
Submitted:
28 June 2023
Posted:
29 June 2023
You are already at the latest version
Abstract
Fibrosis is a prevalent and detrimental condition associated with various diseases with a high impact on global morbidity and mortality rates. Despite its diverse causes and affected organs, common underlying mechanisms drive the development and progression of the disease. These mechanisms include an exaggerated inflammatory response, excessive activation of fibroblasts, and abnormal tissue remodeling following severe or repetitive tissue injury. Although significant advancements have been achieved to enhance our understanding of fibrosis, there is still a gap between identifying potential antifibrotic targets and successfully translating them into effective clinical interventions. Novel approaches that target specific cellular and molecular processes involved in fibrosis hold promise for reducing the pathological consequences of the disease. Understanding the pathogenesis and clinical implications of fibrotic diseases is crucial for developing effective therapeutic strategies and improving patient outcomes. In this review, we introduce the concept of fibrosis, discuss the mechanisms by which it arises, and explore existing and emerging therapeutic approaches in development.
Keywords:
fibrosis
; inflammatory
; fibroblasts
; extracellular matrix
; therapeutic strategy
1. Introduction
Fibrosis is a widely recognized cause of disease-related morbidity and mortality worldwide [1]. Despite the diverse array of etiologies and affected organs, common mechanisms underlying the development and progression of fibrosis exist [2]. Specifically, these include a hyperactive inflammatory response, excessive activation of fibroblasts, and aberrant tissue remodeling following severe or repetitive tissue injury [3]. Such damage may occur in response to a variety of stimuli, such as persistent infections, prolonged exposure to irritating non-infectious agents (e.g., silica, radiation), autoimmune conditions, mechanical injury, or damage to components involved in normal regulation of tissue regeneration [4, 5]. Regardless of the underlying cause, fibrosis can occur in any solid organ or tissue and is a key underlying feature in numerous chronic diseases [6]. Altogether, fibrosis has been implicated in up to 45% of deaths in the developed world [7].
As a pervasive pathological repair process, fibrosis commonly occurs in various organs in response to tissue damage, leading to the substitution of functional tissue with non-functional connective tissue [8]. Despite the widespread prevalence of tissue fibrosis across numerous disease states in diverse organs, the existing therapeutic approaches aimed at preventing or reducing fibrosis are severely limited in terms of both quantify and efficacy. Unfortunately, there is currently no effective cure available to address the structural and functional damage induced by fibrosis-related disorders. In fibrosis, excessive deposition of extracellular matrix (ECM) components occurs [9]. While the deposition of collagen by fibroblasts, specifically activated α-smooth muscle actin+ myofibroblasts, is an essential step in physiological wound repair, dysregulation of this process can lead to ECM accumulation and disruption of normal tissue architecture [10]. Over time, this accumulation contributes to the development of fibrotic diseases, ultimately resulting in compromised organ function or even organ failure [10].
Understanding the pathogenesis and clinical implications of fibrotic disorders is paramount for developing effective therapeutics and improving patient outcomes. In this review, we aim to both introduce the concept of fibrosis and discuss mechanisms by which it may arise. We will then explore several diseases to illustrate the impact of fibrosis on organ function, paying attention to how the common mechanisms that result in fibrosis can have such varied clinical effects. Finally, we will examine the current therapeutic landscape and discuss existing treatments, with a look down the pipeline at what new therapies are currently in development.
1. Pathogenesis of Fibrosis
1.1. Overview of Wound Healing
Repair of soft tissues following injury is a highly coordinated physiologic response that involves three overlapping, but functionally distinct stages: hemostasis and inflammation, cell regeneration, and tissue remodeling [11] [Figure 1].
Almost immediately following injury, damaged epithelial cells, endothelial cells, and fibroblasts release inflammatory mediators that induce vasoconstriction and platelet plug formation to minimize blood loss [12]. Activated platelets within this region aggregate and degranulate to release substances including thromboxane A2 and serotonin, which further constrict blood vessels and promote clot formation [12]. The clotting cascade is then initiated to induce formation of a fibrin meshwork over the platelet plug and stabilization of the blood clot [13, 14]. As this hemostatic process is carried out, damaged tissue is simultaneously secreting growth factors, proinflammatory cytokines, and chemokines to stimulate recruitment of leukocytes to the site of damage [15]. Fibroblasts/myofibroblasts, epithelial cells, and endothelial cells also secrete matrix metalloproteinases (MMPs) to degrade the basement membrane and allow access for infiltrating cells [16]. Neutrophils and macrophages are the first to arrive on the scene and respond by phagocytosing tissue debris and secreting additional factors that induce epithelial regrowth and angiogenesis [17, 18]. Multiple phenotypes of CD4+ T cells (i.e., TH1, TH2, Treg) are then recruited to the wound site where they help mediate later phases of wound repair through regulation of inflammation, stimulation of fibroblast activity, and promotion of tissue remodeling [19, 20]. Several studies have demonstrated that a delicate balance between TH1-induced collagen degeneration and TH2-induced collagen synthesis is necessary for proper ECM remodeling, and that an over-representation of the TH2 phenotype is associated with abnormal fibrogenesis [20, 21].
The proliferative phase involves the formation of new tissue to fill the wound gap [22]. The primary mediators at this stage are the myofibroblasts, which play vital roles in secreting new ECM, inducing wound contraction, and stimulating angiogenesis and granulation tissue formation[23, 24]. The pool of cells contributing to the generation of myofibroblasts is vast and includes resident mesenchymal cells, as well as local epithelial and endothelial cells in a process termed the epithelial/endothelial-mesenchymal transition (EMT/EndoMT) [25]. Synthesis of ECM components including collagen, proteoglycans, and fibronectin provides structural support to the healing tissue [26]. Angiogenic factors induce endothelial cell proliferation and migration towards the wound site, where they will ultimately organize into capillaries that supply oxygen, nutrients, and additional immune cells to aid with the healing process [27]. This stage is also characterized by the proliferation of epithelial cells at the wound edges, allowing them to form a new layer of skin overlying the wound in a process known as reepithelialization [28]. Finally, the newly regenerated tissue undergoes extensive remodeling meant to restore original strength and function. Specifically, collagen fibers are reorganized and mature in a process that increases the tensile strength of the healing tissue[29]. At the same time, excess collagen that was laid down in the regenerative phase begins to be enzymatically digested by MMPs secreted by epithelial cells and activated myofibroblasts [30]. The rate of collagen turnover is regulated by the opposing activity of MMPs and tissue inhibitors of metalloproteinases (TIMPs), among other inhibitors [31].
Throughout this process, various growth factors and signaling molecules are involved in regulating cell migration, proliferation, and ECM synthesis. Transforming growth factor-beta (TGF-β), platelet-derived growth factor (PDGF), and fibroblast growth factors (FGFs) produced from a variety of local and infiltrating cells are among the key drivers of tissue regeneration and remodeling [32]. Importantly, the balance between pro- versus anti-inflammatory mediators and collagen synthesis versus degradation represents an essential component of proper tissue repair [33]. Imbalance of these processes is an important pathological factor seen with deficiencies in tissue repair, chronic inflammatory diseases, and fibrosis [34].
1.2. How Dysregulation of Wound Healing Leads to Fibrosis
With an understanding of the wound healing process, one can appreciate how there are many avenues by which dysregulation can lead to abnormal physiologic effects. Relevant to this review, dysregulation of wound repair and chronic inflammation are the pathogenic mechanisms leading to fibrosis via a complex series of cellular and molecular events [35]. Fibrosis, which describes the excessive deposition of ECM components in an organ or tissue, can result from dysfunction at various stages of the repair process [36].
In the early stages of repair, cells that have been damaged or that are near the site of damage act to induce a proinflammatory environment that culminates with the recruitment of leukocytes, including TH1 and TH2 CD4+ T cells [37, 38]. The importance of such inflammation is that it triggers clearing of debris, pathogens, and damaged cells, and allows for new cells and ECM to fill in the evacuated space [39]. Critically, wound repair involves coordinated transitions between TH1- and TH2-phenotypes to allow for proper regeneration [40]. In instances where this transition is polarized towards a TH2-phenotype, excessive secretion of profibrotic factors such as IL-4 and IL-13 stimulates collagen deposition and tissue scarring [41]. Alternatively, Hoffmann and colleagues found that when IFN-gamma-producing TH1 CD4+ lymphocytes dominate, the development of tissue fibrosis is greatly attenuated [42].
Improper activity of fibroblasts themselves is also a key cellular mechanism underlying the development and progression of fibrotic disorders [43]. Fibroblasts are the main cells responsible for producing, remodeling, and maintaining the extracellular matrix [44]. Fibroblasts also play a role in wound contraction by maturing into collagen-secreting, α-SMA+ myofibroblasts in response to profibrotic signaling via TGF-β, PDGF, and connective tissue growth factor (CTGF) [45]. While this maturation process is important for functional repair, dysregulation can result in overactivation of myofibroblast phenotype and excessive ECM deposition [46].
Matrix metalloproteinases (MMPs) have also been implicated in the development of fibrosis [47]. Normal ECM remodeling depends on a balance between these MMPs and the activity of endogenous protease inhibitors, TIMPs [48]. Pathologically increased activity of TIMPs can disturb this careful balance to favor excessive buildup of extracellular collagen and the disruption of normal tissue architecture [49]. Additionally, conditions that otherwise decrease production or enhance degradation of certain MMPs can have similar effects, whereby ECM components accumulate to levels that result in organ dysfunction. Paradoxically, overexpression of some MMPs (specifically, MMP-2, -3, -8, among others) can also have profibrotic effects, suggesting that the interaction between MMPs and TIMPs is more complex than would be expected [50].
