
Submitted:
15 July 2023
Posted:
17 July 2023
You are already at the latest version
Abstract
Patients with end stage liver disease (ESLD) undergoing liver transplantation are prone to thromboses both while on the waiting list or in the perioperative period. This hypercoagulability is associated with significant endothelial dysfunction (ED) due to nitric oxide dysregulation. ED in combination with increased thrombin generation are the main factors responsible for this hypercoagulability. Sepsis itself can significantly change the coagulation profile, but in combination with ESLD, sepsis, and septic shock is responsible for very complex changes in the patients coagulation making both assessment and management of coagulation in patients with ESLD very challenging. Viscoelastic testing (VET) is a preferred method of coagulation management in patients with cirrhosis because, as with standard laboratory testing, VET can assess the entire coagulation system including the interaction between both pro- and anticoagulants and platelets.
Keywords:
cirrhosis
; hemostasis
; sepsis
; visco-elastic tests
Introduction
End stage liver disease (ESLD) is associated with significant changes in a patient’s coagulation profile. These changes are unique in that they involve all branches of the coagulation system. Despite of significantly reduced concentration of coagulation factors and platelets, patients with ESLD may have provoked or unprovoked bleeding or clotting events. When sepsis is added to the mix, the entire coagulation profile changes more significantly with bleeding/clotting events becoming less predictable. In this review, we discuss several aspects of coagulation management in patients with ESLD.
Hypercoagulability in end stage liver disease: pathophysiology and susceptible populations
End Stage Liver disease can be associated with both severe bleeding and clotting. The majority of coagulation and anticoagulation factors are produced in the liver. Despite a reduction in the production of most of these factors in ESLD and defects in primary secondary, and tertiary coagulation, the coagulation system is most often stable. Some of coagulation factors produced extrahepatic and this is one of the main reasons why hepatic failure is not necessarily associated only with spontaneous unprovoked bleeding, but with clotting as well [1]. One of the first publications demonstrating that patients with ESLD are not naturally anticoagulated but prone to venous thromboembolism (VTE) was published in 2009 by Northup et al [2]. The authors found that the incidence of VTE in hospitalized cirrhotic patients was 0.5%. Most of these patients also had a low serum albumin level (most likely a surrogate marker for circulating anticoagulants). In an evaluation (2009) based on the Danish National Register, Søgaard et al. demonstrated that both patients with cirrhosis and not-cirrhotic chronic liver disease had a high risk of developing VTE (relative risk for thromboses 2.06 and 2.10 respectively) in comparison to matched controls from the general population [3]. An association between chronic liver disease and VTE was later confirmed in several publications with an incidence of between 0.5% and 8% [4,5,6,7].
If patients with ESLD require a LT, the concern for perioperative thrombotic or bleeding complications is even higher. In fact, different types of thromboses have been described during each stage of transplant surgery.
Portal vein thromboses (PVT) are more frequently seen preoperatively with a prevalence of between 1% and 16%, occurring more frequently in patients with decompensated disease [8,9]. An association between preoperative PVT and increased postoperative mortality has been demonstrated [10,11,12].
Intraoperative thrombotic complications with clinical presentation occur with a frequency of 1% and 6% [13,14]. However, if transesophageal echocardiography is routinely used during surgery, a variety of clots have been seen in almost half of all cases with the majority not needing any treatment [15]. Symptomatic clots, in form of pulmonary emboli (PE) or intracardiac thromboses, can occur at each stage of LT surgery, but are most frequently described at the time of graft reperfusion and are associated with significant hemodynamic instability and a high mortality rate [16].
Different types of thrombotic complications occur postoperatively. It has been demonstrated that preoperative PVT is associated with postoperative thrombotic complications [10,17,18]. Postoperative thromboses can manifest in the form of PVT (2%) [17], hepatic artery thromboses (HAT) (3%-6% and over 8% in children),[19,20] and VTE (5%-10%) [21]. Many of these complications are associated with increased mortality. The incidence of PE after LT is about 4% [16] with an associated one-year mortality rate as high as 12% [22]. Early (within 90 days after LT) HAT is associated with graft failure and requires retransplantation with a rate above 50% for adults and above 60% in children [17,19]. The development of postoperative thromboses (especially HAT) is significantly affected by surgical technique and perioperative management.
Although the cause of hypercoagulability in ESLD is multifactorial, endothelial dysfunction (ED) is most likely the main driving mechanism of clotting in cirrhotic patients. The pressure gradient between the systemic and portal circulation results in increasing intravascular shear forces resulting in activation of nitric oxide (NO) production and the development of ED [23]. Increased NO levels lead to significant vasodilation, primarily in the portal circulation, resulting in a “steal effect” in the systemic circulation. Low systemic pressure is responsible for a dramatic decrease in perfusion of the intestines which is associated with increased mucosal permeability, release of endotoxins, and secondary activation of the NO pathway resulting in an even higher degree of systemic hypotension and ED [24]. Other factors contributing to the development of ED in cirrhotic patients are increased inflammatory and oxidative stress [25]. Endotoxemia itself is associated with increased thrombin generation [26] which is one of the major factors in hypercoagulability. Additionally, ED in patients with ESLD is also associated with resistance to thrombomodulin (TM) (a membrane protein expressed on the surface of endothelial cells) that serves as a co-factor for protein C activation with subsequent impairment of the anticoagulation pathway and increase in thrombin generation [27,28]. Additionally, both hepatic production of proteins C and S, and antithrombin III (ATIII), and their activity, are significantly reduced in ESLD [29]. Another ED-related factor contributing to hypercoagulability in cirrhosis is the significantly increased production of endothelium liver-independent coagulation or anticoagulation factors such as Factor (F) FVIII, von Willebrand factor (vWF), as well as plasminogen activator inhibitor 1 (PAI-1) [30,31,32]. VWF is likely one of the key components of hypercoagulability in cirrhosis. Despite thrombocytopenia and impaired platelet function in vitro [33], in vivo, platelet function is likely normal or even increased due to a high concentration of vWF (not just because of increased production but also because of decreased destruction by cleaving protease ADAMTS13 synthetized in the liver) [29,34].
FVIII is one of the targets of activated protein C. In ESLD, the ratio between FVIII and activated protein C (which indicates the severity of cirrhosis) can reach 5.0. This imbalance is an additional factor responsible for the impairment of thrombin generation inhibition [35,36].
In addition to primary and secondary coagulation, tertiary coagulation is significantly affected in cirrhosis. An increase in PAI-1 levels in combination with reduced levels of tissue plasminogen activator is responsible for decreased fibrinolysis in patients with ESLD [37,38]. It has been also shown that patients with ESLD have increased clot stability due to diminished permeability and reduced lysis [39,40].
