Submitted:
11 May 2023
Posted:
12 May 2023
You are already at the latest version
Abstract
The preparation of transition metal salt ammine complexes having oxidizing anions is a great challenge due to the reaction between the ammonia and oxidizing anions during the synthesis of these materials. However, they have an important role in the development of new oxidants in organic chemistry and especially as precursors in the preparation of mixed metal oxides and alloys in controlled temperature thermal decomposition reactions. Therefore, the synthetic procedures to prepare the complexes of transition metal permanganate, pertechnetate, and perrhenate (VIIB group tetraoxometallates) salts with ammonia as ligand or co-ligand have been comprehensively reviewed. The available data about the structure and spectroscopic properties of these compounds, including the hydrogen bonds that act as redox reaction centers under their thermal decomposition are given and evaluated. Their thermal decomposition processes and products have been summarized. The available pieces of information about their role in organic oxidation reactions like the oxidation of benzyl alcohols, and regeneration of oxo-compounds from oximes and phenylhydrazones including the kinetics of these processes have also been collected.
Keywords:
Ammonia
; ammine
; crystal structure
; synthesis
; spectroscopy
; hydrogen bond
; oxidation
; thermal decomposition
; spinel
1. Introduction
The temperature-controlled decomposition of transition metal complexes containing redox-active central atoms, ligands, and anions in a solid-phase quasi-intramolecular redox reaction become a widely used method to prepare nanosized oxides, nitrides, carbides, or metals and their alloys [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19]. The reducing ligands (pyridine, urea or ammonia) and oxidizing anions containing precursors frequently gave amorphous decomposition intermediates, which can be transformed into controlled-sized crystalline materials by heat treatment [7,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28]. Permanganate complexes and other tetraoxometallates result in the formation of various nanosized mixed oxides, especially MAMB2O4 spinels including the ones with various ratio of MA and MB cations at the tetrahedral or octahedral sites of the spinel lattice (MA is the cationic and MB is the anionic metal component) [29]. There are many possibilities to adjust the ratio of MA and MB metals in the formed oxides/spinels. For example, the variation of MA and MB cations ratio as in the case of [Co(NH3)6](MnO4)3 (Co:Mn=1:3), [Co(NH3)5Cl](MnO4)2 (Co:Mn=1:2), or [Co(NH3)6]Cl2MnO4 and [Co(NH3)4CO3]MnO4 (Co:Mn=1:1) with using inner or outer-sphere co-anions decomposes into gases. Another possibility is, the use of (MA1,MA2…..)((MB1O4,MB2O4,…..)n solid solutions or the changes in the valence of transition metals (e.g. CoII/CoIII, FeII/FeIII) or the charge or polymerization degree of the oxidizing anions (MnO4-/MnO42-, CrO42-/Cr2O72-). The isomorph solid solutions with metal-free tetraoxometallates anions (e.g., perchlorate, sulfate), which decompose into gaseous products [30,31,32,33] ensure the preparation of mixed crystals with partial substitution of the metal-containing anion with metal-free ones [34]. Two monovalent anions containing compounds may be isomorphic with compounds containing a divalent cation ad a neutral component, e.g. [M(NH3)4](XO4)2 (M=Zn, Cd, X=Cl, Mn) complexes are isomorphic with [Zn(NH3)4]MoO4.H2O [35,36,37].
The redox activity of each component (cations, anions, ligands) in these complexes, however, does not follow the redox potential order found for these species in aqueous solutions. These special oxidation properties of these complexes have enormous importance in organic synthesis as mild oxidants in organic media [11,14,38,39,40,41,42,43,44,45,46,47,48,49] and strong oxidants in an acidic aqueous environment [50,51,52,53,54].
One of the most promising groups of the abovementioned complexes are the ammonia complexes [34,55,56], especially the complexes with tetraoxometallate anions of VIIB group (Mn, Tc, Re). These complexes show wide variations in coordination number and geometry, which have influence on the temperature-initiated redox reactions in these complexes found not only between the anion and ligand [28,29,37,54,57,58] but in some cases between the oxidizing central ion (e.g., cobalt(III)) and ammonia as well [59,60]. The in-situ ammonia formation, e.g., from urea complexes [11], shows similar reactions as the ammonia complexes decompose with ammonia releasing together with the oxidation reactions. Therefore, in this review, the preparation, properties, thermal decomposition, and organic oxidation reactions of complexes formed from transition metal tetraoxometallate (XO4-, X=Mn, Tc, and Re) and ammonia are reviewed.
2. Discussion
2.1. General consideration on the synthesis of ammonia complexed transition metal tetraoxometallates (XO4-, X=Mn, Tc, Re).
In the preparation of compounds having redox active components, namely, transition metal complexes with ammonia ligands and oxidizing anions like permanganate ions, the main challenge is the preparation of the title compounds without oxidation of the ligand with the anion during the synthesis. Some of the easy synthesis routes, e.g., reactions of manganese heptoxide, permanganic acid or aluminum permanganate with basic precursors may not be used for ammonia complexes, due to the strong oxidation ability of the Mn2O7 or permanganate reactants [63,64,65]. The metathesis reactions using barium permanganate and sulfate compounds [64,66] are not advantageous, because the ammonia complexes of transition metal permanganates generally do not dissolve well in water, and only dilute solutions can be prepared at neutral or not controlled pH of the formed salt. The metathesis reactions of well-soluble transition metal ammine complexes, and water-soluble permanganates as sodium or potassium permanganate, however, ensures a chance for easy control of the oxidation potential of permanganate ion in the aqueous solutions used during the synthesis. The oxidation ability of pertechnetate and perrhenate ions much lower under analog conditions than that of permanganate ions, therefore the conditions is applicable for the synthesis of permanganates may expect to be useful for the synthesis of pertechnetate or perrhenate complexes. Therefore, our discussion is limited to permanganate ion: The Nernst equation can be written as
E=E0-(RT/nF)ln([RED]/[OX])
which unambiguously shows that the concentration of the oxidant and the temperature are key factors in these reactions. The pH controls the number of electrons (n) in the permanganate oxidations, namely, a permanganate oxidizes with five (acidic medium), three (neutral medium), or only one electron (basic medium), thus the pH control is one of the most important factors in these synthesis reactions. Therefore, the key factors in avoid of ammonia ligand oxidation with permanganate (or other tetraoxometallates) anions during the synthesis of the transition metal ammine-complex sats are, especially if we use metathesis reactions between the water-soluble salts of transition metal ammine complexes and soluble permanganate salts (K, Na or Ba salts [65]) are the following:
- 1)
- The low concentration of permanganate ion in the solution is advantageous and can easily reach if the permanganate complex prepared is sparingly soluble. In this case, the decreasing of solubility with salting out effect by using the excess of the most soluble salt (chloride, nitrate) of the complex cation and decreasing the temperature is advantageous. Decreasing the temperature is also advantageous. As starting salt in metathesis reaction with permanganates (transition metal ammonia complex salt, e.g., chloride, nitrate, etc.) is to be selected among the most soluble salts and used as a concentrated solution as possible.
- 2)
- The temperature decrease diminishes the oxidation ability of permanganate ions and generally decreases the solubility, thus acting as a main driving force in the removal of the oxidizing anion from the solution. Therefore, the typical synthesis can be done at and below room temperature, with immediate cooling of the solution until freezing.
- 3)
- The ammonia excess increases the pH and increases the concentration of the ammonia-complexed cations in the solution, thus using the ammonia excess is also a key step in these syntheses. It is the most important factor in decreasing the oxidation ability of permanganate ion.
Keeping these conditions, the majority of the known permanganate [65], pertechnetate and perrhenate [67] salts of transition metal ammonia complexes can easily be prepared. The other possible reaction route is when solid transition metal tetraoxometallates are reacted with gaseous or liquid ammonia in the absence of water. The coordination number of transition metal toward ammonia, however, depends on the nature of the metal and the synthesis conditions, and in general, the solid transition metal salts with ammonia without solvent resulted in higher coordination number complexes [68].
2.2. Diammine complexes
Only several silver(I), furthermore, diammine complexes of Zn, Cd and Ni have been known until now. Two diamminesilver permanganates and the hydrated form of [Ag(NH3)2]MnO4, furthermore, the perrhenate complexes of Ag(I), Zn(II), Cd(II), and Ni(II) [69] were described until now. No diammine complexes of metal pertechnetates have been described until now.
2.2.1. Preparation and properties of diamminesilver(I) permanganate. [Ag(NH3)2]MnO4
The first complex permanganate salt prepared in chemical history was the diamminesilver(I) permanganate. It was isolated by Klobb [70] in the reaction of an ammoniacal solution of silver nitrate with potassium permanganate at 10 °C. In an analog way, Scagliari and Marangoni [71] prepared a compound described as monohydrate, AgMnO4.2NH3.H2O. Bruni and Levi repeated this experiment, however, they found that the anhydrous salt was formed [68]. The [Ag(NH3)2]MnO4 explodes by percussion and gradually decomposes in air by losing ammonia. It forms violet rhombic plate-like crystals, sparingly soluble in cold, and better soluble in warm water [70] Its solubility is 3.6 g/100 mL of water at 20 °C [65]. However, in hot water, it easily hydrolyses [72,73] with ammonia evolution. Heating of the aqueous solution of [diamminesilver(I)] permanganate to remove the ammonia did not result in the expected AgMnO4, instead that ammonium permanganate and silver(I) oxide precipitate was formed. The dissociation of the complex cation with ammonia formation resulted in protonation of the liberated ammonia with ammonium and hydroxide ion formation, and the alter forms insoluble Ag2O, and ammonium ions are accumulated in the solution. During standing or heating of the solution, the permanganate ion concentration did not change, however, the cation concentration decreased[71,72].
The detailed study of Kótai et al. showed that half of the ammonia is evolved as gaseous ammonia, and the other part is protonated and left in the solution as ammonium ion [71,72,74].
2[Ag(NH3)2MnO4 + H2O = 2NH4MnO4 + Ag2O + 2NH3
The removal of the excess ammonia from the equilibrium with heating or using a vacuum fastens the process, whereas leaving the natural evaporation on standing at room temperature with a low ammonia vapor pressure above the solution, monocrystals of ammonium permanganate could be grown [74]. This reaction, because of the equilibrium between the dissociated-dissolved-evaporated ammonia keeps a constant high pH due to the presence of ammonia liberation and stabilizes the ammonium permanganate solution, thus a slow crystallization process could be done without oxidation of ammonia or ammonium ions in the solution. The other methods to prepare ammonium permanganate [65] resulted in ammonium permanganate in the reactions done at not alkaline conditions thus fast precipitation was used to remove that from the solution. The simplest preparation way, used acidic ammonium chloride [75], leading to mixed (K,NH4)MnO4 [65], which repeated recrystallization resulted in the enrichment of K-content [65,78,79].
Fogaca et al studied the earlier published synthesis routes and found that all methods gave the same diamminesilver(I) permanganate without crystalline water [34]. The synthesis of pure material was performed with the reaction of [Ag(NH3)2]NO3 and NaMnO4 solutions at room temperature. The anhydrous [Ag(NH3)2]MnO4 has two monoclinic polymorphs (a=7.9095 Å, b=6.0205 Å, c=12.6904 Å, β=98.056, V=598.34 Å3, d=2.896 P2/m, and a=7.8112 Å, b=6.0682 Å, c=13.1260 Å, β=96.4388, V=618.25 Å3, d=2.803, I2/m, for the low and high-temperature modifications, respectively. The phase transition temperature is 162.3 K, and the heat of transformation was found to be 1.107 kJ/mol.
Scagliari and Marangoni defined isomorphism between the diamminesilver(I) permanganate and perchlorate (both compounds were supposed to be monohydrate), because of forming purple mixed crystals with varying colors depending on the ratio of components [71]. The diamminesilver(I) perchlorate, however, has a monoclinic low-temperature (Tm-o=225.7 K) and an orthorhombic room temperature form. The low-temperature polymorphs of the permanganate and the perchlorate complex are isomorphous [30,34], however, the room-temperature polymorphs (monoclinic and orthorhombic) are different, although the orthorhombic cell is a special case of monoclinic one, with b=90°. Since the cell volumes are close to each other [30,34], the formation of continuous solid solutions could be observed according to the followings:
The reaction of [Ag(NH3)2]NO3 with (K,Na)(MnO4,ClO4)-containing solutions with smaller than 3:7 MnO4−/ClO4− molar ratio resulted in purple precipitates even at room temperature, whereas the solution with 1:1 MnO4−/ClO4− molar ratio requested cooling to obtain any crystalline product. The solid solution products with MnO4−/ClO4− >3 molar ratios (solution phase) could be prepared with the use of highly soluble NaMnO4 [65]. Increasing the MnO4−/ClO4− molar ratio in the solutions resulted in a continuous increase of the MnO4−/ClO4− ratio in the formed solid solutions. Monoclinic and orthorhombic solid solutions were isolated, without miscibility gap and the phase transformation occurred with ∼28 mol % permanganate content. The spectroscopic and XRD characterization of solid solutions are also described, but the thermal decomposition was checked only for the pure [Ag(NH3)2]ClO4 [80], which shows that the reaction routes are very different, thus the studies on the thermal decomposition of solid solutions is expected to give exciting new results.
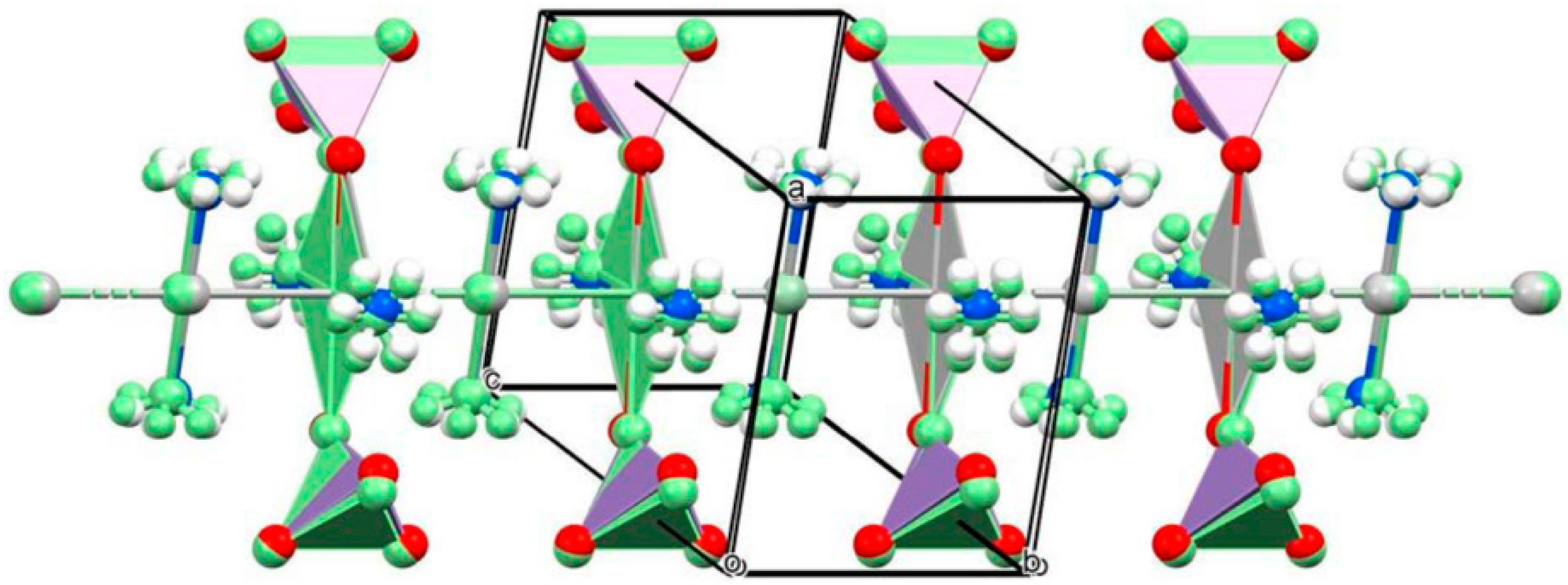
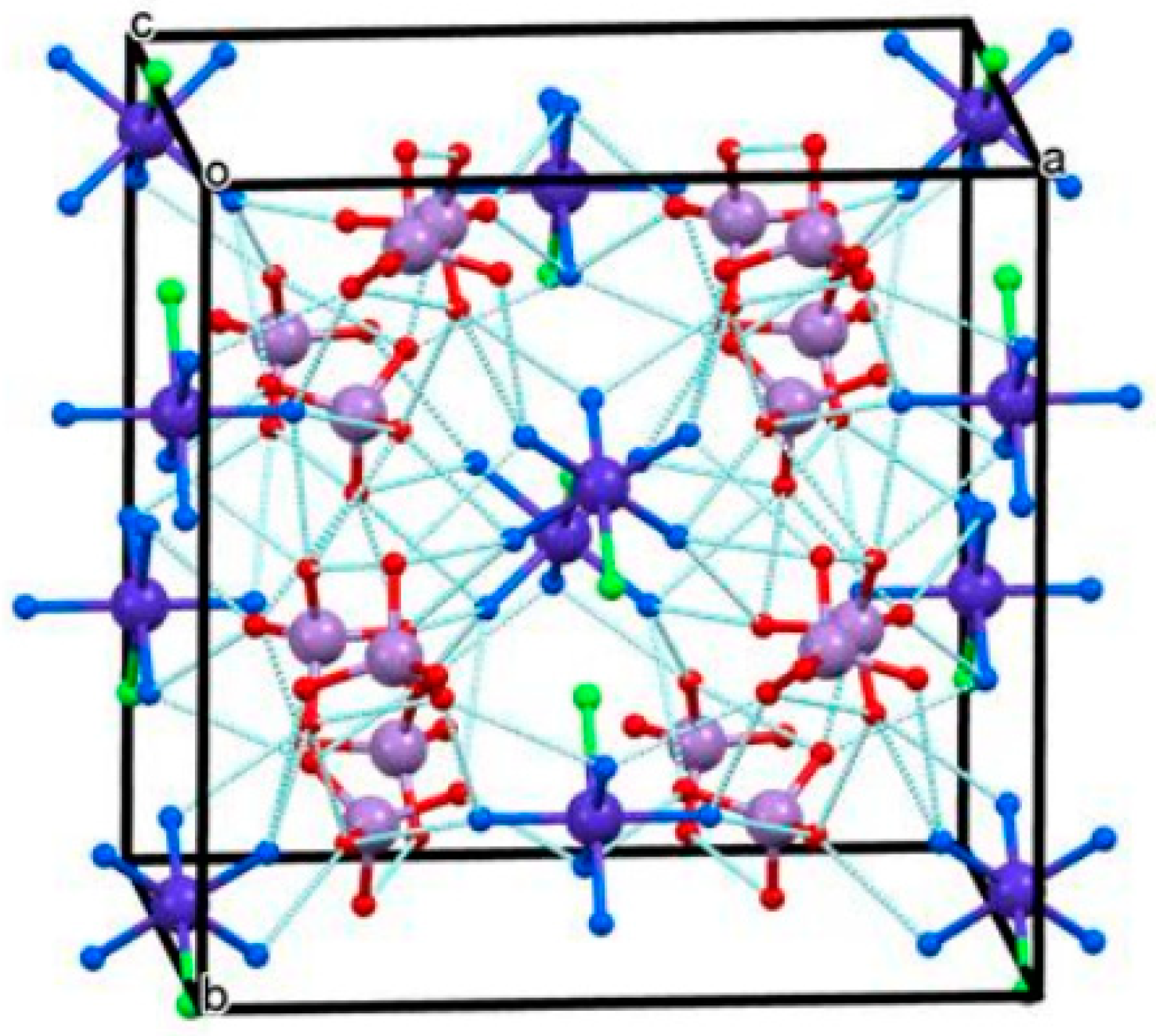
The asymmetric unit of the low- and high-temperature modification contains four quarters Ag, four halves of NH3, two halves of permanganate, and two-quarters of Ag, two halves of NH3, and a half of permanganate, respectively, due to higher symmetry of the high-temperature modification. A unique 3D coordination network is built up in both polymorphs with the formation of chain-like with Ag-Ag bonds, and the permanganate anions were coordinated by every second silver cation. The geometry around to every second Ag cation is octahedral (2-2 neighboring silver(argentophilic interactions), two permanganates, and two NH3 molecules. The axial silver cations have SP-4 geometry (two octahedral silver ions via argentophilic interactions and two ammonia ligands) (Figure 1). The ammonia ligands, the permanganates, and the silver cations are in all-trans arrangements. The high-temperature modification contains more distorted coordination polyhedrons than the low-temperature one.
The coordinated ammonia molecules are disordered in both structures. The infinite ladder-like chains of silver ions are parallel to the b-axis and coincide with the 2-fold rotation axes in both structures. The Ag− Ag distances are half of the length of the b-axis, and the Ag ions sit on the inversion centers in each polymorph. A detailed structural evaluation is given for both polymorphs [34]. Four and two crystallographically different permanganate ion environments were defined and evaluated in the correlation analysis of the low and high-temperature polymorphs, respectively. Their IR and Raman bands were assigned completely [34]. The hydrogen bond parameters and their influence on the IR and Raman spectroscopical features were also discussed in detail [34].
Figure 1.
The crystal structure of [Ag(NH3)2MnO4]. Reproduced from [34].
Figure 1.
The crystal structure of [Ag(NH3)2MnO4]. Reproduced from [34].