Yet another cellular process implicated in the development of fibrosis is the EMT [51]. As has been discussed previously, fibroblasts and myofibroblasts are the main cell type responsible for the deposition of ECM during physiological and pathological tissue regeneration and remodeling. The fibroblasts that participate in this process are those present near the site of injury and those recruited from the bone marrow [52]. Importantly, an additional source includes local epithelial cells that lose characteristic apical-basal polarity and tight junction expression as they transition to a mesenchymal phenotype in a process mediated by TGF-β signaling pathways [53]. EMT has been studied extensively as it relates to fibrogenesis and has been demonstrated to be a driving force in fibrotic disorders of various organs including the lungs, liver, and kidneys, among others [54].
While the specific mechanisms contributing to fibrosis vary depending on the type of injury, the tissue involved, and the underlying pathological conditions, an understanding of these various mechanisms is vital as it relates to the development of novel treatments. Through the development of pharmaceutical and biological agents that target each of these processes, it may be possible to slow, prevent, or even reverse the damages resulting from excessive ECM deposition.
2. Organ-Specific Fibrosis
2.1. Pulmonary Fibrosis
Pulmonary fibrosis is a pathological finding seen in a wide variety of clinical settings and represents a major cause of morbidity and mortality [55]. It describes the process of scarring and thickening of lung parenchyma, ultimately resulting in the disruption of normal tissue architecture, impaired gas exchange, and respiratory failure [56]. It is a common finding in many systemic connective tissue disorders such as systemic sclerosis (scleroderma) or rheumatoid arthritis and can sometimes represent the initial manifestation of these diseases [57]. Furthermore, pulmonary fibrosis may also appear without any apparent etiology, as is the case in idiopathic pulmonary fibrosis (IPF), as well as in response to certain infections, air pollutants, chemotherapies, or radiation therapy [58]. Altogether, conditions in which scarring of the lungs is a prominent feature are grouped together under the label of interstitial lung diseases (ILD) [59]. While the underlying cause may not always be known, the distinction between these various ILD subtypes has clinical implications in that treatments may differ, suggesting diversity in pathophysiological mechanisms [60]. However, regardless of how the disease progresses, fibrotic scarring universally occurs following some initial injury to pulmonary tissue [61]. In situations where pulmonary fibrosis is a complication of systemic autoimmune disorders (i.e., scleroderma, rheumatoid arthritis, Sjogrens, polymyositis/dermatomyositis, etc.), triggering injury results from a chronic inflammatory reaction against self-antigens which leads to excessive ECM deposition and tissue scarring [62]. Accordingly, treatment with corticosteroids and immunomodulators (cyclophosphamide, azathioprine, rituximab, among others) has been shown to be effective in limiting progression and improving respiratory function [60]. Alternatively, while it was once thought that IPF represented another example of immune-driven fibrosis, Selman et. al., reported how there was little supporting evidence for this paradigm, and instead suggested that the disease was primarily related to abnormal fibroblast activation during wound healing [63]. Subsequent reports have provided supporting evidence to this claim, including: a lack of inflammatory cells in fibrotic lesions of IPF patients, disjunction of inflammatory laboratory markers and disease severity, and a lack of efficacy of anti-inflammatory treatment methods [64, 65]. In total, the heterogeneous nature of ILD emphasizes the importance of a holistic approach, whereby conditions involving pulmonary fibrosis are distinguished from each other to provide the best clinical management.
2.2. Hepatic Fibrosis
Similarly, hepatic fibrosis arises due to excessive ECM accumulation in the liver. The driving factor in this case is chronic liver injury as is seen with viral hepatitis infections, autoimmune hepatitis, alcoholic liver disease, non-alcoholic fatty liver disease (NAFLD), or drug-induced liver damage [66]. Acute injury triggers the release of damage-associated molecular patterns (DAMPs) which drive induction of an inflammatory response meant to clean up cellular debris and initiate wound repair [67]. One of the primary effects of this immune response is the activation of normally quiescent hepatic stellate cells (HSCs) and the transformation of the HSCs into collagen-producing myofibroblasts in a process analogous to EMT [68]. These HSCs constitute the main effectors of fibrogenesis, contributing approximately 90% of the ECM-producing cells involved in tissue regeneration [69]. In a normal response, the inflammatory environment would withdraw with resolution of the noxious stimuli and the liver would see a return to homeostasis [70]. However, in the setting of chronic liver injury, a continuous source of DAMPs triggers an extended immune response that increases HSC activity and collagen secretion. Ultimately, this results in the replacement of normal hepatocytes with fibrotic scar tissue that, over time, will act to interrupt normal liver function [71]. Importantly (and in contrast to pulmonary fibrosis), hepatic fibrosis is a reversible process if the underlying cause is treated, and the injury allowed to repair, before it progresses too far [72]. However, if connective tissue builds up to sufficient levels, it can lead to the development of cirrhosis, portal hypertension, and liver failure that are irreversible and often necessitate transplantation [73]. Therefore, aside from earlier detection of hepatic fibrosis, one of the primary ways in which morbidity from this disease can be decreased is through the development of therapeutics that can reverse ECM deposition and restore proper organ function once the progression to cirrhosis has occurred.
2.3. Cardiac Fibrosis
Cardiac fibrosis, also known as myocardial fibrosis, describes the abnormal deposition of ECM in the place of myocardial tissue [74]. It represents a common feature of many cardiovascular pathologies and can have significant implications in the development of structural disease and loss of normal heart function [75]. Myocardial fibrosis is unique in that, unlike fibrotic disorders of other organs, this process may paradoxically confer some cardioprotective functions that help to maintain relatively normal activity. Given that the heart lacks significant regenerative capacity, the generation of a fibrotic scar in conditions that result in large amounts of cardiomyocyte death represents a reparative process [76]. Mechanistically, the process is the same as other disorders previously discussed. Specifically, myocardial damage induces an immune response which culminates with the activation of myofibroblasts and excessive secretion of collagen until a scar is formed. Even though this scar may lack the conductive and contractive function of healthy cardiomyocytes, it nevertheless plays a crucial role in the maintenance of structural integrity within the heart [77]. Reinforcement provided by this scar tissue can serve to prevent catastrophic structural failures, such as cardiac rupture following a transmural myocardial infarction (MI) [78, 79]. Alternatively, other scenarios such as prolonged exposure to excessive pressures or volumes, metabolic dysfunction, or aging involve deposition of fibrotic tissue without a corresponding loss of cardiomyocytes [80]. In these cases, resulting thickening of the myocardium can increase wall stiffness, reduce ventricular compliance, impair diastolic function, and ultimately lead to the development of heart failure [81]. Such structural remodeling can also disrupt electrical conduction pathways within the myocardium, leading to arrhythmias such as atrial fibrillation, heart block, or ventricular tachycardia [82]. In summary, cardiac fibrosis is unique in that reversal of ECM deposition will not always be the most therapeutic option. Thus, treatment regiments must be designed depending on the underlying context of the disease, and the end goal may not always be the disruption of fibrotic scarring.
Many other organ-specific fibrotic diseases exist, including renal fibrosis and skin fibrosis which each have a wide range of etiologies. Despite the diverse array of pathologies in which fibrosis is involved, each can be traced to a collection of common mechanisms. While current antifibrotic therapeutics have mixed success at improving patient outcomes, further exploration of the mechanisms underlying fibrosis generation is key in the search for new molecular targets and treatment strategies.
3. Existing Fibrosis Treatments
Several organ systems have demonstrated the dynamic nature of fibrosis, highlighting its potential implications for therapeutic interventions aimed at utilizing this inherent adaptability. Single cell genomics have deepened our understanding of human fibrotic disease and generated comprehensive cellular and molecular profiles of healthy and fibrotic tissue, demonstrating heterogeneity and functional diversity within lung, liver, and GI fibroblasts [83,84]. Thus, the application of single-cell genomics techniques, such as single-cell RNA sequencing (scRNAseq), has revealed significant diversity within the fibroblast population during organ fibrosis [85]. This discovery holds pivotal implications for the design of targeted therapies aimed at treating organ fibrosis. However, despite considerable advancements in our comprehension of the mechanisms of fibrosis, there continues to be a significant translational divide between the identification of potential antifibrotic targets and their successful translation into effective therapeutic strategies in patients.
3.1. Antifibrotic Agents
Currently, efficient therapeutic approaches available for treatment of fibrotic diseases are limited. One aspect of fibrosis therapies involves systemic delivery of antifibrotic agents to decrease levels of growth factors or cytokines [86]. Moreover, cardioselective drugs, such as β-blockers, renin-angiotensin-aldosterone system (RAAS) inhibitors, ivabradine, loop diuretics, and sildenafil, have been investigated for their potential antifibrotic properties in cardiac tissue [87]. However, the outcomes of these investigations have yielded divergent results, indicating a lack of consensus regarding the efficacy of these drugs in cardiac fibrosis. Other investigations have showed promising findings regarding the potential of statins in reducing fibrosis in multiple organs, including kidney, liver, heart, and skin, as evidenced by animal models [88]. Contrasting studies emphasized the controversial role of statins and found insufficient evidence for its use in antifibrotic treatments [89].
