
Submitted:
23 April 2023
Posted:
24 April 2023
You are already at the latest version
Abstract
Recent studies have revealed that several cancer cell types can upregulate inducible nitric oxide synthase (iNOS) and iNOS-derived nitric oxide (NO) after a moderate photodynamic challenge sensitized by 5-aminolevulinic acid (ALA)-induced protoporphyrin-IX. The NO signaled for cell resistance to photokilling as well as greater growth and migration/invasion aggressiveness of surviving cells. On this basis, it was predicted that diffusible NO from PDT-targeted cells in a tumor might enhance growth, migration, and invasiveness of non- or poorly PDT-targeted bystander cells. This was tested using a novel approach in which ALA-PDT targeted cancer cells on a culture dish were initially segregated from non-targeted bystander cells of the same type via impermeable silicone-rimmed rings. Several hours after LED irradiation, the rings were removed, and both cell populations analyzed in the dark for various responses. After a moderate extent of targeted cell killing (~25%), bystander proliferation and migration were evaluated, and both were found to be significantly enhanced. Enhancement corelated with iNOS/NO upregulation in surviving PDT-targeted cancer cells in the following cell type order: PC3 > MDA-MB-231 >U87 > BLM. If occurring in an actual PDT-challenged tumor, such bystander effects might compromise treatment efficacy by stimulating tumor growth and/or metastatic dissemination. Possible mitigation of these negative effects by using pharmacologic inhibitors of iNOS expression or activity as PDT adjuvants will be discussed.
Keywords:
PDT
; Nitric oxide
; iNOS/NO induction
; Bystander effects
1. Introduction
When cancer cells in a given population are targeted by Ionizing radiation, many cells may elude or survive the challenge. However, instead of retaining preexisting characteristics, these so-called “bystander” cells often exhibit more aggressive behavior in terms of more rapid proliferation, migration, and invasion [1,2,3]. The possibility of off-target bystander effects in conjunction with non-ionizing photodynamic therapy (PDT) for cancer has been recognized for many years, but much less is known about these effects than those induced by ionizing radiation. In considering the limitations for optimal PDT, (e.g. irregularities in photosensitizer-delivering tumor vasculature, non-uniform light delivery, light scattering, etc.), the authors of this review recognized that not all cells in a given tumor would be uniformly targeted during a PDT challenge. Non-uniform vascular delivery of the photosensitizing agent could be at least partially responsible for this. Moreover, because of vascular variability, not all tumor cells would be exposed to O2 to the same extent. Thus, O2 deficiency could also spare some sections of a tumor. From these considerations, we hypothesized [4] that cells experiencing significant PDT stress could send pro-survival/expansion signals to non-stressed counterparts (bystanders). We knew that targeted cells surviving a PDT challenge typically overexpress inducible nitric oxide synthase (iNOS) and that resulting NO stimulates proliferative and migratory aggressiveness [5,6,7,8]. From this knowledge, we postulated that this targeted cell NO, by diffusing to naïve bystander cells, would also make them more aggressive [4]. In this review, we discuss such bystander responses, their negative implications for PDT efficacy, and how the latter might be mitigated by specific pharmacologic interventions.
2. Nitric oxide and its relevance to cancer
Nitric oxide is a short-lived free radical molecule (τ <2 sec in H2O) which diffuses freely on its own in aqueous media and, like O2, tends to partition freely into hydrophobic regions of cells, e.g. mitochondrial and lysosomal membranes [9,10,11]. Naturally occurring NO is produced by enzymes of the nitric oxide synthase (NOS) family, including the neuronal (nNOS/NOS1), endothelial (eNOS/NOS3), and inducible (iNOS/NOS2) isoforms [12]. All three enzymes catalyze the 5-electron oxidation of L-arginine to L-citrulline and NO, NADPH and O2 serving as co-substrates [12]. Once formed, NO can react with abundant O2 to give nitrous anhydride or dinitrogen trioxide (N2O3), this being one important reason for NO’s short lifetime. NO is pleiotropic in nature and is involved in many different normo-physiologic as well as patho-physiologic processes. Because of its instability, NO must be continuously generated to reach biologically meaningful steady state levels. For example, eNOS-generated NO in the 1-10 nM range stimulates cyclic-GMP formation in vascular smooth muscle, leading to blood pressure reduction via vasodilation. However, iNOS-derived NO at much higher levels (1 µM or greater), as produced by vascular macrophages in response to infection, is cytotoxic and potentially oncogenic if it causes DNA damage [9,11]. In addition to relaxing vascular smooth muscle, low level NO can act as a cytoprotective antioxidant by intercepting free radical intermediates (oxyl and peroxyl radicals) that arise during propagative (chain) lipid peroxidation in membranes and lipoproteins [13,14,15]. Thus, NO in the micromolar range tends to be cytotoxic and potentially mutagenic, whereas in the low-to medium nanomolar range it can support survival as well as proliferative and migratory aggressiveness of tumor cells. NO-mediated bystander effects induced by anti-tumor PDT are one example of this acquired aggressiveness, as will be discussed in subsequent sections.
Many cancer cells, including those derived from human breast, prostate, colon, and brain tumors, express significant constitutive levels of iNOS/NO, which are often implicated in tumor persistence and progression [16,17,18]. Knockdown of pre-existing iNOS using siRNA or shRNA methodology has been shown to attenuate growth and expansion of malignancies [16,17,18], thereby substantiating iNOS/NO’s tumor-supporting role. iNOS level in resected tumors from cancer patients is now considered a reliable prognostic indicator, patients with the highest levels given the poorest survival chances [19]. Although constitutive or pre-existing iNOS may provide a survival/growth advantage in some tumors, the level of NO produced might still be limiting. One approach for investigating this is to determine whether low dose NO from a chemical NO donor might stimulate cancer cell proliferation or resistance to therapeutic challenges. In one early example, it was shown that the chemical donor of NO, spermine-NONOate (SPNO) in sub-toxic doses dramatically increased the resistance of human breast cancer COH-BR1 cells to oxidative killing by photodynamic stress [20]. Each of following were shown to contribute to this response: (a) inhibition of pro-apoptotic JNK and p38α activation; (b) inhibition of pro-apoptotic Bax and Bid expression; and (c) inhibition of anti-apoptotic Bcl-xL down-regulation [20]. Thus, these pro-survival signaling effects of NO were well-coordinated, i.e. they inhibited apoptosis promoters on the one hand while stimulating apoptosis inhibitors on the other. Although not identified in this study [20], it is likely that these NO effects occurred via S-nitrosation of the indicated effector proteins or associated regulators. There is also evidence that wild type tumor suppressor p53 can either block iNOS transcription or inactivate iNOS by binding to it [21,22]. On the other hand, it has been reported that NO can reduce p53′s pro-apoptotic activity by chemically modifying it and altering its conformation [23]. Interestingly, human carcinoma cells with dysfunctional (mutated) p53 were found to express higher levels of iNOS than wild-type controls, resulting in more rapid proliferation and upregulation of angiogenic factors [24]. As illustrated by these and many other examples, it is now clear that constitutive low level endogenous iNOS/NO can support the persistence and progression of a variety of human cancers. As we now realize and will discuss next, these factors can be dramatically enhanced in cancer cells that can resist and survive an oxidative photodynamic challenge.
3. iNOS/NO antagonism to anti-tumor photodynamic therapy
Anti-tumor PDT is a unique, minimally invasive therapy involving a photosensitizing agent (PS), molecular oxygen, and PS-exciting light in the far visible-to-near infrared wavelength range [25,26]. PDT is based on cytotoxic photochemical reactions that occur at/near PS localizing sites, i.e. tumor cells per se or proximal vascular cells, e.g. endothelial cells or fibroblasts. These reactions are mediated by PDT-generated reactive oxygen species (ROS), the most prominent of which is singlet molecular oxygen (1O2) [26]. Most PSs are innocuous until photoactivated, and PDT has few (if any) negative side effects unlike chemo- or radiotherapy [26]. The PS can be administered as such, a prime example being Photofrin®, (oligomeric hematoporphyrin), the first PS to receive FDA approval for clinical PDT [25]. Alternatively, a pro-sensitizer like 5-aminolevulinic acid (ALA) can be used [27,28]. Upon entering tumor cells via an amino acid receptor, ALA is metabolized via the heme biosynthetic pathway to the functional PS, protoporphyrin IX (PpIX) [27,28]. This pathway is typically more active in rapidly dividing malignant cells than normal counterparts, which increases selectivity for the former in this approach. PpIX accumulates initially in mitochondria, making these organelles the most prominent early targets of ALA-PDT damage. Besides high tumor cell selectivity, another advantage of this approach is that fluorescence of ALA-induced PpIX under low light intensity can be used for fluorescence-guided tumor surgery (FGS) [29]. This approach is now widely used in order to minimize excision of normal tissue around the tumor area. FGS is often followed up by PDT at higher light doses to eliminate any tumor tissue that is inaccessible to FGS.
