022, 126, 18623–18635. [Google Scholar] [CrossRef]
- Li, L.-F.; Li, Y.-F.; Liu, Z.-P. Oxygen evolution activity on NiOOH catalysts: Four-coordinated Ni cation as the active site and the hydroperoxide mechanism. ACS Catal. 2020, 10, 2581–2590. [Google Scholar] [CrossRef]
- Wang, X.; Xi, S.; Huang, P.; Du, Y.; Zhong, H.; Wang, Q.; Borgna, A.; Zhang, Y.-W.; Wang, Z.; Wang, H.; et al. Pivotal role of reversible NiO6 geometric conversion in oxygen evolution. Nature 2022, 611, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, S. Light inducing the geometric conversion of NiO6 to trigger a faster oxygen evolution reaction pathway: The coupled oxygen evolution mechanism. Energy Environ. Mater. 2023, 6, e12558. [Google Scholar] [CrossRef]
- Jiang, J.; Sun, F.; Zhou, S.; Hu, W.; Zhang, H.; Dong, J.; Jiang, Z.; Zhao, J.; Li, J.; Yan, W.; Wang, M. Atomic-level insight into super-efficient electrocatalytic oxygen evolution on iron and vanadium co-doped nickel (oxy)hydroxide. Nat. Commun. 2018, 9, 2885. [Google Scholar] [CrossRef]
- Rao, R.R.; Corby, S.; Bucci, A.; García-Tecedor, M.; Mesa, C.A.; Rossmeisl, J.; Giménez, S.; Lloret-Fillol, J.; Stephens, I.E.L.; Durrant, J.R. Spectroelectrochemical analysis of the water oxidation mechanism on doped nickel oxides. J. Am. Chem. Soc. 2022, 144, 7622–7633. [Google Scholar] [CrossRef]
- Li, T.; Zhang, L.; Wang, J.; Zhang, X.; Zhang, L.; Wang, M.; Yan, C.; Tao Qian, T. Facilitating reconstruction of the hetero interface electronic structure by the enriched oxygen vacancy for the oxygen evolution reaction. Inorg. Chem. 2023, 62, 10504–10512. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, J.; Wang, Z.; Yuan, H.; Lu, Z.; Luo, B.; Tian, E.; Waterhouse, G.I.N. Structural and electronic engineering of Ir-doped Ni-(oxy)hydroxide nanosheets for enhanced oxygen evolution activity. ACS Catal. 2021, 11, 5386–5395. [Google Scholar] [CrossRef]
- Tian, B.; Shin, H.; Liu, S.; Fei, M.; Mu, Z.; Liu, C.; Pan, Y.; Sun, Y.; Goddard III, W.A.; Ding, M. Double-exchange-induced in situ conductivity in nickel-based oxyhydroxides: An effective descriptor for electrocatalytic oxygen evolution. Angew. Chem. Int. Ed. 2021, 60, 16448–16456. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; He, P. A review on electrocatalysis for alkaline oxygen evolution reaction (OER) by Fe-based catalysts. J. Mater.Sci. 2023, 58, 2041–2067. [Google Scholar] [CrossRef]
- Righi, G.; Plescher, J.; Schmidt, F.-P.; Campen, R.K.; Fabris, S.; Knop-Gericke, A.; Schlögl, R.; Jones, T.E.; Teschner, D.; Piccinin, S. On the origin of multihole oxygen evolution in haematite photoanodes. Nature Catal. 2022, 5, 888–899. [Google Scholar] [CrossRef]
- Adak, M.K.; Mallick, L.; Samanta, K.; Chakraborty, B. Slow O–H dissociation in the first-order oxygen evolution reaction kinetics on polycrystalline γ-FeO(OH). J. Phys. Chem. C 2023, 127, 154–168. [Google Scholar] [CrossRef]
- Gao, L.; Tang, C.; Liu, J.; He, L.; Wang, H.; Ke, Z.; Li, W.; Jiang, C.; He, D.; Cheng, L.; Xiao, X. Oxygen vacancy-induced electron density tuning of Fe3O4 for enhanced oxygen evolution catalysis. Energy Environ. Mater. 2021, 4, 392–398. [Google Scholar] [CrossRef]
- Ouyang, Q.; Cheng, S.; Yang, C.; Lei, Z. Ni, Co, and Yb cation Co-doping and defect engineering of FeOOH nanorods as an electrocatalyst for the oxygen evolution reaction. Inorg. Chem. 2023, 62, 1719–1727. [Google Scholar] [CrossRef]
- Li, C.; Chen, M.; Xie, Y.; Wang, H.; Jia, L. Boosting photoelectrochemical water splitting of bismuth vanadate photoanode via novel co-catalysts of amorphous manganese oxide with variable valence states. J. Colloid. Interf. Sci. 2023, 636, 103–112. [Google Scholar] [CrossRef]
- Mousazade, Y.; Mohammadi, M.R.; Chernev, P.; Bikas, R.; Song, Z.; Lis, T.; Dau, H.; Najafpour, M.M. Water oxidation by a manganese–potassium cluster: Mn oxide as a kinetically dominant “true” catalyst for water oxidation. Catal. Sci. Technol., 2018, 8, 4390–4398. [Google Scholar] [CrossRef]
- Seo, H.; Park, S.; Cho, K.H.; Choi, S.; Ko, C.; Randriamahazaka, H.; Nam, K.T. Complex impedance analysis on charge accumulation step of Mn3O4 nanoparticles during water oxidation. ACS Omega 2021, 6, 18404–18413. [Google Scholar] [CrossRef]
- Yoon, S.; Seo, H.; Jin, K.; Kim, H.G.; Lee, S.-Y.; Jo, J.; Cho, K.H.; Ryu, J.; Yoon, A.; Kim, Y.-W.; Zuo, J.-M.; Kwon, Y.-K.; Nam, K.T.; Kim, M. Atomic reconstruction and oxygen evolution reaction of Mn3O4 nanoparticles. J. Phys. Chem. Lett. 2022, 13, 8336–8343. [Google Scholar] [CrossRef]
- Wang, Y.; Sharma, A.; Duong, T.; Arandiyan, H.; Zhao, T.; Zhang, D.; Su, Z.; Garbrecht, M.; Beck, F.J.; Karuturi, S.; Zhao, C.; Catchpole, K. Direct solar hydrogen generation at 20% efficiency using low-cost materials. Adv. Energ. Mat. 2021, 11, 2101053. [Google Scholar] [CrossRef]
- Zhang, J.; Winkler, J.R.; Gray, H.B.; Hunter, B.M. Mechanism of nickel−iron water oxidation electrocatalysts. Energy Fuels 2021, 35, 19164–19169. [Google Scholar] [CrossRef]
- Yang, Y.; Du, X.; Wang, S.; Zhao, K.; Wang, L.; Qi, Z.; Yang, W.; Hao, J.; Shi, W. Cation transport effect on nickel iron oxyhydroxide electrodes in the oxygen evolution reaction. Ind. Eng. Chem. Res. 2022, 61, 16702–16710. [Google Scholar] [CrossRef]
- Acharya, P.; Manso, R.H.; Hoffman, A.S.; Bakovic, S.I.P.; Kékedy-Nagy, L.; Bare, S.R.; Chen, J.; Greenlee, L.F. Fe coordination environment, Fe-incorporated Ni(OH) phase, and metallic core are key structural components to active and stable nanoparticle catalysts for the oxygen evolution reaction. ACS Catal. 2022, 12, 1992–2008. [Google Scholar] [CrossRef]
- Qian, H.; Wei, J.; Yu, C.; Tang, F.; Jiang, W.; Xia, D.; Gan, L. In situ quantification of the active sites, turnover frequency, and stability of Ni–Fe (oxy)hydroxides for the oxygen evolution reaction. ACS Catal. 2022, 12, 14280–14289. [Google Scholar] [CrossRef]
- Avcı, Ö. N.; Sementa, L.; Fortunelli, A. Mechanisms of the oxygen evolution reaction on NiFe2O4 and CoFe2O4 inverse-spinel oxides. ACS Catal. 2022, 12, 9058–9073. [Google Scholar] [CrossRef]
- Deng, H.; Jiang, H.; Wang, K.; Wang, Z.; Wang, B.; Zhou, Z.; Li, J. Coupling the vanadium-induced amorphous/crystalline NiFe2O4 with phosphide heterojunction toward active oxygen evolution reaction catalysts. Nanotechnol. Rev. 2022, 11, 3165–3173. [Google Scholar] [CrossRef]
- Schoen, M.A.W.; Calderon, O.; Randell, N.M.; Jimenez-Villegas, S.J.; Daly, K.M.; Chernikov, R.; Trudel, S. Local structural changes in polyamorphous (Ni,Fe)O electrocatalysts suggest a dual-site oxygen evolution reaction mechanism. J. Mater. Chem. A 2021, 9, 13252–13262. [Google Scholar] [CrossRef]
- Sugawara, Y.; Kamata, K.; Ishikawa, A.; Tateyama, Y.; Yamaguchi, T. Efficient oxygen evolution electrocatalysis on CaFe2O4 and its reaction mechanism. ACS Appl. Energy Mater. 2021, 4, 3057–3066. [Google Scholar] [CrossRef]
- Zheng, J.; Peng, X.; Xu, Z.; Gong, J.; Wang, Z. Cationic defect engineering in spinel NiCo2O4 for enhanced electrocatalytic oxygen evolution. ACS Catal. 2022, 12, 10245–10254. [Google Scholar] [CrossRef]
- Li, A.; Kong, S.; Guo, C.; Ooka, H.; Adachi, K.; Hashizume, D.; Jiang, Q.; Han, H.; Xiao, J.; Nakamura, R. Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid. Nat. Catal. 2022, 5, 109–118. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, L.; Cao, Z.; Kozlov, S.M.; de Arquer, F.P.G.; Dinh, C.T.; Li, J.; Wang, Z.; Zheng, X.; Zhang, L.; Wen, Y.; et al. High-valence metals improve oxygen evolution reaction performance by modulating 3d metal oxidation cycle energetics. Nat. Catal. 2020, 3, 985–992. [Google Scholar] [CrossRef]
- Simondson, D.; Chatti, M.; Bonke, S.A.; Tesch, M.F.; Golnak, R.; Xiao, J.; Hoogeveen, D.A.; Cherepanov, P.V.; Gardiner, J.L.; Tricoli, A.; MacFarlane, D.R.; et al. Stable acidic water oxidation with a cobalt–iron–lead oxide catalyst operating via a cobalt-selective self-healing mechanism. Angew. Chem. Int. Ed. 2021, 60, 15821–15826. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.V.; Hassani, A.; Eghbali, P.; Nidheesh, P.V. Nanostructured modified layered double hydroxides (LDHs)-based catalysts: A review on synthesis, characterization, and applications in water remediation by advanced oxidation processes. Curr. Opinion Solid State and Mater. Sci. 2022, 26, 100965. [Google Scholar] [CrossRef]
- Xu, S.-M.; Pan, T.; Dou, Y.-B.; Yan, H.; Zhang, S.-T.; Ning, F.-Y.; Shi, W.-Y.; Wei, M. Theoretical and experimental study on MIIMIII -layered double hydroxides as efficient photocatalysts toward oxygen evolution from water. J. Phys. Chem. C 2015, 119, 18823–18834. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, L.; Feng, R.; Wang, S.; Hsu, C.-S.; Ashfaq, Y.N.; Zhang, A.C.; Wu, H.; Chen, H.-M.; Zhang, W.; Li, Y.; Liu, P.; Song, F. Oxygen vacancies unfold the catalytic potential of NiFe-layered double hydroxides by promoting their electronic transport for oxygen evolution reaction. ACS Catal. 2023, 13, 6000–6012. [Google Scholar] [CrossRef]
- Kang, J.; Qiu, X.; Hu, Q.; Zhong, J.; Gao, X.; Huang, R.; Wan, C.; Liu, L.-M.; Duan, X.; Guo, L. Valence oscillation and dynamic active sites in monolayer NiCo hydroxides for water oxidation. Nat. Catal. 2021, 4, 1050–1058. [Google Scholar] [CrossRef]
- Wang, Z.; Goddard III, W.A.; Xiao, H. Potential-dependent transition of reaction mechanisms for oxygen evolution on layered double hydroxides. Nat. Commun. 2023, 14, 4228. [Google Scholar] [CrossRef]
- Xu, J.; Li, Z.; Chen, D.; Yang, S.; Zheng, K.; Ruan, J.; Wu, Y.; Zhang, H.; Chen, J.; Xie, F.; Jin, Y.; Wang, N.; Meng, H. Active electrocatalyst in the oxygen evolution reaction and flexible zinc–air batteries. ACS Appl. Mater. Interfaces, 2021, 13, 48774–48783. [Google Scholar] [CrossRef]