3.2. Antifibrotic Drugs: Nintedanib and Pirfenidone
Between 2011 and 2014, two drugs, Nintedanib and pirfenidone, were approved for the treatment of IPF. The prognosis for untreated patients with IPF is poor, with an average life expectancy ranging from 3 to 5 years in the absence of antifibrotic therapy [90]. Both drugs have been shown to reduce mortality and slow the progression of IPF, prolong patient survival, and reduce respiratory-related hospitalizations [91]. However, the mechanisms of these two drugs have not been well characterized. Nintedanib exerts its inhibitory effects on lung fibroblasts by targeting PDGF, VEGF, and FGF receptor tyrosine kinases, inhibiting pathways of proliferation, migration, and maturation of lung fibroblasts [92]. On the other hand, the precise molecular mechanisms of pirfenidone still remain unclear [93]. However, both nintedanib and pirfenidone share the ability to inhibit the self-assembly of collagen fibrils by downregulating collagen V expression and directly interfering with the formation of extracellular fibril through interaction with the triple-helical collagen [93].
In 2020, the U.S. Food and Drug Administration (FDA) granted approval to Nintedanib as a therapeutic option for the management of progressive phenotype chronic fibrosing interstitial lung diseases, marking the first FDA-approved treatment for this subset of deteriorating fibrosing lung diseases. Ongoing clinical trials are investigating the antifibrotic effects of both Nintedanib and Pirfenidone [Table 1]. Additionally, Nintedanib and Pirfenidone has been used to treat consequential pulmonary fibrosis following SARS-CoV-2 infection. Combination therapy of Nintedanib and Pirfenidone was also found to be more effective than monotherapy of the drugs [94]. While reviews have underscored the therapeutic benefits of these medications for both monotherapy and combination therapy, further clinical investigations are needed to assess the role of these drugs in the treatment of post-COVID lung fibrosis. Currently, a phase IV trial is investigating the efficacy and safety of both Nintedanib and Pirfenidone for use in treating fibrotic lung disease post-COVID [95][Table 1]. Moreover, it is important to consider the non-immunosuppressive nature of these drugs, giving them an advantage over the administration of corticosteroids during the acute phase of the disease [96]. Thus, the potential option of transitioning to antifibrotic therapy warrants further investigation, particularly for patients with increased susceptibility to infections during COVID treatment.
4. Novel Anti-fibrotic Therapies
Recent advancements in anti-fibrotic treatments have been directed towards targeting the activation of myofibroblasts, focusing on factors that play in the role in the progression or inhibition of fibrosis.
4.1. Antifibrotic Role of BMP7
The role of bone morphogenetic protein-7 (BMP7) has been extensively studied and widely accepted as an important aspect in the regulation of genes influencing organ homeostasis and antifibrotic mechanisms [100]. Overall, it is well established that BMP7 serves as an antagonist to TGF-β-mediated profibrogenic signaling, and there is consistence evidence indicating downregulation of BMP7 activity in fibrotic tissues [101]. In early studies, the antifibrotic effect of BMP7 and its involvement in the control of EMT transition and TGF-β profibrogenic signaling in the kidney, heart, liver, and lung were demonstrated through the study of BMP7 deficient mice [102]. Downregulation of BMP7 expression was also observed in renal fibrosis, implying the potential involvement of BMP7 in the progression of renal fibrosis and potentially extending to other forms of organ fibrosis as well [103]. However, while numerous studies highlight the antifibrotic effects of BMP7 on multiple organs, there is conflicting evidence regarding the protective effects of BMP7 and the specific organs that it influences. A study found that BMP7 was effective exclusively in renal fibrosis, demonstrating no antifibrotic effects in lung or skin fibrosis, either in vitro or in vivo [104]. BMP7 was also found to have no effect in hepatocyte or breast epithelial cells [105]. Additionally, it was also discovered that elevated levels of BMP7 promoted fibrogenesis in patients with chronic liver disease [106]. While these studies raise doubts regarding the protective effects of BMP7, additional research focused on organ-specific mechanisms of BMP7 could enhance our understanding its effects on fibrosis.
The integration of genetic engineering techniques with BMP7 therapy has also given promising results. The transduction of BMP7 with bone marrow mesenchymal stem cells (MSCs) provides a novel strategy to enhance its antifibrotic properties and augment therapeutic efficacy, leading to notable mitigation of pulmonary fibrosis in rat models [107]. Furthermore, the in vivo delivery of BMP7 gene via nanoparticles has demonstrated compelling outcomes and resulted in reduced fibrosis in corneal tissues in animal models [108] [109]. These findings emphasize the potential implications of targeted gene delivery approaches for specifically addressing organ fibrosis and warrants further investigation to explore potential clinical applications.
4.2. miRNAs
MicroRNAs (miRNAs) are a class of short non-coding RNAs that exert regulatory control over messenger RNA (mRNA) expression, serving as important regulators of cell differentiation, development, and homeostasis [110]. The observation of altered miRNA expression levels in disease states compared to normal conditions underscores the potential use of miRNAs as diagnostic of therapeutic targets with studies revealing distinct miRNA profiles that are frequently tissue or cellular type specific [111].
Many miRNAs have been linked to fibrosis for both systemic and organ-specific manifestations, identifying miRNA biomarkers as potential targets for antifibrotic therapy [118][Table 2]. Currently, a few antisense oligonucleotides (ASO) are under investigation in the clinical trial phases I and II [Table 2]. In cardiac and renal fibrosis, ASO-mediated inhibition of miR-21 expression has shown attenuation of the disease state in vivo and in vitro [119]. Additionally, administration of RG-012, a miR-21 inhibitor, leads to slowed progression of renal fibrosis [112]. In other studies, the upregulation of certain cardiac miRNA biomarkers was found to have a protective role with reduced collagen production and fibrosis. Tanshinone IIA, a cardioprotective drug, was found to have antifibrotic effects in postinfarct cardiac fibrosis through the upregulation of miR-29b expression [113]. More recently, the antifibrotic effects of miR-101a overexpression were leveraged in conjunction with the regenerative properties of MSCs, utilizing miR-101a-loaded MSC extracellular nanovesicles to effectively reduce cardiac fibrosis by inhibiting TGF-β and collagen production [114]. Recent research efforts have also placed significant emphasis on investigating the role of miRNAs in other organ-specific fibrosis, such as liver, kidney, and lungs. Novel therapeutic approaches, such as coadministration of miRNAs, have focused on synergistic roles of profibrotic factors in liver fibrogenesis [120]. Remarkably, MRG-229, a lung-targeted miR-29 mimic, exhibited notable reduction in pulmonary fibrosis in animal models and human precision cut lung slices, demonstrating compelling in vivo therapeutic potential in humans [116]. Overall, the evolving research highlights the strong potential of miRNA targeting as a therapeutic approach in addressing fibrotic diseases, underscoring the importance of further investigations in this domain.
4.3. Relaxin
Relaxin, a peptide hormone produced by the corpus luteum of the ovary during pregnancy, has gained significant recognition for its established antifibrotic properties in several organs, and consequently, has become a promising therapeutic option for the treatment of fibrosis [121]. The nonreproductive significance of relaxin was highlighted using relaxin-null mouse model, wherein mice exhibited spontaneous fibrosis in various non-reproductive organs, including the lung, heart, kidney, and skin [122,123]. Relaxin has been shown to reduce pathological collagen production by impeding its synthesis and secretion from myofibroblasts, which are crucial fibrosis-generating cells derived from various organs [124]. Additionally, relaxin also modulates the activity of matrix metalloproteinases (MMPs), resulting in the breakdown of extracellular matrix (ECM) and collagen [125]. These mechanisms enable the attenuation of fibrotic diseases.
The investigation of relaxin as a prospective therapeutic approach for fibrotic diseases dates back to the mid-20th century when initial studies centered on utilizing porcine ovary-derived relaxin for treatment of scleroderma (EV 1959). In more recent years, considerable attention has been directed towards serelaxin, a recombinant human relaxin, which has become a focal point in preclinical and clinical trials [127]. In preclinical studies, serelaxin has been shown to reduce cardiac fibrosis in vivo via the inhibition of EMT transition [128]. However, despite initial expectations, a subsequent phase III trial investigating the application of serelaxin in cardiac tissues yielded unsuccessful outcomes [129]. Similarly, another phase III trial exploring serelaxin in scleroderma had demonstrated limited efficacy [130]. Researchers have also directed their efforts towards the development of novel relaxin analogous and agonists, including ML290 and B7-33, which have exhibited promising outcomes in reducing fibrosis in heart and kidney of rodent models [131]. While preclinical in vivo models provide compelling evidence for potential therapeutic application of serelaxin and relaxin mimetics in fibrotic diseases, the underlying mechanisms of these findings remain unclear. Furthermore, the successful translation of these findings into clinical trials involving serelaxin or mimetics is essential to ascertain their safety profile and efficacy in the treatment of fibrotic disease in humans.
4.4. Reprogramming as a Treatment for Fibrosis
Advancements in the reduction of fibrosis and scar formation through cell reprogramming has been a significant highlight in recent research with broad implications. It has been a main topic of discussion in cardiac fibrosis due to the low regenerative capacity of cardiomyocytes [132]. Previous studies have focused on progenitor cell therapy in hopes of restoration of cardiac capacity; however, clinical studies have proven largely unsuccessful [133] [134]. In in vitro studies, cardiac reprogramming factors have been identified to induce fully reprogrammed functional cardiomyocytes from cardiac fibroblasts (CFs) to reverse fibrosis [135]. The combination of three developmental transcription factors, Gata4, Mef2c, and Tbx5 (GMT), to be an effective strategy in reprogramming cardiac fibroblasts to adult-cardiomyocyte-like (CM-like) cells in vitro [136]. Fibroblasts were then shown to convert into CM-like cells in vivo following local delivery of GMT by retroviral-mediated gene transfer [137]. However, the GMT combination, which demonstrated efficacy in mice, showed little effectiveness in reprogramming human fibroblasts [138].
