
Submitted:
02 August 2023
Posted:
04 August 2023
You are already at the latest version
Abstract
Bimetallic (or multimetallic) catalysis has emerged as a powerful tool in modern chemical synthesis, offering improved reaction control and versatility. This review focuses on the recent de-velopments in bimetallic sequential catalysis for the synthesis of nitrogen heterocycles, essential building blocks in pharmaceuticals and fine chemicals. The cooperative action of two (and sometimes more) different metal catalysts enables intricate control over reaction pathways, enhancing selectivity and efficiency of N-heterocyclic compounds synthesis. By activating less reactive substrates, this multimetal catalytic strategy opens new synthetic possibilities for challenging compounds. The use of catalytic materials in bimetallic systems reduces waste and improves atom efficiency, aligning with green chemistry principles. With a diverse range of metal combinations and reaction conditions, bimetallic catalysis provides access to a broad array of N-heterocyclic compounds with various functionalities. This paper highlights the significant progress made in the past decade in this topic, emphasizing the promising potential of bimetallic catalysis in drug discovery and the fine chemical industries.
Keywords:
Bimetallic catalysis
; multimetallic catalysis
; sequential catalysis
; N-heterocycles
; transition metals
; green chemistry
1. Introduction
Catalysis plays a vital role in modern chemical synthesis, efficiently converting simple starting materials into valuable complex compounds. Bimetallic catalysis, involving two different metal species working together synergistically, has emerged as a powerful tool for various chemical reactions [1,2,3,4]. Sequential bimetallic catalysis represents a cutting-edge approach, enabling the synthesis of valuable compounds with higher efficiency, selectivity, and atom economy. It opens new synthetic possibilities for more complex compounds by activating and transforming substrates that may be unreactive or challenging for a single-metal catalyst, aligning with green chemistry principles, and reducing the formation of unwanted by-products [5].
Pioneering studies on transition-metal-catalyzed cross-coupling reactions, such as Suzuki, Heck, and Negishi reactions, laid the foundation for bimetallic catalysis by demonstrating selective coupling of different organic fragments [6]. Early investigations into tandem catalysis, performing consecutive reactions without intermediate isolation, inspired the concept of sequential bimetallic catalysis for increased efficiency and shorter reaction times. The cooperative action of two metal catalysts to activate a substrate showcased the potential of combining multiple metals for improved reactivity and selectivity. Cascade reactions, where bond-forming events occur in a one-pot manner, influenced the design of sequential bimetallic catalysis, promoting multiple cyclical reactions for complex molecule synthesis [3,7].
One particularly promising area of bimetallic catalysis is the synthesis of N-heterocycles, essential building blocks in numerous biologically active compounds and pharmaceuticals [8]. Traditional synthetic routes for N-heterocycles have some drawbacks, involving multiple steps, harsh conditions, and generating significant waste. Therefore, bimetallic catalysis offers several key advantages in N-heterocycle synthesis. In particular, the use of two different metal catalysts in tandem allows for intricate control over the reaction pathways, enhancing selectivity and improving yields of the desired N-heterocyclic products. This is particularly important in complex molecules where traditional methods may lead to competing side reactions.
Moreover, bimetallic catalysis facilitates the activation of less reactive substrates, enabling the synthesis of N-heterocycles that were previously challenging or unattainable through mono-metallic catalysis. This expands the synthetic toolbox, providing access to a broader range of N-heterocyclic structures. In addition, the efficient use of bimetallic catalysis reduces waste and increases atom efficiency, making the process more environmentally friendly and economically viable [3,7].
As the field continues to evolve, this review presents an overview of the recent developments concerning bimetallic sequential catalysis for the synthesis of N-heterocycles, which represents a promising avenue in catalysis.
2. Bimetallic Approaches to N,O-Aminals and Related Spiro N,O-Heterocycles
N,O-aminals are an interesting class of substituted molecules bearing a geminally N,O-substituted (stereogenic) carbon center, and have been recently recognized as an important class of building blocks in organic synthesis [9]. In the conventional approach, N,O-aminals are derived either from the corresponding α-amido sulfones via nucleophilic substitution, replacing the sulfonyl group with an alkoxy moiety [10,11,12,13,14], or from the corresponding imines by means of nucleophilic addition, introducing an alkoxy group [15,16,17,18,19,20,21,22,23].
While several methods exist for synthesizing these compounds, enantioselective examples are still limited, particularly for the construction of chiral tetrasubstituted carbon centers [21]. Moreover, some of these methods do come with certain disadvantages such as the requirement of harsh reaction conditions, such as the use of strong acids or high temperatures, which may lead to side reactions or limited substrate compatibility. Additionally, the formation of unwanted by-products and the potential for racemization can pose challenges, particularly in the synthesis of chiral N,O-aminals. Despite these drawbacks, researchers continue to explore new strategies to overcome these limitations and develop more sustainable and selective routes for N,O-aminal synthesis, where bimetallic catalysis has shown important perspectives [24].
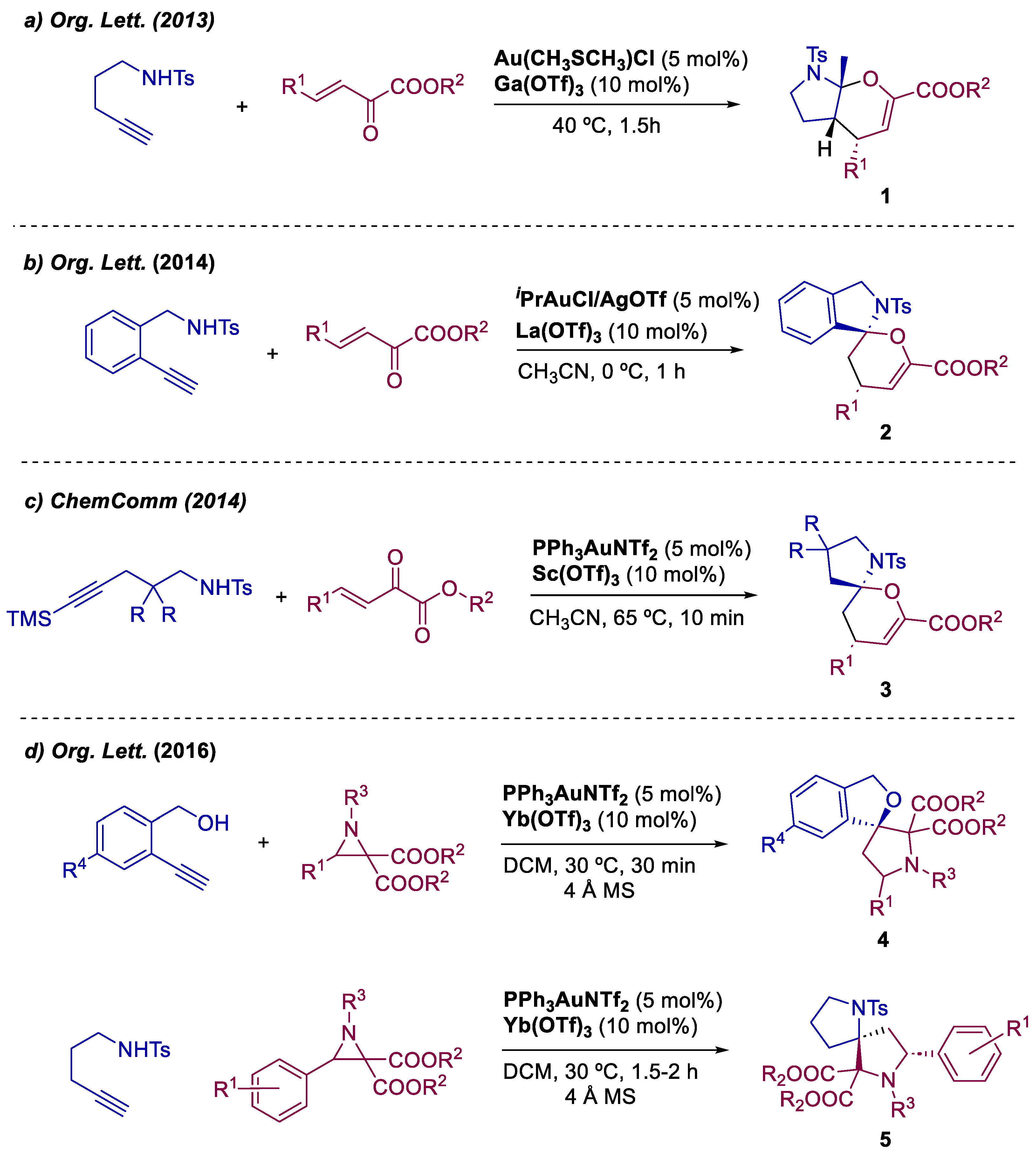
Xu and co-workers has been actively developing synthetic processes for fused bicyclic N,O-aminals and spiro-N,O-aminals. The authors focused on bimetallic catalysis under mild conditions, utilizing Au(I) as a catalyst and a metallic Lewis acid as a co-catalyst. In 2013, the group successfully achieved the synthesis of aromatic and allyl-substituted fused bicyclic aminals 1 through a Au(I)/Ga(III)-catalyzed [4+2] cycloaddition cascade reaction, with 13 examples scope and yields up to 90% (Scheme 1a) [25]. Subsequently, in 2014, the same group disclosed the synthesis of spiro-N,O-aminals 2 by employing a Au(I)/La(III)-catalyzed [4+2] cycloaddition bimetallic approach (Scheme 1b) [24]. To prevent inward isomerization of the generated enamide, a fused aromatic ring was introduced in the same alkyne amine, enabling the reaction with activated electrophiles and yielding spirocyclic products in 12 examples scope with yields up to 90%. In the same year, Xu's group successfully synthesized aromatic and allyl-substituted spiro-aminals 3 using a bimetallic Au(I)/Sc(III)-catalyzed [4+2] cycloaddition process (Scheme 1c) [26]. In this work, trimethylsilyl (TMS) was employed as a traceless controlling group to stabilize the derived enamide and inhibit its isomerization, resulting in a scope of 14 examples and yields up to 89%. Additionally, in 2016, Xu and co-workers reported the preparation of spiro-heterocycles through a Au(I)/Yb(III)-catalyzed diastereoselective [3+2] cycloaddition between aziridines and alkynyl substrates (Scheme 1d) [27]. Various N-protecting groups and aliphatic and aromatic alkynyl substrates were tested, resulting in a scope of substituted spiro-N,O-heterocycles 4 with 16 examples and yields up to 99%. Furthermore, by switching from alkynyl alcohols to alkynyl amides, a similar reaction with aziridines afforded aromatic-substituted spiro-N,N-heterocycles 5 with a scope of 9 examples and yields up to 80%.
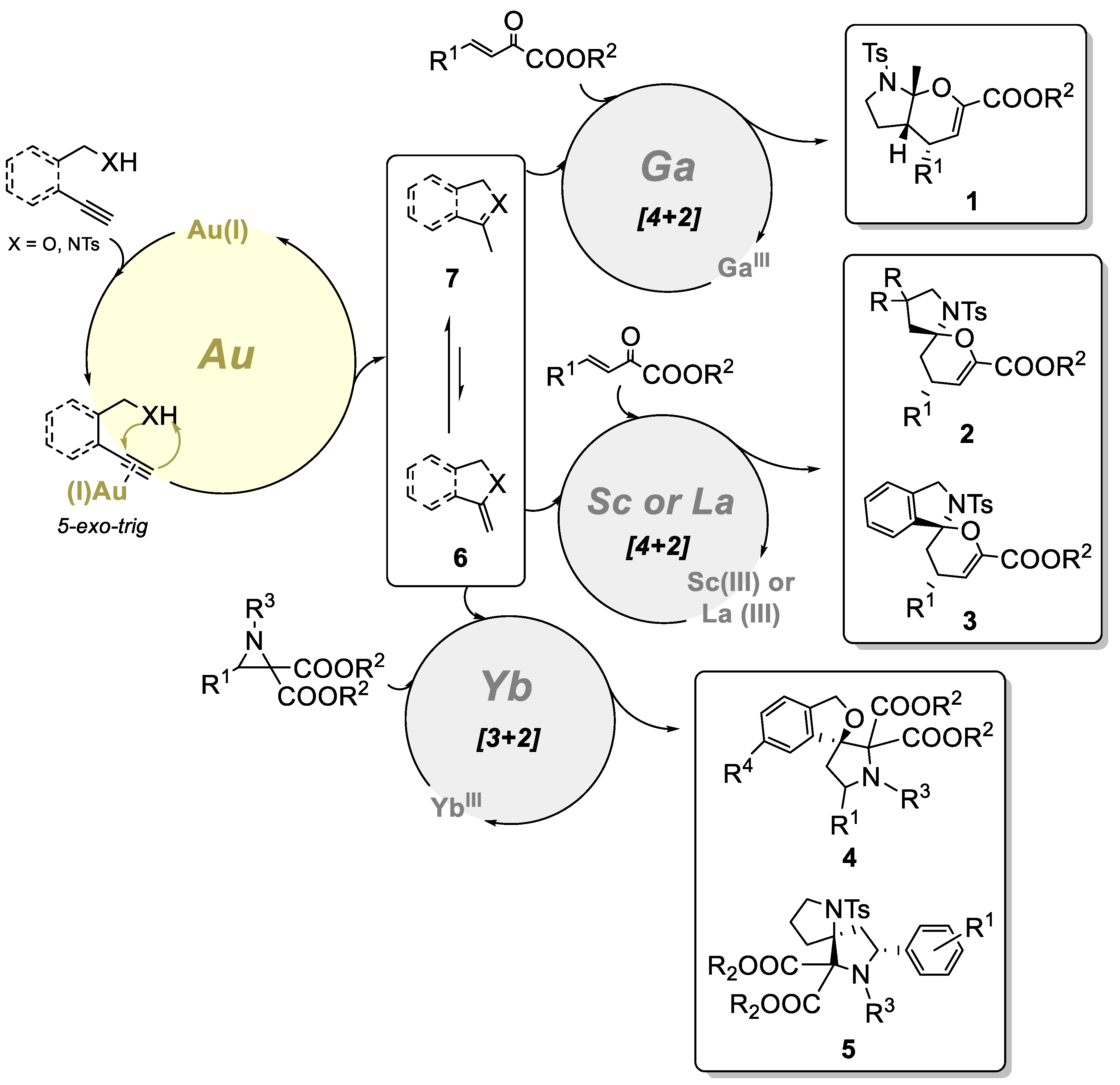
To gain insights into the reactions’ mechanisms (Scheme 2), Xu and co-workers conducted deuteration experiments that showed that the alkynyl substrate undergoes a Au(I)-catalyzed 5-exo-dig cyclization affording the enamide 6, followed by isomerization into another enamide 7. Although 7 is more stable, depending on the reaction conditions and the type or loading of the Lewis acid, either spiro or fused aminals can be afforded by a [4+2] cycloaddition. Thus, the Lewis acid activates the electrophile and generates the final product. When in contact with aziridines, enamide 6 can also undergo a Lewis acid-catalyzed [3+2] cycloaddition.
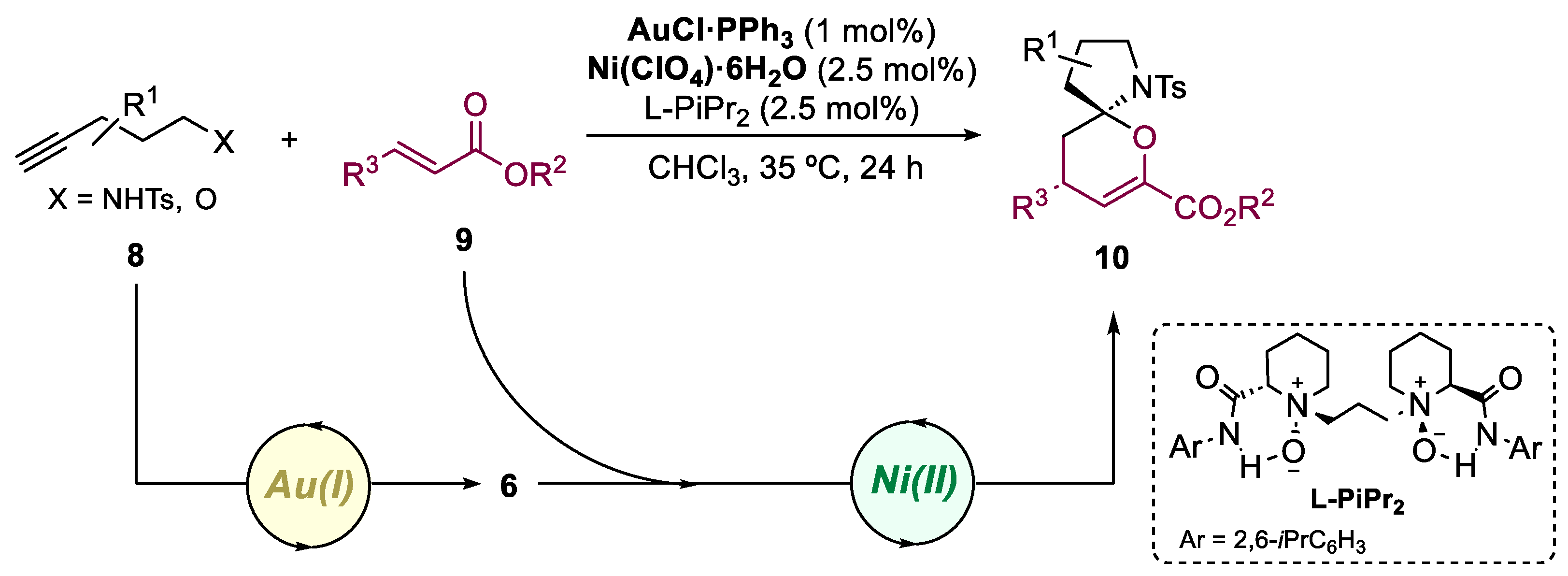
In 2016, Li and co-workers described an innovative asymmetric cascade reaction between alkynyl amides 8 and keto esters 9, employing a bimetallic catalytic system of achiral π-acid Au(I) and chiral Lewis acid N,N'-dioxide Ni(II) complex (Scheme 3) [28]. This approach enabled the synthesis of spiroaminals 10 with high yields (up to 99%), excellent enantioselectivity (over 99% ee), and moderate to high diastereoselectivity (19:1 d.r.) under mild reaction conditions. The bimetallic catalytic system facilitated the sequential activation of the carbonyl and alkyne moieties, with the N,N'-dioxide ligand playing a crucial role.
In 2016, another interesting bimetallic relay catalytic system was developed by the group of Xu, enabling the synthesis of oxazole derivatives from readily available N-(propargyl)-aryl amides and aldehydes under mild reaction conditions (Scheme 4) [29]. The system consists of Zn(OTf)2 and Sc(OTf)3, which act as a π acid and a σ acid, respectively. The reaction proceeds through a cascade of reactions, beginning with an intramolecular 5-exo-dig cyclization of alkynyl amide 11 catalyzed by Zn(OTf)2. Simultaneously, Sc(OTf)3 coordinates with the carbonyl group of the aldehyde 12, promoting the subsequent carbonyl-ene reaction with the oxazoline intermediate 13, yielding the desired oxazole product 14. This method showed a great atom-economy and straightforwardness, possessing the potential for applications in organic synthesis and medicinal chemistry.