- Sun, Z.; Lin, L.; He, J.; Ding, D.; Wang, T.; Li, J.; Li, M.; Liu, Y.; Li, Y.; Yuan, M.; Huang, B.; Li, H.; Sun, G. Regulating the spin state of FeIII enhances the magnetic effect of the molecular catalysis mechanism. J. Am. Chem. Soc. 2022, 144, 8204–8213. [Google Scholar] [CrossRef]
- Qiao, C.; Usman, Z.; Wei, J.; Gan, L.; Hou, J.; Hao, Y.; Zhu, Y.; Zhang, J.; Cao, C. Efficient O–O coupling at catalytic interface to assist kinetics optimization on concerted and sequential proton–electron transfer for water oxidation. ACS Nano, 2023, 17, 12278–12289. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Han, X.; Zhou, X.; Chen, J.; Wang, J.; Chen, Y.; Yu, L.; Zhang, N.; Li, J.; Wang, S.; Jin, H. Electronic modulation with Pt-incorporated NiFe layered doublehydroxide for ultrastable overall water splitting at 1000 mA cm-2. App. Catal. B: Environ. 2023, 331, 122683. [Google Scholar] [CrossRef]
- Gao, Z.-W.; Liu, J.-Y.; Chen, X.-M.; Zheng, X.-L.; Mao, J.; Liu, H.; Ma, T.; Li, L.; Wang, W.-C.; Du, X.-W. Engineering NiO/NiFe LDH intersection to bypass scaling relationship for oxygen evolution reaction via dynamic tridimensional adsorption of intermediates. Adv. Mater. 2019, 31, 1804769. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wu, Y.; Wu, D.; Huang, C.; Xiao, D.; Chen, H.; Zheng, S.; Chu, P.K. NiFe-layered double hydroxide synchronously activated by heterojunctions and vacancies for the oxygen evolution reaction. ACS Appl. Mater. Interfaces, 2020, 12, 42850–42858. [Google Scholar] [CrossRef]
- Zhai, P.; Wang, C.; Zhao, Y.; Zhang, Y.; Gao, J.; Sun, L.; Hou, J. Regulating electronic states of nitride/hydroxide to accelerate kinetics for oxygen evolution at large current density. Nat. Commun. 2023, 14, 1873. [Google Scholar] [CrossRef]
- Liang, X.; Li, Y.; Fan, H.; Deng, S.; Zhao, X.; Chen, M.; Pan, G.; Xiong, Q.; Xia, X. Bifunctional NiFe layered double hydroxide@Ni3S2 hetero structure as efficient electrocatalyst for overall water splitting. Nanotechnology, 2019, 30, 484001. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, X.; Li, H.; Sun, Z.; Cao, M.; Li, Z.; Fang, C.; Zhou, J.; Cao, C.; Dong, J.; Zhao, S.; Chen, Z. A semiconductor-electrocatalyst nano interface constructed for successive photoelectrochemical water oxidation. Nat. Commun. 2023, 14, 2574. [Google Scholar] [CrossRef]
- Guru, S.; Rao, G.R. Review—Strategic design of layered double hydroxides and graphitic carbon nitride heterostructures for photoelectrocatalytic water splitting applications. J. Electrochem. Soc. 2022, 169, 046515. [Google Scholar] [CrossRef]
- Singh, B.; Singh, A.; Yadav, A.; Indra, A. Modulating electronic structure of metal-organic framework derived catalysts for electrochemical water oxidation. Coord. Chem. Rev. 2021, 447, 214144. [Google Scholar] [CrossRef]
- Li, C.; Wang, G.; Li, K.; Liu, Y.; Yuan, B.; Lin, Y. FeNi-based coordination crystal directly serving as efficient oxygen evolution reaction catalyst and its density functional theory insight on the active site change mechanism. ACS Appl. Mater. Interfaces 2019, 11, 20778–20787. [Google Scholar] [CrossRef]
- Gu, M.; Wang, S.-C.; Chen, C.; Xiong, D.; Yi, F.-Y. Iron-based metal–organic framework system as an efficient bifunctional electrocatalyst for oxygen evolution and hydrogen evolution reactions. Inorg. Chem. 2020, 59, 6078–6086. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Li, Z.; Chu, H.; Zhou, W. Insight into the surface-reconstruction of metal–organic framework-based nanomaterials for the electrocatalytic oxygen evolution reaction. Coord. Chem. Rev. 2023, 484, 215117. [Google Scholar] [CrossRef]
- Zhang, W.; Li, F.; Fu, Z.; Dai, S.; Pan, F.; Li, J.; Zhou, L. Co-MOF nanosheets etched by FeCl2 solution for enhanced electrocatalytic oxygen evolution. Energy Fuels 2022, 36, 4524–4531. [Google Scholar] [CrossRef]
- Sunil, J.; Narayana, C.; Kumari, G.; Jayaramulu, K. Raman spectroscopy. An ideal tool for studying the physical properties and applications of metal–organic frameworks (MOFs). Chem. Soc. Rev. 2023, 52, 3397–3437. [Google Scholar] [CrossRef]
- Yu, R.; Wang, C.; Liu, D.; Wang, X.; Yin, J.; Du, Y. Self-reconstruction of Fe-doped Co-metal–organic frameworks boosted electrocatalytic performance for oxygen evolution reaction. Inorg. Chem. 2023, 62, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Huanga, W.-H.; Lia, Q.-H.; Yub, D.-Y.; Tanga, Y.-H.; Linc, D.-Y.; Wanga, F.; Zhanga, J. Hybrid zeolitic imidazolate frameworks for promoting electrocatalytic oxygen evolution via dual-site relay mechanism. Inorg. Chem. 2021, 60, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
- Jayaramulu, K.; Masa, J.; Morales, D.M.; Tomanec, O.; Ranc, V.; Petr, M.; Wilde, P.; Chen, Y.-T.; Zboril, R.; Schuhmann, W.; Fischer, R.A. Ultrathin 2D cobalt zeolite-imidazole framework nanosheets for electrocatalytic oxygen evolution. Adv. Sci. 2018, 5, 1801029. [Google Scholar] [CrossRef] [PubMed]
- Benkó, T.; Lukács, D.; Li, M.; Pap, J.S. Redox-active ligands in artificial photosynthesis: A review. Environ. Chem. Lett. 2022, 20, 3657–3695. [Google Scholar] [CrossRef]
- Li, M.; Liao, R.-Z. Water oxidation catalyzed by a bioinspired tetranuclear manganese complex: Mechanistic study and prediction. ChemSusChem, 2022, 15, e202200187. [Google Scholar] [CrossRef]
- Chen, Q.-F.; Guo, Y.-H.; Yu, Y.-H.; Zhang, M.-T. Bioinspired molecular clusters for water oxidation. Coord. Chem. Rev. 2021, 448, 214164. [Google Scholar] [CrossRef]
- Zhang, Q.; Guan, J. Mono-/multinuclear water oxidation catalysts. ChemSusChem, 2019, 12, 3209–3235. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, P. Transition metal based coordination complexes as catalysts for water oxidation. Resonance, 2022, 27, 1185–1209. [Google Scholar] [CrossRef]
- Masaya, M.; Kondo, M.; Kuga, R.; Kurashige, Y.; Yanai, T.; Hayami, S.; Praneeth, V.K.K.; Yoshida, M.; Yoneda, K.; Kawata, S.; Masaoka, S. A pentanuclear iron catalyst designed for water oxidation. Nature, 2016, 530, 465–468. [Google Scholar] [CrossRef]
- Chen, G.; Lam, W.W.Y.; Lo, P.K.; Man, W.-L.; Chen, L.; Lau, K.-C.; Lau, T.-C. Mechanism of water oxidation by ferrate(VI) at pH 7–9. Chem. Eur. J. 2018, 24, 18735–18742. [Google Scholar] [CrossRef] [PubMed]
- den Boer, D.; Konovalov, A.I.; Siegler, M.A.; Hetterscheid, D.G.H. Unusual water oxidation mechanism via a redox-active copper polypyridyl complex. Inorg. Chem. 2023, 62, 5303–5314. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Y.; Wang, X.-Y.; Li, H.-J.; Chen, J.-Y.; Kou, Y.-H.; Li, X.; Wang, Y. Theoretical study on the mechanism of water oxidation catalyzed by a mononuclear copper complex: important roles of a redox non-innocent ligand and HPO42− anion. RSC Adv. 2023, 13, 8352. [Google Scholar] [CrossRef]
- Iqbal, S.; Safdar, B.; Hussain, I.; Zhang, K.; Chatzichristodoulou, C. Trends and prospects of bulk and single-atom catalysts for the oxygen evolution reaction. Adv. Energy Mater. 2023, 13, 2203913. [Google Scholar] [CrossRef]
- Kumar, P.; Kannimuthu, K.; Zeraati, A.S.; Roy, S.; Wang, X.; Wang, X.; Samanta, S.; Miller, K.A.; Molina, M.; Trivedi, D.; et al. High-density cobalt single-atom catalysts for enhanced oxygen evolution reaction. J. Am. Chem. Soc. 2023, 145, 8052–8063. [Google Scholar] [CrossRef]
- Cipriano, L.A.; Liberto, G.D.; Pacchioni, G. Superoxo and peroxo complexes on single-atom catalysts: Impact on the oxygen evolution reaction. ACS Catal. 2022, 12, 11682–11691. [Google Scholar] [CrossRef]
- Barlocco, I.; Cipriano, L.A.; Liberto, G.D.; Pacchioni, G. Does the oxygen evolution reaction follow the classical OH*, O*, OOH* path on single atom catalysts? J. Catal. 2023, 417, 351–359. [Google Scholar] [CrossRef]
- He, Y.; Zhou, X.; Jia, Y.; Li, H.; Wang, Y.; Liu, Y.; Tan, Q. Advances in transition-metal-based dual-atom oxygen electrocatalysts. Small, 2023, 19, 2206477. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Zhou, J.; Zhang, L.; Wan, W.; Ding, Y.; Sun, X. Synergy of dual-atom catalysts deviated from the scaling relationship for oxygen evolution reaction. Nat. Commun. 2023, 14, 4449. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H. Effect of carbonate salt addition on the photocatalytic decomposition of liquid water over Pt–TiO2 catalyst. J. Chem. Soc., Faraday Trans. 1997, 93, 1647–1654. [Google Scholar] [CrossRef]
- Kusama, H.; Kodera, M.; Yamashita, K.; Sayama, K. Insights into the carbonate effect on water oxidation over metal oxide photocatalysts/photoanodes. Phys. Chem. Chem. Phys. 2022, 24, 5894–5902. [Google Scholar] [CrossRef]
- Yang, C.; Laberty-Robert, C.; Batuk, D.; Cibin, G.; Chadwick, A.V.; Pimenta, V.; Yin, W.; Zhang, L.; Tarascon, J.-M.; Grimaud, A. Phosphate ion functionalization of perovskite surfaces for enhanced oxygen evolution reaction. J. Phys. Chem. Lett. 2017, 8, 3466–3472. [Google Scholar] [CrossRef]
- Tsuneda, T.; Ten-no, S. Water–oxidation mechanism of cobalt phosphate co-catalyst in artificial photosynthesis: a theoretical study. Phys. Chem. Chem. Phys., 2022, 24, 4674–4682. [Google Scholar] [CrossRef]
- Liu, M.; Li, N.; Wang, X.; Zhao, J.; Zhong, D.-C.; Li, W.; Bu, X.-H. Photosystem II inspired NiFe-based electrocatalysts for efficient water oxidation via second coordination sphere effect. Angew. Chem. Int. Ed. 2023, 62, e202300507. [Google Scholar] [CrossRef]
- Gao1, Y.; Wu, C.; Sen, S.; Tan, Y. Hexadecyltrimethylammonium hydroxide promotes electrocatalytic activity for the oxygen evolution reaction. Commun. Chem. 2020, 3, 154. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
S-state cycle in photosystem II (PS II) with time-constant values (in parenthesis were newly determined). Reproduced from Ref. [16] under the license of CCA4.0 from Natsure.com.
Figure 1.
S-state cycle in photosystem II (PS II) with time-constant values (in parenthesis were newly determined). Reproduced from Ref. [16] under the license of CCA4.0 from Natsure.com.
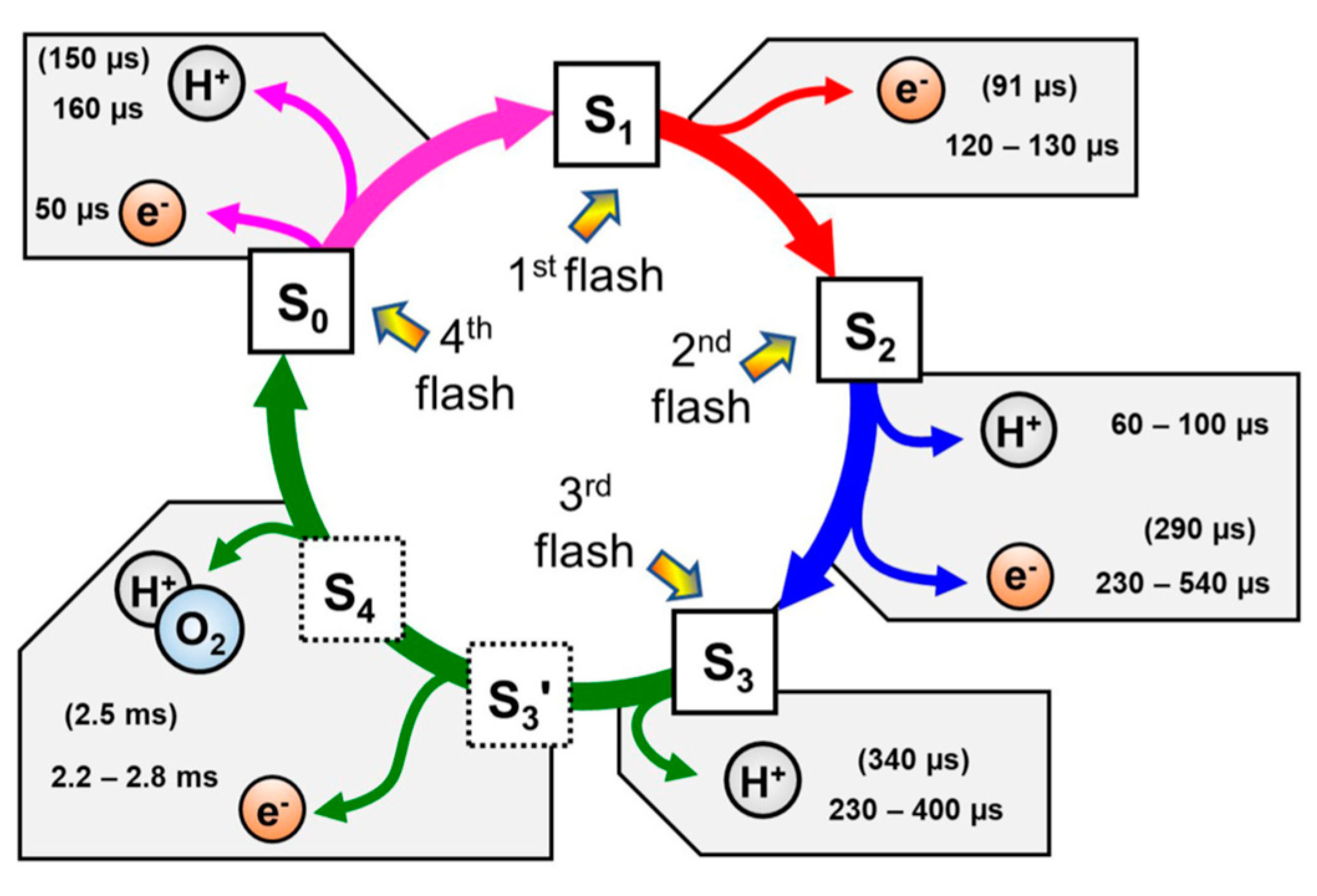
Figure 2.
