Submitted:
11 July 2023
Posted:
13 July 2023
You are already at the latest version
Abstract
We present a case of a combination of two rare hereditary disorders: Аdrenal insufficiency, obesity, and red hair syndrome (OBAIRH) and Duchenne muscular dystrophy (DMD) in a boy. Both diseases were diagnosed during the first year of life. OBAIRH was suggested based on the ethnicity and family history of the patient, while DMD – based on an extreme increase in transaminase and CK levels during biochemical analysis of his blood.
Keywords:
OBAIRH
; DMD
; double trouble
; POMC
1. Introduction
A large number of hereditary disorders are congenital, noticeable immediately after birth, some of them – even during ultrasound examination at various terms of pregnancy; however, most diseases have no distinct phenotypical features. The diagnosis is often established after the manifestation of pronounced clinical symptoms, when therapy is ineffective. Some hereditary disorders are included in the mass neonatal screening programmes, which allow to form a risk group, confirm the diseases, and start therapy before the manifestation of the symptoms. The inclusion of a disorder into such programmes is considered only in case of a certain frequency of the disease in the population and availability of effective therapy. Therefore, rare genetic disorders are often left undiagnosed during the infant’s first year of life. Cases of diagnosing two rare hereditary disorders, which are not included in the screening, in one patient on the first year of life are extremely rare. We present a case of a combination of two rare hereditary disorders: Аdrenal insufficiency, obesity, and red hair syndrome (OBAIRH) and Duchenne muscular dystrophy (DMD) in a boy. Both diseases were diagnosed during the first year of life. OBAIRH was suggested based on the patient’s ethnicity and family history, while DMD – based on the extreme increase in transaminase and CK levels during biochemical blood analysis.
2. Results
Patient D. was initially referred to a geneticist at the age of five months. He was hospitalized in the pediatric unit of the "REGIONAL CHILDREN'S CLINICAL HOSPITAL", Perm Russia, with an atonic seizure and cyanosis as a complication of a feverless viral infection. The anamnesis shows that in the early neonatal period the patient had a seizure because of hypoglycemia down to 1.1 mmol/l without any registered epileptic activity. The seizures did not repeat until the present episode: in the hospital, during the preparation for brain MRI, as a result of prolonged fasting, the patient had another atonic seizure with the glucose level decreasing to 1.1 mmol/l.
The child was born from second pregnancy, first birth, at the term of 40 weeks, weight was 3630 g, length 51 cm, head circumference 36 cm, Apgar score 6/8. His parents are healthy, the father is 35 years old, and the mother is 21 years old, consanguinity denied. Both parents belong in an ethnographic group of Perm’ Tatar – a Tatar group included in the Kazan’ Tatar group and inhabiting the territory of the Perm’ ethnocultural region [1]. The Perm’ Tatar group is the second-largest ethnic group of the Perm’ Krai [2].
The results of the examination at the age of five months are unavailable; at the age of one year the height was 84 cm (greater than the 97th percentile), weight 16.5 kg (greater than the 97th percentile), head circumference 54 cm (greater than the 97th percentile), thoracic circumference 69.5 cm (greater than the 97th percentile). The skin was clean, the hair was red, no pigmentation defects were detected. Thoracic shape was normal, joints unaltered, abdomen was soft and painless, liver and spleen size was normal, stool and diuresis normal. Male genitals, microgenitalism, testicles not in the srotum. The patient did not sit up on his own but sat independently while seated, stood and walked with support, babbled, and followed toys with his eyes.
