Submitted:
02 August 2023
Posted:
03 August 2023
You are already at the latest version
Abstract
Among phosphorylated derivatives, phosphinates occupy a prominent place due to their ability to be bioisosteres of phosphates and carboxylates. These properties imply the necessity to develop efficient methodologies leading to phosphinate scaffolds. For the past years our team have explored the nucleophilic potential of silylated phosphonite towards various electrophiles. In this paper, we propose to extend our study over other electrophiles. We describe here the implementation of a cascade reaction between (trimethylsilyl)imidates and hypophosphorous acid mediated by a Lewis acid allowing the synthesis of aminomethylenebisphosphinate derivatives.
Keywords:
bisphosphinates
; phosphonite
; Lewis acid
; methodological development
1. Introduction
The synthesis of phosphorylated molecules still represents a major challenge for organic chemists to propose new drugs.[1,2,3,4] In the midst of them, phosphinate derivatives (R2R3PO2R1) have gained attention in medicinal chemistry for their potential as bioactive compounds and drug candidates thanks to their ability to mimic phosphate or carboxylate function. Indeed, the presence of P-C bond imparts chemical stability towards hydrolysis, whether it occurs through chemical or enzymatic processes.[2,4]
Hence, the development of efficient methodologies is crucial to access phosphorylated scaffolds. The formation of the P-C bond can be managed by several pathways such as transition metal catalysis, radical reactions, nucleophilic additions or substitutions.[5] Among these methods, the use of silylated phosphonite II represents a versatile tool operating in a smooth way and thus compatible with functionalized molecules. Moreover, they are easily accessible by reaction between H-phosphinate I and a silylated agent like HMDS, TMSCl or bis(trimethylsilyl)acetamide (BSA) depicted in Figure 1. The sila-Arbuzov reaction of silylated phosphonites II on alkyl halides as electrophiles can provide substituted alkyl phosphinates. Aldehydes, ketones and imines can also undergo the nucleophilic attack of silylated phosphonites via Abramov reaction to give various α-hydroxy- and α-aminophosphinates respectively. In addition, the Michael addition on α,β-unsaturated ketones can selectively take place to form functionalized substituted phosphinates.[6]
Our group have contributed to the use of the simplest silylated phosphonite: bis(trimethylsilyl)phosphonite II (R1= TMS, R2= H: BTSP) obtained starting from hypophosphorous acid (H3PO2) and BSA as silylating agent. First, we have demonstrated that only 2 equivalents of BSA were required to fully transform H3PO2 into BTSP despite large excesses of silylated agents were previously employed in the literature.[6] Then, the subsequent addition on aldehydes and ketones provided various α-hydroxyphosphinates as sodium salts in good to excellent yields.[7]
Moreover, we have also performed the successive double nucleophilic addition of BTSP onto trivalent electrophiles as acyl chlorides which enabled the formation of hydroxymethylenebisphosphinates (HMBPi) via silylated α-ketophosphinates in good to excellent yields and short reaction times.[8,9] Thereafter, this easily handled methodology was successfully used on other trivalent electrophiles, like anhydrides and activated esters, which led to more functionalized HMBPi derivatives in good yields.[10]
This method has been subsequently transposed to synthesize hydroxymethylene(phosphinyl)phosphonate derivatives (HMPPs) which have consisted of adding BTSP on in situ pre-formed α-ketophosphonates starting from trimethylphosphite and acyl chlorides. This one-pot procedure allowed the preparation of original HMPPs in which no purification of intermediate species was required (Figure 1, (a)).[11]
Besides, these methodologies were applied to the synthesis of aminoalkyl-substituted HMBPi and HMPPs which are analogues of hydroxymethylenebisphosphonates (HMBPs) currently used in clinics to treat bone diseases such as osteoporosis, solid tumor metastases or myeloma bone disease.[12,13,14,15,16,17,18,19] Moreover, HMBPs have shown interesting antitumor properties on in vitro and in vivo models of soft tissue primary tumor. As a result, the antiproliferative activities of these newly synthesized HMBPi and HMPPs have been evaluated on various cancer cell lines and encouraging results were obtained especially on A549 cells (Figure 1, (b)).[11]
Additionally, several α-aminomethylenebisphosphonates (AMBPs) exhibit biological activities which include antiparasitic,[20,21] antibacterial,[22] herbicidal [23,24] and bone resorption inhibitor [14,25] (Figure 2, (a)). The access to AMBPs is well documented in the literature. [26] Indeed, several approaches display, the double phosphonylation of amides and nitriles mediated by various Lewis acids,[27,28,29,30] a three-component reaction of amines with orthoformate and phosphites,[31,32,33,34,35,36,37,38,39,40] and a Beckmann transposition of oximes in the presence of phosphites [41,42] (Figure 2, (b)). Alternatively, only limited examples were reported for the synthesis of aminomethylenebisphosphinates (AMBPi) and their biological activities remain unknown to date.[43,44] Here, the strategy usually consists of adding in situ pre-formed BTSP onto ethyl formimidate hydrochloride or onto substituted amides in the presence of TMSOTf, respectively (Figure 2, (c)). In this case, only few N-substituted aminomethylenebisphosphinates (AMBPi) were synthesized.
As a result of these works, our team has decided to pursue exploring the nucleophilic potential of BTSP towards less reactive trivalent electrophiles such as nitriles.
In this case, aminomethylenebisphosphinate (AMBPi) scaffolds will be formed through the successive double addition of BTSP on nitriles. However, the lack of nitrile reactivity should require the use of a Lewis acid as was demonstrated in AMBP series.
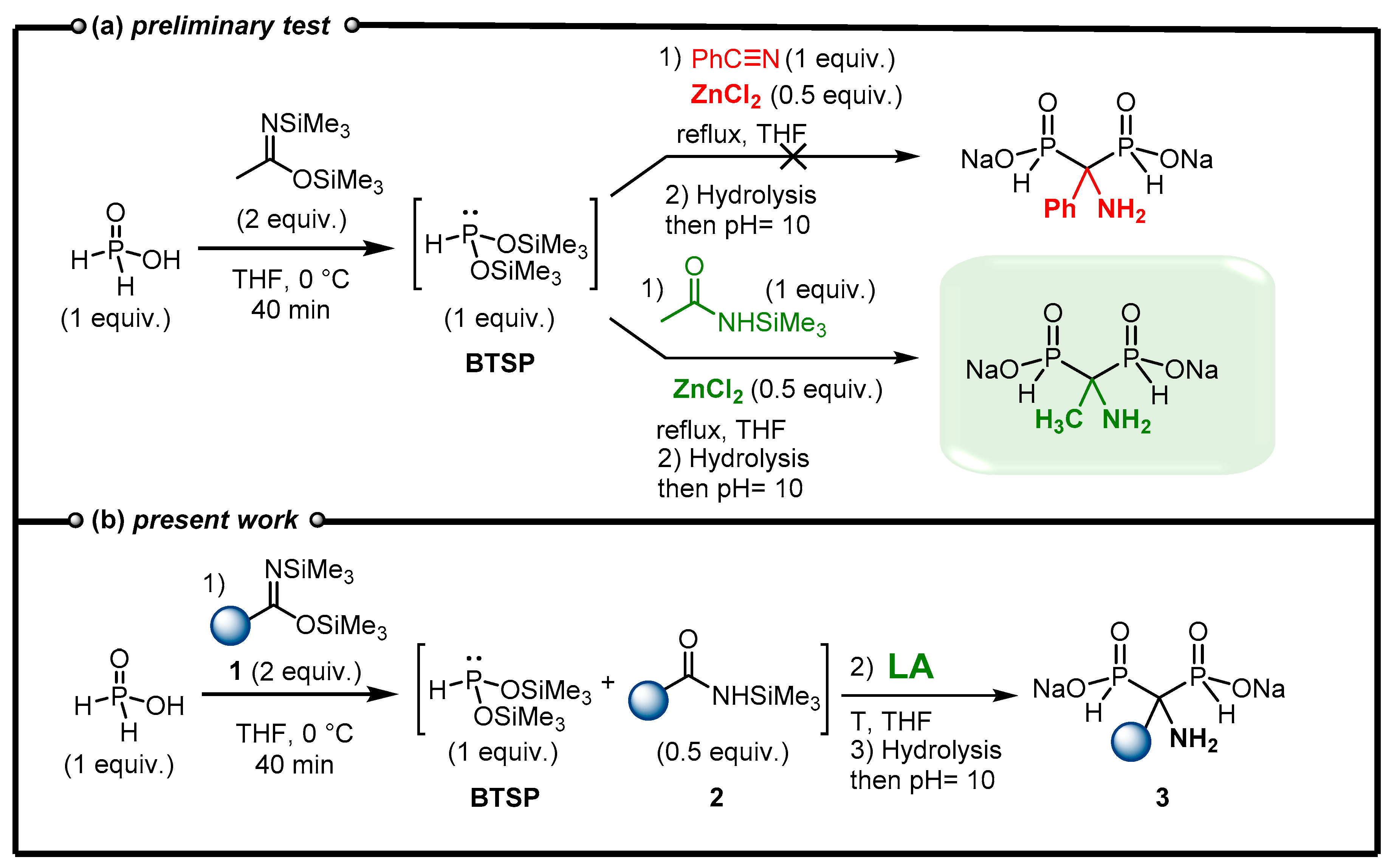
Our initial experiment consisted of the in situ formation of BTSP by silylation of H3PO2 in the presence of BSA in THF followed by the addition of benzonitrile and ZnCl2 as Lewis acid (Figure 3, (a)). Finally, the reaction mixture was stirred under reflux as no conversion was observed at room temperature. The reaction evolution was monitored by 31P and 31P{1H} NMR experiments. After refluxing 6 h, the complete conversion of BTSP was observed and the following methanolysis conducted to an AMBPi derivative. However, the careful analysis of the 1H, 13C spectra and mass spectroscopy indicates the formation of an α-aminomethylenebisphosphinate including a methyl substituent instead of the expected phenyl group. Consequently, the reaction did not occur on nitrile but on the N-silylacetamide generated during the silylation step in the presence of bis(trimethylsilyl)acetamide which is in accordance with some reported works previously mentioned.[30] N-Silylacetamide appears to be a better electrophile than benzonitrile towards the attack of the nucleophilic BTSP.
