Submitted:
22 May 2023
Posted:
24 May 2023
You are already at the latest version
Abstract
Microglia together with other permanent macrophages in central nervous system (CNS) are responsible for regulating the innate immune response of the brain and spinal cord. Upon activation, microglia triggers the release of inflammatory mediators such as cytokines, chemokines, and other proteins related to neuro-inflammation. Elevated levels of neuro-inflammation have been linked to a decline in cognitive performance manifested in Alzheimer’s Disease (AD). There are mounting evidence in the literature to suggest that microglia are responsible for a substantial amount of the synaptic damage seen in AD. Most importantly, scientific studies have suggested that overexpression of microglia-derived neuro-inflammation elevates amyloid beta (Aβ) plaque formation, and hyperactivation of tau protein; the two main pathological characteristic features of AD. Alternatively, Aβ and tau formation further activate microglia to sustain the neuro-inflammation triggering a vicious cascade of neurodegeneration in AD. Here in this review, we discussed the role of microglia associated neuroinflammation for the pathogenesis of AD.
Keywords:
Microglia
; Neuroinflammation
; Alzheimer’s Disease
1. Introduction
Alzheimer's disease (AD) is an age-related progressive neurodegenerative disorder. It manifests as one of the most prevalent forms of dementia predominantly affecting the older generation. According to the Alzheimer's Disease International assessment, there were roughly 46 million individuals living with dementia in the world in 2015 (Prince et al., 2015). It is anticipated that this number would rise to 131 million by the year 2050 (Prince et al., 2015, Bajwa and Klegeris, 2022). Accumulation of amyloid plaques and neurofibrillary tangles inside the brain vasculature are the classic biological hallmarks of AD (Chen and Mobley, 2019). These pathological conditions are neurotoxic which causes acute neuroinflammation, further potentiates AD progression. Various studies have reported that complications of AD are elevated by lifestyle related disorders, such as obesity, hypertension, and Type 2 Diabetes mellitus (T2DM) (Barnes and Yaffe, 2011). Some studies have also suggested that the neuroinflammation observed in AD brains may be the result of peripheral inflammation. Over time this peripheral inflammation leads to inflammation of the brain leading to AD (McKenzie et al., 2017, Newcombe et al., 2018, Bajwa and Klegeris, 2022). Lesions created by chronic inflammation, amyloid plaque and tangles are related to synaptic dysfunction and cognitive decline during AD (Mishra and Brinton, 2018). In addition, evidence of microgliosis surrounding amyloid plaque formation has become one of the interesting aspects of AD research to find out the plausible connection among microglia derived neuroinflammation and AD pathophysiology in recent years (Mishra and Brinton, 2018). In this mini review, we summarized the contribution of microglia associated neuroinflammation and other pathological and molecular factors for the development of AD. Moreover, how microglia driven neuroinflammatory pathway could be a potential target for the treatment of the AD in future.
2. Neuroinflammation and Microglia
Neuroinflammation can be defined as an inflammatory process involving the brain or spinal cord (DiSabato et al., 2016). Apart from the neurons, three distinct glial cells named microglia, oligodendrocytes, and astrocytes comprise central nervous system (CNS). These cells are protected from the remaining portion of the whole nervous system by the tight junction of the blood-brain barrier (BBB) (Daneman and Prat, 2015). Due to the presence of this impermeable BBB structure, many pathogens, leucocytes and other immunoglobins are impeded to enter the brain (Daneman and Prat, 2015). Consequently, CNS has a more diverse immunological defense mechanism than the peripheral tissues (Hilzendeger et al., 2014). This different immune mechanism is mainly regulated by microglial cells, which constitute around 5-20% of immune cells in an adult brain (Ginhoux et al., 2013). These cells are considered as the sentry of the brain’s first line defense system which prevents invasive microorganisms, and toxic materials; and thereby prevents injury to the brain (Xu et al., 2016).
Microglia, the innate immune cells of the CNS, originate from erythromyeloid progenitor cells in the embryonic yolk sac and migrate into the brain around embryonic day 10.5 in mice, after which they propagate, spread, and ramify throughout the brain parenchyma (Ginhoux et al., 2013).
Microglial cells maintain homeostatic balance inside the brain by engulfing cellular debris and waste materials by phagocytosis. This phagocytic mechanism of microglial cells is named as microglial activation (Fu et al., 2014). In this step, ramified microglial cells undergo a structural transformation to amoeboid form (Kettenmann et al., 2011). After transformation, these microglial cells are readily prepared for moving outside the brain and provide an immunological response to the injured site. Although, short term microglial activation is beneficial, chronic and sustained microglial activation might be a potential cause of neuro-degeneration (Cunningham, 2013). Activated microglia are responsible for the release of a number of neurotoxins (nitric oxide, oxidative free radicals and pro-inflammatory cytokines) (Qian et al., 2006). In addition, these neurotoxins further activates microglia to release toxic free radicals and cytokines. (Lull and Block, 2010). This cyclic event eventually causes progressive damage to the neurons in the long run (Lull and Block, 2010). Moreover, redox signaling during microglial activation is suggested to play a pivotal role in chronic neuroinflammation that further initiates pro-inflammatory response by enhancing cytokine production (Lull and Block, 2010). Taken together, hyper-activation of microglia results in neurodegeneration leading to AD in future.
