Submitted:
28 November 2023
Posted:
29 November 2023
You are already at the latest version
Abstract
The manuscript presents the comprehensive integrative theory of the etiology and pathogenesis of Alzheimer’s disease - the amyloid degradation toxicity hypothesis - and describes the logic that underlies it.The analysis of amyloid biomarkers and stable-isotope label kinetics (SILK) studies suggest that AD diagnosis is associated with higher cellular uptake of beta-amyloid. Uptake of beta-amyloid by cells is needed for its cytotoxicity, so the uptake rate should correlate with the rate of neurodegeneration. Also, the initial step in forming extracellular aggregates cannot occur in the interstitial fluid due to the extremely low concentration of beta-amyloid but can occur intralysosomally. Therefore, the density of extracellular aggregates should positively correlate with the rate of cellular amyloid uptake. The model, which considers that both cytotoxicity and aggregation of beta-amyloid are defined by cellular uptake, successfully reproduces the probability distribution of AD diagnosis in the population. Cellular uptake of beta-amyloid is mediated by endocytosis. Endocytosed beta-amyloid induces lysosomal permeabilization that occurs without plasma membrane damage. Lysosomal permeabilization explains ion disturbances, such as an accumulation of intracellular calcium, caused by cell exposure to extracellular beta-amyloid. Some amyloid fragments, produced from beta-amyloid by lysosomal proteases, can form membrane channels in lysosomal membranes, which are large enough to leak cathepsins to the cytoplasm. Appearance of proteases in the cytoplasm results in necrosis and/or initiation of apoptosis. If the cell survives, the damage of lysosomes leads to autophagy failure and slow recycling of mitochondria, promoting the production of reactive oxygen species and potentiating cell damage.Considering the above, the integrative theory of AD etiology and pathogenesis can be formulated. The etiology of AD is the membrane channel formation by amyloid fragments produced in lysosomes. The pathogenesis includes lysosomal permeabilization by giant membrane channels, which leak lysosomal proteases into the cytoplasm. The correlation between the density of amyloid aggregates and the probability of AD appears because the intensity of cellular uptake defines both aggregation rates in vivo and cytotoxicity of beta-amyloid.The amyloid degradation toxicity hypothesis is the integrative theory of Alzheimer’s disease (AD). It successfully interprets multiple phenomena and paradoxes associated with AD pathobiology at various levels, from molecular and cellular to biomarkers. The hypothesis explains the limitations of currently used biomarkers of AD and proposes etiology-related parameters. These parameters could be measured in humans and become novel diagnostic and prognostic clinical tools. Based on the proposed framework, we foresee the development of effective medications to treat, stall the progression of, or prevent disease development.
Keywords:
Alzheimer's disease
; beta-amyloid toxicity
; amyloid depositions
; cellular uptake
; lysosome
Introduction
It was said multiple times that the absence of effective treatments to slow down or prevent Alzheimer’s di sea se results from the lack of understanding of the etiology and pathogenesis of this illness. Billions of dollars were spent on studies in basic research and clinical settings, and the research community accumulated multiple facts about this disease. Nevertheless, no integrative theory emerged. By integrative theory, we consider the theory which describes the disease’s origins and progression starting at the molecular level. Integrative theory links these molecular changes with disturbances at the organelle and cell levels and extends all the way to the population analysis of disease prevalence.
Even though most scientists only hope that such a theory will emerge soon, we discovered sufficient data to formulate a framework for an integrative theory of AD. In this manuscript, we present the supporting data and the structure of the hypothesis, which we call the “amyloid degradation toxicity hypothesis.”
Dr. Alzheimer first described a certain kind of demen tia [1], which was later named in his name (Alzheimer’s disease, AD) [2]. Utilizing his skills in histological analysis, he discovered and described extracellular senile plaques and intracellular neurofibrillary tangles as specific to the disease. As was found later, the main component of the extracellular deposits is beta-amyloid protein (Aβ) [3,4]. Considering that Aβ is toxic to cells in vitro [5,6,7] and that histopathological changes are more likely to occur in cells in close proximity to the plaques [8,9,10], it is understandable why, for a long time, the scientific community had associated the etiology and pathogenesis of AD with the Aβ plaques themselves. The positron emission tomography (PET) technique, which allowed for the measurement of the density of amyloid deposits in live subjects, brought more data that seemingly supported the potential role of Aβ deposits in AD pathogenesis [11]. First, in consensus with data observed in postmortem pathology, patients diagnosed with AD had significantly denser amyloid deposits. Even more importantly, patients with a significant density of amyloid deposits were more prone to decline cognitively compared to amyloid-negative individuals [12].
The strong correlation between the density of amy lo id deposits and the probability of diagnosis was the pri mary reason for the overwhelming dominance of amyloid- centric theories of AD. The most attractive among them was the amyloid cascade hypothesis, which capitalizes on the fact that beta-amyloid can aggregate spontaneously. This hypothesis claims that the deposits somehow drive neurotoxicity [13], so targeting the accumulation of insoluble beta-amyloid could lead to the treatment of the disease or, at least, to the prevention of the disease progression. Correspondingly, the ability to measure the density of amyloid aggregates in living patients and the possibility of dissolving extracellular amyloid deposits attracted most pharmaceutical investments in the last two decades.
Unfortunately for human patients, many experimental studies in animal models supported this pharmaco logical approach. However, recent clinical trials to dissolve amyloid deposits largely failed or showed relatively low effectiveness [14]. The best effect of confirmed “dissolving” amyloid deposits is estimated as slowing the progression of cognitive decline by 30%, very far from prevention or cure, which can be expected if amyloid deposits are, in fact, the driver of neuro degeneration. However, the low effectiveness of such treatments is in line with the fact that beta-amyloid aggregates are non-toxic by themselves, even though soluble Aβ is cytotoxic [5,6,7]. The very fact that not any successful removal of plaques slows down the progression of AD underscores the irrelevance of plaques as such to the progression of the disease. Failures of clinical trials force the research community to critically assess the validity of assumptions under lying amyloidocentric theories of AD. Even though the wave of current anti-amyloid pressure has its reasons, the integrative theory of AD (whatever it would be) needs to explain the correlation between the accumulation of these non-toxic deposits and the diagnosis of AD. Correlation does not mean causation: neuronal death and the accumulation of Aβ deposits could be two consequences of a third process. In any case, it is logical to start by reviewing extensive data on available amyloid biomarkers.

The path to build amyloid degradation toxicity hypothesis
The manuscript describes step-by-step logic that leads to the comprehensive integrative theory of Alzheimer’s disease. This section presents the road map of how the goal of building such a theory was achieved, starting with the conclusions made from the analysis of amyloid biomarkers (Figure 1).
First, the analysis of amyloid biomarkers suggests that patients with AD have a higher intensity of intra brain aggregation-independent removal of beta-amylo id, mainly through proteolytic digestion. Proteases are primarily located intracellularly, so the probability of AD diagnosis positively correlates with the rate of cellular uptake of beta-amyloid. Stable-isotope label kinetics (SILK) studies support this conclusion.
Second, there is no known molecular mechanism of beta-amyloid acting on the plasma membrane in physiological concentrations, which can initiate cyto toxicity. However, treating cells with beta-amyloid induces lysosomal permeabilization without damaging plasma membranes, while Alzheimer’s disease is associated with lysosomal failure. Cells accumulate beta-amyloid in lysosomes through endocytosis. Therefore, the uptake of beta-amyloid by cells is needed for its cytotoxicity.
Third, extracellular amyloid aggregation cannot be initiated spontaneously due to very low concentration in the interstitial fluid. However, cells take beta-amyloid by endocytosis and concentrate it inside lysosomes. In cells in vitro, endocytosed beta-amyloid can be ob ser ved intracellularly for several days. In concentrations that match intralysosomal conditions, aggregation can occur within several days. The intralysosomal aggre gates can be exocytosed and grow extra cellularly. In this scenario, the accumulation of extra cellular aggre gates depends on the intensity of cellular uptake of beta-amyloid.
Fourth, the mathematical model, in which both cytotoxicity and aggregation are defined by cellular uptake, reproduces the distribution of probabilities of AD diagnosis in the population as the function of the density of amyloid deposits and the concentration of beta-amyloid in the cerebrospinal fluid. The model interprets a strong correlation between the probability of AD in a patient and the density of amyloid aggre gates, which are non-cytotoxic by themselves. Also, this model explains why higher concentrations of soluble beta-amyloid (which is cytotoxic, unlike its aggregated form) in biological fluids correlate with a lower probability of AD diagnosis.
Fifth, in vitro exposure of a cell to the added beta-amyloid results in permeabilization of lysosomal membranes, which occurs without plasma membrane damage. Even though beta-amyloid is concentrated inside lysosomes, the intralysosomal concentrations are below the micromolar range. Therefore, perme abilization cannot be explained by direct damage of lipid bilayers by beta-amyloid.
Sixth, beta-amyloid can form membrane channels. Membrane amyloid channels are formed by multiple molecules of peptide, are non-selective, and, based on electrophysiological data on their conductance, can pass macromolecules. The channels are formed by some short amyloid fragments but not by full-length beta-amyloid (either 40- or 42-amino acid long). Channel formation requires a negative surface charge of membranes. Lysosomal membranes carry a signifi cant surface negative charge, while lysosomes are organelles that can digest endocytosed beta-amyloid and produce channel-forming peptide fragments. The formation of membrane amyloid channels explains how treating cells with extracellular beta-amyloid results in lysosomal permeabilization, which occurs without damage to plasma membranes. The leakage of lysosomal proteases to the cytoplasm explains beta-amyloid cytotoxicity, which can occur through both necrosis and apoptosis (the latter is activated by lysosomal proteases).
By summarizing this logic, the amyloid degradation toxicity hypothesis, the integrative theory of AD patho biology, can be formulated. The cells accumulate beta-amyloid intralysosomally by endocytosis. Peptidases digest beta-amyloid into fragments, some of which can form giant membrane channels in lysosomal membra nes. Amyloid membrane channels leak intralysosomal content, including lysosomal enzymes, into the cytoplasm. Leaking cathepsins either induce necrosis or activate apoptosis. Also, the initiation of aggregation requires cellular uptake of beta-amyloid. Therefore, the intensity of cellular uptake correlates with both aggre gation and cytotoxicity of beta-amyloid. For that reason, the presence of amyloid aggregates, which are not cytotoxic by themselves, correlates with the probability of AD diagnosis and can serve as a biomarker. Both aggregation of soluble beta-amyloid on existing aggregates (the density of which is higher in AD patients) and increased cellular uptake (a significant factor defining the development of AD) decrease the concentration of soluble beta-amyloid in the interstitial fluid and, correspondingly, in the cerebrospinal fluid. Therefore, a lower concentration of beta-amyloid in the cerebrospinal fluid is a biomarker associated with neurodegeneration and AD diagnosis.
The amyloid degradation toxicity hypothesis successfully interprets many phenomena and paradoxes associated with AD pathobiology, such as lysosomal failure, increase of reactive oxygen species formation, accumulation of intracellular beta-amyloid, and activation of apoptosis in brain tissues. Importantly, it interprets two central paradoxes of any amyloido centric hypothesis of AD: the progression of the disease is associated with a lower concentration of toxic soluble beta-amyloid but with the accumulation of non-toxic amyloid aggregates.
1. Beta-amyloid biomarkers
1.1. Two major beta-amyloid biomarkers: concentration of Aβ42 in the CSF and the density of amyloid aggregates in the brain
In the original manuscript, Dr. Alzheimer described se nile plaques and neurofibrillary tangles as histo patho logical findings [1]. For most of the twentieth century, the data from the brains of patients with AD could be obtained only postmortem. With the discovery that the main component of the extracellular deposits is beta-amyloid protein (Aβ) and advancing analytical techni ques to measure the concentration of Aβ, it be ca me possible to monitor the concentration of this pep tide in biological fluids, including cerebrospinal fluid [15, 16].
It was found that despite the form of beta-amyloid with 40-amino acids (Aβ40) being predominant, 42-amino acids long peptide (Aβ42) has a better corre lation with the diagnosis of AD [13]. Specifically, the CSF of patients with AD contains significantly less Aβ42 [16,17]. It is reasonable to assume that a higher density of existing amyloid aggregates would promote the aggregation of newly synthesized Aβ42. Faster aggregation of soluble form on existing aggregates would decrease the amount of Aβ42, which can be cleared to the CSF. The sink effect looks like a reasonable explanation for lower CSF-Aβ42 when the density of existing aggregates is high.
The next methodological breakthrough was the ability to measure the density of amyloid deposits in living patients. Compounds such as florbetapir and florbeta ben bind to and accumulate in the senile plaques. Due to the presence of positron-emitting fluorine in their structure, the concentration of these compounds in various brain structures can be measured by positron emission tomography (PET). The sensitivity of the tech nique is extraordinary, considering that a total of 5 mg of beta-amyloid [17,18] (an equivalent to only 10% of the weight of a water drop) is spread in the brain of an amyloid-positive patient (which has a volume of 1-2 L). The studies unequivocally confirmed that patients with cognitive decline due to Alzheimer’s disease have significantly more dense amyloid deposits during life [11] and that amyloid-positive patients have faster cognitive decline than amyloid-negative patients [12].
Both the concentration of Aβ42 in the CSF and the density of amyloid deposits (measured by PET) are amyloid biomarkers of AD and are interconnected. Specifically, high levels of amyloid deposits are associated with significantly decreased Aβ42 levels in the CSF [19,20,21,22]. As stated above, both parameters have a strong negative correlation, which the presence of the sink effect can interpret. For that reason, the overall diagnostic accuracy of CSF-Aβ42 and PET measurements appears similar, even though PET is considered to have higher specificity [11].
The most comprehensive dataset on amyloid biomarkers is available through the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (http://adni.loni.usc.edu/). The ADNI was launched in 2003 as a public-private partnership led by Principal Investigator Michael W. Weiner, MD, and over the years, it has attracted more than $200 million. The primary goal of ADNI has been to test whether serial magnetic resonance imaging, positron emission tomo graphy (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and Alzheimer’s disease (AD). The study protocol for ADNI was approved by the local ethical committees of all participating institutions, and all participants signed informed consent, which included consent for de-identified data being shared with the general scientific community for research purposes [23]. The researchers, who were not involved in data collection but wish to use their expertise in the analysis, can receive de-identified data after approval by ADNI.
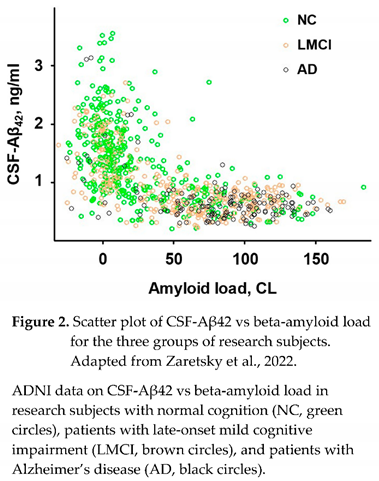
The authors of this manuscript are members of one of the collaborative research projects doing so. In studies published by us previously [24,25], we used depersonalized data from participants in the Alzhei mer’s Disease Neuroimaging Initiative (ADNI) for whom the ascertainment of normal cognition (NC) or an AD diagnosis, as well as cerebrospinal fluid (CSF) collection, were done within one year from a PET scan identifying brain amyloidosis [26]. The distribution of data points in this dataset (Figure 2), which was used in multiple studies [20,24,25,27], is visually similar to other published data [28].

1.2. Characteristic distribution of beta-amyloid biomarkers can be described by a compartment kinetic model
The graph shown in Figure 2 includes all ADNI participants (as of the time of manuscript preparation) for whom the ascertainment of cognition status, AD diagnosis, and CSF collection were made within one year from a PET scan identifying brain amyloidosis. Given the use of two different amyloid PET tracers, the measured endpoint (SUVR) was converted to centiloids (CL) for each patient using the specific equation provided by ADNI for each tracer. The number of research subjects who had been diagnosed with AD (AD), had normal cognition (NC), or were diagnosed with mild cognitive impairment (MCI), either late-onset MCI (LMCI) or early-onset MCI (EMCI) was 143, 416, 340, and 476, respectively.
Most data points from subjects with normal cognition are around zero amyloid loads, while most AD patients are amyloid positive (amyloid load exceeds 30 centiloids). The patients with late-onset cognitive impairment are relatively homogenously distributed along the amyloid load axis. Reflecting a strong negative correlation between amyloid deposits and CSF-Aβ42, most subjects with normal cognition have a higher concentration of Aβ42 in the CSF and low values of PET signal, most patients with AD have low CSF-Aβ42 and high PET signal, and low and high CSF-Aβ42 and PET signal can be found in patients with LMCI with equal probability. Considering the shape of a complete dataset, it is reasonable to test what relationship can be found between the values of CSF-Aβ42 and the density of amyloid deposits (measured by PET).
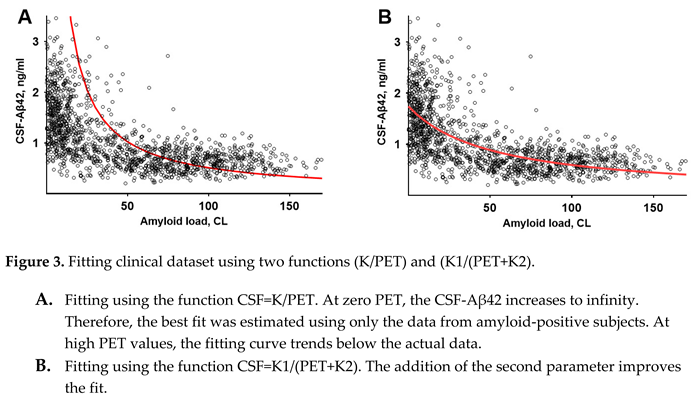
At a first (non-mathematical) glance, it may seem that the dependence can be described by a function , where is a constant. There is no doubt that the best fit for the function , shown in Figure 3, A, is far from reproducing data. In comparison, the function , where and are constants, describes the dataset much better (Figure 3, B).