In view of this unexpected result, we decided to explore the feasibility of developing a cascade reaction in which a bis(trimethylsilyl)imidate 1 could silylate H3PO2 to simultaneously generate nucleophilic BTSP and an electrophilic N-silylamide 2. (Figure 3, (b)). Subsequently and in the presence of a Lewis acid, these products could react together to enable the formation of AMBPi derivatives. Herein, we present our endeavors to develop an efficient cascade process promoted by a Lewis acid to furnish α-aminomethylenebisphosphinates 3 (AMBPi).
2. Results & discussion
2.1. Synthesis of bis(silyl)imidates 1
First, we focused on the synthesis of N,O- bis(trimethylsilyl)imidates 1 [45,46] which could be achieved by silylation of amides [47,48] or by adding LiHMDS on acyl chlorides [45,49,50,51,52] (Scheme 1).
In both cases, the reaction allows to form the N,N-bis(silyl)amides 4 which instantly tautomerizes to the more stable N,O-bis(trimethylsilyl)imidates 1.[45,51]. Indeed, the reaction monitoring by 13C NMR enabled to only detect the quaternary carbon of 1 at ~160-165 ppm. The signal at ~147 ppm corresponding to N,N-bis(silyl)amides 4 was only observed when 2.2 equivalents of TMSOTf/Et3N were sequentially added in two portions to the corresponding acetamide. In our study, the reaction between amides and TMSOTf/Et3N was selected due to its ease of implementation and higher efficiency.
2.2. Optimization of the reaction between N,O-bis(trimethylsilyl)imidates and phosphorous acid mediated by Lewis acid
The reaction was firstly carried out between hypophosphorous acid and commercially available N,O-bis(trimethylsilyl)acetamide 1a (R= Me) (Table 1). The silylation was monitored by 31P NMR and was completed after 40 minutes at 0 °C.
Thereafter, various Lewis acids were screened for the second reaction between trimethylsilylacetamide 2a and BTSP (Table 1, entries 1-4). In the presence of zinc halides, AMBPi 3a was similarly obtained in good conversions and isolated yields after purification (Table 1, entries 1, 2). However, it was noted that the reaction rate is higher with ZnI2 than with ZnCl2, as the former allowed to complete the reaction after only 1.5 hours, whereas the latter took 18 hours. When TMSOTf was used as Lewis acid, the reaction was able to proceed at 0 °C after only 30 minutes and furnished AMBPi 3a in 75 % yield (Table 1, entry 3). In contrast, no conversion was observed in the presence of BF3.OEt2 regardless of the temperature and reaction time (Table 1, entry 4).
Then, the same reactions were performed with freshly prepared N,O-bis(trimethylsilyl)acetamide 1a (R= Me) in the presence of ZnX2 or TMSOTf (Table 1 : entries 5-7 versus 1-3). In these cases, the reactions provided the same results independently of the Lewis acids as expected.
To explore the potential range of the reaction, additional N,O-bis(trimethylsilyl)imidates 1b (R= Pr) and 1c (R= Ph) were initially combined with H3PO2, and the resulting blend was subsequently subjected to various Lewis acids (Table 1, entries 8-13).
Concerning the use of N,O-bis(trimethylsilyl)butanimidate 1b, the silylation of H3PO2 was completed after 40 minutes at 0 °C. The consequently double addition of BTSP on the corresponding silylamide 2b successfully ensued in the presence of ZnX2 to furnish the expected AMBPi 3b after methanolysis and purification (Table 1, entries 8,9). As previously observed, the reaction rate is higher for ZnI2 than for ZnCl2. However, we noted a dramatic drop of the conversion into 2b in the presence of TMSOTf, as a major disproportionation of BTSP was observed (Table 1, entry 10).
Upon investigating the reactivity of aromatic bis(trimethylsilyl)amide (Table 1, entries 11-13), it was revealed that among the various Lewis acids tested, zinc iodide uniquely mediated the attack of BTSP onto 2c, leading to the proper formation of AMBPi 3c (Table 1, entry 12). Indeed, no reaction took place in the presence of TMSOTf (Table 1, entry 13); moreover, the use of zinc chloride resulted in the formation of α-aminophosphinate 5c and a major disproportionation of BTSP into silylated phosphorus derivatives (Table 1, entry 11). Furthermore, the reactivity of zinc chloride seems inadequate to promote the sila-Arbuzov reaction. Additionally, AMBPi 3c may not be stable enough and seems to lead to the formation of 5c, as previously described in the literature.[43]
Finally, the optimization of the cascade reaction showed the potent use of various commercially available and freshly prepared aliphatic and aromatic N,O-bis(trimethylsilyl)imidates 1a-c as silylating agent. In addition, the Lewis acid screening highlighted zinc iodide as the best compromise in terms of reactivity and reaction time.
2.2. NMR monitoring and purification details
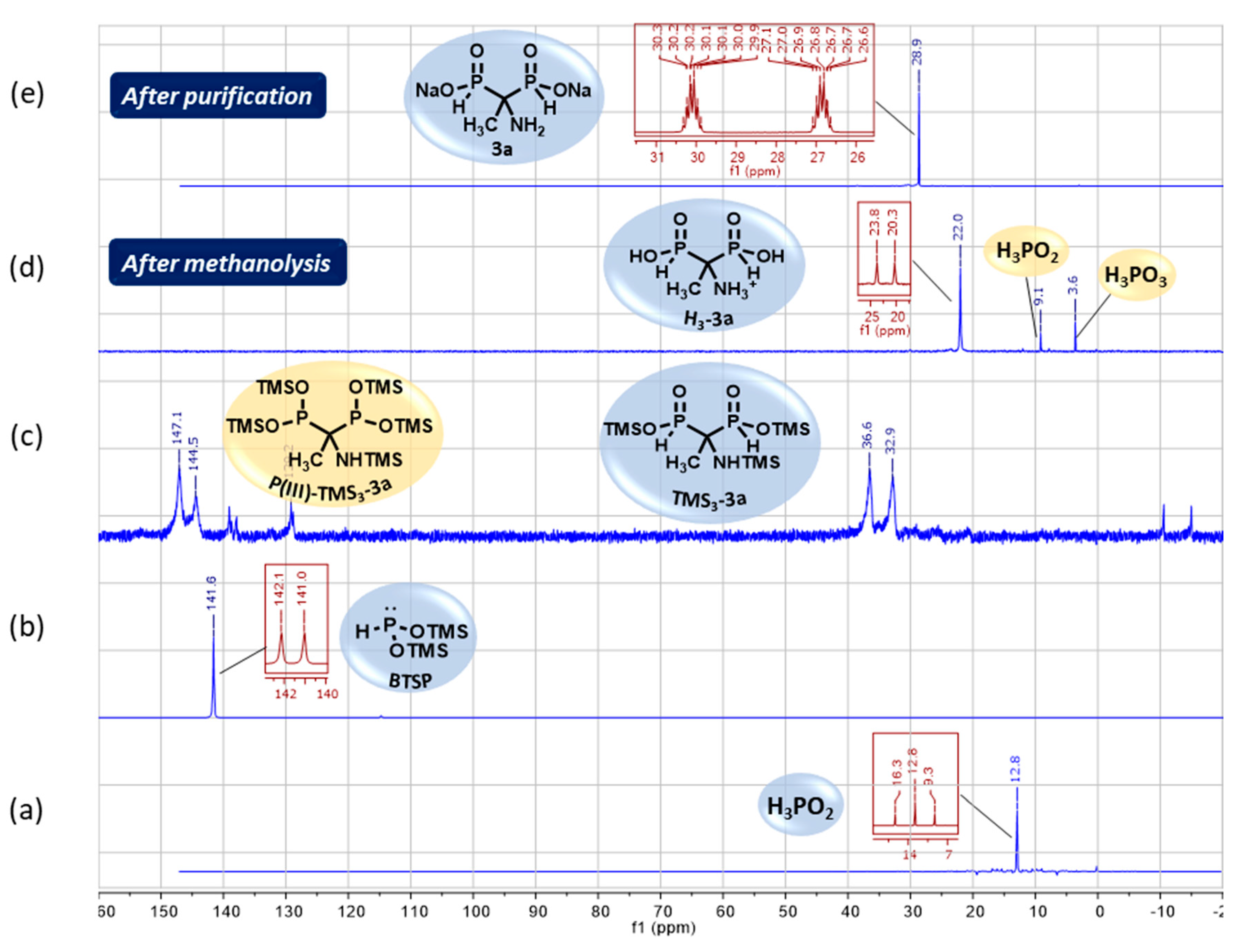
As mentioned earlier, 31P and 31P {1H} NMR experiments are routinely performed to follow the course of the reactions implying phosphorus derivatives. Figure 4 displays the optimized cascade reaction monitoring of the various phosphorus intermediate species.
The rapid silylation of H3PO2 was observed by the disappearance of its signal at 12.8 ppm for the benefit of a new signal at 141.6 ppm in the trivalent phosphorus region that confirmed the formation of BTSP as expected (Figure 4, spectra (b) versus (a)). After refluxing 1.5 hours, the NMR monitoring indicated the complete conversion of BTSP by its missing signal at 141.6 ppm and the appearance of several peaks in the P(III)/P(V) regions (142-147 ppm and 33-36 ppm) related to P(III)-TMS3-3a and TMS3-3a respectively (Figure 4, spectra (c) versus (b)). After methanolysis, a major signal remained at 22.0 ppm which matches the acidic form H3-3a of AMBPi 3a (Figure 4, spectra (d) versus (c)). It was noted that small amounts of H3PO2 and H3PO3 were also generated. The pH adjustment at 10 conducted to AMBPi 3a as a disodium salt and concomitantly the partial precipitation of the zinc salt which was eliminated by centrifugation. Then, successive washes by ethyl acetate (with 0-10% ethanol) and methanol enabled to recover the excess of amide, and to discard both NaH2PO2 and Na2HPO3 respectively. The residual zinc salts were removed thanks to a cation-exchange resin.
The effectiveness of zinc elimination was verified by ICP-AES analysis. Finally, the disodium salt of AMBPi 3a was isolated after lyophilization as a pure form (Figure 4, spectra (e) versus (d)).