Many cancers exhibit an innate or acquired resistance to various types of chemotherapy or radiotherapy [30], and it is now clear that resistance mechanisms also exist for PDT. How endogenous NO might affect PDT efficacy was first investigated ca. 25 years ago by two groups using various mouse-borne syngeneic tumors sensitized with Photofrin® [31,32]. They found that PDT-induced tumor suppression could be much improved when non-specific inhibitors of NOS activity (e.g L-NNA or L-NAME) were present during irradiation. Tumors with the highest constitutive NO output exhibited the greatest sensitivity to NOS inhibition [32]. More recent work with ALA/light-treated tumors produced similar findings [33]. The mechanistic reasoning for all these early findings was that vasodilation caused by low level NO was counteracting PDT’s anti-tumor vasoconstrictive effects [31,32,33]. However, at least three questions remained unanswered: (i) whether constitutive or basal NOS/NO is sufficient for hyper-resistance; (ii) whether stress-induced NOS/NO upregulation is necessary; and (iii) which of the three NOS isoforms is most important. In addressing these questions, Bhowmick et al. [5,6,7] found that the iNOS isoform was the principal source of NO involved in PDT resistance. They referred to this particular PDT as photodynamic “treatment” rather than “therapy” because the experiments were carried out in vitro using cultured human breast and prostate cancer cells. Importantly, the observed hyper-resistance was found to be mainly due to PDT-upregulated iNOS rather than preexisting/constitutive enzyme. At the time, such a finding about iNOS/NO was unprecedented for any type of cancer therapy. ALA-induced PpIX was the PS used in the above studies [5,6,7], so any PpIX export via the ABCG2 transporter [34] could have also imparted some resistance. In this case, however, no photodynamic stress would have been involved, in contrast to NO-induced resistance. Recognition of iNOS/NO involvement in PDT resistance was based on findings such as the following: (i) strong mitigation by a NO scavenger (cPTIO) or by specific inhibitors of iNOS activity (1400W, GW274150), apoptotic cell death increasing in response; (ii) prevention by siRNA-based iNOS knockdown, apoptosis being elevated in response; (iii) substantial “rescue” of iNOS-knockdown cells with spermine-NONOate (SPNO), a chemical NO donor [5,6].
In 2017, the above in vitro findings were replicated and substantiated at the in vivo level. Immunodeficient (SCID) female mice bearing human breast MDA-MB-231 tumor xenografts were sensitized with ALA-induced PpIX and irradiated with 630 nm LED light [35]. This caused a significant slowdown in tumor growth over a 12-day post-irradiation period. Administration of 1400W or GW274150 during this period slowed growth even further, consistent with iNOS/NO-dependent resistance [35]. Immunoblot and NO analyses on post-PDT tumor samples revealed a progressive increase in iNOS expression and NO output (each reaching 5-6-fold over starting level at 6 h post-PDT). A light-only control was unresponsive to 1400W, indicating that overexpressed, but not basal iNOS/NO, was promoting tumor growth/persistence after a PDT challenge [35]. Consistently, anti-apoptotic Bcl-xL, Survivin, and S100A4 underwent 1400W-inhibitable upregulation after the challenge, whereas pro-apoptotic Bax was down-regulated [35]. Collectively, this in vivo evidence was the first of its kind for an anti-tumor therapy that is opposed by the iNOS/NO it induces.
In addition to resisting eradication, certain tumor cells that survive PDT stress have been found to exhibit more aggressive behavior than non-stressed controls. For example, when human prostate cancer PC3 cells remaining alive (attached) after an ALA/light challenge were continuously monitored beyond 24 h, a strikingly (~3-fold) increase in proliferation rate was observed relative to dark (ALA-only) controls [8]. This growth spurt was abolished by 1400W or cPTIO, signifying iNOS/NO involvement. Of added significance was the discovery that surviving PC3 cells were more motile, as manifested by increased migration and invasion rates, iNOS/NO again playing a key driving role [8]. Enhanced iNOS/NO-mediated resistance as well as growth and migratory aggressiveness has also been observed for human glioblastoma U87 cells that can withstand PDT stress [36]. For example, in addition to resisting mitochondriia-initiated apoptosis, PDT-surviving U87 cells underwent a strong growth and invasiveness spurt which, as with PC3 cells, was 1400W-inhibitable, demonstrating iNOS/NO-dependency [36]. Importantly, this dependency was on stress-upregulated iNOS (>3-fold at 24 h post-PDT) rather than background iNOS, which was not significantly affected by 1400W. Matrix metalloproteinases (MMPs) are known to play a key role in cancer cell invasiveness and metastasis. ALA/light stress markedly increased MMP-9 activity in ALA/light-stressed PC3 and U87 cells [8,36]. In each case, inhibition of this activity elevation by 1400W signified iNOS/NO dependency [8,36]. Moreover, expression of TMP-1, which inhibits MMP-9 was progressively reduced, demonstrating cooperative responses that promoted surviving cell migration/invasion [8].
Considerable evidence about the signaling events that drive iNOS upregulation under PDT stress has also been obtained. In at least two human cancer lines, breast COH-BR1 and glioblastoma U87, iNOS induction required initial activation of transcription factor NF-κB. For ALA/light-challenged COH-BR1 cells, Bay11-7082, an inhibitor of phosphorylation and proteasomal removal of NF-κB regulatory subunit, IκB: (i) prevented translocation of NF-κB subunit p65 to the nucleus to initiate transcription, (ii) suppressed iNOS upregulation after the photochallenge, and (iii) increased the extent of cell photokilling via apoptosis [37]. Key signaling events upstream of NF-κB have also been identified. For example, in ALA/light-challenged COH-BR1 and U87 cells, the pro-tumor kinases PI3K and Akt were rapidly hyper-activated via phosphorylation [38]. PI3K inhibitors such as Wortmannin and LY294002 strongly suppressed Akt activation, NF-κB activation, and iNOS upregulation while stimulating apoptosis [37,38]. Thus, a PI3K → Akt → NF-κB → iNOS signaling sequence was in operation. For COH-BR1 cells, ODQ, an inhibitor of soluble guanylate cyclase (sGC), failed to enhance photostress-induced apoptosis, thereby arguing against NO/sGC/cGMP-mediated activation of protein kinase-G, a cancer cell pro-survival/expansion effector. However, pro-apoptotic Bax was upregulated and anti-apoptotic Bcl-xL was downregulated, 1400W or cPTIO accentuating these responses, consistent with NO playing a strong pro-survival/anti-apoptotic role [38]. Additional evidence on upstream signaling revealed that for ALA/light-challenged U87 cells, transacetylase p300 underwent greater: (i) Akt-dependent activation, and (ii) interaction with NF-κB subunit p65, which exhibited hyper-acetylation at lysine-310 (acK310) [38]. Moreover, photostressed U87 cells exhibited greater inactivating disulfide bond formation in tumor suppressor PTEN. This would have favored activation of p300 and Akt, leading to greater iNOS transcription via interaction of p65-acK310 with bromodomain protein Brd4 [39]. In addition to this, deacetylase Sir1 was down-regulated by photostress, which would have favored the observed increase in p65-acK310 level and hence greater iNOS transcription [38]. These findings with two different human cancer lines, COH-BR1 and U87, provided important mechanistic insights on how these, and presumably other cancer cells, may not only resist a PDT challenge, but respond more aggressively if capable of withstanding it.. When obtained, these findings [37,38,39] were unique in the overall field of anti-tumor PDT biology/biochemistry.
Certain chemo and radiotherapeutic strategies based on oxidative stress are also known to be antagonized by iNOS/NO. For chemotherapy, cisplatin (CDDP) is the most widely recognized drug to elicit antagonism. For example, one study involving CDDP-treated melanoma cells showed that two key pro-apoptotic proteins were inhibited by S-nitrosation: caspase-3 and prolyl hydroxylate-2 (PHD2), which otherwise targets pro-survival HIF-1α for proteosomal degradation [40]. Another example pertains to tumor-associated macrophages (TAMS), specifically M2-TAMS which play an important role in tumor survival, expansion, and drug resistance. A recent study showed that iNOS-derived NO from human and murine glioma M2-TAMs induced CDDP resistance by inhibiting acid sphingomyelinase (A-SMase), which otherwise stimulates apoptosis via death receptor CD95 [41]. Interestingly, the antitherapeutic NO in this case, unlike that described above for PDT models [35,36,37,38,39], was not emitted by stressed tumor cells per se, but rather by TAMs in the tumor microenvironment. With regard to ionizing radiation, a recent study showed that immortalized as well as patient-derived glioblastoma cells responded to fractionated radiation (2 Gy of γ-rays over 3 days) by expanding stem-like cell (GSC) populations with upregulated iNOS/NO [42]. Post-radiation GSC expansion was resistant to higher dose radiation (10 Gy), or CDDP treatment. However, prior iNOS knockdown prevented all responses, thus confirming iNOS/NO instigation. As another example, pancreatic ductal adenocarcinoma (PDAC), like glioblastoma, is difficult to treat and highly lethal. Low level iNOS-derived NO was found to play a key role in PDAC persistence and progression after radiation exposure [43]. Another highly relevant study showed that iNOS/NO was strongly upregulated in PDAC-associated fibroblasts (CAFs) after moderate exposure to ionizing radiation [44]. When treated with these CAFs, PANC cells exhibited a striking upregulation of iNOS/NO, which stimulated cell proliferation and migration [44]. Enhancing their significance, these findings were subsequently validated at the in vivo level using orthotopic PDAC tumors in mice [44].