There are number of subpopulations of patients with ESLD predisposed to developing thromboses. It has been demonstrated that patients with nonalcoholic steatohepatitis (NASH) have an increased prevalence of DVT, PE, and PVT [41,42,43]. The reason for this is inflammation and increased oxidative stress that occurs with NASH at a higher degree than in patients with other types of ESLD [37,44].. Other subpopulations prone to thrombotic complications include patients with autoimmune conditions (due to chronic inflammation, cytokine release, and high fibrinogen concentration [45,46]) and patients with hepatitis C (due to chronic inflammation and production of autoantibodies such anticardiolipins and increased thrombin generation) [47,48].
Several genetic mutations associated with thromboses (mostly VTE and PVT) are frequently seen in patients with ESLD. These include FV Leiden [49], Prothrombin G20210A mutation (leads to high prothrombin level)[50], high plasma homocysteine level [51], a mutation in the methylenetetrahydrofolate reductase gene [51], JAK2V617F mutations [52], as well as myeloproliferative neoplasms (MPN) [53]. Due to chronic hypercoagulability associated with ESLD, these mutations make patients with cirrhosis more susceptible to thromboses than the general population.
Monitoring of Hemostasis in Patients with End-Stage-Liver-Disease
Although standard laboratory tests (SLTs) have never been formally evaluated for predicting bleeding or for treatment, these tests are mainly used for managing patients with severe trauma, cardiac surgery, and liver-related hemostasis disorders. Tests such as the prothrombin time (PT), activated partial thrombin time (aPTT), international normalized ratio (INR), fibrinogen, and platelet count have significant limitations with regard to evaluating clot formation and kinetics as well as clot strength [54].
SLTs do not accurately reflect the coagulation status of patients with ESLD. Platelet number do not correlate with platelets function, and fibrinogen levels do not accurately reflect structurally normal fibrinogen or dysfibrinogenemia (compounds with impaired fibrinogen polymerization) that are significantly increased in ESLD [55,56]. Another significant limitation of SLTs is the time necessary to complete the analysis. While the turnaround time for viscoelastic testing (VET) is 7-10 minutes, the turnaround time for SLTs depends on local facilities and can take between 60 and 90 minutes [57,58].
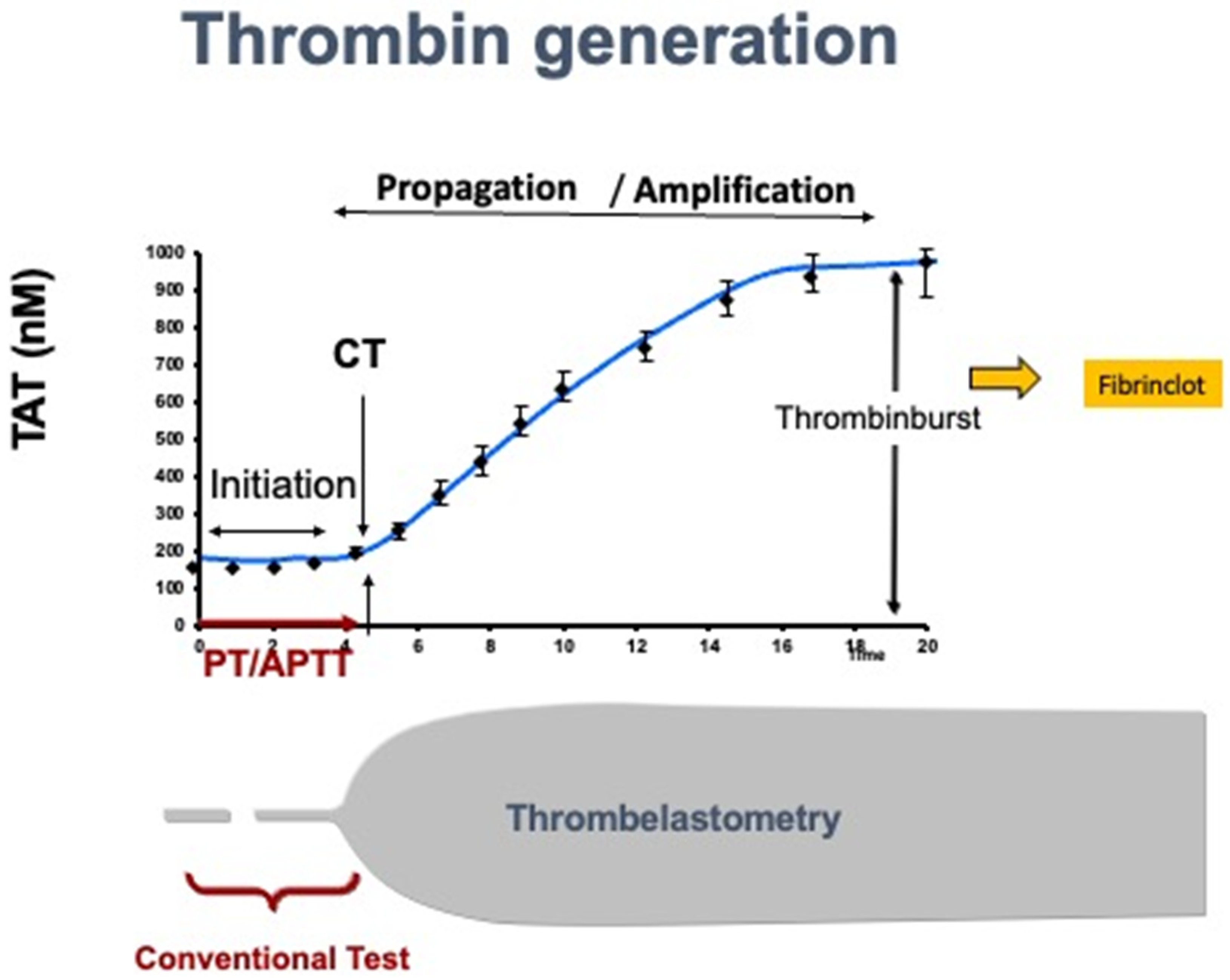
In patients with ESLD, SLTs only provide a limited reflection of the current coagulation concept. Since 2001, a cell-based instead of a cascade-based coagulation model has been accepted as the standard concept [59]. According to the cell-based system, coagulation is divided into three phases: activation, propagation, and amplification. The activation phase begins with tissue factor release from a variety of cells which interacts with FVIIa resulting in subsequent activation of FIX, FX, and prothrombin (FII). Prothrombin release is followed by fibrin production. In the propagation phase, prothrombin activates platelets that used as a matrix where all metabolic processes can then occur. This is followed by activation of FV and FVIII to FVIIIa, F IXa, and F Va/Xa which leads to further release of thrombin. In the amplification phase, the release of thrombin is repeated as a positive feedback mechanism until thrombin achieves a 1000-fold higher concentration compared to baseline (thrombin burst). After the thrombin burst, a fibrin clot is formed (Figure 1). SLTs (such as PT and aPTT) assess only the initiation phase but not the other phases of coagulation and cannot give a complete picture of the status of coagulation (Figure 2).