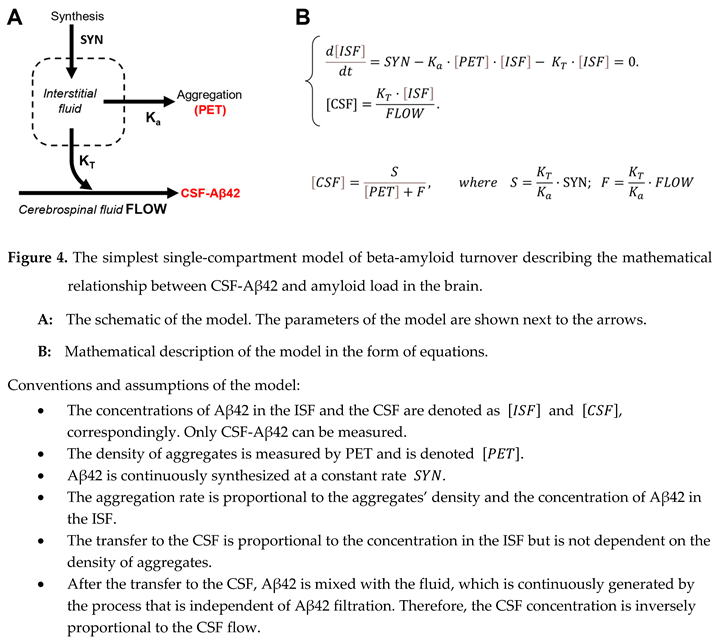
This hyperbolic distribution shape is consistent with the concept that synthesized Aβ42, which is released to the interstitial fluid, is either aggregated or transferred to the CSF. In line with previous modeling [15,16,17], such a concept can be formalized by a kinetic model of Aβ42 turnover, which includes synthesis, filtration to the CSF, and aggregation into non-soluble plaques (Figure 4, A).
The corresponding differential equation describes momentary changes in interstitial concentration as a function of synthesis (SYN), aggregation, and removal to the CSF (Figure 4, B). In the model, the rate of aggregation is the product of aggregation coefficient , the density of amyloid deposits, and interstitial concentration of Aβ42: ). The rate of removal to the CSF is the product of transfer coefficient and interstitial concentration of Aβ42 [ISF]: . Finally, all soluble Aβ42, which is transferred to the CSF, is homogenously distributed in the CSF, so CSF-Aβ42 ([CSF]) can be calculated by dividing the rate of Aβ42 transfer () by the intensity of CSF flow. At the time scale of measurements, the system is in relative equilibrium, so the interstitial concentration (as well as CSF-Aβ42) is not changing, therefore, the derivative is equal to zero. The analytical solution of this system () is the hyperbole which was mentioned above (). The selection of symbols for the parameters and was defined by the fact that the first one () is proportional to synthesis, while the second () is proportional to the rate of the CSF flow.

1.3. The distributions of beta-amyloid biomarkers in subjects with normal cognition and patients with AD: the evidence of increased intrabrain beta-amyloid removal rate
While patients with AD have the distribution of beta-amyloid biomarkers dramatically shifted towards high density of amyloid deposits, the boundaries of distributions in healthy subjects and patients with various degrees of cognitive impairment are overlapping (see Figure 2). The distribution of amyloid biomarkers in research subjects with normal cognition and patients with AD, if considered separately, appears to have similar shape even though most data points in each population is shifted towards opposite sides of the cloud (Figure 5 A, B). Considering that the distribution can be described by a compartment model characterized by the synthesis and removal rates, we tested the hypothesis that the synthesis and removal rates for beta-amyloid are different between groups of research subjects [18]. In this study, all research subjects in the ADNI database were separated into four groups according to the diagnosis: subjects with normal cognition (NC), patients with AD, patients with late-onset mild cognitive impairment (LMCI), and patients with early-onset mild cognitive impairment (EMCI). For each group, we estimated parameters and (according to the model shown in Figure 4), which provide the best fit, and calculated the confidence regions for these parameters. Hyperbolas corresponding to the best-fit values in the NC and AD groups are shown in Figure 5 A, B. It is essential to mention that the statistical method used to process data provides confidence region in 2D space: even if each parameter is not different between groups, pairs of parameters characterizing groups could be statistically different.
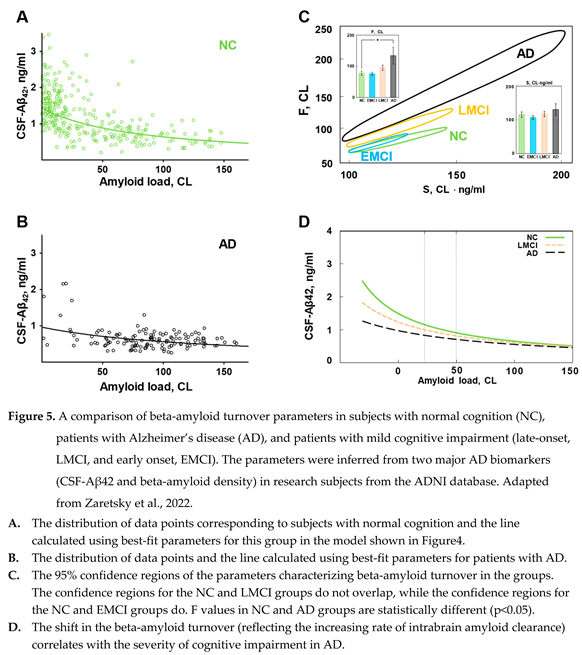
A separate analysis of these four subpopulations reveals that even though the parameters of hyperbolas describing the distribution of biomarkers are close between groups, they are statistically different. Confidence regions for the pairs of values of S and F, which characterize each subpopulation, have ellipsoid shapes and are shown in Figure 5, C. The confidence regions for the three subpopulations (NC, LMCI, and AD) are different, which means that these three subpopulations are different. However, the only statistical difference in the value of individual parameters appears to be in the F value between NC and AD groups. It is obvious that the EMCI group is not statistically different from the NC group, even when both S and F parameters are considered together. This suggests that the pathogenesis of early- and late-onset cognitive impairment is mediated by different mechanisms, while data points corresponding to subjects from the LMCI group have an intermediate distribution between AD and NC groups.
As stated earlier, parameter S is proportional to the synthesis rate. The absence of difference in beta-amyloid synthesis between AD and NC groups was demonstrated earlier using the SILK technique [17,19]. SILK technique estimates synthesis in individual patients, while our analysis confirms this finding using an independent populational approach.
Correspondingly, the rate of removal of beta-amyloid from the brain is the main difference between the AD and NC groups (the value of parameter F is statistically higher in the AD group compared to the NC group). In contrast to aggregation, which removes beta-amyloid from turnover but keeps it inside the brain, parameter F is aggregation-independent removal and was considered by us to represent physical removal from the brain. In the model design, we assumed that the removal with cerebrospinal flow is the predominant way of physically clearing the brain from beta-amyloid. Is there any data supporting a higher rate of removal with the CSF to match the increased value of F in the AD group?
In the model, this removal rate is proportional to the CSF flow. However, the CSF flow in patients with AD is either unchanged or lower than in subjects with normal cognition [20,21]. The resolution of this controversy could be in modifying the model: while removal with the CSF can be the predominant way to clear the brain from beta-amyloid, it is not the only one. It is known that some beta-amyloid is metabolized inside brain tissues, resulting in physical removal from the brain. The removal rate which is found in the modeling is the rate of total removal of beta-amyloid through all aggregation-independent pathways - with and without physical removal from the brain.
Intrabrain removal of beta-amyloid is well-known to researchers. The proteolytic degradation of beta-amyloid is known to participate in the turnover of the peptide, and inhibitors of amyloid-degrading proteases were historically suspected to be promoting the progression of AD [22] precisely for the reason of slowing the removal of beta-amyloid from the brain. To be metabolized by proteases in the brain, the peptide needs to be taken by brain cells. In modeling terms, the removal from the interstitial fluid abolishes the peptide’s ability to be transferred to the CSF. For that reason, we can consider that intrabrain removal reflects cellular uptake by brain cells. Correspondingly, the model can be modified, as shown in Figure 6.
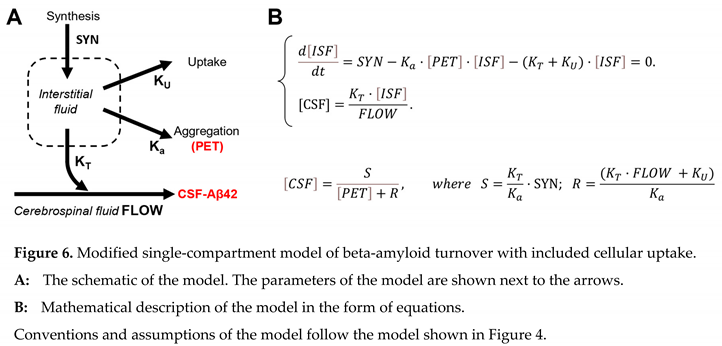
In this model, the parameter F (the rate of removal by CSF flow) is replaced by the parameter R (the rate of removal from the brain, which is the sum of removal with the CSF flow and intrabrain removal). All statistical analysis that was applied to the previous model remains. Therefore, S is not different between the NC and AD groups, while R is statistically higher in the AD group compared to the NC group. Unfortunately, within this model’s framework, it is impossible to distinguish the ratio of removal with the CSF and intrabrain removal because only the sum of them is used in the equations.
However, based on independent estimates, we can consider that the rate of removal with the CSF is either the same or even lower [20,21]. This means that the rate of intrabrain removal is higher. If we assume that the removal with the CSF is predominant and that the rate of removal with the CSF can be either the same or lower (but definitely, it is not higher!), to observe a statistically significant increase in the total removal rate, the increase of intrabrain removal rate should be quite dramatic. As we estimated in the original manuscript, such an increase can be several-fold [18].
The finding of increased clearance of Aβ42 directly contradicts the currently dominating concept that the accumulation of beta-amyloid results from slower re moval of this peptide from the brain of patients with AD. The first studies that attempted to estimate the para meters of amyloid turnover in the brain using the SILK technique seemingly demonstrated slower clearance [19]. However, further analysis by the same research group found that the conclusion about slower removal results from oversimplification of the underlying com partment model and studying synthesis and elimination separately. A more comprehensive analysis of the complete curve of the accumulation and clearance of various beta-amyloid forms directly shows increased intrabrain clearance while also pointing to the increa sed time of intrabrain presence of freshly synthesized Aβ42 before it is eliminated through the CSF [15,17]. Importantly, changed turnover parameters were found only for Aβ42 but not for Aβ40 and Aβ38. Clinical data obtained using the SILK technique and their relevance to the amyloid degradation toxicity hypothesis will be analyzed in more detail in a separate section below.

2. Amyloid biomarkers of AD: two biomarkers – two paradoxes
As it was unequivocally established, the brains of AD patients have a higher density of amyloid deposits. After it became possible to estimate the density of amyloid deposits in the brains of living patients, the data confirmed that the density of deposits in the patient’s brain positively correlates with the probability that this patient has AD (Figure 7, A). This correlation is robust, and for that reason, the density of amyloid deposits is considered the biomarker of AD. Due to the much higher propensity of Aβ42 to aggregate compared to other forms of beta-amyloid, the senile plaques mainly consist of this form, so it would be reasonable to say that what is measured by PET labels is the density of Aβ42 aggregates. Therefore, the density of Aβ42 aggregates is the biomarker of AD.
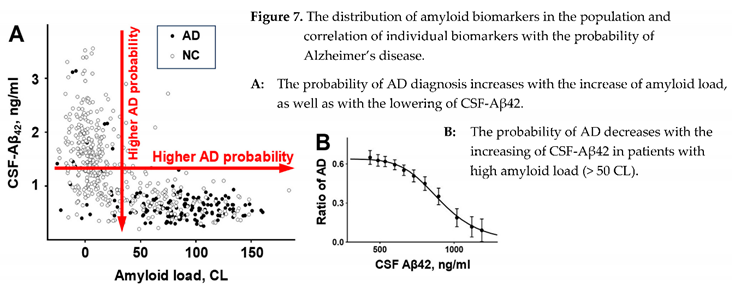
Similar to senile plaques, the beta-amyloid form in the CSF, which is associated with the AD diagnosis, is Aβ42, even though its concentration is approximately ten times lower than the concentration of Aβ40 [17]. However, in contrast to the density of senile plaques, the concentration of Aβ42 in the CSF is negatively correlated with the probability of AD diagnosis (Figure 7, A). The negative correlation is strong; therefore, CSF-Aβ42 is also considered the biomarker of AD.
To summarize the information revealed by amyloid biomarkers of AD: a higher probability of AD diagnosis is linked either to a higher density of amyloid aggre gates or a lower concentration of Aβ42 in the CSF. Unsurprisingly, there is a strong negative correlation between these two biomarkers. This correlation can be easily identified in the distribution of biomarker pairs in the population (Figure 7, A), and due to its strength, these two biomarkers appear to have similar diagnostic power when considered separately [11].
Most of the correlation is linked to the sink effect of ag gre gation of soluble beta-amyloid on existing amy lo id aggregates: higher density of aggregates – more freshly synthesized beta-amyloid is aggregated on the existing seeds – less beta-amyloid reaches the CSF. Hence, the lower concentration of Aβ42 in the CSF.
However, Sturchio et al. demonstrated that even after adjustment for multiple independent variables (inclu ding PET signal), lower CSF-Aβ42 levels are associ ated with a higher probability of AD [23]. In simpler terms, a higher probability of AD is observed in patients with a lower concentration of beta-amyloid in the CSF, even in subgroups with the same density of amyloid aggregates (measured by PET). The trend is not obvious in the distribution to the naked eye. Still, even simple statistical rolling averaging of amyloid-positive patients reveals that the probability of AD diagnosis falls from 0.6 in patients with the lowest levels of CSF-Aβ42 to virtually zero in patients with the highest CSF-Aβ42 (Figure 7, B). Notably, the highest CSF-Aβ42 levels in amyloid-positive patients are approximately twice lower than even the average CSF-Aβ42 in amyloid-negative research subjects. The presence of an amyloid-load-independent negative correlation between CSF-Aβ42 levels and the probability of AD explains the independent predictive powers of these two amyloid biomarkers of Alzheimer’s disease [24].
Before moving forward, we need to underscore that studying the connection between aggregation of beta-amyloid and the progression of AD was prevented by the paradox. First, as extremely large polymers, senile plaques are inert and appear non-toxic to cells in vitro [5]. The absence of cytotoxicity of aggregated beta-amyloid is clearly in conflict with the concept that a higher density of aggregated amyloid in patients’ brains is by itself the etiology of AD. Independent of whether it is amyloid-centric or not, as we stated in the Introduction, any comprehensive theory of AD is required to inter pret the high positive correlation of the presence of a non-toxic compound with the probability of the disease.
However, the concept that CSF-Aβ42 is another major amyloid biomarker of AD also includes a paradox. In contrast to aggregated beta-amyloid, its soluble form is toxic to cells [5]. However, there is no doubt that AD diagnosis is associated with a lower con centration of soluble beta-amyloid in the CSF [25,26].
Therefore, there are two major paradoxes in the relevance of amyloid biomarkers to AD, which any theory of etiology and pathophysiology of AD needs to resolve. These two paradoxes are summarized in Table 1. Despite the extreme popularity of amyloido centric hypotheses, these two paradoxes were not addressed appropriately.
To address the first paradox, the amyloid cascade hypothesis straightforwardly assumes that aggregated amyloid is toxic through an unidentified mechanism despite no strong in vitro evidence of such a phenomenon.
One of the avenues for interpretation is that aggre gated amyloid is in equilibrium with the soluble one, even though the balance is dramatically shifted towards aggregation. When beta-amyloid is not delivered by other routes, senile plaques could be a source of constant low levels of toxic soluble beta-amyloid. The key is that aggregates can be a source of toxic soluble beta-amyloid only if it is not delivered through other routes, including synthesis. However, there is no known data on the pulsatile synthesis of beta-amyloid. In contrast, direct evidence (data generated by the SILK technique) demonstrates continuous delivery of various freshly synthesized beta-amyloid peptides [17,19]. In the presence of constant peptide synthesis, the senile plaques would decrease (!) the concentration of toxic soluble beta-amyloid. For that reason, we consider it highly unlikely that senile plaques can be the source of toxic soluble beta-amyloid due to the presence of aggregation equilibrium.
Alternative explanations include the possibility that senile plaques can selectively accumulate toxic com ponents. For example, metals, such as iron, which can induce the toxicity to surrounding cells by catalyzing the production of reactive oxygen species (ROS).
As for the second paradox, historically, the drop in soluble Aβ42 was associated with an increased density of aggregates, which can serve as a sink for freshly synthesized peptide. However, more detailed analysis revealed that the levels of soluble Aβ42 provide infor mation about neurodegeneration, which is independent of the density of amyloid deposits [24]. The authors of the cited manuscript provided only the results of multi correlation analysis using two biomarkers in the ADNI dataset and used the Akaike information criterion to test if the models that used the combination of CSF and PET had better predictive power for clinical outcomes over models that used only one of the biomarkers. Mattson et al. (2015) suggested that combining CSF and PET in diagnostics may improve the prediction of clinical and pathological aspects of Alzheimer’s disease but did not specify how to do that. They suggested, though, that reduced CSF-Aβ42 may represent an earlier signal than amyloid accumulation detected by PET and that the PET signal may be a more sensitive marker of AD progression [24]. Nevertheless, looking at the correlation coefficients provided in various tables in this manuscript, it is easy to discover that after excluding the effect of PET, worse cognitive status correlates with lower CSF-Aβ42 levels, while higher CSF-Aβ42 levels are observed in patients with better clinical outcomes.
This finding was formulated as a statement only se veral years later by Sturchio et al. [23]. Using the same dataset (ADNI), they directly tested the hypothesis that higher CSF-Aβ42 levels correspond to better cognitive status of patients, even after excluding the effect of the density of deposits. To interpret this, it was hypothe sized that soluble Aβ42 has some important biological function, so low levels of soluble Aβ42 cause AD [23,27,28,29]. Unfortunately, this interpretation has difficulty explaining why the levels of soluble Aβ42 in healthy patients with high density of deposits are the same as the levels of soluble Aβ42 in patients with AD with low density of deposits – the fact which can be found in the figures and was confirmed by us during statistical ana lysis of ADNI dataset. Suppose we follow the proposed framework that Aβ42 has some “good” properties. In that case, we need to accept that low levels of soluble Aβ42 can support normal cognition in the presence of amyloid deposits. However, if the deposits are absent, the same low levels of soluble Aβ42 are insufficient to support the “healthy” status of the brain, so AD ensues.