2.1. Immunoregulatory and neuro-regulatory substances released from microglia: Cytokines and Chemokines.
Activated microglia is responsible for producing a plethora of immunoregulatory mediators named chemokines and cytokines that take part in the pathogenesis of neuroinflammation. Some of the mediators are presented in Table 1.
Table 1: List of neuro-inflammatory mediators involved in neuro-degeneration. IL=Interleukin, IFN=Interferons, TNF=Tumor necrosis factor, TLR=Toll like receptor.
3. Mechanism of microglia induced neuroinflammation triggering the progression of AD.
Pathology of AD is triggered by formation of the amyloid beta (Aβ) plaque and neurofibrillary tangles (NFTs) (Gouras et al., 2015). These structures are accompanied by neuron fragments, stimulate pro-inflammatory mediators as well as inflammatory proteins by activating glial cells. Although these glial cells are considered as neuroprotective at low concentration, a high concentration of glial cells inside the CNS can be a crucial factor for neuro-degeneration (Gleichman and Carmichael, 2020). Especially, studies on reactive microglia have revealed that microglia can perform the role of antigen representing cell upon activation during the commencing stage of adaptive immune reaction (Prinz et al., 2019, Schetters et al., 2018). Another study revealed that activated microglia can secret different neurotoxic elements including nitrogenous substances, matrix metalloproteinase, cathepsins, and L-glutamate (Lindhout et al., 2021). Moreover, when microglia is adversely activated, it releases cytokines, complement proteins and chemokines. In addition, reactive microglia stimulates astrocytes, which again aid microglia in recruiting monocytes and T-cells into the brain parenchyma. All these mechanisms contribute to the development of the chronic neuroinflammation. Ultimately, a vicious cycle of neuroinflammation and microglial activation establishes leading to the progression of AD. (Figure 1) (Liddelow and Barres, 2017, Bachiller et al., 2018, Fakhoury, 2018, A McKenzie et al., 2017, Di Benedetto et al., 2019, Burgaletto et al., 2020, Scheltens et al.).
3.1. Age related changes in microglia and AD assimilation
The incidence of AD is most commonly associated with old age. The term "immunosenescence" refers to the decline in both the peripheral and central immune system components that occurs in old age (Aiello et al., 2019). Several immune-related genes have been found to be differently regulated in different parts of the aging human brain. Upregulation of these immune-related genes in old age have been associated with AD (Cribbs et al., 2012). At the cellular level, microglia have been found to undergo significant phenotypic changes as people age.
Genome-wide transcriptional profiling study in mice performed by Grabert and colleagues demonstrated an uneven and region-specific effects of microglia on aging (Grabert et al., 2016). They showed that with age changes occur in genes that are involved in immune response and bioenergetics. As a result, most of the variations in microglial cells proliferation occur across different geographical location (Grabert et al., 2016).
Compared to microglia isolated from young subjects, microglia from old subject exhibits a reduced expression of receptors on the cell surface and actin assembly (Galatro et al., 2017). Aged human cerebral cortex microglia morphologically portray some deformities, including process fragmentation, de-ramification, gnarling, and spheroid formation (Figure 2) (Newcombe et al., 2018). The BBB becomes more permeable with age, allowing various neuroinflammatory mediators to access into the brain. The contribution of neuroinflammatory mediators for the development of AD has been discussed earlier. Multiple studies have indicated that AD associated neurodegenerative process begins with the breakdown of BBB integrity (Montagne et al., 2015, Walker et al., 2017), leading to hippocampal atrophy (Takechi et al., 2017) and cognitive impairment in later stage of life (Villeda et al., 2011).
3.2. Relationship between microglial activation, inflammasome and gut microbiomes.
Inflammasomes are mainly complexes of large cytosolic multi proteins that assimilates due to infection or stress which in turn activates Casepase-1-related inflammatory responses such as release of pro-inflammatory cytokines IL-1β and IL-18 (de Zoete et al., 2014). Many inflammasomes are responsible for neurodegenerative diseases but among them pyrin domain containing 3 (NLRP3) inflammasome is considered to play a crucial role in amyloid plaque formation and tau protein hyper phosphorylation (Hanslik and Ulland, 2020). Interestingly, NLRP3 inflammasome has tremendous impact on aging and microglial activation (Hu et al., 2019, O’Neil et al., 2018). Genetic study revealed that NLRP3 gene expression was significantly higher in young compared to aged microglia(Youm et al., 2013).
Furthermore, during aging, microglial cells tends to accumulate elevated amount of lipofuscin. These accumulated lipofuscin together with increased oxidative stress contributes to the age-related neuro-degeneration (Kushwaha et al., 2018, Brown, 2009).
In recent years some emerging evidence have highlighted that there is a connection among microglial modulation of inflammasome and gut microbiomes (Ma et al., 2019). For instance, a very recent in-vivo mice model study on transplanted gut microbiota from AD affected brain has reported that gut microbiomes can regulate pathophysiology of AD by i) activation of NLRP3 inflammasome in the intestinal tract and ii) elevation of pro-inflammatory cytokines IL-6 and IL-1β in peripheral blood (Shen et al., 2020). Taken together, all these studies indicate that there is a close relationship between gut microbiomes, NLRP3 inflammasome and microglial mediated neuroinflammation for the potential pathophysiologic manifestation of the AD.