In 2022, the first study demonstrated reversal of fibrosis in vivo in chronic myocardial infarction through cardiac reprogramming by converting profibrotic CFs to an antifibrotic state [139]. Another study demonstrated successful reprogramming of human fibroblasts into cardiac-like myocytes through the forced expression of three or four cardiac transcription factors or muscle-specific miRNA [140]. Notably, a fraction of these reprogrammed cells showed spontaneous contractility, indicating their potential for functional cardiac regeneration [140]. More recently, studies have shown cardiac reprogramming of cardiac pericytes into functional cardiac cells to improve heart function and reduce fibrosis [141]. While most reprogramming studies have focused on cardiac fibrosis, advancements have been made in approaches for other organ-specific fibrosis. In vivo investigations have specifically explored reprogramming of profibrotic macrophages as a potential therapeutic strategy for pulmonary fibrosis [142]. Despite promising results observed in in vivo studies, the translation of these findings to human cells has been challenging, as human cells have shown greater resistance to reprogramming compared to mouse cells.
5. Future Direction and Conclusion
Despite significant progress in our understanding of the pathophysiology of fibrotic diseases, there continues to be a pressing demand for effective therapeutic interventions targeting organ-specific tissue fibrosis. The approval of Nintedanib and Pirfenidone was indeed a breakthrough in treating fibrosis. However, there are currently no generic alternatives, creating a significant financial barrier for use of these therapies in Europe and the United States [143] [144]. Additionally, research into reprogramming, particularly for cardiac fibrosis, is progressing towards clinical application. Recent research has also focused on advancing the development of safe and efficient viral vectors for human applications. Notably, an in vivo study utilizing Sendai virus vectors has demonstrated promising results. Following direct administration of Sendai virus vectors into infarcted hearts, cardiac reprogramming was initiated within one week, and significant improvement in cardiac function was observed one month after gene delivery [145]. Remarkably, the viral vectors specifically targeted cardiac fibroblasts, the desired cell population for cardiac reprograming, rather than cardiomyocytes [145].
The therapeutic options currently available for fibrosis are limited, lacking effective drug treatments for widespread use. Therefore, exploring preventative strategies for fibrosis comes increasingly significant. Research has shown that MMP-1 and MMP-10 limit fibrotic responses following tissue injury [146]. Additionally, it has been demonstrated that mechanical load stimulates a substantial increase in the rate of collagen proteolysis mediated by MMP-1 [147]. Therefore, investigating the potential of exercise-induced mechanical load as a preventative measure for fibrosis warrants further research. Continued multidisciplinary research efforts will continue to be essential in addressing the needs of patients and improving outcomes in fibrotic diseases.
Author Contributions
JCK, JR, and YL contributed to conception and design of this review. JCK and JR wrote sections of the manuscript. YL supervised and structured drafting of the manuscript. All authors contributed to manuscript revision, in addition to reading and approving the submitted version.
Funding
This review is partially supported by a pioneer grant from the Western Michigan University Homer Stryker M.D. School of Medicine.
Acknowledgments
N/A.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Thannickal VJ, Zhou Y, Gaggar A, Duncan SR (2014) Fibrosis: ultimate and proximate causes. Journal of Clinical Investigation 124:4673–4677. [CrossRef]
- Wynn T (2008) Cellular and molecular mechanisms of fibrosis. J Pathol 214:199–210. [CrossRef]
- Henderson NC, Rieder F, Wynn TA (2020) Fibrosis: from mechanisms to medicines. Nature 587:555–566. [CrossRef]
- Mukherjee A, Epperly Mw, Fisher R, Hou W, Shields D, Wang H, Greenberger Js, Ortiz La (2021) Silica Induced Lung Fibrosis Is Associated With Senescence, Fgr, and Recruitment of Bone Marrow Monocyte/Macrophages. In Vivo (Brooklyn) 35:3053–3066. [CrossRef]
- Weiskirchen R, Weiskirchen S, Tacke F (2019) Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol Aspects Med 65:2–15. [CrossRef]
- Wynn TA, Ramalingam TR (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18:1028–40. [CrossRef]
- Jun J-I, Lau LF (2018) Resolution of organ fibrosis. J Clin Invest 128:97–107. [CrossRef]
- Eckes B, Eming SA (2017) Tissue fibrosis: a pathomechanistically unresolved challenge and scary clinical problem. Exp Dermatol 26:135–136. [CrossRef]
- Shinde A, V. , Humeres C, Frangogiannis NG (2017) The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1863:298–309. [CrossRef]
- Wynn TA (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. Journal of Clinical Investigation 117:524–529. [CrossRef]
- Broughton G, Janis JE, Attinger CE (2006) The Basic Science of Wound Healing. Plast Reconstr Surg 117:12S-34S. [CrossRef]
- Heng MCY (2011) Wound healing in adult skin: aiming for perfect regeneration. Int J Dermatol 50:1058–1066. [CrossRef]
- Laurens N, Koolwijk P, De Maat Mpm (2006) Fibrin structure and wound healing. Journal of Thrombosis and Haemostasis 4:932–939. [CrossRef]
- Yun S-H, Sim E-H, Goh R-Y, Park J-I, Han J-Y (2016) Platelet Activation: The Mechanisms and Potential Biomarkers. Biomed Res Int 2016:1–5. [CrossRef]
- Wang P-H, Huang B-S, Horng H-C, Yeh C-C, Chen Y-J (2018) Wound healing. Journal of the Chinese Medical Association 81:94–101. [CrossRef]
- Parks WC, Wilson CL, López-Boado YS (2004) Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 4:617–29. [CrossRef]
- Kim SY, Nair MG (2019) Macrophages in wound healing: activation and plasticity. Immunol Cell Biol 97:258–267. [CrossRef]
- Gantwerker EA, Hom DB (2011) Skin: Histology and Physiology of Wound Healing. Facial Plast Surg Clin North Am 19:441–453. [CrossRef]
- Li J, Tan J, Martino MM, Lui KO (2018) Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front Immunol 9:. [CrossRef]
- Short WD, Wang X, Keswani SG (2022) The Role of T Lymphocytes in Cutaneous Scarring. Adv Wound Care (New Rochelle) 11:121–131. [CrossRef]
- Wynn TA (2004) Fibrotic disease and the TH1/TH2 paradigm. Nat Rev Immunol 4:583–594. [CrossRef]
- Reinke JM, Sorg H (2012) Wound Repair and Regeneration. European Surgical Research 49:35–43. [CrossRef]
- Hinz B (2007) Formation and Function of the Myofibroblast during Tissue Repair. Journal of Investigative Dermatology 127:526–537. [CrossRef]
- Barker TH (2011) The role of ECM proteins and protein fragments in guiding cell behavior in regenerative medicine. Biomaterials 32:4211–4214. [CrossRef]
- Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. Journal of Clinical Investigation 119:1420–1428. [CrossRef]
- Diller RB, Tabor AJ (2022) The Role of the Extracellular Matrix (ECM) in Wound Healing: A Review. Biomimetics 7:87. [CrossRef]
- Tonnesen MG, Feng X, Clark RAF (2000) Angiogenesis in Wound Healing. Journal of Investigative Dermatology Symposium Proceedings 5:40–46. [CrossRef]
- Rousselle P, Braye F, Dayan G (2019) Re-epithelialization of adult skin wounds: Cellular mechanisms and therapeutic strategies. Adv Drug Deliv Rev 146:344–365. [CrossRef]
- Gonzalez AC de O, Costa TF, Andrade Z de A, Medrado ARAP (2016) Wound healing - A literature review. An Bras Dermatol 91:614–620. [CrossRef]
- Caley MP, Martins VLC, O’Toole EA (2015) Metalloproteinases and Wound Healing. Adv Wound Care (New Rochelle) 4:225–234. [CrossRef]
- Kandhwal M, Behl T, Singh S, Sharma N, Arora S, Bhatia S, Al-Harrasi A, Sachdeva M, Bungau S (2022) Role of matrix metalloproteinase in wound healing. Am J Transl Res 14:4391–4405.
- DiPietro LA, Polverini PJ (1993) Role of the macrophage in the positive and negative regulation of wound neovascularization.