The presence of these paradoxes and the fact that the dissolving of amyloid deposits in clinical trials did not dramatically slow down the progression of AD raises the concern that amyloidocentric hypotheses are wrong to start with. Nevertheless, even if so, the phenomena described by the two paradoxes are based on clinical data. Therefore, any integrative theory of AD needs to interpret them.

3. Aggregation and cytotoxicity of beta-amyloid require cellular uptake
3.1. How are amyloid deposits formed?
Before analyzing both paradoxes, we must address one more question typically overlooked in AD studies: “How can amyloid deposits be formed in the brain?” Usually, it is assumed that existing deposits grow by aggregating soluble beta-amyloid. However, forming initial aggregates is the critical and slowest step in the appearance of a noticeable density of amyloid aggrega tes. The delay would be especially pronounced in diluted solutions. Surprisingly, the question “How are the initial aggregation seeds formed in vivo?” did not attract appropriate attention. It is usually overlooked that the concentration of beta-amyloid in biological tissues is not just low – it is extremely low. Most agg regation studies use micromolar concentrations, while the beta-amyloid concentration in biological fluids is in the range of single nanomoles per liter or even lower.
There are two primary reasons to study amyloid agg regation in solutions with concentrations in micromolar but not in the nanomolar range. First, aggregation is dramatically slower at low concentrations. Even in micromolar concentration, the aggregation process takes many hours. Second, methods to observe aggregation, such as fluorescent techniques using Thioflavin T, require relatively large protein concent ra tions. Finding aggregation monitoring techniques with higher sensitivity would most likely be possible. Still, extremely long times to perform a single experiment (multiple days or weeks) would be a serious obstacle in designing a productive research program. Also, study ing aggregation is generally highly dependent on addi tional factors, which are frequently difficult to control in serial experiments. For example, if any possibility to initiate aggregation was not avoided during preparation (for instance, during lyophilization) or the peptide was not completely disaggregated, the aggregation seeds could be present even before the experiment was ini tiated. Also, aggregation can be promoted by the impu ri ties in the solution; for example, the addition of artifi cial lipid vesicles (liposomes) promotes aggregation [30]. Even minimal contaminants, which could serve as aggregation seeds, would dramatically affect the reproducibility of experiments with low peptide concentrations. It appears that the reproducibility of aggregation data increases with protein concentration and can explain why most experimental studies do not include a physiological range of concentrations.
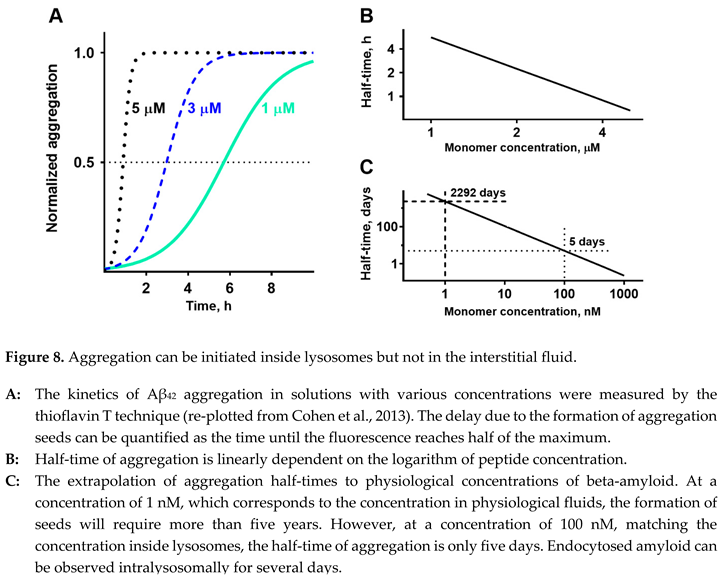
Fortunately, it is possible to extrapolate the time needed to form aggregation seeds at concentrations that are in a physiological range. The kinetics of the for mation of aggregation seeds can be characterized by the time required to aggregate a predetermined ratio of total peptide content in the solution. For simplicity and considering the highest reproducibility due to a sigmoid shape of the curve, it is usually measured at a 50% level (Figure 8, A replots the data from [31]). If we plot half-times of aggregation at various concentrations in loga rithmic coordinates, the dependence appears to be a straight line (data from Figure 8, A are shown in Figure 8, B). Linearization in logarithmic coordinates is consistent with the simplest mathematical model that considers that the aggregation rate is proportional to the probability of two molecules hitting each other in the solution. Experimental data for the study, shown in Figure 8, A, were collected in the micromolar range. Due to the linear dependence, it is possible to extrapolate the range to nanomolar concentrations (Figure 8, C). Decreasing the concentration of Aβ42 to 100 nM lowers the half-time of aggregation to five days. At a physiolo gically relevant concentration of 1 nM, it would take more than five years to aggregate half of the peptide. The problem is that while large polymeric senile plaques are physically stuck in the intercellular space, the ever-present movement of biological fluids will flash oligomeric aggregates, which are still soluble, out of the brain. To initiate a senile plaque, the aggregate in the interstitial fluid should be already large enough to be an insoluble particle, which cannot be physically removed by cleaning mechanisms in the brain tissue. Therefore, to become a senile plaque, the aggregation seed needs to appear in the interstitial space as a sizeable insoluble particle that has already passed the stage of a soluble oligomer. How can this be possible?
Looking at beta-amyloid turnover in the brain is necessary to answer this question. The peptide has several isoforms formed after sequential cleavage of the amyloid precursor protein (APP), a transmembrane glycoprotein with an undetermined function. APP can be cleaved by the proteolytic enzymes α-, β- and γ-secretase (see Figure 9). Aβ peptides, which are the focus of most current AD studies, are generated by a successive action of the β- and γ-secretases.

The γ-secretase, which produces the C-terminal end of the Aβ peptide, cleaves within the transmembrane region of APP and can generate multiple isoforms with various lengths. The isoform dominant in the cerebro spinal fluid is Aβ40, while senile plaques consist mainly of Aβ42. The reason why senile plaques have a compo sition that is different from the composition of interstitial fluid can be explained by the significantly higher pro pensity of Aβ1-42 for aggregation, which in turn is depen dent on the presence of two additional hydrophobic amino acids at the C-terminal of the peptide.
As noted, Aβ molecules could theoretically aggregate in the interstitial fluid to form soluble oligomers, which can continue growing. However, the rates are insufficient for this process to create large insoluble seeds before oligomers, which are still soluble, are moved away by interstitial fluid flow. It is logical that to have time for growth, freshly forming oligomers need to be in a place that is protected from removal to the CSF and still have an opportunity to grow by aggregating more beta-amyloid molecules.
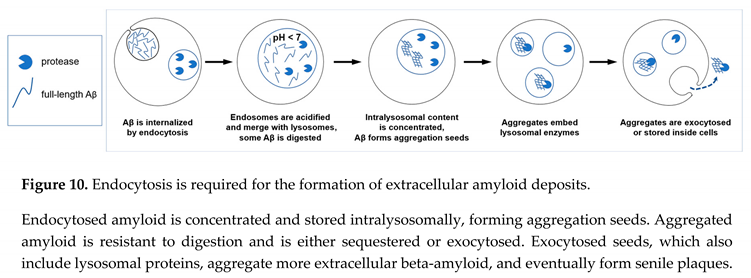
Such a place is widely described in multiple studies. The cells take beta-amyloid by endocytosis – the cell consumes a drop of extracellular fluid by surrounding it with a plasma membrane. This membrane vesicle, which is filled with extracellular fluid, contains nutrients and compounds that can be needed by the cell but are not synthesized locally and cannot cross the plasma membrane by diffusion or with the help of membrane transporters. For example, after being absorbed in the gut or synthesized in the liver, cholesterol, which is insoluble in water, is delivered to the neurons through a complex system of carrier proteins and transcellular transport. Similarly, iron is moved around the organism, tightly bound by transporting proteins, because it is cytotoxic as a free ion. Neurons consume both cholesterol and iron through endocytosis. Also, along with taking elementary units such as amino acids or glucose, cells receive nutrients by absorbing extracellular proteins and digesting them. To accomp lish that, cells merge endosomes with lysosomes, which contain digestive enzymes such as proteases. This process is accompanied by removing water from lysosomes, facilitating the digestion of trapped nutrients by concentrating intralysosomal content.
The most important here is that beta-amyloid is taken by endocytosis, and amyloid-containing endosomes are merged with lysosomes. The first thought could be that endocytosed beta-amyloid is quickly digested, and resulting amino acids are absorbed for the cell to use. But it is not precisely so. The absorbed beta-amyloid can be observed in the lysosomal compartment for several days [32]. Also, it is frequently overlooked how much it is concentrated during intralysosomal storage – according to several independent estimates, the intralysosomal concentration of beta-amyloid in cells in vitro is up to 100 times higher than the concentration of extracellular peptide [32,33]. If the extracellular concentration is 1 nM, the intralysosomal concentration can reach 100 nM. According to the extrapolation shown in Figure 7, C, at this concentration, the half-time of aggregation is only five days. Considering that intralyso so mal peptide is significantly concentrated and is stored for several days, endocytosis and following lyso somal storage of beta-amyloid provide appropriate conditions for the formation of aggregation seeds.
Aggregated amyloid is resistant to proteolysis [34,35]. Importantly, if lysosomes cannot digest their content, lysosomal cargo can either be sequestered (hence intracellular accumulation of aggregated beta-amyloid [45]) or exocytosed. In the latter case, formed aggregation seeds will be moved from the cells to the extracellular fluid. This explains why the presence of cells promotes the aggregation of exogenous beta-amyloid [33]. Importantly, this results in the appearance of already aggregated amyloid seeds, which could be large enough to be insoluble and remain stuck in the extracellular space. The described sequence of events is illustrated in the schematic in Figure 10.
The concept that aggregation seeds can absorb cathepsins, while beta-amyloid is stored intralysosomally, fits the well-described phenomenon that (extracellular) senile plaques can be effectively stained for lysosomal (intracellular) proteases [36,37].

3.2. Is cellular uptake of beta-amyloid relevant to its cytotoxicity?
As it was shown above, the diagnosis of Alzheimer’s disease is associated with an increase in the cellular uptake of beta-amyloid. The progression of AD is defined by neuronal death. Considering that beta-amyloid is cytotoxic, is it reasonable to assume that the cellular uptake of beta-amyloid is linked to cell death?
Mechanisms of cytotoxicity induced by exposure to beta-amyloid are still debated. Not surprisingly, various laboratories tend to stress the importance of pheno mena that they discovered. At the molecular level, most frequently are cited loss of membrane potential, intra cellular ion disturbances in general, or disruption of cellular calcium balance, which are caused either by effects on membrane proteins/receptors or by pore formation in cell membranes. At the cellular level, the exposure of cells in vitro to beta-amyloid can promote apoptosis, cause synaptic loss, and disrupt the cyto skeleton and axonal/dendrite transport – the pheno mena that can also be observed in neurons in the brains of patients with AD. Also, Aβ-induced disbalance of calcium homeostasis by itself can lead to calcium over load, which in turn damages mitochondria and activa tes apoptosis. Finally, treating cells in vitro with beta-amyloid leads to lysosomal permeabilization, which corresponds to autophagy failure, which by itself is one of the most prominent and well-established hallmarks of AD.
Almost all observed molecular disturbances asso ciated with exposure to beta-amyloid can cause each other. The variety of these consequences and the fact of their intertwining makes it virtually impossible to establish the primary molecular insult just by looking at the list. Without starting from some reasonable origina tion point in the logic, finding the pathophysiological pathway of amyloid-induced cytotoxicity could be impossible.
For that reason, the first question is – can the action of beta-amyloid on the plasma membrane be the primary molecular event in the amyloid-induced cytotoxicity? In fact, amyloid cytotoxicity can be observed when the peptide is added to the cultured cells in vitro. In these conditions, plasma membrane is the first organelle exposed to the peptide. Due to Aβ size and charges, the peptide cannot evoke any intracellular reactions leading to cell death by crossing the membrane by diffusion.
If the action on the plasma membrane is considered, the first logical possibility is the action on the lipid bilayer, while acting on some membrane proteins is the alternative. Currently, there is no known to us interaction between beta-amyloid and any membrane protein that can initiate a pathway leading to cell death. We consider the interaction with receptors at the plasma membrane as an unlikely molecular mechanism leading to cell death for another reason – the consequences of cellular exposure to beta-amyloid are developing very slowly. Cell death occurs after prolonged exposure, at least multiple hours, even in experiments involving high peptide concentrations. Intracellular ion disturbances also do not develop immediately; there is an apparent delay of at least several minutes. Importantly, proper techniques of ion concentration measurement (which avoid buffering properties of intracellular probes) reveal that intra cellular disturbances occur in waves – deflection from the equilibrium is followed by recovery, which is often complete. This phenomenon resembles repeated opening-closing of membrane ion channels, the formation of which by beta-amyloid molecules was described electrophysiologically. However, as we reported in [38], the observed phenomena are incompatible with the properties of amyloid membrane channels if they are located in the plasma membrane.
It can be suggested that the primary molecular mechanism of amyloid toxicity could be non-specific disruptions of lipid bilayer structure by peptides that have both non-polar and electrostatically charged sequences. However, such disruptions occur only in micromolar concentrations. In contrast, extracellular concentrations of beta-amyloid are in the nanomolar range, and even intralysosomal concentrations should be no more than 0.1 µM. Therefore, direct destruction of the barrier function of cellular membranes in vivo is unlikely.
Although beta-amyloid cannot cross the plasma membrane by diffusion, it is actively accumulated by cells through endocytosis. Endosomes are merged with lysosomes, and virtually all internalized beta-amyloid is co-labelled with lysosomal markers [32]. Before the mechanisms of cytotoxicity of endocytosed beta-amyloid will be discussed in the following sections, it is critical to state that the exposure of cells in vitro to extracellular beta-amyloid results in permeabilization of amyloid-laden lysosomes [39,40]. Lysosomal perme abilization occurs before the damage to plasma memb ranes is observed. Such a robust consequence of beta-amyloid uptake allows for a hypothesis that there is a molecular cytotoxicity mechanism linking the interna lization of amyloid to cell death, and, as a corollary, the rate of uptake is one of the major factors defining the rate of amyloid-induced neurodegeneration.

3.3. The dependence of both toxicity and aggregation of beta-amyloid on cellular uptake: relevance to clinical data on amyloid biomarkers
As was discussed in two previous sections, both toxicity and aggregation of beta-amyloid depend on this peptide’s cellular uptake. Such dependence could be the reason for the correlation between the probability of AD diagnosis and the density of amyloid deposits in the brains of patients. Such reasoning was tested by comparing clinical data with the output of a mathematical model, which considers the relationship between beta-amyloid turnover and cytotoxicity (and linking the latter to the AD diagnosis) [41].
We extended the model described above (Figure 6) by introducing the connection between Aβ endocytosis and the accumulation of aggregated beta-amyloid in the brain (Figure 11, A). In this case, the amount of aggregated beta-amyloid increases by two mechanisms – exocytosis of aggregated intracellular beta-amyloid and growth of existing oligo/polymers by aggregating more soluble peptide. Under these assumptions, the system of ordinary differential equations describing the dynamics of beta-amyloid concentrations in the ISF and CSF over time can be written as shown in Figure 11, B. Similarly to the less complex model, the system is in relative equilibrium at the time scale of measurements. Hence, the derivatives of the concentrations of Aβ42 in the interstitial fluid and the density of amyloid aggregates measured by PET are equal to zero.
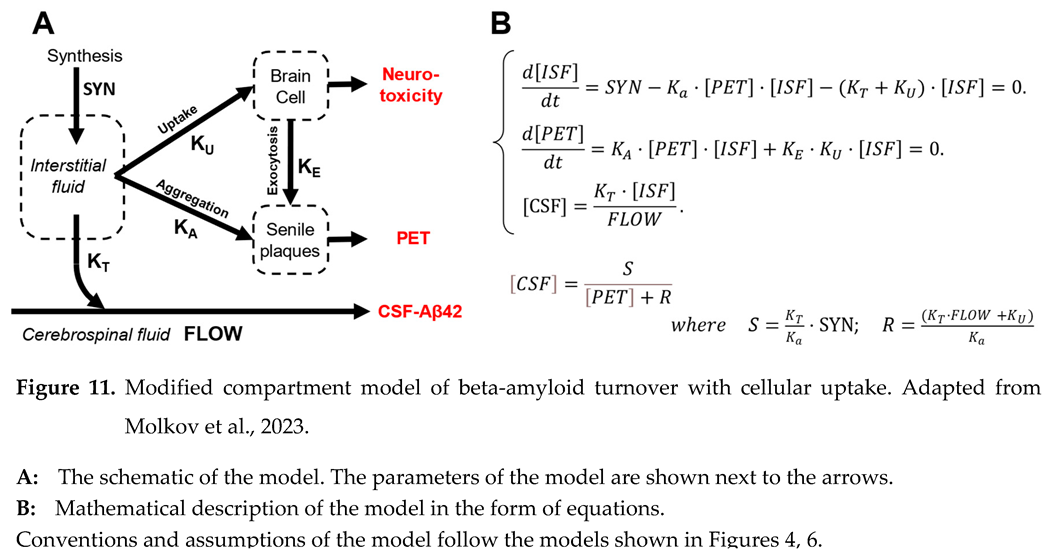
The power of cytotoxic insult (current toxicity) is proportional to the uptake intensity (the product of the uptake rate and concentration of Aβ42 in the interstitial fluid). Neural damage accumulates over time and depends on accumulated toxicity, which is the integral of current toxicity over the duration of illness. We assumed that a subject will be diagnosed with AD if their accumulated toxicity exceeds some threshold, which is a characteristic of an individual. We further assumed that the probability distribution of the toxicity thresholds in the population is normal with a specific mean and variance.
Fitting procedures, described in the original manuscript [41], allowed us to reproduce the experimental data at a 95% confidence level. Figure 12, A shows the probability of an AD diagnosis, calculated using these parameter values, as a function of CSF-Aβ42 and amyloid load (PET signal).