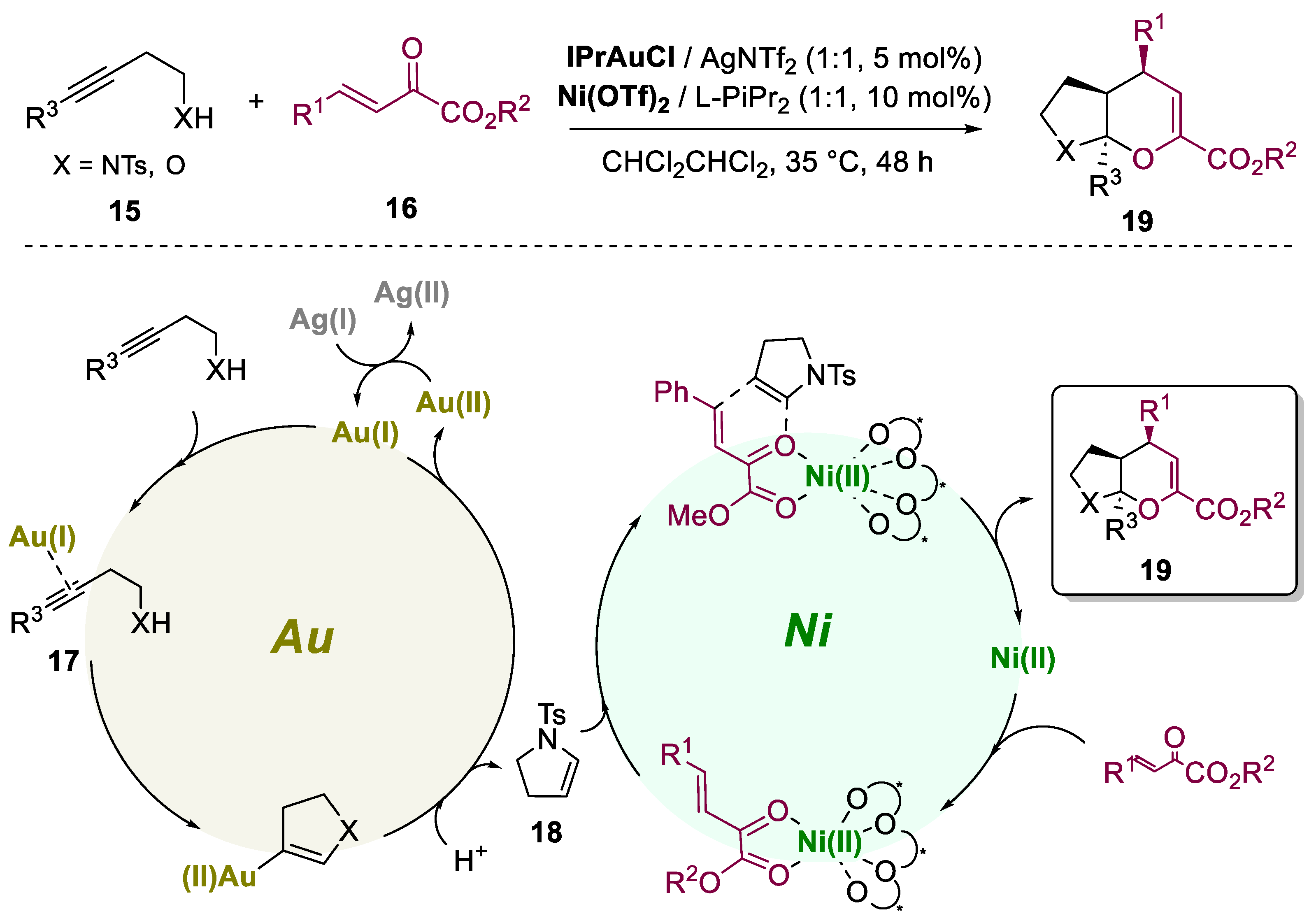
A similar work was reported by Feng's group in 2018 [30]. In this work, a similar efficient catalytic asymmetric cyclization reaction (Scheme 5) was conducted by a bimetallic system that involved an achiral Au(I) catalyst and the same chiral N,N'-dioxide ligand/Ni(II) catalyst. This reaction also involved the cyclization of alkyl amides or alcohols 15 with, β,γ-unsaturated α-ketoesters 16 leading to the formation of fused bicyclic N,O-acetals or O,O-acetals. The researchers employed a 5-endo-dig cyclization process based on Baldwin's rules, resulting in a cycloalkene intermediate capable of reacting with β,γ-unsaturated α-ketoesters to form the desired fused bicyclic products through an inverse-electron-demand hetero-Diels-Alder (IEDHDA) reaction. The optimal conditions for the reaction were determined, yielding products with good yields (77-99%) and excellent enantioselectivities (96-99% ee). The reaction tolerated various substrates, including those with 2-naphthyl and heteroaromatic groups. The substrate scope was expanded to alkynyl alcohols and alkynyl amines, yielding the desired fused bicyclic N,O-acetals with excellent yield and enantioselectivity. Based on control experiments, a reaction mechanism is proposed: initially, the gold catalyst coordinates with the alkynyl substrate 15 to form a p-gold-alkyne complex 17 and, in situ, generated the key cyclic intermediate 18. On the other hand, chiral Lewis acid Ni(II)/L-PiPr2 activated b,g-unsaturated a-ketoester 16 reacts with 18, giving the fused bicyclic acetal 19.
3. Bimetallic Approaches Involving Indole Scaffolds
Indoles and indolines are of great importance in medicinal chemistry, pharmaceuticals, and natural product synthesis due to their involvement in various biological processes and structural significance. Several common methods for synthesizing indoles, including the Fischer indole synthesis, Bischler-Möhlau indole synthesis, Madelung indole synthesis, Reissert indole synthesis, and Buchwald-Hartwig amination, have been widely employed [31]. However, some traditional routes suffer from drawbacks such as harsh reaction conditions, multistep processes, limited substrate scope, and regioselectivity issues, which can lead to the formation of unwanted by-products or the need for expensive catalysts. Thus, bimetallic catalytic routes that involve the formation of an indole intermediate or product has emerged as a desirable approach.
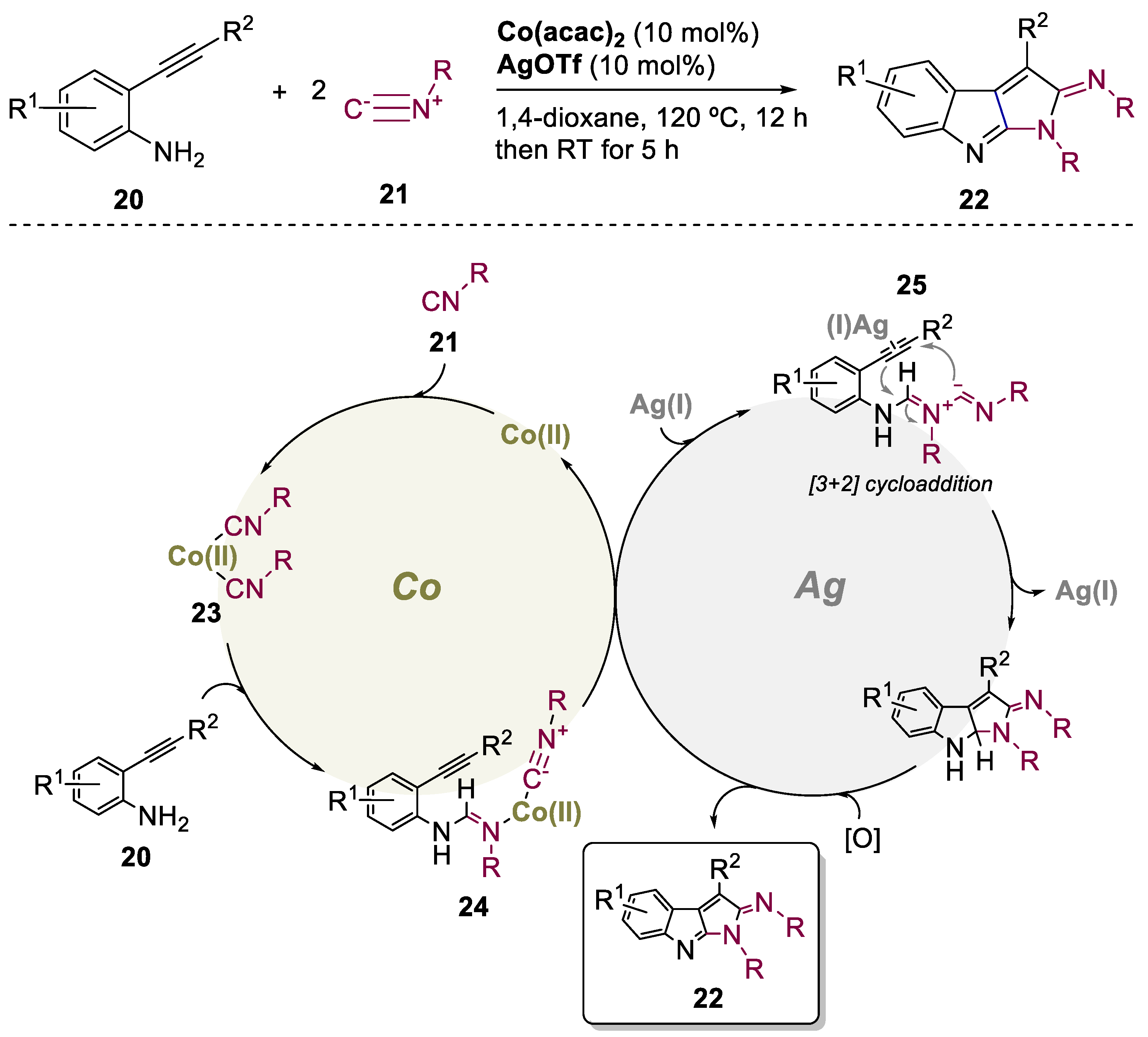
The direct formation of pyrrolo[2,3-b]indoles via catalyzed bicycloaddition reactions is a very attractive yet challenging process. Isocyanides happen to be a simple group and, taking advantage of their electrophilicity, can be inserted either by Lewis acid [32], or transition metal catalysis, undoubtedly with palladium as the most popular choice [33,34,35]. In contrast, Gao and co-workers, in 2015, explored the preparation of pyrrolo[2,3-b]indoles by an inexpensive Co(II)-enabled process (Scheme 6) [36]. The authors demonstrated that the combination of Co(acac)2 and AgOTf promoted a bimetallic relay catalysis reaction between 2-ethynylanilines 20 and isocyanides 21, allowing access to new densely functionalized pyrrolo[2,3-b]indoles 22. Overall 26 examples were reported, in yields up to 86%. The authors suggested that the reaction pathway involves a Co(II)-catalyzed double isocyanide insertion followed by a Ag(I)-catalyzed 1,3-dipolar cycloaddition. A suitable mechanism for the formation of pyrrolo[2,3-b]indoles can be described by a ligand exchange, affording the intermediate complex 23, which activates the electrophile isocyanides, a key step in this process. It then undergoes an N-H insertion to obtain the enyne-imine species 24, detected using GC-MS. Then, a second migratory insertion affords the 1,3-dipole 25 and regenerates the Co(II) catalyst. The presence of Ag(I) allows an intramolecular 1,3-dipolar cycloaddition followed by dehydrogenation under air conditions, that leads to the desired thermodynamically stable pyrrolo[2,3-b]indole 22.
Scheme 6.
General conditions and a plausible mechanism for the preparation of pyrrolo[2,3-β]indoles by a Co(II)/Ag(I)-catalyzed bimetallic approach.
Scheme 6.
General conditions and a plausible mechanism for the preparation of pyrrolo[2,3-β]indoles by a Co(II)/Ag(I)-catalyzed bimetallic approach.
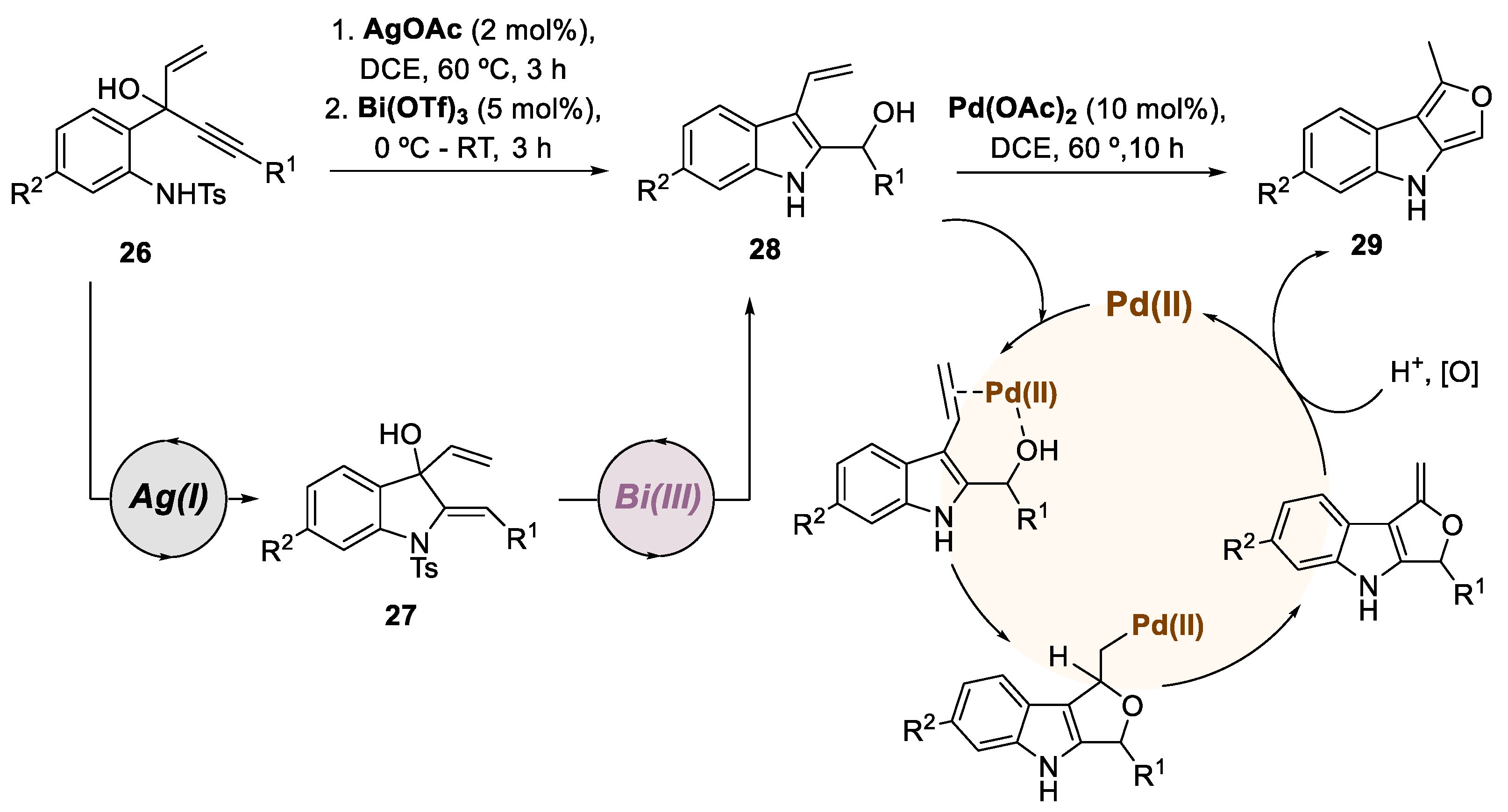
In 2016, Ramasastry and co-workers published two research works involving indole synthesis and subsequent transformation through a trimetallic catalytic system. In the first study, the authors reported a successful three orthogonal metal relay catalytic system for preparing furo[3,4-b] indoles (Scheme 7) [37]. This approach utilized a sequential Ag(I)/Bi(III)/Pd(II) trimetallic catalysis, demonstrating its versatility with 10 examples and yields up to 57%. The preparation of cyclopenta[b]indoles was also investigated using a one-pot Ag(I)/Brønsted acid catalysis from 3-(2-aminophenyl)-4-pentenyn-3-ols, although not involving a multimetallic catalytic approach. The reaction mechanism involved Ag(I)-catalyzed 5-exo-dig cyclization of substrate 26 to generate intermediate 27, followed by Bi(III)-catalyzed 1,3-allylic alcohol isomerization leading to 38. Finally, a Pd(II)-catalyzed intramolecular etherification through a 5-exo-trig cyclization under oxidative conditions resulted in the desired furo[3,4-b]indole 29.
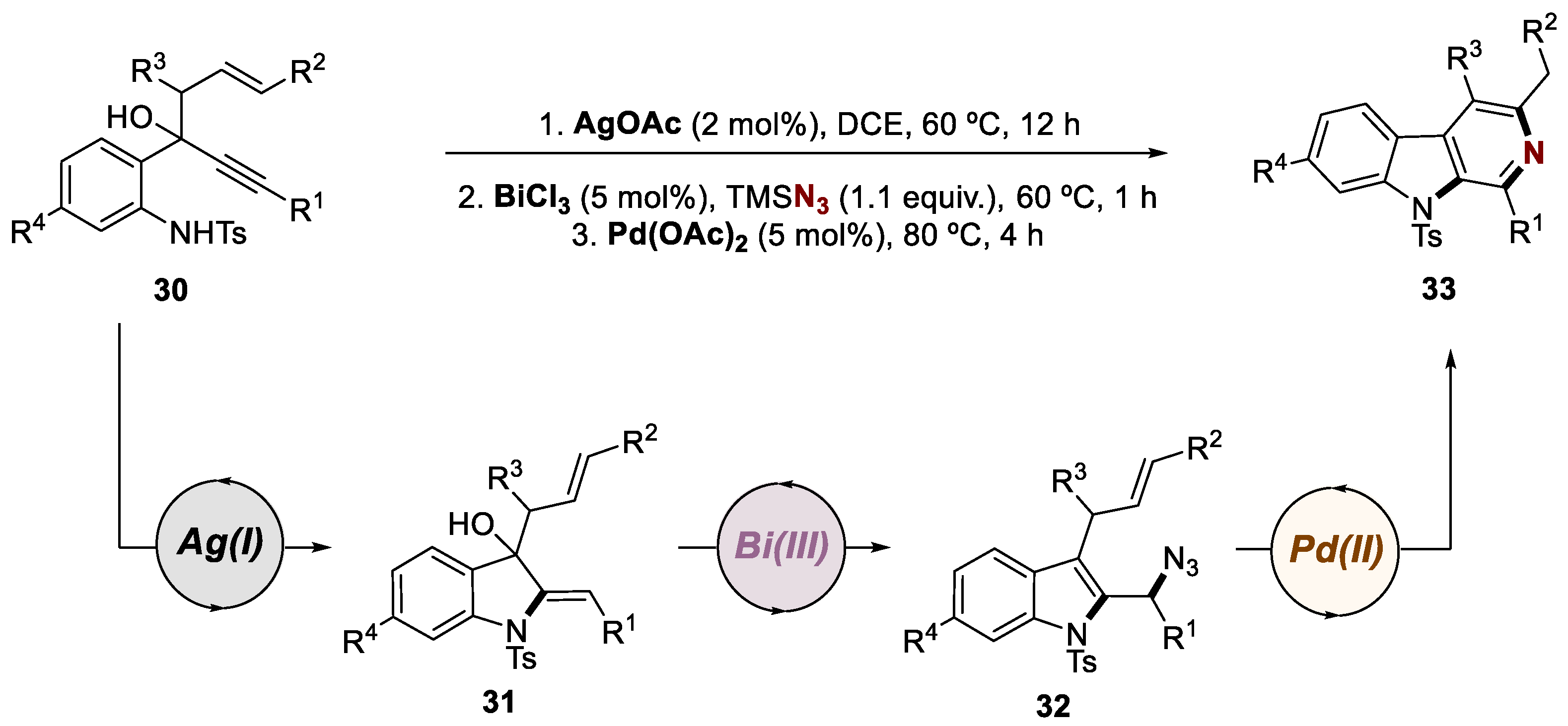
In the second work of 2016, Ramasastry and co-workers presented a similar one-pot triple-orthogonal-metal relay catalysis strategy for synthesizing 1,3-di- and 1,3,4-trisubstituted β-carbolines, also employing silver, bismuth, and palladium catalysts in a sequential manner (Scheme 8) [38]. The synthetic pathway included intramolecular hydroamination, Friedel–Crafts-type dehydrative azidation, and a unique annulation step leading to the formation of the pyridine ring. Starting from 30, they achieved indoline intermediate 31 through a Ag(I)-catalyzed 5-exo-dig cyclization and protodemetalation. A subsequent Bi(III)-promoted cascade reaction involving 1,3-allylic alcohol isomerization and nucleophilic azidation led to the formation of azide 32. This intermediate underwent a Pd(II)-mediated aziridine formation, followed by deprotonation, ring opening of the aziridine, and aromatization, resulting in the desired substituted N-heterocycle 33. This innovative approach provided access to a diverse range of distinct β-carbolines, offering new possibilities for the synthesis of these valuable compounds.