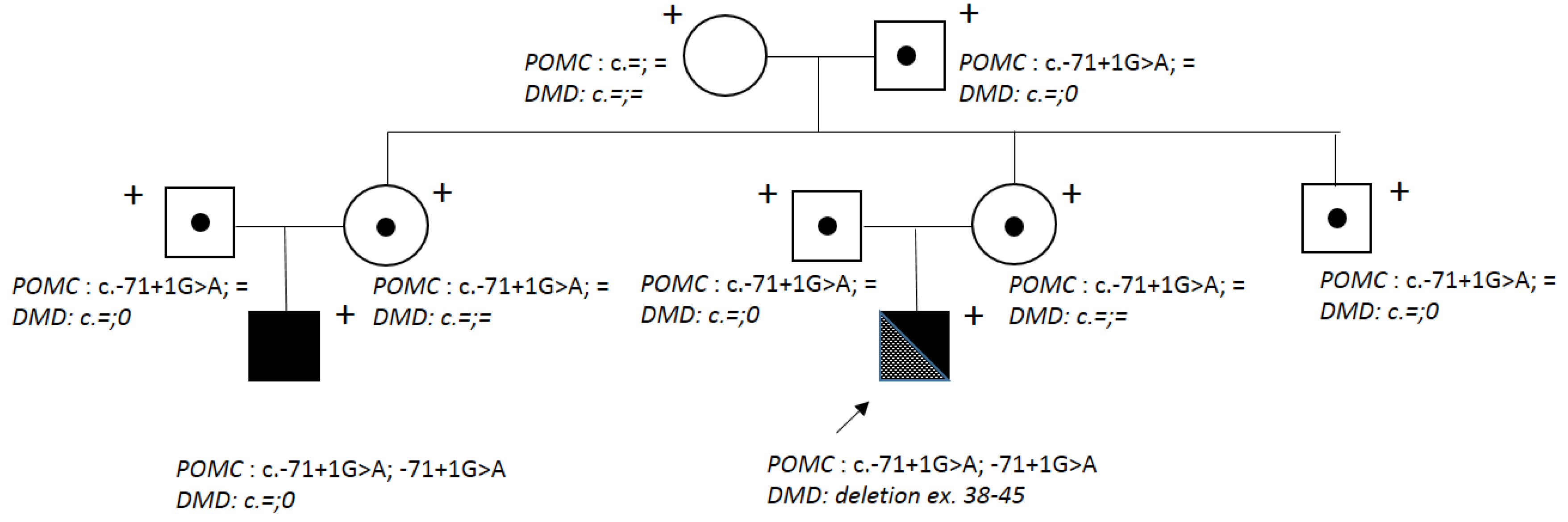
Genealogical analysis showed that the proband’s cousin on the mother’s side is affected by the Obesity, adrenal insufficiency, and red hair syndrome (OMIM 609734) due to POMC deficiency caused by a homozygous ethnic c.-71+1G>A mutation in the regulatory region of the POMC gene (Figure 1).
Blood hormone level analysis at the age of five months showed a decrease in cortisol levels to < 13.8 nmol/l (reference levels 101.2 – 535.7 nmol/l), while ACTH, insulin, TSH, and T4 levels were normal.
Biochemical examination at the age of five months detected a significant increase in transaminase levels AST – 374 МU/l (reference < 40 МU/l), ALT – 424 МU/l (reference < 40 МU/l), CК – 14 333 U/l (reference 24-190 U/l), CK-МВ – 261 U/l (reference 0-24 U/l), LDH – 1594 U/l (reference 225-937 U/l); total protein, albumins, creatinine, urea, alkaline phosphatase, CRP, bilirubin, glucose (without a seizure), and electrolytes were normal. At the age of one year AST – 228 МU/l (reference < 40 МU/l), ALT – 197 МU/l (reference < 40 МU/l), CК – 10 005 U/l (reference 24-190 U/l), CK-МВ – 253 U/l (reference 0-24 U/l), LDH – 1633 U/l (reference 225-937 U/l); total protein, albumins, creatinine, urea, alkaline phosphatase, CRP, bilirubin, glucose, and electrolytes were normal
Ultrasound examination of the abdominal cavity at the age of five months and one year showed no structural alterations in the organs. Brain MRI at the age of five months detected no focal structural alterations in the brain; however, the subarachnoid space was expanded. Ultrasound imaging of the heart showed a functioning oval window.
Based on the genealogical analysis results, the patient’s phenotype (excessive body mass and height, red hair), hypoglycemia episodes, decrease in cortisol level, as well as the ethnicity of Perm’ Tatar, patient D. was diagnosed with OBAIRH, which was confirmed by molecular genetic analysis – Sanger sequencing of the regulatory region of the POMC gene (NM_001035256.3): a pathogenic c.-71+1G>A variant was detected in a homozygous state.
However, biochemical blood analysis for the proband’s cousin with the same symptoms and genotype did not show any alterations: AST, ALT, and CK levels were within the reference values.
Seeing that patient D. did not have hepatic pathology, a primary muscle lesion was suggested. At the age of ten months, electromyography of the shoulder girdle and lower limbs was carried out. It detected a significant decrease in MUAP duration and amplitude, which may signify muscular lesion type; however, the examination was not completed due to the patient’s anxiety.