First, the model allows for interpreting the shape of biomarker distribution in the human population (Figure 12, B). The region of possible PET and CSF-Aβ42 values has an upper boundary representing a zero-uptake rate (). The lower boundary is defined by the highest biologically present rate of uptake . At the highest rate, cellular uptake from the interstitial fluid is higher than the aggregation-mediated removal of soluble Aβ42, so the bottom boundary looks inde pen dent on amyloid load (solid green line is almost parallel to X-axis). The right boundary is defined either by dramatic neurodegeneration with the probability of AD approaching one or by the length of the disease, which is needed to generate an amyloid load above 150 CL.
Notably, the model interprets both paradoxes related to amyloid biomarkers described above. The dependence of both amyloid aggregation and amyloid-induced cytotoxicity on the cellular uptake of the peptide results in a strong correlation between the probability of AD diagnosis (the sequela of neurotoxicity) and accumulation of amyloid aggregates. This phenomenon is reproduced by the model not only qualitatively but also quantitatively (Figure 12, C)
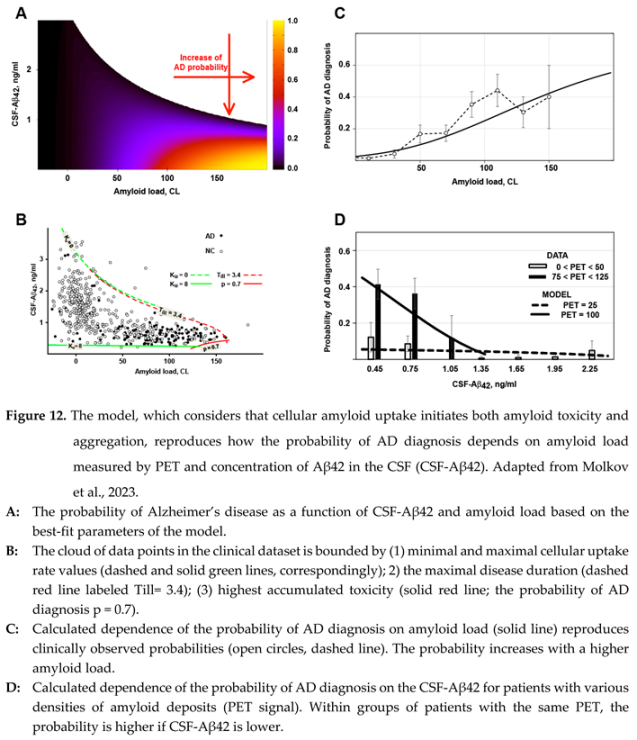
On the other hand, increased cellular uptake of Aβ42 results in the decrease of the interstitial concentration of the peptide and, consequently, in the reduction of CSF-Aβ42. Even though the interstitial concentration is lower, a higher uptake rate still leads to increased total amyloid uptake because the decrease in interstitial concentration is lower than the increase in uptake rate, so the product of the two parameters is higher. Consequently, if all other parameters (including the density of aggregated amyloid) are equal, the increased cellular uptake rate results in increased accumulated uptake and higher accumulated cytotoxicity. This explains why lower CSF-Aβ42, which is the consequence of increased cellular uptake, is associated with increased neurodegeneration, mani festing in a higher probability of AD diagnosis. In terms of multivariate regression analysis, it means that the negative correlation between CSF-Aβ42 and the proba bility of AD diagnosis has an aggregation-independent component, which is mediated by increased cellular uptake of beta-amyloid. The model also reproduces this phenomenon quantitatively (Figure 12, D).
One of the essential observations coming from the fitting of clinical data is that the threshold for accumulated neurotoxicity in the patient, which is needed to be reached for being diagnosed with AD, has very wide variability in the population: the mean value of the threshold is 170 CL with the standard deviation of 87 CL. Such variability explains why many subjects with extremely high amyloid load (and corresponding high accumulated amyloid uptake and resulting accumulated toxicity) retain normal cognitive functions. These patients probably have a high threshold of neurotoxicity for AD. In other words, these patients have high resistance to amyloid-induced cytotoxicity, so even high levels of beta-amyloid cellular uptake do not result in significant neurodegeneration.

4. Molecular and cellular mechanisms of beta-amyloid-induced cytotoxicity
4.1. Lysosomal permeabilization by beta-amyloid – the mystery of a reality
As we are looking for a mechanism of amyloid-induced cytotoxicity related to cellular uptake, it is reasonable to skip any hypotheses focusing on plasma membrane damage as the primary molecular process. Considering that it is well-known and universally accepted that beta-amyloid is endocytosed, and endosomes merge with lysosomes, it is reasonable to check if anything is known about the status of lysosomes in neurons of patients with AD. Unsurprisingly, it is widely accepted that lysosomal dysfunction is one of the hallmarks of AD [42,43,44,45,46,47], but the correlation does not explain if this phenomenon is the reason or the consequence of the disease.
Among many actions of beta-amyloid on cells, amyloid-induced lysosomal permeabilization [39,40] stands out as a mysteriously powerful phenomenon.
The phenomenon itself can be observed visually. Some fluorescent dyes, such as Lucifer Yellow (MW 444, Figure 13, A), are water-soluble and not membrane-permeable due to the presence of multiple charged moieties in their structure. For that reason, if they are taken by cells together with extracellular fluid – are endocytosed – they cannot leak to the cytoplasm. If cells have an intense endocytic process and are in cubated in the medium containing Lucifer Yellow, they accumulate endosomes filled with the liquid containing this fluorescent compound. After such cells are washed from the fluorescent dye, the dye remains intracellularly and can be visualized as a punctate pattern (Figure 13, B) [39,40]. Due to the merging of endosomes with lyso somes, the fluorescence of Lucifer Yellow is co-labelled with lysosomal markers [32]. The brightness of fluorescent puncta is contrasted by a concentrating of intralysosomal content – up to 100-fold [32].
However, in cells exposed to Aβ42, the fluorescence of intracellular Lucifer Yellow becomes diffuse and covers all areas of the cell [39,40], even though some bright dots can still be observed (Figure 13, C) [40]. The fact that lysosomes become permeabilized is confirmed by the appearance in the cytoplasm of macromolecules, such as the lysosomal enzyme β-hexosaminidase (M.W. 150 kDa) [39].
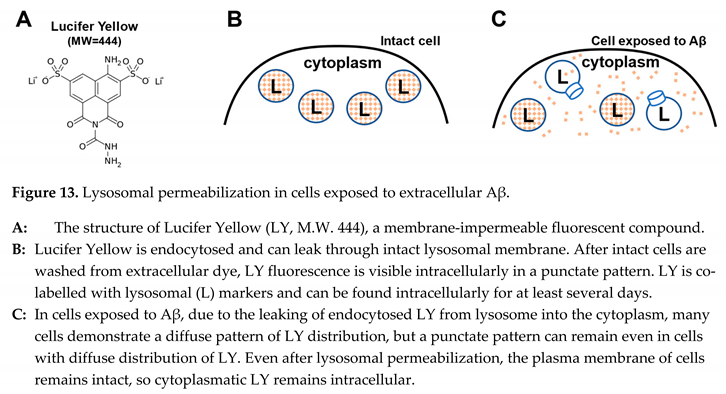
It can be suggested that lysosomes become perme abilized due to the cell’s death. If that is the case, the plasma membrane should become permeable, so Lucifer Yellow, which leaked from the lysosomes, would leak from the cytoplasm, too. And no diffuse intracellular fluorescence would be observed. The authors of the initial report addressed this possibility by showing that dying cells change their shape, while lysosomal permeabilization was observed in cells that retained the typical shape of a living cell [39].
The fact that in cells exposed to extracellular (!) Aβ42, lysosomal permeabilization was observed while the plasma membrane still retains its barrier function, was probably looking like an artifact. Even though the phenomenon was described independently by various laboratories, it did not attract the overwhelming attention of researchers. One of the reasons for such “ignorance” could be the mystery of how adding extracellular peptide to cells in culture can cause profound damage to the intracellular membranes without affecting the plasma membrane.
However, this phenomenon is clearly relevant to a vast swath of literature focused on the importance of lysosomal permeabilization to the initialization of apoptosis [48]. Cathepsins were shown to activate the apo ptotic pathway. Permeabilization of lysosomal membranes to macromolecules as large as β-hexos aminidase (M.W. 150 kDa) should be accompanied by the leakage of cathepsins (most of which have M.W. less than 50kDa) to the cytoplasm. In fact, cathepsin D was visualized in the cytoplasm of cells treated with beta-amyloid [39]. Cathepsins can directly activate caspases, initiating apoptosis – programmed cell death. Importantly, increased apoptotic activity in the brain is well-known to be associated with Alzheimer’s disease. The literature on the role of lysosomal permeabilization in apoptosis is still non-conclusive on the potential mechanisms of such permeabilization, which will be discussed in the next section.
Importantly, lysosomal permeabilization is relevant not only to the turnover of macromolecules but also to ion metabolism. Since early studies of Aβ-mediated cytotoxicity, it was known that exposure of cells to beta-amyloid results in the accumulation of intracellular cal cium. It was demonstrated that calcium accumulation is linked to oligomeric forms of beta-amyloid, while monomeric and fibrillated beta-amyloid do not affect intracellular calcium [5]. The major problem with the interpretation of measurements of intracellular calcium is the buffering capacity of intracellular calcium-sensitive probes such as Fura-2 [49]. Due to high intracellular concentration of sensing probe, calcium changes are frequently smoothed, so fine dynamics remain non-visible. However, with proper precautions, fast responses can be observed.
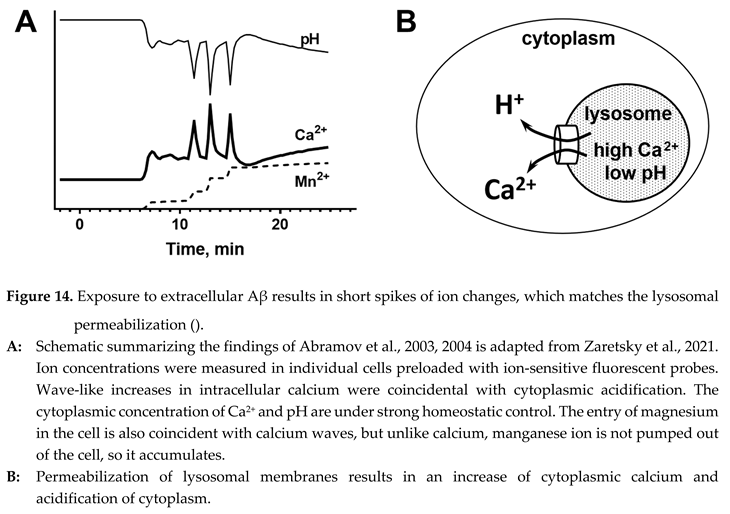
Specifically, Abramov et al. demonstrated that cal cium accumulation occurs not as a monotonic increase but in short waves [50,51]. In these studies, which were performed on individual cells in culture, the concent rations of several ions (sodium, potassium, calcium, and protons) were recorded by fluorescent microscopy. Several critical findings can be summarized as follows (see also the schematic in Figure 14, A).
- Intracellular concentrations of multiple ions change as synchronous waves.
- Waves of cytoplasmic acidification mirror calcium waves without any temporal offset.
- Waves are short, and the cytoplasmic con cen trations are typically reversed to pre-wave level.
- Ion responses to Aβ exposure are initiated with a noticeable delay of at least several minutes.
The first question is if the permeabilization of the plasma membrane can explain the observed changes. It appears that the changes in cytoplasmic concent ra tions of sodium and potassium were opposite to the potential effects of permeabilization of plasma membranes [50,52]. Also, it is difficult to explain why the changes in plasma membrane permeability result in wave-like oscillations of intracellular concentrations of various ions [52].
In contrast, the observations fit the hypothesis that the changes in intracellular ion concentrations are explained by lysosomal permeabilization (Figure 14, B). Most importantly, lysosomes are known to be a calcium storage and have acidic content. Permeabilization of lysosomes would release both calcium and protons into the cytoplasm and explain the synchronous and coun ter-directional changes of cytoplasmic calcium and pH. Short-lived fluctuations would be logical because a small volume of lysosomes would restrict the amplitude of consequences of each permeabilization event. After the content of the lysosome is leaked completely, the recovery will be swift due to the presence of homeo sta tic mechanisms controlling intracellular pH and calcium concentrations. Correspondingly, for ions that are en docytosed and reach lysosomes but are not controlled by homeostatic mechanisms, such as manganese, the intracellular concentration will increase synchronously to the rising front of calcium waves (as shown in Figure 14, A) but would not return to baseline [51].
The presence of multiple waves can be reasonably connected to the permeabilization of numerous lysosomes, which occurs at different times. Critically, there should be a delay before any ion wave occurs because beta-amyloid must be endocytosed and reach lysosomes. Finally, lysosomal permeabilization explains why all these changes can be observed without plasma membrane damage.
Now, we come to the critical question – how does the permeabilization occur? This question was asked before. In the study’s design, Abramov et al. hypothesized that intracellular ion disturbances arise from the appearance of non-selective membrane ion channels formed by beta-amyloid [51]. The formation of such channels was indisputably described by multiple laboratories starting in 1993 [53,54,55,56,57,58,59,60,61,62,63]. In short, adding beta-amyloid to lipid membranes results in forming a non-selective pore, which can pass virtually any cation, including sodium, potassium, and calcium. With previously demonstrated amyloid-induced intracellular calcium accumulation, channel formation was a reasonable hypothesis of the molecular mechanism responsible for amyloid-induced cytotoxicity [60]. It was unsurprising that amyloid channel theory attracted attention as a potential AD theory.
Correspondingly, in elegant microscopic studies, Abramov et al. successfully demonstrate not only wave-formed intracellular synchronous fluctuations of multiple ions but also that each wave originates from a focal point, spreads through the cell, and fades on its way. The decrease of the amplitude of the wave of ion concentration with the distance contrasts with the propagation of action potential, which involves multiple membrane ion channels. However, it matches the concept of a single amyloid membrane channel. Abramov et al. assumed that such a channel is formed in the plasma membrane because the cell is exposed to extracellular peptide, which cannot cross the plasma membrane due to the size and presence of electrostatic charges [50,51].
There is no doubt that the formation of even a single giant amyloid channel in a plasma membrane would result in extremely fast dissipation of any trans memb rane gradient [63], while amyloid-induced cell death develops over multiple hours even though apoptotic markers become increased much earlier [64]. This discrepancy can explain why researchers concentrated on amyloid channels as the mechanism mediating calcium overload [65], which would initiate a relatively slow pathway leading to cell death. However, the channels are non-selective and pass sodium and potassium with an effectiveness similar to their ability to transport calcium. It is not apparent why calcium overload should be viewed as a significant problem for cells if gradients of all other ions, including sodium and potassium, are disappearing.
Prediction of fast equilibration of transmembrane gradients was based on electrophysiological properties of amyloid channels [63]. However, the recordings of intracellular ion concentrations did not reveal complete equilibration of intracellular and extracellular concen trations of ions, but were counterintuitive – the changes of sodium and potassium were opposite to the direction of changes which can be expected from perme abilization of the plasma membrane [50,51]. Finally, it is difficult to explain why, in the thirty years since the discovery of amyloid channels, no convincing electrophysiological data supports the possibility of channel formation in plasma membranes of living cells.
Our hypothesis that lysosomal permeabilization is the culprit in ion disturbances caused by Aβ exposure allows for interpreting all these contradictions [52]. However, the question “How are lysosomal membranes permeabilized?” remains. Importantly, this question requires an answer concerning both the transfer of small cations and how lysosomes of Aβ-exposed cells can leak macromolecules.

4.2. Beta-amyloid oligomers permeabilize membranes by forming channels
Non-specific damage to lipid membranes induced by beta-amyloid (absorption of a large amphiphilic compound) can be explained by a detergent-like property of the peptide, which destroys the barrier function of a lipid membrane [66,67]. However, this can be observed only in extremely high peptide concen trations (micromolar concentrations), which were never observed in any (patho)physiological condition.
This brings us back to the question of membrane channel formation. Can such a channel be formed in a lysosome? Can such channels explain the perme abilization to macromolecules? Why is membrane permeabilization observed only in lysosomes (or was it missed in other cellular membranes)?
First, it is critical to understand what was known about membrane channel formation until recently. The experimental design typically used to study channels included studying electric currents evoked by various potentials applied across the lipid membrane, which is exposed to Aβ solutions. The most important aspect of channel formation in such conditions is that despite the presence of trillions of amyloid molecules, the technique records single channels. Such rare events would be difficult to use in the interpretation of cell death in cell cultures that have thousands of cells. It is evident that to die, each cell needs to receive at least one channel. Also, the channels are extremely large, with the conductance measured in hundreds of picosiemens compared with typical membrane ion channels, which have the conductance measured in tens of picosiemens. If such a channel is formed in a cell’s plasma membrane, it will kill the cells quickly, while Aβ-exposed cells in culture die over many hours.
To study the quantitative aspect of membrane channel formation, we developed the flowmetric techni que to detect liposomes (artificial lipid vesicles), which were permeabilized [68]. The liposomes are extruded to contain a membrane-impermeable calcium-sensing fluorescent probe (e.g., Fluo-3) and have a calcium chelator. The size of liposomes is less than the wave length of UV light, so scattering cannot be used to identify vesicles in the flow. For that reason, we needed to add a fluorescent membrane probe (e.g., DiD) to identify the liposomes when they passed the interrogation point of the laser during measurements. The intensity of DiD fluorescence reflects the amount of membrane material in the recorded vesicle. Because calcium does not cross intact lipid membranes, liposomes do not have significant fluorescence of the calcium-sensing probe even after calcium is added to the extravesicular solution (Figure 15, A). However, if some liposomes become permeabilized, they become fluorescent (Figure 15, B). The reason for the distribution was discussed in the original manuscript [68]. The technique was validated by experiments using various concentrations of extravesicular calcium, the addition of calcium ionophore, and negative controls (in the presence of a calcium chelator and/or in the absence of a calcium-sensing probe). It is essential that within relatively low concentrations of channel-forming fragments, the number of permeabilized liposomes can serve as a measure of the number of channel-forming units in the sample.