4. Targeting inflammation and modification of microglial activation can be an effective therapeutic target for treating AD
4.1. Nonsteroidal anti-inflammatory drugs for the treatment of AD
In epidemiological studies, nonsteroidal anti-inflammatory drugs (NSAIDs) have been shown to reduce the prevalence of AD progression in the typical aging population. (McGeer et al., 2016, Fu et al., 2018). Eicosanoid lipid mediators including prostaglandins, thromboxane, and leukotriene B4 are made when fatty acids like arachidonic acid are oxidized by cyclooxygenases (COX-1 and COX-2) (Fu et al., 2018). These eicosanoid lipid mediators play a role in AD pathology by triggering the inflammatory response. It is widely believed that NSAIDs exert their effects by inhibiting the expression of cyclooxygenase (COX). (Fu et al., 2018). In addition, AD transgenic mouse models exhibited elevated levels of arachidonic acid, cyclooxygenase 2, and prostaglandins (Hein and O’Banion, 2009, Sanchez-Mejia et al., 2008, Fu et al., 2018). Some NSAIDs (e.g., ibuprofen and indomethacin) modulate γ-secretase activity to inhibit Aβ42 peptide production and microglial activation (Zhang et al., 2021), Puhl et al., 2015). Having said that, most of the NSAIDs that were examined in clinical trials for the treatment of AD in advanced stage, were ineffective at alleviating AD symptoms, and some actually made symptoms worse (Miguel-Alvarez et al., 2015). According to recent evaluations, neuroinflammation begins in the initial stages of AD, long before the onset of obvious cognitive decline (Fu et al., 2018) . As a result, there is hope that giving NSAIDs to those with AD in the pre-symptomatic or the early stage could improve the outcome (Fu et al., 2018).
4.2. AD treatment strategy by modification of inflammatory response by microglia
Microglia mediated NLRP3 inflammasome activation plays a central role in AD pathology and direct or indirect inhibition of this inflammasome can prevent microglial pathogenesis in AD (Thawkar and Kaur, 2019). Hence, many studies are conducted to find effective NLRP3 inhibitors, and some inhibitors are under preclinical stage (Dempsey et al., 2017, Lučiūnaitė et al., 2020, Yin et al., 2018). Edaravone, a promising free radical scavenging agent which is used as a treatment options for cerebral infarction, has remarkable microglial inflammation modifying activity through inhibition of NLRP3 inflammasome activity (Wang et al., 2017). Moreover, edaravone can substantially lower amyloid beta plaque formation and is documented as neuroprotective (Wang et al., 2017). Thus, edaravone, can be effective in AD treatment by inhibiting NLRP3 activation and enhancing cognitive function (Parikh et al., 2018).
5. Future prospects of neuroinflammation as a potential therapeutic target to treat AD
A very recent study has demonstrated the effectiveness of targeting neuroinflammation as a potential target for treating AD (Liu et al., 2022). Several immune components, including cytokines and complement factors, have been investigated for their potential pathogenic roles for the development of AD (Heneka et al., 2015). Multiple transgenic animal model studies demonstrated that blocking IL-10 or IL-12/IL-23 signaling, or suppressing NLRP3 inflammasome activation lowers inflammatory responses and amyloid deposition and promotes learning and memory function in AD (Fu et al., 2016, Guillot-Sestier et al., 2015, Heneka et al., 2013, Venegas et al., 2017, Fu et al., 2018, Golde, 2019). Taken together, it is rational to think that preventing the neuroinflammatory pathology could be a therapeutic potential for the treatment of the AD.
6. Conclusion and future direction
Despite the fact that inflammation has a number of beneficial effects, such as pathogen elimination and phagocytosis during tissue regeneration, unrestricted inflammation can have negative consequences, such as the production of neurotoxic factors that worsen pathological characteristics of neurodegeneration. There is plethora of cytokines, chemokines, and their inflammatory mediators involved in this complicated immune response. As we discussed in this review, there are mounting evidence indicating that neuroinflammation serves as one of the vital players in the pathogenesis of AD and hence neuroinflammation can be a potential target for the treatment of the AD.
References
- A MCKENZIE, J. , J SPIELMAN, L., B POINTER, C., R LOWRY, J., BAJWA, E., W LEE, C. & KLEGERIS, A. 2017. Neuroinflammation as a common mechanism associated with the modifiable risk factors for Alzheimer's and Parkinson's diseases. Current aging science, 10, 158-176.
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Bajwa, E. Neuroinflammation as a mechanism linking hypertension with the increased risk of Alzheimer’s disease. Neural Regen. Res. 2022, 17, 2342–2346. [Google Scholar] [CrossRef] [PubMed]
- Brinton, R.D.; Yao, J.; Yin, F.; Mack, W.J.; Cadenas, E. Perimenopause as a neurological transition state. Nat. Rev. Endocrinol. 2015, 11, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R. Role of Microglia in Age-Related Changes to the Nervous System. Sci. World J. 2009, 9, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- BURGALETTO, C. , MUNAFÒ, A., DI BENEDETTO, G., DE FRANCISCI, C., CARACI, F., DI MAURO, R., BUCOLO, C., BERNARDINI, R. & CANTARELLA, G. 2020. The immune system on the TRAIL of Alzheimer’s disease. Journal of Neuroinflammation, 17, 1-11.