- Landén NX, Li D, Ståhle M (2016) Transition from inflammation to proliferation: a critical step during wound healing. Cellular and Molecular Life Sciences 73:3861–3885. [CrossRef]
- Xue M, Jackson CJ (2015) Extracellular Matrix Reorganization During Wound Healing and Its Impact on Abnormal Scarring. Adv Wound Care (New Rochelle) 4:119–136. [CrossRef]
- El Ayadi A, Jay JW, Prasai A (2020) Current Approaches Targeting the Wound Healing Phases to Attenuate Fibrosis and Scarring. Int J Mol Sci 21:. [CrossRef]
- Jones RE, Foster DS, Hu MS, Longaker MT (2019) Wound healing and fibrosis: current stem cell therapies. Transfusion (Paris) 59:884–892. [CrossRef]
- Koh TJ, DiPietro LA (2011) Inflammation and wound healing: the role of the macrophage. Expert Rev Mol Med 13:e23. [CrossRef]
- Yussof SJMohd, Omar E, Pai DR, Sood S (2012) Cellular events and biomarkers of wound healing. Indian Journal of Plastic Surgery 45:220–228. [CrossRef]
- Diegelmann RF (2004) Wound healing: an overview of acute, fibrotic and delayed healing. Frontiers in Bioscience 9:283. [CrossRef]
- Gause WC, Wynn TA, Allen JE (2013) Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol 13:607–614. [CrossRef]
- Gieseck RL, Wilson MS, Wynn TA (2018) Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol 18:62–76. [CrossRef]
- Hoffmann KF, Cheever AW, Wynn TA (2000) IL-10 and the Dangers of Immune Polarization: Excessive Type 1 and Type 2 Cytokine Responses Induce Distinct Forms of Lethal Immunopathology in Murine Schistosomiasis. The Journal of Immunology 164:6406–6416. [CrossRef]
- Kendall RT, Feghali-Bostwick CA (2014) Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol 5:. [CrossRef]
- Gomes RN, Manuel F, Nascimento DS (2021) The bright side of fibroblasts: molecular signature and regenerative cues in major organs. NPJ Regen Med 6:43. [CrossRef]
- Desmouliere A, Darby IA, Laverdet B, Bonté F (2014) Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol 301. [CrossRef]
- Klingberg F, Hinz B, White ES (2013) The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol 229:298–309. [CrossRef]
- Giannandrea M, Parks WC (2014) Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech 7:193–203. [CrossRef]
- Rohani MG, Parks WC (2015) Matrix remodeling by MMPs during wound repair. Matrix Biology 44–46:113–121. [CrossRef]
- Arpino V, Brock M, Gill SE (2015) The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biology 44–46:247–254. [CrossRef]
- Chuliá-Peris L, Carreres-Rey C, Gabasa M, Alcaraz J, Carretero J, Pereda J (2022) Matrix Metalloproteinases and Their Inhibitors in Pulmonary Fibrosis: EMMPRIN/CD147 Comes into Play. Int J Mol Sci 23:6894. [CrossRef]
- Liu L, Sun Q, Davis F, Mao J, Zhao H, Ma D (2022) Epithelial–mesenchymal transition in organ fibrosis development: current understanding and treatment strategies. Burns Trauma 10:. [CrossRef]
- Grande MT, López-Novoa JM (2009) Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat Rev Nephrol 5:319–328. [CrossRef]
- Rout-Pitt N, Farrow N, Parsons D, Donnelley M (2018) Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res 19:136. [CrossRef]
- Di Gregorio J, Robuffo I, Spalletta S, Giambuzzi G, De Iuliis V, Toniato E, Martinotti S, Conti P, Flati V (2020) The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front Cell Dev Biol 8:. [CrossRef]
- Savin IA, Zenkova MA, Sen’kova A V. (2022) Pulmonary Fibrosis as a Result of Acute Lung Inflammation: Molecular Mechanisms, Relevant In Vivo Models, Prognostic and Therapeutic Approaches. Int J Mol Sci 23:14959. [CrossRef]
- Noble PW, Barkauskas CE, Jiang D (2012) Pulmonary fibrosis: patterns and perpetrators. Journal of Clinical Investigation 122:2756–2762. [CrossRef]
- Vij R, Strek ME (2013) Diagnosis and Treatment of Connective Tissue Disease-Associated Interstitial Lung Disease. Chest 143:814–824. [CrossRef]
- Harari S, Caminati A (2010) IPF: new insight on pathogenesis and treatment. Allergy 65:537–553. [CrossRef]
- Antoniou KM, Margaritopoulos GA, Tomassetti S, Bonella F, Costabel U, Poletti V (2014) Interstitial lung disease. European Respiratory Review 23:40–54. [CrossRef]
- Kreuter M, Müller-Ladner U, Costabel U, Jonigk D, Heußel CP (2021) The Diagnosis and Treatment of Pulmonary Fibrosis. Dtsch Arztebl Int. [CrossRef]
- Shao T, Shi X, Yang S, Zhang W, Li X, Shu J, Alqalyoobi S, Zeki AA, Leung PS, Shuai Z (2021) Interstitial Lung Disease in Connective Tissue Disease: A Common Lesion With Heterogeneous Mechanisms and Treatment Considerations. Front Immunol 12:. [CrossRef]
- Wilson MS, Wynn TA (2009) Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol 2:103–121. [CrossRef]
- Selman M, King TE, Pardo A (2001) Idiopathic Pulmonary Fibrosis: Prevailing and Evolving Hypotheses about Its Pathogenesis and Implications for Therapy. Ann Intern Med 134:136. [CrossRef]
- Gifford AH, Matsuoka M, Ghoda LY, Homer RJ, Enelow RI (2012) Chronic inflammation and lung fibrosis: pleotropic syndromes but limited distinct phenotypes. Mucosal Immunol 5:480–484. [CrossRef]
- Bringardner BD, Baran CP, Eubank TD, Marsh ClayB (2008) The Role of Inflammation in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Antioxid Redox Signal 10:287–302. [CrossRef]
- Bataller R, Brenner DA (2005) Liver fibrosis. Journal of Clinical Investigation 115:209–218. [CrossRef]
- Han H, Desert R, Das S, Song Z, Athavale D, Ge X, Nieto N (2020) Danger signals in liver injury and restoration of homeostasis. J Hepatol 73:933–951. [CrossRef]
- Zhang C-Y, Yuan W-G, He P, Lei J-H, Wang C-X (2016) Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J Gastroenterol 22:10512. [CrossRef]
- Hoffmann C, Djerir NEH, Danckaert A, Fernandes J, Roux P, Charrueau C, Lachagès A-M, Charlotte F, Brocheriou I, Clément K, Aron-Wisnewsky J, Foufelle F, Ratziu V, Hainque B, Bonnefont-Rousselot D, Bigey P, Escriou V (2020) Hepatic stellate cell hypertrophy is associated with metabolic liver fibrosis. Sci Rep 10:3850. [CrossRef]
- Seki E, Schwabe RF (2015) Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 61:1066–1079. [CrossRef]
- Tanwar S, Rhodes F, Srivastava A, Trembling PM, Rosenberg WM (2020) Inflammation and fibrosis in chronic liver diseases including non-alcoholic fatty liver disease and hepatitis C. World J Gastroenterol 26:109–133. [CrossRef]
- Ismail M, Pinzani M (2009) Reversal of liver fibrosis. Saudi Journal of Gastroenterology 15:72. [CrossRef]
- Tan Z, Sun H, Xue T, Gan C, Liu H, Xie Y, Yao Y, Ye T (2021) Liver Fibrosis: Therapeutic Targets and Advances in Drug Therapy. Front Cell Dev Biol 9:. [CrossRef]
- Frangogiannis NG (2021) Cardiac fibrosis. Cardiovasc Res 117:1450–1488. [CrossRef]
- Jiang W, Xiong Y, Li X, Yang Y (2021) Cardiac Fibrosis: Cellular Effectors, Molecular Pathways, and Exosomal Roles. Front Cardiovasc Med 8:. [CrossRef]
- Talman V, Ruskoaho H (2016) Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res 365:563–581. [CrossRef]
- Gao X-M, White DA, Dart AM, Du X-J (2012) Post-infarct cardiac rupture: Recent insights on pathogenesis and therapeutic interventions. Pharmacol Ther 134:156–179. [CrossRef]
- van den Borne SWM, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J (2010) Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol 7:30–37. [CrossRef]
- Shinde A V., Frangogiannis NG (2014) Fibroblasts in myocardial infarction: A role in inflammation and repair. J Mol Cell Cardiol 70:74–82. [CrossRef]
- Berk BC, Fujiwara K, Lehoux S (2007) ECM remodeling in hypertensive heart disease. Journal of Clinical Investigation 117:568–575. [CrossRef]
- Schimmel K, Ichimura K, Reddy S, Haddad F, Spiekerkoetter E (2022) Cardiac Fibrosis in the Pressure Overloaded Left and Right Ventricle as a Therapeutic Target. Front Cardiovasc Med 9:. [CrossRef]
- SCHIAU C, LEUCUȚA D-C, DUDEA SM, MANOLE S (2021) Myocardial Fibrosis as a Predictor of Ventricular Arrhythmias in Patients With Non-ischemic Cardiomyopathy. In Vivo (Brooklyn) 35:1677–1685. [CrossRef]
- Xie T, Wang Y, Deng N, Huang G, Taghavifar F, Geng Y, Liu N, Kulur V, Yao C, Chen P, Liu Z, Stripp B, Tang J, Liang J, Noble PW, Jiang D (2018) Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep 22:3625–3640. [CrossRef]
- Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, Wallenborg K, Picelli S, Zagorska A, Pendem S V, Hudson TE, Wu MM, Budas GR, Breckenridge DG, Harrison EM, Mole DJ, Wigmore SJ, Ramachandran P, Ponting CP, Teichmann SA, Marioni JC, Henderson NC (2019) Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep 29:1832-1847.e8. [CrossRef]
- Peyser R, MacDonnell S, Gao Y, Cheng L, Kim Y, Kaplan T, Ruan Q, Wei Y, Ni M, Adler C, Zhang W, Devalaraja-Narashimha K, Grindley J, Halasz G, Morton L (2019) Defining the Activated Fibroblast Population in Lung Fibrosis Using Single-Cell Sequencing. Am J Respir Cell Mol Biol 61:74–85. [CrossRef]
- Ortiz La, Lasky J, Gozal E, Ruiz V, Lungarella G, Cavarra E, Brody Ar, Friedman M, Pardo A, Selman M (2001) Tumor Necrosis Factor Receptor Deficiency Alters Matrix Metalloproteinase 13/Tissue Inhibitor of Metalloproteinase 1 Expression in Murine Silicosis. Am J Respir Crit Care Med 163:244–252. [CrossRef]
- Fang L, Murphy AJ, Dart AM (2017) A Clinical Perspective of Anti-Fibrotic Therapies for Cardiovascular Disease. Front Pharmacol 8:186. [CrossRef]
- Dolivo DM, Reed CR, Gargiulo KA, Rodrigues AE, Galiano RD, Mustoe TA, Hong SJ (2023) Anti-fibrotic effects of statin drugs: A review of evidence and mechanisms. Biochem Pharmacol 214:115644. [CrossRef]
- Kim JW, Barrett K, Loke Y, Wilson AM (2021) The effect of statin therapy on disease-related outcomes in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Respir Med Res 80:100792. [CrossRef]
- Fernández Fabrellas E, Peris Sánchez R, Sabater Abad C, Juan Samper G (2018) Prognosis and Follow-Up of Idiopathic Pulmonary Fibrosis. Medical Sciences 6:51. [CrossRef]
- Finnerty JP, Ponnuswamy A, Dutta P, Abdelaziz A, Kamil H (2021) Efficacy of antifibrotic drugs, nintedanib and pirfenidone, in treatment of progressive pulmonary fibrosis in both idiopathic pulmonary fibrosis (IPF) and non-IPF: a systematic review and meta-analysis. BMC Pulm Med 21:411. [CrossRef]
- Hostettler KE, Zhong J, Papakonstantinou E, Karakiulakis G, Tamm M, Seidel P, Sun Q, Mandal J, Lardinois D, Lambers C, Roth M (2014) Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res 15:157. [CrossRef]
- Epstein Shochet G, Wollin L, Shitrit D (2018) Fibroblast-matrix interplay: Nintedanib and pirfenidone modulate the effect of IPF fibroblast-conditioned matrix on normal fibroblast phenotype. Respirology 23:756–763. [CrossRef]
- Huh J-Y, Lee JH, Song JW (2021) Efficacy and safety of combined use of pirfenidone and nintedanib in patients with idiopathic pulmonary fibrosis. Idiopathic interstitial pneumonias PA468. [CrossRef]
- Pirfenidone vs. Nintedanib for Fibrotic Lung Disease After Coronavirus Disease-19 Pneumonia (PINCER). https://clinicaltrials.gov/ct2/show/NCT04856111. Accessed 14 Jun 2023.