One of the first studies which challenged the use of SLTs for predicting bleeding was published in 1981. Ewe et al. attempted to determine if the bleeding time was dependent on platelet number and prothrombin time (PT) during a laparoscopic procedure in 200 cirrhotic patients having a liver biopsy performed with a 1.8 mm Meninghi needle [60]. The authors found no correlation between bleeding time and PT or platelet number. Two patients had a bleeding time of > 12 minutes (mean 4 minutes 37 sec.) although their SLTs hemostatic values (PT and platelet count) were normal.
One of the most important papers contributing to our understanding of coagulation in patients with ESLD was published by Tripodi et al [1]. The authors evaluated endogenous thrombin generation in cirrhotic patients and healthy volunteers. The level of thrombin generation in healthy volunteers was significantly higher compared to cirrhotic patients. This difference, however, disappeared when TM was added to the assay. When thrombin binds to the TM receptor, protein C is activated. This then inhibits Factor Va and Factor VIIIa. These data have demonstrated that in cirrhotic patients, both pro- and anticoagulants are reduced.
SLTs are not designed to assess levels of anticoagulants. This is the main reason why these tests are able to show a potential predisposition to coagulopathy, but not a hypercoagulable condition. Haas et al. performed a met-analysis regarding using SLTs to predict bleeding events [61]. The authors were able to identify 11 guidelines and 64 studies evaluating the use of SLTs to predict bleeding. There were only 3 prospective studies with a total of only 108 patients included with no clear results. The authors concluded that SLTs were unsuitable for this purpose.
In contrast to SLTs, VET can overcome most of these disadvantages. In comparison to SLTs, VET dynamically assesses hemostasis in whole blood, not only in plasma, and includes the interaction between pro-, anticoagulants, and platelets. VET assesses the entire coagulation process to identify which part is disrupted which allows for targeted therapy. Dr. Kang (working in Pittsburgh at this time) was the first to report the advantages of VET for perioperative management of LT [62]. In 1985, he reported that transfusion requirements were reduced by 33% if VET was used. In a randomized controlled trial (RCT), De Pietrie et al. evaluated cirrhotic patients who underwent invasive procedures. Sixty patients were 1:1 randomized to guide hemostasis management based on VET or SLTs [63]. Patients in the VET group received significantly fewer RBC, FFP, and platelet transfusions compared to the SLT group. Post-intervention hemoglobin in the VET group was considerably higher compared to the SLT group. Bleeding events were not significantly different between groups. Similar results were reported by Kumar et al [64]. These authors were able to demonstrate that VET- guided hemostasis management in cirrhotic patients with non-variceal bleeding could significantly reduce the number of transfused blood products without increasing the incidence of bleeding events. Similar findings were shown in children who underwent invasive procedures where VET-guided hemostasis management was associated with a significantly reduced rate of blood product transfusion [65].
Currently, two VET devices are available, TEG and ROTEM. Hemostasis management with these two systems was assessed by Wikkelsø et al. in a Cochrane Database Systematic Review [66]. They enrolled 17 studies with a total of 1493 patients primarily having cardiac surgical procedures. Their analysis demonstrated reduced mortality (3.9% vs. 7.4%, RR = 0.52, 95% CI: 0.28-0.95) and significantly reduced transfusion rate of any blood product (red blood cells (RBC), fresh frozen plasma (FFP), or platelets) when TEG or ROTEM were used.
Karkouti et al. performed an RCT on patients having cardiac surgery and demonstrated reduced transfusion requirement when VET was used [67]. Serraino et al. compared SLTs and VET in 8737 patients undergoing cardiac surgery and also demonstrated a decreased need for RBC and platelet transfusion. However, no difference in mortality, prevalence of stroke, time on a ventilator, hospital stay, or incidence of bleeding could not be demonstrated [68]. This report sparked a discussion among experts. In this study, the relative risk for mortality was 0.55 with a 95% CI of 0.28-1.1. If, however, the number of patients would have been larger, differences in mortality would have been significant [69]. It was also emphasized that reducing the rate of RBC and platelet transfusion was associated with a significant decrease in acute renal failure (RR= 0.42, 95% CI 0.2-0.86) and correlated with a better outcome [70].
Although the advantages of VET were already shown in 1985, and since then, several evaluations and metanalysis have demonstrated the beneficial effects of VET in reducing blood product transfusion as well as for decreasing morbidity and mortality, many LT centers are still reluctant to use of VET and rely on SLTs.
Hemostasis in Cirrhotic Patients with Sepsis
Patients with cirrhosis present with a persistent systemic inflammatory state and with different degrees of both innate and acquired immune dysfunction that is influenced by the severity of the liver disease. Portal hypertension, increased permeability of the intestine, and altered gut microbiota lead to intestinal bacteria translocation, endotoxemia, and worsening of systemic inflammation which is the typical pathophysiology of severe infections in patients with cirrhosis [71,72,73]. It has been previously demonstrated that sepsis is diagnosed in one-third of hospitalized cirrhotic patients [74]. Although sepsis is frequent in patients with ESLD, these patients have been underrepresented in sepsis and septic shock trials. The early diagnosis of organ dysfunction induced by sepsis is challenging in cirrhotic patients because, at the time of diagnosis, they have already hematologic changes caused by chronic liver disease. Coagulation disturbances in septic patients with cirrhosis are the result of coagulation activation induced by sepsis superimposed on an already altered hemostasis due to hepatic dysfunction. These changes in coagulation are dependent on the severity of both liver disease and infection and are not adequately evaluated by SLTs. VET and thrombin generation assay are global tests that can better reflect the hemostatic changes induced by sepsis or severe infection in patients with cirrhosis.
Unfortunately, there are not many studies assessing coagulation changes in septic cirrhotic patients, and some of these were published more than 20 years ago [75,76,77]. Taking into account the heterogeneity of patients with liver disease and concomitant severe infections, sepsis, or septic shock, it is difficult to have a complete picture of the hemostatic system in this patient population.
In patients without liver disease, severe infections result in coagulation activation mainly through the extrinsic pathway, with inhibition of natural anticoagulants and hypofibrinolysis [78,79].