Molecular sketches of atomic and electronic rearrangements leading to peroxide formation corresponding to the S3’ to S4 states in Figure 1. Reproduced from Ref. [16] under the license of CCA4.0 from Natsure.com.
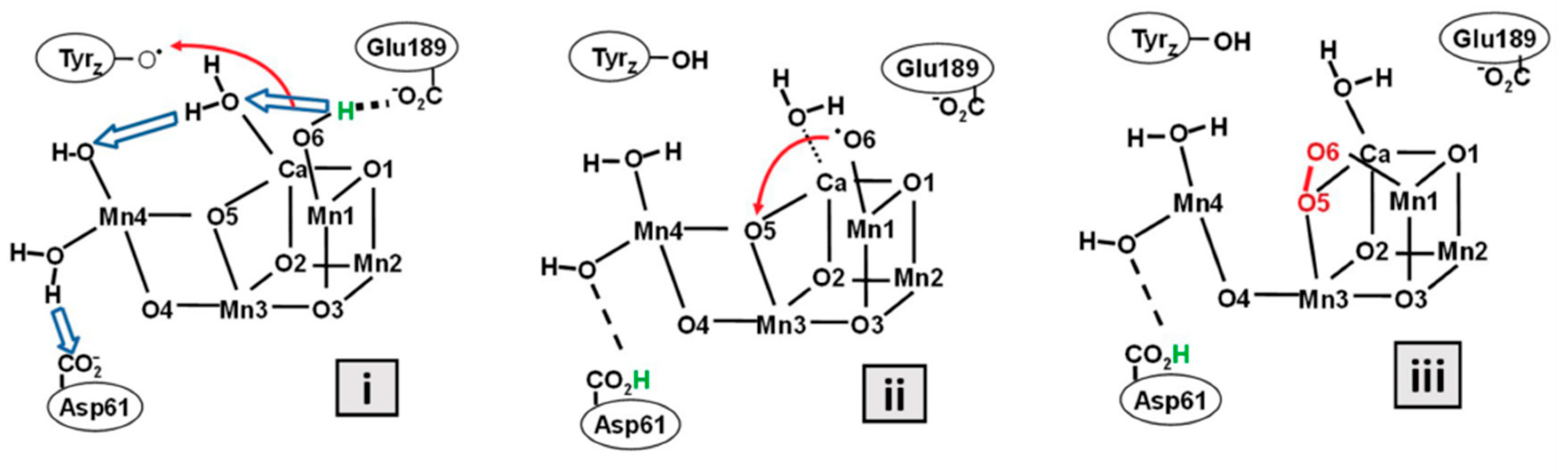
Figure 3.
Schematic illustration of simplified OER mechanisms; (a) adsorbate evolution mechanism (AEM), (c) oxide pair mechanism (OPM), and (d) one of the lattice oxygen mechanism (LOM), (b) schematic free energy change at each oxidation step in AEM OER process.
Figure 3.
Schematic illustration of simplified OER mechanisms; (a) adsorbate evolution mechanism (AEM), (c) oxide pair mechanism (OPM), and (d) one of the lattice oxygen mechanism (LOM), (b) schematic free energy change at each oxidation step in AEM OER process.
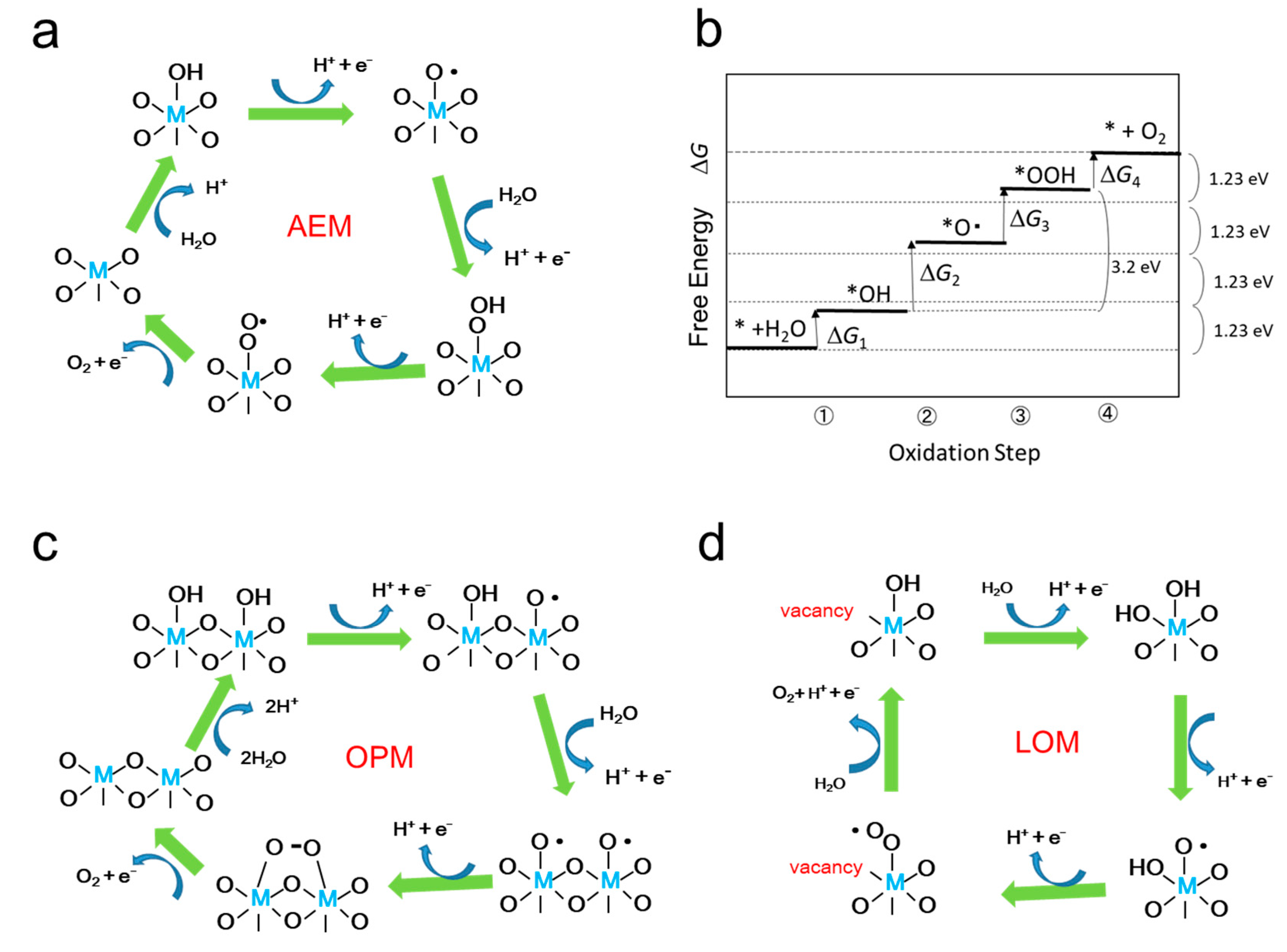
Figure 4.
Various LOM processes; (a) OVSM = oxygen-vacancy site mechanism, (b) SMSM = single-metal-site mechanism), and (c) DMSM = double-metal-site mechanism. Reproduced from Ref. [112] with permission from the Royal Society of Chemistry.
Figure 4.
Various LOM processes; (a) OVSM = oxygen-vacancy site mechanism, (b) SMSM = single-metal-site mechanism), and (c) DMSM = double-metal-site mechanism. Reproduced from Ref. [112] with permission from the Royal Society of Chemistry.
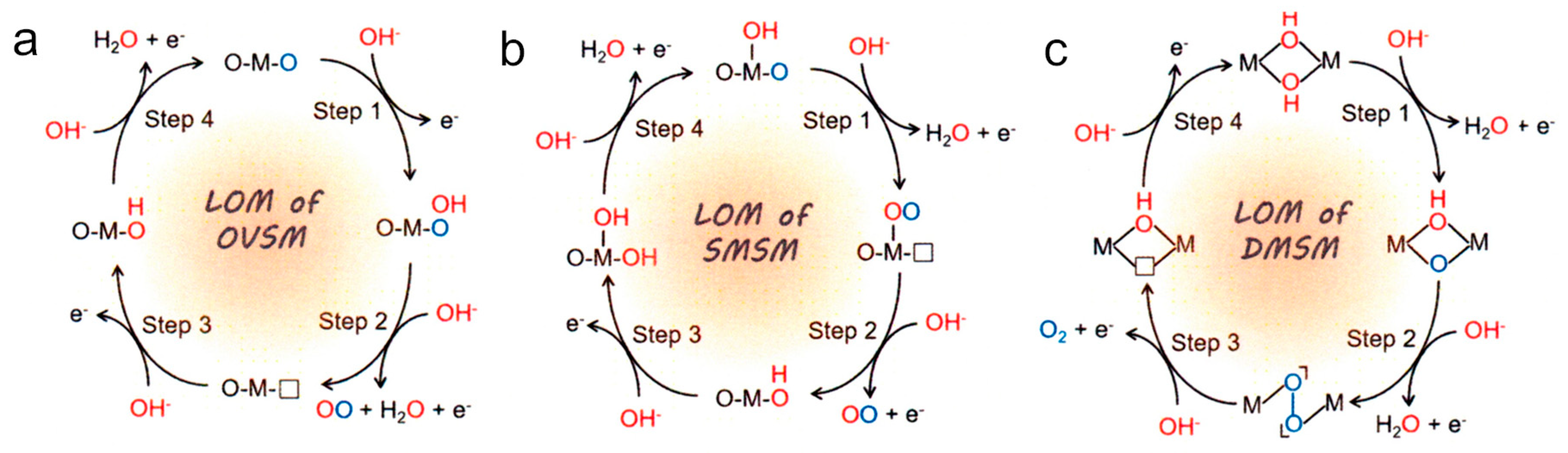
Figure 5.
Key steps in water oxidation deduced from a DFT calculation for the model molecule of rutile TiO2(011) surface. Reproduced from Ref. [113] under the license of CC BY 4.0 from MDPI publishers.
Figure 5.
Key steps in water oxidation deduced from a DFT calculation for the model molecule of rutile TiO2(011) surface. Reproduced from Ref. [113] under the license of CC BY 4.0 from MDPI publishers.
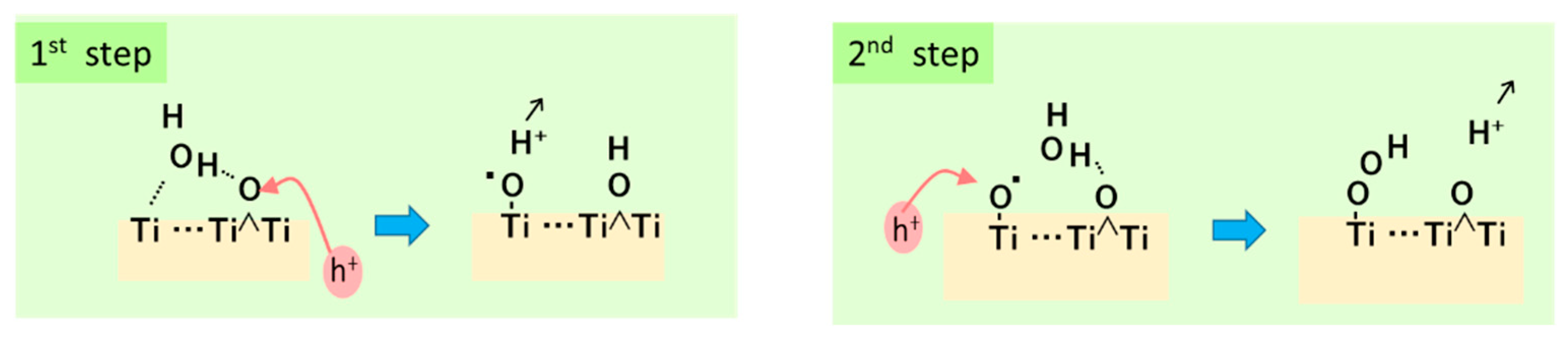
Figure 6.