- Chen, X.-Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Csölle, C.; Sperlágh, B. Peripheral origin of IL-1β production in the rodent hippocampus under in vivo systemic bacterial lipopolysaccharide (LPS) challenge and its regulation by P2X7 receptors. J. Neuroimmunol. 2010, 219, 38–46. [Google Scholar] [CrossRef]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2012, 61, 71–90. [Google Scholar] [CrossRef]
- DANEMAN, R. & PRAT, A. 2015. The blood–brain barrier. Cold Spring Harbor perspectives in biology, 7, a020412.
- DE ZOETE, M. R. , PALM, N. W., ZHU, S. & FLAVELL, R. A. 2014. Inflammasomes. Cold Spring Harbor perspectives in biology, 6, a016287.
- Dempsey, C.; Rubio-Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O'Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain, Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef]
- DEZFULIAN, M. 2018. A new Alzheimer's disease cell model using B cells to induce beta amyloid plaque formation and increase TNF alpha expression. International Immunopharmacology, 59, 106-112.
- Di Benedetto, G.; Burgaletto, C.; Carta, A.R.; Saccone, S.; Lempereur, L.; Mulas, G.; Loreto, C.; Bernardini, R.; Cantarella, G. Beneficial effects of curtailing immune susceptibility in an Alzheimer’s disease model. J. Neuroinflammation 2019, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: the devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef] [PubMed]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a Major Cytokine in the Central Nervous System. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef]
- FAKHOURY, M. 2018. Microglia and astrocytes in Alzheimer's disease: implications for therapy. Current neuropharmacology, 16, 508-518.
- Ferrari, D.P.; Bortolanza, M.; Del Bel, E.A. Interferon-γ Involvement in the Neuroinflammation Associated with Parkinson’s Disease and L-DOPA-Induced Dyskinesia. Neurotox. Res. 2021, 39, 705–719. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef]
- Fu, A.K.Y.; Hung, K.-W.; Yuen, M.Y.F.; Zhou, X.; Mak, D.S.Y.; Chan, I.C.W.; Cheung, T.H.; Zhang, B.; Fu, W.-Y.; Liew, F.Y.; et al. IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc. Natl. Acad. Sci. 2016, 113, E2705–E2713. [Google Scholar] [CrossRef]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of Microglia in the Central Nervous System Diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef]
- FU, W.-Y. , WANG, X. & IP, N. Y. 2018. Targeting neuroinflammation as a therapeutic strategy for Alzheimer’s disease: mechanisms, drug candidates, and new opportunities. ACS chemical neuroscience, 10, 872-879.
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Gameiro, C.M.; Romão, F.; Castelo-Branco, C. Menopause and aging: Changes in the immune system—A review. Maturitas 2010, 67, 316–320. [Google Scholar] [CrossRef]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef]
- GLEICHMAN, A. J. & CARMICHAEL, S. T. 2020. Glia in neurodegeneration: Drivers of disease or along for the ride? Neurobiology of Disease, 142, 104957.
- Golde, T.E. Harnessing Immunoproteostasis to Treat Neurodegenerative Disorders. Neuron 2019, 101, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-amyloid Peptides and Amyloid Plaques in Alzheimer's Disease. Neurotherapeutics 2015, 12, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region−dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef]
- GUILLOT-SESTIER, M.-V. , DOTY, K. R., GATE, D., RODRIGUEZ JR, J., LEUNG, B. P., REZAI-ZADEH, K. & TOWN, T. 2015. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron, 85, 534-548.
- HANSLIK, K. L. & ULLAND, T. K. 2020. The role of microglia and the Nlrp3 inflammasome in Alzheimer's disease. Frontiers in Neurology, 11, 570711.
- Hein, A.M.; O’banion, M.K. Neuroinflammation and Memory: The Role of Prostaglandins. Mol. Neurobiol. 2009, 40, 15–32. [Google Scholar] [CrossRef] [PubMed]
- HENEKA, M. T. , CARSON, M. J., EL KHOURY, J., LANDRETH, G. E., BROSSERON, F., FEINSTEIN, D. L., JACOBS, A. H., WYSS-CORAY, T., VITORICA, J. & RANSOHOFF, R. M. 2015. Neuroinflammation in Alzheimer's disease. The Lancet Neurology, 14, 388-405.