Severe infections occurring in patients with compensated liver disease affect their hemostatic balance which often results in bleeding or thrombotic complications. In patients with decompensated cirrhosis, severe infections usually lead to a deterioration in SLTs, platelet count, and parameters of VET indicating hypocoagulability [75,77,80]. Resolving infection is associated with improvement in coagulopathy, while in patients continuing to deteriorate, hemostasis worsens [75]. In a study using thrombelastography, viscoelastic parameters reflecting clotting initiation and propagation (r, k, alpha) were more useful than SLTs to identify significant hemostatic changes in patients with persistent infection [75]. This reinforces the fact that VET is more sensitive for coagulation assessment in cirrhotic patients. When compared to cirrhotic patients without infections, or having mild infections, cirrhotic patients with sepsis have lower levels of pro- and anticoagulant factors, lower platelet counts, and increased soluble fibrin, independent of the severity of the liver disease [81]. These findings suggest that inflammation-induced coagulation activation leads to the consumption of coagulation factors and platelets in cirrhotic patients with severe infections or sepsis [81].
Sepsis causes 25-30% of acute-on-chronic liver failure (ACLF). About 50% of patients with ACLF from other causes will develop sepsis in the course of the disease [82]. A more recent study has demonstrated that patients with ACLF, presenting with an already decompensated hemostatic system, have further aggravation of coagulopathy by sepsis [80]. When compared to ACLF in patients without sepsis, patients with sepsis have been shown to have worse SLTs, but the differences were not statistically significant [80]. When compared to non-septic patients with ACLF presenting with hypocoagulability (confirmed by VET), patients with sepsis had more severely disturbed hemostasis manifested by longer clotting times and lower clot amplitude on thrombelastography [80]. Measurement of individual coagulation factors demonstrate that patients with ACLF have decreased levels of protein C and AT III, as well as increased levels of von vWF, FVIII, and tissue factor activity. All these parameters worsened significantly in patients who developed sepsis and this deterioration correlated with increased mortality at 28 days [80].
A recent study comparing patients with decompensated cirrhosis with and without infections revealed decreased levels of natural anticoagulants (protein C, S and, ATIII), FVII, and prolonged INR in infected patients, that normalized with infection resolution [83]. However, in this study the decrease of anticoagulants was not correlated with a higher degree of thrombin generation, which was similar in infected and non-infected patients [83].
In non-cirrhotic patients, sepsis leads to defects in platelet aggregation [84]. Similarly, patients with decompensated cirrhosis and bacterial infections present with decreased whole blood platelet aggregation when compared to cirrhotic patients without infections [83] This could be explained by the increased release of NO and prostacyclin induced by bacterial endotoxins in infected patients with cirrhosis, contributing to further impairment of primary hemostasis [71,85]. As opposite to secondary hemostasis which improved after infection resolution, a further impairment of platelet aggregation was noted in cirrhotic patients that recovered from infection independently of baseline platelet count and agonist used [83].
It is known that severe infections and sepsis in patients without liver cirrhosis are usually accompanied by hypofibrinolysis. Infected patients with decompensated cirrhosis present mixed fibrinolytic changes, with lower plasminogen levels, higher levels of tissue plasminogen activator, and similar levels of plasmin-antiplasmin complexes compared to non-infected patients [83].
Bacterial infection is associated with a significant heparin-like effect in chronic liver disease, impairing coagulation due to the release of endogenous heparinoids from endothelial surfaces, and possibly due to activation of mast cells with subsequent release of heparin and other mediators [76,77]. In their study, Montalto et al, demonstrated the presence of a heparin-like effect due to endogenous heparinoids using heparanase-modified thrombelastography in cirrhotic patients with severe infections, but not in non-infected cirrhotic patients or in infected patients without cirrhosis [77]. This heparin-like effect disappeared after the resolution of the infection [77]. These results were confirmed by Zambruni et al. who were able to demonstrate higher anti-Xa levels in infected compared to non-infected cirrhotic patients [76]. Endotoxin or cytokines are involved in the release of heparinoids from endothelial surfaces in a dose-dependent manner. This may be the reason why this effect was not shown in non-infected cirrhotic patients (having lower endotoxin levels). In non-cirrhotic patients with infections, this effect was not demonstrated because, with unaltered liver function, clearance of heparinoids is not affected [77]. These findings are important for explaining the increased bleeding risk in cirrhotic patients with severe infections.
Sepsis is the most common cause of disseminated intravascular coagulation (DIC). In DIC, both coagulation factors and platelet counts are decreased mainly due to consumption. This, in combination with increased fibrinolytic activity, represents the routine diagnostic criteria for DIC [86,87]. Diagnosing DIC in the presence of chronic liver disease is challenging due to the similarities in coagulation tests with both conditions. The problem of diagnosing DIC becomes even more complicated in patients with decompensated cirrhosis and ACLF. In these cases, DIC is often over-diagnosed with the usual DIC scoring systems. It is not surprising then that the diagnosis of DIC is often missed in patients with sepsis and ACLF, as the coagulation markers in ACLF patients can be severely altered even before they develop an infection.
In conclusion, patients with ESLD are prone to hypercoagulability on the level of primary, secondary, and tertiary hemostasis. Despite decreases in both coagulation and anticoagulation factors in the liver, these patients have an imbalanced system prone to both bleeding and clotting. ED dysfunction and increase thrombin generation play a central role in hypercoagulability associated with ESLD. SLTs do not accurately reflect the coagulation profile of patients with ESLD. VET is more reliable than SLTs for managing coagulation in this patient population. VET is particularly helpful in patients with ESLD complicated by sepsis, a situation with very complex coagulation disturbances.
Author Contributions
Conceptualization, FHS, DB, and ES; Methodology, FHS and DB; Formal Analysis, AF and KW; Investigation, FHS, DB, ES, and DCB; Writing-original draft preparation, FHS, DB, and ES.; Writing-FHS, DB, and ES; Visualization, DCB. All authors have read and agreed to the final version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Review article: no IRB statement needed.
Informed Consent Statement
Review report - no informed consent required.
Data Availability Statement
No own dataset.
Conflicts of Interest
FS has served as a speaker for CSL Behring, Werfen, Biotest, and Merz Pharmaceuticals and ES has served as a speaker for CSL Behring. Neither have conflicts of interest. The remaining authors declare no competing interests.