- El-Saber Batiha G, Al-Gareeb AI, Saad HM, Al-Kuraishy HM (2022) COVID-19 and corticosteroids: a narrative review. Inflammopharmacology 30:1189–1205. [CrossRef]
- Wells AU, Flaherty KR, Brown KK, Inoue Y, Devaraj A, Richeldi L, Moua T, Crestani B, Wuyts WA, Stowasser S, Quaresma M, Goeldner R-G, Schlenker-Herceg R, Kolb M, Abe S, Aburto M, Acosta O, Andrews C, Antin-Ozerkis D, Arce G, Arias M, Avdeev S, Barczyk A, Bascom R, Bazdyrev E, Beirne P, Belloli E, Bergna MA, Bergot E, Bhatt N, Blaas S, Bondue B, Bonella F, Britt E, Buch K, Burk J, Cai H, Cantin A, Castillo Villegas DM, Cazaux A, Cerri S, Chaaban S, Chaudhuri N, Cottin V, Crestani B, Criner G, Dahlqvist C, Danoff S, Dematte D’Amico J, Dilling D, Elias P, Ettinger N, Falk J, Fernández Pérez ER, Gamez-Dubuis A, Giessel G, Gifford A, Glassberg M, Glazer C, Golden J, Gómez Carrera L, Guiot J, Hallowell R, Hayashi H, Hetzel J, Hirani N, Homik L, Hope-Gill B, Hotchkin D, Ichikado K, Ilkovich M, Inoue Y, Izumi S, Jassem E, Jones L, Jouneau S, Kaner R, Kang J, Kawamura T, Kessler R, Kim Y, Kishi K, Kitamura H, Kolb M, Kondoh Y, Kono C, Koschel D, Kreuter M, Kulkarni T, Kus J, Lebargy F, León Jiménez A, Luo Q, Mageto Y, Maher TM, Makino S, Marchand-Adam S, Marquette C, Martinez R, Martínez M, Maturana Rozas R, Miyazaki Y, Moiseev S, Molina-Molina M, Morrison L, Morrow L, Moua T, Nambiar A, Nishioka Y, Nunes H, Okamoto M, Oldham J, Otaola M, Padilla M, Park JS, Patel N, Pesci A, Piotrowski W, Pitts L, Poonyagariyagorn H, Prasse A, Quadrelli S, Randerath W, Refini R, Reynaud-Gaubert M, Riviere F, Rodríguez Portal JA, Rosas I, Rossman M, Safdar Z, Saito T, Sakamoto N, Salinas Fénero M, Sauleda J, Schmidt S, Scholand MB, Schwartz M, Shapera S, Shlobin O, Sigal B, Silva Orellana A, Skowasch D, Song JW, Stieglitz S, Stone H, Strek M, Suda T, Sugiura H, Takahashi H, Takaya H, Takeuchi T, Thavarajah K, Tolle L, Tomassetti S, Tomii K, Valenzuela C, Vancheri C, Varone F, Veeraraghavan S, Villar A, Weigt S, Wemeau L, Wuyts W, Xu Z, Yakusevich V, Yamada Y, Yamauchi H, Ziora D (2020) Nintedanib in patients with progressive fibrosing interstitial lung diseases—subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med 8:453–460. [CrossRef]
- Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR (2014) Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. New England Journal of Medicine 370:2071–2082. [CrossRef]
- Behr J, Prasse A, Kreuter M, Johow J, Rabe KF, Bonella F, Bonnet R, Grohe C, Held M, Wilkens H, Hammerl P, Koschel D, Blaas S, Wirtz H, Ficker JH, Neumeister W, Schönfeld N, Claussen M, Kneidinger N, Frankenberger M, Hummler S, Kahn N, Tello S, Freise J, Welte T, Neuser P, Günther A, Behr J, Kreuter M, Johow J, Rabe KF, Bonella F, Bonnet R, Grohe C, Held M, Wilkens H, Hammerl P, Koschel D, Blaas S, Wirtz H, Ficker JH, Neumeister W, Schönfeld N, Claussen M, Kneidinger N, Frankenberger M, Hummler S, Kahn N, Tello S, Freise J, Welte T, Neuser P, Günther A, Schade-Brittinger C, Aminossadati B, Nasemann C, Yahiaoui S, Dupuy Backofen C, Hahmann M, Wittenberg M, Drakopanagiotakis F, von der Beck D, Ghofrani S, Heinemann S, Krauss E, Rethorn H, Koch A, Leuschner G, Matthes S, Neurohr C, Veit T, Milger-Kneidinger K, Herth F, Benstz J, Hummler S, Bahmer T, Biller H, Waschki B, Apel R-M, Costabel U, Börner E, Wessendorf T, Arnrich M, Ilie L, Wald A, Seyfarth H-J, Reinhardt C, Cinar A, Vogler M, Huhn SM, Richter J, Neff U, Blum TG, Vesenbeckh S, Boch C, Semper H, Wilke A, Pfeifer M, Schweda A, Krill A, Lensch C, Joa F, Schröder B, Plaßmeier A, Baron S, Froehling KP, Waschki B (2021) Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med 9:476–486. [CrossRef]
- Aluganti Narasimhulu C, Singla DK (2020) The Role of Bone Morphogenetic Protein 7 (BMP-7) in Inflammation in Heart Diseases. Cells 9:. [CrossRef]
- Nguyen TQ, Goldschmeding R (2008) Bone Morphogenetic Protein-7 and Connective Tissue Growth Factor: Novel Targets for Treatment of Renal Fibrosis? Pharm Res 25:2416–2426. [CrossRef]
- Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R (2007) Fibroblasts Derive from Hepatocytes in Liver Fibrosis via Epithelial to Mesenchymal Transition. Journal of Biological Chemistry 282:23337–23347. [CrossRef]
- Biyikli NK, Tugtepe H, Cakalagaoglu F, Ilki A, Alpay H (2005) Downregulation of the expression of bone morphogenetic protein 7 in experimental pyelonephritis. Pediatric Nephrology 20:1230–1236. [CrossRef]
- Murray LA, Hackett TL, Warner SM, Shaheen F, Argentieri RL, Dudas P, Farrell FX, Knight DA (2008) BMP-7 Does Not Protect against Bleomycin-Induced Lung or Skin Fibrosis. PLoS One 3:e4039. [CrossRef]
- Valcourt U, Kowanetz M, Niimi H, Heldin C-H, Moustakas A (2005) TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell 16:1987–2002. [CrossRef]
- Tacke F, Gäbele E, Bataille F, Schwabe RF, Hellerbrand C, Klebl F, Straub RH, Luedde T, Manns MP, Trautwein C, Brenner DA, Schölmerich J, Schnabl B (2007) Bone Morphogenetic Protein 7 is Elevated in Patients with Chronic Liver Disease and Exerts Fibrogenic Effects on Human Hepatic Stellate Cells. Dig Dis Sci 52:3404–3415. [CrossRef]
- Li X, An G, Wang Y, Liang D, Zhu Z, Lian X, Niu P, Guo C, Tian L (2017) Anti-fibrotic effects of bone morphogenetic protein-7-modified bone marrow mesenchymal stem cells on silica-induced pulmonary fibrosis. Exp Mol Pathol 102:70–77. [CrossRef]
- Tandon A, Sharma A, Rodier JT, Klibanov AM, Rieger FG, Mohan RR (2013) BMP7 Gene Transfer via Gold Nanoparticles into Stroma Inhibits Corneal Fibrosis In Vivo. PLoS One 8:. [CrossRef]
- Midgley AC, Wei Y, Zhu D, Gao F, Yan H, Khalique A, Luo W, Jiang H, Liu X, Guo J, Zhang C, Feng G, Wang K, Bai X, Ning W, Yang C, Zhao Q, Kong D (2020) Multifunctional natural polymer nanoparticles as antifibrotic gene carriers for CKD therapy. Journal of the American Society of Nephrology 31:2292–2311. [CrossRef]
- Gebert LFR, MacRae IJ (2019) Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 20:21–37. [CrossRef]
- Condrat CE, Thompson DC, Barbu MG, Bugnar OL, Boboc A, Cretoiu D, Suciu N, Cretoiu SM, Voinea SC (2020) miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 9:. [CrossRef]
- Gomez IG, MacKenna DA, Johnson BG, Kaimal V, Roach AM, Ren S, Nakagawa N, Xin C, Newitt R, Pandya S, Xia T-H, Liu X, Borza D-B, Grafals M, Shankland SJ, Himmelfarb J, Portilla D, Liu S, Chau BN, Duffield JS (2015) Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest 125:141–56. [CrossRef]
- Yang F, Li P, Li H, Shi Q, Li S, Zhao L (2015) microRNA-29b Mediates the Antifibrotic Effect of Tanshinone IIA in Postinfarct Cardiac Remodeling. J Cardiovasc Pharmacol 65:456–464. [CrossRef]
- Wang J, Lee CJ, Deci MB, Jasiewicz N, Verma A, Canty JM, Nguyen J (2020) MiR-101a loaded extracellular nanovesicles as bioactive carriers for cardiac repair. Nanomedicine 27:102201. [CrossRef]
- Gallant-Behm CL, Piper J, Lynch JM, Seto AG, Hong SJ, Mustoe TA, Maari C, Pestano LA, Dalby CM, Jackson AL, Rubin P, Marshall WS (2019) A MicroRNA-29 Mimic (Remlarsen) Represses Extracellular Matrix Expression and Fibroplasia in the Skin. Journal of Investigative Dermatology 139:1073–1081. [CrossRef]
- Chioccioli M, Roy S, Newell R, Pestano L, Dickinson B, Rigby K, Herazo-Maya J, Jenkins G, Ian S, Saini G, Johnson SR, Braybrooke R, Yu G, Sauler M, Ahangari F, Ding S, DeIuliis J, Aurelien N, Montgomery RL, Kaminski N (2022) A lung targeted miR-29 mimic as a therapy for pulmonary fibrosis. EBioMedicine 85:104304. [CrossRef]