Oxygen generation mechanism suggested by Nakamura, et al. Blue arrow was added by the present author. Reproduced from Ref. [120] with permission from the American Chemical Society.
Figure 6.
Oxygen generation mechanism suggested by Nakamura, et al. Blue arrow was added by the present author. Reproduced from Ref. [120] with permission from the American Chemical Society.
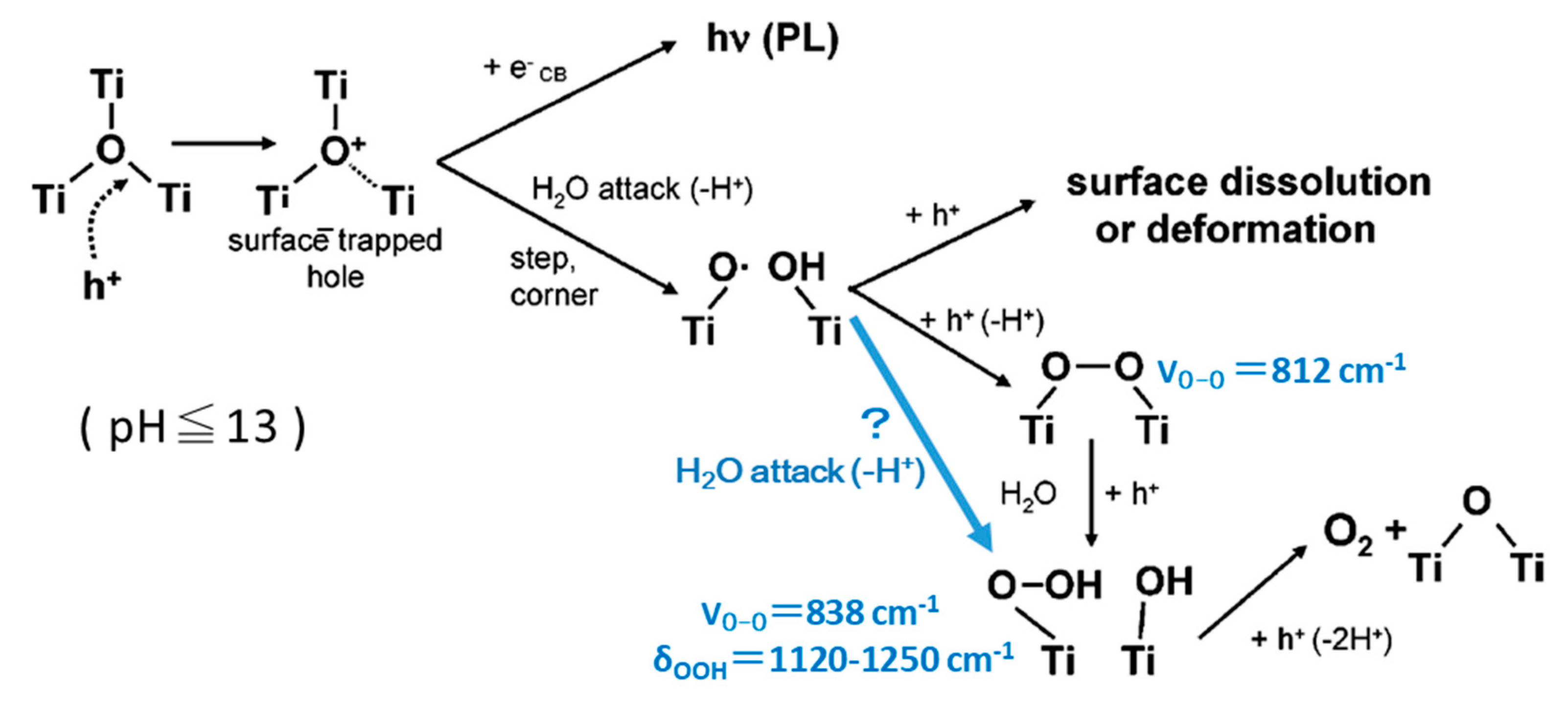
Figure 7.
Suggested pathways deduced from DFT calculations for oxygen evolution reaction (OER) and hydrogenperoxide evolution reaction (HPER). Reproduced from Ref. [135] with permission from the American Chemical Society.
Figure 7.
Suggested pathways deduced from DFT calculations for oxygen evolution reaction (OER) and hydrogenperoxide evolution reaction (HPER). Reproduced from Ref. [135] with permission from the American Chemical Society.
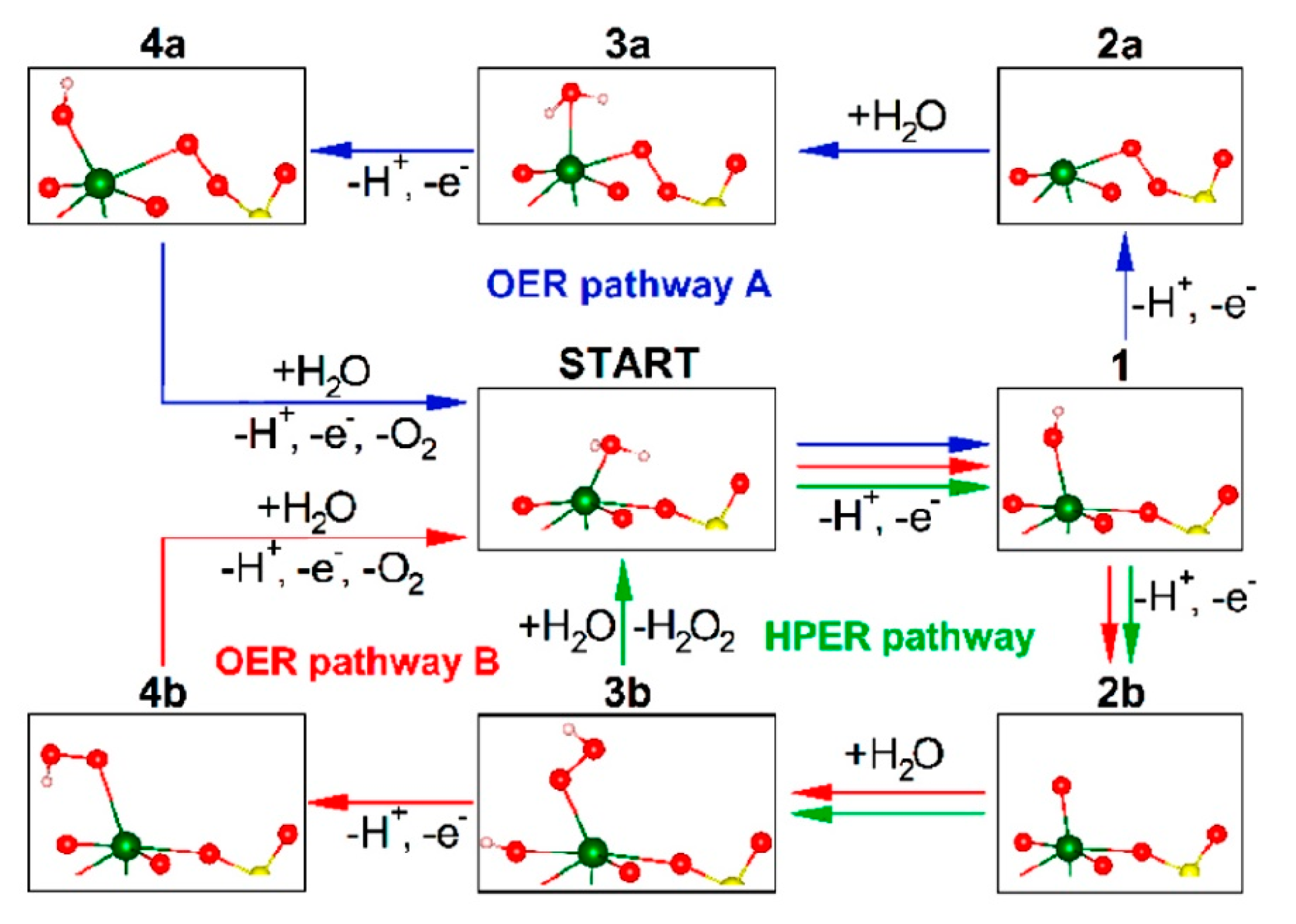
Figure 8.
ATR-FTIR spectra of BiVO4 photoanode under light irradiation. Compiled from the figures in Ref [138]. Reproduced from Ref. [138] with permission from the American Chemical Society.
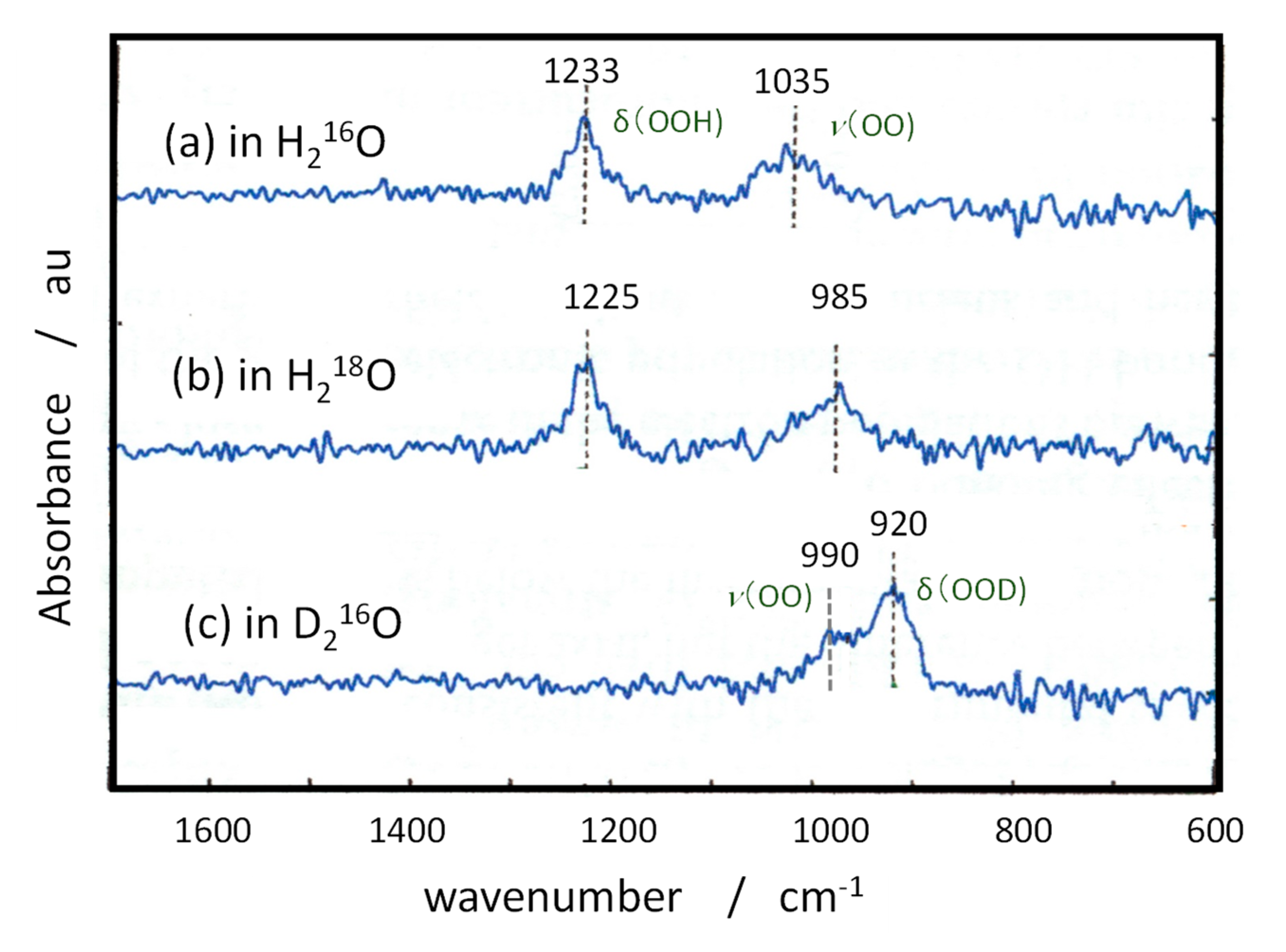
Figure 9.
Initial step of photocatalytic OER at SrTiO3/water interface. (a) OH vibration energy relaxed by H-bond network. (b) Proposed reaction diagram of forming two kinds of surface radicals. Reproduced from Ref. [143] with permission from the American Chemical Society.
Figure 9.
Initial step of photocatalytic OER at SrTiO3/water interface. (a) OH vibration energy relaxed by H-bond network. (b) Proposed reaction diagram of forming two kinds of surface radicals. Reproduced from Ref. [143] with permission from the American Chemical Society.