References
- Tripodi, A.; Salerno, F.; Chantarangkul, V.; Clerici, M.; Cazzaniga, M.; Primignani, M.; Mannucci, P.M. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology 2005, 41, 553–558. [Google Scholar] [CrossRef]
- Northup, P.G.; McMahon, M.M.; Ruhl, A.P.; E Altschuler, S.; Volk-Bednarz, A.; Caldwell, S.H.; Berg, C.L. Coagulopathy Does Not Fully Protect Hospitalized Cirrhosis Patients from Peripheral Venous Thromboembolism. Am. J. Gastroenterol. 2006, 101, 1524–1528. [Google Scholar] [CrossRef]
- Søgaard, K.K.; Horváth-Puhó, E.; Grønbæk, H.; Jepsen, P.; Vilstrup, H.; Sørensen, H.T. Risk of Venous Thromboembolism in Patients With Liver Disease: A Nationwide Population-Based Case–Control Study. Am. J. Gastroenterol. 2008, 104, 96–101. [Google Scholar] [CrossRef]
- Søgaard, K.K.; Horváth-Puhó, E.; Montomoli, J.; Vilstrup, H.; Sørensen, H.T. Cirrhosis is Associated with an Increased 30-Day Mortality After Venous Thromboembolism. Clin. Transl. Gastroenterol. 2015, 6, e97. [Google Scholar] [CrossRef]
- Wu, H.; Nguyen, G.C. Liver Cirrhosis Is Associated With Venous Thromboembolism Among Hospitalized Patients in a Nationwide US Study. Clin. Gastroenterol. Hepatol. 2010, 8, 800–805. [Google Scholar] [CrossRef]
- Dabbagh, O.; Oza, A.; Prakash, S.; Sunna, R.; Saettele, T.M. Coagulopathy Does Not Protect Against Venous Thromboembolism in Hospitalized Patients With Chronic Liver Disease. Chest 2010, 137, 1145–1149. [Google Scholar] [CrossRef]
- Gulley, D.; Teal, E.; Suvannasankha, A.; Chalasani, N.; Liangpunsakul, S. Deep Vein Thrombosis and Pulmonary Embolism in Cirrhosis Patients. Dig. Dis. Sci. 2008, 53, 3012–3017. [Google Scholar] [CrossRef]
- Mantaka, A.; Augoustaki, A.; Kouroumalis, E.A.; Samonakis, D.N. Portal vein thrombosis in cirrhosis: diagnosis, natural history, and therapeutic challenges. Ann Gastroenterol. 2018, 31, 315–329. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, B.-Y.; Wang, X.-B.; Zheng, X.; Huang, Y.; Chen, J.; Meng, Z.-J.; Gao, Y.-H.; Qian, Z.-P.; Liu, F.; et al. Prevalence and Clinical Significance of Portal Vein Thrombosis in Patients With Cirrhosis and Acute Decompensation. Clin. Gastroenterol. Hepatol. 2020, 18, 2564–2572. [Google Scholar] [CrossRef]
- Stine, J.G.; Pelletier, S.J.; Schmitt, T.M.; Porte, R.J.; Northup, P.G. Pre-transplant portal vein thrombosis is an independent risk factor for graft loss due to hepatic artery thrombosis in liver transplant recipients. HPB 2015, 18, 279–286. [Google Scholar] [CrossRef]
- Stine, J.G.; Shah, P.M.; Cornella, S.L.; Rudnick, S.R.; Ghabril, M.S.; Stukenborg, G.J.; Northup, P.G. Portal vein thrombosis, mortality and hepatic decompensation in patients with cirrhosis: A meta-analysis. World J. Hepatol. 2015, 7, 2774–80. [Google Scholar] [CrossRef]
- Chardot, C.; Herrera, J.M.; Debray, D., et al. Portal vein complications after liver transplantation for biliary atresia. Liver transplantation and surgery: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society 1997, 3, 351–358. [CrossRef]
- Gologorsky, E.; De Wolf, A.M.; Scott, V.; Aggarwal, S.; Dishart, M.; Kang, Y. Intracardiac thrombus formation and pulmonary thromboembolism immediately after graft reperfusion in 7 patients undergoing liver transplantation. Liver Transplant. 2001, 7, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.B.; Sundar, E.; Mahmood, F.; Sarge, T.; Hanto, D.W.; Panzica, P.J. Four Cases of Cardiopulmonary Thromboembolism During Liver Transplantation Without the Use of Antifibrinolytic Drugs. Obstet. Anesthesia Dig. 2005, 101, 1608–1612. [Google Scholar] [CrossRef]
- Shillcutt, S.K.; Ringenberg, K.J.; Chacon, M.M.; Brakke, T.R.; Montzingo, C.R.; Lyden, E.R.; Schulte, T.E.; Porter, T.R.; Lisco, S.J. Liver Transplantation: Intraoperative Transesophageal Echocardiography Findings and Relationship to Major Postoperative Adverse Cardiac Events. J. Cardiothorac. Vasc. Anesthesia 2016, 30, 107–114. [Google Scholar] [CrossRef]
- Sakai, T.; Matsusaki, T.; Dai, F.; Tanaka, K.A.; Donaldson, J.B.; Hilmi, I.A.; Marsh, J.W.; Planinsic, R.M.; Humar, A. Pulmonary thromboembolism during adult liver transplantation: incidence, clinical presentation, outcome, risk factors, and diagnostic predictors. Br. J. Anaesth. 2011, 108, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Duffy, J.P.; Hong, J.C.; Farmer, D.G.; Ghobrial, R.M.; Yersiz, H.; Hiatt, J.R.; Busuttil, R.W. Vascular Complications of Orthotopic Liver Transplantation: Experience in More than 4,200 Patients. J. Am. Coll. Surg. 2009, 208, 896–903. [Google Scholar] [CrossRef]
- Bezinover, D.; Iskandarani, K.; Chinchilli, V.; McQuillan, P.; Saner, F.; Kadry, Z.; Riley, T.R.; Janicki, P.K. Autoimmune conditions are associated with perioperative thrombotic complications in liver transplant recipients: A UNOS database analysis. BMC Anesthesiol. 2015, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bekker, J.; Ploem, S.; de Jong, K.P. Early hepatic artery thrombosis after liver transplantation: a systematic review of the incidence, outcome and risk factors. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2009, 9, 746–757. [Google Scholar] [CrossRef]
- Iida, T.; Kaido, T.; Yagi, S.; Hori, T.; Uchida, Y.; Jobara, K.; Tanaka, H.; Sakamoto, S.; Kasahara, M.; Ogawa, K.; et al. Hepatic arterial complications in adult living donor liver transplant recipients: a single-center experience of 673 cases. Clin. Transplant. 2014, 28, 1025–1030. [Google Scholar] [CrossRef]