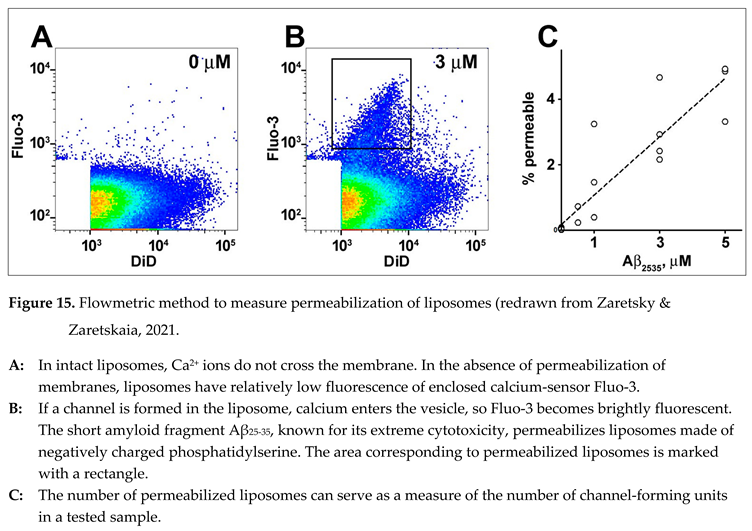
- Thousands of liposomes are permeabilized by short fragment Aβ25-35, known for its neurotoxicity, in a fashion that matches the formation of membrane channels.
- Aβ25-35 permeabilizes the membranes made of phosphatidylserine (acidic, negatively charged phospholipid) but not in the membranes made of phosphatidylcholine (neutral phospholipid, which does not carry a net electric charge).
- No permeabilization was observed by Aβ42 (full-length beta-amyloid, which is relevant to senile plaque formation and is the isoform that correlates with the progression of AD).
- Other short fragments, Aβ22-35 and Aβ31-35, do not permeabilize lipid membranes.
Our data match the observations from other peer-reviewed publications:
- In planar lipid bilayers, adding Aβ25-35, but not full-length beta-amyloid, resulted in effective channel formation [54]. In studies on lipid bilayers, full-length peptides usually produced only single channels [62,63]. In some studies using full-length peptides, the formation of channels was unreproducible [67].
- Permeabilization was observed only in nega tively charged membranes [70].
Considering this match, we formulated a concept of how amyloid channels are formed:
- Only some amyloid fragments, but not full-length beta-amyloid, can form membrane channels.
- The formation of single channels in experiments on planar lipid bilayers using full-length beta-amyloid could result from contamination with fragments. Synthetic peptides usually have a purity of less than 99%, so it is statistically possible that there was a sufficient concentration of channel-forming fragments. The dependence of results on the contaminants can explain why some laboratories could not reproduce the data.
- The fragment that can effectively form memb rane channels (Aβ25-35) is mostly lipophilic and carries a single positive charge, while the nega tively charged fragment cannot create channels.
- Positively charged fragments can form channels only in negatively charged membranes. Electrostatic inter action between the membrane and channel-forming unit is most likely needed to incorporate into the membrane.
Within this concept of membrane permeabilization by amyloid channels, one of the possible pathways to the formation of membrane channels in a cell can be summarized in the schematic (Figure 16). Freshly synthe sized beta-amyloid does not have any conformation. Intralysosomally, it is cut into fragments, degraded into amino acids, and utilized by the cell as nutrients. However, some fragments, which carry a positive charge and can form membrane channels, aggregate in the beta-sheet. The coaster-like structure, which consists of several peptide molecules, can roll in a barrel shape by creating a beta-sheet between peptide chains at the opposite sides of the coaster. Incorpo ra tion of a positively charged barrel into the negatively charged membrane results in the formation of memb rane pore (channel). Since the linear size of the coaster that rolls into the barrel is large, the pore is giant by the standards of membrane ion channels and is not selective.
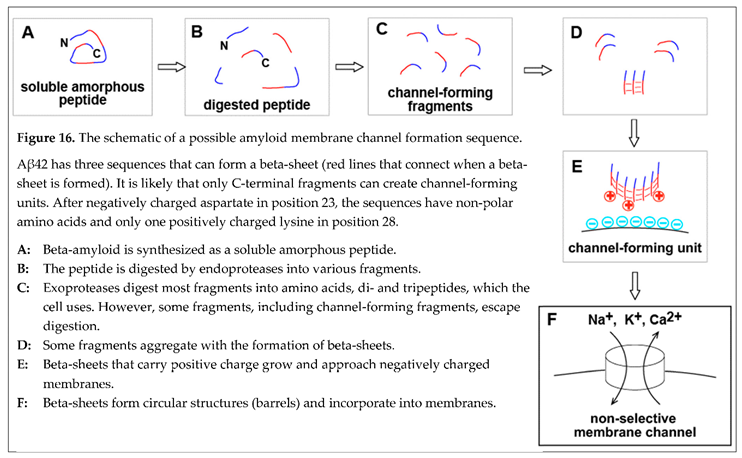
Considering that this is not the only possible way to form an amyloid channel in the cell membrane is essen tial. One of the alternatives includes the absorption of full-length beta-amyloid on the membrane, which could significantly affect the proteolysis (for example, by changing the availability of sensitive to cleavage peptide bonds). In this case, membranes would also promote the aggregation of protected fragments. Also, aggregation can occur first. Peptide sequences, which are needed for channel formation, would be forming beta-sheet among the first. Therefore, these sequences would become more protected from proteolysis, while the rest of the beta-amyloid molecule will be cut from the oligomeric coaster-like structure. The sequence of events in vitro and in vivo can be different, and this will be critical for finding ways to control this process pharmacologically.
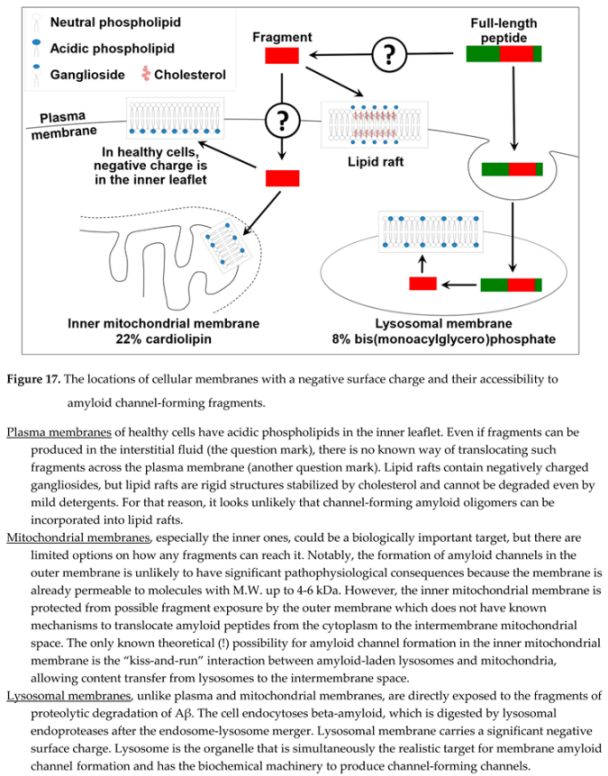
Assuming that channel formation is relevant to amyloid cytotoxicity, it would be logical to assess where amyloid channel-forming fragments can be produced by proteolysis and have access to cellular membranes with a negative charge. Three cellular organelles have membranes with negative surface charge: plasma membranes, mitochondrial membranes, and lysosomal membranes (Figure 17). Amyloid fragments are not produced near plasma membranes and mitochondrial membranes (except for the delivery from lysosomes to the intermembrane space of mitochondria by the kiss-and-run mechanism [71,72]). However, lysosomes are organelles that produce channel-forming fragments, while lysosomal membranes have the properties needed to incorporate amyloid membrane channels.
How does lysosomal permeabilization lead to cell death? Ion disturbances are local and are limited by the small size of the lysosome. Homeostatic mechanisms quickly recover ion gradients. One of the key charac teristics of lysosomes is the acidification of content. Low pH is critical for the optimal function of acidic proteases. Dysfunction of lysosomes due to the dissipation of pH gradient at the lysosomal membrane could be one of the mechanisms explaining lysosomal dysfunction, which is characteristic of the brains of AD patients. Can amyloid channels dissipate the pH gradient? To our knowledge, the only experiments demonstrating such a possibility are those of Abramov et al., which showed pH waves synchronous with waves of intracellular calcium [50,51]. Considering that the channels are extremely large and non-selective and pass all tested cations with a single charge [62], while protons are the smallest ions, it is reasonable to predict that amyloid membrane channels would effectively dissipate the pH gradient. However, direct evidence is not available yet.
Importantly, lysosomal dysfunction is observed in living cells, so even if it is one of the pathophysiological processes that underlie the pathobiology of AD, most likely, it is not the primary mechanism of cell death itself. For a long time, the destabilization of lysosomal membranes was hypothesized as a mechanism that allows for the leakage of lysosomal proteases to the cytoplasm [73,74]. It was demonstrated that once lyso somal cathepsins appear in the cytoplasm, they can activate the apoptotic pathway of cell death through direct action on caspases. The cytoplasm contains protease inhibitors, which protect the cell from such development. Still, if the protection becomes insuffi cient, apoptosis (also known as programmed cell death) would be a reasonable mechanism of neuronal loss. The increased levels of apoptotic markers are one of the well-described features of AD brains, and apoptosis is activated in cells exposed to beta-amyloid [75]. The leakage of lysosomal cathepsins requires the permeabilization of lysosomal membranes to macro molecules with M.W. exceeding 50 kDa. While “destabilization of lysosomal membranes” was consi dered for decades, there was no confirmed molecular mechanism that beta-amyloid could initiate through a known pathway. Can the formation of amyloid memb rane channels mediate this destruction of the barrier function of lysosomal membrane and be the mecha nism of “destabilization of lysosomal membranes”?
No direct data supports either positive or negative answers to this question. However, critical estimates can be made based on the electrophysiological data [53]. As we stated, membrane channels are formed by short amyloid fragments such as Aβ25-35. Therefore, the most relevant dataset can be found in the electrophysiological data obtained using this fragment [53,54]. Typical membrane ion channels have conductance in the range of tens of picosiemens and usually have relatively narrow variability. In contrast, amyloid membrane channels have extremely wide conductance variability, which could exceed 1000 picosiemens [53]. Arispe et al. used the term “giant” in the original manuscript to underscore the extreme conductance values [63]. The channel size distribution depends on the solution’s acidity and salinity [53].
Without direct data on the permeability of amyloid channels to macromolecules, we attempted to estimate the size of molecules that can be transferred through the largest channels. Our first step was to convert the conductance to channel properties relevant to the topic of interest – membrane permeabilization [38]. In the modeling study, we described a membrane channel as a cylinder with a length equal to the thickness of a phospholipid bilayer, filled with saline, and located in a non-conductive membrane. Calculating the diameter of a model channel from the conductance value is possible. Next, we considered that the largest sphere, which a channel can pass, has the same diameter as the channel. It is possible to calculate the molecular weight of an object that has the density of a globular protein (1.37 g/ml) and is a sphere with a particular diameter. In this model, a pore with the conductance of one nanosiemens would allow the passage of a protein with an M.W. of approximately 10 kDa (Figure 18, A). Considering that we assumed that the calculation estimates the molecular weight of the largest protein that can pass the channel, we called this estimate the molecular weight cut-off (MWCO).
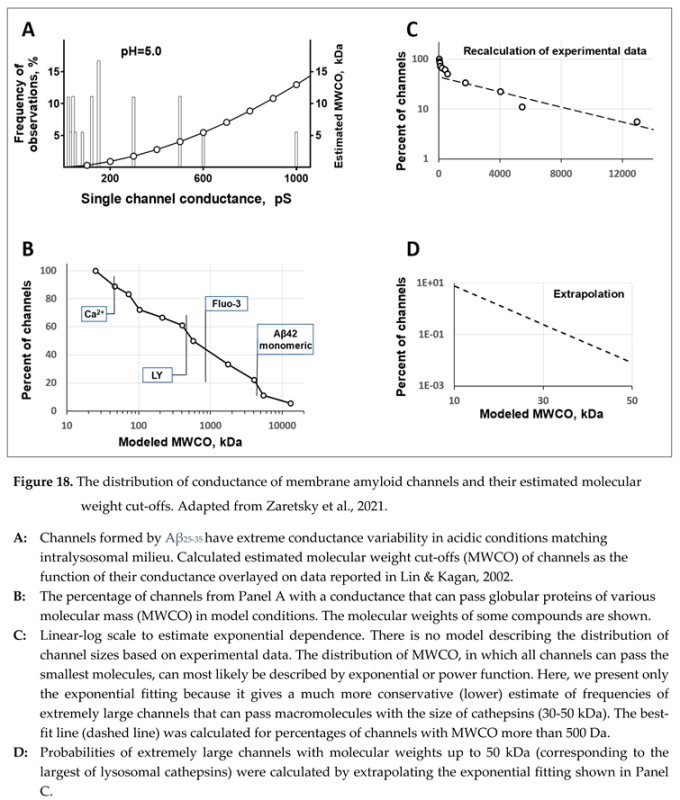
Even a glance at the distribution of amyloid channel conductance gives the impression that while gigantic channels are less frequent than relatively small ones (Figure 18, B), the probability is not approaching zero fast and does not allow to conclude that there is a clear limit of channel size (Figure 18, C). The availability of the distri bution allowed us to extrapolate the probabilities of channels of various conductances in the range excee ding experimental observations [38]. The extrapolation results are sensitive to the choice of the function. Still, even a conservative estimate using the exponential function shows that even rare, gigantic channels that can pass proteins of interest – such as lysosomal cathepsins, which mostly have a molecular weight in the range of 30-50 kDa – can be formed with a non-zero probability (Figure 18, D). The results of the modeling do not leave any doubt that amyloid membrane channels can mediate the leakage of lysosomal proteases to the cytoplasm.

5. Independent clinical evidence of increased beta-amyloid uptake in AD
While there are no ready-to-use techniques to measure cellular amyloid uptake in the brain, the stable isotope labeling kinetic (SILK) technique could be one of the platforms [17], even though it is labor-intensive and invasive. The technique uses the ability of high-performance liquid chromatography (HPLC) combined with mass spectrometry (MS) to measure the concent rations of beta-amyloid isoforms. In the case of Aβ studies, the patient is infused with 13C-labelled leucine. While the heavier isotope of carbon naturally exists and represents approximately 1% of total carbon in the environment, the chance of random appearance of leucine molecule, which contains all six carbon atoms as 13C, is negligible (0.016 or 10-12). Mass-spectrometry can distinguish leucine molecules with six 13C atoms from leucine molecules with only 12C atoms or just one 13C atom. Therefore, if a patient is infused with 13C6-leucine, any Aβ molecule containing this heavy amino acid was synthesized after the infusion started. In this method, HPLC separates the compounds of interest (for example, separates Aβ42 from Aβ40 and Aβ38) while following mass-spectrometry measures separately the total concentration of the peptide of interest and how much of that total concentration is represented by the freshly synthesized peptide.
For SILK studies, it is advantageous that the metabolism of leucine is high, so the equilibrium between labeled and unlabeled amino acids is established within the first hour after the initial bolus, followed by continuous infusion. Labeled leucine is infused for several hours, which allows observing the Aβ accumulation phase, and after the infusion is stopped, the washout of labeled proteins can be monitored. Blood and cerebrospinal fluid are repeatedly collected from the patient [17].
The analysis of beta-amyloid concentrations kinetics in the CSF is performed using a multi-compartment kinetic model described by differential equations [15], which is similar to our approach. However, SILK allows to study Aβ turnover in a single patient, while we needed the dataset collected from a cohort of patients.
There are two significant observations made by the SILK technique, which are not just relevant but critically important for the proposed hypothesis. First, the model has a rate of intrabrain irreversible removal v42, which is calculated explicitly [17]. This rate is significantly increased (approximately two-fold) in patients with AD, which supports our hypothesis of increased cellular uptake, which ends with proteolytic digestion of Aβ. Notably, the increase was found in both amyloid-positive and amyloid-negative patients, which makes doubtful the interpretation that demonstrated by the SILK technique increased intrabrain irreversible removal is only due to the aggregation on existing amyloid seeds.
Second, the authors discovered that the model cannot properly reproduce data unless an additional compartment is introduced into the schematic – the compartment with a reversible exchange. Essentially, in patients with AD, the model needs to have some intrabrain compartment that takes the peptide from the ISF but returns the peptide after some time. The modeling itself does not allow us to say what this compartment represents. Still, this observation fits the concept of cellular uptake and temporary intracellular Aβ storage, which is linked to AD pathophysiology.
Common sense guides that cellular uptake would lead to both irreversible loss and reversible exchange. The portion of the peptide that is degraded represents an irreversible loss. However, if some content is returned to the ISF, it will mean a reversible exchange.
In healthy subjects, the portion of Aβ42 consumed by cells is relatively small. The endocytosed peptide is probably mostly degraded, while the percentage of endocytosed peptide returned to the ISF is small enough that its addition to the model cannot be justified statistically. In contrast, in patients with AD, the irreversible removal is higher, which means that more of the peptide is taken by cells and digested. But, even more critical, the endocytosed peptide is stored intracellularly in amounts that are significant enough to be noticeable to justify the addition of an extra compartment to the model. In the original manuscript [17], authors suggested that aggregated beta-amyloid could be the physical representation of this compartment due to reversible aggregation of the peptide at the surface of senile plaques. However, reversible exchange was observed in patients who were amyloid-negative. We suggest that the concept of cellular uptake and storage is a more biologically reasonable interpretation of the same clinical observations.
Within the amyloid degradation toxicity hypothesis framework, the more prolonged intracellular presence of beta-amyloid would promote both the accumulation of extracellular deposits and neurodegeneration. As was discussed in the section on how aggregation seeds are formed, more extended storage promotes the formation of aggregation seeds. Considering that this compartment represents “reversible exchange, " the content returns to the ISF. It means that beta-amyloid, including its oligomerized form, is returned to the ISF. Large particles can become the core of future senile plaques. From this perspective, the appearance of a significant reversible exchange pool could be the initial point leading to the accumulation of deposits. Also, considering that beta-amyloid is not quickly di ges ted and remains intralysosomally for a long time, prolonged intralysosomal presence of peptide provides more opportunities for endopeptidases to produce channel-forming fragments. More intralysosomal beta-amyloid would lead to more chances of lysosomal permeabilization and cytotoxicity.