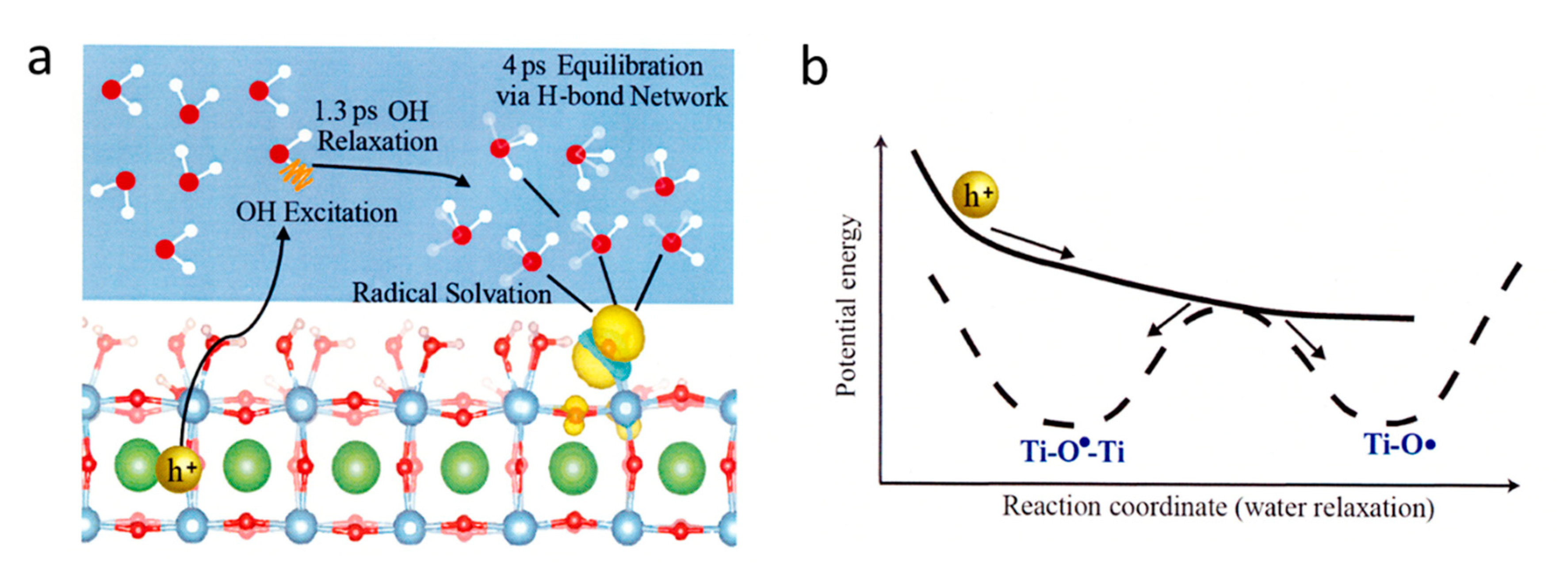
Figure 10.
Electrocatalytic OER activities on noble metal (Ir, Ru, Pt ) based catalysts under acidic conditions. Reproduced from Ref. [172] with permission from the Royal Society of Chemistry.
Figure 10.
Electrocatalytic OER activities on noble metal (Ir, Ru, Pt ) based catalysts under acidic conditions. Reproduced from Ref. [172] with permission from the Royal Society of Chemistry.
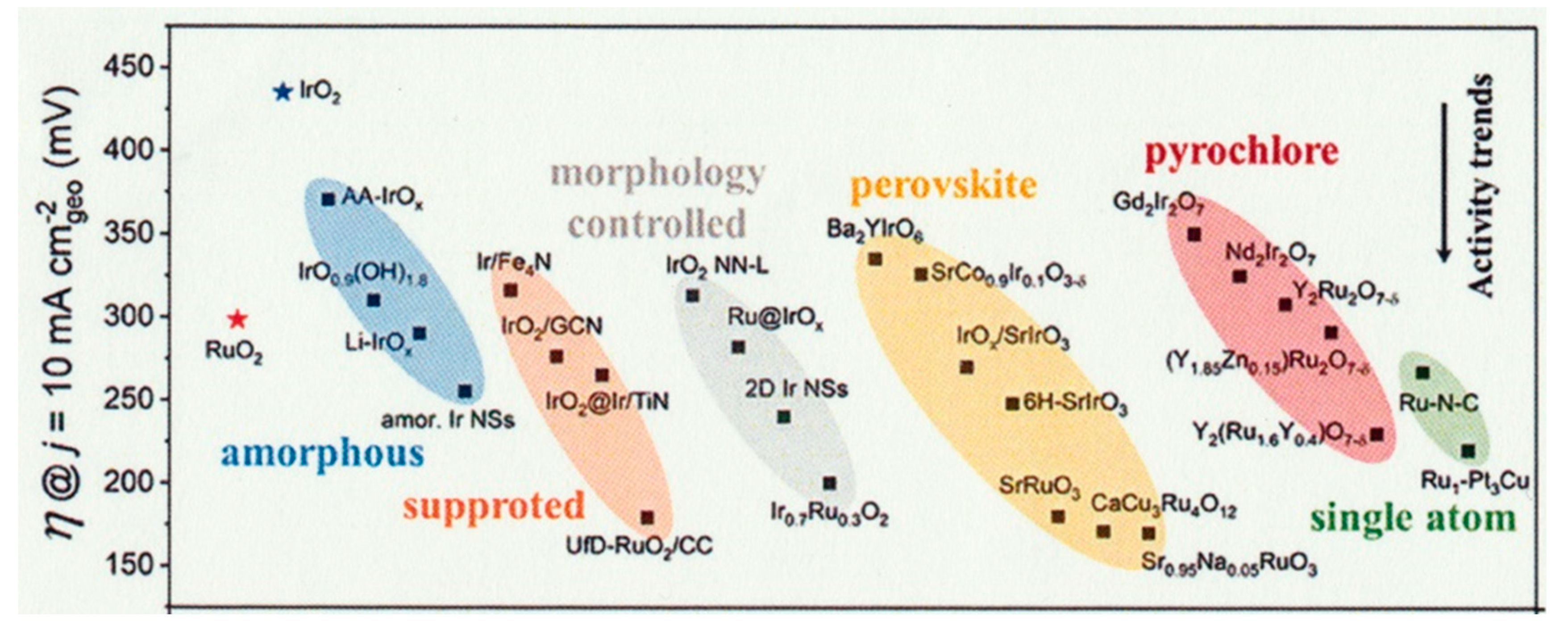
Figure 11.
DFT calculation of HION, IrO2, and IrOOH. (a,b,c) Gibbs free energy diagrams of H4IrO4, IrO2, and IrOOH for OER at Ir sites. (d,e,f) Charge density difference induced by oxygen adsorption of H4IrO4, IrO2, and IrOOH at Ir sites. Reproduced from Ref. [177] with permission from the American Chemical Society.
Figure 11.
DFT calculation of HION, IrO2, and IrOOH. (a,b,c) Gibbs free energy diagrams of H4IrO4, IrO2, and IrOOH for OER at Ir sites. (d,e,f) Charge density difference induced by oxygen adsorption of H4IrO4, IrO2, and IrOOH at Ir sites. Reproduced from Ref. [177] with permission from the American Chemical Society.
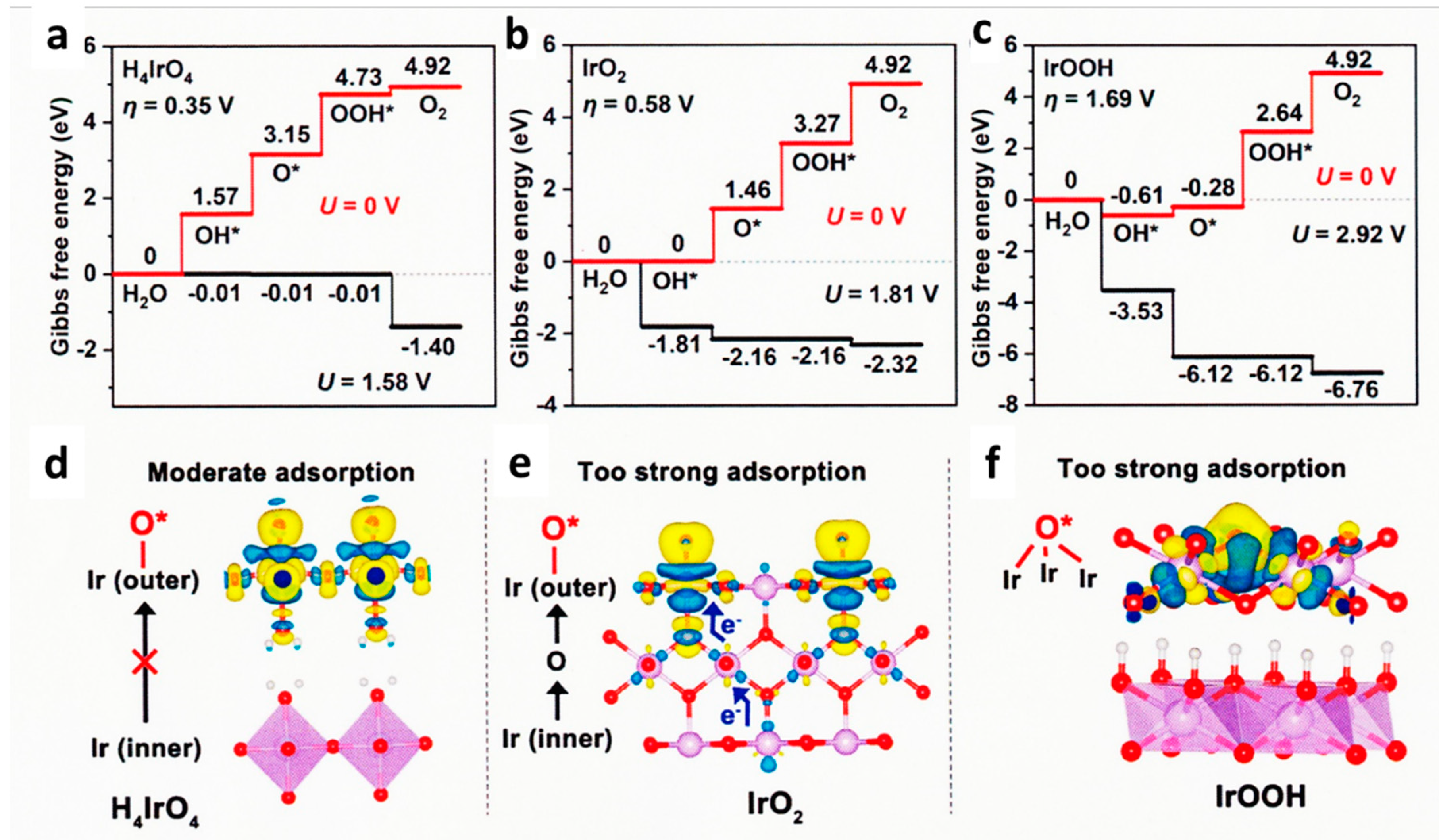
Figure 12.
Proposed reaction mechanisms of the OER for (a) Co3O4 and (b) Vo-Co3O4. Oxygen vacancy (Vo) prepared by NaBH4 reduction was represented by the red dotted boxes. WNA=water nucleophilic attack, INA=intramolecular nucleophilic attack, and I2M=interaction between the two metal-O unit. Reproduced from Ref. [191] with permission from the American Chemical Society.
Figure 12.
Proposed reaction mechanisms of the OER for (a) Co3O4 and (b) Vo-Co3O4. Oxygen vacancy (Vo) prepared by NaBH4 reduction was represented by the red dotted boxes. WNA=water nucleophilic attack, INA=intramolecular nucleophilic attack, and I2M=interaction between the two metal-O unit. Reproduced from Ref. [191] with permission from the American Chemical Society.
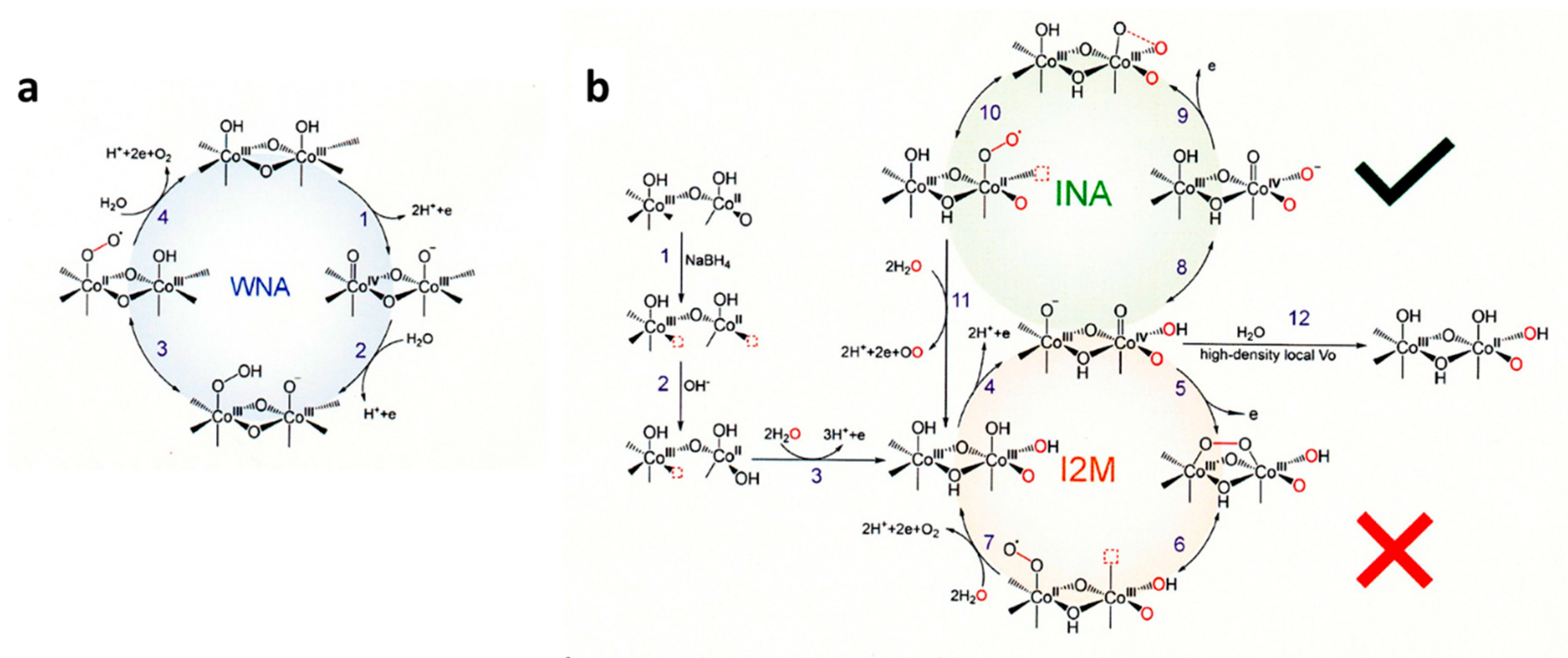
Figure 13.