Exothermic decomposition of the complex can be observed at 85 °C with the liberation of one mol of ammonia, then a second decomposition step occurs at 204 °C. Water also forms in both steps, which shows that the ammonia and permanganate ions reacted with each other even in the first reaction step (the only hydrogen and oxygen sources for water formation may only be the ammonia and permanganate ions, respectively). The formal main decomposition step can be written as follows:
2[Ag(NH3)2]MnO4.H2O = 2[AgMnO2] + 3H2O + NH4NO3 + 2NH3
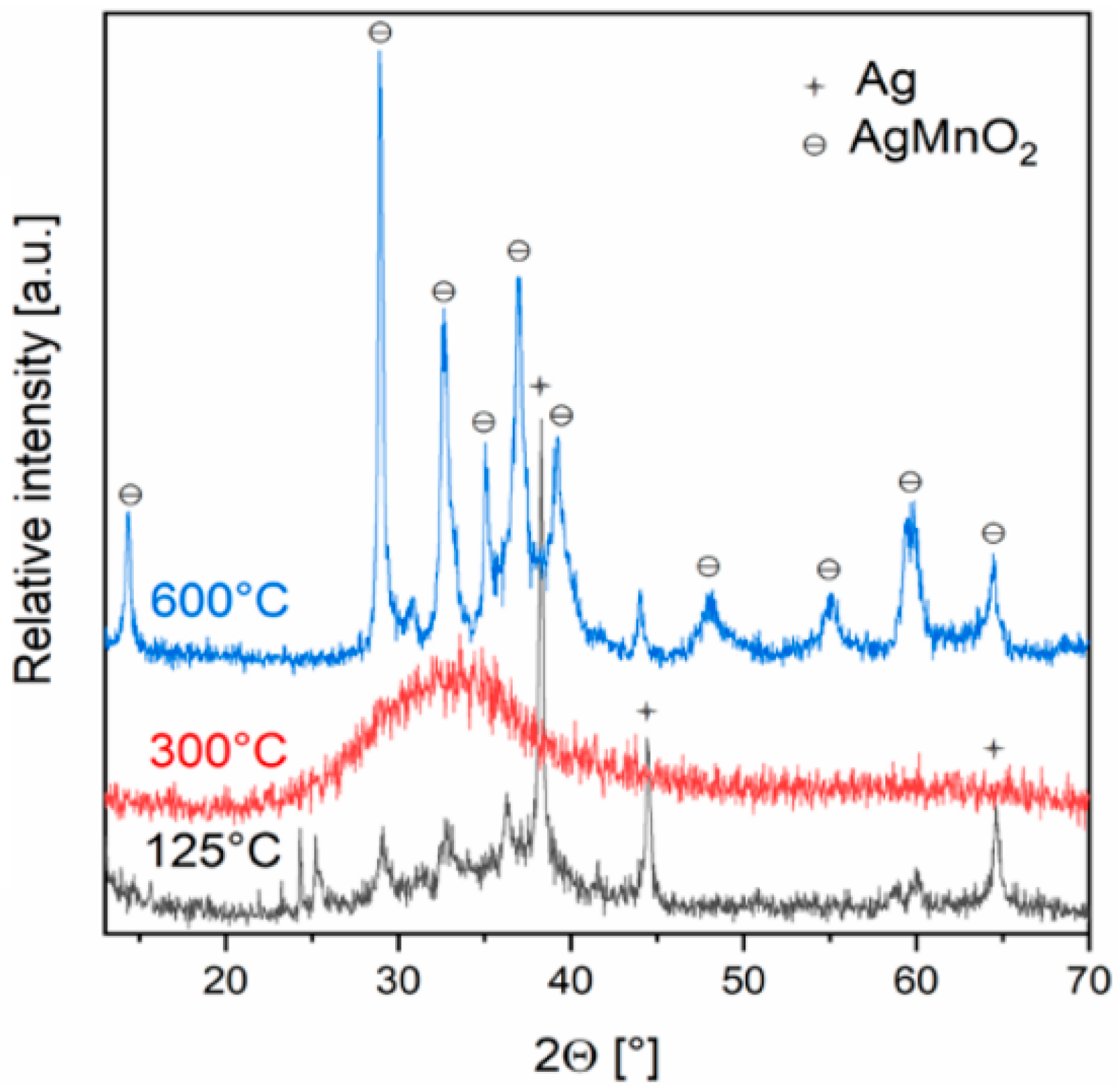
As can be seen, roughly ¼ part of ammonia is oxidized, ¼ parts turn into ammonium ion, and half part eliminate as ammonia gas. The low decomposition temperature of the NH4NO3 formed (the second decomposition step) was attributed to the catalytic effect of the silver-manganese oxide on the decomposition of NH4NO3. Based on the weight loss, the most probable amorphous decomposition product has an “AgMnO2” composition. Although the crystalline AgMnO2 is a known and stable compound of the silver-manganese-oxygen system [81,82], the thermal decomposition of [Ag(NH3)2]MnO4 gave a finely dispersed elementary silver and amorphous MnOx compounds mixture, together with H2O, N2 and NO as gases. The annealing of the solid primary decomposition product at 573 K, the metallic silver reacted with the manganese oxides and resulted in the formation of amorphous silver manganese oxides, which started to crystallize only at 773 K and completely transformed into AgMnO2 at 873 K (Figure 2) [34].
Figure 2.
Powder XRD of the decomposition product that formed from [Ag(NH3)2MnO4]. Reproduced from [34].
Figure 2.
Powder XRD of the decomposition product that formed from [Ag(NH3)2MnO4]. Reproduced from [34].
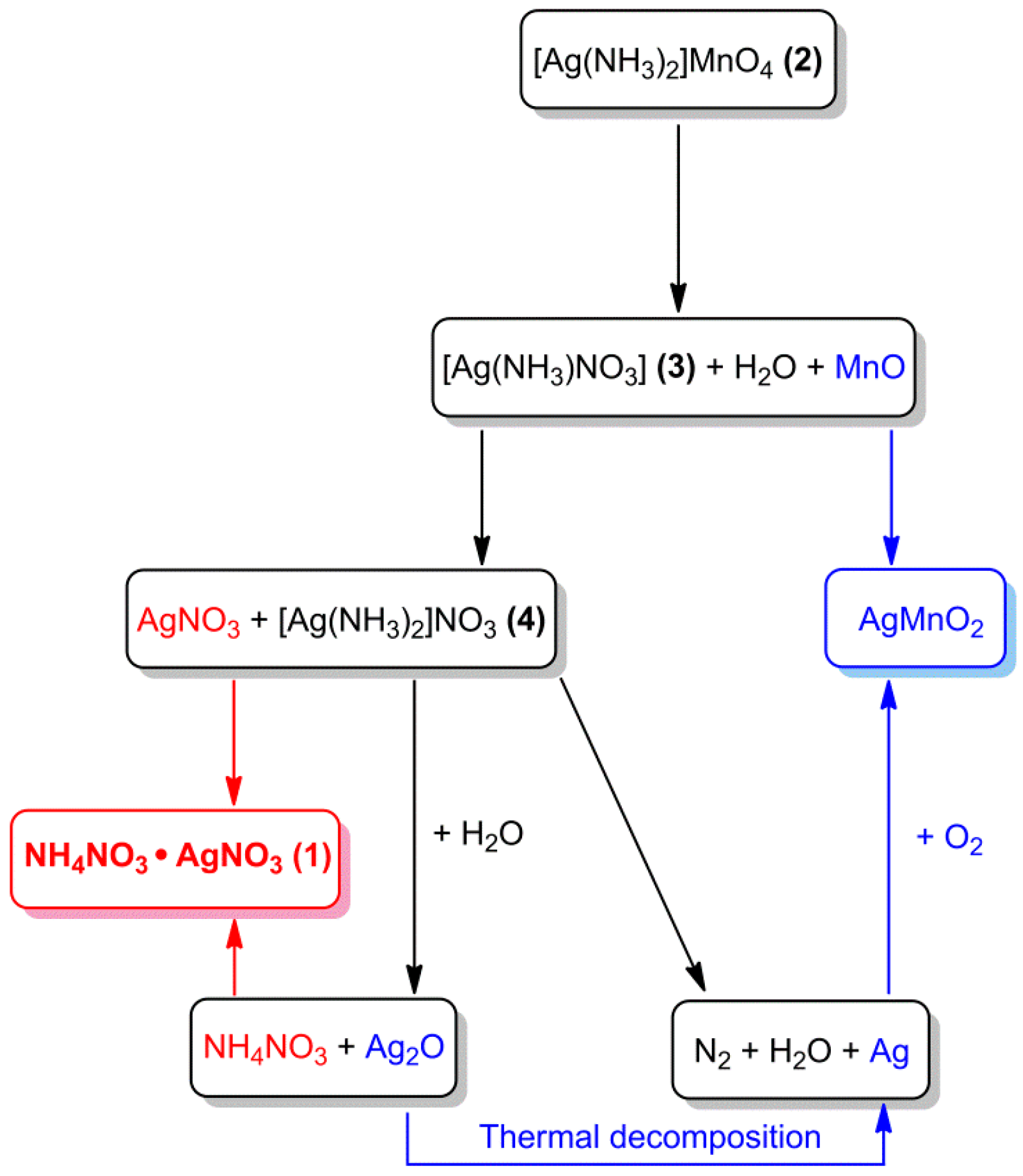
The mechanism of the thermal decomposition of [Ag(NH3)2]MnO4 is a complicated process. The overall oxygen amount in the starting complex taking into consideration of AgMnO2 formation is only 2 O atoms/mol. In the first decomposition step, a solid-phase intermediate was the unstable [Ag(NH3)NO3]. This compound decomposes into Ag and MnOx under further heating, but in the presence of water (aqueous leaching), this intermediate disproportionated into AgNO3 and [Ag(NH3)2]NO3. The hydrolysis of [Ag(NH3)2]NO3 during evaporation of the solvent resulted in insoluble Ag2O and soluble NH4NO3 [72,73], and the AgNO3 and NH4NO3 formed the double salt AgNO3.NH4NO3 isolated in the crystalline form[34,83]. The thermal decomposition process and hydrolysis of the decomposition intermediate are summarized in Scheme 1.
Scheme 1.
Decompositon scheme of [Ag(NH3)2]MnO4. Reproduced from [83].
Scheme 1.
Decompositon scheme of [Ag(NH3)2]MnO4. Reproduced from [83].
2.2.2. Diamminesilver(I) perrhenate
The diamminesilver(I) perrhenate was prepared first by Wilke-Dörfurt in 1933, in the reaction of silver perrhenate with ammonia in an aqueous solution and formed on cooling as colorless prisms [69]. The crystals are monoclinic and less sensitive to light than the silver perrhenate, its solubility in ammonia solution (d=0.930) at 20 °C was found to be 16.18 g/L. Its density in the solid state is 3.901 g/mL, molar volume is 100.5 cm3/mol [69]. No spectroscopic, structural, or thermal decomposition investigations have been done on this compound until now.
2.2.3. [Diamminezinc(II)], [diamminecadmium(II)], [diamminecopper(II)] and [diamminenickel(II)] perrhenates, [M(NH3)2](ReO4)2 (M=Zn, Cd, Ni)
The [M(NH3)2](ReO4)2 (M=Zn, Cd, Cu, Ni) complexes were prepared as air-stable non-hygroscopic white (Zn, Cd) and green (Cu, Ni) crystalline mass with the isotherm heating of the appropriate [tetraamminemetal(II)] perrhenate at 90° (Zn) or 150 °C (Cd, Cu, Ni). These complexes are practically insoluble in water and common organic solvents [84,85,86].
The IR and Raman spectroscopic studies on the complexes showed the appearance of the symmetric Re-O stretching modes and the splitting of the antisymmetric Re-O stretching modes, which led to the conclusion of the coordinated nature of the perrhenate ion in these complexes. The singlet symmetric stretching mode appears as two bands in the Raman spectra of Zn and Cd-complexes and the most probably coordination mode of perrhenate ion in these complexes is unidentate with C3v symmetry [86]. However, the well-separated doublet (20 cm-1) nature of the antisymmetric Re-O stretching mode in the Cu-complex [84] suggests unidentate or bridging perrhenate anion bonding [84]. The triplet and doublet nature of the antisymmetric Re-O bands of perrhenate ion in the IR and Raman spectra of [Ni(NH3)2](ReO4)2, respectively, were also taken as evidence of the symmetry lowering and coordinated nature of perrhenate ion [85].
The kinetic studies on the non-isothermal decomposition reaction of Zn complex between 230 and 275 ˘C resulting in [Zn(ReO4)2 showed the reaction order of 0.45 with 97.9 kJ/mol activation energy [86]. The Cu complex decomposition resulted in anhydrous copper perrhenate between 285 and 325 °C, with the reaction order of 0.47 and 44.3 kJ/mol activation energy [84]. The thermal decomposition of the cadmium complex follows a single mechanism, the activation energy values were found to be 103.1 and 102.7 kJ/mol calculated by the Kissinger-Akahira-Sunose (KAS) and Kissinger methods, respectively [61]. The decomposition of the nickel complex between 529 and 593 K shows a fluctuation of activation energy in the middle of a conversion, which suggests multiple mechanisms Increasing the conversion degree, a single mechanism was observed, and the activation energy values were found to be 369.66, and 365.66 kJ/mol, calculated by KAS and Kissinger methods, respectively. The decomposition product was nickel(II) perrhenate [62].
The measured magnetic moment of Cu-complex, 1.81 B.M, and its UV spectrum shows a tetragonal Cu(II) environment (14 000 cm-1) [84]. The value of magnetic moment for the Ni-complex (meff=3.07 BM) and that’s temperature independence shows similar hexacoordinated nickel(II) environment as was found in the [tetraamminenickel(II)] perrhenate. The d-d transitions in its UV-Vis spectrum are also similar to that of [tetraamminenickel(II)] perrhenate and show a tetragonal distortion (10400 and 12800 cm-1 (3B1g-->3B2g or 3B1g-->3Eg), 14300 and 16700 cm-1 (3B1g-->3A2g), 24400 (3B1g-->3Eg or 3B1g-->3A2g) [85].
2.3. Triammine complexes
Preparation and properties of [triamminesilver(I) permanganate, [Ag(NH3)3]MnO4
The reaction of solid silver permanganate with gaseous ammonia at 10 °C resulted in the triammine complex, [Ag(NH3)3]MnO4 in 72 h [68]. Klobb could not prepare any ammonia complex in the reaction of AgMnO4 and ammonia [70], because this compound does not exist at atmospheric pressure at room temperature [87]. The dissociation curve of [Ag(NH3)3]MnO4 was determined, and the dissociation pressure values at -21, 0, and 10 °C (synthesis temperature) were found to be 97, 330, and 670 mmHg, respectively [88].
2.4. Tetraammine complexes
Among the [M(NH3)4](XO4)2 complexes, only the permanganates and perrhenates of Cu, Zn, Cd, Pd(II), the pertechnetate of Pt(II), and the perrhenates of Pt(II), Ni(II), Co(II) have been prepared until now. There are known other complexes with four ammonia and further other ligands to form an octahedral environment around the central atom, e.g. [Co(NH3)4CO3]MnO4 and [Ru(NO)(OH)(NH3)4](ReO4)2.
2.4.1. Tetraamminecopper(II) permanganate
[Tetraamminecopper(II)] permanganate was prepared first in the reaction of an ammoniacal copper sulfate solution cooled to 8 °C with potassium permanganate pre-cooled to the same temperature [89,90]. Single crystals could be prepared in this reaction in an ice-cooled bath [91], but the ammonia excess at room temperature resulted in contamination [89]. Starting from [tetraamminecopper(II)] sulfate and potassium permanganate solution, a slower deposition rate or crystals were observed [89]. In order to prepare pure [tetraamminecopper(II)] permanganate, the best way is the reaction of [tetraamminecopper(II)] sulfate and sodium or potassium permanganate solutions with a 5/2 °C temperature gradient [76]. The reaction product formed by mixing the reactants at room temperature led to NH4MnO4 contaminated product due to the temperature-dependent hydrolysis of the complex cation [72,73,76] because of the followings: the saturated aqueous [Cu(NH3)4](MnO4)2 solution has a pH value of 9.60 indicates the partial dissociation of the complex cation into Cu2+, NH3 and [Cu(NH3)n]2+, n=1-3 species. The dissociation rate increases with increasing temperature. The released ammonia in the solution is partly protonated by the water to form NH4+ and OH- ions. Increasing the hydroxide ion concentration causes precipitation of the Cu2+ ions formed in complex dissociation equilibrium, removing the Cu2+ and OH- ions from the solution due to the low solubility product of Cu(OH)2 (L=4.8∙10-20). It shifts the complex decomposition and ammonia protonation equilibrium with the accumulation of free ammonium and permanganate ions, which due to the low solubility of NH4MnO4 [65] results in co-deposition of NH4MnO4 together with [Cu(NH3)4](MnO4)2 (the solubility product of [Cu(NH3)4](MnO4)2 is L = 7.81∙10-3 [73,76])
[Cu(NH3)4](MnO4)2 + 2H2O = Cu(OH)2 + 2NH3 + 2NH4MnO4
This equilibrium can be completely shifted to the right with the removal of the ammonia from equilibrium. The ammonia vapor pressure is higher than that of the water, furthermore, depends on the temperature, thus the pressure decreasing (vacuum) or temperature increasing (heating) can complete the hydrolysis process. This reaction route was used to prepare single crystals of highly pure alkali metal-free ammonium permanganate [73,76]. The remaining ammonium and permanganate ions were crystallized out as ammonium permanganate [74]
[Tetraamminecopper(II)] permanganate is a purple-black crystalline powder [89] or violet crystalline material [91], slightly soluble in water and in its aqueous solutions gradually decomposes and the initially beautiful purple color of the solution disappears, and manganese oxide is deposited [89]. It is soluble without similar decomposition in diluted sulfuric acid [89]. It also dissolves in polar organic solvents such as DMF and Ac2O but is insoluble in non-polar solvents like hydrocarbons and chlorinated solvents [76]. In its DMF solution, [tetraamminecopper(II)] permanganate dissociates completely into cation and anion (the antisymmetric stretching Mn-O mode of permanganate ion appeared as a sharp intensive singlet band at 900 cm-1), whereas the Cu-N region of the IR spectrum showed a very wide band system due to the solvation/ligand exchange of the complex cation with the DMF solvent [76]. The DMF solution of [Cu(NH3)4](MnO4)2 has an intense purple color which disappears within half an hour [76], and MnO2 formation was observed. In DMSO, it dissolves with the formation of a green compound that decomposes fast on standing [76].
It is stable in the dry state for several weeks, but on longer standing in a wet or impure state it easily decomposes with MnO2 formation, especially above 10 °C, and the sunlight increases the decomposition rate [89,91]. It detonates under the shock of the hammer; heating or crushing in a mortar [89,90], fusing with releasing ammonia, and producing a cloud of very finely divided oxides and voluminous lightweight and contoured ash [89].
Prismatic twinned single crystals were grown by slow evaporation of a saturated aqueous solution over concentrated H2SO4 at ~278 K, dexp(flotation) = 2.39 g/mL [91]. Its monoclinic elementary cell (Table 1) contains two isolated [Cu(NH3)4(MnO4)2] units with distorted octahedral copper(II) (4+2) coordination with four ammonia nitrogen atoms in the equatorial and two permanganate oxygen atoms in the axial sites [91].

Table 1.
Crystallographic data of [M(NH3)4](XO4)2 [tetraamminemetal(II)] permanganates, pertechnetates and perrhenates.
Table 1.
Crystallographic data of [M(NH3)4](XO4)2 [tetraamminemetal(II)] permanganates, pertechnetates and perrhenates.
| Compound | T, K | a,b,c, Å | a,b,g, ° | Space group | Z | V, Å3 | Dcalcd, g/mL | Ref. |
|---|---|---|---|---|---|---|---|---|
| M=Cu, X=Mn | 298 | 5.413 9.093 10.749 |
96.18 |
P21/m | 2 | 526.0 | 2.33 | [91] |
| M=Cu, X=Re | 150 | 6.5167; 6.7790; 7.4627 | 67.336; 80.004 70.687 |
P-1 | 1 | 3.661 | [92] | |
| M=Zn, X=Mn | 298 | 10.335 | F-43m | 4 | [37] | |||
| M=Zn, X=Re | 10.53 | F4-3m | 4 | 3.60 | [90] | |||
| 10.66 | F4-3m | 4 | [93] | |||||
| M=Cd,X=Mn | 298 | 10.432 | F-43m | 4 | [58] | |||
| 10.44 | 2.41 | [90] | ||||||
| M=Cd, X=Re | 10.53 | F4-3m | 4 | [94] | ||||
| 10.54 | [93] | |||||||
| 10.67 | 3.71 | [90] | ||||||
| M=Ni, X=Re | 9.2 5.2 6.7 |
1 | 3.22 | [85] | ||||
| M=Co, X=Re | 10.54 | F4-3m | 4 | 3.56 | [95] | |||
| M=Pd, X=Mn | 5.1746 7.5861 7.7217 |
69.313 78.872 76.883 |
P1 | 1 | 274.1 | 2.50 | [96] | |
| M=Pd, X=Re | 5.1847 7.7397 7.9540 |
69.531 79.656 77.649 |
P-1 | 1 | 290.19 | 4.37 | [96] | |
| M=Pt, X=Tc | 5.179 7.725 7.935 |
69.33 79.74 77.41 |
P-1 | 1 | 3.396 | [97] | ||
| M=Pt, X=Re | 298 | 5.1847 7.7397 7.9540 |
69.531 79.656 77.649 |
P1 | 1 | 290.19 | 4.370 | [98] |
| 12.70 8.91 5.09 |
104.1 | C2/m or Cm | 2 | 4.55 | [99] |
Due to the two crystallographically nonequivalent permanganate ions the IR and Raman bands are doubled, although among the 2x3 antisymmetric Raman bands only a quintet can be seen due to the overlapping (Figure 3).
Figure 3.
Raman spectrum of [Cu(NH3)4](MnO4)2. Reproduced from [76].
Figure 3.
Raman spectrum of [Cu(NH3)4](MnO4)2. Reproduced from [76].