4. Bystander effects of PDT-upregulated iNOS/NO
When exposed to physical or chemical perturbations, cancer cells often send stress signals to unperturbed neighboring cells (bystanders), which may cause the latter to respond in diverse ways, ranging from viability loss to stimulated migration and proliferation, depending on signal intensity. This phenomenon, referred to as a “bystander effect”, was first recognized as an unexpected side effect of ionizing radiation [1,2,3]. When such radiation (e.g. X-rays, γ-rays, α-particles) is used for anti-tumor therapy, not all cells in any given solid tumor will be targeted uniformly due to many technical limitations [3]. However, well-exposed cells are known to elicit a variety of stress responses in non- or minimally exposed bystanders, ranging from DNA damage, faulty damage repair, and apoptotic cell death to faster proliferation and migration [1,2,3]. Stress signals can be sent by at least two different mechanisms: (i) via gap junctions between cells, or (ii) via the extracellular medium, i.e. without actual physical contact between cells [2,3]. The latter mechanism is illustrated schematically in Figure 1A.
For ionizing radiation, a variety of signaling molecules capable of traversing aqueous media from targeted to bystander cells have been identified, e.g. (i) cytokines such as TGF-β and TNF [45,46], (ii) ROS such as H2O2 [47], and (iii) NO or NO-derived species like nitrous anhydride (N2O3) [48,49]. Like H2O2, continuously generated NO diffuses rapidly in aqueous media,, but unlike H2O2, NO can partition into low polarity lipoprotein or membrane sites. In addition, NO is not susceptible to any known enzymatic scavenging, which distinguishes it from H2O2; Thus, enzymatic scavenging could not impose limitations on NO’s sphere of activity, including potential bystander activity.
Bystander effects of non-ionizing PDT were first described over twenty years ago [50], but intercellular signaling mechanisms were not defined in rigorous biochemical terms. Subsequent studies with cancer cells involved (i) a plasma membrane-bound PS with trans-well inserts to separate targeted from bystander cells [51] or (ii) time-lapse fluorescence microscopy to monitor two non-contacting cell groups on a culture dish after either one was PDT-stressed. [52]. The only endpoint monitored in these studies was ROS-induced death of targeted or bystander cells. There was no consideration of a possible iNOS/NO role in the observed responses or whether any photostress-surviving cells might divide or migrate more aggressively.
These questions were directly addressed by Bazak et al. [53] about six years ago. The investigators devised a novel approach based on silicone-rimmed rings for initially separating photodynamically-targeted cancer cells from non-targeted bystanders. Figure 1B illustrates a typical experimental arrangement with two rings in place on a large culture dish. Human prostate cancer PC3 cells were used in initial studies [53]. Target cells (outside rings) were sensitized with ALA-induced PpIX while PC3 bystanders (inside rings) saw no ALA. The cells were then exposed to ~1 J/cm2 of LED irradiation. After a suitable dark delay, e.g. 2 h, the rings were carefully removed, allowing diffusible stress-upregulated molecules like NO to flow from targeted to bystander zones. As anticipated from previous findings [5,6,7,8], surviving targeted cells exhibited a very robust upregulation of iNOS, which reached ~12-fold the control level 4h after irradiation, and remained there for at least another 20 h (Figure 2A). Importantly, iNOS was also upregulated in non-stressed bystander cells, albeit more slowly and less extensively, reaching 4-5-times its starting level after 24 h (Figure 2B) [53]. Target cell controls (ALA alone or light alone) showed no significant increase in iNOS above background for either of the two cell populations. Unlike iNOS, nNOS and eNOS exhibited no change from their low initial levels in PC3 cells after the photodynamic challenge. Thus, iNOS appeared to be unique in this capacity, as substantiated with several other cancer lines [4,35,36]. Moreover, iNOS knockdown in target PC3 cells markedly suppressed the enzyme’s induction in bystanders, suggesting that iNOS-derived NO from the former was essential [53]. This was confirmed by showing that the NO trap, cPTIO, strongly inhibited bystander iNOS induction, thus revealing that diffusible NO generated continuously by targeted cells was the active mediator. In agreement with this, conditioned medium from these cells had no significant effect [53], thus indicating that relatively long-lived agents such as hydroperoxides or cytokines could not have been involved. Also ruled out were relatively stable byproducts of NO such as N2O3 and nitrite (NO2-). In addition to iNOS, at least three other proteins were either upregulated or hyperactivated in targeted and bystander cells. Cyclooxygenase-2 (COX-2), which is expressed in many different tumors and plays a pro-survival (anti-apoptotic) role, was upregulated in ALA/light-targeted and bystander cells, though more slowly in the latter, after irradiation [53]. Both cell responses were strongly attenuated by NO scavenger, cPTIO, implying NO-dependency. In contrast to COX-2, protein kinases Akt and ERK-1/2 were not overexpressed, but became more enzymatically active, as indicated by greater phosphorylation in Western blot bands [53]. Phosphorylation of Akt and ERK-1/2 was nullified by the NO trap, cPTIO, again signifying NO-dependency. In accord with these responses, a striking increase in proliferative and migratory aggressiveness was observed for bystander PC3 cells and, once again, this was dependent on stress-upregulated NO released from targeted cells [53]. For the experiment represented in Figure 3A, bystander cells were dividing at least 50% faster than controls at 39 h after irradiation. Likewise, at 21 h after irradiation, bystanders were migrating at least 30% faster than their control cells (Figure 3B). Thus, bystander cells were replicating the enhanced aggressiveness exhibited by cells that remained alive and active in the PDT-targeted compartment.
In follow-up work, Bazak et al. [54] compared the bystander behavior of prostate PC3 cells with that of three other human cancer lines: melanoma BLM, glioblastoma U87, and breast MDA-MB-231. ALA treatment was the same for all four lines, but subsequent light fluences were varied such that a uniform targeted cell kill was attained for all, i.e. ~25% at 24 h after irradiation [54]. Targeted cell iNOS levels cells were assessed by immunoblotting at various post-irradiation times up to 24 h. Enhanced bystander cell proliferation or migration was tracked as a function of maximal iNOS upregulation. As shown in Figure 4A, greater proliferation rate of bystander cells increased exponentially with maximal targeted iNOS induction for the four cancer lines examined, BLM cells showing the smallest change and PC3 cells the greatest. A similar exponential increase in elevated migration rate was observed for the four bystander lines (Figure 4B), PC3 cells with maximal iNOS induction showing, again, the maximal effect. Thus, the extent to which growth and migration of bystander cells was escalated depended on the degree to which iNOS/NO was boosted in PDT-targeted cells [54]. Aa initially indicated, surviving targeted cells also exhibited greater growth/migration aggressiveness (Sect. 3); however, this was self-promoted, i.e. directly due to upregulated iNOS/NO in the targeted population. Since NO from targeted cells stimulated iNOS upregulation in bystanders (Figure 2B), it appears that a “feed-forward field effect” was in operation whereby the signaling effects of NO from PDT-challenged cells were “broadcasted” to non-challenged bystanders, thereby disseminating pro-growth/migration stimuli. When reported, these observations [4,53,54} were unprecedented in the context of PDT. Their significance was enhanced by another group’s recent studies which described differences in bystander behavior for prostate cancer cells of varying malignancy grade [55].
The mechanism by which elevated NO diffusing from targeted to bystander cells stimulates iNOS expression in the latter (see Figure 2) is unknown. Recall that upregulation of targeted cell iNOS requires PI3K and Akt activation followed by NF-κB activation with nuclear transfer and acetylation of p65 (Sect 3). However, this sequence of events was set off by photooxidative stress signaling in targeted cells [53,54], whereas no oxidative pressure was imposed on naïve bystanders. Cytokines such as interferon-γ (INF-γ), interferon-β (INF-β), and tumor necrosis factor-α (TNF-α) are known to stimulate iNOS expression in mammalian cells [56,57]. It is possible that NO continuously emanating from targeted cells somehow activated one or more of these cytokines, which in turn could have stimulated iNOS formation. More comprehensive PDT-based studies are needed in order to deduce the precise mechanism of bystander induction by targeted cell-derived NO.