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Hilzendeger, A.M.; Shenoy, V.; Raizada, M.K.; Katovich, M.J. Neuroinflammation in Pulmonary Hypertension: Concept, Facts, and Relevance. Curr. Hypertens. Rep. 2014, 16, 1–7. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Hu, M.; Lin, Y.; Zhang, B.; Lu, D.; Lu, Z.; Cai, W. Update of inflammasome activation in microglia/macrophage in aging and aging-related disease. CNS Neurosci. Ther. 2019, 25, 1299–1307. [Google Scholar] [CrossRef]
- Kamada, M.; Irahara, M.; Maegawa, M.; Yasui, T.; Yamano, S.; Yamada, M.; Tezuka, M.; Kasai, Y.; Deguchi, K.; Ohmoto, Y.; et al. B cell subsets in postmenopausal women and the effect of hormone replacement therapy. Maturitas 2001, 37, 173–179. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Kushwaha, S.S.; Patro, N.; Patro, I.K. A Sequential Study of Age-Related Lipofuscin Accumulation in Hippocampus and Striate Cortex of Rats. Ann. Neurosci. 2018, 25, 223–233. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Lindhout, I.A.; Murray, T.E.; Richards, C.M.; Klegeris, A. Potential neurotoxic activity of diverse molecules released by microglia. Neurochem. Int. 2021, 148, 105117. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Sun, Y.; Peng, G. Neuroinflammation as a Potential Therapeutic Target in Alzheimer’s Disease. Clin. Interv. Aging 2022, ume 17, 665–674. [Google Scholar] [CrossRef]
- Lučiūnaitė, A.; McManus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem. 2019, 155, 650–661. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef]
- Ma, J.; Choi, B.-R.; Chung, C.; Min, S.S.; Jeon, W.K.; Han, J.-S. Chronic brain inflammation causes a reduction in GluN2A and GluN2B subunits of NMDA receptors and an increase in the phosphorylation of mitogen-activated protein kinases in the hippocampus. Mol. Brain 2014, 7, 33–33. [Google Scholar] [CrossRef]
- Ma, Q.; Xing, C.; Long, W.; Wang, H.Y.; Liu, Q.; Wang, R.-F. Impact of microbiota on central nervous system and neurological diseases: the gut-brain axis. J. Neuroinflamm. 2019, 16, 1–14. [Google Scholar] [CrossRef]
- Manly, J.; Merchant, C.; Jacobs, D.; Small, S.; Bell, K.; Ferin, M.; Mayeux, R. Endogenous estrogen levels and Alzheimer’s disease among postmenopausal women. Neurology 2000, 54, 833–837. [Google Scholar] [CrossRef]
- MCGEER, P. L. , ROGERS, J. & MCGEER, E. G. 2016. Inflammation, antiinflammatory agents, and Alzheimer’s disease: the last 22 years. Journal of Alzheimer's Disease, 54, 853-857.
- Mejias, N.H.; Martinez, C.C.; Stephens, M.E.; de Rivero Vaccari, J.P. Contribution of the inflammasome to inflammaging. J. Inflamm. 2018, 15, 1–10. [Google Scholar] [CrossRef]
- Miguel-Álvarez, M.; Santos-Lozano, A.; Sanchis-Gomar, F.; Fiuza-Luces, C.; Pareja-Galeano, H.; Garatachea, N.; Lucia, A. Non-Steroidal Anti-Inflammatory Drugs as a Treatment for Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Treatment Effect. Drugs Aging 2015, 32, 139–147. [Google Scholar] [CrossRef] [PubMed]
- MISHRA, A. & BRINTON, R. D. 2018. Inflammation: bridging age, menopause and APOEε4 genotype to Alzheimer’s disease. Frontiers in aging neuroscience, 10, 312.
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Berti, V.; Guyara-Quinn, C.; McHugh, P.; Petrongolo, G.; Osorio, R.S.; Connaughty, C.; Pupi, A.; Vallabhajosula, S.; Isaacson, R.S.; et al. Perimenopause and emergence of an Alzheimer’s bioenergetic phenotype in brain and periphery. PLOS ONE 2017, 12, e0185926. [Google Scholar] [CrossRef] [PubMed]
- MULLER, G. C. , GOTTLIEB, M. G. V., CORREA, B. L., GOMES FILHO, I., MORESCO, R. N. & BAUER, M. E. 2015. The inverted CD4: CD8 ratio is associated with gender-related changes in oxidative stress during aging. Cellular immunology, 296, 149-154.
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflammation 2018, 15, 1–26. [Google Scholar] [CrossRef] [PubMed]
- O’neil, S.M.; Witcher, K.G.; McKim, D.B.; Godbout, J.P. Forced turnover of aged microglia induces an intermediate phenotype but does not rebalance CNS environmental cues driving priming to immune challenge. Acta Neuropathol. Commun. 2018, 6, 1–20. [Google Scholar] [CrossRef]
- Parikh, A.; Kathawala, K.; Li, J.; Chen, C.; Shan, Z.; Cao, X.; Wang, Y.-J.; Garg, S.; Zhou, X.-F. Self-nanomicellizing solid dispersion of edaravone: part II: in vivo assessment of efficacy against behavior deficits and safety in Alzheimer’s disease model. Drug Des. Dev. Ther. 2018, ume 12, 2111–2128. [Google Scholar] [CrossRef]
- PRINCE, M. J. , WIMO, A., GUERCHET, M. M., ALI, G. C., WU, Y.-T. & PRINA, M. 2015. World Alzheimer Report 2015-The Global Impact of Dementia: An analysis of prevalence, incidence, cost and trends.
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Puhl, A.C.; Milton, F.A.; Cvoro, A.; Sieglaff, D.H.; Campos, J.C.; Bernardes, A.; Filgueira, C.S.; Lindemann, J.L.; Deng, T.; Neves, F.A.; et al. Mechanisms of Peroxisome Proliferator Activated Receptor γ Regulation by Non-steroidal Anti-inflammatory Drugs. Nucl. Recept. Signal. 2015, 13, e004. [Google Scholar] [CrossRef]
- QIAN, L. , HONG, J.-S. & FLOOD, P. 2006. Role of microglia in inflammation-mediated degeneration of dopaminergic neurons: neuroprotective effect of interleukin 10. Parkinson’s Disease and Related Disorders. Springer.