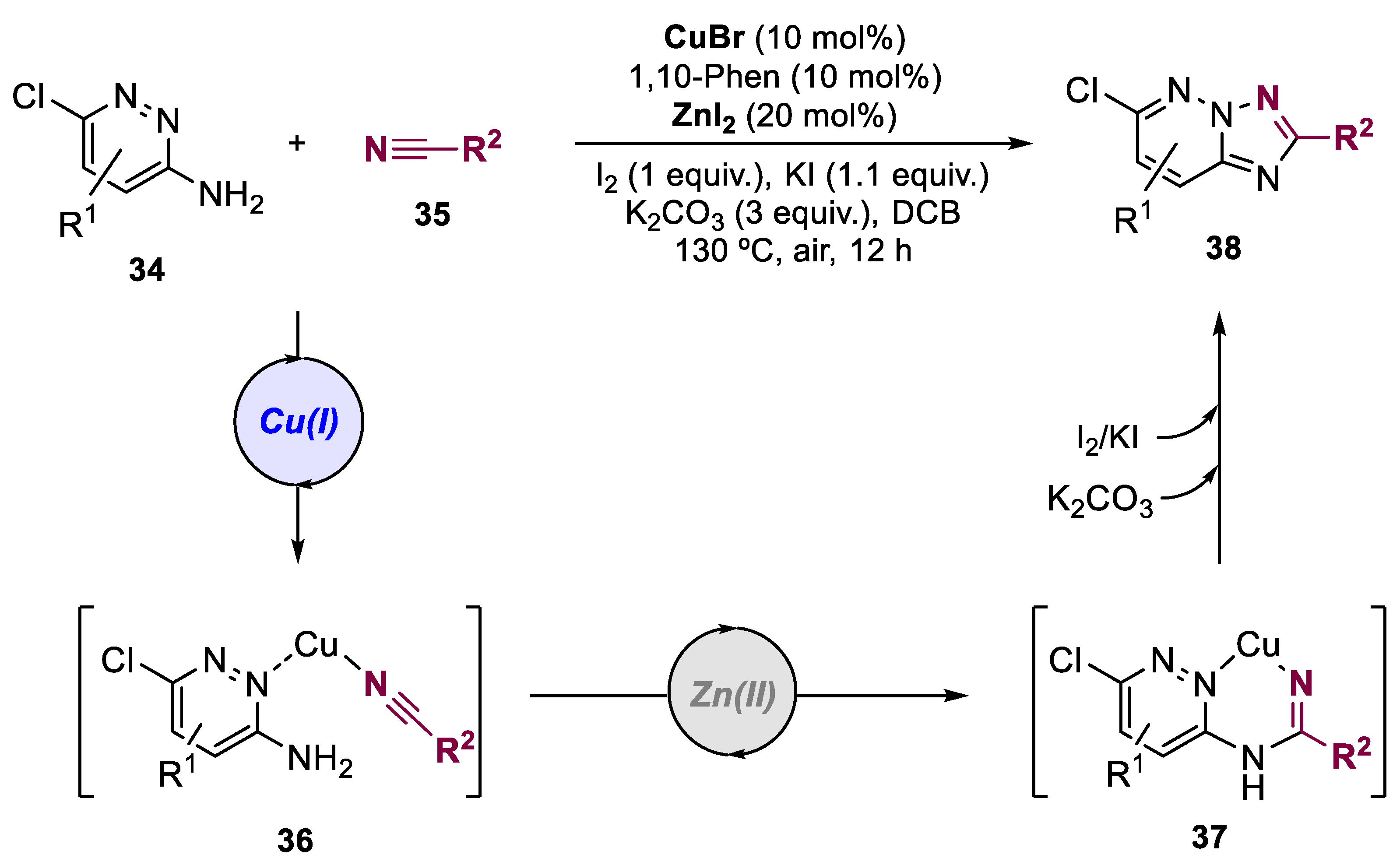
In 2017, Mu and co-workers reported a new and efficient protocol for the preparation of chlorine-containing 1,2,4-triazolo[1,5-b]pyridazine scaffolds (Scheme 9) [39]. The authors developed a one-pot oxidative cycloaddition reaction of 3-aminopyridazine derivatives 34 and nitriles 35, involving cooperative Cu(I) and Zn(II)-catalyzed tandem C-N addition to achieve intermediate 36 followed by the amidine 37. Then, a I2/KI-mediated intramolecular oxidative N-N bond formation affords the final 1,2,4-triazolo[1,5-b] pyridazine derivatives 38.
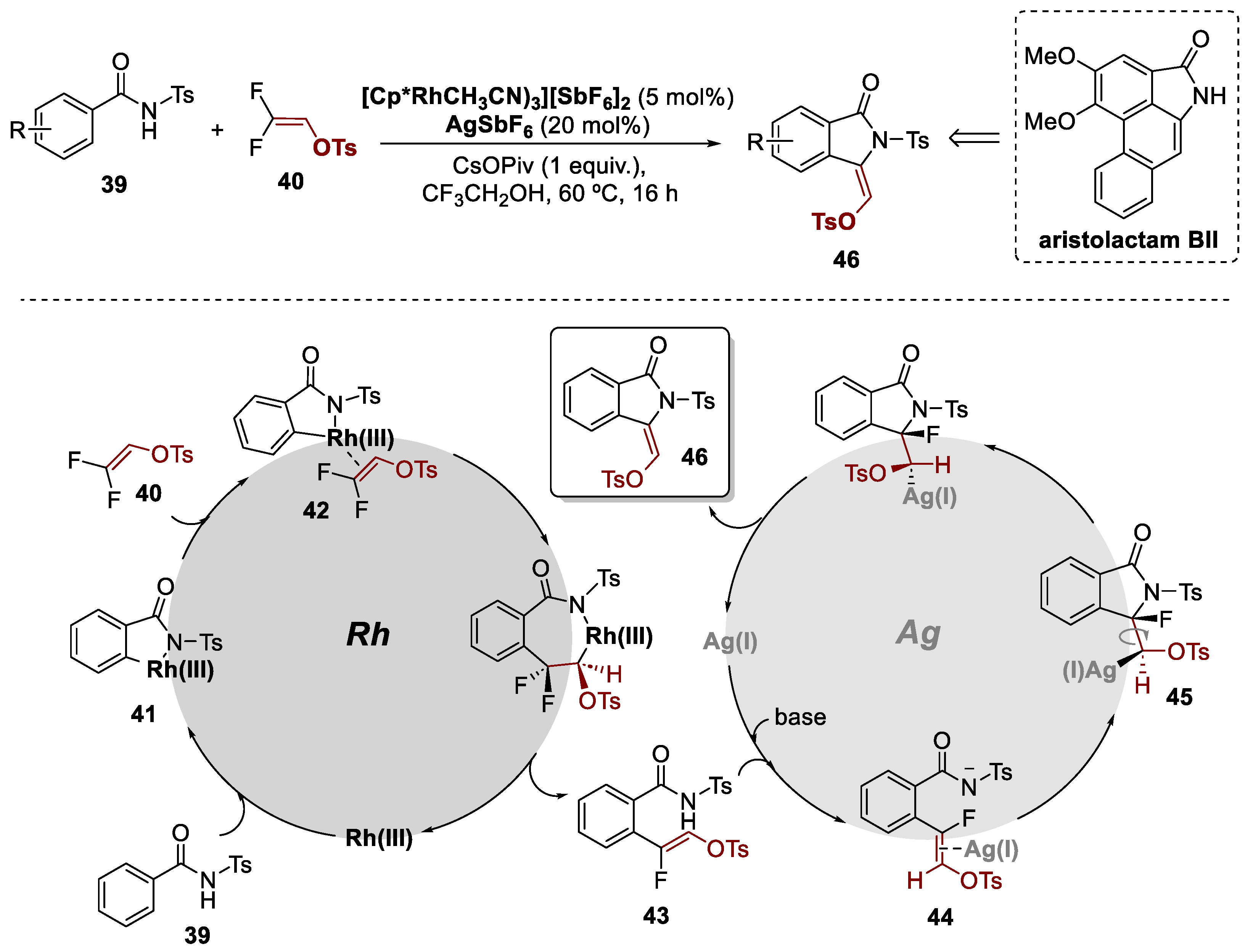
In 2017, Wang and co-workers reported the construction of 3-alkylidene isoindolinones. This was efficiently achieved through a redox-neutral bimetallic Rh(III)/Ag(I) relay catalysis between N-tosyl benzamides 39 and 2,2-difluorovinyl tosylate 40 (Scheme 10) [40]. The Rh(III) catalyst facilitates the C-H monofluoroalkenylation reaction, while the Ag(I) salt acts as an activator for the subsequent cyclization step. According to the authors, the mechanism starts with the Rh(III)-catalyzed C-H activation in N-tosylbenzamide 39, assisted by the NTs group, generating intermediate 41. The subsequent coordination of 40, that leads to intermediate 42, underwent a regioselective olefin insertion, followed by an anti-coplanar β-F elimination, resulting in the Z-type monofluoroalkenylation product 43 with notable stereoselectivity. The Ag(I) salt was suggested to act as a π acid, promoting the activation of the olefin (44) and facilitating the intramolecular cyclization reaction. Consequently, the anti-addition to the double bond induced 5-exo cyclization, resulting in the formation of intermediate 45. The selective attack at the α-position of the fluorine atom was attributed to the low-lying LUMO with a significant coefficient at this position. Finally, a stereospecific formation of the E-type 3-alkylidene isoindolinone product 46 was achieved through an anti-coplanar β-F elimination process. The methodology described in this study was used to rapidly synthesize aristolactam BII, a natural product with potential pharmaceutical applications. The results demonstrate the potential of difluorovinyl tosylate and Rh(III)/Ag(I) relay catalysis for the efficient synthesis of a variety of biologically active compounds.
X. Feng and co-workers reported a highly efficient asymmetric cascade reaction of alkenyloxindoles with pyridines and diazoacetates via a bimetallic iron(III)/chiral N,N′-dioxide-scandium(III) complex catalyst [41]. Tetrahydroindolizines were obtained with good to excellent diastereo- and enantioselectivities.
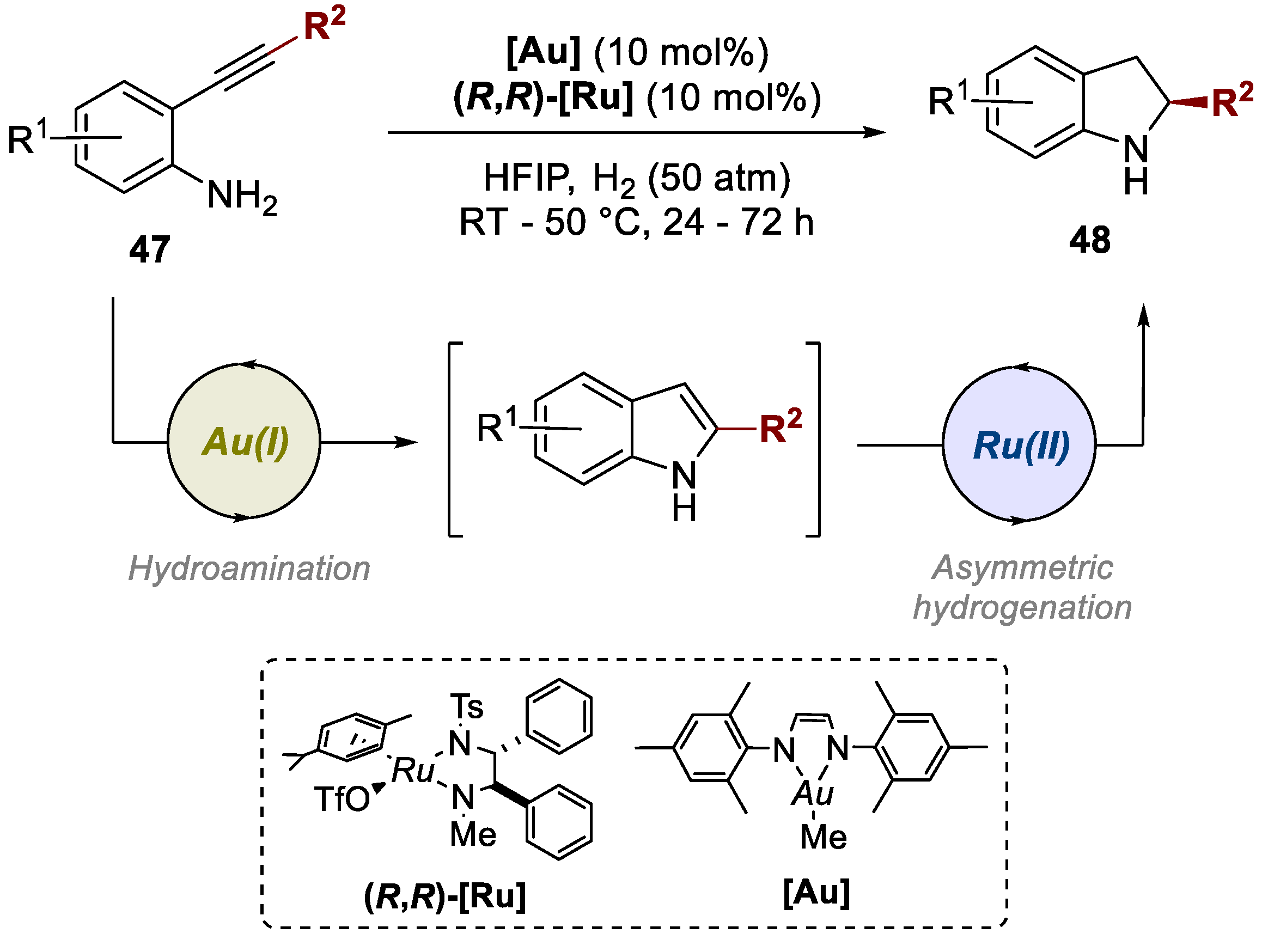
In 2019, Fan and co-workers published a one-pot cascade reaction strategy to afford functionalized indolines. This process involved a sequential Au(I)-catalyzed intramolecular hydroamination followed by a Ru(II)-catalyzed asymmetric hydrogenation of various anilino-alkynes 47 (Scheme 11) [42]. This enabled the access to chiral indolines 48. Optimal reaction conditions were determined, and the reactions proceeded smoothly, achieving full conversions, with high yields (up to 98%) and moderate to excellent enantioselectivities (up to 97% ee). This work was also extended to chiral 1,2,3,4-tetrahydroquinolines, although only requiring one ruthenium catalyst, and not a bimetallic approach.
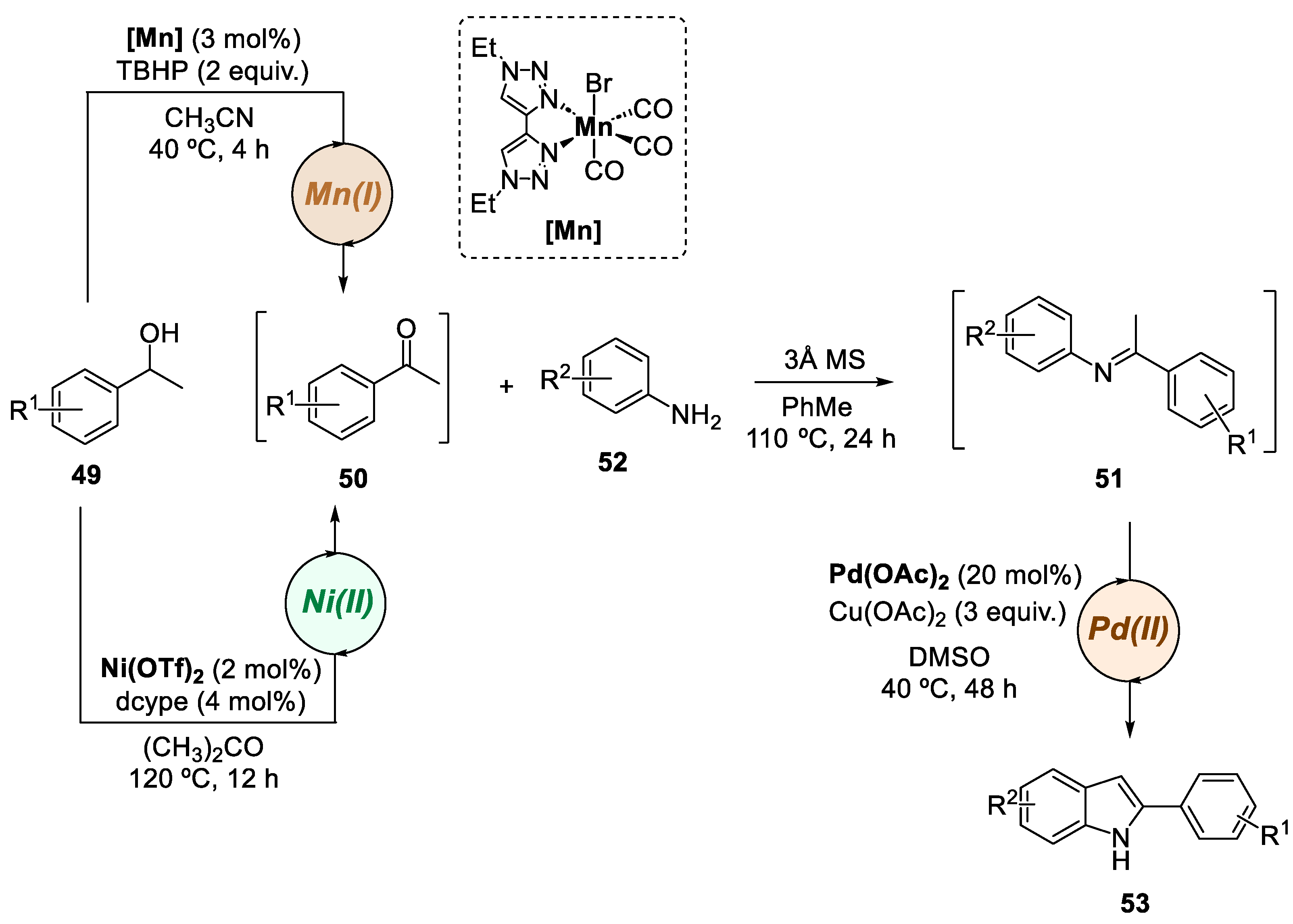
Marques and co-workers have recently disclosed a novel one-pot bimetallic catalytic approach for the synthesis of indole derivatives, using secondary alcohols and anilines as starting materials (Scheme 12) [8]. A commercially available nickel catalyst combined with a simple phosphine was investigated for the dehydrogenation of alcohol 49, while a phosphine-free manganese complex was also synthesized to achieve this oxidation step. Both systems were studied to obtain the desired ketone 50, which was subsequently converted to an imine 51 through condensation with an aniline 52, followed by an in-situ palladium-catalyzed oxidative cyclization. This system achieved several 2-arylindoles 53, with a 3-step synthetic pathway and overall yields of up to 45%. This process had the advantage of avoiding the isolation of sensitive intermediates and presented a sustainable pathway for the preparation of functionalized indoles.
4. Bimetallic Approaches Involving Lactam Scaffolds
The β-lactam is the common core structure of clinically used drugs such as penicillin, cephalosporin, and monocyclic antibiotics such as aztrenam [43,44,45,46]. Development of novel methods to access new β-lactams is important in further studies of this heterocyclic system, including structure-activity relationships and the discovery of new antibiotics, and is thus important in global health problems. Synthetic methods that construct the β-lactam ring have been developing exorbitantly over the last century from almost every imaginable set of synthons. Yet, there is still a necessity for innovation and improvement in the field, even within each category of well-established reactions [47]. Despite all of the synthetic methods to date for obtaining achiral or racemic lactams, asymmetric methodologies remain largely limited to chiral auxiliary-based systems [48]. This review presents below two examples of asymmetric bimetallic catalytic approaches to afford substituted β-lactams.