2.3. Viability considerations of the cascade reaction
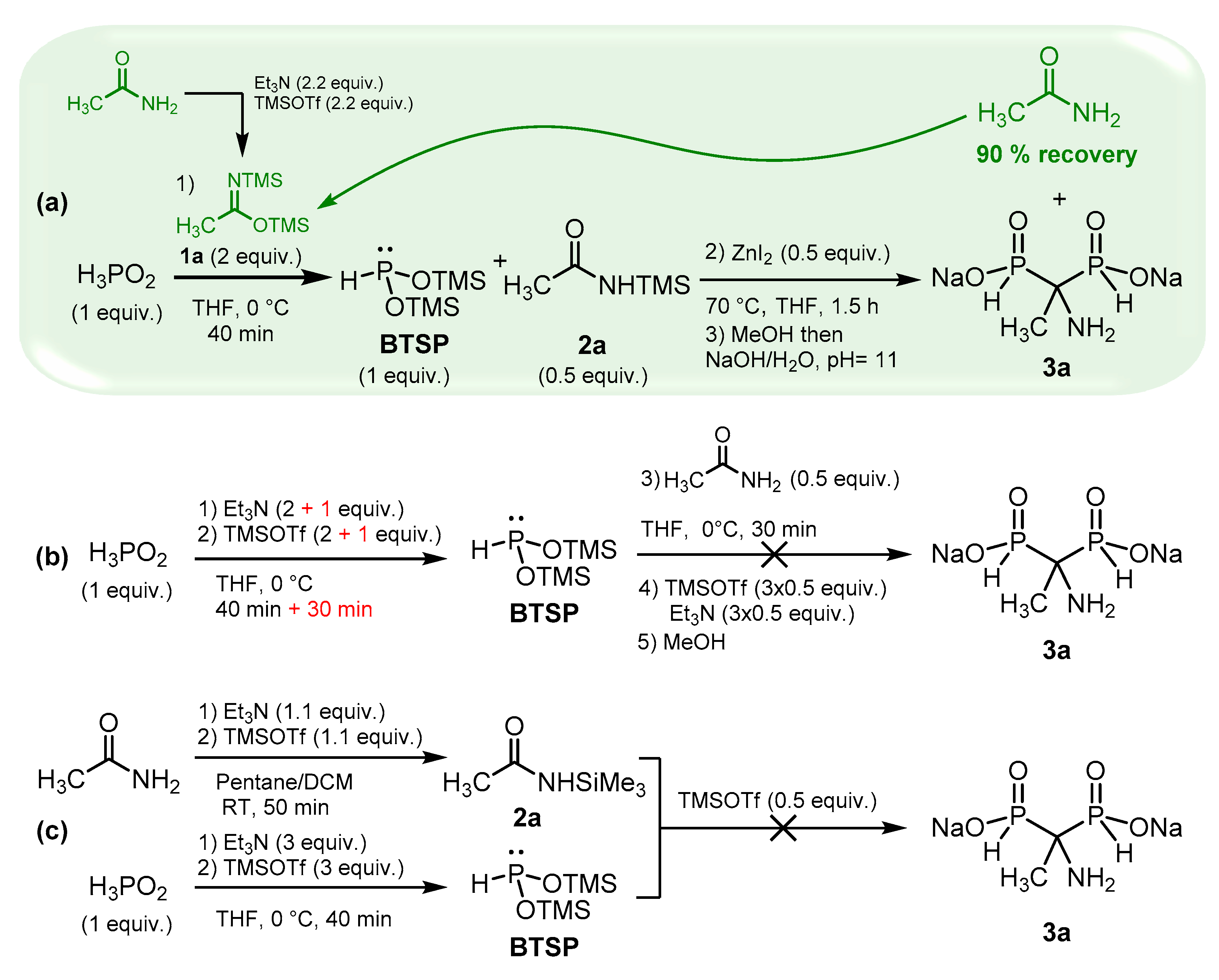
The cascade reaction appears to be an interesting and straightforward method for accessing AMBPi scaffolds; however, 2 equivalents of silylimidates 1 are required for the silylation step, but only 0.5 equivalent of the silylamide generated in the first step participates in the sila-Arbuzov reaction. To overcome this significant drawback, we focused on retrieving the excess of amides initially used for the formation of the silylimidates (Scheme 2, (1)). Several attempts enabled us to finally recover up to 90 % of the resulting amides during the purification procedure which could be reused for the same reaction.
We also wondered if the reaction could proceed via a direct silylation of H3PO2 in the presence of Et3N/TMSOTf followed by the addition of an amide in the presence of TMSOTf. Here, TMSOTf will both play the role of silylating agent and Lewis acid (Scheme 2, (b)). First, hypophosphorous acid, and 2 equivalents of triethylamine/TMSOTf were mixed at 0 °C. Unfortunately, the silylation was partial even after an extended period. To achieve completion, an additional equivalent of Et3N/TMSOTf was added, leading to a 30-minute reaction time. However, this approach resulted in a significant amount of HP(O)(OTMS)2 due to the oxidation of BTSP. In the second step, acetamide and TMSOTf were subsequently introduced at 0 °C.
Unfortunately, no AMBPi derivative was formed, and the reaction only resulted in the disproportionation of BTSP, giving hypophosphorous and phosphorous acids.
As a final attempt, we independently synthesized 2a and BTSP in the presence of triethylamine and TMSOTf, which were subsequently mixed together (Scheme 2, (c)). Alas, the reaction did not occur under these conditions.
These assays validated the viability of the cascade reaction we proposed. Moreover, the excess of amide can be successfully recovered, thus limiting its impact on the reaction implementation.
2.3. Scope of the cascade reaction
The scope of the cascade reaction was carried out in the presence of various prepared aliphatic and aromatic bis(trimethylsilyl)imidates 1a-l, hypophosphorous acid and zinc iodide as Lewis acid (Scheme 3). As a general trend, all imidates enabled to promote the silylation of H3PO2 into BTSP efficiently.
We were pleased to observe that the reaction was successful with aliphatic imidates bearing a longer chain 1b and 1d. In these cases, the corresponding AMBPi 3b and 3d were obtained in good conversions and isolated yields after purification. Although bis(trimethylsilyl)trifluoroacetimidate 1e can properly promote the silylation of H3PO2, the sila-Arbuzov reaction did not happen. It was only noted the oxidation of BTSP.
Concerning the use of aromatic bis(trimethylsilyl)imidate derivatives, the reactivity of para-substituted aromatic imidate derivatives was evaluated under the previous optimized conditions. The substitution at the para- position by a methoxy- group had little influence on the course of the reaction which conducted to AMBPi 3f in similar yield than 3c. When the reaction was performed with a para-substituted methyl moiety on silylamide 2i, the yield for the formation of 3i surprisingly decreased.
However, meta-substituted methyl aromatic amide 2j properly underwent the sila- Arbuzov in good conversion and isolated yield for 3j. The reaction was also carried out with electrowithdrawing para-fluoro and para-trifluoromethyl substituted groups on amides 2g and 2h. Although the conversion into 3g reached 60 %, the isolated yield dropped to 29 % due to its oxidation during the purification. Moreover, 3 h was not produced as only the disproportionation of BTSP took place.
The introduction of ortho- substituted methyl group and heteroaromatic moiety on aromatic imidates 2k and 2l was also considered. In these cases, only AMBPi 3l was generated in good conversion and isolated yield, probably due to steric hindrance of substrate 2k.
It was noted that α-aminophosphinates 5c,f,g,i,j were detected after methanolysis. According to NMR spectra, these compounds represented 10 to 15 % proportion (31P NMR) of crude products. This observation could justify the lower yields obtained for these AMBPi 3c,f,g,i,j.
Even if the results were moderate for some substituted aromatic AMBPi, this cascade reaction represents the first example of AMBPi which displays alkyl or aromatic groups at the methylene carbon on α-aminomethylenebisphosphinates.
3. Materials and Methods
3.1. General informations
Reagents were purchased from usual commercial suppliers (Sigma-Aldrich, Alfa Aesar, Acros Organics) and used as delivered. Triethylamine was distillated and stored over KOH under argon. All solvents were extra-dried grade prior used. N,O-bis(trimethylsilyl)acetamide (BSA) was purchased from Alfa Aesar (batch number: 10186753). Anhydrous H3PO2 was dehydrated from commercially available aqueous solution of H3PO2 (50% w/w) according to the procedure reported by Montchamp et al [53] Reactions requiring inert conditions were carried out in flame-dried glassware under an argon atmosphere. The solvents were degassed by argon bubbling for 30 minutes.
NMR spectra were recorded at 20 °C on a Bruker Avance-III-400 spectrometer (1H: 400 MHz, 13C: 101 MHz, 31P: 162 MHz, 19F: 377 MHz). Chemical shifts (δ) were given in ppm, the number of protons (n) for a given resonance was indicated by n H and coupling constants J in Hz. 1H NMR spectra were calibrated on non-deuterated solvent residual peak (H2O: 4.79 ppm) while H3PO4 (85% in water) was used as an external standard for 31P NMR. The following abbreviations were used for 1H, 13C, 31P and 19F NMR spectra to indicate the signal multiplicity: s (singlet), d (doublet), t (triplet), dd (doublet of doublets), dm (doublet of multiplet), m (multiplet), dq (doublet of quartets) and ddq (doublet of doublets of quartets). All 13C NMR spectra were measured with 1H decoupling while 31P and 19F NMR spectra were measured with 1H coupling and 1H decoupling. 1H experiments with water presaturation were performed with D1 = 2 s and 128 scans. The reactions were followed by 31P and 31P{1H} NMR experiments (the spectra were recorded without lock and shims). All NMR peak assignments were performed thanks to 2D NMR COSY, HMQC and HMBC experiments. High-resolution mass spectra (HRMS) were performed on a Bruker maXis mass spectrometer in negative (ESI-) mode (ESI) by the "Fédération de Recherche" ICOA/CBM (FR2708) platform. MS analyses were performed using a QTOF Impact HD mass spectrometer equipped with the electrospray (ESI) ion source (Bruker Daltonics). The instrument was operated in the negative mode with an ESI source on a Q-TOF mass spectrometer with an accuracy tolerance of 2 ppm. Samples were diluted with acetonitrile and water (15:85) and were analyzed by mass spectrometry in continuous infusion using a syringe pump at 200 µL/min. The mass profiles obtained by ESI-MS were analyzed using Data Analysis software (Bruker Daltonics). ICP-AES analyses were performed by “plateforme Analytiques des Inorganiques” IPHC UMR7178 on Varian 720ES.