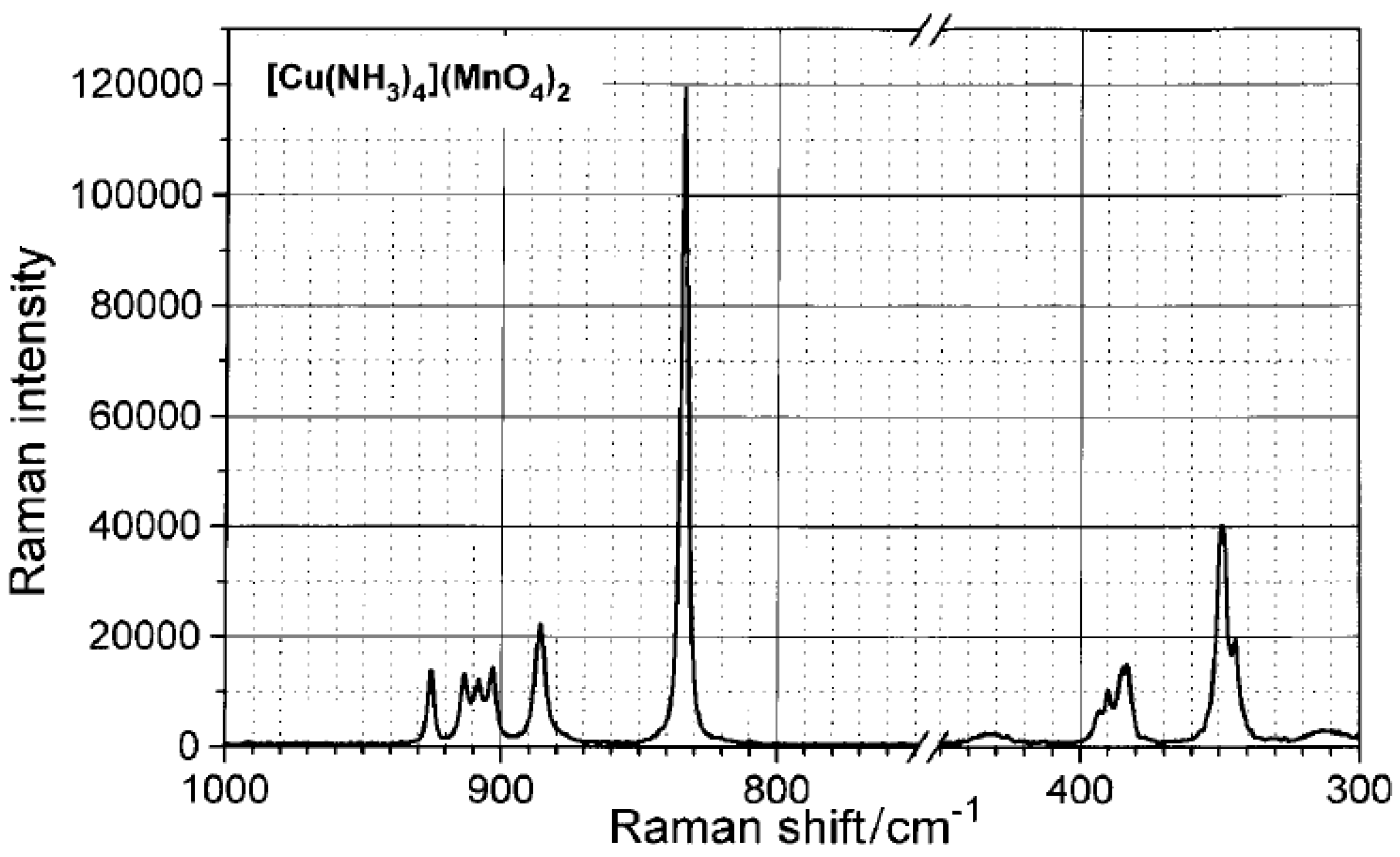
All of the IR and Raman bands belonging to the cation and anion modes were completely assigned. The shift and splitting of the hydrogen-bond sensitive rocking NH3 mode (ρr(NH3)) in the IR spectrum of [tetraamminecopper(II)] permanganate indicate the presence of weak N-H…O-Mn hydrogen bonds [76]. The ESR g-factors (gzz=2.273, gxx=gyy=2.090) are typical according to the O-ligation with a square-planar Cu environment. The sharpness of the parallel and perpendicular ESR bands of [Cu(NH3)4](MnO4)2, and the lack of Cu hyperfine structure show that the exchange interactions between the magnetically equivalent copper centers are stronger than the dipole couplings [76].
[Tetraamminecopper(II)] permanganate has unique thermal properties. Its heat of formation is 355 kcal/mol, and burns easily under oxygen pressure according to the equation [100]:
[Cu(NH3)4](MnO4)2 = Cu(liq.)+0.66Mn3O4+5.34H2O+0.44NH3+1.78N2
The heat of combustion is 152 kcal/mol, and the calculated combustion temperature is 1500 K. It starts to burn at 8 technical atm pressure with a dark red glow. It is a fast-burning material, um=16 g/cm2.s.gauge atm, tdel=<1 s at 280 °C [100].
The preliminary TG studies in air showed that [tetraamminecopper(II)] permanganate explodes at ~363 K giving a mixture of Cu and Mn oxides. It decomposes under N2 in at least two steps. Based on the weight losses, the decomposition mechanism was summarized as follows[91].
[Cu(NH3)4](MnO4)2 → Cu(MnO4)2 → CuMn2O4 –
-4NH3, 3013-423 K -2O2, 423-773 K
The intermediate phase Cu(MnO4)2 was given as amorphous whereas the CuMn2O4 was found to be crystalline [91]. Further studies, that used combined methods (TG-MS, DSC,) pointed out a much more complicated decomposition mechanism [76].
DSC studies showed a strongly exothermic decomposition reaction (the ammonia ligand loss is expected to be endothermic) in both steps, and the IR results of the decomposition intermediates did not show the presence of permanganate ion, whereas the IR spectrum of the intermediate formed at 250 °C contained a sharp unexpected peak at ~2200 cm-1. The shift of the Mn-O antisymmetric stretching modes to the low wavenumber region showed a strong decrease in the oxidation number of manganese [76]. These results completely coincide with the formation of [Cu(NH3)2](MnO4)2 and Cu(MnO4)2 intermediates given previously.
TG-MS and TG-gas titrimetric studies showed that in the two well-defined decomposition steps of [tetraamminecopper(II)] permanganate, 2 mol of ammonia, water, and N2O are formed without oxygen evolution. The two ammonia molecules were formed in the first step, together with H2O evolution, and in the second step, N2O and H2O could be detected as main products, but without any ammonia evolution. The decomposition residue is amorphous, its formula corresponds to the CuMn2O4+x stoichiometry. It does not dissolve in nitric acid; thus, it is not the stoichiometric mixture of CuO and Mn-oxides. Heating of the amorphous decomposition residue until 500 °C resulted in cubic CuMn2O4 spinel. The IR spectra of the decomposition intermediate showed the presence of ammonium nitrate confirmed by the XRD and IR of the evaporation residue crystallized out from the aqueous leachate. The formation of N2O and H2O in the 2nd step is attributed to the decomposition of NH4NO3, and the sharp peak in the IR of the decomposition intermediate belongs to the gas-inclusion of N2O [76]. The two-step process was described as follows [76]:
[Cu(NH3)4](MnO4)2 = CuMn2O4 + 2NH3 + NH4NO3 + H2O
NH4NO3 = N2O + 2H2O
In the first decomposition step, the oxidation of one ammonia ligand into nitrate and H2O is a strongly exothermic process and its reaction heat overflows the endothermicity of the two residual ammonia ligand loss. The reaction starts at 65 °C, which is lower than the temperature of the ammonia ligand loss of [tetraamminecopper(II)] cation, thus the ammonia-permanganate redox reaction takes place in solid state [76].
The temperature-controlled decomposition of [tetraamminecopper(II)] permanganate in CHCl3 and CCl4 at 61 and 77 ◦C, respectively, resulted in the formation of amorphous copper manganese oxide and ammonium nitrate mixtures. The oxygen surplus in the CuMn2O4+x oxides varied between x=0 and 0.35. The formation of (Cu,Mn)2O3 and (Cu,Mn)T-4(Cu,Mn)OC-62O4 oxides, their crystallite size and catalytic activity strongly depend on the heating temperature (CHCl3 or CCl4), or the removal of ammonium nitrate (aq. washing or heat treatment (Scheme 2) [28]).
Scheme 2.
Temperature-controlled decomposition scheme of [tetraamminecopper(II) permanganate]. Reproduced from [28].
Scheme 2.
Temperature-controlled decomposition scheme of [tetraamminecopper(II) permanganate]. Reproduced from [28].
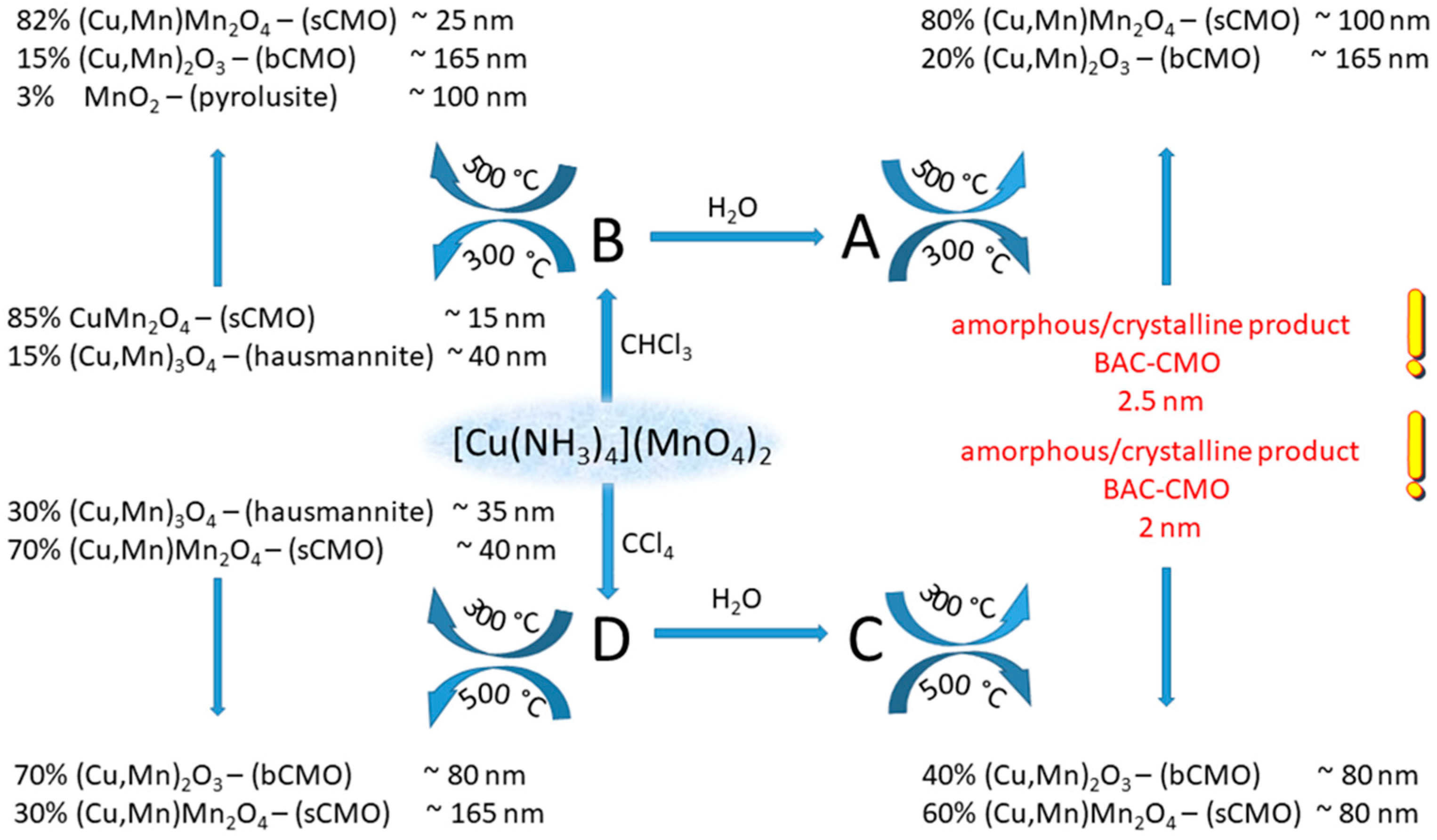
These copper manganese oxides were proved to be catalytically active in CO oxidation, thus [tetraamminecopper(II) permanganate] is a potential candidate in the preparation of Hopcalite-like catalysts [28].
The oxidative deoximation of aldoximes and ketoximes and oxidative regeneration of phenylhydrazones by [tetraamminecopper](II) permanganate in 1:1 aqueous acetic acid resulted in the appropriate oxo-compounds [91,101]. Benzaldehyde and acetophenone oximes are deoximated at room temperature with 82 and 81 % yield, respectively. The oxidative regeneration of acetaldolphenylhydrazone resulted in a three-electron reduction of [tetraamminecopper(II)] permanganate with the formation of MnIV. No influence of acrylonitrile on the reaction, and no acrylonitrile polymerization (lack of free radicals) was observed [55,56].
The kinetics of the oxidative regeneration of oximes (278-298 K) and phenylhydrazones (288-318 K) of H-C(=O)-R1 aldoximes (R1=H, Me, Et, Pr, i-Pr, ClCH2, Ph) and R1-C(=O)-R2 ketoximes (R1=Me, R2=Me, Et, Ph and R1=R2=Et) were studied in 1:1 aq. acetic acid. The reactions are first order for the organic components and oximes and [tetraamminecopper(II)] permanganate as well. The oxidation rate of keto-derivatives is slower than that of aldehyde-derived compounds. The substituent-dependent regeneration reaction rates can be described by the Pavelich-Taft dual substituent-parameter equation. The low positive values found for the polar reaction constants indicate a nucleophilic attack by a permanganate-oxygen on the double-bond carbon atom. The low activation enthalpy values indicate that the bond cleavages and bond formations are almost synchronous. The large negative values of activation entropies support the formation of a rigid cyclic activated complex. The alkyl group’s steric hindrance has an influence on the reaction. The reaction rate-determining step is the formation of an acyclic intermediate [55,56].
The [tetraamminecopper(II)] permanganate oxidizes benzyl alcohol in CHCl3 solution into benzaldehyde with 67% yield in 3 h reflux, but only 4 % benzonitrile (4%) was formed. Increasing the reflux temperature (CCl4) increased the nitrile yield to 14% in 3 h. This indicates that the ammonia could not be liberated easily from the coordination sphere to react with the aldehyde formed. The addition of coordinating solvents to substitute ammonia in the coordination sphere of the complex cation (DMF or CH3CN) suppressed the nitrile formation due to the stable solvated ammine complex formation [65]. It shows that the stability of the NH3 source is a key factor in the ammoxidation reactions of benzyl alcohol into benzonitrile, because the ammonium permanganate [102], or [hexakis(urea)iron(III)] permanganate (urea acts as ammonia precursor) [11] results in much more benzonitrile formation than the [tetraamminecopper(II)] permanganate [65].
The oxidation of benzyl alcohol and its ortho (Me, OMe, NO2, COOMe, F, Cl, Br, I, CN, NHAc, SMe, CF3), meta (Me, OMe, F, Cl, NO2, CF3, COOMe, Br, NHAc, CN, SMe) and para (Me, OMe, Cl, Br, F, NO2, COOMe, CF3, CN, SMe, NHAc, NMe2) monosubstituted benzyl alcohols by [tetraamminecopper(II)] permanganate in aqueous acetic acid resulted in the formation of the corresponding benzaldehydes. The kinetics of these reactions were measured between 288 and 318 K, moreover, the rate constants and activation parameters were calculated. The reactions are first order regarding the oxidant, substrates, and hydrogen ions. The oxidation of PhCD2OH exhibits a substantial temperature-dependent kinetic isotope effect (kH/kD at 298 K was found to be 5.83). The reaction rate increases with an increase with the polarity of the solvent. The oxidation rates of meta- or para, and the ortho-substituted benzyl alcohols correlated in terms of Charton’s triparametric LDR and tetraparametric LDRS equations, respectively. The oxidation of para-substituted benzyl alcohols is more sensitive to the delocalization effect than that of the ortho- and meta-substituted derivatives, which show a great dependence on the field effect. The positive h values suggest the presence of the electron-deficient reaction centers in the rate-determining step. The ortho substituents show steric acceleration [77].
2.4.2. Tetraamminezinc(II) and [tetraamminecadmium(II)] permanganates
[Tetraamminezinc(II)] and [tetraamminecadmium(II)] permanganate were prepared first by Klobb as a fine purple powder in the reaction of 0.2 M zinc and cadmium sulfate, ammonium hydroxide, and a saturated potassium permanganate solution at 10 °C. To avoid contamination the Zn-complex that formed must be filtered very quickly [89,90]. The Cd-complex cannot be dried over lime or sulfuric acid without decomposition, but 48 h drying over P2O5 resulted in black crystals/purple [tetraamminecadmium(II)] permanganate in 48 h [89].
Dark purple microcrystalline [M(NH3)4](MnO4)2 (M=Zn, Cd) complexes were prepared in a pure state in the reaction of the saturated aqueous [tetraamminezinc(II)] or [tetraamminecadmium(II)] sulfate and KMnO4 solutions with +5/+2 °C temperature gradient [37,58,90]. [Tetraamminezinc(II)] and [tetraamminecadmium(II)] permanganates are very fine violet crystalline solids [89,90], dexp=2.27 g/mL (M=Zn) [90]. They explode on rubbing or crushing in a mortar [89,90], under the shock of the hammer; or on heating, fuses with releasing ammonia and producing a cloud of very finely divided oxides. They give a brown insoluble powder after 1-2 h (Zn) or a few days (M=Cd) storage at room temperature[89]. In a wet form, the Zn-complex easily decomposes if exposed to light [89] but is stable in dry conditions at room temperature [37].
Both complexes are slightly soluble in water (0.91 g/100 ml H2O at 19 °C for the Zn compound [37]), but their aqueous solutions lose their beautiful purple color and quickly manganese oxide is deposited [58,89]. Their hydrolysis-reaction proceeds in aq. solutions, with the formation of ammonium permanganate and zinc(II) or cadmium(II) hydroxide [58]. They dissolve in diluted sulfuric acid and no sign of similar decomposition could be occurred [89].
The powder X-ray data of [tetraamminezinc(II)] and [tetraamminecadmium(II)] permanganates (d values and their relative intensities), including Müller indices are given [90]. Single crystal studies showed some additional weak reflections indicating a face-centered cubic supercell with Z=32 and a=20.62 Å (Zn) and a=20.88 Å (Cd) values. These large cells show a slight distortion of the tetrahedral units since the intensities of the weak additional reflections were calculated to be zero, assuming an exactly tetrahedral environment. The space group for the structures with a larger cell could not be determined exactly, probably is Td2-F4-3m or F4-3c-Td [90]. Due to the lack of good-quality single crystals their structures were solved from PXRD data with Rietveld refinement using the atomic coordinates of the isostructural [Zn(NH3)4](ClO4)2 [37,58]. [Tetraamminezinc(II)] and [tetraamminecadmium(II)] permanganates crystallize in a closely packed cubic structure (Table 1) and built up a three-dimensional M-N-H....O-Mn hydrogen-bonded network with block-like structural motifs of four M(NH3)42+ and four MnO4- ions (M=Zn, Cd). Only one of the two permanganates of each complex takes part in the building up of the 3D network, the second kind of permanganate ion is captured in the cavities enclosed by the tetramer building blocks of the network [37,58]. There are significant differences between the strength of the N-H...O-Mn hydrogen bonds of the 3D network forming and the cavity-embedded permanganate ions. The hydrogen bonds between the ammonia hydrogens and the cavity-embedded permanganate ions hinder the free rotation of the embedded permanganate ion, although its rotational freedom is even higher than that of the network-fixed permanganate ion [37,58].
The IR and Raman bands of [tetraamminezinc(II)] and [tetraamminecadmium(II)] permanganates were completely assigned, and the cation and anion modes are listed. The splitting of antisymmetric Mn-O stretching bands (F2) could not be explained well by simple site-symmetry considerations based on Td2 space-group, therefore dynamic effects through the interaction of neighboring ions in a Z=4 unit cell (factor group splitting neglected in site symmetry considerations) are noticeable [90].
Due to the presence of two crystallographically different tetrahedral permanganate ions in the lattice, the IR and the Raman bands belonging to the Mn-O modes are doubled. The MN4 skeleton (M=Zn, Cd) of the complex cation also has tetrahedral geometry. The factor group analysis resulted that the degeneration of the antisymmetric modes (F2) in this crystallographic environment due to the symmetry relations should not be ceased, and the symmetric IR modes should not appear. The appearance of the IR forbidden symmetric (νs and δE) modes and the splitting of the triply degenerated (F2) antisymmetric modes (νas and δas) was attributed to the orientation effects of cavity embedded permanganate ion, which strengthened with decreasing the temperature [37,58]. The increase of the νas(Mn-O)/νs(Mn-O) integrated intensity ratio values by decreasing the temperature from 293 to 173 K indicates that the permanganate-ion orientation is frozen, whereas the dynamic lattice distortion due to slowing down the anisotropic thermal motions would cause change with an opposite sign under cooling. The IR bands belonging to cavity-embedded permanganate were identified [37,58].
The thermal decomposition of [tetraamminezinc(II)] and [tetraamminecadmium(II)] permanganates under an inert atmosphere proceeds in 2+1 steps with the formation of an amorphous decomposition product with an MMn2O4+x (x=0-0.35) formula (M=Zn, Cd), which transforms at 500 °C. The peak temperatures of the first decomposition step strongly depend on the heating rate and vary between 100 and 130 °C. Two molecules of ammonia are released only in the first decomposition steps at 100 °C, the other two transform into ammonium nitrate and water. No O2 evolution occurs. In the second decomposition step, the ammonium nitrate decomposition could be observed, which is catalyzed by the presence of MMn2O4 (M=Zn and Cd) spinels at 230 and 224 °C, respectively.
The main decomposition processes are the following:
[M(NH3)4](MnO4)2 = MMn2O4 + NH4NO3 + 2NH3 + H2O (M=Zn, Cd)
NH4NO3 = N2O+ 2H2O
All decomposition steps are exothermic, the overall reaction heats are ∆H = -169 and -318 kJ/mol, which shows that the reaction heats of the ammonia-permanganate reactions in these complexes are higher than the energy demands of the partial de-ammoniation reactions [37,58].
In toluene as heat-convection media, the reaction temperature is controlled at 110 °C by the evaporation of toluene (reflux). Leaching of the amorphous decomposition residue with water (removal of NH4NO3) resulted in amorphous MMn2O4+x (x=0-0.35) (M=Zn, Cd) compounds, and the leaching solutions evaporation gave crystalline ammonium nitrate [37,58].
2.4.3. [Tetraamminecopper(II)], [tetraamminezinc(II)] and [tetraamminecadmium(II)] perrhenates
[Tetraamminecopper(II)] perrhenate was prepared first by Briscoe et al. [103] by adding aq. concentrated ammonium hydroxide or gaseous ammonia to a concentrated hot copper(II) perrhenate solution until the formed precipitate completely re-dissolved and then the deep blue crystals were crystallized out after cooling the solution [103]. The copper(II) perrhenate could be prepared in situ from CuCO3 and HReO4, and the concentrated solution that formed was mixed directly with concentrated ammonia [84]. [Tetraamminecopper(II)] perrhenate was also be prepared in the metathesis reaction of ammoniacal copper sulfate with ammonium perrhenate or perrhenic acid [104] between 20 and 60 °C. The ammonia added to copper sulfate solutions caused appearing a deep blue color, which changes into dark violet on adding perrhenic acid. Neither the ammonia nor the perrhenic acid excess caused the formation of any other insoluble phase. The optimal concentration of each reactant to prepare the maximal yield of [Cu(NH3)4](ReO4)2 has been determined[104]. It was also prepared from hot aq. solution of copper(II) acetate mixed with 1:1 aq. ammonia to get pH 11-12, with 2 equivalent of concentrated aq. solution of sodium perrhenate. On cooling, a dark-lilac crystalline precipitate formed with a yield of ~77% [92].