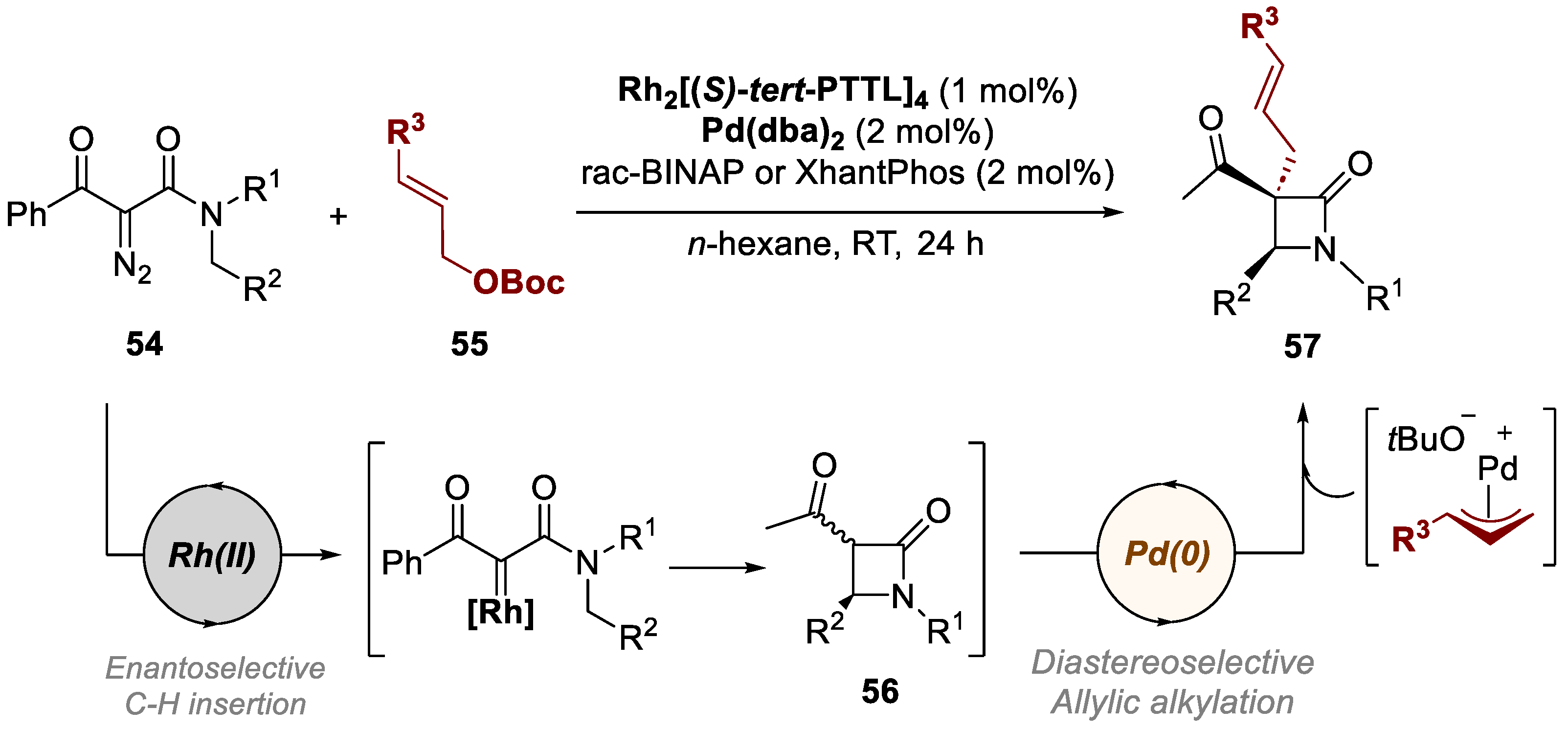
In 2018, Lee and co-workers developed a novel asymmetric dual Rh(II)/Pd(0) relay catalysis for synthesizing α-quaternary allylated chiral β-lactams, by reacting N-benzyl α-diazoamides 54 and allyl tert-butyl carbonates 55. (Scheme 13) [49]. The experiments conducted in this work supported a relay reaction with the formation of β-lactam intermediate 56 that resulted on the desired α-quaternary allylated chiral β-lactams 57. Optimization showed that nonhalogenated and nonpolar solvents yielded superior results compared to halogenated solvents. Different electron-donating and withdrawing groups on the phenyl ring were well-tolerated. The position of substituents on the aromatic ring affected the reaction, with ortho substituents exhibiting reduced reactivity due to steric hindrance. Heteroaromatic rings and naphthyl rings demonstrated favorable performance. This method provided a wide scope, achieving yields up to 99% and with good diastereomeric ratios (up to >91:1 dr) and enantioselectivities (up to 98% ee). Furthermore, by varying the allylic substrates, given the widespread availability of the diazo compounds and allyl carbonates, this asymmetric dual relay catalysis strategy may be a cornerstone for many new reactions exploiting multimetallic transformations of Rh(II) carbenoids and π-allyl Pd(II) complexes.
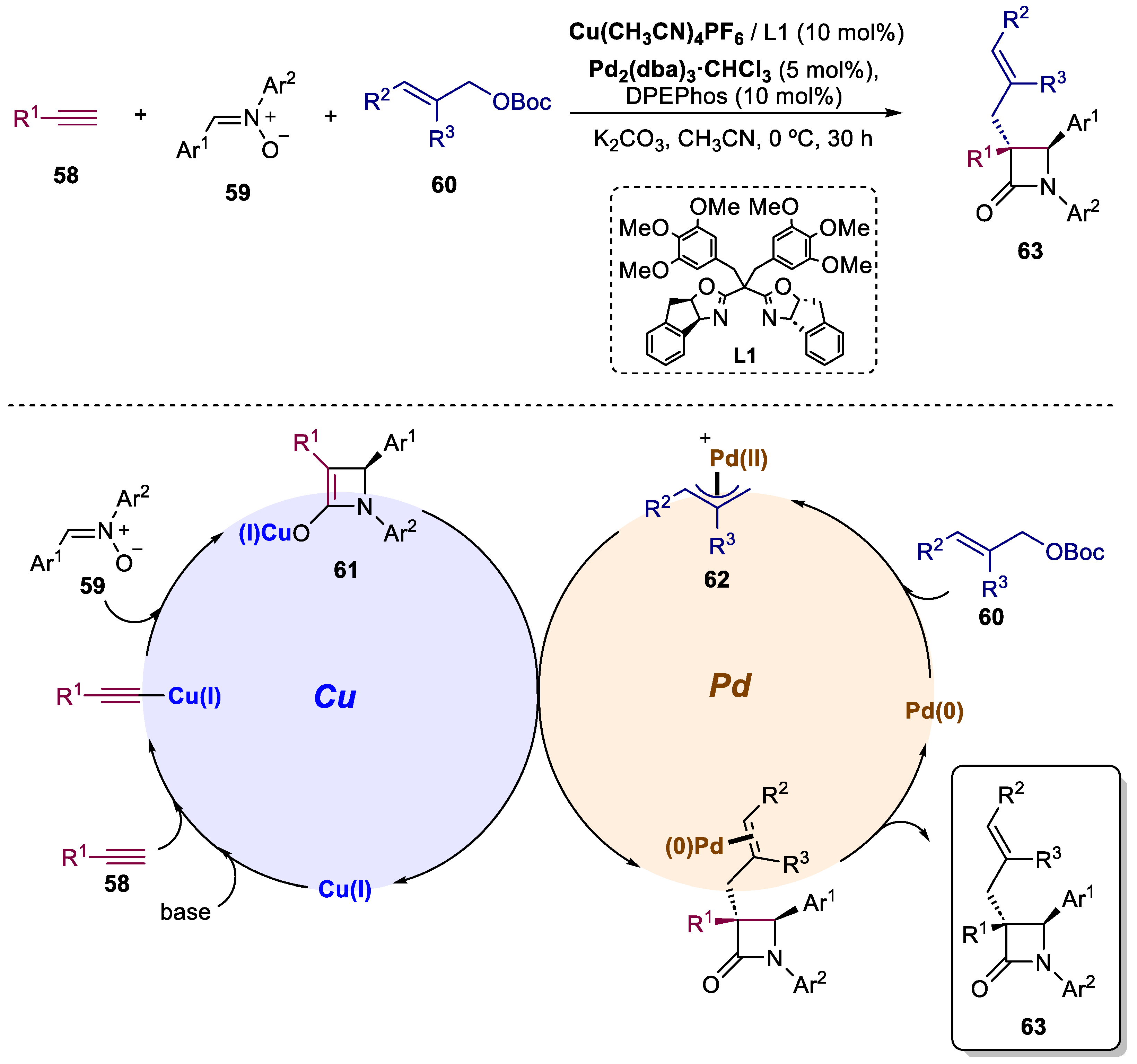
Xu and co-workers have successfully disclosed a groundbreaking asymmetric multicomponent reaction: the interrupted Kinugasa allylic alkylation (Scheme 14) [50]. This methodology synergistically merges a copper-catalyzed Kinugasa system with a palladium-catalyzed allylic alkylation. This remarkable reaction enables the synthesis of α-quaternary chiral β-lactams from simple and readily available alkynes 58, nitrones 59, and allylic carbonates 60, with high yield (up to 87%) and excellent stereoselectivity (up to 96:4 er). The most plausible mechanism can be initiated with the cycloaddition of Cu(I) acetylide and nitrones, a pivotal chiral four-membered enolate Cu(I) intermediate (61) is formed. Simultaneously, the palladium catalyst reacts with the allylic electrophile, resulting in the creation of an allylic palladium intermediate (62). Subsequent stereo-controlled allylic substitution between 61 and 62 leads to the desired α-quaternary chiral β-lactams 63, while concurrently regenerating both the Cu(I) and Pd(0) catalysts. This one-pot approach is distinguished by a well-programmed reaction sequence, highly efficient formation of multiple bonds in asymmetric multicomponent reactions, and the construction of medicinally important α-quaternary chiral β-lactams, thus anticipating its utility in the synthesis of other biologically attractive molecules. Furthermore, in 2023, the same research group reported a novel bimetallic system, substituting the palladium with an iridium catalyst, capable of accomplishing the synthesis of the same type of compounds [51].
5. Bimetallic Approaches Involving Triazole and Tetrazole Scaffolds
Triazoles and their derivatives are important N-heterocyclic scaffolds in medicinal chemistry, presenting significant biological properties such as antimicrobial, antiviral, antitubercular, anticancer, antioxidant and anti-inflammatory activities, among others [52]. The wide range of bioactivity displayed by these N-heterocycles has stimulated the development of many synthetic strategies.
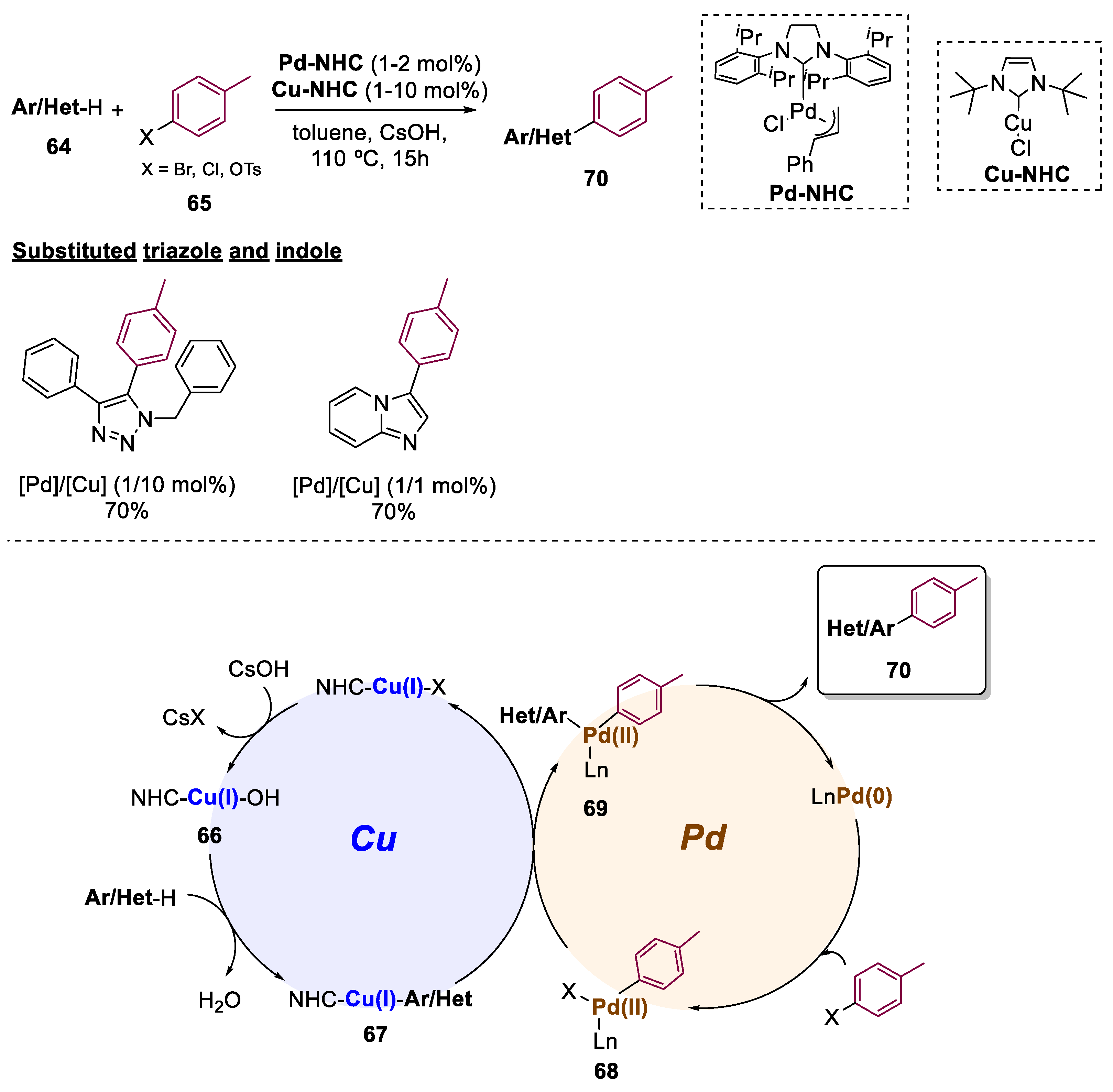
In comparison to the more established cross-coupling reactions, C−H activation is becoming a hot topic in all areas of chemistry, dismissing the pre-functionalization of both coupling partners [53]. Considering the inferior reactivity of arenes, when compared to aryl halides, their selective and direct arylation remains a challenge. These issues can be addressed by the presence of directing groups on the arene substrate, however, may provide a limited scope since the directing group strictly facilitates the activation in the ortho-positioned C–H bond. In this context, in 2014, Cazin and co-workers developed a process that promotes the construction of C–C bonds through an intermolecular direct arylation, that eliminated the need for directing groups (Scheme 15) [54]. The scope of this reaction included the functionalization of N-heterocycles, respectively, triazole and indole rings. The authors reported a novel Cu/Pd bimetallic catalytic system to promote C-H activation from arenes or heteroarenes 64 using aryl halides 65. Both Pd and Cu complexes were composed of imidazole-based ligands (Pd/Cu-NHC), with a scope constituted by 20 examples and yields of up to 98%. The authors performed mechanistic studies that allowed them to propose a catalytic cycle, firstly including the formation of the hydroxide [Cu(OH)(NHC)] (66) by transmetallation involving CsOH. Then, an acid-base reaction promoted the C–H activation of the heteroaryl, producing 67 and H2O. The transmetallation between 67 and the Pd(II) catalyst 68 leads to the regeneration of the Cu(I) catalyst and the Pd(II) intermediate 69. Finally, product 70 is released after reductive elimination and the Pd(0) catalyst is regenerated, thus completing the catalytic cycle.
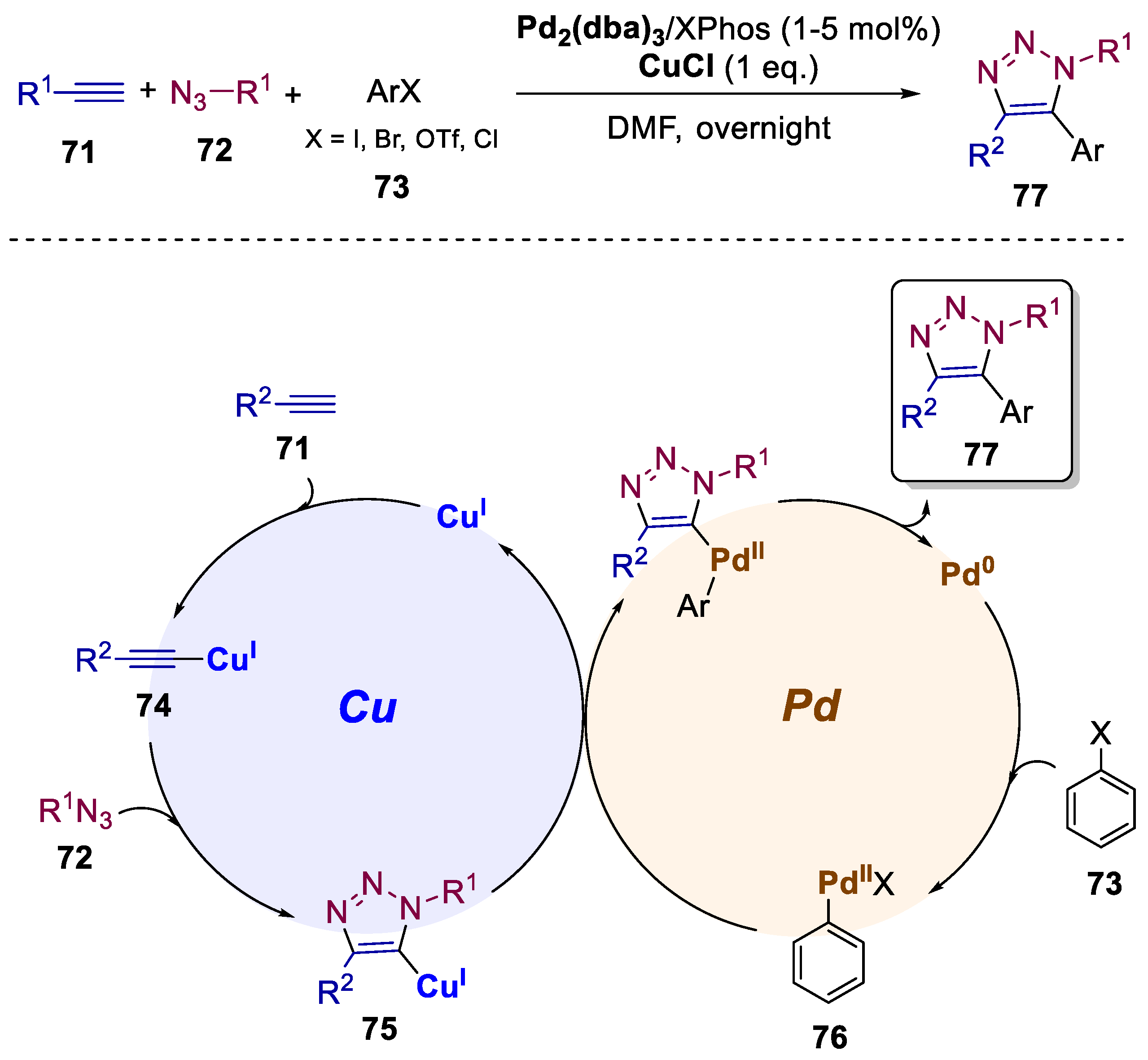
A crucial limitation of the famous click reaction remains the difficulty to obtain fully substituted 1,2,3-triazoles from internal alkynes as substrates, owing to the increased energy barrier and difficulty in regiocontrol, particularly for intermolecular reactions [55]. Nevertheless, in 2015, Xu and coworkers successfully synthesized 1,4,5-trisubstituted 1,2,3-triazoles by relying on a Cu/Pd transmetallation relay catalysis, a modular synthesis that afforded trisubstituted triazoles from the reaction between alkynyl substrates 71 with azides 72 and aryl halides 73 (Scheme 16) [56]. This reaction makes it possible to freely install three different substituents onto the triazole ring in one step, with a total of 33 examples with yields up to 98%. The most plausible mechanism starts with the cycloaddition of Cu(I) acetylide 74 with azide and generates cuprate−triazole intermediate 75. Simultaneously, oxidative addition of aryl halide to Pd(0) catalyst forms the palladium intermediate 76. The transmetalation reaction between 75 and 76, followed by reductive elimination produced the target trisubstituted triazole 77 and regenerate the Pd(0) catalyst.
Imidazo[1,2-a]pyridine constitutes a valuable skeleton for a variety of pharmaceuticals [57,58]. Hence, different strategies for the preparation of these scaffolds have been implemented [59,60,61]. Although there is already a report bearing a Cu-catalyzed process to obtain 1,2,3-triazole-fused imidazo[1,2-a]pyridines, it relies on brominated imidazo[1,2-a]pyridines as substrates [62]. To optimize the atom economy and environmental aspects of this process, Fan and co-workers reported, in 2016, an efficient one-pot synthesis of 1,2,3-triazole/quinoline-fused imidazo[1,2-a]pyridines starting from 2-(2-bromophenyl)imidazo[1,2-a]pyridines 78, alkynes 79, and sodium azide (Scheme 17) [63]. This involved a cascade one-pot bimetallic Cu/Pd relay-catalyzed process combining azide-alkyne cycloaddition, C-N coupling, and cross-dehydrogenative C-C coupling. They built a scope of different alkynes and 2-(2-bromophenyl)imidazo[1,2-a]pyridines with a total of 24 examples and yields up to 74%. Fan suggested that the mechanism started with a Cu(I)-catalyzed azide−alkyne cycloaddition to afford intermediate 80. 81 is then formed by a copper-hydrogen exchange. C−N coupling between 78 and 81 results in the formation of the key intermediate 82. In the second phase of this cascade process, aromatic palladation of 82 by a sequence of C-H bond cleavage yields the seven-membered palladacycle 83. Finally, reductive elimination affords the final 1,2,3-triazole/quinoline-fused imidazo[1,2-a]pyridine 84 and Pd(0) is re-oxidized into Pd(II) by Cu(II)/atmospheric O2.