3.2. General procedure for the cascade synthesis of Aminomethylenebisphosphinates 3a-l
To a dry and argon flushed 100 mL three-necked flask, equipped with a thermometer, an argon inlet and a septum, were successively introduced the corresponding amide 1a-l (15.00 mmol, 1.00 equiv.), anhydrous pentane (34.00 mL), anhydrous dichloromethane (1.50 mL) and triethylamine (33.00 mmol, 5.58 mL, 2.20 equiv.). Trimethylsilyltrifloromethanesulfonate (33.00 mmol, 5.73 mL, 2.20 equiv.) was added dropwise at 0 °C and the mixture was stirred for 30 minutes at room temperature. The lower phase obtained during the process was eliminated. Then, the solvent was evaporated under reduced pressure. The imidates 2a-l were used in the next step without further purification.
To another dry and argon flushed 25 mL three necked flask equipped with a thermometer, a reflux condenser with an argon inlet and a septum was added anhydrous hypophosphorous acid (5.00 mmol, 0.330 g, 0.50 equiv.) and anhydrous tetrahydrofuran (1.00 mL) under argon atmosphere. The synthesized imidates 2a-l (10.00 mmol, 2.00 equiv.) were added dropwise at 0 °C and the mixture was stirred for 40 minutes. The reaction conversion was monitored by 31P NMR. A solution of zinc iodide (2.50 mmol, 0.750 g, 0.50 equiv.) in anhydrous tetrahydrofuran (4.00 mL) was added dropwise at 0°C and the mixture was stirred under reflux condition. The reaction conversion was also monitored by 31P NMR upon completion. Then, anhydrous methanol (3.00 mL) was added dropwise at 0°C. The solvent was evaporated, and the crude compound was dissolved in minimum of water (2.00 mL) and an aqueous solution of sodium hydroxide (0.50 M, 8.00 mL) was added carefully to adjust pH to 10.00. The mixture was centrifugated to partially discard precipitated zinc salts. The filtrate was then washed with ethyl acetate (5 x 5.00 mL) (with 0-10 % ethanol) and methanol (10 x 2.00 mL) to eliminate the excess of amides 1a-l and phosphorous acid respectively. In addition, a cation-exchange resin was used to eliminate the residual zinc salts. Finally, the solution was lyophilized to afford the pure AMBPi 3 as a disodium salt.
3.2. Spectral data of aminomethylenebisphosphinates 3a-l
1-aminoethane-1,1-bis(H-phosphinate) disodium salts 3a. White powder. 425 mg, 79 % yield. 31P {1H} NMR (162 MHz, D2O) δ 28.5 (s). 31P NMR (162 MHz, D2O) δ 28.5 (dm, 1JP-H= 524.9 Hz). 1H NMR (400 MHz, D2O) δ 6.80 (dt, 1JP-H= 525.3 Hz, 2J= 11.7 Hz, 2H), 1.19 (t, 2JP-H = 15.8 Hz, 3H). 13C NMR (101 MHz, D2O) δ 52.2 (t, 1JP-C= 89.4 Hz), 15.0. MS (ESI-) m/z 171.99 [M-H]-, 193.97 [M-2H+Na]-, 153.98 [M-H-H2O]-. HRMS (ESI-) m/z: [M-H]- Calcd. for [C2H8NO4P2]: 171.9934, found: 171.9934.
1-amino-1-propylmethane-1,1-bis(H-phosphinate) disodium salts 3b. White powder. 359 mg, 60 % yield. 31P {1H} NMR (162 MHz, D2O) δ 28.1 (s). 31P NMR (162 MHz, D2O) δ 28.1 (dp, 1JP-H= 523.6 Hz, 2J= 13.4 Hz). 1H NMR (400 MHz, D2O) δ 6.84 (dt, 1JP-H= 523.5 Hz, 2J= 12.1 Hz, 2H), 1.72-1.55 (m, 2H), 1.54-1.39 (m, 2H), 0.88 (t, J= 7.2 Hz, 3H). 13C NMR (101 MHz, D2O) δ 55.3 (t, 1JP-C= 89.0 Hz, 33.1, 16,6 (t, 2JP-C = 6.8 Hz), 14.3. MS (ESI-) m/z 200.02 [M-H]-, 222.00 [M-2H+Na]-, 182.01 [M-H-H2O]-, 134.04 [M-H-H3PO2]-. HRMS (ESI-) m/z: [M-H]- Calcd. for [C4H12NO4P2]: 200.0247, found: 200.0247.
1-amino-1-phenylmethane-1,1-bis(H-phosphinate) disodium salts 3c. White powder. 500 mg, 72 % yield. 31P {1H} NMR (162 MHz, D2O) δ 26.8 (s). 31P NMR (162 MHz, D2O) δ 26.8 (dt, 1JP-H= 538.6 Hz, J= 12.1 Hz). 1H NMR (400 MHz, D2O) δ 7.43 (d, 3JP-H = 8.1 Hz, 2H), 7.32 (t, 4JP-H= 7.6 Hz, 2H), 7.23 (t, 1JP-H= 7.6 Hz, 1H), 6.87 (dt, 1JP-H= 539.0 Hz,2J = 10.3 Hz, 2H). 13C NMR (101 MHz, D2O) δ 164.0, 128.5, 127.0, 126.0, 60.5 (t, 1JP-C= 86.0 Hz. MS (ESI-) m/z 234.00 [M-H]-, 215.99 [M-H-H2O]-, 170.04 [M-H-HPO2]-. HRMS (ESI-) m/z: [M-H]- Calcd. for [C7H10NO4P2]: 234.0090, found: 234.0090.
1-amino-1-butylmethane-1,1-bis(H-phosphinate) disodium salts 3d White powder, 417 mg, 65 % yield. 31P {1H} NMR (162 MHz, D2O) δ 27.9 (s). 31P NMR (162 MHz, D2O) δ 27.9 (dp, 1JP-H= 523.8 Hz, 2J= 13.2 Hz). 1H NMR (400 MHz, D2O) δ 6.85 (dt, 1JP-H= 524.3 Hz, 2J= 11.9 Hz, 2H), 1.73-1.62 (m, 2H), 1.48-1.40 (m, 2H), 1.28 (hex., J= 7.3 Hz, 2H), 0.86 (t, J= 7.3 Hz, 3H). 13C NMR (101 MHz, D2O) δ 55.2 (t, 1JP-C= 88.6 Hz), 30.45 (C2), 25.1 (t, 3JP-C = 6.7 Hz), 23.0, 13.1. MS (ESI-) m/z 214.04 [M-H]-, 236.02 [M-2H+Na]-, 196.03 [M-H-H2O]-.HRMS (ESI-) m/z: [M-H]- Calcd. for [C5H14NO4P2]: 214.0403, found: 214.0403.
1-amino-1-(4-methoxyphenyl)methane-1,1-bis(H-phosphinate) disodium salts 3f. White powder. 0.483 mg, 65 % yield. 31P {1H} NMR (162 MHz, D2O) δ 26.8 (s). 31P NMR (162 MHz, D2O) δ 26.8 (dt, 1JP-H= 538.7 Hz, 2J= 12.4 Hz). 1H NMR (400 MHz, D2O) 7.47 (d, 3J= 8.9 Hz, 2H), 7.02 (d, 3JP-H= 8.5 Hz, 2H), δ 6.95 (dt, 1JP-H= 537.9 Hz, 2J= 11.1 Hz, 2H), 3.81 (s, 3H). 13C NMR (101 MHz, D2O) δ 157.8 (t, J = 2.4 Hz), 128.2, 127.4 (t, 3JP-C= 4.9 Hz), 114.0, 59.8 (t, 1JP-C= 86.8 Hz), 55.3. MS (ESI-) m/z 264.02 [M-H]-, 286.00 [M-2H+Na]-, 246.01 [M-H-H2O]-, 200.05 [M-H-HPO2]-. HRMS (ESI-) m/z: [M-H]- Calcd. for [C8H12NO5P2]: 264.0196, found: 200.0204.
1-amino-1-(4-fluorophenyl)methane-1,1-bis(H-phosphinate) disodium salts 3g. White powder. 220 mg, 29 % yield. 31P {1H} NMR (162 MHz, D2O) δ 26.4 (s). 31P NMR (162 MHz, D2O) δ 26.4 (dt, 1JP-H= 538.6 Hz, J= 11.1 Hz). 19P NMR (377 MHz, D2O) δ 116.8 (m). 1H NMR (400 MHz, D2O) δ 7.57-7.46 (m, 2H), 7.14 (t, 4JP-H= 8.8 Hz, 2H), 6.96 (dt, 1JP-H= 537.4 Hz, 1JP-H = 10.8 Hz, 2H). 13C NMR (101 MHz, D2O) δ 160.7 (dt, 1JC-F= 243.2 Hz, 4JP-C= 2.8 Hz), 131.6-131.5 (m), 127.8-127.7 (m), 115.2, 115.0, 60.0 (t, 1JP-C= 86.0 Hz). MS (ESI-) m/z 252.00 [M-H]-, 273.98 [M-2H+Na]-, 233.99 [M-H-H2O]-, 188.03 [M-H-HPO2]-. HRMS (ESI-) m/z: [M-H]- Calcd. for [C7H9FNO4P2]: 251.9996, found: 251.9996.