As indicated In Sect. 3, there are many examples of cancer cells that exploit NO to divide and migrate more aggressively after withstanding a chemo- or radiotherapeutic attack. Existing evidence for ionizing- and PDT-induced bystander effects [1,2,3,4] suggests that similar effects might occur during chemotherapeutic treatments based on imposed oxidative stress. In support of this idea is recent preliminary evidence which showed that exposing PC3 cells to cisplatin (CDDP), also elicited hyper-aggressiveness in non-exposed bystander cells. Bystander iNOS was overexpressed ~3-fold in target cells that were incubated with 10 μM CDDP for 12 h, after which there was a strong increase in NO-mediated (GW274150- or 1400W-inhibitable) bystander proliferation rate [JM Fahey, AW Girotti, unpublished findings]. This suggests that NO-mediated hyper-aggressiveness of bystander cells is not limited to PDT, but could extend to many oxidative chemotherapies as well. We look forward to comprehensive follow-up studies dealing with this aspect.
5. Pharmacologic suppression of iNOS/NO anti-therapeutic effects
If occurring during clinical PDT (or even chemotherapy or radiotherapy), the iNOS/NO effects described could have negative effects on treatment outcomes, particularly if significant numbers of tumor cells can withstand the challenge or lie in NO-accessible bystander regions. Pharmacologic inhibition of iNOS activity has been advocated for tumors that significantly rely on iNOS-derived NO for promotion of growth and metastatic dissemination. Activity inhibitors such as L-NAME, and L-NNA have been tested in in vitro and in vivo model systems [2,4,5,6]. Such inhibitors could be used alone or in combination with PDT treatment. However, these compounds are not specific for iNOS and might also inhibit eNOS or nNOS. Thus, the possibility of compromising normal physiologic functions such as blood pressure regulation and bacterial infections would be a concern. At least two activity inhibitors, L-NIL and GW274150, which are highly specific for iNOS, have been safely tested in clinical trials, although these had no relationship to cancer or cancer therapeutics [58,59]. Such agents in appropriate doses and with sufficiently long post-administration lifetimes might improve the efficacy of PDT for cancer patients, and we look forward to their clinical testing for this purpose.
Another possibility for mitigating the negative effects of overexpressed iNOS on PDT would be to suppress its expression at the transcriptional level. Recent studies [38,39] have shown that accelerated growth and migration of PDT-surviving glioblastoma cells in vitro could be prevented by JQ1, an inhibitor of the bromo/extra-terminal (BET) domain of Brd4, an epigenetic “reader” protein [60,61]. Brd4, along with acetylated p65 subunit of NF-κB, was found to be essential for iNOS transcription in these cells [38,39]. At 100-fold lower concentration than 1400W or GW274150, JQ1 was much more effective in suppressing post-PDT hyper-aggressiveness than either of these other inhibitors. This was the first known evidence that a BET inhibitor, by blocking iNOS expression could strongly diminish cancer cell aggressiveness [39]. JQ1 may have also interfered with transcription of other pro-tumor proteins such as Bcl-xL, MMP-9, and Survivin. If their levels were reduced, however, this might have been an indirect effect of JQ1 on iNOS because NO is known to signal for induction of these proteins [39]. BET inhibitors like JQ1 have been shown to be highly effective against several cancers at the in vitro, animal model, and clinical trial levels. Hence there is great promise in their being used as adjuvants for anti-tumor PDT. Thus, there is good reason to begin clinical trials on BET inhibitors as PDT adjuvants as soon as possible.
6. Summary and outlook
There is now substantial evidence that preexisting and/or overexpressed iNOS/O can compromise anti-tumor PDT and possibly stimulate disease progression if the extent of tumor eradication is not great enough. Sensitizer or pro-sensitizer uptake by cells in any given tumor is not expected to be uniform throughout, nor is light delivery or O2 availability. As a result, some cells will be more heavily stressed by photodynamic action than others, some of which could be relatively unaffected bystanders. Using in vitro model systems, the authors and coworkers have shown that surviving targeted cancer cells strongly upregulate iNOS and that its continuously generated NO promotes cell survival and expansion [62]. Moreover, diffusion of this NO to bystander cells induces iNOS/NO there, and this also stimulates cell growth and migratory aggressiveness. Metastatic dissemination of malignant cells would be a particularly unfortunate outcome of such enhanced aggressiveness. These effects have the overall appearance of a “relay-type” process whereby NO overproduced by targeted cells stimulates NO generation in nearby bystanders, which can then propagate it to other naïve cells; see Figure 5 for an illustration.
This is an example of a “feed-forward” process, a term first used for describing the bystander effects of ionizing radiation [1,2,3]. Similar propagation could occur in the targeted cell population. However, it was only through evaluation of separated cell populations that this phenomenon came to be realized [4,53]. In the genre of radiation biology this process has also been represented as a “NO feed-forward field effect” [3]. As documented for targeted cells that survive a PDT challenge, enhanced proliferative and migratory/invasive aggressiveness of bystander cells is likely to promote tumor growth and metastatic expansion. These negative side effects could be mitigated by introducing inhibitors of iNOS activity as PDT adjuvants. Two candidates in this regard are L-NIL and GW274150, which have already been tested in clinical trials, although these were not cancer- or PDT-related [58,59]. A more promising approach for suppressing iNOS/NO-enhanced aggressiveness of PDT-surviving or bystander cells would be to administer a BET inhibitor like JQ1. This is supported by the following observation. At a far lower concentration than GW274150, JQ1 was much more effective in inhibiting accelerated proliferation of PDT-surviving glioblastoma cells [39]. JQ1 should also act as a strong suppressor of the enhanced aggressiveness of post-PDT bystander cells. However, direct experimental evidence for the latter effect has not yet been obtained. On their own, JQ1 and other BET inhibitors are very effective against a variety of malignancies [60,61]. Consequently, administering such inhibitors as PDT adjuvants holds great promise for limiting the negative effects of upregulated iNOS/NO (62). Thus, we look forward to the first clinical testing of BET inhibitors for this purpose.
Acknowledgements
Studies by the authors were supported by the following grants: USPHS Grant CA70832 from the National Cancer Institute (to AWG) and Grant UMO-2017/27/B/NZ5/02620 from the (to WK).
Conflicts of Interest
The authors have no conflicts of interest to declare.
References
- Azzam, E.I.; de Toledo, S.M.; Little, J.B. Stress signaling from irradiated to non-irradiated cells. Curr. Cancer Drug Targets 2006, 4, 53–64. [Google Scholar] [CrossRef]
- Baskar, R. Emerging role of radiation-induced bystander effects: cell communications and carcinogenesis. Genome Integr. 2010, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Hei, T.K.; Zhow, H.; Chai, Y.; Ponaiya, B.; Ivanov, V.N. Radiation-induced non-targeted response: mechanism and potential clinical implications. Curr. Mol. Pharmacol. 2011, 4, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Bazak, J.; Fahey, J.M.; Wawak, K.; Korytowski, W.; Girotti, A.W. Bystander effects of nitric oxide in anti-tumor photodynamic therapy. Cancer Cell & Microenvironment 2017, 4, e1511. [Google Scholar]
- Bhowmick, R.; Girotti, A.W. Cytoprotective induction of nitric oxide synthase in a cellular model of 5-aminolevulinic acid-based photodynamic therapy. Free Radic. Biol. Med. 2010, 48, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Girotti, A.W. Rapid upregulation of cytoprotective nitric oxide in breast tumor cells subjected to a photodynamic therapy-like oxidative challenge. Photochem. Photobiol. 2011, 87, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Girotti, A.W. Pro-survival and pro-growth effects of stress-induced nitric oxide in a prostate cancer photodynamic therapy model. Cancer Lett. 2014, 343, 115–122. [Google Scholar] [CrossRef]
- Fahey, J.M.; Girotti, A.W. Accelerated migration and invasion of prostate cancer cells after a photodynamic therapy-like challenge: role of nitric oxide. Nitric Oxide, 2015, 49, 47–55. [Google Scholar] [CrossRef]
- Wink, D.A.; Mitchell, J.B. ; Chemical biology of nitric oxide; insights into the regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 1998, 25, 434–456. [Google Scholar] [CrossRef]
- Thomas, D.D.; Liu, X.; Kantrow, S.P.; Lancaster, J.R., Jr. The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA, 2001, 98, 355–360. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Thomas, D.D.; Donzelli, S.; Espey, M.G.; Roberts, D.D.; Wink, D.A. et al. The biphasic nature of nitric oxide responses in tumor biology. Antioxid. Redox. Signal. 2006, 8, 1329–1337.