In 2018, Sawant and colleagues developed a fast and efficient method to produce aminotetrazoles (Scheme 18) [64]. The authors used aryl azides 85, isocyanides 86, and TMSN3 in a sequential Pd(0)/Fe(III) catalyzed reaction. The process involved a Pd-catalyzed reaction to generate carbodiimide 87 in situ, which then reacts with TMSN3 in the presence of FeCl3, all in one pot, and finally yields the respective aminotetrazole 88. This approach has advantages over traditional methods that use toxic Hg and Pb salts in large amounts. With the optimized conditions in hand, they investigated the reaction's versatility showing various aryl azides with different substituents, including electron-donating and electron-withdrawing groups, which reacted well with different isocyanides and TMS-N3 to produce the corresponding 5-amino-1H-tetrazoles. Substituents at all three positions of the aryl azides were well-tolerated, even ortho-substituents that usually pose steric hindrance. Alkyl-, cycloalkyl-, and aryl-substituted isocyanides reacted successfully under the optimized conditions, obtaining a scope with 19 examples and yields of up to 90%, although aryl isocyanides with electron-donating groups and aliphatic azides did not react under the standard conditions.
6. Bimetallic Approaches Involving Pyridine, Pyrimidine, and Related Scaffolds
Isoquinolinones can be prepared via metal-catalyzed C-H activation [65,66,67]. However, this process bears the requirement of internal/external oxidants and, in the case of 3,4-disubstituted isoquinolinones, poor diastereoselectivity is encountered. To overcome this limitations, in 2013, Wang and co-workers disclosed the first redox-neutral ReI/MgII cocatalyzed [4+2] annulation of benzamides 89 and alkynes 90 via C-H/N-H functionalization to afford both cis- and trans-3,4-dihydroisoquinolinones (91 and 92, respectively) in a highly diastereoselective fashion (Scheme 19) [68]. This was accessed in a simply by subtle tuning of reaction conditions, which adds further values to this bimetallic catalyst system. The scope for the formation of both cis- and trans-disubstituted scaffolds involved a total of 44 examples, affording yields up 90% and isomer ratios up to 36:1 and 16:1, respectively. Mechanistic experiments were conducted and allowed the authors to formulate a plausible reaction mechanism. With the aid of PhMgBr, via amido-magnesium 93, amido-rhenium 94 is initially formed, which then undergoes a deprotonative cyclorhenation affording rhenacycle 95. The ensuing coordination and insertion of an alkyne gives rise to seven-membered rhenacycle 96, which further leads to intermediate 97 upon protonation. Transmetalation results into a new amido-magnesium intermediate 98 which undergoes an intramolecular nucleophilic addition/cyclization generating the cis product, or further leading to the trans product via intermediate 99. Protonation of these species affords the final products and regenerates the amido-magnesium 93, thus closing the entire Re/Mg bimetallic tandem catalytic cycles.
Cross-Ullmann couplings are amongst the most common procedures to obtain biaryls. Nevertheless, a crucial challenge of cross-Ullmann reactions remains the achievement of selectivity for the hetero-coupling product over the homo-coupling [69]. For this matter, in 2015, Ackerman and co-workers developed a method to couple aryl halides 100 with aryl triflates 101 and affording hetero-coupled bi(hetero)aryls 102 by directly by a Ni/Pd bimetallic-catalyzed cross-Ullmann (Scheme 20) [70]. The selectivity was envisioned by the orthogonal reactivity of the two catalysts and the relative stability of the two arylmetal intermediates. Initially, each catalyst formed less than a 5% yield of the cross-coupled product. Yet, a total of 20 examples were described achieving yields of up to 94%. This new method can obtain biaryls, heteroaryls, dienes, and, specifically, N-heterocycles by functionalization of the pyridine ring, affording 2,3’-bipyridine and 2-arylpyridine. The authors suggest that the mechanism of this cross-Ullmann reaction undergoes a bimetallic approach, where the Ni catalyst reacts preferentially with aryl bromides to form a transient, reactive intermediate 103, while the Pd catalyst reacts preferentially with aryl triflates to afford a persistent intermediate 104. When each of the two catalysts activates only one of the two substrates, transmetallation occurs from nickel to palladium (105), which affords the final product by reductive elimination, thus regenerating the Pd0 catalyst. Ni0 catalyst is reobtained with the help of a Zn0 reductant.
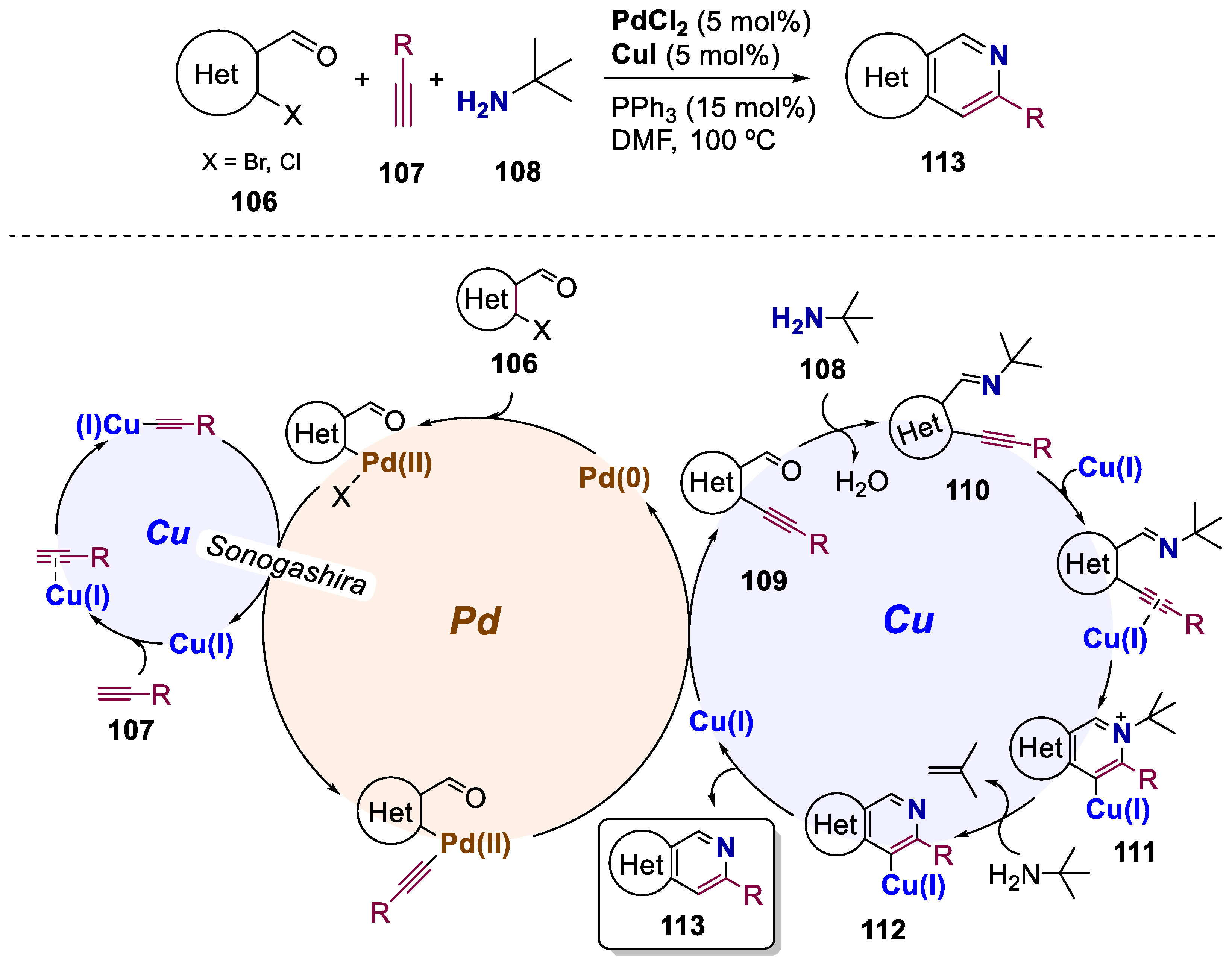
Copper-catalyzed electrophilic cyclization is highly attractive due to its low cost, easy availability, and high tolerance towards diverse functional groups. Nevertheless, there are relatively few synthetic studies on the Cu-catalyzed three component tandem reaction for the synthesis of polyheterocycles. Moreover, there has been no report which shows the dual behaviour of a Cu(I) catalyst, as well as tert-butylamine, for the synthesis of polyheterocycles. For this matter, in 2016, Verma’s group reported the synthesis of heterocyclic scaffolds starting from substrates 106 and alkynyls 107 via Pd(II)/Cu(I)-catalyzed Sonogashira coupling followed by a Cu(I)-catalyzed 6-endo-dig cyclization, with tert-butylamine 108 as a nitrogen source (Scheme 21) [71]. The scope of this process included the preparation of naphthyridines, isoquinolines, benzothieno- and benzofuropyridines with a total of 33 examples and yields up to 83%. To give insights into the mechanism, an array of preliminary control experiments was performed and revealed the dual role of Cu: to enhance the rate of Sonogashira coupling and to assist electrophilic cyclization. The catalytic system involves the formation of C–C and C–N bonds via Sonogashira coupling and electrophilic cyclization, respectively. The authors suggest that, initially, the ortho-halo aldehyde reacts with terminal alkynes under Sonogashira coupling conditions generating the ortho-alkynyl aldehyde intermediate 109. The latter reacts with tert-butylamine and leads to the formation of imine 110. π-Complexation between 110 and Cu(I) facilitates the electrophilic cyclization and affords 111. The presence of a tert-butyl group enhances the formation of intermediate 112 by the elimination of an isobutylene fragment. Finally, protodemetalation yields the desired cyclized pyridine-containing heterocycle 113.
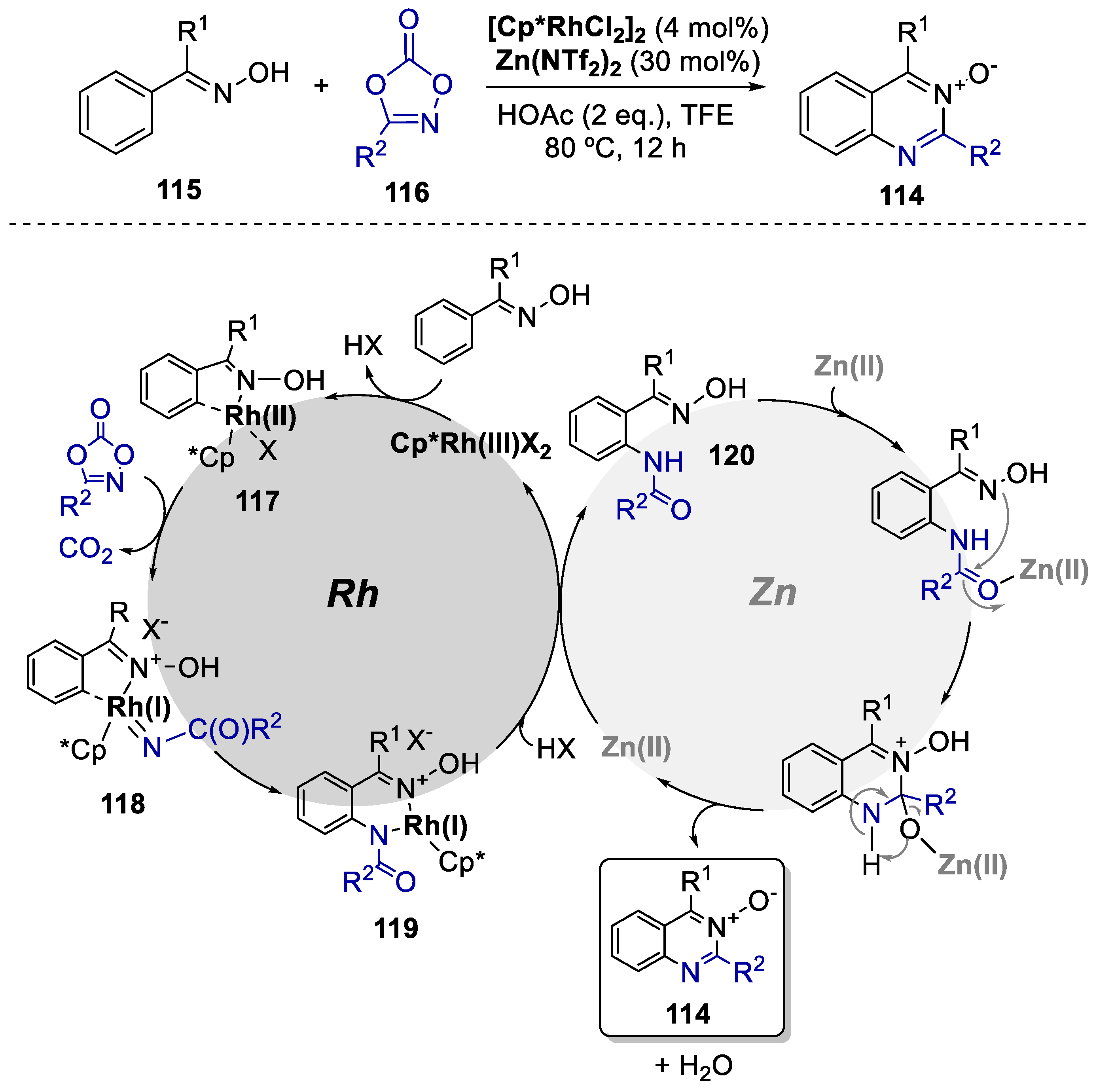
Until nowadays, the preparation of N-oxides of azacycles via direct C−H activation of arenes remains a challenge and highly underexplored, with only a few reports on the synthesis of N-oxides of isoquinoline [72,73]. For this reason, in 2016, Li and co-workers prepared quinazoline-N-oxides 114 via a single-step C−H activation approach by a Rh(III)/Zn(II) bimetallic-catalyzed process. They selected simple substrates, such as ketoximes 115 and 1,4,2-dioxazol-5-ones 116 (Scheme 22) [74]. This annulation system proceeded with high efficiency under mild conditions with H2O and CO2 as the coproducts, obviating any need for oxidants. A total of 32 examples of both omixes and dioxazolones was obtained in yields of up to 95%. Preliminary mechanistic studies were constructed to gain insight into the mechanism of this annulation reaction) and concluded that Rh(III) participated in the C−H activation-amidation of the ketoximes and Zn(II) in the cyclization. The authors suggest that, first, an active rhodium catalyst Cp*RhX2 (where X = NTf2 or OAc) is generated from the anion exchange between [RhCp*Cl2]2 and ZnNTf2 or HOAc. Next, the oxime reagent undergoes a ciclometalation to afford the rhodacyclic intermediate 117 and an acid via a concerted metalation−deprotonation mechanism. Coordination of dioxazolone is followed by a decarboxylation with CO2 elimination, yielding a nitrenoid species 118, and subsequent migratory insertion of the Rh-aryl bond produces the amidate 119. Protonolysis of the latter releases the amidated intermediate 120 and regenerates the Rh(III) complex. Zn(II) will then catalyze the cyclization and condensation of 120, furnishing the final quinazoline N-oxide 114.
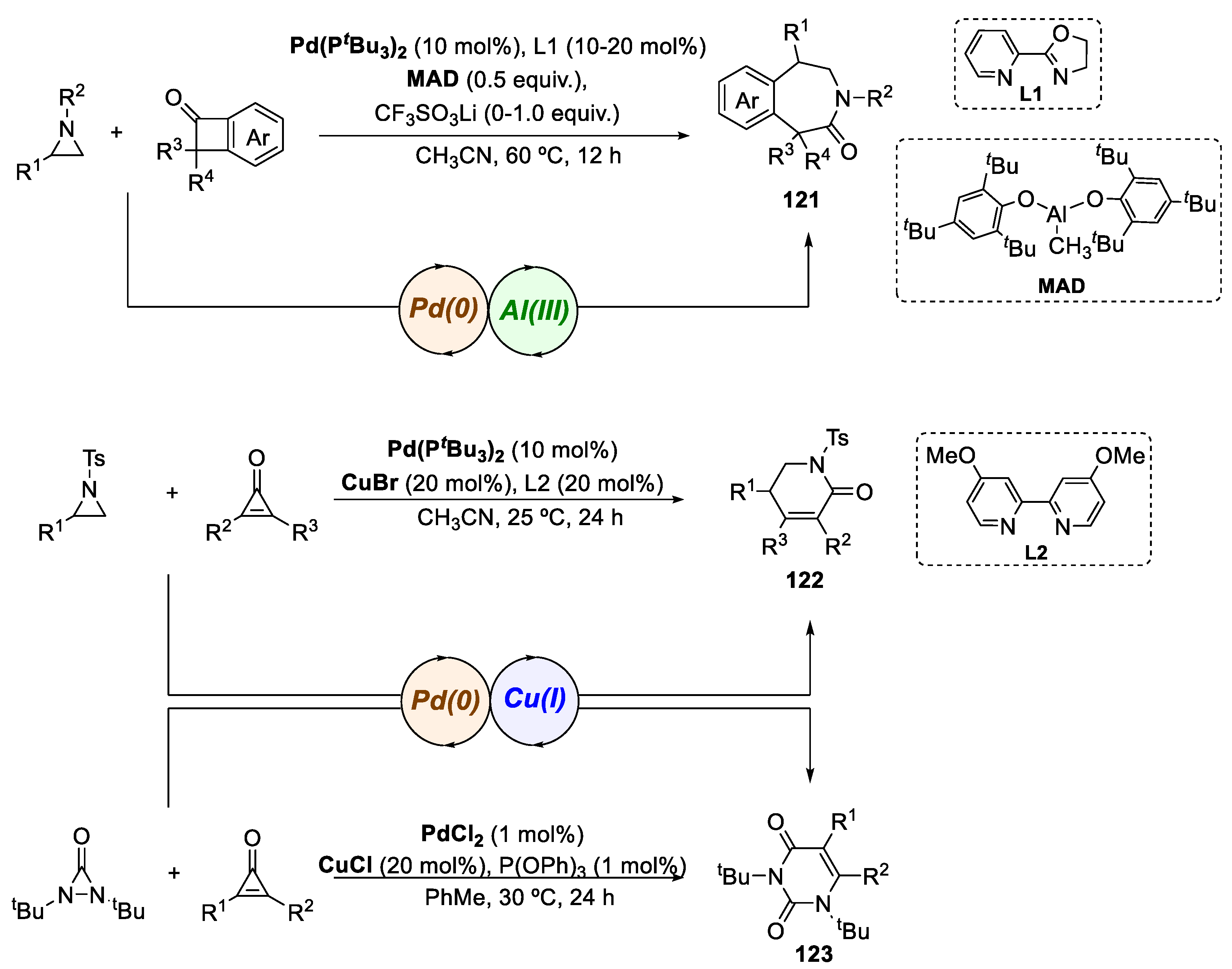
In 2021, Zhao and co-workers developed a general ring expansion strategy that enables the efficient and scalable synthesis of diverse N-heterocycles (Scheme 23), such as 3-benzazepinones 121 (up to 90% yield), dihydropyridinones 122 (up to 91% yield) and uracils 123 (up to 98% yield) [75]. The designed concept is based on the use of synergistic bimetallic catalysis to promote a formal cross-dimerization reaction between three-membered aza-heterocycles and three- and four-membered-ring ketones. The authors present a novel methodology that combines strain-release-induced oxidative C-C bond cleavage and C-N bond cleavage, effectively expanding the scope for stereospecific N-heterocycle synthesis. In this route, the palladium complex serves as the main catalyst for the reactions, although aluminum or copper, which function as Lewis acids, are also highlighted as critical components in this pathway. This approach provides a versatile and reliable method for the synthesis of 3-benzazepinones, dihydropyridinones and uracils, showcasing its flexibility and significant potential in the synthesis of complex molecules through transition-metal-catalyzed formal cross-dimerization of cyclic compounds.