Decrease in the binding energy from Zn to Mn doped nickel oxides showed a volcano type OER activity by changing the rate determining step from •O formation to O-O coupling. Reproduced from Ref. [200] with permission from the American Chemical Society.
Figure 13.
Decrease in the binding energy from Zn to Mn doped nickel oxides showed a volcano type OER activity by changing the rate determining step from •O formation to O-O coupling. Reproduced from Ref. [200] with permission from the American Chemical Society.
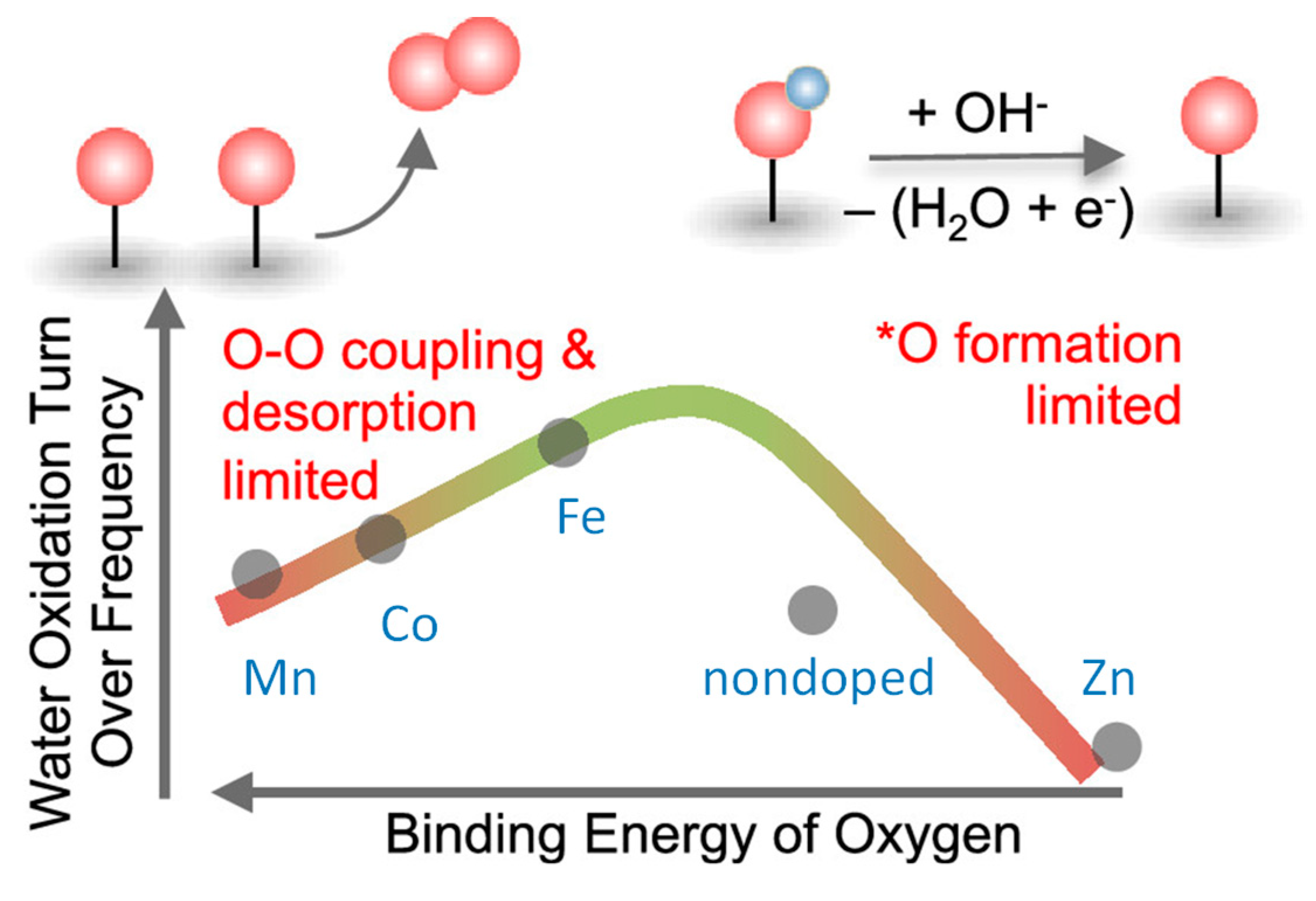
Figure 14.
In photoelectrochemical water oxidation on -Fe2O3, the OER mechanism switches from WNA to I2M at a strong alkaline solution. Reproduced from Ref. [89] with permission from the American Chemical Society.
Figure 14.
In photoelectrochemical water oxidation on -Fe2O3, the OER mechanism switches from WNA to I2M at a strong alkaline solution. Reproduced from Ref. [89] with permission from the American Chemical Society.
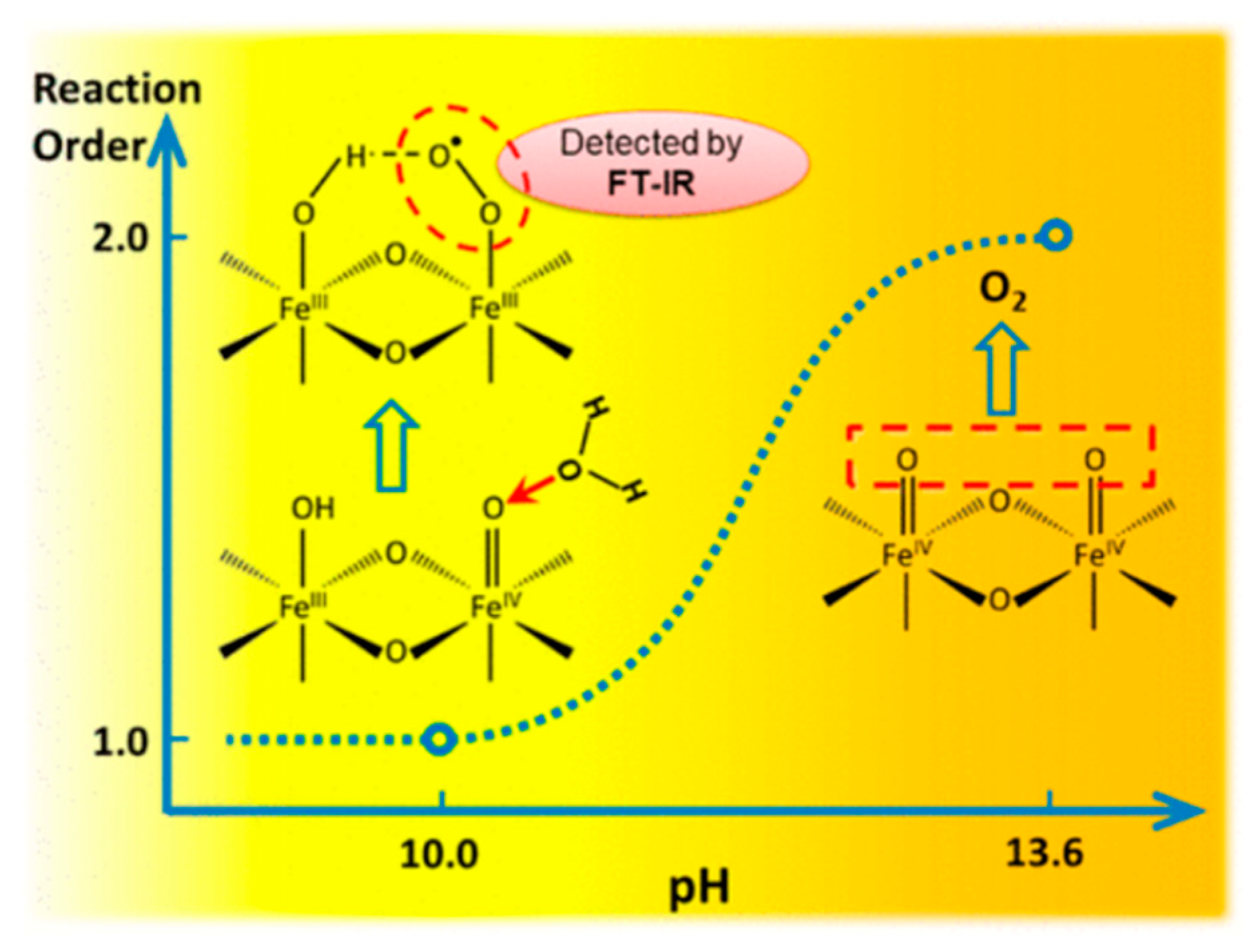
Figure 15.
Proposed mechanism in MnOx-catalyzed OER. Reproduced from Ref. [68] (Elsevier) under the license of CC BY 4.0.
Figure 15.
Proposed mechanism in MnOx-catalyzed OER. Reproduced from Ref. [68] (Elsevier) under the license of CC BY 4.0.
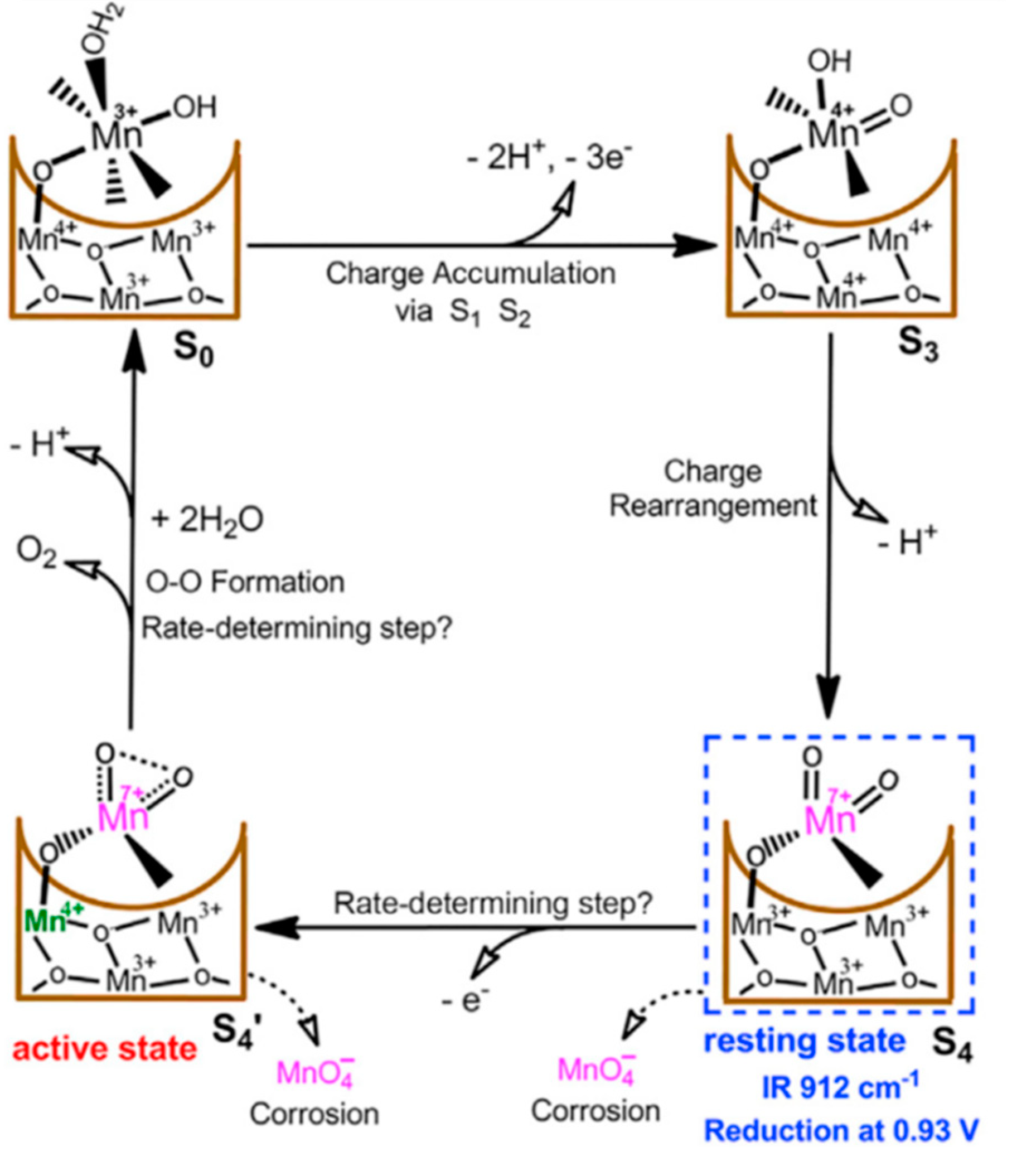
Figure 16.