1-amino-1-(4-tolyl)methane-1,1-bis(H-phosphinate) disodium salts 3i. White powder. 250 mg, 35 % yield. 31P {1H} NMR (162 MHz, D2O) δ 26.8 (s). 31P NMR (162 MHz, D2O) δ 26.8 (dt, 1JP-H= 539.8 Hz, J= 12.0 Hz). 1H NMR (400 MHz, D2O) δ 7.46-7.40 (m, 2H), 7.26-7.22 (m, 2H), 6.96 (dt, 1JP-H= 538.2 Hz, 2J= 10.8 Hz, 2H), 2.30 (s, 3H). 13C NMR (101 MHz, D2O) δ 137.0, 132.6, 129.1, 126.05 (t, 3JP-C= 4.8 Hz), 60.2 (t, 1JP-C= 86.5 Hz), 20.1. MS (ESI-) m/z 248.02 [M-H]-, 270.00 [M-H-H2O]-, 184.05 [M-H-HPO2]-. HRMS (ESI-) m/z: [M-H]- Calcd. for [C8H12FNO4P2]: 248.0247, found: 248.0247.
1-amino-1-(3-tolyl)phenyl)methane-1,1-bis(H-phosphinate) disodium salts 3j. White powder. 400 mg, 57 % yield. 31P {1H} NMR (162 MHz, D2O) δ 26.8 (s). 31P NMR (162 MHz, D2O) δ 26.8 (dt, 1JP-H= 542.3 Hz, J= 11.5 Hz). 1H NMR (400 MHz, D2O) δ 7.36 (s, 1H), 7.30-7.29 (m, 2H), 7.15 (s, 1H), 6.96 (dt, 1JP-H= 538.9 Hz, 2J = 11.8 Hz, 2H), 2.32 (s, 3H). 13C NMR (101 MHz, D2O) δ 138.4, 135.8 (t, 4JP-C = 2.3 Hz), 128.4, 127.6, 126.7 (t, 3JP-C = 4.9 Hz), 123.0 (t, 3JP-C = 5.0 Hz), 60.5 (t, 1JP-C = 86.1 Hz), 20.7. MS (ESI-) m/z 248.02 [M-H]-, 270.01 [M-2H+Na]-, 230.01 [M-H-H2O]-, 184.05 [M-H-HPO2]-. HRMS (ESI-) m/z: [M-H]- Calcd. for [C8H12NO4P2]: 248.0247, found: 248.0247.
1-amino-1-(2-thienyl)ethane-1,1-bis(H-phosphinate) disodium salts 3l. White powder. 437 mg, 59 % yield. 31P {1H} NMR (162 MHz, D2O) δ 26.5 (s). 31P NMR (162 MHz, D2O) δ 26.5 (dm, 1JP-H= 531.6 Hz). 1H NMR (400 MHz, D2O) δ 7.28-7.27 (m, 1H, H6), 6.99-6.97 (m, 2H), 6.83 (dt, 1JP-H = 530.6 Hz, 2J= 11.8 Hz, 2H), 3.26 (t, J= 12.7 Hz, 2H). 13C NMR (101MHz, D2O) δ 136.9 (t, 3JP-C = 9.1 Hz), 128.5, 126.9, 125.1, 55.1 (t, 1JP-C = 89.3 Hz), 29.8. MS (ESI-) m/z 253.98 [M-H]-, 275.96 [M-2H+Na]-, 253.97 [M-H-H2O]-. HRMS (ESI-) m/z: [M-H]- Calcd. for [C6H10NO4P2S]: 253.9811, found: 253.9811.
4. Conclusions
In this study, we have established a cascade reaction involving the silylation of hypophosphorous acid by a N,O-bis(trimethylsilyl)imidate, leading to the formation of bis(trimethylsilyl)phosphonite (BTSP) and a N-silylamide. The latter can subsequently undergo nucleophilic attack of BTSP through a sila-Arbuzov reaction which is mediated by zinc iodide as Lewis acid. This approach relies on an unexpected result as our initial attempt was to investigate the reactivity of nitriles in the presence of BTSP and a Lewis acid. We present a detailed methodology to propose a novel access to AMBPi scaffolds which have been understudied in the literature. The screening of Lewis acid has highlighted zinc iodide as the best promoter for the sila-Arbuzov reaction. Consequently, we successfully synthesized various AMBPi 3a-l in moderate to good yields. Better results were obtained in aliphatic series and will enable us to extend this method to more functionalized AMBPi, analogous to aminomethylenebisphosphonates which have demonstrated relevant biological activities.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. 1H-, 13C-, 31P- spectra of AMBPi are available online.
Author Contributions
Conceptualization, J.D., T.L., M.L.; methodology, J.D., T.L., N.A., A.D., J.D.-G.; validation, J.D., T.L., M.L.; formal analysis, E.M.-G., M.M., T.B.A.; investigation, N.A., A.D., J.D.-G.; writing—original draft preparation, J.D.; writing—review and editing, J.D., T.L., M.L., N.A., A.D., J.D.-G. E.M.-G., M.M., T.B.A. and supervision, J.D., T.L., M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Université Sorbonne Paris Nord (USPN), Centre National de la Recherche Scientifique (CNRS), Ministère de l’Enseignement Supérieur et de la Recherche (MESR), by the Hubert Curien “Utique” partnership N° 46347XD of the French Ministry of Europe and Foreign Affairs and N° 21G1207 of the Tunisian Ministry of Higher Education and Scientific Research and GDR Phosphore 2008 (CNRS).
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
The data presented in this study are available in article or Supplementary Materials.
Acknowledgments
We acknowledge the NMR-PF facility (Université Sorbonne Paris Nord-USPN, the authors would like to thank Cyril Colas from the "Fédération de Recherche" ICOA/CBM (FR2708)" for HRMS analysis and Anne Boos from “Plateforme Analytiques des Inorganiques” IPHC UMR7178 for ICP-AES analysis.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 3a-l are available from the authors.
References
- Horsman, G. P.; Zechel, D. L. Phosphonate Biochemistry. Chem. Rev. 2017, 117, 5704–5783. [Google Scholar] [CrossRef] [PubMed]
- Virieux, D.; Volle, J. N.; Bakalara, N.; Pirat, J. L. Synthesis and biological applications of phosphinates and derivatives. Top. Curr. Chem. 2015, 360, 39–114. [Google Scholar] [CrossRef] [PubMed]
- Pradere, U.; Garnier-Amblard, E. C.; Coats, S. J.; Amblard, F.; Schinazi, R. F. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev. 2014, 114, 9154–9218. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yang, H.; Shi, E.; Tang, W. Development and Clinical Application of Phosphorus-Containing Drugs. Med. Drug. Discov. 2020, 8, 100063. [Google Scholar] [CrossRef]
- Montchamp, J.-L. Challenges and solutions in phosphinate chemistry. Pure Appl. Chem. 2019, 91, 113–120. [Google Scholar] [CrossRef]
- Montchamp, J.-L. Recent advances in phosphorus–carbon bond formation: synthesis of H-phosphinic acid derivatives from hypophosphorous compounds. J. Organomet. Chem. 2005, 690, 2388–2406. [Google Scholar] [CrossRef]
- Dussart, J.; Deschamp, J.; Monteil, M.; Gager, O.; Migianu-Griffoni, E.; Lecouvey, M. A General Protocol for the Synthesis of H --Hydroxyphosphinates. Synthesis 2019, 51, 421–432. [Google Scholar] [CrossRef]
- Guedeney, N.; Dussart, J.; Deschamp, J.; Ouechtati, M.; Migianu-Griffoni, E.; Lecouvey, M. A convenient one-pot synthesis of 1-hydroxymethylene-1,1-bisphosphinic acids. Phosphorus, Sulfur Silicon Relat. Elem. 2019, 194, 323–325. [Google Scholar] [CrossRef]
- Dussart, J.; Guedeney, N.; Deschamp, J.; Monteil, M.; Gager, O.; Legigan, T.; Migianu-Griffoni, E.; Lecouvey, M. A convenient synthetic route towards H-bisphosphinates. Org. Biomol. Chem. 2018, 16, 6969–6979. [Google Scholar] [CrossRef]
- Dussart-Gautheret, J.; Deschamp, J.; Monteil, M.; Gager, O.; Legigan, T.; Migianu-Griffoni, E.; Lecouvey, M. Formation of 1-Hydroxymethylene-1,1-bisphosphinates through the Addition of a Silylated Phosphonite on Various Trivalent Derivatives. J. Org. Chem. 2020, 85, 14559–14569. [Google Scholar] [CrossRef]
- Dussart-Gautheret, J.; Deschamp, J.; Legigan, T.; Monteil, M.; Migianu-Griffoni, E.; Lecouvey, M. One-Pot Synthesis of Phosphinylphosphonate Derivatives and Their Anti-Tumor Evaluations. Molecules 2021, 26, 7609. [Google Scholar] [CrossRef]
- Barbosa, J. S.; Almeida Paz, F. A.; Braga, S. S. Bisphosphonates, Old Friends of Bones and New Trends in Clinics. J. Med. Chem. 2021, 64, 1260–1282. [Google Scholar] [CrossRef]
- Dussart, J.; Deschamp, J.; Migianu-Griffoni, E.; Lecouvey, M. From Industrial Method to the Use of Silylated P(III) Reagents for the Synthesis of Relevant Phosphonylated Molecules. Org. Process Res. Dev. 2020, 24, 637–651. [Google Scholar] [CrossRef]
- Reszka, A. A.; Rodan, G. A. Nitrogen-Containing Bisphosphonate Mechanism of Action. Mini Rev. Med. Chem. 2004, 4, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Ebetino, F. H.; Hogan, A. M.; Sun, S.; Tsoumpra, M. K.; Duan, X.; Triffitt, J. T.; Kwaasi, A. A.; Dunford, J. E.; Barnett, B. L.; Oppermann, U.; Lundy, M. W.; Boyde, A.; Kashemirov, B. A.; McKenna, C. E.; Russell, R. G. The relationship between the chemistry and biological activity of the bisphosphonates. Bone 2011, 49, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Clézardin, P. Bisphosphonates' antitumor activity: An unravelled side of a multifaceted drug class. Bone 2011, 48, 71–79. [Google Scholar] [CrossRef]
- Rogers, M. J.; Frith, J. C.; Luckman, S. P.; Coxon, F. P.; Benford, H. L.; M̈onkk̈onen, J.; Auriola, S.; Chilton, K. M.; Russell, R. G. G. Molecular mechanisms of action of bisphosphonates. Bone 1999, 24, 73S–79S. [Google Scholar] [CrossRef]
- Lin, J. H. Bisphosphonates: A review of their pharmacokinetic properties. Bone 1996, 18, 75–85. [Google Scholar] [CrossRef]
- Mimura, M.; Hayashida, M.; Nomiyama, K.; Ikegami, S.; Iida, Y.; Tamura, M.; Hiyama, Y.; Ohishi, Y. Synthesis and Evaluation of (Piperidinomethylene)bis(phosphonic acid) Derivatives as Anti-osteoporosis Agents. Chem. Pharm. Bull. 1993, 41, 1971–1986. [Google Scholar] [CrossRef]