7. Bimetallic Approaches Involving Other N-heterocyclic Scaffolds
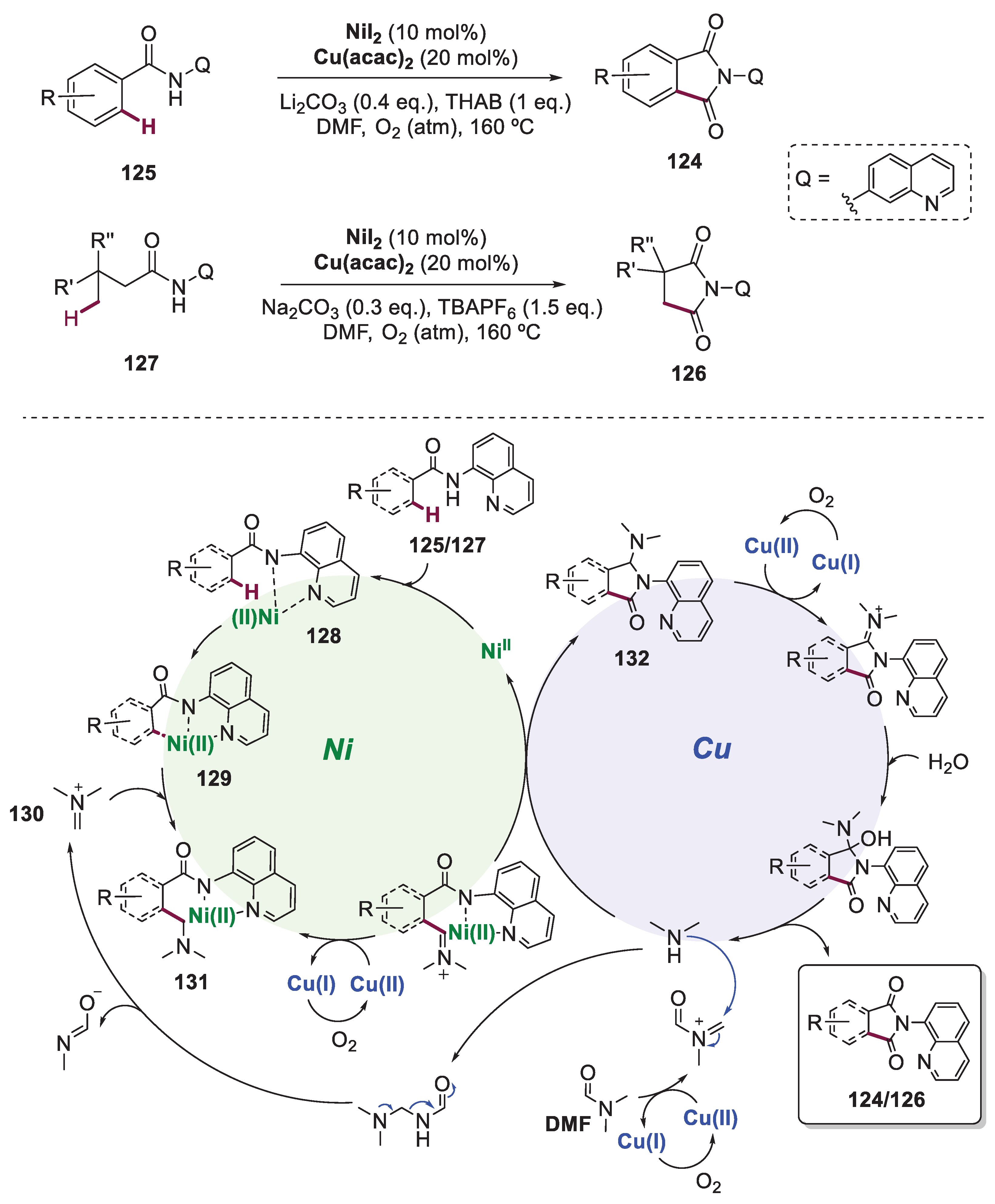
Whitin the metal-catalyzed C−H functionalization reaction’s class, direct carbonylation has attracted considerable attention in recent years due to the prevalent presence of the carbonyl group in organic molecules. The direct carbonylation of C(sp3)−H bonds has been demonstrated by transition metal-catalyzed processes, however, mainly rely on the use of toxic CO gas at high pressure [76,77,78]. To overcome this limitation, Ge and co-workers described, in 2015, the direct carbonylation of aromatic sp2 and unactivated sp3 C−H bonds of amides via a Ni/Co bimetallic catalysis with N,N-dimethylformamide (DMF) as the carbonyl source (Scheme 24) [79]. The reactions are performed under atmospheric O2 and the substrates are constituted by a bidentate directing group (Q). The authors provided a scope of aryl-substituted phthalimides 124, from aromatic amides 125 achieving yields up to 90% with the optimized conditions, as well as 3,3’-disubstituted succinimides 126, from aliphatic amides 127, with yields up to 81%. Preliminary control experiments elucidated that both nickel and copper catalysts are required for this process, suggesting that this reaction is performed via synergistic catalysis. The authors suggested that the process starts with the coordination of amide 125/127 to Ni(II) via a ligand exchange under basic conditions, forming complex 128. Then, cyclometalation of 128 occurs via either sp2 or sp3 C−H bond activation to generate the intermediate 129, keeping in mind that sp2 C−H bond cleavage is a reversible step while sp3 is irreversible. Electrophile 130 is then inserted into the catalytic cycle, generated in situ from DMF. This suffers decarboxylation or an elimination process via Cu(II)/O2. These intermediates react through a nucleophilic addition and sequential decarbonylation. The nucleophilic addition of the intermediate C to the iminium ion intermediate 130 provides 131. Oxidation of the latter followed by intramolecular nucleophilic addition gives rise to the intermediate 132 which then produces the product 124 or 126 via oxidation by Cu(II) and hydrolysis.
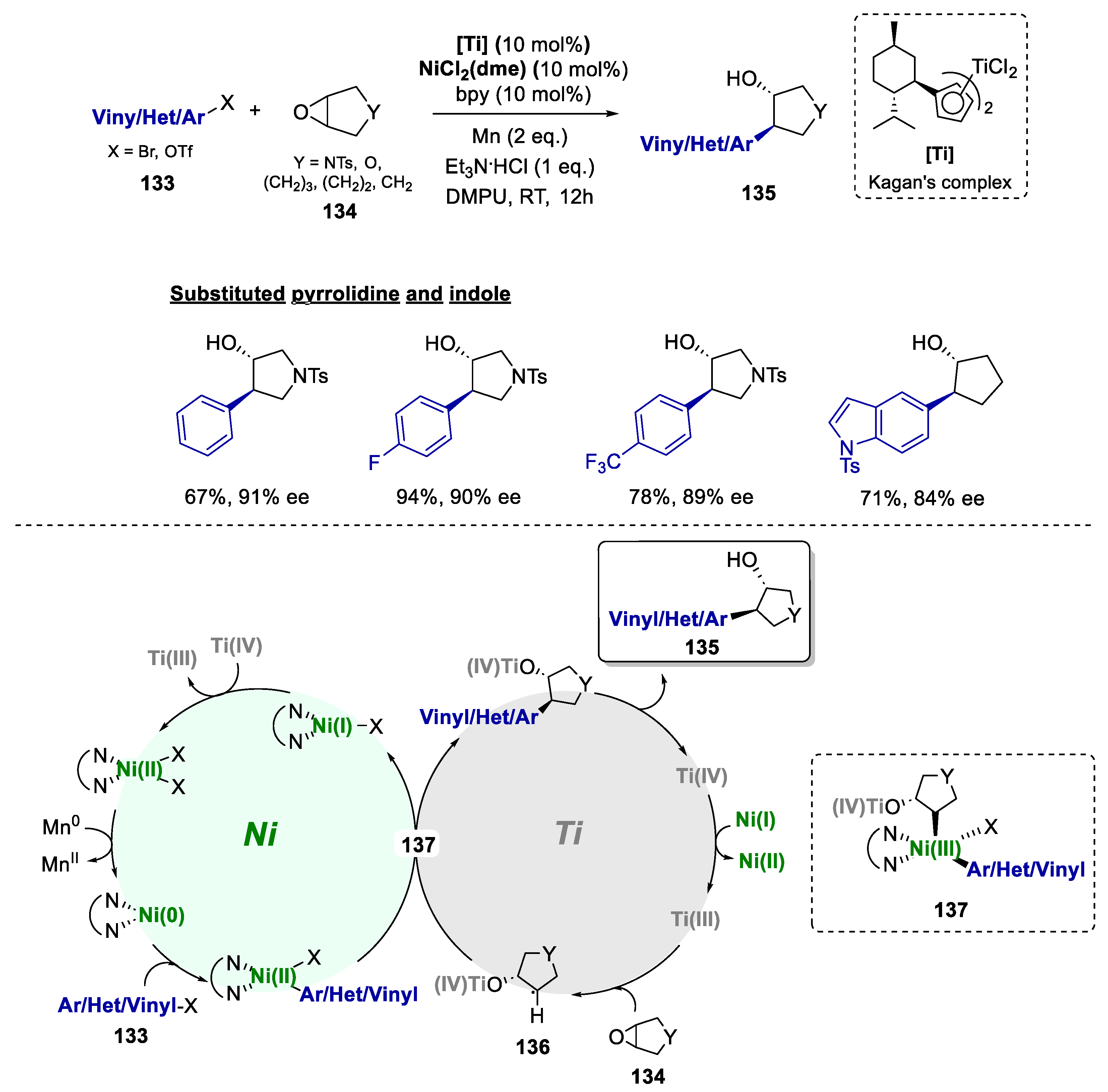
The enantioselective coupling of aryl and vinyl nucleophiles with meso-epoxides is considered to be highly challenging, with the best results to date resorting to aryl lithium reagents and chiral ligands [80,81]. In this context, in 2015, Zhao and Weix reported an enantioselective cross-electrophile coupling of aryl halides 133 with meso-epoxides 134 to form trans-β-arylcycloalkanols 135, from a novel bimetallic Ni(II)/Ti(III) catalytic system (Scheme 25) [82]. The reaction was catalyzed by a combination of (bpy)NiCl2 and a chiral titanocene under reducing conditions. Different titanocenes were tested, and the one first reported by Cesarotti and co-workers [83] showed the highest yield and enantioselectivity . This combination enantioselectively coupled aryl, heteroaryl, or vinyl halides with meso-epoxides with a scope constituted by 28 examples. It also included examples on the functionalization of pyrrolidine scaffolds and one example for indole, with yields up to 94% and enantiomeric excesses up to 91% ee. These authors suggested that the coupling mechanism (Scheme 25) would be initiated by the enantioselective formation of a β-titanoxy carbon radical from the meso-epoxide (136). Then, the oxidative addition of a β-titanoxy carbon radical to an arylnickel(II) intermediate would form a diorganonickel(III) species ( 137), and the reductive elimination of the product (135). Finally, the reduction of both catalysts closes the catalytic cycle.
8. Conclusion
Bimetallic catalysis offers the potential for synergistic effects and dual activation of reactants, leading to enhanced reactivity and selectivity. By employing two different metal catalysts in a sequential or concurrent manner, researchers have unlocked new synthetic pathways and achieved more efficient and selective transformations. In recent years, bimetallic catalysis has shown promising results in various organic transformations.
The diversity of metal combinations and reaction conditions available in bimetallic catalysis offers a versatile platform to access a wide array of N-heterocyclic compounds with diverse functionalities. This benefits not only the pharmaceutical industry, where these compounds play a crucial role in drug discovery but also the fine chemical industries, where N-heterocycles serve as important intermediates in the synthesis of various key chemicals.
Author Contributions
All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research was funded by Fundação para a Ciência e Tecnologia (FCT, PTDC/QUI-QOR/0712/2020), and fellowships PD/BD/05960/2020 (D.R.) and 2022.12365.BD (N.V.) .
Data Availability Statement
Does not apply
Acknowledgments
The authors also thank the support by the Laboratório Associado para a Química Verde (LAQV), which is financed by national funds from FCT/Ministério da Ciência, Tecnologia e Ensino Superior (FCT/MCTES, UIDB/50006/2020, UIDP/50006/2020, and LA/P/0008/2020).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huo, X.; Li, G.; Wang, X.; Zhang, W. Bimetallic Catalysis in Stereodivergent Synthesis. Angew. Chemie - Int. Ed. 2022, 61. [CrossRef]
- Mankad, N.P. Selectivity Effects in Bimetallic Catalysis. Chem. - A Eur. J. 2016, 22, 5822–5829. [CrossRef]
- Pye, D.R.; Mankad, N.P. Bimetallic Catalysis for C-C and C-X Coupling Reactions. Chem. Sci. 2017, 8, 1705–1718. [CrossRef]
- Park, J.; Hong, S. Cooperative Bimetallic Catalysis in Asymmetric Transformations. Chem. Soc. Rev. 2012, 41, 6931–6943. [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [CrossRef]
- Reetz, M.T.; Breinbauer, R.; Wanninger, K. Suzuki and Heck Reactions Catalyzed by Preformed Palladium Clusters and Palladium/Nickel Bimetallic Clusters. Tetrahedron Lett. 1996, 37, 4499–4502. [CrossRef]
- Gupta, G.R.; Shah, J.; Vadagaonkar, K.S.; Lavekar, A.G.; Kapdi, A.R. Hetero-Bimetallic Cooperative Catalysis for the Synthesis of Heteroarenes. Org. Biomol. Chem. 2019, 17, 7596–7631. [CrossRef]
- Ferro, R.; Viduedo, N.; Santos, A.S.; Silva, A.M.S.; Royo, B.; Marques, M.M.B. Bimetallic Catalyzed Synthesis of 2-Arylindoles. Synthesis (Stuttg). 2023. [CrossRef]
- Huang, Y.Y.; Cai, C.; Yang, X.; Lv, Z.C.; Schneider, U. Catalytic Asymmetric Reactions with N,O-Aminals. ACS Catal. 2016, 6, 5747–5763. [CrossRef]
- Katritzky, A.R.; Pernak, J.; Fan, W.Q.; Saczewski, F. N-(1-Benzotriazol-1-Ylalkyl)Amides, Versatile α-Amidoalkylation Reagents. 1. α-Amidoalkylation of CH Acids. J. Org. Chem. 1991, 56, 4439–4443. [CrossRef]
- Katritzky, A.R.; Fan, W.Q.; Black, M.; Pernak, J. N-[1-(Benzotriazol-1-Yl)Alkyl]Amides, Versatile Amidoalkylation Reagents. 5. A General and Convenient Route to N-(α-Alkoxyalkyl)Amides. J. Org. Chem. 1992, 57, 547–549. [CrossRef]
- Gizecki, P.; Ait Youcef, R.; Poulard, C.; Dhal, R.; Dujardin, G. Diastereoselective Preparation of Novel Tetrahydrooxazinones via Heterocycloaddition of N-Boc, O-Me-Acetals. Tetrahedron Lett. 2004, 45, 9589–9592. [CrossRef]
- Harding, K.E.; Todd Coleman, M.; Liu, L.T. Stereoselective Reactions of N-Acyl-N,O-Acetals with 2-(Trimethysilyloxy)Furan. Tetrahedron Lett. 1991, 32, 3795–3798. [CrossRef]
- Yin, B.; Zhang, Y.; Xu, L.W. Recent Applications of α-Amido Sulfones as in Situ Equivalents of Activated Imines for Asymmetric Catalytic Nucleophilic Addition Reactions. Synthesis (Stuttg). 2010, 3583–3595. [CrossRef]
- Li, H.; Belyk, K.M.; Yin, J.; Chen, Q.; Hyde, A.; Ji, Y.; Oliver, S.; Tudge, M.T.; Campeau, L.C.; Campos, K.R. Enantioselective Synthesis of Hemiaminals via Pd-Catalyzed C-N Coupling with Chiral Bisphosphine Mono-Oxides. J. Am. Chem. Soc. 2015, 137, 13728–13731. [CrossRef]
- Zhang, W.Z.; Chu, J.C.K.; Oberg, K.M.; Rovis, T. Enantioselective Rhodium-Catalyzed Isomerization of 4-Iminocrotonates: Asymmetric Synthesis of a Unique Chiral Synthon. J. Am. Chem. Soc. 2015, 137, 553–555. [CrossRef]
- Wang, T.; Yu, Z.; Hoon, D.L.; Phee, C.Y.; Lan, Y.; Lu, Y. Regiodivergent Enantioselective γ-Additions of Oxazolones to 2,3-Butadienoates Catalyzed by Phosphines: Synthesis of α,α-Disubstituted α-Amino Acids and N,O-Acetal Derivatives. J. Am. Chem. Soc. 2016, 138, 265–271. [CrossRef]
- Kim, H.; Rhee, Y.H. Stereodefined N,O-Acetals: Pd-Catalyzed Synthesis from Homopropargylic Amines and Utility in the Flexible Synthesis of 2,6-Substituted Piperidines. J. Am. Chem. Soc. 2012, 134, 4011–4014. [CrossRef]
- Rueping, M.; Antonchick, A.P.; Sugiono, E.; Grenader, K. Asymmetric Brønsted Acid Catalysis: Catalytic Enantioselective Synthesis of Highly Biologically Active Dihydroquinazolinones. Angew. Chemie - Int. Ed. 2009, 48, 908–910. [CrossRef]
- Vellalath, S.; Čorić, I.; List, B. N-Phosphinyl Phosphoramide-A Chiral Brønsted Acid Motif for the Direct Asymmetric N,O-Acetalization of Aldehydes. Angew. Chemie - Int. Ed. 2010, 49, 9749–9752. [CrossRef]
- Li, G.; Fronczek, F.R.; Antilla, J.C. Catalytic Asymmetric Addition of Alcohols to Imines: Enantioselective Preparation of Chiral N,O-Aminals. J. Am. Chem. Soc. 2008, 130, 12216–12217. [CrossRef]
- Li, T.Z.; Wang, X.B.; Sha, F.; Wu, X.Y. Catalytic Enantioselective Addition of Alcohols to Isatin-Derived N-Boc Ketimines. Tetrahedron 2013, 69, 7314–7319. [CrossRef]
- Hiramatsu, K.; Honjo, T.; Rauniyar, V.; Toste, F.D. Enantioselective Synthesis of Fluoro-Dihydroquinazolones and -Benzooxazinones by Fluorination-Initiated Asymmetric Cyclization Reactions. ACS Catal. 2016, 6, 151–154. [CrossRef]
- Wang, X.; Dong, S.; Yao, Z.; Feng, L.; Daka, P.; Wang, H.; Xu, Z. Synthesis of Spiroaminals and Spiroketals with Bimetallic Relay Catalysis. Org. Lett. 2014, 16, 22–25. [CrossRef]
- Wang, X.; Yao, Z.; Dong, S.; Wei, F.; Wang, H.; Xu, Z. Synthesis of Fused Bicyclic Aminals through Sequential Gold/Lewis Acid Catalysis. Org. Lett. 2013, 15, 2234–2237. [CrossRef]
- Zhang, S.; Xu, Z.; Jia, J.; Tung, C.H.; Xu, Z. Synthesis of Spiroaminals by Bimetallic Au/Sc Relay Catalysis: TMS as a Traceless Controlling Group. Chem. Commun. 2014, 50, 12084–12087. [CrossRef]
- Wang, B.; Liang, M.; Tang, J.; Deng, Y.; Zhao, J.; Sun, H.; Tung, C.H.; Jia, J.; Xu, Z. Gold/Lewis Acid Catalyzed Cycloisomerization/Diastereoselective [3 + 2] Cycloaddition Cascade: Synthesis of Diverse Nitrogen-Containing Spiro Heterocycles. Org. Lett. 2016, 18, 4614–4617. [CrossRef]
- Li, J.; Lin, L.; Hu, B.; Lian, X.; Wang, G.; Liu, X.; Feng, X. Bimetallic Gold(I)/Chiral N,N′-Dioxide Nickel(II) Asymmetric Relay Catalysis: Chemo- and Enantioselective Synthesis of Spiroketals and Spiroaminals. Angew. Chemie - Int. Ed. 2016, 55, 6075–6078. [CrossRef]
- Wang, B.; Chen, Y.; Zhou, L.; Wang, J.; Xu, Z. Zn/Sc Bimetallic Relay Catalysis: One Pot Cycloisomerization/Carbonyl-Ene Reaction toward Oxazole Derivatives. Org. Biomol. Chem. 2016, 14, 826–829. [CrossRef]
- Hu, B.; Li, J.; Cao, W.; Lin, Q.; Yang, J.; Lin, L.; Liu, X.; Feng, X. Asymmetric Synthesis of Fused Bicyclic N,O- and O,O-Acetals via Cascade Reaction by Gold(I)/N,N′-Dioxide-Nickel(II) Bimetallic Relay Catalysis. Adv. Synth. Catal. 2018, 360, 2831–2835. [CrossRef]
- Taber, D.F.; Tirunahari, P.K. Indole Synthesis: A Review and Proposed Classification. Tetrahedron 2011, 67, 7195–7210. [CrossRef]
- Tobisu, M.; Ito, S.; Kitajima, A.; Chatani, N. GaCI3- and TiCl4-Catalyzed Insertion of Isocyanides into a C-S Bond of Dithioacetals. Org. Lett. 2008, 10, 5223–5225. [CrossRef]
- Kong, W.; Wang, Q.; Zhu, J. Synthesis of Diversely Functionalized Oxindoles Enabled by Migratory Insertion of Isocyanide to a Transient σ-Alkylpalladium(II) Complex. Angew. Chemie - Int. Ed. 2016, 55, 9714–9718. [CrossRef]
- Vlaar, T.; Cioc, R.C.; Mampuys, P.; Maes, B.U.W.; Orru, R.V.A.; Ruijter, E. Sustainable Synthesis of Diverse Privileged Heterocycles by Palladium-Catalyzed Aerobic Oxidative Isocyanide Insertion. Angew. Chemie - Int. Ed. 2012, 51, 13058–13061. [CrossRef]
- Liu, B.; Yin, M.; Gao, H.; Wu, W.; Jiang, H. Synthesis of 2-Aminobenzoxazoles and 3-Aminobenzoxazines via Palladium-Catalyzed Aerobic Oxidation of o -Aminophenols with Isocyanides. J. Org. Chem. 2013, 78, 3009–3020. [CrossRef]
- Gao, Q.; Zhou, P.; Liu, F.; Hao, W.J.; Yao, C.; Jiang, B.; Tu, S.J. Cobalt(II)/Silver Relay Catalytic Isocyanide Insertion/Cycloaddition Cascades: A New Access to Pyrrolo[2,3-b]Indoles. Chem. Commun. 2015, 51, 9519–9522. [CrossRef]
- Manisha; Dhiman, S.; Mathew, J.; Ramasastry, S.S.V. One-Pot Relay Catalysis: Divergent Synthesis of Furo[3,4-: B] Indoles and Cyclopenta [b] Indoles from 3-(2-Aminophenyl)-1,4-Enynols. Org. Biomol. Chem. 2016, 14, 5563–5568. [CrossRef]
- Dhiman, S.; Mishra, U.K.; Ramasastry, S.S. V. One-Pot Trimetallic Relay Catalysis: A Unified Approach for the Synthesis of β-Carbolines and Other [ c ]-Fused Pyridines. Angew. Chemie 2016, 128, 7868–7872. [CrossRef]
- Mu, Q.C.; Lv, J.Y.; Chen, M.Y.; Bai, X.F.; Chen, J.; Xia, C.G.; Xu, L.W. Bimetallic Copper and Zinc-Catalyzed Oxidative Cycloaddition of 3-Aminopyridazines and Nitriles: A Direct Synthesis of 1,2,4-Triazolo[1,5-: B] Pyridazines via C-N and N-N Bond-Forming Process. RSC Adv. 2017, 7, 37208–37213. [CrossRef]
- Ji, W.W.; Lin, E.; Li, Q.; Wang, H. Heteroannulation Enabled by a Bimetallic Rh(III)/Ag(i) Relay Catalysis: Application in the Total Synthesis of Aristolactam BII. Chem. Commun. 2017, 53, 5665–5668. [CrossRef]
- Zhang, D.; Lin, L.; Yang, J.; Liu, X.; Feng, X. Asymmetric Synthesis of Tetrahydroindolizines by Bimetallic Relay Catalyzed Cycloaddition of Pyridinium Ylides. Angew. Chemie - Int. Ed. 2018, 57, 12323–12327. [CrossRef]
- Xu, C.; Feng, Y.; Li, F.; Han, J.; He, Y.M.; Fan, Q.H. A Synthetic Route to Chiral Benzo-Fused N-Heterocycles via Sequential Intramolecular Hydroamination and Asymmetric Hydrogenation of Anilino-Alkynes. Organometallics 2019, 38, 3979–3990. [CrossRef]
- Von Nussbaum, F.; Brands, M.; Hinzen, B.; Weigand, S.; Häbich, D. Antibacterial Natural Products in Medicinal Chemistry - Exodus or Revival? Angew. Chemie - Int. Ed. 2006, 45, 5072–5129. [CrossRef]
- R. B. Sykes, C. M. Cimarusti, D. P. Bonner, K. Bush, D. M. Floyd, N. H. Georgopapadakou, W. H. Koster, W. C. Liu, W. L. Parker, P. a. Principe, M. L. Rathnum, W. a. Slusarchyk, W.H.T.& J.S.W. Monobactams.Pdf. Nat. Rev. Microbiol. 1981, 489–491.