[Tetraamminezinc(II)] and [tetraamminecadmium(II)] perrhenates were synthesized from in situ-prepared concentrated metal perrhenate (M=Zn, Cd, from zinc or cadmium carbonate and 0.004 M perrhenic acid) solution and concentrated aqueous ammonia solution [61,69,94]. Zagorodnyaya et al prepared [tetraamminezinc(II)] perrhenate with the addition of zinc sulfate then 0.025-0.1 M ammonium perrhenate solutions to 20 % excess of aq. ammonia at room temperature [105]. [Tetraamminecadmium(II)] perrhenate and its deuterium and 116/110Cd isotope-containing samples were prepared in a similar way from Cd(ReO4)2 and 110/116Cd(ReO4)2 (prepared by the reaction of CdCl2, 110/116CdCl2 and AgReO4, respectively) with aq. ammonia or deuterated ammonia in heavy water [106]. It also formed during the processing of rhenium-containing lead dust, when rhenium generally follows the cadmium during the extraction of Zn and Cd chloride and sulfate-containing solutions with trialkyl amines with subsequent re-extraction with aq. ammonia [107]. The bluish violet (Cu) and colorless (Zn, Cd) microcrystalline substances were purified by recrystallization from warm concentrated ammonia [69,84].
[Tetraamminecopper(II)] perrhenate is a blue [90] or bluish violet[94,103], the Zn and Cd-complexes are white powder or colorless crystals [69,86,90]. They are non-hygroscopic, insoluble in water and the common organic solvent [69,86,105]. The Cu-complex is stable in air until 100 °C [103], starts to decompose at 140 °C, and on further heating in air turns into pale green materials and then melts with darkening and completely decomposes [84,103]. It forms regular prismatic anisotropic violet-blue single crystals with medium to high birefringence. Distinct pleochroism is from pale blue to dark purple, the optical phase is biaxial and negative. Refractive indices: np=1.635-1.64; ng=1.655-1.660 [104].
Both the Zn and Cd-complexes consist of regular cubes, but the Zn-complex forms octagons or rectangles as well [86,105]. The single crystalline Zn-complex is isotropic (refractive index is n=1.627) [105], its density and molar volume are d=3.608 g/mL and 175.75, respectively [69,90].
There is controversial information about the solubility of [tetraamminezinc(II)] and cadmium perrhenates, which are given as insoluble material in water [86], but Zagorodnyaya gave the solubility of Zn-complex as 6.23 g/100 g water [105]. The solubilities of the Zn and Cd-complexes in ammonia (d=0.930) at 20 °C is 1.852 g/L [69] (increases from 0.128 to 0.359 g/100g solution increasing the ammonia concentration from 1.2 to 12.0 M [105]) and at 11 °C is 0.37 g/L, respectively [69]. The pycnometric density and molar volume of the Cd-complex are d=3.714 (25 °C) g/mL [69] or 3.72 g/mL[90] and 183.5 [69].
Powder X-ray studies showed that [tetraamminezinc(II)] perrhenate is isomorphous with the analog Cd and Co-complexes [95]. The powder-X-ray diffractogram of the Cu, Cd, and Zn-complexes, including the d-values and intensity data were discussed [84,90,95,104,105]. The Miller indices were given only for the Zn and Cd complexes [90,95]. Their single crystal studies showed some weak additional reflections showing a face-centered cubic supercell with a=21.06 Å, Z=32, Td2-F4-3m (Zn, Cd). This large cell indicated a slight distortion of the tetrahedral MN4 (M=Zn, Cd) and ReO4 units since the intensities of the weak additional reflections were calculated to be zero, assuming exactly tetrahedral environments [90]. All three complexes have face-centered cubic lattices (Table 1). The lattice constant of the Cd-complex agrees satisfactorily with the calculated one from the experimental density (d254 = 3.714, a0 = 10.66 Å,) when four molecules are taken to the unit [94]. Both the Zn and Cd-complexes are isostructural with [M(NH3)4](MnO4)2 (M=Zn, Cd) complexes as well [106]. The cadmium complex has a first order order-disorder type phase transition at 368 K with 2.0 kJ/mol enthalpy change and 8 K hysteresis during the DSC measurements [61].
The IR bands of the [tetraamminecopper(II)] perrhenate and their assignments were discussed in detail The forbidden symmetric stretching Re-O mode of perrhenate ion appears in the IR spectrum together with a well-separated doublet (10 cm-1) of the antisymmetric Re-O stretching mode suggesting unidentate or bridging perrhenate anion bonding[84,104]. The magnetic moment value is μeff=1.84 B.M and the UV spectrum shows bands characteristic of a tetragonal Cu environment (16000 and 14000 cm-1) [84].
The IR bands of [tetraamminezinc(II)] and [tetraamminecadmium(II)] permanganates were assigned completely [90,107], including measurements on 14N/15N and 110/116Cd isotope substituted derivatives to assign the ZnN4 and CdN4 skeletons, and on H/D isotope substituted derivatives to the unambiguous assignation of the ammonia ligand modes [106,108]. The wavenumber shifts due to 14N/15N isotope substitution were varied between 2.5-9.0 cm-1, the highest shifts were found for antisymmetric Zn-N stretching (9 cm-1) and for the symmetric HNH deformation modes (7 cm-1) [106]. The 110/116Cd isotope substitution does not cause isotope shift in the Cd-N symmetric stretching and bending modes in the Raman spectra, whereas the H/D substitution caused 27 and 16.5 cm-1 shift in the IR, and 6 cm-1 shift in the Raman spectra. The antisymmetric stretching and bending modes showed 2.0 and 0.5 cm-1 (110/116Cd) and 23 or 12.5 cm-1 shifts (H/D) isotope shifts in the IR spectra, respectively [108].
The symmetric Re-O stretching mode of perrhenate-ion is absent and the antisymmetric Re-O stretching mode is not split in the IR spectra of the Zn and Cd-complexes, which indicates the non-coordinating nature of perrhenate ion in this complex [86], however, Müller et al. [90] found splitting of antisymmetric Re-O stretching bands (F2) in both IR spectra, which could not be explained by simple site-symmetry considerations based on Td2 space-group, therefore dynamic effects through the interaction of neighboring ions in a Z=4-unit cell (factor group splitting neglected in site symmetry considerations) are noticeable [90]. Experimental IR and Raman spectra were measured and compared with the results of quantum-chemical calculations [61]. The complete assignments of the IR and Raman bands have been given [61]. The low-temperature IR and Raman scattering measurements revealed that the coordinated ammonia ligands perform fast reorientational motions below 368 K. This motion is slowed down at around 40 K. The estimated activation energy for this motion were found to be ~4 kJ mol−1 from both the IR and Raman measurements. It was confirmed by quasi-elastic neutron scattering measurements which confirmed that the ammonia ligands reorientate even in low temperatures as well. These motions are probably jumps around a 3-fold symmetry axis [61].
The crystal structure of [tetraamminecopper(II)] perrhenate was determined at 150 K [92], it is isostructural with [MA(NH3)4](MBO4)2 (MA = Pt, Pd; MB = Re, Mn) (Table 1). The Cu atom (in the symmetry center) is coordinated with four N atoms located at the vertices of a square. The perrhenate anion is slightly distorted. There are N–H…O-Re hydrogen bonds, the shortest one being 2.179 Å. Translation sublattice isolation technique resulted in sub-cell parameters ac = 6.52 Å, bc= 5.14 Å, cc = 3.90 Å, showing that it can be conventionally considered hexagonal, and it was identified hexagonal layers formed by metal atoms, which are repeated with cc = 3.90 Å [92].
[Tetraamminecopper(II)], [tetraamminezinc(II)], and [tetraamminecadmium(II)] per perrhenates hydrolyze in their aqueous solutions with the formation of a blue precipitate (Cu), and the appropriate M(OH)2 hydroxides (M=Cd, Zn), respectively. The blue crystalline phase that formed from the Cu-complex, is not the expected Cu(OH)2 as that was found in the case of [Cu(NH3)4] (MnO4)2 [76]. This phase is orthorhombic, its lattice parameters are a=11.276 Å, b=15.460 Å, c=16.865 Å. The intensities of IR bands are weak compared to the OH bands found at 704 cm-1 (it is located at 696 cm-1 in Cu(OH)2). The heating of the solution containing this phase causes appearing CuO at 50 °C. The compound contains copper (59.5 %) and a small amount of rhenium (1.75% and ammonia (0.7%). The bands at 1500, 1464, and 1372 cm-1 might belong to N-O or N-H bond-containing species as well [104].
The hydrolysis process of the complexes is completed at 100 °C as in the case of permanganate and perchlorate complexes (M=Cu, Zn, Cd) with the formation of M(OH)2 (M=Cu, Zn, Cd) precipitates and simple ammonia liberation [72,73,76,105,107,109]. Increasing the temperature, [tetraamminecopper(II)] perrhenate also follows this kind of hydrolysis process, because above 50 °C, CuO (as a dehydration product of Cu(OH)2) appears as the main reaction product.
M[NH3)4(ReO4)2+ 4H2O = M(OH)2+ 2NH4OH + 2NH4ReO4
M(OH)2 = MO +H2O, M=Cu, Zn, Cd
The presence of ammonia prevents the hydrolysis equilibrium, and [tetraamminecopper(II)] perrhenate dissolves in 1.2-12 M ammonia solutions without decomposition. Its solubility is between 0.898-3.11 g/100 g solution. The hydrolysis of [tetraamminezinc(II)] and [tetraamminecadmium(II)] perrhenates can be prevented even in 1.2 M ammonia solutions and the original salts can be crystallized out [105,109]. Removing ammonia from the hydrolysis equilibrium [73] with mineral acids (HCl, H2SO4, or HNO3) or acetic acid shifts the hydrolysis equilibrium into the direction of ammonium perrhenate formation. The acid concentrations above 0.1 M cause complete decomposition [104,105,109]:
[M(NH3)4](ReO4)2 + 2HX = MX2 + 2NH4ReO4 + 2NH4X
X=Cl, OAc, NO3 or 1/2SO4 M=Cu, Zn: Cd
The effect of temperature and acid concentrations were optimized including adding an excess of ammonium sulfate as a salting-out agent both for the aqueous (100 °C) hydrolysis and sulfuric acid neutralization processes [104,105,109].
The thermal decomposition of the [M(NH3)4](ReO4)2 (M=Cu, Zn, Cd) complexes in an inert atmosphere show a decomposition step at 175-225, 1 50-195 and 100-150 °C with the formation of [M(NH3)2](ReO4)2 compounds with 0.37, 1.27 and 0.42 reaction order 67.2 kJ/mol, 47.3 kJ/mol and 28.9 kJ/mol activation energy, respectively [84,86]. Hetmanczyk et al. [61] determined the kinetic parameters for the first decomposition step of the cadmium complex resulting in the diamminecadmium(II) perrhenate. The decomposition step follows a single mechanism, the activation energy values were found to be 97.7 and 101.7 kJ/mol calculated by Kissinger-Akahira-Sunose and Kissinger methods, respectively. [61].
The thermal decomposition of the complexes in air atmosphere, however, looks more complicated. The decomposition between 130-245, 129-360, and 250-360 °C with the formation of ammonium perrhenate and hydrated copper, zinc, and cadmium perrhenates, respectively. The next decomposition steps belong to the decomposition of ammonium perrhenate and metal perrhenates, with elimination and redox reactions of rhenium heptoxide with ReO2 and ReO3 formation, and with their re-oxidation into Re2O7 [110,111,112]. The final decomposition residues are CuO, ZnO and CdO [104,105,107,110,113].
2.4.4. [Tetraamminenickel(II)] and [tetraamminecobalt(II)] perrhenates, [M(NH3)4](ReO4)2 (M=Ni, Co)
[Tetraamminenickel(II)] perrhenate was isolated as a decomposition intermediate of [hexaamminenickel(II)] perrhenate on standing in air [62,114], whereas the cobalt complex was precipitated from a hot aq. cobalt(II) perrhenate solution by ammonia gas passing into that as a bright violet cube [69,103]. Excess of ammonia resulted in the formation of a brown precipitate [103]. Under an inert atmosphere, the concentrated solution of cobalt perrhenate containing a small amount of hydroxylamine hydrochloride and ammonia gas resulted in a bright red precipitate formed, which turned into a magnificent, crimson-colored regular tetrahedron containing crystalline mass upon shaking (d=3.428 g/mL; mol-volume is 183 cm3/mol) without formation of the brown oxidation by-product [69].
The [tetraamminenickel(II)] perrhenate is stable at 100 C°, but on strong heating in air decomposes with nickel oxide formation [103]. It is non-hygroscopic and insoluble in water or organic solvents. The violet crystals of [Co(NH3)4](ReO4)2 are stable in air and can be washed with ammonia-containing water without decomposition. However, its treatment with ammonia-free water resulted in an insoluble bright green material (probably a basic perrhenate) [103].
The powder XRD of [Ni(NH3)4](ReO4)2 shows an orthorhombic lattice (Table 1), the experimental density was found to be 2.96 g/mL [85]. [Co(NH3)4(ReO4)2] is cubic (Table 1), dexp=3.43 g/mL [95]. It is isostructural with the analog [M(NH3)4](XO4)2 and [M(NH3)4](OSO3N)2 (M=Cd, Zn, X=Re, Mn) compounds. Single-crystal diffraction measurements showed additional weak reflexes, which were not measurable in the powder records. It shows the presence of a possible cubic flat-centered superstructure with a =21.08 Å, Z=32, and Td2-F4-3c [95].
The triplet and doublet nature of the antisymmetric Re-O band of perrhenate ion in [Ni(NH3)4](ReO4)2 and [Co(NH3)4](ReO4)2, respectively, were taken as evidence of the perrhenate ion coordination [85]. Since the ν3(Co-N) mode did not split in the IR spectrum of the Co-complex (the ν3(Re-O) is a doublet), the distortion of the ReO4 tetrahedron was declared to be higher than that of the CoN4 tetrahedron in [Co(NH3)4](ReO4)2 [95]. Complete assignment of vibrational bands including the far-IR region have been given in [62]. Five δ(N-H) bands were observed in the IR spectrum of [Ni(NH3)4](ReO4)2 between 1345–1100 cm-1 (instead of the singlet one given in [85]) and splitting of ρ(NH3) at ~680 cm-1 was also observed together with two shoulders at 645 and 580 cm-1. The far-IR bands show that the ReO4 units are not isolated tetrahedrons but rather are joined to the complex cations to form a polymeric chain. The presence of the Ni–O–Re bond is characterized by the band observed at 219 cm-1 [62].
The magnetic moment value (μeff=3.08 BM for Ni complex) and that’s temperature independence show a hexacoordinated nickel(II) environment. The UV-Vis spectra of [Ni(NH3)4](ReO4)2 shows tetragonal distortion (10600 cm-1 (3B1g->3B2g or 3B1g->3Eg),16100 cm-1 (3B1g->3A2g), 21300 cm-1 (no assignation), 27000 and 25600 cm-1 (3B1g->3Eg or 3B1g->3A2g), although the band assignations due to overlapping is hard in the case of similar distorted low-symmetry structures [85].
The CoII ion ground state is 4F and the next quartet-state (4P) is located at 11 kK higher energy. The ground term splits in the tetrahedral field (4A2 (ground state), 4T2 and 4T1), and according to this, the possible transitions are 4A2-4T2, 4A2-4T1(F) and 4A2-4T1(P). The first transition cannot be seen but the other two were clearly observed at 10.0 and 18.5 kK [95]. Detailed evaluation of the electronic spectrum shows a tetrahedral Co(II) environment and weak Co-N dative bond in this compound confirmed by the temperature-dependent magnetic measurements (meff=4.50-4.54 between 88 and 306 K) [85].
[Tetraamminenickel(II)] perrhenate has a phase transition at 188 K with 0.307 kJ/mol enthalpy change. The low hysteresis and entropy values found by the DSC measurement suggests a one mechanism second-order phase transition. The stepwise decomposition of [tetraamminenickel(II)] perrhenate was followed by thermal analysis methods [85]. Non-isotherm heating between 160-195 °C [95]/141-210 °C [62]) resulted in losing two ammonia molecules and the formation of [Ni(NH3)2](ReO4)2 [62,85]. The activation energy of the decomposition step was found to be 100.62 and 98.52 kJ/mol with Kissinger-Akahira-Sunose and Kissinger methods, respectively [62].
[Tetraamminecobalt(II)] perrhenate decomposes on strong heating in air resulting in cobalt oxide as the final product [95,103]. The thermal decomposition shows three separated endotherm steps, with 2 ammonia and Re2O7 loss in the first two steps each and in the last step, at 130-190, 190-250 and 600-800 °C range, respectively [95].
2.4.5. [Co(NH3)4CO3]MnO4
[Carbonatotetraamminecobalt(II)] permanganate is a key precursor for the preparation of Co-Mn spinel oxide nanocomposite used as a catalyst in Fischer-Tropsch synthesis [19,20]. One equiv. of solid KMnO4 was added to the aq. solution of [Co(NH3)4CO3]NO3, and the [Co(NH3)4CO3]MnO4 precipitate was collected in 82 % yield [19,20]. The IR spectrum of [Co(NH3)4CO3]MnO4 contains bands at 3301 and 1621 cm-1 that belong to the stretching frequencies of the υ(N-H) and υ(C=O) modes of the coordinated ammonia and carbonate ligands, respectively. The band located at 899 cm-1 is assigned to the antisymmetric υ3(Mn–O) stretching mode. The powder X-ray diffractogram and TG/DSC curves of [Co(NH3)4CO3]MnO4 are given. It decomposes at 250 °C into Co-Mn-oxides. The TGA data of [Co(NH3)4CO3]MnO4 show the existence of two thermal decomposition steps, the first around 130 °C (removal of water)1 and the second stage at 150-210 °C belongs to disruption of the complex structure. No weight loss was observed above 250 °C. The DSC curve shows one endothermic peak between 80 and 130 °C, whereas the decomposition has two exothermic effects between 130 and 250 °C [20].
2.4.6. [Tetraamminemetal(II)] permanganates, pertechnetates, and perrhenates of platinum group metals , [M(NH3)4](XO4)2 (M=Pt, Pd, X= Mn, Tc, Re) and [Ru(NO)(OH)(NH3)4](ReO4)2
[Tetraamminepalladium(II)] permanganate and perrhenate, or [tetraammineplatinum(II)] pertechnetate were prepared first in the reaction of ice-cooled concentrated aqueous solutions of [M(NH3)4]Cl2 (M=Pd, Pt) complexes and a stoichiometric amount of NaMnO4 (Pd) or NaReO4 (Pd, Pt) in aq. soln. were mixed, when after slow evaporation in air, needle-shaped single crystals were obtained in 75-80 % yield [96,99]. [Tetraammineplatinum(II)] perrhenate was also synthesized with the use of solid silver perrhenate with 40 min boiling in 80 % yield [99]. The analogous [tetraammineplatinum(II)] pertechnetate(VII) was synthesized as colorless platelet single crystals by adding 2 equivalents of NH4TcO4 in its aqueous solution at room temperature for 2-4 days [97].
The [tetraamminepalladium(II)] permanganate and perrhenate are colorless triclinic crystals (Table 1), but they are not isomorphic, because the crystal symmetry of permanganate complex is lower (P1) than that of the analogous perrhenate complex (P-1). [96]. The [Pd(NH)4](ReO4)2 is isomorphic with the colorless triclinic [Pt(NH3)4](XO4)2 (X=Tc, Re) complexes (Table 1). A monoclinic polymorph of [tetraammineplatinum(II)] perrhenate was also described (Table 1) as colorless, non-hygroscopic plates, which are isometric in the plan, but there are also prismatic elongated varieties. The double-sided crystals exhibit oblique extinction towards the prismatic faces and the pinacoids. The optical sign is minus, there is no pleochroism. There is no refractive index dispersion, Ng=1.715, Nm=1.714, Np=1.676 [99]. This polymorph is slightly soluble in water [99]. The triclinic compounds (Pd and Re, or Pt and Tc or Re) consist of two tetrahedral XO4- anions and a square [M(NH3)4]2+ cations are linked by Re-O···H–N hydrogen bonds. The polyhedral complex cations form hexagonal layers in the y-z plane. Every Pd or Pt atom is surrounded by twelve Re atoms giving hexagonal prisms [97,99]. The IR spectrum of [Pt(NH3)4](ReO4)2 did not show a specific influence on the energetical states of complex constituents, although the IR forbidden symmetric stretching (υ1) mode of perrhenate ion can be shown at 971 cm-1 [99].
The thermal decomposition of [Pd(NH3)4](MnO4)2 was studied both in H2 and He atmospheres. A thermal explosion occurred at ~200 °C; the products were X-ray amorphous and annealing at 200–400 °C in an inert or reducing atmosphere did not improve their crystallinity [96]. The thermolysis of [Pd(NH3)4](ReO4)2 in helium atmosphere started at 210 °C according to the following equation:
[Pd(NH3)4](ReO4)2 = Pd + 2NH4ReO4 + 4/3NH3↑ + 1/3N2↑
The next decomposition step corresponds to the NH4ReO4 decomposition resulting in a mixture of metallic Pd and X-ray amorphous Re oxides [96]. The thermal decomposition in an H2 atmosphere at temperatures above 300 °C resulted in the formation of single-phase powder with a hexagonal lattice. This product is a solid solution based on a rhenium structure with the composition Pd0.33Re0.67 [96].
[Tetraammineplatinum(II)] perrhenate decomposing starts at 370 °C in air, which ends at 444 °C. The decomposition residue is metallic Pt, and the rhenium is released as Re2O7 [99].
2 [Pt(NH3)4](ReO4)2 +5O2 =2Pt + 2Re2O7 +4N2 +12H2O
In a hydrogen atmosphere, the decomposition starts even at 200 °C with the formation of NH4ReO4 and finely dispersed Pt in 45 min. Further annealing of the decomposition residue in H2 at 250 °C for 3 h gave ReO3 and Re in addition to NH4ReO4 and Pt, whereas after 5 h annealing, only Pt and Re metal phases could be found [98]. If [Pt(NH3)4](ReO4)2 was heated under hydrogen at 200 °C for 45 min then subsequently at 600 °C for 3 h, Pt0.35Re0.65 Re monophase solid solution was formed. A two-phase ReO3 -containing Pt-Re alloy was formed at 700 °C in 3 h due to the oxidation of Re by air which contacted the sample during taking out from the furnace. On heating of [Pt(NH3)4](ReO4)2 at 900 °C in a hydrogen atmosphere for 7 h, the product was a Pt0.33Re0.67 solid solution (alloy) [98].
[Ru(NO)(OH)(NH3)4](ReO4)2 was prepared from [Ru(NO)(OH)(NH3)4]Cl2 and NH4ReO4. It is orthorhombic, space group Pbca, Z = 8. The NO ligand is trans to the hydroxide-ion ligand, and the ReO4- anion is H-bonded to the complex cation [115].