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and Chemokines at the Crossroads of Neuroinflammation, Neurodegeneration, and Neuropathic Pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef]
- SANCHEZ-MEJIA, R. O. , NEWMAN, J. W., TOH, S., YU, G.-Q., ZHOU, Y., HALABISKY, B., CISSÉ, M., SCEARCE-LEVIE, K., CHENG, I. H. & GAN, L. 2008. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer's disease. Nature neuroscience, 11, 1311-1318.
- SCHELTENS, P. , DE STROOPER, B., KIVIPELTO, M., HOLSTEGE, H., CHÉTELAT, G. & TEUNISSEN, C. van der Flier, WM (2021, ). Alzheimer’s disease. Lancet, 397, 1577-1590. 24 April.
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef]
- SHEN, H. , GUAN, Q., ZHANG, X., YUAN, C., TAN, Z., ZHAI, L., HAO, Y., GU, Y. & HAN, C. 2020. New mechanism of neuroinflammation in Alzheimer's disease: the activation of NLRP3 inflammasome mediated by gut microbiota. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 100, 109884.
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [PubMed]
- Takechi, R.; Lam, V.; Brook, E.; Giles, C.; Fimognari, N.; Mooranian, A.; Al-Salami, H.; Coulson, S.H.; Nesbit, M.; Mamo, J.C.L. Blood-Brain Barrier Dysfunction Precedes Cognitive Decline and Neurodegeneration in Diabetic Insulin Resistant Mouse Model: An Implication for Causal Link. Front. Aging Neurosci. 2017, 9, 399–399. [Google Scholar] [CrossRef] [PubMed]
- THAWKAR, B. S. & KAUR, G. 2019. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. Journal of neuroimmunology, 326, 62-74.
- Uri-Belapolsky, S.; Shaish, A.; Eliyahu, E.; Grossman, H.; Levi, M.; Chuderland, D.; Ninio-Many, L.; Hasky, N.; Shashar, D.; Almog, T.; et al. Interleukin-1 deficiency prolongs ovarian lifespan in mice. Proc. Natl. Acad. Sci. 2014, 111, 12492–12497. [Google Scholar] [CrossRef] [PubMed]
- VENEGAS, C. , KUMAR, S., FRANKLIN, B. S., DIERKES, T., BRINKSCHULTE, R., TEJERA, D., VIEIRA-SAECKER, A., SCHWARTZ, S., SANTARELLI, F. & KUMMER, M. P. 2017. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature, 552, 355-361.
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A.; et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94. [Google Scholar] [CrossRef] [PubMed]
- VON BERNHARDI, R. , EUGENÍN-VON BERNHARDI, L. & EUGENÍN, J. 2015. Microglial cell dysregulation in brain aging and neurodegeneration. Frontiers in aging neuroscience, 7, 124.
- WALKER, K. A. , HOOGEVEEN, R. C., FOLSOM, A. R., BALLANTYNE, C. M., KNOPMAN, D. S., WINDHAM, B. G., JACK, C. R. & GOTTESMAN, R. F. 2017. Midlife systemic inflammatory markers are associated with late-life brain volume: the ARIC study. Neurology, 89, 2262-2270.
- Wang, H.-M.; Zhang, T.; Huang, J.-K.; Xiang, J.-Y.; Chen, J.-J.; Fu, J.-L.; Zhao, Y.-W. Edaravone Attenuates the Proinflammatory Response in Amyloid-β-Treated Microglia by Inhibiting NLRP3 Inflammasome-Mediated IL-1β Secretion. Cell. Physiol. Biochem. 2017, 43, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Coe, C.L.; Birdsill, A.C.; Bendlin, B.B.; Colman, R.J.; Alexander, A.L.; Allison, D.B.; Weindruch, R.H.; Johnson, S.C. Interleukin-8 and interleukin-10, brain volume and microstructure, and the influence of calorie restriction in old rhesus macaques. AGE 2013, 35, 2215–2227. [Google Scholar] [CrossRef]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2015, 53, 6709–6715. [Google Scholar] [CrossRef]
- Yin, F.; Yao, J.; Sancheti, H.; Feng, T.; Melcangi, R.C.; Morgan, T.E.; Finch, C.E.; Pike, C.J.; Mack, W.J.; Cadenas, E.; et al. The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity. Neurobiol. Aging 2015, 36, 2282–2295. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 55, 1977–1987. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, Z.; Hu, H.; Zhao, M.; Sun, L. Microglia in Alzheimer’s Disease: A Target for Therapeutic Intervention. Front. Cell. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Microglia, playing a central role in vicious cycle of neuroinflammation and related neuro-degeneration in AD.
Figure 1.
Microglia, playing a central role in vicious cycle of neuroinflammation and related neuro-degeneration in AD.