Also, longer intracellular storage could result in more digestion of the peptide even if the activity of enzymes remains the same. The result of such prolongation of intracellular storage would be increased irreversible loss of peptide, which was also observed.
Potential pathophysiological consequences of slow lysosomal turnover bring the chicken or the egg causality dilemma. Is lysosomal dysfunction the consequence of lysosomal damage induced by endocytosed beta-amyloid? Or is it the initial slowdown of lysosomal turnover that leads to the cytotoxicity and accumulation of extracellular amyloid deposits?

6. Cornerstones of the amyloid degradation hypothesis of Alzheimer’s disease
The amyloid degradation toxicity hypothesis of Alzhei mer’s disease is based on multiple experimental disco veries or conclusions made from these discoveries.
- 1)
- Amyloid membrane channels are formed by fragments of Aβ (such as Aβ25-35 but most likely other fragments as well), not full-length beta-amyloid (such as Aβ42 or Aβ40).
- 2)
- Incorporating amyloid channel-forming oligomers into lipid membranes requires that the membranes have a negative surface charge.
- 3)
- Lysosomal membranes are one of three cellular membranes with a negative surface charge (along with the inner leaflet of plasma membrane and mitochondrial membranes).
- 4)
- Lysosomes provide both the biochemical machinery, which creates channel-forming fragments from endocytosed beta-amyloid, and the membranes permeabilized by these channels.
- 5)
- Any (including the smallest) amyloid channels dissipate the lysosomal membrane’s pH gradient, resulting in lysosomal dysfunction due to the inhibition of acidic proteases.
- 6)
- Lysosomal dysfunction prevents normal quality control of organelles (mitochondria are the most sensitive to this disturbance due to high meta bolic damage associated with their function).
- 7)
- Largest amyloid channels can pass macro molecules, including cathepsins, which either directly mediate necrosis or activate apoptosis. Both necrosis and apoptosis result in cell death.
- 8)
- Membrane amyloid channel formation can be considered the primary molecular mechanism of beta-amyloid toxicity.
- 9)
- Membrane amyloid channels mediate lysosomal permeabilization.
- 10)
- Neuronal death is the cellular mechanism of cognitive impairment and dementia in AD.
- 11)
- The degree of cell death depends on the accu mulated Aβ toxicity, so the disease progresses over time.
- 12)
- The accumulated toxicity of Aβ, and as a result, the degree of neuronal death, has a positive correlation with the intensity of Aβ endocytosis.
- 13)
- The initial step of amyloid aggregation occurs intralysosomally. Therefore, the accumulation of amyloid aggregates positively correlates with the intensity of endocytosis of beta-amyloid.
- 14)
- Both cytotoxicity and intrabrain aggregation of beta-amyloid positively correlate with the intensity of endocytosis, which explains why the accumulation of extracellular amyloid aggregates is the biomarker of AD.
- 15)
- Both aggregation on existing amyloid aggregates and cellular uptake decrease interstitial concent rations of soluble Aβ, so a low concentration of soluble Aβ in the CSF serves as AD biomarker.
- 16)
- Higher cellular uptake results in faster neuro degeneration and lowers concentration of soluble Aβ independent of aggregation of existing aggregates, so lower soluble Aβ has a negative correlation with the progression of AD, which is independent of the density of deposits (measured by PET).
- 17)
- The severity of cognitive impairment in patients with late-onset AD positively correlates with the intensity of cellular Aβ uptake.
- 18)
- There is wide variability of sensitivity to amyloid endocytosis: patients with similar uptake can have significantly different levels of neuronal death.
- 19)
- The sensitivity to the endocytosed beta-amyloid depends on the balance between the rates of production and degradation of channel-forming fragments, the activity of biochemical pathways mediating neuronal death induced by lysosomal permeabilization, and cellular protection against consequences of lysosomal permeabilization.
- 20)
- Increased sensitivity to the endocytosed beta-amyloid could be one of the alternative mechanisms of AD: patients with early-onset AD do not have an increased Aβ uptake.
7. The description of the amyloid degradation toxicity hypothesis of Alzheimer’s disease at various levels
For each level of organization, the logic is presented through a series of statements and conclusions identified by indentation and highlighting/shading. References were provided above and omitted here for brevity. The description of each level is summarized in a figure which follows the corresponding table. Figures are numbered with the prefix “S” (Summary”).
7.1. Molecular level
| Beta-amyloid is cleaved from a long precursor molecule (amyloid precursor peptide, APP) by proteases called secretases. |
| The predominant form is the 40-amino acid-long peptide (Aβ40). However, senile plaques in the brain, which are histological hallmarks of AD, contain mostly 42-amino acid-long Aβ42. Both amyloid forms are produced by consecutive action of beta-secretase and gamma-secretase. |
| Immediately after synthesis, beta-amyloid has no secondary structure and is soluble. |
| With time, the peptide can form a pleated beta-sheet (one of two major secondary protein structures) within the molecule. The formation of multimolecular beta-sheets produces beta-amyloid oligomers. |
| Elongation of oligomers and interaction between multiple oligomeric complexes form insoluble aggregates. Senile plaques are amyloid polymers that include other proteins. |
| Polymerization of beta-amyloid results in the formation of insoluble aggregates. Among beta-amyloid isoforms, Aβ42 is most prone to aggregation. |
| Soluble beta-amyloid is toxic to healthy cells in vitro, while aggregated beta-amyloid is non-toxic. |
| The oligomeric form of beta-amyloid mediates the peptide’s cytotoxicity. |
| Senile plaques are the hallmark of AD but are not the etiology of the disease. |
| No known receptor or ion channel can be activated by beta-amyloid peptide(s) in a way that such activation would result in cell death. |
| Absorption of beta-amyloid on lipid membranes can cause membrane damage only in concentrations exceeding 1 µM, while extracellular concentrations of beta-amyloid are in the nanomolar range. |
| Beta-amyloid peptide(s) can form membrane channels. |
| The formation of amyloid membrane channels is the only known primary molecular mechanism that can explain the cytotoxic action of endogenous beta-amyloid. |
| Some amyloid fragments form membrane channels much more effectively than full-length peptides. |
| Not all fragments have channel-forming ability. |
| Channels are formed in membranes carrying negative surface charge. |
| Multiple molecules of peptide form each channel (channels are oligomers). |
| Amyloid membrane channels are non-selective. |
| Channels are giant (with conductance up to several nanosiemens) and have various sizes. |
| The largest channels (conductance higher than 1 nS) can pass macromolecules. |
|
Beta-amyloid can induce cell damage if: 1. There is a production of channel-forming fragments 2. These fragments can target membranes carrying negative surface charge |
| Alzheimer’s disease is developing over multiple years. |
| The conductance of a single channel is sufficient to destroy the barrier function of the membrane of an organelle or whole cell. |
| In physiological conditions, the formation of amyloid channels, which can cause cell death, is rare. It is likely that not every instance of channel formation leads to cell death. |
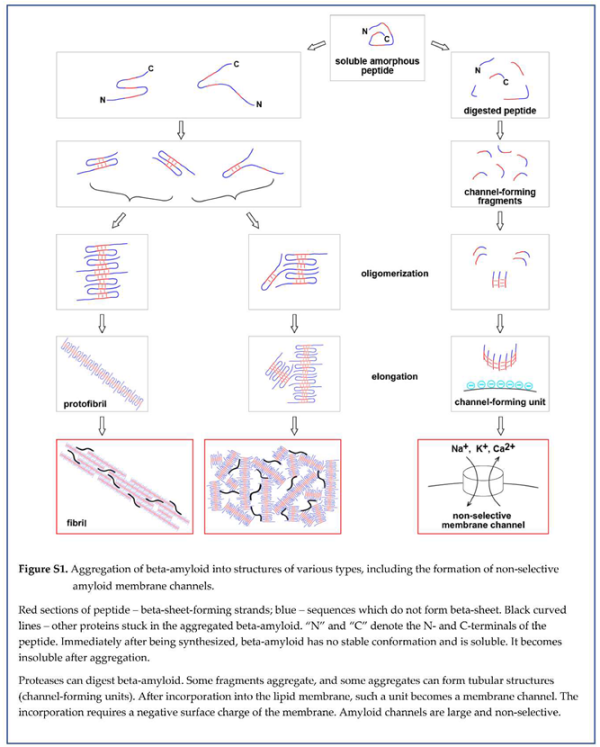
7.2. Organelle level
| The outer leaflet of the plasma membrane of healthy cells is not negatively charged. |
| No reliable experimental electrophysiological data supports the formation of giant amyloid channels in the plasma membrane of cultured cells in vitro. |
| Amyloid membrane channels are not formed in the cellular plasma membrane. |
| The inner mitochondrial membrane (IMM) is negatively charged, but known pathways require the involvement of other organelles (such as endosomes) to deliver channel-forming beta-amyloid fragments to the IMM. |
| The mitochondrial membrane is not a likely target for permeabilization by amyloid membrane channels. |
| Beta-amyloid is endocytosed by cells and is accumulated in lysosomes. |
| The presence of various lysosomal proteases suggests that at least some of them can generate channel-forming fragments. |
| Lysosomal membranes have a strong negative surface charge, which is the prerequisite for forming membrane amyloid channels. |
| Lysosomes are both the producers of channel-forming fragments and the targets of these toxic amyloid species. |
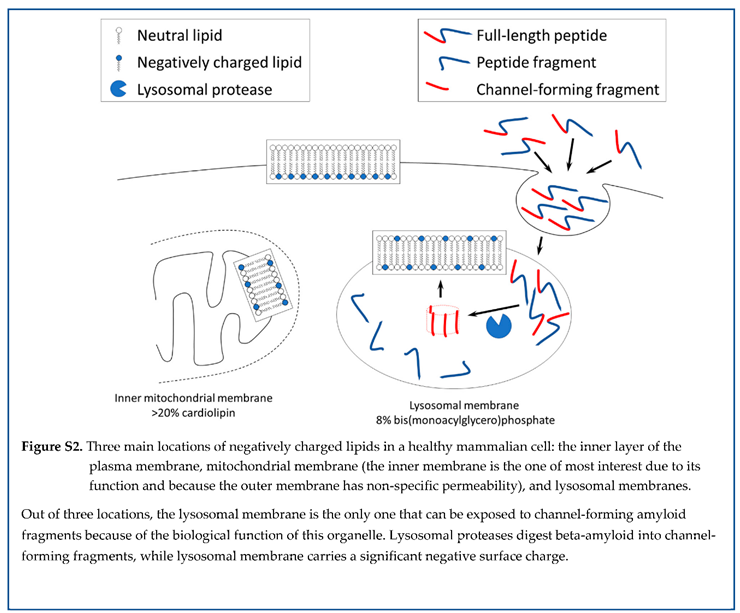
7.3. Cellular level
| Cells exposed to the Aβ42 demonstrate synchronous oscillations of concentrations of several intracellular ions (calcium, sodium, potassium, pH) that match the concept of a non-selective membrane channel opening. |
| The frequency of channel conductance oscillations observed electrophysiologically (millisecond range) does not match the time scale of oscillations in intracellular ion concentrations (seconds to minutes). |
| The conductance of amyloid membrane channels is very high. If a channel is formed in the plasma membrane, intracellular and extracellular ion concentrations will equilibrate fast. |
| The cellular ion concentrations return to their baseline after each wave induced by exposure to beta-amyloid. |
| The direction of ion disturbances and rapid recovery does not support the concept that channels are formed in the plasma membrane. |
| Amyloid membrane channels are not formed in the cellular plasma membrane. |
| Observed oscillations in intracellular ion concentra tions (an increase in calcium and potassium and a decrease in pH and sodium) match the ion distur bances that the permeabilization of lysosomes could cause. |
| The small volume of individual lysosomes allows for quick restoration of normal ion balance in the cell following the damage to a single lysosome. This results in the ion disturbance being observed as a wave. |
| The oscillations are caused by the permeabilization of multiple lysosomes. |
| Channels in lysosomal membranes explain intracellular ion changes induced by exposure to beta-amyloid. |
| Cells exposed to the Aβ42 demonstrate the content leakage from multiple lysosomes (but not all) without signs of plasma membrane damage. |
| Cells exposed to the Aβ42 demonstrate lysosomal content leakage before cell death. |
| In cells exposed to the beta-amyloid, lysosomes leak not only small molecules (such as Lucifer Yellow, M.W.444 or ethidium bromide, M.W.394) but also larger molecules such as cathepsin D and enzymes with M.W. 150 kDa. |
| The electrophysiological data suggests that the largest amyloid channels can pass macromolecules. |
| Channels in lysosomal membranes explain lysosomal permeabilization induced by exposure to beta-amyloid. |
| Channels in lysosomal membranes explain lysosomal permeabilization that occurs without damage to the plasma membrane. |
| Even a single channel is enough to equilibrate ion concentrations inside the entire organelle with the cytoplasm. |
| Amyloid membrane channel, regardless of its size, is permeable to protons and can cause lysosomal content to become non-acidic. |
| Neutralization of lysosomal content inactivates acidic proteases, prevents normal digestion of lysosomal cargo, and results in lysosomal failure. |
| Improper cellular recycling due to lysosomal failure prevents the removal of damaged lysosomes. |
| Channels formed in lysosomal membranes can explain lysosomal failure, which is the hallmark of AD. |
| Though rare, the largest amyloid channels (with a con ductance of several nS) can transport macro mo lecules equal to or larger than lysosomal cathepsins. |
| The lysosomal cathepsins released into the cytoplasm can induce either necrosis or activate caspa ses, leading to apoptosis. In both cases, cell death occurs. |
| Lysosomal permeabilization due to the formation of channels in the lysosomal membrane can lead to cell death. |
| After exposure to beta-amyloid, the pathway leading to cell death is a multistep process that includes endocytosis, intralysosomal proteolysis, and the leakage of cathepsins into the cytoplasm. |
| The formation of a giant amyloid channel is a rare event. Only a tiny percentage of amyloid channels can kill the cell. |
| It could require prolonged exposure to beta-amyloid to produce a sufficient number of cytotoxic channels in a significant proportion of cells. |
| Cell death does not occur immediately after exposure to beta-amyloid. |
| The formation of small channels is a more frequent event, but it does not directly lead to cell death. |
| Cells with lysosomes carrying small amyloid channels survive but damaged lysosomes remain dysfunctional. |
| After exposure to beta-amyloid, cell death is primarily mediated by the formation of extremely large (giant) channels. |
| Lysosomal failure in AD is observed because many damaged neurons survive but cannot recycle damaged lysosomes. |
| The symptoms of AD depend on the number and functional status of remaining neurons. |
| Neurons that are damaged by the formation of small amyloid channels can recover. |
| The recovery requires the prevention of the formation of new channels. |
| It is feasible that some AD symptoms (specifically those that appear due to the dysfunction of surviving neurons) could be reversed if the formation of new amyloid channels is prevented. |
| The progression of AD depends on the rate of neuronal death. |
| Closing small amyloid channels is an ineffective way to prevent amyloid-induced neuronal death. |
| To slow the progression of AD, the cyto toxicity induced by the formation of giant amyloid channels should be prevented. |
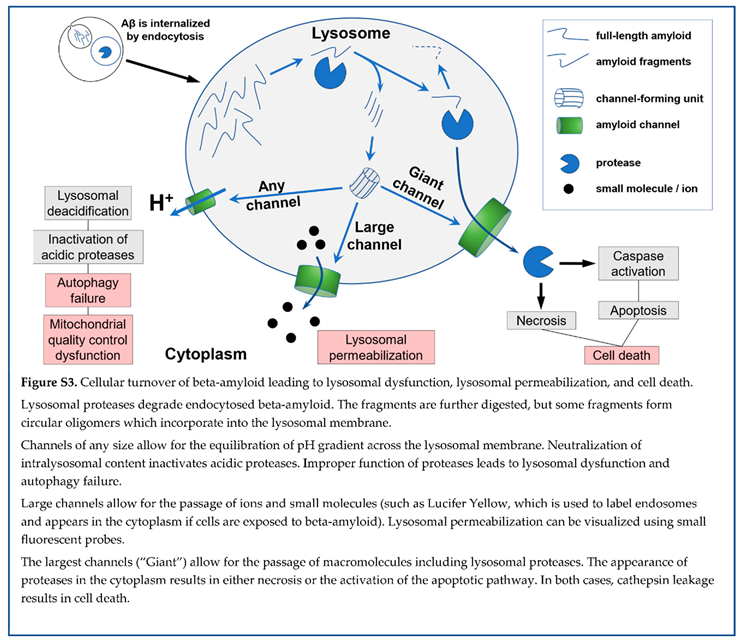
7.4. Tissue/Organ level
| Neurons are responsible for 70–80% of total brain energy consumption. | |
| Neuronal loss is a major contributor to lower brain metabolic activity in patients with AD. | |
| Lysosomal failure leads to improper recycling of various cell organelles. | |
| Mitochondria are the most sensitive to improper recycling due to the high rate of metabolic damage. | |
| Accumulation of damaged mitochondria results in an increased generation of reactive oxygen species, amplifying the damage to other organelles. | |
| Damaged but not recycled organelles occupy intracellular space and prevent normal mitogenesis. | |
| Mitophagy/mitogenesis disbalance in cells exposed to beta-amyloid explains higher production of toxic ROS and could be another contributor to lower brain metabolic activity in patients with AD. | |
| The aggregation of Aβ42 cannot spontaneously initiate in the interstitial fluid due to a very low peptide concentration (around 1 nanoM). | |
| Endocytosed Aβ42 is concentrated inside lysosomes up to 100-fold and can be observed intracellularly for more than 48 hours. | |
| Estimated intralysosomal Aβ42 concentration and the length of Aβ42 presence inside lysosomes allow for the initiation of amyloid aggregation. | |
| Aggregated Aβ42 is resistant to proteolytic digestion and can be sequestered inside cells. | |
| Intracellular amyloid aggregates are typical in AD brains. | |
| As an alternative to intracellular sequestration, the aggregated peptide is exocytosed along with remaining non-digested soluble Aβ42. | |
| When exposed to Aβ42, cultured cells in vitro promote beta-amyloid aggregation. | |
| Extracellular aggregation seeds are initially formed intralysosomally. | |
| Higher endocytosis intensity increases aggregation seeds’ production to the interstitial fluid. | |
| Faster accumulation of amyloid deposits will be observed in patients with a higher rate of Aβ42 endocytosis. |
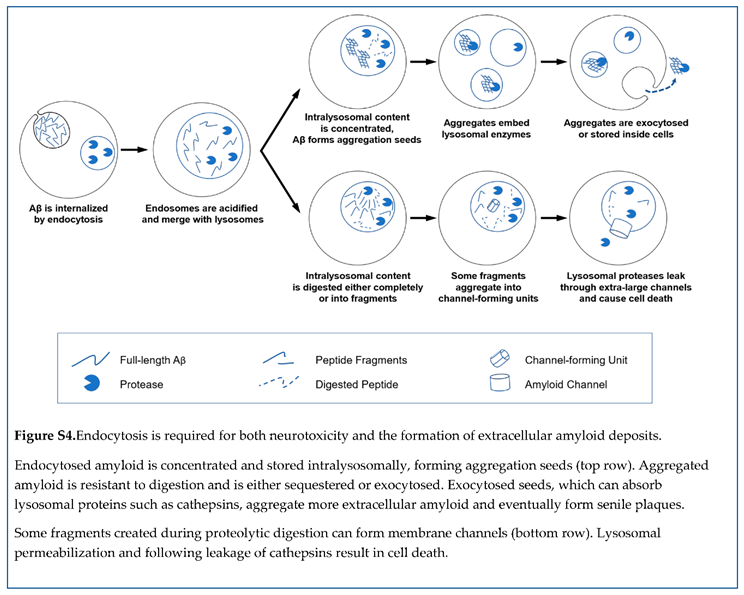
7.5. Organism level
| The cytotoxicity and the appearance of senile plaques are both the consequence of the same initial process - endocytosis of Aβ42. |