- Alberton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases in mammals. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Rubbo, H.; Radi, R.; Trujillo, M.; Telleri, R.; Kalyanaraman, B.; Barnes, S. et al. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation; formation of novel nitrogen-containing oxidized lipid derivatives. J. Biol. Chem. 1994, 269, 26066–26075. [Google Scholar] [CrossRef] [PubMed]
- Rubbo, H.; Parthasarathy, S.; Barnes, S.; Kirk, M.; Kalyanaraman, B.; Freeman, B. Nitric oxide inhibition of lipoxygenase-dependent liposome and low-density lipoprotein oxidation termination of radical chain propagation reactions and formation of nitrogen-containing oxidized lipid derivatives. Arch. Biochem. Biophysics 1995, 324, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Kelley, E.E.; Wagner, B.A.; Buettner, G.R.; Burns, C.P. Nitric oxide inhibits iron-induced lipid peroxidation in HL-60 cells. Arch. Biochem. Biophys. 1999, 370, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Kostorou, V.; Cartwright, J.E.; Johnstone, A.P.; Boult, J.K.R.; Cullis, E.R.; Whitley, G.S.J. The role of tumour-derived iNOS in tumor progression and angiogenesis. Br. J. Cancer 2011, 104, 83–90. [Google Scholar] [CrossRef]
- Heinecke, J.L.; Ridnour, L.A.; Cheng, R.Y.; Switzer, C.H.; Lizardo, M.M.; Khanna, C. Tumor microenvironment-based feed-forward regulation of NOS2 in breast cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, 6323–6328. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Heinecke, J.L.; Ridnour, L.A.; Cheng, R.Y.; Kesarwala, A.H.; Switzer, C.H. Signaling and stress: the redox landscape in NOS2 biology. Free Radic. Biol. Med. 2015, 87, 204–225. [Google Scholar] [CrossRef]
- Glynn, S.A.; Boersma, B.J.; Dorsey, T.H.; Yi, M.; Yfantis, H.G.; Ridnour, L.A. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. J. Clin. Invest. 2010, 120, 3843–3854. [Google Scholar] [CrossRef]
- Bhowmick, R.; Girotti, A.W. Signaling events in apoptotic photokilling of 5-aminolevulinic acid-treated tumor cells; inhibitory effects of nitric oxide. Free Radic. Biol. Med. 2009, 47, 731–740. [Google Scholar] [CrossRef]
- Forrester, K.; Ambs, S.; Lupold, S.E.; Kapust, R.B.; Spillare, E.A.; Weinberg, W.C.; et al. Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc. Natl. Acad. Sci. USA 1996, 93, 2442–2447. [Google Scholar] [CrossRef]
- Ambs, S.; Ogunfusika, M.O.; Merriam, W.G.; Bennett, W.P.; Billiar, R.R.; Harris, C.C. Upregulation of inducible nitric oxide synthase expression in cancer-prone p53 knockout mice. Proc. Natl. Acad. Sci. USA 1998, 95, 8823–8828. [Google Scholar] [CrossRef]
- Calmels, S.; Hainaut, P.; Ohshima, H. Nitric oxide induces conformational and functional modifications of wild-type p53 tumor suppressor protein. Cancer Res. 1997, 57, 3365–3369. [Google Scholar]
- Ambs, S.; Merriam, W.G.; Ogunfusika, M.O.; Bennett, W.P.; Ishibe, N.; Hussain, S.P. p53 and vascular endothelial growth factor regulate tumor growth of NOS2-expressing human carcinoma cells. Nat. Med. 1998, 4, 137101376. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.G.; Gollnick, S.O. Photodynamic therapy of cancer: an update. CA Cander J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Kennedy, J.C.; Pottier, R.H. Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy. J. Photochem. Photobiol. 1992, B14, 275–292. [Google Scholar] [CrossRef]
- Peng, Q.; Berg, J.; Moan, J.; Kongshaug, M.; Nesland, J.M. 5-Aminolevulinic acid-based photodynamic therapy; principles and experimental research. Photochem. Photobiol. 1997, 65, 235–251. [Google Scholar] [CrossRef]
- DeLong, J.C.; Hoffman, R.M.; Bouvet, M. Current status and future perspectives of fluorescence-guided surgery for cancer. Expert Rev. Anticancer Ther. 2016, 16, 71–81. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Dudas, J.; Skvortsov, S.; Ganswindt, U.; Riechelmann, H.; Skvortsov, I.I. Therapy resistance mediated by cancer stem cells. Semin. Cancer Biol. 2018, 53, 156–157. [Google Scholar] [CrossRef]
- Henderson, B.W.; Sitnik, T.M.; Vaughan, L.A. Potentiation of photodynamic therapy antitumor activity in mice by nitric oxide synthase inhibition is fluence rate-dependent. Photochem. Photobiol. 1999, 70, 64–71. [Google Scholar] [CrossRef]
- Korbelik, M.; Parkins, C.S.; Shibuya, H.; Cecic, I.; Stratford, R.M.L.; Chaplin, D.J. Nitric oxide production by tumor tissue: impact on the response to photodynamic therapy. Br. J. Cancer 2000, 82, 1835–1843. [Google Scholar] [CrossRef]
- Reeves, K.J.; Reed, M.W.R.; Brown, N.J. Is nitric oxide important in photodynamic therapy? J. Photochem. Photobiol. B. 2009, 95, 141–147. [Google Scholar] [CrossRef]
- Palasuberniam, P.; Yang, X.; Kraus, D.; Jones, P.; Myers, K.A.; Chen, B. ABCG2 transporter inhibitor restores the sensitivity of triple negative breast cancer cells to aminolevulinic acid-mediated photodynamic therapy. Sci. Rep. 2015, 5, 13298. [Google Scholar] [CrossRef]
- Fahey, J.M.; Girotti, A.W. Nitric oxide-mediated resistance to photodynamic therapy in a human breast tumor xenograft: improved outcome with NOS2 inhibitors. Nitric Oxide 2017, 62, 52–61. [Google Scholar] [CrossRef]
- Fahey, J.M.; Emmer, J.V.; Korytowski, W.; Hogg, N.; Girotti, A.W. Antagonistic effects of endogenous nitric oxide in a glioblastoma photodynamic therapy model. Photochem. Photobiol. 2016, 92, 842–853. [Google Scholar] [CrossRef]
- Bhowmick, R.; Girotti, A.W. Cytoprotective signaling associated with nitric oxide upregulation in tumor cells subjected to photodynamic therapy-like oxidative stress. Free Radic. Biol. Med. 2013, 57, 39–48. [Google Scholar] [CrossRef]
- Fahey, J.M.; Korytowski, W.; Girotti, A.W. Upstream signaling events leading to elevated production of pro-survival nitric oxide in photodynamically-challenged glioblastoma cells. Free Radic. Biol. Med. 2019, 137, 37–48. [Google Scholar] [CrossRef]
- Fahey, J.M.; Stancill, J.S.; Smith, B.C.; Girotti, A.W. Nitric oxide antagonism to glioblastoma photodynamic therapy and mitigation thereof by BET bromodomain inhibitor JQ1. J. Biol.Chem. 2018, 393, 5345–5359. [Google Scholar] [CrossRef]
- Godoy, L.C.; Anderson, C.T.M.; Chowdury, R.; Trudel, L.J.; Wogan, G.N. Endogenously produced nitric oxide mitigates sensitivity of melanoma cells to cisplatin. Proc. Natl. Acad. Sci. USA. 2012, 109, 21373–30378. [Google Scholar] [CrossRef]
- Perotta, C.; Cervia, D.; DiRenzo, L.; Moscheni, C.; Bassi, M.T.; Campana, L.; et al. Nitric oxide generated by tumor-associated macrophages is responsible for cancer resistance to cisplatin and correlated with syntaxin-4 and acid sphingomyelinase inhibition. Front. Immunol. 2018, 9, 1186. [Google Scholar] [CrossRef]