- Szajnman, S. H.; Ravaschino, E. L.; Docampo, R.; Rodriguez, J. B. Synthesis and biological evaluation of 1-amino-1,1-bisphosphonates derived from fatty acids against Trypanosoma cruzi targeting farnesyl pyrophosphate synthase. Bioorg. Med. Chem. Lett. 2005, 15, 4685–4690. [Google Scholar] [CrossRef] [PubMed]
- Kotsikorou, E.; Song, Y.; Chan, J. M. W.; Faelens, S.; Tovian, Z.; Broderick, E.; Bakalara, N.; Docampo, R.; Oldfield, E. Bisphosphonate Inhibition of the Exopolyphosphatase Activity of the Trypanosoma brucei Soluble Vacuolar Pyrophosphatase. J. Med. Chem. 2005, 48, 6128–6139. [Google Scholar] [CrossRef] [PubMed]
- Leon, A.; Liu, L.; Yang, Y.; Hudock, M. P.; Hall, P.; Yin, F.; Studer, D.; Puan, K.-J.; Morita, C. T.; Oldfield, E. Isoprenoid Biosynthesis as a Drug Target: Bisphosphonate Inhibition of Escherichia coli K12 Growth and Synergistic Effects of Fosmidomycin. J. Med. Chem. 2006, 49, 7331–7341. [Google Scholar] [CrossRef]
- Occhipinti, A.; Berlicki, L.; Giberti, S.; Dziedziola, G.; Kafarski, P.; Forlani, G. Effectiveness and mode of action of phosphonate inhibitors of plant glutamine synthetase. Pest. Manag. Sci. 2010, 66, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Kafarski, P.; Lejczak, B.; Forlani, G. Herbicidally active aminomethylenebisphosphonic acids. Heteroat. Chem. 2000, 11, 449–453. [Google Scholar] [CrossRef]
- Simoni, D.; Gebbia, N.; Invidiata, F. P.; Eleopra, M.; Marchetti, P.; Rondanin, R.; Baruchello, R.; Provera, S.; Marchioro, C.; Tolomeo, M.; Marinelli, L.; Limongelli, V.; Novellino, E.; Kwaasi, A.; Dunford, J.; Buccheri, S.; Caccamo, N.; Dieli, F. Design, Synthesis, and Biological Evaluation of Novel Aminobisphosphonates Possessing an in Vivo Antitumor Activity Through a γδ-T Lymphocytes-Mediated Activation Mechanism. J. Med. Chem. 2008, 51, 6800–6807. [Google Scholar] [CrossRef]
- Chmielewska, E.; Kafarski, P. Synthetic Procedures Leading towards Aminobisphosphonates. Molecules 2016, 21. [Google Scholar] [CrossRef]
- Hong, Y. C.; Ye, J. L.; Huang, P. Q. One-Pot Synthesis of alpha-Amino Bisphosphonates from Nitriles via Tf(2)O/HC(OR)(3)-Mediated Interrupted Ritter-Type Reaction. J. Org. Chem. 2022, 87, 9044–9055. [Google Scholar] [CrossRef]
- Kaboudin, B.; Esfandiari, H.; Moradi, A.; Kazemi, F.; Aoyama, H. ZnCl2-Mediated Double Addition of Dialkylphosphite to Nitriles for the Synthesis of 1-Aminobisphosphonates. J. Org. Chem. 2019, 84, 14943–14948. [Google Scholar] [CrossRef] [PubMed]
- Islas, R. E.; García, J. J. Nickel-Catalyzed Hydrophosphonylation and Hydrogenation of Aromatic Nitriles Assisted by Lewis Acid. ChemCatChem 2019, 11, 1337–1345. [Google Scholar] [CrossRef]
- Wang, A. E.; Chang, Z.; Sun, W. T.; Huang, P. Q. General and chemoselective bisphosphonylation of secondary and tertiary amides. Org. Lett. 2015, 17, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Prishchenko, A. A.; Alekseyev, R. S.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Petrosyan, V. S. Synthesis of new functionalized aryl and pyridyl aminomethylenebisphosphonic acids and their derivatives via silicon-assisted methodology. J. Organomet. Chem. 2020, 912, 121177. [Google Scholar] [CrossRef]
- Prishchenko, A. A.; Alekseyev, R. S.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Petrosyan, V. S. Silicon-assisted synthesis of new aminomethylenebisphosphonic acids with quinolines moieties. J. Organomet. Chem. 2020, 917, 121286. [Google Scholar] [CrossRef]
- Prishchenko, A. A.; Alekseyev, R. S.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Petrosyan, V. S. Organosilicon based synthesis of new functionalized aminomethylenediphosphonates with moieties of amino acids. J. Organomet. Chem. 2018, 871, 36–39. [Google Scholar] [CrossRef]
- Prishchenko, A. A.; Alekseyev, R. S.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Petrosyan, V. S. Tris(trimethylsilyl) phosphite as key synthon for convenient synthesis of new organosilicon(phosphorus)-containing N-heterocycles. J. Organomet. Chem. 2018, 867, 149–154. [Google Scholar] [CrossRef]
- Prishchenko, A. A.; Alekseyev, R. S.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Terenin, V. I.; Petrosyan, V. S. Synthesis of new functionalized mono- and diphosphonic acids with five-membered aza-heterocycles moieties. Heteroat. Chem. 2017, 28, e21353. [Google Scholar] [CrossRef]
- Prishchenko, A. A.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Ershov, I. S.; Petrosyan, V. S. Synthesis of new types of aminomethylenediphosphorus-containing acids and their derivatives. Russ. J. Gen. Chem. 2015, 85, 370–379. [Google Scholar] [CrossRef]
- Prishchenko, A. A.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Ershov, I. S.; Petrosyan, V. S. Synthesis of the New Types ofN-Unsubstituted Aminomethylenebisorganophosphorus Acids and Their Derivatives. Heteroat. Chem. 2015, 26, 101–105. [Google Scholar] [CrossRef]
- Minaeva, L. I.; Patrikeeva, L. S.; Kabachnik, M. M.; Beletskaya, I. P.; Orlinson, B. S.; Novakov, I. A. Synthesis of novel aminomethylenebisphosphonates and bisphosphonic acids, containing adamantyl fragment. Heteroat. Chem. 2011, 22, 55–58. [Google Scholar] [CrossRef]
- Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. Insight into the mechanism of three component condensation leading to aminomethylenebisphosphonates. J. Organomet. Chem. 2009, 694, 3806–3813. [Google Scholar] [CrossRef]
- Kaboudin, B.; Alipour, S. A microwave-assisted solvent- and catalyst-free synthesis of aminomethylene bisphosphonates. Tetrahedron Lett. 2009, 50, 4243–4245. [Google Scholar] [CrossRef]
- Wu, M.; Chen, R.; Huang, Y. Simple, Efficient and One-Pot Method for Synthesis of Aminomethylene gem-Diphosphonic Acid Derivatives from Ketones via Beckmann Rearrangement. Synthesis 2004, 2441–2444. [Google Scholar] [CrossRef]
- Yokomatsu, T.; Yoshida, Y.; Nakabayashi, N.; Shibuya, S. Simple and Efficient Method for Preparation of Conformationally Constrained Aminomethylene gem-Diphosphonate Derivatives via Beckmann Rearrangement. J. Org. Chem. 1994, 59, 7562–7564. [Google Scholar] [CrossRef]
- David, T.; Procházková, S.; Kotek, J.; Kubíček, V.; Hermann, P.; Lukeš, I. Aminoalkyl-1,1-bis(phosphinic acids): Stability, Acid-Base, and Coordination Properties. Eur. J. Inorg. Chem. 2014, 4357–4368. [Google Scholar] [CrossRef]
- Prishchenko, A. A.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Ershov, I. S.; Petrosyan, V. S. Synthesis and Reactivity of the New Trimethylsilyl Esters of Aminomethylenebisorganophosphorus Acids. Heteroat. Chem. 2013, 24, 355–360. [Google Scholar] [CrossRef]
- Samples, M. S.; Yoder, C. H. The structure of bis(organosily1) amides containing the dimethylsilyl and bis( dimethylsilyl) ethylene groups. J. Organomet. Chem. 1987, 332, 69–73. [Google Scholar] [CrossRef]
- Klebe, J. F.; Finkbeiner, H.; White, D. M. Silylations with Bis(trimethylsilyl)acetamide, a Highly Reactive Silyl Donor. J. Am. Chem. Soc. 1966, 88, 3390–3395. [Google Scholar] [CrossRef]
- iodolactamization: 8-exo-iodo-2-azabicyclo[3.3.0]octan-3-one. Org. Synth. 1992, 70, 101. [CrossRef]
- Knapp, S.; Levorse, A. T. Synthesis and reactions of iodo lactams. J. Org. Chem. 1988, 53, 4006–4014. [Google Scholar] [CrossRef]
- Hanada, S.; Motoyama, Y.; Nagashima, H. Hydrosilanes Are Not Always Reducing Agents for Carbonyl Compounds but Can Also Induce Dehydration: A Ruthenium-Catalyzed Conversion of Primary Amides to Nitriles. Eur. J. Org. Chem. 2008, 4097–4100. [Google Scholar] [CrossRef]
- Dwak, B.; Lasocki, Z. Structure and tautomerism of cyclic silylamides I. Disiloxane derivatives of acetamide and benzamides. J. Organomet. Chem. 1983, 246, 151–158. [Google Scholar] [CrossRef]
- Bassindale, A. R.; Posner, T. B. The Structure of Silylated Amides-. N-Methyl-NTrimethylsilyltrifluoroacetamide, a Reassignment of Structure. J. Organomet. Chem. 1979, 175, 273–284. [Google Scholar] [CrossRef]
- Itoh, K.; Katsuda, M.; Ishii, Y. Reactions of Group IV Organometallic Compounds. Part X1X.l Substituent Effects on the Rate of Trimethylsilyl Migration in Substituted N,O-Bis(trimethylsilyl)benzimidates. J. Chem. Soc. (B) 1970, 302–304. [Google Scholar] [CrossRef]
- Deprèle, S.; Montchamp, J. L. Triethylborane-Initiated Room Temperature Radical Addition of Hypophosphites to Olefins: Synthesis of Monosubstituted Phosphinic Acids and Esters. J. Org. Chem. 2001, 66, 6745–6755. [Google Scholar] [CrossRef] [PubMed]
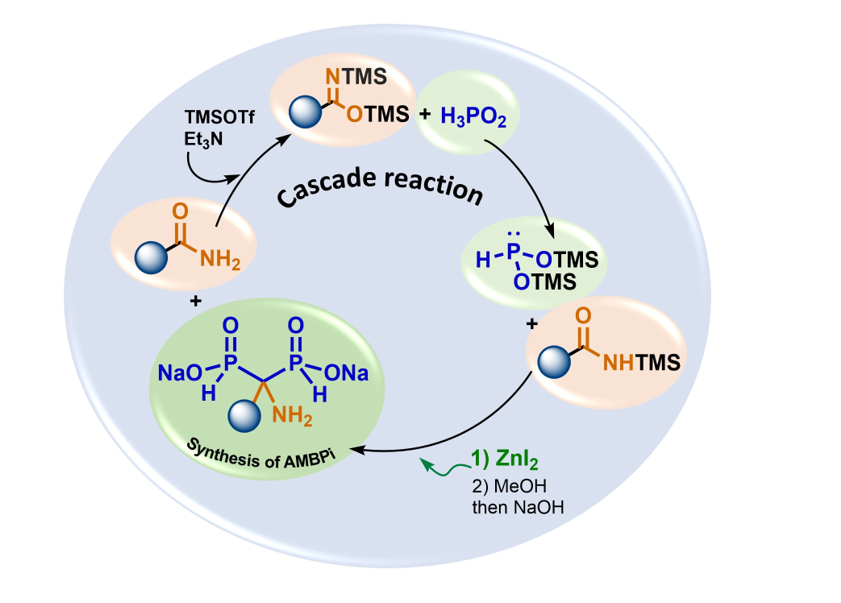
Figure 1.