| The density of amyloid aggregates in the brain positively correlates with the level of neurodegeneration. |
| The density of amyloid deposits can be measured in living subjects by positron emission tomography (PET) using appropriate positron-emitting probes. |
| While the density of amyloid aggregates is relevant to AD pathophysiology, it is an indirect biomarker of AD. |
| In patients with AD, the concentration of Aβ42 in the CSF is lower than in the subjects with normal cognition. |
| Synthesis of Aβ42 in patients with AD is not differ rent from that of subjects with normal cognition. |
| In patients with AD, CSF flow is either the same or lower than in subjects with normal cognition. |
| Neither lower synthesis nor higher CSF flow explains the lower concentration of Aβ42 in the CSF in patients with AD. |
| Existing amyloid aggregates serve as aggregation centers for soluble Aβ42. |
| If the density of existing aggregates is high, more freshly synthesized Aβ42 aggregates. |
| If a higher ratio of freshly synthesized Aβ42 aggregates, the concentration of Aβ42 in the interstitial fluid decreases, so less Aβ42 is transported to the CSF. |
| Patients with AD have a higher density of amyloid deposits. |
| AD patients have lower levels of Aβ42 in the CSF due to the increased aggregation of Aβ42 on existing amyloid plaques. |
| The density of amyloid deposits, which is the biomarker of AD, is the primary determinant of the level of Aβ42 in the CSF. |
| The concentration of Aβ42 in the CSF is relevant to AD pathophysiology, but similarly to the density of amyloid deposits, it also is the indirect biomarker of AD. |
| The concentration of Aβ42 in the CSF as the standalone biomarker has a diagnostic value similar to PET measurements. |
| According to multivariate correlation analysis, CSF-Aβ42 levels and PET imaging data have independent predictive powers for AD diagnostics, even though these two biomarkers are highly correlated. |
| Combining two biomarkers (PET data and the concentration of Aβ42 in the CSF) provides more information than each biomarker alone. |
| If we compare the concentration of Aβ42 in the CSF of patients with AD and subjects with normal cognition with the same density of amyloid deposits, the Aβ42 concentration in patients with AD is significantly lower. |
| There is some aggregation-independent process that affects the concentration of Aβ42 in the interstitial fluid and has a rate that is different between patients with AD and subjects with normal cognition. |
| The rate of removal with CSF is not higher in patients with AD compared with subjects with normal cognition, so this additional process occurs inside the brain. |
| The synthesis rate of Aβ42 is not different between patients with AD and subjects with normal cognition. |
| The additional process is the aggregation-independent intrabrain removal of Aβ42. |
| A high ratio of patients with a high density of amyloid aggregates also have advanced neurodegeneration, but some of them have no cognitive deficiencies at all. |
| A maximal concentration of Aβ42 in the CSF can be observed for each given density of amyloid deposits. |
| Subjects with the highest possible concentrations of Aβ42 in the CSF for a given amyloid deposit density have a very low AD probability. |
| Higher aggregation-independent intrabrain removal of Aβ42 corresponds to the decrease of Aβ42 concentration in the CSF. |
| The aggregation-independent intrabrain removal of Aβ42 is more intense in the brains of AD patients. |
| Most of intrabrain Aβ42 removal is mediated by proteolytic degradation. |
| Most proteases are located intracellularly. |
| To be degraded by proteases, Aβ42 needs to be internalized by cells. |
| The primary way of Aβ42 cellular uptake is endocytosis. |
| Aβ42 endocytosis is increased in the brains of AD patients. |
| Endocytosis is the initial step of beta-amyloid neurotoxicity. |
| Increased Aβ42 endocytosis is involved in higher neurodegeneration in AD patients. |
| The stable isotope labeling kinetics (SILK) technique enables the investigation of beta-amyloid turnover in the brains of living patients. |
| SILK studies showed increased irreversible intrabrain removal of Aβ42 in AD patients, which can be observed even in the absence of existing plaques. |
| The data generated by the studies of beta-amyloid turnover supports the hypothesis about higher Aβ42 endocytosis in patients with AD. |
| SILK studies also demonstrated that patients with mutation-induced AD are characterized by the pre sence of an “exchange pool” of freshly synthesized Aβ42. In subjects with normal cognition, the size of such a pool is not statistically different from zero. |
| Increased endocytosis of Aβ42 is associated with the exocytosis of non-aggregated intralysosomal Aβ42, which is exocytosed along with amyloid aggregation seeds. |
| Dramatically increased endocytosis of Aβ42 fits the concept of “exchange pool.” |
| Increased endocytosis explains both increased irreversible intrabrain removal of Aβ42 and the larger size of its “exchange pool.” |
| Increased cellular uptake of Aβ42 is one of the reasons for the increased production of channel-forming fragments, but there could be other reasons. |
| Increased activity of proteases producing channel-forming fragments and/or decreased activity of proteases degrading fragments would result in higher concentrations of toxic fragments. Such disbalance would increase beta-amyloid toxicity without increased uptake. |
| Cellular Aβ42 uptake in patients with early-onset AD is not different from uptake in subjects with normal cognition. The early-onset AD is most likely characterized by higher toxicity of endocytosed Aβ42. |
| The amyloid degradation toxicity hypothesis provides the framework for the pathophysiology-based classification of various AD forms. |
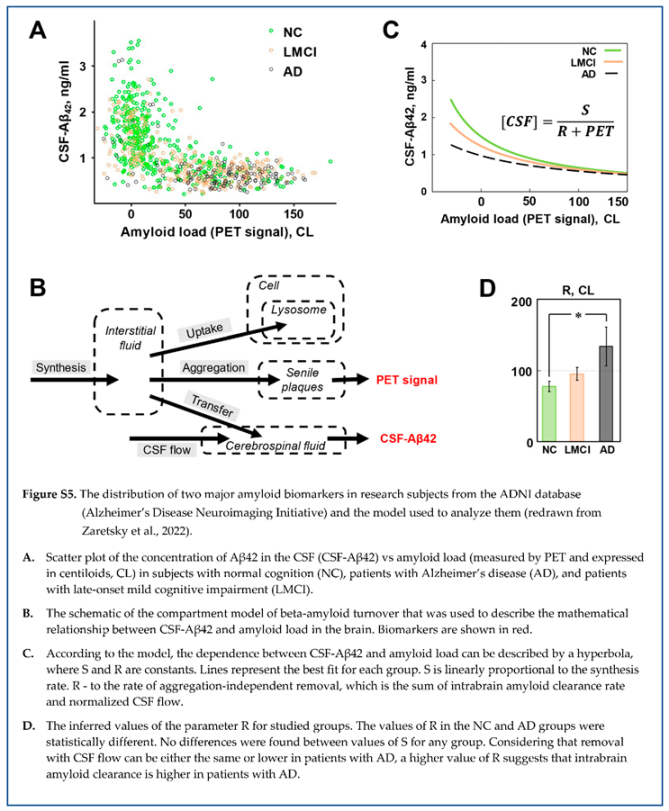
7.6. Population level
| The probability of the disease in a particular patient increases with increased accumulated cellular uptake of Aβ42. |
| The probability of AD increases with age. |
| Cellular uptake defines the toxic insult made by beta-amyloid. Still, the level of neuronal death, the severity of clinical symptoms, and eventually the diagnosis of AD depends on the individual relative toxicity of endocytosed beta-amyloid. |
| The ability of lysosomal proteases to produce and degrade channel-forming fragments defines the relative toxicity of endocytosed amyloid. Also, the ability of cells to resist the consequences of lysosomal permeabilization has individual variability (for example, due to different levels of cytoplasmatic protease inhibitors). |
| There is a wide variability of sensitivity (resistance) to the toxic action of beta-amyloid. |
| Patients with a high rate of endocytosis have a higher probability of advanced neurodegeneration and AD diagnosis, but some of them can have no cognitive deficiencies. |
| High endocytosis of beta-amyloid leads to increased accumulation of amyloid aggregates (higher amyloid load, higher amyloid PET signal) |
| On average, patients with high amyloid load have a higher rate of neurodegeneration and a higher probability of AD diagnosis. Still, some patients with high values of amyloid PET signal can have normal cognition. |
| The same process - endocytosis of Aβ42 – initiates both neurotoxicity and amyloid aggregation. |
| Both accumulated cytotoxicity and the density of senile plaques are increasing over time (and, correspondingly, with age). |
| The model, which describes the turnover of Aβ42 as the interaction between synthesis, transfer to the CSF, aggregation on existing plaques, and cellular uptake of the peptide, can be expressed as a system of differential equations. |
| In modeling, the probability of AD diagnosis was approximated as a sigmoid function of accumulated endocytosed beta-amyloid. At low accumulated uptakes, until some threshold is reached, the neurotoxicity can be compensated, so the probability of AD diagnosis is close to zero. At the highest accumulated uptakes, neuronal death reaches levels when the probability of AD diagnosis approaches 100% and cannot be increased anymore. |
| The clinical dataset, which contains values of amyloid deposits density and levels of Aβ42 in the CSF in populations of patients with AD and subjects with normal cognition (ADNI database), was used to infer the parameters of the model. |
| The model reproduces the distribution of late-onset AD in the population if the best-fit set of parameters is used. |
| A goodness-of-fit test confirms that the model reproduces clinical data accurately. |
| The distribution of two major amyloid biomarkers in the human population supports the amyloid degradation toxicity hypothesis of Alzheimer’s disease. |
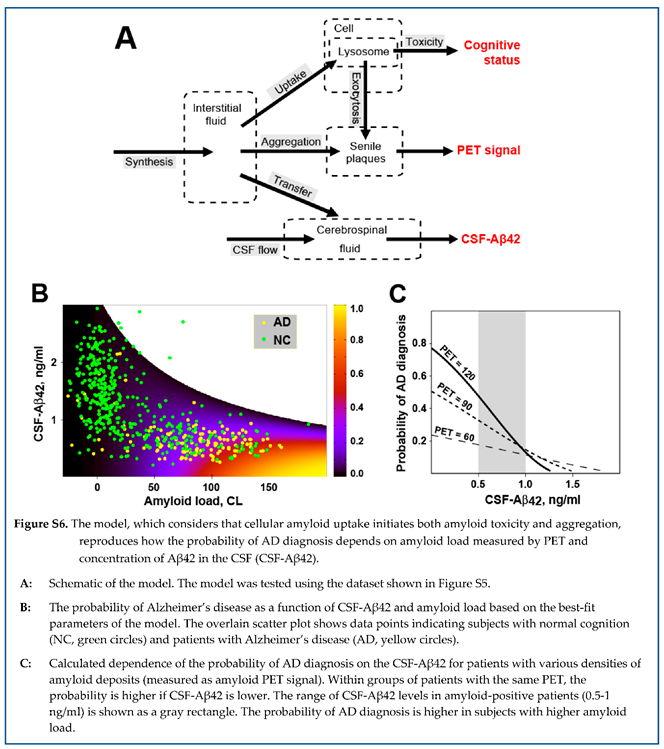
7.7. Multiscale summary of amyloid degradation toxicity hypothesis
| Molecular Level |
| Beta-amyloid is the neurotoxin that causes neurodegeneration in AD. The primary molecular interaction underlying beta-amyloid cytotoxicity is the formation of non-selective giant membrane channels. Amyloid membrane channels are formed by amyloid fragments with a positive charge in membranes carrying a negative surface charge. Spontaneous aggregation of beta-amyloid cannot be initiated in the intercellular space because of extremely low concentration, but existing amyloid aggregates grow extracellularly by absorbing soluble beta-amyloid. |
| Organelle Level |
| Lysosomes are the primary target in the cytotoxic action of beta-amyloid. Intralysosomal digestion of beta-amyloid produces channel-forming amyloid fragments. Lysosomal membrane carries a significant negative surface charge required for the formation of membrane amyloid channels. |
| Cellular Level |
| Endocytosis of beta-amyloid is the first step required for cytotoxicity. Membrane channels of any size cause lysosomal dysfunction, while extremely large channels allow for the leakage of lysosomal proteins to the cytoplasm. Leaked cathepsins induce either necrosis or apoptosis. Lysosomal permeabilization and lysosomal failure are the origins of other cellular hallmarks of AD – mitochondrial insufficiency, increased production of reactive oxygen species, the appearance of dysfunctional lysosomes, tau-protein accumulation, intracellular ion disturbances, etc. Beta-amyloid, which is stored intralysosomally, is the origin of aggregation seeds. Seeds of aggregated beta-amyloid are exocytosed and then grow extracellularly. Endocytosis of beta-amyloid is required for the appearance of senile plaques. |
| Tissue/Organ Level |
| Cellular death explains gross brain pathology in AD (especially in advanced stages of the dise ase): parenchymal atrophy, low metabolism, etc. The common origin of both aggregation and cytotoxicity – cellular uptake of beta-amyloid - explains why the presence of senile plaques is the hallmark of AD. Therefore, the density of amyloid deposits is a pathophysiology-relevant but indirect biomarker of AD. |
| Organism Level |
| Patients with late-onset AD have higher cellular uptake of Aβ42 than subjects with normal cogni tion. As both neurodegeneration and beta-amyloid aggregation are initiated by the same process - cellular amyloid uptake - higher amyloid uptake is the reason for faster neurodegeneration and high er density of amyloid deposits in patients with AD. The concentration of Aβ42 in the CSF is predominantly defined by the aggregation of this toxic soluble peptide on existing amyloid depo sits, while the density of deposits correlates with neurodegeneration. For that reason, CSF-Aβ42 can serve as a standalone AD biomarker, which is similar in value to the density of amyloid deposits. Therefore, CSF-Aβ42 is also the pathophysiology-relevant but indirect biomarker of AD. However, in patients with the same density of beta-amyloid deposits, a higher cellular uptake rate results in lower CSF-Aβ42. At a predefined density of deposits, lower CSF-Aβ42 indicates higher cellular amyloid uptake and faster neurodegeneration. Therefore, CSF-Aβ42 has a diagnostic value independent of the diagnostic value of the density of amyloid deposits. Using two major amyloid biomarkers together is advantageous until direct pathophysiology-relevant biomarkers are developed. |
| Population Level |
| The probability of AD diagnosis in a particular patient depends on the accumulated neurotoxicity of Aβ42, which in turn depends on the accumu lated amount of endocytosed Aβ42. However, the neurotoxicity of endocytosed Aβ42 also depends on the relative toxicity of this pep tide. Relative toxicity can be different between patients. The mathematical model that considers all these conditions accurately reproduces clinical data on the distribution of biomarkers and the probability of AD in the population. Currently used biomarkers (the density of amyloid deposits and the concentration of Aβ42 in the CSF) are related to AD pathobiology and correlate well with the disease. However, their non-direct nature limits the ability to use them to diagnose and, more importantly, predict disease progression.Methods that allow for the estimation of cellular amyloid uptake (such as the stable isotope labeling kinetics technique, SILK) would be more optimal for developing biomarkers and predictors of AD. Methods to estimate and modulate the relative neurotoxicity of beta-amyloid in patients are also the way to address the unmet need in the treatment of Alzheimer’s disease. |
7.5. The amyloid degradation toxicity hypothesis is the integrative theory of Alzheimer’s disease
| The hypothesis was described at all levels of bio logical organization – from molecular to population. |
| The hypothesis suggests the etiology and pathophysiology of the disease. |
| The amyloid degradation toxicity hypothesis is an integrative theory. |
There are two major paradoxes in any amyloid-centric theory of AD:
|
| AD is characterized by lysosomal failure, lysosomal permeabilization, activation of apoptosis, accumulation of intracellular amyloid aggregates, mitochondrial dysfunction, increased reactive oxygen species production, lower brain metabolism, accumulation of tau protein, etc. |
| The general human population is characterized by a particular distribution of amyloid biomarkers. |
| The amyloid degradation toxicity hypo thesis interprets multiple phenomena and paradoxes associated with AD. |
| The hypothesis explains why currently used amyloid-based biomarkers of AD (brain amyloid load and level of Aβ42 in the CSF) are associated with the pathophysiology of AD. |
| Both biomarkers provide indirect measures of parameters relevant to the etiology of AD. |
| The hypothesis interprets why there is a significant overlap between two amyloid biomarkers and what independent information can be found if these two biomarkers are considered together. |
7.8. Amyloid degradation toxicity hypothesis provides a framework for future research
| The hypothesis identifies processes that are relevant to AD pathophysiology and can be quantitatively characterized in a patient. |
| The rate of cellular amyloid uptake is likely to be the most informative parameter. There are tested approaches to measure this rate in clinical practice. |
| The hypothesis suggests novel biomarkers that are directly related to AD pathophysiology. |
| The hypothesis identifies the etiology of AD and involved biochemical and cellular pathways. |
| The hypothesis explains why currently used treatments do not prevent disease progression. |
| The hypothesis explains why dissolving amyloid deposits did not result in a reliable clinical effect. |
| The etiology and pathways involved in AD contain druggable targets (for example, proteases can be inhibited). |
| The hypothesis provides the framework for the development of novel medica tions to treat, slow the progression, or prevent the development of Alzheimer’s disease. |
8. Summary: etiology and pathogenesis of Alzheimer’s disease
The integrative amyloid degradation toxicity hypothesis of Alzheimer’s disease can be summarized in a set of statements.