- Salami, A.; Qureshi, W.; Kuriakose, P.; Moonka, D.; Yoshida, A.; Abouljoud, M. Frequency and Predictors of Venous Thromboembolism in Orthotopic Liver Transplant Recipients: A Single-Center Retrospective Review. Transplant. Proc. 2013, 45, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Næss, I.A.; Christiansen, S.C.; Romundstad, P.; Cannegieter, S.C.; Rosendaal, F.R.; Hammerstrøm, J. Incidence and mortality of venous thrombosis: a population-based study. J. Thromb. Haemost. 2007, 5, 692–699. [Google Scholar] [CrossRef]
- Mookerjee, R.P.; Vairappan, B.; Jalan, R. The puzzle of endothelial nitric oxide synthase dysfunction in portal hypertension: The missing piece? Hepatology 2007, 46, 943–946. [Google Scholar] [CrossRef]
- Harrison, P.; Wendon, J.; Williams, R. Evidence of increased guanylate cyclase activation by acetylcysteine in fulminant hepatic failure. Hepatology 1996, 23, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Clapp, B.R.; Hingorani, A.D.; Kharbanda, R.K.; Mohamedali, V.; Stephens, J.W.; Vallance, P.; MacAllister, R.J. Inflammation-induced endothelial dysfunction involves reduced nitric oxide bioavailability and increased oxidant stress. Cardiovasc. Res. 2004, 64, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Ferro, D.; Basili, S.; Lionetti, R.; Rossi, E.; Merli, M.; Riggio, O.; Bezzi, M.; Capocaccia, L. Ongoing Prothrombotic State in the Portal Circulation of Cirrhotic Patients. Thromb. Haemost. 1997, 77, 044–047. [Google Scholar] [CrossRef]
- Tripodi, A. Hemostasis in Acute and Chronic Liver Disease. Semin Liver Dis. 2017, 37, 28–32. [Google Scholar] [CrossRef]
- Tripodi, A.; Anstee, Q.M.; Sogaard, K.K.; Primignani, M.; Valla, D.C. Hypercoagulability in cirrhosis: causes and consequences. J. Thromb. Haemost. 2011, 9, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Raparelli, V.; Basili, S.; Carnevale, R.; Napoleone, L.; Del Ben, M.; Nocella, C.; Bartimoccia, S.; Lucidi, C.; Talerico, G.; Riggio, O.; et al. Low-grade endotoxemia and platelet activation in cirrhosis. Hepatology 2016, 65, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Saner, F.H.; Gieseler, R.K.; Akız, H.; Canbay, A.; Görlinger, K. Delicate Balance of Bleeding and Thrombosis in End-Stage Liver Disease and Liver Transplantation. Digestion 2013, 88, 135–144. [Google Scholar] [CrossRef]
- Hollestelle, M.J.; Geertzen, H.G.M.; Straatsburg, I.H.; van Gulik, T.M.; van Mourik, J.A. Factor VIII expression in liver disease. Thromb. Haemost. 2004, 91, 267–275. [Google Scholar] [CrossRef]
- Tripodi, A.; Mannucci, P.M. The Coagulopathy of Chronic Liver Disease. New Engl. J. Med. 2011, 365, 147–156. [Google Scholar] [CrossRef]
- Laffi, G.; Cominelli, F.; Ruggiero, M.; Fedi, S.; Chiarugi, V.P.; La Villa, G.; Pinzani, M.; Gentilini, P. Altered platelet function in cirrhosis of the liver: Impairment of inositol lipid and arachidonic acid metabolism in response to agonists. Hepatology 1988, 8, 1620–1626. [Google Scholar] [CrossRef] [PubMed]
- Lisman, T.; Bongers, T.N.; Adelmeijer, J.; Janssen, H.L.; de Maat, M.P.; de Groot, P.G.; Leebeek, F.W. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology 2006, 44, 53–61. [Google Scholar] [CrossRef]
- Tripodi, A.; Primignani, M.; Chantarangkul, V.; Dell’Era, A.; Clerici, M.; de Franchis, R.; Colombo, M.; Mannucci, P.M. An Imbalance of Pro- vs Anti-Coagulation Factors in Plasma From Patients With Cirrhosis. Gastroenterology 2009, 137, 2105–2111. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Primignani, M.; Lemma, L.; Chantarangkul, V.; Dell’Era, A.; Iannuzzi, F.; Aghemo, A.; Mannucci, P.M. Detection of the imbalance of procoagulant versus anticoagulant factors in cirrhosis by a simple laboratory method. Hepatology 2010, 52, 249–255. [Google Scholar] [CrossRef]
- Kotronen, A.; Joutsi-Korhonen, L.; Sevastianova, K.; Bergholm, R.; Hakkarainen, A.; Pietiläinen, K.H.; Lundbom, N.; Rissanen, A.; Lassila, R.; Yki-Järvinen, H. Increased coagulation factor VIII, IX, XI and XII activities in non-alcoholic fatty liver disease. Liver Int. 2010, 31, 176–183. [Google Scholar] [CrossRef]
- Cigolini, M.; Targher, G.; Agostino, G.; Tonoli, M.; Muggeo, M.; De Sandre, G. Liver Steatosis and Its Relation to Plasma Haemostatic Factors in Apparently Healthy Men - Role of the Metabolic Syndrome. Thromb. Haemost. 1996, 76, 069–073. [Google Scholar] [CrossRef]
- Hugenholtz, G.C.; Macrae, F.; Adelmeijer, J.; Dulfer, S.; Porte, R.J.; Lisman, T.; Ariëns, R.A.S. Procoagulant changes in fibrin clot structure in patients with cirrhosis are associated with oxidative modifications of fibrinogen. J. Thromb. Haemost. 2016, 14, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Nowakowski, T.; Cieśla-Dul, M.; Sadowski, J. Abnormal plasma fibrin clot characteristics are associated with worse clinical outcome in patients with peripheral arterial disease and thromboangiitis obliterans. Atherosclerosis 2011, 215, 481–486. [Google Scholar] [CrossRef]
- Stine, J.G.; Shah, N.L.; Argo, C.K.; Pelletier, S.J.; Caldwell, S.H.; Northup, P.G. Increased risk of portal vein thrombosis in patients with cirrhosis due to nonalcoholic steatohepatitis. Liver Transplant. 2015, 21, 1016–1021. [Google Scholar] [CrossRef]
- Stine, J.G.; Argo, C.K.; Pelletier, S.J.; Maluf, D.G.; Caldwell, S.H.; Northup, P.G. Advanced non-alcoholic steatohepatitis cirrhosis: A high-risk population for pre-liver transplant portal vein thrombosis. World J. Hepatol. 2017, 9, 139–146. [Google Scholar] [CrossRef]