Molecular genetic analysis of the DMD gene (NM_004006.3) using quantitative MLPA detected a hemizygous deletion spanning exons 38 to 45 of this gene and leading to a frameshift.
Molecular genetic analysis was also carried out for the proband’s relatives. His mother, grandmother, and sister did not carry this deletion. A heterozygous POMC (NM_001035256.3) variant c.-71+1G>A was detected in the proband’s parents, aunt and her husband (parents of the patient’s cousin with OBAIRH), grandfather, and uncle. The grandmother did not have the c.-71+1G>A variant.
When the diagnosis was established, replacement therapy using glucocorticosteroids was implemented: the dose for this age was 1.25 mg three times per day. Fractional nutrition was prescribed every 2-3 hours with the exception of night time. The glucose level with constant monitoring was within the norm (3.5 – 4.5 mmol/l) and did not decrease after night sleep.
The medication for obesity therapy in case of proopiomelanocartin deficiency (Setmelanotide) cannot be prescribed until the age of six months. For patients with DMD caused by a deletion of exons 38 – 45 of the DMD gene, pathogenetic and etiotropic therapy is unavailable; however, corticosteroids are proven to be effective in patients with this type of muscular dystrophy [3] as supportive therapy during the wait for new genotherapeutic medication for this disorder [4].
3. Discussion
The OBAIRH syndrome is an extremely rare hereditary disorder with autosomal recessive inheritance type [5]. However, patient D. belongs to an ethnic group of Perm’ Tatar, for which the accumulation of this disease, caused by the c.-71+1G>A variant spread in this group due to the founder effect [6], was shown; aside from that, there was a case of this syndrome in the proband’s cousin. The accumulation of ethnically specific recessive mutations caused by the founder effect was described for various populations in the Russian Federation: osteopetrosis caused by the c.807+5G>A mutation in the TCIRG1 gene, as well as erythrocytosis caused by the c.598C>T variant in the VHL gene [7,8,9] in Chuvash and Mari El populations; hyperphenylalaninemia caused by the p.Arg261* variant in the PAH gene and type 1 tyrosinemia caused by the c.1025C>T (Pro342Leu) mutation in the HT1 gene in North Caucasian populations [10]; methemoglobinemia caused by the с.806С>T variant in the CYB5R3 gene in the Yakut group [11], as well as other disorders [12]. Thus, during medical genetic counseling, the information about the patients’ ethnic origin and studies on the accumulation of rare genetic variants in certain ethnic groups are crucial. This information allows to shorten the patient’s path to the definitive diagnosis and corresponding adequate therapy. The c.-71+1G>A variant in the POMC gene (NM_001035256.3) is located in the splicing region of a non-coding exon; however, despite the fact that the mechanism of its pathogenicity is not connected to the formation of aberrant RNA forms, it still leads to a drastic decrease in RNA and POMC protein levels, which could be connected to the decrease in transcription levels, or to nuclear degradation of pre-mRNA [6].
The combination of two genetic disorders is quite rare, and usually one of the diseases has a high prevalence in the population. Duchenne muscular dystrophy is very common: according to various sources, the frequency is 1 in 3500 - 10000 males [13,14]. Some studies describe combinations of DMD caused by deletions in the DMD gene with skeletal dysplasia: osteogenesis imperfecta (mutations in the COL1A1 gene) and pseudoachondroplasia (mutation in the COMP gene) [15]. A Czech patient had a phenotype caused by the combination of a point mutation in the DMD gene and a duplication of the PMP22 gene causing CMT1A [16], and a Chinese patient – a duplication of the PMP22 gene and a DMD deletion [17]. In both cases, CMT1A was inherited in the families from generation to generation, and DMD was caused by de novo mutations. There are described cases of combinations of various muscular dystrophy forms – Becker muscular dystrophy inherited from a carrier mother and facioscapulohumeral muscular dystrophy inherited from the father [18].
Duchenne muscular dystrophy is usually diagnosed in male patients at the age of three to five years, when neurologists note progressive muscular weakness and gait impairments [3]. Detecting DMD at such a young age (one year) was possible due to the diagnostic apprehension of the doctors, who noted additional symptoms not characteristic of OBAIRH. A constant increase in transaminase levels, which was not detected in the proband’s relative with POMC deficiency, was a reason to search for a second disorder. As a result, we detected a de novo deletion of exons 38 – 45 of the DMD gene leading to a frameshift, which is characteristic of a more severe disease form – Duchenne muscular dystrophy.