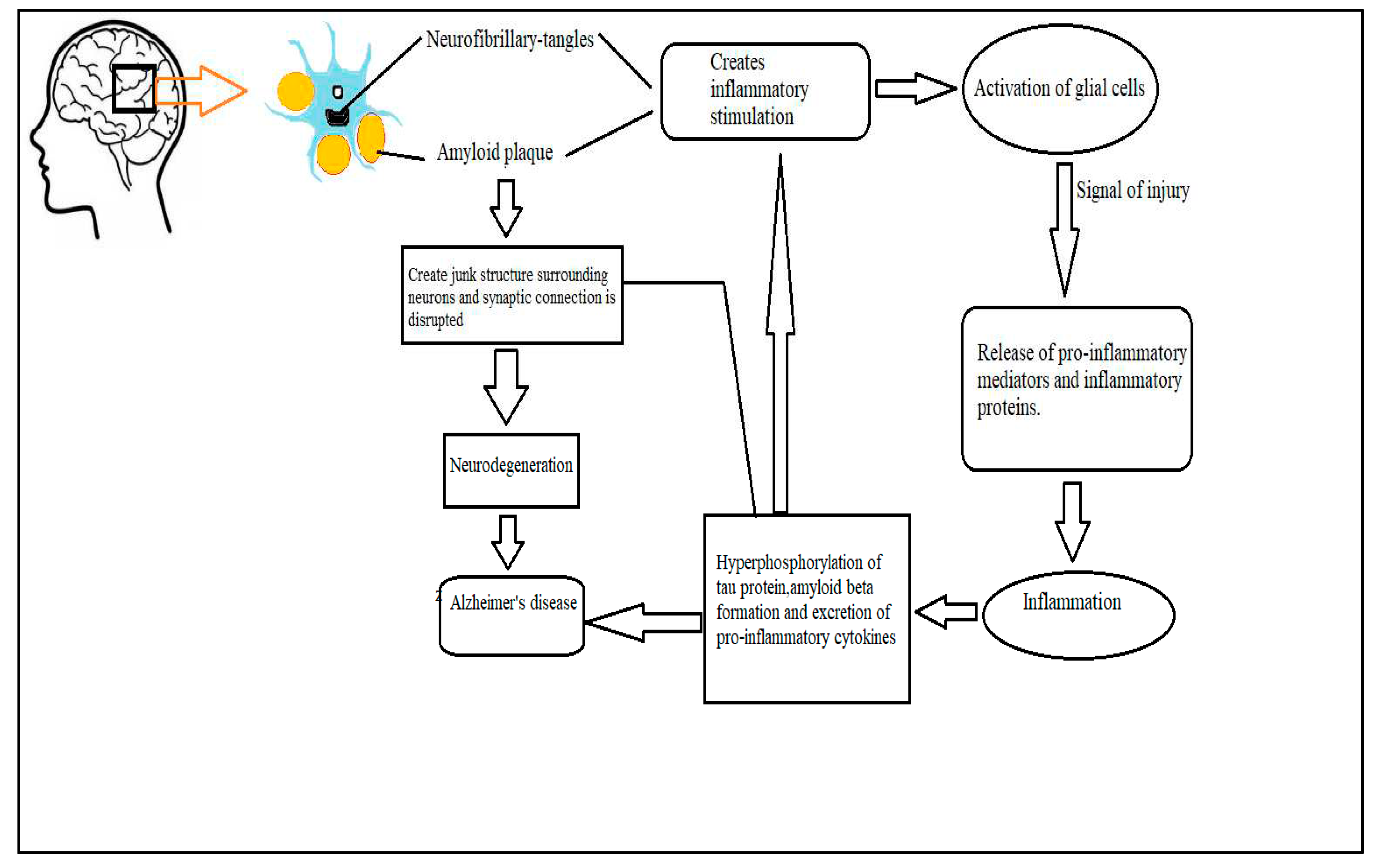
Figure 2.
Difference in characteristics of blood brain barrier of young and old brain inflammation (Newcombe et al., 2018).Young brains have intact BBBs with tight junctions, endothelial cells, pericytes and astrocytes (blue). Microglia (orange) consists of extended processes during homeostasis. During an inflammatory event, microglia prepare for phagocytosis to eliminate the inflammatory stimulus, such as a bacterial infection after an injury. On the other hand, monocytes can cross the BBB to enhance phagocytosis and debris clearance in the brain. Young brain microglia can return to surveillance after an inflammatory event. Age leaks the BBB structure continuously. Therefore, in aged brain, microglia reacts quickly to inflammatory stimuli but lose phagocytosis and infection clearing activity. If microglia can not remove the infection, it releases inflammatory mediators leading to increased inflammatory cell migration via the BBB which becomes permeable during disease (Newcombe et al., 2018).
Figure 2.
Difference in characteristics of blood brain barrier of young and old brain inflammation (Newcombe et al., 2018).Young brains have intact BBBs with tight junctions, endothelial cells, pericytes and astrocytes (blue). Microglia (orange) consists of extended processes during homeostasis. During an inflammatory event, microglia prepare for phagocytosis to eliminate the inflammatory stimulus, such as a bacterial infection after an injury. On the other hand, monocytes can cross the BBB to enhance phagocytosis and debris clearance in the brain. Young brain microglia can return to surveillance after an inflammatory event. Age leaks the BBB structure continuously. Therefore, in aged brain, microglia reacts quickly to inflammatory stimuli but lose phagocytosis and infection clearing activity. If microglia can not remove the infection, it releases inflammatory mediators leading to increased inflammatory cell migration via the BBB which becomes permeable during disease (Newcombe et al., 2018).