- Kim, R.-K.; Suh, Y.; Cui, Y.-H.; Hwang, E.; Lim, E.-J.; Yoo, K.-C.; et., al. Fractionated radiation-induced nitric oxide promotes expansion of glioma stem-like cells. Cancer Sci. 1177. [Google Scholar] [CrossRef]
- Tonini, V.; Zanni, M. Pancreatic cancer in 2021: what you need to know to win. World J. Gastroenterol. 2021, 27, 2851–2889. [Google Scholar] [CrossRef]
- Pereira, P.M.R.; Edwards, K.J.; Komel, M.; Carter, L.M.; Escorcia, F.E.; Compesato, L.F. et. al. et. al. iNOS regulates the therapeutic response of pancreatic cancer cells to radiotherapy. Cancer Res. 2020, 80, 1681–1692. [Google Scholar] [CrossRef]
- Shao, C.; Folkard, M.; Prise, K.M. Role of TGF-beta and nitric oxide in the bystander response of irradiated glioma cells. Oncogene 2008, 27, 434–440. [Google Scholar] [CrossRef]
- Kearney, C.J.; Vervoort, S.J.; Hogg, S.J.; Ramsbottom, K.M.; Freeman, A.J.; Lalaoui, N. Tumor immune evasion arises through loss of TNF sensitivity. Sci. Immunol. 2019, 3, eaar345. [Google Scholar] [CrossRef]
- Pelle, E.; Mammone, T.; Maes, D.; Frenkel, K. Keratinocytes act as a source of reactive oxygen species by transferring hydrogen peroxide to melanocytes. J. Investig. Dermatol. 2005, 124, 793–797. [Google Scholar] [CrossRef]
- Matsumoto, H.; Hayashi, S.; Hatashita, M.; Ohnishi, K.; Shioura, H.; Ohtsubo, T. Induction of radioresistance by nitric oxide-mediated bystander effect. Radiat. Res. 2001, 155, 387–396. [Google Scholar] [CrossRef]
- Shao, C.; Stewart, V.; Fokard, M.; Michael, B.D.; Prise, K.M. Nitric oxide -mediated signaling in the bystander response of individually targeted glioma cells. Cancer Res. 2003, 63, 8437–8442. [Google Scholar]
- Dahle, J.; Bagdonas, S.; Kaalhus, O.; Olsen, G.; Seen, H.B.; Moan, J. The bystander effect in photodynamic inactivation of cells. Biochim. Biophys. Acta 2000, 1475, 273–280. [Google Scholar] [CrossRef]
- Chakraborty, A.; Held, K.D.; Prise, K.M.; Liber, H.L.; Redmond, R.W. Bystander effects induced by diffusing mediators after photodynamic stress. Radiat. Res. 2009, 172, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Rubio, N.; Rajadurai, A.; Held, K.D.; Prise, K.M.; Liber, H.L.; Redford, R.W. Real-time imaging of novel spatial and temporal responses to photodynamic stress. Free Radic. Biol. Med. 2009, 47, 283–290. [Google Scholar] [CrossRef]
- Bazak, J.; Fahey, J.M.; Wawak, K.; Korytowski, W.; Girotti, A.W. Enhanced aggressiveness of bystander cells in an anti-tumor photodynamic therapy model: role of nitric oxide produced by targeted cells. Free Radic. Biol. Med. 2017, 102, 111–121. [Google Scholar] [CrossRef]
- Bazak, J.; Korytowski, W.; Girotti, A.W. Bystander effects of nitric oxide in cellular models of anti-tumor photodynamic therapy. Cancers 2019, 11, 1674. [Google Scholar] [CrossRef]
- Gani, M.; Xodo, L.E.; Rapozzi, V. Bystander effects in photosensitized prostate cancer cells with a different grade of malignancy: the role of nitric oxide. Nitric Oxide, 2022, 128, 25–36. [Google Scholar] [CrossRef]
- Kleinert, H.; Pautz, N.; Linker, K.; Schwarz, P.M. Regulation of the expression of inducible nitric oxide synthase. European J. Pharmacol. 2004, 500, 255–266. [Google Scholar] [CrossRef]
- Burke, S.J.; Updegraff, B.L.; Bellich, R.M.; Goff, M.R.; Lu, D.; Minkin, S.C. Regulation of iNOS gene transcription by IL-1B and IFN-γ requires a coactivator exchange mechanism. Mol Endocrinol. 2013, 27, 1724–1742. [Google Scholar] [CrossRef]
- Hansel, T.T.; Kharitonov, S.A.; Donnelly, L.E.; Erin, E.M.; Currie, M.G.; Moore, W.M. A selective inhibitor of inducible nitric oxide synthase inhibits exhaled breath nitric oxide in healthy volunteers and asthmatics. FASEB J. 2003, 17, 1298–1317. [Google Scholar] [CrossRef]
- Singh, D.; Richards, D.; Knowles, R.C.; Schwartz, S.; Woodcock, A.; Langley, S. Selective inducible nitric oxide synthase inhibition has no effect on allergen challenge in asthma. Am. J. Respir. Crit. Care Med. 2007, 176, 988–993. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Federov, O. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, P. Targeting bromodomains: epigenetic readers of lysing acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Girotti, A.W.; Fahey, J.M.; Korytowski, W. Role of nitric oxide in hyper-aggressiveness of tumor cells that survive various anti-cancer therapies. Crit. Rev. Oncol. Hematol. 2022, 179, 103805. [Google Scholar] [CrossRef]
Figure 1.
PDT-induced bystander effects and how they can be assessed. Panel A: Schematic showing a population of cancer cells, some of which (targeted) are exposed to a photodynamic challenge while others (bystanders) receive pro-division/migration signals from targeted counterparts. Panel B: View of an experimental setup for initially separating targeted and bystander cell populations on a large (13.5-cm. diam.) culture dish. Two silicone-rimmed rings segregate the two populations, targeted cells lying outside the rings and bystanders inside. At some point after a challenge, the rings are removed and effects of signaling molecules diffusing from outside to inside are examined.
Figure 1.
PDT-induced bystander effects and how they can be assessed. Panel A: Schematic showing a population of cancer cells, some of which (targeted) are exposed to a photodynamic challenge while others (bystanders) receive pro-division/migration signals from targeted counterparts. Panel B: View of an experimental setup for initially separating targeted and bystander cell populations on a large (13.5-cm. diam.) culture dish. Two silicone-rimmed rings segregate the two populations, targeted cells lying outside the rings and bystanders inside. At some point after a challenge, the rings are removed and effects of signaling molecules diffusing from outside to inside are examined.
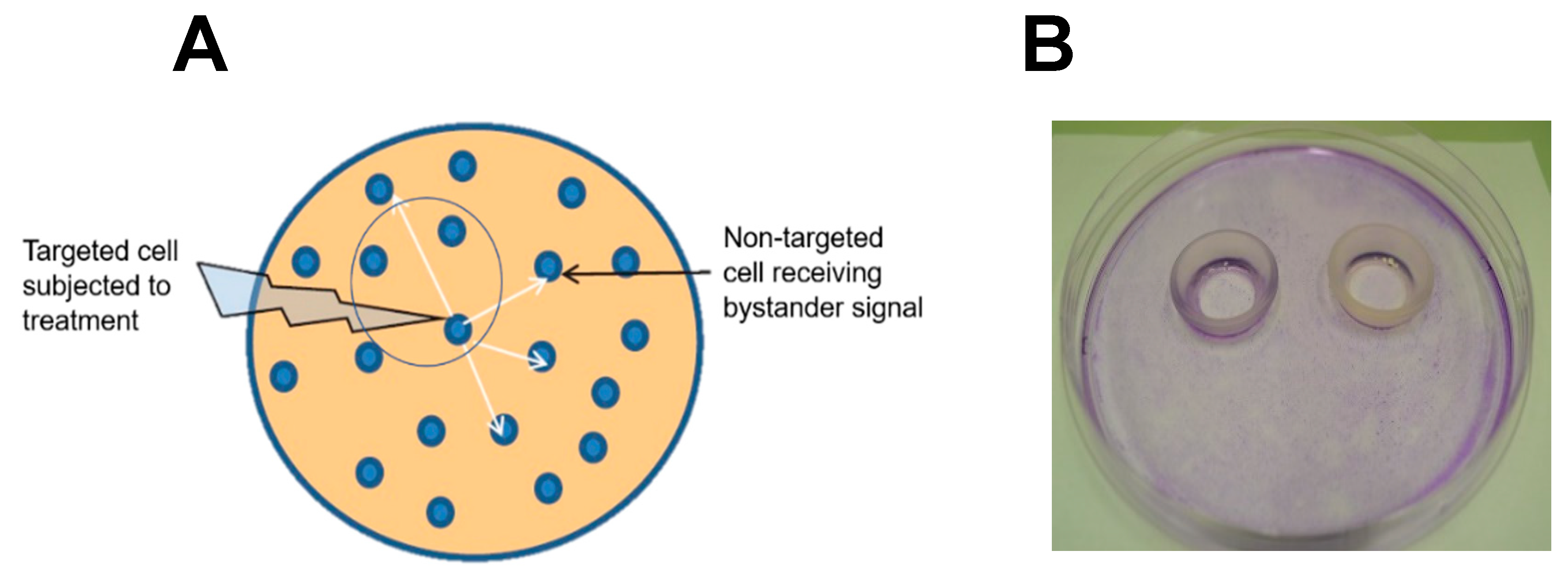
Figure 2.
Western blot-assessed iNOS upregulation in photodynamically-targeted PC-3 cells (A) and bystander counterparts (B). Target cells were dark-incubated with 1 mM ALA for 40 min in serum- and phenol red-free RPMI medium. After switching to ALA-free medium, cells were exposed to ~1 J/cm2 of LED light. Non-sensitized bystander cells within two silicone rings apart from targeted cells were irradiated simultaneously. At 2 h post-irradiation, rings were removed and medium switched to 10% serum in RPMI. After the indicated periods of additional dark incubation, targeted and bystander cells were recovered for iNOS and β-actin immunoblot analysis at 30 µg total cell protein per lane. Plotted values indicate iNOS band intensity relative to β-actin and normalized to time-0, means ± SEM (n=3). No difference was seen between the indicated time-0 values and a target cell dark control (ALA-only).
Figure 2.