2.5. Pentaammine complexes
One permanganate compound, [(chlorido)pentaaminecobalt(III)] permanganate, and seven perrhenate complexes, [(aquo)(pentaammine)cobalt(III)] perrhenate, [M(NH3)5Cl](ReO4)2 (M=Co, Cr, Ru, Rh and Ir) complexes and [Pt(NH3)5Cl](ReO4)3 have been known until now. Four coordinated perrhenate containing complexes of cobalt(III), [Co(NH3)5ReO4]X2 (X=ReO4, ClO4, Cl and NO3) were also described. The crystallographic data of [M(NH3)5Y](XO4)n compounds are given in Table 2.
Table 2.
Crystallographic parameters of [Co(NH3)5Y](XO4)2 compounds.
| Compound | T, K | a,b,c | a,b,g | Space group | Z | V, Å3 | Dcalcd., g/mL | Ref. |
|---|---|---|---|---|---|---|---|---|
| M=Co, Y=H2O, X=Re, n=3, , x2H2O | 150 | 9.9797 12.6994 14.7415 |
102.870 |
C2/c | 4 | 1821.35 | 3.456 | [116] |
| M=Co, Y=Cl, X=Re, n=2, x2H2O | 14.9446 14.6562 12.2434 |
Cmc21 | 8 | 2681.68 | 3.368 | [116] | ||
| M=Co, Y=Cl, X=Re, n=2; x0.5H2O | 293 | 8.0086 12.9839 17.5122 |
91.858 |
P21/n | 4 | 1820.01 | 3.462 | [116] |
| M=Co, Y=Cl, X=Re, n=2 | 293 | 14.974 14.688 12.2434 |
2708.5 | 3.33 | [25] | |||
| M=Rh, Y=Cl, X=Re, n=2 | 293 | 15.0740 14.7290 12.3470 |
Cmc21 | 1430.5 | 3.19 | [25] | ||
| M=Cr, Y=Cl, X=Re, n=2 | 293 | 15.071 14.806 12.439 |
2775.7 | 3.30 | [25] | |||
| M=Ru, Y=Cl, X=Re, n=2 | 293 | 15.053 14.793 12.445 |
2741.3 | 3.54 | [25] | |||
| M=Rh, Y=Cl, X=Re, n=2 | 293 | 15.033 14.723 12.331 |
2729.2 | 3.55 | [25] | |||
| M=Ir, Y=Cl, X=Re, n=2 | 293 | 15.059 14.718 12.359 |
2739.2 | 3.98 | [25] | |||
| M=Co, Y=Cl, X=Mn, n=2 | 14.2753 14.2816 12.2342 |
Cmc21 | 8 | 2494.24 | 2.216 | [60] | ||
| M=Pt, X=Cl, X=Re, n=3, x2H2O | 10.3476 12.8955 14.3536 |
P21/n | 4 | 1847.94 | 3.962 | [117] |
The [M(NH3)5Cl](ReO4)2 complexes (X=Co, Cr, Ir, Rh, Ru, Co) are isomorphic with each other and the [Co(NH3)5Cl](MnO4)2 (Table 2).
2.5.1. [(Chlorido)pentamminecobalt(III)] permanganate
[Chlorido(pentaammine)cobalt(III)] permanganate was prepared first by Franguelli et al [60] in the reaction of [Co(NH3)5Cl]Cl2 and excess of concentrated NaMnO4 as an orthorhombic (Table 1) reddish-purple crystalline mass with a 57% yield [60]. Previously Krestov and Yatsimirksii calculated its theoretical thermodynamical properties [118]. Its solubility in water is 3.71 g/L and 9.39 g/L at 0 ◦C and 25 ◦C, respectively. It is insoluble in organic solvents like aliphatic hydrocarbons, chloroform, dichloromethane, or benzene, and soluble in DMF but decomposes in DMSO [60].
The Co3+ cation is surrounded by five ammonia and one coordinated chloride ion in a distorted octahedral arrangement. The asymmetric unit contains two half cations and two permanganate anions (Figure 4). The cobalt ion, the chloride ion, and a part of ammonia ligands sit on a mirror plane (have half-site occupancy), thus the hydrogens of these ammonia ligands are disordered over two mirrored positions. The axial Co-N bond distance is shorter and longer than that of the equatorial ones in cation B and cation A, respectively. The Co-Cl bonds were almost equal in cations A and B. Layer of the cations in the bc plane was embedded between permanganate layers [60].
Figure 4.
Packing arrangement of [Co(NH3)5Cl](MnO4)2 and hydrogen bonds in light blue (hanging contacts are omitted for clarity) between the ions. Reproduced from [60].
Figure 4.
Packing arrangement of [Co(NH3)5Cl](MnO4)2 and hydrogen bonds in light blue (hanging contacts are omitted for clarity) between the ions. Reproduced from [60].
The [Co(NH3)5Cl]2+ cation in [Co(NH3)5Cl](MnO4)2 has five hydrogen donor groups with 3x5 donor sites and one hydrogen acceptor group (chloride-ion). The cations are arranged in the crystal lattice in such a way that all ammonia hydrogens could form at least one acceptor (chloride or permanganate oxygen) atom [60]. Detailed spectroscopic analysis (correlation analysis, low-temperature IR, Raman, and far-IR ) spectroscopic studies were done and all vibrational bands appeared in the IR and Raman spectra of [Co(NH3)5Cl](MnO4)2 were assigned [60]. The singlet nature symmetric stretching mode of permanganate ion appears as a twin peak in the low-temperature Raman spectra, which confirms the presence of two crystallographically and spectroscopically different permanganate ions [60]. The ρ(NH3) rocking mode is sensitive to the presence of hydrogen bonds in ammonia complexes, but the ρ(NH3) IR band of [Co(NH3)5Cl](MnO4)2 is coinciding with its νs(Mn-O) mode. Therefore, deuteration experiments were done to assign the ρ (ND3) band position (646 cm−1), and with the use of the ρ(NH3)/ρ(ND3) wavenumber ratio found for [Co(NH3)5X]X2 (X=halogen) compounds [119], the position of ρ(NH3) could be calculated (818 cm−1). It showed that the strength of the hydrogen bonds in the structure of [Co(NH3)5Cl](MnO4)2 lies between that of in [Co(NH3)5Br]Br2 (ρ(NH3) = 830 cm−1) and [Co(NH3)5I]I2 (ρ(NH3) = 810 cm−1) [60,119].
No important differences were found between the thermal decomposition characteristics of [(chlorido)pentaamminecobalt(III)] permanganate in argon and air atmospheres. The decomposition product formed above 400 °C was CoMn2O4 (average crystallite size of 16.8 nm) [60]. The thermal decomposition of [(chlorido)pentaamminecobalt(III)] permanganate starts at 121 °C. The ammonia molecules are bonded with various strengths (ε = 0.80 and 0.88 [120], which predicted stepwise ammonia loss. The TG-MS study showed that the releasing material in the first decomposition step is not ammonia. Either N2 or NOx compounds were not formed in this decomposition step. Since [(chlorido)pentaamminecobalt(III)] permanganate is anhydrous, and the decomposition was done in an inert atmosphere, and water as the reaction product could be formed in the first step only as a product of a solid-phase redox reaction between ammonia ligand and permanganate ion (the only hydrogen and oxygen sources are ammonia and permanganate ion, respectively). As a result of this, no permanganate ion was found in the amorphous reaction product of the first decomposition step [121].
The decomposition was performed in a refluxing toluene solvent, since in this case the decomposition temperature could not be exceeded the boiling point of toluene, furthermore, the liberated reaction heat was turned to evaporate the toluene. The amorphous product before heating was washed out with water when the soluble phases were identified as NH4NO3, [Co(NH3)5Cl]Cl2, and NH4Cl. The solid residue based on the elementary analysis and material balance was a cobalt manganese oxide containing ammonia and formed according to the equation [121]:
3[Co(NH3)5Cl](MnO4)2 = [Co(NH3)5Cl]Cl2 + 3H2O + 3NH4NO3 + {Co2(NH3)4Mn6O12}
Further studies showed that the ammonia content is non-coordinated but it is in ammonium-ion form, and the real formula of the amorphous solid can be written as [NH4)4Co2Mn6O12] [59]. This composition is very similar to the todorokite mineral group members. The amorphous decomposition product phase contains the water-soluble and insoluble components that were heated above 400 °C which resulted in the formation of CoMn2O4 and gaseous (N2, H2O, NH3, NH4Cl) products. The crystallite size and photocatalytic activity of the stoichiometric CoMn2O4 products in degradation or organic dyes strongly depended on the heating time and temperature [60]. Heating of the amorphous (NH4)4Co2Mn6O16 also gave spinel-like decomposition product, but with Co:Mn=1:3 stoichiometry [121].
2.5.2. [Pentamminechlorometal(III)] perrhenates, [M(NH3)5Cl](ReO4)2 (M=Co, Cr, Ru, Rh, Ir)
Dilute solutions of chloropentaamminemetal dichloride ([M(NH3)5Cl]Cl2, M=Co, Cr, Ru, Rh, Ir, 0.01 M) and NaReO4 (0.01 M ) were stirred at room temperature when almost immediately a needle-like pink (M=Co), light brown (M=Cr), dark yellow (M=Ru), light yellow (M=Rh, Ir) crystalline precipitate formed in ~80 % yield [25,116]. The single crystals of the cobalt complex hemihydrate were obtained by interdiffusion in agarose gel in a U-tube at room temperature (one end of the tube was filled with a diluted aqueous solution of [Co(NH3)5Cl]Cl2 and another one was filled with 2 equivalent of NaReO4 in a few days [116]. The single crystals of Rh-complex were obtained by slowly evaporating a dilute aqueous solution of [Rh(NH3)5Cl]Cl2 and 2 equiv. of NaReO4 at room temperature [25]. The lattice parameters of [pentaamminechlorometal(III)] perrhenates, [M(NH3)5Cl](ReO4)2 (M=Co, Cr, Ru, Rh, Ir) and the [Co(NH3)5Cl](MnO4)2 complexes are given in Table 2.
These complexes are anhydrous, but the Co-complex has hemi and dihydrate forms as well. The dihydrate consisting of distorted octahedral [Co(NH3)5Cl]2+ cations occupy two crystallographically independent sites oriented to symmetry elements in a different way, and there are two crystallographically independent perrhenate anions [116]. The cationic and anionic layers along the x-axis are bound inside and between themselves by weak hydrogen bonds (N–H…O-Re and N–H…Cl). Water molecules are located in the cation layers: the minimum Сo…Сo distance is 5.968 Å in the cation layer; the shortest Ow…N-H contact is 3.02 Å. In the perrhenate anion layer, the shortest Re…Re distances are 4.224-4.457 Å [116].
The structure of the rhodium-complex (and the other isomorphic ones) is an island type and consists of [Rh(NH3)5Cl]2+ octahedra and ReO4-tetrahedra with two crystallographically independent cationic and two anionic units. The one kind (1) of rhodium ions lie on a mirror plane; the Cl and one of the ammonia N atoms from the Rh environment are in the m plane, whereas other N atoms are pairwise related by this plane; the Cl and three N atoms from the other (2 rhodium environment lie in the m plane, and two N atoms are related by this plane [25]. Each [Rh(NH3)5Cl]2+ cation is surrounded by twelve perrhenate ions forming a distorted hexahedral prism. The Rh…Rh distances in the distorted prism lie between 4.921 and 6.391 Å; the Re…Re distances are between 4.25 and 4.48 Å. The 2nd coordination sphere of the perrhenate anion involves six cations with Rh…Re distances between 5.372 and 6.336 Å. Along the ɏ axis layers of [Rh(NH3)5Cl]2+ cations and perrhenate anions with intra- and interlayer hydrogen bonds (N–H…O-Re and N–H…Cl of 2.91-3.29 Å and 3.33-3.53 Å, respectively) are observed. The shortest interionic contacts Re-O…O-Re and Re-O…Cl are 3.10 Å and 3.36 Å [25].
The thermal decomposition of [M(NH3)5Cl](ReO4)2 (M=Co, Cr, Ru, Rh, Ir) in a helium atmosphere resulted in coinciding decomposition steps. The decomposition products are amorphous/multiphase oxide materials. In a hydrogen atmosphere at 600 °C, however, the thermal decomposition product is a monophase binary alloy, M0.33Re0.67, (M=Co, Ru, Ir, Rh) which are ultrafine metal powders (solid solutions) derived from hexagonal close packing of rhenium [25]. In the case of the chromium complex, in a hydrogen atmosphere, multiphase decomposition products are formed consisting of only one crystalline compound characterized by the Cr0.003Re0.997 formula [25].
The rhodium and iridium complexes decompose in an analogous way. The thermal decomposition of [chloropentaamminerhodium(III)] perrhenate in a step-by-step quenching under an H2 atmosphere showed that at 190 °C (1st step) both the cation and anion are partially reduced resulting in appearing of a very broad diffuse maximum at the angles corresponding to the most intense peaks of the metallic rhenium (100, 002, 101) and rhodium (111, 200). The reflections of the [Rh(NH3)5Cl](ReO4)2 have completely vanished at 200 °C and the peaks belonged to NH4ReO4, NH4Cl and [Rh(NH3)5Cl]Cl2 have emerged. At 220 °C, in the third stage, only a small amount of NH4ReO4 and the broad peaks of the Rh0.70Re0.30 solid solution with hcp lattice were found. It shows, that the metallic solid solution formed at the first stage is definitely rhenium-deficient. At the fourth stage between 220 and 280 ºC, the NH4ReO4 content was completely decomposed and the formed rhenium penetrated into the lattice of the solid solution resulting in gradually increasing the lattice parameters corresponding to the composition Rh0.30Re0.70. The vacuum annealing of this solid solution at 950 °C for 400 h leads to an increase in crystallite sizes [26].
The in situ synchrotron X-ray studies on thermal decomposition products under an H2 atmosphere showed a very wide band appears at the decomposition stage at ~200 °C, whereas further heating resulted in the formation of the solid solution with Rh0.40Re0.60 composition with hcp lattice of rhenium [27]. Further raising of temperature up to 340ºC was accompanied by an extension of the unit cell dimensions and the formation of the solid solution with Rh0.33Re0.67 composition. In the first stage of the decomposition reaction metallic rhodium develops as an amorphous non-diffracting phase, whereas rhenium is formed in the second stage of the process and penetrates into nanosized rhodium particles. The Rh0.40Re0.60, Rh0.35Re0.65, and Rh0.34Rh0.66 alloys were formed at 206, 230 and 250 °C, respectively, whereas above 280 °C only the final product, Rh0.33Re0.67 could be detected [27]. Contrary to the experiments done by step-by-step quenching of the reaction products, no Rh0.30Re0.70 and other intermediates such as NH4ReO4, NH4Cl and [Rh(NH3)5Cl]Cl2 were observed [27].
2.5.3. [Pentammine(aquo)cobalt(III)] perrhenate dihydrate
A 10 % ammonia solution was added to a 0.01 M aqueous [Co(NH3)5Cl]Cl2 solution and heated in a water bath for 10 min under constant stirring until the dark red solution formed. A 0.01 M NaReO4 solution was added until complete precipitation of [Co(NH3)5H2O](ReO4)3.2H2O. The yield was 90%. Its single crystals were grown in 3 days in a U-tube filled with a diluted aqueous [Co(NH3)5Cl]Cl2 and 2 equiv. of NaReO4 (0.41 mmol) [116]. With the use of a 5.4 M HReO4 solution and an aqueous solution of [Co(NH3)5OH2](ClO4)3, the pink [Co(NH3)5(H2O)](ReO4)3.2H2O was formed in 30 min in >95 % yield [122]. A 18OReO3 labeled sample was prepared in an analogous way, from NaReO*4 isotope labeled perrhenate salt. Its dehydration at 50-60° and at 0.001 Hgmm vacuum for 3 h resulted in complete loss of the lattice water and the formation of pink [Co(NH3)5(H2O)](ReO4)3 [122]. No isotope exchange was observed between the perrhenate ion and crystallization water during the dehydration process [122].
The complex cation in [Co(NH3)5Н2О](ReO4)3.2H2O has a slightly distorted octahedral structure. The cis angle deviations are not much than 1.9°. The length of the Co–N bond located in the trans-position towards water is shorter only on average by 0.03 Å than the other bonds [116]. The structural units are hydrogen bonded involving the crystalline water molecules. The intermolecular interactions in the Оw…О-Re linkages are stronger than in the Оw…N-H linkages. Each cation is surrounded by 12 perrhenate anions; the Co…Re distances between the centers are 5.297-5.875 Å; the shortest Re…Re distances in the perrhenate layers are 4.307-4.885 Å [116]. The IR spectrum in the perrhenate normal modes region was also recorded and analyzed[122].
2.5.4. [Perrhenatopentamminecobalt(III)] perrhenate, [Co(NH3)5ReO4](ReO4)2
Dehydration of [Co(NH3)5(H2O)](ReO4)3 between 95 and 130° at 0.001 mmHg pressure resulted in a quantitative loosing of the coordination water in a period of 2-5 h. No ammonia evolution was detected. The solid residual product is [Co(NH3)5(OReO3)](ReO4)2. This salt is purple and quite insoluble in water [122]. The 18O-perrhenate labeled compound was treated in a vacuum at 110-120 °C when only 2 % of isotope exchange was observed between the perrhenate ion and the coordinated water [122]. The aquation of [(NH3)5Co-OReO3]2+ ion in the aqueous solution of [Co(NH3)5ReO4](ReO4)2 and on the fixed form on ion-exchange resins were followed at different pH values. The O18 isotopic exchange of H2O with [(NH3)5Co(OH2)]3+ and ReO4- were also studied. The aquation of [(NH3)5CoOReO3]2+ (H2O-ReO4- exchange) was faster in the acidic region and slower in the basic region. A new term of the rate equation was introduced when ReO4- was on the resin [123]. The IR spectra in the perrhenate normal modes region were recorded [122].
2.5.5. [Co(NH3)5ReO4]X2 (X=Cl, Cl, ClO4, NO3)
A Dowex 1-X4 ion exchanger resin was converted to its chloride, nitrate, or perchlorate form and washed with 0.001 M 2,6-lutidine (to retard hydrolysis of Co-complex) solution, then the slightly soluble [Co(NH3)5OReO3](ReO4)2 was ground with the resin and water at 10-15°, and the solution was separated. The addition of saturated LiCl, LiNO3, and LiClO4 solutions at 0° resulted in crystalline [Co(NH3)5OReO3]X2 compounds in 60-80 % yield. The violet-red chloride salt is not stable for a long time even in the solid state, an internal replacement takes place with the chloride ion resulting in some [Co(NH3)5Cl]2+ derivative [122,123,124]. The violet-red perchlorate salt, however, is stable in the solid state over a period of 6 months [122,123,124]. Both salts are soluble in water and hydrolyze with the formation of [Co(NH3)5(H2O)]2+ solution with >95 % yield [122]. The kinetics of these reactions in acidic, alkaline, and neutral solutions were analyzed in detail [122]. The first-order rate constant, the kinetic parameters, and their temperature dependences are given. They show an absorption maximum of 530 nm [122]. The hydrolysis kinetics of the nitrate complex was studied in more detail, using an 18O-perrhenate-containing sample, which was prepared in an analogous way as the non-labeled nitrate salt. The first-order rate constant and the kinetic parameters and their temperature dependence is given, including the results of acetic acid/acetate ion catalysis in the hydrolysis reaction between pH= 4 and 7, which proceeds with 80-95 % conversion [122]. It has an absorption maximum of 530 nm [122]. The isotope-labeled [Co(NH3)5ReO3](NO3)2 was dissolved in water at the appropriate pH and buffered with 2,6-lutidine and HNO3. The isotope exchange between the isotope-labeled perrhenate-ion and water was fast and complete [122]. The rhenium-oxygen fission is shown to be the primary process in the hydrolysis catalyzed by acids and bases and the isotope transfer experiments. The rate constants of aquation with Co-OReO3 fission show that this complex aquation mechanism is different from some other analog [acido-pentaamminecobalt(III)] complexes. The dissociation at pH 4-5 occurs predominantly by Co-O fission [122].
2.5.6. [Pentaamminechloroplatinum(IV)] perrhenate
[Pt(NH3)5Cl](ReO4)3.2H2O was synthesized from aq. solution of [Pt(NH3)5Cl]Cl3.H2O and excess of hot aq. NaReO4 solution. Colorless crystals as elongated monoclinic platelets appear in 1 h, the yield was 75-80 % [117]. Its structure consists of a [Pt(NH3)5Cl]3+ cation with three and two crystallographically independent ReO4- anions and crystallization water, respectively. The cations and anions are in general positions. The bond angles of the tetrahedral anion and the octahedral cation deviate from ideal values. An extended network of hydrogen bonds involving the cation, anion, and molecules of crystallization water is built up. The shortest intermolecular contacts involving water are H2O…Cl= 3.35 Å, H2O…N =2.86 Å, H2O…O-Re=2.75 Å. The [Pt(NH3)5Cl]3+ cations are surrounded by 12 ReO4- anions, whereas the anions have four cationic neighbors and build up an almost regular tetrahedron. The packing of the ions is perpendicular to the [101] direction. The perrhenate anions form a hexagonal network, which is alternated with the hexagonal layer of [Pt(NH3)5Cl]3+ cations. The crystallization waters occupy the tetrahedral voids within the anionic framework. The cationic sublattice approach was used to determine the M…M distances. The Re…Re distances fall within 4.38-4.80 Å, the Pt…Pt distances at 7.68, whereas the Pt…Re distances between 5.32-5.64 Å, and in general, the average distances between metal atoms correspond to the edge lengths of the distinguished rhombohedra [117].
Its decomposition in an inert atmosphere proceeded with the elimination of crystalline water at 110-140 °C, followed by a decomposition step starting at 280 and ending at 500 °C. The decomposition product consists of two solid solutions, one Pt and another Re-based one. In an H2 atmosphere, a Re0.75Pt0.25 alloy was formed with Re-structure [117].
2.6. Hexammine complexes
Table 3.
Crystallographic data of [M(NH3)6](XO4)n [hexaamminemetal] permanganates, pertechnetates and perrhenates.
Table 3.