- Stine, J.G.; Niccum, B.A.; Zimmet, A.N.; Intagliata, N.; Caldwell, S.H.; Argo, C.K.; Northup, P.G. Increased risk of venous thromboembolism in hospitalized patients with cirrhosis due to non-alcoholic steatohepatitis. Clin. Transl. Gastroenterol. 2018, 9, e140. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Fracanzani, A.L.; Primignani, M.; Chantarangkul, V.; Clerici, M.; Mannucci, P.M.; Peyvandi, F.; Bertelli, C.; Valenti, L.; Fargion, S. Procoagulant imbalance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2014, 61, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Biagini, M.R.; Tozzi, A.; Marcucci, R.; Paniccia, R.; Fedi, S.; Milani, S.; Galli, A.; Ceni, E.; Capanni, M.; Manta, R.; et al. Hyperhomocysteinemia and hypercoagulability in primary biliary cirrhosis. World J. Gastroenterol. 2006, 12, 1607–1612. [Google Scholar] [CrossRef]
- Zöller, B.; Li, X.; Sundquist, J.; Sundquist, K. Autoimmune diseases and venous thromboembolism: a review of the literature. Am. J. Cardiovasc. Dis. 2012, 2, 171–183. [Google Scholar]
- Prieto, J.; Yuste, J.R.; Beloqui, O., et al. Anticardiolipin antibodies in chronic hepatitis C: implication of hepatitis C virus as the cause of the antiphospholipid syndrome. Hepatology 1996, 23, 199–204. [CrossRef]
- Violi, F.; Ferro, D.; Basili, S.; Artini, M.; Valesini, G.; Levrero, M.; Cordova, C. Increased rate of thrombin generation in hepatitis C virus cirrhotic patients. Relationship to venous thrombosis. J. Investig. Med. 1995, 43, 550–554. [Google Scholar]
- Dahlbäck, B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood 2008, 112, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.P. Portal vein thrombosis: what is the role of genetics? Eur J Gastroenterol Hepatol. 2005, 17, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Amitrano, L.; Brancaccio, V.; Guardascione, M.A.; Margaglione, M.; Iannaccone, L.; D’Andrea, G.; Marmo, R.; Ames, P.R.; Balzano, A. Inherited coagulation disorders in cirrhotic patients with portal vein thrombosis. Hepatology 2000, 31, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Yang, Z.; Bai, M.; Shi, X.; Han, G.; Fan, D. Meta-analysis: the significance of screening for JAK2V617F mutation in Budd-Chiari syndrome and portal venous system thrombosis. Aliment. Pharmacol. Ther. 2011, 33, 1087–1103. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Li, H.; Liu, X.; Yao, H.; Han, G.; Hu, F.; Shao, L.; Guo, X. Novel insights into the development of portal vein thrombosis in cirrhosis patients. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1421–1432. [Google Scholar] [CrossRef]
- Saner, F.H.; Abeysundara, L.; Hartmann, M.; Mallett, S.V. Rational approach to transfusion in liver transplantation. Minerva Anestesiol. 2018, 84, 378–388. [Google Scholar] [CrossRef]
- Saner, F.H.; Bezinover, D. Assessment and management of coagulopathy in critically-ill patients with liver failure. Curr. Opin. Crit. Care 2019, 25, 179–186. [Google Scholar] [CrossRef]
- Palascak, J.E.; Martinez, J. Dysfibrinogenemia Associated with Liver Disease. J. Clin. Investig. 1977, 60, 89–95. [Google Scholar] [CrossRef]
- Reed, M.J.; Nimmo, A.F.; McGee, D., et al. Rotational thrombolelastometry produces potentially clinical useful results within 10 min in bleeding emergency department patients: the DEUCE study. Eur. J. Emerg. Med. 2013, 20, 160–166. [CrossRef]
- Haas, T.; Spielmann, N.; Mauch, J.; Madjdpour, C.; Speer, O.; Schmugge, M.; Weiss, M. Comparison of thromboelastometry (ROTEM(R)) with standard plasmatic coagulation testing in paediatric surgery. Br. J. Anaesth. 2012, 108, 36–41. [Google Scholar] [CrossRef]
- Monroe, D.; Hoffman, M. A Cell-based Model of Hemostasis. Thromb. Haemost. 2001, 85, 958–965. [Google Scholar] [CrossRef]
- Ewe, K. Bleeding after liver biopsy does not correlate with indices of peripheral coagulation. Dig. Dis. Sci. 1981, 26, 388–393. [Google Scholar] [CrossRef]
- Haas, T.; Fries, D.; Tanaka, K.A.; Asmis, L.; Curry, N.S.; Schochl, H. Usefulness of standard plasma coagulation tests in the management of perioperative coagulopathic bleeding: is there any evidence? Br J Anaesth. 2015, 114, 217–224. [Google Scholar] [CrossRef]
- Kang, Y.G.; Martin, D.J.; Marquez, J.; Lewis, J.H.; Bontempo, F.A.; Shaw, B.W.; Starzl, T.E.; Winter, P.M. Intraoperative Changes in Blood Coagulation and Thrombelastographic Monitoring in Liver Transplantation. Obstet. Anesthesia Dig. 1985, 64, 888–896. [Google Scholar] [CrossRef]
- De Pietri, L.; Bianchini, M.; Montalti, R.; De Maria, N.; Di Maira, T.; Begliomini, B.; Gerunda, G.E.; di Benedetto, F.; Garcia-Tsao, G.; Villa, E. Thrombelastography-guided blood product use before invasive procedures in cirrhosis with severe coagulopathy: A randomized, controlled trial. Hepatology 2015, 63, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ahmad, J.; Maiwall, R.; Choudhury, A.; Bajpai, M.; Mitra, L.G.; Saluja, V.; Agarwal, P.M.; Bihari, C.; Shasthry, S.M.; et al. Thromboelastography-Guided Blood Component Use in Patients With Cirrhosis With Nonvariceal Bleeding: A Randomized Controlled Trial. Hepatology 2019, 71, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Maria, A.; Lal, B.B.; Khanna, R.; Sood, V.; Mukund, A.; Bajpai, M.; Alam, S. Rotational thromboelastometry-guided blood component use in cirrhotic children undergoing invasive procedures: Randomized controlled trial. Liver Int. 2022, 42, 2492–2500. [Google Scholar] [CrossRef]
- Wikkelsø, A.; Wetterslev, J.; Møller, A.M.; Afshari, A. Thromboelastography (TEG) or thromboelastometry (ROTEM) to monitor haemostatic treatment versus usual care in adults or children with bleeding. Cochrane Database Syst. Rev. 2016, 2018, CD007871. [Google Scholar] [CrossRef]
- Karkouti, K.; Callum, J.; Wijeysundera, D.N., et al. Point-of-Care Hemostatic Testing in Cardiac Surgery: A Stepped-Wedge Clustered Randomized Controlled Trial. Circulation 2016, 134, 1152–1162. [CrossRef] [PubMed]
- Serraino, G.F.; Murphy, G.J. Routine use of viscoelastic blood tests for diagnosis and treatment of coagulopathic bleeding in cardiac surgery: updated systematic review and meta-analysis. Br. J. Anaesth. 2017, 118, 823–833. [Google Scholar] [CrossRef]