Western blot-assessed iNOS upregulation in photodynamically-targeted PC-3 cells (A) and bystander counterparts (B). Target cells were dark-incubated with 1 mM ALA for 40 min in serum- and phenol red-free RPMI medium. After switching to ALA-free medium, cells were exposed to ~1 J/cm2 of LED light. Non-sensitized bystander cells within two silicone rings apart from targeted cells were irradiated simultaneously. At 2 h post-irradiation, rings were removed and medium switched to 10% serum in RPMI. After the indicated periods of additional dark incubation, targeted and bystander cells were recovered for iNOS and β-actin immunoblot analysis at 30 µg total cell protein per lane. Plotted values indicate iNOS band intensity relative to β-actin and normalized to time-0, means ± SEM (n=3). No difference was seen between the indicated time-0 values and a target cell dark control (ALA-only).
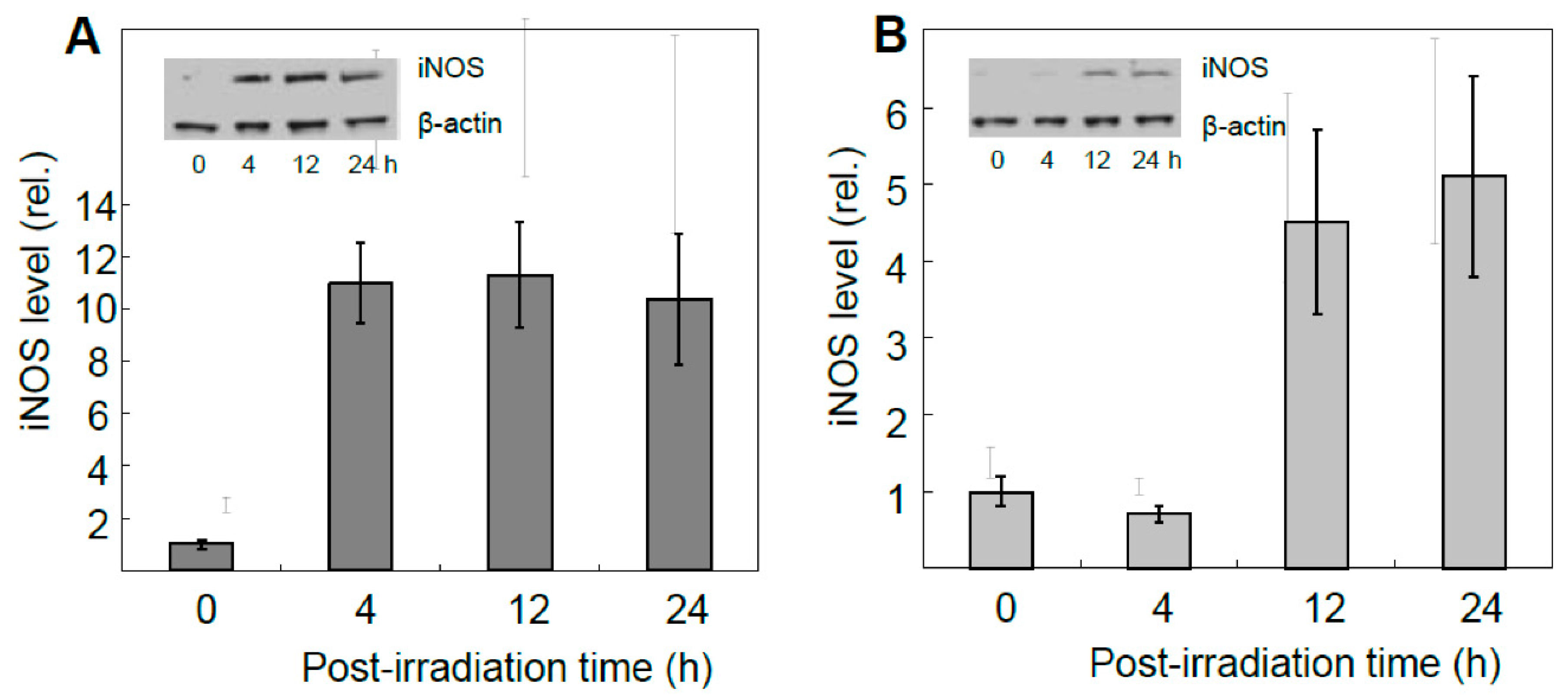
Figure 3.
Accelerated proliferation and migration of PC3 bystander cells after exposure to photodynamically-targeted counterparts. Target cells separated from ring-enveloped bystanders were incubated with 1mM ALA for 40 min, then switched to ALA-free medium and irradiated as described in Figure 2. A non-ALA-treated control was irradiated alongside. (A) Bystander proliferation during post-irradiation incubation as assessed by Image-J analysis; means ± SEM (n=6), *P<0.05 vs. control. (B) Bystander migration determined by gap-closure (“wound-healing”) assay; means ± SEM (n=5), *P<0.05 vs. control.
Figure 3.
Accelerated proliferation and migration of PC3 bystander cells after exposure to photodynamically-targeted counterparts. Target cells separated from ring-enveloped bystanders were incubated with 1mM ALA for 40 min, then switched to ALA-free medium and irradiated as described in Figure 2. A non-ALA-treated control was irradiated alongside. (A) Bystander proliferation during post-irradiation incubation as assessed by Image-J analysis; means ± SEM (n=6), *P<0.05 vs. control. (B) Bystander migration determined by gap-closure (“wound-healing”) assay; means ± SEM (n=5), *P<0.05 vs. control.
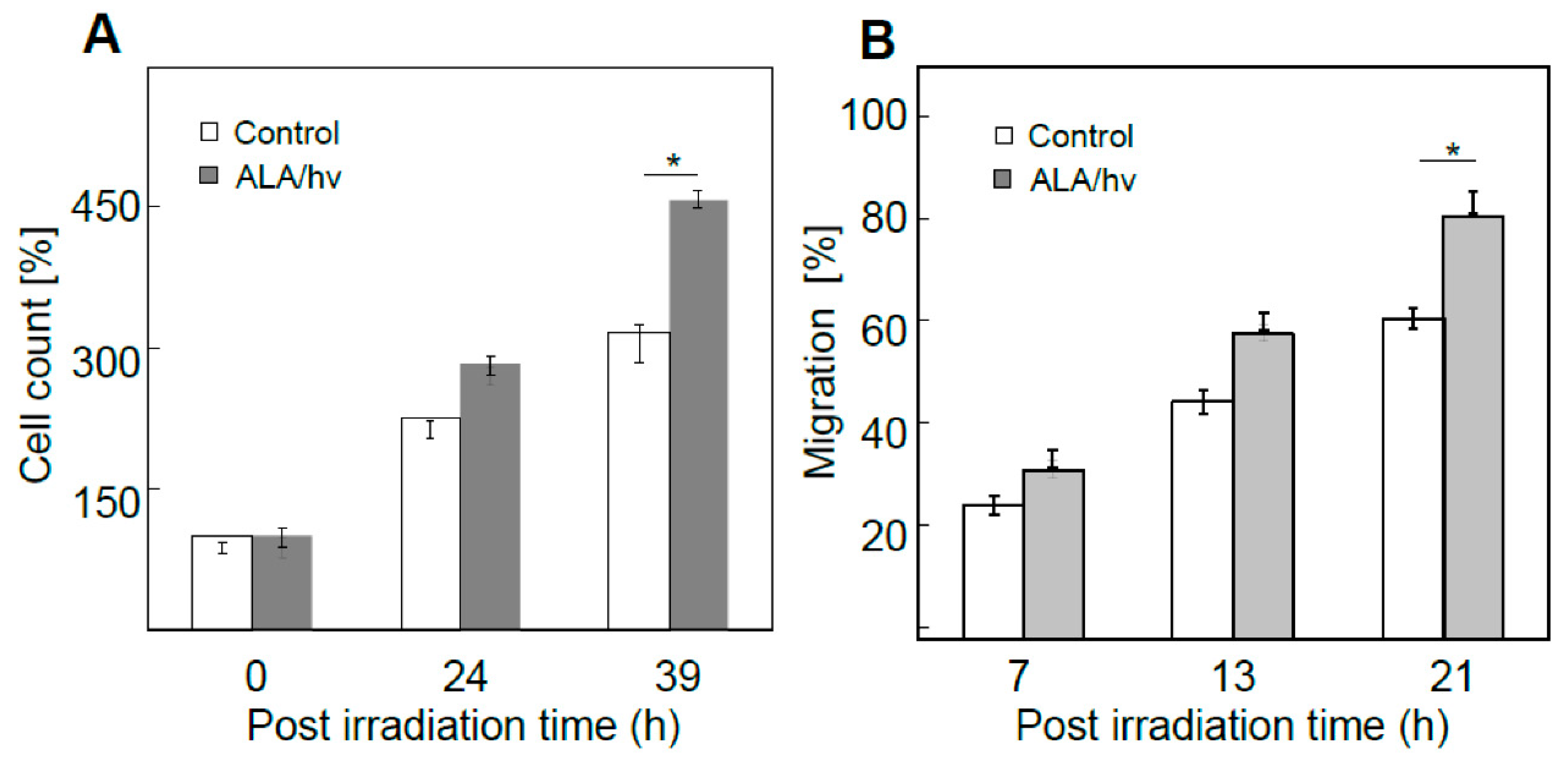
Figure 4.