Crystallographic data of [M(NH3)6](XO4)n [hexaamminemetal] permanganates, pertechnetates and perrhenates.
| Compound | T, K | a,b,c | a,b,g | Space group | Z | V, Å3 | Dcalcd., g/mL | Ref. |
|---|---|---|---|---|---|---|---|---|
| M=Co, X=Re, n=3, x2H2O | 293 | 14.9446 14.6562 12.2434 |
Cmc21 | 8 | 2681.68 | 3.368 | [116] | |
| M=Co, X=Mn, n=3 | 11.39 | Td2--F4-3m | 4 | 1477.6 | 2.33 | [114] | ||
| 11.39 | 4 | 1477.65 | [125] | |||||
| M=Co, X=Tc, n=3, x2H2O | 8.0266 12.6275 17.6438 |
91.320 | P121/n | [101] | ||||
| M=Cr, X=Mn, n=3 | 11.45 | Td2-F4-3m | 4 | 1501.1 | 2.26 | [114] |
2.6.1. [Ni(NH3)6](MnO4)2
The reaction of nickel(II) sulfate solution with ammonia and saturated potassium permanganate solution below 5° resulted in a blackish small crystal deposit in twenty to thirty minutes, declared to be the dihydrate of [hexaamminenickel(II)] permanganate [89]. It is soluble without decomposition in diluted sulfuric acid, but from the aqueous solutions which are initially beautiful purple in color, quickly manganese oxide is deposited. It detonates under the shock of the hammer; heating or rubbing/crushing in a mortar, fuse with releasing ammonia and producing a cloud of very finely divided oxides. After five to six days it has partially decomposed. This salt later was found to be impure, because in spite of using the chilled nickel salt and potassium permanganate solutions, on adding the ammonia solution, the reaction heat causes partial decomposition. However, adding an excess of aqueous ammonia to the nickel nitrate solution and cooling the mixture with ice, and adding 2 equiv. of ice-cold potassium permanganate solution, the [hexamminenickel(II)] permanganate was found to be very pure and anhydrous [126]. It can be dried at temperatures below 100 °C or in a vacuum over caustic soda in the dark. The [hexamminenickel(II)] permanganate can also be prepared in the reaction of the [hexaamminenickel(II)] chloride and potassium permanganate under ice-cooling [114]. It consists of isotropic octahedra with violet-black color. Its aqueous solution is violet in color. In contrast to the salt described by Klobb, it is fairly stable, at least in the dark. Its complete decomposition occurs only after several months. It forms violet mixed crystals with the analog tetrafluoroborate. These mixed crystals were more stable than the pure permanganate compound, no decomposition was started even after six months of storage, and dissolved in water with purple color without residue. [126]. The [Ni(NH3)6](MnO4)2 cannot be indexed as cubic material, although the corresponding perchlorate is cubic. The powder X-ray data including the d-values and intensities are given [114]. The IR band assignment for the cationic and anionic parts is given and evaluated in detail. The presence of hydrogen bonds has an influence on the rocking and symmetric deformation N-H modes [114].
2.6.2. Ni(NH3)6](ReO4)2
[Hexaamminenickel(II)] perrhenate was prepared in the reaction of the [hexaamminenickel(II)] chloride and sodium perrhenate. Wilke Dürfurt et al. prepared it from in situ synthesized nickel(II) perrhenate solution (dissolving nickel carbonate in 0.004 M perrhenic acid). Hetmaczyk et al. used basic nickel carbonte and 75-80 % perrhenic acid solution [62]. Evaporation of the solution in a water bath, then the addition of ammonia and cooling with an ice-sodium chloride mixture resulted in violet [hexaamminenickel(II)] perrhenate [69]. [Hexaamminenickel(II)] perrhenate has needle-shaped crystals with light-purple color. It shows oblique extinction and is believed to be a triclinic prism. It easily decomposes with ammonia releasing [114], and on standing in air gradually loses its ammonia and turns into a light blue material (fast at 100 °C [103]), which was identified by Wilke-Dörfurt et al as [Ni(NH3)4](ReO4)2 [69]. It can be stored under an ammonia atmosphere for a long period of time without any decomposition [62]. Its solubility in aq. ammonia solution of specific gravity 0.930 at 26 °C: 33.4 g/L, its density at 25 °C is d= 3.000 g/mL, mol-vol 220.5 [69]. No phase transitions of the Ni-complex could be detected [62]. The powder X-ray data including the d-values and intensities and the IR bands assignment for the cationic and anionic parts are given. The presence of hydrogen bonds has an influence on the position and splitting of the rocking and symmetric deformation N-H modes [114], which was confirmed with the complete assignment given on the basis of quantum chemical calculations [62]. The appearance of the IR inactive υ1(Re-O) IR band suggests distorted tetrahedral geometry of the perrhenate ion [62].
The thermal decomposition of [Ni(NH3)6](ReO4)2 is a four-step process, but the first thermal decompositon step at 300-368 K resulted in [tetraamminenickel(II)] perrhenate, without any redox reaction, thus the further decomposition steps belong to the decomposition steps of [tetraamminenickel(II)], [diamminenickel(II)] and nickel(II) perrhenates, respectively [62]. The kinetic studies showed 76.66 and 78.21 kJ/mol activation energy with KAS and Kissinger methods, respectively. The thermal decomposition of [Ni(NH3)6](ReO4)2 is involved with one mechanism [62].
2.6.3. [Hexaamminemetal(III)] permanganate, pertechnetate and perrhenate, [M(NH3)6](XO4)3, (M=Cr, X=Mn, Re), M=Co, X=Tc, Re)
[Hexaamminechromium(III)] permanganate and the dihydrates of [hexaamminecobalt(III)] perrhenate and pertechnetate were prepared in the reaction of the [hexaamminemetal(III)] chloride (M=Cr, Co) and potassium permanganate, ammonium pertechnetate, and sodium perrhenate, as purple (M=Cr, X=Mn), lemon-yellow (M=Cr, X=Re), orange (M=Co, X=Tc) or yellowish orange (M=Co, X=Re) crystalline materials [101,114,116,126]. The yields of Co-complexes with pertechnetate and perrhenate anions were found to be 56 and 80 %, respectively.
The reaction of a warm solution of [hexaamminechromium(III)] nitrate and ~10 equivalent of potassium permanganate also resulted in violet-black octahedrons of [hexamminechromium(III)] permanganate, which is in a thin layer and at the edges transmit the light with a violet color. The analogous reaction with perrhenic acid resulted in the perrhenate dihydrate concentrating and cooling the solution. The formed precipitate was recrystallized from an aq. solution containing a small amount of perrhenic acid. [69], with subsequent drying over calcium chloride. When the crystals were dried for several hours over P2O5 in a vacuum, the anhydrous perrhenate salt was formed [69].
The needle-like of [Co(NH3)6](ReO4)3.2H2O appeared immediately, and its single crystals were grown by interdiffusion in agarose gel in a U-tube at room temperature (one end of the tube was filled with a diluted aqueous solution of [Co(NH3)6]Cl3 and another one was filled with 2 equivalent of NaReO4 in a few days [116]. If a concentrated aq. solution of [hexaamminecobalt(III)] chloride was reacted with an excess of an equally concentrated aqueous solution of perrhenic acid, and the solution was concentrated and cooled, [Co(NH3)6](ReO4)3.2H2O was crystallized out. The recrystallization from the water was performed in the presence of a small amount of perrhenic acid [69]. The crystals dried over calcium chloride was proved to be dihydrate, which transforms into anhydrous salt in several hours over P2O5 in a vacuum [69], as that was found for the analog [Cr(NH3)6](ReO4)3.2H2O/[Cr(NH3)6](ReO4)3 pair as well.
Among the chromium complexes, the permanganate salt is sparingly soluble in water and forms mixed crystals with the perchlorate and the tetrafluoroborate. On heating, it deflagrates violently, emitting a puff of dark smoke [126] and explodes under rubbing [114]. It is isostructural with the analog Co-complex, and crystallizes in a cubic face-centered lattice. Its pycnometric density was found to be 2.21 g/mL. The hexamminechromium(III) perrhenate dihydrate is lemon-yellow, its small prisms showing straight eruptions [69,114]. Its solubility in water at 20 °C is 0.684 g/L, and the density and the molar volume are 3.280 g/mL and 287.5 cm3/mol, respectively, whereas the same values for the anhydrous salt are 3.408 g/mL and 265.5 cm3/mol [69]. The perrhenate does not belong to the regular system and is less soluble than other similar salts with tetrahedral anions (perchlorate, permanganate, fluorosulfonate, tetrafluoroborate) [69].
The powder X-ray data including d-values and Miller indexes and the IR bands assignment for the cationic and anionic parts for both Cr-complexes were given and evaluated in detail.
The cobalt-pertechnetate complex can be kept in air for months without decomposition. Its solubility in water at room temperature is 6.15 × 10-4 M. The cobalt perrhenate complex consists of small prisms, which show straight extinction as birefringent crystals [69,114]. Its solubility in water at 20 °C was found to be 0.469 g/L, and the density and molar volume is 3.506 g/mL and 260 cm3/mol, whereas the same values for the dihydrate are 3.329 g/mL and 285 cm3/mol [69], respectively. It does not form crystals belonging to the regular system and its solubility is lower than that of some similar salt with tetrahedral anions (perchlorate, permanganate, fluorosulfonate, tetrafluoroborate) [69]. The powder X-ray data including the d-values and intensities are given [114].
Monoclinic single crystals of the cobalt pertechnetate salt were grown from a 1:1 water:methanol mixture at -15 °C for a week [101]. It is monclinic, but not isostructural with [Co(NH3)6](ReO4)3.2H2O or [Co(NH3)6](MnO4)3 [101]. The asymmetric unit of the pertechnetate salt consists of 3 crystallographically independent TcO4- anions, 1 [Co(NH3)6]3+ cation, and 2 water molecules. The TcO4- tetrahedra (C3v symmetry) and the octahedral cation are slightly distorted [101]. [Co(NH3)6][TcO4]3 2H2O exhibits a layered structure. No van der Waals interactions within the cationic layer (the shortest Co-Co and N-N distances between 2 distinct cationic units are 7.797 and 4.091 Å, respectively). The cationic and anionic layers are bound by hydrogen bonds, their thicknesses are 4.5 and 4.41 Å, respectively. Each cation is connected to twelve pertechnetate anions, and the shortest Co-N-H….O-Tc distances vary between 2.031 and 2.089 Å. The shortest Tc-Tc separation in the anionic layer is 4.326 Å, and the pertechnetate anion are connected with each other via hydrogen bonds mediated by water molecules. The shortest Tc-O…H-O-H distances are 1.948 and 2.085 Å [101].
The centrosymmetric cobalt cation in the [hexaamminecobalt(III)] perrhenate coordinates six ammonia molecules, forming a practically regular octahedron. The average Сo–N distances are 1.963 Å; bond angle deviations from 90° at the Сo atom do not exceed 1.8° [116]. It contains two crystallographically independent perrhenate anions, the Re atom in one of them is located on the twofold axis [116]. It has layered packing having hydrogen bond interactions. The water molecules are bound with perrhenate anions via О–Н…О-Re bonds. In the anion layers, the Re…Re distances between the nearest perrhenate anions are 4.357-5.949 Å; the minimum Сo…Сo distance in the cation layer was found to be 7.927 Å. The cations and anions are also bound by weak hydrogen bonds of the N–H…О-Re type. The shortest distances between the centers of the complex ions and anions are Сo…Rе = 5.394-5.651 Å [116].
2.6.4. [Hexammincobalt(III)] permanganate
The [hexaamminecobalt(III)] permanganate was prepared in the reaction of the [hexaamminecobalt(III)] chloride and potassium permanganate [70,114,125,127]. The interaction of hot concentrated solutions of [hexaamminecobalt(III)] chloride with 2 equiv. of potassium permanganate at a temperature that must not exceed 60 °C resulted in a mixture of the almost insoluble [hexaamminecobalt(III)] permanganate and a by-product consisting of hexagonal plates on cooling. The latest compound forms in higher proportion if not taken with a large excess of potassium permanganate. The cold water dissolves well the hexagonal salt and that can be washed out. Recrystallization from water at 60 ° resulted in black octahedra [70,127]. The use of high excess potassium permanganate at 55 °C resulted in the product in 98 % purity [125]. It is a purple crystalline material, that explodes under rubbing [114]. [Co(NH3)6](MnO4)3 forms brilliant crystals, which are very sparingly soluble in cold water (1 part requires 1388 parts of water at 0 °C), more soluble in hot water but partially decomposes. It explodes quite strongly on heating and under the impact of the hammer as well. It turns into manganese(II) chloride and luteocobaltic chloride by treating with concentrated hydrochloric acid [70,127].
It is isostructural with the analog [hexaamminechromium(III)] permanganate [125] and the [hexaamminecobalt(III)] perchlorate as well [114], crystallizes in a cubic face-centered lattice, its pycnometric density was given as 2.26 g/mL [114] or 2.16 g/mL [125]. Its powder X-ray diffractogram could be indexed according to a cubic cell [114].
All N-H and permanganate bands were unambiguously identified in the IR spectrum of [Co(NH3)6](MnO4)3. The two bands of antisymmetric Mn-O stretching mode were at 913 and 897 cm-1. The two bands may belong to the two components of the triple degenerate υ3 modes or may belong to two kinds of permanganate ions that are located in two different positions [125]. The IR band assignment for the cationic and anionic parts and the influence of the presence of hydrogen bonds in rocking and symmetric deformation N-H modes were also evaluated [114].
The thermal decomposition of [hexaamminechromium(III)] permanganate starts with a very faint exothermic peak at 100 °C, followed by an endothermic one at 103 °C(probably the physisorbed water) and a further exothermic peak is at 107 °C, and from 108 °C it begins to decompose very quickly, which ends explosively at 116 °C [125].
2.6.5. [Hexaamminecobalt(III)] dichloride permanganate and [hexammincobalt(III)] dibromide permanganate
The [hexaamminecobalt(III)] dichloride permanganate and dibromide permanganate, [Co(NH3)6]X2(MnO4) (X=Cl, Br) were formed by a direct combination of 1 equiv. of powdered [Co(NH3)6](MnO4)3 and 8 equiv. of [Co(NH3)6]X3 (X=Cl, Br) in a small amount of hot water. On cooling, small strips of black crystals are deposited which are washed with water quantity just sufficient and dried at 50 °C [127] The chloride compound was prepared at room temperature which resulted in the same product in 16.4% yield due to the solubility of [hexaammincobalt(III)] dichloride permanganate in water at room temperature (7.89 g/100 mL) [59]. They were also prepared directly from [hexaamminecobalt(III)] salts with 1.5 equivalent of potassium permanganate solutions leaving the solution to stay for 6 h [127].
The chloride salt is not very stable and decomposes in its aq. solution with releasing of chloride ion completely, but dissolves well in luteocobaltic chloride solution without decomposition. The bromide salt could be dried at 50 °C without decomposition. It has shiny hexagonal blades, similar to the chloride analog, but the bromide compound is much more stable. Water does not seem to split that even on boiling. The chloride salt is insoluble in aliphatic and aromatic hydrocarbons, acetone, and chlorinated solvents such as CCl4, chloroform, or dichloromethane, but it is soluble in DMF (0.848 g/100 mL) and decomposes in DMSO immediately. It also decomposes in a wet state in a day but when it is dry and in the absence of light, it can be stored for several days [59]. Borh compounds detonate on heating with ammonia releasing, but they are not sensitive to the impact of a hammer [127].
The chloride salt forms red platelets, monoclinic, space group P21/c, a=13.6133 Å, b=7.3658 Å, c=12.3682 Å; β=108.547°, Z=4, Dcalcd.=1.983 g/mL, T=163 K, V=1175.78 Å3 [59]. The elementary cell contains four formula units, whereas the asymmetric unit contains two halves of the complex cation, two chloride ions, and one permanganate ion (Figure 5). There are two differently distorted octahedral cations (labeled as A and B), which bonded to the permanganate oxygens with hydrogen bonds with different lengths. No direct metal–metal interactions were found.
Figure 5.
Crystal packing of [hexaamminecobalt(III)] dichloride permanganate. Reproduced from [59].
Figure 5.
Crystal packing of [hexaamminecobalt(III)] dichloride permanganate. Reproduced from [59].
There are two cationic layers in the structure, cation A is placed together with Cl anions, whereas cation B is placed in a different kind of layer without any chloride ions. The 2nd type of chloride ion is pushed into the anionic layer formed by the permanganate ions. The permanganate ion layers are in close contact with the 2nd (B) cation layers, and they are further from the 1st (A) cationic layers in which the chloride ions shadow the positive charges. A total number of 25 and mostly strong hydrogen bonds occur between the complex cations and the two types of anions. Each ammonia has 3-5 hydrogen bonds with the anions. The chlorides involve 5 and 6 hydrogen bonds, which are weaker than that made by permanganate ions. The intermolecular interactions of the two crystallographically independent cations were compared by a Hirshfeld surface analysis and 2D fingerprint plots are generated and shown in Figure 6.
Figure 6.
Hirshfeld surface analysis and 2D fingerprint plots of [hexaamminecobalt(III)] dichloride permanganate. Reproduced from [59].
Figure 6.
Hirshfeld surface analysis and 2D fingerprint plots of [hexaamminecobalt(III)] dichloride permanganate. Reproduced from [59].
The fingerprint plots for the two cations show marked differences. The strongest H−bonds are formed with cation B (with a permanganate ion), which is indicated by the much lower de and di values. The spike of the N–H···Cl interaction for cation B appears at higher de values, and the spike for the N–H···O interactions for cation A is missing. It shows that the interactions between the cation A and the permanganate ions are slightly loose, whereas for the cation B, the N–H···O spike is pronounced. The number of the N–H···Cl interactions of cation A is higher than that for cation (indicated by red) [59].
The vibrational spectra (IR and Raman) and factor group analysis were done for the chloride salt, and all the normal modes for the cation and anion have been assigned. Using a relative bond strength parameter (ε) for the ammonia molecules in ammine complexes [120] defined by Grinberg was found to be 0.94 and 0.90 for the coordinated ammonia which shows that the average strength of the hydrogen bonds in this compound is between the strength of an average hydrogen bond strengths in [Co(NH3)6]Cl3 [128] and [Co(NH3)6](MnO4)3 [59,125]. Its solid phase UV−VIS spectrum at room temperature consists of strongly overlapping bands of four possible d–d transitions of the [Co(NH3)6]3+ cation and CT bands of the permanganate anion. The 1A1→1T1 and 1A1→1T2 transitions of the octahedral CoIII cation are spin-allowed. The distortion due to hydrogen bonds results in trigonal distortion (compression) [59].
The chloride salt behaves as an explosive on heating, but a slow heating rate (2 °C/min) resulted in stepwise decomposition reactions. The first decomposition step was observed at 107 and 129 °C in inert and air atmospheres, respectively. The DSC peak temperatures were the same in O2 and N2 atmospheres (109 °C). The second decomposition step was observed at 134 °C in both atmospheres. The reaction heat of each step (−107.1 and −260.8 kJ/mol in the first and −90.3 and −64.5 kJ/mol in the 2nd decomposition step, in O2 and N2 atmospheres, respectively) was found to be different. Although the outer oxygen does not play a direct role in the starting of the decomposition reaction, however, an indirect influence via the formation of endothermic nitrogen oxides - such as NO or N2O detected by TG-MS [59] was found on the reaction heat. Two other decomposition steps were observed at 382 and 441 °C under N2 [59], whereas, in air, the oxidizable residues were completely oxidized and disappeared at 378 °C [59]. TG−MS measurements show that the first three decomposition steps consist of redox reactions, because H2O, NO, and N2O redox products were formed. The first redox reaction is a reaction between the ammonia ligands and permanganate ions. Water and N2 formed in all three, whereas N2O only in the first and third, and NO in the second and third steps. Ammonia is also evolved because it is not enough permanganate to oxidize all ammonia molecules. There was no oxygen evolution in any step.
With the use of toluene as a heat-absorbing medium to avoid local overheating due to the exothermicity of the first decomposition step, an isotherm-controlled-temperature decomposition was performed at 110 °C. The reaction temperature could not be exceeded the boiling point (110 °C) of toluene until liquid toluene is present. The CoIII centers and permanganate ion are oxidants, whereas the chloride ions and ammonia act as reducing agents. The amorphous decomposition was leached with water, and the aqueous extract contained [Co(NH3)6]Cl3, [Co(NH3)6]Cl2, NH4NO3, and NH4Cl, whereas the water-insoluble product was the same todorokite like phase as in the case of [Co(NH3)5Cl](MnO4)2 [59], (NH4)4Co2Mn6O12. Based on magnetic measurements, cobalt is in trivalent high-spin whereas the manganese is in the divalent or trivalent high-spin state, thus the todorokite-like compound probably contains MnII4MnIII2O1210− manganese oxide network with square-shape channels [59].
Heating of the decomposition products in toluene with (removal a part of Co in soluble compounds form) and without (keeping the Co:Mn=1:1 ratio) aqueous leaching at 500 °C resulted in Co-Mn spinels with cubic and tetragonal structures and various Co:Mn ratios depending on the reaction conditions (Scheme 3) [59].
Scheme 3.
The thermal decomposition of [Co(NH3)5Cl](MnO4)2. Reproduced from [59].
Scheme 3.
The thermal decomposition of [Co(NH3)5Cl](MnO4)2. Reproduced from [59].