- Ranucci, M. Bank blood shortage, transfusion containment and viscoelastic point-of-care coagulation testing in cardiac surgery. Br. J. Anaesth. 2017, 118, 814–815. [Google Scholar] [CrossRef]
- Kozek, S.; Schoechl, H.; Gratz, J. Response to: Routine use of viscoelastic blood tests for diagnosis and treatment of coagulopathic bleeding in cardiac surgery: updated systematic review and meta-analysis. Br. J. Anaesth. 2017, 119, 543–544. [Google Scholar] [CrossRef]
- Thalheimer, U.; Triantos, C.K.; Samonakis, D.N.; Patch, D.; Burroughs, A.K. Infection, coagulation, and variceal bleeding in cirrhosis. Gut 2005, 54, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Zazueta, G.; de León-Garduño, L.A.P.; Aguirre-Valadez, J.; Torre-Delgadillo, A. Bacterial infections in cirrhosis: Current treatment. Ann. Hepatol. 2019, 19, 238–244. [Google Scholar] [CrossRef]
- Wong, F.; Bernardi, M.; Balk, R.; Christman, B.; Moreau, R.; Garcia-Tsao, G.; Patch, D.; Soriano, G.; Hoefs, J.; Navasa, M. Sepsis in cirrhosis: report on the 7th meeting of the International Ascites Club. Gut 2005, 54, 718–725. [Google Scholar] [CrossRef]
- Safi, W.; Elnegouly, M.; Schellnegger, R.; Umgelter, K.; Geisler, F.; Reindl, W.; Saugel, B.; Hapfelmeier, A.; Umgelter, A. Infection and Predictors of Outcome of Cirrhotic Patients after Emergency Care Hospital Admission. Ann. Hepatol. 2018, 17, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoridis, G.V.; Patch, D.; Webster, G.J.; Brooker, J.; Barnes, E.; Burroughs, A.K. Infection and hemostasis in decompensated cirrhosis: A prospective study using thrombelastography. Hepatology 1999, 29, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Zambruni, A.; Thalheimer, U.; Coppell, J., et al. Endogenous heparin-like activity detected by anti-Xa assay in infected cirrhotic and non-cirrhotic patients. Scandinavian journal of gastroenterology 2009, 39, 830–836. [CrossRef]
- Montalto, P.; Vlachogiannakos, J.; Cox, D.J.; Pastacaldi, S.; Patch, D.; Burroughs, A.K. Bacterial infection in cirrhosis impairs coagulation by a heparin effect: a prospective study. J. Hepatol. 2002, 37, 463–470. [Google Scholar] [CrossRef]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N Engl J Med. 2013, 369, 840–851. [Google Scholar] [CrossRef]
- Levi, M. The coagulant response in sepsis and inflammation. Hamostaseologie 2010, 30, 10–16. [Google Scholar] [CrossRef]
- Premkumar, M.; Saxena, P.; Rangegowda, D.; Baweja, S.; Mirza, R.; Jain, P.; Bhatia, P.; Kumar, G.; Bihari, C.; Kalal, C.; et al. Coagulation failure is associated with bleeding events and clinical outcome during systemic inflammatory response and sepsis in acute-on-chronic liver failure: An observational cohort study. Liver Int. 2019, 39, 694–704. [Google Scholar] [CrossRef]
- Plessier, A.; Denninger, M.-H.; Consigny, Y.; Pessione, F.; Francoz, C.; Durand, F.; Francque, S.; Bezeaud, A.; Chauvelot-Moachon, L.; Lebrec, D.; et al. Coagulation disorders in patients with cirrhosis and severe sepsis. Liver Int. 2003, 23, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Dong, V.; Karvellas, C.J. Acute-on-chronic liver failure: Objective admission and support criteria in the intensive care unit. JHEP Rep. 2019, 1, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zanetto, A.; Campello, E.; Bulato, C.; Gavasso, S.; Saggiorato, G.; Shalaby, S.; Burra, P.; Angeli, P.; Senzolo, M.; Simioni, P. Global hemostatic profiling in patients with decompensated cirrhosis and bacterial infections. JHEP Rep. 2022, 4, 100493. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.-L.; Yagushi, A.; Pradier, O. Platelet function in sepsis. Crit. Care Med. 2002, 30, S313–S317. [Google Scholar] [CrossRef]
- Kujovich, J.L. Coagulopathy in liver disease: a balancing act. Hematology 2015, 2015, 243–249. [Google Scholar] [CrossRef]
- Iba, T.; Levi, M.; Levy, J.H. Sepsis-Induced Coagulopathy and Disseminated Intravascular Coagulation. Semin. Thromb. Hemost. 2019, 46, 089–095. [Google Scholar] [CrossRef]
- Taylor, F.B.; Jr Toh, C.H.; Hoots, W.K.; Wada, H.; Levi, M. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 2001, 86, 1327–1330. [Google Scholar] [CrossRef]
Figure 1.
Cell-based coagulation process. The waterfall cascade hemostasis model was replaced 2001 with a cell-based model [59]. The coagulation process begins with release of Tissue Factor from endothelial cells, monocytes, and macrophages. Tissue Factor then starts with the initiation phase with F VIIa. Propagation and amplification follow ending with a thrombin burst. The fibrin net is then built and stabilized with F XIII.
Figure 1.
Cell-based coagulation process. The waterfall cascade hemostasis model was replaced 2001 with a cell-based model [59]. The coagulation process begins with release of Tissue Factor from endothelial cells, monocytes, and macrophages. Tissue Factor then starts with the initiation phase with F VIIa. Propagation and amplification follow ending with a thrombin burst. The fibrin net is then built and stabilized with F XIII.

Figure 2.
Standard laboratory tests assess only 5-10% of the coagulation process, while viscoelastic tests, like Rotem, assess the whole coagulation process. TAT: Thrombin-Anti-Thrombin; PT: Prothrombin time; aPTT: activated partial thromboplastin time.
Figure 2.
Standard laboratory tests assess only 5-10% of the coagulation process, while viscoelastic tests, like Rotem, assess the whole coagulation process. TAT: Thrombin-Anti-Thrombin; PT: Prothrombin time; aPTT: activated partial thromboplastin time.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