Accelerated proliferation and migration of PC3 bystander cells compared with BLM, U87, and MDA-MD-231 bystander cells as a function of maximal post-irradiation iNOS upregulation in corresponding targeted cells of each type. (A) Plot of increased proliferative potency vs. maximal targeted cell iNOS induction for the different cell lines. (B) Plot of increased migration potency vs. maximal targeted cell iNOS induction for the different lines. All data points are means ± SEM (n=3-6). Adapted from Ref. 54.
Figure 4.
Accelerated proliferation and migration of PC3 bystander cells compared with BLM, U87, and MDA-MD-231 bystander cells as a function of maximal post-irradiation iNOS upregulation in corresponding targeted cells of each type. (A) Plot of increased proliferative potency vs. maximal targeted cell iNOS induction for the different cell lines. (B) Plot of increased migration potency vs. maximal targeted cell iNOS induction for the different lines. All data points are means ± SEM (n=3-6). Adapted from Ref. 54.
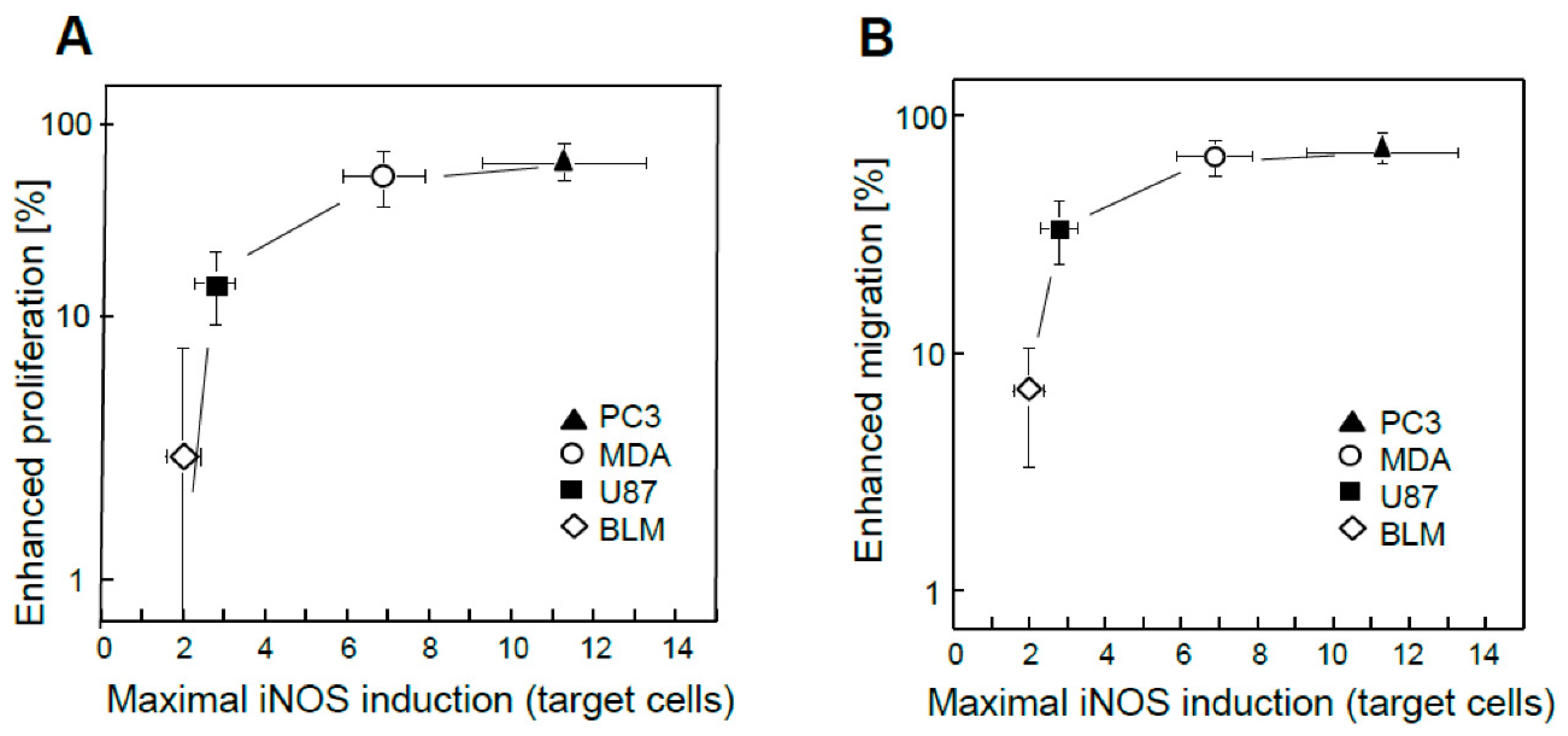
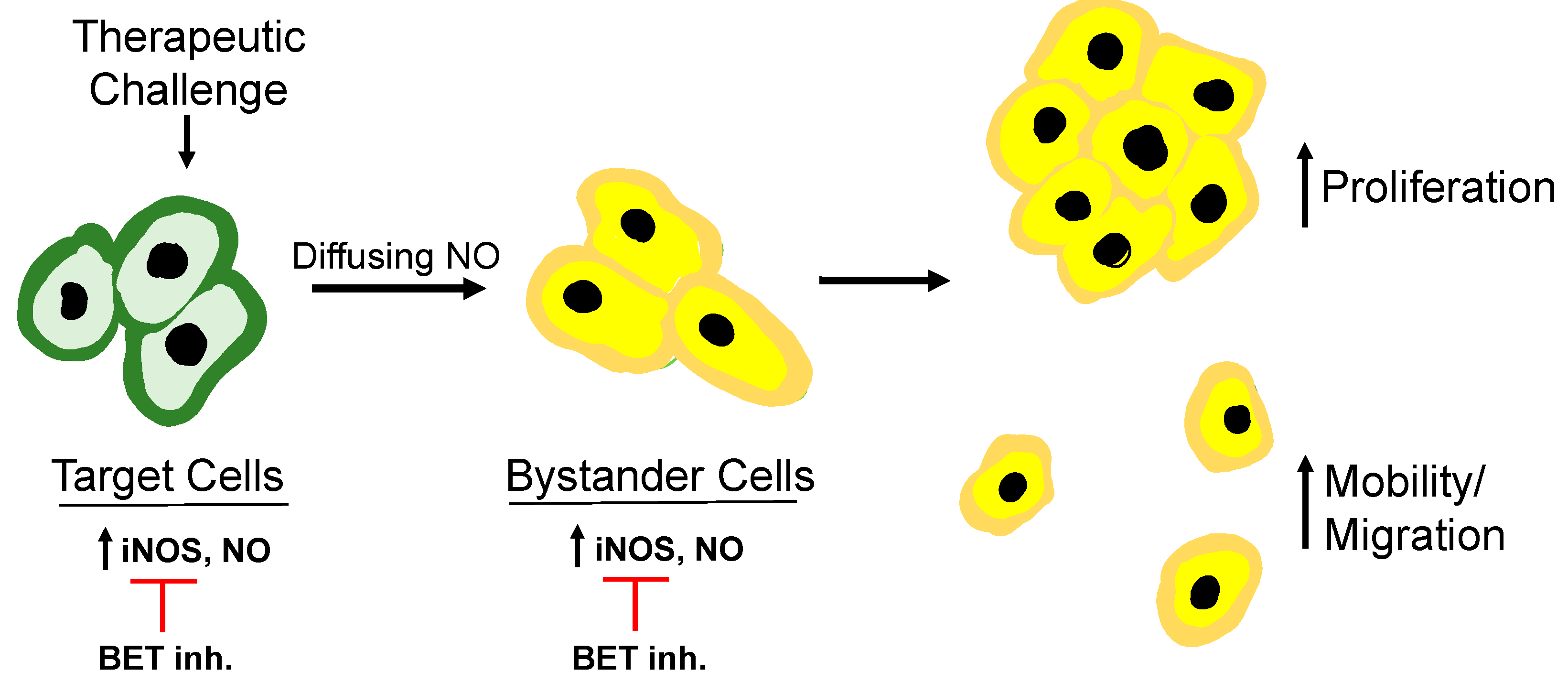
Figure 5.
Schematic depicting (a) upregulated iNOS in cancer cells directly targeted by a PDT oxidative challenge, (b) diffusion of the resulting NO to naïve bystander cells, (c) iNOS/NO upregulation in the latter cells, which increases proliferative and migratory aggressiveness, and (d) suppression by BET inhibitors .
Figure 5.
Schematic depicting (a) upregulated iNOS in cancer cells directly targeted by a PDT oxidative challenge, (b) diffusion of the resulting NO to naïve bystander cells, (c) iNOS/NO upregulation in the latter cells, which increases proliferative and migratory aggressiveness, and (d) suppression by BET inhibitors .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