4. Materials and Methods
DNA was extracted from whole blood samples using a Wizard® Genomic DNA Purification Kit (Promega, Madison, Wisconsin, USA) according to the manufacturer’s protocol.
Automated Sanger sequencing was carried out using an ABIPrism 3500xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s protocol. Primer sequences were chosen according to the POMC (NM_001035256.3) reference sequence. POMC fragment sequences were amplified using the patient’s and relatives’ genomic DNA as a template with the following primers: POMCF:TAGGGCAAGCGGCGGCGAAGGAGG and POMCR:TTCGCACGATCTCGGCATCTTCCAG.
Quantitative analysis was carried out using the SALSA MLPA Probemix P034-DMD1 and P035-DMD2 (MRC-Holland, Amsterdam, Netherlands).
This study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of the Research Center for Medical Genetics, Moscow, Russia (approval number 2018-3/4). The probands gave informed consent to the genetic testing and the publication of anonymized data.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, O.S. and V.K.; methodology, O.S., V.K. V.P,; software, A.P.; validation, O.S., V.K, A.P., S.E. and V.P..; formal analysis, V.P..; investigation O.S., V.K, A.P., S.E, E.Z..; resources, A.P..; data curation, A.P., E.Z.; writing—original draft preparation, O.S., A.P..; writing—review and editing, A.P., S.E..; supervision, A.P..; project administration, O.S..; funding acquisition, A.P. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This work was supported by a state assignment of the Ministry of Science and Higher Education of the Russian Federation for RCMG.
Institutional Review Board Statement
This study was approved by the local ethics committee of the Research Centre for Medical Genetics (approval number 2018-3/4).
Informed Consent Statement
All procedures performed in studies involving human participants were in accordance with the ethical standards of the WMA DECLARATION OF HELSINKI “ETHICAL PRINCIPLES FOR MEDICAL RESEARCH INVOLVING HUMAN SUBJECTS” (https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (accessed on 05 June 2023)). This article does not contain any studies with animals performed by any of the authors. Written informed consent was obtained for genetic examination and publication with anonymity from all patients or their legal representa-tives.
Data Availability Statement
We can provide them or conduct a reanalysis upon request by mail schagina@med-gen.ru.
Conflicts of Interest
The authors declare no conflict of interest. Raw data cannot be posted due to patient confidentiality.
References
- Khudiakova, E. S Components of Ethnic Identity of Tatars of Perm Territory (According to Spontaneous Texts). Components of Ethnic Identity of Tatars of Perm Territory (according to Spontaneous Texts) 2020, 0, 190–205. [Google Scholar] [CrossRef]
- Rosstat — All-Russian Population Census 2020. Available online: https://rosstat.gov.ru/vpn_popul (accessed on 23 June 2023).
- Weber, F.J.; Latshang, T.D.; Blum, M.R.; Kohler, M.; Wertli, M.M. Prognostic Factors, Disease Course, and Treatment Efficacy in Duchenne Muscular Dystrophy: A Systematic Review and Meta-Analysis. Muscle Nerve 2022, 66, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Zhang, J.; Shi, K.; Liu, Z. Drug Development Progress in Duchenne Muscular Dystrophy. Front Pharmacol 2022, 13, 950651. [Google Scholar] [CrossRef] [PubMed]
- Graves, L.E.; Khouri, J.M.; Kristidis, P.; Verge, C.F. Proopiomelanocortin Deficiency Diagnosed in Infancy in Two Boys and a Review of the Known Cases. Journal of Paediatrics and Child Health 2021, 57, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Viakhireva, I.; Kalinchenko, N.; Vasilyev, E.; Chistousova, G.V.; Filatova, A.; Marakhonov, A.; Rubtsov, P.M.; Skoblov, M.; Tiulpakov, A. A Founder Mutation in the POMC 5′-UTR Causes Proopiomelanocortin Deficiency Through Splicing-Mediated Decrease of MRNA. The Journal of Clinical Endocrinology & Metabolism 2022, 107, e3654–e3660. [Google Scholar] [CrossRef]