For Co-NiFe-LDH nanosheet, weakening alkaline concentration changes rate-determining step (RDS) from the release of O2 to the oxo radical formation. Reproduced from Ref. [233] with permission from the American Chemical Society.
Figure 16.
For Co-NiFe-LDH nanosheet, weakening alkaline concentration changes rate-determining step (RDS) from the release of O2 to the oxo radical formation. Reproduced from Ref. [233] with permission from the American Chemical Society.
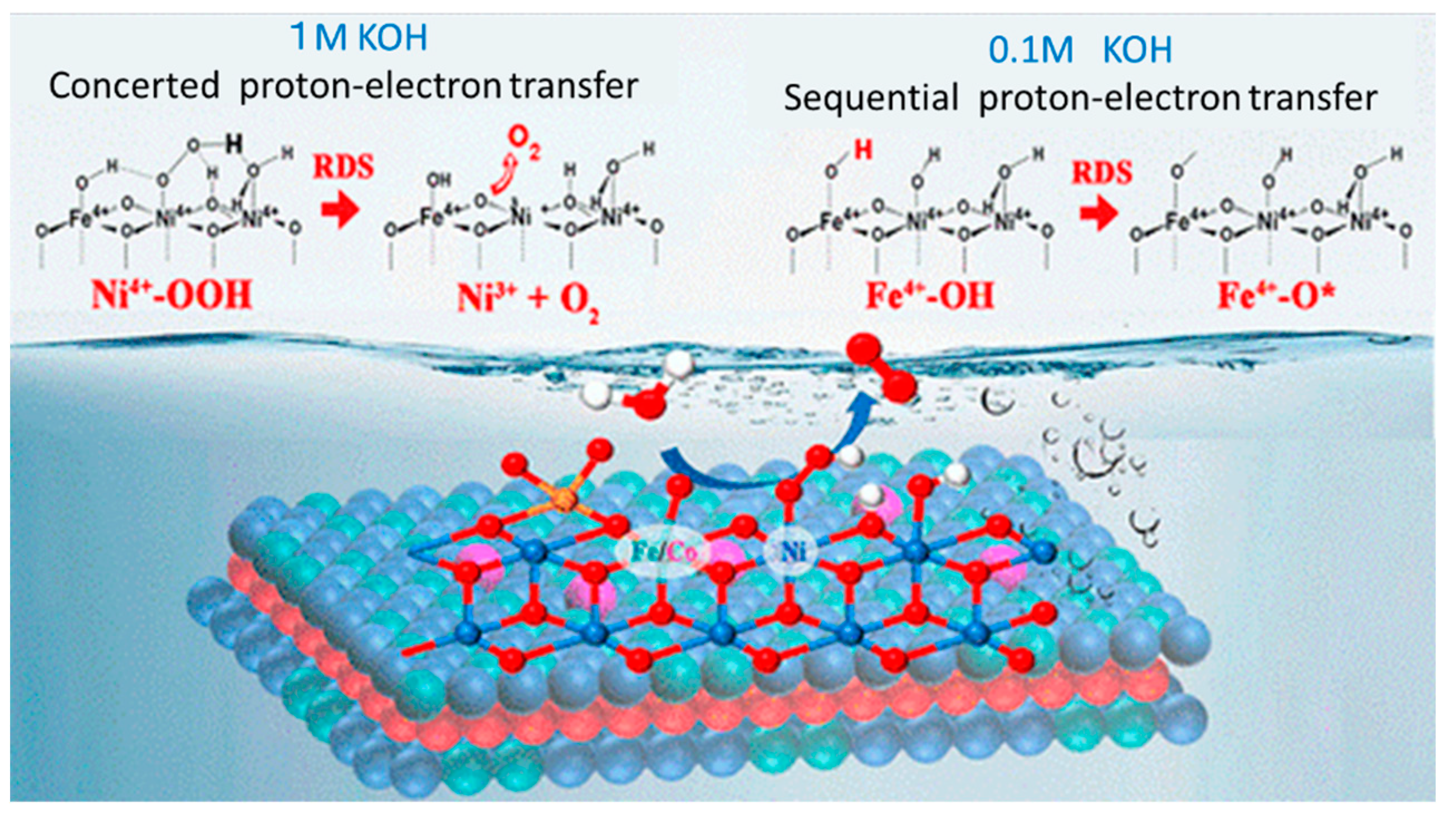
Figure 17.
Proposed scheme of the nature of the active site of the exfoliated Co-ZIF-9(III) 2D catalyst and the underlying mechanism of the OER. Reproduced from Ref. [249] under the license of CC BY 4.0 of John Wiley & Sons, Inc.
Figure 17.
Proposed scheme of the nature of the active site of the exfoliated Co-ZIF-9(III) 2D catalyst and the underlying mechanism of the OER. Reproduced from Ref. [249] under the license of CC BY 4.0 of John Wiley & Sons, Inc.
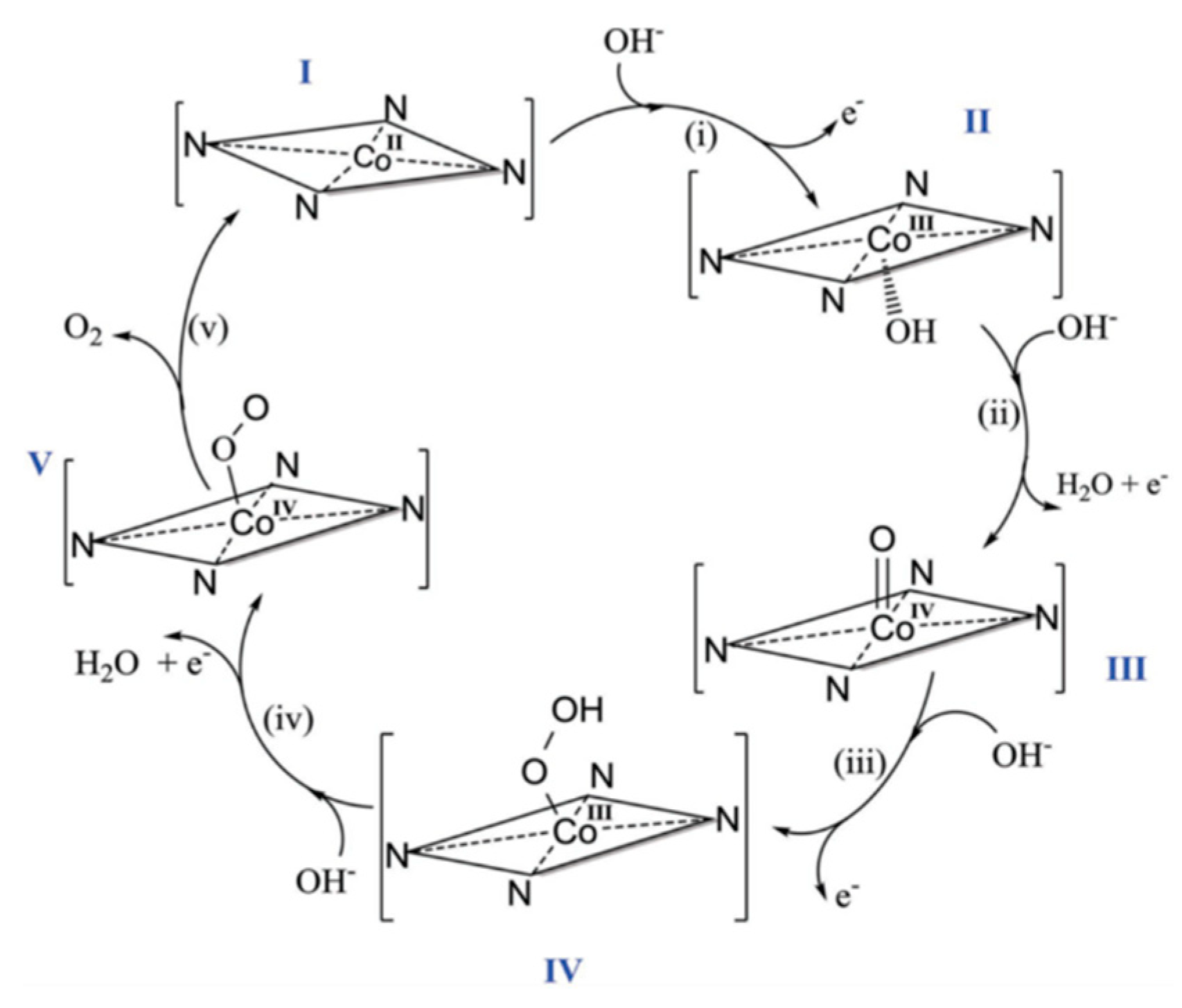
Figure 18.
(a) Schematic representation of the Cu complex used in the calculation. (b) Proposed catalytic cycle for water oxidation catalyzed by the Cu complex. The O-O bond formation process is shown at the inset. Reproduced from Ref. [258] with permission from the Royal Society of Chemistry.
Figure 18.
(a) Schematic representation of the Cu complex used in the calculation. (b) Proposed catalytic cycle for water oxidation catalyzed by the Cu complex. The O-O bond formation process is shown at the inset. Reproduced from Ref. [258] with permission from the Royal Society of Chemistry.
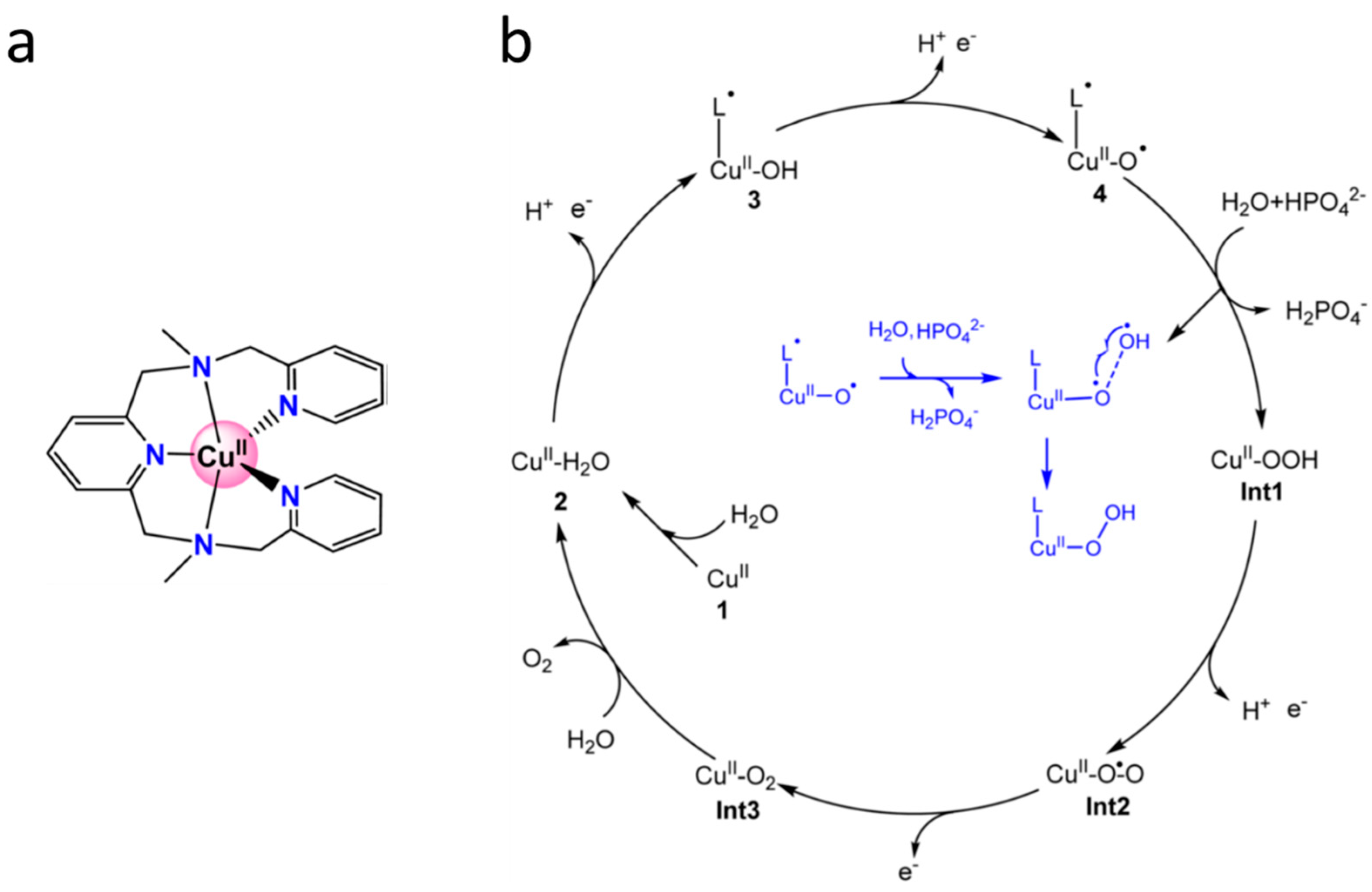
Figure 19.