- Imada, A.; Kitano, K.; Kintaka, K.; Muroi, M.; Asai, M. Sulfazecin and Isosulfazecin, Novel β-Lactam Antibiotics of Bacterial Origin. Nature 1981, 289, 590–591.
- Galletti, P.; Giacomini, D. Monocyclic β-Lactams: New Structures for New Biological Activities. Curr. Med. Chem. 2011, 18, 4265–4283. [CrossRef]
- Pitts, C.R.; Lectka, T. Chemical Synthesis of β-Lactams: Asymmetric Catalysis and Other Recent Advances. Chem. Rev. 2014, 114, 7930–7953. [CrossRef]
- France, S.; Weatherwax, A.; Taggi, A.E.; Lectka, T. Advances in the Catalytic, Asymmetric Synthesis of β-Lactams. Acc. Chem. Res. 2004, 37, 592–600. [CrossRef]
- Huang, L.Z.; Xuan, Z.; Jeon, H.J.; Du, Z.T.; Kim, J.H.; Lee, S.G. Asymmetric Rh(II)/Pd(0) Relay Catalysis: Synthesis of α-Quaternary Chiral β-Lactams through Enantioselective C-H Insertion/Diastereoselective Allylation of Diazoamides. ACS Catal. 2018, 8, 7340–7345. [CrossRef]
- Qi, J.; Wei, F.; Tung, C.H.; Xu, Z. Modular Synthesis of α-Quaternary Chiral β-Lactams by a Synergistic Copper/Palladium-Catalyzed Multicomponent Reaction. Angew. Chemie - Int. Ed. 2021, 60, 13814–13818. [CrossRef]
- Qi, J.; Song, T.; Yang, Z.; Sun, S.; Tung, C.H.; Xu, Z. Simultaneous Dual Cu/Ir Catalysis: Stereodivergent Synthesis of Chiral β-Lactams with Adjacent Tertiary/Quaternary/Tertiary Stereocenters. ACS Catal. 2023, 13, 2555–2564. [CrossRef]
- Matin, M.M.; Matin, P.; Rahman, M.R.; Ben Hadda, T.; Almalki, F.A.; Mahmud, S.; Ghoneim, M.M.; Alruwaily, M.; Alshehri, S. Triazoles and Their Derivatives: Chemistry, Synthesis, and Therapeutic Applications. Front. Mol. Biosci. 2022, 9, 1–8. [CrossRef]
- Dalton, T.; Faber, T.; Glorius, F. C-H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 7, 245–261. [CrossRef]
- Lesieur, M.; Lazreg, F.; Cazin, C.S.J. A Cooperative Pd-Cu System for Direct C-H Bond Arylation. Chem. Commun. 2014, 50, 8927–8929. [CrossRef]
- Spiteri, C.; Moses, J.E. Copper-Catalyzed Azide-Alkyne Cycloaddition: Regioselective Synthesis of 1,4,5-Trisubstituted 1,2,3-Triazoles. Angew. Chemie - Int. Ed. 2010, 49, 31–33. [CrossRef]
- Wei, F.; Li, H.; Song, C.; Ma, Y.; Zhou, L.; Tung, C.H.; Xu, Z. Cu/Pd-Catalyzed, Three-Component Click Reaction of Azide, Alkyne, and Aryl Halide: One-Pot Strategy toward Trisubstituted Triazoles. Org. Lett. 2015, 17, 2860–2863. [CrossRef]
- Verma, Y.K.; Reddy, B.S.; Pawar, M.S.; Bhunia, D.; Sampath Kumar, H.M. Design, Synthesis, and Immunological Evaluation of Benzyloxyalkyl-Substituted 1,2,3-Triazolyl α-GalCer Analogues. ACS Med. Chem. Lett. 2016, 7, 172–176. [CrossRef]
- Meldal, M.; Tomøe, C.W. Cu-Catalyzed Azide - Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [CrossRef]
- Xiao, X.; Xie, Y.; Bai, S.; Deng, Y.; Jiang, H.; Zeng, W. Transition-Metal-Free Tandem Chlorocyclization of Amines with Carboxylic Acids: Access to Chloroimidazo[1,2-α]Pyridines. Org. Lett. 2015, 17, 3998–4001. [CrossRef]
- Monir, K.; Bagdi, A.K.; Ghosh, M.; Hajra, A. Regioselective Oxidative Trifluoromethylation of Imidazoheterocycles via C(Sp2)-H Bond Functionalization. J. Org. Chem. 2015, 80, 1332–1337. [CrossRef]
- Cai, Q.; Yan, J.; Ding, K. A CuAAC/Ullmann C-C Coupling Tandem Reaction: Copper-Catalyzed Reactions of Organic Azides with N -(2-Iodoaryl)Propiolamides or 2-Iodo- N -(Prop-2-Ynyl)Benzenamines. Org. Lett. 2012, 14, 3332–3335. [CrossRef]
- Pericherla, K.; Jha, A.; Khungar, B.; Kumar, A. Copper-Catalyzed Tandem Azide-Alkyne Cycloaddition, Ullmann Type C-N Coupling, and Intramolecular Direct Arylation. Org. Lett. 2013, 15, 4304–4307. [CrossRef]
- Wang, Z.; Li, B.; Zhang, X.; Fan, X. One-Pot Cascade Reactions Leading to Pyrido[2′,1′:2,3]Imidazo[4,5-c][1,2,3]Triazolo[1,5-a]Quinolines under Bimetallic Relay Catalysis with Air as the Oxidant. J. Org. Chem. 2016, 81, 6357–6363. [CrossRef]
- Pathare, R.S.; Ansari, A.J.; Verma, S.; Maurya, A.; Maurya, A.K.; Agnihotri, V.K.; Sharon, A.; Pardasani, R.T.; Sawant, D.M. Sequential Pd(0)/Fe(III) Catalyzed Azide-Isocyanide Coupling/Cyclization Reaction: One-Pot Synthesis of Aminotetrazoles. J. Org. Chem. 2018, 83, 9530–9537. [CrossRef]
- Wang, H.; Grohmann, C.; Nimphius, C.; Glorius, F. Mild Rh(III)-Catalyzed C-H Activation and Annulation with Alkyne MIDA Boronates: Short, Efficient Synthesis of Heterocyclic Boronic Acid Derivatives. J. Am. Chem. Soc. 2012, 134, 19592–19595. [CrossRef]
- Guimond, N.; Gorelsky, S.I.; Fagnou, K. Rhodium(III)-Catalyzed Heterocycle Synthesis Using an Internal Oxidant: Improved Reactivity and Mechanistic Studies. J. Am. Chem. Soc. 2011, 133, 6449–6457. [CrossRef]
- Hyster, T.K.; Rovis, T. An Improved Catalyst Architecture for Rhodium(Iii) Catalyzed C-H Activation and Its Application to Pyridone Synthesis. Chem. Sci. 2011, 2, 1606–1610. [CrossRef]
- Tang, Q.; Xia, D.; Jin, X.; Zhang, Q.; Sun, X.Q.; Wang, C. Re/Mg Bimetallic Tandem Catalysis for [4+2] Annulation of Benzamides and Alkynes via C-H/N-H Functionalization. J. Am. Chem. Soc. 2013, 135, 4628–4631. [CrossRef]
- Kang, K.; Huang, L.; Weix, D.J. Sulfonate Versus Sulfonate: Nickel and Palladium Multimetallic Cross-Electrophile Coupling of Aryl Triflates with Aryl Tosylates. J. Am. Chem. Soc. 2020, 142, 10634–10640. [CrossRef]
- Ackerman, L.K.G.; Lovell, M.M.; Weix, D.J. Multimetallic Catalysed Cross-Coupling of Aryl Bromides with Aryl Triflates. Nature 2015, 524, 454–457. [CrossRef]
- Kumar, S.; Saunthwal, R.K.; Aggarwal, T.; Kotla, S.K.R.; Verma, A.K. Palladium Meets Copper: One-Pot Tandem Synthesis of Pyrido Fused Heterocycles: Via Sonogashira Conjoined Electrophilic Cyclization. Org. Biomol. Chem. 2016, 14, 9063–9071. [CrossRef]
- Shi, Z.; Koester, D.C.; Boultadakis-Arapinis, M.; Glorius, F. Rh(III)-Catalyzed Synthesis of Multisubstituted Isoquinoline and Pyridine N-Oxides from Oximes and Diazo Compounds. J. Am. Chem. Soc. 2013, 135, 12204–12207. [CrossRef]
- Phatake, R.S.; Patel, P.; Ramana, C. V. Ir(III)-Catalyzed Synthesis of Isoquinoline N-Oxides from Aryloxime and α-Diazocarbonyl Compounds. Org. Lett. 2016, 18, 292–295. [CrossRef]
- Wang, Q.; Wang, F.; Yang, X.; Zhou, X.; Li, X. Rh(III)- and Zn(II)-Catalyzed Synthesis of Quinazoline N-Oxides via C-H Amidation-Cyclization of Oximes. Org. Lett. 2016, 18, 6144–6147. [CrossRef]
- Li, R.; Li, B.; Zhang, H.; Ju, C.W.; Qin, Y.; Xue, X.S.; Zhao, D. A Ring Expansion Strategy towards Diverse Azaheterocycles. Nat. Chem. 2021, 13, 1006–1016. [CrossRef]
- Gao, K.; Yoshikai, N. Low-Valent Cobalt Catalysis: New Opportunities for C-H Functionalization. Acc. Chem. Res. 2014, 47, 1208–1219. [CrossRef]
- Xie, P.; Xie, Y.; Qian, B.; Zhou, H.; Xia, C.; Huang, H. Palladium-Catalyzed Oxidative Carbonylation of Benzylic C-H Bonds via Nondirected C(Sp 3)-H Activation. J. Am. Chem. Soc. 2012, 134, 9902–9905. [CrossRef]
- Hasegawa, N.; Shibata, K.; Charra, V.; Inoue, S.; Fukumoto, Y.; Chatani, N. Ruthenium-Catalyzed Cyclocarbonylation of Aliphatic Amides through the Regioselective Activation of Unactivated C(Sp3)-H Bonds. Tetrahedron 2013, 69, 4466–4472. [CrossRef]
- Wu, X.; Zhao, Y.; Ge, H. Direct Aerobic Carbonylation of C(Sp2)-H and C(Sp3)-H Bonds through Ni/Cu Synergistic Catalysis with DMF as the Carbonyl Source. J. Am. Chem. Soc. 2015, 137, 4924–4927. [CrossRef]
- Vrancken, E.; Alexakis, A.; Mangeney, P. Organolithium/Chiral Lewis Base/BF3: A Versatile Combination for the Enantioselective Desymmetrization of Meso-Epoxides. European J. Org. Chem. 2005, 1354–1366. [CrossRef]
- Oguni, N.; Miyagi, Y.; Itoh, K. Highly Enantioselective Arylation of Symmetrical Epoxides with Phenyllithium Promoted by Chiral Schiff Bases and Salens. Tetrahedron Lett. 1998, 39, 9023–9026. [CrossRef]
- Zhao, Y.; Weix, D.J. Enantioselective Cross-Coupling of Meso -Epoxides with Aryl Halides. J. Am. Chem. Soc. 2015, 137, 3327–3340. [CrossRef]
- Cesarotti, E.; Kagan, H.B.; Goddard, R.; Krüger, C. Synthesis of New Ligands for Transition Metal Complexes: Menthyl- and Neomenthyl-Cyclopentadienes. J. Organomet. Chem. 1978, 162, 297–309. [CrossRef]
Scheme 1.
Xu’s contributions towards the bimetallic synthesis of fused bicyclic- and spiroaminals.
Scheme 2.
A plausible mechanism of the Au/Lewis acid bimetallic synthesis of fused bicyclic- and spiroaminals by Xu’s group.
Scheme 2.
A plausible mechanism of the Au/Lewis acid bimetallic synthesis of fused bicyclic- and spiroaminals by Xu’s group.
Scheme 3.
General conditions for the preparation of spiroaminals by a Au(I)/Ni(II)-catalyzed bimetallic approach.
Scheme 3.
General conditions for the preparation of spiroaminals by a Au(I)/Ni(II)-catalyzed bimetallic approach.
Scheme 4.
General conditions for the preparation of oxazoles by a Zn(II)/Sc(III)-catalyzed bimetallic approach.
Scheme 4.
General conditions for the preparation of oxazoles by a Zn(II)/Sc(III)-catalyzed bimetallic approach.
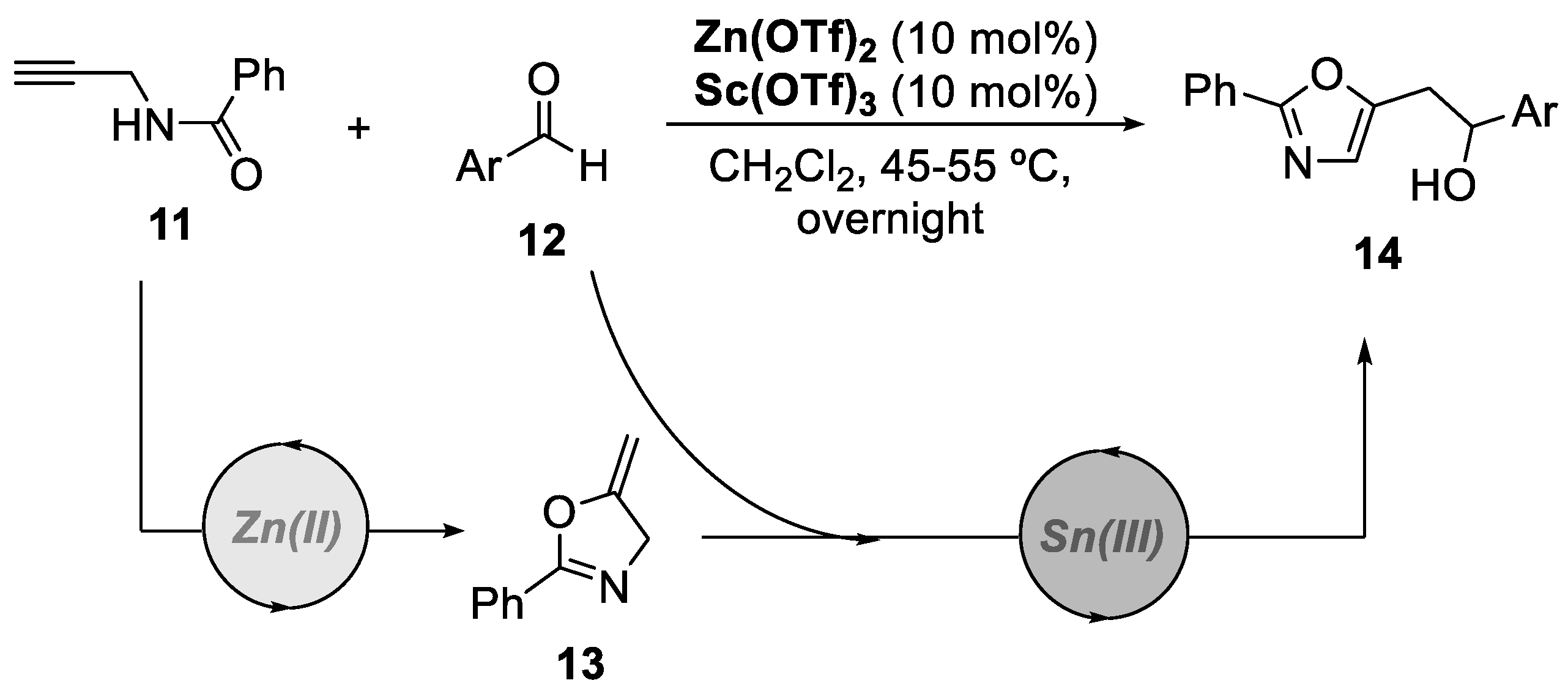
Scheme 5.
General conditions and a plausible mechanism for the preparation of fused bicyclic acetals or aminals by a Au(I)/Ni(II)-catalyzed bimetallic approach.
Scheme 5.
General conditions and a plausible mechanism for the preparation of fused bicyclic acetals or aminals by a Au(I)/Ni(II)-catalyzed bimetallic approach.
Scheme 7.
General conditions and a plausible mechanism for the preparation of furo[3,4-]indoles by a Ag(I)/Bi(III)/Pd(II)-catalyzed trimetallic approach.
Scheme 7.
General conditions and a plausible mechanism for the preparation of furo[3,4-]indoles by a Ag(I)/Bi(III)/Pd(II)-catalyzed trimetallic approach.
Scheme 8.
General conditions and a plausible mechanism for the preparation of 1,3-di- and 1,3,4-trisubstituted β-carbolines by a Ag(I)/Bi(III)/Pd(II)-catalyzed trimetallic approach.
Scheme 8.
General conditions and a plausible mechanism for the preparation of 1,3-di- and 1,3,4-trisubstituted β-carbolines by a Ag(I)/Bi(III)/Pd(II)-catalyzed trimetallic approach.
Scheme 9.
General conditions and a plausible mechanism for the preparation of 1,2,4-triazolo[1,5-b] pyridazines by a Cu(I)/Zn(II)-catalyzed bimetallic approach.
Scheme 9.
General conditions and a plausible mechanism for the preparation of 1,2,4-triazolo[1,5-b] pyridazines by a Cu(I)/Zn(II)-catalyzed bimetallic approach.
Scheme 10.
General conditions and a plausible mechanism for the preparation of 3-alkylidene isoindolinones by a Rh(III)/Ag(I)-catalyzed bimetallic approach.