(a) synthesis and uses of silylated phosphonites II in the presence of various electrophiles (b) structures of HMBPs and their analogues in bisphosphonate and phosphinylphosphonate series.
Figure 1.
(a) synthesis and uses of silylated phosphonites II in the presence of various electrophiles (b) structures of HMBPs and their analogues in bisphosphonate and phosphinylphosphonate series.
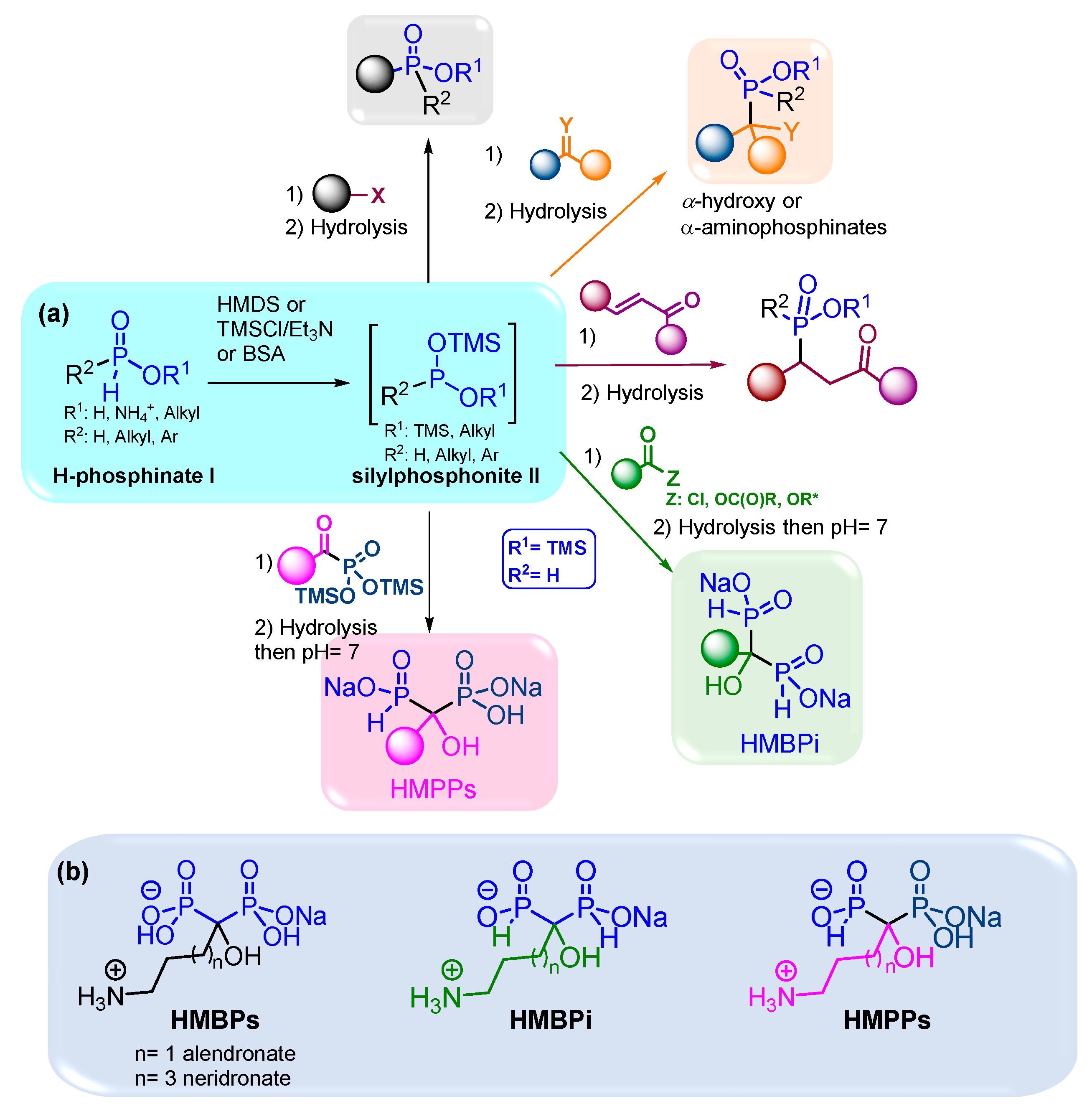
Figure 2.
(a) Representative bioactive AMBPs. (b) Synthetic pathways to AMBPs. (c) Synthetic pathways to AMBPi.
Figure 2.
(a) Representative bioactive AMBPs. (b) Synthetic pathways to AMBPs. (c) Synthetic pathways to AMBPi.
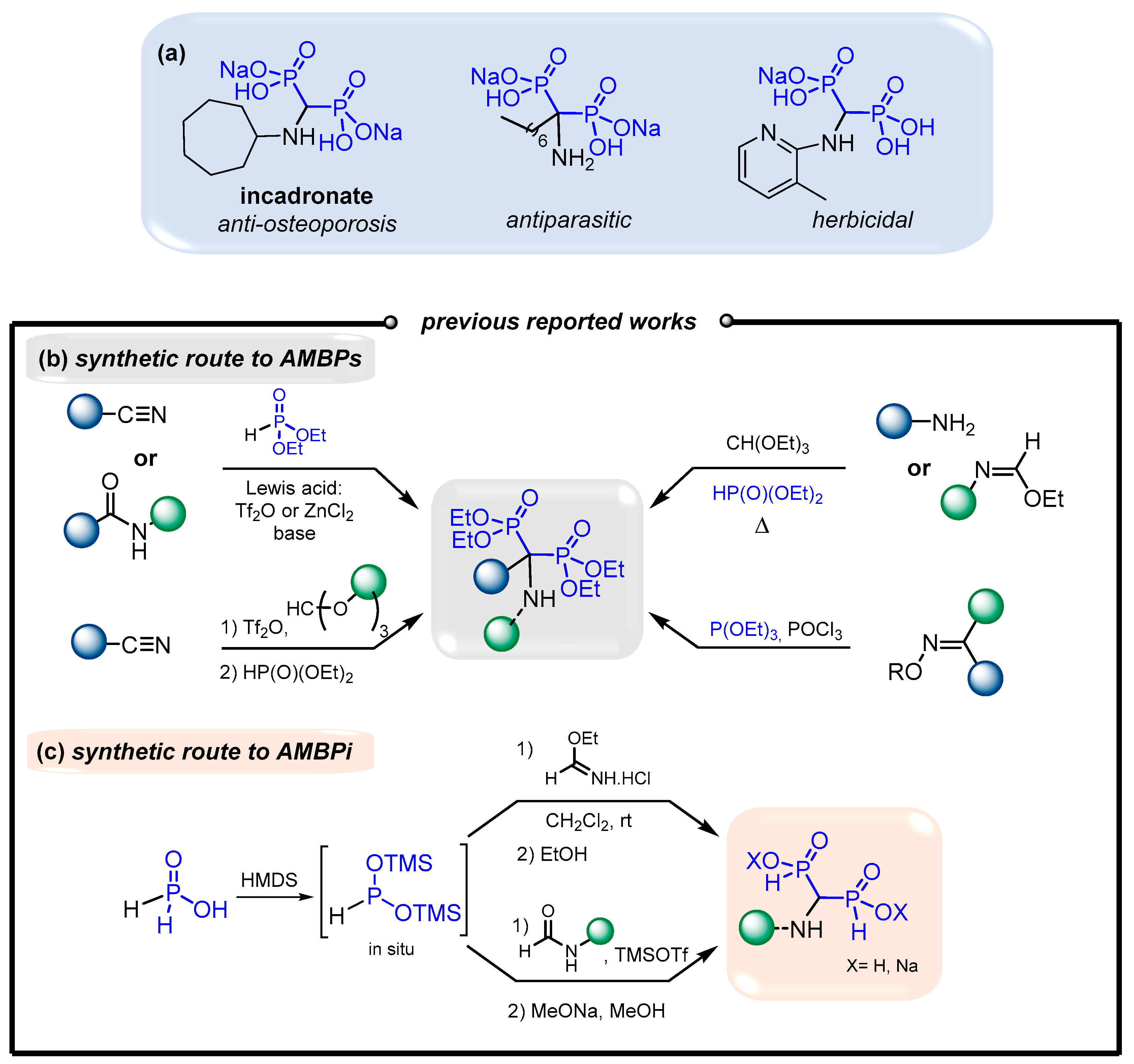
Figure 3.