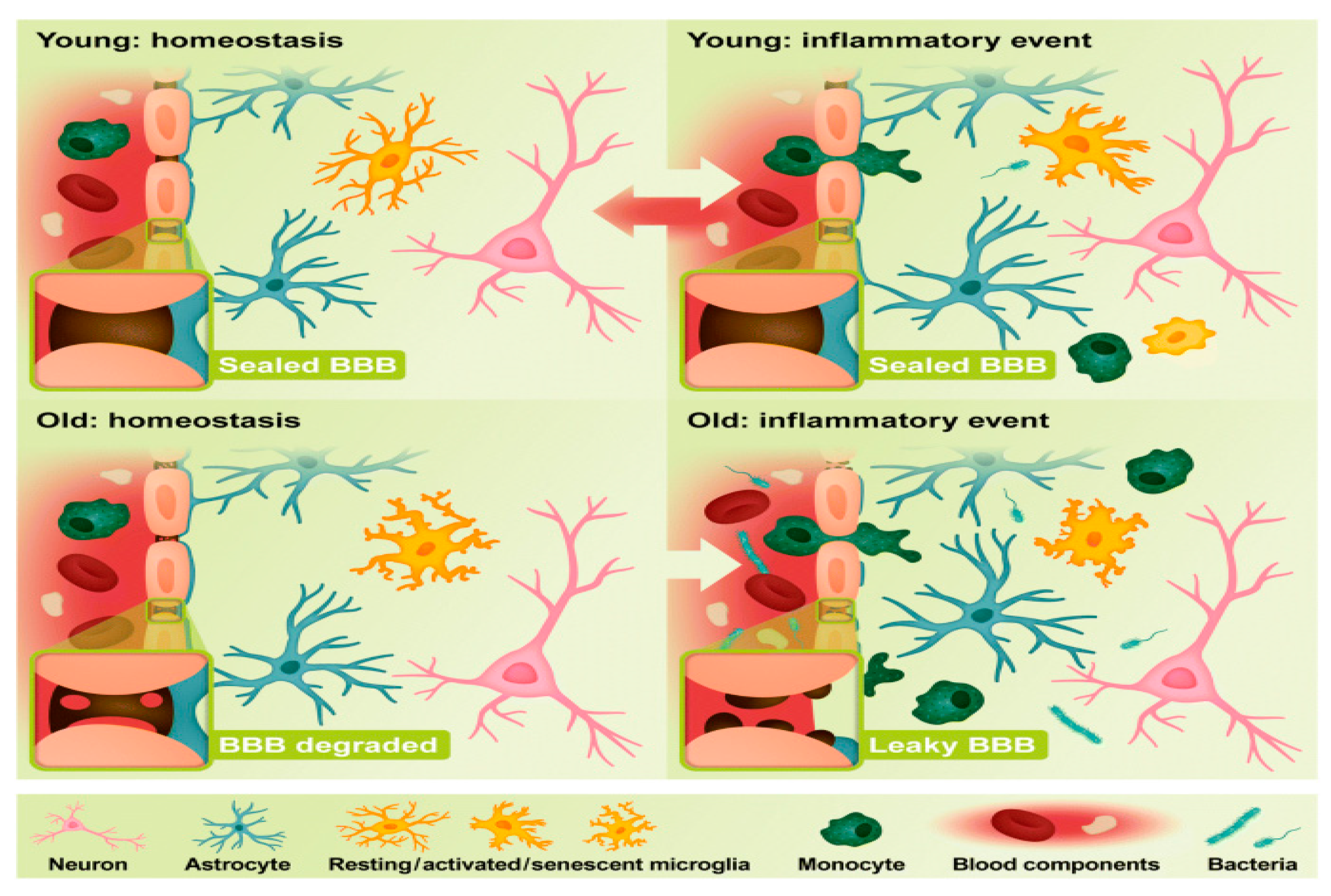

Table 1.
Neuroinflammatory mediators and their effects on neuro-disorders.
| Neuro-regulatory factors | Group | Name | Origin | Effects | Reference |
|---|---|---|---|---|---|
| Cytokines | Interleukins | IL-6 | IL-6 is produced by activated astrogliosis when neurons are injured. | Interleukins increase neuronal survival but are involved in several brain diseases | (Erta et al., 2012) |
| IL-1β | IL-1β is induced by Bacterial endotoxin | Causes neurotoxicity by producing excess glutamate | (Csölle and Sperlagh, 2010) | ||
| Type-IIInterferon | IFN-γ | IFN-γ is activated by natural killer cell (NK) .T-Cell may also induce IFN-γ. | Activates microglia and causes neuro-toxicity in Parkinson disease. | (Ferrari et al., 2021) | |
| The tumor necrosis factor, adipokine | TNF-α | TNF-α is generated by macrophages during systemic inflammation. | Involves in amyloid plaque production during Alzheimer’s disease. | (Dezfulian, 2018) | |
| Chemokines | Interleukin | IL-8R | Macrophages | Causes neurotoxicity by amyloid-beta formation. | (Willette et al., 2013) |
| Toll like receptor | TLR-4 | Extracellular factors induce Toll-like receptor | Mediates activation of microglial cell. TLR-4 reduces tau hyper-phosphorylation but induce amyloid beta production. | (Fiebich et al., 2018) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