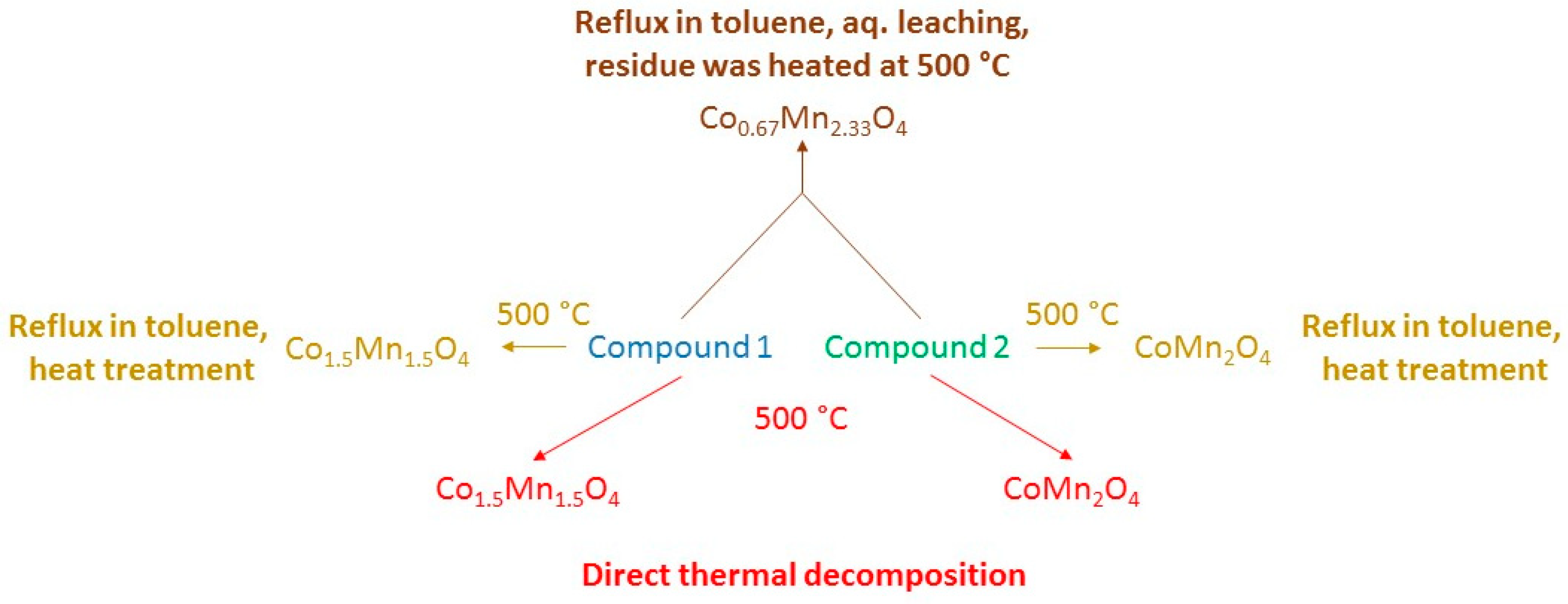
The heat treatment of the solid phase decomposition residue at 500 °C resulted in cubic spinel with Co1.5Mn1.5O4 (MnCo2O4 type) composition. The toluene-derived material without aqueous leaching resulted in tetragonal Co1.5Mn1.5O4, whereas after aq. leaching, a Co0.67Mn2.33O4 phase was obtained [59]. The photocatalytic activity of these spinel-like phases were observed in the degradation of harmful organic dyes as Congo red depending on their composition and synthesis conditions [59]
2.6.6. Potassium [hexaamminecobalt(III)] dichloride dipermanganate
Klobb analyzed the hexagonal crystalline by-product formed during the synthesis of [hexaamminecobalt(III)] permanganate and found that the compound contains chloride and potassium as well. This compound is soluble in water and crystallizes slowly at high concentrations only. It forms pretty crystals with very clear contours in a beautiful violet color. This salt can be prepared in a pure state by mixing a concentrated cold solution of 1 equivalent of [hexaamminecobalt(III)] chloride with 1.5 equivalent of potassium permanganate. The purple salt crystallizes in several hours [127].
The double salt can also be prepared with the dissolution of [hexaamminecobalt(III)] dichloro permanganate in a concentrated solution of KCl, together with [hexaamminecobalt(III)] chloride as a by-product:
2Co(NH3)6Cl2MnO4 + KCl = Co(NH3)6Cl3 + K[Co(NH3)6Cl2(MnO4)2]
[Hexaamminecobalt(III)] permanganate also reacts with potassium chloride:
Co(NH3)6(MnO4)3 + 2KCl = KMnO4 + K[Co(NH3)6Cl2(MnO4)2]
The double salt forms small violet or black crystals, according to the thickness, often with a greasy luster. It is very well soluble in water with partial decomposition. Evaporation of its aq. solutions in cold leaves back a residue in which KCl, [Co(NH3)6](MnO4)3 and [Co(NH3)6]Cl3 could be identified. It is insensitive to the impact of a hammer but detonates on heating with ammonia releasing [127]. Klobb declares that similar reactions of KBr and NH4Cl resulted in hexagonal blades under analog conditions, which were supposed to be the analog bromide or ammonium compounds, and analysis has not been performed [127].
3. Conclusions
The available data about permanganate, perrhenate, and pertechnetate salts of transition metal ammonia complexes, including the mono-, di-, tri-, tetra-, penta- and hexa-ammine derivatives have been classified and reviewed.
The simple ammine and mixed ligand complexes have been evaluated. The synthesis, spectral and thermal properties and structural features of the complexes have been given comprehensively.
Funding
Please add: This research received no external funding
| 1 | The authors declare the leaving of physisorbed and crystallization water, however, the compound was given as anhydrous. |
Conflicts of Interest
The author declare no conflict of interest.
References
- Lazarenko, G. A.; Neokladnova, L. N.; Shablovskaya, S. F. Effect of metals on the thermal decomposition of cobalt(III) hexaammine complexes with a metal-containing anion. Koordinatsionnaya Khimiya 1981, 7, 1485–1488. [Google Scholar]
- Lagunova, V.I.; Filatov, E.Y.; Plyusnin, P.E.; Korenev, S.V. In Situ and Ex Situ Studies of Tetrammineplatinum(II) Chromate Thermolysis. Russ. J. Inorg. Chem. 2020, 65, 1566–1570. [Google Scholar] [CrossRef]
- Serebrennikova, P.S.; Komarov, V.Y.; Sukhikh, A.S.; Khranenko, S.P.; Zadesenets, A.V.; Gromilov, S.A.; Yusenko, K.V. [Nien3](MoO4)0.5(WO4)0.5 co−crystals as single−source precursors for ternary refractory Ni–Mo–W alloys. Nanomaterials 2021, 11, 3272–3284. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.G.; Mogk, J.; Conrad, M.; Kraus, F. Octaammine EuII and YbII Azides and Their Thermal Decompositions to the Nitrides. Eur. J. Inorg. Chem. 2016, 26, 4162–4169. [Google Scholar] [CrossRef]
- Domonov, D.P.; Pechenyuk, S.I.; Semushina, Y.P.; Yusenko, K.V. Solid−state transformations in inner coordination sphere of [Co(NH3)6][Fe(C2O4)3]·3H2O as a route to access catalytically active Co−Fe materials. Materials 2019, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Yusenko, K.V.; Pechenyuk, S.I.; Vikulova, E.S.; Semushina, Y.P.; Baidina, I.A.; Filatov, E.Y. Isostructurality and Thermal Properties in the Series of Double Complex Salts [M1(NH3)6][M2(C2O4)3]·3H2O (M1=Co, Ir, M2 = Fe, Cr). J. Struct. Chem. 2019, 60, 1062–1071. [Google Scholar] [CrossRef]
- Farhadi, S.; Pourzare, K.; Bazgir, S. Co3O4 nanoplates: Synthesis, characterization and study of optical and magnetic properties. J. Alloys Compd. 2014, 587, 632–637. [Google Scholar] [CrossRef]
- Garkul’, I.A.; Zadesenets, A.V.; Plyusnin, P.E.; Filatov, E.Y.; Asanova, T.I.; Kozlov, D.V.; Korenev, S.V. Zinc(II) and Manganese(II) Oxalatopalladates as Precursors of Bimetallic Nanomaterials. Russ. J. Inorg. Chem. 2020, 65, 1571–1576. [Google Scholar] [CrossRef]
- Ilyushin, M.A.; Shugalei, I.V.; Tverjanovich, A.S.; Smirnov, A.V. Influence of the Mechanism of the Initial Stages of the Ligand Decomposition on the Initiating Ability of Cobalt(III) Ammine Tetrazolate Complexes. Russ. J. Inorg. Chem. 2020, 90, 640–647. [Google Scholar] [CrossRef]
- Béres, K.A.; Szilágyi, F.; Homonnay, Z.; Dürvanger, Zs.; Bereczki, L.; Trif, L.; Petruševski, V.M.; Farkas, A.; Bayat, N.; Kótai, L. Structural, Spectroscopic, and Thermal Decomposition Features of [Carbonatotetraamminecobalt(III)] Iodide—Insight into the Simultaneous Solid-Phase Quasi-Intramolecular Redox Reactions. Inorganics 2023, 11, 68–89. [Google Scholar] [CrossRef]
- Béres, K.A.; Homonnay, Z.; Kvitek, L.; Dürvanger, Z.; Kubikova, M.; Harmat, V.; Szilágyi, F.; Czégény, Z.; Németh, P.; Bereczki, L.; Petruševski, V.M. Béres, K.A.; Homonnay, Z.; Kvitek, L.; Dürvanger, Z.; Kubikova, M.; Harmat, V.; Szilágyi, F.; Czégény, Z.; Németh, P.; Bereczki, L.; Petruševski, V.M.; Pápai, M; Farkas, A.; Kótai, L. Thermally Induced Solid-Phase Quasi-Intramolecular Redox Reactions of [Hexakis(urea-O)iron(III)] Permanganate: An Easy Reaction Route to Prepare Potential (Fe,Mn)Ox Catalysts for CO2 Hydrogenation. Inorg. Chem. 2022, 61, 14403–14418. [Google Scholar] [PubMed]
- Franguelli, F.P.; Béres, K.A; Kótai, L. Pyridinesilver Tetraoxometallate Complexes: Overview of the Synthesis, Structure, and Properties of Pyridine Complexed AgXO4 (X = Cl, Mn, Re) Compounds. Inorganics 2021, 9, 79–92. [Google Scholar] [CrossRef]
- Holló, B.B.; Petruševski, V.M.; Kovács, G.B.; Franguelli, F.P.; Farkas, A.; Menyhárd, A.; Lendvay, Gy.; Sajó, I.E.; Nagy-Bereczki, L.; Pawar, R.P.; Szilágyi, I.M.; Bódis, E.; Kótai, L. Thermal and spectroscopic studies on a double-salt-type pyridine–silver perchlorate complex having κ1-O coordinated perchlorate ions. J. Therm. Anal. Calorim. 2019, 138, 1193–1205. [Google Scholar] [CrossRef]
- Kovacs, G.B.; May, N.V.; Bombicz, P.A.; Klébert, S.; Németh, P.; Menyhárd, A.; Novodárszki, G.; Petrusevski, V.; Franguelli, F.P.; Magyari, J.; Béres, K.; Szilágyi, I.M.; Kótai, L. An unknown component of a selective and mild oxidant: structure and oxidative ability of a double salt-type complex having κ1O-coordinated permanganate anions and three- and four-fold coordinated silver cations. RSC Advances 2019, 9, 28387–28398.
- Kocsis, T.; Magyari, J.; Sajo, I.E.; Pasinszki, T.; Homonnay, Z.; Szilagyi, I.M; Farkas, A.; May, Z.; Effenberger, H.; Szakall, S.; Pawar, R.P.; Kótai, L. Evidence of quasi-intramolecular redox reactions during thermal decomposition of ammonium hydroxodisulfitoferriate(III), (NH4)2[Fe(OH)(SO3)2]·H2O. J. Therm. Anal. Calorim 2018, 132, 493–502. [Google Scholar] [CrossRef]
- Sajó, I.E.; Kovács, G.B.; Pasinszki, T.; Bombicz, A.P.; May, Z.; Szilágyi, I.M.; Jánosity, A.; Banerji, K.K.; Kant, R.; Kótai, L. The chemical identity of ‘[Ag(py)2]MnO4‘ organic solvent soluble oxidizing agent and new synthetic routes for the preparation of [Ag(py)n]XO4 (X= Mn, Cl, Re, n=2-4) complexes. J. Coord. Chem. 2018, 71, 2884–2904. [Google Scholar] [CrossRef]
- Korenev, S.V.; Venediktov, Yu.V; Shubin, Yu.V.; Gromilov, S.A.; Yusenko, K.V. Synthesis and Structure of Binary Complexes of Platinum Group Metals — Precursors of Metallic Materials. J. Struct. Chem. 2003, 44, 45–59. [Google Scholar] [CrossRef]
- Chakravorti, M.C. The chemistry of coordinated perrhenateperrhenate (ReO4-). Coord. Chem. Revs. 1990, 106, 205–25. [Google Scholar] [CrossRef]
- Mansouri, M.; Atashi, H.; Tabrizi, F.F.; Mirzaei, A.A.; Mansouri, G. Kinetics studies of nano−structured cobalt−manganese oxide catalysts in Fischer−Tropsch synthesis. J. Ind. Eng. Chem. 2013, 19, 1177–1183. [Google Scholar] [CrossRef]
- Mansouri, G.; Mansouri, M. Synthesis and characterization of Co−Mn nanocatalyst prepared by thermal decomposition for Fischer−Tropsch reaction. Iran. J. Chem. Eng. 2018, 37, 1–9. [Google Scholar]
- Béres, K.A.; Homonnay, Z.; Barta Holló, B.; Gracheva, M.; Petruševski, V. M.; Farkas, A.; Dürvanger, Zs.; Kótai, L. Synthesis, structure, and Mössbauer spectroscopic studies on the heat-induced solid-phase redox reactions of hexakis(urea-O)iron(III) peroxodisulfate. J. Mater. Res. 2022. [Google Scholar] [CrossRef]
- Sajo, I.E.; Bakos, L.P.; Szilagyi, I.M.; Lendvay, G.; Magyari, J.; Mohai, M.; Szegedi, A.; Farkas, A.; Janosity, A.; Klebert, S.; Kótai, L. Unexpected Sequential NH3/H2O Solid/Gas Phase Ligand Exchange and Quasi-Intramolecular Self-Protonation Yield [NH4Cu(OH)MoO4], a Photocatalyst Misidentified before as (NH4)2Cu(MoO4)2. Inorg. Chem. 2018, 57, 13679–13692. [Google Scholar] [CrossRef] [PubMed]
- Kótai, L.; Fodor, J.; Jakab, E.; Sajó, I.E.; Szabó, P.; Lónyi, F.; Valyon, J.; Gács, I.; Argay, G.Y.; Banerji, K.K. A thermally induced low-temperature intramolecular redox reaction of bis(pyridine)silver(I) permanganate and its hemipyridine solvate. Transit. Met. Chem. 2006, 31, 30–34. [Google Scholar] [CrossRef]
- Kótai, L.; Sajó, I.; Fodor, J.; Szabó, P.; Jakab, E.; Argay, G.Y.; Holly, S.; Gács, I.; Banerji, K.K. Reasons for and consequences of the mysterious behaviour of newly prepared hemipyridine solvate of bis(pyridine)silver(I) permanganate, Agpy2MnO4 0.5py. Transit. Met. Chem. 2005, 30, 939–943. [Google Scholar] [CrossRef]
- Shubin, Yu.V.; Filatov, E.Yu.; Baidina, I.A.; Yusenko, K.V.; Zadesenetz, A.V.; Korenev, S.V. Synthesis of [M(NH3)5Cl](ReO4)2 (M = Cr, Co, Ru, Rh, Ir) and investigation of thermolysis products. crystal structure of [Rh(NH3)5Cl](ReO4)2. J. Struct. Chem. 2006, 47, 1103–1110. [Google Scholar] [CrossRef]
- Filatov, E.Yu.; Shubin, Yu.V.; Korenev, S.V. Study of nanoalloys formation mechanism from single-source precursors [M(NH3)5Cl](ReO4)2, M = Rh, Ir. Z. Kristallogr. Suppl. 2007, 26, 283–288. [Google Scholar] [CrossRef]
- Filatov, E.; Shubin, Yu.; Sharafutdinov, M. In situ synchrotron X-ray diffraction study of formation mechanism of Rh0.33Re0.67 nanoalloy powder upon thermal decomposition of complex precursor. Z. Kristallogr. Suppl. 2008, 27, 185–192. [Google Scholar] [CrossRef]
- Solt, H.E.; Németh, P.; Mohai, M.; Sajó, I.E.; Klébert, Sz.; Franguelli, F.P.; Fogaca, L.A; Pawar, R.P.; Kótai, L. Temperature-Limited Synthesis of Copper Manganites along the Borderline of the Amorphous/Crystalline State and Their Catalytic Activity in CO Oxidation. ACS OMEGA 2021, 6, 1523–1533. [Google Scholar] [CrossRef]
- Kótai, L.; Petruševski, V.M.; Bereczki, L.; Béres, K.A. Catalytic Properties of the Spinel-Like CuxMn3−xO4 Copper Manganese Oxides—An Overview. Catalysts 2023, 13, 129–160. [Google Scholar] [CrossRef]
- Nockemann, P.; Meyer, G. [Ag(NH3)2]ClO4: Kristallstrukturen, Phasenumwandlung, Schwingungsspektren. Z. Anorg. Allg. Chem. 2002, 628, 1636–1640. [Google Scholar] [CrossRef]
- Górska, N.; Mikuli, E.; Kótai, L. Spectroscopic, Structural and Thermal Characterization of Crystalline [Cr(OC(NH2)2)6]X3 (X = CIO4, BF4 and CI) Complexes. Eur. Chem. Bull. 2014, 3, 474–481. [Google Scholar]
- Bereczki, L.; Fogaca, L.A.; Durvanger, Z.; Harmat, V.; Kamaras, K.; Nemeth, G.; Hollo, B.B.; Petrusevski, V.M.; Bodis, E.; Farkas, A.; Szilágyi, I.M.; Kótai, L. Dynamic disorder in the high-temperature polymorph of bis[diamminesilver(I)] sulfate-reasons and consequences of simultaneous ammonia release from two different polymorphs, J. Coord. Chem., 2021, 74, 2144–2162. [Google Scholar] [CrossRef]
- Franguelli, F.P.; Barta-Holló, B.; Petruševski, V.M.; Sajó, I.E.; Klébert, Sz.; Farkas, A.; Bódis, E.; Szilágyi, I.M.; Pawar, R.P.; Kótai, L. Thermal decomposition and spectral characterization of di[carbonatotetraamminecobalt(III)] sulfate trihydrate and the nature of its thermal decomposition products. J. Therm. Anal. Calorim. 2021, 145, 2907–2923. [Google Scholar] [CrossRef]
- Fogaca, L.; Éva, K.; Németh, G.; Kamarás, K.; Béres, K.A.; Németh, P.; Petruševski, V.; Bereczki, L.; Barta-Holló, B.; Sajó, I.E.; Klébert, S.; Farkas, A.; Szilagyi, I.M.; Kótai, L. Solid-Phase Quasi-Intramolecular Redox Reaction of [Ag(NH3)2]MnO4: An Easy Way to Prepare Pure AgMnO2. Inorg. Chem. 2021, 60, 3749–3760. [Google Scholar] [CrossRef] [PubMed]
- Hillebrecht, H.; Thiele, G.; Koppenhöfer, A.; Vahrenkamp, H. Kristallstruktur und Schwingungsspektren von Zn(NH3)4(ClO4)2/Crystal Structure and Vibrational Spectra of Zn(NH3)4(ClO4)2. Z. Naturforsch. B 1994, 49, 1163–1168. [Google Scholar] [CrossRef]
- Béres, K.A.; Sajó, I.E.; Lendvay, Gy.; Trif, L.; Petruševski, V.M.; Barta-Holló, B.; Korecz, L.; Franguelli, F.P.; László, K.; Szilágyi, I.M.; Kótai, L. Solid-Phase “Self-Hydrolysis” of [Zn(NH3)4MoO4@2H2O] Involving Enclathrated Water—An Easy Route to a Layered Basic Ammonium Zinc Molybdate Coordination Polymer. Molecules 2021, 26, 4022–4041. [Google Scholar] [CrossRef] [PubMed]
- Sajó, I.E.; Kótai, L.; Keresztury, G.; Gács, I.; Pokol, GY.; Kristóf, J.; Soptrayanov, B.; Petrusevski, V.M.; Timpu, D.; Sharma, P.K. Studies on the chemistry of tetraamminezinc(II) dipermanganate ([Zn(NH3)4](MnO4)2). Low-temperature synthesis of the manganese zinc oxide (ZnMn2O4) catalyst precursor. Helv. Chim. Acta 2008, 91, 1646–1658. [Google Scholar] [CrossRef]
- Firozabadi, H.; Sardarian, A. R. An Efficient Conversion of Oximes to Their Corresponding Carbonyl Compounds with Bispyridinesilver Permanganate Under Mild Conditions. Synth. Commun. 1983, 13, 863–866. [Google Scholar] [CrossRef]
- Gulevskaya, A.L.; Pozharskii, A.F.; Lomachenkova, L.V. , Chichibabin amination of 1,3-dimethyllumazine, Khim. Geterotriskl. Soed. 1990, 1575. [Google Scholar]
- Besedin, D.V.; Gulevskaya, A.V.; Pozharskii, A. F. Reaction of 6,8-dimethylpyrimido[4,5-c]pyridazine-5,7(6H,8H)-dione with α,ω-diamines as the first example of tandem nucleophilic substitution in neutral azines. Mendeleev Commun. 2000, 10, 150–151. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Vessal, B.; Naderi, M. Bispyridinesilver permanganate[Ag(C5H5N)2]MnO4: an efficient oxidizing reagent for organic substrates. Tetrahedron Lett. 1982, 23, 1847–1850. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Sardarian, A.R. Facile Oxidation of Polycyclic Arenes and Acetylenic Hydrocarbons with Bis(pyridine)silver Permanganate and Bis(2,2′-bipyridyl)copper(II) Permanganate Under Mild and Neutral Conditions. Synthesis 1986, 11, 946–948. [Google Scholar] [CrossRef]
- Gulevskaya, A.V. , Maes, B.U.W.; Meyers, C. A Novel Synthetic Route to 2-Amino and 2-Alkylamino-1,3,5-Triazines Based on Nucleophilic Aromatic Substitution of Hydrogen: The First Reactions of 1,3,5-Triazine with Nucleophiles without Ring Decomposition. SYNLETT 2007, 1, 71–74. [Google Scholar]
- Chandra, R.; Sarkar, A. A Facile Oxidation of 3,4-Diphenylthiophene-2,5-Dimethanol and Furan-2,5-Dimethanol to 3,4-Diphenylthiophene-2,5-Dicarboxylic Acid using Bis(pyridine)silver permanganate, Proc. Ind. Nat. Sci. Acad. 1994, 60A, 465. [Google Scholar]
- Anjaneyuu, A.S.R.; Umasundari, P.; Sastry, Ch.V.M. Synthetic Experiments in Lignans. Part 10. Use of Bis (pyridine) silver Permanganate as a New Reagent for Synthesis of 1-Phenylnaphthalene Lactones. Ind. J. Chem. 1986, B25, 955. [Google Scholar]
- Poonam, A.; Rao, A.; Sharma, G.; Kotai, L.; Sajo, I.E.; Sharma, P.K. Kinetics and mechanism of the oxidation of aliphatic secondary alcohols by tetrakis(pyridine)silver dichromate. Int. J. Chem. 2015, 4, 13–21. [Google Scholar]
- Panwar, S.; Soni, U.; Goswami, G.; Kotai, L.; Sajo, I.E.; Sharma, P.K. Structure-Reactivity Correlation in the Oxidation of Substituted Benzyl Alcohols by Tetrakis (pyridine) Silver Dichromate. Int. J. Chem. 2014, 3, 288–299. [Google Scholar]
- Prasad, R.L.; Kushwaha, A.; Kumar, R.; Szilágyi, I.M.; Kótai, L. Solid-state thermal degradation behaviour of 1-D coordination polymers of Ni(II) and Cu(II) bridged by conjugated ligand. J. Therm. Anal. Calorim. 2013, 114, 653–664. [Google Scholar] [CrossRef]
- Meena, A.K; Daiya, A.; Sharma, A.; Banerji, J.; Sajo, S.; Kótai, L.; Sharma, V. Oxidation of some organic sulficles by tetrakis (pyridine) silver dichromate: A kinetic & mechanistic approach, Int. J. Chem. 2012, 1, 55–65. [Google Scholar]
- Banerji, J.; Kótai, L.; Sharma, P.K.; Banerji, K.K. Kinetics and mechanism of the oxidation of substituted benzaldehydes with bis(pyridine) silver permanganate. Eur. Chem. Bull. 2012, 1, 135–140. [Google Scholar]
- Purohit, T.; Banerji, J.; Kotai, L.; Sajo, I.; Banerji, K.K.; Sharma, P.K. Kinetics and mechanism of the oxidation of substituted benzaldehydes with bis(pyridine)silver permanganate. Indian J. Chem. Sect. A 2012, 89, 1045–1052. [Google Scholar]
- Banerji, J.; Kótai, L.; Banerji, K.K. Kinetics and mechanism of oxidation of formic and oxalic acids by bis(pyridine) silver permanganate. J. Indian Chem. Soc. 2009, 48, 797–800. [Google Scholar]
- Banerji, J.; Banerji, K.K.; Kotai, L.; Sharma, D.; Sharma, P.K. Kinetics and mechanism of the oxidation of organic sulfides with bis(pyridine)silver permanganate. J. Indian Chem. Soc. 2011, 88, 1879–1886. [Google Scholar]
- Meena, A.K.; Daiya, A.; Sharma, A.; Banerji, J.; Kotai, L.; Sharma, V. Oxidation of some vicinal and non-vicinal diols by tetrakis(pyridine)silver dichromate: A kinetic and mechanistic study. J. Indian Chem. Soc. 2011, 88, 1887–1893. [Google Scholar]
- Kumar, A.; Mishra, P.; Kotai, L.; Banerji, K.K. Kinetics and mechanism of the oxidative regeneration of carbonyl compounds from oximes by tetraamminecopper (II) permanganate. Indian J. Chem. Sect. A. 2003, 42, 72–74. [Google Scholar]
- Shukla, R .; Kotai, L.; Sharma, P.K.; Banerji, K.K. Kinetics and mechanism of the oxidative regeneration of carbonyl compounds from phenylhydrazones by tetramminecopper(2+)bis-(permanganate). J. Chem. Res. Synop. 2003, 4, 184–185.