Scheme 10.
General conditions and a plausible mechanism for the preparation of 3-alkylidene isoindolinones by a Rh(III)/Ag(I)-catalyzed bimetallic approach.
Scheme 11.
General conditions for the preparation of asymmetrically-substituted indolines by a Au(I)/Ru(II)-catalyzed bimetallic approach.
Scheme 11.
General conditions for the preparation of asymmetrically-substituted indolines by a Au(I)/Ru(II)-catalyzed bimetallic approach.
Scheme 12.
General conditions for the preparation of 2-arylindoles by a Mn(I)/Pd(II)- or Ni(II)/Pd(II)- catalyzed bimetallic approaches.
Scheme 12.
General conditions for the preparation of 2-arylindoles by a Mn(I)/Pd(II)- or Ni(II)/Pd(II)- catalyzed bimetallic approaches.
Scheme 13.
General conditions and plausible mechanism for the preparation of allylated chiral β-lactams by a Rh(II)/Pd(0)-catalyzed bimetallic approach.
Scheme 13.
General conditions and plausible mechanism for the preparation of allylated chiral β-lactams by a Rh(II)/Pd(0)-catalyzed bimetallic approach.
Scheme 14.
General conditions and plausible mechanism for the preparation of α-quaternary chiral β-lactams by a Cu(I)/Pd(0)-catalyzed bimetallic approach.
Scheme 14.
General conditions and plausible mechanism for the preparation of α-quaternary chiral β-lactams by a Cu(I)/Pd(0)-catalyzed bimetallic approach.
Scheme 15.
General conditions and plausible mechanism for the preparation of functionalized N-heterocycles, including triazole and indole scaffolds by a Cu(I)/Pd(II)-catalyzed bimetallic approach.
Scheme 15.
General conditions and plausible mechanism for the preparation of functionalized N-heterocycles, including triazole and indole scaffolds by a Cu(I)/Pd(II)-catalyzed bimetallic approach.
Scheme 16.
General conditions and plausible mechanism for the preparation of 1,4,5-trisubstituted 1,2,3-triazoles by a Cu(I)/Pd(0)-catalyzed bimetallic approach.
Scheme 16.
General conditions and plausible mechanism for the preparation of 1,4,5-trisubstituted 1,2,3-triazoles by a Cu(I)/Pd(0)-catalyzed bimetallic approach.
Scheme 17.
General conditions and plausible mechanism for the preparation of 1,2,3-triazole/quinoline-fused imidazo[1,2-a]pyridines by a Cu(I)/Pd(II)-catalyzed bimetallic approach.
Scheme 17.
General conditions and plausible mechanism for the preparation of 1,2,3-triazole/quinoline-fused imidazo[1,2-a]pyridines by a Cu(I)/Pd(II)-catalyzed bimetallic approach.
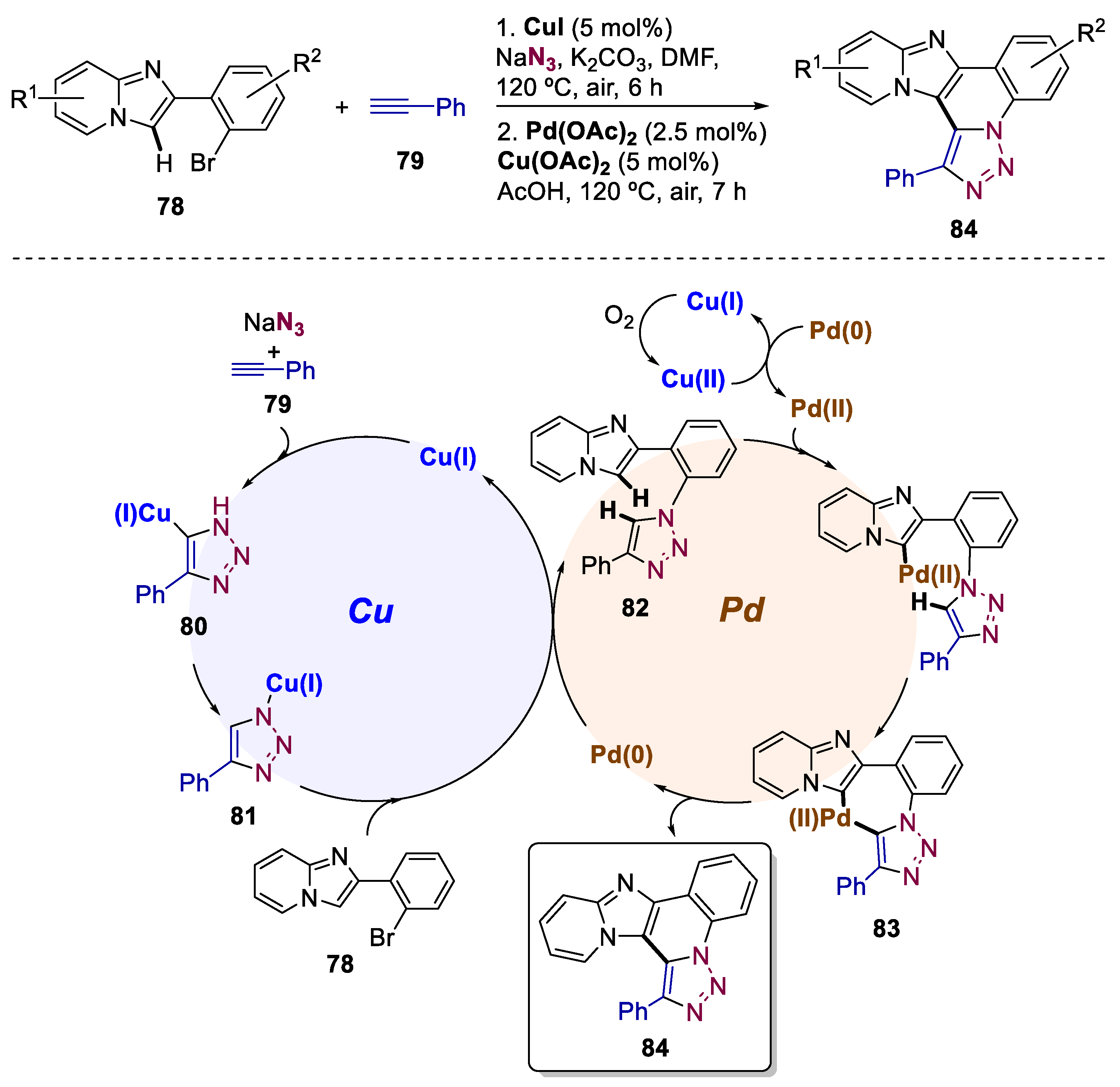
Scheme 18.
General conditions and plausible mechanism for the preparation of aminotetrazoles by a Pd(0)/Fe(III)-catalyzed bimetallic approach.
Scheme 18.
General conditions and plausible mechanism for the preparation of aminotetrazoles by a Pd(0)/Fe(III)-catalyzed bimetallic approach.
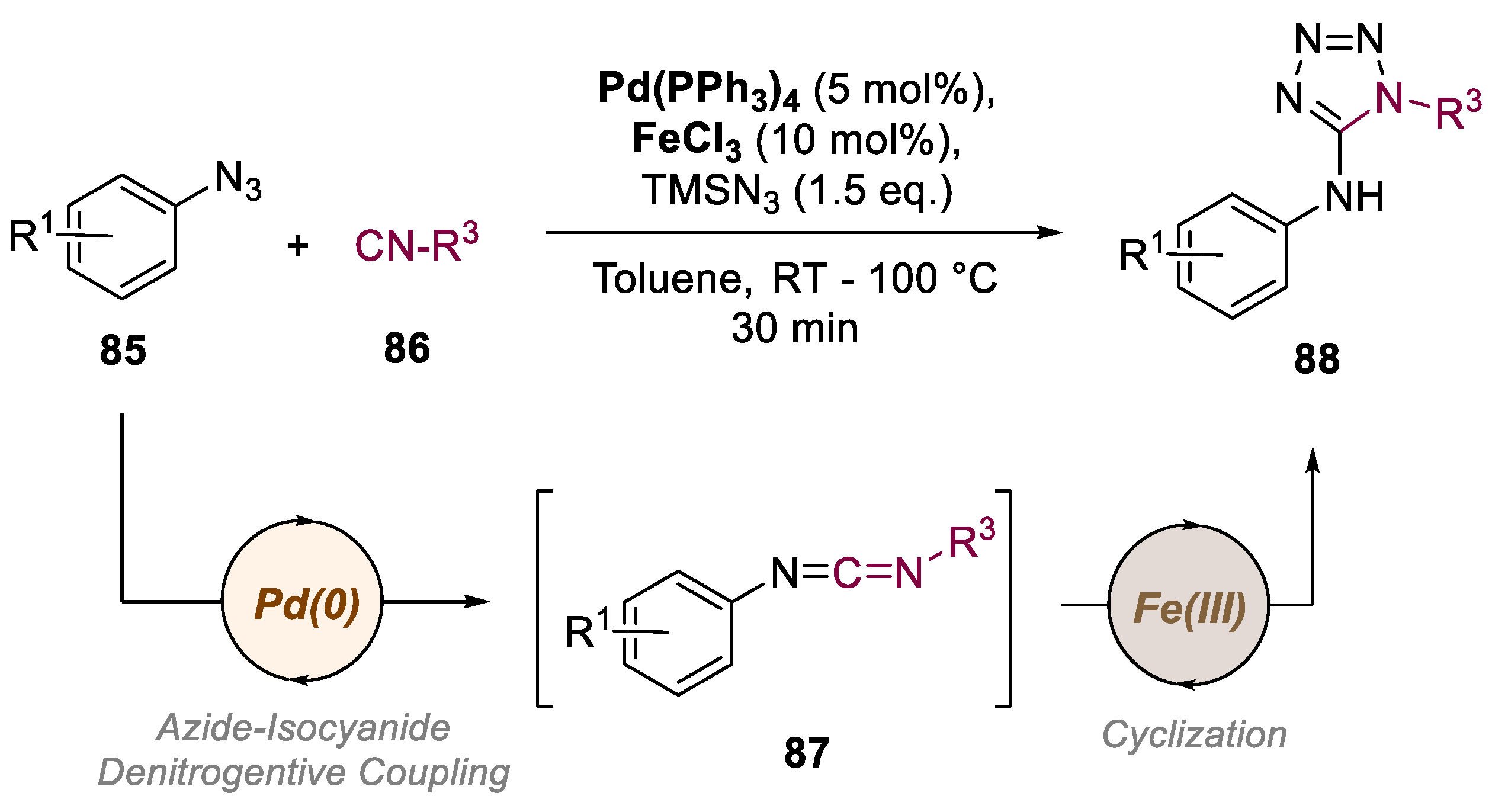
Scheme 19.
General conditions and plausible mechanism for the preparation of cis- and trans-3,4-dihydroisoquinolinones by a Re(I)/Mg-catalyzed bimetallic approach.
Scheme 19.
General conditions and plausible mechanism for the preparation of cis- and trans-3,4-dihydroisoquinolinones by a Re(I)/Mg-catalyzed bimetallic approach.
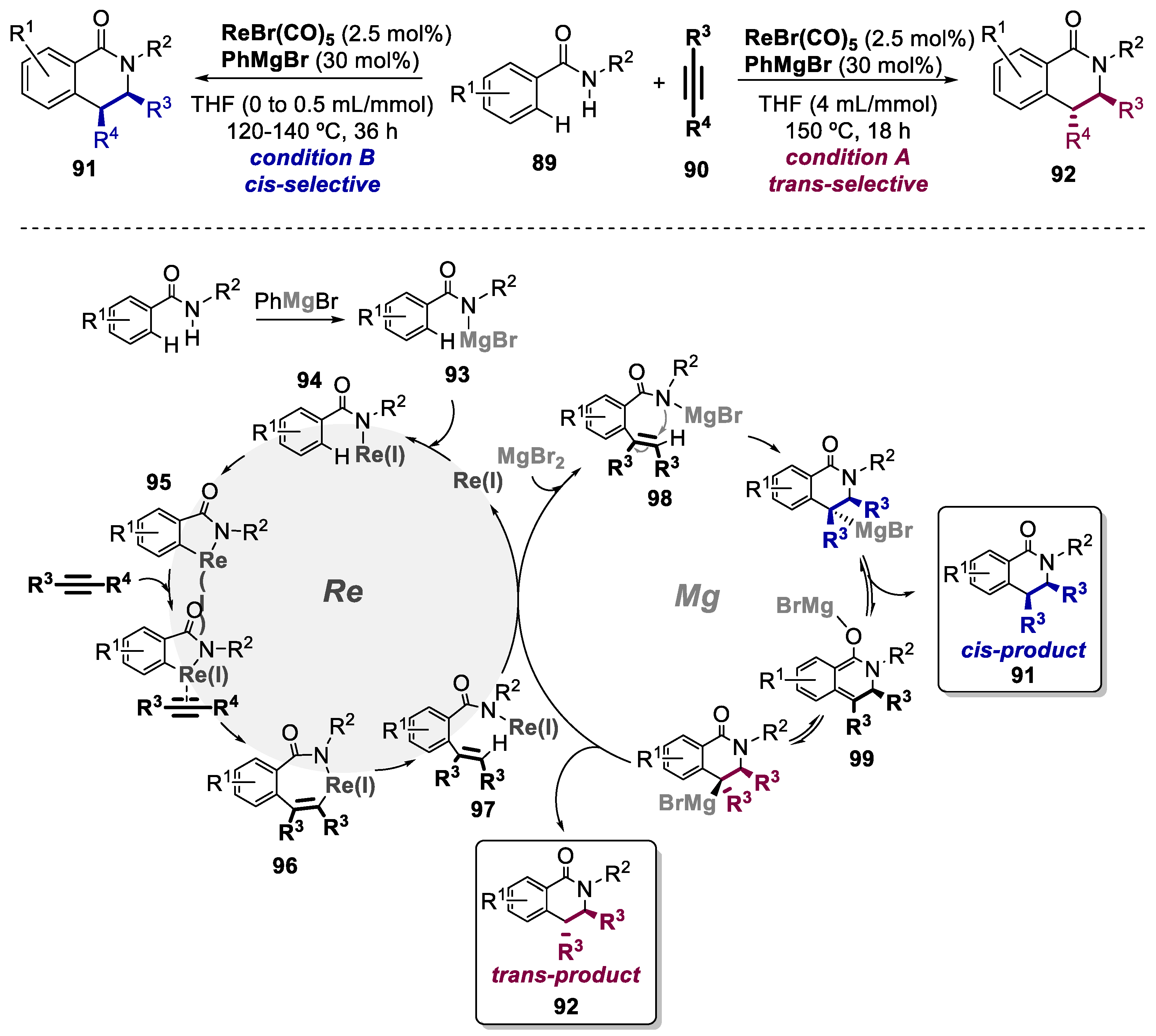
Scheme 20.
General conditions and plausible mechanism for the preparation of hetero-coupled bi(hetero)aryls by a Ni(II)/Pd(II)-catalyzed bimetallic approach.
Scheme 20.
General conditions and plausible mechanism for the preparation of hetero-coupled bi(hetero)aryls by a Ni(II)/Pd(II)-catalyzed bimetallic approach.
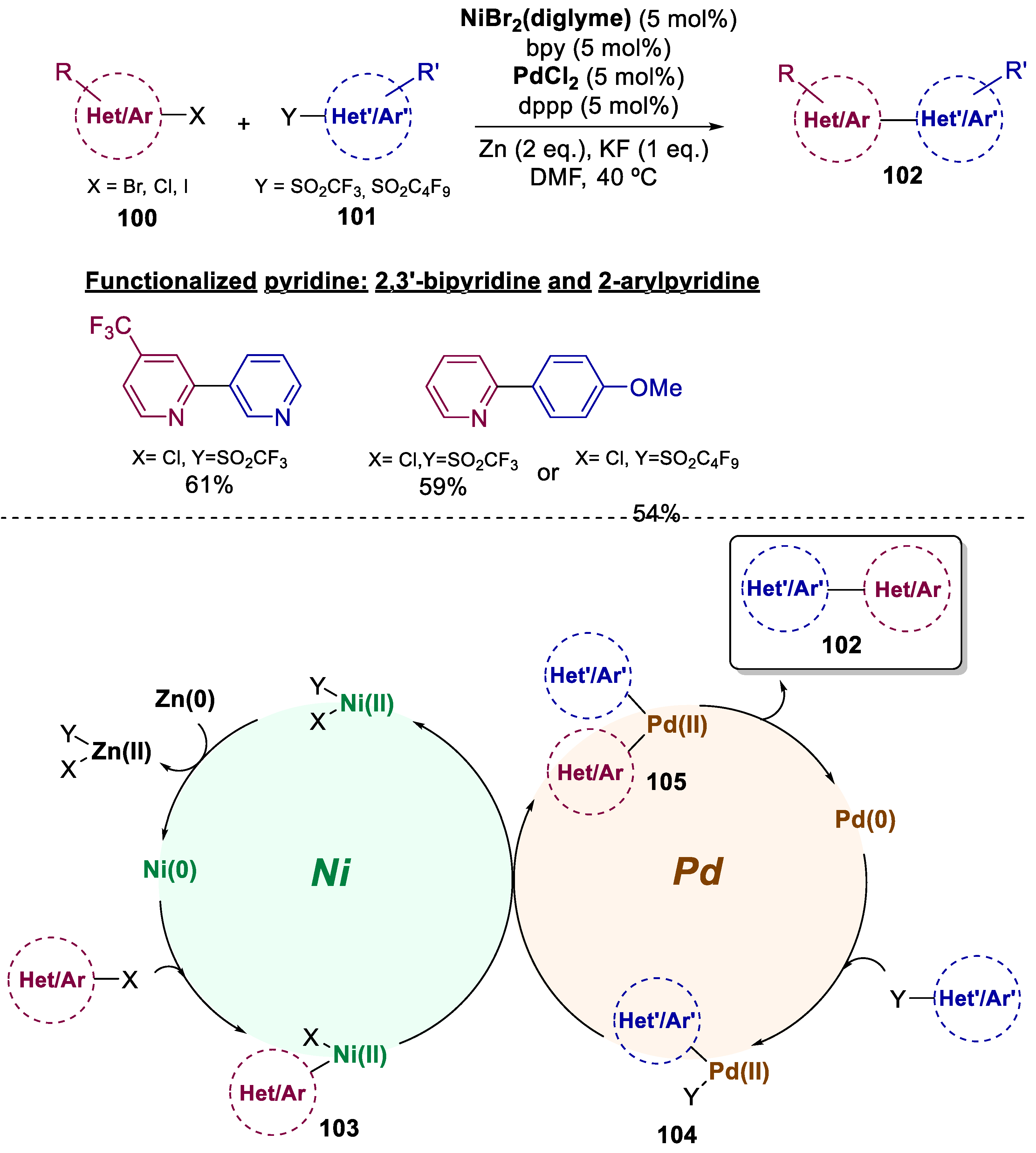
Scheme 21.
General conditions and plausible mechanism for the preparation of polyheterocycles by a Cu(I)/Pd(II)-catalyzed bimetallic approach.
Scheme 21.
General conditions and plausible mechanism for the preparation of polyheterocycles by a Cu(I)/Pd(II)-catalyzed bimetallic approach.
Scheme 22.
General conditions and plausible mechanism of the preparation of quinazoline N-oxides by a Rh(III)/Zn(II)-catalyzed bimetallic approach.
Scheme 22.
General conditions and plausible mechanism of the preparation of quinazoline N-oxides by a Rh(III)/Zn(II)-catalyzed bimetallic approach.
Scheme 23.
General conditions for the preparation of 3-benzazepinones by a Pd(0)/Al(III)-catalyzed bimetallic approach and dihydropyridinones and uracils by a Pd(0)/Cu(I)-catalyzed bimetallic approach.
Scheme 23.
General conditions for the preparation of 3-benzazepinones by a Pd(0)/Al(III)-catalyzed bimetallic approach and dihydropyridinones and uracils by a Pd(0)/Cu(I)-catalyzed bimetallic approach.
Scheme 24.
General conditions and plausible mechanism of the preparation of phthalimides and 3,3’-disubstituted succinimides by a Ni(II)/Cu(I)-catalyzed bimetallic approach.
Scheme 24.
General conditions and plausible mechanism of the preparation of phthalimides and 3,3’-disubstituted succinimides by a Ni(II)/Cu(I)-catalyzed bimetallic approach.
Scheme 25.
General conditions and plausible mechanism of the preparation of trans-β-arylcycloalkanols by a Ni(II)/Ti(IV)-catalyzed bimetallic approach.
Scheme 25.
General conditions and plausible mechanism of the preparation of trans-β-arylcycloalkanols by a Ni(II)/Ti(IV)-catalyzed bimetallic approach.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