- Sergeyeva, A.; Gordeuk, V.R.; Tokarev, Y.N.; Sokol, L.; Prchal, J.F.; Prchal, J.T. Congenital Polycythemia in Chuvashia. Blood 1997, 89, 2148–2154. [Google Scholar] [CrossRef] [PubMed]
- Bliznetz, E.A.; Tverskaya, S.M.; Zinchenko, R.A.; Abrukova, A.V.; Savaskina, E.N.; Nikulin, M.V.; Kirillov, A.G.; Ginter, E.K.; Polyakov, A.V. Genetic Analysis of Autosomal Recessive Osteopetrosis in Chuvashiya: The Unique Splice Site Mutation in TCIRG1 Gene Spread by the Founder Effect. Eur J Hum Genet 2009, 17, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; Sergueeva, A.I.; Miasnikova, G.Y.; Okhotin, D.; Voloshin, Y.; Choyke, P.L.; Butman, J.A.; Jedlickova, K.; Prchal, J.T.; Polyakova, L.A. Congenital Disorder of Oxygen Sensing: Association of the Homozygous Chuvash Polycythemia VHL Mutation with Thrombosis and Vascular Abnormalities but Not Tumors. Blood 2004, 103, 3924–3932. [Google Scholar] [CrossRef]
- Gundorova, P.; Zinchenko, R.A.; Kuznetsova, I.A.; Bliznetz, E.A.; Stepanova, A.A.; Polyakov, A.V. Molecular-Genetic Causes for the High Frequency of Phenylketonuria in the Population from the North Caucasus. PLOS ONE 2018, 13, e0201489. [Google Scholar] [CrossRef] [PubMed]
- Galeeva, N.M.; Voevoda, M.I.; Spiridonova, M.G.; Stepanov, V.A.; Poliakov, A.V. [Population frequency and age of c. 806C > T mutation in CYB5R3 gene as cause of recessive congenital methemoglobinemia in Yakutia]. Genetika 2013, 49, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Zinchenko, R.A.; Ginter, E.K.; Marakhonov, A.V.; Petrova, N.V.; Kadyshev, V.V.; Vasilyeva, T.P.; Alexandrova, O.U.; Polyakov, A.V.; Kutsev, S.I. Epidemiology of Rare Hereditary Diseases in the European Part of Russia: Point and Cumulative Prevalence. Frontiers in Genetics 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nat Rev Dis Primers 2021, 7, 1–19. [Google Scholar] [CrossRef]
- Guiraud, S.; Chen, H.; Burns, D.T.; Davies, K.E. Advances in Genetic Therapeutic Strategies for Duchenne Muscular Dystrophy. Exp Physiol 2015, 100, 1458–1467. [Google Scholar] [CrossRef]
- Donkervoort, S.; Schindler, A.; Tesi-Rocha, C.; Schreiber, A.; Leach, M.E.; Dastgir, J.; Hu, Y.; Mankodi, A.; Wagner, K.R.; Friedman, N.R.; et al. ‘Double Trouble’: Diagnostic Challenges in Duchenne Muscular Dystrophy in Patients with an Additional Hereditary Skeletal Dysplasia. Neuromuscular Disorders 2013, 23, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Vondracek, P.; Hermanova, M.; Sedlackova, J.; Fajkusova, L.; Stary, D.; Michenkova, A.; Gaillyova, R.; Seeman, P.; Mazanec, R. Charcot-Marie-Tooth Neuropathy Type 1A Combined with Duchenne Muscular Dystrophy. Eur J Neurol 2007, 14, 1182–1185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cui, F.; Chen, D.; Pu, C.; Chen, Z.; Yang, F.; Wu, H.; Huang, X. Coexistence of Peripheral Myelin Protein 22 and Dystrophin Mutations in a Chinese Boy: CMT Type 1A Comorbid with DMD. Muscle Nerve 2013, 48, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Schöneborn, S.; Weis, J.; Kress, W.; Häusler, M.; Zerres, K. Becker’s Muscular Dystrophy Aggravating Facioscapulohumeral Muscular Dystrophy – Double Trouble as an Explanation for an Atypical Phenotype. Neuromuscular Disorders 2008, 18, 881–885. [Google Scholar] [CrossRef]
Figure 1.
The proband’s family tree. The arrow points at patient D. Plus signs indicate the examined and genotyped family members; dots - carriers of the c.-71+1G>A variant in the POMC gene; black colour – patients with OBAIRH. .
Figure 1.
The proband’s family tree. The arrow points at patient D. Plus signs indicate the examined and genotyped family members; dots - carriers of the c.-71+1G>A variant in the POMC gene; black colour – patients with OBAIRH. .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