- The toxicity of soluble beta-amyloid is the driving force in the development of Alzheimer’s disease.
- Amyloid membrane channel formation is the primary molecular mechanism of beta-amyloid toxicity and can be considered the etiology of AD.
- Lysosomal permeabilization due to membrane amyloid channel formation is the initial cellular damage.
- Lysosomes digest endocytosed Aβ, producing fragments, some of which can form membrane channels, while lysosomal membranes have characteristics required to incorporate channel-forming amyloid oligomers.
- Neuronal death is the result of activation of necrosis and/or apoptosis due to the leakage of lysosomal proteases through extremely large amyloid membrane channels, which are formed with low, but non-zero, probability.
- Dissipation of the pH gradient on the lysosomal membrane through amyloid membrane channels of any size results in lysosomal dysfunction and corresponding autophagy failure.
- The progression of AD includes functional (potentially reversible) cellular changes and cell death (irreversible, so the disease progresses over time).
- The degree of neuronal death depends on the accumulated Aβ toxicity and, therefore, on the intensity of Aβ endocytosis and time.
- Humans have very wide variability of sensitivity to the endocytosed Aβ because the pathway from Aβ endocytosis to cell death includes multiple steps.
- The sensitivity to endocytosed Aβ can be high, which can explain why some forms of AD are not associated with increased cellular amyloid uptake.
Acknowledgments
The research reported in this publication did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Abbreviations
AD – Alzheimer’s disease; NC – normal cognition; Aβ – beta-amyloid; Aβ40 – Aβ1-40; Aβ42 – Aβ1-42; CSF – cerebrospinal fluid; CSF- Aβ42 – concentration of Aβ42 in the CSF; PET – positron emission tomography.
References
- Alzheimer, A. über eigenartige Krankheitsfälle des späteren Alters. Zeitschrift für die gesamte Neurologie und Psychiatrie 1911, 4, 356. [Google Scholar] [CrossRef]
- Graeber, M.B.; Mehraein, P. Reanalysis of the first case of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249 (Suppl. 3), 10–13. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G. Amyloid beta protein and the basis for Alzheimer’s disease. Prog. Clin. Biol. Res. 1989, 317, 857–868. [Google Scholar] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium Dysregulation and Membrane Disruption as a Ubiquitous Neurotoxic Mechanism of Soluble Amyloid Oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [PubMed]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. JAD 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Knafo, S.; Alonso-Nanclares, L.; Gonzalez-Soriano, J.; Merino-Serrais, P.; Fernaud-Espinosa, I.; Ferrer, I.; DeFelipe, J. Widespread changes in dendritic spines in a model of Alzheimer’s disease. Cereb. Cortex 2009, 19, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Grutzendler, J.; Duff, K.; Gan, W.B. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 2004, 7, 1181–1183. [Google Scholar] [CrossRef]
- Bittner, T.; Fuhrmann, M.; Burgold, S.; Ochs, S.M.; Hoffmann, N.; Mitteregger, G.; Kretzschmar, H.; LaFerla, F.M.; Herms, J. Multiple Events Lead to Dendritic Spine Loss in Triple Transgenic Alzheimer’s Disease Mice. PLoS ONE 2010, 5, e15477. [Google Scholar] [CrossRef]
- Mattsson, N.; Insel, P.S.; Landau, S.; Jagust, W.; Donohue, M.; Shaw, L.M.; Trojanowski, J.Q.; Zetterberg, H.; Blennow, K.; Weiner, M. Diagnostic accuracy of CSF Ab42 and florbetapir PET for Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2014, 1, 534–543. [Google Scholar] [CrossRef]
- Ong, K.T.; Villemagne, V.L.; Bahar-Fuchs, A.; Lamb, F.; Langdon, N.; Catafau, A.M.; Stephens, A.W.; Seibyl, J.; Dinkelborg, L.M.; Reininger, C.B.; et al. Aβ imaging with 18F-florbetaben in prodromal Alzheimer’s disease: a prospective outcome study. J. Neurol. Neurosurg. Psychiatry 2015, 86, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Elbert, D.L.; Patterson, B.W.; Bateman, R.J. Analysis of a compartmental model of amyloid beta production, irreversible loss and exchange in humans. Math. Biosci. 2015, 261, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Patterson, B.W.; Elbert, D.L.; Mawuenyega, K.G.; Kasten, T.; Ovod, V.; Ma, S.; Xiong, C.; Chott, R.; Yarasheski, K.; Sigurdson, W.; et al. Age and amyloid effects on human central nervous system amyloid-beta kinetics. Ann. Neurol. 2015, 78, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Potter, R.; Patterson, B.W.; Elbert, D.L.; Ovod, V.; Kasten, T.; Sigurdson, W.; Mawuenyega, K.; Blazey, T.; Goate, A.; Chott, R.; et al. Increased in vivo amyloid-β42 production, exchange, and loss in presenilin mutation carriers. Sci. Transl. Med. 2013, 5, 189ra177. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, D.V.; Zaretskaia, M.V.; Molkov, Y.I. Patients with Alzheimer’s disease have an increased removal rate of soluble beta-amyloid-42. PLoS ONE 2022, 17, e0276933. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Silverberg, G.D.; Heit, G.; Huhn, S.; Jaffe, R.A.; Chang, S.D.; Bronte-Stewart, H.; Rubenstein, E.; Possin, K.; Saul, T.A. The cerebrospinal fluid production rate is reduced in dementia of the Alzheimer’s type. Neurology 2001, 57, 1763–1766. [Google Scholar] [CrossRef]
- Fishman, R.A. The cerebrospinal fluid production rate is reduced in dementia of the Alzheimer’s type. Neurology 2002, 58, 1866. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.R. Potential roles of protease inhibitors in Alzheimer’s disease. Neurobiol. Aging 1989, 10, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Sturchio, A.; Dwivedi, A.K.; Young, C.B.; Malm, T.; Marsili, L.; Sharma, J.S.; Mahajan, A.; Hill, E.J.; Andaloussi, S.E.L.; Poston, K.L. High cerebrospinal amyloid-β 42 is associated with normal cognition in individuals with brain amyloidosis. EClinicalMedicine 2021, 100988. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Insel, P.S.; Donohue, M.; Landau, S.; Jagust, W.J.; Shaw, L.M.; Trojanowski, J.Q.; Zetterberg, H.; Blennow, K.; Weiner, M.W. Independent information from cerebrospinal fluid amyloid-β and florbetapir imaging in Alzheimer’s disease. Brain 2015, 138, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Motter, R.; Vigo-Pelfrey, C.; Kholodenko, D.; Barbour, R.; Johnson-Wood, K.; Galasko, D.; Chang, L.; Miller, B.; Clark, C.; Green, R.; et al. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann. Neurol. 1995, 38, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, N.; Hesse, C.; Davidsson, P.; Minthon, L.; Wallin, A.; Winblad, B.; Vanderstichele, H.; Vanmechelen, E.; Blennow, K. Cerebrospinal Fluid β-Amyloid(1-42) in Alzheimer Disease: Differences Between Early- and Late-Onset Alzheimer Disease and Stability During the Course of Disease. Arch. Neurol. 1999, 56, 673–680. [Google Scholar] [CrossRef]
- Espay, A.J.; Herrup, K.; Kepp, K.P.; Daly, T. The Proteinopenia Hypothesis: Loss of Aβ42 and the Onset of Alzheimer’s Disease. Ageing Res. Rev. 2023, 102112. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; Sturchio, A.; Schneider, L.S.; Ezzat, K. Soluble Amyloid-β Consumption in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2021. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; Lafontant, D.-E.; Poston, K.L.; Caspell-Garcia, C.; Marsili, L.; Cho, H.R.; McDaniel, C.; Kim, N.; Coffey, C.S.; Mahajan, A.; et al. Low soluble amyloid-β 42 is associated with smaller brain volume in Parkinson’s disease. Park. Relat. Disord. 2021, 92, 15–21. [Google Scholar] [CrossRef]
- Terakawa, M.S.; Yagi, H.; Adachi, M.; Lee, Y.H.; Goto, Y. Small liposomes accelerate the fibrillation of amyloid β (1-40). J. Biol. Chem. 2015, 290, 815–826. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [PubMed]
- Wesén, E.; Jeffries, G.D.M.; Matson Dzebo, M.; Esbjörner, E.K. Endocytic uptake of monomeric amyloid-β peptides is clathrin- and dynamin-independent and results in selective accumulation of Aβ(1-42) compared to Aβ(1-40). Sci. Rep. 2017, 7, 2021. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.-M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [Google Scholar] [CrossRef] [PubMed]
- Knauer, M.F.; Soreghan, B.; Burdick, D.; Kosmoski, J.; Glabe, C.G. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/beta protein. Proc. Natl. Acad. Sci. USA 1992, 89, 7437–7441. [Google Scholar] [CrossRef] [PubMed]
- Burdick, D.; Kosmoski, J.; Knauer, M.F.; Glabe, C.G. Preferential adsorption, internalization and resistance to degradation of the major isoform of the Alzheimer’s amyloid peptide, A beta 1-42, in differentiated PC12 cells. Brain Res. 1997, 746, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Thayer, C.Y.; Bird, E.D.; Wheelock, T.R.; Nixon, R.A. Lysosomal proteinase antigens are prominently localized within senile plaques of Alzheimer’s disease: evidence for a neuronal origin. Brain Res. 1990, 513, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Li, J.; Du, P.; Jin, W.; Gao, G.; Cui, D. Senile plaques in Alzheimer’s disease arise from Aβ- and Cathepsin D-enriched mixtures leaking out during intravascular haemolysis and microaneurysm rupture. FEBS Lett. 2023, 597, 1007–1040. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, D.V.; Zaretskaia, M.V.; Molkov, Y.I. Membrane channel hypothesis of lysosomal permeabilization by beta-amyloid. Neurosci. Lett. 2021, 136338. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.J.; Chandswangbhuvana, D.; Margol, L.; Glabe, C.G. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J. Neurosci. Res. 1998, 52, 691–698. [Google Scholar] [CrossRef]
- Ji, Z.S.; Miranda, R.D.; Newhouse, Y.M.; Weisgraber, K.H.; Huang, Y.; Mahley, R.W. Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. J. Biol. Chem. 2002, 277, 21821–21828. [Google Scholar] [CrossRef]
- Molkov, Y.I.; Zaretskaia, M.V.; Zaretsky, D.V. Towards The Integrative Theory of Alzheimer’s Disease: Linking Molecular Mechanisms of Neurotoxicity, Beta-Amyloid Biomarkers, And The Diagnosis. Curr. Alzheimer Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Mamun, A.A.; Labu, Z.K.; Hidalgo-Lanussa, O.; Barreto, G.E.; Ashraf, G.M. Autophagic dysfunction in Alzheimer’s disease: Cellular and molecular mechanistic approaches to halt Alzheimer’s pathogenesis. J. Cell. Physiol. 2019, 234, 8094–8112. [Google Scholar] [CrossRef] [PubMed]
- Peric, A.; Annaert, W. Early etiology of Alzheimer’s disease: tipping the balance toward autophagy or endosomal dysfunction? Acta Neuropathol. 2015, 129, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, D.M.; Lee, J.H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Cataldo, A.M. A.; Cataldo, A.M. Lysosomal system pathways: genes to neurodegeneration in Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2006, 9, 277–289. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Kavčič, N.; Pegan, K.; Turk, B. Lysosomes in programmed cell death pathways: from initiators to amplifiers. Biol. Chem. 2017, 398, 289–301. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.M.; Jackson, M.B. An Inconvenient Truth: Calcium Sensors Are Calcium Buffers. Trends Neurosci. 2018, 41, 880–884. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Calcium signals induced by amyloid β peptide and their consequences in neurons and astrocytes in culture. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2004, 1742, 81–87. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 5088–5095. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, D.V.; Zaretskaia, M.V. Intracellular ion changes induced by the exposure to beta-amyloid can be explained by the formation of channels in the lysosomal membranes. Biochim. Biophys. Acta. Mol. Cell Res. 2021, 119145. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-c.A.; Kagan, B.L. Electrophysiologic properties of channels induced by Abeta25-35 in planar lipid bilayers. Peptides 2002, 23, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Mirzabekov, T.; Lin, M.C.; Yuan, W.L.; Marshall, P.J.; Carman, M.; Tomaselli, K.; Lieberburg, I.; Kagan, B.L. Channel formation in planar lipid bilayers by a neurotoxic fragment of the beta-amyloid peptide. Biochem. Biophys. Res. Commun. 1994, 202, 1142–1148. [Google Scholar] [CrossRef]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef]
- Kourie, J.I.; Culverson, A.L.; Farrelly, P.V.; Henry, C.L.; Laohachai, K.N. Heterogeneous amyloid-formed ion channels as a common cytotoxic mechanism: implications for therapeutic strategies against amyloidosis. Cell Biochem. Biophys. 2002, 36, 191–207. [Google Scholar] [CrossRef]
- Kagan, B.L.; Hirakura, Y.; Azimov, R.; Azimova, R.; Lin, M.C. The channel hypothesis of Alzheimer’s disease: current status. Peptides 2002, 23, 1311–1315. [Google Scholar] [CrossRef]
- Bhatia, R.; Lin, H.; Lal, R. Fresh and globular amyloid beta protein (1-42) induces rapid cellular degeneration: evidence for AbetaP channel-mediated cellular toxicity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2000, 14, 1233–1243. [Google Scholar]
- Lin, H.; Zhu, Y.J.; Lal, R. Amyloid beta protein (1-40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesicles. Biochemistry 1999, 38, 11189–11196. [Google Scholar] [CrossRef]
- Pollard, H.B.; Arispe, N.; Rojas, E. Ion channel hypothesis for Alzheimer amyloid peptide neurotoxicity. Cell. Mol. Neurobiol. 1995, 15, 513–526. [Google Scholar] [CrossRef]
- Pollard, H.B.; Rojas, E.; Arispe, N. A new hypothesis for the mechanism of amyloid toxicity, based on the calcium channel activity of amyloid beta protein (A beta P) in phospholipid bilayer membranes. Ann. N. Y. Acad. Sci. 1993, 695, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Pollard, H.B.; Rojas, E. Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membranes. Proc. Natl. Acad. Sci. USA 1993, 90, 10573–10577. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Good, T. A rapid method to measure beta-amyloid induced neurotoxicity in vitro. J. Neurosci. Methods 2007, 161, 1–10. [Google Scholar] [CrossRef]
- Arispe, N.; Pollard, H.B.; Rojas, E. beta-Amyloid Ca(2+)-channel hypothesis for neuronal death in Alzheimer disease. Mol. Cell Biochem. 1994, 140, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Valincius, G.; Heinrich, F.; Budvytyte, R.; Vanderah, D.J.; McGillivray, D.J.; Sokolov, Y.; Hall, J.E.; Lösche, M. Soluble amyloid beta-oligomers affect dielectric membrane properties by bilayer insertion and domain formation: implications for cell toxicity. Biophys. J. 2008, 95, 4845–4861. [Google Scholar] [CrossRef]
- Sokolov, Y.; Kozak, J.A.; Kayed, R.; Chanturiya, A.; Glabe, C.; Hall, J.E. Soluble amyloid oligomers increase bilayer conductance by altering dielectric structure. J. Gen. Physiol. 2006, 128, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, D.V.; Zaretskaia, M.V. Flow cytometry method to quantify the formation of beta-amyloid membrane ion channels. Biochim. Biophys. Acta. Biomembr. 2020, 183506. [Google Scholar] [CrossRef]
- Zaretsky, D.V.; Zaretskaia, M. Degradation Products of Amyloid Protein: Are They The Culprits? Curr. Alzheimer Res. 2020, 17, 869–880. [Google Scholar] [CrossRef]
- Alarcon, J.M.; Brito, J.A.; Hermosilla, T.; Atwater, I.; Mears, D.; Rojas, E. Ion channel formation by Alzheimer’s disease amyloid beta-peptide (Abeta40) in unilamellar liposomes is determined by anionic phospholipids. Peptides 2006, 27, 95–104. [Google Scholar] [CrossRef]
- Das, A.; Nag, S.; Mason, A.B.; Barroso, M.M. Endosome-mitochondria interactions are modulated by iron release from transferrin. J. Cell Biol. 2016, 214, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, A.; Roshan, T.M.; Kahawita, T.M.; Mason, A.B.; Sheftel, A.D.; Ponka, P. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2859–2867. [Google Scholar] [CrossRef] [PubMed]
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2012, 1824, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Stoka, V.; Rozman-Pungercar, J.; Cirman, T.; Droga-Mazovec, G.; Oresić, K.; Turk, V. Apoptotic pathways: involvement of lysosomal proteases. Biol. Chem. 2002, 383, 1035–1044. [Google Scholar] [CrossRef]
- Goel, P.; Chakrabarti, S.; Goel, K.; Bhutani, K.; Chopra, T.; Bali, S. Neuronal cell death mechanisms in Alzheimer’s disease: An insight. Front. Mol. Neurosci. 2022, 15, 937133. [Google Scholar] [CrossRef]
Figure 1.
The chart of the logic that leads to the amyloid degradation toxicity hypothesis of Alzheimer’s disease.
Figure 1.
The chart of the logic that leads to the amyloid degradation toxicity hypothesis of Alzheimer’s disease.


| Biomarker | Toxicity of amyloid form | Concentration in patients with AD |
| Aggregated Aβ | inert | increased |
| Soluble Aβ42 | toxic | decreased |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