Representative Gibbs energy profile of OER over a single atom catalyst (SAC). Reproduced from Ref. [261] with permission from the American Chemical Society.
Figure 19.
Representative Gibbs energy profile of OER over a single atom catalyst (SAC). Reproduced from Ref. [261] with permission from the American Chemical Society.
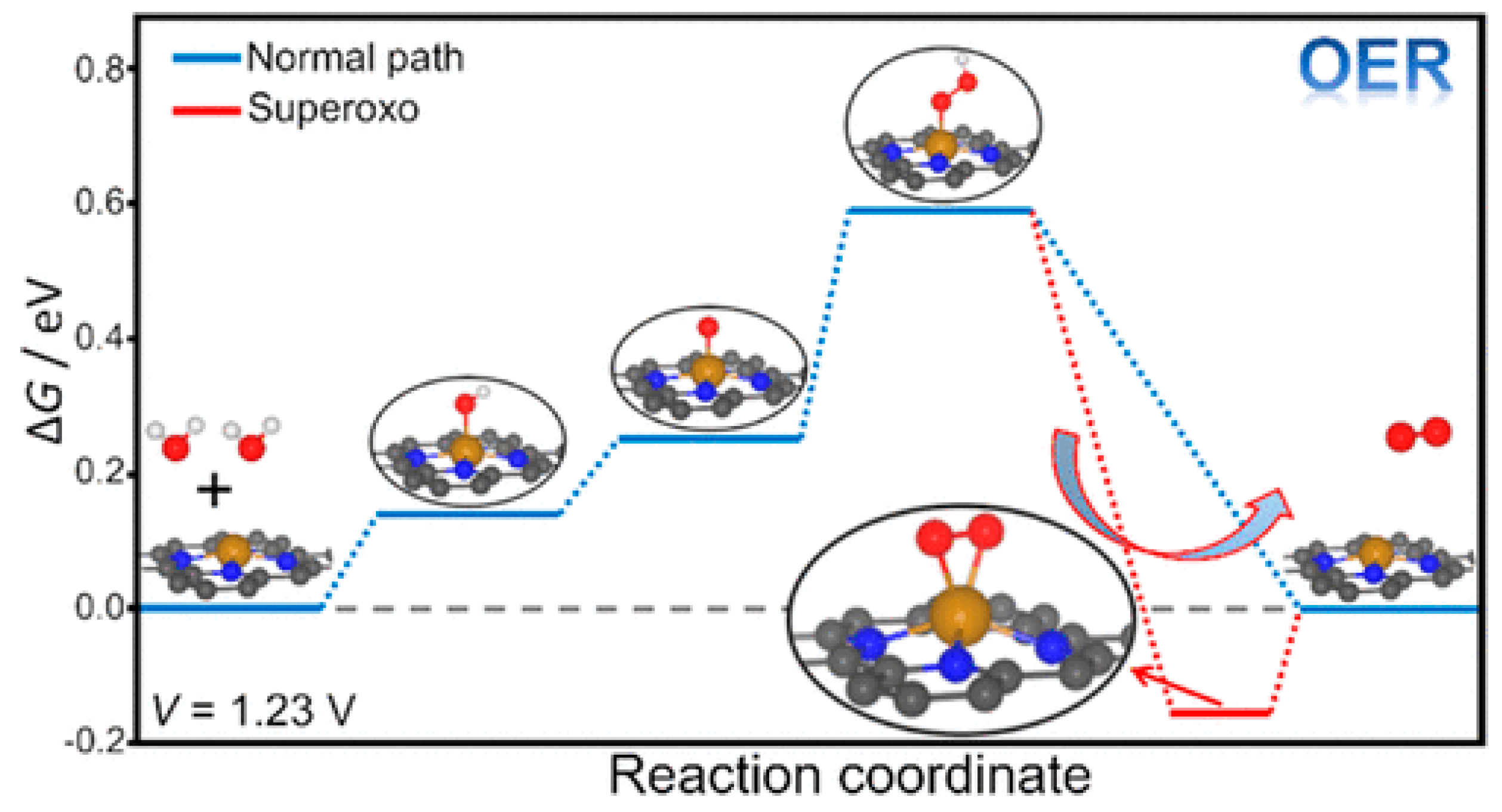
Figure 20.
Proposed mechanisms for improving the OER activity of perovskite catalysts. Reproduced from Ref. [267] with permission from the American Chemical Society.
Figure 20.
Proposed mechanisms for improving the OER activity of perovskite catalysts. Reproduced from Ref. [267] with permission from the American Chemical Society.
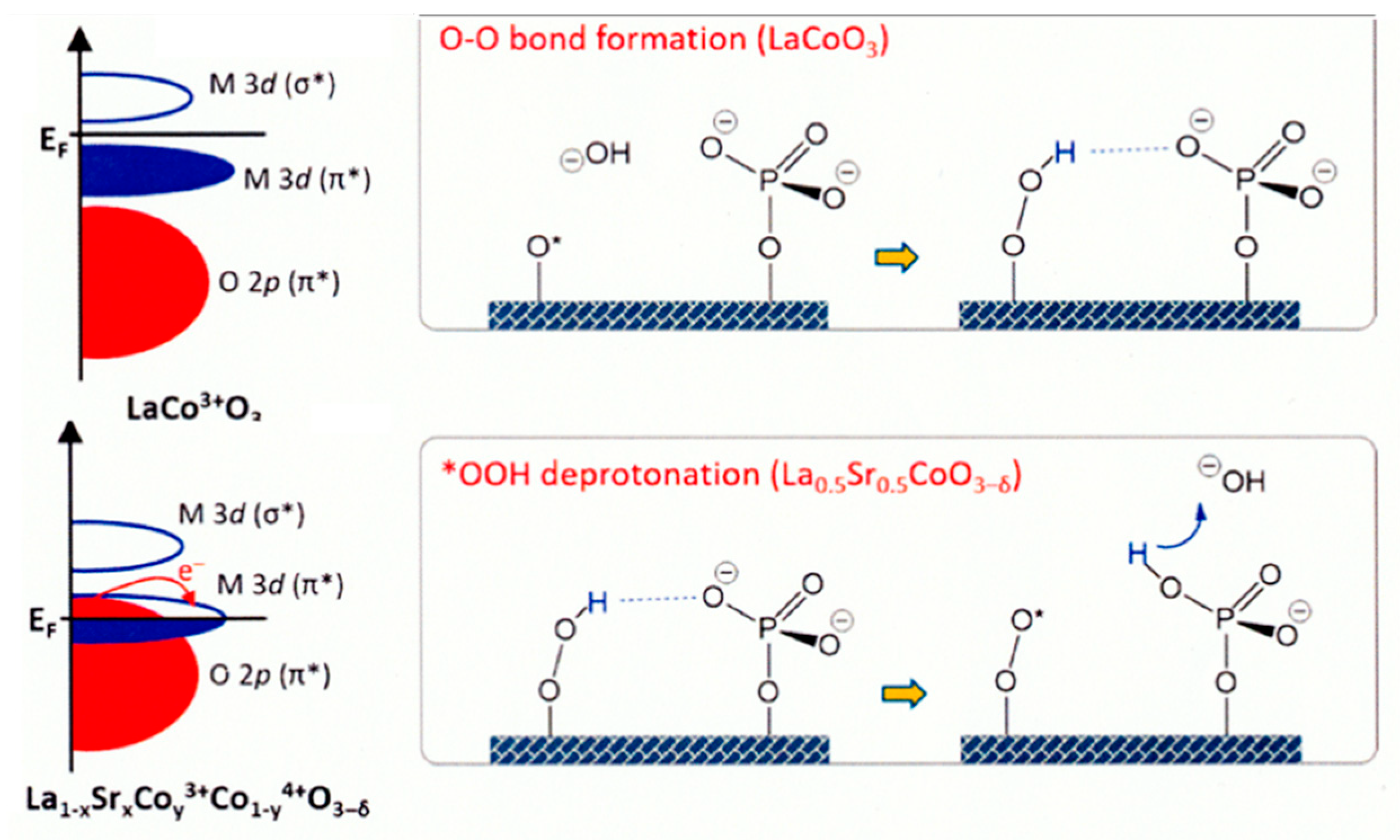

Table 1.
FTIR peak position of the intermediate spaces in the OER processes.
| Oxidation step | /cm-1 | mode | sample | [Reference] year | |
|---|---|---|---|---|---|
| 1 | Co(IV)=O | 840 | νMO | Co3O4 | [64] 2014 |
| Ni-O∙ | 908 | Ni-NC | [65] 2020 | ||
| Ni-O∙ | 916 | Ni-Vac | [65] 2020 | ||
| Ni-O∙ | 1170-1330 | NiV-MOF | [66] 2021 | ||
| Mn(IV)=O | 712–755 | Mn complex | [67] 2001 | ||
| Mn(V)=O | 970–981 | Mn complex | [67] 2001 | ||
| Mn(VII)=O | 912 | NiFe-LDH | [68] 2018 | ||
| Fe(IV)=O | 898 | α-Fe2O3 | [69] 2016 | ||
| Ti(IV)-O∙ | 795 | n-SrTiO3 | [70] 2016 | ||
| Ir-O | 784 | Atomic Ir | [71] 2015 | ||
| Ir(VI)=O | 946 | CaIrO2 | [63] 2022 | ||
| 2 | Mn(IV)=O2 | 810 | Mn complex | [72] 2021 | |
| Li-OOH | 1170 | Li in DMSO | [73] 2017 | ||
| Ti-OOH | 837 | νOO | Ti-SiO2 | [74] 2002 | |
| Ti-OOH | 838 | νOO | TiO2 | [75] 2003 | |
| Ti-OOH | 1120-1250 | δOOH | TiO2 | [75] 2003 | |
| Ni-OOH | 1060 | Ni-Vac | [76] 2022 | ||
| Ni(IV)-OOD | 1048 | NiFe MOF | [77] 2019 | ||
| Ni-OO- | 850-1200 | NiOOH | [78] 2019 | ||
| Cu-OO- | 1180 | Cu complex | [79] 2021 | ||
| Ru-OOH | 986 | RuO2 | [80] 2021 | ||
| Ga-OOH | 978 | Ga2O3 | [81] 2021 | ||
| Ir (VI)-OO- | 870 | νOO | CaIrO2 | [63] 2022 | |
| Ir(III)-OOH | 830 | νOO | IrO2 | [82] 2011 | |
| Ir-OOH | 1055 | Atomic Ir | [71] 2021 | ||
| Ir-OOH | 1065 | Ir/Co3O4 | [83] 2023 | ||
| Au-OOH | 1268 | Au electrode | [84] 2005 | ||
| Co-OO-Co | 833 | νOO | Co3O4 | [85] 2020 | |
| Co-OO-Co | 1075 | νOO | CoOOH | [86] 2020 | |
| Li-OO-Li | 780, 830 | Li in DMSO | [73] 2017 | ||
| Ti-OO-Ti | 812 | TiO2 | J75[ 2003 | ||
| Fe(VI)(=O)2 | 840, 856 | NiFe-LDH | [87] 3018 | ||
| Fe-OO-Fe | 1145 | Fe3O4/Au | [88] 2021 | ||
| Ni-OO-Fe | 1076 | Ni3FeOx/Au | [88] 2021 | ||
| M-OO-M | 1089 | Ru/MnO2 | [80] 2021 | ||
| 3 | Co(III)-OO∙(HO) | 1013 | Co3O4 | [64] 2014 | |
| Fe-OO∙(HO) | 1096 | α-Fe2O3 | [89] 2018 | ||
| Li-OO∙ | 1127-1139 | Li in DMSO | [89] 2018 | ||
| M-OO | 1128 | Ru/MnO2 | [80] 2021 | ||
| Pt-OO∙ | 1005-1016 | Pt in Alkaline | [90] 2006 | ||
| Mn-OO | 1080 | Mn3O4 | [91] 2018 |
Table 2.
FTIR peak position of hydroxyperoxo coordinated to TiO2 and the isotope shifts.
| mode | Ti-OOH | Ti-O18OH | Ti-18O18OH | Ti-OOD | |
|---|---|---|---|---|---|
| Experimental a | δ(OOH) | 1120-1250 cm-1 | Δ =-220 --320 cm-1 | ||
| ν(OO) | 838 cm-1 | Δ =-19 cm-1 | Δ =-45 cm-1 | Δ =-67 --97 cm-1 | |
| DFT calculation b | δ(OOH) | 1257 cm-1 | Δ =-2 cm-1 | Δ =-8 cm-1 | Δ =-299 cm-1 |
| ν(OO) | 881 cm-1 | Δ =-23 cm-1 | Δ =-49 cm-1 | Δ =-22 cm-1 |
Table 3.
FTIR peak position of hydroxyperoxo group coordinated to BiVO4.
| mode | Bi-OOH | Bi-O18OH | Bi-18O18OH | Bi-OOD | |
|---|---|---|---|---|---|
| BiVO4 Experiment a | δ(OOH) | 1233 cm-1 | Δ = -8 cm-1 | Δ = -313 cm-1 | |
| ν(OO) | 1030 cm-1 | Δ = -45 cm-1 | Δ = -65 cm-1 | ||
| BiOOH DFT calc. b | δ(OOH) | 1257 cm-1 | Δ = -2 cm-1 | Δ = -8 cm-1 | Δ = -299 cm-1 |
| ν(OO) | 881 cm-1 | Δ = -23 cm-1 | Δ = -50 cm-1 | Δ = -22 cm-1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