- Kótai, L.; Németh, P.; Kocsis, T.; Sajó, I.E.; Pasinszki, T.; Szilágyi, M.A.; Kant, R.; Pawar, R.P.; Sharma, P.K. A new route to synthesize controlled-size MMn2O4-type transition metal (M=Cd, Zn, Cu) nanomanganites. Nano Studies, 2016, 13, 7–13. [Google Scholar]
- Kótai, L.; Sajó, I.E.;, Jakab, E.; Keresztury, G; Németh, C.; Gács, I.; Menyhárd, A.; Kristóf, J.; Hajba, L.; Petrusevski, V.M.; Timpu, D.; Sharma P.K. Studies on the Chemistry of [Cd(NH3)4](MnO4)2. A Low Temperature Synthesis Route of the CdMn2O4+x Type NOx and CH3SH Sensor Precursors. Z. Anorg. Allg. Chem. 2012, 638, 177–186.
- Bereczki, L.; Petruševski, V.M.; Franguelli, F.P.; Béres, K.A.; Farkas, A.; Holló, B.B.; Czégény, Z.; Szilágyi, I.M.; Kótai, L. [Hexaamminecobalt(III)] Dichloride Permanganate—Structural Features and Heat-Induced Transformations into (CoII,MnII)(CoIII,MnIII)2O4 Spinels. Inorganics 2022, 10, 252–278. [Google Scholar] [CrossRef]
- Franguelli, F.P.; Kováts, É.; Czégény, Zs.; Bereczki, L.; Petruševski, V.M.; Holló, B.B.; Béres, K.A; Farkas, A.; Szilágyi, I.M.; Kótai, L. Multi-Centered Solid-Phase Quasi-Intramolecular Redox Reactions of [(Chlorido)Pentaamminecobalt(III)] Permanganate—An Easy Route to Prepare Phase Pure CoMn2O4 Spinel. Inorganics 2022, 10, 18–40. [Google Scholar] [CrossRef]
- Hetmanczyk, L.; Hetmanczyk, J. Phase transition, thermal dissociation and dynamics of NH3 ligands in [Cd(NH3)4](ReO4)2. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2016, 164, 24–32. [CrossRef] [PubMed]
- Hetmanczyk, L.; Hetmanczyk, J. Comparison of vibrational dynamics, thermal behaviour, and phase transition in [Ni(NH3)4](ReO4)2 and [Ni(NH3)6](ReO4)2. J. Therm. Anal. Calorim. 2015, 119, 1415–1428. [Google Scholar] [CrossRef]
- Kótai, L.; Banerji, K.K. An improved method for the preparation of high-purity permanganate salts. Synth. React. Inorg. Met. Org. Chem. 2001, 31, 491–495. [Google Scholar] [CrossRef]
- Kótai, L.; Sajó, I.E.; Gács, I.; Sharma, P.K.; Banerji, K.K. Convenient routes for the preparation of barium permanganate and other permanganate salts. Z. Anorg. Allg. Chem. 2007, 633, 1257–1260. [Google Scholar] [CrossRef]
- Kótai, L.; Gács, I.; Sajó, I.E.; Sharma, P.K.; Banerji, K.K. Beliefs and facts in permanganate chemistry - An overview on the synthesis and the reactivity of simple and complex permanganates. Trends in Inorganic Chemistry 2009, 11, 25–104. [Google Scholar] [CrossRef]
- Kótai, L.; Keszler, Á.; Pató, J.; Holly, S.; Banerji, K.K. The reaction of barium manganate with acids and their precursors. Indian J. Chem. Sect. A. 1999, 38, 966–968. [Google Scholar]
- Franguelli, F.P.; Béres, K.A; Kótai, L., Pyridinesilver Tetraoxometallate Complexes: Overview of the Synthesis, Structure, and Properties of Pyridine Complexed AgXO4 (X = Cl, Mn, Re) Compounds. In Cornerstones in Contemporary Inorganic Chemistry, 1st ed.; Duncan, H.G., MDPI: Basel, Switzerland, 2022; pp. 251–263.
- Bruni, G.; Levi, G. Gli ammoniacati del sali d’argento. Gazz. Chim. Ital. 1916, 46, 17–42. [Google Scholar]
- Wilke-Dörfurt, E.; Gunzert, Th. Über Neue Salze der Perrheniumsäure. Z. Anorg. Allg. Chem. 1933, 215, 369–387. [Google Scholar] [CrossRef]
- Klobb, T. Combinaisons de l’ammoniaque avec les permanganates metalliques. Compt. Rend. Hebd. Seanc. Acad. Sci. ́ 1886, 103, 384–385. [Google Scholar]
- Scagliari, G.; Marangoni, A. , Isomorfismo fra perclorati e permanganate, Gazz. Chim. Ital. 1915, 45, 42–44. [Google Scholar]
- Kótai, L.; Gács, I.; Kazinczy, B.; Sajó, I.E.; Sreedhar, B. Quasi-intramolecular acid-base interactions in aqueous solutions of metal-complexes of basic ligands I. Generalized theoretical considerations on the deammoniation of [MLm]Xn type ammonia complexes. Transit. Met. Chem. 2003, 28, 292–295. [Google Scholar] [CrossRef]
- Kótai, L.; Horváth, T.; Szentmihályi, K.; Keszler, Á. Evidence for quasi-intramolecular acid-base reactions in solutions of transition metal ammine complexes. Transit. Met. Chem. 2000, 25, 293–294. [Google Scholar] [CrossRef]
- Kotai, L.; Argay, G.; Holly, S.; Keszler, A.; Pukanszky, B.; Banerji, K.K. Study on the Existence of Hydrogen Bonds in ammonium permanganate. Z. Anorg. Allg. Chem. 2001, 627, 114–118. [Google Scholar] [CrossRef]
- Christensen, O.T. Untersuchungen über Manganverbindungen. I. Über Ammoniumpermanganat, Z. Anorg. Allgem. Chem. 1900, 24, 203–2019. [Google Scholar]
- Kótai, L.; Banerji, K.K.; Sajó, I.; Kristóf, J.; Sreedhar, B.; Holly, S.; Keresztury, G.; Rockenbauer, A.; An unprecedented-type intramolecular redox reaction of solid tetraamminecopper (2+) bis (permanganate) ((Cu(NH3)4)(MnO4)2) - A low-temperature synthesis of copper dimanganese tetraoxide-type (CuMn2O4) nanocrystalline catalyst precursors. Helv. Chim. Acta 2002, 85, 2316–2327.
- Sharma, M.; Songara, U.; Swami, P.; Purohit, P.; Vyas, S.; Sharma, P. K. Structure-reactivity correlation in oxidation of substituted benzyl alcohols by tetramminecopper(II) bis(permanganate, Int. J. Chem. 2016, 5, 74–82. [Google Scholar]
- Radwan, F.M.; Abd El-Hameed, A.M.; Mahmoud, M.R.; Fahim, R. B. Thermal decomposition of ammonium permanganate. J. Therm. Anal. 1987, 32, 883–884. [Google Scholar] [CrossRef]
- Bircumshaw, L.L.; Taylor, F.M. The thermal decomposition of ammonium permanganate. J. Chem. Soc., 1950, 3674–3678. [Google Scholar] [CrossRef]
- Fogaça, L.A.; Bereczki, L.; Petruševski, V.M.; Barta-Holló, B.; Franguelli, F.P.; Mohai, M.; Béres, K.A.; Sajó, I.E.; Szilágyi, I.M.; Kotai, L. A Quasi-Intramolecular Solid-Phase Redox Reaction of Ammonia Ligands and Perchlorate Anion in Diamminesilver(I) Perchlorate. Inorganics 2021, 9, 38–57. [Google Scholar] [CrossRef]
- Mahroua, O.; Alili, B.; Ammari, A.; Bellal, B.; Bradai, D.; Trari, M. On the physical and semiconducting properties of the crednerite AgMnO2 prepared by sol-gel auto-ignition. Ceram. Int. 2019, 45, 10511–10517. [Google Scholar] [CrossRef]
- Koriche, N.; Bouguelia, A.; Mohammedi, M.; Trari, M. Synthesis and physical properties of new oxide AgMnO2. J. Mater. Sci. 2007, 42, 4778–4784. [Google Scholar] [CrossRef]
- Béres, K.A.; Petruševski, V.M.; Barta-Holló, B.; Németh, P.; Fogaça, L.A.; Franguelli, F.P.; Farkas, A.; Menyhárd, A.; Szilágyi, I.M.; Kótai, L. AgNO3·NH4NO3 - an enigmatic double-salt type "decomposition intermediate" of diamminesilver(I) permanganate. Z. Anorg. Allg. Chem. 2021, 647, 1166–1174. [Google Scholar] [CrossRef]
- Chakravorti, M.C.; Sarkar, M.B. Rhenium. Part XVII : A Few Complexes of Copper Containing Coordinated Perrhenate, [ReO4]. J. Indian Chem. Soc. 1983, 60, 617–714. [Google Scholar]
- Chakravorti, M.C.; Sarkar, M.B.; Bharadwaj, P.K. Rhenium part XV. Tetragonal complexes of nickel(II) containing coordinated perrhenate [ReO4]−. Transit. Met. Chem. 1981, 6, 211–214. [Google Scholar] [CrossRef]
- Chakravorti, M.C.; Sarkar, M.B. Rhenium. Part XVI. Ammine and pyridine compounds of zinc and cadmium containing coordinated perrhenate. Transit. Met. Chem. 1982, 7, 19–22. [Google Scholar] [CrossRef]
- Ephraim, F. Ueber die Natur der Nebenvalenzen XIX. Ammoniakate des Silbers. Ber. Dtsch. Chem. Ges. 1918, 51, 706–710. [Google Scholar] [CrossRef]
- Ephraim, F. Ueber die Natur der Nebenvalenzen XIX. Ammoniakate des Silbers. Ber. Dtsch. Chem. Ges. 1918, 51, 706–710. [Google Scholar] [CrossRef]
- Klobb, T. Combinaisons de l’ammoniaque avec les permanganates métalliques. Bull. Soc. Chim. Fr. 1890, 3, 508. [Google Scholar]
- Müller, A.; Böschen, I.; Baran, E.J.; Aymonino, P.J. Über Tetramminmetallchalkogenometallate. Monatsh. Chem. 1973, 104, 836–847. [Google Scholar] [CrossRef]
- Seferiadis, N.; Dubler, E.; Oswald, H.R. . Structure of Tetraamminecopper(II) Dipermanganate. Acta Cryst. 1986, 42, 942–945. [Google Scholar] [CrossRef]
- Khranenko, S.P.; Shusharina, E.A.; Gromilov, S.A.; Smolentsev, A.I. Crystal structure of [Cu(NH3)4](ReO4)2. J. Struct. Chem. 2009, 50, 1201–1203. [Google Scholar] [CrossRef]
- Müller, A.; Böschen, I.; Sievert, W. Notizen: Zur Kristallstruktur von [Zn(NH3)4](OsO3N)2 und [Cd(NH3)4](OsO3N)2. Z. Naturfosch. 1970, 25, 311–312. [Google Scholar] [CrossRef]
- Pitzer, K.S. The Crystal Structure of Tetramminocadmium Perrhenate, Cd(NH3)4(ReO4). Z. Krist. 1935, 92, 131–135. [Google Scholar] [CrossRef]
- Müller, A.; Christophliemk, P.; Tossidiss, I. Struktur thermisches verhalten und eigenschaften von tetramminkobalt(II)-perrhenat [Co(NH3)4](ReO4)2; elektronen-und schwingungsspektre. J. Mol. Struct. 1973, 15, 289–299. [Google Scholar] [CrossRef]
- Zadesenets, A.V.; Khranenko, S.P.; Shubin, Yu.V.; Baidina, I.A.; Korenev, S.V. Complex Salts [Pd(NH3)4](ReO4)2 and [Pd(NH3)4](MnO4)2: Synthesis, Structure, and Thermal Properties. Russ. J. Coord. Chem. 2006, 32, 374–379. [Google Scholar] [CrossRef]
- Rochon, F.D.; Kong, P.C.; Melanson, R. [Tetraammineplatinum(ll) Bislpertechnetate(VIl)]. Acta Cryst. 1990, 46, 8–10. [Google Scholar]
- Korolkov, I.V.; Zadesenets, A.V.; Gromilov, S.A.; Yusenko, K.V.; Baidina, I.A.; Korenev, S.V.; Metal solid solutions obtained by thermolysis of Pt and Re salts. Crystal structure of [Pt(NH3)4](ReO4)2. J. Struct. Chem. 2006, 47, 489–498. [Google Scholar] [CrossRef]
- Pechenyuk, S.I.; Kuznetsov, V.Ya.; Popova, R.A.; Zalkind, O.A. Synthesis and study of tetraammineplatinum(II) perrhenate. Zh. Neorg Khim. 1979, 24, 3306–3308. [Google Scholar]
- Gorbunov, V.V.; Shmagin, L.F. Burning of copper (II) tetramine salts. Combust. Explos. Shock Waves 1972, 8, 429–431. [Google Scholar] [CrossRef]
- Poineau, F.; Mausolf, E.; Kerlin, W.; Czerwinski, K. Hexaammine-cobalt(III) pertechnetate: preparation, structure and solubility. J. Radioanal. Nucl. Chem. 2016, 311, 775–778. [Google Scholar] [CrossRef]
- Kotai, L.; Kazinczy, B.; Keszler, Á.; Holly, S.; Gács, I.; Banerji, K.K. Three Reagents in One: Ammonium Permanganate in the Oxidation of Benzyl Alcohol. Z. Naturforsch. 2001, 56, 823–825. [Google Scholar] [CrossRef]
- Briscoe, H.V.A.; Robinson, P.L.; Rudge, A.J. Perrhenates of copper, nickel and cobalt and the ammines of these compounds. J. Chem. Soc. 1931, 1, 2211–13. [Google Scholar] [CrossRef]
- Zagorodnyaya, A.N.; Abisheva, Z.S.; Aitekeyeva, S.N.; Bukurov, T.N.; Sapukov, I.A. Synthesis and properties of tetraamminecopper diperrhenate, Komplex Ispolz. Miner. Sirya 2003, 1, 16–23. [Google Scholar]
- Zagorodnyaya, A.N.; Abisheva, Z.S. Rhenium recovery from ammonia solutions. Hydrometallurgy 2002, 65, 69–76. [Google Scholar] [CrossRef]
- Tellez, C. Espectro infrarrojo de [Zn(NH3)4](ReO4)2 con substitucion isotopica 14N/15N. Semina 1983, 4, 410–411. [Google Scholar]
- Zagordnyaya, A.N.; Abisheva, Z.S.; Sadikanova, S.E; Kvyatkovskaya, M.N.; Kokoveshnikova, T.A.; Sapukov, I.A. hermal decomposition of tetraamminecadmium diperrhenate. Izv. Nats. Akad. Nauk Respub. Kazakh. Ser. Khim. 2005, 1, 54–60. [Google Scholar]
- Tellez, C. Metal-ligand vibrations of [Cd(NH3)4](ReO4)2 with 110/116Cd and H/D isotope substitution. Semina 1980, 6, 73–74. [Google Scholar]
- Zagordnyaya, A.N.; Ponomareva, E.I; Abisheva, Z.S., Extraction technology for rhenium recovery from chloride-sulfate zinc-cadmium solutions. In Solvent extraction in the process industries: ISEC93. 1993rd ed. Logsdail, D.H.; Slater, M.J. Elsevier Applied Science: Amsterdam, Netherlands, 1993; pp. 167-174.
- Zagorodnyaya, A.N.; Abisheva, Z.S.; Kvyatkovskaya, M.N.; Kokoveshnikova, T.A.; Kasymova, A.S.; Sadykanova, S.E. Thermal decomposition of tetraamminecopper diperrhenate. Dokl. Nats. Akad. Nauk Respub. Kazakh. 2004, 3, 89–96. [Google Scholar]
- Bibikova, V.I.; Vasilevskaya, I.I.; Vasilyeva, A.G.; Niselson, L.A. Thermal decomposition of ammonium perrhenate to obtain rhenium heptoxide. J. Appl. Chem. (Zh. Prikl. Khim.) 1973, 43, 1115–1116. [Google Scholar]
- Ratner, Yu.E.; Tsvetkov, Yu.V.; Berezkina, L.G. Thermal dissociation and reduction of ammonium perrhenate. Zh. Neorg. Khim. 1968, 13, 1516–1519. [Google Scholar]
- Zagorodnyaya, A.N.; Abisheva, Z.S.; Kvyatkovskaya, M.N.; Kokoveshnikova, T.A.; Kasymova, A.S.; Sadykanova, S.E.; Bochevskaya, E.G. Thermal decomposition of tetraamminzinc diperrhenate. Komplex Ispolz. miner. Sirya 2005, 1, 22–29. [Google Scholar]
- Müller, A.; Böschen, I.; Baran, E.J. Uber Hexamminmetallchalkogenometallate. Monatsh. Chem. 1973, 104, 821–835. [Google Scholar] [CrossRef]
- Minacheva, L.Kh.; Kokunova, V.N.; Sergienko, V.S.; Kokunov, Yu.V. Crystal structure of hydroxonitrosotetraamineruthenium(IV) perrhenate [Ru(NO)(OH)(NH)](ReO4)2. Zh. Neorg. Khim. 2001, 46, 1293–1296. [Google Scholar]
- Baidina, I.A.; Filatov, E.Yu.; Makotchenko, E.V.; Smolentsev, A.I. Synthesis and structure investigation of Co(III) complex salts with the perrhenate anion. J. Struct. Chem. 2012, 53, 112–118. [Google Scholar] [CrossRef]
- Yusenko, K.V.; Baidina, I.A.; Gromilov, S.A.; Korenev, S.V. nvestigation of pentaamminechloroplatinum(IV) perrhenate dihydrate. J. Struct. Chem. 2007, 48, 578–582. [Google Scholar] [CrossRef]
- Krestov, G.A.; Yatsimirsikii, K.B. Thermodynamical characteristics of (chloro)(pentaamine)cobalt type complex compounds. Zh. Neorg. Khim. 1961, 6, 2294–2303. [Google Scholar]
- Sacconi, B.L.; Sabatini, A.; Cans, P. Infrared Spectra from of Some Metal-Ammine Complexes. Inorg. Chem. 1964, 3, 1772–1774. [Google Scholar] [CrossRef]
- Grinberg, A.A.; Varshavskii, Y.S. The frequency of coordinated ammonia deformation mode and its relationship with the chemical properties of transition metal ammonia complexes. Primen. Molekul. Spektr. Khim. 1966, 1, 104–107. [Google Scholar]
- Petrushevski, V.M.; Béres, K.A.; Bombicz, P.; Farkas, A.; Kótai, L.; Bereczki, L. Structural and Raman Spectroscopic Characterization of Tetrapyridinesilver(I) Perrhenate, [Agpy4]ReO4. Maced. J. Chem. Chem. 2022, 41, 37–46. [Google Scholar] [CrossRef]
- Lenz, E.; Murmann, K.R. The Preparation and Kinetics of Hydrolysis of Pentaammineperrhenatocobalt(III) Ion. Inorg. Chem. 1968, 7, 1880–1885. [Google Scholar] [CrossRef]
- Liss, I.B.; Murmann, R.K. Complex Ion Kinetics. Reaction Rates on Ion-Exchange Resins Compared to Those in Water, Inorg. Chem. 1975, 14, 2314–2317. [Google Scholar]
- Lenz, E.; Murmann, R. K. Pentaammineperrhenatocobalt(III) salts. Inorg. Synth. 1970, 12, 214–18. [Google Scholar]
- Baran, E.J.; Aymonino, P.J. Über Hexamminkobalt(lll)-permanganat. Z. Anorg. Allgem. Chem. 1968, 362, 215–219. [Google Scholar] [CrossRef]
- Willke-Dörfurt, E.; Balz, G.; Weinhard, A. Beitrag zur Kenntnis der Fluorsulfonsäure. Z. Anorg. Allgem. Chem. 1929, 185, 417–424. [Google Scholar] [CrossRef]
- Klobb, T. De quelques permanganates nouveaux. Bull. Soc. Chim. 1887, 47, 240–244. [Google Scholar]
- Schmidt, K.H.; Müller, A. Vibrational Spectra and Force Constants of [Cr(NH3)6]3+, [Co(NH3)6]3+, [Cu(NH3)]42+, and [Pd(NH3]42+ with 50Cr/53Cr, 63Cu/65Cu, 104Pd/110Pd, and H/D isotopic substitution. J. Mol. Struct. 1974, 22, 343–352. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