- Chahal J, Gebert LFR, Camargo C, MacRae IJ, Sagan SM (2021) miR-122–based therapies select for three distinct resistance mechanisms based on alterations in RNA structure. Proceedings of the National Academy of Sciences 118:. [CrossRef]
- O’Reilly S (2016) MicroRNAs in fibrosis: opportunities and challenges. Arthritis Res Ther 18:11. [CrossRef]
- Zhong X, Chung ACK, Chen H-Y, Meng X-M, Lan HY (2011) Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J Am Soc Nephrol 22:1668–81. [CrossRef]
- Chen X, Zhang D, Wang Y, Chen K, Zhao L, Xu Y, Jiang H, Wang S (2020) Synergistic antifibrotic effects of miR-451 with miR-185 partly by co-targeting EphB2 on hepatic stellate cells. Cell Death Dis 11:402. [CrossRef]
- Jelinic M, Marshall SA, Stewart D, Unemori E, Parry LJ, Leo CH (2018) Peptide hormone relaxin: from bench to bedside. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 314:R753–R760. [CrossRef]
- Samuel CS, Zhao C, Yang Q, Wang H, Tian H, Tregear GW, Amento EP (2005) The Relaxin Gene Knockout Mouse: A Model of Progressive Scleroderma. Journal of Investigative Dermatology 125:692–699. [CrossRef]
- Sarwar M, Du X-J, Dschietzig TB, Summers RJ (2017) The actions of relaxin on the human cardiovascular system. Br J Pharmacol 174:933–949. [CrossRef]
- Samuel CS, Unemori EN, Mookerjee I, Bathgate RAD, Layfield SL, Mak J, Tregear GW, Du X-J (2004) Relaxin Modulates Cardiac Fibroblast Proliferation, Differentiation, and Collagen Production and Reverses Cardiac Fibrosis in Vivo. Endocrinology 145:4125–4133. [CrossRef]
- Kanai AJ, Konieczko EM, Bennett RG, Samuel CS, Royce SG (2019) Relaxin and fibrosis: Emerging targets, challenges, and future directions. Mol Cell Endocrinol 487:66–74. [CrossRef]
- EVANS JA (1959) Relaxin (Releasin) Therapy in Diffuse Progressive Scleroderma. AMA Arch Derm 79:150. [CrossRef]
- Ng HH, Shen M, Samuel CS, Schlossmann J, Bennett RG (2019) Relaxin and extracellular matrix remodeling: Mechanisms and signaling pathways. Mol Cell Endocrinol 487:59–65. [CrossRef]
- Cai J, Chen X, Chen X, Chen L, Zheng G, Zhou H, Zhou X (2017) Anti-Fibrosis Effect of Relaxin and Spironolactone Combined on Isoprenaline-Induced Myocardial Fibrosis in Rats via Inhibition of Endothelial–Mesenchymal Transition. Cellular Physiology and Biochemistry 41:1167–1178. [CrossRef]
- Metra M, Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Pang PS, Ponikowski P, Voors AA, Adams KF, Anker SD, Arias-Mendoza A, Avendaño P, Bacal F, Böhm M, Bortman G, Cleland JGF, Cohen-Solal A, Crespo-Leiro MG, Dorobantu M, Echeverría LE, Ferrari R, Goland S, Goncalvesová E, Goudev A, Køber L, Lema-Osores J, Levy PD, McDonald K, Manga P, Merkely B, Mueller C, Pieske B, Silva-Cardoso J, Špinar J, Squire I, Stępińska J, Van Mieghem W, von Lewinski D, Wikström G, Yilmaz MB, Hagner N, Holbro T, Hua TA, Sabarwal S V., Severin T, Szecsödy P, Gimpelewicz C (2019) Effects of Serelaxin in Patients with Acute Heart Failure. New England Journal of Medicine 381:716–726. [CrossRef]
- Khanna D, Clements PJ, Furst DE, Korn JH, Ellman M, Rothfield N, Wigley FM, Moreland LW, Silver R, Kim YH, Steen VD, Firestein GS, Kavanaugh AF, Weisman M, Mayes MD, Collier D, Csuka ME, Simms R, Merkel PA, Medsger TA, Sanders ME, Maranian P, Seibold JR, Relaxin Investigators and the Scleroderma Clinical Trials Consortium (2009) Recombinant human relaxin in the treatment of systemic sclerosis with diffuse cutaneous involvement: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 60:1102–11. [CrossRef]
- Hossain MA, Kocan M, Yao ST, Royce SG, Nair VB, Siwek C, Patil NA, Harrison IP, Rosengren KJ, Selemidis S, Summers RJ, Wade JD, Bathgate RAD, Samuel CS (2016) A single-chain derivative of the relaxin hormone is a functionally selective agonist of the G protein-coupled receptor, RXFP1. Chem Sci 7:3805–3819. [CrossRef]
- Vagnozzi RJ, Kasam RK, Sargent MA, Molkentin JD (2021) Cardiac Cell Therapy Fails to Rejuvenate the Chronically Scarred Rodent Heart. Circulation 144:328–331. [CrossRef]
- Makkar RR, Kereiakes DJ, Aguirre F, Kowalchuk G, Chakravarty T, Malliaras K, Francis GS, Povsic TJ, Schatz R, Traverse JH, Pogoda JM, Smith RR, Marbán L, Ascheim DD, Ostovaneh MR, Lima JAC, DeMaria A, Marbán E, Henry TD (2020) Intracoronary ALLogeneic heart STem cells to Achieve myocardial Regeneration (ALLSTAR): a randomized, placebo-controlled, double-blinded trial. Eur Heart J 41:3451–3458. [CrossRef]
- Liu C, Han D, Liang P, Li Y, Cao F (2021) The Current Dilemma and Breakthrough of Stem Cell Therapy in Ischemic Heart Disease. Front Cell Dev Biol 9:. [CrossRef]
- Adams E, McCloy R, Jordan A, Falconer K, Dykes IM (2021) Direct Reprogramming of Cardiac Fibroblasts to Repair the Injured Heart. J Cardiovasc Dev Dis 8:72. [CrossRef]
- Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D (2010) Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142:375–86. [CrossRef]
- Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu J, Srivastava D (2012) In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485:593–8. [CrossRef]
- Wada R, Muraoka N, Inagawa K, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Kaneda R, Suzuki T, Kamiya K, Tohyama S, Yuasa S, Kokaji K, Aeba R, Yozu R, Yamagishi H, Kitamura T, Fukuda K, Ieda M (2013) Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc Natl Acad Sci U S A 110:12667–72. [CrossRef]
- Tani H, Sadahiro T, Yamada Y, Isomi M, Yamakawa H, Fujita R, Abe Y, Akiyama T, Nakano K, Kuze Y, Seki M, Suzuki Y, Fujisawa M, Sakata-Yanagimoto M, Chiba S, Fukuda K, Ieda M (2023) Direct Reprogramming Improves Cardiac Function and Reverses Fibrosis in Chronic Myocardial Infarction. Circulation 147:223–238. [CrossRef]
- Nam Y-J, Song K, Luo X, Daniel E, Lambeth K, West K, Hill JA, DiMaio JM, Baker LA, Bassel-Duby R, Olson EN (2013) Reprogramming of human fibroblasts toward a cardiac fate. Proc Natl Acad Sci U S A 110:5588–93. [CrossRef]
- Avolio E, Katare R, Thomas AC, Caporali A, Schwenke D, Carrabba M, Meloni M, Caputo M, Madeddu P (2022) Cardiac pericyte reprogramming by MEK inhibition promotes arteriologenesis and angiogenesis of the ischemic heart. Journal of Clinical Investigation 132:. [CrossRef]
- Zhang F, Ayaub EA, Wang B, Puchulu-Campanella E, Li Y, Hettiarachchi SU, Lindeman SD, Luo Q, Rout S, Srinivasarao M, Cox A, Tsoyi K, Nickerson-Nutter C, Rosas IO, Low PS (2020) Reprogramming of profibrotic macrophages for treatment of bleomycin-induced pulmonary fibrosis. EMBO Mol Med 12:. [CrossRef]
- Dempsey TM, Thao V, Moriarty JP, Borah BJ, Limper AH (2022) Cost-effectiveness of the anti-fibrotics for the treatment of idiopathic pulmonary fibrosis in the United States. BMC Pulm Med 22:18. [CrossRef]
- Rinciog C, Diamantopoulos A, Gentilini A, Bondue B, Dahlqvist C, Froidure A, Wuyts WA, Soulard S (2020) Cost-Effectiveness Analysis of Nintedanib Versus Pirfenidone in Idiopathic Pulmonary Fibrosis in Belgium. Pharmacoecon Open 4:449–458. [CrossRef]
- Yamakawa H, Ieda M (2021) Cardiac regeneration by direct reprogramming in this decade and beyond. Inflamm Regen 41:20. [CrossRef]
- Craig VJ, Zhang L, Hagood JS, Owen CA (2015) Matrix Metalloproteinases as Therapeutic Targets for Idiopathic Pulmonary Fibrosis. Am J Respir Cell Mol Biol 53:585–600. [CrossRef]
- Adhikari AS, Chai J, Dunn AR (2011) Mechanical Load Induces a 100-Fold Increase in the Rate of Collagen Proteolysis by MMP-1. J Am Chem Soc 133:1686–1689. [CrossRef]
Figure 1.