(a) Preliminary test on the addition of BTSP onto nitrile. (b) New methodology leading to AMBPi 3.
Figure 3.
(a) Preliminary test on the addition of BTSP onto nitrile. (b) New methodology leading to AMBPi 3.
Scheme 1.
Synthesis of N,O-bis(silyl)imidates 1 .
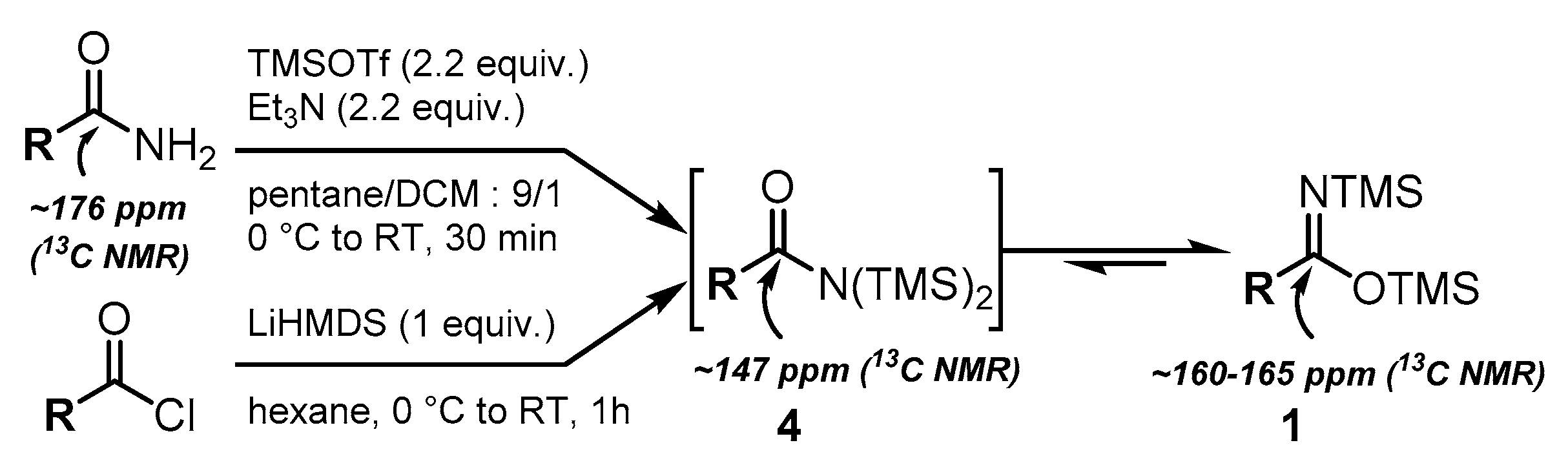
Figure 4.
31P (dark red) and{1H}31P (blue)NMR monitoring of the optimized cascade reaction between phosphorous acid and N,O-bis(trimethylsilylacetamide 1a.
Figure 4.
31P (dark red) and{1H}31P (blue)NMR monitoring of the optimized cascade reaction between phosphorous acid and N,O-bis(trimethylsilylacetamide 1a.
Scheme 2.
Complementary tests to validate the cascade reaction.
Scheme 3.
Scope of the cascade reaction between various bis(trimethylsilyl)imidates 2a-l and hypophosphorous acid in the presence of zinc iodide.
Scheme 3.
Scope of the cascade reaction between various bis(trimethylsilyl)imidates 2a-l and hypophosphorous acid in the presence of zinc iodide.
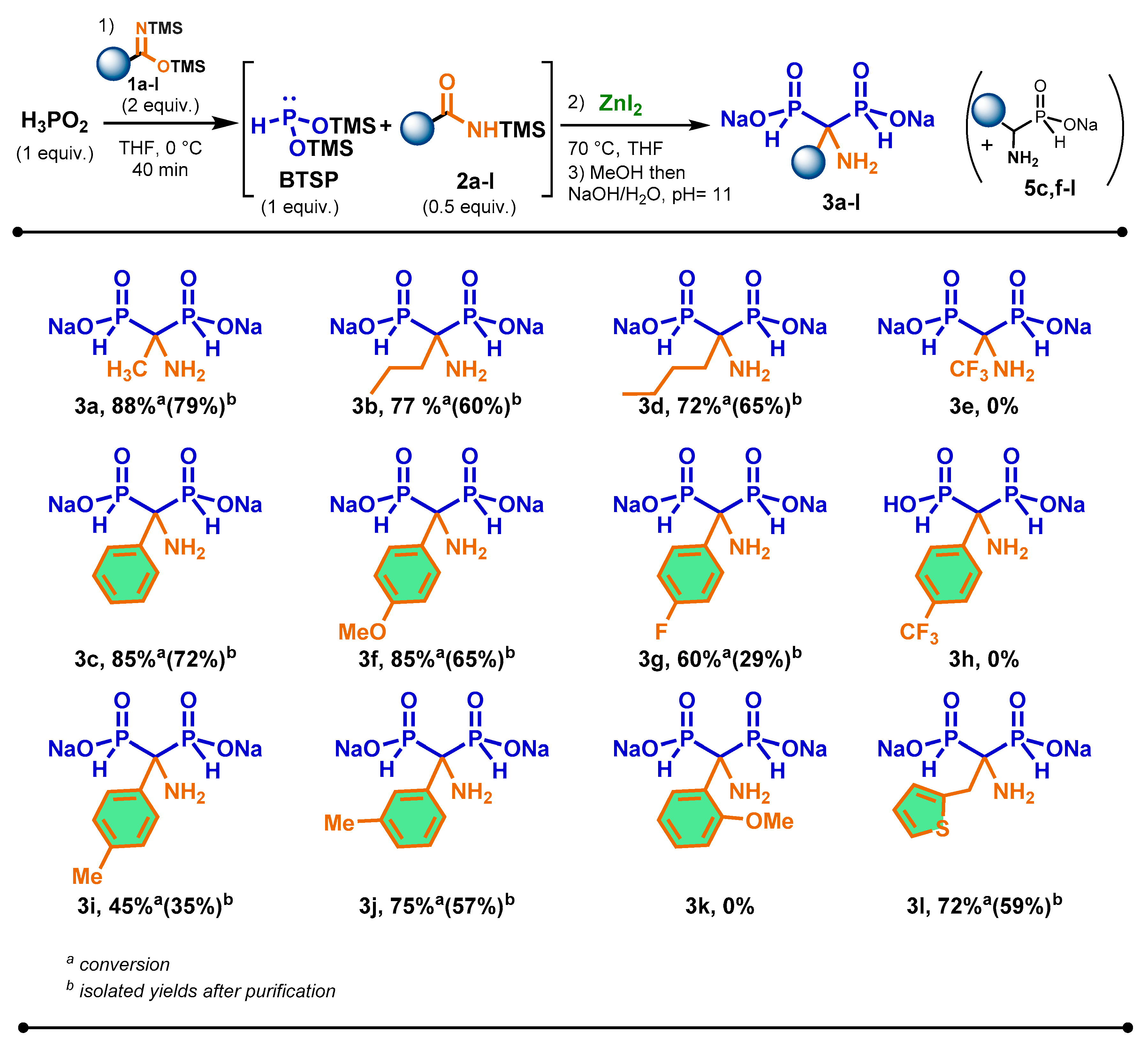

Table 1.
Optimizations of reaction parameters.
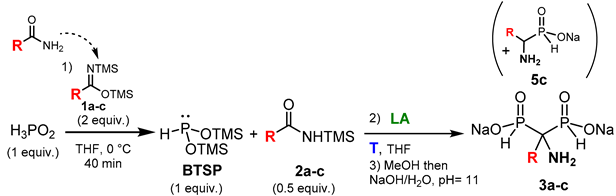 | ||||||
| Entry | 2a-c | R | LA | T, °C | Time, hour | 3a-c/5c, Yield (%)1 |
| Entry 1 | 2a | Me5 | ZnCl2 | 70 | 18 | 3a, 902(75)3 |
| Entry 2 | 2a | Me5 | ZnI2 | 70 | 1.5 | 3a, 882(83)3 |
| Entry 3 | 2a | Me5 | TMSOTf | 0 | 0.5 | 3a, 902(75)3 |
| Entry 4 | 2a | Me5 | BF3.OEt2 | 0-70 | 18 | - |
| Entry 5 | 2a | Me6 | ZnCl2 | 70 | 15 | 3a, 902(75)3 |
| Entry 6 | 2a | Me6 | ZnI2 | 70 | 1.5 | 3a, 882(79)3 |
| Entry 7 | 2a | Me6 | TMSOTf | 0 | 0.5 | 3a, 902(75)3 |
| Entry 8 | 2b | Pr | ZnCl2 | 70 | 18 | 3b, 772(60)3 |
| Entry 9 | 2b | Pr | ZnI2 | 70 | 2 | 3b, 772(60)3 |
| Entry 10 | 2b | Pr | TMSOTf | 0 | 0.5 | 3b, 64 |
| Entry 11 | 2c | Ph | ZnCl2 | 70 | 18 | 5c, 154 |
| Entry 12 | 2c | Ph | ZnI2 | 70 | 1 | 3c, 852(72)3 |
| Entry 13 | 2c | Ph | TMSOTf | 0 | 0.5 | - |
1 The reaction evolution was monitored by 31P NMR. 2 The conversions were determined by 31P NMR after methanolysis. 3 Isolated yields after purification. 4 Proportion determined by 31P NMR after methanolysis in the crude mixture. 5 Commercially available BSA was used. 6 BSA was synthesized according to literature procedure.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