Mechanisms of wound healing and fibrosis development. The normal wound healing process proceeds through three overlapping, yet independent phases: hemostasis and inflammation, regeneration, and remodeling. At each stage, dysregulation can lead to the development of fibrosis. A dominance of the TH2 CD4+ T cell phenotype during the inflammatory phase leads to the overproduction of profibrotic cytokines that induce excessive ECM deposition. Hyperactivity of fibroblasts/myofibroblasts, either though chronic activation, decreased programmed apoptosis, or oversupply via EMT, also results in accumulation of collagen and fibrotic scarring. Abnormal remodeling may also lead to buildup of collagen through improper degradation in a process involving MMPs and TIMPs. Created with BioRender.com.
Figure 1.
Mechanisms of wound healing and fibrosis development. The normal wound healing process proceeds through three overlapping, yet independent phases: hemostasis and inflammation, regeneration, and remodeling. At each stage, dysregulation can lead to the development of fibrosis. A dominance of the TH2 CD4+ T cell phenotype during the inflammatory phase leads to the overproduction of profibrotic cytokines that induce excessive ECM deposition. Hyperactivity of fibroblasts/myofibroblasts, either though chronic activation, decreased programmed apoptosis, or oversupply via EMT, also results in accumulation of collagen and fibrotic scarring. Abnormal remodeling may also lead to buildup of collagen through improper degradation in a process involving MMPs and TIMPs. Created with BioRender.com.
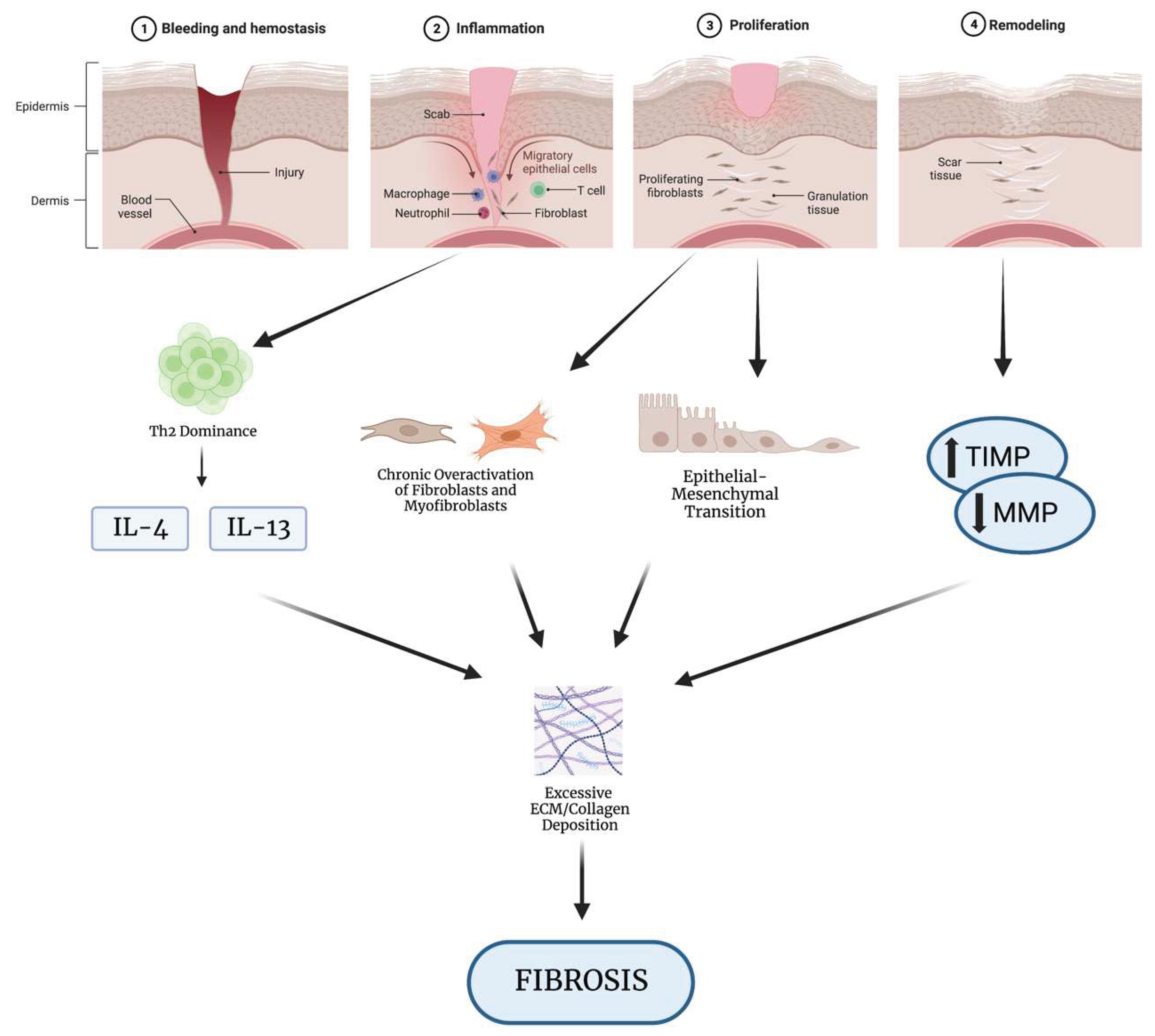

Table 1.
Major antifibrotic clinical trials for Nintedanib and Pirfenidone.
| Trial Name | Intervention | Phase | Study Design | Reference |
|---|---|---|---|---|
| INBUILD (NCT02999178) |
Nintedanib: 150 mg twice daily vs placebo | III | Randomized, double-blind, placebo-controlled, 663 patients with fibrosing lung disease | [97] |
| INPULSIS I (NCT01335464) |
Nintedanib: 150 mg twice daily vs placebo |
III | Randomized, double-blind, placebo-controlled, 515 patients with IPF | [98] |
| INPULSIS II (NCT01335477) |
Nintedanib: 150 mg twice daily vs placebo |
III | Randomized, double-blind, placebo-controlled, 551 patients with IPF | [98] |
| PINCER (NCT04856111) |
Pirfenidone (600 mg daily, escalated by 600 mg daily every 3-7 days up to targeted dose of 2400 mg daily) vs. Nintedanib (150mg twice daily) | IV | Randomized, parallel-assignment, 48 patients with COVID-19 | [95] |
| RELIEF (NCT03099187) |
Pirfenidone: 801mg three times daily vs. placebo |
IIb | Randomized, double-blind, placebo-controlled, 253 patients with unclassifiable progressive fibrosing ILD | [99] |
Table 2.
Overview of current miRNA therapies, targets, expression levels, and clinical trial status. .
Table 2.
Overview of current miRNA therapies, targets, expression levels, and clinical trial status. .
| Fibrotic Disease | Drug/Therapy | Target | Target Expression Level | Phase | Study |
|---|---|---|---|---|---|
| Renal | RG-012 | miR-21 | Down-regulation | I/II | [112] |
| Cardiac | Tanshinone IIA | miR-29b | Up-regulation | -- | [113] |
| Cardiac | miR-101a-loaded MSC extracellular nanovesicles | miR-101a | Up-regulation | -- | [114] |
| Lung | Remlarsen/MRG-201 | miR-29 | Down-regulation | II | [115] |
| Lung | MRG-229 | miR-29 | Down-regulation | -- | [116] |
| Liver | Miravirsen | miR-122 | Down-regulation | II | [117] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



